Syndromes with Craniosynostosis
Evaluation and Treatment
• Mutations in Craniosynostosis and Craniosynostosis Syndromes
• Neurologic Aspects of Craniosynostosis
• Effects of Midface Deficiency on the Upper Airway
• Surgical Approach to Craniosynostosis Sydromes
• Staging of Reconstruction for Crouzon Syndrome
General Considerations
Craniosynostosis, which is the premature fusion of cranial sutures, affects approximately 1 in 2500 children. Patients may present with a wide range of phenotypic and functional deformities.94 Virchow’s law from 1851 states that the premature fusion of a cranial vault suture results in growth arrest perpendicular to the affected suture and compensatory growth parallel to the fused suture.16,164,269 This principle is generally applicable to single sutures; multisutural synostosis—such as the combination of sagittal and lambdoid synostosis (coup de sabre) or a cloverleaf skull—can assume more complex patterns of cranial morphology.43,46,47 In addition to the type of affected suture, the eventual head shape depends on the timing and order of cranial suture fusion. Craniosynostosis may be of prenatal or perinatal onset, or it may occur during infancy.14,74 The earlier synostosis occurs, the more dramatic the effect on subsequent cranial growth and development; the later synostosis occurs, the less effect on cranial growth and development.44
Active pediatric neurosurgical services generally see a characteristic frequency, gender predilection, and phenotypic presentation for sutural involvement.6,15,55,125,133,154,161,193,232,238,243,278,283 Sagittal synostosis occurs most commonly, with a higher incidence among boys than girls. Patients typically present with a boat-shaped skull (i.e., scaphocephaly) in combination with midline ridging in the posterior half of the skull. Metopic synostosis is currently the second most frequent form of synostosis. Presentations occur along a spectrum that ranges from metopic ridging to the presence of a triangular-shaped skull (i.e., trigonocephaly). It should be noted that metopic ridging is common during infancy and childhood and that it is not necessarily associated with a diagnosis of craniosynostosis. Coronal synostosis occurs next most frequently; girls slightly predominate or, in some surveys, boys and girls have equivalent frequency.46 Unicoronal synostosis is marked by frontal plagiocephaly, whereas bicoronal synostosis is characterized by a short and wide skull (i.e., brachycephaly). Multiple synostoses of various types are less common than coronal synostosis. Lambdoid synostosis occurs with least frequency, and it is marked by occipital plagiocephaly.44
Craniosynostosis is etiologically heterogeneous and pathogenetically variable. Overall, 8% of cases have a familial component, and this is generally marked by an autosomal dominant inheritance pattern. Specifically, pedigrees are familial in 14.4%, 6%, and 5.6% of coronal, sagittal, and metopic synostoses, respectively.44 Although some pedigrees show synostosis of a specific suture, others may show fusion of different sutures in affected relatives of the same family. Different chromosomal aberrations have been linked to craniosynostosis, with variable penetrance. In some conditions the penetrance is high, such as with dup(3q), del(7p), del(9p), and del(11q). In other cases, penetrance can be very low, such as with dup(5p), del(6)(22q22.2-q23.1), del(8q), and dup(15q).44
Many syndromes are associated with craniosynostosis, and well over 100 are known.44 It is important to accurately diagnose syndromal patients for three reasons.22 First, most syndromes with craniosynostosis affect not only the cranial vault but also the cranial base and the midface. Deficiencies in these skeletal sites, which are variable in degree yet commonly serious, must be addressed as part of the staged reconstructive approach. Second, syndromes are often genetic and familial, thus necessitating proper counseling. Third, depending on the experience of the particular surgeon, the molecular diagnosis should be made. Often, the patient may be referred for molecular diagnosis and counseling to avoid the possibility of litigation.37,43,46,47
Primary forms of craniosynostosis are most common and include single- and multi-sutural synostosis. Some conditions, however, result in secondary synostosis. These include hyperthyroidism; rickets; mucopolysaccharidoses, such as Hurler syndrome and Morquio syndrome; hematologic disorders, such as thalassemia and sickle cell anemia; teratogens, such as diphenylhydantoin, retinoids, valproate, and aminopterin; and certain malformations, such as holoprosencephaly and microcephaly.44
Mutations in Craniosynostosis and Craniosynostosis Syndromes
Mutations responsible for craniosynostosis have been identified in FGFR1, FGFR2, FGFR3, TWIST, MSX2, EFNB1, RAB23, EFNA4, POR, and ALPL.44 These include Apert syndrome (two common FGFR2 mutations; Ser252Trp, Pro253Arg), Crouzon syndrome (more than 30 FGFR2 mutations), Pfeiffer syndrome (more than 30 FGFR2 mutations), are known and at least 6 of them are the same as those found with Crouzon syndrome; one Pfeiffer syndrome (mutation on FGFR1).* In addition, there are some cases of FGFR2 mutations with isolated coronal synostosis as well as with some other forms of non-syndromic synostosis. A single known FGFR2 mutation is known for Jackson-Weiss syndrome.1,41,182 Although this disorder is distinctly uncommon, affected families are often large, with variable phenotypic expression. Beare-Stevenson cutis gyrate syndrome involves one of two possible mutations in the transmembrane domain of FGFR2.37,42
Muenke syndrome is the most common craniosynostosis syndrome that is currently known. It involves a single mutation on FGFR3, and it is characterized most commonly by unilateral coronal synostosis and less commonly by bicoronal synostosis. About 6% of affected patients have macrocephaly without synostosis, and cloverleaf skull has been reported in some instances.37,42 Therefore, all patients with coronal synostosis should be tested for this very common FGFR3 mutation; if they are negative, they should be tested for FGFR2.
Crouzonodermoskeletal syndrome is characterized by one specific FGFR3 mutation. The name indicates its characteristic features: either a Crouzonoid or cloverleaf skull appearance; acanthosis nigricans; and a decreased interpediculate distance that is radiographically present but not clinically significant. Serious decreased interpediculate distance is a feature of the most common mutation for achondroplasia, which is only 11 amino acids away from the FGFR3 mutation for Crouzonodermoskeletal syndrome. To call this disorder “Crouzon syndrome with acanthosis” is unwarranted, because only one FGFR3 mutation is responsible for all cases; this is in contrast with Crouzon syndrome, which involves more than 30 different FGFR2 mutations.38
Rarely, patients with achondroplasia and hypochondroplasia (both of which involve mutations on FGFR3) may also have craniosynostosis (there have been three reported cases of hypochondroplasia and two reported cases of achondroplasia). Saethre–Chotzen syndrome has TWIST mutations inside or outside of the coding region that result in haploinsufficiency.48 Craniofrontonasal syndrome is caused by heterozygous loss-of-function mutations in EFNB1. EFNA4 mutations rarely cause non-syndromal coronal synostosis. Carpenter syndrome, which is an autosomal recessive disorder, is caused by RAB23 mutations. Boston-type craniosynostosis, which is an autosomal dominant disorder caused by MSX2 mutations, presents with variable expressivity (coronal synostosis, frontal depression, or cloverleaf skull). Antley–Bixler syndrome is caused by POR mutations, and infantile hypophosphatasia is caused by ALPL mutation.44,184
Complex Craniosynostosis
Background
Complex craniosynostosis, which involves the fusion of multiple cranial sutures, occurs in about 5% of non-syndromic cases.47 Two thirds of cases involve two affected sutures, whereas one third of cases involve more than two affected sutures.203 As the number of affected sutures increases, so does the risk of intracranial hypertension and associated mental deficiency. As demonstrated by Renier and colleagues, the incidence of intracranial hypertension rises from 14% to 47% when going from single to several affected sutures.228,226,227,229,230 In addition, increasing sutural involvement can be accompanied by worsened phenotypic severity. In a series reported by Chumas and colleagues,34 59% of cases of sagittal and bilateral lambdoid synostosis had an acute angulation of the posterior skull, which is known in French as coup de serpe and means “cut with a scythe.” In a similar fashion, marked anterior dysmorphism with frontal bone hypoplasia was found in 23% of cases of metopic and bilateral coronal synostosis.
Cloverleaf skulls, which represent the extremes of phenotypic severity, are pathogenetically variable (Fig. 30-1).30,204 Synostosis may involve the coronal, lambdoid, and metopic sutures, which are marked by bulging of the cerebrum through an open sagittal suture or, in some cases, through patent squamosal sutures. Synostosis of the sagittal and squamosal sutures with cerebral eventration through a widely patent anterior fontanel may also be observed. A trilobular skull shape may also occur, with complete synostosis of all cranial sutures in some cases or with widely patent sutures and no craniosynostosis at birth in other instances.36,51 In addition to being pathogenetically variable, cloverleaf skulls are also etiologically heterogeneous. This condition most commonly occurs with type 2 thanatophoric dysplasia (i.e., in about 40% of cases) with a specific FGFR3 mutation, but stillborn status or an early demise during very early infancy precludes treatment. Isolated cloverleaf skull occurs in about 20% of cases. It also occurs in about 15% of cases of type 3 Pfeiffer syndrome, and, although the prognosis is guarded, these patients can be treated aggressively.45,48
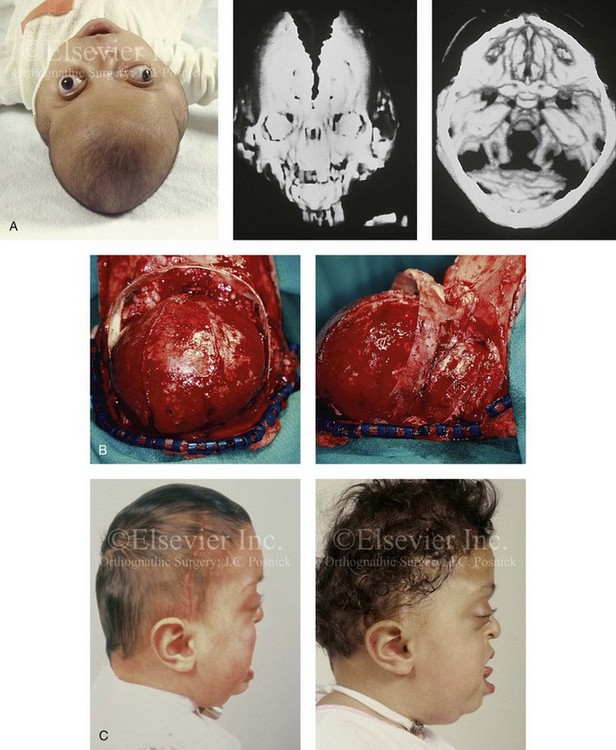
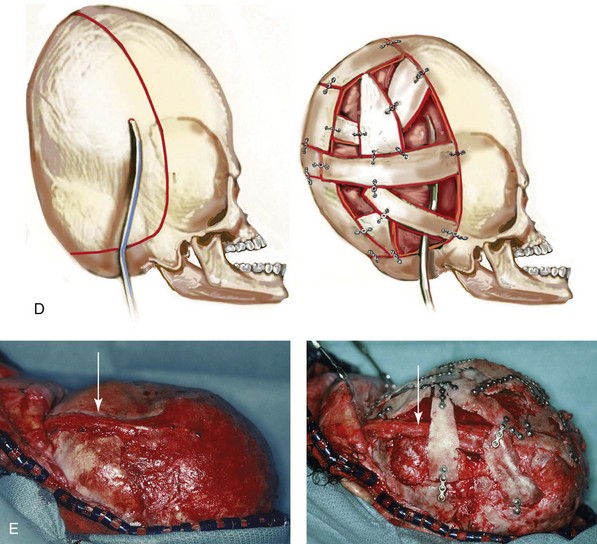
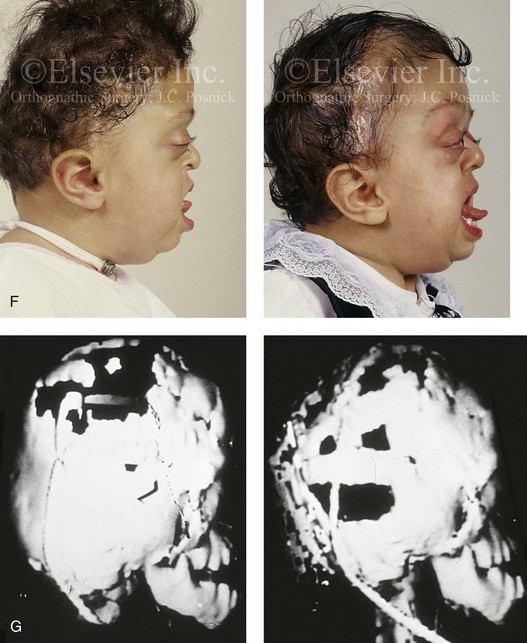
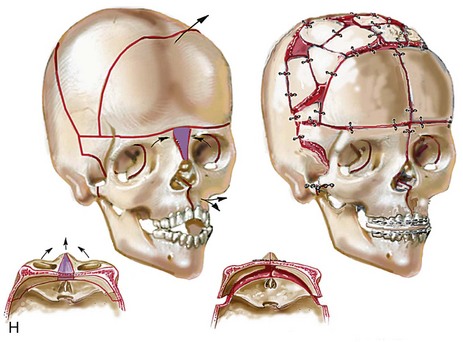
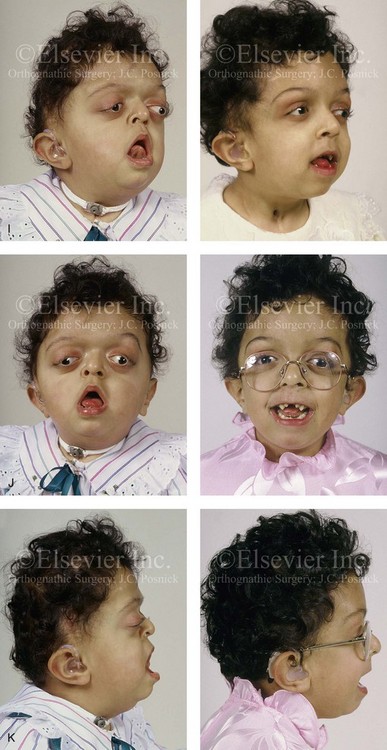
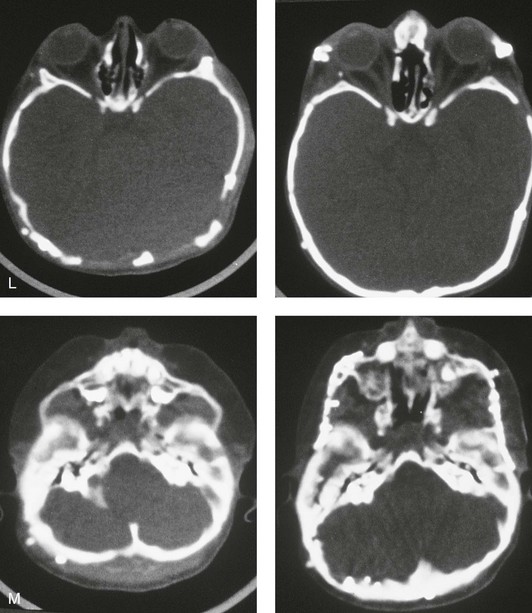
Figure 30-1 A child with a severe form of cloverleaf skull anomaly. At 10 months of age, she was referred to this surgeon and underwent first-stage cranial vault and upper orbital decompression with reshaping. She then required a ventriculoperitoneal shunt for the management of hydrocephalus. When the patient was  years old, posterior cranial vault decompression with reshaping to increase the intracranial volume was performed. When she was
years old, posterior cranial vault decompression with reshaping to increase the intracranial volume was performed. When she was  years old, facial bipartition osteotomies in combination with anterior cranial vault reshaping and advancement were carried out. After the cranial vault and facial bipartition procedures, the patient’s airway improved, and it was possible to remove the tracheostomy tube. The cranial vault reshaping expanded the intracranial volume, thereby providing more space for the brain. The midface advancement improved the eye proptosis as well as the patient’s ability to chew and articulate speech. As part of the staged reconstruction, she will require orthognathic surgery in combination with orthodontic treatment at the time of early skeletal maturity. A, Frontal facial and computed tomography (CT) scan views at 10 months of age. B, Intraoperative views at 10 months of age after craniotomy and the removal of the cranial vault and the upper orbits. The “bandeau” is in place (3 cm advancement) before the reconstruction of the overlying cranial vault. C, Profile views at
years old, facial bipartition osteotomies in combination with anterior cranial vault reshaping and advancement were carried out. After the cranial vault and facial bipartition procedures, the patient’s airway improved, and it was possible to remove the tracheostomy tube. The cranial vault reshaping expanded the intracranial volume, thereby providing more space for the brain. The midface advancement improved the eye proptosis as well as the patient’s ability to chew and articulate speech. As part of the staged reconstruction, she will require orthognathic surgery in combination with orthodontic treatment at the time of early skeletal maturity. A, Frontal facial and computed tomography (CT) scan views at 10 months of age. B, Intraoperative views at 10 months of age after craniotomy and the removal of the cranial vault and the upper orbits. The “bandeau” is in place (3 cm advancement) before the reconstruction of the overlying cranial vault. C, Profile views at  and then at
and then at  years of age with flattened posterior cranial vault and severe midface deficiency. D, Illustration of flattened posterior cranial vault with the ventriculoperitoneal shunt in place. Illustration after posterior cranial vault reshaping is also shown. E, Intraoperative side view with the patient in the prone position before and after the craniotomy and the reshaping of the posterior cranial vault. Arrows point to the ventriculoperitoneal shunt, which remains intact and deep to the skull reconstruction. F, Profile view at
years of age with flattened posterior cranial vault and severe midface deficiency. D, Illustration of flattened posterior cranial vault with the ventriculoperitoneal shunt in place. Illustration after posterior cranial vault reshaping is also shown. E, Intraoperative side view with the patient in the prone position before and after the craniotomy and the reshaping of the posterior cranial vault. Arrows point to the ventriculoperitoneal shunt, which remains intact and deep to the skull reconstruction. F, Profile view at  years of age, before and after posterior cranial vault reconstruction. G, CT scan views before and just after posterior cranial vault reshaping. H, Illustration before facial bipartition. The second illustration indicates the planned reconstruction. I, Oblique facial views before and after facial bipartition reconstruction. The tracheostomy has been removed. J, Facial views before and after facial bipartition reconstruction. K, Profile views before and after facial bipartition reconstruction. L, Axial CT views through the midorbits before and after reconstruction. M, Axial CT views through the zygomatic arches before and after reconstruction. A, B, C (right), D, E, F, H (left), I, K, L, M, From Posnick JC: The craniofacial dysostosis syndromes: secondary management of craniofacial disorders, Clin Plast Surg 24:429-446, 1997.
years of age, before and after posterior cranial vault reconstruction. G, CT scan views before and just after posterior cranial vault reshaping. H, Illustration before facial bipartition. The second illustration indicates the planned reconstruction. I, Oblique facial views before and after facial bipartition reconstruction. The tracheostomy has been removed. J, Facial views before and after facial bipartition reconstruction. K, Profile views before and after facial bipartition reconstruction. L, Axial CT views through the midorbits before and after reconstruction. M, Axial CT views through the zygomatic arches before and after reconstruction. A, B, C (right), D, E, F, H (left), I, K, L, M, From Posnick JC: The craniofacial dysostosis syndromes: secondary management of craniofacial disorders, Clin Plast Surg 24:429-446, 1997.
Plagiocephaly is defined as the asymmetric distortion of the skull. Well-known types include synostotic anterior plagiocephaly (unilateral coronal synostosis), synostotic posterior plagiocephaly (unilateral lambdoid synostosis), deformational anterior plagiocephaly, and deformational posterior plagiocephaly.146,148,268 Among these types, deformational posterior plagiocephaly is common, whereas unilateral lambdoid synostosis is rare. Deformational posterior plagiocephaly can be caused by intrauterine factors such as hypotonia, fetal positioning, and prematurity. This can produce asymmetric flattening of the occiput that becomes favored by infants sleeping on their backs, thereby exaggerating the plagiocephaly. Deformational posterior plagiocephaly has also increased dramatically since the 1992 “Back to Sleep” campaign by the American Academy of Pediatrics, which calls for supine infant sleeping to reduce the risk of sudden infant death syndrome.44 Finally, rare forms of plagiocephaly can be produced by unilateral frontosphenoidal synostosis and unilateral frontozygomatic synostosis.44
Apert Syndrome
Apert syndrome is characterized by craniosynostosis, midface deficiency, symmetric syndactyly of the hands and feet, and other abnormalities* (Figs. 30-2 through 30-5). Most cases are associated with brachycephaly secondary to bicoronal synostosis. Megalencephaly and increased cranial height are characteristic of Apert syndrome; affected children typically display head circumference, length, and weight that are above the normal 50th percentile. Benign distortion ventriculomegaly is also a feature of Apert syndrome, and patent sutures and synchondroses (except for coronal synostosis) are present as well. The frequency of progressive hydrocephalus is much lower than in it is with Crouzon and Pfeiffer syndromes. Central nervous system anomalies may include hypoplasia of the corpus callosum, agenesis of the corpus callosum, agenesis of the septum pellucidum, cavum septum pellucidum, dorsally displaced hippocampi and hippocampal gyri, hypoplastic white matter, and heterotopic gray matter.46,48
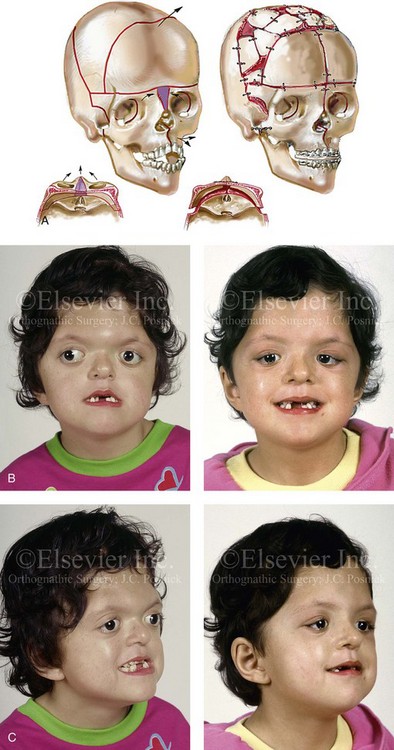
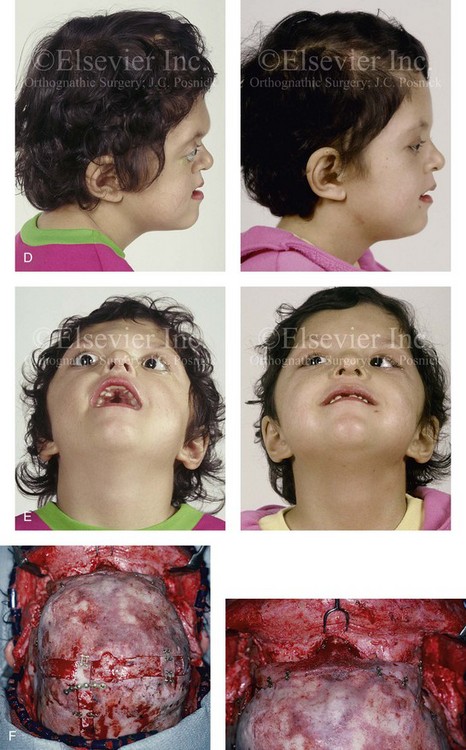
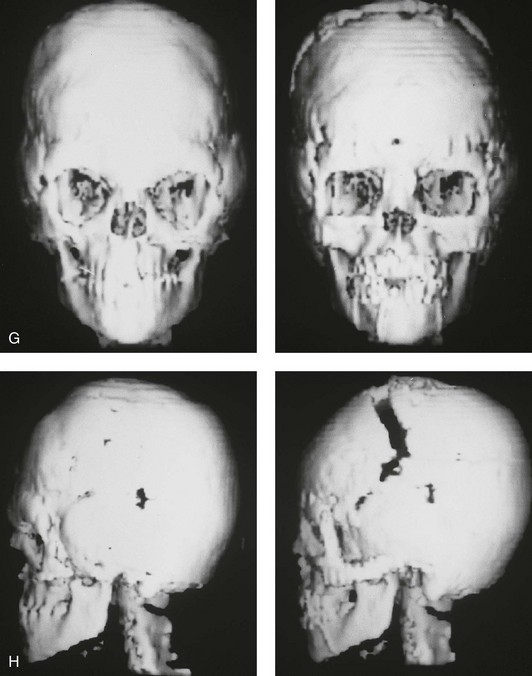
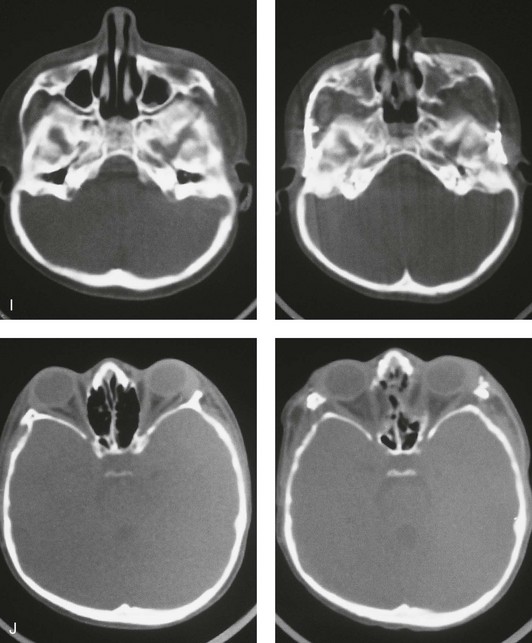
Figure 30-2 A child who was born with Apert syndrome underwent a bilateral lateral canthal advancement procedure when she was 6 weeks old; this was carried out by a neurosurgeon working independently. When she was 18 months old, she returned with turricephaly and a constricted anterior cranial vault that required further cranio–orbital decompression and reshaping. When she was 5 years old, she underwent anterior cranial vault and facial bipartition osteotomies with reshaping. As part of the staged reconstruction, she will require orthognathic surgery and orthodontic treatment during her teenage years. A, Illustrations of craniofacial morphology before and after the facial bipartition osteotomies and reconstruction. B, Frontal views before and after facial bipartition reconstruction. C, Oblique facial views before and after facial bipartition reconstruction. D, Profile views before and after facial bipartition reconstruction. E, Worm’s-eye views before and after facial bipartition reconstruction. F, Intraoperative views after facial bipartition and anterior cranial vault reconstruction. G and H, Computed tomography scan views before and early after reconstruction. I, Axial computed tomography scan views through the zygomas before and after reconstruction. J, Axial computed tomography scan views through the midorbits before and after reconstruction indicating relief of proptosis.
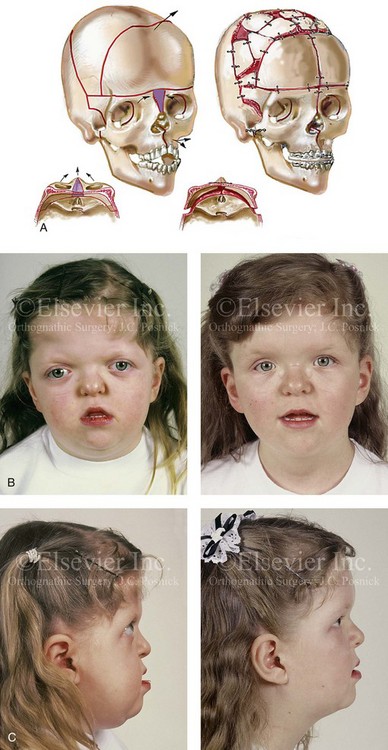
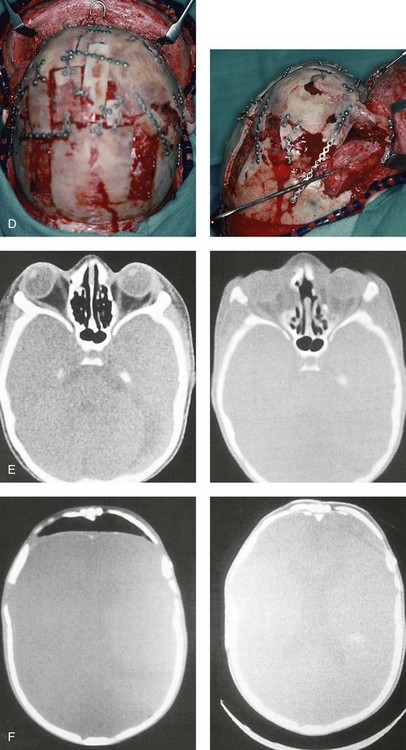
Figure 30-3 A 5-year-old girl with Apert syndrome who underwent lateral canthal advancements when she was 6 months old and in the care of another surgeon. She then presented to this surgeon with residual craniofacial deformities that required anterior cranial vault and facial bipartition osteotomies with reshaping. She will require orthognathic surgery and orthodontic treatment during her teenage years to complete the reconstruction. A, Illustrations of preoperative craniofacial morphology. The planned cranial vault and facial bipartition osteotomies and the planned reshaping are also shown. B, Frontal facial views before and after anterior cranial vault and facial bipartition reconstruction. C, Profile views before and after facial bipartition reconstruction. D, Intraoperative views after anterior cranial vault and facial bipartition reconstruction. E, Axial computed tomography scan views through the midorbits before and after reconstruction that demonstrate improvement in orbital hypertelorism and orbital depth with diminished eye proptosis. F, Axial computed tomography scan views through the cranial vault 1 week after facial bipartition (note the dead space in the retrofrontal region) and at 1 year (note that the initial retrofrontal dead space has been resolved by brain expansion). A (right), C (left), E, From Posnick JC: Craniofacial dysostosis. Staging of reconstruction management of the midface deformity, Neurosurg Clin North Am 2:683-702, 1991.
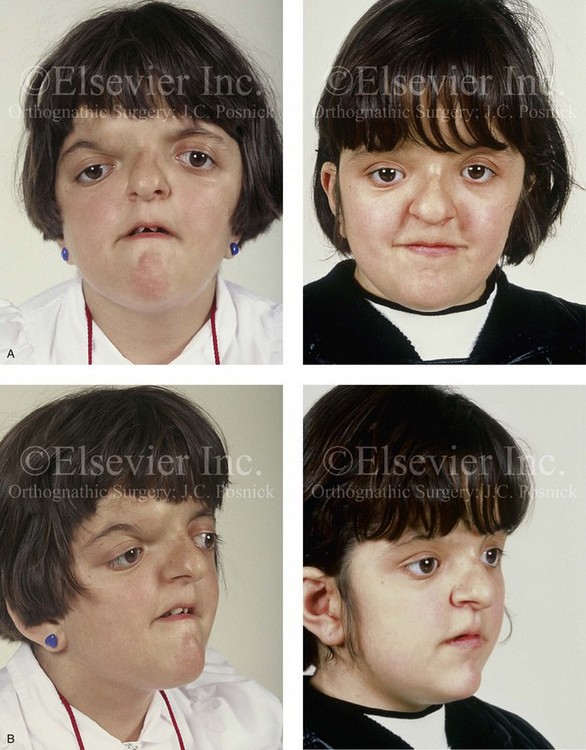
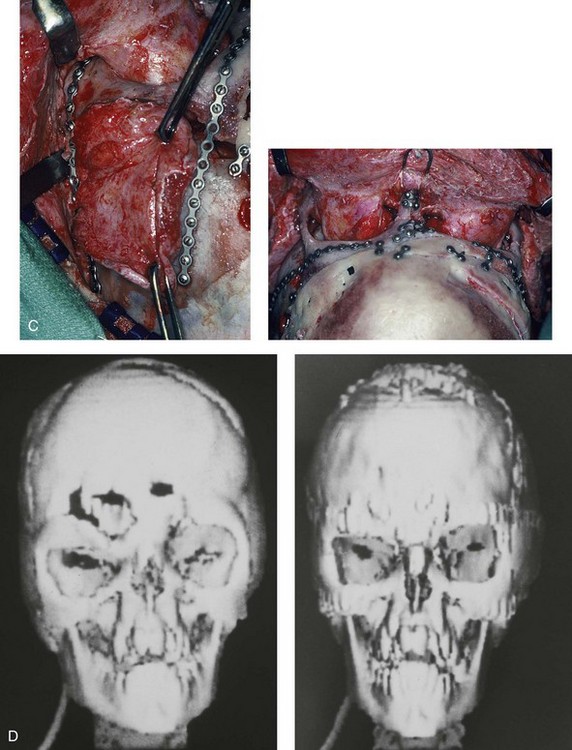
Figure 30-4 An 8-year-old girl who was born with Apert syndrome. She had undergone cranio–orbital surgery on two occasions and an attempted Le Fort III osteotomy at another institution. The previous procedures had resulted in complications, including infection and traumatic encephalocele. She was referred to this surgeon with cranial vault dysplasia, orbital dystopia, hypertelorism, midface deficiency, and skull defects. She then underwent anterior cranial vault, facial bipartition, and additional segmental orbital osteotomies with reshaping and reconstruction. She will require orthodontic treatment and orthognathic surgery during her teenage years to complete the reconstruction. A, Frontal views before and after anterior cranial vault and facial bipartition reconstruction. B, Oblique facial views before and after facial bipartition reconstruction. C, Intraoperative views after anterior cranial vault and facial bipartition reconstruction. Encephalocele and skull defects have also been repaired. D, Computed tomography scan views before and early after reconstruction. From Posnick JC: Craniosynostosis: surgical management in infancy. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1839.
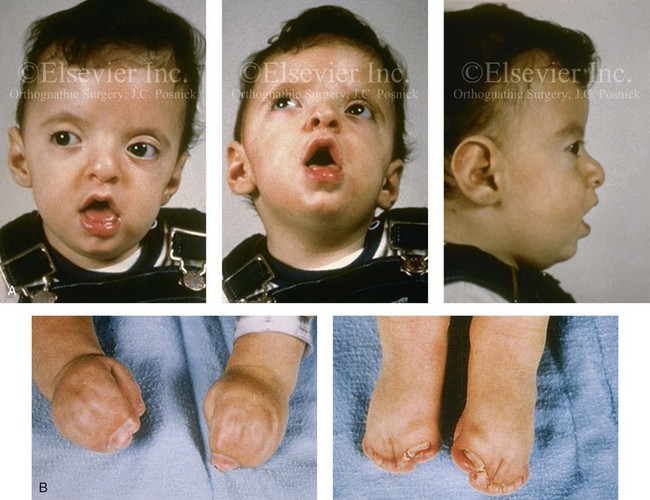
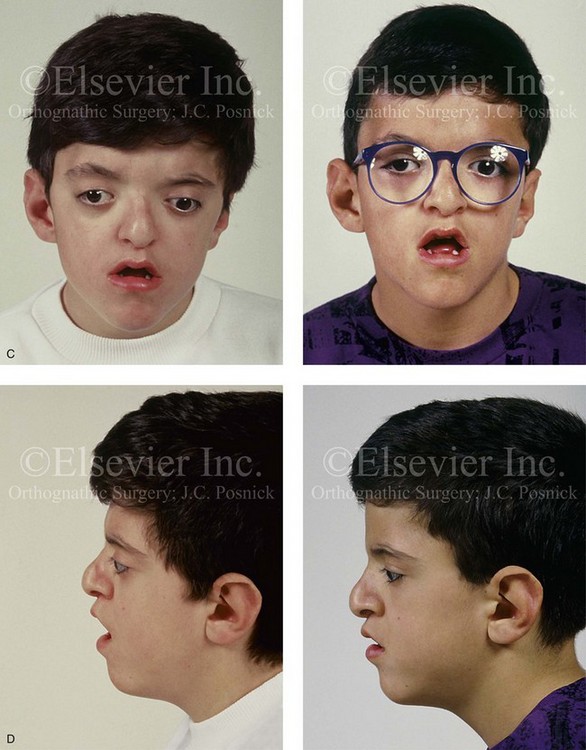
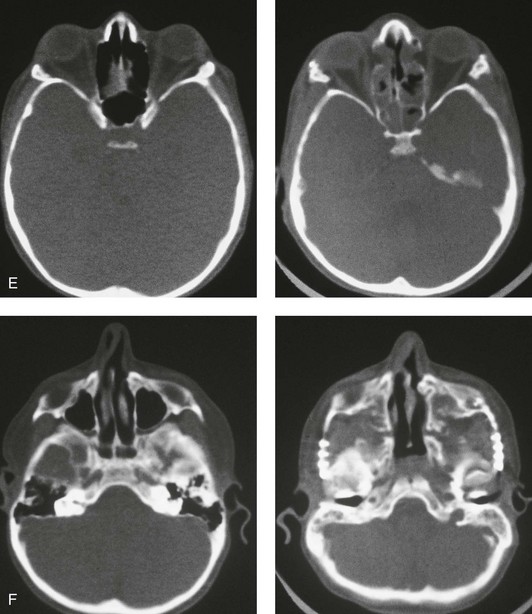
Figure 30-5 A child born with a mild form of Apert syndrome. When he was 10 years old, he was referred to this surgeon and underwent anterior cranial vault and monobloc osteotomies with advancement. He will require orthognathic surgery combined with orthodontic treatment to complete the reconstruction. A, Facial views during infancy. B, Views of the hands and feet that indicate complex compound syndactyly. C, Frontal facial views before and after anterior cranial vault and monobloc osteotomies with advancement. D, Profile views before and after monobloc reconstruction. E, Axial computed tomography scan views through the midorbits before and after reconstruction indicating relief of proptosis. F, Axial computed tomography scan views through the zygomatic arches before and after reconstruction.
Skeletal abnormalities with Apert syndrome include symmetric syndactyly of the hands and feet; abnormal shoulders with significant limitation of motion; and abnormalities of the elbows, hips, knees, rib cage, and spine.46 It should be carefully noted that the radiologic interpretation of the hands, feet, and cervical spine as being affected by so-called “progressive fusion” is very misleading. In actuality, there is a failure of cartilage segmentation during embryonic and fetal life, so these abnormalities are preset (ab initio). These failures in cartilage segmentation cannot be readily observed radiographically at birth. When they do become evident radiographically after birth, they are said to be a “progressive abnormality,” which they are not. The same can be said for the cervical vertebrae,46 as 68% of these patients have cervical abnormalities, particularly involving C5 and C6. Before considering a surgical procedure, magnetic resonance imaging of the brain, radiography of the cervical spine, and the assessment of cardiovascular (10% of patients) and genitourinary (9.6% of patients) anomalies should be carried out.44,46,48
Patients with Apert syndrome also suffer from reduced nasopharyngeal dimensions and reduced patency of the posterior choanae; these features pose the risks of respiratory embarrassment, obstructive sleep apnea (OSA), cor pulmonale, and even sudden death. During infancy, patients may be trialed on a nasopharyngeal airway, but tracheostomy may be the only definitive treatment. Clinicians must always maintain a high index of suspicion for OSA and monitor patients for snoring or an unusual amount of daytime somnolence. When either of these is apparent, patients should be referred to a sleep center for proper diagnosis and treatment. It should be carefully noted that, although patients with Apert syndrome very commonly have OSA, the excessive sweating that occurs with this syndrome is not a sign of OSA but rather a consequence of the increased number of sweat and sebaceous glands that accompany the syndrome. Even those patients who have surgical advancement of the midface in combination with continuous positive airway pressure (CPAP) will still have excessive sweating of the head at night and of the hands all the time. Another consequence of an increased number of sweat and sebaceous glands is a high frequency of conglobate acne during adolescence, often with extension to the forearm, the thighs, and the buttocks. The acne vulgaris of Apert syndrome suggests exquisite end-organ responsiveness to steroid hormones.46
Ocular findings include hypertelorism, proptosis (often asymmetric), and down-slanting palpebral fissures. The presence of V-pattern strabismus, which is marked by divergent upgaze and esotropic downgaze, is common and secondary to structural abnormalities of the extraocular muscles (i.e., the absence of the superior rectus muscle). Hyperopia, myopia, and astigmatism can also be present. Strabismus and significant refractive errors can sometimes cause amblyopia. Proptosis in Apert syndrome may require tarsorrhaphies to prevent exposure keratitis.131
Hearing loss occurs in 90% of these patients, with 80% of cases resulting from conductive pathology. Inner-ear anomalies are found in all patients; these most commonly present as dilated vestibules, malformed semicircular canals, and cochlear dysplasia.285 Otitis media is common, and it is possibly related to the presence of cleft palate and accompanying eustachian tube dysfunction.
Oral and maxillofacial anomalies can also be present. The palate can be highly arched and constricted with a median furrow in up to 94% of patients. The hard palate is often shorter than normal, whereas the soft palate is longer and thicker than normal. Clefting of the soft palate occurs in 41% of patients, and a bifid uvula is present in 35%. Dental anomalies include severely delayed eruption (68%), ectopic eruption (50%), and shovel-shaped incisors (30%). Dental crowding is more severe in the maxilla than in the mandible. Common dental deformities include anterior open bite (73%), posterior crossbite (63%), and mandibular overjet (81%).46,48
Crouzon Syndrome
Crouzon syndrome is characterized by craniosynostosis, maxillary hypoplasia, shallow orbits, and ocular proptosis.* (Figs. 30-6 through 30-11). Most cases are associated with brachycephaly secondary to bicoronal synostosis; however, trigonocephaly, scaphocephaly, and cloverleaf skull have also been noted. Cranial malformation depends on the order and rate of progression of the sutural synostosis. The cranial volume of patients with Crouzon syndrome is much smaller than that found in patients with Apert syndrome. Central nervous system findings include shunted hydrocephalus (25%), chronic tonsillar herniation (72%), jugular foramen stenosis with venous obstruction (30%), seizures (10.5%), frequent headaches (29%), and mental deficiency (3%).46,48 As a result of severe maxillary deficiency, patients should be monitored for snoring and excess daytime somnolence. If this is severe during infancy, tracheostomy may be the only option. Older children should be initially worked up with a polysomnographic study. Treatment options range from CPAP therapy to midface advancement on the basis of the severity level.46
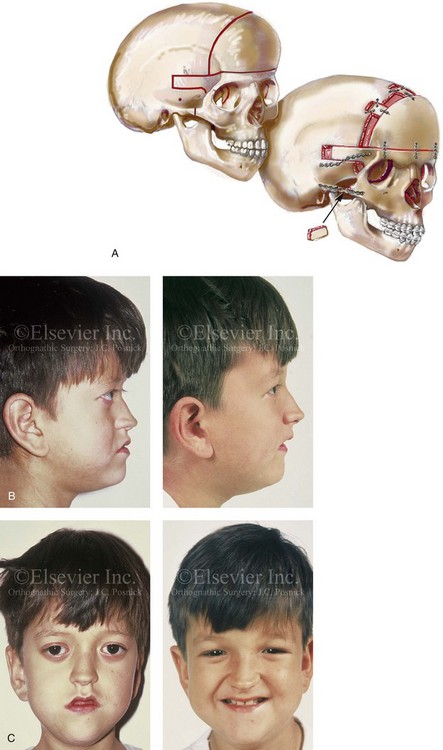
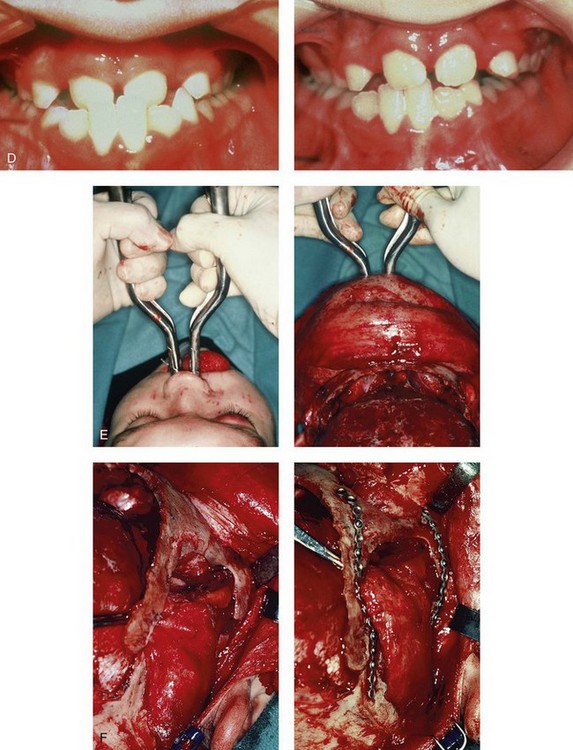
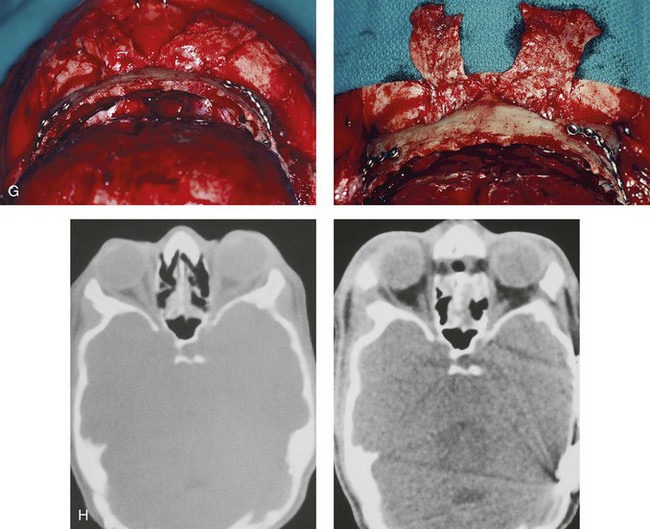
Figure 30-6 An 8-year-old boy who was born with Crouzon syndrome and who underwent a first-stage cranio–orbital procedure when he was 6 weeks old by a neurosurgeon who was working independently. He was referred to this surgeon and then underwent anterior cranial vault and monobloc osteotomies with advancement. A, Illustration of craniofacial morphology before and after anterior cranial vault and monobloc osteotomies with advancement. B, Profile views before and after reconstruction. C, Frontal views before and after anterior cranial vault and monobloc reconstruction. D, Occlusal views before and after reconstruction. Note the improvement but without a full correction of the malocclusion; this will await orthognathic correction during the patient’s teenage years. E, Intraoperative view of disimpaction after monobloc osteotomy with the use of nasomaxillary forceps. F, Intraoperative view of monobloc showing tenon extension and zygomatic arch advancement before and after plate and screw fixation. G, After monobloc advancement and fixation, dead space in the retrofrontal region with communication into the nasal cavity can be seen. Pericranial flaps are elevated from under the surface of the scalp for the closure of cranio–nasal communication. H, Axial computed tomography scan views through the midorbits before and after reconstruction. Reconstruction resulted in increased intraorbital depth and decreased proptosis. A, B, C,D, E, H, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1889.
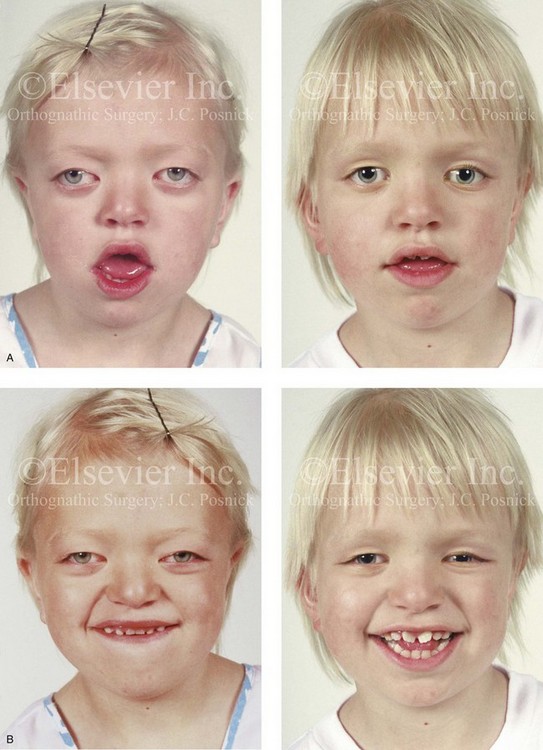
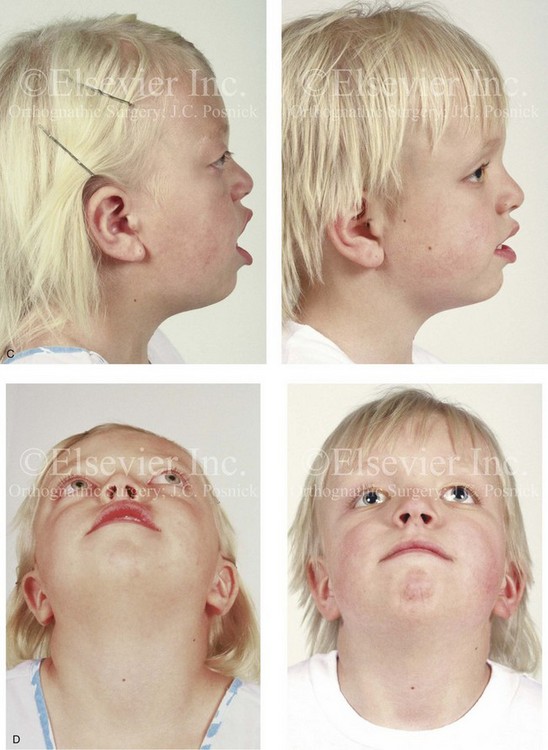
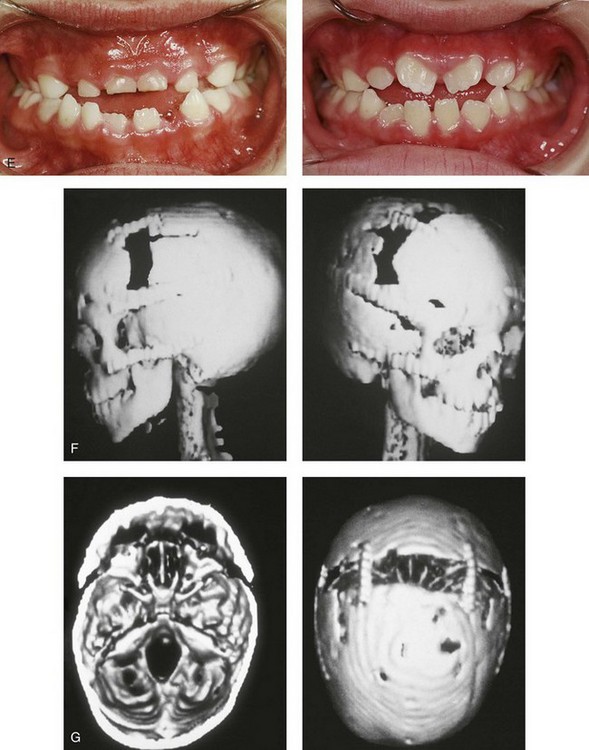
Figure 30-7 A 6-year-old girl who was born with Crouzon syndrome and who underwent anterior cranial vault and monobloc osteotomies with advancement. A, Frontal facial views in repose before and after anterior cranial vault and monobloc reconstruction. B, Frontal views with smile before and after monobloc reconstruction. C, Profile views before and after monobloc reconstruction. D, Worm’s-eye views before and after monobloc reconstruction. E, Occlusal views before and after reconstruction. Note the improvement but without the correction of the malocclusion; this will await orthognathic surgery during the patient’s teenage years. F and G, Computed tomography scan views immediately after reconstruction that indicate advancement with increased volume in the anterior cranial vault and orbits. A, C, D, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
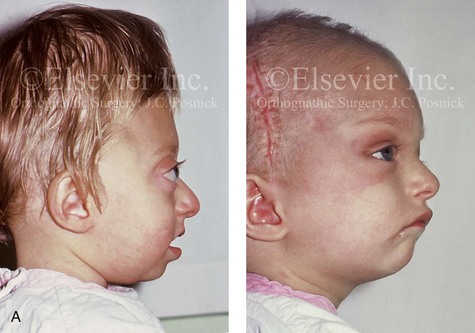
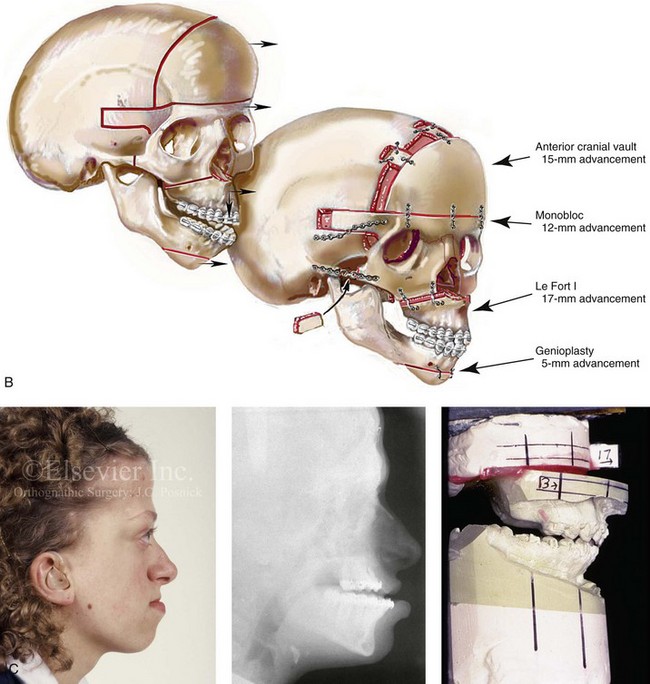
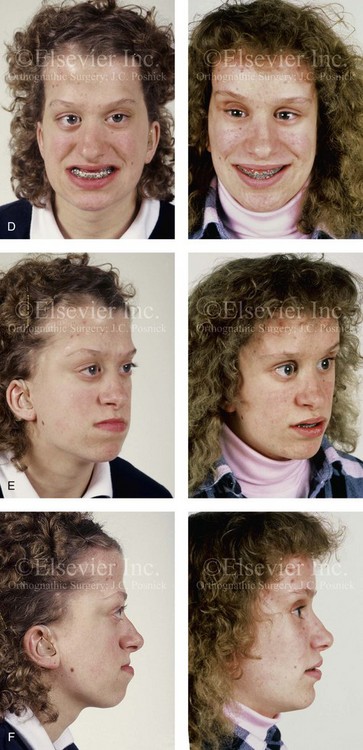
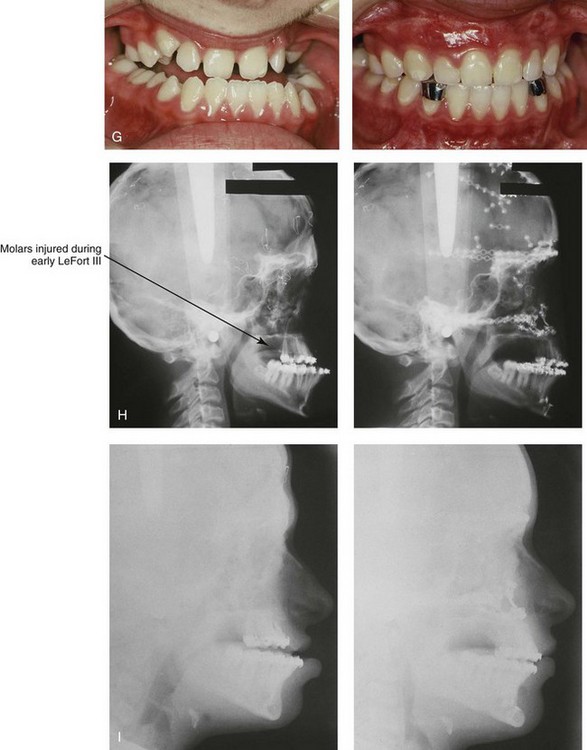
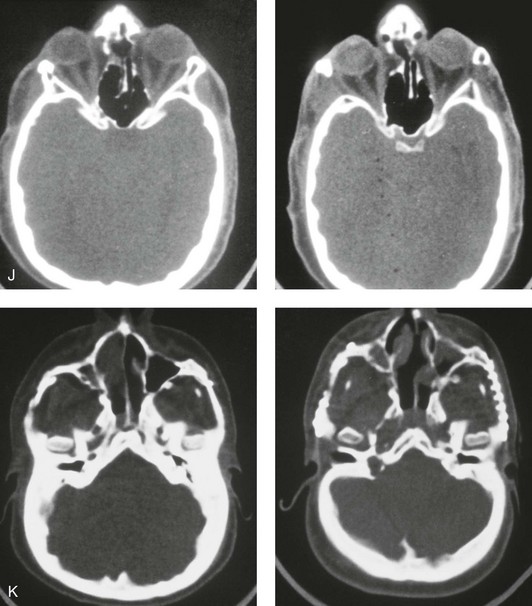
Figure 30-8 A child who was born with Crouzon syndrome and who underwent bilateral coronal suture release when she was 3 months old. Additional craniotomy and cranial vault reshaping were completed when she was 9 months old. When the patient was 2 years old, she underwent a Le Fort III midface osteotomy and forehead advancement procedure through an intracranial approach. She presented to this surgeon when she was 14 years old with residual deformity for which she underwent simultaneous anterior cranial vault, monobloc, Le Fort I, and chin osteotomies with differential advancements. A, Profile facial views at 1 year of age and at 2 years of age after Le Fort III and anterior cranial vault advancement. B, Illustration of planned and completed anterior cranial vault, monobloc, Le Fort I, and chin osteotomies. C, Profile view and lateral cephalometric radiograph before surgery. Articulated dental casts after model planning that indicate that 17-mm advancement is required at the occlusal level. The amount of advancement required at the supraorbital ridge level was just 12 mm. Note the absence of permanent molars in maxilla; this is the result of the Le Fort III osteotomy carried out when the patient was 2 years old. The pterygomaxillary disjunction destroyed the developing teeth. D, Frontal facial views before and after reconstruction. E, Oblique facial views before and after reconstruction. F, Profile views before and after reconstruction. G, Occlusal views before and after reconstruction and orthodontic treatment. H, Lateral cephalometric radiographs before and after reconstruction. I, Soft-tissue lateral cephalometric radiographs before and after reconstruction. J, Axial computed tomography scan views through the midorbits before and after reconstruction confirm relief of proptosis. K, Axial computed tomography scan views through the zygomatic arches before and after reconstruction. A, B (right), D, G, H (left), I, J, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
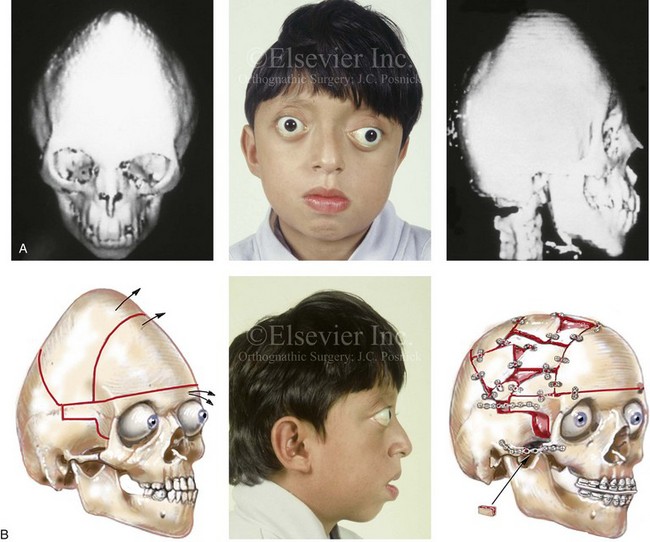
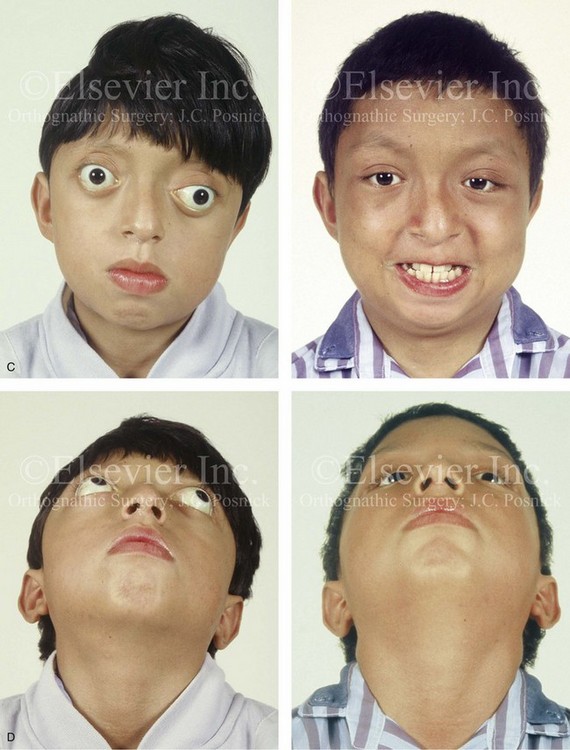
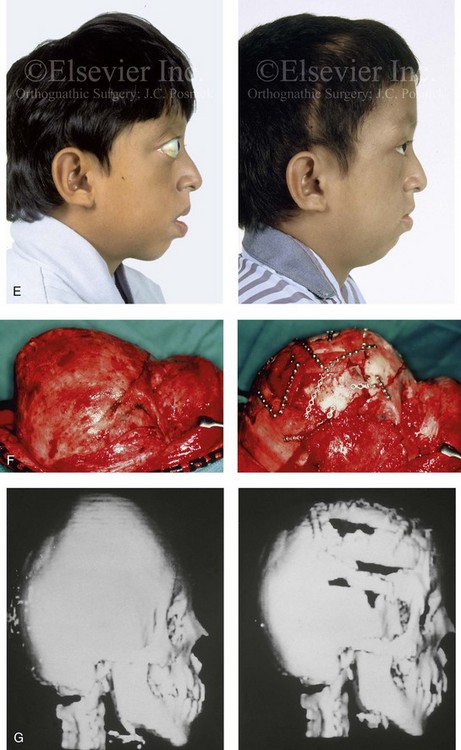
Figure 30-9 A 12-year-old boy with unrepaired Crouzon syndrome was referred to this surgeon for evaluation. He underwent total cranial vault and monobloc osteotomies with reshaping and advancement. A, Facial and computed tomography scan views before surgery. B, Profile facial view before surgery. Illustrations of the patient’s craniofacial morphology with planned osteotomy locations indicated. A second illustration is shown after the proposed osteotomies were completed, with reshaping and advancement. C, Frontal facial views before and after reconstruction. D, Worm’s-eye views before and after reconstruction. E, Profile views before and after reconstruction. F, Intraoperative views before and after total cranial vault and monobloc osteotomy with reshaping and advancement. G, Computed tomography scan views before and immediately after reconstruction. A (right), B (left, right), C (right), D (right), E (right), F, G, From Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.
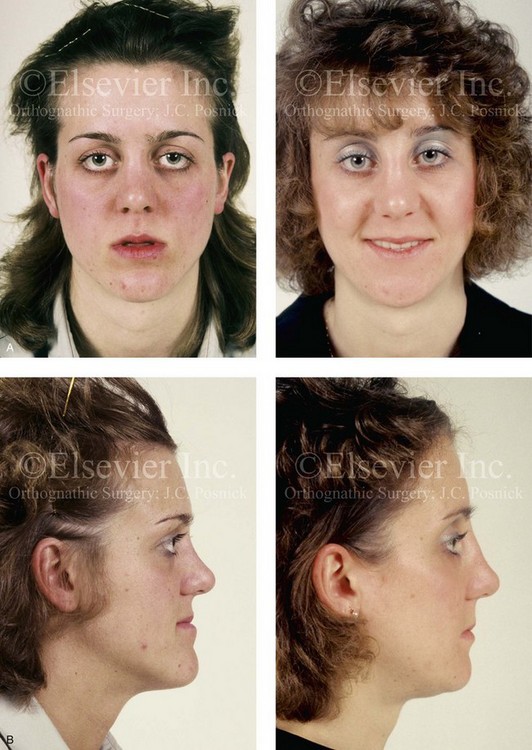
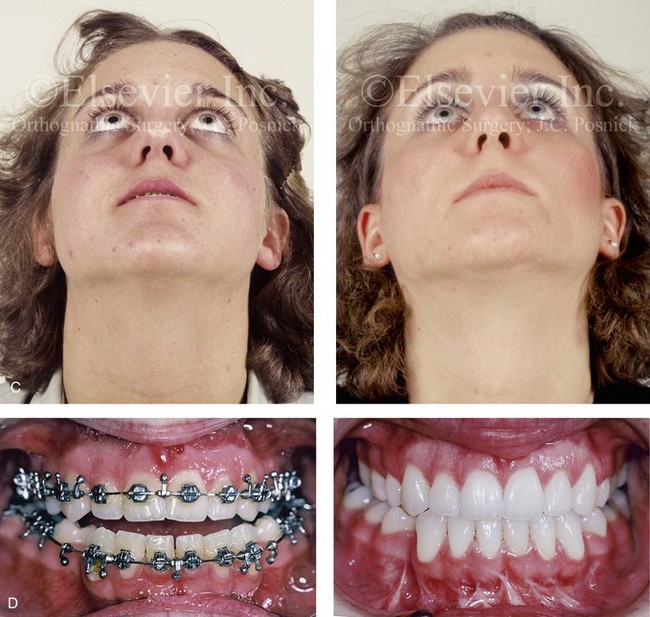
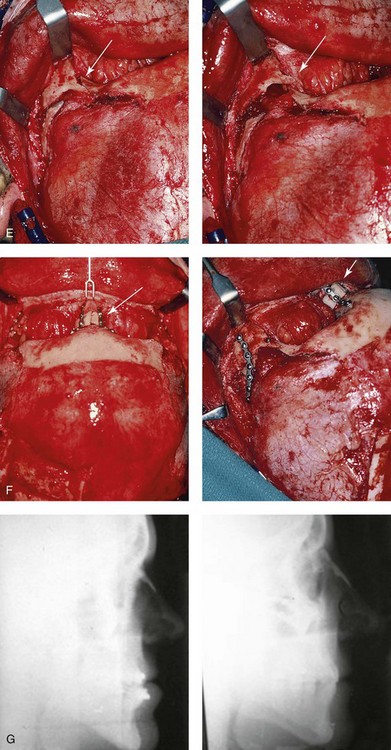
Figure 30-10 A 15-year-old girl with Crouzon syndrome that is characterized by mild to moderate midface deficiency with retrusion of the infraorbital rims, the zygomatic buttresses, and the maxilla. The midface hypoplasia results in increased scleral show, nasal obstruction, and malocclusion. The cranial vault and the upper orbits have normal morphology. Orthodontic camouflage treatment was in progress, including mandibular bicuspid extractions with retraction. The patient was referred to this surgeon for evaluation and treatment. She then underwent a Le Fort III extracranial osteotomy with advancement. A, Frontal facial views before and after reconstruction. B, Profile views before and after reconstruction. C, Worm’s-eye views before and after reconstruction. D, Occlusal views before and after reconstruction. E, Intraoperative view. Note the potential for unsightly step-off at the lateral orbital rim after advancement. F, Cranial bone graft is placed in the frontonasal region. Note the unavoidable lengthening of the nose and the potential for the flattening of the nasofrontal angle that occurs as part of the Le Fort III advancement. G, Lateral cephalometric radiographs before and after reconstruction. A, B (right), C, D, E, F (left), G, from Posnick JC: Craniosynostosis: surgical management of the midface deformity. In Bell WH, ed: Orthognathic and reconstructive surgery, vol 3, Philadelphia, 1992, W.B. Saunders, p 1888.

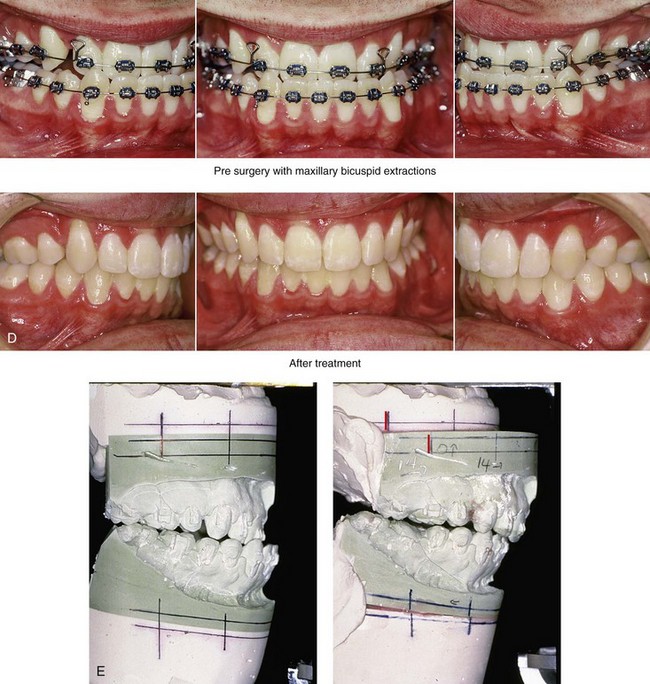
Figure 30-11 A 19-year-old boy who was born with Crouzon syndrome. When he was 11 years old, he underwent a Le Fort III osteotomy with advancement via an extracranial approach by another surgeon. He presented to this surgeon during his late teenage years with asymmetric and dystopic orbits, zygomatic asymmetry, a retrusive upper jaw, an asymmetric lower jaw, and a vertically long chin. He underwent a combined orthodontic (upper bicuspid extractions) and surgical approach. The procedures included Le Fort I osteotomy (horizontal advancement); bilateral sagittal split ramus osteotomies (correction of asymmetry); and osseous genioplasty (vertical reduction and horizontal advancement). He also underwent the reopening of a coronal scalp incision with the harvesting of split cranial grafts and the recontouring and augmentation of the orbits and the zygomas. A, Frontal views before and after reconstruction. B, Oblique facial views before and after reconstruction. C, Profile views before and after reconstruction. D, Occlusal views before and after reconstruction. E, Articulated dental casts that indicate analytic model planning.
Cervical vertebral anomalies are found in 25% of patients with Crouzon syndrome, and all involve C2 and C3. Anomalies should be assessed before the patient undergoes anesthesia for any craniofacial procedure. Patients with Crouzon syndrome already have problematic airways as a result of their relatively inflexible necks; thus, cervical anomalies will compound the issue.46
Ophthalmologic abnormalities include hypertelorism, pronounced ocular proptosis, exotropia, exposure keratitis, poor vision, and optic atrophy. Luxation of the eye globes may be observed in some instances. Although emergency reduction followed by tarsorrhaphies may be necessary, some patients can luxate and reduce all by themselves.131
Oral findings include lateral palatal swellings (50%), obligatory mouth breathing (32%), bifid uvula (9%), and cleft palate (9%). The maxillary dental arch is shortened and associated with a posterior crossbite, dental crowding, and ectopic eruption of the maxillary first molars. Anterior open bite, mandibular overjet, and crowding of the mandibular anterior teeth are also common.46
Pfeiffer Syndrome
Pfeiffer syndrome is characterized by craniosynostosis; midface deficiency; broad thumbs, great toes, or both; brachydactyly; variable soft-tissue syndactyly; and other anomalies (Figs. 30-12 through 30-15).188,231 Three clinical subtypes have been proposed. Type 1 is representative of the aforementioned description. Type 2 is characterized by cloverleaf skull, elbow ankylosis or synostosis, and a cluster of unusual anomalies in addition to the Type 1 characteristics. Type 3 involves a very short cranial base, severe ocular proptosis, elbow ankylosis or synostosis, and an assortment of unusual anomalies. Types 2 and 3 also include an increased risk for neurodevelopmental difficulties. A favorable outcome can be achieved in some cases with aggressive medical and surgical management; however, normal outcome is not the rule, and neurodevelopmental outcome and life expectancy prognoses remain guarded in most cases.44 Although these subtypes are useful, they have limited nosologic status, and some clinical overlap does occur.
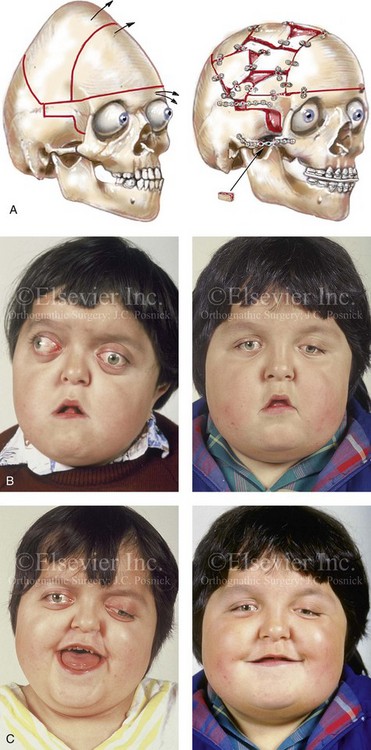
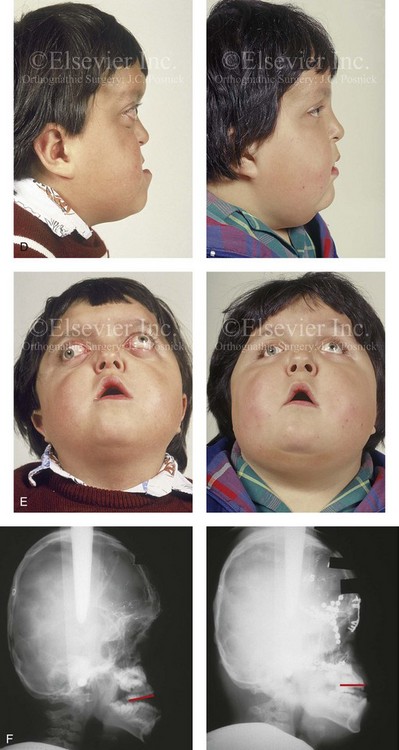
Figure 30-12 A 7-year-old boy who was born with Pfeiffer syndrome was referred for evaluation. He previously had undergone bilateral lateral canthal advancements when he was 3 months old; these were performed by a neurosurgeon who was working independently. The patient then required the placement of a ventriculoperitoneal shunt. He presented to this surgeon with total cranial vault dysplasia, orbital dystopia, and midface deficiency. He underwent cranial vault and monobloc osteotomies with reshaping and advancement. A, An illustration of the proposed cranial vault and monobloc osteotomies and reconstruction is also shown. B, Frontal facial views in repose before and after cranial vault and monobloc reconstruction. C, Frontal facial views with smile before and after reconstruction. D, Profile views before and after reconstruction. E, Worm’s-eye views before and after reconstruction. F, Lateral cephalometric radiographs before and after reconstruction. A (left), B, D, from Posnick JC: Craniofacial dysostosis: staging of reconstruction and management of the midface deformity, Neurosurg Clin North Am 2:683-702, 1991.
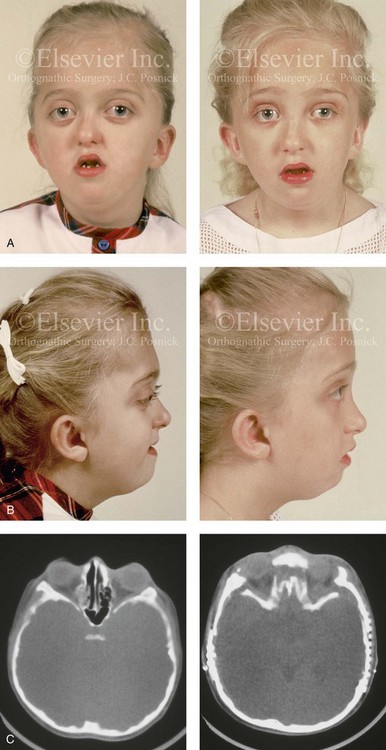
Figure 30-13 A 6-year-old girl who was born with Pfeiffer syndrome. She had undergone cranio–orbital reshaping earlier during her childhood. She presented to this surgeon with a constricted anterior cranial vault, orbital dystopia, and midface deficiency. She underwent anterior cranial vault and monobloc osteotomies with reshaping and advancement. A, Frontal facial views before and after reconstruction. B, Profile views before and after reconstruction. She still requires orthodontic treatment and orthognathic surgery, which are planned for her teenage years. C, Axial computed tomography scan views through the midorbits before and after reconstruction.

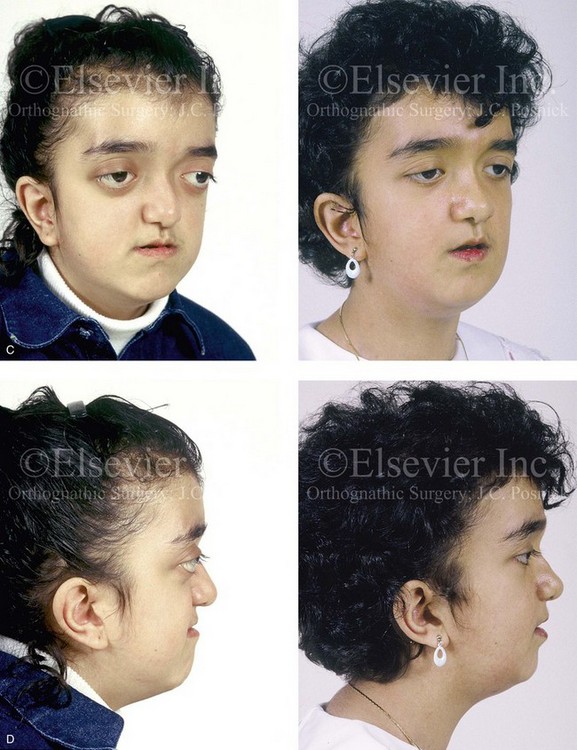
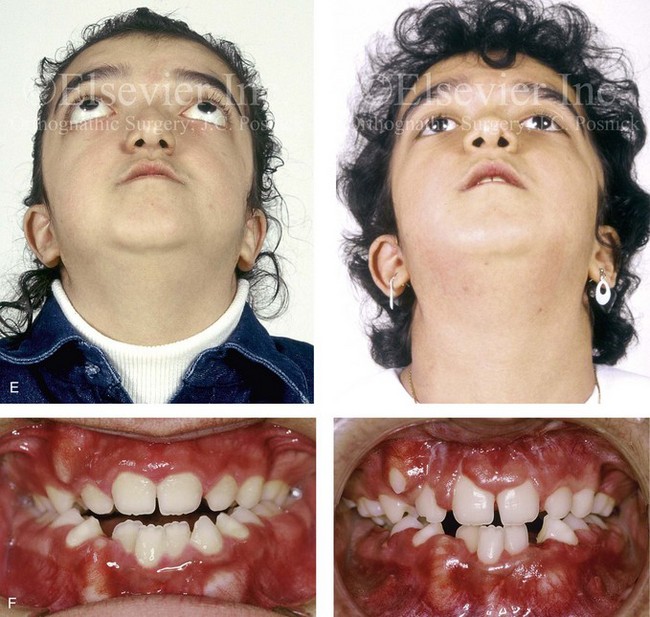
Figure 30-14 A 12-year-old girl who was born with Pfeiffer syndrome was referred to this surgeon for evaluation. She underwent anterior cranial vault and facial bipartition osteotomies with reshaping. She is shown before and after reconstruction. Quantitative analysis of the presenting deformity on the basis of computed tomography scan views confirmed the abnormal cranial vault length (85% of normal), medial orbital wall length (68% of normal), and zygomatic arch length (83% of normal) as well as the extent of globe protrusion (140% of normal). All measurements demonstrated horizontal deficiency in the upper face and the midface. In addition, the anterior interorbital distance (136% of normal), the mid-interorbital distance (129% of normal), the lateral orbital distance (121% of normal), and the intertemporal distance (132% of normal) all indicated a degree of upper-face hypertelorism. The patient then underwent anterior cranial vault and facial bipartition osteotomies for reconstruction. She achieved improved horizontal facial depth, and the upper face hypertelorism was also improved. A, Frontal facial views in repose before and after reconstruction. B, Frontal facial views with smile before and after reconstruction. C, Oblique facial views before and after reconstruction. D, Profile views before and after reconstruction. E, Worm’s-eye views before and after reconstruction. F, Occlusal views before and after reconstruction. Note that the occlusion is improved but that the patient will require orthognathic surgery and orthodontic treatment during her teenage years. From Posnick JC, Waitzman A, Armstrong D, Pron G: Monobloc and facial bipartition osteotomies: quantitative assessment of presenting deformity and surgical results based on computed tomography scans, J Oral Maxillofac Surg 53:358-367, 1995.
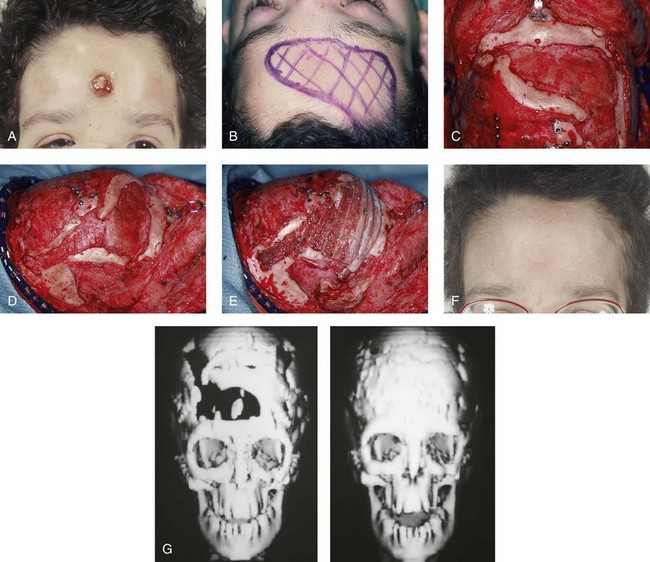
Figure 30-15 A 12-year-old boy with Noonan syndrome. When he was 5 years old, he underwent bilateral total orbital osteotomies with medial translocation for the correction of orbital hypertelorism; this was carried out by another surgeon. When he was 12 years old, he presented to this surgeon with a retruded irregular forehead, shallow orbits, residual proptosis, and midface deficiency. He underwent anterior cranial vault and monobloc osteotomies with horizontal advancement. An occult cerebral spinal fluid leak with drainage through the nose occurred. Eventually, there was erosion through the mid-forehead skin and resorption of a segment of the anterior and lateral cranial vault. One year after the monobloc procedure, the patient was taken back to the operating room for the repair and closure of the cerebral spinal fluid fistula and a cranioplasty that consisted of autogenous split-rib grafts. The cranial vault reconstruction was successful, without further infection or long-term sequelae. A, Frontal view of the upper face 6 months after the initial surgery, with a cerebral spinal fluid fistula through central forehead. B and C, Intraoperative views of the anterior cranial vault documenting the region of bone loss. D, Intraoperative lateral view that shows the area of bone loss. E, Intraoperative lateral view after autogenous split-rib graft reconstruction. F, Frontal view of the upper face 1 year after dura repair and the split-rib reconstruction of the skull defects. G, Computed tomography scan views before and after successful split-rib graft cranioplasty.
Craniofacial features include brachycephaly as a result of bicoronal synostosis, hypertelorism, ocular proptosis, and midface deficiency.44 Distortion ventriculomegaly, progressive hydrocephalus, and cerebellar herniation are common. Progressive hydrocephalus is much more common among patients with Pfeiffer and Crouzon syndromes than it is among those with Apert syndrome. Gyral abnormalities have been observed in some cases.44 Vertebral anomalies occur in 70% of patients, particularly at C2 and C3, but such anomalies may involve other cervical vertebrae as well as the lumbar vertebrae in some cases. 46 As in Apert and Crouzon syndrome, OSA can affect patients with Pfeiffer syndrome. For a further understanding of diagnosis and therapy, please refer to the relevant discussions in the sections of this chapter about the Apert and Crouzon syndromes. Other anomalies have been noted in some cases, including cardiovascular defects, gastrointestinal issues, and genitourinary anomalies.44
Saethre–Chotzen Syndrome
Saethre–Chotzen syndrome is characterized by a heterogeneous phenotypic presentation that involves craniosynostosis, a low-set frontal hairline, facial asymmetry, ptosis of the eyelids, a deviated nasal septum, brachydactyly, partial soft-tissue syndactyly of the second and third fingers, and various skeletal anomalies.4,33,40,234 Craniosynostosis is facultative and not obligatory (i.e., some patients do not have it). When it is present, the time of onset and the degree of craniosynostosis can be quite variable. Brachycephaly is found most commonly, and this is followed by synostotic anterior plagiocephaly. Frontal bossing, parietal bossing, and occipital flattening can also accompany the cranial deformity. Large late-closing fontanelles and large parietal foramina have also been reported. Ptosis of the eyelids, hypertelorism, and strabismus are common. The ears may be low set, small, and posteriorly angulated, and mild conductive hearing loss is common. Oral anomalies include a narrow or highly arched palate, a cleft palate on occasion, malocclusion, and supernumerary teeth.40
Carpenter Syndrome
Carpenter syndrome is characterized by craniosynostosis (commonly, but not always); preaxial polydactyly of the feet; brachydactyly, clinodactyly, or both; congenital heart defects (these are seen in 33% of patients), such as ventricular septal defect, atrial septal defect, patent ductus arteriosus, pulmonary stenosis, and tetralogy of Fallot; short stature, with a height that is usually below the 25th centile; obesity; and mental deficiency in some cases.28,39 Craniosynostosis involves the sagittal and lambdoid sutures first, with the coronal suture being the last to close. The head is usually broad, but it is variable in shape, and asymmetry may be a feature. Other features include dystopia canthorum, down-slanting palpebral fissures, epicanthal folds, low-set ears, and a short neck. The mandible may be hypoplastic, and the palate may be narrow or highly arched.39
Neurologic Aspects of Craniosynostosis
Generally, craniosynostosis tends to restrict intracranial volume. The earlier synostosis occurs, the more dramatic the effect on subsequent cranial growth and development. The later synostosis occurs, the less the effect on cranial growth and development. Growth restriction also worsens with increasing sutural involvement; complex craniosynostosis with two or more affected sutures demonstrates higher growth restriction as compared with single sutural involvement. There are two exceptions to this observation: scaphocephaly and Apert syndrome. With scaphocephaly, mechanical forces produce a large head circumference; with Apert syndrome, brain weights have been shown to be above the 97th centile, regardless of age.46,48
Mental deficiency is correlated with the number of fused sutures, and it occurs more frequently with two or more sutural involvements than with single-suture synostosis. In addition, mental deficiency occurs more commonly with coronal synostosis than with other single-suture craniosynostoses. With single-suture craniosynostoses (other than coronal), mental deficiency may be more attributed to a brain malformation rather than to growth restriction. This may occur in patients with trigonocephaly and even in some cases of sagittal synostosis.25
In the section about Apert syndrome, a distinction was made between benign megalencephaly or distortion megalencephaly and progressive hydrocephalus. The mechanism of hydrocephalus may vary. Although communicating hydrocephalus with obstruction at the level of the basal cisterns is most common, aqueductal stenosis is fairly frequent. Chronic tonsillar herniation occurs in 73% of patients with Crouzon syndrome as compared with only 1.9% of patients with Apert syndrome.62 Premature synostosis of the lambdoid suture may be responsible for this finding. It has also been suggested that the obstruction of the venous drainage of the brain may play a role by narrowing the jugular foramina. Some patients are able to compensate well for their hydrocephalus.25
Clinicians must maintain a high index of suspicion for hydrocephalus in all patients with craniosynostosis, particularly those with complex or syndromic types.77,91,108,170,174 Among the syndromic craniosynostoses, shunted hydrocephalus is found in 26.6% of patients with Crouzon syndrome and in 27.8% of patients with Pfeiffer syndrome but in only 6.5% to 8% of patients with Apert syndrome.46 Increased intracranial pressure in craniosynostosis may be the result of a mismatch in cranial volume to brain growth or of hydrocephalus. Increased intracranial pressure is found most commonly in patients with multiple suture synostoses.9,26,49,58,122,191,222,226,227–230,245,266 However, recent studies have indicated that increased pressure may also occur in patients with single-suture synostosis. In one report, 3 out of 18 children with scaphocephaly had increased intracranial pressure during 12- to 24-hour recordings.265
When pressure is low grade, clinical symptoms can be subtle or absent. Headache is the classic symptom that is associated with increased intracranial pressure of any cause. However, children seem to experience this inconsistently, and infants are not developed enough to communicate their symptoms. The most common physical finding is papilledema, although this may not be as apparent during infancy or early childhood, when bulging may occur at the anterior fontanel instead. Longstanding papilledema may result in optic atrophy and eventual blindness.25
Regardless of the cause, increased intracranial pressure tends to rise during rapid eye movement sleep, presumably as a result of increased cerebral blood flow. Indeed, plateaus of increased intracranial pressure that last approximately 10 to 120 minutes have been observed during sleep. Conversely, intracranial pressure may be normal while the patient is awake or between periods of rapid eye movement sleep. Thus, the diagnosis of increased intracranial pressure may be missed by a single lumbar puncture measurement. Reasonably safe ways of long-term, direct measurement of increased intracranial pressure are possible with the use of an epidural pressure transducer to record pressure for at least 12 hours. In this way, the significance of increased intracranial pressure as a contributing factor to mental deficiency can be examined.25
With these considerations in mind, the neurologic assessment of patients with craniosynostosis can be performed as follows. The medical history should concentrate on genetic factors as well as the on child’s development. The physical examination should include a careful search for associated anomalies, with scrupulous attention paid to each of the cranial nerves (and particularly to cranial nerves II and VIII). Head circumference should be plotted on the standard growth curve. Investigations should include a skull radiograph, head computed tomography (CT) scan, brain magnetic resonance imaging, initial assessment for hydrocephalus, and formal assessment for hearing. An ophthalmologic examination is mandatory. Depending on the findings, a 12- to 24-hour pressure recording should be considered. Whenever possible, a formal developmental or newborn psychologic battery should be administered before surgery. The family should be assessed for psychosocial problems, especially when a complicated course of surgical interventions is contemplated. Follow up by a pediatrician or pediatric neurologist at regular intervals is essential. Assessments at monthly intervals for the first 12 months, every 3 months during the second year, and every 6 months until 6 years of age seem appropriate.25
Effects of Midface Deficiency on the Upper Airway
Sleep apnea may be central, obstructive, or of mixed origin; proper workup and assessment are crucial to the establishment of the correct diagnosis and treatment.* Central apnea may result from intracranial hypertension. If so, the condition should improve upon brain decompression by appropriate cranio–orbital or posterior cranial vault expansion or when effective shunting of the hydrocephalus is accomplished.
OSA has already been addressed in the sections about the Apert, Crouzon, and Pfeiffer syndromes.105,138,149,190 OSA in a child who is affected by one of these conditions is frequently treated with tracheostomy. It may also be treated by adenoidectomy, tonsillectomy, midface advancement, or CPAP. If left untreated or ineffectively treated, OSA will result in disabilities that include failure to thrive, recurrent upper respiratory infection, cognitive dysfunction, developmental delay, cor pulmonale, and sudden death.173 Midface hypoplasia in the setting of a craniosynostosis syndrome is often a primary cause of OSA. If feasible, surgical midface advancement is the preferred biologic treatment approach; even with such treatment, some patients will still require a tracheostomy or CPAP therapy.
Al-Saleh and colleagues completed a retrospective review of children with syndromal craniosynostosis who were referred to the Hospital for Sick Children in Toronto, Canada, between 1996 and 2008 for initial polysomnography to rule out sleep-related disordered breathing.2 The research confirmed a high prevalence of this type of breathing in children with Crouzon, Apert, Pfeiffer, and Saethre–Chotzen syndromes (N = 35). Despite a spectrum of surgical and non-surgical interventions having been carried out, the complete resolution of the sleep-related disordered breathing could not be routinely achieved.
Ishii and colleagues used lateral cephalometric radiographs to evaluate the nasopharyngeal airway after Le Fort III advancement in patients with Apert or Crouzon syndrome and OSA.114 The researchers documented improvement in the nasopharyngeal airway space after successful midface advancement surgery. Arnaud and colleagues reviewed respiratory improvement in patients with syndromic craniosynostosis and OSA after monobloc (MB) advancement.8 Eighty-eight percent of patients (n = 16) showed improvement as measured by oxygen level during sleep; 4 of 6 patients underwent successful tracheostomy decannulation. In one patient, a tracheostomy was required 6 months after midface advancement for severe and recurrent OSA.
Twenty-four months after MB advancement, Witherow and colleagues documented the significant improvement of airway obstruction on polysomnography in all patients who had been diagnosed with Apert, Crouzon, or Pfeiffer syndrome.280 Before MB advancement, 14 patients were managed with tracheostomy or CPAP therapy for severe OSA. Of these 14 patients who were undergoing MB advancement for severe OSA, only 43% (n = 6) had resolution of their OSA; 8 of 14 patients (57%) remained dependent on tracheostomy or CPAP despite MB advancement.
Nelson and colleagues studied 18 patients with craniosynostosis syndromes who also had coronal synostosis and midface deficiency.172 Eighty-three percent (16 of 18 patients) were treated with tracheostomy or CPAP for known OSA before midface advancement. After the midface advancement procedures, five of the tracheostomy patients were decannulated; in six others, CPAP was no longer required. This represents a 73% (11 of 15 patients) initial success rate for the relief of OSA. The mean postsurgical follow up was 3 years.
Morphologic Considerations
Examination of the patient’s entire craniofacial region should be meticulous and systematic. The skeleton and the soft tissues are assessed in a standard way to identify all normal and abnormal anatomy.65,66,68,202,205,206,211,212,216,217,272,273 Specific findings tend to occur in particular malformations, but each patient is unique. The achievement of symmetry, normal proportions, and the reconstruction of specific aesthetic units is essential to forming an unobtrusive face in a child who is born with one of the craniosynostosis syndromes.
Surgical Approach to Craniosynostosis Syndromes
Historical Perspectives
As early as 1890, Lannelogue134 and then Lane133 described a surgical approach to the treatment of craniosynostosis. Lannelogue’s aim was to remove the fused suture (i.e., strip craniectomy) in the hope of controlling the problem of brain compression within a congenitally small cranial vault. By the turn of the century, Harvey Cushing, who was the most prominent neurosurgeon of his day, suggested that surgical intervention for the problem of craniosynostosis was misdirected and that more attention should instead be given to the schooling of these children.55 Shillito disagreed with Cushing and enthusiastically supported the concept of surgical intervention to improve the outlook for these children.243 He believed that the linear “strip” craniectomy of the fused sutures would “release” the skull and allow the cranium to reshape itself and continue to grow in a normal and symmetric fashion. The strip craniectomy procedures were supposed to allow for a new suture line at the site of the previous synostosis. With the realization that this goal was not biologically feasible, attempts were made to remove portions of the cranial vault surgically and leave large open areas or to use the removed segments as free grafts to refashion the cranial vault shape. Problems with these methods included uncontrolled postoperative skull molding, reossification into dysmorphic configurations, and the occurrence of large residual skull defects.
The concept of the simultaneous release of the fused suture in combination with more meticulous cranial vault reshaping in infants was initially suggested by Rougerie and colleagues232 and then refined by Hoffman and Mohr in 1976 for children who were born with unilateral coronal synostosis.107 In 1977, Whitaker and colleagues proposed a more formal anterior cranial vault and orbital reshaping procedure for unilateral coronal synostosis.279 Marchac and Renier published their experience with a “floating forehead” technique to manage craniosynostosis during infancy; this consisted of simultaneous unilateral coronal and bilateral coronal suture release and forehead and upper orbital osteotomies with advancement.144 Unfortunately, the “floating forehead” technique resulted in unpredictable reossification of the open cranial vault areas.145 In addition, bitemporal constrictions and bulging of the skull concavities were frequent occurrences as reossification occurred. In any case, the hope for midface growth did not materialize.
During the 1950s, Gillies and Harrison reported their experience with an extracranial Le Fort III osteotomy to improve the anterior projection of the midface in an adult with Crouzon syndrome. The initial Gillies procedure was actually carried out in 1942.89 Gillies mobilized the midface via a variety of osteotomies performed through skin incisions directly over each osteotomy site. The midface was mobilized and advanced, and intermaxillary fixation was applied. After the removal of the intermaxillary fixation (i.e., 2 weeks after the operation) a metal cast-cap dental splint was attached to a plaster headcap and maintained for 3 weeks. There was no evidence that Gillies used bone grafts to bridge the surgical gaps that were created. The early enthusiasm for this technique later turned to discouragement when the patient’s facial skeleton relapsed to its preoperative status. Relapse occurred at the maxillary incisor level and also resulted in ocular proptosis.
Dr. Tessier was aware of the previous work of Gillies and its accompanying difficulties. In 1967, Tessier described a new intracranial–cranial base approach to the management of Crouzon syndrome.242 This work was first presented in France at a meeting in Montpellier in 1966 and then again the following year at the International Plastic Surgery Meeting in Rome.252–254 Tessier’s landmark presentations and publications were the beginning of modern craniofacial surgery (see chapter 2). 252–264 To overcome the earlier problems encountered by Gillies, Tessier developed an innovative basic surgical approach that included new locations for the Le Fort III osteotomy, a combined intracranial–extracranial (cranial base) approach, the use of a coronal skin incision to expose the upper facial bones, and the use of fresh autogenous bone graft. He also applied an external fixation device to help maintain bony stability until healing had occurred.
In 1971, Tessier described a single-stage frontofacial advancement in which the fronto–orbital bandeau was advanced as a separate element in conjunction with the Le Fort III complex below and the frontal bones above.256 Seven years later, Ortiz-Monasterio and colleagues refined the MB osteotomy to advance the orbits and midface as one unit; this was combined with frontal bone (anterior cranial vault) repositioning to correct the upper and midface deficiency associated with Crouzon syndrome.178,179 In 1979, Van der Meulen described the medial fasciotomy for the correction of midline facial clefting.267 Van der Meulen split the MB osteotomy vertically in the midline, removed central nasal and ethmoid bone, and then moved the two halves of the facial skeleton together for the correction of the orbital hypertelorism. To correct the midface dysplasia and the associated orbital hypertelorism in patients with Apert syndrome, Tessier refined the vertical splitting and reshaping of the midline split MB segments, thereby correcting the midface deformity in three dimensions via a procedure that is now known as facial bipartition. Wolfe and colleagues,281,282 Kawamoto,19 and Posnick and colleagues198,207,208,220 have all independently documented the advantages of Tessier’s facial bipartition technique for the correction of the upper and midface abnormalities associated with Apert syndrome. The widespread use of autogenous cranial bone grafting has virtually eliminated the need for rib and hip grafts when bone replacement is required during cranio–orbito–zygomatic procedures.213 This represents another of Tessier’s contributions to craniofacial surgery.255
In 1968, Luhr introduced the use of small metal (vitalium) plates and screws to stabilize maxillofacial fractures and then osteotomies (see Chapter 2). In current practice, the use of internal miniplate and microplate and screw (titanium) fixation of various sizes is the preferred form of rigid fixation when the stability and three-dimensional craniomaxillofacial reconstruction of multiple osteotomized bone segments and grafts are required. In infants and young children, resorbable materials are now generally used for the stabilization of non–load-bearing osteotomy segments (e.g., the cranial vault).139,196,199,221 This avoids the issues or uncertainty regarding growth restriction and brain trauma from retained non-resorbable hardware.
Philosophy Regarding the Timing of Surgery
To limit impairment while simultaneously achieving long-term preferred facial aesthetics and head and neck function, the surgeon must ask an essential question, “during the course of craniofacial development, does the operated-on facial skeleton of a child with a craniosynostosis syndrome tend to grow abnormally, resulting in further distortions and dysmorphology, or are the initial positive skeletal changes achieved (at operation) maintained during ongoing growth?” Unfortunately, the proposed theory that craniofacial procedures carried out in early infancy will “unlock growth” has not been documented through the scientific method.194,200–206,218,219
Final reconstruction of the upper midface deformities in those born with a craniosynostosis syndrome can be managed as early as 7 to 10 years of age. By this age, the cranial vault and orbits normally attain approximately 85% to 90% of their adult size. Whenever feasible, waiting until the maxillary first molars have erupted is also preferred. When the “upper midface” reconstruction is carried out after this age, the objective is to attain adult morphology in the cranio-orbito-zygomatic region with the expectation of a stable result (no longer influenced by growth) once healing has occurred. Psychosocial considerations also support the age 7 to 10 years time frame for the upper midface procedure. When a successful reconstruction is achieved at this age, the child may progress through school with an opportunity for a healthy body image and self-esteem.*
Management of the Upper Midface Deformity in Children and Young Adults
Incision Placement and Soft-Tissue Management
For the exposure of the craniofacial skeleton above the Le Fort I level, the approach used is the coronal skin incision. This allows for relatively camouflaged access to the anterior and posterior cranial vault, the orbits, the nasal dorsum, the zygomas, the upper maxilla, the pterygoid fossa, and the temporomandibular joints. For added cosmetic advantage, the placement of the coronal incision more posteriorly on the scalp and with postauricular rather than preauricular extensions is useful.214 When the exposure of the maxilla at the Le Fort I level is required, a circumvestibular maxillary intraoral incision is used. Unless complications occur that warrant unusual exposure, no other incisions are required to manage any aspect of reconstruction in patients with a craniosynostosis syndrome. These incisions (i.e., coronal [scalp] and maxillary [circumvestibular]) may be reopened as needed to further complete the individual’s staged reconstruction.
Management of the Ventricular System
In the patient with a craniosynostosis syndrome who is to undergo intracranial volume expansion with the concurrent management of hydrocephalus as part of the reconstruction, the potential for morbidity increases.25,77,91,108,170,174 Complications may arise from either excessive cerebrospinal fluid drainage (overshunting) or inadequate shunting that leads to increased intracranial pressure. With overshunting there is decreased central nervous system mass to fill any surgically created retrofrontal dead space. Uncontrolled hydrocephalus may result in raised intracranial pressure, which leads to its own set of problems. In either situation, fronto–facial advancement via an intracranial approach to the midface or isolated cranial vault expansion procedures should be thoughtfully staged with ventriculoperitoneal shunt management. We believe that the presence or absence of a ventriculoperitoneal shunt is not in itself a major factor in the success of a fronto–facial advancement procedure. An important aspect is the satisfactory physiologic function of the ventricular system, with or without the placement of a shunt. Ultimately, the decision regarding the need for and sequencing of shunting is based on the patient’s neurologic findings and the neurosurgeon’s judgment. In a patient with a ventriculoperitoneal shunt in place before the surgery, experienced neurosurgical evaluation, including imaging studies of the ventricular system, is carried out to confirm satisfactory physiologic function.
Upper Midface Reconstruction Options
The study by McCarthy confirms that the Le Fort III osteotomy is not effective as an aesthetic option to manage the upper midface deformity in the majority of craniosynostosis syndrome patients (see section later in this chapter concerning Le Fort III option).276 By anatomic design, the Le Fort III prevents management of the whole orbital aesthetic unit during one operative setting. Therefore a major aesthetic shortcoming of the Le Fort III osteotomy, when its indications do not fit the presenting dysmorphology, is the creation of irregular step-offs in the lateral orbital rims. This will occur even when only a moderate Le Fort III advancement is carried out. These lateral orbital step-offs will be visible to the casual observer as unattractive at conversational distance and surgical attempts at modification performed later will be suboptimal. Another problem with the Le Fort III osteotomy is the difficulty in judging an ideal orbital depth. This frequently results in either residual proptosis or enophthalmos. Simultaneous correction of upper face (orbital) hypertelorism and the concave midface arc-of-rotation typical in Apert syndrome is also not possible with the Le Fort III procedure. Excessive lengthening of the nose, accompanied by flattening of the nasofrontal angle, will also occur if the Le Fort III osteotomy is selected when the skeletal dysmorphology favors an MB or facial bipartition. Unfortunately, it is not possible to later correct the elongated nose or the flattened naso-frontal angle. To avoid these short comings, it is not a matter of becoming more proficient at the Le Fort III osteotomy or simply “managing the overlying soft tissues” in a different way (i.e., canthopeties or midface lift). The Le Fort III osteotomy is not consistent with the presenting dysmorphology in most craniosynostosis syndrome patients and, therefore, will not provide the opportunity to achieve the desired aesthetic result. Nevertheless, the Le Fort III is often considererd the “go to” approach by surgeons as it is: 1) an extracranial procedure; 2) requires less surgeon skill/experience; 3) is less likely to result in significant blood loss; and 4) is less likely to result in perioperative complications (i.e., cranionasal fistula, intracranial abscess, bone resorption).
Monobloc and Facial Bipartition Osteotomies (Intracranial Approach to the Orbits and the Midface)
Cranial vault reconstruction in the child with a craniosynostosis syndrome should provide space for the compressed brain to expand. Immediately after completing cranial vault and monobloc (MB) or facial bipartition (FB) osteotomies with advancement, extradural (retrofrontal) dead space remains in the anterior cranial fossa above the skull base gap created by the osteotomy.208 The skull base gap is in direct communication with the nasal cavity. Therefore, the postoperative recovery may be complicated by cerebrospinal fluid leakage across the skull base gap followed by infection, fistula formation, and subsequent bone resorption in the glabella region. After fronto–facial advancement by either MB or FB, the communication between the nasal cavity and the cranial fossa must be managed to limit these potential complications. The most effective method to do so remains unclear, but all agree that it is a critical aspect for successful reconstruction. Technical aspects of management include the following: 1) gentle tissue handling; 2) achieving good hemostasis; 3) effective repair of dural tears; 4) avoidance of over expansion of the cranial vault; 5) maximum separation of dura and nasal mucosal tissue planes (i.e., interposing tissue such as bone grafts, tissue sealants, and flaps); 6) rigid plate and screw fixation of the osteotomies and bone segments; 7) avoidance of pressure gradients across the opening to facilitate nasal mucosa healing; and 8) prevention of too much or too little ventriculoperitoneal shunting.
After cranio–orbital reconstruction in the infant with a craniosynostosis syndrome, rapid filling of the expanded intracranial volume by the previously compressed frontal lobes of the brain has been documented.208 This has also been shown to occur after MB and FB advancement in children and young adults.249 More gradual filling of the space is thought to occur in older adults. At the time of the MB or FB osteotomies, the nasal cavity can be sealed from the cranial fossa by doing the following: 1) inserting pericranial tissue; 2) placing bone grafts to bridge the osteotomy gaps; and 3) using tissue sealants. This provides time for the re-epithelialization (i.e., healing) of the nasal mucosa. Until the torn nasal mucosa heals, communication between the nasal cavity and the anterior cranial fossa may result in the transfer of air, fluid, and bacteria followed by infection and naso–cranial fistula formation. The postoperative continuation of nasotracheal intubation for several days and nasopharyngeal tube placement after extubation have also proven useful to limit pressure gradients across the communication. The avoidance of positive pressure ventilation, the enforcement of sinus precautions, and the restriction of nose blowing further limits the reflux of air, fluid, and bacteria early after surgery.
Posnick and colleagues studied the issues of retrofrontal dead space; communication across the skull base osteotomy gap; and associated morbidity in a consecutive series of children in the mixed dentition and young adults in the permanent dentition (n = 23) who were undergoing either MB or FB osteotomies in combination with cranial vault expansion.208 The procedures were carried out by a single craniofacial surgeon (Posnick) and one of three neurosurgeons at a single tertiary care hospital from 1987 to 1991. The extradural (retrofrontal) dead space was measured from consistent CT scan images at specific postoperative intervals (immediate, 6 to 8 weeks, and 1 year). The study confirmed the presence of an immediate retrofrontal dead space that generally filled in with the expanding brain and dura by 6 to 8 weeks after surgery. Specific intraoperative maneuvers were undertaken by the surgeons to close the nasofrontal communication, including flaps, fibrin glue, bone grafts, and Gelfoam. After surgery, care was taken to limit a pressure gradient across the communication (via the repair of dural tears, sinus precautions, and nasal stenting), with the objective of providing time for nasal mucosa healing. The infection rate in this study group was limited to 2 of 23 patients (9%). In both patients who developed an infection, a retrofrontal (extradural) fluid collection was noted, with drainage across the residual nasofrontal communication into the nose. Both patients healed without significant comorbidity (i.e., brain or eye injury) but did require further reconstruction of the resorbed portions of the anterior cranial vault and the supraorbital ridges (see Fig. 30-15).
Wolfe completed a critical analysis of 81 MB advancements carried out over a 27-year period.281 This was a retrospective chart analysis of a series of patients who underwent either MB (frontofacial) advancement (MFFA) or FB (MFFA plus FB). The procedures were carried out at seven different craniofacial centers and included 49 MFFA and 32 MFFA plus FB. The MFFA and MFFA plus FB osteotomies were either placed in their preferred location in the operating room (standard approach) or gradually distracted (DO technique) with internal or external devices. Complications included two deaths (cardiac arrest in one patient and complications arising from hypovolemia in the other). One case was aborted as a result of a large-volume blood loss; there were three infections or sequestrations and one persistent cerebrospinal fluid leak (no meningitis). There were significant complications documented in the DO group and fewer in the non-DO group. Blood loss and operative time were equivalent for both standard and DO techniques. Interestingly, the incidence of infection and cerebrospinal fluid leaks was not diminished with the use of the alternative distraction DO approach. The author also concluded that, for the majority of patients, the standard approach offered improved morphologic results. The author then compared the morphologic results of the MMFA and MMFA plus FB with those of the extracranial Le Fort III option and concluded that the Le Fort III was less favorable. Regardless of the technique used, all patients required orthognathic surgery (at the Le Fort I level) to complete the reconstruction.
Bradley and colleagues completed a single-center retrospective study that compared differences in morbidity in a series of patients who were born with a craniosynostosis syndrome and who then underwent MB osteotomy for the correction of upper and midface anomalies or hypoplasia.19 The authors describe three different sequential treatment groups that were followed during a period of 23 years. Group I patients (1979 to 1989; n = 12) underwent MB osteotomies without any special attention paid to the retrofrontal dead space or the communication through the skull base between the anterior cranial fossa and the nasal cavity. Group II patients (1989 to 1995; n = 11) underwent MB osteotomies with various attempts at closure of the skull base gap with pericranial flaps and fibrin glue. Group III (1995 to 2002; n = 24) patients underwent MB osteotomies without immediate advancement. An internal distraction device was placed across the osteotomized zygomatic arch on each side. After a 7-day latency period, the advancement of the MB and forehead unit was initiated at 1 mm per day for approximately 2 to 4 weeks. The infection rate for Group III patients was significantly lower (2 of 24 [8%]) than it was for those in Groups I and II. Neither of the two infections in Group III resulted in bone loss. Group I patients had the greatest morbidity likely as a result of the limited fixation performed during the 1970s and 1980s.
As described by Bradley and colleagues, the DO technique does allow more time for the brain to expand into the retrofrontal dead space after the completion of an MB osteotomy (i.e. delayed for 7 days) before advancing the upper midface.19 In theory, this should facilitate early nasal mucosa healing and thereby limit the communication of fluid, air, and bacteria across the surgically created skull base gap. This likely explains the drop in infection rate in the Group III patients as compared with the Group I and II patients. The rate of infection in Bradley’s Group III patients, who were treated with a DO approach, essentially matches that of the patients described by Posnick and colleagues who were treated with a standard approach (8% and 9%, respectively).
Bradley and colleagues also described greater advancement in Group III (DO approach) patients as compared with Group I and II patients.19 Confounding variables may explain these differences, including increased surgical experience during the later years of the study (i.e., group III patients) and the impact of complications during the earlier years of the study (i.e., extremely high rates of infection in Groups I and II), which likely resulted in relapse and limited the long-term midface advancement. More importantly, there was no correlation between the number of millimeters of MB advancement and either greater functional gains or enhanced facial aesthetics.
 COMMENT: With an MB osteotomy, as much aesthetic damage is done by overadvancement (enophthalmos) as by underadvancement (residual eye proptosis). In addition, the achievement of a “normal” occlusion is not usually a treatment objective at the time of MB advancement. Accomplishing an ideal occlusion without creating enophthalmos generally requires a separate Le Fort I osteotomy to differentially reposition the maxilla.
COMMENT: With an MB osteotomy, as much aesthetic damage is done by overadvancement (enophthalmos) as by underadvancement (residual eye proptosis). In addition, the achievement of a “normal” occlusion is not usually a treatment objective at the time of MB advancement. Accomplishing an ideal occlusion without creating enophthalmos generally requires a separate Le Fort I osteotomy to differentially reposition the maxilla.A fourth clinical study sheds further light on this subject. Ahmad and colleagues described a series of 12 children who were born with a craniosynostosis syndrome who also had multiple functional problems, including the following: 1) raised intracranial pressure as a result of a diminished cranial vault volume; 2) exposure of the eyes (i.e., corneal irritation) as a result of shallow orbits; 3) airway obstruction from a reduced upper airway space (i.e. OSA); and 4) feeding difficulties.3 Each of the study patients underwent fronto–facial (MB) advancement with the use of DO techniques. The mean age at operation was 18 months (range, 4 to 30 months). The mean advancement was 16.6 mm at the forehead level and 17 mm at the midface level. Ocular protection and the reduction of intracranial pressure (when raised) was achieved in all children. At least a degree of airway improvement was achieved in all but one child. The authors subjectively state that there was marked improvement in every patient’s appearance. Complications included cerebrospinal fluid leaks (2 of 12 patients [16.6%]); pin-site infections (3 of 12 patients [25%]); external DO device frame slippage that required replacement (2 of 12 patients [16.6%]); and overadvancement with resulting enophthalmos (1 of 12 patients [8.4%]). In addition, one patient died 9 months after the procedure in conjunction with tracheal reconstruction. The published article was discussed by Hopper,111 who recommends that clinicians consider a less invasive approach and accept limited aesthetics objectives in an attempt to lower perioperative morbidity. The approach that he suggests includes the following: 1) either anterior cranial or posterior cranial vault expansion without simultaneous midface advancement to relieve intracranial pressure; 2) extracranial Le Fort III advancement without simultaneous cranial vault expansion to open the upper airway; 3) tarsorrhaphies to protect the eyes from exposure rather than orbital expansion; and 4) continued tracheostomy for airway management rather than midface advancement.
 COMMENT: The four studies reviewed and the discussion by Hopper that followed demonstrate the wide variation of opinions on how best to reconstruct individuals with a craniosynostosis syndrome. In this author’s opinion, for the majority of these children, only the MB or facial bipartition osteotomies offer a realistic opportunity to achieve close to normal morphology and the opportunity to develop healthy self-esteem. Favorable craniofacial function is expected to follow the enhanced facial form. This includes cranial vault expansion (i.e., relief of ICP); orbital expansion (i.e., relieve of proptosis and eye protection); and upper airway expansion (i.e., relief of OSA and improved breathing during the day). Gains in mastication, swallow mechanism, speech articulation, breathing, vision, and cognitive function are also expected.
COMMENT: The four studies reviewed and the discussion by Hopper that followed demonstrate the wide variation of opinions on how best to reconstruct individuals with a craniosynostosis syndrome. In this author’s opinion, for the majority of these children, only the MB or facial bipartition osteotomies offer a realistic opportunity to achieve close to normal morphology and the opportunity to develop healthy self-esteem. Favorable craniofacial function is expected to follow the enhanced facial form. This includes cranial vault expansion (i.e., relief of ICP); orbital expansion (i.e., relieve of proptosis and eye protection); and upper airway expansion (i.e., relief of OSA and improved breathing during the day). Gains in mastication, swallow mechanism, speech articulation, breathing, vision, and cognitive function are also expected.1. The ability to remove, segment, reshape and then stabilize the cranial vault
2. The ability to separate the orbits and midface as a unit MB from the skull base
3. The ability to segment and reshape the upper orbits of the MB including interposing bone grafts as needed to reconstruct each orbital aesthetic unit during a single operative setting.
4. The ability to separate the MB into halves (facial bipartition) and then three-dimensionally reposition and stabilize (plate and screw fixation) the two facial halves to achieve the most favorable morphology in all three planes (i.e., pitch, role, yaw orientation). This often requires increasing the maxillary width (i.e., arch expansion) and decreasing the upper face width (i.e., correction of hypertelorism of the orbits, zygomas, and bitemporal regions). Facial bipartition also provides the ability to correct the transverse facial arc of rotation. An example is changing the concave facial arc of rotation (i.e., yaw orientation) in Apert syndrome.
Le Fort III Osteotomy (Extracranial Approach to the Midface)
Shetye and colleagues set out to examine long-term (10-year) midface skeletal stability and the potential for maxillary growth after extracranial Le Fort III advancements in children with craniosynostosis syndromes.242 Their study group included 25 children (10 had Crouzon syndrome, 9 had Apert syndrome, and 6 had Pfeiffer syndrome) who underwent extracranial Le Fort III advancement before they were 11 years old (mean, 5.8 years; range, 3.8 to 10.9 years). Two different forms of fixation were used: Group 1’s treatment included intraosseous wires, bone grafts, suspension wires, dental splint, and 8 weeks of intermaxillary fixation; Group 2’s treatment involved titanium plates and screws, bone grafts, dental splints, and elastics. Good stability of the midface was documented in both groups at 1 year, 5 years, and 10 years. As has been found with previously published studies, no significant horizontal growth occurred in the midface after surgery. The form of fixation used was not a factor (i.e., plate and screw fixation did not increase incidence of growth restriction). The mandible was measured at the pogonion and found to advance 5.72 mm and 7.32 mm at the 5- and 10-year time points, respectively. This study confirms that, when the Le Fort III advancement is carried out at a young age, the occurrence of jaw disharmony with ongoing growth is inevitable and will require further staged reconstruction.
In a second publication, Shetye and colleagues evaluated a larger group of syndromal craniosynostosis patients who also underwent extracranial Le Fort III advancement between the years of 1973 and 2006.238 Three different stabilization techniques were sequentially used over the study period: Group 1 (n = 20) was treated with intraosseous wires, suspension wires, bone grafts, dental splinting, and 8 weeks of intermaxillary fixation; Group 2 (n = 20) was treated with titanium plates and screws, bone grafts, dental splinting, and elastics; and Group 3 (n = 20) was treated with the extended use of an external DO device during 3 months of healing. The stability of midface advancement was assessed with the use of measurements from 1- and 5-year postoperative lateral cephalometric radiographs. Patients in Groups 1 and 3 showed equal levels of skeletal stability, whereas Group 2 showed slightly less stability at the A point. At the orbitale, all three groups showed similar levels of stability. Interestingly, all three methods of fixation for the extracranial Le Fort III advancements showed good levels of stability. The study also confirmed that all study subjects who were in the mixed dentition demonstrated progressive facial disharmony as a result of ongoing mandibular growth, thereby necessitating further skeletal reconstruction by the late teenage years (i.e., different forms of fixation had no bearing on the incidence of growth restriction).
McCarthy and colleagues recently reviewed the long-term midface growth and periorbital/malar (upper midface) aesthetic results in individuals who were born with a craniosynostosis syndrome and who had undergone Le Fort III advancement.276 The study represented a follow-up period of more than 20 years for four identified patients (one with Apert syndrome and three with Crouzon syndrome) who were 4, 6, 14, and 20 years old at the time of the Le Fort III advancement. Thus, this study represented two young children and two skeletally mature individuals who underwent Le Fort III midface advancement. The authors’ review confirmed several points: 1) a Le Fort III advancement carried out during early childhood results in no further horizontal growth of the midface; 2) a Le Fort III advancement carried out during early childhood results in further vertical growth of the midface with clockwise rotation of the mandible; and 3) a Le Fort III advancement carried out in individuals with either Crouzon or Apert syndrome—whether during childhood or adulthood—does not effectively correct the presenting zygomatic, orbital, and nasal malformations. The upper midface aesthetic results as viewed through the overlying soft-tissue envelope (i.e., the eyelid–cheek–malar region) were suboptimal after Le Fort III advancement. Furthermore, with age, the upper midface aesthetic results became even more unfavorable.
McCarthy’s findings of continued vertical growth of the midface after Le Fort III osteotomy carried out during early childhood is consistent with the inability to achieve adequate nasal airflow in the operated patient. In addition, even if adequate nasal airflow is accomplished, at least some individuals will have limited masticatory muscle tone. In either case, when a child continues with an open-mouth protrusive tongue posture, hypereruption (i.e., vertical growth) of the maxillary dentition through the alveolar bone with clockwise rotation of the mandible is expected (see Chapter 4). A lack of horizontal growth of the maxilla after either a Le Fort I, Le Fort III, or MB osteotomy is carried out during childhood has been documented by a number of independent investigators.31,50,75,76,102,103,112,192 This results from both intrinsic growth limitations of the malformation and injury to the sutural growth centers from the osteotomies that are carried out.
Monobloc and Facial Bipartition Osteotomies: Step-by-Step Description of the Surgical Technique
This section provides the surgeon with a step-by-step technical description for the MB and FB osteotomies as this author has routinely performed them. The detailed operative technique is illustrated through the case of a 12-year-old Persian girl who was born with Pfeiffer syndrome and who had never undergone surgery. The patient then underwent FB osteotomies in combination with anterior cranial vault reshaping (see Fig. 30-14). A review of the October 1992 Plastic Surgery Education Foundation Teleplast Conference: Monobloc and Facial Bipartition for Reconstruction of Craniofacial Malformations may serve as an additional educational tool to complete the surgeon’s understanding of the technical aspects of the procedures that will be described ( Video 15).
Video 15).
Monobloc and Facial Bipartition Osteotomies: Step-by-Step Approach
Step 1. Satisfactory airway management in a patient who is undergoing an MB/FB osteotomy is essential. A method that I have used effectively involves an orotracheal tube that is secured adjacent to the cusp edge of the mandibular incisors with a circummandibular wire (Fig. 30-16, A). After the MB/FB osteotomies and disimpaction are completed, a nasotracheal tube is placed, and the orotracheal tube is removed (Fig. 30-16, B). Through this controlled approach, endotracheal tube injury at the completion of the osteotomies and dislodgement during disimpaction are prevented. In addition, direct contact between the maxillary and mandibular teeth can be achieved for the improved control of the occlusion. The nasotracheal tube remains in place at the end of the procedure to allow for nasal mucosal stenting during the initial postoperative phase. Other approaches to managing the airway have been described and used effectively (e.g., tracheostomy, submental intubation, orotracheal intubation without exchange).
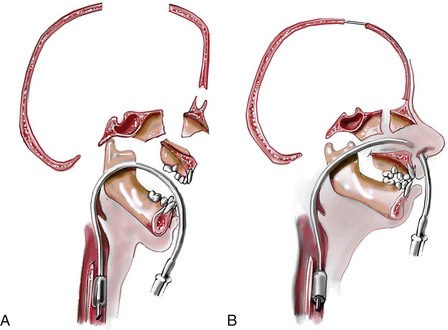
Figure 30-16 A and B, Step 1. Illustrations of a 12-year-old girl with Pfeiffer syndrome (see Fig. 30-14) before and after facial bipartition. The step-by-step approach for the surgical technique that was carried out is described in the text. From Posnick JC: Craniofacial dysostosis syndromes: a staged reconstructive approach. In Turvery JA, Vig TWL, Fonseca RJ, eds: Facial clefts and craniosynostosis: principles and management, vol 26, Philadelphia, 1995, W.B. Saunders, p 645.
Step 2. When feasible, I recommend the placement of Crawford tubes to protect the nasolacrimal apparatus during surgery (Fig. 30-17). The puncta are dilated, and a probe is inserted through each punctum to confirm entrance into the nose. The Crawford tube is inserted through each punctum and pulled through the nose. Within the nose, the Silastic sheeting is stripped; the tubing is tied in the nose, and the excess is cut in the nose with scissors.
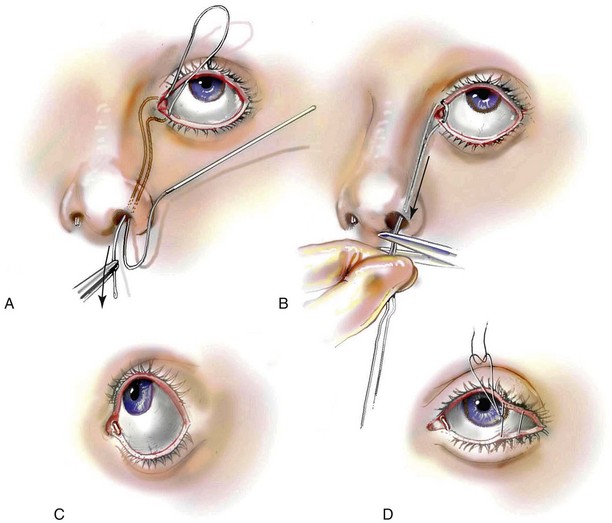
Step 3. Surgical arch wires are generally applied (Fig. 30-18). A throat pack is placed, and the mouth is cleansed. Erich arch bars are applied to the maxillary and mandibular teeth. Circum-mandibular wires are placed to further stabilize the mandibular arch bar, and the orotracheal tube is secured with a wire to the symphyseal region of the surgical arch bar.
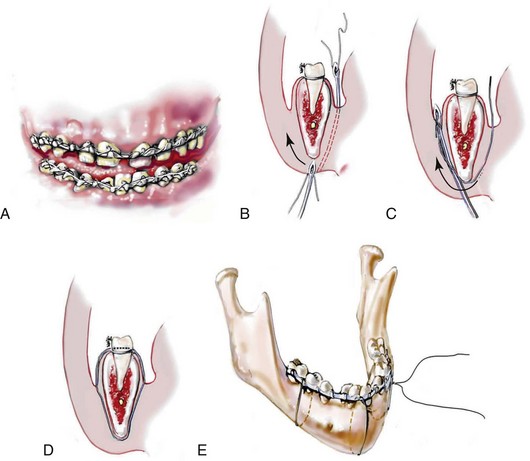
Step 4. The patient is prepared and draped. The patient’s head is placed in a Mayfield (horseshoe-shaped) head holder in the neutral neck position. Cleansing of the entire scalp is performed with povidone–iodine (Betadine) soap; the soap is then washed out with sterile water. Next, the application of povidone–iodine solution to the scalp, face, and neck is carried out. The surgical field is draped to expose the neck down to the clavicles; the full face, including the external ears; and the anterior scalp back to the planned incision. The separation of the mouth/nose from the eyes/forehead is achieved with an additional sterile towel drape; this is done to limit the contamination of the intracranial cavity with oral or nasal flora.
Step 5. A coronal (scalp) skin incision is completed (Fig. 30-19). The incision site is postauricular and posterior in the scalp. Other incision modifications have been described (e.g., Z-plasty in the temporal regions). Lidocaine with epinephrine is injected to facilitate hemostasis.
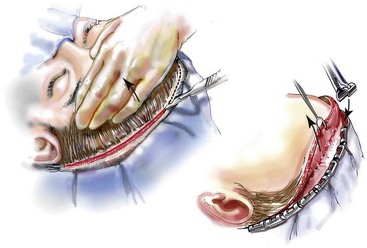
Step 6. The anterior scalp flap is then elevated by remaining deep to the superficial layer of the deep temporal fascia over the temporalis muscles; subperiosteal over the mid-forehead region; subperiosteal down the lateral orbital rims; subperiosteal with exposure of the anterior maxilla; subperiosteal for exposure of the zygomatic aches; and subperiosteal over the dorsum of the nose. Elevation of the temporalis muscles off of the squamous temporal bones is then carried out (Fig. 30-20).
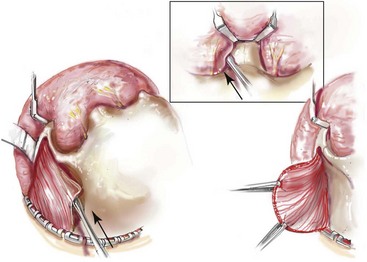
Step 7. Bifrontal craniotomy is completed (Fig. 30-21, A). The craniotomy lines are drawn with a sterile pencil, and bur holes are completed with a perforator as needed. The craniotomies are completed with a craniotome. The frontal bones are separated from the underlying dura and then removed. The frontal and temporal lobes of the brain are adequately retracted to safely accomplish skull base osteotomies. The brain is protected with cottonoid pledgets (Fig. 30-21, B).
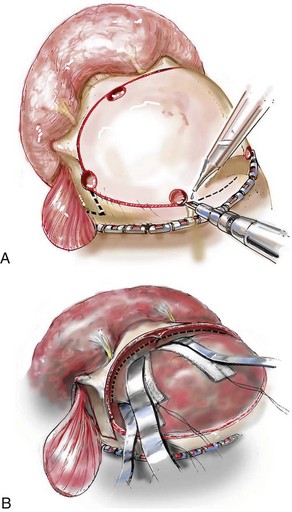
Step 8. Zygomatic arch osteotomies are performed (Fig. 30-22). Retractors are placed, and an osteotomy is completed through the midzygomatic arch on each side with a sagittal (reciprocating) saw.
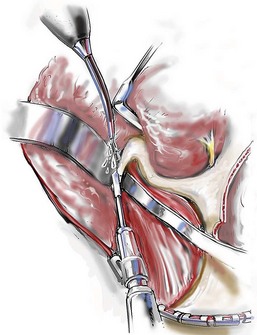
Step 9. With the use of the sagittal saw, the lateral orbital wall osteotomy is initiated into the inferior orbital fissure. The osteotomy is extended superiorly through the lateral orbital wall (Fig. 30-23).
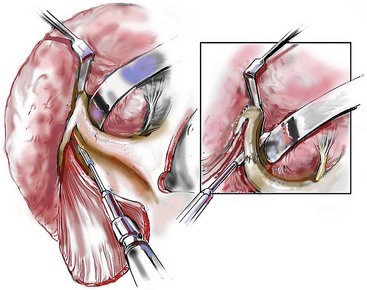
Step 10. With the continued use of the sagittal saw, the lateral tenon extension of the osteotomy through the squamous temporal bone and skull base is carried out (Fig. 30-24).
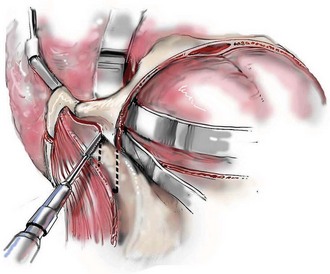
Step 11. The orbital roof osteotomy is completed with the sagittal saw through the anterior skull base (Fig. 30-25).
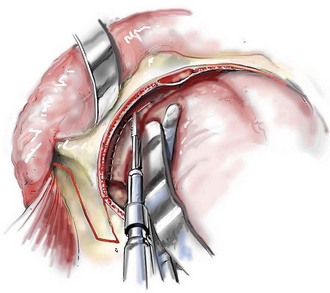
Step 12. The orbital roof osteotomy continues laterally through the sphenoid wing. This osteotomy will join up with the previous tenon extension on each side (Fig. 30-26).
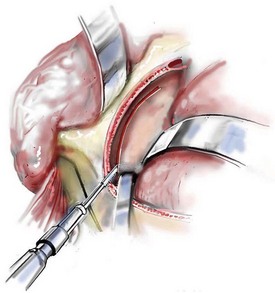
Step 13. A thin chisel placed through the anterior skull base is used to confirm the completion of the sphenoid wing osteotomy and continuity with the tenon extension (Fig. 30-27).
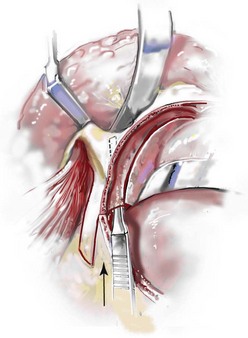
Step 14. With a thin chisel and working through the skull base, the medial orbital wall osteotomy is completed posterior to the medial canthus and nasolacrimal apparatus and inferiorly into the inferior orbital fissure (Fig. 30-28).
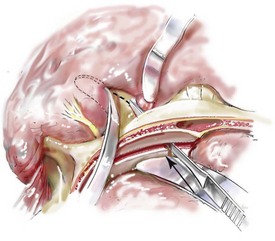
Step 15. The anterior aspect of the nasal septum is separated from the cranial base. A straight 15-mm wide chisel is placed through the cranial base just anterior to the crista galli and used to complete this osteotomy and to further separate the midface from the base of the skull (Fig. 30-29).
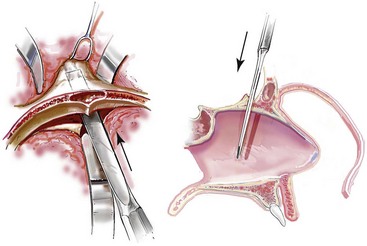
Step 16. The separation of the pterygomaxillary sutures is completed. A long 10-mm wide chisel is placed through the coronal incision and the infratemporal fossa to the pterygomaxillary suture. One double-gloved hand is placed in the patient’s mouth, and the other hand is used to place the chisel through the coronal incision and into the infratemporal fossa (Fig. 30-30, A). A mallet is then used to separate the pterygomaxillary suture with the chisel. The success of the separation is confirmed with the pterygomaxillary spreader forceps (Fig. 30-30, B).
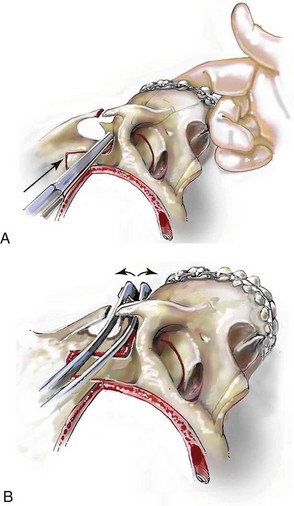
Step 17. The midface (MB) is disimpacted with the use of two nasomaxillary forceps that are placed in the nose and the mouth. Pterygomaxillary spreader forceps are simultaneously placed through the coronal incision on each side. The midface is then disimpacted and stretched forward to confirm adequate advancement at the occlusal level (Fig. 30-31). Next, the endotracheal airway exchange is completed (see Fig. 30-16, B). Additional sterile drapes are placed over the scalp and the face and neck regions, and the throat pack is removed. The surgeon places the nasotracheal tube through the nose and into the oropharynx. The anesthesiologist then removes the orotracheal tube and completes the insertion of the nasotracheal tube through the larynx using the direct or GlideScope laryngoscopic technique.
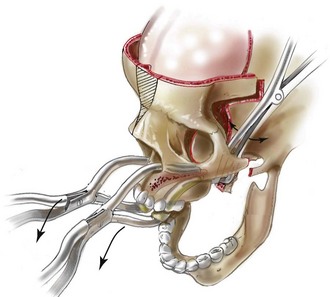
Step 18. For FB, a midnasal osteotomy (i.e., an ostectomy) is completed. Working through the coronal incision, the surgeon marks out the proposed midnasal osteotomy with calipers and a pencil and then completes it with a sagittal saw. When the ostectomy is completed, the removal of portions of the underlying cartilaginous nasal septum is also accomplished (Fig. 30-32).
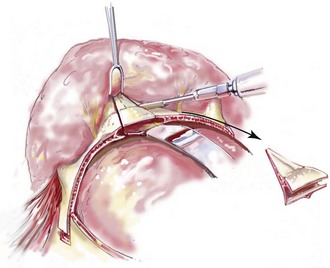
Step 19. For FB, separate sterile oral instruments are used to split the maxilla into two segments. An intraoral maxillary vestibular incision is made, and this is followed by the subperiosteal exposure of the anterior maxilla, the anterior nasal spine, and the nasal floor (Fig. 30-33, A). With a sagittal saw, a midline osteotomy is completed between the central incisors and then parasagittally down the hard palate; the palatal mucosa is left undisturbed (Fig. 30-33, B). The segmental separation is completed with a thin 5-mm wide chisel placed between the central incisors (Fig. 30-33, C). The Erich arch bar is also cut between the incisors. Further separation of the posterior maxilla is completed with a spreader forceps as needed (Fig. 30-33, D). The oral wound is closed, the intraoral instruments are discarded, and fresh gloves are put on the hands.
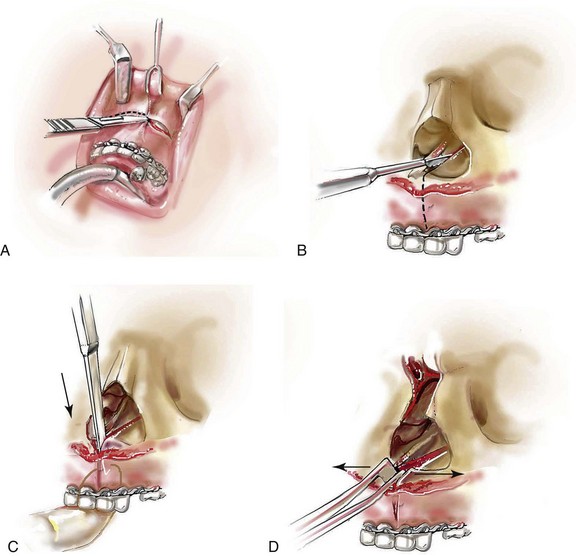
Step 20. For FB, stabilization of the upper orbits and the nasal bones is completed next (Fig. 30-34, A, and B). Repositioning of the facial halves medially with correction of the midface arc of rotation is completed; this will require refinement with a rotary drill (Fig. 30-34, A). The upper orbits and the nasal bones are fixed with a titanium plate and screws (Fig. 30-34, B). A rotary drill is also used to cranialize the frontal sinus, if needed (Fig. 30-34, C).
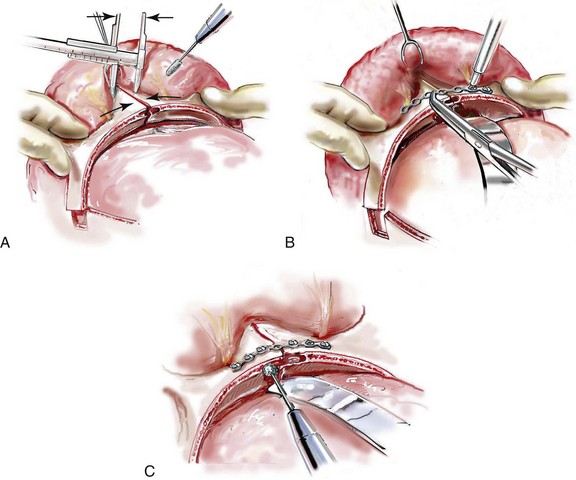
Step 21. Midface advancement at the level of the maxillary dentition is accomplished (Fig. 30-35). Working through the coronal scalp incision, the primary surgeon advances the midface. With the use of separate sterile oral instruments, the assistant to the surgeon wires the jaws together into the planned occlusion.
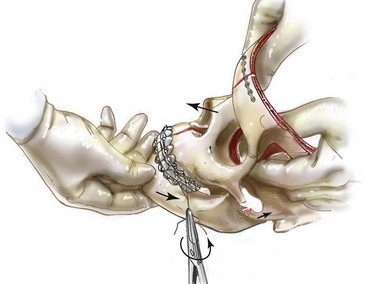
 NOTE: The patient is rarely placed in an ideal occlusion. The mid face advancement at the occlusal level is of secondary importance. The advancement should be based on the most favorable depth of the orbits and position of the zygomas.
NOTE: The patient is rarely placed in an ideal occlusion. The mid face advancement at the occlusal level is of secondary importance. The advancement should be based on the most favorable depth of the orbits and position of the zygomas.Step 22. Upper midface advancement is established at the zygomatic arches (Fig. 30-36). The amount of advancement at each zygomatic arch is measured with a caliper. A titanium plate is conformed to extend from the anterior maxilla across the arch and surgical gap to the posterior zygoma on each side. The plate is secured with titanium screws.
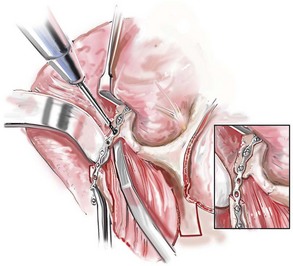
Step 23. Additional segmental osteotomies of the orbits are often required. Occasionally, the lateral superior orbits have further dysplasia and segmental osteotomies with reshaping for reconstruction is necessary (Fig. 30-37, A). If so, the lateral orbital segments are removed with a reciprocating saw. Additional segmental orbital osteotomies are then completed with a reciprocating saw (Fig. 30-37, B). The lateral orbital rim and superior orbital rim segments are further reshaped with a rotary drill (Fig. 30-37, C). The segments are fixed with titanium plates and screws (Fig. 30-37, D).
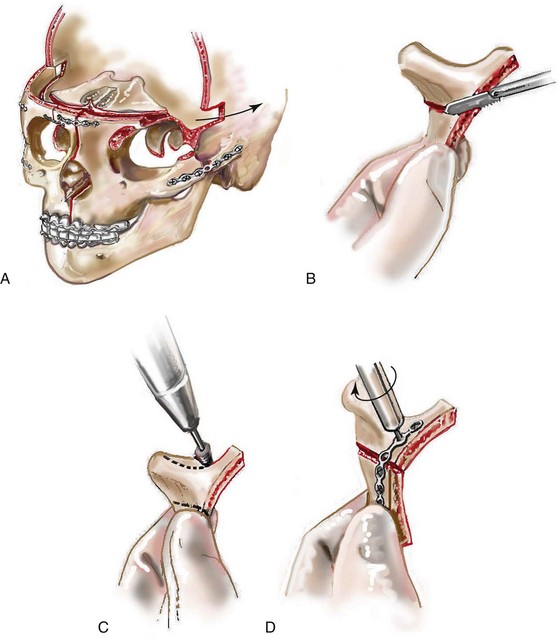
Step 24. Stabilization of the midface advancement at the upper orbital tenon extension is accomplished (Fig. 30-38). The desired advancement is measured with a caliper; a miniplate is adapted to bridge the gap between the tenon extension and the posterior cranial vault. Titanium screws are used for plate stabilization.
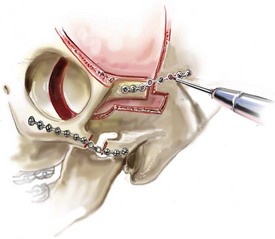
Step 25. Hyperplastic ethmoidal air cells are debrided as needed in patients with orbital hypertelorism (Fig. 30-39). With visualization through the anterior cranial base, rongeurs are used to debride hyperplastic ethmoidal air cells and to reduce midorbital hypertelorism.
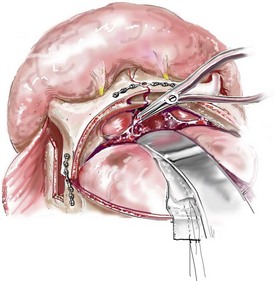
Step 26. The opening between the anterior cranial fossa and the nasal cavity is managed (Fig. 30-40, A and B). The surgeon irrigates and suctions the intranasal cavity through the anterior cranial base exposure. My usual approach is to then apply a sheet of Gelfoam to separate the opening between the two cavities (Fig. 30-40, A). I then inject fibrin glue over the Gelfoam to seal the separation (Fig. 30-40, B). Other methods of managing the separation between the nasal cavity and the anterior cranial fossa may be used, depending on the clinical circumstances (e.g., cranial bone grafts, soft-tissue flaps).
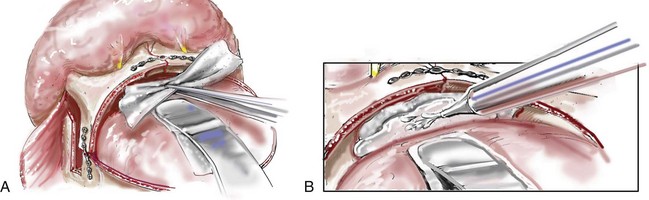
Step 27. The anterior cranial vault is reshaped, advanced, and secured in place (Fig. 30-41, A, B, and C). With the use of a reciprocating saw, osteotomies of the removed anterior cranial vault are completed (Fig. 30-41, A). Rotary drill recontouring is also accomplished to achieve the desired shape (Fig. 30-41, B). Split- or full-thickness cranial bone graft is harvested and used as needed to fill in defects, and fixation is accomplished with plates and screws. Split-thickness cranial bone graft is also placed and fixed in the midzygomatic arch segmental defects (Fig. 30-41, C).
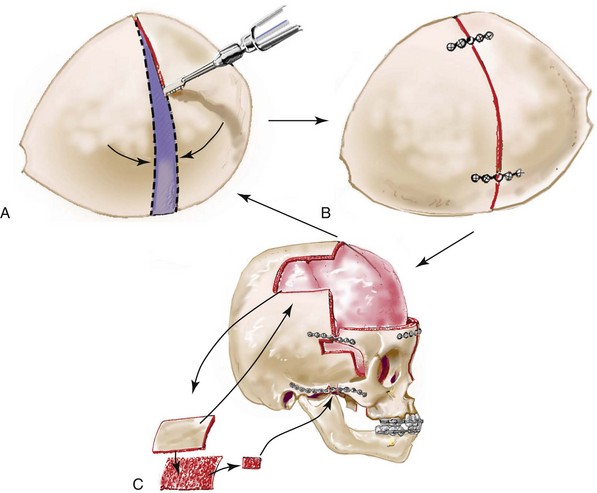
Step 28. Lateral canthopexies are completed (Fig. 30-42). Two holes are drilled at each new frontozygomatic suture region. The lateral canthi are identified with a skin hook through the coronal incision. A figure-of-eight wire suture is placed through each lateral canthus through the coronal incision. Each lateral canthus is fixed by passing the wire through the drill holes in the frontozygomatic suture.
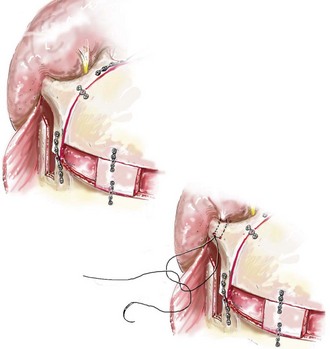
Step 29. Each temporalis muscle is resecured to bone (Fig. 30-43). The temporalis muscles are repositioned anteriorly and secured to the lateral orbital rims and the temporal bones with interrupted sutures.
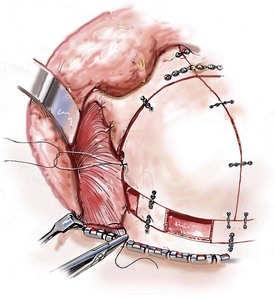
Step 30. The scalp wound is closed (Fig. 30-44). Suction drains are placed through the posterior scalp flap; one is placed on each side. One drain is placed under the anterior flap, and the other is placed below the posterior flap. The galea closure is completed with interrupted sutures, and the skin layer closure is completed with staples or resorbable sutures in accordance with surgeon preference.
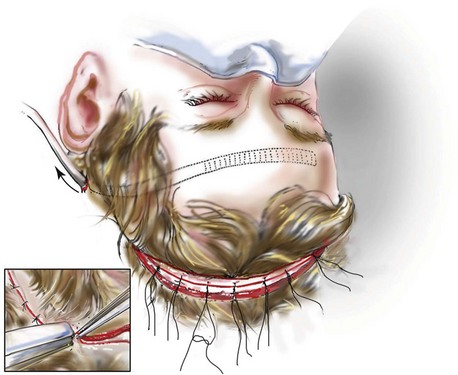
An overview of the skeletal morphology is shown before (Fig. 30-45, A) and after (Fig. 30-45, B) Facial Bipartition (FB) osteotomies and anterior cranial vault reshaping, repositioning, and stabilization. The locations of the proposed osteotomies are indicated.
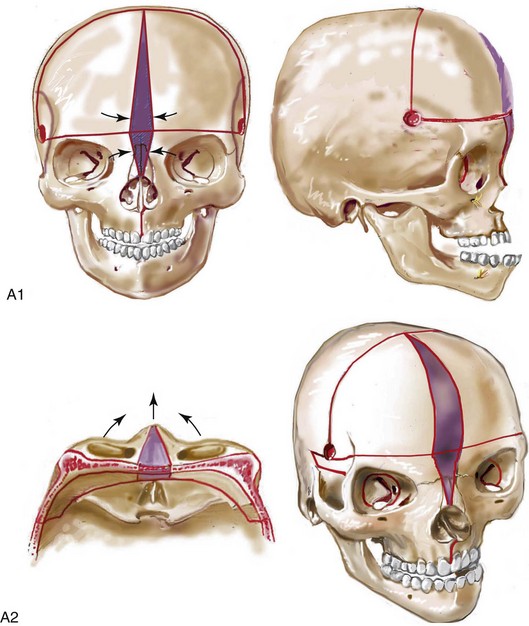
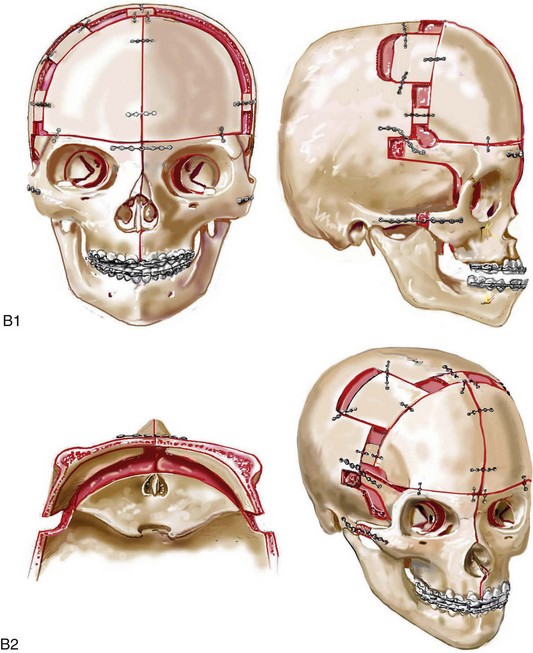
Staging of Reconstruction for Crouzon Syndrome
Primary Brain Decompression: Cranio–Orbital Reshaping during Infancy
The initial surgery for a child with a craniosynostosis syndrome generally requires cranial suture release, simultaneous anterior cranial vault and upper orbital osteotomies, and reshaping and advancement for brain decompression.200,203,205,216,274 There continues to be a search for minimally invasive approaches to the surgical decompression of the brain in the infant with syndromal craniosynostosis; the goal is to limit morbidity while achieving morphologic correction or at least improvement. Current minimally invasive techniques that are under consideration include endoscopic suture release; spring-mediated cranioplasty; and DO procedures. These techniques must be compared with the standard “open” approach of cranio–orbital reshaping carried out by an experienced craniofacial team; this is described later in this chapter. In addition, the preferred timing for brain decompression (i.e., <6 months; 6 to 12 months, >12 months) remains undetermined. Current studies support the 6- to 12-month time frame for first-stage brain decompression whenever feasible. Operating at an earlier age is more likely to result in the need for early reoperation and may involve increased perioperative risk.
In the infant with Crouzon syndrome who presents with bicoronal synostosis, my preference is to carry out the primary cranio–orbital reshaping when the child is 9 to 12 months old unless clear signs of increased intracranial pressure are identified earlier during his or her life.153,200,203,205,216 The standard technique of reshaping of the upper three quarters of the orbits and the tenon extensions is geared toward decreasing the bitemporal and anterior cranial base width while achieving anteroposterior advancement. This increases the depth of the anterior cranial base and the upper orbits with at least some improvement of ocular proptosis. The anterior cranial vault is then reconstructed in accordance with the patient’s morphologic needs. The goals at this stage are to provide increased space in the anterior fossa for the brain; to increase orbital volume for better eye protection; and to improve the morphology of the forehead and the upper orbits. By allowing additional growth to occur before surgery (i.e., by waiting until the child is 9 to 12 months old), the reconstructed cranial vault and the upper orbital shape are better maintained with less need for repeat craniotomy procedures during early childhood.
Further Craniotomy for Brain Decompression during Childhood
After the initial suture release and the reshaping of the cranio–orbital region during infancy, the child is observed clinically at set intervals by the craniofacial surgeon, the pediatric neurosurgeon, the pediatric ophthalmologist, and the developmental specialist; he or she also undergoes interval CT scanning. Should signs of increased intracranial pressure develop, urgent brain decompression via further cranial vault expansion and reshaping is performed.64,87,137,200,203,205,216 When increased intracranial pressure occurs, the suspected location of brain compression influences the region of the skull for which further expansion and reshaping is planned. If progressive brain compression is judged to be anterior, then additional forehead and upper orbital osteotomies with reshaping for expansion are carried out. The technique is similar to that described earlier in this chapter. If the problem is posterior, then expansion of the occipital region with the patient in the prone position is required. The repeat craniotomy is often complicated by brittle irregular cortical bone, which lacks a diploic space and which may contain sharp spicules that pierce the dura; by the presence of previously placed fixation hardware in the operative field; and by convoluted thin dura that is compressed against or herniated into the inner table of the skull. This results in a greater potential for dural tears during the calvarectomy than would normally occur during a primary procedure.
Management of the Upper Midface Deformity during Childhood
The approach selected to manage upper midface deficiencies and anomalies and residual cranial vault dysplasia should depend on the extent and location of dysmorphology.200,203,205 A main objective of this phase of reconstruction is to normalize the orbits, the zygomas, and the cranial vault. The correction of the maxillomandibular deformity will require orthognathic surgery that includes a separate Le Fort I osteotomy. The selection of either an MB osteotomy (with or without additional orbital segmentation), an FB osteotomy (with or without additional orbital segmentation), or an extracranial Le Fort III osteotomy to manage the basic horizontal, transverse, and vertical upper and midface deficiencies and anomalies will depend on the original malformation, the previous procedures that have been carried out, and the effects of ongoing growth. The final reconstruction of the upper midface deformities in a patient with Crouzon syndrome can be managed when he or she is as young as 7 to 10 years old. (See previous sections in this chapter concerning upper midface reconstruction options).
Orthognathic Procedures to Achieve Facial Balance, an Improved Airway, and Definitive Occlusal Correction
Although the mandible has a normal basic growth potential in a patient with Crouzon syndrome, the maxilla does not. An Angle class III malocclusion caused by deficient maxillary growth is to be expected. A segmented Le Fort I osteotomy for maxillary horizontal advancement, transverse widening, and vertical adjustment is generally required in combination with an osseous genioplasty (vertical reduction and horizontal advancement) to further correct the lower face dysmorphology.200,203,205 Secondary deformity of the mandible is also frequent and requires simultaneous sagittal ramus osteotomies. If correction of chronic nasal obstruction is required, then septoplasty, inferior turbinate reduction, the widening of the pyriform rims, and nasal floor recontouring are simultaneously completed (see Chapter 10). The definitive orthognathic and intranasal procedures are carried out in conjunction with orthodontic treatment that is planned for completion at the time of early skeletal maturity (i.e., ≈13 to 15 years in girls and ≈14 to 16 years in boys).
Assessment of Results in the Patient with Crouzon Syndrome
CT scans have provided the quantitative assessment of baseline craniofacial abnormalities in patients with Crouzon syndrome as well as of surgical results after first-stage cranio–orbital reconstruction.216 The purposes of the quantitative assessment of the craniofacial complex—whether by CT scan or by anthropometric, cephalometric, or model analysis—are to help predict growth patterns, to confirm or refute clinical impressions, to help with treatment planning, and to provide a framework for the objective assessment of the immediate and long-term reconstructive results.
Quantitative Assessment of Presenting Crouzon Deformity and Surgical Results on the Basis of Computed Tomography Scan Analysis after First-Stage Cranio–Orbital Reconstruction
Waitzman and colleagues developed a method of analysis based on CT scan measurements and that allows for a quantitative assessment of the upper and midface skeleton in both the horizontal and transverse planes.272,273 This method of quantitative CT scan analysis was used to document the differences in the cranio–orbito–zygomatic region between children with Crouzon syndrome who had not yet undergone reconstruction and age-matched controls.29 Morphologic results that were achieved in those children 1 year after undergoing standard suture release and anterior cranial vault and upper orbital procedures to reshape these regions were also evaluated.216
The preoperative CT scan measurements of the children with untreated Crouzon syndrome confirmed a widened anterior cranial vault at 108% of normal and a cranial length that averaged only 92% of normal.29 In comparison with age-matched controls, orbital measurements revealed a widened anterior interorbital width at 122% of normal, an increased intertemporal width at 121% of normal, globe protrusion at 119% of normal, and a short medial orbital wall length at only 86% of normal. The distance between the zygomatic buttresses and the interarch widths were found to be increased at 106% and 103% of normal, respectively. The zygomatic arch lengths were substantially shortened at only 87% of the values of age-matched controls. These findings confirmed clinical observations of brachycephalic anterior cranial vaults with shallow and frequently hyperteloric orbits and globe proptosis. In general, the upper midface in patients with Crouzon syndrome is horizontally retrusive and transversely wide, which is reflected in wide and shortened zygomas. The same quantitative CT scan assessment was carried out in the children with Crouzon syndrome more than 1 year after they underwent anterior cranial vault and upper orbital osteotomies with reshaping to compare their values to the new age-matched control values.216 No significant improvement in the cranio–orbito–zygomatic measurements was demonstrated.
Quantitative Intracranial Volume Measurements Before and After Cranio–Orbital Reshaping in Children with Crouzon Syndrome
In a previous study, the intracranial volumes in children with Crouzon syndrome before and after standard cranio–orbital reshaping procedures was documents.209 The intracranial volumes were also compared with those of an age- and gender-matched cohort, and the rate of cranial expansion with growth was reviewed. The study included 13 children who presented sequentially with Crouzon syndrome and who subsequently underwent a standard first-stage cranio–orbital reconstruction by the author (Posnick) in conjunction with a pediatric neurosurgeon. The average age at the time of operation was 13 months (range, 6 to 46 months). Postoperative clinical follow up ranged from 12 to 60 months at the time of the study’s completion. Of the children with Crouzon syndrome who were evaluated preoperatively, 12 out of 13 had intracranial volume values that were greater than the mean. When comparing postoperative volumes with the normative data, all 13 maintained volumes that were at or greater than the mean. When reviewing the cranial capacity of each patient with Crouzon syndrome over time, 5 of the 13 patients approximated the normal growth curve, whereas 6 of the 13 patients exceeded it. According to the findings of the study, for the majority of children who were born with Crouzon syndrome, the cranial capacity will exceed the mean early during life and expand at a rapid rate in conjunction with cranio–orbital decompression and reconstruction. The biologic explanation for these unexpected findings remains unclear. It should be noted that other researchers have studied the intracranial volume of children who were born with craniosynostosis.18,20,21,24,60,64,67,73,80,85–87,93,137,140,141,210,246,247
Staging of Reconstruction for Apert Syndrome
Primary Brain Decompression: Cranio–Orbital Reshaping during Infancy
The initial craniofacial procedure for patients with Apert syndrome generally includes cranial suture release as well as simultaneous anterior cranial vault and upper three-quarter orbital osteotomies, with reshaping and advancement for brain decompression 202 This author’s preference is to carry out these procedures when the child is 9 to 12 months old, unless signs of increased intracranial pressure are identified earlier during the child’s life. The main goals at this stage are to decompress the brain by providing increased space in the anterior cranial vault and to increase the orbital volume for improved eye protection. The fronto–orbital surgical technique is similar to that described for Crouzon syndrome (see the section about Crouzon syndrome earlier in this chapter).
Further Craniotomy for Brain Decompression during Childhood
As described previously for Crouzon syndrome, the initial suture release, brain decompression, and cranio–orbital reshaping are carried out during infancy (i.e., 9 to 12 months of age).202 The child is observed clinically at intervals by the craniofacial surgeon, the pediatric neurosurgeon, the pediatric ophthalmologist, and the developmental pediatrician, and interval CT scanning is also performed. Should signs of increased intracranial pressure develop, further brain decompression with reshaping of the cranial vault to expand the intracranial volume is performed. In patients with Apert syndrome, the posterior cranial vault more commonly requires expansion at this stage. The technique is similar to what has been previously described (see the section about Crouzon syndrome earlier in this chapter).
Management of the Upper Midface Deformity during Childhood
For almost all patients with Apert syndrome, facial bipartition osteotomies in combination with further cranial vault reshaping allow for a better correction of the dysmorphology than can be achieved through any other upper midface procedure (i.e., MB or Le Fort III).202 When using FB osteotomies, the correction of the concave midface arc of rotation is also possible. This further reduces the stigmata of the “flat, wide, and retrusive” facial appearance associated with Apert syndrome. The FB allows the orbits and the zygomas as units to shift to the midline (i.e., correction of hypertelorism) while the maxillary arch is simultaneously widened (i.e., relief of the V-shaped upper midface). Horizontal advancement of the reassembled upper midface complex is then possible to improve orbital depth and zygomatic length. The forehead is generally flat, tall, and retruded, with a constricting band just above the supraorbital ridge. The reshaping of the anterior cranial vault is also simultaneously carried out. A Le Fort III osteotomy is virtually never adequate for an ideal correction of the residual upper midface anomalies documented among patients with Apert syndrome (see earlier section in this chapter concerning upper midface reconstruction options).
Orthognathic Procedures to Achieve Facial Balance, an Improved Airway, and Definitive Occlusal Correction
The mandible has normal basic growth potential in patients with Apert syndrome, but it is generally secondarily deformed. The extent of maxillary deficiency will result in an Angle Class III anterior open-bite malocclusion. A segmented Le Fort I osteotomy is required for horizontal advancement, transverse widening, and vertical adjustment. This is often in combination with an osseous genioplasty and sagittal split ramus osteotomies of the mandible. Septoplasty and inferior turbinate reduction are generally carried out to improve nasal breathing, and the recontouring of the nasal floor and the pyriform rims is also required (see Chapter 10). The definitive orthognathic and intranasal surgery is carried out in conjunction with orthodontic treatment and planned for completion at the time of early skeletal maturity (i.e., ≈13 to 15 years of age).202
Assessment of Results in the Patient with Apert Syndrome
Quantitative Assessment of Presenting Apert Deformity and Surgical Results on the Basis of Computed Tomography Scan Analysis after First-Stage Cranio–Orbital Reconstruction
In a previously published study, a method of quantitative CT scan analysis was applied to document the differences in the cranio–orbito–zygomatic region between children with Apert syndrome who had not been operated on and age-matched controls.29 Eight consecutive infants and young children with Apert syndrome who underwent a standard cranio–orbital procedure by a single craniofacial surgeon (Posnick) in conjunction with one of three pediatric neurosurgeons over a 4-year period were reviewed 1 year after surgery.217 The series included seven girls and one boy, with an average age at surgery of 12 months (range, 9 to 23 months). The average postoperative follow-up period was 34 months (range, 12 to 48 months) at the close of the study. Preoperative and postoperative (>1 year) CT scans were compared with those of age-matched controls, and percentages of normal were then compared for significant differences. Two of the children had clear evidence of hydrocephalus and required ventriculoperitoneal shunting before the cranio–orbital reconstruction at 3 and 5 months of age. A third child had mildly increased ventricular size, but clinical correlation did not suggest the need for shunting.
Significant preoperative morphologic findings included a wide anterior cranial vault at 110% of normal, a maximum cranial length that averaged only 90% of normal, a substantially widened anterior interorbital width at 117% of normal, an increased lateral interorbital distance at 112% of normal, and a widened bitemporal width at 122% of normal.217 Globe protrusion was significant at 121% of normal, and the medial orbital wall length was less than normal at 92%. In the upper midface zygomatic region, both the width between the zygomatic buttresses and the interarch width were found to be increased at 109% of normal, whereas the zygomatic arch lengths were substantially shortened at 79% of normal. The measurements confirmed the clinical observations of brachycephalic and hyperteloric anterior cranial vaults, orbits, and zygomas accompanied by globe proptosis and midface deficiency. Results of surgical reconstruction as documented by CT scan measurements demonstrated that, at more than 1 year after surgery, none of the craniofacial measurements had significantly improved (P < .05) as compared with those of the new age-matched controls.
Quantitative CT scan measurements of the cranio–orbito–zygomatic region confirmed the clinical findings in the unoperated children to be brachycephalic anterior cranial vaults and upper face hypertelorism of the orbits and the zygomas,) with ocular proptosis and a flat midface.217 It was found that cranial vault and upper orbital reshaping before 1 year of age does not achieve or maintain a corrected shape in the craniofacial skeleton in children with Apert syndrome. Although the cranio–orbito–zygomatic dysmorphology did not worsen when analyzed at least 1 year after surgery, values remained far from those of the new age-matched controls, thereby confirming the need for a staged reconstructive approach.
Quantitative Intracranial Volume Measurements Before and After Cranio–Orbital Reshaping in Children with Apert Syndrome
In published studies, a proven method for obtaining intracranial volumes with the use of CT scan measurement was applied to a consecutive series of children with Apert syndrome before any craniofacial procedures were performed.209 A standard cranio–orbital operation was performed for each child, and this was followed by longitudinal follow up and the remeasurement of the intracranial volume at least 1 year later. The patients’ intracranial volumes were also compared with those of an age-matched cohort, and each patient’s cranial growth velocity was reviewed. The study included six girls and two boys with an average age at operation of 12 months (range, 9 to 23 months). The average postoperative follow-up duration at the close of the study was 34 months (range, 12 to 48 months).209
Preoperative intracranial volume in the patients with Apert syndrome ranged from 393 mL in a 2-month-old girl to 1715 mL in a 28-month-old girl.209 A comparison of the preoperative intracranial volume in patients with Apert syndrome with those of the age- and gender-matched cohort group showed that six of the eight patients had values of at least two standard deviations above the mean. Interestingly, the other two were infants who underwent ventriculoperitoneal shunting for hydrocephalus earlier in life. Their measured preoperative intracranial volumes were two standard deviations below the mean. When the patients’ postoperative intracranial volumes were compared with those of the cohort group, all eight achieved values of at least two standard deviations above the mean. The majority of the measured preoperative and postoperative intracranial volume values of the patients with Apert syndrome followed a growth curve that greatly exceeded the rate expected for normal children. In three of the patients, cranial vault growth velocity seemed to match closely that expected for a normal child but with a starting point determined by their preoperative values.
The findings confirmed that untreated patients with Apert syndrome are generally macrocephalic early during their lives, that standard cranio–orbital procedures carried out during childhood do not alter this trend, and that continued cranial volume expansion often exceeds the mean. The ability to develop “normal” intracranial volume standards and to identify variations from normal in specific syndromes and in individual patients before and after surgery continues to be elusive.18,20,21,24,60,73,80,85,86,93,96,140,141,210,246,247
Quantitative Assessment of Presenting Deformity during the Mixed Dentition in Children with Apert Syndrome and Surgical Results after Facial Bipartition Osteotomy on the Basis of Computed Tomography Scan Analysis
In published studies, children with Apert syndrome were assessed during their middle childhood years with the use of quantitative CT scan measurements.220 Many of these children’s measurements varied from normal as compared with those of age-matched controls. The orbital measurements showed a substantially increased anterior interorbital width (123% of normal), an increased mid-interorbital width (122% of normal), and an increased intertemporal width (126% of normal). The globe protrusion beyond the sagittal plane of the lateral orbital walls was excessive (142% of normal). There was also a short medial orbital wall length (85% of normal). The width between the lateral orbital walls was excessive at 111% of normal. Zygomatic arch lengths were substantially shortened at 83% of normal.
After undergoing FB osteotomies with three-dimensional repositioning in combination with cranial vault reshaping, CT scan measurements were again taken both early after the operation and 1 year later.220 An analysis of the measurements showed an improvement toward the normal range. As compared with the values of age-matched controls, the orbital measurements reflected the correction of the hypertelorism; the anterior interorbital width early after operation was 106% of normal and later 105% of normal. The mid-interorbital width initially improved to 106% of normal and later to 100% of normal. The width between the lateral orbital walls stabilized at 108% of normal, and the intertemporal width was 115% of normal, which was an improvement over the preoperative value of 126% of normal. The zygomatic arch length was initially overcorrected at 110% of normal and then stabilized at 103% of normal.
Further studies were carried out to evaluate the presence of extradural (retrofrontal) dead space after the FB osteotomy to reconstruct the upper midface deformity associated with Apert syndrome.208 Seven patients with Apert syndrome (mean age, 8 years) underwent FB osteotomies with advancement. Extradural (retrofrontal) dead space was measured from a reproducible axial CT scan slice for each patient at standard postoperative intervals (1 to 2 weeks, 6 to 8 weeks, and 1 year). An initial extradural (retrofrontal) dead space was identified early after surgery in each patient. There was good resolution of the dead space by the 6- to 8-week postoperative interval through expansion of the dura and the frontal lobes of the brain. The dead space was confirmed to be closed in all patients at the 1-year postoperative interval. The morbidity of the same consecutive series of patients was then reviewed. For the seven children, there were no deaths, cardiopulmonary sequelae, injuries to the brain or eyes, new seizure activity, or central nervous system problems.
References
1. Ades, LC, Mulley, JC, Senga, IP, et al. Jackson-Weiss syndrome: Clinical and radiological findings in a large kindred and exclusion of the gene from 7p21 and 5qter. Am J Med Genet. 1994; 51:121.
2. Al-Saleh, A, Riekstins, A, Forrest, CR, et al. Sleep-related disordered breathing in children with syndromic craniosynostosis. J Craniomaxillofac Surg. 2011; 39:153–157.
3. Ahmad, F, Cobb, AR, Mills, C, et al. Frontofacial monoblock distraction in the very young: A review of 12 consecutive cases. Plast Reconstr Surg. 2012; 129:488E.
4. Anderson, PJ, Hall, CM, Evans, RD, et al. The cervical spine in Saethre–Chotzen syndrome. Cleft Palate Craniofac J. 1997; 34:79.
5. Anderson, PJ, Netherway, DJ, Cox, TC, et al. Do craniosynostosis syndrome phenotypes with both FGFR2 and TWIST mutations have a worse clinical outcome? J Craniofac Surg. 2006; 17:166.
6. Anderson, PM, Geiger, I. Craniosynostosis: A survey of 204 cases. J Neurosurg. 1956; 22:229.
7. Apert, E. De l’acrocephalosyndactlie. Bull Mem Soc Med Hop Paris. 1906; 23:1310.
8. Arnaud, E, Marchac, D, Renier, D. Reduction of morbidity of the frontofacial monobloc advancement in children by the use of internal distraction. Plast Reconstr Surg. 2007; 120:1009–1026.
9. Atkins, FRB. Hereditary craniofacial dysostosis or Crouzon disease. Med Press Circ. 1937; 195:118.
10. Bachmayer, DI, Ross, RB. Stability of Le Fort III advancement surgery in children with Crouzon, Apert, and Pfeiffer syndromes. Cleft Palate J. 1986; 23(Suppl 1):69–74.
11. Bachmayer, DI, Ross, RB, Munro, IR. Maxillary growth following Le Fort III advancement surgery in Crouzon, Apert, and Pfeiffer syndromes. Am J Orthod Dentofacial Orthop. 1986; 90:420–430.
12. Baldwin, JL. Dysostosis craniofacialis of Crouzon: A summary of recent literature and case reports with involvement of the ear. Laryngoscope. 1968; 78:1660.
13. Barr, M, Jr., Kreiborg, S. The cervical spine in the Apert syndrome. Am J Med Genet. 1992; 43:704.
14. Bernard, JP, Levaillant, JM. Prenatal diagnosis of craniosynostosis. Neurochirurgie. 2006; 52:246.
15. Berrada, A, Erode sur les craniostenoses en milieum marocain: A propos de 87 observations (these medicale). 1963-1964.
16. Bertelsen, TI. The premature synostosis of the cranial sutures. Acta Ophthalmol. 1958; 51(Suppl):87.
17. Bigot, C. L’acrocephalo-syndactylie (these pour le doctorat en medicine). Paris, Faculte de Medicine. 1922.
18. Blinkov, SM, Glezer, II, Haigh, B. The human brain: A quantitative handbook. New York: Basic Books; 1968.
19. Bradley, JP, Gabbay, JS. Monobloc advancement by distraction osteogenesis decreases morbidity and relapse. Plast Reconstr Surg. 2006; 118:1585.
20. Bray, PF, Shields, WD, Wolcott, GJ, et al. Occipitofrontal head circumference: An accurate measure of intracranial volume. J Pediatr. 1969; 75:303.
21. Breiman, RS, Beck, JW, Korobkin, M, et al. Volume determinations using computed tomography. Am J Roentgenol. 1982; 138:329.
22. Broder, HL. Psychological research of children with craniofacial anomalies: Review, critique and implications for the future. Cleft Palate Craniofac J. 1997; 34:402.
23. Bu, BH, Kaban, LB, Vargervik, K. Effect of Le Fort III osteotomy on mandibular growth in patients with Crouzon and Apert syndrome. J Oral Maxillofac Surg. 1989; 47:666.
24. Buda, FB, Reed, JC, Rabe, EF. Skull volume in infants. Am J Dis Child. 1975; 129:1171.
25. Camfield, PR, Camfield, CS, Cohen, M, Jr. Neurologic aspects of craniosynostosis. In: Cohen MM, Jr., MacLean RE, eds. Craniosynostosis: Diagnosis, evaluation and management. ed 2. New York: Oxford University Press; 2000:177–183.
26. Campbell, JW, Albright, AL, Losken, HW, et al. Intracranial hypertension after cranial vault decompression for craniosynostosis. Pediatr Neurosurg. 1995; 22:270.
27. Carinci, F, Pezzetti, F, Locci, P, et al. Apert and Crouzon syndromes: Clinical findings, genes and extracellular matrix. J Craniofac Surg. 2005; 16:361.
28. Carpenter, G. Two sisters showing malformations of the skull and other congenital abnormalities. Rep Soc Study Dis Child (London). 1901; 1:110.
29. Carr, M, Posnick, JC, Pron, G, Armstrong, D. Cranio-orbito-zygomatic measurements from standard CT scans in unoperated Crouzon and Apert infants: Comparison with normal controls. Cleft Palate Craniofac J. 1992; 29(2):129–136.
30. Chen, CP, Lin, SP, Su, YN, et al. A cloverleaf skull associated with Crouzon syndrome. Arch Dis Child Fetal Neonatal Ed. 2006; 91:98.
31. Chen, P, Por, Y, Liou, EJ, Chang, FC. Maxillary distraction osteogenesis in the adolescent cleft patient: Three-dimensional computed tomography analysis of linear and volumetric changes over five years. Cleft Palate Craniofac J. 2011; 40:445–454.
32. Chin, M, Toth, BA. Le Fort III advancement with gradual distraction using internal devices. Plast Reconstr Surg. 1997; 100:819.
33. Chotzen, F. Eine eigenartige familiare entwicklungsstorung. (Akrocephalosyndaktylie, dystosis craniofacialis und hypertelorismus). Monatschr Kinderheilkd. 1932; 55:97.
34. Chumas, PD, Cinalli, G, Arnaud, E, et al. Classification of previously unclassified cases of craniosynostosis. J Neurosurg. 1997; 86:177–181.
35. Clinical practice guideline: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics. 2002; 109:704–712.
36. Coccaro, PJ, McCarthy, JG, Epstein, FJ, et al. Early and late surgery in craniofacial dysostosis: A longitudinal cephalometric study. Am J Orthod. 1980; 77:421–436.
37. Cohen, MM, Jr. Perspectives on craniosynostosis. West J Med. 1980; 132:508.
38. Cohen, MM, Jr. Let’s call it “Crouzonodermoskeletal syndrome” so we won’t be prisoners of our own conventional terminology. Am J Med Genet. 1999; 84:74.
39. Cohen, MM, Jr. Carpenter syndrome. In: Cohen MM, Jr., MacLean RE, eds. Craniosynostosis: diagnosis, evaluation and management. ed 2. New York: Oxford University Press; 2000:377–379.
40. Cohen, MM, Jr. Saethre-Chotzen syndrome. In: Cohen MM, Jr., MacLean RE, eds. Craniosynostosis: Diagnosis, evaluation and management. ed 2. New York: Oxford University Press; 2000:374–376.
41. Cohen, MM, Jr. Jackson-Weiss syndrome. Am J Med Genet. 2001; 100:325–329.
42. Cohen, MM, Jr. FGFs/FGFRs and associated disorders. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, eds. Inborn errors of development. New York: Oxford University Press; 2004:380–400.
43. Cohen, MM, Jr. Invited comment: Perspectives on craniosynostosis. Am J Med Genet. 2005; 136A:313–326.
44. Cohen, MM, Jr., Craniofacial abnormalities. Potter’s pathology of the fetus, infant, and child. ed 2. Gilbert-Barness, E, eds. Potter’s pathology of the fetus, infant, and child; vol I. Mosby/Elsevier, Philadelphia, 2007:885–918.
45. Cohen, MM, Jr. Cloverleaf skull: Etiologic heterogeneity and pathogenetic variability. J Craniofac Surg. 2009; 20(Suppl):652–656.
46. Cohen, MM, Jr., Apert, Crouzon, and Pfeiffer syndromes. Monographs in human genetics. Craniosynostosis: Molecular genetics, diagnosis, and treatment. Muenke, M, Kress, W, Collmann, H, Solomon, BD, eds. Craniosynostosis: Molecular genetics, diagnosis, and treatment; 19. 2011:67–88.
47. Cohen, MM, Jr. No man’s craniosynostosis: The arcane of sutural knowledge. J Craniofac Surg. 2012; 23:338–348.
48. Cohen, MM, Jr., MacLean, RE. Craniosynostosis: Diagnosis, evaluation and management, ed 2. New York: Oxford University Press; 2000.
49. Connolly, JP, Gruss, J, Seto, ML, et al. Progressive postnatal craniosynostosis and increased intracranial pressure. Plast Reconstr Surg. 2004; 113:1313.
50. Correa Normando, AD, da Silva Filho, OG, Capelozza Filho, L. Influence of surgery on maxillary growth in cleft lip and/or palate patients. J Craniomaxillofac Surg. 1992; 20:111.
51. Crouzon, O. Une nouvelle famille atteinte de dysostose craniofaciale hereditaire. Arch Med Enfant. 1915; 18:540.
52. Crouzon, O. Sur la dysostose cranio-faciale hereditaire et sur les rapports avec l’acrocephalosyndactylie. Bull Mem Soc Med Hop Paris. 1932; 48:1568.
53. Crouzon, O. Les dysostose prechordales. Bull Acad Med. 1936; 115:696.
54. Cruveiller, J. La Maladie d’Apert-Crouzon (these medicale). Paris. 1954.
55. Cushing, H. Neurosurgery. In: Keen WW, ed. Surgery: Its principles and practice. Philadelphia: WB Saunders; 1908:254.
56. David, DJ, Cooter, RD. Craniofacial infections in 10 years of transcranial surgery. Plast Reconstr Surg. 1987; 80:213.
57. David, DJ, Sheen, R. Surgical correction of Crouzon syndrome. Plast Reconstr Surg. 1990; 85:344.
58. David, LR, Velotta, E, Weaver, RG, Jr., et al. Clinical findings precede objective diagnostic testing in the identification of increased ICP in syndromic craniosynostosis. J Craniofac Surg. 2002; 13:676.
59. DeGunten, P. Contribution a l’etude des malformations de la face et des maxillaires dans la dysostose craniofaciale. Ann Otolaryngol. 1938; 57:1056.
60. Dekaban, AS. Tables of cranial and orbital measurements, cranial volume and derived indexes in males and females from 7 days to 20 years of age. Ann Neurol. 1977; 2:485.
61. Devine, P, Bhan, I, Feingold, M, et al. Completely cartilaginous trachea in a child with Crouzon syndrome. Am J Dis Child. 1984; 138:40.
62. Dickerman, RD, Lefkowitz, M, Arinsburg, SA, Schneider, SJ. Chiari malformation and odontoid panus causing craniovertebral stenosis in a child with Crouzon syndrome. J Clin Neurosci. 2005; 12:963.
63. Diner, PA. Le Fort III advancement with gradual distraction using internal devices [discussion]. Plast Reconstr Surg. 1997; 100:831.
64. Disler, DG, Marr, DS, Rosenthal, DI. Accuracy of volume measurements of computed tomography and magnetic resonance imaging phantoms by three-dimensional reconstruction and preliminary clinical application. Invest Radiol. 1994; 29:739.
65. Farkas, LG, Posnick, JC. Growth and development of regional units in the head and face based on anthropometric measurements. Cleft Palate Craniofac J. 1992; 29:301–302.
66. Farkas, LG, Posnick, JC, Hreczko, T. Anthropometric growth study of the head. Cleft Palate Craniofac J. 1992; 29:303–307.
67. Farkas, LG, Posnick, JC, Hreczko, T. Growth patterns in the orbital region: A morphometric study. Cleft Palate Craniofac J. 1992; 29:315–317.
68. Farkas, LG, Posnick, JC, Hreczko, T. Growth patterns of the face: a morphometric study. Cleft Palate Craniofac J. 1992; 29:308–314.
69. Fearon, JA. The Le Fort III osteotomy: To distract or not to distract? Plast Reconstr Surg. 2001; 107:1091–1103.
70. Fearon, JA. Halo distraction of the Le Fort III in syndromic craniosynostosis: A long-term assessment. Plast Reconstr Surg. 2005; 115:1524.
71. Fearon, JA, Whitaker, LA. Complications with facial advancement: A comparison between the Le Fort III and monobloc advancements. Plast Reconstr Surg. 1993; 91:990.
72. Fearon, JA, Yu, J, Bartlett, SP, et al. Infections in craniofacial surgery: A combined report of 567 procedures from two centers. Plast Reconstr Surg. 1997; 100:862.
73. Fernandez, F, Moule, N, Singhi, S. Roentgenographic skull volume in Jamaican children between the ages of 1 month and 5 years. West Indian Med J. 1984; 33:227.
74. Ferreira, JC, Carter, SM. Second-trimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstet Gynecol. 1999; 14:426.
75. Figueroa, AA, Polley, JW, Friede, H, et al. Long-term skeletal stability after maxillary advancement with distraction osteogenesis using a rigid external distraction device in cleft maxillary deformities. Plast Reconstr Surg. 2004; 114:1382.
76. Figueroa, AA, Polley, JW, Ko, EW. Maxillary distraction for the management of cleft maxillary hypoplasia with a rigid external distraction system. Semin Orthod. 1999; 5:46.
77. Fishman, MA, Hogan, GR, Dodge, PR. The concurrence of hydrocephalus and craniosynostosis. J Neurosurg. 1971; 34:621.
78. Flippen, JH, Jr. Craniofacial dysostosis of Crouzon: Report of a case in which the malformation occurred in four generations. Pediatrics. 1950; 5:90.
79. Flores, RL, Shetye, PR, Zeitler, D, et al. Airway changes following Le Fort III distraction osteogenesis for syndromic craniosynostosis: A clinical and cephalometric study. Plast Reconstr Surg. 2009; 124:590–601.
80. Fok, H, Jones, BM, Gault, DG, et al. Relationship between intracranial pressure and intracranial volume in craniosynostosis. Br J Plast Surg. 1992; 45:394.
81. Freitas, EC, Nascimento, SR, de Mello, MP, Gil-da-Silva-Lopes, VL. Q289P mutation in FGFR2 gene causes Saethre-Chotzen syndrome: some considerations about familial heterogeneity. Cleft Palate Craniofac J. 2006; 43:142.
82. Fujisawa, H, Hasegawa, M, Kida, S, Yamashita, J. A novel fibroblast growth factor receptor 2 mutation in Crouzon syndrome associated with Chiari type I malformation and syringomyelia. J Neurosurg. 2002; 97:396.
83. Funato, N, Nohtomi-Ohyama, J, Ohyama, K. Monozygotic twins concordant for Crouzon syndrome. Am J Med Genet A. 2005; 133:225.
84. Garcin, M, Thurel, R, Rudeaux, P. Sur en cas asole de dysostose craniofaciale (maladie de Crouzon) avec extradactylie. Bull Soc Med Hop. 1932; 56:1458.
85. Gault, DT, Brunelle, F, Renier, D, et al. The calculation of intracranial volume using CT scans. Childs Nerv Sys. 1988; 4:271.
86. Gault, DT, Renier, D, Marchac, D, et al. Intracranial volume in children with craniosynostosis. J Craniofac Surg. 1990; 1:1.
87. Gault, DT, Renier, D, Marchac, D, Jones, BM. Intracranial pressure and intracranial volume in children with craniosynostosis. Plast Reconstr Surg. 1992; 90:230–271.
88. Genest, P, Mortezai, MA, Tremblay, M. Le syndrome d’Apert (acrocephalosyndactyly). Arch Fr Pediatr. 1966; 23:887.
89. Gillies, HD, Harrison, SH. Operative correction by osteotomy of recessed malar maxillary compound in case of oxycephaly. Br J Plast Surg. 1950; 3:123.
90. Golabi, M, Chierici, G, Ousterhout, DK, et al. Radiographic abnormalities of Crouzon syndrome: A survey of 23 cases. Proc Greenwood Genet Center. 1984; 3:102.
91. Golabi, M, Edwards, MSB, Ousterhout, DK. Craniosynostosis and hydrocephalus. Neurosurgery. 1987; 21:63.
92. Goldberg, S, Shatz, A, Picard, E, et al. Endoscopic findings in children with obstructive sleep apnea: Effects of age and hypotonia. Pediatr Pulmonol. 2005; 40:205–210.
93. Gordon, IRS. Measurement of cranial capacity in children. Br J Radiol. 1966; 39:377.
94. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
95. Gorry, MC, Preston, RA, White, GJ, et al. Crouzon syndrome: Mutations in two splice forms of FGFR2 and a common point mutation shared with Jackson-Weiss syndrome. Hum Mol Genet. 1995; 4:1387.
96. Gosain, AK, McCarthy, JG, Glatt, P, et al. A study of intracranial volume in Apert syndrome. Plast Reconstr Surg. 1995; 95:284.
97. Gosain, AK, Santoro, TD, Havlik, RJ, et al. Midface distraction following Le Fort III and monobloc osteotomies: Problems and solutions. Plast Reconstr Surg. 2002; 109:1797.
98. Gray, TL, Casey, T, Selva, D, et al. Ophthalmic sequelae of Crouzon syndrome. Ophthalmology. 2005; 112:1129.
99. Green, SM. Pathological anatomy of the hands in Apert syndrome. J Hand Surg. 1982; 7:450.
100. Grenet, H, Leveuf, J, Issac, G. Etude anatomique de la maladie de Crouzon. Bull Soc Pediatr. 1934; 32:343.
101. Gulleminault, C, Lee, JH, Chan, A. Pediatric obstructive sleep apnea syndrome. Arch Pediatr Adolesc Med. 2005; 159:775–785.
102. Harada, K, Baba, Y, Ohyama, K, et al. Maxillary distraction osteogenesis for cleft lip and palate children using an external, adjustable, rigid distraction device: A report of 2 cases. J Oral Maxillofac Surg. 2001; 59:1492.
103. Harada, K, Sato, M, Omura, K. Long-term maxillomandibular skeletal and dental changes in children with cleft lip and palate after maxillary distraction. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006; 102:292.
104. Harris, V, Beligere, N, Pruzansky, S. Progressive generalized bony dysplasia in Apert syndrome. Birth Defects. 1977; 14:175.
105. Hoeve, HL, Joosten, KF, van den Berg, S. Management of obstructive sleep apnea syndrome in children with craniofacial malformation. Int J Pediatr Otorhinolaryngol. 1999; 49(Suppl 1):S59–S61.
106. Hoeve, HL, Pijpers, M, Joosten, KF. OSAS in craniofacial syndromes: An unresolved problem. Int J Pediatr Otorhinolaryngol. 2003; 67(Suppl 1):111–113.
107. Hoffman, HJ, Mohr, G. Lateral canthal advancement of the supraorbital margin: A new corrective technique in the treatment of coronal synostosis. J Neurosurg. 1976; 45:376.
108. Hogan, GR, Bauman, ML. Hydrocephalus in Apert syndrome. J Pediatr. 1971; 79:782.
109. Hogeman, KE, Willmar, K. On Le Fort III osteotomy for Crouzon disease in children: Report of a four-year follow-up in one patient. Scand J Plast Reconstr Surg. 1974; 8:169–172.
110. Hollier, L, Kelly, P, Babigumira, E, et al. Minimally invasive Le Fort III distraction. J Craniofac Surg. 2002; 13:44.
111. Hopper, RA. Frontofacial monobloc distraction in the very young: A review of 12 consecutive cases. Plast Reconstr Surg. 2012; 129(3):498e–501e.
112. Huang, CS, Harikrishnan, P, Liao, YF, et al. Long-term follow-up after maxillary distraction osteogenesis in growing children with cleft lip and palate. Cleft Palate Craniofac J. 2007; 44:274.
113. Iannetti, G, Fadda, T, Agrillo, A, et al. Le Fort III advancement with and without osteogenesis distraction. J Craniofac Surg. 2006; 17:536–543.
114. Ishii, K, Kaloust, S, Ousterhout, DK, Vargervik, K. Airway changes after Le Fort III osteotomy in craniosynostosis syndromes. J Craniofac Surg. 1996; 7:363–370.
115. Ito, S, Sekido, K, Kanno, H, et al. Phenotypic diversity in patients with craniosynostoses unrelated to Apert syndrome: The role of fibroblast growth factor receptor gene mutations. J Neurosurg. 2006; 102(1 Suppl):23.
116. Jabs, EW, Li, X, Scott, AF, et al. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994; 8:275–279.
117. Jensen, JN, McCarthy, JG, Grayson, BH, et al. Bone deposition/generation with Le Fort III (midface) distraction. Plast Reconstr Surg. 2007; 119:298–307.
118. Juberg, RC, Chambers, SR. An autosomal recessive form of craniofacial dysostosis (the Crouzon syndrome). J Med Genet. 1973; 10:89.
119. Kaban, LB, Conover, M, Mulliken, J. Midface position after Le Fort III advancement: A long-term follow-up study. Cleft Palate J. 1986; 23(Suppl):75–77.
120. Kaban, LB, West, B, Conover, M, et al. Midface position after Le Fort III advancement. Plast Reconstr Surg. 1984; 73:758–767.
121. Kaloust, S, Ishii, K, Vargervik, K. Dental development in Apert syndrome. Cleft Palate Craniofac J. 1997; 34:117.
122. Kapp-Simon, KA, Figueroa, A, Jocher, CA, et al. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction. Plast Reconstr Surg. 1993; 92:831.
123. Kasser, J, Upton, J. The shoulder, elbow, and forearm in Apert syndrome. Clin Plast Surg. 1991; 18:381.
124. Khong, JJ, Anderson, P, Gray, TL, et al. Ophthalmic findings in Apert syndrome prior to craniofacial surgery. Am J Ophthalmol. 2006; 142:328.
125. Kirkpatrick, WN, Koshy, CE, Waterhouse, N, et al. Pediatric transcranial surgery: A review of 114 consecutive procedures. Br J Plast Surg. 2002; 55:561.
126. Kolar, JC, Munro, IR, Farkas, LG. Patterns of dysmorphology in Crouzon syndrome: An anthropometric study. Cleft Palate J. 1988; 25:235–244.
127. Kreiborg, S. Crouzon syndrome: A clinical and roentgencephalometric study. Scand J Plast Reconstr Surg. 1981; 18(Suppl):1.
128. Kreiborg, S, Barr, M, Jr., Cohen, M, Jr. Cervical spine in the Apert syndrome. Am J Med Genet. 1992; 43:704.
129. Kreiborg, S, Cohen, MM, Jr. The infant Apert skull. Neurosurg Clin North Am. 1991; 2:551–554.
130. Kreiborg, S, Cohen, MM, Jr. The oral manifestations of the Apert syndrome. J Craniofac Genet Dev Biol. 1992; 12:41.
131. Kreiborg, S, Marsh, JL, Cohen, MM, Jr., et al. Comparative three-dimensional analysis of CT scans of the calvaria and cranial base in Apert and Crouzon syndromes. J Craniomaxillofac Surg. 1993; 21:181–188.
132. Kreiborg, S, Prydsoe, U, Dahl, E, Fogh-Anderson, P. Calvarium and cranial base in Apert syndrome: An autopsy report. Cleft Palate J. 1976; 13:296–303.
133. Lane, LC. Pioneer craniectomy for relief of mental imbecility due to premature sutural closure and microcephalus. JAMA. 1892; 18:49.
134. Lannelongue, M. De la craniectomie dans la microcephalie. Compte Rendu Acad Sci. 1890; 110:1382.
135. Lauritzen, C, Lilja, J, Jarlstedt, J. Airway obstruction and sleep apnea in children with craniofacial anomalies. Plast Reconstr Surg. 1986; 77:1–6.
136. Lee, Y, Kim, WJ. How to make the blockage between the nasal cavity and intracranial space using a four-layer sealing technique. Plast Reconstr Surg. 2006; 117:233.
137. Lichtenberg, R. Radiographic du crane de 226 enfants normaux de la naissance a 8 ans: Impressions digitformes, capacite: Angles et indices (thesis). Paris: University of Paris; 1960.
138. Lo, LJ, Chen, YR. Airway obstruction in severe syndromic craniosynostosis. Ann Plast Surg. 1999; 43:258–264.
139. Lo, LJ, Marsh, JL, Yoon, J, et al. Stability of fronto-orbital advancement in non-syndromic bilateral coronal synostosis: A quantitative three-dimensional computed tomographic study. Plast Reconstr Surg. 1996; 98:393.
140. Mackinnon, IL. The relation of the capacity of the human skull to its roentgenological length. Am J Radiol. 1955; 74:1026.
141. Mackinnon, IL, Kennedy, JA, Davies, TV. The estimation of skull capacity from roentgenological measurements. Am J Radiol. 1956; 76:303.
142. Mah, J, Kasser, J, Upton, J. The foot in Apert syndrome. Clin Plast Surg. 1991; 18:391.
143. Mantilla-Capacho, JM, Arnaud, L, Diaz-Rodriguez, M, Barros-Nunez, P. Apert syndrome with preaxial polydactyly showing the typical mutation Ser252Trp in the FGFR2 gene. Genet Couns. 2005; 16:403.
144. Marchac, D. Radical forehead remodeling for craniosynostosis. Plast Reconstr Surg. 1978; 62:335–338.
145. Marchac, D, Renier, D, Jones, BM. Experience with the “floating forehead. ”. Br J Plast Surg. 1988; 41:1–15.
146. Marsh, JL, Gado, M. Surgical anatomy of the craniofacial dysostoses: Insights from CT scans. Cleft Palate J. 1982; 19:212–221.
147. Marsh, JL, Galic, M, Vannier, MW. Surgical correction of craniofacial dysmorphology of Apert syndrome. Clin Plast Surg. 1991; 18:251.
148. Marsh, JL, Vannier, MW. The “third” dimension in craniofacial surgery. Plast Reconstr Surg. 1983; 71:759–767.
149. Mathijssen, I, Arnaud, E, Marchac, D, et al. Respiratory outcome of midface advancement with distraction: A comparison between Le Fort III and frontofacial monobloc. J Craniofac Surg. 2006; 17:880–882.
150. Matsumoto, K, Nakanishi, H, Koizumi, Y, et al. Segmental distraction of the midface in a patient with Crouzon syndrome. J Craniofac Surg. 2002; 13:273.
151. Mavili, ME, Tuncbilek, G, Vargel, I. Rigid external distraction of the midface with direct wiring of the distraction unit in patients with craniofacial dysplasia. J Craniofac Surg. 2003; 14:783.
152. McCarthy, JG, Coccaro, PJ, Eptstein, F, Converse, JM. Early skeletal release in the infant with craniofacial dysostosis: The role of the sphenozygomatic suture. Plast Reconstr Surg. 1978; 62:335–346.
153. McCarthy, JG, Epstein, FJ, Sadove, M, et al. Early surgery for craniofacial synostosis: An 8-year experience. Plast Reconstr Surg. 1984; 73:521.
154. McCarthy, JG, Glasberg, SB, Cutting, CB, et al. Twenty-year experience with early surgery for craniosynostosis: I. Isolated craniofacial synostosis—results and unsolved problems. Plast Reconstr Surg. 1995; 96:272.
155. McCarthy, JG, Grayson, B, Bookstein, F, et al. Le Fort III advancement osteotomy in the growing child. Plast Reconstr Surg. 1984; 74:343.
156. McCarthy, JG, La Trenta, GS, Breitbart, AS, et al. The Le Fort III advancement osteotomy in the child under 7 years of age. Plast Reconstr Surg. 1990; 86:633–646.
157. Meazzini, MC, Mazzoleni, F, Caronni, E, Bozzetti, A. Le Fort III advancement osteotomy in the growing child affected by Crouzon and Apert syndromes: Presurgical and postsurgical growth. J Craniofac Surg. 2005; 16:369.
158. Meling, TR, Hans-Erik, H, Per, S, Due-Tonnessen, BJ. Le Fort III distraction osteogenesis in syndromal craniosynostosis. J Craniofac Surg. 2006; 17:28.
159. Meyers, GA, Day, D, Goldberg, R, et al. FGFR2 exon III and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: Evidence for missense changes, insertions and a deletion due to alternative RNA splicing. Am J Hum Genet. 1996; 58:491.
160. Meyers, GA, Orlow, SJ, Munro, IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995; 11:462.
161. Montaut, J, Stricker, M. Dysmorphies craniofaciales: Les synostoses prematurees (craniostenoses et facio-stenoses). Neurochirurgie. 1977; 23(Suppl 2):1.
162. Moore, MH. Upper airway obstruction in the syndromal craniosynostoses. Br J Plast Surg. 1993; 46:355–362.
163. Moretti, G, Sraeffen, J. Dysostose craniofaciale de Crouzon et syringomylie: Association chez le frere et la soeur. Presse Med. 1959; 67:376.
164. Moss, ML. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel). 1959; 37:351–370.
165. Muenke, M, Gripp, KW, McDonald-McGinn, DM, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997; 60:555.
166. Muenke, M, Schell, U. Fibroblast-growth-factor receptor mutations in human skeletal disorders. Trends Genet. 1995; 11:308.
167. Muenke, M, Schell, U, Hehr, A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994; 8:269.
168. Muenke, M, Schell, U, Robin, NH, et al. Variable clinical spectrum in Pfeiffer syndrome: Correlation between phenotype and genotype. Proc Green Genet Ctr. 1995; 15:126.
169. Mulliken, JB, Godwin, SL, Pracharktam, N, et al. The concept of the sagittal orbital-globe relationship in craniofacial surgery. Plast Reconstr Surg. 1996; 97:700.
170. Murovic, JA, Posnick, JC, Drake, JM, et al. Hydrocephalus in Apert syndrome: A retrospective review. Pediatr Neurosurg. 1993; 19(3):151–155.
171. Murray, JE, Swanson, LT. Midface osteotomy and advancement for craniosynostosis. Plast Reconstr Surg. 1968; 41:299–306.
172. Nelson, TE, Mulliken, JB, Padwa, BL. Effect of midfacial distraction on the obstructed airway in patients with syndromic bilateral coronal synostosis. J Oral Maxillofac Surg. 2008; 66:2318–2321.
173. Nixon, GM, Brouillette, RT. Sleep. 8: Pediatric obstructive sleep apnea. Thorax. 2005; 60511–60516.
174. Noetzel, MJ, Marsh, JL, Palkes, H, et al. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr. 1985; 107:885.
175. Nout, E, Bouw, FP, Veenland, JF, et al. Three-dimensional airway changes after Le Fort III advancement in syndromic craniosynostosis patients. J Plast Reconstr Surg. 2010; 126:564–571.
176. Nout, E, Cesteleyn, LL, Van der Wal, KG, et al. Advancement of the midface, from conventional Le Fort III osteotomy to Le Fort III distraction: Review of the literature. Int J Oral Maxillofac Surg. 2008; 37:781–789.
177. Oldridge, M, Wilkie, AOM, Slaney, SF, et al. Mutations in the third immunoglobulin domain of the fibroblast growth factor receptor-2 gene in Crouzon syndrome. Hum Mol Genet. 1995; 4:1077.
178. Ortiz-Monasterio, F, Fuente del Campo, A. Refinements on the monobloc orbitofacial advancement. In: Caronni EP, ed. Craniofacial surgery. Boston: Little, Brown; 1985:263.
179. Ortiz-Monasterio, F, Fuente del Campo, A, Carillo, A. Advancement of the orbits and the midface in one piece, combined with frontal repositioning for the correction of Crouzon syndrome. Plast Reconstr Surg. 1978; 61:507–516.
180. Ousterhout, DK, Vargervik, K, Clark, S. Stability of the maxilla after Le Fort III advancement in craniosynostosis syndromes. Cleft Palate J. 1986; 23(Suppl 1):91–101.
181. Padwa, B. Effects of midface distraction osteogenesis on obstructive sleep apnea in patients with syndromic craniosynostosis. J Oral Maxillofac Surg. 2006; 64:49.
182. Park, WJ, Meyers, GA, Li, X, et al. Novel FGFR2 mutations in Crouzon and Jackson-Weiss syndromes show allelic heterogeneity and phenotypic variability. Hum Mol Genet. 1995; 4:1229.
183. Park, WJ, Theda, C, Maestri, NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995; 57:321–328.
184. Paznekas, WA, Cunningham, ML, Howard, TD, et al. Genetic heterogeneity of Saethre-Chotzen syndrome due to TWIST and FGFR mutations. Am J Hum Genet. 1998; 62:1370.
185. Perkins, JA, Sie, KC, Milczuk, H, et al. Airway management in children with craniofacial anomalies. Cleft Palate Craniofac J. 1997; 34:135.
186. Peterson, SJ, Pruzansky, S. Palatal anomalies in the syndromes of Apert and Crouzon. Cleft Palate J. 1974; 11:394.
187. Peterson-Falzone, SJ, Pruzansky, S, Purris, P, et al. Nasopharyngeal dysmorphology in the syndromes of Apert and Crouzon. Cleft Palate J. 1981; 18:237.
188. Pfeiffer, RA. Dominant erbliche akrocephalosyndaktylie. Z Kinderheilkd. 1964; 90:301–320.
189. Phillips, JH, George, AK, Tompson, B. Le Fort III osteotomy or distraction osteogenesis imperfecta: Your choice. Plast Reconstr Surg. 2006; 117:1255–1260.
190. Pijpers, M, Poels, PJ, Vaandrager, JM, et al. Undiagnosed obstructive sleep apnea syndrome in children with syndromal craniofacial synostosis. J Craniofac Surg. 2004; 15:670–674.
191. Pollack, IF, Losken, HW, Biglan, AW. Incidence of increased intracranial pressure after early surgical treatment of syndromic craniosynostosis. Pediatr Neurosurg. 1996; 24:202.
192. Polley, JW, Figueroa, AA. Management of severe maxillary deficiency in childhood and adolescence through distraction osteogenesis with an external, adjustable, rigid distraction device. J Craniofac Surg. 1997; 8:181–185.
193. Pope, AW, Ward, J. Self-perceived facial appearance and psychosocial adjustment in preadolescents with craniofacial anomalies. Cleft Palate Craniofac J. 1997; 34:396.
194. Posnick, JC. Craniofacial dysostosis: Staging of reconstruction and management of the midface deformity. Neurosurg Clin North Am. 1991; 2:683–702.
195. Posnick, JC. The craniofacial dysostosis syndromes: Current reconstructive strategies. Clin Plast Surg. 1994; 21(4):585–598.
196. Posnick, JC. The effects of rigid fixation on the craniofacial growth of the rhesus monkeys [discussion]. Plast Reconstr Surg. 1994; 93:11.
197. Posnick, JC. Craniofacial dysostosis syndromes: A staged reconstructive approach. In: Turvey TA, Vig KWL, Fonseca RJ, eds. Facial clefts and craniosynostosis: Principles and management. Philadelphia: WB Saunders, 1996.
198. Posnick, JC. Monobloc and facial bipartition osteotomies: A step-by-step description of the surgical technique. J Craniofac Surg. 1996; 7(3):229–250.
199. Posnick, JC. Stability of fronto-orbital advancement in non-syndromic bilateral coronal synostosis: A quantitative three-dimensional computed tomographic study [discussion]. Plast Reconstr Surg. 1996; 98:406.
200. Posnick, JC. Crouzon syndrome: Basic dysmorphology and staging of reconstruction. Techniques in Neurosurgery. 1997; 3:216–229.
201. Posnick, JC. The craniofacial dysostosis syndromes: Staging of reconstruction and management of secondary deformities. Clin Plast Surg. 1997; 24(3):429–446.
202. Posnick, JC. Apert syndrome: Evaluation and staging of reconstruction. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:308–342.
203. Posnick, JC. Brachycephaly: Bilateral coronal synostosis without midface deficiency. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:249–268.
204. Posnick, JC. Cloverleaf skull anomalies: Evaluation and staging reconstruction. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:354–366.
205. Posnick, JC. Crouzon syndrome: Evaluation and staging of reconstruction. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:271–307.
206. Posnick, JC. Pfeiffer syndrome: Evaluation and staging of reconstruction. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:343–353.
207. Posnick, JC. The monobloc and facial bipartition osteotomies: A step-by-step description of the surgical technique. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:367–388.
208. Posnick, JC, Al-Qattan, MM, Armstrong, D. Monobloc and facial bipartition osteotomies reconstruction of craniofacial malformations: A study of extradural dead space. Plast Reconstr Surg. 1996; 97(6):1118–1128.
209. Posnick, JC, Armstrong, D, Bite, U. Crouzon and Apert syndrome: Intracranial volume measurements prior to and after cranio-orbital reshaping in childhood. Plast Reconstr Surg. 1995; 96(3):539–548.
210. Posnick, JC, Bite, NP, Nakano, P, Davis, J, Armstrong, D. Indirect intracranial volume measurements using CT scans: Clinical applications for craniosynostosis. Plast Reconstr Surg. 1992; 89(1):34–45.
211. Posnick, JC, Farkas, LG. The application of anthropometric surface measurements in craniomaxillofacial surgery. In: Farkas LG, ed. Anthropometry of the head and face. New York: Raven Press, 1994.
212. Posnick, JC, Farkas, LG. Anthropometric surface measurements in the analysis of craniomaxillofacial deformities: Normal values and growth trends. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:55–79.
213. Posnick, JC, Goldstein, JA, Armstrong, D, Rutka, JT. Reconstruction of skull defects in children and adolescents by the use of fixed cranial bone grafts: Long-term results. Neurosurgery. 1993; 32(5):785–791.
214. Posnick, JC, Goldstein, JA, Clokie, C. Advantages of the postauricular coronal incision. Ann Plast Surg. 1992; 29(2):114–116.
215. Posnick, JC, Goldstein, JA, Clokie, C. Refinements in pterygomaxillary dissociation for total midface osteotomies: Instrumentation, technique and CT scan analysis. Plast Reconstr Surg. 1993; 91(1):167–172.
216. Posnick, JC, Lin, KY, Jhawar, BJ, Armstrong, D. Crouzon syndrome: Quantitative assessment of presenting deformity and surgical results based on CT scans. Plast Reconstr Surg. 1993; 92(6):1027–1037.
217. Posnick, JC, Lin, KY, Jhawar, BJ, Armstrong, D. Apert syndrome: Quantitative assessment in presenting deformity and surgical results after first-stage reconstruction by CT scan. Plast Reconstr Surg. 1994; 93:489–497.
218. Posnick, JC, Ruiz, R. The craniofacial dysostosis syndromes: Current surgical thinking and future directions. Cleft Palate Craniofac J. 2000; 37(5):433.
219. Posnick, JC, Ruiz, R, Tiwana, P. The craniofacial dysostosis syndromes: Stages of reconstruction. Oral Maxillofac Surg Clin North Am. 2004; 475–491.
220. Posnick, JC, Waitzman, A, Armstrong, D, Pron, G. Monobloc and facial bipartition osteotomies: Quantitative assessment of presenting deformity and surgical results based on computer tomography scans. J Oral Maxillofac Surg. 1995; 53(4):358–367.
221. Posnick, JC, Yaremchuk, M. The effect of non-resorbable internal fixation devices placed on and within a child’s cranial vault: Brain function, morbidity and growth restrictions [editorial]. Plast Reconstr Surg. 1995; 96:966.
222. Pugeaut, R. Le probleme neuro chirurgical des craniostenoses. Cahier Med Lyon. 1968; 44:3343.
223. Reardon, W, Winter, RM, Rutland, P, et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994; 8:98.
224. Reddy, BSN. An unusual association of acanthosis nigricans and Crouzon disease. J Dermatol. 1985; 12:85.
225. Regnault, F, Crouzon, O. Etude sur un cas de dysostose craniofaciale hereditaire. Ann Med Enfant. 1927; 43:676.
226. Renier, D. Intracranial pressure in craniosynostosis: Pre- and postoperative recordings—correlation with functional results. In: Persing JA, Edgerton MT, Jane JA, eds. Scientific foundations and surgical treatment of craniosynostosis. Baltimore: Williams & Wilkins; 1989:263–269.
227. Renier, D, Arnaud, E, Cinalli, G, et al. Prognosis for mental function in Apert syndrome. J Neurosurg. 1996; 85:66.
228. Renier, D, Marchac, D. Intracranial pressure recordings: Analysis of 300 cases. In: Marchac D, ed. Craniofacial surgery: Proceedings of the First International Congress on Craniomaxillofacial Surgery. Heidelberg: Springer-Verlag, 1988.
229. Renier, D, Marchac, D. Longitudinal assessment of mental development in infants with nonsyndromic craniosynostosis with and without cranial release and reconstruction [discussion]. Plast Reconstr Surg. 1993; 92:840.
230. Renier, D, Sainte-Rose, C, Marchac, D, et al. Intracranial pressure in craniosynostosis. J Neurosurg. 1982; 57:370.
231. Robin, NH, Scott, JA, Arnold, JE, et al. Favorable prognosis for children with Pfeiffer syndrome types 2 and 3: Implications for classification. Am J Med Genet. 1998; 75:240.
232. Rougerie, J, Derome, P, Anquez, L. Craniostenosis et dysmorphies craniofaciales. Principes d’un nouvelle technique de traitement et ses resultats. Neurochirurgie. 1972; 18:429.
233. Rutland, P, Pulley, LJ, Reardon, W. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet. 1995; 9:173–176.
234. Saethre, H. Ein beitrag zum turmschadelproblem (pathogenese, erblichkeit und symptomatologie). Deutsche Zeitschrift fur Nervenheilkunde. 1931; 117:533.
235. Satoh, K, Mitsukawa, N, Hosaka, Y. Dual midfacial distraction osteogenesis: Le Fort III minus I and Le Fort I for syndromic craniosynostosis. Plast Reconstr Surg. 2003; 111:1019.
236. Schell, U, Hehr, A, Feldman, GJ, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995; 4:323.
237. Schiller, JG. Craniofacial dysostosis of Crouzon: A case report and pedigree with emphasis on hereditary. Pediatrics. 1959; 23:107.
238. Seruya, M, Oh, A, Boyajian, M, et al. Long-term outcomes of primary craniofacial reconstruction for craniosynostosis: A 12-year experience. Plast Reconstr Surg. 2011; 127:2397.
239. Shetye, PR, Boutros, S, Grayson, BH, et al. Midterm follow-up of midface distraction for syndromic craniosynostosis: A clinical and cephalometric study. Plast Reconstr Surg. 2007; 120:1621–1632.
340. Shetye, PR, Davidson, EH, Sorkin, M, et al. Evaluation of three surgical techniques for advancement of the midface in the growing children with syndromic craniosynostosis. Plast Reconstr Surg. 2010; 126:982–994.
241. Shetye, PR, Grayson, BH, McCarthy, JG. Le Fort III distraction: Controlling position and path of the osteotomized midface segment on a rigid platform. J Craniofac Surg. 2010; 21:1118–1121.
242. Shetye, PR, Kadadia, H, Grayson, BH, McCarthy, JG. A 10-year study of skeletal stability and growth of the mid face following Le Fort III advancement in syndromic craniosynostosis. Plast Reconstr Surg. 2010; 126:973–981.
243. Shillito, J, Jr., Matson, DD. Craniosynostosis: A review of 519 surgical patients. Pediatrics. 1968; 41:829.
244. Shin, JH, Duncan, CC, Persing, J. Monobloc distraction: Technical modification and considerations. J Craniofac Surg. 2003; 14:763.
245. Siddiqi, SN, Posnick, JC, Buncic, R, et al. The detection and management of intracranial hypertension after initial suture release and decompression for craniofacial dysostosis syndromes. Neurosurgery. 1995; 36(4):703–708.
246. Singal, VK, Mooney, MO, Burrows, AM, et al. Age-related changes in intracranial volume in rabbits with craniosynostosis. Plast Reconstr Surg. 1997; 100:1121.
247. Singhi, S, Walia, BNS, Singhi, P, et al. A simple nonroentgenographic alternative for skull volume measurements in children. Ind J Med Res. 1985; 82:150.
248. Slaney, SF, Oldridge, M, Hurst, JA, et al. Differential effects of FGFR2 mutations on syndactyly and cleft plate in Apert syndrome. Am J Med Genet. 1996; 58:923–932.
249. Spinelli, HM, Irizarry, D, McCarthy, JG, et al. An analysis of extradural dead space after fronto-orbital surgery. Plast Reconstr Surg. 1994; 93:1372.
250. Steinberger, D, Mulliken, JB, Muller, U. Predisposition for cysteine substitutions in the immunoglobulin-like chain of FGFR2 in Crouzon syndrome. Hum Genet. 1995; 96:113.
251. Stevens, CA, Roeder, ER. Ser351Cys mutation in the fibroblast growth factor receptor 2 gene results in severe Pfeiffer syndrome. Clin Dysmorphol. 2006; 15:187.
252. Tessier P: Demonstration operations at Hopital Foch, Paris-Suresnes, Nov 1967.
253. Tessier, P. Total facial osteotomy. Crouzon syndrome, Apert syndrome: Oxycephaly, scaphocephaly, turricephaly [French]. Ann Chir Plast. 1967; 12:273–286.
254. Tessier, P. Dysostosis cranio-faciales (syndromes de Crouzon et d’Apert) osteotomies totales de la face. Transactions of the Fourth International Congress of Plastic and Reconstructive Surgery. Rome: Excerpta Medica Foundation, Amsterdam; 1969.
255. Tessier, P. Autogenous bone grafts taken from the calvarium for facial and cranial applications. Plast Reconstr Surg. 1971; 48:224.
256. Tessier, P. The definitive plastic surgical treatment of the severe facial deformities of craniofacial synostosis: Crouzon and Apert diseases. Plast Reconstr Surg. 1971; 48:419.
257. Tessier, P. Total osteotomy of the middle third of the face for faciostenosis or for sequelae of the Le Fort III fractures. Plast Reconstr Surg. 1971; 48:533.
258. Tessier, P. Traitement des dysmorphies faciales propres aux dysostoses craniofaciales (DGF), maladies de Crouzon et d’Apert. Neurochirurgie. 1971; 17:295.
259. Tessier, P. Recent improvement in the treatment of facial and cranial deformities in Crouzon disease and Apert syndrome. In: Symposium of Plastic Surgery of the Orbital Region. St Louis, MO: CV Mosby Co; 1976:271.
260. Tessier, P. Relationship of craniosynostosis to craniofacial dysostosis and to faciosynostosis: A study with therapeutic implications. Clin Plast Surg. 1982; 9:531.
261. Tessier, P. Apert syndrome: Acrocephalosyndactyly, type I. In: Caronni P, ed. Craniofacial surgery. Boston: Little, Brown, 1985.
262. Tessier, P. Craniofacial surgery in syndromic craniosynostosis: Craniosynostosis, diagnosis, evaluation and management. New York: Raven Press; 1986.
263. Tessier, P. The monobloc and frontofacial advancement: Do the pluses outweigh the minuses? [discussion]. Plast Reconstr Surg. 1993; 91:988.
264. Tessier, P, Guiot, G, Rougerie, J, et al. Osteotomies cranio-naso-orbito-faciales, hypertelorisme. Chir Plast. 1967; 12:103.
265. Thompson, DN, Harkness, W, Jones, B, et al. Subdural intracranial pressure monitoring in craniosynostosis: Its role in surgical management. Childs Nerv Syst. 1995; 11:269–275.
266. Tuite, GF, Chong, WK, Evanson, J, et al. The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurgery. 1996; 38:272.
267. Van der Meulen, JC. Medial faciotomy. Br J Plast Surg. 1979; 32:339–342.
268. Vannier, MW, Pilgram, TK, Marsh, JL, et al. Craniosynostosis: Diagnostic imaging with three-dimensional CT presentation. AJNR Am J Neuroradiol. 1994; 15:1861–1869.
269. Virchow, R. Uber den cretinismus, nametlich in Franken, under uber pathologische. Schadelformen Verk Phys Med Gessellsch Wurszburg. 1851; 2:230–271.
270. Vogt, A. Dyskephalie (dysostosis craniofacialis, maladie de Crouzon 1912) und eine neuartige kombination dieser krankheir mit syndaktylie der 4 extremitaeten (dyskephlodaktylie). Klin Monatsbl Augenheilk. 1933; 90:441.
271. Vuilliamy, DG, Normandale, PA. Craniofacial dysostosis in a Dorset family. Arch Dis Child. 1966; 41:275.
272. Waitzman, AA, Posnick, JC, Armstrong, D, Pron, GE. Craniofacial skeletal measurements based on computed tomography: Part I. Accuracy and reproducibility. Cleft Palate Craniofac J. 1992; 29:112–117.
273. Waitzman, AA, Posnick, JC, Armstrong, D, Pron, GE. Craniofacial skeletal measurements based on computed tomography. Part II. Normal values and growth trends. Cleft Palate Craniofac J. 1992; 29:118–128.
274. Walia, HK, Sodhi, JS, Gupta, BB, et al. Roentgenologic determination of the cranial capacity in the first four years of life. Indian J Radiol Imaging. 1972; 26:250.
275. Ward, SL, Marcus, CL. Obstructive sleep apnea in infants and young children. J Clin Neurophysiol. 1996; 13:198–207.
276. Warren, SM, Shetye, PR, Obaid, SI, et al. Long-term evaluation of midface position after Le fort III advancement: A 20-plus-year follow-up. Plast Reconstr Surg. 2012; 129:234–242.
277. Whitaker, LA, Bartlett, SP, Schut, L, et al. Craniosynostosis: An analysis of the timing, treatment and complications in 164 consecutive patients. Plast Reconstr Surg. 1987; 80:195.
278. Whitaker, LA, Munro, IR, Sayler, KE, et al. Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg. 1979; 64:198.
279. Whitaker, LA, Schut, L, Kerr, LP. Early surgery for isolated craniofacial dysostosis. Plast Reconstr Surg. 1977; 60:575.
280. Witherow, H, Dunaway, D, Evans, R, et al. Functional outcomes in monobloc advancement by distraction using the rigid external distractor device. Plast Reconstr Surg. 2008; 121:1311–1322.
281. Wolfe SA: Critical analysis of 81 monobloc frontofacial advancements over a 27-year period: Should they all be distracted, or not? Rancho Mirage, Calif, 2009, Abstract Presentation, 88th Annual Meeting, American Association of Plastic Surgeons. pp 200–201.
282. Wolfe, SA, Morrison, G, Page, LK, et al. The monobloc and frontofacial advancement: Do the pluses outweigh the minuses? Plast Reconstr Surg. 1993; 91:977.
283. Wong, GB, Kakulis, EG, Mulliken, JB. Analysis of fronto-orbital advancement for Apert, Crouzon, Pfeiffer, and Saethre-Chotzen syndromes. Plast Reconstr Surg. 2000; 105:2314.
284. Xu, HS, Mu, XZ, Yu, ZY, et al. Changes of different section area at different parts of upper-airway after Le Fort III osteotomy [Chinese]. Zhonghua Zheng Xing Wai Ke Za Zhi. 2008; 24:181–183.
285. Zhou, G, Schwartz, LT, Gopen, Q. Inner ear anomalies and conductive hearing loss in children with Apert syndrome: An overlooked otologic aspect. Otol Neurotol. 2009; 30:184–189.
*References 5, 42, 45, 81, 82, 95, 115, 116, 131, 143, 159, 160, 165–168, 177, 183, 223, 233, 236, 248, 250, 251
*References 7, 13, 17, 54, 88, 98, 99, 104, 121, 123, 124, 128–130, 132, 142, 147, 152, 186
*References 9, 12, 27, 51–53, 59, 61, 78, 83, 84, 90, 95, 98, 100, 118, 126, 127, 130, 163, 224, 225, 237, 238, 270, 271
*References 2, 35, 79, 92, 101, 105, 106, 114, 135, 141, 162, 172, 173, 175, 181, 185, 187, 190, 275, 284
*References 8, 10, 11, 23, 32, 36, 56, 57, 63, 69, 70–72, 89, 97, 109, 110, 113, 117, 120, 119, 136, 149–151, 155–158, 169, 171, 176, 180, 189, 192, 195, 197, 215, 235, 239, 241, 244, 277