8 Systemic Inflammation
Numerous advances in perioperative care have allowed increasingly high-risk patients to safely undergo cardiac surgery. Although mortality rates of 1% are quoted for “low-risk” cardiac surgery, results from large series of patients older than 65 years suggest that mortality rates are actually more substantial.1 For example, Birkmeyer et al1 reviewed a large number (n = 474,108) of “all-comers” undergoing coronary artery bypass surgery (CABG) or aortic valve surgery in the Medicare Claims Database. Notably, 30-day all-cause mortality was in the range of 4.0% to 5.4% after CABG and 6.5% to 9.1% after aortic valve replacement. The patient population studied, although elderly (age > 65), would not be considered to be particularly high risk by today’s standards. These data do not point to the cause of death. Nevertheless, they indicate that outcome after routine cardiac surgery is poor for many patients. Outcome after these procedures is even worse if the extent of postoperative complications is considered. Postoperative morbidity is common,2 and complications include atrial fibrillation, poor ventricular function requiring inotropic agents, and noncardiac-related causes such as infection, gastrointestinal dysfunction, acute lung injury, stroke, and renal dysfunction. For example, in Rady et al’s3 large series of patients ≥ 75 years of age undergoing cardiac surgery (n = 1,157), the mortality rate was 8%. The rate of serious complications, however, exceeded 50%.
Many postoperative complications appear to be caused by an exaggerated systemic proinflammatory response to surgical trauma.4–6 The most severe form of this inflammatory response leads to multiple organ dysfunction syndrome and death.5,6 Milder forms of a proinflammatory response cause less severe organ dysfunction, which does not lead to admission to an intensive care unit (ICU), but nevertheless causes suffering, increased hospital length of stay, and increased cost. The cause and clinical relevance of systemic inflammation after cardiac surgery are poorly understood. Systemic inflammation is a multifactorial process and has profound secondary effects on both injured and normal tissues. Proinflammatory mediators can have beneficial as well as deleterious effects on multiple organ systems. According to most theories, tissue injury, endotoxemia, and contact of blood with the foreign surface of the cardiopulmonary bypass (CPB) circuit are some of the major factors postulated to initiate a systemic inflammatory response. Nevertheless, controversy surrounds the cause and pathogenesis of inflammation in the perioperative period.
Terminology
The terminology of inflammation is confusing and has hampered effective communication among scientists and clinicians. Despite attempts to standardize the terminology, variation in usage still exists in the scientific literature, as well as the clinical setting.7 Much of the confusion relates to the term inflammation, defined as “a fundamental pathologic process consisting of a dynamic complex of cytologic and chemical reactions that occur in the affected blood vessels and adjacent tissues in response to an injury or abnormal stimulation caused by a physical, chemical, or biologic agent, including (1) the local reactions and resulting morphologic changes, (2) the destruction or removal of the injurious material, (3) the responses that lead to repair and healing.”8 This definition acknowledges the potential role of noninfectious causative factors; that is, infection is not a prerequisite for the development of inflammation. The American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference has developed definitions for terms related to inflammation (Table 8-1). Figure 8-1 demonstrates the possible interrelations among many of these terms.
| Infection = microbial phenomenon characterized by an inflammatory response to the presence of microorganisms or the invasion of normally sterile host tissue by those organisms. |
| Bacteremia = the presence of viable bacteria in the blood. |
| Systemic inflammatory response syndrome (SIRS) = the systemic inflammatory response to a variety of severe clinical insults. The response is manifested by two or more of the following conditions: (1) temperature > 38°C or < 36°C; (2) heart rate > 90 beats per minute; (3) respiratory rate > 20 breaths per minute or PaCO2 < 32 mm Hg; and (4) white blood cell count > 12,000/mm3, < 4000/mm3, or > 10% immature (band) forms. |
| Sepsis = the systemic response to infection, manifested by two or more of the following conditions as a result of infection: (1) temperature > 38°C or < 36°C; (2) heart rate > 90 beats per minute; (3) respiratory rate > 20 breaths per minute or PaCO2 < 32 mm Hg; and white blood cell count > 12,000/mm3, < 4000/mm3, or > 10% immune (band) forms. |
| Severe sepsis = sepsis associated with organ dysfunction, hypoperfusion, or hypotension. Hypoperfusion and perfusion abnormalities may include, but are not limited to, lactic acidosis, oliguria, or an acute alteration in mental status. |
| Septic shock = sepsis-induced with hypotension despite adequate fluid resuscitation together with the presence of perfusion abnormalities that may include, but are not limited to, lactic acidosis, oliguria, or an acute alteration in mental status. Patients who are receiving inotropic or vasopressor agents may not be hypotensive at the time that perfusion abnormalities are measured. |
| Sepsis-induced hypotension = a systolic blood pressure < 90 mm Hg or a reduction of ≥ 40 mm Hg from baseline in the absence of other causes for hypotension. |
| Multiple organ dysfunction syndrome (MODS) = presence of altered organ function in an acutely ill patient such that homeostasis cannot be maintained without intervention. |
From Bone RC, Balk RA, Cerra FB, et al: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis: ACCP/SCCM consensus conference. Chest 101:1644–1655, 1992.
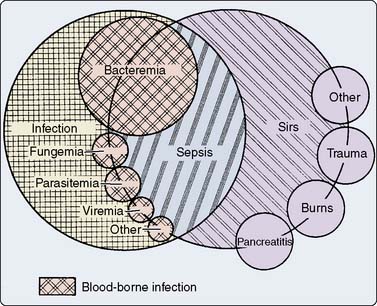
Figure 8-1 The interrelations among systemic inflammatory response syndrome (SIRS), sepsis, and infection.
(From Bone RC, Balk RA, Cerra FB, et al: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis: ACCP/SCCM Consensus Conference. Chest 101:1644–1655, 1992.)
In the surgical population, the use of the phrase and definition for SIRS has generated some controversy.9 This controversy relates to the fact that almost all patients after major surgery fulfill the criteria for SIRS. Most patients, however, clearly do not develop clinically significant organ dysfunction from their systemic inflammation. Critics may argue that the use of the term SIRS in cardiac surgery patients, therefore, is meaningless because it does not differentiate patients who will have a benign versus a complicated postoperative course. For these reasons, SIRS is used more commonly by investigators than by practicing physicians. Use of the phrase SIRS, nevertheless, has the benefit of increasing awareness regarding the many noninfectious causes of inflammation.
The acute-phase response to tissue injury and infection is characterized by leukocytosis, fever, increased vascular permeability, a negative nitrogen balance, changes in plasma steroid and metal concentrations, and increased synthesis of hepatic acute-phase proteins. Examples of these proteins include haptoglobin, fibrinogen, C-reactive protein (CRP), complement factors (C3, factor B), serum amyloid A, α1-acid glycoprotein, and α1-antichymotrypsin.10 The terms acute-phase response and systemic inflammation often are used interchangeably.
A common misconception relates to the terms bacteremia and endotoxemia. Whereas bacteremia refers to the presence of viable bacteria in the blood, endotoxemia refers to the presence of endotoxin in the blood. Endotoxin, also known as lipopolysaccharide (LPS), is a component of the cell membranes of gram-negative bacteria, and hence its presence does not require the existence of viable organisms. In fact, it has been clearly established that cardiac surgical patients have a high incidence of intraoperative endotoxemia despite simultaneously exhibiting a low incidence of culture-proven bacteremia. This observation is consistent with the observation that “sterile” instruments and solutions, including intravenous fluids and the CPB circuit, may be contaminated with endotoxin.11
Systemic Inflammation and Cardiac Surgery
The systemic inflammatory response after cardiac surgery is multifactorial. As described earlier, the term SIRS is not particularly helpful in clarifying the pathophysiology of inflammation in cardiac surgery.9 A schematic of the inflammatory process is depicted in Figure 8-2. There does not appear to be much disagreement with the statement that all of these processes may happen and may be responsible for causing complications in cardiac surgical patients. Tissue injury, endotoxemia, and contact of blood with the foreign surface of the CPB circuit are thought to initiate a systemic inflammatory response after cardiac surgery. What is least understood and of most controversy is the issue of which of these many processes is the most clinically relevant. It appears as if major surgery is an important cause of systemic inflammation, and that CPB further exacerbates the elaboration of proinflammatory mediators. Various causes and mediators of inflammation are reviewed in the subsequent sections.
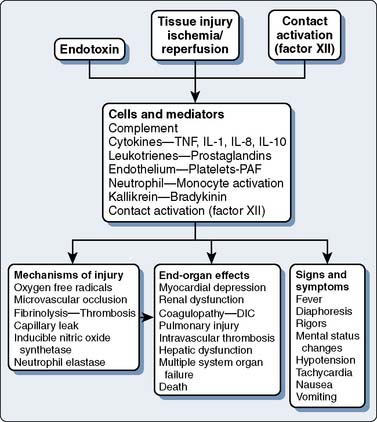

Mechanisms of Inflammation-Mediated Injury
It is not entirely clear how inflammation ultimately damages cells and organ systems. Activation of neutrophils and other leukocytes is central to most theories regarding inflammation-induced injury.6,12–15 Neutrophil activation leads to the release of oxygen radicals, intracellular proteases, and fatty acid (i.e., arachidonic acid) metabolites. These products, as well as those from activated macrophages and platelets, can cause or exacerbate tissue injury.
In localized areas of infection, oxygen free radicals liberated by activated neutrophils aid in the destruction of pathogens.16 Complement, in particular, C5a, results in activation of leukocytes and oxygen free radical formation.17 These activated neutrophils liberate toxic amounts of oxygen free radicals such as hydrogen peroxide, hydroxyl radicals, and superoxide anion. Oxygen free radicals are thought to cause cellular injury, ultimately through damage to the lipid membrane.18–20 Increased levels of lipid peroxidation products, that is, products of oxidation of membrane lipids such as malondialdehyde, are thought to reflect the severity of free radical cellular damage.21 Consistent with this model of injury, Royston et al22 demonstrated increased levels of peroxidation products in cardiac surgical patients. In another study, oxygen free radicals were found to be increased in 21 patients undergoing cardiac surgery; however, the clinical relevance of these changes was not studied.21
A related mechanism of injury results from the degranulation of neutrophils. Activated neutrophils release granules that contain myeloperoxidase, as well as other toxic digestive enzymes such as neutrophil elastase, lactoferrin, β-glucuronidase, and N-acetyl-β-glucosaminidase.23–26 Release of these intracellular enzymes not only causes tissue damage but also reduces the number of cells that can participate in bacterial destruction. In one study, cardiac surgical patients who developed splanchnic hypoperfusion, a possible cause of inflammation, demonstrated increased neutrophil degranulation and increased plasma neutrophil elastase concentrations.26
Another mechanism of inflammation-mediated injury involves microvascular occlusion. Activation of neutrophils leads to adhesion of leukocytes to endothelium and formation of clumps of inflammatory cells, that is, microaggregates.14,27 Activated leukocytes have less deformable cell membranes, which affects their ability to pass through capillaries.28 Microaggregates can cause organ dysfunction through microvascular occlusion and reductions in blood flow and oxygen at the local level.22,28,29 After the disappearance of these microaggregates and restoration of microvascular flow, reperfusion injury may occur.

Physiologic Mediators of Inflammation
Cytokines
Cytokines are believed to play a pivotal role in the pathophysiology of acute inflammation associated with cardiac surgery.30,31 Cytokines are proteins released from activated macrophages, monocytes, fibroblasts, and endothelial cells, which have far-reaching regulatory effects on cells.32 They are small proteins that exert their effects by binding to specific cell-surface receptors. Many of these proteins are called interleukins because they aid in the communication between white blood cells (leukocytes).
Tumor Necrosis Factor
Endotoxemia unequivocally results in initiation of proinflammatory pathways, most likely through stimulation of TNF.33–36 Michie et al33 administered endotoxin intravenously to human volunteers and detected peak levels of TNF 90 to 180 minutes later. Peak concentrations of TNF correlated with increased temperature and heart rate (HR), as well as circulating levels of adrenocorticotropic hormone and epinephrine. In this and other studies, TNF levels soon appear after a proinflammatory stimulus and disappear quickly, which helps explain a common finding from clinical studies. TNF levels are often not increased when measured in patients with systemic inflammation, probably because test samples are obtained long after exposure to the primary inflammatory stimulus. This issue of sampling time may partially account for the fact that some cardiac surgical studies have detected increased TNF levels, whereas others have not.37–53
Interleukins
After the appearance of TNF, levels of IL-1 increase in cardiac surgical patients.47,50,52,54 Measured levels are low and may peak within several hours after CPB.54 Others have demonstrated maximum levels 1 day after cardiac surgery, which may explain the inability of some investigators to detect IL-1 during the intraoperative period.50 IL-1 may decrease systemic vascular resistance after CPB through induction of nitric oxide synthesis in vascular endothelial cells.55 Although IL-1 appears to be important in the initiation and propagation of the inflammatory cascade, it is not clear whether IL-1 levels cause deleterious effects or even serve as a marker for patients who will develop organ dysfunction after cardiac surgery. Some of the reported effects of IL-1 may be due instead to other cytokines, in particular, TNF, which are detected at the same time.
IL-8 is also believed to be an important component of the proinflammatory cascade. It is a potent chemoattractant of neutrophils to the site of injury or infection. IL-8 also is responsible for the activation, priming, and degranulation of neutrophils.56,57 The relevance of increases in IL-8 levels to outcome after cardiac surgery has not been established.42–44,46,47,51,53,58,59 Rothenburger et al60,61 observed a significant association between prolonged mechanical ventilation and postoperative IL-8 levels but not IL-6 levels.
IL-6 levels have been shown to increase in the setting of cardiac surgery, although this is not a universal finding.10,42,43,45–48,51–54,58,62–65 Peak levels of this cytokine appear after maximum values for TNF and IL-1. For example, Steinberg et al54 measured plasma cytokine levels in 29 patients undergoing CPB. IL-6 levels peaked at 3 hours after separation from CPB and remained increased 24 hours after surgery. No association was found between IL-6 levels and hemodynamic parameters or postoperative pulmonary function.
Anti-inflammatory Cytokines
The regulation of inflammation is complex and involves a balance between proinflammatory and anti-inflammatory cytokines. IL-10 is a potent inhibitor of the synthesis of TNF, IL-1, IL-6, and IL-8, and increases in the perioperative period.44,66–68 McBride et al68 obtained blood samples perioperatively from 20 patients undergoing cardiac surgery. Before and during CPB, increases were observed in the proinflammatory cytokines TNF, IL-1, and IL-8. At the same time that proinflammatory cytokine levels began to decrease, increases in the anti-inflammatory cytokines IL-10 and IL-1ra were observed. The authors suggest that the balancing effects of these two types of cytokines may determine whether a patient suffers from the effects of excessive systemic inflammation (i.e., postoperative organ dysfunction) or the effects of inadequate immune system enhancement (i.e., postoperative infection and poor wound healing). Using this theory to improve outcome has not been translated yet into a clinical trial involving surgical patients. One concern related to potentially deleterious effects of inhibiting proinflammatory mediators has been borne out in sepsis trials in which mortality was increased in the group given an anti-inflammatory agent.69 An understanding of the interaction between proinflammatory and anti-inflammatory mediators may result in the development of an effective and safe approach to reducing complications related to excessive systemic inflammation.
Complement System
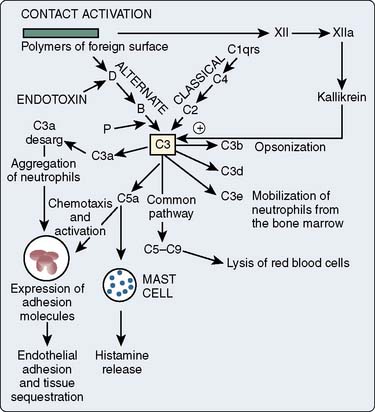
The complement cascade is illustrated in Figure 8-3. The complement cascade can be triggered by either the classic pathway or the alternate pathway. In the alternate pathway, C3 is activated by contact of complement factors B and D with complex polysaccharides, endotoxin, or exposure of blood to foreign substances such as the CPB circuit. Contact activation (Figure 8-4) describes contact of blood with a foreign surface with resulting adherence of platelets and activation of factor XII (Hageman factor). Activated factor XII has numerous effects, including initiation of the coagulation cascade through factor XI and conversion of prekallikrein to kallikrein. Kallikrein leads to generation of plasmin, which is known to activate the complement and the fibrinolytic systems. Kallikrein generation also activates the kinin-bradykinin system.
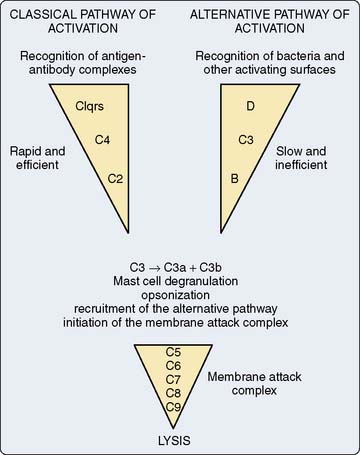
Figure 8-3 Simplified components of the complement system.
(From Haynes BF, Fauci AS: Introduction to clinical immunology. In Braunwald E, Isselbacher KJ, Petersdorf RG, et al [eds]: Harrison’s principles of internal medicine, 11th ed. New York: McGraw-Hill, 1987, pp 328–337.)
The classic pathway involves the activation of C1 by antibody-antigen complexes. In the case of cardiac surgery, there are two likely mechanisms for the activation of the classic pathway. Endotoxin can be detected in the serum of almost all patients undergoing cardiac surgery. Endotoxin forms an antigen-antibody complex with antiendotoxin antibodies normally found in serum, which can then activate C1. The administration of protamine after separation from CPB has been reported to result in heparin/protamine complexes, which also can activate the classic pathway70,71 (see Chapters 28 through 31). Others, however, have not observed this effect.72 Contact activation leads to activation of factor XII, which results in the generation of plasmin. Plasmin is capable of activating complement factors C1 and C3. Table 8-2 is a summary of the physiologic effects of the complement system.
TABLE 8-2 Biologically Significant Effects of the Various Complement-Split Products
| Biologic Effect | Complement-Split Products |
|---|---|
| Mast cell degranulation, contraction of smooth muscle, increased vascular permeability | C3a, C5a |
| Chemotaxis of neutrophils | C5a, C5a des Arg |
| Neutrophil aggregation | C5a, C5a des Arg |
| Lysosomal enzyme release | C5a, C3b |
| Leukocytosis | C3e |
| Immune adherence/opsonization | C3b, C4b |
| Membrane lysis | C5b-9 (membrane attack complex) |
From Knudsen F, Anderson LW: Immunological aspects of cardiopulmonary bypass. J Cardiothorac Anesth 4:245, 1990.
Although some elements of complement activation have been elucidated, clinicians are only now learning about the clinical relevance of this process to patients undergoing cardiac surgery. Several studies have reported increased complement levels during cardiac surgery.38,58,73–78 Chenoweth et al73 measured plasma C3a and C5a levels at different time points in 15 adults undergoing cardiac surgery with CPB. Although C3a levels were not affected by surgical stimulation, complement activation increased significantly during CPB (Figure 8-5). This and other studies did not test the association between increased complement levels and adverse postoperative outcome. Thus, they do not provide any evidence that complement activation causes clinically significant systemic inflammation. Kirklin et al.75 measured plasma C3a levels in 116 patients undergoing cardiac surgery with CPB and 12 patients undergoing operations without CPB. In this study, an increase of complement activation during CPB was associated with postoperative morbidity. Patients undergoing procedures without CPB did not demonstrate increases in complement. This result suggests that a factor unique to CPB causes activation of complement. This study, however, did not pinpoint the clinical relevance of complement activation or of the CPB circuit, in part because even patients without postoperative morbidity had increased complement levels. Furthermore, confounding factors capable of causing SIRS, such as endotoxin, were not measured and accounted for in this study.
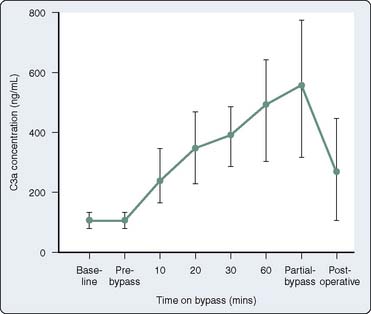
Figure 8-5 Plasma levels of C3a in patients undergoing cardiopulmonary bypass.
(From Chenowith DE, Cooper SW, Hugli TE, et al: Complement activation during cardiopulmonary bypass: Evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med 304:497, 1981. Copyright 1981 Massachusetts Medical Society. All rights reserved.)
The results from several large, randomized clinical trials in which complement activation was selectively blocked have become available.79–81 These studies indicate that attenuation of complement activation results in less myocardial injury; however, there did not appear to be an impact on complications such as pulmonary and renal dysfunction and severe vasodilation. These results suggest that complement activation may not play as large a role in the development of systemic inflammation-mediated morbidity as previously thought. These trials are discussed in more detail later in this chapter.
Endotoxin
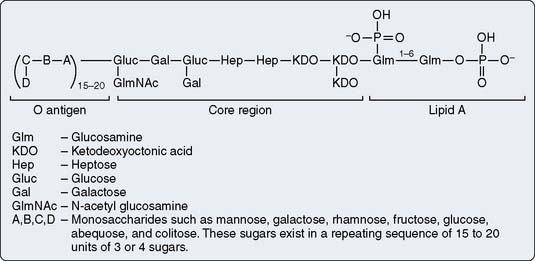
Endotoxin, also called LPS, is a component of the cell membrane of gram-negative bacteria. It is a potent activator of complement and cytokines, and appears to be one of the initial triggers of systemic inflammation, as summarized in Figure 8-2.12,82–84 Although the LPS constituent varies from one bacterial species to another, it generally may be described with reference to Figure 8-6 as consisting of three structural regions: (a) lipid A, (b) core, and (c) O-polysaccharide outer region. The lipid region of lipid A is embedded in the outer leaflet of the outer membrane. The oligosaccharide core region is positioned between lipid A and the O-polysaccharide outer region. Lipid A has the same basic structure in practically all gram-negative bacteria and is the toxic component of endotoxin. The LPS core region shows a high degree of similarity among various bacteria. It usually consists of a limited number of sugars. For example, the inner core region is constituted of heptose and 3-deoxy-d-manno-2-octulosonate (KDO) residues, whereas the outer core region comprises galactose, glucose, or N-acetyl-d-glucosamine residues displayed in various manners depending on the strain. The O-polysaccharide outer region (also called O-specific antigen or O-specific side chain) is highly variable and is composed of one or more oligosaccharide repeating units characteristic of the serotype.
Endotoxemia
Endotoxemia refers to the presence of endotoxin in the blood. Endotoxemia is common in cardiac surgical patients.10,11,38,41,60,64,65,85–95 It is not surprising that some investigators have failed to detect endotoxemia during cardiac surgery given its transient and intermittent nature, although differences in endotoxin-assaying techniques used also may contribute to this discrepancy.51,52,96,97 Andersen et al11 measured circulating endotoxin levels in 10 patients undergoing cardiac surgery. All preoperative blood samples were free of endotoxin; however, substantial levels of endotoxin were detected intraoperatively. Blood endotoxin levels from eight typical patients undergoing cardiac surgery are presented in Figure 8-7.85 Although endotoxin can be found in sterile fluids administered to patients, it is believed that the majority of endotoxin arises through a patient’s impaired gut barrier.11 Rothenburger et al60 studied the association of endotoxin levels with prolonged mechanical ventilation in 78 cardiac surgical patients. Endotoxin levels were three times greater in patients with a postoperative mechanical ventilation time longer than 24 hours (n = 13) compared with patients with ventilator time less than 24 hours.
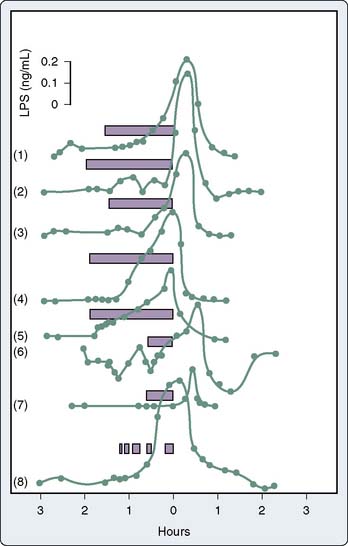
Normally, intestinal flora contain a large amount of endotoxin from gram-negative microorganisms.98 The average human colon contains approximately 25 billion nanograms of endotoxin, which is an enormous quantity when 300 ng endotoxin is toxic to humans.33,34 The leakage of live bacterial cells into the bloodstream can result in infection as these viable bacteria multiply.99 However, many of the bacteria in the intestine are dead, and thus endotoxin also can enter the bloodstream contained within cell membrane fragments of dead bacteria. In this case, infection per se does not develop. Instead, endotoxin may initiate a systemic inflammatory response through potent activation of macrophages and other proinflammatory cells.100 A plasma endotoxin concentration of only 1 ng/mL has been reported to be lethal in humans.101
On entry to the bloodstream, endotoxin forms complexes with numerous intravascular compounds including high-density lipoprotein, LPS-binding protein, and endotoxin-specific immunoglobulins. Endotoxin has been linked to dysfunction in every organ system of the body and may be the key initiating factor in the development of systemic inflammation.12,82–84,93

Normal Host Defenses against Endotoxemia
Early Tolerance
If endotoxemia is deleterious to patients, it would be logical to assume that patients have defense mechanisms against this ubiquitous toxin. Tolerance to endotoxin was studied extensively by Greisman and Hornick in the early 1970s.102 Two distinct types of tolerance to endotoxin exist and are classified as early tolerance and late tolerance.102 Early tolerance to endotoxin represents a reduction in the proinflammatory effects of LPS when administered several hours after a prior infusion of LPS.103 It appears to be due to an LPS-induced refractory state of macrophages in which they release less TNF in response to endotoxin. This early refractive state shows no LPS specificity and can be overcome with increased doses of endotoxin. The degree of this tolerance is directly proportional to the dose, and hence intensity of the initial LPS-induced inflammatory state. Early tolerance begins within hours of LPS exposure and decreases almost to baseline within 2 days. It cannot be transferred with plasma. Early tolerance may protect the host from lethal systemic inflammation after an overwhelming exposure of LPS.
Late Tolerance
Late tolerance to endotoxin is due to the synthesis of immunoglobulins, that is, antibodies, directed against the offending LPS.102 Late tolerance begins approximately 72 hours after exposure to LPS, which correlates with the appearance of the early-appearing IgM class of antibodies. This form of tolerance persists for at least 2 weeks and correlates with the presence of serum immunoglobulins. In contrast with early tolerance, the late response is not proportional to the intensity of the initial LPS-induced inflammatory response but is related to the immunogenicity of the initial LPS. Furthermore, late tolerance does not generally protect against a subsequent challenge with a dissimilar type of LPS. In other words, late tolerance is most pronounced when the same, that is, homologous, LPS serotype is used for both the initial and the subsequent challenge. It is not definitively understood how antiendotoxin antibodies responsible for late tolerance confer protection from LPS-induced systemic inflammation. Proposed mechanisms include increased clearance of endotoxin into the reticuloendothelial system, as well as direct neutralization through binding.
Understanding of the host’s normal humoral defense against endotoxin is further complicated because of the numerous serotypes of endotoxin.98 Serotype-specific antibodies, that is, antibodies synthesized in response to a particular LPS, exhibit high-affinity binding to and protection from the specific serotype of endotoxin. These serotype, that is, O-specific, antiendotoxin antibodies, however, do not recognize the many possible variations of endotoxin O-polysaccharide side chains, and thus are ineffective at conferring protection against the numerous serotypes of endotoxin likely to be encountered in the clinical setting. Antibodies, however, directed against the well-conserved inner core structure of endotoxin should theoretically be cross-reactive against many clinically relevant serotypes of endotoxin. Cardiac surgical patients are exposed to a wide variety of endotoxin types. For example, at least 164 O-antigens exist for Escherichia coli, the gram-negative bacteria most commonly isolated in high-risk surgical and ICU patients.104,105

Criticisms of Endotoxin as a Causative Factor
There are several criticisms of the theory that endotoxemia is an important cause of postoperative morbidity. A common criticism relates to the low incidence of culture-proven bacteremia in surgical and ICU patients.106–109 Endotoxemia, however, is clearly prevalent in these patients and usually exists in the setting of negative blood cultures.10,11,24,38,41,64,65,85–95 In fact, studies attempting to detect endotoxemia probably underestimate its incidence given its intermittent nature.
The failure of two anti-lipid A monoclonal antibodies (HA-1A, Centocor, Malvern, PA; and E5, Xoma, Berkeley, CA) to improve outcome on an “intention-to-treat” basis in ICU patients with established sepsis also has been used to suggest that endotoxemia is not clinically relevant.110,111 These monoclonal antibodies may not bind to endotoxin with high affinity, which may explain, in part, their lack of demonstrable efficacy.112 In addition, they were tested in patients with established sepsis and organ failure, which is an entirely different setting from elective surgical patients, who are more likely to benefit from prophylaxis with endotoxin-neutralizing drugs. Arguing against the clinical relevance of endotoxin is the negative result from a trial of prophylactic administration of a lipid A antagonist (E5564) in cardiac surgery.113
Splanchnic Perfusion
Splanchnic hypoperfusion appears to be an important cause of systemic inflammation.114–117 The gut is one of the most susceptible organs to hypoperfusion during conditions of trauma or stress.117–119 In the 1960s, Price et al118 removed 15% of the blood volume from healthy volunteers, causing a 40% reduction in splanchnic blood volume. In this study, cardiac output (CO), blood pressure (BP), and HR did not change from baseline. A study was conducted by Hamilton-Davies et al, in which 25% of the blood volume was removed from six healthy volunteers.120 Gastric mucosal perfusion, as measured by saline tonometry, was the first variable to decline (in five of the six subjects). Stroke volume (SV) also decreased; however, routinely measured cardiovascular variables such as HR, BP, and CO did not change significantly enough from baseline values to cause suspicion of a hypovolemic state. Based on these types of studies, the Advanced Trauma Life Support (ATLS) course teaches that a 15% blood loss (Class I hemorrhage) rarely results in changes in HR, BP, or urine output.121 Significant decreases in systolic BP are a late sign of shock, which typically occurs after Class III hemorrhage (30–40% blood loss).
These studies suggest that during periods of hypovolemia, the gut vasoconstricts, thus shunting blood toward “more vital organs” such as the heart and brain.117–119 In addition to hypovolemia, endogenously released vasoconstrictors during CPB, such as angiotensin II, thromboxane A2, and vasopressin, also may result in decreased splanchnic perfusion.122–125 Vasoconstrictors, such as phenylephrine, are routinely administered by anesthesiologists and perfusionists to increase BP and are likely to further reduce gut perfusion. Oudemans-van Straaten et al89 measured intestinal permeability and endotoxin levels in 23 patients during cardiac surgery. Intestinal leak was measured by the amount of orally administered cellobiose present in the patients’ urine. Intestinal permeability increased during surgery and correlated with circulating endotoxin levels. Administration of ephedrine, low central venous pressures, and less fluid balance during surgery also were associated with intestinal permeability, confirming the theory that gut perfusion is reduced by vasoconstrictors, as well as hypovolemia. There is also evidence that systemic endotoxemia may worsen intestinal permeability, thus exacerbating splanchnic hypoperfusion and initiating a vicious circle.126
Several studies have observed a high incidence of splanchnic hypoperfusion during cardiac surgery, with some showing an association between abnormal gut perfusion during cardiac surgery and postoperative complications.10,87,88,127–130 Fiddian-Green and Baker127 used saline tonometry to measure gastric mucosal perfusion in 85 cardiac surgical patients. Half (49%) of these patients developed evidence of abnormal perfusion, and all serious postoperative complications (eight patients, including five deaths) developed in this group. Gastric tonometry was shown in this and in two other studies to be a more sensitive predictor of adverse postoperative outcome compared with more routinely used global measures such as CO, BP, HR, and urine output.128,130 A study using air tonometry demonstrated an increased gastric mucosal Pco2 in 52% of cardiac surgical patients. Thirty-five percent of these patients with abnormal perfusion developed postoperative complications, in contrast with 5% in the group without evidence of hypoperfusion.130
Studies that have failed to demonstrate an association between splanchnic hypoperfusion and adverse postoperative outcome are limited, in part, by small sample size, insensitive measures of postoperative morbidity, and deviation from validated methodology of tonometry.87,88 Tonometric measurements of gastric mucosal perfusion during hypothermic CPB have not been validated in terms of their ability to predict postoperative morbidity.
Postoperative Complications Attributable To Inflammation

Types of Complications
Many postoperative complications appear to be caused by an exaggerated systemic proinflammatory response to surgical trauma. A common misunderstanding relates to the types of postoperative complications that may be attributable to systemic inflammation and, in particular, splanchnic hypoperfusion. Many of the complications that are thought to be linked to splanchnic hypoperfusion do not involve the gastrointestinal system. Because splanchnic hypoperfusion may cause injury through a systemic inflammatory response, it would be expected that every organ system of the body potentially would be involved.131 For example, endotoxin has been reported to have adverse effects on the pulmonary, renal, cardiac, and vascular systems.41,46,83,84,132–135 It affects the coagulation system and may be both antihemostatic, potentially explaining bleeding, and prothrombotic.83,132,136 Prothrombotic effects may account for some cases of postoperative stroke, deep venous thrombosis, and pulmonary emboli. There is also circumstantial evidence that systemic inflammation may worsen neurologic injury.137 Activation of inflammatory cascades has been shown to worsen neurologic injury in numerous animal models.
Infections are common after cardiac surgery and increase hospital length of stay and cost.107,138,139 Infecting bacteria may arise from translocation across the patient’s gastrointestinal tract.107,114,116 Surgical wounds (sternum and lower extremity) and the respiratory tract are common sources of postoperative infection.140 Infections of prosthetic heart valves are less common but represent a devastating complication.141 Infections are probably not caused by the direct effects of inflammation, but instead through secondary effects on host immunity.142
Widespread activation of the complement system results in depletion of complement factors, which are crucial to the effective opsonization of bacterial pathogens.143,144 Systemic activation and degranulation of neutrophils render these cells less capable of destroying bacteria via phagocytosis. CPB leads to reductions in immunoglobulin levels through denaturation of these and other proteins.144–148 Antibody production by B lymphocytes (plasma cells) is depressed after cardiac surgery.149 Cell-mediated immunity, revealed by decreased T-lymphocyte function, appears to be impaired after cardiac surgery.150 Thus, reduced antibody levels, as well as reduced B- and T-cell function in the post-CPB period, may lead to increased infection rates after cardiac surgery.

Incidence of Complications
The “low” mortality rate after cardiac surgery also has been cited as evidence that splanchnic hypoperfusion, endotoxemia, and systemic inflammation only result in “rare” complications. As discussed earlier, it is clear that mortality1 and morbidity3,151 are still significant problems after cardiac surgery. In addition, many studies only report frank organ failures or catastrophes and do not take into account less severe forms of organ dysfunction, which do not lead to admission to an ICU but nevertheless cause suffering and increase hospital length of stay.
For example, a series by Huddy et al152 of 4473 cases involving CPB demonstrated a very low incidence rate (0.78%) of “gastrointestinal complications.” In a series of 3129 patients, Christenson et al153 reported an incidence rate of 2.3% for major gastrointestinal complications after coronary artery bypass graft surgery. The low incidence of major gastrointestinal complications (i.e., perforation, necrotic bowel, major gastrointestinal bleed) is often used to call into question the clinical relevance of splanchnic hypoperfusion. There is growing evidence, however, that less severe forms of splanchnic dysfunction (e.g., ileus, nausea, anorexia, and abdominal distention) are clinically relevant and increase hospital length of stay.
Some series of postoperative complications have broadened their scope. In a series of 572 patients, Corwin et al154 reported the incidence of renal failure requiring dialysis (1%), as well as the incidence of renal dysfunction not requiring dialysis (6.3%). This study demonstrates that organ dysfunction is common after surgery, despite a low incidence of organ failure.
Similarly, the incidence rate of acute respiratory distress syndrome after cardiac surgery has been reported to be very low (< 2%) in several series.155,156 The incidence, however, of less severe pulmonary dysfunction appears to be much greater, with as many as 7% of patients requiring supplemental oxygen 11 days after surgery.157 Furthermore, a high incidence of postoperative pulmonary dysfunction, as measured by diagnostic tests, gives further support to the hypothesis that many patients have abnormal pulmonary physiology potentially attributable to systemic inflammation.158–160

Potential Therapies for the Prevention of Inflammation-Related Complications
Steroid Administration
Several attempts have been made to prevent increases in proinflammatory cytokines and complement activation with steroids during cardiac surgery.37,86,161–169 In a randomized, double-blind study of 25 cardiac surgical patients, dexamethasone administration (1 mg/kg on induction of anesthesia) prevented increases in TNF, as well as reduced postoperative hyperthermia and hypotension.162 There was a trend toward improved outcome in the treatment group; however, the small sample size prevented any conclusions from being drawn regarding clinically relevant outcomes.
Inaba et al163 randomized 17 patients undergoing cardiac surgery to placebo or methylprednisolone, 30 mg/kg given immediately before the initiation of CPB. In this study, glucocorticoid administration minimized intraoperative increases in both plasma endotoxin and IL-6 levels; however, no improvement in postoperative outcome was reported.
Cavarocchi et al161 randomized 91 patients undergoing CPB to a bubble oxygenator without methylprednisolone (Group 1), bubble oxygenator with 30 mg/kg methylprednisolone (Group 2), or membrane oxygenator (Group 3). C3a levels increased in all three groups during CPB but were greater in Group 1 than in the other two groups. This study suggests that complement activation may be reduced by specific medical interventions. Andersen et al86 randomized 16 patients to receive, at induction of anesthesia, either methylprednisolone (30 mg/kg) or placebo. This study demonstrated a reduction of complement activation in the protocol group.
Randomized clinical trials by Chaney et al170,171 evaluated the effects of methylprednisolone (30 mg/kg) or placebo in cardiac surgical patients. Patients randomized to the steroid groups exhibited statistically significantly prolonged extubation times and received more vasoconstrictors. The investigators also observed significantly more hyperglycemia in the postoperative period in the methylprednisolone-treated patients. In another study, cardiac surgical patients randomized to steroid administration did not show benefit compared with placebo; however, a third study arm randomized to ultrafiltration without steroids did show a reduction in time to extubation.172 A 2008 meta-analysis of 44 randomized trials involving 3,205 patients demonstrated a reduction of new-onset atrial fibrillation, postoperative bleeding, and ICU length of stay.173 There were less pronounced reductions in hospital length of stay and mortality, and no safety concerns were identified. Therefore, although there are conflicting data on this issue, there is some evidence that there may be some benefit to steroid administration.
Role of Cardiopulmonary Bypass Technique
Although heparin-coated circuits have many theoretic advantages, there is little evidence that their use during cardiac surgery results in fewer clinically significant adverse complications. Steinberg et al54 found no difference in cytokine levels or markers of complement activation between patients randomized to a heparin-coated circuit or to a traditional circuit. Borowiec et al,25 however, observed lower levels of myeloperoxidase and lactoferrin (markers of inflammation) in patients undergoing CPB with a heparin-coated circuit. Other investigators have reported reduced plasma levels of cytokines and/or neutrophil proteases in patients subjected to CPB using heparin-coated circuits; however, no improvement in outcome was observed in these small studies23,53,174,175 (see Chapters 28 and 29). A meta-analysis involving 3,434 patients from 41 randomized trials demonstrated reductions in blood transfusion and durations of mechanical ventilation, ICU and hospital length of stays, which provides some support for this intervention.176
Centrifugal vortex blood pumping has been shown to result in reduced complement and neutrophil activation, as well as reduced hemolysis during cardiac surgery compared with standard roller blood pumping.177,178 Centrifugal vortex blood pumping, however, did not significantly prevent increases in cytokines in 17 pediatric patients randomized to this bypass technique.58
A randomized study of 15 patients suggested that pulsatile flow CPB may result in less endotoxemia than CPB involving nonpulsatile flow.94 Levine et al125 randomized 20 patients to pulsatile versus nonpulsatile flow and observed a less marked increase in vasopressin levels (an endogenous vasoconstrictor) in patients perfused with pulsatile flow. Taylor et al122 demonstrated increased levels of the endogenously produced vasoconstrictor angiotensin II in patients (N = 24) randomized to nonpulsatile flow CPB as compared with pulsatile flow. Watkins et al124 observed fewer marked alterations in thromboxane B2 and prostacyclin levels in patients (N = 16) randomized to pulsatile CPB. These studies evaluating pulsatile flow suggest that splanchnic perfusion may be better preserved with pulsatile flow because of less endogenously mediated vasoconstriction. Quigley et al,52 however, reported a lack of endotoxemia and pathologic cytokinemia in an uncontrolled study of patients who underwent nonpulsatile CPB. They claimed that the use of “adequate flow and perfusion pressures” during CPB accounted for their findings.
The role of membrane oxygenators as a means of reducing systemic inflammation-related complications also is controversial. Less complement activation has been observed with the use of membrane oxygenators; however, other studies have found no difference.62,74,161,179–183 The use of membrane oxygenators was associated with better pulmonary function as compared with the use of a bubble oxygenator; however, it is unclear whether the difference observed reflected reduced systemic inflammation in the protocol group.181 Butler et al62 randomized 20 patients undergoing cardiac surgery to either a membrane or bubble oxygenator. IL-6 levels peaked 4 hours after surgery, yet there were no significant differences between groups in IL-1 or IL-6 levels, or in intrapulmonary shunting. This study failed to show a difference in postoperative outcome, possibly because of short CPB durations (< 1 hour), as well as the small sample size, which makes detecting a clinically significant difference in postoperative complications unlikely. Host defenses may be better maintained with the use of membrane oxygenators.182
There also is controversy whether hypothermia during CPB worsens systemic inflammation.40,51,76,184–186 Hypothermia has been shown to reduce markers of complement activation.76 Another study demonstrated reduced markers of inflammation, such as TNF and IL-6, as well as reduced neutrophil activation in the hypothermia group.185 In contrast, another study randomized 30 cardiac surgical patients to either normothermic or hypothermic CPB.40 They found no association between CPB temperature and plasma TNF levels at any time point in the perioperative period, suggesting a limited role for temperature as an independent cause of proinflammatory cytokine release. In one of the largest randomized trials to date, 300 elective CABG patients were randomized to either normothermic (35.5°C to 36.5°C) or hypothermic (28°C to 30°C) CPB.187 No differences were seen in either short-term or longer term outcome, suggesting no benefit to intraoperative hypothermia. There is evidence, however, that perioperative hyperthermia may be detrimental to the brain.188 These investigators also observed an association between greater levels of the proinflammatory cytokine IL-6 and postoperative hyperthermia, suggesting a potential role of inflammation in the increases in temperature commonly observed after major surgery.
Finally, current data suggest that the use of CPB for cardiac surgery may not in and of itself be more deleterious than cardiac surgery without the use of CPB. Results from initial randomized clinical trials did not suggest that outcomes were substantially different in patients undergoing on- versus off-pump CABG surgery.189–193 Given the importance of this question, 2203 cardiac surgical patients were enrolled at 18 Veterans Affairs medical centers from 2002–2008 and randomized to on- or off-pump CABG. No benefit was found with regard to outcomes such as duration of mechanical ventilation, lengths of stay in the ICU or hospital, renal failure, or a composite end point of complications.194 Systemic inflammation was not specifically studied in this important trial, but these data suggest that systemic inflammation attributable to CPB may have a more modest role in determining clinical outcome than previously thought. Another plausible conclusion, however, is that hemodynamic instability and use of potent vasoactive agents associated with the off-pump technique may be an equivalent insult to the use of CPB.
Complement Inhibition
The results from several large, randomized clinical trials in which complement activation is selectively blocked have become available.79–81 For example, in the largest randomized, double-blind clinical trial conducted to date, 3099 adults undergoing CABG surgery at 205 hospitals in North America and Western Europe were enrolled.80 Patients were randomized to placebo or to a 24-hour infusion of pexelizumab, which is a recombinant, humanized, single-chain antibody fragment that binds to human C5 complement and prevents its activation. The administration of pexelizumab resulted in rapid and complete inhibition of complement activation. The primary outcome variable (death or myocardial infarction within 30 days) did not achieve statistical significance (p = 0.07). The subset of patients undergoing CABG plus valve did demonstrate a statistically significant difference (p = 0.03) in this outcome. This finding is somewhat contradictory to the previous phase IIb trial (n = 914) that enrolled patients undergoing CABG and/or valve surgery and did not show a significant difference with regard to the end point of death or myocardial infarction.79 This previous trial did achieve significance in the subset undergoing isolated CABG, which was the apparent justification for studying isolated CABG surgeries in the larger phase III trial. These studies indicate that attenuation of complement activation results in less myocardial injury; however, there did not appear to be an impact on complications such as pulmonary and renal dysfunction and severe vasodilation. These results suggest that complement activation may not play as large a role in the development of systemic inflammation-mediated morbidity as previously thought.
Ultrafiltration
Removal of excess fluid with ultrafiltration has been proposed as a method for removing proinflammatory mediators during cardiac surgery, particularly in the pediatric population.46,195 It is unclear in studies performed thus far whether beneficial effects of ultrafiltration are due to one or some combination of the following factors: prevention of initiation of inflammation, removal of inflammatory mediators, or removal of excessive fluid alone. In one study, Journois et al196 randomized 20 pediatric cardiac surgical patients to either a control group or to high-volume, zero-fluid balance ultrafiltration. Measured TNF, IL-1, IL-10, myeloperoxidase, and C3a levels were lower in the protocol group compared with the control group. The authors suggested that hemofiltration may have some beneficial effects that are not due to water removal alone. Patients in the ultrafiltration group had less postoperative fever, reduced perioperative blood loss, reduced time to extubation, and reduced postoperative alveolar-arterial oxygen gradient, which suggests, yet does not prove, a causal relation between proinflammatory cytokinemia and several clinically meaningful end points. The small sample size of this study precludes any conclusions from being made regarding other outcomes such as the incidence of multiple organ dysfunction syndrome or hospital length of stay.
In an interesting study, 192 cardiac surgical patients were randomized to placebo (no steroids or ultrafiltration), steroid administration without ultrafiltration, or hemofiltration without steroids.172 The study arm randomized to ultrafiltration without steroids showed a reduction in time to extubation; however, steroid administration was not effective compared with placebo. These data are promising, but the small number of subjects per arm and limited number of positive outcomes reported make it unclear whether ultrafiltration should be used more routinely.
Leukocyte Depletion
Removal of leukocytes during CPB with an inline leukocyte filter has been proposed as a method for reducing the concentration of activated leukocytes. This, in turn, may prevent inflammatory-mediated postoperative complications. This technology has been nicely reviewed by Warren et al,197 who, in their review of 63 studies, concluded that there may be some modest benefits, but that the low quality of evidence from the predominantly small trials precluded any definitive conclusions on this matter. For example, patients randomized to leukocyte depletion (n = 20) had better oxygenation after CPB; however, there were no differences in other outcomes measured.198 A prospective randomized study of patients (N = 50) receiving inline leukocyte filtration demonstrated decreased leukocytes; however, postoperative arterial blood gases, pulmonary vascular resistance, ventilator time, and hospital length of stay were no different between groups.199 Another study demonstrated no difference in postoperative complications or in the plasma levels of neutrophil proteases in patients undergoing leukocyte depletion with an inline filter.200
Davies et al201 used another method of leukocyte depletion in which they removed platelets and leukocytes by plasmapheresis from patients before cardiac surgery. These patients, compared with the control group, demonstrated reduced postoperative thoracic drainage, reduced allogeneic blood product administration, and improved pulmonary function. This technique is not in widespread clinical use because of a lack of studies confirming its findings, as well as the time and cost involved in performing plasmapheresis (see Chapters 28 through 31).
The techniques described earlier differ from the issue of administration of leukocyte-reduced packed red blood cells. This method of minimizing a patient’s exposure to leukocytes involves the filtering of the collected blood either at the time of donation (fresh filtered) or before its release by the blood bank (stored filtered).202 Van de Watering and colleagues203 reported results from a large (n = 914) randomized clinical trial in which patients undergoing cardiac surgery were randomized to receive allogeneic red cells without buffy coat, fresh-filtered allogeneic red cells, or stored filtered units. Patients randomized to either filtration group experienced a significant reduction in postoperative mortality (P = 0.015); however, this effect was most robust in patients administered more than three transfusions. It should be noted that differences between groups in the incidence of infection did not achieve statistical significance (P = 0.13). No differences between groups were found in ICU or hospital length of stay in those patients in the overall study population or in the subset administered more than three units. These data are from a randomized trial and, therefore, should be more heavily weighted. Data from some retrospective cohort studies have shown no benefit to leukocyte reduction.204,205

Aprotinin and Other Serine Protease Inhibitors
Aprotinin, a 58-amino-acid serine protease inhibitor isolated from bovine lung, has been shown in numerous studies to decrease bleeding associated with cardiac surgery. It antagonizes numerous proteolytic enzymes including plasmin and kallikrein, and may have some anti-inflammatory effects,14,206 although a recent meta-analysis found no beneficial effect of aprotinin on systemic markers of inflammation.207 Aprotinin’s blood-sparing effects were apparently discovered serendipitously while it was being evaluated as an anti-inflammatory agent in cardiac surgical patients. Despite more than 45 randomized clinical trials conducted to date, there are little data to support the hypothesis that aprotinin administration reduces postoperative complications attributable to excessive systemic inflammation. In these trials, numerous surrogate markers of postoperative morbidity, such as the duration of postoperative tracheal intubation, ICU stay, and hospital length of stay, were not reported to be improved in aprotinin-treated patients. A large trial was recently completed in 2,331 cardiac surgical patients at 19 Canadian centers.208 Patients were randomized to aprotinin, aminocaproic acid, or tranexamic acid. No benefit of aprotinin was observed with regard to complications such as respiratory failure, renal failure, or multisystem organ failure, and the study was terminated early because of an increase in mortality in aprotinin-treated patients. The findings from this trial, as well as several previous observational studies, led to market withdrawal of aprotinin.
It is worth noting that several novel drugs are under development that are postulated to reduce inflammation and blood loss, while being free of potential adverse effects associated with aprotinin. These include the synthetic serine protease inhibitors CU-2010 and CU-2020209 and DX-88/ecallantide, a plasma kallikrein inhibitor.210
Tumor Necrosis Factor Antagonists
Soluble TNF receptor proteins antagonize the toxic effects of LPS-induced lethality in mice.211 These agents are ineffective in the treatment of sepsis/septic shock but have not been tested in the setting of cardiac surgery.212 Anti-TNF monoclonal antibodies have been studied in septic ICU patients as well; however, they have not yet been tested prophylactically in cardiac surgical patients.69 A study involving prophylactic administration would allow for the antibody to be present before the TNF and thus determine whether TNF has overall harmful or beneficial effects. If TNF and other cytokines are essential to the healing process, complete inhibition of their effects may result in worse rather than improved postoperative outcome. Well-designed, large clinical trials could resolve these controversial issues.
E5564
E5564 is a synthetically derived lipid A analog that is a potent Toll-like receptor 4–directed endotoxin antagonist.213 It does not have lipid A agonist properties, and even in high doses does not cause signs or symptoms of endotoxemia or systemic inflammation in humans and animals. Healthy volunteers were administered E5564 before a standard challenge dose of reference endotoxin (4 ng/kg). Single E5564 doses of 50 to 250 μg blocked or attenuated all of the effects of LPS in a dose-dependent manner. All E5564 dose groups had statistically significant reductions in increased temperature, HR, CRP levels, white blood cell count, and cytokine levels (TNF-α and IL-6), compared with placebo (P < 0.01).213 This drug has shown promising results in critically ill patients with sepsis; however, results from a phase II trial in cardiac surgical patients were disappointing.113 In this trial, 152 cardiac surgical patients at 9 U.S. centers were randomized to receive placebo or ascending doses of E5564. Blocking lipid A with eritoran did not result in any overt beneficial effects on markers of systemic inflammation (IL-6, IL-8, or CRP) or measures of organ injury. These results call into question the potential clinical relevance of lipid A in this setting.
Pentoxifylline
In an initial study, Hoffman et al214 randomized 40 patients with an Acute Physiology and Chronic Health Enquiry (APACHE) II score ≥ 19 after cardiac surgery to placebo or pentoxifylline (1.5 mg/kg/hr for 48 hours). In this study, patients administered pentoxifylline had significantly fewer days on mechanical ventilation, less need for hemofiltration, and a shorter ICU length of stay. In a historic control study, Thabut et al215 administered pentoxifylline to 23 consecutive patients undergoing lung transplantation. Compared with historic controls, patients administered pentoxifylline experienced less allograft dysfunction and a significant reduction in 60-day mortality was noted. These findings need to be confirmed in a prospective, randomized clinical trial.
Boldt et al216,217 randomized 30 elderly (> 80 years) patients undergoing cardiac surgery to placebo or pentoxifylline (300 mg bolus administered immediately after induction of general anesthesia followed by a continuous infusion of 1.5 mg/kg/hr for 48 hours). In this study, pentoxifylline administration minimized intraoperative and postoperative increases in plasma CRP, polymorphonuclear elastase, IL-6, and IL-8 levels. Duration of mechanical ventilation was significantly lower in patients randomized to pentoxifylline. This study was not powered to detect differences in rare but serious complications. Other small studies also suggested possible benefit218,219; however, these results have not been confirmed in a large, multicenter trial.
Ethyl pyruvate
This is a novel anti-inflammatory agent.220,221 It has been shown to protect the intestinal mucosa from mesenteric ischemia and reperfusion in rats and improve survival in murine models of acute endotoxemia and bacterial peritonitis. Results from a phase II trial in cardiac surgical patients were disappointing.222 In this trial, 102 high-risk cardiac surgical patients were randomized to receive placebo or ethyl pyruvate at 13 U.S. centers. Administration of ethyl pyruvate did not result in any overt beneficial effects on markers of systemic inflammation (TNF-α, IL-6, or CRP) or measures of organ injury.
Statins
Statins are routinely used to reduce cholesterol levels in patients at risk for cardiovascular disease; however, their anti-inflammatory effects have received significant attention.223 It has been speculated that prophylactic statin administration before surgery may have beneficial effects. A recently published trial randomized 497 vascular surgical patients to placebo or fluvastatin daily from randomization to 30 days after surgery.224 Patients randomized to the statin exhibited lower levels of the inflammatory markers IL-6 and CRP, and also reduced myocardial ischemia (P = 0.01). All-cause mortality was lower in statin-treated patients (2.4% vs. 4.9%); however, this difference did not achieve statistical significance (P = 0.14). There are no similar large trials in cardiac surgery, but a meta-analysis was recently completed of 8 trials involving a total of 638 such patients.225 This analysis showed that statin use decreased levels of IL-6, IL-8, CRP, and TNF-α; however, no improvement in clinical outcomes was reported. At this point, the use of statins in cardiac surgery must be investigational until additional data are obtained to support this indication.
N-acetylcysteine
This is used to prevent radiocontrast-induced nephropathy and as an antidote for acetaminophen overdose. Its anti-inflammatory and antioxidant properties have been studied in the ICU setting and in cardiac surgical patients, with mixed results.A meta-analysis of N-acetylcysteine to ameliorate postoperative morbidity was recently published and included 1,338 patients from 13 trials.226 This analysis suggested that N-acetylcysteine may have a beneficial effect with regard to postoperative atrial fibrillation but did not appear to be beneficial with regard to other postoperative complications.
Other Potential Antiendotoxin or Anti-Inflammatory Agents
Other potential approaches to preventing endotoxin-related complications involve the use of either synthetic or naturally occurring antiendotoxin compounds. Bactericidal/permeability-increasing protein (BPI) is a neutrophil granule protein and has been shown to have endotoxin-neutralizing and bactericidal activity in animal models. A human recombinant version, rBPI21, neutralized endotoxin-mediated toxicity in humans. A recombinant version of an antiendotoxin factor, endotoxin-neutralizing protein, is another agent that has been shown to protect animals from endotoxin-mediated toxicity.227 Reconstituted high-density lipoprotein (rHDL) neutralized some of endotoxin’s toxic effects during an experimental model of human endotoxemia.228 Polymyxin B neutralizes the toxic effects of endotoxin, although toxicity has prevented prophylactic intravenous use.229 Dextran-polymyxin B is a variation of polymyxin B that has been reported to have antiendotoxin properties, as well as minimal toxicity in animal models. Soluble TNF receptor proteins antagonize the toxic effects of LPS-induced lethality in mice. This agent was not effective in the treatment of sepsis/septic shock but has not been tested in the setting of cardiac surgery.211

Role of Anesthetic Agents and Vasoactive Agents
Anesthetic agents, defined here as drugs that induce hypnosis, amnesia, muscle relaxation, or regional anesthesia, have not been shown to result in clinically meaningful reductions in systemic inflammation after cardiac surgery. Numerous studies have evaluated the effect of these agents on the immune system with varied results; however, no studies have reported a difference in outcome with one technique versus another. Ketamine is a promising agent that has been studied largely as an adjunct to reduce postoperative pain in noncardiac surgery. In an initial study in cardiac surgery, administration of a low dose (0.25 mg/kg) of ketamine before CPB prevented an increase in IL-6 for 7 days after surgery.230 In addition, ketamine administration inhibited TNF production and leukocyte adherence in animal models and suppressed oxygen radical production in vitro. These results were confirmed in another small study of patients undergoing CPB.231 However, there are no outcome data from large outcome trials, so it is unknown whether this intervention reduces clinically relevant complications.
All general anesthetics can reduce splanchnic perfusion indirectly through a depression of myocardial function and a reduction in CO, and hence oxygen delivery to the splanchnic mucosa.232,233 Isoflurane theoretically may be better than halothane, enflurane, or propofol because of its vasodilating properties, which may preserve splanchnic blood flow and blood volume.232–235 A prospective randomized study of cardiac surgical patients demonstrated better splanchnic perfusion in patients maintained with isoflurane in contrast with propofol or enflurane.236
Although not definitively supported yet, there is evidence that splanchnic hypoperfusion and endotoxin-induced inflammation can be prevented in the operating room by strategies familiar to clinicians. Strategies involve the use of fluid loading to maximize SV,129 as well as the use of adequate levels of vasodilating volatile anesthetics. Inodilating agents, such as milrinone, amrinone, dopexamine, and dobutamine, may be more protective of splanchnic perfusion than inoconstricting agents such as epinephrine, norepinephrine, and dopamine. Patients randomized to enoximone administration during cardiac surgery demonstrated lower endotoxin levels, suggesting a beneficial effect on the barrier function of the gut.91 Endotoxemia is probably a more sensitive marker of loss of barrier function than gastric mucosal hypoperfusion because these patients still demonstrated decreases in calculated gastric mucosal pH (pHi). When tested in vitro, amrinone was a potent inhibitor of endotoxin-induced TNF production at clinically relevant drug concentrations, suggesting an additional advantage to the use of this phosphodiesterase inhibitor.237 Dopamine is often touted as preserving splanchnic blood flow; however, responses to this agent are unpredictable, with vascular resistance increasing in some patients at low doses (3–5 μg/kg/min).

Selective Digestive Decontamination
Selective digestive decontamination represents a possible approach to limiting the incidence and severity of systemic inflammation. The technique attempts to reduce the total amount of endotoxin exposure by reducing the reservoir of endotoxin normally contained within the gut. Martinez-Pellús et al65 conducted a prospective, open, randomized, controlled trial in 80 cardiac surgical patients. Patients were randomized to either a control group or up to 3 days of preoperative selective digestive decontamination accomplished with the administration of oral nonabsorbable antibiotics (polymyxin E, tobramycin, amphotericin B). Patients in the protocol group demonstrated much lower gut bacterial counts, as well as lower blood levels of endotoxin and the proinflammatory cytokine IL-6 in the operating room and the postoperative unit. The study was not designed with sufficient power to determine whether this technique affects outcomes such as mortality and morbidity. Nevertheless, it is interesting to note that there was a trend toward improved outcome (mortality, hospital length of stay) in the protocol group. In contrast, Bouter et al238 found no beneficial effects of selective digestive decontamination on clinical outcome or blood levels of TNF-α, IL-6, or IL-10 in 78 cardiac surgical patients.

1 Birkmeyer J.D., Stukel T.A., Siewers A.E., et al. Surgeon volume and operative mortality in the United States. N Engl J Med. 2003;349(22):2117-2127.
2 Hammermeister K.E., Burchfiel C., Johnson R., Grover F.L. Identification of patients at greatest risk for developing major complications at cardiac surgery. Circulation. 1990;82(Suppl 5):IV380-IV389.
3 Rady M.Y., Ryan T., Starr N.J. Perioperative determinants of morbidity and mortality in elderly patients undergoing cardiac surgery. Crit Care Med. 1998;26(2):225-235.
4 Goris R.J., te Boekhorst T.P., Nuytinck J.K., Gimbrere J.S. Multiple-organ failure. Generalized autodestructive inflammation? Arch Surg. 1985;120(10):1109-1115.
5 Bone R.C., Balk R.A., Cerra F.B., et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644-1655.
6 Schlag G., Redl H., Hallstrom S. The cell in shock: The origin of multiple organ failure. Resuscitation. 1991;21(2–3):137-180.
7 American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20(6):864-874.
8 Stedman’s Medical Dictonary. ed 26. 1995. Williams & Wilkins. Baltimore
9 Vincent J.L. Dear SIRS, I’m sorry to say that I don’t like you. Crit Care Med. 1997;25(2):372-374.
10 Berendes E., Mollhoff T., Van Aken H., et al. Effects of dopexamine on creatinine clearance, systemic inflammation, and splanchnic oxygenation in patients undergoing coronary artery bypass grafting. Anesth Analg. 1997;84(5):950-957.
11 Andersen L.W., Baek L., Degn H., et al. Presence of circulating endotoxins during cardiac operations. J Thorac Cardiovasc Surg. 1987;93(1):115-119.
12 Doran J.E. Biological effects of endotoxin. Curr Stud Hematol Blood Transfus. 1992;59:66-99.
13 Herskowitz A., Mangano D.T. Inflammatory cascade. A final common pathway for perioperative injury? Anesthesiology. 1996;85(5):957-960.
14 Royston D. Preventing the inflammatory response to open-heart surgery: The role of aprotinin and other protease inhibitors. Int J Cardiol. 1996;53(Suppl):S11-S37.
15 Miller B.E., Levy J.H. The inflammatory response to cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1997;11(3):355-366.
16 Weiss S.J. Tissue destruction by neutrophils. N Engl J Med. 1989;320(6):365-376.
17 Webster R.O., Hong S.R., Johnston R.B.Jr, Henson P.M. Biologial effects of the human complement fragments C5a and C5ades Arg on neutrophil function. Immunopharmacology. 1980;2(3):201-219.
18 Freeman B.A., Crapo J.D. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47(5):412-426.
19 Meerson F.Z., Kagan V.E., Kozlov Yu P., et al. The role of lipid peroxidation in pathogenesis of ischemic damage and the antioxidant protection of the heart. Basic Res Cardiol. 1982;77(5):465-485.
20 Halliwell B., Guiteridge M. Oxygen radical and tissue damage. Bologna, Italy: Cooperativa Libraria Iniversitavia Editrice, 1982.
21 Prasad K., Kalra J., Bharadwaj B., Chaudhary A.K. Increased oxygen free radical activity in patients on cardiopulmonary bypass undergoing aortocoronary bypass surgery. Am Heart J. 1992;123(1):37-45.
22 Royston D., Fleming J.S., Desai J.B., et al. Increased production of peroxidation products associated with cardiac operations. Evidence for free radical generation. J Thorac Cardiovasc Surg. 1986;91(5):759-766.
23 Fosse E., Moen O., Johnson E., et al. Reduced complement and granulocyte activation with heparin-coated cardiopulmonary bypass. Ann Thorac Surg. 1994;58(2):472-477.
24 Kharazmi A., Andersen L.W., Baek L., et al. Endotoxemia and enhanced generation of oxygen radicals by neutrophils from patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1989;98(3):381-385.
25 Borowiec J., Thelin S., Bagge L., et al. Heparin-coated circuits reduce activation of granulocytes during cardiopulmonary bypass. A clinical study. J Thorac Cardiovasc Surg. 1992;104(3):642-647.
26 Mythen M.G., Purdy G., Mackie I.J., et al. Postoperative multiple organ dysfunction syndrome associated with gut mucosal hypoperfusion, increased neutrophil degranulation and C1- esterase inhibitor depletion. Br J Anaesth. 1993;71(6):858-863.
27 Bjork J., Hugli T.E., Smedegard G. Microvascular effects of anaphylatoxins C3a and C5a. J Immunol. 1985;134(2):1115-1119.
28 Liu B., Belboul A., al-Khaja N., et al. Effect of high-dose aprotinin on blood cell filterabiltiy in association with cardiopulmonary bypass. Coron Artery Dis. 1992;3:129.
29 Blauth C., Arnold J., Kohner E.M., Taylor K.M. Retinal microembolism during cardiopulmonary bypass demonstrated by fluorescein angiography. Lancet. 1986;2(8511):837-839.
30 Sheeran P., Hall G.M. Cytokines in anaesthesia. Br J Anaesth. 1997;78(2):201-219.
31 Tonnesen E., Christensen V.B., Toft P. The role of cytokines in cardiac surgery. Int J Cardiol. 1996;53(Suppl):S1-S10.
32 Baumann H., Gauldie J. The acute phase response. Immunol Today. 1994;15(2):74-80.
33 Michie H.R., Manogue K.R., Spriggs D.R., et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318(23):1481-1486.
34 Michie H.R., Spriggs D.R., Manogue K.R., et al. Tumor necrosis factor and endotoxin induce similar metabolic responses in human beings. Surgery. 1988;104(2):280-286.
35 Tracey K.J., Lowry S.F., Fahey T.J.3rd, et al. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obstet. 1987;164(5):415-422.
36 Hesse D.G., Tracey K.J., Fong Y., et al. Cytokine appearance in human endotoxemia and primate bacteremia. Surg Gynecol Obstet. 1988;166(2):147-153.
37 Jansen N.J., van Oeveren W., van Vliet M., et al. The role of different types of corticosteroids on the inflammatory mediators in cardiopulmonary bypass. Eur J Cardiothorac Surg. 1991;5(4):211-217.
38 Jansen N.J., van Oeveren W., Gu Y.J., et al. Endotoxin release and tumor necrosis factor formation during cardiopulmonary bypass. Ann Thorac Surg. 1992;54(4):744-747. discussion 747–748
39 Abe K., Nishimura M., Sakakibara T. Interleukin-6 and tumour necrosis factor during cardiopulmonary bypass. Can J Anaesth. 1994;41(9):876-877.
40 Tonz M., Mihaljevic T., von Segesser L.K., et al. Normothermia versus hypothermia during cardiopulmonary bypass: A randomized, controlled trial. Ann Thorac Surg. 1995;59(1):137-143.
41 te Velthuis H., Jansen P.G., Oudemans-van Straaten H.M., et al. Myocardial performance in elderly patients after cardiopulmonary bypass is suppressed by tumor necrosis factor. J Thorac Cardiovasc Surg. 1995;110(6):1663-1669.
42 Hennein H.A., Ebba H., Rodriguez J.L., et al. Relationship of the proinflammatory cytokines to myocardial ischemia and dysfunction after uncomplicated coronary revascularization. J Thorac Cardiovasc Surg. 1994;108(4):626-635.
43 Wan S., Marchant A., DeSmet J.M., et al. Human cytokine responses to cardiac transplantation and coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1996;111(2):469-477.
44 Seghaye M., Duchateau J., Bruniaux J., et al. Interleukin-10 release related to cardiopulmonary bypass in infants undergoing cardiac operations. J Thorac Cardiovasc Surg. 1996;111(3):545-553.
45 Deng M.C., Dasch B., Erren M., et al. Impact of left ventricular dysfunction on cytokines, hemodynamics, and outcome in bypass grafting. Ann Thorac Surg. 1996;62(1):184-190.
46 Millar A.B., Armstrong L., van der Linden J., et al. Cytokine production and hemofiltration in children undergoing cardiopulmonary bypass. Ann Thorac Surg. 1993;56(6):1499-1502.
47 Furunaga A. Measurement of cytokines at cardiopulmonary-bypass. Nippon Kyobu Geka Gakkai Zasshi. 1994;42(12):2200-2206.
48 Kawamura T., Wakusawa R., Okada K., Inada S. Elevation of cytokines during open heart surgery with cardiopulmonary bypass: Participation of interleukin 8 and 6 in reperfusion injury. Can J Anaesth. 1993;40(11):1016-1021.
49 Markewitz A., Faist E., Lang S., et al. Regulation of acute phase response after cardiopulmonary bypass by immunomodulation. Ann Thorac Surg. 1993;55(2):389-394.
50 Haeffner-Cavaillon N., Roussellier N., Ponzio O., et al. Induction of interleukin-1 production in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1989;98(6):1100-1106.
51 Frering B., Philip I., Dehoux M., et al. Circulating cytokines in patients undergoing normothermic cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1994;108(4):636-641.
52 Quigley R.L., Caplan M.S., Perkins J.A., et al. Cardiopulmonary bypass with adequate flow and perfusion pressures prevents endotoxaemia and pathologic cytokine production. Perfusion. 1995;10(1):27-31.
53 Steinberg B.M., Grossi E.A., Schwartz D.S., et al. Heparin bonding of bypass circuits reduces cytokine release during cardiopulmonary bypass. Ann Thorac Surg. 1995;60(3):525-529.
54 Steinberg J.B., Kapelanski D.P., Olson J.D., Weiler J.M. Cytokine and complement levels in patients undergoing cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1993;106(6):1008-1016.
55 Kilbourn R.G., Belloni P. Endothelial cell production of nitrogen oxides in response to interferon gamma in combination with tumor necrosis factor, interleukin-1, or endotoxin. J Natl Cancer Inst. 1990;82(9):772-776.
56 Finn A., Naik S., Klein N., et al. Interleukin-8 release and neutrophil degranulation after pediatric cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1993;105(2):234-241.
57 Huber A.R., Kunkel S.L., Todd R.F.3rd, Weiss S.J. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254(5028):99-102.
58 Ashraf S.S., Tian Y., Cowan D., et al. Proinflammatory cytokine release during pediatric cardiopulmonary bypass: Influence of centrifugal and roller pumps. J Cardiothorac Vasc Anesth. 1997;11(6):718-722.
59 Kawahito K., Kawakami M., Fujiwara T., et al. Proinflammatory cytokine levels in patients undergoing cardiopulmonary bypass. Does lung reperfusion influence the release of cytokines? ASAIO J. 1995;41(3):M775-M778.
60 Rothenburger M., Soeparwata R., Deng M.C., et al. Prediction of clinical outcome after cardiac surgery: The role of cytokines, endotoxin, and anti-endotoxin core antibodies. Shock. 2001;16(Suppl 1):44-50.
61 Rothenburger M., Tjan T.D., Schneider M., et al. The impact of the pro- and anti-inflammatory immune response on ventilation time after cardiac surgery. Cytometry. 2003;53B(1):70-74.
62 Butler J., Chong G.L., Baigrie R.J., et al. Cytokine responses to cardiopulmonary bypass with membrane and bubble oxygenation. Ann Thorac Surg. 1992;53(5):833-838.
63 Almdahl S.M., Waage A., Ivert T., Vaage J. Release of bioactive interleukin-6 but not of tumor necrosis factor-alpha after elective cardiopulmonary bypass. Perfusion. 1993;8:233.
64 Cremer J., Martin M., Redl H., et al. Systemic inflammatory response syndrome after cardiac operations. Ann Thorac Surg. 1996;61(6):1714-1720.
65 Martinez-Pellús A.E., Merino P., Bru M., et al. Can selective digestive decontamination avoid the endotoxemia and cytokine activation promoted by cardiopulmonary bypass? Crit Care Med. 1993;21(11):1684-1691.
66 de Waal Malefyt R., Abrams J., Bennett B., et al. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174(5):1209-1220.
67 Bogdan C., Vodovotz Y., Nathan C. Macrophage deactivation by interleukin 10. J Exp Med. 1991;174(6):1549-1555.
68 McBride W.T., Armstrong M.A., Crockard A.D., et al. Cytokine balance and immunosuppressive changes at cardiac surgery: Contrasting response between patients and isolated CPB circuits. Br J Anaesth. 1995;75(6):724-733.
69 Fisher C.J.Jr, Agosti J.M., Opal S.M., et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334(26):1697-1702.
70 Best N., Sinosich M.J., Teisner B., et al. Complement activation during cardiopulmonary bypass by heparin-protamine interaction. Br J Anaesth. 1984;56(4):339-343.
71 Kirklin J.K., Chenoweth D.E., Naftel D.C., et al. Effects of protamine administration after cardiopulmonary bypass on complement, blood elements, and the hemodynamic state. Ann Thorac Surg. 1986;41(2):193-199.
72 Chiu R.C., Samson R. Complement (C3, C4) consumption in cardiopulmonary bypass, cardioplegia, and protamine administration. Ann Thorac Surg. 1984;37(3):229-232.
73 Chenoweth D.E., Cooper S.W., Hugli T.E., et al. Complement activation during cardiopulmonary bypass: Evidence for generation of C3a and C5a anaphylatoxins. N Engl J Med. 1981;304(9):497-503.
74 Hammerschmidt D.E., Stroncek D.F., Bowers T.K., et al. Complement activation and neutropenia occurring during cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1981;81(3):370-377.
75 Kirklin J.K., Westaby S., Blackstone E.H., et al. Complement and the damaging effects of cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1983;86(6):845-857.
76 Moore F.D.Jr, Warner K.G., Assousa S., et al. The effects of complement activation during cardiopulmonary bypass. Attenuation by hypothermia, heparin, and hemodilution. Ann Surg. 1988;208(1):95-103.
77 Riegel W., Spillner G., Schlosser V., Horl W.H. Plasma levels of main granulocyte components during cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1988;95(6):1014-1019.
78 Bonser R.S., Dave J.R., Davies E.T., et al. Reduction of complement activation during bypass by prime manipulation. Ann Thorac Surg. 1990;49(2):279-283.
79 Shernan S.K., Fitch J.C., Nussmeier N.A., et al. Impact of pexelizumab, an anti-C5 complement antibody, on total mortality and adverse cardiovascular outcomes in cardiac surgical patients undergoing cardiopulmonary bypass. Ann Thorac Surg. 2004;77(3):942-949. discussion 949–950
80 Verrier E.D., Shernan S.K., Taylor K.M., et al. Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: A randomized trial. JAMA. 2004;291(19):2319-2327.
81 Fitch J.C., Rollins S., Matis L., et al. Pharmacology and biological efficacy of a recombinant, humanized, single-chain antibody C5 complement inhibitor in patients undergoing coronary artery bypass graft surgery with cardiopulmonary bypass. Circulation. 1999;100(25):2499-2506.
82 Morrison D.C., Cochrane C.G. Direct evidence for Hageman factor (factor XII) activation by bacterial lipopolysaccharides (endotoxins). J Exp Med. 1974;140(3):797-811.
83 Morrison D.C., Ryan J.L. Endotoxins and disease mechanisms. Annu Rev Med. 1987;38:417-432.
84 Natanson C., Eichenholz P.W., Danner R.L., et al. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. J Exp Med. 1989;169(3):823-832.
85 Rocke D.A., Gaffin S.L., Wells M.T., et al. Endotoxemia associated with cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1987;93(6):832-837.
86 Andersen L.W., Baek L., Thomsen B.S., Rasmussen J.P. Effect of methylprednisolone on endotoxemia and complement activation during cardiac surgery. J Cardiothorac Anesth. 1989;3(5):544-549.
87 Andersen L.W., Landow L., Baek L., et al. Association between gastric intramucosal pH and splanchnic endotoxin, antibody to endotoxin, and tumor necrosis factor-alpha concentrations in patients undergoing cardiopulmonary bypass. Crit Care Med. 1993;21(2):210-217.
88 Riddington D.W., Venkatesh B., Boivin C.M., et al. Intestinal permeability, gastric intramucosal pH, and systemic endotoxemia in patients undergoing cardiopulmonary bypass. JAMA. 1996;275(13):1007-1012.
89 Oudemans-van Straaten H.M., Jansen P.G., Hoek F.J., et al. Intestinal permeability, circulating endotoxin, and postoperative systemic responses in cardiac surgery patients. J Cardiothorac Vasc Anesth. 1996;10(2):187-194.
90 Oudemans-van Straaten H.M., Jansen P.G., Velthuis H., et al. Endotoxaemia and postoperative hypermetabolism in coronary artery bypass surgery: The role of ketanserin. Br J Anaesth. 1996;77(4):473-479.
91 Loick H.M., Mollhoff T., Berendes E., et al. Influence of enoximone on systemic and splanchnic oxygen utilization and endotoxin release following cardiopulmonary bypass. Intensive Care Med. 1997;23(3):267-275.
92 te Velthuis H., Jansen P.G., Oudemans-van Straaten H.M., et al. Circulating endothelin in cardiac operations: Influence of blood pressure and endotoxin. Ann Thorac Surg. 1996;61(3):904-908.
93 Bowles C.T., Ohri S.K., Klangsuk N., et al. Endotoxaemia detected during cardiopulmonary bypass with a modified Limulus amoebocyte lysate assay. Perfusion. 1995;10(4):219-228.
94 Watarida S., Mori A., Onoe M., et al. A clinical study on the effects of pulsatile cardiopulmonary bypass on the blood endotoxin levels. J Thorac Cardiovasc Surg. 1994;108(4):620-625.
95 Nilsson L., Kulander L., Nystrom S.O., Eriksson O. Endotoxins in cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1990;100(5):777-780.
96 Myles P., Buckland M., Cannon G., et al. The association among gastric mucosal pH, endotoxemia, and low systemic vascular resistance after cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 1996;10(2):195-200.
97 Imai T., Shiga T., Saruki N., et al. Change in plasma endotoxin titres and endotoxin neutralizing activity in the perioperative period. Can J Anaesth. 1996;43(8):812-819.
98 Lebek G., Cottier H. Notes on the bacterial content of the gut. Curr Stud Hematol Blood Transfus. 1992;59:1-18.
99 Fink M.P. Effect of critical illness on microbial translocation and gastrointestinal mucosa permeability. Semin Respir Infect. 1994;9(4):256-260.
100 Daniel M. Response of man to endotoxin. Immunobiology. 1993;187:403.
101 Rubin J., Robbs J.V., Gaffin S.L., Wells M.T. Plasma lipopolysaccharide increase after aortic aneurysm resection. S Afr Med J. 1988;74(4):193.
102 Greisman S.E., Hornick R.B. Mechanism of endotoxin tolerance with special reference to man. J Infect Dis. 1973;128(S):265.
103 Astiz M.E., Rackow E.C., Still J.G., et al. Pretreatment of normal humans with monophosphoryl lipid A induces tolerance to endotoxin: A prospective, double-blind, randomized, controlled trial. Crit Care Med. 1995;23(1):9-17.
104 Luderitz O., Staub A.M., Westphal O. Immunochemistry of O and R antigens of Salmonella and related Enterobacteriaceae. Bacteriol Rev. 1966;30(1):192-255.
105 Volk W.B.D.K.R. Medical microbiology. Philadelphia: Lippincott, 1986;396-398.
106 DeCamp M.M., Demling R.H. Posttraumatic multisystem organ failure. JAMA. 1988;260(4):530-534.
107 Ford E.G., Baisden C.E., Matteson M.L., Picone A.L. Sepsis after coronary bypass grafting: Evidence for loss of the gut mucosal barrier. Ann Thorac Surg. 1991;52(3):514-517.
108 Moore F.A., Moore E.E., Poggetti R., et al. Gut bacterial translocation via the portal vein: A clinical perspective with major torso trauma. J Trauma. 1991;31(5):629-636. discussion 636–628
109 Rush B.F.Jr, Sori A.J., Murphy T.F., et al. Endotoxemia and bacteremia during hemorrhagic shock. The link between trauma and sepsis? Ann Surg. 1988;207(5):549-554.
110 Ziegler E.J., Fisher C.J.Jr, Sprung C.L., et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991;324(7):429-436.
111 Greenman R.L., Schein R.M., Martin M.A., et al. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA Sepsis Study Group. JAMA. 1991;266(8):1097-1102.
112 Warren H.S., Amato S.F., Fitting C., et al. Assessment of ability of murine and human anti-lipid A monoclonal antibodies to bind and neutralize lipopolysaccharide. J Exp Med. 1993;177(1):89-97.
113 Bennett-Guerrero E., Grocott H.P., Levy J.H., et al. A phase II, double-blind, placebo-controlled, ascending-dose study of Eritoran (E5564), a lipid A antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth Analg. 2007;104(2):378-383.
114 Deitch E.A., Berg R. Bacterial translocation from the gut: a mechanism of infection. J Burn Care Rehabil. 1987;8(6):475-482.
115 Deitch E.A. Bacterial translocation of the gut flora. J Trauma. 1990;30(Suppl 12):S184-S189.
116 Deitch E.A. The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch Surg. 1990;125(3):403-404.
117 Mythen M.G., Webb A.R. The role of gut mucosal hypoperfusion in the pathogenesis of post-operative organ dysfunction. Intensive Care Med. 1994;20(3):203-209.
118 Price H.L., Deutsch S., Marshall B.E., et al. Hemodynamic and metabolic effects of hemorrhage in man, with particular reference to the splanchnic circulation. Circ Res. 1966;18(5):469-474.
119 Lundgren O. Physiology of intestinal circulation. In: Martson A.F-G.R., editor. Splanchnic ischemia and multiple organ failure. St. Louis: Mosby; 1989:29-40.
120 Hamilton-Davies C., Mythen M.G., Salmon J.B., et al. Comparison of commonly used clinical indicators of hypovolaemia with gastrointestinal tonometry. Intensive Care Med. 1997;23(3):276-281.
121 American College of Surgeons. Advanced Trauma Life Support. Chicago: American College of Surgeons, 1993.
122 Taylor K.M., Bain W.H., Russell M., et al. Peripheral vascular resistance and angiotensin II levels during pulsatile and no-pulsatile cardiopulmonary bypass. Thorax. 1979;34(5):594-598.
123 Richardson P.D., Withrington P.G. The effects of intraportal injections of noradrenaline, adrenaline, vasopressin and angiotensin on the hepatic portal vascular bed of the dog: Marked tachyphylaxis to angiotensin. Br J Pharmacol. 1977;59(2):293-301.
124 Watkins W.D., Peterson M.B., Kong D.L., et al. Thromboxane and prostacyclin changes during cardiopulmonary bypass with and without pulsatile flow. J Thorac Cardiovasc Surg. 1982;84(2):250-256.
125 Levine F.H., Philbin D.M., Kono K., et al. Plasma vasopressin levels and urinary sodium excretion during cardiopulmonary bypass with and without pulsatile flow. Ann Thorac Surg. 1981;32(1):63-67.
126 Fink M.P., Antonsson J.B., Wang H.L., Rothschild H.R. Increased intestinal permeability in endotoxic pigs. Mesenteric hypoperfusion as an etiologic factor. Arch Surg. 1991;126(2):211-218.
127 Fiddian-Green R.G., Baker S. Predictive value of the stomach wall pH for complications after cardiac operations: Comparison with other monitoring. Crit Care Med. 1987;15(2):153-156.
128 Mythen M.G., Webb A.R. Intra-operative gut mucosal hypoperfusion is associated with increased post-operative complications and cost. Intensive Care Med. 1994;20(2):99-104.
129 Mythen M.G., Webb A.R. Perioperative plasma volume expansion reduces the incidence of gut mucosal hypoperfusion during cardiac surgery. Arch Surg. 1995;130(4):423-429.
130 Bennett-Guerrero E., Panah M.H., Bodian C.A. Automated detection of gastric luminal partial pressure of carbon dioxide during cardiovascular surgery using the Tonocap. Anesthesiology. 2000;92(1):38-45.
131 Martich G.D., Boujoukos A.J., Suffredini A.F. Response of man to endotoxin. Immunobiology. 1993;187(3–5):403-416.
132 Braude A.I., Douglas H., Davis C.E. Treatment and prevention of intravascular coagulation with antiserum to endotoxin. J Infect Dis. 1973;128(Suppl):157-164.
133 Suffredini A.F., Fromm R.E., Parker M.M., et al. The cardiovascular response of normal humans to the administration of endotoxin. N Engl J Med. 1989;321(5):280-287.
134 Cunnion R.E., Parrillo J.E. Myocardial dysfunction in sepsis. Crit Care Clin. 1989;5(1):99-118.
135 Hollenberg S.M., Cunnion R.E., Parrillo J.E. The effect of tumor necrosis factor on vascular smooth muscle. In vitro studies using rat aortic rings. Chest. 1991;100(4):1133-1137.
136 Clauss M., Ryan J., Stern D. Modulation of endothelial cell hemostatic properties by TNF: Insights into the role of endothelium in the host response to inflammatory stimuli. In: Beutler B., editor. Tumor necrosis factors: The molecules and their emerging role in medicine. New York: Raven; 1992:49-63.
137 Arvin B., Neville L.F., Barone F.C., Feuerstein G.Z. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20(3):445-452.
138 Loop F.D., Lytle B.W., Cosgrove D.M., et al. J. Maxwell Chamberlain memorial paper. Sternal wound complications after isolated coronary artery bypass grafting: Early and late mortality, morbidity, and cost of care. Ann Thorac Surg. 1990;49(2):179-186. discussion 186–177
139 Weintraub W.S., Jones E.L., Craver J., et al. Determinants of prolonged length of hospital stay after coronary bypass surgery. Circulation. 1989;80(2):276-284.
140 Sarr M.G., Gott V.L., Townsend T.R. Mediastinal infection after cardiac surgery. Ann Thorac Surg. 1984;38(4):415-423.
141 Miholic J., Hudec M., Domanig E., et al. Risk factors for severe bacterial infections after valve replacement and aortocoronary bypass operations: Analysis of 246 cases by logistic regression. Ann Thorac Surg. 1985;40(3):224-228.
142 Kress H.G., Scheidewig C., Engelhardt W., et al. Prediction and prevention, by immunological means, of septic complications after elective cardiac surgery. Prog Clin Biol Res. 1989;308:1031-1035.
143 Collett B., Alhaq A., Abdullah N.B., et al. Pathways to complement activation during cardiopulmonary bypass. Br Med J (Clin Res Ed). 1984;289(6454):1251-1254.
144 Parker D.J., Cantrell J.W., Karp R.B., et al. Changes in serum complement and immunoglobulins following cardiopulmonary bypass. Surgery. 1972;71(6):824-827.
145 van Oeveren W., Kazatchkine M.D., Descamps-Latscha B., et al. Deleterious effects of cardiopulmonary bypass. A prospective study of bubble versus membrane oxygenation. J Thorac Cardiovasc Surg. 1985;89(6):888-899.
146 Hairston P., Manos J.P., Graber C.D., Lee W.H.Jr. Depression of immunologic surveillance by pump-oxygenation perfusion. J Surg Res. 1969;9(10):587-593.
147 Lee W.H.Jr, Krumhaar D., Fonkalsrud E.W., et al. Denaturation of plasma proteins as a cause of morbidity and death after intracardiac operations. Surgery. 1961;50:29-39.
148 van Velzen-Blad H., Dijkstra Y.J., Schurink G.A., et al. Cardiopulmonary bypass and host defense functions in human beings: I. Serum levels and role of immunoglobulins and complement in phagocytosis. Ann Thorac Surg. 1985;39(3):207-211.
149 Eskola J., Salo M., Viljanen M.K., Ruuskanen O. Impaired B lymphocyte function during open-heart surgery. Effects of anaesthesia and surgery. Br J Anaesth. 1984;56(4):333-338.
150 Markewitz A., Faist E., Lang S., et al. Successful restoration of cell-mediated immune response after cardiopulmonary bypass by immunomodulation. J Thorac Cardiovasc Surg. 1993;105(1):15-24.
151 Welsby I.J., Bennett-Guerrero E., Atwell D., et al. The association of complication type with mortality and prolonged stay after cardiac surgery with cardiopulmonary bypass. Anesth Analg. 2002;94(5):1072-1078. table of contents
152 Huddy S.P., Joyce W.P., Pepper J.R. Gastrointestinal complications in 4473 patients who underwent cardiopulmonary bypass surgery. Br J Surg. 1991;78(3):293-296.
153 Christenson J.T., Schmuziger M., Maurice J., et al. Gastrointestinal complications after coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1994;108(5):899-906.
154 Corwin H.L., Sprague S.M., DeLaria G.A., Norusis M.J. Acute renal failure associated with cardiac operations. A case-control study. J Thorac Cardiovasc Surg. 1989;98(6):1107-1112.
155 Fowler A.A., Hamman R.F., Good J.T., et al. Adult respiratory distress syndrome: Risk with common predispositions. Ann Intern Med. 1983;98(5 Pt 1):593-597.
156 Messent M., Sullivan K., Keogh B.F., et al. Adult respiratory distress syndrome following cardiopulmonary bypass: Incidence and prediction. Anaesthesia. 1992;47(3):267-268.
157 Bennett-Guerrero E: Unpublished data
158 Geha A.S., Sessler A.D., Kirklin J.W. Alveolar-arterial oxygen gradients after open intracardiac surgery. J Thorac Cardiovasc Surg. 1966;51(5):609-615.
159 el-Fiky M.M., Taggart D.P., Carter R., et al. Respiratory dysfunction following cardiopulmonary bypass: Verification of a non-invasive technique to measure shunt fraction. Respir Med. 1993;87(3):193-198.
160 Turnbull K.W., Miyagishima R.T., Gerein A.N. Pulmonary complications and cardiopulmonary bypass: A clinical study in adults. Can Anaesth Soc J. 1974;21(2):181-194.
161 Cavarocchi N.C., Pluth J.R., Schaff H.V., et al. Complement activation during cardiopulmonary bypass. Comparison of bubble and membrane oxygenators. J Thorac Cardiovasc Surg. 1986;91(2):252-258.
162 Jansen N.J., van Oeveren W., van den Broek L., et al. Inhibition by dexamethasone of the reperfusion phenomena in cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1991;102(4):515-525.
163 Inaba H., Kochi A., Yorozu S. Suppression by methylprednisolone of augmented plasma endotoxin-like activity and interleukin-6 during cardiopulmonary bypass. Br J Anaesth. 1994;72(3):348-350.
164 Niazi Z., Flodin P., Joyce L., et al. Effects of glucocorticosteroids in patients undergoing coronary artery bypass surgery. Chest. 1979;76(3):262-268.
165 Miranda D.R., Stoutenbeek C., Karliczek G., Rating W. Effects of dexamethason on the early postoperative course after coronary artery bypass surgery. Thorac Cardiovasc Surg. 1982;30(1):21-27.
166 Jorens P.G., De Jongh R., De Backer W., et al. Interleukin-8 production in patients undergoing cardiopulmonary bypass. The influence of pretreatment with methylprednisolone. Am Rev Respir Dis. 1993;148(4 Pt 1):890-895.
167 Tabardel Y., Duchateau J., Schmartz D., et al. Corticosteroids increase blood interleukin-10 levels during cardiopulmonary bypass in men. Surgery. 1996;119(1):76-80.
168 Tennenberg S.D., Bailey W.W., Cotta L.A., et al. The effects of methylprednisolone on complement-mediated neutrophil activation during cardiopulmonary bypass. Surgery. 1986;100(2):134-142.
169 Toledo-Pereyra L.H., Lin C.Y., Kundler H., Replogle R.L. Steroids in heart surgery: A clinical double-blind and randomized study. Am Surg. 1980;46(3):155-160.
170 Chaney M.A., Durazo-Arvizu R.A., Nikolov M.P., et al. Methylprednisolone does not benefit patients undergoing coronary artery bypass grafting and early tracheal extubation. J Thorac Cardiovasc Surg. 2001;121(3):561-569.
171 Chaney M.A. Corticosteroids and cardiopulmonary bypass: A review of clinical investigations. Chest. 2002;121(3):921-931.
172 Oliver W.C.Jr, Nuttall G.A., Orszulak T.A., et al. Hemofiltration but not steroids results in earlier tracheal extubation following cardiopulmonary bypass: A prospective, randomized double-blind trial. Anesthesiology. 2004;101(2):327-339.
173 Whitlock R.P., Chan S., Devereaux P.J., et al. Clinical benefit of steroid use in patients undergoing cardiopulmonary bypass: A meta-analysis of randomized trials. Eur Heart J. 2008;29(21):2592-2600.
174 Weerwind P.W., Maessen J.G., van Tits L.J., et al. Influence of Duraflo II heparin-treated extracorporeal circuits on the systemic inflammatory response in patients having coronary bypass. J Thorac Cardiovasc Surg. 1995;110(6):1633-1641.
175 Ovrum E., Mollnes T.E., Fosse E., et al. Complement and granulocyte activation in two different types of heparinized extracorporeal circuits. J Thorac Cardiovasc Surg. 1995;110(6):1623-1632.
176 Mangoush O., Purkayastha S., Haj-Yahia S., et al. Heparin-bonded circuits versus nonheparin-bonded circuits: An evaluation of their effect on clinical outcomes. Eur J Cardiothorac Surg. 2007;31(6):1058-1069.
177 Jakob H., Hafner G., Iversen S., et al. Reoperation and the centrifugal pump? Eur J Cardiothorac Surg. 1992;6(Suppl 1):S59-S63.
178 Driessen J.J., Dhaese H., Fransen G., et al. Pulsatile compared with nonpulsatile perfusion using a centrifugal pump for cardiopulmonary bypass during coronary artery bypass grafting. Effects on systemic haemodynamics, oxygenation, and inflammatory response parameters. Perfusion. 1995;10(1):3-12.
179 Tamiya T., Yamasaki M., Maeo Y., et al. Complement activation in cardiopulmonary bypass, with special reference to anaphylatoxin production in membrane and bubble oxygenators. Ann Thorac Surg. 1988;46(1):47-57.
180 Jones H.M., Matthews N., Vaughan R.S., Stark J.M. Cardiopulmonary bypass and complement activation. Involvement of classical and alternative pathways. Anaesthesia. 1982;37(6):629-633.
181 Byrick R.J., Noble W.H. Postperfusion lung syndrome. Comparison of Travenol bubble and membrane oxygenators. J Thorac Cardiovasc Surg. 1978;76(5):685-693.
182 van Oeveren W., Dankert J., Wildevuur C.R. Bubble oxygenation and cardiotomy suction impair the host defense during cardiopulmonary bypass: A study in dogs. Ann Thorac Surg. 1987;44(5):523-528.
183 Videm V., Fosse E., Mollnes T.E., et al. Complement activation with bubble and membrane oxygenators in aortocoronary bypass grafting. Ann Thorac Surg. 1990;50(3):387-391.
184 Croughwell N.D., Newman M.F., Lowry E., et al. Effect of temperature during cardiopulmonary bypass on gastric mucosal perfusion. Br J Anaesth. 1997;78(1):34-38.
185 Menasche P., Peynet J., Lariviere J., et al. Does normothermia during cardiopulmonary bypass increase neutrophil-endothelium interactions? Circulation. 1994;90(5 Pt 2):II275-279.
186 Ohata T., Sawa Y., Kadoba K., et al. Normothermia has beneficial effects in cardiopulmonary bypass attenuating inflammatory reactions. ASAIO J. 1995;41(3):M288-M291.
187 Grigore A.M., Mathew J., Grocott H.P., et al. Prospective randomized trial of normothermic versus hypothermic cardiopulmonary bypass on cognitive function after coronary artery bypass graft surgery. Anesthesiology. 2001;95(5):1110-1119.
188 Grocott H.P., Mackensen G.B., Grigore A.M., et al. Postoperative hyperthermia is associated with cognitive dysfunction after coronary artery bypass graft surgery. Stroke. 2002;33(2):537-541.
189 van Dijk D., Nierich A.P., Jansen E.W., et al. Early outcome after off-pump versus on-pump coronary bypass surgery: Results from a randomized study. Circulation. 2001;104(15):1761-1766.
190 Puskas J.D., Williams W.H., Mahoney E.M., et al. Off-pump vs conventional coronary artery bypass grafting: Early and 1-year graft patency, cost, and quality-of-life outcomes: A randomized trial. JAMA. 2004;291(15):1841-1849.
191 Nathoe H.M., van Dijk D., Jansen E.W., et al. A comparison of on-pump and off-pump coronary bypass surgery in low-risk patients. N Engl J Med. 2003;348(5):394-402.
192 Khan N.E., De Souza A., Mister R., et al. A randomized comparison of off-pump and on-pump multivessel coronary-artery bypass surgery. N Engl J Med. 2004;350(1):21-28.
193 Racz M.J., Hannan E.L., Isom O.W., et al. A comparison of short- and long-term outcomes after off-pump and on-pump coronary artery bypass graft surgery with sternotomy. J Am Coll Cardiol. 2004;43(4):557-564.
194 Shroyer A.L., Grover F.L., Hattler B., et al. On-pump versus off-pump coronary-artery bypass surgery. N Engl J Med. 2009;361(19):1827-1837.
195 Andreasson S., Gothberg S., Berggren H., et al. Hemofiltration modifies complement activation after extracorporeal circulation in infants. Ann Thorac Surg. 1993;56(6):1515-1517.
196 Journois D., Israel-Biet D., Pouard P., et al. High-volume, zero-balanced hemofiltration to reduce delayed inflammatory response to cardiopulmonary bypass in children. Anesthesiology. 1996;85(5):965-976.
197 Warren O., Alexiou C., Massey R., et al. The effects of various leukocyte filtration strategies in cardiac surgery. Eur J Cardiothorac Surg. 2007;31(4):665-676.
198 Gu Y.J., de Vries A.J., Boonstra P.W., van Oeveren W. Leukocyte depletion results in improved lung function and reduced inflammatory response after cardiac surgery. J Thorac Cardiovasc Surg. 1996;112(2):494-500.
199 Lust R.M., Bode A.P., Yang L., et al. In-line leukocyte filtration during bypass. Clinical results from a randomized prospective trial. ASAIO J. 1996;42(5):M819-M822.
200 Mihaljevic T., Tonz M., von Segesser L.K., et al. The influence of leukocyte filtration during cardiopulmonary bypass on postoperative lung function. A clinical study. J Thorac Cardiovasc Surg. 1995;109(6):1138-1145.
201 Davies G.G., Wells D.G., Mabee T.M., et al. Platelet-leukocyte plasmapheresis attenuates the deleterious effects of cardiopulmonary bypass. Ann Thorac Surg. 1992;53(2):274-277.
202 Vamvakas E.C. Meta-analysis of randomized controlled trials investigating the risk of postoperative infection in association with white blood cell-containing allogeneic blood transfusion: The effects of the type of transfused red blood cell product and surgical setting. Transfus Med Rev. 2002;16(4):304-314.
203 van de Watering L.M., Hermans J., Houbiers J.G., et al. Beneficial effects of leukocyte depletion of transfused blood on postoperative complications in patients undergoing cardiac surgery: A randomized clinical trial. Circulation. 1998;97(6):562-568.
204 Llewelyn C.A., Taylor R.S., Todd A.A., et al. The effect of universal leukoreduction on postoperative infections and length of hospital stay in elective orthopedic and cardiac surgery. Transfusion. 2004;44(4):489-500.
205 Dzik W.H., Anderson J.K., O’Neill E.M., et al. A prospective, randomized clinical trial of universal WBC reduction. Transfusion. 2002;42(9):1114-1122.
206 Soeparwata R., Hartman A.R., Frerichmann U., et al. Aprotinin diminishes inflammatory processes. Int J Cardiol. 1996;53(Suppl):S55-S63.
207 Brown J.R., Toler A.W., Kramer R.S., Landis R.C. Anti-inflammatory effect of aprotinin: A meta-analysis. J Extra Corpor Technol. 2009;41(2):79-86.
208 Fergusson D.A., Hebert P.C., Mazer C.D., et al. A comparison of aprotinin and lysine analogues in high-risk cardiac surgery. N Engl J Med. 2008;358(22):2319-2331.
209 Szabo G., Veres G., Radovits T., et al. Effects of novel synthetic serine protease inhibitors on postoperative blood loss, coagulation parameters, and vascular relaxation after cardiac surgery. J Thorac Cardiovasc Surg. 2010;139(1):181-188. discussion 188
210 Lehmann A. Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin Biol Ther. 2008;8(8):1187-1199.
211 Lesslauer W., Tabuchi H., Gentz R., et al. Recombinant soluble tumor necrosis factor receptor proteins protect mice from lipopolysaccharide-induced lethality. Eur J Immunol. 1991;21(11):2883-2886.
212 Abraham E., Glauser M.P., Butler T., et al. p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 Study Group. JAMA. 1997;277(19):1531-1538.
213 Lynn M., Rossignol D.P., Wheeler J.L., et al. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis. 2003;187(4):631-639.
214 Hoffmann H., Markewitz A., Kreuzer E., et al. Pentoxifylline decreases the incidence of multiple organ failure in patients after major cardio-thoracic surgery. Shock. 1998;9(4):235-240.
215 Thabut G., Brugiere O., Leseche G., et al. Preventive effect of inhaled nitric oxide and pentoxifylline on ischemia/reperfusion injury after lung transplantation. Transplantation. 2001;71(9):1295-1300.
216 Boldt J., Brosch C., Lehmann A., et al. Prophylactic use of pentoxifylline on inflammation in elderly cardiac surgery patients. Ann Thorac Surg. 2001;71(5):1524-1529.
217 Boldt J., Brosch C., Piper S.N., et al. Influence of prophylactic use of pentoxifylline on postoperative organ function in elderly cardiac surgery patients. Crit Care Med. 2001;29(5):952-958.
218 Heinze H., Rosemann C., Weber C., et al. A single prophylactic dose of pentoxifylline reduces high dependency unit time in cardiac surgery: A prospective randomized and controlled study. Eur J Cardiothorac Surg. 2007;32(1):83-89.
219 Cagli K., Ulas M.M., Ozisik K., et al. The intraoperative effect of pentoxifylline on the inflammatory process and leukocytes in cardiac surgery patients undergoing cardiopulmonary bypass. Perfusion. 2005;20(1):45-51.
220 Fink M.P. Ringer’s ethyl pyruvate solution: A novel resuscitation fluid for the treatment of hemorrhagic shock and sepsis. J Trauma. 2003;54(Suppl 5):S141-S143.
221 Fink M.P. Ethyl pyruvate: A novel anti-inflammatory agent. Crit Care Med. 2003;31(Suppl 1):S51-S56.
222 Bennett-Guerrero E., Swaminathan M., Grigore A.M., et al. A phase II multicenter double-blind placebo-controlled study of ethyl pyruvate in high-risk patients undergoing cardiac surgery with cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2009;23(3):324-329.
223 Devaraj S., Rogers J., Jialal I. Statins and biomarkers of inflammation. Curr Atheroscler Rep. 2007;9(1):33-41.
224 Schouten O., Boersma E., Hoeks S.E., et al. Fluvastatin and perioperative events in patients undergoing vascular surgery. N Engl J Med. 2009;361(10):980-989.
225 Morgan C., Zappitelli M., Gill P. Statin prophylaxis and inflammatory mediators following cardiopulmonary bypass: A systematic review. Crit Care. 2009;13(5):R165.
226 Baker W.L., Anglade M.W., Baker E.L., et al. Use of N-acetylcysteine to reduce post-cardiothoracic surgery complications: A meta-analysis. Eur J Cardiothorac Surg. 2009;35(3):521-527.
227 Nelson D., Kuppermann N., Fleisher G.R., et al. Recombinant endotoxin neutralizing protein improves survival from Escherichia coli sepsis in rats. Crit Care Med. 1995;23(1):92-98.
228 Pajkrt D., Doran J.E., Koster F., et al. Antiinflammatory effects of reconstituted high-density lipoprotein during human endotoxemia. J Exp Med. 1996;184(5):1601-1608.
229 Palmer J.D., Rifkind D. Neutralization of the hemodynamic effects of endotoxin by polymyxin B. Surg Gynecol Obstet. 1974;138(5):755-759.
230 Roytblat L., Talmor D., Rachinsky M., et al. Ketamine attenuates the interleukin-6 response after cardiopulmonary bypass. Anesth Analg. 1998;87(2):266-271.
231 Bartoc C., Frumento R.J., Jalbout M., et al. A randomized, double-blind, placebo-controlled study assessing the anti-inflammatory effects of ketamine in cardiac surgical patients. J Cardiothorac Vasc Anesth. 2006;20(2):217-222.
232 Debaene B., Goldfarb G., Braillon A., et al. Effects of ketamine, halothane, enflurane, and isoflurane on systemic and splanchnic hemodynamics in normovolemic and hypovolemic cirrhotic rats. Anesthesiology. 1990;73(1):118-124.
233 Stoelting R.K. Pharmacology and physiology in anesthetic practice. Philadelphia: JB Lippincott, 1991.
234 Gelman S., Fowler K.C., Smith L.R. Regional blood flow during isoflurane and halothane anesthesia. Anesth Analg. 1984;63(6):557-565.
235 Conzen P.F., Peter K. Volatile anesthetics and organ blood flow. In: Torri G., Damin G., editors. Update on modern inhalation anesthetics. New York: Worldwide Medical Communications; 1989:29-35.
236 Mythen M.G.W., Webb A.R. There is no correlation between gastric mucosal perfusion (tonometer pHi) and arterial hemoglobin concentration during major surgery. Med Intensiva. 1993;17:S44.
237 Giroir B.P., Beutler B. Effect of amrinone on tumor necrosis factor production in endotoxic shock. Circ Shock. 1992;36(3):200-207.
238 Bouter H., Schippers E.F., Luelmo S.A., et al. No effect of preoperative selective gut decontamination on endotoxemia and cytokine activation during cardiopulmonary bypass: A randomized, placebo-controlled study. Crit Care Med. 2002;30(1):38-43.