Chapter 44 Sustained Ventricular Tachycardia with Heart Disease
The definition of sustained VT has undergone evolution. Although no formal definition has been adopted for spontaneous sustained VT compared with VT induced by programmed stimulation, two versions are in common use. In the Electrophysiologic Study Versus Electrocardiographic Monitoring (ESVEM) trial, a 15-second duration was considered sustained VT.1 Alternatively, many physicians prefer to use 30-second duration or evidence of hemodynamic compromise warranting earlier termination to define VT, as in the case of induced VT. This chapter provides an overview of the epidemiology, pathology, electrophysiology, and management of sustained VT in patients with structural heart disease. In such patients, VT can be conveniently divided into (1) VT caused by, or related to, a “fixed arrhythmogenic substrate” and (2) VT in which the substrate is present intermittently during transient abnormalities in cardiac electrophysiological structure and function (e.g., with changes in the neurohormonal milieu).
Etiology and Pathologic Anatomy
In general, the majority of hearts in patients with a previous history of VT reveal varying types of pathologic changes, with areas of healthy myocardium interspersed among the pathologic processes.2–4 The most common cause of VT is coronary artery disease (CAD), in either its acute phase or chronic phase. During the acute phase, VT is believed to be caused by altered physiologic, biochemical, or metabolic states. In the chronic phase, a re-entry circuit is created at the junctional areas of the healthy myocardium and scar tissue. The infarct zone is surrounded by varying degrees of border zone with surrounding but damaged myocardial fibers. In general, the infarct size is quite large in these patients and may be related to the development of a ventricular aneurysm.2–4
VT also frequently occurs in cardiomyopathy. Any disease state that affects the heart in a chronic process can eventually lead to a cardiomyopathy. Thus VT is expected to occur in advanced cardiomyopathy during the end stage of heart diseases such as CAD, myocarditis, valvular disease, hypertensive heart disease, familial types of cardiomyopathy, and in many other disease states. This cardiomyopathy can have several etiologies; however, the most common type is an idiopathic form. In some patients, “idiopathic” cardiomyopathy may be the end result of previous viral myocarditis. Pathologically, in most cases of cardiomyopathy, the ventricular myocardial cells show varying types of histologic features such as hypertrophy of cells and varying stages of degenerative changes in the cells and fibrosis, with or without the presence of chronic inflammatory cells. Varying degrees of myocardial fiber damage can provide a substrate for a re-entry circuit at the junctional areas with the healthy myocardium.3
Myocarditis of any type, in the acute phase or the chronic phase, may cause VT that may result in SCD. The heart may be normal at the gross anatomic level, but microscopic examination may reveal an interstitial type of a myocarditis in the ventricular myocardium, including the bundle branches. In many patients, clinical history may be essentially unremarkable, or a history of a mild attack of influenza several weeks before death may be present.2–4
Fibrotic scars in the ventricular myocardium are often seen in young victims of SCD who otherwise have a normal heart. They may be associated with pathologic changes in the peripheral conduction system such as the branching atrioventricular (AV) bundle and bundle branches. The focal scars in the ventricular myocardium, surrounded by healthy myocardium, may form an anatomic substrate for ventricular arrhythmias that promotes re-entry or abnormal automaticity. The etiology of the fibrotic scars in the ventricular myocardium and the beginning of the bundle branches currently is unknown. One hypothesis is that these may represent the end result of an autoimmune reaction or an allergic state that may or may not be related to a silent form of a previous myocarditis.2–4
Uncommon Types of Cardiomyopathy
Less commonly, VT is seen in mitral valve prolapse. Several pathologic abnormalities in cases of mitral valve prolapse are associated with VT.2–5 A variety of pathologic findings such as a right-sided AV bundle, fibrotic scars in the ventricular septum, degenerative changes in the conduction system, and arteriolosclerosis, may be seen in these patients.
In arrhythmogenic right ventricular dysplasia, the anterior wall of the right ventricle can be partially or completely replaced by fibro-fatty tissue. Some intact myocardial cells may be scattered within the fatty tissue. Similar findings extend to the ventricular septum and the left ventricular myocardium. This often is associated with necrosis of cells and mononuclear cell infiltration. Varying amounts of degenerative changes in the myocardium are present in the right and left ventricles. A segmented and looping left-sided AV bundle has been reported. Small-vessel disease of the ventricular septum may also be present. Thus, in addition to acquired pathologic changes, congenital abnormalities of the conduction system in the sinoatrial, AV node, and AV bundle also occur.2–46
In hypertrophic cardiomyopathy, the heart is hypertrophied and enlarged. Hypertrophy of the interventricular septum is seen in varying degrees. The AV node may be partly or mostly situated within the central fibrous body and occasionally partly embedded in the tricuspid valve annulus or at the aortic-mitral annulus. The AV nodal artery is usually thickened and narrowed. The sinoatrial (SA) and AV nodes frequently are infiltrated with fat. The AV bundle may be on the right side of the ventricular septum with loop formation and fibrosis of the branching bundle. Focal, fibrotic scars in the ventricular myocardium are associated with myocardial fiber disarray and arteriolosclerosis in the summit of the ventricular septum.2–47
Infiltrative Diseases of the Myocardium
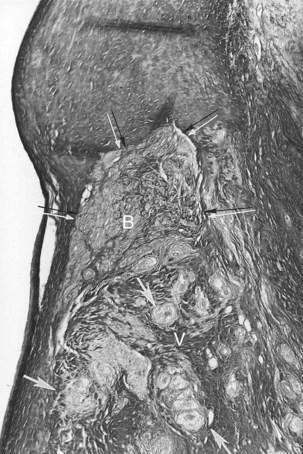
VT may occur in sarcoidosis and amyloidosis. An infiltrative disease of the myocardium such as sarcoidosis may affect the branching AV bundle and the bundle branches as well as the ventricular myocardium. Likewise, amyloidosis, in either the primary or the secondary form, may involve the ventricular myocardium, including the bundle branches. In 70% of cases of primary amyloidosis, the heart is involved, often with disruption to the entire conduction system. The infiltration of amyloid in the heart eventually results in a restrictive cardiomyopathy, and SCD is commonly seen.2–48 The heart is affected in approximately 13% to 25% of cases of sarcoidosis. Cardiac involvement often is associated with lymph node and lung involvement. Sarcoidosis has a predilection to affect the posterior wall of the left ventricle with aneurysm formation. Sarcoid granulomas may be present in the conduction system and the surrounding myocardium (Figure 44-1). In general, infiltrative lesions can cause conduction disturbances and provide a substrate for re-entrant or automatic VT.2–48
Primary Electrical Diseases
Supraventricular arrhythmias are well known to occur in pre-excitation syndrome. However, ventricular arrhythmias also can occur in this entity. Pathologically, the accessory pathway is obvious on either the left or right side, associated with cardiomyopathy and fibroelastosis of the left ventricle, along with hypertrophy and degenerative changes of the myocardium.2–49
Familial Q-T interval prolongation is predominantly an autosomal dominant disorder associated with syncopal episodes, VT, and SCD. Recently, abnormal genes have been identified on several chromosomes in patients with congenital prolonged Q-T interval. Genetic heterogeneity has been documented in linkage studies, in which loci on several chromosomes have been identified. Furthermore, mutations in ion channel genes on chromosomes 3, 7, and others have been identified as related to other forms of long QT syndrome.2–410 Pathologically, there are marked fatty infiltration in the approaches to the AV node; a lobulated AV bundle with or without loop formation; arteriolosclerosis; and focal fibrosis of the summit of the ventricular septum seen predominantly on the right side. In addition, fibrotic changes are seen to a varying degree in the AV bundle and bundle branches with chronic inflammatory cells in the ventricular myocardium. Of note, pathologic findings do exist in congenital long QT syndrome leading to SCD.2–410
Skeletal Muscle Disorders
The conduction system frequently is affected in congenital myotonic dystrophy. This progressive, generalized disease is characterized by typical atrophy of skeletal muscles with associated myotonia. Various types of arrhythmias are known to occur in this disease. Pathologically, degenerative changes in the smooth muscles of the cardiac vessels in the left atrium and the aorta with fatty infiltration in the approaches to the AV node, fibrosis of the bundle branches, summit of the ventricular septum, and varying degenerative changes in the myocardium are present. These could form a substrate for AV block and VT.3,11
Kearns-Sayre syndrome is characterized by progressive external ophthalmoplegia, retinitis pigmentosa, and AV block rather than VT. Progressive, degenerative changes affect the entire heart. The conduction system is replaced by fibrotic destruction of the bundle branches with fibro-fatty replacement. Some muscle cells may reveal hypertrophy; others become atrophied with perineural and perivascular fibrosis, eventually leading to cardiomyopathy. Although AV block frequently occurs in this disease, electrophysiological studies have demonstrated that the disease affects the entire His-Purkinje system. These findings in the conduction system and ventricular myocardium may form an anatomic basis for VT.3,12
Tumors and Parasitic Diseases of the Heart
Any tumor, either primary or secondary, can produce ventricular arrhythmias, as can parasitic infiltration. Hydatid cyst infiltration can result in fibrosis in the perimeter tissues around the cyst. This can permit re-entry and may result in VT.3,13
Aneurysm and Diverticulum of the Heart
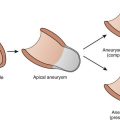
Aneurysm and diverticulum of the heart may occur in either the right or left ventricle of an otherwise normal heart, and VT may be the first clinical manifestation.3,13 Pathologic anatomy reveals a large, wide-mouthed aneurysm, but its microscopic appearance can vary with the etiology.
Ventricular Tachycardia in Postoperative Patients with Congenital Heart Disease
Ventricular arrhythmias are known to occur in postoperative patients with congenital heart diseases such as tetralogy of Fallot, aortic stenosis, or other congenital cardiac anomalies. VT can appear many years after the surgery. Fibrotic scars along with healthy myocardium are seen, for example, at the outflow tract of the right ventricle or in any other area in the heart. This substrate can result in re-entry circuits leading to VT.2–4,14,15 In aortic stenosis, cardiomegaly may occur with myocardial fibrosis.16
Iatrogenic Disorders
Antiarrhythmic drugs may alter the physiological, metabolic, and biochemical states of the myocardium, the conduction system, or both and give rise to VT, which may or may not be transient. Likewise, the various resuscitative techniques used and catheter ablation of the myocardium may alter myocardial tissue, leading to formation of fibro-fatty scar and chronic inflammation. This may become a future substrate for VT.2–4,13,17
Familial Ventricular Tachycardia
Pathologically, the conduction system and the ventricular myocardium may show degenerative changes with mononuclear cell infiltration and fat to varying degrees. In other individuals, atrophy of the branching part of the AV bundle, with almost complete absence of both right and left bundle branches, may be present. These findings suggest a genetic abnormality of the conduction system that leads to degenerative changes, inflammatory phenomena, and susceptibility to ventricular arrhythmias that cause SCD.18,19 The anatomic substrate may originate from the myocardial disarray that can be present in either the ventricular septal myocardium or the conduction system. Left or right ventricular septal hypertrophy may also cause degeneration of the AV node, AV bundle, and the main left bundle branch (Figure 44-2).3,4,20
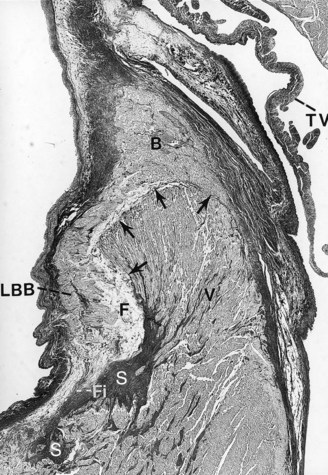
 -year-old boy with a history of exercise-related syncope died suddenly while swimming. He had a family history of sudden cardiac death involving three consecutive generations, including a brother. The electrocardiogram and cardiac catheterization results were normal. During electrophysiological studies, with extrastimulus testing, he had polymorphic nonsustained ventricular tachycardia; during stage 5 of the Bruce protocol, he had a run of nonsustained ventricular tachycardia. Photomicrograph of the branching atrioventricular bundle being compressed by the right ventricular septal muscle at the region of the posterior radiation of left bundle branch (Weigert-van Gieson stain ×22.5). B, Branching bundle; F, fatty metamorphosis; Fi, fibrosis and linear change of left bundle branch; LBB, posterior radiation of left bundle branch; S, increased sclerosis on the mid-septal area on the left; V, summit of the ventricular septum. Arrows point to the pressure of the right ventricular septal hypertrophy on the atrioventricular bundle at the level of the posterior radiation of left bundle branch.
-year-old boy with a history of exercise-related syncope died suddenly while swimming. He had a family history of sudden cardiac death involving three consecutive generations, including a brother. The electrocardiogram and cardiac catheterization results were normal. During electrophysiological studies, with extrastimulus testing, he had polymorphic nonsustained ventricular tachycardia; during stage 5 of the Bruce protocol, he had a run of nonsustained ventricular tachycardia. Photomicrograph of the branching atrioventricular bundle being compressed by the right ventricular septal muscle at the region of the posterior radiation of left bundle branch (Weigert-van Gieson stain ×22.5). B, Branching bundle; F, fatty metamorphosis; Fi, fibrosis and linear change of left bundle branch; LBB, posterior radiation of left bundle branch; S, increased sclerosis on the mid-septal area on the left; V, summit of the ventricular septum. Arrows point to the pressure of the right ventricular septal hypertrophy on the atrioventricular bundle at the level of the posterior radiation of left bundle branch.Changes on the Right Side of the Ventricular Septum Related to Premature Aging
Premature aging occurs in the summit of the ventricular septum in some individuals, with degenerative changes of the branching bundle and the right bundle branch. These changes are usually associated with arteriolosclerosis of the summit of the ventricular septum and often are seen in SCD in teenagers.3,4,21
Idiopathic Ventricular Tachycardia Caused by Congenital Abnormalities of the Conduction System
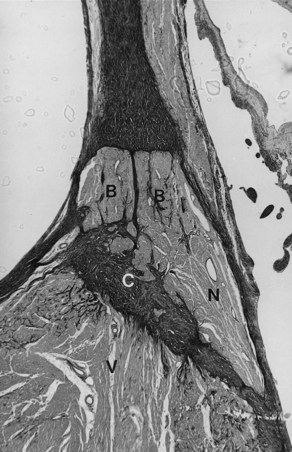
Chronic recurrent right VT with QRS morphology of a left bundle branch block (LBBB) pattern in a normal heart, normal coronary arteries, and normal cardiac catheterization findings has been well documented, especially in young patients. However, an anatomic substrate for this abnormality has only been occasionally reported.22 In one report, a 13-year-old boy with a history of recurrent VT died suddenly. A right-sided, markedly septated AV bundle was found at autopsy. The AV node formed the AV bundle, and the node-bundle junction was ill defined (Figure 44-3). The penetrating AV bundle cells were not well defined as typical cells of the AV bundle, and the latter remained on the right side and eventually became the right bundle branch. In addition, patchy fibrotic scars were present in the right ventricular myocardium. A right-sided, markedly septated, undifferentiated AV bundle that eventually continues as the right bundle branch has been hypothesized to cause recurrent right VT in an otherwise normal heart.2,3,22
Basic Electrophysiology
Ischemia and Ventricular Tachyarrhythmias
The ventricular arrhythmias caused by myocardial ischemia and infarction occur in several distinct phases. A ventricular arrhythmia can be induced by acute ischemia and reperfusion. It usually occurs between 2 and 30 minutes after acute coronary artery occlusion, when the changes caused by ischemia are still reversible. Arrhythmias associated with the development of myocardial infarction can be categorized as delayed arrhythmias, usually occurring between 5 and 48 hours, late arrhythmias occurring after days to weeks, and chronic arrhythmias occurring months to years later.23,24 Delayed arrhythmias such as slow VT and accelerated idioventricular rhythms rarely degenerate into VF and are caused by abnormal automaticity of Purkinje fibers overlying the infarct.
Data obtained in 4- to 5-day-old canine infarcts show that the healing infarct undergoes structural and functional changes. The surviving epicardial cells overlying the infarct have abnormal action potentials with diminished upstrokes with loss of the plateau and shorter action potential duration. The density and kinetics of a number of ion channels are altered, and sodium ion and calcium ion currents are reduced, as are transient outward potassium ion currents and the delayed and inward rectifying potassium ion currents.25 During this stage, re-entrant VT in the so-called epicardial border zone (the layer of surviving cells overlying the infarct) can easily be induced by premature stimuli. Both the cellular abnormalities, as well as a re-distribution of intercellular gap junctions, play a role in determining the “substrate” for re-entry.26 These VTs may degenerate into VF, especially in the presence of a high sympathetic tone, but this is uncommon.24
In the following few weeks, transmembrane action potentials of the surviving cells gradually return to normal as most of the ion channels recover; by 2 months, action potential configuration in both canine and human infarcts is completely normal (Figure 44-4).27,28 When the healing phase of myocardial infarction is over and when the fully healed phase begins are difficult points to ascertain. The electrophysiological substrate for VT likely develops gradually over several weeks and remains stable from several months to 15 to 20 years.29
The Substrate for Re-entrant Tachycardia in the Human Heart with a Healed Infarct
In the prethrombolytic era, the incidence of sustained monomorphic VT after discharge from the hospital in patients surviving a myocardial infarction (MI) has been reported to be approximately 3% and that of nonsustained VT as 10% to 20%.30 Although thrombolysis has drastically reduced arrhythmic events early after MI, with an incidence of sustained VT lower than 1%, post-infarction arrhythmias have certainly not become irrelevant.31 With improved therapy in the acute stage of MI, more patients survive, but many survivors have left ventricular dysfunction. Ventricular dysfunction is known to be the most important risk factor for SCD, as verified in the European Myocardial Infarct Amiodarone Trial (EMIAT). The combination of arrhythmic death and resuscitated cardiac arrest occurred in 8.6% of patients.32 How many of these patients developed sustained VT that degenerated into VF is unknown, as is how many had an episode of acute ischemia that induced the lethal arrhythmia. Still, despite intensive therapy, arrhythmic events in patients who have had MI, especially in the presence of ventricular dysfunction, remain an important cause of death.
Reconstruction of the re-entrant circuits and the mechanism of slow conduction in the human heart with a healed infarct is based on observations made during mapping-guided surgery in patients with infarct-related VT as well as on studies on isolated, Langendorff-perfused human hearts and isolated papillary muscles from patients with infarcts undergoing cardiac transplantation.28,33–35 Figure 44-5 shows the activation map of one beat of a VT induced by programmed electrical stimulation in an isolated, Langendorff-perfused heart. The endocardial surface of the left ventricle is schematically depicted with the left ventricle folded out by making a vertical cut along the left anterior descending artery. The black zone indicates the infarcted area.
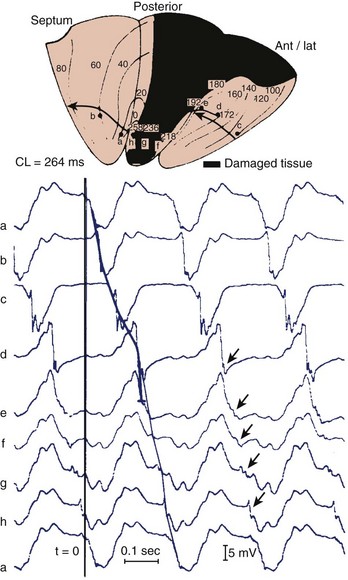
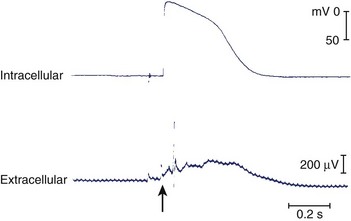
Selected extracellular electrograms, recorded simultaneously from the endocardium by a balloon electrode inserted into the ventricular cavity, are shown in Figure 44-5 as well. The cycle length of this tachycardia was 264 ms. The site of earliest activation during this tachycardia was in a small area near the apex, on the border of the septum and the posterior wall, at the margin of the infarcted region (the area encircled by the 0-ms isochrone). Activation spread from this area to the left in the figure (from a to b) and continued to the anterior wall (from b to c to d to e) to reach the other margin of the infarct after 192 ms (near site e). Although activation at this margin seems to have died out, because of the slow positive deflection recorded at site e, the presence of small deflections indicated by the arrows over the infarcted zone suggests that the spread of activation continued via tracts of surviving muscle within the infarct, through sites f, g, and h, to reach the opposite side of the infarct after 258 ms, re-exciting the area of the 0-ms isochrone and completing a large re-entrant circuit around the circumference of the ventricle.
Subsequent histologic studies of this region confirmed the presence of surviving myocardial muscle bundles, several millimeters below the endocardial surface that connected both margins of the infarct, completing the re-entrant circuit. Figure 44-6 shows superimposed drawings of many histologic sections of this region, where viable muscle is in black and connective tissue in white. Similar tracts were found in other hearts, embedded within the scar of the healed infarct. Such tracts could be localized in the subendocardium, the subepicardium, and intramurally. The myocardial fibers were sometimes arranged in a parallel fashion along the long axis of the tract, allowing relatively rapid transmission of the re-entrant impulse from one side of the infarct to the other. However, in other hearts the fibers were oriented transverse to the direction of impulse propagation; in these cases, transmission through the infarct was very slow.

Figures 44-7 and 44-8 show the mechanism of such “slow” conduction. Figure 44-7 shows selected electrograms from an infarcted human papillary muscle to illustrate the highly fragmented waveforms, typical when connective tissue is intermingled with viable myocardium. In this muscle, the delay between the deflections at the extreme electrode terminals of the multiple electrode (distance, 1.4 mm) was 45 ms, which would correspond to an overall conduction velocity on the order of 3 cm/sec. As illustrated in Figure 44-8, this apparent slow conduction was caused by “zigzag” conduction in small muscle bundles separated by collagenous septa. Activity in the muscle tracts proceeded both toward the site of stimulation A and away from it. Many tracts were dead-end pathways. The actual pathway of activation is plotted in panel B. Although the shortest distance between A and B was 1.2 mm, the length of the zigzag pathway was 25.2 mm. In 10 papillary muscles, conduction velocity parallel to the fiber orientation was, on average, 79 cm/sec, which indicates that both the active electrical properties (action potential amplitude and upstroke velocity) and passive electrical properties (longitudinal coupling resistance) were normal. Conduction velocity at the bifurcation points was only 49 cm/sec, indicating that in addition to the pathway length, impedance mismatch at bifurcations also contributed to activation delay.
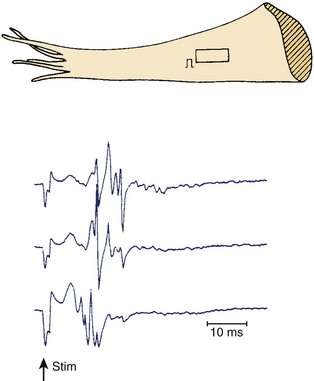
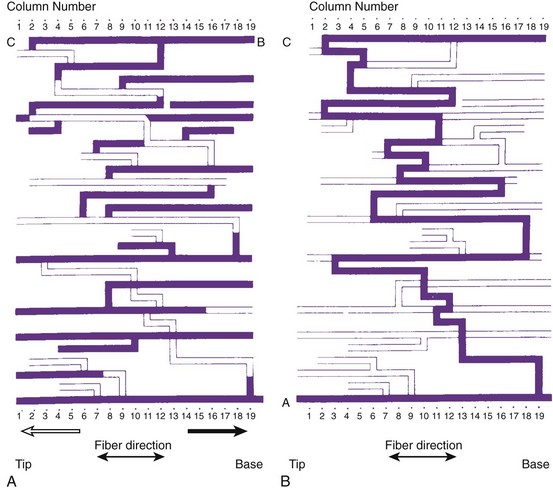
FIGURE 44-8 Map illustrating the spread of activation of the rectangular area shown in the inset of Figure 44-4. In the solid tracts of A, activation moves away from the site of stimulation (site A); in the open tracts, activation moves toward the site of stimulation. B, The tortuous route that activation followed to reach site B.
(Modified from De Bakker JMT, van Capelle FJL, Janse MJ, et al: Slow conduction in the infarcted human heart: “Zigzag” course of activation, Circulation 88:915–926, 1993.)
Thus, in the human heart with a healed infarct, the substrate for re-entry is formed by the surviving fibers within the infarct. In addition to a substrate, a trigger is needed to initiate the tachycardia. This trigger likely occurs in the myocardium remote from the infarct. In the presence of progressive left ventricular dysfunction, these myocardial zones can undergo arrhythmogenic remodeling. Furthermore, the properties of the noninfarcted myocardium can determine whether VT degenerates into VF. Dispersion of refractoriness in the noninfarcted myocardium is three times larger in patients who have had an MI and subsequently develop VF than in those in whom the VT remains monomorphic and hemodynamically stable.36
Role of Cardiac Remodeling in Ventricular Tachycardia
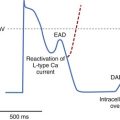
The heart may respond to a variety of abnormal environmental stimuli by altering gene expression or the functional properties of proteins. This ultimately leads to both functional and structural cardiac alterations that may be arrhythmogenic. These processes are referred to as cardiac remodeling. Common to all studies on the hypertrophic and failing ventricular myocardium is a prolonged action potential, especially at slow heart rates.37 This may be considered an adaptive process. In the setting of a prolonged action potential, the intracellular calcium transport is increased, causing enhanced force of myocardial contraction. It can be argued that a prolonged action potential may also protect the heart against re-entrant excitation. However, prolonging repolarization may be arrhythmogenic. Drugs, both cardiac and noncardiac, can prolong the action potential. A risk of development of early afterdepolarizations (EADs) and torsades de pointes (TdP) does exist in this situation.38 An EAD-induced premature beat generally causes re-entry only in the presence of a marked increase in dispersion of repolarization.38 In the hypertrophic and failing myocardium, increased dispersion in repolarization (or refractoriness) has been reported.36,39–41 In hearts with a healed infarct, repolarization of normal, lateral border and infarct-zone cells is nonuniform, with myocardial cells showing different degrees of action potential disturbance and prolongation. Many of these cells, especially those close to the infarct border, show post-repolarization refractoriness.25 Several reasons for the increased dispersion in repolarization exist: unequal distribution of remodeled ion channels, decreased expression of gap junctional connexins and an altered distribution of gap junctions with the development of fibrosis, and changes in autonomic innervation.41–46
Changes in ionic currents contributing to repolarization are seen. The most consistent finding is a reduction in the transient outward current, Ito.37,42,43 Although this current is very important in determining action potential duration in small mammals such as the rat, its downregulation probably does not have much effect on action potential duration in the hearts of large mammals. It does change the level of the plateau phase of the action potential, and it can affect other currents activated later during the action potential. Both rapid and slow components of the delayed rectifier, Ikr and Iks, are reduced in rabbits with pacing-induced heart failure.47 The inward rectifier current, IK1, can decrease, remain unchanged, or increase.37 If IK1 is reduced, it will lead to an unstable resting potential; when it is unregulated, pacemaker activity could result.48,49 The L-type calcium current is either unchanged or decreased, and it is unlikely that this current contributes to action potential prolongation.50 A role may exist for the late sodium current, which is increased.51 For most of these remodeled currents, data on regional differences are insufficient. However, in the presence of enhanced β-adrenergic activity, transmural dispersion in repolarization increases because of augmentation of residual Iks in the epicardial and endocardial layers, resulting in action potential shortening.52 But this does not occur in mid-mural M cells, in which Iks is intrinsically weak.52 A decrease in electrical cell-to-cell coupling, either by a decrease in expression of connexins or by the development of microscopic fibrosis in hypertrophied myocardium, reduces electronic current flow and will unmask intrinsic differences in action potential duration. The well-coupled myocardium will attenuate these differences.
Ischemia and infarction result in both afferent and efferent parasympathetic and sympathetic dysfunction in regions apical to the area of infarction. The denervated but otherwise normal myocardium develops adrenergic supersensitivity, and therefore the response to circulating catecholamines is exaggerated.45 The hypothesis of nerve sprouting in ventricular arrhythmias and SCD states that myocardial infarction results in nerve injury followed by sympathetic nerve sprouting and regional, heterogeneous myocardial hyperinnervation, which, together with electrical remodeling, leads to heterogeneous distribution of repolarization and ventricular arrhythmias.46
It is generally agreed that intracellular calcium handling is compromised in heart failure, but reports on virtually all components involved in calcium homeostasis are contradictory.37,53–55 It is generally accepted that adenosine triphosphate–dependent calcium accumulation by the sarcoplasmic reticulum is decreased. In the presence of a prolonged action potential and altered calcium homeostasis, both EAD and delayed afterdepolarization may occur.43,49 In hearts with an infarct, in which an anatomic re-entrant circuit may be present, either premature ventricular depolarizations caused by these afterdepolarizations or salvos of triggered activity may initiate sustained re-entrant tachycardias. In hypertrophied and failing hearts without an infarct, with fibrosis and increased dispersion of repolarization, these triggers may equally initiate re-entrant tachycardias. In animal models of heart failure (e.g., rabbits), combined volume and pressure overload resulted in nonsustained VT developing in more than 50% of animals, and SCD was common.56 However, it is not certain whether SCD is always caused by VT degenerating into VF. In humans with end-stage heart failure as well as in rabbit models of heart failure, bradycardia, asystole, and electromechanical dissociation have been documented to cause SCD.57–59
Clinical Presentation
The symptoms associated with sustained VT can range from an asymptomatic patient to cardiovascular collapse resulting in circulatory arrest and unconsciousness. Clinical literature dating to the early twentieth century has documented case reports of sustained VT without symptoms or minimal symptoms. Remarkably, some of these episodes may last weeks or even months. These patients may have only minimal or no palpitations, that, when present, can be regarded as an insignificant symptom. Figure 44-9 shows a patient with incessant VT who, in 1981, had palpitations with mild dyspnea during a single sustained slow VT episode that lasted 1 week, despite the presence of severe left ventricular systolic dysfunction. The most common symptoms of sustained VT are palpitations, dyspnea, angina, hypotension, and near or frank syncope. The severity of symptoms is related to several factors, including tachycardia rate, morphology, severity of left ventricular dysfunction, preload, and coexisting diseases such as CAD. The symptom complex often is defined by the hemodynamic impact of the arrhythmia. In an early study, Saksena and colleagues proved that VT resulted in impaired left ventricular relaxation and subsequently systolic dysfunction and decline in negative and positive dP/dt.60 Tachycardia rate and pre-existing LV dysfunction were important variables, as was preload. Faster VT rates and low preload predisposed to hypotension and syncope. Hamer noted similar associations with syncope during VT.61 In a study of defibrillator recipients, presyncope or syncope preceding delivery of implantable cardioverter defibrillator (ICD) therapy was determined by faster events in the “ventricular fibrillation” zone.62 However, the absence of hemodynamic compromise does not exclude the diagnosis of VT. An axiom holds that wide QRS tachycardia in the presence of history or electrocardiogram (ECG) evidence of myocardial infarction is due to VT until proven otherwise by subsequent investigation.
Electrocardiography
Electrocardiographic Diagnosis of Ventricular Tachycardia
Twelve-lead ECGs are particularly useful to differentiate supraventricular tachycardia (SVT) from VT. Distinguishing these arrhythmias is more difficult when aberrant intraventricular conduction occurs during SVT, but several criteria have been proposed to improve diagnostic accuracy.63–69 AV dissociation is the most specific ECG criterion for the diagnosis of VT in recordings of wide QRS complex tachyarrhythmias. Certain rare types of SVT (e.g., AV node re-entry with retrograde block, junctional tachycardia using a nodo-ventricular fiber with retrograde atrial block) may mimic this finding. AV dissociation is seen in only 21% of ECG recordings with VT and may be difficult to identify with absolute certainty.65 When AV dissociation is not apparent, other criteria that depend on whether the QRS resembles LBBB or RBBB must be used.
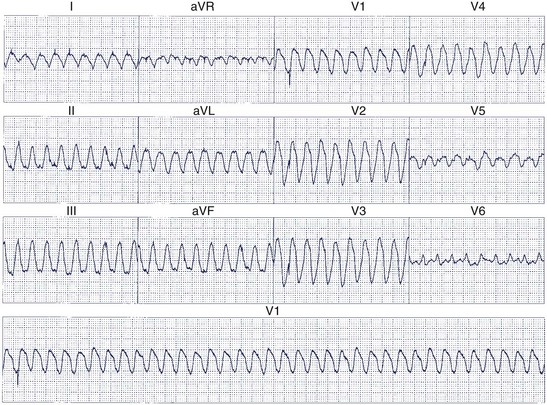
When the ventricles are structurally normal, QRS morphology is an excellent marker of the arrhythmia origin. VT with an LBBB configuration in lead V1 can have its origin in the right ventricle or the intraventricular septum (either right or left side of the septum) (Figure 44-10). A QRS frontal-plane axis directed inferiorly (dominant R waves in leads II, III, and aVF) indicates an origin in the cranial aspect of the heart (e.g., anterior wall of the left ventricle or the right ventricular outflow tract). A QRS frontal plane axis directed superiorly (dominant S waves in leads II, III, and aVF) indicates initial depolarization arising in the inferior wall of the left or right ventricle. Dominant R waves in leads V3 to V4 favor a location of the focus nearer the base of the heart than the apex. Dominant S waves in these leads favor a more apical location. VT with a typical LBBB or RBBB QRS configuration suggests bundle branch re-entry as a mechanism. When areas of ventricular scarring are present, the QRS morphology is less reliable and is sometimes quite misleading.
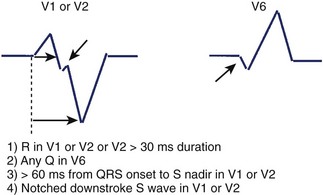
Kindwall observed several characteristic patterns found in patients with VT complexes resembling LBBB and evaluated four criteria to distinguish VT with LBBB pattern from SVT with aberrant conduction.66 All patients in the study had a predominantly negative QRS in V1 and a QRS duration greater than 120 ms. Four electrocardiographic criteria were evaluated in this study: (1) R wave in lead V1 or V2 of greater than 30-ms duration, (2) any Q wave in lead V6, (3) a duration of greater than 60 ms from the onset of the QRS to the nadir of S wave in lead V1 or V2, and (4) notching on the downstroke of the S wave in those leads. Table 44-1 summarizes the sensitivity and predictive accuracy of these criteria. None of the criteria, by itself, was very sensitive, but all patients with VT had at least one of these criteria. The specificity remained high (89%) when the combined criteria were used, and the predictive accuracy was excellent (Figure 44-11). Left-axis deviation was of no value in distinguishing VT from SVT in this study. These criteria are easily measured and provide a practical approach to differentiating VT from SVT.
Table 44-1 Sensitivity and Predictive Accuracy of Electrocardiographic Criteria to Distinguish VT Resembling LBBB from SVT with LBBB Aberration
| PREDICTIVE CRITERIA* | SENSITIVITY (%) | ACCURACY (%) |
|---|---|---|
| R > 30 ms in lead V1 or V2 | 36 | 100 |
| Any Q in V6 | 55 | 98 |
| Duration >60 ms from QRS to S nadir in lead V1 or V2 | 63 | 98 |
| Notched downstroke S wave | 36 | 97 |
| Any of the above present | 100 | 96 |
LBBB, Left bundle branch block; SVT, supraventricular tachycardia; VT, ventricular tachycardia.
* Diagnostic criteria for VT with a morphology resembling left bundle branch block.
From Kindwall KE, Brown J, Josephson ME: Electrocardiographic criteria for ventricular tachycardia in wide complex left bundle branch block morphology tachycardias, Am J Cardiol 61:1279–1283, 1988.
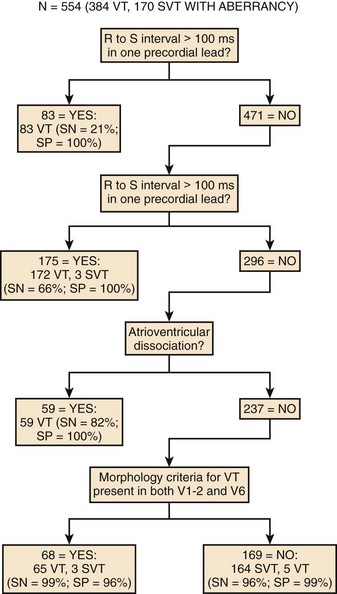
Brugada prospectively analyzed a series of wide QRS tachycardias that included morphologies resembling both LBBB and RBBB.67 The criteria used to differentiate VT from SVT included (1) the presence of an RS complex in at least one precordial lead, (2) an R-S interval in any precordial lead greater than 100 ms, (3) the detection of AV dissociation, and (4) morphology criteria for VT present in leads V1 to V2 and V6. The morphology criteria used for the diagnosis of VT are summarized in Table 44-2. These criteria were present in leads V1 to V2 and V6 in 61% of patients with VT. Unfortunately, only one third of VT recordings fulfill these criteria because of discordance in the morphology criteria in different ECG leads. An RS complex was present in at least one precordial lead in all SVT recordings, but only 26% of the VT recordings did not have an RS complex in any precordial lead. None of the SVTs had an R-S interval greater than 100 ms, but this criterion was met only in 52% of VTs. From these observations the authors concluded that an RS complex in all precordial leads or an R-S interval greater than 100 ms in any precordial lead was highly specific for VT.

To address the sensitivity and specific concerns, Brugada7 used a stepwise approach to differentiate SVT from VT and prospectively tested this algorithm (Figure 44-12).67 The algorithm was both sensitive (99%) and specific (96%). The advantage of the stepwise approach is that it guides the analyst to the correct diagnosis. If an RS complex is not present in any precordial lead, the diagnosis of VT is confirmed without the need for further analysis. If an RS complex is present and the longest R-S interval in any precordial lead exceeds 100 ms, then the diagnosis of VT is confirmed. If the R-S interval is less than 100 ms, then an examination for AV dissociation must be performed. The presence of this criterion helps diagnose VT. If AV dissociation is not detected, then the diagnosis of VT depends on the presence of morphology criteria in both leads V1 and V6.
Electrocardiographic Localization of the Origin of Ventricular Tachycardia
The morphology of monomorphic VT documented by a 12-lead ECG is also useful in predicting the exit site of re-entrant VT on the basis of studies in patients being considered for catheter ablation or surgical excision of their arrhythmias. Josephson analyzed the QRS morphology during VT in patients selected for electrophysiological mapping.68 VT morphologies can be separated into RBBB and LBBB patterns on the basis of the QRS morphology in lead V1 (Figure 44-13). The RBBB pattern includes monophasic, biphasic, or triphasic R waves in lead V1 or a qR complex in that lead. The LBBB pattern was defined by a QS, rS, or qrS recorded in lead V1. Sixteen VTs with LBBB pattern were studied in patients with CAD. The QRS morphology in lead V1 showed an rS pattern in 10, QS pattern in 4, and qrS pattern in 2. All but one of these arose from sites on or immediately adjacent to the intraventricular septum. Gross electrocardiographic features such as the bundle branch block pattern or QRS axis did not reliably localize the origin of the tachycardia.
Kuchar analyzed QRS configurations observed during left ventricular pacing in 22 patients to construct an algorithm to localize the origin of VT, which was then prospectively tested in a second group of 44 episodes of VT.67 As shown in Figure 44-14, apical sites versus basal sites were differentiated by QRS configurations in leads V4 and aVR; anterior sites versus inferior sites were differentiated by QRS configuration in leads II, II, and V6; and septal sites versus lateral sites were distinguished by QRS configuration in leads I, aVL, and V1. Anterior sites were correctly identified in 83%, inferior sites in 84%, septal sites in 90%, and lateral sites in 82%. Apical and basal sites were accurately distinguished in 70%, but the algorithm was not reliable for intermediate sites. Overall, the site of origin was localized in 39%, and in an additional 36% the site of origin was immediately adjacent to the predicted site.
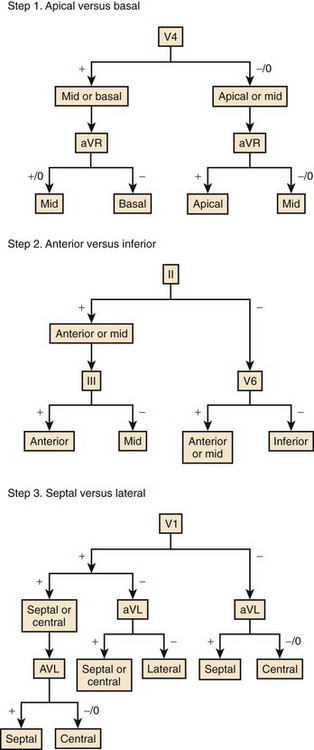
FIGURE 44-14 Algorithm for the localization of the origin of ventricular tachycardia.
(Modified from Kuchar DL, Ruskin JN, Garan H: Electrocardiographic localization of the site of origin of ventricular tachycardia in patients with prior myocardial infarction, J Am Coll Cardiol 13:893–903, 1989.)
Sippensgroenewegen compared 62-lead body surface QRS integral maps and scalar 12-lead ECGs obtained during pacemapping in patients with prior MI and VT to determine the accuracy of localizing the origin of VT with these two methods.70 An evaluation of the localization results obtained during pacemapping was performed to compare 12-lead ECGs with body surface mapping. The site of origin, as determined by endocardial mapping, was compared with the location suggested by pacemapping. With pacemapping used in combination with body surface maps, the site of origin was identified within 2 cm of the origin in 80% of VTs. The remaining 20% were localized to an adjacent (2 to 4 cm) or disparate (>4 cm) site. Results obtained with standard 12-lead ECGs were less accurate. Correct localization (<2 cm) was achieved in only 18%. The localization site was adjacent to the site of origin in 55% and disparate in 27%. The size of the corresponding endocardial area was much smaller with body surface mapping (6.0 ± 4.5 cm) than with the 12-lead ECG (15.1 ± 12.0 cm). These results are concordant with a study by Josephson, which showed that pacemapping with a 12-lead ECG allows localization to a relatively large area of 20 to 25 cm.2,71
Further study is needed to determine the usefulness of body surface mapping for ablation of VT; however, results obtained by Sippensgroenewegen indicated that this approach is relatively accurate.72 QRS integral maps (62 leads) were compared with activation mapping data acquired in 64 episodes of VT. The maps were compared with a previously generated infarct-specific reference database of paced QRS integral maps. Each pattern of the database corresponded to 1 of 18 to 22 segments of the left ventricle. Electrocardiographic localization was compared with intraoperative or catheter endocardial activation mapping. Body surface mapping identified the correct segment of origin in 62%, an adjacent segment in 30%, and disparate segments in 8%.
Clinical Electrophysiology
Programmed Stimulation for Initiation of Ventricular Tachycardia
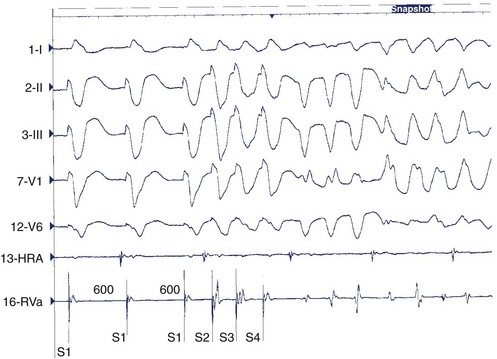
Most laboratories proceed through a stepwise stimulation protocol for initiation of sustained VT. After ventricular pacing for 8 to 15 beats, a single extrastimulus is used to scan electrical diastole until it encounters refractoriness or reaches a very short coupling interval (<200 ms). Second, third, and, in some cases, fourth stimuli are then added. Two or more different basic pacing rates (drive cycles) typically are used (e.g., 100 and 150 beats/min) before premature stimulus delivery. If pacing at the right ventricular apex fails to induce VT, pacing at a second right ventricular site (e.g., the right ventricular outflow tract) generally is used. Some laboratories also use pacing from the left ventricle. The number of stimuli, basic drive cycles, and ventricular pacing sites varies among laboratories. In general, pacing with up to three extrastimuli at two cycle lengths and two sites induces VT in approximately 90% of patients who have had this arrhythmia spontaneously after MI.73 The addition of rapid burst pacing, left ventricular stimulation, and programmed stimulation during isoproterenol infusion further increases sensitivity.
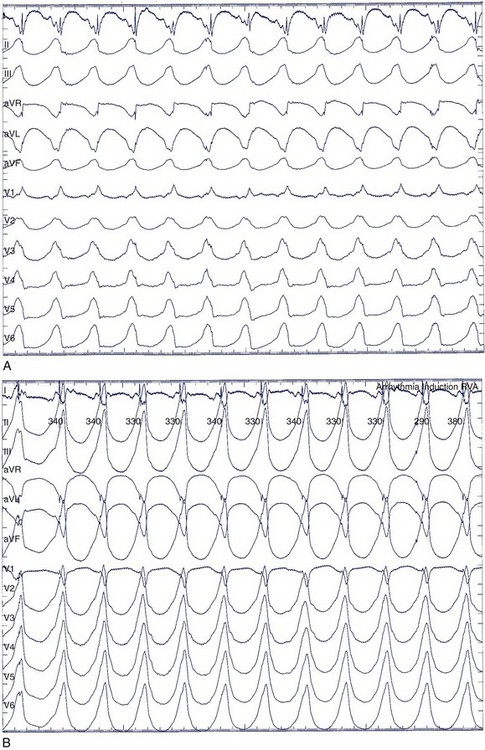
As the number of extrastimuli increases, the risk of initiating nonspecific, polymorphic VT (Figure 44-15) or VF increases. Limiting the closest coupling interval to greater than 200 ms reduces the risk of initiating ventricular fibrillation.73 Although stimulation protocols are relatively sensitive for detecting an inducible VT, the precise number of required stimuli and stimuli-coupling intervals often varies over time such that a change in the number of extrastimuli required for initiation of VT is not necessarily a reliable indication that susceptibility to VT has changed.74,75 VT initiated by isoproterenol infusion or with rapid burst pacing during isoproterenol administration, but not in response to coupled extrastimuli, is believed to be more likely caused by abnormal automaticity rather than by re-entry. Rarely, monomorphic VT may be induced by such measures in genetically based cardiac arrest syndromes.
Confirming the Diagnosis
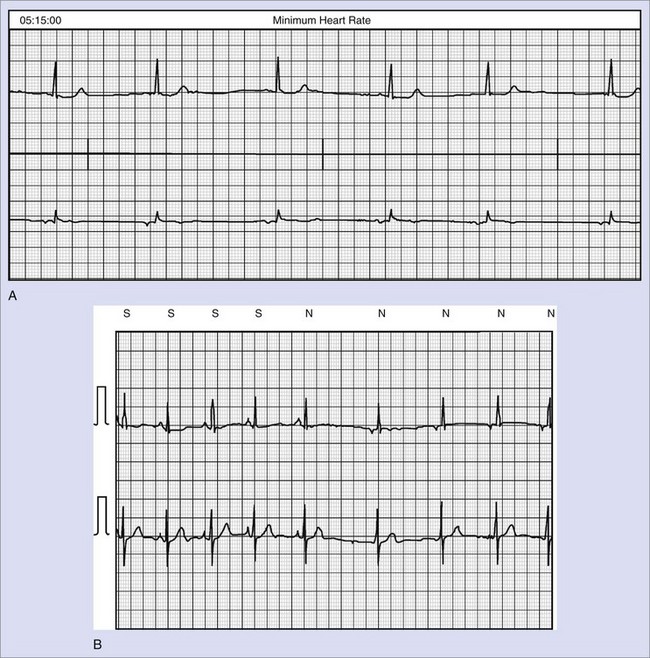
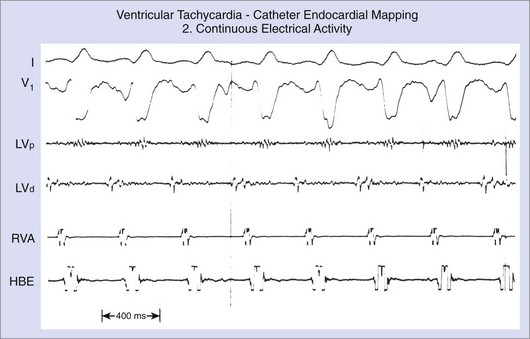
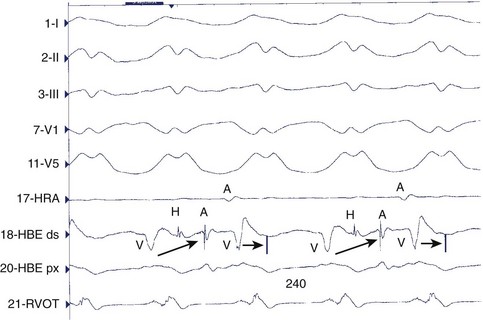
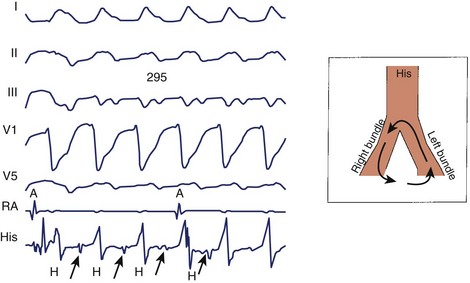
In contrast to ECGs, in which atrial activity may be difficult to detect, direct recordings from the atrium always allow clear delineation of whether AV dissociation is present (Figure 44-16). When AV dissociation is present, the diagnosis is VT, with the rare exception of junctional ectopic tachycardia with ventriculoatrial (VA) block or rare forms of SVT without atrial tissue participation. The former arrhythmia, which occurs most commonly in the early postoperative period after repair of congenital heart disease in children, is rare in adults. When AV dissociation is present and depolarization of the bundle of His does not precede each QRS complex, the diagnosis of VT is unequivocal (see Figure 44-16). However, diagnostic dilemmas occasionally are encountered. If each QRS complex is followed by an atrial depolarization, VT with 1:1 conduction in a retrograde manner through the His-Purkinje system and the AV node must be distinguished from antidromic AV re-entry using an accessory pathway (see Chapters 25 and 43).
Polymorphic Ventricular Tachycardia
Polymorphic VT (see Figure 44-15) indicates a continually changing ventricular activation sequence. Spontaneous polymorphic VT is most commonly caused by myocardial ischemia or TdP associated with Q-T interval prolongation. TdP is not reproducibly inducible in the electrophysiology laboratory, and electrophysiological studies are not warranted in the presence of active ischemia. Polymorphic VT also occurs in idiopathic VF, Brugada syndrome, and hypertrophic cardiomyopathy as well as other cardiomyopathies.
During ventricular stimulation, the significance of initiating polymorphic VT is often difficult to interpret. When sustained, polymorphic VT usually quickly deteriorates to VF. Nonsustained polymorphic VT is a nonspecific response to programmed stimulation that can be observed in patients who do not have a susceptibility to arrhythmias. Sustained polymorphic VT (lasting >30 seconds or requiring termination) is initiated less commonly in normal hearts and usually requires aggressive stimulation (three or more extrastimuli and relatively short stimulus coupling intervals of <200 ms). Initiation of polymorphic VT may be a marker of electrical instability in some situations. It is induced in approximately one third of patients who have been resuscitated from VF associated with depressed ventricular function and CAD.76 In patients with prior infarction and inducible polymorphic VT, sustained monomorphic VT may become inducible after administration of IV procainamide.74 Stabilization of a re-entry circuit by an antiarrhythmic drug is a possible mechanism, but the clinical relevance of this observation is unclear.
Initiation of polymorphic VT is of possible relevance in patients with suspected Brugada syndrome. Polymorphic VT, often induced with two or fewer extrastimuli, is induced in approximately 80% of patients with Brugada syndrome who have been resuscitated from cardiac arrest.77 In a patient with other features of the syndrome, inducible polymorphic VT further supports the diagnosis. In patients with hypertrophic cardiomyopathy, sustained VT (usually polymorphic) is inducible in 66% of those who had a prior cardiac arrest compared with 23% of patients without a history of cardiac arrest or syncope.78 The prognostic significance of inducible polymorphic VT in hypertrophic cardiomyopathy and other cardiomyopathies is controversial.79
Sustained Monomorphic Ventricular Tachycardia
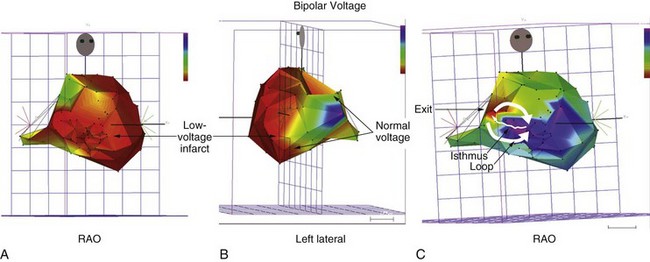
During sustained monomorphic VT, each QRS complex resembles the preceding and following QRS (see Figure 44-16). The ventricles are repetitively depolarized in the same sequence. In contrast to polymorphic VT, which can occasionally be induced in the absence of a cardiac abnormality, spontaneous or inducible sustained monomorphic VT indicates the presence of either an abnormal region supporting re-entry or a focus of automaticity. Re-entry through regions of ventricular scar from MI is the most common cause. Other causes of scar-related VT include arrhythmogenic right ventricular cardiomyopathy, sarcoidosis, scleroderma, Chagas disease, ventricular incisions after cardiac surgery, or other cardiomyopathic processes.78 Areas of scarring can often be identified as regions of low electrogram voltage during catheter mapping (Figure 44-17). On occasion, an incessant, idiopathic VT consistent with automaticity will cause tachycardia-induced cardiomyopathy.80
Bundle Branch Re-entry Ventricular Tachycardia
A bundle of His deflection is consistently present before each QRS during an uncommon type of VT caused by re-entry through the bundle branches (Figure 44-18). The re-entry wavefront circulates up the left bundle branch, down the right bundle branch, and then through the intraventricular septum to re-enter the left bundle.81 Ventricular depolarization proceeds from the right bundle, giving rise to a tachycardia with an LBBB configuration. Rarely, the circuit revolves in the opposite direction (down the left bundle and back up the right bundle) or is confined to the fascicles of the left bundle branch system, giving rise to a tachycardia with an RBBB configuration. The diagnosis is confirmed by showing that the atria can be dissociated but that the bundle of His depolarization is closely linked to the circuit. Bundle branch re-entry should be particularly suspected when sustained monomorphic VT occurs in the absence of a large region of ventricular scar or infarction, such as in patients with valvular heart disease, cardiomyopathy, or muscular dystrophy and in association with evidence of His-Purkinje system disease (intraventricular conduction delay) during sinus rhythm.82,83 Most patients have an LBBB QRS pattern on the resting ECG during sinus rhythm and a history of dilated cardiomyopathy.
QRS Morphology of Induced Ventricular Tachycardia
The QRS morphology reflects the ventricular activation sequence and often provides an indication of the likely location of the arrhythmogenic region (Figure 44-19). For automatic or focal VT, activation spreads away from one arrhythmia focus; the QRS morphology is an excellent indicator of the location of the focus. The QRS morphology is less reliable as an indicator of the circuit location for scar-related VT but is still useful.
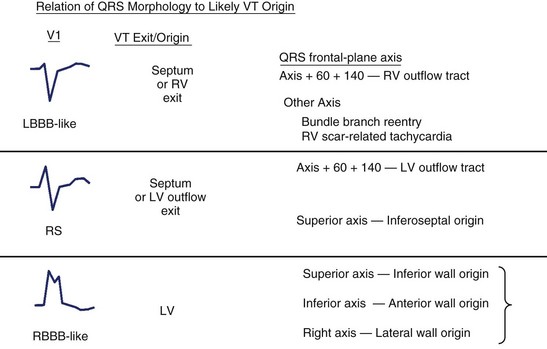
FIGURE 44-19 The QRS morphology suggests the likely origin and often the type of heart disease and ventricular tachycardia (VT). The morphology of lead V1 provides an initial indication of the likely ventricle of origin. Further refinement is based on the frontal-plane QRS axis, as shown at right. LBBB, Left bundle branch block; LV, left ventricle; RBBB, right bundle branch block; RV, right ventricle. See also Figure 44-14.
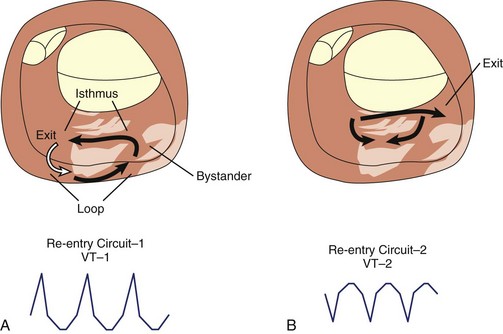
Scar-related re-entry circuits can be modeled as having surviving bundles of myocytes in the region of the scar. The depolarization of these strands is not detected in the surface ECG (Figure 44-20; also see Figure 44-17). These bundles may form narrow isthmuses in the re-entry circuit that are desirable targets for ablation. Recordings from these regions often reveal multiple low-amplitude potentials (Figure 44-21). During VT, the QRS is inscribed after the re-entry wavefront emerges from an isthmus at its exit and propagates across the ventricles. The QRS morphology indicates the location of the re-entry circuit exit (see Figure 44-20). Patients with scar-related VTs often have multiple morphologies of inducible VT; typically three or more are observed in patients referred for catheter ablation (Figure 44-22; also see Figure 44-20).84 VT that has been observed to occur spontaneously is often referred to as clinical VT. Other VTs may arise from the same region of the scar or from anatomically separate regions. Some “nonclinical” VTs are subsequently observed to occur spontaneously or are initiated by anti-tachycardia pacing from an implanted defibrillator during an attempt to terminate a clinical VT.
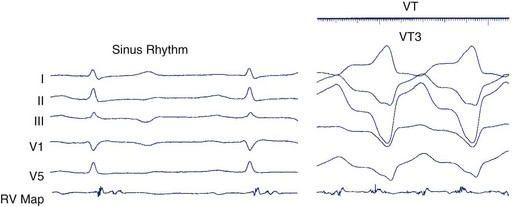
Pacing for Ventricular Tachycardia Termination
Monomorphic VT can be reliably terminated by electrical cardioversion, which presumably depolarizes the re-entry circuit in advance of the circulating wavefront, which then collides with refractory tissue, extinguishing re-entry. Many monomorphic VTs can also be terminated by pacing that is faster than the tachycardia (overdrive pacing). The stimulated wavefront propagates to the re-entry circuit and then splits into wavefronts traveling in the same direction as the re-entry circuit wavefronts (orthodromic) and a second wavefront traveling in the opposite direction (antidromic). The antidromic wavefront collides with the next re-entry wavefront, and both are extinguished. The stimulated orthodromic wavefront continues through the circuit and may reset the circuit so that tachycardia continues (known as resetting or entrainment). If the orthodromic wavefront encounters refractory tissue and is extinguished, the tachycardia terminates. Pacing during tachycardia also has the possibility to accelerate the tachycardia or initiate VF (see Figure 44-22). Thus the capability for prompt defibrillation must be present when anti-tachycardia pacing is used.
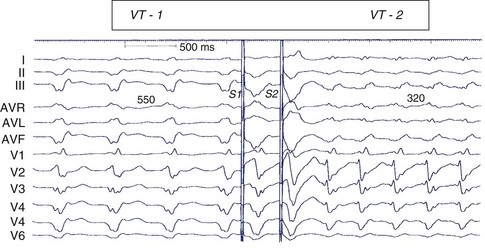
Catheter Mapping of Ventricular Tachycardia
Pacing during tachycardia can be used to determine the re-entry circuit location (entrainment mapping). Pacing trains that capture, but have no effect on, tachycardia often indicate that the pacing site is not in the tachycardia circuit. Stimulated wavefronts that propagate to the circuit, enter the circuit, and propagate through the circuit will reset the re-entry circuit in a characteristic manner. Whether this occurs depends on the timing of the stimulus relative to activation in the circuit. The wavefront must reach a portion of the circuit after that area has recovered excitability from the preceding wavefront. A single pacing stimulus may reset the tachycardia while a train of several stimuli continually resets the tachycardia, a response known as entrainment.84
Management
Principles of Practice
Sustained monomorphic VT occurs in patients with prior left ventricular scarring (most commonly from prior MI); most often arises without a specific predisposing event, often at rest or with modest activity; and generally is not preceded by symptomatic, electrocardiographic, or enzymatic evidence for myocardial ischemia or MI.85 Sustained VT is observed in a circadian pattern similar to that of SCD and MI, with a predilection for the morning hours (at least in patients with implanted defibrillators).86,87 It tends to occur in clusters, suggesting that neurohumoral, in particular sympathetic, activation may contribute indirectly to the instantaneous probability of VT occurring in a susceptible patient.87,88 On occasion, sustained VT occurs many times within a brief period, a syndrome commonly termed electrical storm. It is often arbitrarily defined as more than two or three episodes of sustained VT in a 24-hour period. Patients with this syndrome are perceived to have a very poor prognosis, although aggressive therapy, particularly β-blockers and amiodarone, may allow a prognosis similar to that for patients who do not develop electrical storm, as has been shown in observational studies.88,89 A related syndrome is that of polymorphic VT in patients with severe ventricular scarring, occurring most often in the setting of a generalized cardiomyopathy such as dilated idiopathic cardiomyopathy, or that associated with hypertrophic cardiomyopathy, valvular disease, or hypertensive cardiomyopathy. Although the precipitating events for this type of VT are also not clearly understood, they may be more likely to occur during physical exercise, elevated sympathetic tone, or episodes of worsening heart failure. The management of these patients, in addition to antiarrhythmic therapy, may also be aggressively directed toward improving myocardial function and decreasing sympathetic tone, for example, with afterload reduction, preload reduction, and sympathetic blockade.
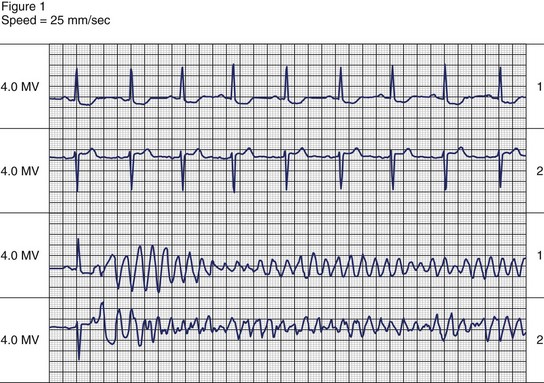
A particular pattern of sustained ventricular arrhythmias can be observed in patients with myocardial ischemia, acute MI, or severe CAD. In these patients, VT arises more frequently under conditions of physical or psychological stress; it may be preceded by chest pain or other symptoms of myocardial ischemia; and the ECG before or after treatment of the VT frequently shows sinus tachycardia, ST-segment depression, or signs of MI. The electrocardiographic signature of this arrhythmia (Figure 44-23) typically involves a relatively short Q-T interval as well as the initiating beat of tachycardia being closely coupled to the last normal beat, which immediately ushers in polymorphic VT that may rapidly degenerate to VF. Patients observed to have this pattern should be immediately and thoroughly investigated for myocardial ischemia; treatment directed at ischemia, rather than only at the arrhythmia, is often sufficient to prevent recurrence.90,91
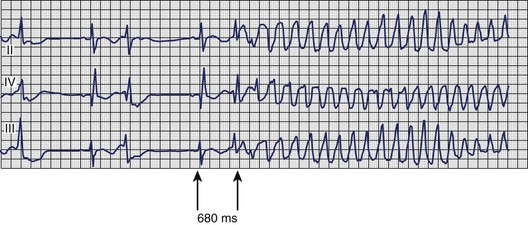
The syndrome of polymorphic VT associated with prolonged repolarization is discussed in detail in other sections of this book. However, it should be noted that sustained VT in association with delayed repolarization (TdP) may manifest as relatively monomorphic VT, especially at its outset (Figure 44-24). The electrocardiographic clues to the etiology as being related to abnormal and delayed repolarization rather than to an underlying “arrhythmogenic scar” are also to be found in the initiating sequence of tachycardia. Delayed repolarization–related VT is generally associated with Q-T interval prolongation, a pause-dependent initiating sequence, and a relatively long coupling interval between the last normal beat and the first beat of tachycardia. This beat typically falls on the late portion of the T wave or T-U complex of a beat with prolonged repolarization. These arrhythmias should be carefully distinguished from those caused by an arrhythmogenic scar because they can be effectively treated with IV magnesium and by repletion of potassium, as indicated, by increasing the heart rate, and by treating the underlying disturbance leading to prolonged repolarization, which is often drug therapy that prolongs repolarization, causes bradycardia, or both. In patients with structural heart disease, the precipitating causes are usually antiarrhythmic drugs that prolong repolarization, hypokalemia or hypomagnesemia, or bradycardia.
Acute Treatment of Sustained Ventricular Tachycardia
The acute management of VT should first be directed at restoring effective circulation, as outlined in the International Guidelines for Advanced Cardiac Life Support.92 In brief, all patients with substantial hemodynamic compromise, including diminished level of consciousness, hypotension, and clinical signs suggesting cerebral or vital organ hypoperfusion, heart failure, or signs or symptoms of myocardial ischemia, should be treated with urgent synchronized cardioversion. Of note, carefully performed electrical cardioversion is almost completely effective for the treatment of VT, if it can be provided promptly, and is associated with extremely low risk. The perceived need for anesthesia is often seen as a limitation to acute cardioversion but can generally be performed by conscious sedation without intubation or serious anesthetic risk. If drug therapy is chosen either as initial treatment of VT or therapy to prevent recurrences immediately after electrical cardioversion, IV procainamide and IV amiodarone are recommended.92 Careful monitoring for hypotension and bradycardia, which may be caused by the antiarrhythmic drug therapy, is required.
A large evidence base of randomized clinical trials is not available to assist the clinician in the treatment of patients immediately following successful conversion from sustained VT to sinus rhythm. The immediate risk of recurrence will vary with the clinical context and be generally relatively low in patients with stable CAD and left ventricular dysfunction with monomorphic VT. It is higher in patients with acute hemodynamic compromise independent of the tachycardia; ongoing myocardial ischemia; or other metabolic disturbances found in postoperative states, sepsis, pneumonia, or hypoxia. In patients with nonischemic cardiomyopathies, therapy specifically directed at blunting the effects of sympathetic activation on the heart with intravenous β-blockers, or stellate ganglion block, is an often neglected but important aspect of therapy.89 Limited, controlled clinical trials suggest that IV amiodarone and bretylium are likely effective at preventing recurrent VT.93 Bretylium, however, is no longer commercially available. In a randomized, multi-center trial in patients with electrical storm (defined as more than two episodes of sustained VT in a 24-hour period [mean, 4.93 episodes] resistant to treatment with IV lidocaine and procainamide), patients were randomized to blinded treatment with IV amiodarone (1000 mg/day), IV bretylium (2.5 g/day), and low-dose amiodarone (125 mg/day).93 Although interpretation of this study has been complicated by the fact that many patients in the low-dose amiodarone group and the bretylium group received open-label treatment with unblinded IV amiodarone, the results suggest that IV amiodarone is efficacious at reducing the likelihood of recurrent VT. The median number of episodes was 0 in the first 12 hours after treatment with amiodarone and 0.48 during the entire study period. The median time to termination of incessant VT was 4.23 hours after the initiation of IV amiodarone therapy. No comparable trials of class I drugs or sotalol in electrical storm have been conducted so far. Extrapolation from clinical trials of prophylactic therapy suggests that class I drugs (sodium channel blockers) should be avoided in patients with frequently recurring VT or VF.92
In the Optimal Pharmacological Therapy in Cardioverter Defibrillator Patients (OPTIC) study in patients with implanted defibrillators, amiodarone was superior to sotalol or β-blockers in preventing appropriate ICD therapies for VT or VF.94
Although no randomized clinical trials of sedation in patients with frequently recurring VT have been performed, anecdotal experience suggests that vigorous sedation is an important adjunct in the treatment of this syndrome. As an acute therapy, IV sotalol, IV flecainide, and IV procainamide have all been reported to be effective in the acute termination of sustained VT.92 Given the negative inotropic and proarrhythmic risks of drugs with sodium channel–blocking activity, it seems prudent to use such drugs with great care, if at all, in patients with structural heart disease and sustained VT, especially in the context of CAD.
Impact of Clinical Trials
A recent explosion of epidemiologic insights and information from randomized trials has changed clinical practice in the treatment of VT (Box 44-1). For several years, the similarity of patient profiles (stratifying by left ventricular ejection fraction [LVEF], previous MI) and risk for sudden cardiac death and sustained VT has been apparent.85 In 1975, Schaffer et al reported on 234 patients after successful resuscitation of cardiac arrest. Over a follow-up period of 51 months, 89 episodes (approximately 38%) of recurrent cardiac arrest or death occurred.95 A similar rate of recurrence was noted by Myerburg et al in 1984—the recurrence rate for cardiac arrest was 10% the first year after arrest and 5% per year in each of the following 3 years.96 In 1993, the Cardiac Arrest in Seattle: Conventional versus Amiodarone Drug Evaluation (CASCADE) study reported an improvement in out-of-hospital cardiac arrest survival in patients randomized to amiodarone (versus conventional antiarrhythmic therapy) of 78% at 2 years and 52% at 4 years.97 Therapeutic options drastically shifted in the 1980s with the emergence of ICDs. Several recent trials have compared the efficacy of antiarrhythmic therapy and ICDs for overall mortality in patients who have sustained a VT event.
Box 44-1 Indications for Implantable Cardioverter Defibrillator Therapy for Patients at Risk of Sustained VT
Class I
Class IIA
Class IIB
Class III
Modified from Epstein AE, DiMarco JP, Ellenbogen KA, et al: ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: American College of Cardiology/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons, J Am Coll Cardiol 51:1–62, 2008.
In 1995, Dutch investigators randomized 60 patients with previous MI, cardiac arrest secondary to VT or VF, and inducible ventricular arrhythmia at electrophysiological study to conventional therapy (class IA, IC, and III drugs) or the ICD.98 The LVEF was approximately 30% in each group. Drug efficacy was assessed by serial drug testing, and nonresponders were given an ICD. An imbalance of coronary revascularization occurred in the conventional therapy group (10%) and the ICD group (26%). Although the numbers of clinical endpoints (death, prolonged syncope with circulatory arrest, and congestive heart failure requiring transplantation) were small, the outcome favored treatment with an ICD (P < .02).
The Cardiac Arrest Study Hamburg (CASH) began enrollment in 1987 and randomized patients to antiarrhythmic therapy (propafenone, metoprolol, or amiodarone) or ICD.99 Entry criteria included survivors of cardiac arrest secondary to VT (83%) or VF (16%). All patients underwent electrophysiological testing. The primary endpoint was total mortality, with secondary endpoints of VT recurrence, need to discontinue medical therapy, recurrence of cardiac arrest, and need for cardiac transplantation. In 1992, the CASH Safety and Monitoring Board discontinued the propafenone randomization arm because of an excess of SCD, recurrent cardiac arrest, and recurrent VT (all secondary endpoints) in the patients taking class Ic drugs compared with the ICD arm (P < .05) despite no differences in the total mortality rate. LVEF averaged for all four groups was more than 40%. The other three treatment arms continued to recruit patients. The final results showed a 36.4% mortality rate in the ICD group and a 44.4% mortality rate in the metoprolol and amiodarone groups combined. A one-sided t test (which would have amplified any benefit of the ICD) had a P value of .081 as the reported significance value.
Two larger studies, having slightly more heterogeneous populations, also address the issue of ICD versus antiarrhythmic therapy in patients with symptomatic VT. The Canadian Implantable Defibrillator Study (CIDS) randomized 659 patients with documented VF, out-of-hospital cardiac arrest, symptomatic VT, or syncope with inducible VT at electrophysiology study to ICD or amiodarone therapy.100 Entry criteria in approximately 50% of each group were VF or cardiac arrest. Mean LVEF was 33% in each group. The primary endpoint was all-cause mortality. An intention-to-treat analysis was used to analyze the data in a one-sided t test. On treatment analysis showed 94% of those randomized to ICD actually received an ICD, and 85% of patients randomized to amiodarone were still on therapy at their 5-year follow-up. Although β-blocker use was a potential confounder (23% in the amiodarone group, 53% in the ICD group), the overall mortality rate (10.2% per year) in the amiodarone group was not significantly higher than that in the ICD group (8.3%, P = .142).
The largest study to date has been the Antiarrhythmics versus Implantable Defibrillator (AVID) study, which randomized 1016 patients with resuscitated VF, sustained VT with syncope, and sustained VT with hemodynamic compromise to antiarrhythmics or to ICD implantation.101 The 6035 patients were screened, with 17% randomized. Of the randomized patients, 45% had VF and 55% had VT at entry. The patients randomized to antiarrhythmics could be further randomized to electrophysiology-guided treatment with sotalol or empiric amiodarone at the discretion of the investigator. LVEF was similar in each group (31% and 32%). An on-treatment analysis showed that 98% of the ICD group received an ICD. In the antiarrhythmic group, 85% received empiric amiodarone, 11% received amiodarone after not responding to sotalol, and 2.6% were treated with sotalol. The study was terminated early in April 1997 because of the recommendation of the Data and Safety Monitoring Board; unlike the CIDS study, the ICD group showed a significant improvement in total mortality rate over an average follow-up of 18 months (15.8% in the ICD group, 24% in the antiarrhythmic group; P = .02).
In summary, the available epidemiologic and clinical trial data evoke several principles applicable to patients with sustained symptomatic VT. The ICD prevents arrhythmic death only. The majority of participants in these trials with low LVEF and structural heart disease have competing modes of death. Antiarrhythmics may have some positive effect on congestive heart failure (amiodarone) or actually worsen arrhythmic death via proarrhythmia (propafenone in the CASH study). Investigator and subject bias is unavoidable in a trial that is not blinded to the treatment arm. Furthermore, a heterogeneous study population (as in the AVID and CIDS studies) may not have the same treatment effect across all strata in the group. Domanski et al reported a differential effect of antiarrhythmics and ICD therapy from the AVID database based on strata of left ventricular EF.91,102 In patients with LVEF greater than 35%, the two therapies showed no difference in survival, suggesting that all the treatment effects came from enrolled patients with LVEF less than 35%. Despite the inherent limitations of the published clinical trials, the largest studies favor ICD therapy in patients whose primary mortality risk is from symptomatic ventricular tachyarrhythmia. Box 44-1 lists current indications and contraindications to ICD therapy.
In summary, the long-term management of patients with sustained VT and heart disease is very similar to that for patients resuscitated from cardiac arrest.103 A retrospective analysis of outcome in patients with “tolerated” VT and VT without cardiac arrest in the AVID trial suggests that their outcome is very similar to that in patients with VF or VT with serious cardiac compromise. It seems reasonable to expect that most patients with sustained symptomatic VT or VF, regardless of the severity of symptoms, have a poor prognosis and should be managed as outlined in the section on evidence-based therapy in VF. However, patients with relatively preserved LV function (EF > 40%) and symptomatic VT without cardiac arrest were not included in the randomized trials of ICD therapy. Whether such patients would have had improved survival with ICD therapy compared with “best medical therapy” (which should almost certainly include β-blockers and amiodarone) is unknown. Subanalyses of the CIDS and AVID studies suggest that a benefit of the ICD over amiodarone, in the medium term, may not be observed in patients with LVEF greater than 35% and symptomatic VT.102,104 These latter patients can be reasonably treated with either an ICD or oral amiodarone and a β-blocker.105 Long-term management of patients with sustained VT without heart disease is detailed in the next section.
Impact of New Device Technology
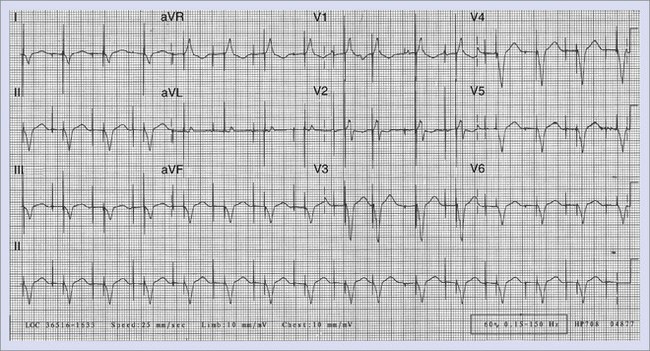
Concurrent with these important secondary and primary prevention clinical trials have been important technologic advances in device technology.106 These are discussed in greater detail in Section IX of this text. Of greatest importance for patients with sustained VT and left ventricular dysfunction is the advent of biventricular pacing. Biventricular pacing has shown benefits in patients with drug-refractory congestive heart failure and coexisting intraventricular and interventricular dyssynchronous wall motion.107 These patients have been largely identified by the presence of prolonged QRS complexes, although other techniques of wall motion analysis are increasingly being applied. Thus patients with bundle branch block, particularly with QRS complexes greater than 0.15 seconds and first-degree AV block, which can further compromise ventricular filling, are candidates for this technique. Atrial fibrillation (AF) has been an important coexisting arrhythmia in many of these sustained VT patients and causes progressive heart failure and increased mortality rates. New AF therapies for prevention and termination are now available in ICD devices. Thus alternative-site pacing such as dual-site atrial pacing, novel preventive pacing algorithms for atrial premature beat suppression, anti-tachycardia pacing for atrial tachycardia termination, and AF therapies are now available. Figure 44-25 shows an ECG from an older patient with refractory persistent AF, sustained VT with syncope, and refractory congestive heart failure. A four-chamber ICD device (Insync III; Medtronic, Minneapolis, MN) with biventricular pacing and dual-site right atrial pacing leads was inserted for the management of these clinical syndromes.
Impact of Catheter Ablation Techniques
Catheter ablation technologies have been applied for ablation of sustained and hemodynamically stable VT in patients with and without organic heart disease who could undergo catheter mapping.108,109 Radiofrequency ablation remains the mainstay of this approach. In addition to ECG morphology, catheter mapping with contact catheter and noncontact three-dimensional mapping techniques has facilitated determination of the exact localization of the VT substrate. (The details of these techniques are reviewed in other chapters in this text.) These methods now permit rapid mapping and accurate geometric localization of these diseased tissues in a few or even one tachycardia cycle. Thus increasingly hemodynamically unstable VT episodes can be treated by ablative interventions.110 Radiofrequency energy delivered by contact electrodes with or without cooled-tip technology can now produce moderate-sized lesions in diseased ventricular myocardium. Thus VT ablation can temporarily suppress, reduce the frequency of, and even eliminate recurrent sustained VT in selected patients. It can reduce the need for ICD therapies.111
However, it is not curative, and VT recurrences often occur during long-term follow-up.112 Thus VT ablation in patients with organic heart disease is a potential management tool in specific clinical scenarios. It is valuable in drug-refractory incessant VT or electrical storm unresponsive to medical therapy. In patients with frequent, recurrent ICD shocks despite antiarrhythmic drug therapy, VT ablation will reduce the need for these therapies. Finally, it can be used in a patient who is intolerant to antiarrhythmic drug therapy when frequent VT recurrences must be addressed. However, VT ablation should be used in conjunction with ICD therapy, whenever possible, to provide backup rescue therapies for recurrent VT or VF.
Impact on Patient Selection
The selection of the first line of therapy in VT management is now defined by the clinical disease, intercurrent factors, and arrhythmia characteristics. Management of the underlying disease and associated conditions such as heart failure and ischemia is an integral part of patient management. Arrhythmia frequency is an important consideration. Frequent or incessant VT requires evaluation for intercurrent precipitating factors such as electrolyte abnormalities, enhanced sympathetic tone, hypoxia, or uncontrolled ischemia. Correction of these factors often converts this intolerable VT frequency to “occasional event” status. The hemodynamic impact of the arrhythmia also determines the urgency of the response. Hemodynamic collapse requires urgent use of both pharmacologic and nonpharmacologic therapies. In more stable VT episodes, drug therapy, mapping and ablation, and anti-tachycardia pacing become competitive options. With reduction of event frequency or in patients with sporadic VT episodes, a longer term management strategy is appropriate for implementation. Prevention of SCD with ICD backup is an essential part of the therapeutic prescription in these patients. Antiarrhythmic drugs and catheter ablation can play a supportive role in this phase of management to reduce VT event rates, make it amenable to pacing therapies, and prevent frequent symptoms related to the arrhythmia and ICD therapies.113
Key References
Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: Meta-analysis of individual data from 6500 patients in randomized trials. Lancet. 1997;350:1417-1424.
The AVID Investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
Bharati S, Lev M. The pathologic aspects of ventricular tachycardia. In: Iwa T, Fontaine G, editors. Cardiac arrhythmias: Recent investigation and management. Amsterdam: Elsevier Science Publishers, BV Biomedical Division, 1988.
Brugada P, Brugada J, Mont L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649-1659.
Connolly SJ, Dorian P, Roberts RS, et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study: A randomized trial. JAMA. 2006;295:165-171.
Connolly S, Gent M, Roberts R, et al. Canadian implantable defibrillator study (CIDS)—a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation. 2000;101:1297-1302.
De Bakker JMT, Van Capelle FJL, Janse MJ, et al. Reentry as a cause of ventricular tachycardia in patients with chronic ischemic heart disease: Electrophysiologic and anatomic correlation. Circulation. 1988;77:589-606.
Delacretaz E, Stevenson WG. Catheter ablation of ventricular tachycardia in patients with coronary heart disease. Part II: Clinical aspects, limitations, and recent developments. Pacing Clin Electrophysiol. 2001;24:1403-1411.
2000 Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care: International consensus on science. Part 6: Advanced cardiac life support; Section 5: Pharmacology I: Agents for arrhythmias. Circulation. 2000;102:I112-I128.
Kowey PR, Levine JH, Herre JM, et al. Randomized, double-blind comparison of intravenous amiodarone and bretylium in the treatment of patients with recurrent, hemodynamically destabilizing ventricular tachycardia or fibrillation. The Intravenous Amiodarone Multicenter Investigators Group. Circulation. 1995;92:3255-3263.
Reddy VY, Neuzil P, Taborsky M, Ruskin JN. Short-term results of substrate mapping and radiofrequency ablation of ischemic ventricular tachycardia using a saline-irrigated catheter. J Am Coll Cardiol. 2003;41:2228-2236.
Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270-283.
Wit AL, Janse MJ. The ventricular arrhythmias of ischemia and infarction: Electrophysiological mechanisms. Mount Kisco, NY: Futura Publishing; 1993.
Zipes DP. Influence of myocardial ischemia and infarction on autonomic innervation of heart. Circulation. 1990;82:1095-1104.
Zipes DP, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines. Circulation. 2006;114:e385-e484.
1 Mason JW. For the Electrophysiologic Study versus Electro cardiographic Monitoring Investigators: A comparison of electrophysiologic testing with Holter monitoring to predict antiarrhythmic drug efficacy for ventricular tachyarrhythmias. N Engl J Med. 1993;329:445.
2 Bharati S. Pathology of the conduction system. In Silver MD, Gotlieb AI, Schoen FJ, editors: Cardiovascular pathology, ed 3, New York: Churchill Livingstone, 2001.
3 Bharati S, Lev M. The pathologic aspects of ventricular tachycardia. In: Iwa T, Fontaine G, editors. Cardiac arrhythmias: Recent investigation and management. Amsterdam: Elsevier Science Publishers BV Biomedical Division, 1988.
4 Bharati S, Lev M. The cardiac conduction system in unexplained sudden death. Mt. Kisco, NY: Futura Publishing; 1990.
5 Bharati S, Granston AS, Liebson PR, et al. The conduction system in mitral valve prolapse syndrome with sudden death. Am Heart J. 1981;101:667-670.
6 Bharati S, Feld AW, Bauernfeind R, et al. Hypoplasia of the right ventricular myocardium with ventricular tachycardia. Arch Pathol Lab Med. 1983;107:249-253.
7 Bharati S, McAnulty JH, Lev M, Rahimtoola SH. Idiopathic hypertrophic subaortic stenosis with split His bundle potentials: Electrophysiologic and pathologic correlations. Circulation. 1980;62:1373-1380.
8 Bharati S, Lev M, Denes P, et al. Infiltrative cardiomyopathy with conduction disease and ventricular arrhythmia: Electrophysiologic and pathologic correlations. Am J Cardiol. 1980;45:163-173.
9 Bharati S, Strasberg B, Bilitch M, et al. Anatomic substrate for preexcitation in idiopathic myocardial hypertrophy with fibroelastosis of the left ventricle. Am J Cardiol. 1981;48:47-58.
10 Bharati S, Driefus L, Bucheleres G, et al. The conduction system in patients with a prolonged QT interval. J Am Coll Cardiol. 1985;6:1110-1119.
11 Bharati S, Bump T, Bauernfeind R, Lev M. Dystrophica myotonia. Chest. 1984;86:444-450.
12 Gallastegui J, Hariman RJ, Handler B, et al. Cardiac involvement in the Kearns-Sayre syndrome. Am J Cardiol. 1987;60:385-388.
13 Bharati S, Lev M. Arrhythmogenic ventricles. Pacing Clin Electrophysiol. 1983;6:1035-1049.
14 Bharati S, Lev M. Sequelae of atriotomy and ventriculotomy on the endocardium, conduction system and coronary arteries. Am J Cardiol. 1982;50:580-586.
15 Bharati S, Lev M. Conduction system in cases of sudden death in congenital heart disease many years after surgical correction. Chest. 1986;90:861-868.
16 Bharati S, Lev M. The pathology of congenital heart disease. A personal experience with more than 6300 congenitally malformed hearts. Armonk, NY: Futura Publishing; 1996.
17 Bharati S, Scheinmann MM, Morady F, et al. Sudden death after catheter-induced atrioventricular junctional ablation. Chest. 1985;88:883-889.
18 Gault JH, Cantwell J, Lev M, Braunwald E. Fatal familial cardiac arrhythmias. Am J Cardiol. 1972;29:548-553.
19 Husson GS, Blackman MS, Rogers MC, et al. Familial congenital bundle branch system disease. Am J Cardiol. 1973;32:365-369.
20 Brookfield L, Bharati S, Denes P, et al. Familial sudden death: Report of a case and review of the literature. Chest. 1988;94:989-993.
21 Bharati S, Lev M. Congenital abnormalities of the conduction system in sudden death in young adults. J Am Coll Cardiol. 1986;8:1096-1104.
22 Bharati S, Bauernfeind R, Scheinman M, et al. Congenital abnormalities of the conduction system in two patients with tachyarrhythmias. Circulation. 1979;59:593-606.
23 Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. 1989;69:1049-1169.
24 Wit AL, Janse MJ. The ventricular arrhythmias of ischemia and infarction: Electrophysiological mechanisms. Mount Kisco, NY: Futura Publishing; 1993.
25 Pinto JMB, Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovasc Res. 1999;42:284-297.
26 Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin 43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988-996.
27 Ursell PC, Gardner PI, Albala A, et al. Structural and electrophysiological changes in the epicardial border zone during infarct healing. Circ Res. 1985;56:436-451.
28 De Bakker JMT, Van Capelle FJL, Janse MJ, et al. Reentry as a cause of ventricular tachycardia in patients with chronic ischemic heart disease: Electrophysiologic and anatomic correlation. Circulation. 1988;77:589-606.
29 Callans DJ, Josephson ME. Ventricular tachycardia associated with coronary artery disease. In Zipes DP, Jalife J, editors: Cardiac electrophysiology: From cell to bedside, ed 2, Philadelphia: WB Saunders, 1995.
30 Willems AR, Tijssen JGP, van Capelle FJL, et al. Determinants of prognosis in symptomatic ventricular tachycardia or ventricular fibrillation late after myocardial infarction. J Am Coll Cardiol. 1990;16:521-530.
31 Hohnloser SH, Franck P, Klingenheben T, et al. Open infarct artery, late potentials and other prognostic factors in patients after acute myocardial infarction in the thrombolytic era. A prospective trial. Circulation. 1994;90:1747-1756.
32 Julian DG, Camm AJ, Frangin G, et al. Randomized trial of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997;349:667-674.
33 De Bakker JMT, Coronel R, Tasseron S, et al. Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: Role of the arrangement of surviving cardiac fibers. J Am Coll Cardiol. 1990;15:1594-1607.
34 De Bakker JMT, van Capelle FJL, Janse MJ, et al. Slow conduction in the infarcted human heart: “Zigzag” course of activation. Circulation. 1993;88:915-926.
35 Maglaveras N, de Bakker JMT, van Capelle FJL, et al. Activation delay in healed myocardial infarction: A comparison between model and experiment. Am J Physiol. 1995;269:H1441-H1449.
36 Ramdat Misier AR, Opthof T, van Hemel NM, et al. Dispersion of “refractoriness” in noninfarcted myocardium of patients with ventricular tachycardia or ventricular fibrillation after myocardial infarction. Circulation. 1995;91:2566-2572.
37 Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270-283.
38 Haverkamp W, Breithardt G, Camm AJ, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: Clinical and regulatory implications. Cardiovasc Res. 2000;47:219-233.
39 McIntosh MA, Cobbe SM, Smith GL. Heterogeneous changes in action potential and intracellular Ca++ in left ventricular myocyte subtypes from rabbits with heart failure. Cardiovasc Res. 2000;45:397-409.
40 Bryant SM, Shipsey SY, Hart G. Regional differences in electrical and mechanical properties of myocytes from guinea-pig hearts with mild left ventricular hypertrophy. Cardiovasc Res. 1997;35:315-323.
41 Keung EC, Aronson RS. Non-uniform electrophysiological properties and electrotonic interaction in hypertrophied myocardium. Circ Res. 1981;49:150-158.
42 Qin D, Zhang ZH, Caret EB, et al. Cellular and ionic basis of arrhythmias in postinfarction remodeled ventricular myocardium. Circ Res. 1996;79:461-473.
43 Peters NS, Cabo C, Wit AL. Arrhythmogenic mechanisms: Automaticity, triggered activity, and reentry. In Zipes DP, Jalife J, editors: Cardiac electrophysiology: From cell to bedside, ed 3, Philadelphia: WB Saunders, 2000.
44 Saffitz JE, Schuessler RB, Yamada KA. Mechanisms of remodeling of gap junction distribution and the development of anatomic substrates of arrhythmias. Cardiovasc Res. 1999;42:309-317.
45 Zipes DP. Influence of myocardial ischemia and infarction on autonomic innervation of heart. Circulation. 1990;82:1095-1104.
46 Cao J-M, Chen LS, KenKnight BH, et al. Nerve sprouting and sudden cardiac death. Circ Res. 2000;86:816-821.
47 Tsuji Y, Opthof T, Kamiya K, et al. Pacing-induced heart failure causes a reduction of delayed rectifier potassium currents along with a decrease in calcium and transient outward current in rabbit ventricle. Cardiovasc Res. 2000;48:300-309.
48 Cerbai E, Pino R, Porciati F, Mugelli A. Characterization of the hyperpolarization-activated current in ventricular myocytes from human failing heart. Circulation. 1998;95:568-571.
49 Vermeulen JT, McGuire MA, Opthof T, et al. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28:1547-1554.
50 Wickenden AD, Kaprielien R, Tiassiri Z, et al. Role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc Res. 1998;37:312-323.
51 Makielski J, Kamp T, Valdivia CR. Late sodium current is increased in ventricular myocytes from failing human heart compared with non-failing hearts, Jpn. J Electrocardiol. 2000;20(Suppl 3):99-100.
52 Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: Effects of beta-adrenergic agonists and sodium channel blockers on transmural dispersion of repolarization and torsades de pointes. Circulation. 1998;98:2314-2322.
53 Hasenfuss G. Alteration of calcium regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279-289.
54 Movsesian M, Schwinger RHG. Calcium sequestration by the sarcoplasmic reticulum in heart failure. Cardiovasc Res. 1998;37:352-359.
55 Phillips RM, Novayam P, Gomez A, et al. Sarcoplasmic reticulum in heart failure: Central players or bystanders? Cardiovasc Res. 1998;7:346-351.
56 Opthof T, Coronel R, Rademaker HME, et al. Changes in sinus node function in a rabbit model of heart failure with ventricular arrhythmias and sudden death. Circulation. 2000;101:2975-2980.
57 Rademaker HME. Arrhythmogenesis during the development of heart failure in rabbits [thesis]. Amsterdam: University of Amsterdam; 1997.
58 Luu M, Stevenson WG, Stevenson LW, et al. Diverse mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation. 1989;80:1675-1680.
59 Stevenson WG, Stevenson LW, Middlekauf HR, Saxon LA. Sudden death prevention in patients with advanced ventricular dysfunction. Circulation. 1993;88:2953-2961.
60 Saksena S, Ciccone J, Craelius W, et al. Studies on left ventricular function during sustained ventricular tachycardia. J Am Coll Cardiol. 1984;4:501-508.
61 Hamer AW, Rubin SA, Peter T, Mandel WJ. Factors that predict syncope during ventricular tachycardia in patients. Am Heart J. 1984;107:997-1005.
62 Krol RB, Saksena S. Clinical trials of antiarrhythmic drugs in recipients of implantable cardioverter-defibrillators. Interventional electrophysiology, ed 2. Futura, Mt. Kisco, NY, 1996.
63 Sandler A, Marriott HJL. The differential morphology of anomalous ventricular complexes of RBBB type in lead V1.Ventricular ectopy versus aberration. Circulation. 1965;31:551-556.
64 Marriott HJL, Sandler IA. Criteria, old and new, for differentiating between ectopic ventricular beats and aberrant ventricular conduction in the presence of atrial fibrillation, Progr. Cardiovasc Dis. 1966;9:18-28.
65 Wellens JHH, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med. 1978;64:27-33.
66 Kindwall KE, Brown J, Josephson ME. Electrocardiographic criteria for ventricular tachycardia in wide complex left bundle branch block morphology tachycardias. Am J Cardiol. 1988;61:1279-1283.
67 Brugada P, Brugada J, Mont L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649-1659.
68 Josephson ME, Horowitz LN, Waxman HL, et al. Sustained ventricular tachycardia: Role of the 12-lead electrocardiogram in localizing the site of origin. Circulation. 1981;64:257-271.
69 Kuchar DL, Ruskin JN, Garan H. Electrocardiographic localization of the site of origin of ventricular tachycardia in patients with prior myocardial infarction. J Am Coll Cardiol. 1989;13:893-903.
70 Sippensgroenewegen A, Spekhorts H, Van Hemel NM, et al. Localization of the site of origin of postinfarction ventricular tachycardia by endocardial pace mapping: Body surface mapping compared with the 12-lead electrocardiogram. Circulation. 1993;88:2290-2306.
71 Josephson ME, Waxman HL, Cain ME, et al. Ventricular activation during ventricular endocardial pacing, II: Role of pace-mapping to localize origin of ventricular tachycardia. Am J Cardiol. 1982;50:11-22.
72 Sippensgroenewegen A, Spekhorst H, Van Hemel NM, et al. Value of body surface mapping in localizing the site of origin of ventricular tachycardia in patients with previous myocardial infarction. J Am Coll Cardiol. 1994;24:1708-1724.
73 Hummel JD, Strickberger SA, Daoud E, et al. Results and efficiency of programmed ventricular stimulation with four extrastimuli compared with one, two, and three extrastimuli. Circulation. 1994;90:2827-2832.
74 Cooper MJ, Koo CC, Skinner MP, et al. Comparison of immediate versus day to day variability of ventricular tachycardia induction by programmed stimulation. J Am Coll Cardiol. 1989;13:1599-1607.
75 Wellens HJ, Brugada P, Stevenson WG. Programmed electrical stimulation of the heart in patients with life-threatening ventricular arrhythmias: What is the significance of induced arrhythmias and what is the correct stimulation protocol? Circulation. 1985;72:1-7.
76 Buxton AE, Josephson ME, Marchlinski FE, et al. Polymorphic ventricular tachycardia induced by programmed stimulation: Response to procainamide. J Am Coll Cardiol. 1993;21:90-98.
77 Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: A marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97:457-460.
78 Fananapazir L, Chang AC, Epstein SE, et al. Prognostic determinants in hypertrophic cardiomyopathy. Prospective evaluation of a therapeutic strategy based on clinical, Holter, hemodynamic, and electrophysiological findings. Circulation. 1992;86:730-740.
79 Turitto G, Ahuja RK, Caref EB, et al. Risk stratification for arrhythmic events in patients with nonischemic dilated cardiomyopathy and nonsustained ventricular tachycardia: Role of programmed ventricular stimulation and the signal-averaged electrocardiogram. J Am Coll Cardiol. 1994;24:1523-1528.
80 Delacretaz E, Stevenson WG, Ellison KE, et al. Mapping and radiofrequency catheter ablation of the three types of sustained monomorphic ventricular tachycardia in nonischemic heart disease. J Cardiovasc Electrophys. 2000;11:11-17.
81 Blanck Z, Dhala A, Deshpande S, et al. Bundle branch reentrant ventricular tachycardia: Cumulative experience in 48 patients. J Cardiovasc Electrophysiol. 1993;4:253-262.
82 Narasimhan C, Jazayeri MR, Sra J, et al. Ventricular tachycardia in valvular heart disease: Facilitation of sustained bundle-branch reentry by valve surgery. Circulation. 1997;96:4307-4313.
83 Merino JL, Carmona JR, Fernandez-Lozano I, et al. Mechanisms of sustained ventricular tachycardia in myotonic dystrophy: Implications for catheter ablation. Circulation. 1998;98:541-546.
84 Stevenson WG, Friedman PL, Sager PT, et al. Exploring postinfarction reentrant ventricular tachycardia with entrainment mapping. J Am Coll Cardiol. 1997;29:1180-1189.
85 Stevenson W, Brugada P, Waldecker B, et al. Clinical, angiographic, and electrophysiologic findings in patients with aborted sudden death as compared with patients with sustained ventricular tachycardia after myocardial infarction. Circulation. 1985;71:1146-1152.
86 Nanthakumar K, Newman D, Paquette M, et al. Circadian variation of sustained ventricular tachycardia in patients subject to standard adrenergic blockade. Am Heart J. 1997;134:752-757.
87 Greene M, Newman D, Geist M, et al. Is electrical storm in ICD patients the sign of a dying heart? Outcome of patients with clusters of ventricular tachyarrhythmias. Europace. 2000;2:263-269.
88 Credner SC, Klingenheben T, Mauss O, et al. Electrical storm in patients with transvenous implantable cardioverter-defibrillators: Incidence, management and prognostic implications. Am Coll Cardiol. 1998;32:1909-1915.
89 Nademanee K, Taylor R, Bailey WE, et al. Treating electrical storm: Sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000;102:742-747.
90 Holmes DRJr, Davis K, Gersh BJ, et al. Risk factor profiles of patients with sudden cardiac death and death from other cardiac causes: A report from the Coronary Artery Surgery Study (CASS). J Am Coll Cardiol. 1989;13:524-530.
91 Garan H, Ruskin JN, DiMarco JP, et al. Electrophysiologic studies before and after myocardial revascularization in patients with life-threatening ventricular arrhythmias. Am J Cardiol. 1983;51:519-524.
92 Guidelines 2000 for cardiopulmonary resuscitation and emergency cardiovascular care: International consensus on science. Part 6: Advanced cardiac life support; Section 5: Pharmacology I: Agents for arrhythmias. Circulation. 2000;102:I112-I128.
93 Kowey PR, Levine JH, Herre JM, et al. Randomized, double-blind comparison of intravenous amiodarone and bretylium in the treatment of patients with recurrent, hemodynamically destabilizing ventricular tachycardia or fibrillation. The Intravenous Amiodarone Multicenter Investigators Group. Circulation. 1995;92:3255-3263.
94 Connolly SJ, Dorian P, Roberts RS, et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: The OPTIC Study: A randomized trial. JAMA. 2006;295:165-171.
95 Schaffer W, Cobb L. Recurrent ventricular fibrillation and modes of death in survivors of out-of-hospital ventricular fibrillation. N Engl J Med. 1975;293:259-262.
96 Myerburg R, Kessler K, Estes D, et al. Long term survival after prehospital cardiac arrest: Analysis of outcome during an 8 year study. Circulation. 1984;70:538-546.
97 The CASCADE Investigators. Randomized antiarrhythmic drug therapy in survivors of cardiac arrest (the CASCADE study). Am J Cardiol. 1993;72:280-287.
98 Wever EFD, Hauer RNW, van Capelle FJI, et al. Randomized study of implantable defibrillator as first-choice therapy versus conventional strategy in postinfarct sudden death survivors. Circulation. 1995;91:2195-2203.
99 Kuck KH, Cappato R, Siebels J, Ruppel R. Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: The Cardiac Arrest Study Hamburg (CASH). Circulation. 2000;102:748-754.
100 Connolly S, Gent M, Roberts R, et al. Canadian implantable defibrillator study (CIDS)—a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation. 2000;101:1297-1302.
101 The AVID investigators. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576-1583.
102 Domanski M, Saksena S, Epstein A, et al. Relative effectiveness of the implantable cardioverter-defibrillator and antiarrhythmic drugs in patients with varying degrees of left ventricular dysfunction who have survived malignant ventricular arrhythmias. J Am Coll Cardiol. 1999;34:1090-1095.
103 Zipes DP, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385-e484.
104 Raj SR, Sheldon RS. The implantable cardioverter-defibrillator: Does everybody need one? Prog Cardiovasc Dis. 2001;44:169-194.
105 Amiodarone Trials Meta-Analysis Investigators. Effect of prophylactic amiodarone on mortality after acute myocardial infarction and in congestive heart failure: Meta-analysis of individual data from 6500 patients in randomized trials. Lancet. 1997;350:1417-1424.
106 Rao BH, Saksena S. Implantable cardioverter-defibrillators in cardiovascular care: Technologic advances and new indications. Curr Opin Crit Care. 2003;9:362-368.
107 Kalinchak DM, Schoenfeld MH. Cardiac resynchronization: A brief synopsis part I: Patient selection and results from clinical trial. J Interv Card Electrophysiol. 2003;9:155-161.
108 Delacretaz E, Stevenson WG. Catheter ablation of ventricular tachycardia in patients with coronary heart disease: Part I: Mapping. Pacing Clin Electrophysiol. 2001;24:1261-1277.
109 Delacretaz E, Stevenson WG. Catheter ablation of ventricular tachycardia in patients with coronary heart disease. Part II: Clinical aspects, limitations, and recent developments. Pacing Clin Electrophysiol. 2001;24:1403-1411.
110 Sra J, Bhatia A, Dhala A, et al. Electroanatomically guided catheter ablation of ventricular tachycardias causing multiple defibrillator shocks. Pacing Clin Electrophysiol. 2001;24:1645-1652.
111 Reddy VY, Neuzil P, Taborsky M, Ruskin JN. Short-term results of substrate mapping and radiofrequency ablation of ischemic ventricular tachycardia using a saline-irrigated catheter. J Am Coll Cardiol. 2003;41:2228-2236.
112 Weinstock J, Wang PJ, Homoud MK, et al. Clinical results with catheter ablation: AV junction, atrial fibrillation and ventricular tachycardia. J Interv Card Electrophysiol. 2003;9:275-288.
113 Camm AJ, Savelieva I. Rationale and patient selection for “hybrid” drug and device therapy in atrial and ventricular arrhythmias. J Interv Card Electrophysiol. 2003;9:207-214.