[level-membership-for-anesthesiology-category]
23 Surgery, Anesthesia, and the Immature Brain
Effects of Anesthetic Exposure on the Developing Brain
Putative Mechanisms for Neurotoxicity
Specific Anesthetic and Sedative Agents
Exposure Time, Dose, and Anesthetic Combinations
Deleterious Effects of Untreated Pain and Stress
Potential Alleviating Strategies
Critical Evaluation of Animal Studies and Interspecies Comparisons
Long-Term Outcome in Children Exposed to Anesthesia and Surgery
Outcome after Exposure to Anesthesia Outside of the Operating Room
MILLIONS OF CHILDREN UNDERGO SURGERY with anesthesia every year.1 During the perioperative period they are exposed to a multitude of stressors capable of interfering with normal brain development. Pain, stress, inflammation, hypoxia, and ischemia have all previously been shown to adversely affect the immature central nervous system. However, recent findings from animal studies have indicated that sedatives and anesthetics—the very drugs used to reduce pain and stress—may themselves undesirably influence brain development by triggering structural and functional abnormalities. There is now an extensive body of work, mostly based on laboratory research, that has defined this phenomenon and explored mechanisms and protective strategies. However, translating these laboratory findings to humans in clinical settings is laden with uncertainties and questions.
Normal Brain Development
In utero and during the early postnatal period, the human brain initially undergoes a rapid growth in size and cell number, and is then pared back to achieve the efficient network of about 100 billion neurons of the adult brain. At birth, the size of the immature brain is one-third that of the adult brain, doubling in size within the first year of life, and reaching 90% of its eventual size by 6 years of age.2 This dramatic growth spurt coincides with a remarkable overabundance of neurons and neuronal connections. In fact, less than half the neurons generated during development survive into adulthood.3,4 Superfluous neurons that lose in the competition for a limited amount of trophic factors are removed by programmed cell death. Also called apoptosis, this cellular suicide program is built into every mammalian cell. Apoptotic cells enter a well-orchestrated, stepwise, and energy-consuming destruction process that involves a cascade of enzymes called caspases and ultimately leads to the breakdown of cellular proteins and DNA.5 This process is heavily used during development, such as during embryonal deletion of the interdigital mesenchymal tissue to separate fingers and toes. Similar to the clotting cascade, the apoptotic cascade remains active throughout life and is held in check by antiapoptotic factors, whereas proapoptotic factors promote its execution. Neurons are protected from apoptosis by neurotrophic factors. Conversely, noxious stimuli, such as pain, hypoxia, and ischemia, can increase the level of proapoptotic factors and activate the apoptotic suicide program, leading to neuronal cell death.
In addition to an overabundance in the total number of neurons generated during early development, the mammalian brain also forms an excess of neuronal connections, or synapses, during this period. Depending on brain region, synaptic density reaches its maximum in infants and young toddlers between 3 and 15 months of age, and will undergo a progressive reduction by about half into adulthood.6 Connections that are active with continued electrical and chemical signals are sustained, whereas those with little or no activity are lost.
In summary, brain architecture changes rapidly and dramatically throughout life. Neuronal density is greatest during fetal life, and excess neurons are eliminated via apoptosis, predominantly in utero, during the neonatal period and throughout infancy.7 Rapid growth of dendrites and synaptic connections occurs during infancy and early childhood, and unneeded dendrites and synapses are trimmed back, predominantly during later childhood and adolescence.6,7
Accordingly, the first several years after birth represent a critical period of development for many brain regions. Recent findings in animals suggest that exposure to anesthetics or sedatives may interfere with proper neuronal development, brain architecture, and subsequent function. Although the exact molecular mechanisms by which anesthetics afford their therapeutic properties of amnesia, analgesia, and immobility are only incompletely understood, their interaction with a wide variety of ion channels, such as sodium, calcium, and potassium channels, as well as several cell membrane proteins, including the receptors for γ-aminobutyric acid (GABA), glycine, glutamate (and N-methyl-d-aspartate [NMDA]), acetylcholine, and serotonin, make it conceivable that anesthetics could interfere with normal electrical and chemical activity in the developing brain. In fact, both GABA and NMDA play critical roles as trophic factors and in regulating neuronal maturation and programmed cell death. During brain development, GABA directs cell proliferation, neuroblast migration, and dendritic maturation.8 Developmental NMDA receptor stimulation fosters survival and maturation of some neurons.9,10 It is therefore not implausible that anesthetics might interfere with these developmental processes.
Effects of Anesthetic Exposure on the Developing Brain
Concerns regarding neurologic abnormalities following general anesthesia in young children were first raised more than half a century ago, when postoperative personality changes were observed following the administration of vinyl ether, cyclopropane, or ethyl chloride for otolaryngologic surgery.11 However, these abnormalities were felt to be psychological in nature because they were alleviated by the timely administration of preoperative sedative drugs.11,12 Approximately two decades later the focus of research shifted to examine the effects of anesthetics in animal models that represented occupational exposure in pregnant healthcare workers.13–16 Delayed synaptogenesis and behavioral abnormalities were observed in neonatal rats born to dams that were chronically exposed to subanesthetic doses of halothane during their entire pregnancy. However, interest in the effects of anesthetics on children did not elicit widespread interest until publications began to appear 10 years ago. In a seminal study in neonatal rat pups, widespread neuronal degeneration was observed after repeated injections of ketamine.17 This led to numerous editorials and review articles, and more than 200 publications in which structural brain abnormalities and/or functional impairment were demonstrated after a wide variety of immature animal species (including chicks, mice, rats, guinea pigs, swine, sheep, and rhesus monkeys) were exposed to almost every sedative and anesthetic in clinical use.18–65 However, no discussion about the effects of drug exposure on the developing brain would be complete without examining the effects of opioid analgesics in the developing brain.54,55 Accordingly, this chapter will examine the various specific effects of sedatives, anesthetics, and analgesics in the immature brain.
Apoptotic Cell Death
The most widely studied deleterious consequence of exposure to sedatives or anesthetics in immature animals is apoptosis, or programmed cell death. Although neuronal apoptosis eliminates approximately 50% to 70% of neurons throughout the brain during development, at any particular time, this natural process affects only a small fraction of cells. Exposure to anesthetics or sedatives briefly, but dramatically, increases the number of apoptotic neurons (Fig. 23-1). Some studies demonstrated up to a 68-fold increase in the density of degenerating neurons after a combination of anesthetics in neonatal rats, compared with control animals, although it remains unclear what fraction of the entire neuronal population these degenerating neurons represent. Unpublished data from one of the authors’ laboratory suggest that a 6-hour exposure to a clinically relevant dose of isoflurane triggers apoptotic cell death in 2% of neurons in the superficial cortex of neonatal mice, whereas less than 0.1% of neurons undergo physiologic apoptosis in this region in unanesthetized litter mates (Loepke, personal communication). Interestingly, dying neurons are immediately adjacent to seemingly unaffected neighboring cells (Fig. 23-2), and the exact mechanism and selectivity of the cell death process remains unknown. Increased neuroapoptosis has also been observed following either in vitro or in vivo, exposure to a wide variety of sedatives and anesthetics, including chloral hydrate, clonazepam, diazepam, midazolam, nitrous oxide, desflurane, enflurane, halothane, isoflurane, sevoflurane, ketamine, pentobarbital, phenobarbital, propofol, and xenon, as well as opioid receptor agonists, in a wide variety of species, including chicks, mice, rats, guinea pigs, piglets, and rhesus monkeys (E-Table 23-1). Cell death–selective stains, such as cupric silver and Fluoro-Jade (EMD Millipore, Billerica, Mass.), have confirmed the cellular demise in neurons that positively stain for activated caspase 3, the central executioner enzyme of the apoptotic cascade.



E-TABLE 23-1 Preclinical Studies Examining Structural and Neurocognitive Effects of Anesthetic Exposure Early in Life


















Apoptosis represents an inherent, energy-consuming process using a cascade of enzymes called caspases. Apoptosis is highly conserved among species and culminates in self-destruction and elimination of cells, even under physiologic conditions, when these cells are functionally redundant or potentially detrimental to the organism.208 Apoptosis involves an orderly breakdown of the cell that includes chromatin aggregation, nuclear and cytoplasmic condensation, and partitioning of cytoplasmic and nuclear material into apoptotic bodies for subsequent phagocytosis, without an extensive inflammatory response. That contrasts with the features observed during necrosis, which include energy failure, cellular swelling, membrane rupture, and release of cytoplasmic content into the extracellular compartment, followed by an inflammatory response.208 Accordingly, apoptosis has also been termed cellular suicide and is extensively used during tissue homeostasis, endocrine-dependent tissue atrophy, and normal embryogenesis (e.g., ablation of tail tissue as part of tadpole metamorphosis in amphibians). Similarly, brain cells are also produced in excess during normal brain development, and up to 50% to 70% of immature neurons are eliminated during normal brain maturation in rodents, nonhuman primates, and humans.3,4 Accordingly, physiologic apoptotic cell death is critical to establish proper brain structure and function, and any disruption of this process can lead to massive brain malformations and intrauterine demise.209 However, in addition to this intrinsic pathway of physiologic, developmental apoptosis, cell death can also be triggered by pathologic, extrinsic factors, such as hypoxia and ischemia.210 It currently remains unknown whether anesthesia-induced neuroapoptosis accelerates physiologic programmed cell death or whether it eliminates cells not destined to die, as in pathologic apoptosis.
Animal studies have identified a narrow window of maximum susceptibility to neuronal cell death induced by several anesthetic drugs, such as the NMDA antagonist ketamine, the GABA agonist isoflurane, or ethanol (a combined NMDA antagonist and GABA agonist). Ketamine-induced neuronal demise occurs in neonatal rodents between 5 and 7 days of age, or before 6 days of age in monkeys, but not in older animals.17,69,139 Similarly, neurotoxicity was not detected after isoflurane anesthesia in 1-day-old animals or in those more than 10 days of age, whereas neuroapoptosis reached a maximum effect at 7 days of age.87 However, preliminary data from the laboratory of one of the authors challenges the notion that anesthetic-induced neuroapoptosis is limited to this age range (Loepke and associates, unpublished data). Conversely, intrauterine exposure to clinical doses of isoflurane in prenatal rats may actually decrease physiologic apoptosis and improve subsequent memory retention,96 whereas only supraclinical doses of isoflurane induced neuroapoptosis in this setting.112
Long-Term Brain Cellular Viability, Neurologic Function, and Behavior
In order to answer the important question whether anesthetics simply hasten physiologic apoptosis or whether they induce pathologic apoptosis, long-term neuronal density and neurologic function have to be assessed in adult animals exposed to anesthesia as neonates. If exposure to anesthetics or analgesics only temporarily accelerated physiologic apoptosis, one would expect normal cell counts and function in adult animals. Conversely, a permanent neuronal cell loss and long-term neurocognitive impairment after anesthetic exposure early in life would suggest that anesthesia-induced neuronal apoptosis may be pathologic in nature. However, this would only be true if the organism was unable to compensate for the neonatal cell loss by increasing neuronal plasticity and repair. To answer these questions, several studies measured neurologic function, assessed behavior, and/or determined neuronal density in adult animals after they were exposed to anesthesia in the neonatal period. Unfortunately, results from these studies are conflicting. Several studies reported long-term neurocognitive or behavioral abnormalities after exposure of neonates to enflurane, halothane, isoflurane, sevoflurane, propofol, or ketamine, or to a combination of isoflurane, nitrous oxide, and midazolam.* Importantly, however, many of these studies only observed abnormalities in very specific tests or subsets of neurocognitive batteries, whereas many other neurobehavioral domains remained intact. For example, a 6-hour exposure to midazolam, isoflurane, and nitrous oxide in neonatal rats led to transient impairment in a water maze learning task in young adulthood and in older animals, whereas, in the same animals, several other tests of behavior and learning, including acoustic startle response, sensorimotor tests, spontaneous behavior in an open field, and learning and memory in the radial arms maze, remained unimpaired.85 Similarly, after a 4-hour exposure to the minimal alveolar concentration (1 MAC) of isoflurane in 7-day-old rats, long-term memory retention was abnormal at two time points, whereas performance at several other time points, as well as in other tests of learning and memory, remained intact.107 Accordingly, the relevance of these limited learning deficits remains uncertain. Similar to humans, performance of rodents and primates in learning tasks depends to a great extent on maternal behavior and rearing conditions, making them strong confounders during neurocognitive testing.105,211–213 Moreover, another obvious and important factor in neurocognitive testing is the verification of similar degrees of motivation when comparing separate groups of animals. For example, a 24-hour exposure to ketamine sedation early in life impaired subsequent performance of rhesus monkeys in learning and memory tests, in addition to decreasing their motivation to perform these tasks.158
Studies of prolonged opioid administration in immature animals have also found evidence for long-term impairment in learning tasks,174–176,179–181 as well as altered pain responses in adult animals after exposure to morphine, fentanyl, heroin, or methadone early in life.†
Conversely, several other investigations have observed no neurologic abnormalities after administration of midazolam, isoflurane, sevoflurane, or ketamine, even when using complex neurologic tests in neonatal animals.* It remains to be determined whether these differential findings are attributable to the anesthetic doses, exposure times, species, or related to the type or timing of the neurologic tests performed. Interestingly, escalating exposure times of isoflurane in neonatal rats caused neuronal apoptosis beginning at 2 hours of anesthesia, but no evidence of long-term neurologic abnormalities until 4 hours of anesthesia.106 In another study, a 6-hour exposure to isoflurane caused significant apoptosis immediately after exposure in neonatal mice, but resulted in no measurable long-term deficit in performance of complex neurologic tests as adult animals.105 Moreover, in brain regions significantly affected by the neonatal neuroapoptosis, adult neuronal density was not diminished compared with unanesthetized litter mates.105 These findings could either suggest that isoflurane may only accelerate physiologic apoptosis or that the developing brain’s plasticity and capacity for repair could compensate for a pathologic insult early in life. Conversely, a study in similarly aged rats observed a permanent elimination of neurons, as well as neurologic abnormalities in adult animals, after exposure to isoflurane, nitrous oxide, and midazolam as neonates, suggesting that either the specific combination of anesthetic drugs (isoflurane alone vs. the combination exposure) or species differences (rats vs. mice) could affect any relationships between neonatal neuroapoptosis and long-term function and neuronal density.97 Alternatively, these conflicting results may be explained by the dissimilar testing environment, because neurocognitive tests are easily transferable among laboratories.214 On the other hand, neonatal apoptosis may not be causally linked to adult neurocognitive performance at all, as evidenced by substantial apoptosis immediately after carbon dioxide–induced hypercarbia in neonatal rats without long-term neurologic sequelae.107
Effects on Neurogenesis and Gliogenesis
The generation of new neurons, or neurogenesis, as well as new astrocytes, or gliogenesis, is most active in the immature brain in utero or soon after birth. Accordingly, anesthetic exposure during this critical period may interfere with neuronal proliferation, or even eliminate neuronal precursor cells, thereby portending permanent reductions in neuronal density and long-term neurologic impairment. To test this relationship, several studies have investigated the effects of anesthetic exposure, primarily isoflurane, on progenitor viability and rates of neurogenesis. Although isoflurane did not cause neural progenitor cell death in vitro, 3.4% isoflurane for 4 hours did decrease the rate of neuronal proliferation and increased neuronal fate selection.108 These findings were recently confirmed in vitro, where 2.8% isoflurane for 6 hours resulted in no effects on neural stem cell viability and larger doses inhibited cell proliferation.121 Similarly, morphine has been found to decrease DNA synthesis in cerebellar neuronal precursors without affecting cell survival.170
Isoflurane also impairs the growth of immature astrocytes and delays their maturation after 24 hours of 3% isoflurane, although this enormous exposure had no effect on cell viability.119 Morphine, although increasing apoptosis in neurons and microglia, did not affect astrocytes in a study involving human fetal cell cultures.173
Alterations in Dendritic Architecture
The immature brain accumulates an overabundance of neuronal connections in infancy, and the number of dendrites and synapses dramatically decreases after the first year of life. Several studies have examined the effects of propofol, isoflurane, sevoflurane, desflurane, midazolam, and ketamine on dendritic arborization and synaptic architecture.* A common theme in these studies is that anesthetics can affect dendritic arborization and synaptic density, and that the direction of this change, whether it is an increase or a decrease in the number of dendritic spines, depends on the age at which the animals were exposed to anesthetics, and therefore the developmental state of the brain. During the first 2 weeks of life, the evidence indicates that anesthetic exposure leads to a decrease in synaptic and dendritic spine density in small rodents, while causing an increase in the number of dendrites beyond that age.110,197 However, the permanence of these changes remains controversial, with some studies only observing a transient effect after ketamine, midazolam, or isoflurane anesthesia early in life.120,153
Decrease in Trophic Factors
Isoflurane- or propofol-based anesthesia in the neonatal animal has been associated with a decrease in brain-derived neurotrophic factor (BDNF),91,109,198 a protein integral to neuronal survival, growth, and differentiation. The cellular mechanism involves a reduction in tissue plasminogen activator and plasmin, which converts proBDNF to BDNF. Accordingly, isoflurane triggered proBDNF/p75NTR (p75 neurotrophin receptor) complex–mediated apoptosis in neonatal mice.109 Moreover, prolonged exposure to opioid receptor agonists early in life has also been found to alter nerve growth factors in the immature brain.66,164
Degeneration of Mitochondria
Ultrastructural morphologic abnormalities have been reported in mitochondria of pyramidal neurons in the subiculum of 7-day-old rats after 6 hours of isoflurane, nitrous oxide, and midazolam.126 A morphometric analysis demonstrated mitochondrial enlargement, impaired structural integrity, and decreased mitochondrial density, indicative of a protracted injury to the mitochondria after anesthetic exposure. Moreover, ultrastructural examination of pyramidal neurons of anesthetized animals by electron microscopy revealed evidence for increased autophagy, a form of cell death.
Abnormal Reentry into Cell Cycle
Ketamine induces reentry of postmitotic neurons into cell cycle in immature rats.155 Neuronal progenitor cells enter cell cycle during proliferation, but mature neurons lose this ability, and if they are forced to reenter cell cycle they will follow a path to apoptotic cell death.
Destabilization of The Cytoskeleton
The integrity of the cellular cytoskeleton is critical for proper neuronal morphology and function. Actin is one of the major components of the cytoskeleton of all eukaryotic cells, and participates in important cellular processes, including cell signaling, motility, and cell division. It is also essential in the formation of dendritic spines. Isoflurane can lead to depolymerization of actin in neurons and astrocytes, initiating cytoskeletal destabilization, impairment of astrocyte morphologic differentiation and maturation, as well as neuronal apoptosis.119,123
Effects on the Developing Spinal Cord
Most animal studies have focused on the effect of general anesthetics and sedatives on the developing brain. However, it is important to also consider the developmental impact of anesthetics on the spinal cord. After 6 hours of 0.75% isoflurane with 75% nitrous oxide in 7-day-old rats, neuroapoptosis increased in the lumbar region of the spinal cord.100 Similar results were reported after 6 hours of isoflurane in a similar model, although 1 hour of isoflurane or spinal bupivacaine resulted in no neuroapotosis.124 Intrathecal ketamine causes neuroapoptosis in the developing spinal cord of 3-day-old rats, but not at 7 days of age.157 Preservative-free ketamine was associated with long-term alterations in spinal cord function and gait disturbances,157 whereas, in a separate study, even high-dose intrathecal morphine produced no signs of spinal cord toxicity.215
Putative Mechanisms for Neurotoxicity
The exact mechanisms that trigger the above responses to anesthetics and sedatives in the immature brain remain unresolved. Elucidating these mechanisms is critical in order to establish the relevance of these findings for pediatric anesthesia and neonatal critical care medicine, as well as to develop mitigating interventions, if necessary. The current, overarching hypothesis is that anesthetics and sedatives interfere with normal GABAA and NMDA receptor–mediated activity, which are the putative targets for unconsciousness, amnesia, and immobility,216 but are also essential for mammalian central nervous system development.9,217 Some have suggested that administering GABAA-receptor agonists and/or NMDA-receptor antagonists may cause abnormal neuronal inhibition during a vulnerable period in brain development, triggering apoptosis in susceptible neurons, which in turn leads to neurocognitive impairment and decreased neuronal density in adults.22,60,85,97 Other lines of evidence suggest that the NMDA receptor–blocking properties of ketamine may upregulate NMDA receptors, rendering the neurons more susceptible to excitotoxic injury caused by endogenous glutamate immediately after ketamine withdrawal.135,139 However, several observations violate both hypotheses; neuronal cell death has been reported during exposure to anesthetics and not only after their discontinuation. Moreover, several anesthetics with minimal NMDA-receptor interaction, such as propofol and barbiturates, have demonstrated robust neurotoxic properties, whereas the neurotoxic potency of the NMDA-antagonist xenon is limited, therefore casting doubt on receptor upregulation being the sole mechanism for anesthetic neurotoxicity. In terms of abnormal neuronal inhibition being the main trigger for apoptosis in developing neurons, GABAA-receptor stimulation indeed decreases neuronal activity in the mature brain; however, it also causes excitation in developing neurons,218 thereby contradicting the inhibition hypothesis. In immature neurons, intracellular chloride (Cl−) concentration is high, thus GABA-induced opening of Cl− channels allows this anion to exit the cell, leading to membrane depolarization. On the other hand, the intracellular Cl− concentration is low in mature neurons. When anesthetics open Cl− channels in mature neurons, ions enter the cell, thereby hyperpolarizing the membrane. This reversal of the cellular Cl− gradient occurs as a result of a switch from the immature Na+-K+-2Cl− cotransporter 1 (NKCC1) to the mature brain form, K+-Cl− cotransporter 2 (KCC2).219 Along these lines, studies in neonatal rats demonstrated excitatory properties in the brain and episodes of epileptic seizures during sevoflurane anesthesia in neonatal rats.204 Isoflurane has also been shown to cause an excessive release of Ca2+ from the endoplasmic reticulum via over-activation of inositol 1,4,5-trisphosphate receptors (InsP3Rs) in neonatal rats in vivo and in vitro.220 A similar mechanism may be linked to the production of Alzheimer-associated increases in β-amyloid protein levels after anesthesia.221 Moreover, whereas xenon and hypothermia cause neuronal inhibition, they do not appear to exacerbate isoflurane-induced neuronal cell death, as expected by cumulative neuronal inhibition, but rather significantly reduce it.94,101,207
Importantly, evidence indicates that equianesthetic concentrations of the three contemporary inhalational anesthetics cause similar degrees of neuroapoptosis, suggesting that it is the anesthetic depth and not the doses of the anesthetics that determines the cytotoxic potency.71 However, other studies have failed to link the anesthetic and the apoptotic mechanisms. Specifically, although racemic ketamine and (S)-ketamine both elicit their anesthetic effects via NMDA-receptor blockade, (S)-ketamine induces up to 80% less cell death in vitro when compared with equipotent doses of racemic ketamine.154 Moreover, concomitant administration of the GABAA-receptor antagonist gabazine did not attenuate neuroapoptosis induced by the GABA agonist isoflurane, whereas the α2-agonist dexmedetomidine did.73 Decreases in anesthetic-induced neuronal activity may therefore be less important than the disruption of the neuronal balance of excitation and inhibition, as demonstrated by a series of studies that examined anesthesia-induced dendritic morphologic changes in mice.70,152 Whereas simultaneous blockade of excitatory and inhibitory activity with tetrodotoxin did not lead to structural changes during synaptogenesis as would have been expected from a causative relationship between neuronal inhibition and structural damage, the administration of either GABAA-agonistic or NMDA-antagonistic compounds alone altered synaptogenesis.152
It is not entirely clear at this time whether cytotoxicity is a direct effect of the anesthetic itself, or of anesthetic byproducts, or by the metabolic acidosis and respiratory derangements that have been observed during anesthesia in small rodents.105,107,222 Hypercarbia can trigger widespread neuroapoptosis, even in unanesthetized neonatal rats exposed to increased partial pressures of carbon dioxide. Whereas apoptotic cell death was quantitatively indistinguishable from neurodegeneration in isoflurane-treated litter mates, which were also hypercarbic, adult neurocognitive impairment was only observed in the isoflurane-treated animals.107
Lastly, experimental models of neurodegeneration have implicated reentry of postmitotic neurons into the cell cycle, leading to cell death. Ketamine exposure has been found to induce aberrant cell cycle reentry, leading to apoptotic cell death in the developing rat brain.155
Specific Anesthetic and Sedative Agents
In order to provide a succinct overview of the available laboratory data, we briefly review the effects of each class of anesthetics separately. Although the effects of some anesthetics, such as ketamine and isoflurane, have been extensively studied on the developing brain, the effects of others, such as xenon and desflurane, have not. However, the current data suggest that all anesthetics exert deleterious effects to some degree, on the developing animal brain (see E-Table 23-1). It is important to appreciate that although the MAC values for inhalational anesthetics are fairly constant across species, equipotent dosing of intravenous (IV) medications is not. That is, the dose of most IV medications to effect sedation or anesthesia in animals is approximately 6- to 10-fold greater than that in humans. The dosing is further complicated by the different routes by which drugs are administered in neonatal animals, which include the subcutaneous and intraperitoneal (IP) routes, as opposed to the oral or IV routes in humans. The possible importance of the interspecies differences in the pharmacology of IV medications on neuroapoptosis and cognitive dysfunction has not been addressed.
Ketamine
Possibly the most frequently studied anesthetic is ketamine, an antagonist of the NMDA and glutamate receptor that also interacts with other cell membrane proteins, such as muscarinic and opioid receptors, as well as voltage-gated calcium channels. Ketamine’s properties, which include potent analgesia, dissociative anesthesia, and relative hemodynamic stability, have made it a popular choice for procedural sedation in children, as well as for induction of anesthesia in children with critical congenital heart disease or pulmonary hypertension.223–225 However, about 10 years ago, a seminal study that examined the effects of repeated IP injections of ketamine on the brain of neonatal rats observed widespread apoptosis.17 Seven injections of 20 mg/kg of ketamine, administered over a 9 hour period in evenly divided intervals, to 7-day-old rat pups caused a 3- to 31-fold increase in degenerating neurons, depending on brain region. This led to speculation that these changes might contribute to neuropsychiatric disorders.17 These initial findings for ketamine have been confirmed in more than fifty studies in small rodents, as well as nonhuman primates, both in vitro and in vivo (see E-Table 23-1). Several of these studies have identified relationships between neurodegeneration and dose, duration of treatment, as well as animal species and age during exposure. Single doses of up to 75 mg/kg or multiple IP injections up to 17 mg/kg/hr for 6 hours were not neurotoxic to neonatal rat brains,128 whereas single doses between 20 and 50 mg/kg were neurotoxic to neonatal mice brains.74,134 Six or seven repeated injections of 20 to 25 mg/kg consistently cause apoptosis in neonatal rat brains.17,128,131,149,150 Although the doses of ketamine in these small-animal models appeared to be excessive and plasma concentrations in the rodents were up to 7 times greater than those in humans,131 these increased doses are consistent with the requirements for IV anesthesia in small animals (refer to later section on interspecies comparison). Coadministration of midazolam, diazepam, propofol, or thiopental compounded the neuronal injury caused by ketamine.74,134,142 Studies in rats, mice, and nonhuman primates suggest that the susceptibility to ketamine-induced neurotoxicity is limited to a brief period after birth, with a maximum impact between 3 and 7 days of age in small rodents and less than 35 days of age in monkeys.17,139 Beyond these ages, ketamine-induced neuroapoptosis subsides. In addition to apoptosis, both small rodents and nonhuman primates that were anesthetized with ketamine have exhibited impaired learning tasks later in life.129,130,158
In summary, ketamine is the most frequently studied anesthetic, in terms to its neurotoxic effects, and has repeatedly been shown to cause widespread apoptosis (an effect that is exacerbated by the coadministration of other anesthetics), as well as neurologic impairment in adult animals exposed early in life. Importantly, long-lasting learning impairment has been demonstrated in nonhuman primates, the closest animal model to humans.158 However, these animals were anesthetized with ketamine for 24 hours to produce this effect, a duration that exceeds the usual clinical scenario. Furthermore, it is unclear whether the observed learning abnormalities could not be explained by a reduction in motivation to perform the learning tasks.158
Inhalational Anesthetics
The second most commonly studied class of drugs is the inhalational anesthetics (see E-Table 23-1). The anesthetics desflurane, sevoflurane, isoflurane, enflurane, and halothane exert their anesthetic properties predominantly by their agonistic effects on the GABAA receptor, but also by differing degrees on glycine, NMDA, acetylcholine, serotonin (5-HT3), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors. Whereas GABA represents the main inhibitory neurotransmitter in the adult central nervous system, it has excitatory properties in the developing brain,218 which may have implications for neurotoxicity, as discussed previously. Most studies of inhalational anesthetics examined either isoflurane alone or a combination of midazolam, isoflurane, and nitrous oxide. This combination of GABA agonists and NMDA antagonists has been repeatedly found to cause widespread increases in brain cell degeneration in neonatal animals.85,87,91,92 In addition to the immediate deleterious effects on brain structure, long-term abnormalities in spatial learning tasks and decreased neuronal cell density in adult rats have been observed after exposure to this anesthetic combination early in life.85,97 One MAC of isoflurane administered as the sole anesthetic for 4 hours to neonatal rats led to neurocognitive deficits in rats when they matured to adults,106,107 whereas up to 0.6 MAC for 6 hours in neonatal mice, while causing widespread neuronal degeneration immediately after exposure, failed to cause neurocognitive deficits or decreases in neuronal density in adulthood.105,115 These inconsistent findings raise the question of whether neonatal neuronal apoptotic cell death is linked directly to behavioral and learning abnormalities in adulthood. Neuronal cell death has also been observed in neonatal rhesus monkeys after 5 hours of 0.75% to 1.5% isoflurane,114 although long-term neurologic studies in this species have not yet been published.
Similar to isoflurane, sevoflurane has also been shown to induce neuroapoptosis in neonatal mice,71,115,202 although the long-term effects on learning and behavior are conflicting.72,115,202 To date, few studies have examined desflurane in this context. Desflurane causes age- and species-dependent neuronal cell death in 7-day-old mice, but not 16-day-old rats.70,71
Few studies have compared the neurotoxicity of contemporary anesthetics. At equianesthetic concentrations of desflurane, isoflurane, and sevoflurane in neonatal mice, the degree of neuronal degeneration in the superficial neocortex, a brain region significantly affected in this model, is similar.71 These results contrast with those from a study of lower concentrations of sevoflurane and isoflurane, in which cell death was less after the former than the latter, but without differences in neurocognitive performance in adult rats.115 In another comparative mouse study, desflurane caused a greater degree of neurodegeneration than isoflurane and sevoflurane, and long-term neurologic impairment only occurred after desflurane.72 The significance of these differential findings remains unknown, but could be related to methodologic differences.
Although largely phased out from clinical pediatric anesthesia practice, halothane and enflurane have been shown to induce brain abnormalities. These anesthetics were studied in models of intermittent or chronic occupational exposure during pregnancy, and caused subsequent delayed synaptogenesis and behavioral and learning abnormalities in rats after fetal exposure.13–1676
Nitrous Oxide
Nitrous oxide, an NMDA antagonist, is the oldest anesthetic in clinical use, although its low potency (MAC of 115% in adult humans) necessitates the coadministration of other anesthetics to provide surgical anesthesia. Anesthetic combinations with nitrous oxide often include the GABA agonist midazolam, and the mixed GABA-agonist/NMDA-antagonist isoflurane.* In rats, nitrous oxide alone did not induce neuronal apoptosis,85,87,94 whereas in an in vitro study, it did cause neuronal cell death in mouse hippocampal slices.94 When administered in combination, however, nitrous oxide has been shown to exacerbate neuronal cell death induced by isoflurane, and also to contribute to long-term neurologic abnormalities in rats when combined with isoflurane and midazolam.85
Xenon
Due to the cost differential between other inhalational anesthetics and this rare, colorless, and odorless noble gas, xenon has not reached widespread clinical anesthesia practice, despite its NMDA-antagonistic, anesthetic properties.226 Xenon has a relatively low anesthetic potency, with a MAC measuring between 65% and 70% in adults227,228; its low blood-gas solubility speeds emergence from anesthesia.229 Xenon’s effects on neuronal apoptosis have been examined by two groups, with slightly differing results. Although 75% xenon for 6 hours did not cause neuronal apoptosis in 7-day-old rat pups,94 70% xenon for 4 hours did increase neuroapoptosis in 7-day-old mice.207 Interestingly, both studies demonstrated that xenon decreased the neurodegeneration induced by isoflurane anesthesia,94,207 which may have relevance to the phenomenon’s putative mechanism, as discussed.
Benzodiazepines
Benzodiazepines, such as clonazepam, diazepam, and midazolam, have been investigated regarding their effects on the immature brain, either alone or in combination with other drugs. These GABA agonists are frequently used in toddlers and older children for preoperative anxiolysis, but infrequently in neonates and infants. Studies have shown that they increase neuronal degeneration in small-animal models, depending on the dose, region of the brain, species, and age of the animal. Although single doses of 5 mg/kg of diazepam or 9 mg/kg of midazolam IP did not increase neuronal cell death in neonatal rats,68,85 5 mg/kg subcutaneous diazepam did cause neuronal cell death in some brain regions in mice, though this was not associated with learning deficits in adults.74 In these studies, the neuroapoptosis associated with diazepam was significantly augmented by the coadministration of other sedatives, such as ketamine.74 Studies have consistently reported increased neuroapoptosis in neonatal rats after diazepam in doses of 10 mg/kg or greater,68,69 an effect that was prevented by the coadministration of the benzodiazepine-antagonist flumazenil in one study.68 Two studies reported no neurocognitive learning disabilities in adult mice after they were sedated with diazepam or midazolam as neonates.74,166
Chloral Hydrate
The sedative chloral hydrate, a chlorination product of ethanol that is a GABA agonist as well as NMDA antagonist, has been largely supplanted by barbiturates and benzodiazepines in pediatric clinical practice. However, it is still being used in doses of up to 120 mg/kg for sedation for radiology studies,230 and its neurotoxic properties have been investigated. Preliminary results indicate that it causes neuroapoptosis in the cerebral cortex and the caudate-putamen complex in immature mouse pups in doses of 100 mg/kg or greater.67 The neurofunctional outcome in adult mice, however, has not as yet been investigated.
Barbiturates
Barbiturates act largely via the GABAA receptor, but also exert effects via nicotinic acetylcholine, AMPA, and kainate receptors. Thiopental alone, in doses of 25 mg/kg subcutaneously in neonatal mice, did not induce apoptosis, although when doses of 5 mg/kg were administered in conjunction with 25 mg/kg of ketamine subcutaneously, neuronal degeneration occurred and was associated with long-term impairment of learning and memory.142 Pentobarbital and phenobarbital have been shown to induce neurodegeneration in mouse and rat pups. Furthermore, after receiving these sedatives as neonates, long-term alterations in brain protein expression and learning and memory have been observed,103,184,186 although in one study these long-term alternations may be attributed in part to hypoxia and hypercarbia during the neonatal sedation.184 Interestingly, estradiol has been shown to attenuate phenobarbital-induced neuroapoptosis.68,185
Propofol
In recent years, propofol has supplanted the barbiturates as the IV induction agent of choice in children. Propofol predominantly acts via GABA and glycine receptor–agonistic properties, but also weakly on nicotinic, AMPA, and NMDA receptors, and has been repeatedly studied regarding its neurotoxic profile, both in vitro and in vivo. The overall consensus of this body of literature is that propofol, in a dose- and exposure time–dependent fashion, has dramatic effects on the developing brain in animals. Propofol has consistently caused neuroapoptosis after doses exceeding 50 mg/kg (subcutaneous or IP) or repeated doses exceeding 20 mg/kg/hr for 4 to 5 hours in neonatal small rodents.142,148,191,196 Interestingly, lithium protected from propofol-induced neuroapoptosis in neonatal mice.148 However, after 24 hours of IV anesthesia with propofol (6 mg/kg/hr) and fentanyl (10 µg/kg/hr), there was no evidence of apoptosis.192 Apart from overt neuronal cell death, propofol decreases the GABAergic enzyme glutamic acid decarboxylase,188 decreases nerve growth factors,194,198 and causes neurite growth cone collapse in tissue culture.190 In addition, propofol alters dendritic spine architecture in developing rats, depending on the age at the time of anesthetic exposure.197 For example, dendritic spine density decreased if the rat was anesthetized during week 1 of life, but it increased spine development if exposure took place during week 3 of life; the mechanism of these differing responses remains elusive.197
Dexmedetomidine
Unlike other anesthetics and sedatives, dexmedetomidine is a sedative and analgesic that does not interact with GABA, NMDA, or opioid receptors, but rather acts by stimulating presynaptic α2-adrenergic receptors. Given this different mechanism of action, it is interesting that dexmedetomidine, administered in three IP injections of 1 to 25 µg/kg at 0, 2, and 4 hours to 7-day-old rat pups, did not cause neuronal apoptosis, but attenuated isoflurane-induced neuroapoptosis. Even a dose of 75 µg/kg dexmedetomidine (75-fold greater than the median effective dose needed for sedation), did not cause apoptosis in this model. Furthermore, dexmedetomidine prevented the long-term cognitive impairment from isoflurane.73
Opioid Analgesics
To date, only one study investigated neurotoxic properties of opioids in relation to an inhalational anesthetic regimen. Mechanically ventilated, neonatal pigs, sedated with an IV bolus of 30 µg/kg fentanyl followed by 15 µg/kg/hr for 4 hours, exhibited significantly less neuroapoptosis in several regions of the brain, compared with an intramuscular injection of 1 mg/kg of midazolam, followed by 4 hours of 0.55% isoflurane and 75% nitrous oxide.79 These initial findings are interesting, although future neurotoxicity studies that include opioid infusions need to also include adjuvants that produce amnesia, as is customary in pediatric anesthesia practice. Moreover, when comparing different anesthetic regimens regarding their neurotoxic potential, equianesthetic potency should be established during the study.
Importantly, however, the long-term consequences of opioid administration to the immature brain needs to be further elucidated before recommending such a regimen as an alternative strategy. Similar to GABA and NMDA receptors, opioid receptors are also intimately involved in early brain development and synaptogenesis,231,232 which would make it plausible that opioids could similarly affect brain development during the critical period of synaptogenesis. Increased neuronal cell death and decreased neuronal density after perinatal exposure to opioid receptor agonists, such as morphine and heroin, have been observed in developing animals.82,83,233 Chronic buprenorphine and methadone treatment early in life have been found to diminish concentrations of nerve growth factors in the immature brain.66,164 Moreover, immediate and permanent reductions in µ-opioid receptor density have been observed after perinatal exposure to morphine,168,169 and may be associated with long-term impairment of memory and cognitive function in small animals,171,174–176,179–181 as well as exaggerated nociceptive responses to a pain challenge later in life.177,182 Stimulation of the κ-opioid receptor may amplify neuronal cell death induced by proapoptotic agents.234 High-dose fentanyl has been shown to significantly exacerbate white-matter brain lesions induced by glutamatergic overstimulation in mice.78 All these animal data suggest that prolonged perinatal exposure to opioids may have immediate and long-term effects on the developing brain and may amplify proapoptotic stimuli, although the effects of a brief or single dose exposure to opioids is not well understood. These data suggest that further studies into the interactions of opioid analgesics and anesthetics in the developing brain are warranted.
Exposure Time, Dose, and Anesthetic Combinations
Neuronal injury depends on the anesthetic dose and/or duration of anesthesia for inhalational anesthetics, and the dose, route of administration, and the number of doses for injectable anesthetics. Moreover, combinations of several anesthetics and sedatives have, in general, caused more neuroapoptosis and long-term cognitive changes than have single drugs. For example, combinations of midazolam, nitrous oxide, and isoflurane cause a much greater degree of neuroapoptosis in neonatal rats than isoflurane alone, even when the latter is administered at a greater inspired concentration.85 In the case of ketamine, its neurodegenerative potency is amplified when coadministered with thiopental or propofol.142 Anesthetic combinations of mixed GABA agonists and NMDA antagonists demonstrate exaggerated effects. This evidence supports the notion that a deeper level of anesthesia increases the neuroapoptotic injury. However, two anesthetics, xenon and dexmedetomidine, which actually attenuate the neurotoxic effects of isoflurane anesthesia,73,94,207 do not lend support to this notion.
Deleterious Effects of Untreated Pain and Stress
Given the evidence for deleterious effects of anesthetics and analgesics on the developing brain and the paucity of complete anesthetics devoid of cytotoxicity, would children fare better by withholding potentially toxic drugs and allowing them to experience the pain and stress of surgery? Of course such a practice would be unethical, and there is a plethora of evidence of the deleterious effects caused by the stressful conditions of exposing the immature nervous system to recurrent painful stimulation. Noxious stimulation in the neonate has been associated with subsequent hyperalgesia or hypoalgesia, depending on the type and severity of injury.235 Repetitive painful skin lacerations for procedures as minor as blood draws in neonates have been found to lead to long-term, local sensory hyperinnervation.236 In addition to these local responses, repetitive, inflammatory pain early in life has been found to result in hyperalgesia and lasting changes in nociceptive circuitry of the adult dorsal horn.237 Repeated painful injections into the paws of rat pups resulted in a generalized thermal hypoalgesia.235 In addition to altered pain processing and sensory perception, repetitive or persistent pain in the neonate alters behavior and cognitive function in adulthood, decreases pain thresholds, and increases vulnerability to stress and anxiety disorders or chronic pain syndromes later in life.141,238–241
In addition to painful stimulation, early adverse emotional experiences can also induce long-lasting abnormalities, such as imbalances of the inhibitory nervous system,242 impairment of normal development of the nociceptive system, long-term behavioral changes,243 and persistent learning impairment.179 Therefore fetuses, neonates, and infants subjected to pain and stresses associated with invasive procedures without adequate anesthesia and analgesia may be at risk for long-term adverse outcomes.
Accordingly, preemptive administration of analgesics and sedatives, such as morphine or ketamine, has been found to ameliorate the deleterious effects of neonatal pain in some of the animal studies.141,179,241 However, either the presence of painful stimulation or 5 days of morphine administered to neonatal mice independently impaired adult rewarded behavior, although the combination did not.179
Clinical reports have also demonstrated that human neonates and infants can mount a metabolic and endocrine response to perioperative stress and painful stimulation, which include surges in catecholamine, cortisol, β-endorphin, insulin, glucagon, and growth hormone levels.244–246 Some of these markers, such as cortisol, remain increased for more than a year after the insult, possibly as a result of cumulative stress related to multiple painful procedures early in life.247 Inhalational anesthetics, opioid analgesics, as well as regional anesthesia, inhibit intraoperative stress and improve postoperative outcomes.245,248,249 Moreover, adequate perioperative anesthesia reduced the incidence of other complications, such as the incidence of sepsis and disseminated intravascular coagulation, that led to a decrease in overall mortality.250 Even less invasive procedures, such as circumcisions performed without analgesia in young boys, can exaggerate responses to painful challenges (e.g., immunizations) later in life.251 Conversely, topical or regional anesthesia for circumcision blunts not only the immediate humoral stress response during the procedure,252 but also pain-induced, long-term hyperalgesia.251 In preterm neonates, painful stimulations early in life have also been associated with subsequent diminished cognition and motor function.253 In a retrospective study of children greater than 1 year of age who were born at less than 32 weeks gestational age without significant neonatal brain injury or major sensorineural impairment, the more skin-breaking procedures from birth to term (including heel sticks, intramuscular injections, chest tube placements, and central line insertions) predicted poorer subsequent cognitive and motor development (as assessed using the Bayley Scales of Infant Development II) when compared with term controls. Importantly, after controlling for severity of illness, duration of morphine administration, and exposure to postnatal dexamethasone, gestational age at birth was not significantly associated with cognitive or motor outcome. These findings suggest that repetitive pain-related stressful experiences and not prematurity per se was responsible for the poor neurodevelopmental outcome.253 Although this study did not examine the effects of anesthetic or analgesic administration during painful stimulation on subsequent outcome, a small, retrospective study suggested an improvement in outcome after administering anesthesia during painful stimulation.254 Painful stimulation during reduction of herniated bowel without anesthesia in infants suffering from gastroschisis tended to more frequently lead to serious adverse events, such as bowel ischemia, need for total parenteral nutrition, and unplanned reoperation, than in infants undergoing the same procedure with general anesthesia.254 However, despite persistently large numbers of painful and stressful procedures performed in vulnerable neonates, data indicate that the majority of these procedures are still not accompanied by adequate analgesia or anesthesia.255
Potential Alleviating Strategies
The sedative dexmedetomidine and the anesthetic xenon possess very limited neurotoxic potencies themselves, but dramatically reduce isoflurane-induced neuroapoptosis.73,94,207 In addition, the coadministration of dexmedetomidine prevents long-term memory impairment after 6-hour isoflurane exposure in rats.73 Interestingly, neuroprotective preconditioning has been demonstrated in pheochromocytoma PC12 cells in which pretreatment with a small concentration of isoflurane prevented the neurotoxic response to a subsequent greater dose.95 In in vitro studies, the inositol triphosphate–receptor antagonist xestospongin C, tissue plasminogen activator, plasmin, inhibition of the neurotrophic receptor p75NTR and the RhoA receptor, or prevention of cytoskeletal depolymerization with either jasplakinolide or TAT-Pep5 significantly attenuated isoflurane-mediated neuroapoptosis.104,109,123 In addition, l-carnitine attenuated neuronal apoptosis after 6 hours of isoflurane and nitrous oxide in 7-day-old rat pups.102 Supplementation with the naturally occurring hormones β-estradiol or melatonin prevented the deleterious effects on neuronal survival of a prolonged exposure to midazolam, isoflurane, and nitrous oxide.87,91 Similarly, coadministration of β-estradiol significantly reduced phenobarbital-induced neuroapoptosis.68,185 The only potentially protective agent that has been unsuccessfully tested in published studies is the GABA antagonist gabazine.73 On the other hand, pilocarpine reduces neuroapoptosis induced by the GABA agonists isoflurane and midazolam, while augmenting the damage after administration of the NMDA antagonist phencyclidine, based on preliminary results from neonatal mice.93 Preliminary data from the same laboratory also suggest that whole-body hypothermia with a targeted brain temperature of less than 30° C may protect from the neuroapoptotic ramifications of a 4-hour exposure to 0.75% isoflurane or to 40 mg/kg of IP ketamine in neonatal mice.41,101 Another therapy that has been successfully tested in mouse pups that received 40 mg/kg of ketamine or 100 mg/kg of propofol was lithium, 3 to 6 mEq/kg subcutaneously, which abolished the anesthetic-induced neuroapoptosis in cortex and the caudate-putamen complex.148
Because the applicability and extent of anesthetic neurotoxicity has not been established in humans, it is obviously premature to recommend any of these protective strategies for children. Moreover, the safety of many of these drugs and interventions has not been established in human neonates and infants. Tissue plasminogen activator and plasmin promote fibrinolysis and may not be first-line treatments during invasive surgical procedures. The sex hormone β-estradiol may not be a feasible adjuvant in prepubescent boys. The safety of pilocarpine in young children may be hampered by its proconvulsant activity observed in animal studies.256,257 Lithium has been labeled harmful to the human fetus and may cause neurocognitive impairment in young children.258–260 Whole body hypothermia below 30° C may not be a clinically feasible modality because even mild perioperative hypothermia, at least in adults, has been causally linked to numerous complications, including increased blood loss and transfusion requirements, morbid myocardial outcomes, prolonged postanesthetic recovery and hospitalization, thermal discomfort, as well as an increased risk of surgical wound infections.261 Therefore hypothermia to treat anesthesia-induced neurotoxicity is unlikely to play a substantive role during routine pediatric anesthesia, but may have a role in infants undergoing hypothermic cardiopulmonary bypass for heart surgery to treat congenital defects. Unfortunately, the latter population often presents with neurocognitive abnormalities before anesthesia.262 Although xenon’s scarcity renders it a very expensive anesthetic, dexmedetomidine’s wider availability and increased usage in pediatric anesthesia makes it a more attractive option for further research into protective strategies.263,264
Anesthetic Neuroprotection
Even relatively brief periods of inadequate oxygen or blood flow to the brain may lead to neuronal injury and long-term neurologic impairment, because of the limited tolerance of the brain to ischemia. Importantly, animal studies have repeatedly confirmed the protective properties of anesthetics when administered during episodes of brain hypoxia-ischemia. However, most of these studies have been conducted in adult animals.265 In immature animal models, anesthetics have also been found to reduce neurologic injury and improve functional outcome after brain ischemia. Desflurane alleviates neuronal cell death and early neurologic dysfunction in a model of the neonatal pig during hypothermic cardiopulmonary bypass and deep hypothermic circulatory arrest.266,267 Isoflurane treatment before hypoxia-ischemia protects the brain and improves survival in neonatal rats and mice.268–270 Both xenon and sevoflurane protect the immature brain during simulated hypoxia-ischemia in an in vitro model.271 Furthermore, sevoflurane may be protective in a neonatal mouse in vivo model.272 These findings in immature animals suggest that critically ill human neonates could potentially benefit from these protective properties during clinical scenarios of neurologic injury, such as cardiopulmonary bypass, neurologic surgery, or perioperative cardiocirculatory arrest; these potential benefits need to be weighed against the potential neurotoxic properties of the current anesthetics.
Critical Evaluation of Animal Studies and Interspecies Comparisons
To determine whether the findings from animal studies should guide clinical practice, it is important to rationally evaluate how well animal studies represent the human clinical scenario.*
Duration of Exposure
In order to elicit toxic effects, the designs of many animal studies include durations of anesthesia from 1 to 24 hours. Accordingly, some of these times may extend well beyond the average time of routine pediatric anesthesia. However, expressing the duration of anesthesia as a fraction of a subject’s life span, thereby equating a 6-hour anesthetic in mice to a 2-week or greater anesthetic in humans, is probably an oversimplification. The duration of neurodevelopment may not be immediately relevant when considering the likelihood of injury at a cellular level. Nonetheless, the extent of the injury and its functional implications may potentially be related to the duration of the entire brain developmental period, even though the mechanism of anesthetic-induced neurodegeneration is unknown. Because human brain development occurs at a much slower pace than in any of the other species, similar exposure times could have different effects on potential susceptibility and the ability for postexposure repair among species. For example, the brain reaches adult size at 20 days of age in rats, 3 years of age in rhesus monkeys, 7 years of age in chimpanzees, and not until 15 years of age in humans.2,279,280 Exposure to anesthesia during a larger proportion of the period of development could be expected to have a greater impact on total development and maturation. Similarly, given the plasticity of the developing brain, it seems conceivable that brains with slower growth rates possess more time for repair.281,282 Conversely, in complex organisms, such as humans, relatively minor degrees of injury in crucial areas and at crucial times during development could have relatively profound long-term effects.
Anesthetic Doses
In all animal studies, doses needed for injectable anesthetics to cause neuronal degeneration were greater, sometimes by orders of magnitude, when compared with weight-based doses commonly used in human clinical practice. However, to produce immobility, small animals also required significantly greater doses of IV anesthetics than larger animals. Due to their smaller size, greater metabolic rate, and shorter physiologic time, drug doses in animals significantly differ from those used in humans.283 Using a process called allometric scaling, which takes these species differences into account, animal drug doses comparable to human doses have been estimated to be approximately three-, six-, or twelvefold greater, respectively, for monkeys, rats, or mice (see also Chapter 6).283,284 Whereas drug doses used in anesthetic neurotoxicity studies frequently still exceed these doses calculated using allometric scaling, they do not include a significant safety margin (Fig. 23-3). Moreover, using allometrically scaled doses, plasma concentrations for ketamine, for example, were approximately 3 to 10 times greater in small rodents and monkeys than those observed during clinical human practice.131,139 This suggests that the neurotoxic properties observed with large doses of injectable anesthetics, such as ketamine, would only have direct applicability to humans if the anesthetic and the neurotoxic effects were based on the same molecular mechanism, which has yet to be verified. Otherwise, animal studies would expose subjects to much greater plasma concentrations of a neurotoxicant than those used during anesthesia in humans, thereby leading to an overestimation of the neurotoxic effects of IV anesthetics in laboratory studies.
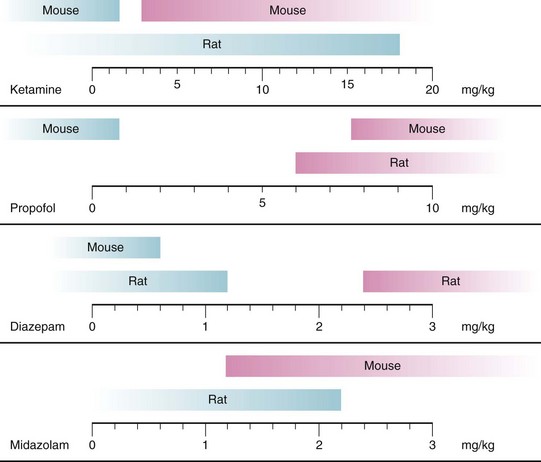
FIGURE 23-3 Neurodegenerative effects of injectable anesthetics in developing animals are dose-dependent. Blue bars represent doses of the respective anesthetics not causing neurodegeneration in rats and mice; pink bars represent doses causing neurodegeneration in animals. Figure includes only studies using single injections of anesthetics and doses used in animals were scaled to doses for children by using an allometric scaling technique, based on calculations outlined in reference 284. Neurotoxic data are based on experiments described in references 17, 68, 69, 74, 85, 128, 131, 134, 139, 142, 151, 194.
Doses for inhaled anesthetics generating immobility in animals, on the other hand, are much closer to clinically used doses. Moreover, similar to human anesthesia, potency of inhaled anesthetics increases with subject age, necessitating larger doses in younger animals,71,222,285 which could suggest closer applicability of laboratory data to humans.
Experimental Versus Clinical Conditions
Another important distinction between laboratory studies in animals and clinical studies in humans is the presence of significant comorbidities, such as the presence of stress and pain associated with the underlying disease that require the administration of anesthetics and analgesics. Very few of the laboratory studies to date have included the presence of surgical stress that may increase or decrease the degree of anesthesia-induced neurotoxicity. Tail clamping or injection of caustic substances are used as a model for surgical stress. Results from these studies varied: one study reported no influence of the painful stimulus on anesthesia-induced apoptosis,206 whereas a second reported that painful stimulation increased anesthetic-induced neuroapoptosis.127 In contrast, therapeutic effects have been demonstrated with concomitant administration of small doses of analgesics or sedatives, compared with no analgesia or sedative when pain was induced.141,179
When studies measured metabolic and respiratory effects of anesthetic exposure in small rodent species, several demonstrated significant differences from pediatric anesthesia practice, such as extensive hypercarbia, metabolic acidosis, and hypoglycemia observed in the small rodents.105,107,222 Tracheal intubation and mechanical ventilation do not seem to completely obviate these abnormalities.222 In stark contrast to anesthesia in children, administering clinical doses of anesthetics for as little as 2 to 4 hours can be lethal for more than 20% of small rodents,105 even when intermittent painful stimuli were applied.107 Moreover, rearing conditions after anesthesia have a profound impact on the brain’s repair mechanisms after injury. Environmental enrichment and exercise dramatically increase neurogenesis in rodents and therefore may facilitate plasticity and repair after anesthesia, compared with regular cage housing conditions.286,287 Children face daily cognitive challenges in their “enriched” environment, different from laboratory animal housing,79,114,139,158 that could attenuate the postulated neurocognitive effects of anesthesia. This is highlighted in a recent study in which environmental enrichment reversed the deleterious effects of anesthesia on subsequent neurologic performance in rats, resulting in performance similar to environmentally enriched, unanesthetized animals and superior to unenriched control and anesthesia-exposed animals.206 This study suggests that neurobehavioral outcome depends on multiple factors and that anesthesia may be only one minor insult compared with the many other more significant events in childhood.
Comparative Brain Development
A major obstacle in translating animal data to humans centers on the difficulties of matching up brain maturational stages in model animals with the equivalent stages of the immature human brain. The ongoing discussion regarding these comparisons is somewhat reminiscent of the cliché of 1 “dog year” being equivalent to 7 “human years.” Because animal studies have suggested that anesthesia-induced neuroapoptosis may be limited to very defined, early stages of development, such as from 3 to 10 days of age in small rodents,17,87 it becomes imperative to identify the equivalent period during human brain development, in order to assess human applicability of the animal data and to adequately plan clinical studies.
Brain architecture and development, however, are not easily compared among mammalian species. Small rodents, such as mice and rats, have a smooth (or lissencephalic) brain surface, whereas humans and monkeys exhibit the typical fissured, gyrencephalic brain surface of gyri and sulci. Overall brain size and number of neurons are vastly different between humans and animal species. Moreover, brain development varies dramatically in terms of timing and duration. Whereas considerable brain development takes place postnatally in rodents, most critical steps in humans occur in utero.273 It has generally been accepted that mice and rats are born at relatively earlier stages of brain maturity compared with humans, but that neurodevelopment in small rodents rapidly catches up with humans, mostly during the first 2 to 3 weeks of the rodent’s life. Older data based on simple estimations of brain cell numbers and degree of myelination have been interpreted to mean that the first week of life in small rodents, when the peak of vulnerability to anesthetic neurotoxicity occurs, equates to an extended time span in humans, from the third gestational trimester all the way to the third year of life.2,22,274 However, more contemporary approaches, including computational models, have approximated the 7-day-old rat to be closer in brain maturity to human fetuses at 20 to 22 weeks gestation, and the immature rhesus monkey to approximate the immature human brain closer to term (Fig. 23-4).275–277288 According to these models, stages of brain maturity equivalent to term human neonates are not reached until after postnatal day 14 in rats or mice (online calculator available at http://www.translatingtime.net, accessed January 23, 2012). Therefore it remains questionable whether rodent models hold relevance to routine pediatric anesthesia in term neonates and infants, or whether they more closely correspond to brain maturational states during fetal surgery in midgestation. Nonhuman primates, on the other hand, exhibit brain maturation more comparable to human brain maturation at birth and could be more applicable to neonatal anesthesia. Interestingly, a recent study in rhesus macaques corroborates the greater fetal susceptibility by demonstrating a higher degree of neuroapoptosis after ketamine anesthesia in utero compared with postnatally.160 However, major differences in brain development still exist between humans and nonhuman primates. In general, brain development progresses at a much slower pace in humans and developmental stages are up to 50% longer. Even on a cellular level, remarkable differences exist between humans and animals; cell cycle duration during cortical neurogenesis is approximately 17 hours in mice, 28 hours in macaque monkeys, and 36 hours in humans.282 All these differences indicate that it is not sensible to directly apply observations of anesthetic effects in the developing animal brain of any species to human clinical anesthesia practice.
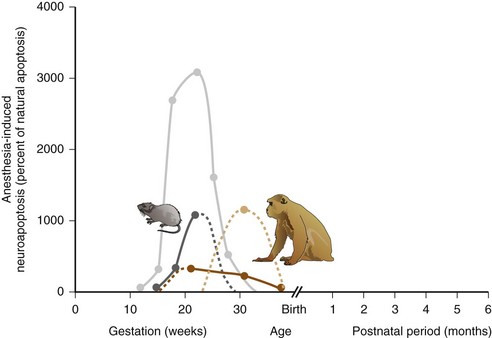
FIGURE 23-4 The degree of neuronal apoptotic cell death following anesthetic exposure is highly dependent on the age of the animal during exposure. Graphs demonstrate the percent increase of apoptotic cell death compared with natural apoptosis following an exposure to an isoflurane/nitrous oxide–based anesthetic in immature rats (dark gray) or very young macaque monkeys (dark brown) or prolonged exposure to ketamine in immature rats (light gray) or macaque monkeys (tan). Solid lines connect the available data points; interrupted lines represent extrapolations of the available data. Neurotoxicity data were derived from references, 17, 91, 114, 139. To infer the potential age of anesthetic vulnerability in humans, respective brain maturity in the animal species during exposure were equated to the corresponding state of the developing human brain, and relative human ages for each data point were plotted accordingly, using the mathematical model outlined in references, 276, 277, 288 and available in the online calculator at http://www.translatingtime.net.
Assessing Neurobehavioral or Cognitive Outcomes
Translating neurodevelopmental outcomes from animals to humans is difficult. Human cognitive performance includes the vast capacity for learning, the ability for abstract thinking, the aptitude for solving complex mathematical equations, and even the capability of inventing and operating complex machinery. Cognition is a complex process that includes such diverse processes as perception, attention, working memory, long-term memory, executive function, language, and social cognition. These brain functions are all difficult to model in animals.278 It is therefore imperative to critically evaluate any animal models attempting to replicate human cognitive performance. Moreover, it is important to assess the validity of these models in the context of the critical period for human brain development that they are trying to represent. Current assessment of neurocognitive performance after developmental exposure largely relies on hippocampal-dependent tests administered to adult animals following anesthetic exposure early in life.85,105–107,158 However, it remains unclear whether these are the same domains that may potentially be targeted in children during anesthesia. Brain regions maximally affected by anesthesia may change with the age of the animal and developmental state of the brain.79,160 Accordingly, subsequent neurobehavioral abnormalities may vary depending on the age at which anesthesia was administered.
This discussion, however, does not entirely discredit the results from small animal studies, but rather seriously limits the generalizability of their findings to clinical pediatric anesthesia practice. More closely resembling clinical pediatric anesthesia practice, several large-animal models have used tracheal intubation and mechanical ventilation.79,114,160 However, none of these large-animal studies included surgical stimulation during the anesthesia.79,114,139,158
Long-Term Outcome in Children Exposed to Anesthesia and Surgery
There is evidence for an association between surgery with anesthesia in early childhood and subsequent altered neurodevelopmental outcome.37 Some human cohort studies have demonstrated an association between major surgery in the neonatal period and poor neurodevelopmental outcome.289 Children born with esophageal atresia had a lower IQ and more frequently suffered from depression, emotional, and behavioral problems compared with the general population.290 Children with congenital diaphragmatic hernia repair also have a high rate of neurologic sequelae.291 Extremely premature, low–birth-weight neonates who underwent laparotomy had poorer neurodevelopmental outcomes compared with matched controls.292 A cohort of infants who underwent major surgery did not perform as well in school as a matched control group of healthy infants or with infants who had major nonsurgical medical conditions.293 In a randomized trial of indomethacin treatment in 426 infants less than 1000 g at birth, neurologic impairment was present in significantly more of the 110 children who had undergone surgery (53%) compared with that in the 316 children who had received medical therapy (34%).294 In a study of extremely preterm infants, the IQ of those who had undergone surgery was lower at 5 years of age and exhibited more sensorineural disability than those who had not undergone surgery.295
Several recent cohort studies have focused primarily on the effects of anesthesia. In an established population–based, retrospective birth cohort, Wilder and colleagues studied the association between anesthetic exposure before 4 years of age and the subsequent development of learning disabilities.296 Regression was used to calculate hazard ratios for anesthetic exposure as a predictor of learning disability, with adjustment for gestational age at birth, sex, and birth weight. Of 5357 children in the cohort, 593 had been exposed to general anesthesia before 4 years of age. Compared with those not exposed to anesthesia, a single exposure was not associated with an increased risk of learning disability (hazard ratio = 1.0; 95% confidence interval [CI] of 0.79 to 1.27). However, children who underwent two separate episodes of anesthesia had an increased risk of a learning disability (hazard ratio = 1.59; 95% CI of 1.06 to 2.37) and those who underwent 3 or more separate episodes of anesthesia had an even greater risk (hazard ratio = 2.60; 95% CI of 1.60 to 4.24). The association between learning disability and multiple episodes of anesthesia remained after adjusting for American Society of Anesthesiologist physical status. The risk for a learning disability also increased according to the cumulative duration of anesthesia. However, this study suffered from several deficiencies. Because the study reported anesthetics administered between 1976 and 1982, the most common anesthetic treatment was halothane and nitrous oxide, and none of the children were monitored with pulse oximetry or capnography. It is not possible to determine in how many of these children excessive hyperventilation or unrecognized desaturation had occurred. Furthermore, the maternal birth histories were not described (e.g., magnesium may cause neuroapoptosis or be neuroprotective). Three different learning disabilities were considered with equipoise in the final analysis, and these disabilities were not tested in all children, but only when a teacher or parent requested testing. These questions limit the external validity of these data.
To reduce the impact of confounding factors, using the same population-based, retrospective birth cohort, the same group conducted further studies with a matched cohort design.297 The researchers matched 350 children exposed to anesthesia before the age of 2 to 700 children not exposed to anesthesia. The matching was based on several known risk factors for learning disabilities: gender, mother’s education, birth weight, and gestational age at birth. Outcomes of interest were: learning disability, need for individualized education program for an emotional or behavioral disorder, and group-administered achievement tests. In the analysis, an adjustment was also performed for burden of illness. The primary finding was that children exposed to two or more occurrences of anesthesia (but not a single occurrence) were at increased risk for having a learning disability (hazard ratio = 2.12 with 95% CI of 1.26 to 3.54), and an amplified need for individualized education programs for speech and language impairment. However, there was not an increased need for a program for emotional or behavioral disorders. The authors also detected an association between multiple exposures to anesthesia and lower mathematical scores. The same criticisms apply to this study as to the earlier study from this institution.296
To investigate a potential association between perinatal exposure to general anesthesia during cesarean delivery and subsequent diagnosis of learning disability,298 the risk of a learning disability was compared in the 193 children delivered via cesarean section under general anesthesia, the 304 delivered via cesarean section under regional anesthesia, and 4823 delivered vaginally without any anesthesia. The association between mode of delivery and learning disability was adjusted for sex, birth weight, gestational age at birth, exposure to anesthesia before 4 years of age, and maternal education. The risk of disability was similar between children delivered by vagina with no anesthesia, and cesarean delivery under general anesthesia, but risk of disability was less in children delivered via cesarean under regional anesthesia than by vagina with no anesthesia (hazard ratio = 0.64, 95% CI of 0.44 to 0.92; P = 0.017). The results implied that brief exposure to general anesthesia during delivery was not associated with subsequent learning disability, but the reason why the risk was less with regional anesthesia compared with no anesthesia is unclear, although this may suggest the possibility of substantial confounding influences. To explore the possibility that the regional blockade was protective, the authors subsequently compared those born without general anesthesia by vaginal delivery, with and without regional analgesia, and found no difference in risk of learning disability.299
Using the same epidemiologic cohort, the same research group detected an increased prevalence of attention deficit hyperactivity disorder (ADHD) in children who had undergone repeated surgical procedures with anesthesia, compared with none or only one exposure.299a Kalkman and associates performed a small pilot study to test the feasibility of using a cohort of children who had urologic surgery, in order to test the association between age at surgery and neurobehavioral outcome (measured with the Child Behavior Check List).300 In their sample of 314 children, they found no evidence for an association between timing of surgery and neurobehavioral outcome.
DiMaggio and colleagues performed a retrospective cohort analysis using the New York State Medicaid records.301 The researchers matched 383 children who underwent hernia repair before 3 years of age to 5050 children who did not undergo inguinal hernia repair. After adjustment for sex, age, and complicating birth conditions (such as low birth weight), children who had hernia repair were more than twice as likely to have a subsequent diagnosis of a developmental or behavioral disorder (hazard ratio = 2.3, 95% CI of 1.3 to 4.1).
To reduce environmental confounding effects, the authors then proceeded to use the data from the New York State Medicaid program to construct a retrospective sibling birth cohort.302 Once again, they assessed the association between exposure to anesthesia in children less than 3 years of age and the subsequent risk of diagnosis of developmental or behavioral disorders. A total of 10,450 siblings were identified; 304 of whom underwent surgery before 3 years of age, without any history of behavioral or developmental disorders before the surgery, and 10,146 children who did not undergo surgery. The association of exposure to anesthesia with subsequent developmental or behavioral disorders was assessed with both proportional hazards modeling, and pair-matched analysis. As with their previous study, they found evidence for an association between surgery and poor neurodevelopmental outcome. The incidence of developmental or behavioral disorders was 128.2 diagnoses per 1000 person-years for those that had surgery and 56.3 diagnoses per 1000 person-years for those that did not. This association persisted when adjusted for sex, history of birth-related medical complications, and clustering by sibling status; the estimated hazard ratio of developmental or behavioral disorders associated with any exposure to anesthesia was 1.6 (95% CI of 1.4 to 1.8). The risk increased from 1.1 (95% CI of 0.8 to 1.4) for one operation to 2.9 (94% CI of 2.5 to 3.1) for two operations and 4.0 (95% CI of 3.5 to 4.5) for three or more operations. The number of siblings available for a matched analysis was relatively small. There were only 138 sibling pairs. In the pair-matched analysis there was no evidence for an association between surgery and poor outcome, with a relative risk of 0.9 (95% CI of 0.6 to 1.4); however, the small numbers may limit the power of this analysis.
Even better than sibling studies, investigations involving identical twins may further reduce confounding environmental and genetic influences. Performing such a study, Bartels and associates examined the possible association between anesthesia exposure before 3 years of age and school performance in 1143 monozygotic twin pairs.303 In the identical twins who were discordant for exposure to anesthesia (one twin was exposed to anesthesia and the other was not), their school performance was identical. This would suggest that surgery with anesthesia may not be the cause of poor school performance. Interestingly, the school performance in both the discordant pairs and the pairs where both twins underwent surgery was poorer than that for concordant twin pairs where neither twin was exposed to anesthesia. This finding could imply that there may exist an unknown genetic factor that increases the risk of both the need for surgery and poor school performance.
The Western Canadian Complex Pediatric Therapies Follow-up Group recently published a prospective follow-up study of 95 infants at 2 years of age who underwent surgical correction for congenital heart disease at the Alberta Children’s Hospital from April 2003 to December 2006.304 A multiple logistic regression analysis of variables associated with developmental delay found that only more days of ventilatory support postoperatively and older age at surgery were associated with significant developmental delay. There was also no evidence for an association between the sedation variables and significant motor delays. However, the use of two neurocognitive assessment tools during the study period precluded an analysis of the cognitive and motor scores as continuous variables, which may have been a more sensitive way to detect subtle impairment.
Hansen and colleagues used a very large Danish birth cohort to compare academic performance in 2689 children that had inguinal hernia repair in infancy with 14,575 children who were randomly selected as an age-matched sample from the general population.305 Those who had hernia repair performed worse at school, but there was no evidence for any association between surgery and school performance after adjustment for likely confounding variables. In contrast to the studies of DiMaggio and Wilder, this study detected no evidence for an association between surgery and poor neurobehavioral outcome.
Lastly, in a recent study using the Western Australian Pregnancy Cohort (Raine), Ing and colleagues found an association between language and abstract reasoning deficits in 10-year-old children who were exposed to one or more anesthetics prior to age 3, compared with previously unexposed children. After adjustment for confounders, any anesthetic before age 3 increased the risk ratio of disability in receptive, expressive, or total language as measured by the Clinical Evaluation of Language Fundamentals (CELF) test, as well as abstract reasoning (Raven’s Colored Progressive Matrices) to between 1.7 and 2.1 of unexposed controls, whereas other tests of vocabulary, behavior, and motor function were unaffected by exposure.305a
Outcome after Exposure to Anesthesia Outside of the Operating Room
The operating room is not the only area where anesthetic neurotoxicity may be relevant. Ketamine and benzodiazepines are frequently administered in the emergency room, and in neonatal and pediatric intensive care units. These drugs may be administered for a protracted period of time, increasing their theoretical risk of neurotoxicity.53,55
Unfortunately, determining the clinical relevance of any neurotoxicity in intensive care patients is even more difficult than for the operating room. The total number of children that can be examined is smaller, they tend to be a very heterogeneous population, and most importantly, these children often have multiple significant confounding comorbidities, including exposure to anesthesia, which could significantly affect neurologic outcome. To date, there is mixed evidence that exposure to anesthetic or sedative drugs is associated with poorer neurobehavioral outcome. A Cochrane review found some evidence for worsened short-term outcome in neonates who had prolonged exposures to midazolam infusions.306 In contrast, after correction for severity of illness, another study (using the EPIPAGE cohort) found no evidence for an association between prolonged sedation exposure and adverse neurologic outcome.307 In that study, however, many children received opioids for sedation rather than benzodiazepines.
Limitations of the Available Clinical Studies
Epidemiologic studies are generally unable to separate the effects of surgery, or the need for surgery, from the potential anesthetic effects. As previously discussed, surgery may result in significant neurohumoral stress and/or inflammatory responses that may influence neurocognitive outcome, in addition to metabolic, hemodynamic, and respiratory events that occur perioperatively. Data collection for several of the epidemiologic studies occurred before continuous pulse-oximetry and capnography monitoring were standard, and at a time when inhalational anesthetics with profound adverse cardiovascular effects, such as halothane, were used.296–297 Prospective studies of capnography and pulse oximetry have demonstrated that the very population considered to be at risk (those under 2 years of age) had the greatest incidence of desaturation events, hypercarbia, and hypocarbia.308–310
Children undergoing surgery or diagnostic procedures early in life may suffer more frequently from concomitant chromosomal and genetic abnormalities or comorbidities, such as prematurity, which have been linked to abnormal neurobehavioral outcomes. For example, children with cyanotic congenital heart disease often have abnormal neurocognitive development before any surgical or anesthesia intervention.262 The indication for surgery or for a diagnostic procedure requiring anesthesia, such as injury or infection, may artificially increase the incidence of neurodevelopmental abnormalities in the anesthetized cohort.262 On the other hand, it has been argued that subjects suffering from underlying abnormalities adversely affecting neurodevelopment that do not receive the required surgery may be introduced into the pool of unanesthetized children, and therefore mask potentially toxic effects of anesthesia.57
Further complicating the interpretation of retrospective data is the substantial gap in time between exposure and neurologic assessment. The advantage of performing neurologic assessments in school-age children includes the increased precision and greater predictive value of such assessments compared with neurobehavioral testing performed for children under 2 years of age, for both prospective and retrospective studies.311 However, especially in retrospective studies that cannot adequately control for confounders, the protracted time interval between surgery and the neurocognitive assessment may introduce even more “noise” into the study by increasing the time for negative influence of other environmental confounders of brain development or, conversely, the positive effects of repair mechanisms and plasticity to affect neurologic outcome.
No consensus exists, even in the animal literature, concerning what duration of anesthesia exposure is required to induce long-lasting injury. Although the anesthetic exposure may represent a much larger fraction of an animal’s life than a humans, at the cellular level apoptosis is likely to be triggered by a more similar duration of exposure. However, cell cycle duration and also the developmental period of the brain are considerably greater in humans than in rodents. It is therefore plausible, if an anesthesia-induced increase in apoptosis does occur in humans during a few hours of exposure, that it may be functionally less important than the same increase in apoptosis over the same period of time in a rodent. Similarly, the plasticity and capacity for recovery may differ between species. On the one hand, it can be argued that the period of development in humans is greater than it is in animals, and hence there may be more time for recovery, but on the other hand, human development is far more complex and humans may therefore be more vulnerable. Moreover, injuries during critical periods of development may have an exaggerated effect compared with the same injury outside of these periods.273,312 Accordingly, studies that find no evidence of association with shorter exposures may not be generalizable to all applications of anesthesia, and studies that find evidence for an association with greater exposure times may not be applicable to short exposures.
Lastly the possible neuroprotective effects of anesthesia complicate this conundrum even further. Infants undergoing surgery who experience major intraoperative adverse events, such as cardiopulmonary arrest, are generally expected to suffer from abnormal neurologic outcomes, irrespective of their anesthetic exposure. Neurologic abnormalities observed after an otherwise uneventful procedure are considered neurotoxic effects of anesthesia. However, because anesthetics may also, at least partially, protect from the harmful metabolic, immunologic, and humoral responses to surgery and pain,245 an alternative explanation may be that inadequate levels of anesthesia, insufficient postoperative pain relief, or unabated inflammation may be the culprit for the observed neurodevelopmental abnormalities. It is also possible that anesthetics may protect in major adverse settings, but may be deleterious in the absence of toxic stimuli, causing injury during minor procedures, but ameliorating injury during major surgery, such as complex heart surgery involving cardiopulmonary bypass.
Recommendations for Clinical Practice
The currently available laboratory data are not sufficient to make recommendations for clinical pediatric anesthesia practice. As outlined above, numerous laboratory studies have unequivocally documented deleterious effects of anesthetic exposure on the developing animal brain. Although these results should not be easily dismissed, the problems in translation from animal to human are substantial. Epidemiologic studies in children are also contradictory and can neither rule out nor confirm anesthetic-related neurotoxicity. Accordingly, the U.S. Food and Drug Administration (FDA) and the International Anesthesia Research Society (IARS) state that “dangers to infants and children from anesthesia remain unproven at this point” (http://www.smarttots.org/familyResourceCenter.html).
Pediatric surgery is never entirely elective, and in neonates may be required in order to preserve life, such as for critical congenital heart disease, necrotizing enterocolitis, or congenital diaphragmatic hernia. Accordingly, calls to postpone infant surgery are not easy to accommodate. Delaying surgery is also problematic, as no vulnerable or safe period has been identified in animal studies to guide this practice. Accordingly, the FDA and IARS have stated, “Currently, there is no scientific basis for delaying essential surgery” (http://www.smarttots.org/familyResourceCenter.html). It might be reasonable to delay purely elective surgeries; however, these only represent a very small fraction of surgeries performed in children.
Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74.
Istaphanous GK, Howard J, Nan X, et al. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology. 2011;114:578–587.
Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882.
Paule MG, Li M, Allen RR, et al. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–230.
Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804.
1 DeFrances CJ, Cullen KA, Kozak LJ. National Hospital Discharge Survey: 2005 annual summary with detailed diagnosis and procedure data. Vital Health Stat. 2007;13:1–209.
2 Dobbing J. The later development of the brain and its vulnerability. In: Davis JA, Dobbing J, eds. Scientific foundations of paediatrics. London: Heinemann Medical; 1991:744–759.
3 Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501.
4 Rakic S, Zecevic N. Programmed cell death in the developing human telencephalon. Eur J Neurosci. 2000;12:2721–2734.
5 Twomey C, McCarthy JV. Pathways of apoptosis and importance in development. J Cell Mol Med. 2005;9:345–359.
6 Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178.
7 Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990;28:517–527.
8 Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28:278–283.
9 de Lima AD, Opitz T, Voigt T. Irreversible loss of a subpopulation of cortical interneurons in the absence of glutamatergic network activity. Eur J Neurosci. 2004;19:2931–2943.
10 Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated “ménage à trois”. Trends Neurosci. 1997;20:523–529.
11 Eckenhoff JE. Relationship of anesthesia to postoperative personality changes in children. AMA Am J Dis Child. 1953;86:587–591.
12 Eckenhoff JE. Preanesthetic sedation of children; analysis of the effects for tonsillectomy and adenoidectomy. AMA Arch Otolaryngol. 1953;57:411–416.
13 Quimby KL, Aschkenase LJ, Bowman RE, Katz J, Chang LW. Enduring learning deficits and cerebral synaptic malformation from exposure to 10 parts of halothane per million. Science. 1974;185:625–627.
14 Quimby KL, Katz J, Bowman RE. Behavioral consequences in rats from chronic exposure to 10 PPM halothane during early development. Anesth Analg. 1975;54:628–633.
15 Uemura E, Bowman RE. Effects of halothane on cerebral synaptic density. Exp Neurol. 1980;69:135–142.
16 Uemura E, Levin ED, Bowman RE. Effects of halothane on synaptogenesis and learning behavior in rats. Exp Neurol. 1985;89:520–529.
17 Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74.
18 Haberny KA, Paule MG, Scallet AC, et al. Ontogeny of the N-methyl-D-aspartate (NMDA) receptor system and susceptibility to neurotoxicity. Toxicol Sci. 2002;68:9–17.
19 Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23:659–668.
20 Olney JW, Wozniak DF, Jevtovic-Todorovic V, et al. Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathol. 2002;12:488–498.
21 Davidson A, Soriano S. Does anaesthesia harm the developing brain—evidence or speculation? Paediatr Anaesth. 2004;14:199–200.
22 Olney JW, Young C, Wozniak DF, Ikonomidou C, Jevtovic-Todorovic V. Anesthesia-induced developmental neuroapoptosis. Does it happen in humans? Anesthesiology. 2004;101:273–275.
23 Olney JW, Young C, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–139.
24 Jevtovic-Todorovic V. General anesthetics and the developing brain: friends or foes? J Neurosurg Anesthesiol. 2005;17:204–206.
25 Perouansky M. Liaisons dangereuses? General anaesthetics and long-term toxicity in the CNS. Eur J Anaesthesiol. 2006:1–9.
26 Bhutta AT. Ketamine: a controversial drug for neonates. Semin Perinatol. 2007;31:303–308.
27 Doherty D, Splinter W. Can modern anesthesia practice harm the developing brain? Paediatr Drugs. 2007;9:213–214.
28 Gascon E, Klauser P, Kiss JZ, Vutskits L. Potentially toxic effects of anaesthetics on the developing central nervous system. Eur J Anaesthesiol. 2007;24:213–224.
29 Kaindl AM, Ikonomidou C. Glutamate antagonists are neurotoxins for the developing brain. Neurotox Res. 2007;11:203–218.
30 Mellon RD, Simone AF, Rappaport BA. Use of anesthetic agents in neonates and young children. Anesth Analg. 2007;104:509–520.
31 Bree B, Gourdin M, De Kock M. Anesthesia and cerebral apoptosis. Acta Anaesthesiol Belg. 2008;59:127–137.
32 Davidson AJ, McCann ME, Morton NS, Myles PS. Anesthesia and outcome after neonatal surgery: the role for randomized trials. Anesthesiology. 2008;109:941–944.
33 Henschel O, Gipson KE, Bordey A. GABAA receptors, anesthetics and anticonvulsants in brain development. CNS Neurol Disord Drug Targets. 2008;7:211–224.
34 Jevtovic-Todorovic V. General anaesthesia and the developing brain. In: Gullo A, ed. Anaesthesia, pain, intensive care and emergency A.P.I.C.E., Proceedings of the 22nd Postgraduate Course in Critical Care Medicine, Venice-Mestre, Italy—November 9-11, 2007. Milan, Italy: Springer; 2008:45–50.
35 Jevtovic-Todorovic V, Olney JW. PRO: Anesthesia-induced developmental neuroapoptosis: status of the evidence. Anesth Analg. 2008;106:1659–1663.
36 Loepke AW, McGowan Jr, FX., Soriano SG. CON: The toxic effects of anesthetics in the developing brain: the clinical perspective. Anesth Analg. 2008;106:1664–1669.
37 Loepke AW, Soriano SG. An assessment of the effects of general anesthetics on developing brain structure and neurocognitive function. Anesth Analg. 2008;106:1681–1707.
38 Perouansky M. General anesthetics and long-term neurotoxicity. Handb Exp Pharmacol. 2008:143–157.
39 Sanders RD, Ma D, Brooks P, Maze M. Balancing paediatric anaesthesia: preclinical insights into analgesia, hypnosis, neuroprotection, and neurotoxicity. Br J Anaesth. 2008;101:597–609.
40 Wang C, Slikker Jr, W. Strategies and experimental models for evaluating anesthetics: effects on the developing nervous system. Anesth Analg. 2008;106:1643–1658.
41 Creeley CE, Olney JW. The young: neuroapoptosis induced by anesthetics and what to do about it. Anesth Analg. 2010;110:442–448.
42 Hansen TG, Flick R. Anesthetic effects on the developing brain: insights from epidemiology. Anesthesiology. 2009;110:1–3.
43 Ikonomidou C. Triggers of apoptosis in the immature brain. Brain Dev. 2009;31:488–492.
44 Istaphanous GK, Loepke AW. General anesthetics and the developing brain. Curr Opin Anaesthesiol. 2009;22:368–373.
45 McCann ME, Soriano SG. Is anesthesia bad for the newborn brain? Anesthesiol Clin. 2009;27:269–284.
46 Perouansky M, Hemmings Jr, HC. Between Clotho and Lachesis: how isoflurane seals neuronal fate. Anesthesiology. 2009;110:709–711.
47 Perouansky M, Hemmings Jr, HC. Neurotoxicity of general anesthetics: cause for concern? Anesthesiology. 2009;111:1365–1371.
48 Sanders RD, Davidson A. Anesthetic-induced neurotoxicity of the neonate: time for clinical guidelines? Paediatr Anaesth. 2009;19:1141–1146.
49 Stratmann G, Sall JW, May LD, Loepke AW, Lee MT. Beyond anesthetic properties: the effects of isoflurane on brain cell death, neurogenesis, and long-term neurocognitive function. Anesth Analg. 2009;110:431–437.
50 Thompson BL, Levitt P, Stanwood GD. Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci. 2009;10:303–312.
51 Blaylock M, Engelhardt T, Bissonnette B. Fundamentals of neuronal apoptosis relevant to pediatric anesthesia. Paediatr Anaesth. 2010;20:383–395.
52 Borsook D, George E, Kussman B, Becerra L. Anesthesia and perioperative stress: consequences on neural networks and postoperative behaviors. Prog Neurobiol. 2010;92:601–612.
53 Durrmeyer X, Vutskits L, Anand KJ, Rimensberger PC. Use of analgesic and sedative drugs in the NICU: integrating clinical trials and laboratory data. Pediatr Res. 2010;67:117–127.
54 Istaphanous GK, Ward CG, Loepke AW. The impact of the perioperative period on neurocognitive development, with a focus on pharmacologic concerns. Best Pract Res Clin Anaesthesiol. 2010;24:433–449.
55 Loepke AW. Developmental neurotoxicity of sedatives and anesthetics: a concern for neonatal and pediatric critical care medicine? Pediatr Crit Care Med. 2010;11:217–226.
56 Sun L. Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth. 2010;105:i61–i68.
57 Wilder RT. Is there any relationship between long-term behavior disturbance and early exposure to anesthesia? Curr Opin Anaesthesiol. 2010;23:332–336.
58 Hays SR, Deshpande JK. Newly postulated neurodevelopmental risks of pediatric anesthesia. Curr Neurol Neurosci Rep. 2011;11:205–210.
59 Hudson AE, Hemmings Jr, HC. Are anaesthetics toxic to the brain? Br J Anaesth. 2011;107:30–37.
60 Jevtovic-Todorovic V. Anesthesia and the developing brain: are we getting closer to understanding the truth? Curr Opin Anaesthesiol. 2011;24:395–399.
61 Rappaport B, Mellon RD, Simone A, Woodcock J. Defining safe use of anesthesia in children. N Engl J Med. 2011;364:1387–1390.
62 Davidson AJ. Anesthesia and neurotoxicity to the developing brain: the clinical relevance. Paediatr Anaesth. 2011;21:716–721.
63 Vutskits L. Anesthetic-related neurotoxicity and the developing brain: shall we change practice? Paediatr Drugs. 2012;14:13–21.
64 Stratmann G. Review article: neurotoxicity of anesthetic drugs in the developing brain. Anesth Analg. 2011;113:1170–1179.
65 Ward CG, Loepke AW. Anesthetics and sedatives: toxic or protective for the developing brain? Pharmacol Res. 2012;65:271–274.
66 Wu VW, Mo Q, Yabe T, Schwartz JP, Robinson SE. Perinatal opioids reduce striatal nerve growth factor content in rat striatum. Eur J Pharmacol. 2001;414:211–214.
67 Cattano D, Straiko MM, Olney JW. Chloral hydrate induces and lithium prevents neuroapoptosis in the infant mouse brain. Anesthesiology. 2008;109:A315.
68 Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. 2002;99:15089–15094.
69 Ikonomidou C, Bittigau P, Ishimaru MJ, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060.
70 Briner A, De Roo M, Dayer A, et al. Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology. 2010;112:546–556.
71 Istaphanous GK, Howard J, Nan X, et al. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology. 2011;114:578–587.
72 Kodama M, Satoh Y, Otsubo Y, et al. Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology. 2011;115:979–991.
73 Sanders RD, Xu J, Shu Y, et al. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology. 2009;110:1077–1085.
74 Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–376.
75 Stefovska VG, Uckermann O, Czuczwar M, et al. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. 2008;64:434–445.
76 Chalon J, Tang CK, Ramanathan S, et al. Exposure to halothane and enflurane affects learning function of murine progeny. Anesth Analg. 1981;60:794–797.
77 Thornton SR, Smith FL. Long-term alterations in opiate antinociception resulting from infant fentanyl tolerance and dependence. Eur J Pharmacol. 1998;363:113–119.
78 Laudenbach V, Calo G, Guerrini R, et al. Nociceptin/orphanin FQ exacerbates excitotoxic white-matter lesions in the murine neonatal brain. J Clin Invest. 2001;107:457–466.
79 Rizzi S, Ori C, Jevtovic-Todorovic V. Timing versus duration: determinants of anesthesia-induced developmental apoptosis in the young mammalian brain. Ann N Y Acad Sci. 2010;1199:43–51.
80 Uemura E, Ireland WP, Levin ED, Bowman RE. Effects of halothane on the development of rat brain: a golgi study of dendritic growth. Exp Neurol. 1985;89:503–519.
81 Yanai J, Steingart RA, Snapir N, et al. The relationship between neural alterations and behavioral deficits after prenatal exposure to heroin. Ann N Y Acad Sci. 2000;914:402–411.
82 Wang Y, Han TZ. Prenatal exposure to heroin in mice elicits memory deficits that can be attributed to neuronal apoptosis. Neuroscience. 2009;160:330–338.
83 Cunha-Oliveira T, Rego AC, Garrido J, et al. Street heroin induces mitochondrial dysfunction and apoptosis in rat cortical neurons. J Neurochem. 2007;101:543–554.
84 Simon W, Hapfelmeier G, Kochs E, Zieglgansberger W, Rammes G. Isoflurane blocks synaptic plasticity in the mouse hippocampus. Anesthesiology. 2001;94:1058–1065.
85 Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882.
86 McClaine RJ, Uemura K, de la Fuente SG, et al. General anesthesia improves fetal cerebral oxygenation without evidence of subsequent neuronal injury. J Cereb Blood Flow Metab. 2005;25:1060–1069.
87 Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–827.
88 Rizzi S, Carter LB, Jevtovic-Todorovic V. Clinically used general anesthetics induce neuroapoptosis in the developing piglet brain. Washington, D.C.: Society for Neuroscience; 2005. 2005 Annual Meeting Program No. 251.7
89 Wise-Faberowski L, Zhang H, Ing R, Pearlstein RD, Warner DS. Isoflurane-induced neuronal degeneration: an evaluation in organotypic hippocampal slice cultures. Anesth Analg. 2005;101:651–657.
90 Wei H, Kang B, Wei W, et al. Isoflurane and sevoflurane affect cell survival and BCL-2/BAX ratio differently. Brain Res. 2005;1037:139–147.
91 Lu LX, Yon JH, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–1615.
92 Yon JH, Carter LB, Reiter RJ, Jevtovic-Todorovic V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–530.
93 Olney JW, Wang H, Qin Y, Labruyere J, Young C. Pilocarpine pretreatment reduces neuroapoptosis induced by midazolam or isoflurane in infant mouse brain. Atlanta: Society for Neuroscience; 2006. 2006 Annual Meeting Program No. 286.15
94 Ma D, Williamson P, Januszewski A, et al. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology. 2007;106:746–753.
95 Wei H, Liang G, Yang H. Isoflurane preconditioning inhibited isoflurane-induced neurotoxicity. Neurosci Lett. 2007;425:59–62.
96 Li Y, Liang G, Wang S, et al. Effects of fetal exposure to isoflurane on postnatal memory and learning in rats. Neuropharmacology. 2007;53:942–950.
97 Nikizad H, Yon JH, Carter LB, Jevtovic-Todorovic V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann N Y Acad Sci. 2007;1122:69–82.
98 Rizzi S, Carter LB, Ori C, Jevtovic-Todorovic V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008;18:198–210.
99 Johnson SA, Young C, Olney JW. Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. J Neurosurg Anesthesiol. 2008;20:21–28.
100 Sanders RD, Xu J, Shu Y, et al. General anesthetics induce apoptotic neurodegeneration in the neonatal rat spinal cord. Anesth Analg. 2008;106:1708–1711.
101 Olney JW, Straiko MM, Cattano D, Young C, Creeley C. Preventing anesthesia-induced developmental neuroapoptosis—temperature regulation approach. Anesthesiology. 2008;109:A1414.
102 Zou X, Sadovova N, Patterson TA, et al. The effects of l-carnitine on the combination of, inhalation anesthetic-induced developmental, neuronal apoptosis in the rat frontal cortex. Neuroscience. 2008;151:1053–1065.
103 Rothstein S, Simkins T, Nunez JL. Response to neonatal anesthesia: effect of sex on anatomical and behavioral outcome. Neuroscience. 2008;152:959–969.
104 Wang QJ, Li KZ, Yao SL, Li ZH, Liu SS. Different effects of isoflurane and sevoflurane on cytotoxicity. Chin Med J. 2008;121:341–346.
105 Loepke AW, Istaphanous GK, McAuliffe JJ, 3rd., et al. The effects of neonatal isoflurane exposure in mice on brain cell viability, adult behavior, learning, and memory. Anesth Analg. 2009;108:90–104.
106 Stratmann G, Sall JW, May LD, et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–848.
107 Stratmann G, May LD, Sall JW, et al. Effect of hypercarbia and isoflurane on brain cell death and neurocognitive dysfunction in 7-day-old rats. Anesthesiology. 2009;110:849–861.
108 Sall JW, Stratmann G, Leong J, et al. Isoflurane inhibits growth but does not cause cell death in hippocampal neural precursor cells grown in culture. Anesthesiology. 2009;110:826–833.
109 Head BP, Patel HH, Niesman IR, et al. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–825.
110 Lunardi N, Ori C, Erisir A, Jevtovic-Todorovic V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox Res. 2010;17:179–188.
111 Olney JW, Brambrink AM, Avidan MS, Farber NB, Back SA. Isoflurane-induced oligoapoptosis in neonatal rhesus macaque brain. Anesthesiology. A1598, 2009.
112 Wang S, Peretich K, Zhao Y, et al. Anesthesia-induced neurodegeneration in fetal rat brains. Pediatr Res. 2009;66:435–440.
113 Zhu C, Gao J, Karlsson N, et al. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J Cereb Blood Flow Metab. 2010;30:1017–1030.
114 Brambrink AM, Evers AS, Avidan MS, et al. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–841.
115 Liang G, Ward C, Peng J, et al. Isoflurane causes greater neurodegeneration than an equivalent exposure of sevoflurane in the developing brain of neonatal mice. Anesthesiology. 2010;112:1325–1334.
116 Yuede CM, Creeley CE, Olney JW, Caffeine augments neuroapoptosis induced in the infant mouse brain by isoflurane. Presented at the International Anesthesia Research Society and SAFEKIDS International Science Symposium, March 20, 2010. Honolulu
117 Kulak A, Duarte JM, Do KQ, Gruetter R. Neurochemical profile of the developing mouse cortex determined by in vivo 1H NMR spectroscopy at 14.1 T and the effect of recurrent anaesthesia. J Neurochem. 2010;115:1466–1477.
118 Shu Y, Patel SM, Pac-Soo C, et al. Xenon pretreatment attenuates anesthetic-induced apoptosis in the developing brain in comparison with nitrous oxide and hypoxia. Anesthesiology. 2010;113:360–368.
119 Lunardi N, Hucklenbruch C, Latham JR, Scarpa J, Jevtovic-Todorovic V. Isoflurane impairs immature astroglia development in vitro: the role of actin cytoskeleton. J Neuropathol Exp Neurol. 2011;70:281–291.
120 Yang G, Chang PC, Bekker A, Blanck TJ, Gan WB. Transient effects of anesthetics on dendritic spines and filopodia in the living mouse cortex. Anesthesiology. 2011;115:718–726.
121 Culley DJ, Boyd JD, Palanisamy A, et al. Isoflurane decreases self-renewal capacity of rat cultured neural stem cells. Anesthesiology. 2011;115:754–763.
122 Campbell LL, Tyson JA, Stackpole EE, et al. Assessment of general anaesthetic cytotoxicity in murine cortical neurones in dissociated culture. Toxicology. 2011;283:1–7.
123 Lemkuil BP, Head BP, Pearn ML, et al. Isoflurane neurotoxicity is mediated by p75NTR-RhoA activation and actin depolymerization. Anesthesiology. 2011;114:49–57.
124 Yahalom B, Athiraman U, Soriano SG, et al. Spinal anesthesia in infant rats: development of a model and assessment of neurologic outcomes. Anesthesiology. 2011;114:1325–1335.
125 Zou X, Liu F, Zhang X, et al. Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol Teratol. 2011;33:592–597.
126 Sanchez V, Feinstein SD, Lunardi N, et al. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology. 2011;115:992–1002.
127 Shu Y, Zhou Z, Wan Y, et al. Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiol Dis. 2012;45:743–750.
128 Hayashi H, Dikkes P, Soriano SG. Repeated administration of ketamine may lead to neuronal degeneration in the developing rat brain. Paediatr Anaesth. 2002;12:770–774.
129 Fredriksson A, Archer T. Hyperactivity following postnatal NMDA antagonist treatment: reversal by d-amphetamine. Neurotox Res. 2003;5:549–564.
130 Fredriksson A, Archer T. Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox Res. 2004;6:435–456.
131 Scallet AC, Schmued LC, Slikker Jr, W., et al. Developmental neurotoxicity of ketamine: morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol Sci. 2004;81:364–370.
132 Rudin M, Ben-Abraham R, Gazit V, et al. Single-dose ketamine administration induces apoptosis in neonatal mouse brain. J Basic Clin Physiol Pharmacol. 2005;16:231–243.
133 Wang C, Sadovova N, Fu X, et al. The role of the N-methyl-d-aspartate receptor in ketamine-induced apoptosis in rat forebrain culture. Neuroscience. 2005;132:967–977.
134 Young C, Jevtovic-Todorovic V, Qin YQ, et al. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br J Pharmacol. 2005;146:189–197.
135 Wang C, Sadovova N, Hotchkiss C, et al. Blockade of N-methyl-d-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol Sci. 2006;91:192–201.
136 Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Effect of ketamine on dendritic arbor development and survival of immature GABAergic neurons in vitro. Toxicol Sci. 2006;91:540–549.
137 Takadera T, Ishida A, Ohyashiki T. Ketamine-induced apoptosis in cultured rat cortical neurons. Toxicol Appl Pharmacol. 2006;210:100–107.
138 Shang Y, Wu Y, Yao S, et al. Protective effect of erythropoietin against ketamine-induced apoptosis in cultured rat cortical neurons: involvement of PI3K/Akt and GSK-3 beta pathway. Apoptosis. 2007;12:2187–2195.
139 Slikker Jr, W., Zou X, Hotchkiss CE, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–158.
140 Vutskits L, Gascon E, Potter G, Tassonyi E, Kiss JZ. Low concentrations of ketamine initiate dendritic atrophy of differentiated GABAergic neurons in culture. Toxicology. 2007;234:216–226.
141 Anand KJ, Garg S, Rovnaghi CR, et al. Ketamine reduces the cell death following inflammatory pain in newborn rat brain. Pediatr Res. 2007;62:283–290.
142 Fredriksson A, Ponten E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-d-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107:427–436.
143 Wang C, Sadovova N, Patterson TA, et al. Protective effects of 7-nitroindazole on ketamine-induced neurotoxicity in rat forebrain culture. Neurotoxicology. 2008;29:613–620.
144 Viberg H, Ponten E, Eriksson P, Gordh T, Fredriksson A. Neonatal ketamine exposure results in changes in biochemical substrates of neuronal growth and synaptogenesis, and alters adult behavior irreversibly. Toxicology. 2008;249:153–159.
145 Rovnaghi CR, Garg S, Hall RW, Bhutta AT, Anand KJ. Ketamine analgesia for inflammatory pain in neonatal rats: a factorial randomized trial examining long-term effects. Behav Brain Funct. 2008;4:35.
146 Boctor SY, Wang C, Ferguson SA. Neonatal PCP is more potent than ketamine at modifying preweaning behaviors of Sprague-Dawley rats. Toxicol Sci. 2008;106:172–179.
147 Vutskits L, De Roo M, Klauser P, Adrian B, Muller D. Anesthetics impair dendritic spine development in the postnatal mouse somatosensory cortex. Anesthesiology. 2008;109:A325.
148 Straiko MM, Young C, Cattano D, et al. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology. 2009;110:862–868.
149 Zou X, Patterson TA, Sadovova N, et al. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol Sci. 2009;108:149–158.
150 Ibla JC, Hayashi H, Bajic D, Soriano SG. Prolonged exposure to ketamine increases brain derived neurotrophic factor levels in developing rat brains. Curr Drug Saf. 2009;4:11–16.
151 Zou X, Patterson TA, Divine RL, et al. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–731.
152 De Roo M, Klauser P, Briner A, et al. Anesthetics rapidly promote synaptogenesis during a critical period of brain development. PLoS ONE. 2009;4:e7043.
153 Tan H, Ren RR, Xiong ZQ, Wang YW. Effects of ketamine and midazolam on morphology of dendritic spines in hippocampal CA1 region of neonatal mice. Chin Med J. 2009;122:455–459.
154 Braun S, Gaza N, Werdehausen R, et al. Ketamine induces apoptosis via the mitochondrial pathway in human lymphocytes and neuronal cells. Br J Anaesth. 2010;105:347–354.
155 Soriano SG, Liu Q, Li J, et al. Ketamine activates cell cycle signaling and apoptosis in the neonatal rat brain. Anesthesiology. 2010;112:1155–1163.
156 Shi Q, Guo L, Patterson TA, et al. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience. 2010;166:852–863.
157 Walker SM, Westin BD, Deumens R, Grafe M, Yaksh TL. Effects of intrathecal ketamine in the neonatal rat: evaluation of apoptosis and long-term functional outcome. Anesthesiology. 2010;113:147–159.
158 Paule MG, Li M, Allen RR, et al. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol Teratol. 2011;33:220–230.
159 Zhang X, Paule MG, Newport GD, et al. MicroPET imaging of ketamine-induced neuronal apoptosis with radiolabeled DFNSH. J Neural Transm. 2011;118:203–211.
160 Brambrink AM, Evers AS, Avidan MS, et al. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology. 2012;116:372–384.
161 Rech RH, Lomuscio G, Algeri S. Methadone exposure in utero: effects on brain biogenic amines and behavior. Neurobehav Toxicol. 1980;2:75–78.
162 Hutchings DE, Zmitrovich AC, Brake SC, Church SH, Malowany D. Prenatal administration of methadone in the rat increases offspring acoustic startle amplitude at age 3 weeks. Neurotoxicol Teratol. 1993;15:157–164.
163 Robinson SE, Maher JR, Wallace MJ, Kunko PM. Perinatal methadone exposure affects dopamine, norepinephrine, and serotonin in the weanling rat. Neurotoxicol Teratol. 1997;19:295–303.
164 Robinson SE. Effect of prenatal opioid exposure on cholinergic development. J Biomed Sci. 2000;7:253–257.
165 Vutskits L, Gascon E, Tassonyi E, Kiss JZ. Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated gamma-aminobutyric acid-positive interneurons. Anesthesiology. 2005;102:970–976.
166 Xu H, Liu ZQ, Liu Y, et al. Administration of midazolam in infancy does not affect learning and memory of adult mice. Clin Exp Pharmacol Physiol. 2009;36:1144–1148.
167 Hammer Jr, RP., Seatriz JV, Ricalde AR. Regional dependence of morphine-induced mu-opiate receptor down-regulation in perinatal rat brain. Eur J Pharmacol. 1991;209:253–256.
168 Tempel A. Visualization of mu opiate receptor downregulation following morphine treatment in neonatal rat brain. Brain Res Dev Brain Res. 1991;64:19–26.
169 Rimanoczy A, Vathy I. Prenatal exposure to morphine alters brain mu opioid receptor characteristics in rats. Brain Res. 1995;690:245–248.
170 Hauser KF, Houdi AA, Turbek CS, Elde RP, Maxson W, 3rd. Opioids intrinsically inhibit the genesis of mouse cerebellar granule neuron precursors in vitro: differential impact of mu and delta receptor activation on proliferation and neurite elongation. Eur J Neurosci. 2000;12:1281–1293.
171 Slamberova R, Schindler CJ, Pometlova M, et al. Prenatal morphine exposure differentially alters learning and memory in male and female rats. Physiol Behav. 2001;73:93–103.
172 Stiene-Martin A, Knapp PE, Martin K, et al. Opioid system diversity in developing neurons, astroglia, and oligodendroglia in the subventricular zone and striatum: impact on gliogenesis in vivo. Glia. 2001;36:78–88.
173 Hu S, Sheng WS, Lokensgard JR, Peterson PK. Morphine induces apoptosis of human microglia and neurons. Neuropharmacology. 2002;42:829–836.
174 Yang SN, Huang LT, Wang CL, et al. Prenatal administration of morphine decreases CREBSerine-133 phosphorylation and synaptic plasticity range mediated by glutamatergic transmission in the hippocampal CA1 area of cognitive-deficient rat offspring. Hippocampus. 2003;13:915–921.
175 Che Y, Sun H, Tan H, et al. The effect of prenatal morphine exposure on memory consolidation in the chick. Neurosci Lett. 2005;380:300–304.
176 McPherson RJ, Gleason C, Mascher-Denen M, et al. A new model of neonatal stress which produces lasting neurobehavioral effects in adult rats. Neonatology. 2007;92:33–41.
177 Zissen MH, Zhang G, Kendig JJ, Sweitzer SM. Acute and chronic morphine alters formalin pain in neonatal rats. Neurosci Lett. 2006;400:154–157.
178 Svensson AL, Bucht N, Hallberg M, Nyberg F. Reversal of opiate-induced apoptosis by human recombinant growth hormone in murine foetus primary hippocampal neuronal cell cultures. Proc Natl Acad Sci U S A. 2008;105:7304–7308.
179 Boasen JF, McPherson RJ, Hays SL, Juul SE, Gleason CA. Neonatal stress or morphine treatment alters adult mouse conditioned place preference. Neonatology. 2009;95:230–239.
180 Niu L, Cao B, Zhu H, et al. Impaired in vivo synaptic plasticity in dentate gyrus and spatial memory in juvenile rats induced by prenatal morphine exposure. Hippocampus. 2009;19:649–657.
181 Lin CS, Tao PL, Jong YJ, et al. Prenatal morphine alters the synaptic complex of postsynaptic density 95 with N-methyl-D-aspartate receptor subunit in hippocampal CA1 subregion of rat offspring leading to long-term cognitive deficits. Neuroscience. 2009;158:1326–1337.
182 Rozisky JR, Medeiros LF, Adachi LS, et al. Morphine exposure in early life increases nociceptive behavior in a rat formalin tonic pain model in adult life. Brain Res. 2011;1367:122–129.
183 Morimoto Y, Nishihira J, Kemmotsu O, et al. Pentobarbital inhibits apoptosis in neuronal cells. Crit Care Med. 2000;28:1899–1904.
184 Tachibana K, Hashimoto T, Kato R, et al. Long-lasting effects of neonatal pentobarbital administration on spatial learning and hippocampal synaptic plasticity. Brain Res. 2011;1388:69–76.
185 Asimiadou S, Bittigau P, Felderhoff-Mueser U, et al. Protection with estradiol in developmental models of apoptotic neurodegeneration. Ann Neurol. 2005;58:266–276.
186 Kim JS, Kondratyev A, Tomita Y, Gale K. Neurodevelopmental impact of antiepileptic drugs and seizures in the immature brain. Epilepsia. 2007;48(Suppl 5):19–26.
187 Katz I, Kim J, Gale K, Kondratyev A. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phenytoin on cell death in the neonatal rat brain. J Pharmacol Exp Ther. 2007;322:494–500.
188 Honegger P, Matthieu JM. Selective toxicity of the general anesthetic propofol for GABAergic neurons in rat brain cell cultures. J Neurosci Res. 1996;45:631–636.
189 Spahr-Schopfer I, Vutskits L, Toni N, et al. Differential neurotoxic effects of propofol on dissociated cortical cells and organotypic hippocampal cultures. Anesthesiology. 2000;92:1408–1417.
190 Al-Jahdari WS, Saito S, Nakano T, Goto F. Propofol induces growth cone collapse and neurite retractions in chick explant culture. Can J Anaesth. 2006;53:1078–1085.
191 Cattano D, Young C, Straiko MM, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106:1712–1714.
192 Gressens P, Dingley J, Plaisant F, et al. Analysis of neuronal, glial, endothelial, axonal and apoptotic markers following moderate therapeutic hypothermia and anesthesia in the developing piglet brain. Brain Pathol. 2008;18:10–20.
193 Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic mechanism of propofol toxicity in immature neurons. J Neurosurg Anesthesiol. 2008;20:233–240.
194 Pesic V, Milanovic D, Tanic N, et al. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27:279–287.
195 Berns M, Seeberg L, Schmidt M, Kerner T. High-dose propofol triggers short-term neuroprotection and long-term neurodegeneration in primary neuronal cultures from rat embryos. J Int Med Res. 2009;37:680–688.
196 Bercker S, Bert B, Bittigau P, et al. Neurodegeneration in newborn rats following propofol and sevoflurane anesthesia. Neurotox Res. 2009;16:140–147.
197 Briner A, Nikonenko I, De Roo M, et al. Developmental stage-dependent persistent impact of propofol anesthesia on dendritic spines in the rat medial prefrontal cortex. Anesthesiology. 2011;115:282–293.
198 Pontén E, Fredriksson A, Gordh T, Eriksson P, Viberg H. Neonatal exposure to propofol affects BDNF but not CaMKII, GAP-43, synaptophysin and tau in the neonatal brain and causes an altered behavioural response to diazepam in the adult mouse brain. Behav Brain Res. 2011;223:75–80.
199 Tu S, Wang X, Yang F, et al. Propofol induces neuronal apoptosis in infant rat brain under hypoxic conditions. Brain Res Bull. 2011;86:29–35.
200 Pearn ML, Hu Y, Niesman IR, et al. Propofol neurotoxicity is mediated by p75 neurotrophin receptor activation. Anesthesiology. 2012;116:352–361.
201 Zhang X, Xue Z, Sun A. Subclinical concentration of sevoflurane potentiates neuronal apoptosis in the developing C57BL/6 mouse brain. Neurosci Lett. 2008;447:109–114.
202 Satomoto M, Satoh Y, Terui K, et al. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–637.
203 Berns M, Zacharias R, Seeberg L, Schmidt M, Kerner T. Effects of sevoflurane on primary neuronal cultures of embryonic rats. Eur J Anaesthesiol. 2009;26:597–602.
204 Edwards DA, Shah HP, Cao W, et al. Bumetanide alleviates epileptogenic and neurotoxic effects of sevoflurane in neonatal rat brain. Anesthesiology. 2010;112:567–575.
205 Lu Y, Wu X, Dong Y, et al. Anesthetic sevoflurane causes neurotoxicity differently in neonatal naive and Alzheimer disease transgenic mice. Anesthesiology. 2010;112:1404–1416.
206 Shih J, May LD, Gonzalez HE, et al. Delayed environmental enrichment reverses sevoflurane-induced memory impairment in rats. Anesthesiology. 2012;116:586–602.
207 Cattano D, Williamson P, Fukui K, et al. Potential of xenon to induce or to protect against neuroapoptosis in the developing mouse brain. Can J Anaesth. 2008;55:429–436.
208 Zimmermann KC, Green DR. How cells die: apoptosis pathways. J Allergy Clin Immunol. 2001;108:S99–103.
209 Kuida K, Zheng TS, Na S, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372.
210 Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007;12:993–1010.
211 Tamaroff MH, Nir Y, Straker N. Children reared in a reverse isolation environment: effects on cognitive and emotional development. J Autism Dev Disord. 1986;16:415–424.
212 Kraemer GW. Psychobiology of early social attachment in rhesus monkeys. Clinical implications. Ann N Y Acad Sci. 1997;807:401–418.
213 Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci. 2000;3:799–806.
214 Wahlsten D, Bachmanov A, Finn DA, Crabbe JC. Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci U S A. 2006;103:16364–16369.
215 Westin BD, Walker SM, Deumens R, Grafe M, Yaksh TL. Validation of a preclinical spinal safety model: effects of intrathecal morphine in the neonatal rat. Anesthesiology. 2010;113:183–199.
216 Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med. 2003;348:2110–2124.
217 Varju P, Katarova Z, Madarasz E, Szabo G. GABA signalling during development: New data and old questions. Cell Tissue Res. 2001;305:239–246.
218 Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: A pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284.
219 Lu J, Karadsheh M, Delpire E. Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol. 1999;39:558–568.
220 Zhao Y, Liang G, Chen Q, et al. Anesthetic-induced neurodegeneration mediated via inositol 1,4,5-trisphosphate receptors. J Pharmacol Exp Ther. 2010;333:14–22.
221 Wei H, Xie Z. Anesthesia, calcium homeostasis and Alzheimer’s disease. Curr Alzheimer Res. 2009;6:30–35.
222 Loepke AW, McCann JC, Kurth CD, McAuliffe JJ. The physiologic effects of isoflurane anesthesia in neonatal mice. Anesth Analg. 2006;102:75–80.
223 Green SM, Coté CJ. Ketamine and neurotoxicity: clinical perspectives and implications for emergency medicine. Ann Emerg Med. 2009;54:181–190.
224 Williams GD, Maan H, Ramamoorthy C, et al. Perioperative complications in children with pulmonary hypertension undergoing general anesthesia with ketamine. Paediatr Anaesth. 2010;20:28–37.
225 Murphy TW, Smith JH, Ranger MR, Haynes SR. General anesthesia for children with severe heart failure. Pediatr Cardiol. 2011;32:139–144.
226 Cullen SC, Gross EG. The anesthetic properties of xenon in animals and human beings, with additional observations on krypton. Science. 1951;113:580–582.
227 Cullen SC, Eger EI, 2nd., Cullen BF, Gregory P. Observations on the anesthetic effect of the combination of xenon and halothane. Anesthesiology. 1969;31:305–309.
228 Nakata Y, Goto T, Ishiguro Y, et al. Minimum alveolar concentration (MAC) of xenon with sevoflurane in humans. Anesthesiology. 2001;94:611–614.
229 Goto T, Saito H, Shinkai M, et al. Xenon provides faster emergence from anesthesia than does nitrous oxide-sevoflurane or nitrous oxide-isoflurane. Anesthesiology. 1997;86:1273–1278.
230 Cortellazzi P, Lamperti M, Minati L, et al. Sedation of neurologically impaired children undergoing MRI: a sequential approach. Paediatr Anaesth. 2007;17:630–636.
231 Cameron HA, Hazel TG, McKay RD. Regulation of neurogenesis by growth factors and neurotransmitters. J Neurobiol. 1998;36:287–306.
232 Wang H, Cuzon VC, Pickel VM. Postnatal development of mu-opioid receptors in the rat caudate-putamen nucleus parallels asymmetric synapse formation. Neuroscience. 2003;118:695–708.
233 Hammer Jr, RP., Ricalde AA, Seatriz JV. Effects of opiates on brain development. Neurotoxicology. 1989;10:475–483.
234 Dawson G, Dawson SA, Goswami R. Chronic exposure to kappa-opioids enhances the susceptibility of immortalized neurons (F-11kappa 7) to apoptosis-inducing drugs by a mechanism that may involve ceramide. J Neurochem. 1997;68:2363–2370.
235 Bhutta AT, Rovnaghi C, Simpson PM, et al. Interactions of inflammatory pain and morphine in infant rats: long-term behavioral effects. Physiol Behav. 2001;73:51–58.
236 Reynolds ML, Fitzgerald M. Long-term sensory hyperinnervation following neonatal skin wounds. J Comp Neurol. 1995;358:487–498.
237 Ruda MA, Ling QD, Hohmann AG, Peng YB, Tachibana T. Altered nociceptive neuronal circuits after neonatal peripheral inflammation. Science. 2000;289:628–631.
238 Anand KJ, Coskun V, Thrivikraman KV, Nemeroff CB, Plotsky PM. Long-term behavioral effects of repetitive pain in neonatal rat pups. Physiol Behav. 1999;66:627–637.
239 Al-Chaer ED, Kawasaki M, Pasricha PJ. A new model of chronic visceral hypersensitivity in adult rats induced by colon irritation during postnatal development. Gastroenterology. 2000;119:1276–1285.
240 LaPrairie JL, Murphy AZ. Female rats are more vulnerable to the long-term consequences of neonatal inflammatory injury. Pain. 2007;132:S124–S133.
241 LaPrairie JL, Johns ME, Murphy AZ. Preemptive morphine analgesia attenuates the long-term consequences of neonatal inflammation in male and female rats. Pediatr Res. 2008;64:625–630.
242 Helmeke C, Ovtscharoff Jr, W., Poeggel G, Braun K. Imbalance of immunohistochemically characterized interneuron populations in the adolescent and adult rodent medial prefrontal cortex after repeated exposure to neonatal separation stress. Neuroscience. 2008;152:18–28.
243 Rokyta R, Yamamotova A, Slamberova R, et al. Prenatal and perinatal factors influencing nociception, addiction and behavior during ontogenetic development. Physiol Res. 2008;57:S79–S88.
244 Anand KJ, Brown MJ, Causon RC, et al. Can the human neonate mount an endocrine and metabolic response to surgery? J Pediatr Surg. 1985;20:41–48.
245 Anand KJ, Sippell WG, Schofield NM, Aynsley-Green A. Does halothane anaesthesia decrease the metabolic and endocrine stress responses of newborn infants undergoing operation? Br Med J. 1988;296:668–672.
246 Anand KJ, Hansen DD, Hickey PR. Hormonal-metabolic stress responses in neonates undergoing cardiac surgery. Anesthesiology. 1990;73:661–670.
247 Grunau RE, Haley DW, Whitfield MF, et al. Altered basal cortisol levels at 3, 6, 8 and 18 months in infants born at extremely low gestational age. J Pediatr. 2007;150:151–156.
248 Anand KJ, Sippell WG, Aynsley-Green A. Randomised trial of fentanyl anaesthesia in preterm babies undergoing surgery: effects on the stress response. Lancet. 1987;1:62–66.
249 Wolf AR, Eyres RL, Laussen PC, et al. Effect of extradural analgesia on stress responses to abdominal surgery in infants. Br J Anaesth. 1993;70:654–660.
250 Anand KJ, Hickey PR. Halothane-morphine compared with high-dose sufentanil for anesthesia and postoperative analgesia in neonatal cardiac surgery. N Engl J Med. 1992;326:1–9.
251 Taddio A, Katz J, Ilersich AL, Koren G. Effect of neonatal circumcision on pain response during subsequent routine vaccination. Lancet. 1997;349:599–603.
252 Stang HJ, Gunnar MR, Snellman L, Condon LM, Kestenbaum R. Local anesthesia for neonatal circumcision. Effects on distress and cortisol response. JAMA. 1988;259:1507–1511.
253 Grunau RE, Whitfield MF, Petrie-Thomas J, et al. Neonatal pain, parenting stress and interaction, in relation to cognitive and motor development at 8 and 18 months in preterm infants. Pain. 2009;143:138–146.
254 Rao SC, Pirie S, Minutillo C, et al. Ward reduction of gastroschisis in a single stage without general anaesthesia may increase the risk of short-term morbidities: results of a retrospective audit. J Paediatr Child Health. 2009;45:384–388.
255 Carbajal R, Rousset A, Danan C, et al. Epidemiology and treatment of painful procedures in neonates in intensive care units. JAMA. 2008;300:60–70.
256 Sankar R, Shin DH, Liu H, et al. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393.
257 Mikati MA, Rizk E, El Dada S, et al. Programmed cell death in the lithium pilocarpine model: evidence for NMDA receptor and ceramide-mediated mechanisms. Brain Dev. 2008;30:513–519.
258 Grandjean EM, Aubry JM. Lithium: updated human knowledge using an evidence-based approach: part III: clinical safety. CNS Drugs. 2009;23:397–418.
259 Pinelli JM, Symington AJ, Cunningham KA, Paes BA. Case report and review of the perinatal implications of maternal lithium use. Am J Obstet Gynecol. 2002;187:245–249.
260 Kozma C. Neonatal toxicity and transient neurodevelopmental deficits following prenatal exposure to lithium: another clinical report and a review of the literature. Am J Med Genet A. 2005;132:441–444.
261 Sessler DI. Non-pharmacologic prevention of surgical wound infection. Anesthesiol Clin. 2006;24:279–297.
262 DiNardo JA. Should what we know about neurobehavioral development, complex congenital heart disease, and brain maturation affect the timing of corrective cardiac surgery? Paediatr Anaesth. 2011;21:781–786.
263 Tobias JD. Dexmedetomidine: applications in pediatric critical care and pediatric anesthesiology. Pediatr Crit Care Med. 2007;8:115–131.
264 Mason KP, Lerman J. Review article: Dexmedetomidine in children: current knowledge and future applications. Anesth Analg. 2011;113:1129–1142.
265 Koerner IP, Brambrink AM. Brain protection by anesthetic agents. Curr Opin Anaesthesiol. 2006;19:481–486.
266 Kurth CD, Priestley M, Watzman HM, McCann J, Golden J. Desflurane confers neurologic protection for deep hypothermic circulatory arrest in newborn pigs. Anesthesiology. 2001;95:959–964.
267 Loepke AW, Priestley MA, Schultz SE, et al. Desflurane improves neurologic outcome after low-flow cardiopulmonary bypass in newborn pigs. Anesthesiology. 2002;97:1521–1527.
268 Zhao P, Zuo Z. Isoflurane preconditioning induces neuroprotection that is inducible nitric oxide synthase-dependent in neonatal rats. Anesthesiology. 2004;101:695–703.
269 McAuliffe JJ, Joseph B, Vorhees CV. Isoflurane-delayed preconditioning reduces immediate mortality and improves striatal function in adult mice after neonatal hypoxia-ischemia. Anesth Analg. 2007;104:1066–1077.
270 McAuliffe JJ, Loepke AW, Miles L, et al. Desflurane, isoflurane, and sevoflurane provide limited neuroprotection against neonatal hypoxia-ischemia in a delayed preconditioning paradigm. Anesthesiology. 2009;111:533–546.
271 Luo Y, Ma D, Leong E, et al. Xenon and sevoflurane protect against brain injury in a neonatal asphyxia model. Anesthesiology. 2008;109:782–789.
272 Loepke AW, McAuliffe JJ, Miles L, et al. Early and long-term protection during severe brain ischemia in a novel neonatal mouse model using sevoflurane. Anesth Analg. 2009;108:S273.
273 Rice D, Barone Jr, S. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–533.
274 Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 1978;4:345–356.
275 Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144.
276 Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17.
277 Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931–937.
278 Keeler JF, Robbins TW. Translating cognition from animals to humans. Biochem Pharmacol. 2011;81:1356–1366.
279 Herndon JG, Tigges J, Anderson DC, Klumpp SA, McClure HM. Brain weight throughout the life span of the chimpanzee. J Comp Neurol. 1999;409:567–572.
280 Malkova L, Heuer E, Saunders RC. Longitudinal magnetic resonance imaging study of rhesus monkey brain development. Eur J Neurosci. 2006;24:3204–3212.
281 Kornack DR, Rakic P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci U S A. 1998;95:1242–1246.
282 Molnar Z, Metin C, Stoykova A, et al. Comparative aspects of cerebral cortical development. Eur J Neurosci. 2006;23:921–934.
283 Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 2009;157:907–921.
284 Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661.
285 Eger EI, 2nd. Age, minimum alveolar anesthetic concentration, and minimum alveolar anesthetic concentration-awake. Anesth Analg. 2001;93:947–953.
286 Komitova M, Perfilieva E, Mattsson B, Eriksson PS, Johansson BB. Effects of cortical ischemia and postischemic environmental enrichment on hippocampal cell genesis and differentiation in the adult rat. J Cereb Blood Flow Metab. 2002;22:852–860.
287 Fabel K, Wolf SA, Ehninger D, et al. Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Front Neurosci. 2009;3:50.
288 Clancy B, Kersh B, Hyde J, et al. Web-based method for translating neurodevelopment from laboratory species to humans. Neuroinformatics. 2007;5:79–94.
289 Walker K, Holland AJ, Winlaw D, Sherwood M, Badawi N. Neurodevelopmental outcomes and surgery in neonates. J Paediatr Child Health. 2006;42:749–751.
290 Bouman NH, Koot HM, Hazebroek FW. Long-term physical, psychological, and social functioning of children with esophageal atresia. J Pediatr Surg. 1999;34:399–404.
291 Hedrick HL, Crombleholme TM, Flake AW, et al. Right congenital diaphragmatic hernia: prenatal assessment and outcome. J Pediatr Surg. 2004;39:319–323.
292 Chacko J, Ford WD, Haslam R. Growth and neurodevelopmental outcome in extremely-low-birth-weight infants after laparotomy. Pediatr Surg Int. 1999;15:496–499.
293 Ludman L, Spitz L, Wade A. Educational attainments in early adolescence of infants who required major neonatal surgery. J Pediatr Surg. 2001;36:858–862.
294 Kabra NS, Schmidt B, Roberts RS, et al. Neurosensory impairment after surgical closure of patent ductus arteriosus in extremely low birth weight infants: results from the Trial of Indomethacin Prophylaxis in Preterms. J Pediatr. 2007;150:229–234.
295 Surgery and the tiny baby: sensorineural outcome at 5 years of age. The Victorian Infant Collaborative Study Group. [No authors listed]. J Paediatr Child Health. 1996;32:167–172.
296 Wilder RT, Flick RP, Sprung J, et al. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804.
297 Flick RP, Katusic SK, Colligan RC, et al. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–e1061.
298 Sprung J, Flick RP, Wilder RT, et al. Anesthesia for cesarean delivery and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;111:302–310.
299 Flick RP, Lee K, Hofer RE, et al. Neuraxial labor analgesia for vaginal delivery and its effects on childhood learning disabilities. Anesth Analg. 2011;112:1424–1431.
299a Sprung J, Flick RP, Katusic SK, et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin Proc. 2012;87:120–129.
300 Kalkman CJ, Peelen L, Moons KG, et al. Behavior and development in children and age at the time of first anesthetic exposure. Anesthesiology. 2009;110:805–812.
301 DiMaggio CJ, Sun LS, Kakavouli A, Byrne MW, Li G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J Neurosurg Anesthesiol. 2009;21:286–291.
302 DiMaggio C, Sun L, Li G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth Analg. 2011;113:1143–1151.
303 Bartels M, Althoff RR, Boomsma DI. Anesthesia and cognitive performance in children: No evidence for a causal relationship. Twin Res Hum Genet. 2009;12:246–253.
304 Guerra GG, Robertson CM, Alton GY, et al. Neurodevelopmental outcome following exposure to sedative and analgesic drugs for complex cardiac surgery in infancy. Paediatr Anaesth. 2011;21:932–941.
305 Hansen TG, Pedersen JK, Henneberg SW, et al. Academic performance in adolescence after inguinal hernia repair in infancy: a nationwide cohort study. Anesthesiology. 2011;114:1076–1085.
305a Ing C, DiMaggio C, Whitehouse A, et al. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics. 2012. in press (doi: 10.1542/peds.2011-3822)
306 Ng E, Taddio A, Ohlsson A. Intravenous midazolam infusion for sedation of infants in the neonatal intensive care unit. Cochrane Database Syst Rev (Online). 2003. CD002052
307 Rozé JC, Denizot S, Carbajal R, et al. Prolonged sedation and/or analgesia and 5-year neurodevelopment outcome in very preterm infants: results from the EPIPAGE cohort. Arch Pediatr Adolesc Med. 2008;162:728–733.
308 Coté CJ, Liu LM, Szyfelbein SK, et al. Intraoperative events diagnosed by expired carbon dioxide monitoring in children. Can Anaesth Soc J. 1986;33:315–320.
309 Coté CJ, Goldstein EA, Coté MA, Hoaglin DC, Ryan JF. A single-blind study of pulse oximetry in children. Anesthesiology. 1988;68:184–188.
310 Coté CJ, Rolf N, Liu LM, et al. A single-blind study of combined pulse oximetry and capnography in children. Anesthesiology. 1991;74:980–987.
311 Hack M, Taylor HG, Drotar D, et al. Poor predictive validity of the Bayley Scales of Infant Development for cognitive function of extremely low birth weight children at school age. Pediatrics. 2005;116:333–341.
312 Cronise K, Marino MD, Tran TD, Kelly SJ. Critical periods for the effects of alcohol exposure on learning in rats. Behav Neurosci. 2001;115:138–145.
*References 13, 14, 74, 76, 85, 103, 106, 107, 113, 118, 129, 130, 142, 144, 158, 196, 198, 202
†References 77, 81, 82, 161, 162, 177
*References 72, 96, 105, 106, 115, 117, 132, 141, 145, 166, 196
*References 70, 109, 110, 120, 136, 140, 152, 153, 165, 197
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
23 Surgery, Anesthesia, and the Immature Brain
Effects of Anesthetic Exposure on the Developing Brain
Putative Mechanisms for Neurotoxicity
Specific Anesthetic and Sedative Agents
Exposure Time, Dose, and Anesthetic Combinations
Deleterious Effects of Untreated Pain and Stress
Potential Alleviating Strategies
Critical Evaluation of Animal Studies and Interspecies Comparisons
Long-Term Outcome in Children Exposed to Anesthesia and Surgery
Outcome after Exposure to Anesthesia Outside of the Operating Room
MILLIONS OF CHILDREN UNDERGO SURGERY with anesthesia every year.1 During the perioperative period they are exposed to a multitude of stressors capable of interfering with normal brain development. Pain, stress, inflammation, hypoxia, and ischemia have all previously been shown to adversely affect the immature central nervous system. However, recent findings from animal studies have indicated that sedatives and anesthetics—the very drugs used to reduce pain and stress—may themselves undesirably influence brain development by triggering structural and functional abnormalities. There is now an extensive body of work, mostly based on laboratory research, that has defined this phenomenon and explored mechanisms and protective strategies. However, translating these laboratory findings to humans in clinical settings is laden with uncertainties and questions.
Normal Brain Development
In utero and during the early postnatal period, the human brain initially undergoes a rapid growth in size and cell number, and is then pared back to achieve the efficient network of about 100 billion neurons of the adult brain. At birth, the size of the immature brain is one-third that of the adult brain, doubling in size within the first year of life, and reaching 90% of its eventual size by 6 years of age.2 This dramatic growth spurt coincides with a remarkable overabundance of neurons and neuronal connections. In fact, less than half the neurons generated during development survive into adulthood.3,4 Superfluous neurons that lose in the competition for a limited amount of trophic factors are removed by programmed cell death. Also called apoptosis, this cellular suicide program is built into every mammalian cell. Apoptotic cells enter a well-orchestrated, stepwise, and energy-consuming destruction process that involves a cascade of enzymes called caspases and ultimately leads to the breakdown of cellular proteins and DNA.5 This process is heavily used during development, such as during embryonal deletion of the interdigital mesenchymal tissue to separate fingers and toes. Similar to the clotting cascade, the apoptotic cascade remains active throughout life and is held in check by antiapoptotic factors, whereas proapoptotic factors promote its execution. Neurons are protected from apoptosis by neurotrophic factors. Conversely, noxious stimuli, such as pain, hypoxia, and ischemia, can increase the level of proapoptotic factors and activate the apoptotic suicide program, leading to neuronal cell death.
In addition to an overabundance in the total number of neurons generated during early development, the mammalian brain also forms an excess of neuronal connections, or synapses, during this period. Depending on brain region, synaptic density reaches its maximum in infants and young toddlers between 3 and 15 months of age, and will undergo a progressive reduction by about half into adulthood.6 Connections that are active with continued electrical and chemical signals are sustained, whereas those with little or no activity are lost.
In summary, brain architecture changes rapidly and dramatically throughout life. Neuronal density is greatest during fetal life, and excess neurons are eliminated via apoptosis, predominantly in utero, during the neonatal period and throughout infancy.7 Rapid growth of dendrites and synaptic connections occurs during infancy and early childhood, and unneeded dendrites and synapses are trimmed back, predominantly during later childhood and adolescence.6,7
Accordingly, the first several years after birth represent a critical period of development for many brain regions. Recent findings in animals suggest that exposure to anesthetics or sedatives may interfere with proper neuronal development, brain architecture, and subsequent function. Although the exact molecular mechanisms by which anesthetics afford their therapeutic properties of amnesia, analgesia, and immobility are only incompletely understood, their interaction with a wide variety of ion channels, such as sodium, calcium, and potassium channels, as well as several cell membrane proteins, including the receptors for γ-aminobutyric acid (GABA), glycine, glutamate (and N-methyl-d-aspartate [NMDA]), acetylcholine, and serotonin, make it conceivable that anesthetics could interfere with normal electrical and chemical activity in the developing brain. In fact, both GABA and NMDA play critical roles as trophic factors and in regulating neuronal maturation and programmed cell death. During brain development, GABA directs cell proliferation, neuroblast migration, and dendritic maturation.8 Developmental NMDA receptor stimulation fosters survival and maturation of some neurons.9,10 It is therefore not implausible that anesthetics might interfere with these developmental processes.
Effects of Anesthetic Exposure on the Developing Brain
Concerns regarding neurologic abnormalities following general anesthesia in young children were first raised more than half a century ago, when postoperative personality changes were observed following the administration of vinyl ether, cyclopropane, or ethyl chloride for otolaryngologic surgery.11 However, these abnormalities were felt to be psychological in nature because they were alleviated by the timely administration of preoperative sedative drugs.11,12 Approximately two decades later the focus of research shifted to examine the effects of anesthetics in animal models that represented occupational exposure in pregnant healthcare workers.13–16 Delayed synaptogenesis and behavioral abnormalities were observed in neonatal rats born to dams that were chronically exposed to subanesthetic doses of halothane during their entire pregnancy. However, interest in the effects of anesthetics on children did not elicit widespread interest until publications began to appear 10 years ago. In a seminal study in neonatal rat pups, widespread neuronal degeneration was observed after repeated injections of ketamine.17 This led to numerous editorials and review articles, and more than 200 publications in which structural brain abnormalities and/or functional impairment were demonstrated after a wide variety of immature animal species (including chicks, mice, rats, guinea pigs, swine, sheep, and rhesus monkeys) were exposed to almost every sedative and anesthetic in clinical use.18–65 However, no discussion about the effects of drug exposure on the developing brain would be complete without examining the effects of opioid analgesics in the developing brain.54,55 Accordingly, this chapter will examine the various specific effects of sedatives, anesthetics, and analgesics in the immature brain.
Apoptotic Cell Death
The most widely studied deleterious consequence of exposure to sedatives or anesthetics in immature animals is apoptosis, or programmed cell death. Although neuronal apoptosis eliminates approximately 50% to 70% of neurons throughout the brain during development, at any particular time, this natural process affects only a small fraction of cells. Exposure to anesthetics or sedatives briefly, but dramatically, increases the number of apoptotic neurons (Fig. 23-1). Some studies demonstrated up to a 68-fold increase in the density of degenerating neurons after a combination of anesthetics in neonatal rats, compared with control animals, although it remains unclear what fraction of the entire neuronal population these degenerating neurons represent. Unpublished data from one of the authors’ laboratory suggest that a 6-hour exposure to a clinically relevant dose of isoflurane triggers apoptotic cell death in 2% of neurons in the superficial cortex of neonatal mice, whereas less than 0.1% of neurons undergo physiologic apoptosis in this region in unanesthetized litter mates (Loepke, personal communication). Interestingly, dying neurons are immediately adjacent to seemingly unaffected neighboring cells (Fig. 23-2), and the exact mechanism and selectivity of the cell death process remains unknown. Increased neuroapoptosis has also been observed following either in vitro or in vivo, exposure to a wide variety of sedatives and anesthetics, including chloral hydrate, clonazepam, diazepam, midazolam, nitrous oxide, desflurane, enflurane, halothane, isoflurane, sevoflurane, ketamine, pentobarbital, phenobarbital, propofol, and xenon, as well as opioid receptor agonists, in a wide variety of species, including chicks, mice, rats, guinea pigs, piglets, and rhesus monkeys (E-Table 23-1). Cell death–selective stains, such as cupric silver and Fluoro-Jade (EMD Millipore, Billerica, Mass.), have confirmed the cellular demise in neurons that positively stain for activated caspase 3, the central executioner enzyme of the apoptotic cascade.
E-TABLE 23-1 Preclinical Studies Examining Structural and Neurocognitive Effects of Anesthetic Exposure Early in Life
Apoptosis represents an inherent, energy-consuming process using a cascade of enzymes called caspases. Apoptosis is highly conserved among species and culminates in self-destruction and elimination of cells, even under physiologic conditions, when these cells are functionally redundant or potentially detrimental to the organism.208 Apoptosis involves an orderly breakdown of the cell that includes chromatin aggregation, nuclear and cytoplasmic condensation, and partitioning of cytoplasmic and nuclear material into apoptotic bodies for subsequent phagocytosis, without an extensive inflammatory response. That contrasts with the features observed during necrosis, which include energy failure, cellular swelling, membrane rupture, and release of cytoplasmic content into the extracellular compartment, followed by an inflammatory response.208 Accordingly, apoptosis has also been termed cellular suicide and is extensively used during tissue homeostasis, endocrine-dependent tissue atrophy, and normal embryogenesis (e.g., ablation of tail tissue as part of tadpole metamorphosis in amphibians). Similarly, brain cells are also produced in excess during normal brain development, and up to 50% to 70% of immature neurons are eliminated during normal brain maturation in rodents, nonhuman primates, and humans.3,4 Accordingly, physiologic apoptotic cell death is critical to establish proper brain structure and function, and any disruption of this process can lead to massive brain malformations and intrauterine demise.209 However, in addition to this intrinsic pathway of physiologic, developmental apoptosis, cell death can also be triggered by pathologic, extrinsic factors, such as hypoxia and ischemia.210 It currently remains unknown whether anesthesia-induced neuroapoptosis accelerates physiologic programmed cell death or whether it eliminates cells not destined to die, as in pathologic apoptosis.
Animal studies have identified a narrow window of maximum susceptibility to neuronal cell death induced by several anesthetic drugs, such as the NMDA antagonist ketamine, the GABA agonist isoflurane, or ethanol (a combined NMDA antagonist and GABA agonist). Ketamine-induced neuronal demise occurs in neonatal rodents between 5 and 7 days of age, or before 6 days of age in monkeys, but not in older animals.17,69,139 Similarly, neurotoxicity was not detected after isoflurane anesthesia in 1-day-old animals or in those more than 10 days of age, whereas neuroapoptosis reached a maximum effect at 7 days of age.87 However, preliminary data from the laboratory of one of the authors challenges the notion that anesthetic-induced neuroapoptosis is limited to this age range (Loepke and associates, unpublished data). Conversely, intrauterine exposure to clinical doses of isoflurane in prenatal rats may actually decrease physiologic apoptosis and improve subsequent memory retention,96 whereas only supraclinical doses of isoflurane induced neuroapoptosis in this setting.112
Long-Term Brain Cellular Viability, Neurologic Function, and Behavior
In order to answer the important question whether anesthetics simply hasten physiologic apoptosis or whether they induce pathologic apoptosis, long-term neuronal density and neurologic function have to be assessed in adult animals exposed to anesthesia as neonates. If exposure to anesthetics or analgesics only temporarily accelerated physiologic apoptosis, one would expect normal cell counts and function in adult animals. Conversely, a permanent neuronal cell loss and long-term neurocognitive impairment after anesthetic exposure early in life would suggest that anesthesia-induced neuronal apoptosis may be pathologic in nature. However, this would only be true if the organism was unable to compensate for the neonatal cell loss by increasing neuronal plasticity and repair. To answer these questions, several studies measured neurologic function, assessed behavior, and/or determined neuronal density in adult animals after they were exposed to anesthesia in the neonatal period. Unfortunately, results from these studies are conflicting. Several studies reported long-term neurocognitive or behavioral abnormalities after exposure of neonates to enflurane, halothane, isoflurane, sevoflurane, propofol, or ketamine, or to a combination of isoflurane, nitrous oxide, and midazolam.* Importantly, however, many of these studies only observed abnormalities in very specific tests or subsets of neurocognitive batteries, whereas many other neurobehavioral domains remained intact. For example, a 6-hour exposure to midazolam, isoflurane, and nitrous oxide in neonatal rats led to transient impairment in a water maze learning task in young adulthood and in older animals, whereas, in the same animals, several other tests of behavior and learning, including acoustic startle response, sensorimotor tests, spontaneous behavior in an open field, and learning and memory in the radial arms maze, remained unimpaired.85 Similarly, after a 4-hour exposure to the minimal alveolar concentration (1 MAC) of isoflurane in 7-day-old rats, long-term memory retention was abnormal at two time points, whereas performance at several other time points, as well as in other tests of learning and memory, remained intact.107 Accordingly, the relevance of these limited learning deficits remains uncertain. Similar to humans, performance of rodents and primates in learning tasks depends to a great extent on maternal behavior and rearing conditions, making them strong confounders during neurocognitive testing.105,211–213 Moreover, another obvious and important factor in neurocognitive testing is the verification of similar degrees of motivation when comparing separate groups of animals. For example, a 24-hour exposure to ketamine sedation early in life impaired subsequent performance of rhesus monkeys in learning and memory tests, in addition to decreasing their motivation to perform these tasks.158
Studies of prolonged opioid administration in immature animals have also found evidence for long-term impairment in learning tasks,174–176,179–181 as well as altered pain responses in adult animals after exposure to morphine, fentanyl, heroin, or methadone early in life.†
Conversely, several other investigations have observed no neurologic abnormalities after administration of midazolam, isoflurane, sevoflurane, or ketamine, even when using complex neurologic tests in neonatal animals.* It remains to be determined whether these differential findings are attributable to the anesthetic doses, exposure times, species, or related to the type or timing of the neurologic tests performed. Interestingly, escalating exposure times of isoflurane in neonatal rats caused neuronal apoptosis beginning at 2 hours of anesthesia, but no evidence of long-term neurologic abnormalities until 4 hours of anesthesia.106 In another study, a 6-hour exposure to isoflurane caused significant apoptosis immediately after exposure in neonatal mice, but resulted in no measurable long-term deficit in performance of complex neurologic tests as adult animals.105 Moreover, in brain regions significantly affected by the neonatal neuroapoptosis, adult neuronal density was not diminished compared with unanesthetized litter mates.105 These findings could either suggest that isoflurane may only accelerate physiologic apoptosis or that the developing brain’s plasticity and capacity for repair could compensate for a pathologic insult early in life. Conversely, a study in similarly aged rats observed a permanent elimination of neurons, as well as neurologic abnormalities in adult animals, after exposure to isoflurane, nitrous oxide, and midazolam as neonates, suggesting that either the specific combination of anesthetic drugs (isoflurane alone vs. the combination exposure) or species differences (rats vs. mice) could affect any relationships between neonatal neuroapoptosis and long-term function and neuronal density.97 Alternatively, these conflicting results may be explained by the dissimilar testing environment, because neurocognitive tests are easily transferable among laboratories.214 On the other hand, neonatal apoptosis may not be causally linked to adult neurocognitive performance at all, as evidenced by substantial apoptosis immediately after carbon dioxide–induced hypercarbia in neonatal rats without long-term neurologic sequelae.107
Effects on Neurogenesis and Gliogenesis
The generation of new neurons, or neurogenesis, as well as new astrocytes, or gliogenesis, is most active in the immature brain in utero or soon after birth. Accordingly, anesthetic exposure during this critical period may interfere with neuronal proliferation, or even eliminate neuronal precursor cells, thereby portending permanent reductions in neuronal density and long-term neurologic impairment. To test this relationship, several studies have investigated the effects of anesthetic exposure, primarily isoflurane, on progenitor viability and rates of neurogenesis. Although isoflurane did not cause neural progenitor cell death in vitro, 3.4% isoflurane for 4 hours did decrease the rate of neuronal proliferation and increased neuronal fate selection.108 These findings were recently confirmed in vitro, where 2.8% isoflurane for 6 hours resulted in no effects on neural stem cell viability and larger doses inhibited cell proliferation.121 Similarly, morphine has been found to decrease DNA synthesis in cerebellar neuronal precursors without affecting cell survival.170
Isoflurane also impairs the growth of immature astrocytes and delays their maturation after 24 hours of 3% isoflurane, although this enormous exposure had no effect on cell viability.119 Morphine, although increasing apoptosis in neurons and microglia, did not affect astrocytes in a study involving human fetal cell cultures.173
Alterations in Dendritic Architecture
The immature brain accumulates an overabundance of neuronal connections in infancy, and the number of dendrites and synapses dramatically decreases after the first year of life. Several studies have examined the effects of propofol, isoflurane, sevoflurane, desflurane, midazolam, and ketamine on dendritic arborization and synaptic architecture.* A common theme in these studies is that anesthetics can affect dendritic arborization and synaptic density, and that the direction of this change, whether it is an increase or a decrease in the number of dendritic spines, depends on the age at which the animals were exposed to anesthetics, and therefore the developmental state of the brain. During the first 2 weeks of life, the evidence indicates that anesthetic exposure leads to a decrease in synaptic and dendritic spine density in small rodents, while causing an increase in the number of dendrites beyond that age.110,197 However, the permanence of these changes remains controversial, with some studies only observing a transient effect after ketamine, midazolam, or isoflurane anesthesia early in life.120,153
Decrease in Trophic Factors
Isoflurane- or propofol-based anesthesia in the neonatal animal has been associated with a decrease in brain-derived neurotrophic factor (BDNF),91,109,198 a protein integral to neuronal survival, growth, and differentiation. The cellular mechanism involves a reduction in tissue plasminogen activator and plasmin, which converts proBDNF to BDNF. Accordingly, isoflurane triggered proBDNF/p75NTR (p75 neurotrophin receptor) complex–mediated apoptosis in neonatal mice.109 Moreover, prolonged exposure to opioid receptor agonists early in life has also been found to alter nerve growth factors in the immature brain.66,164
Degeneration of Mitochondria
Ultrastructural morphologic abnormalities have been reported in mitochondria of pyramidal neurons in the subiculum of 7-day-old rats after 6 hours of isoflurane, nitrous oxide, and midazolam.126 A morphometric analysis demonstrated mitochondrial enlargement, impaired structural integrity, and decreased mitochondrial density, indicative of a protracted injury to the mitochondria after anesthetic exposure. Moreover, ultrastructural examination of pyramidal neurons of anesthetized animals by electron microscopy revealed evidence for increased autophagy, a form of cell death.
Abnormal Reentry into Cell Cycle
Ketamine induces reentry of postmitotic neurons into cell cycle in immature rats.155 Neuronal progenitor cells enter cell cycle during proliferation, but mature neurons lose this ability, and if they are forced to reenter cell cycle they will follow a path to apoptotic cell death.
Destabilization of The Cytoskeleton
The integrity of the cellular cytoskeleton is critical for proper neuronal morphology and function. Actin is one of the major components of the cytoskeleton of all eukaryotic cells, and participates in important cellular processes, including cell signaling, motility, and cell division. It is also essential in the formation of dendritic spines. Isoflurane can lead to depolymerization of actin in neurons and astrocytes, initiating cytoskeletal destabilization, impairment of astrocyte morphologic differentiation and maturation, as well as neuronal apoptosis.119,123
Effects on the Developing Spinal Cord
Most animal studies have focused on the effect of general anesthetics and sedatives on the developing brain. However, it is important to also consider the developmental impact of anesthetics on the spinal cord. After 6 hours of 0.75% isoflurane with 75% nitrous oxide in 7-day-old rats, neuroapoptosis increased in the lumbar region of the spinal cord.100 Similar results were reported after 6 hours of isoflurane in a similar model, although 1 hour of isoflurane or spinal bupivacaine resulted in no neuroapotosis.124 Intrathecal ketamine causes neuroapoptosis in the developing spinal cord of 3-day-old rats, but not at 7 days of age.157 Preservative-free ketamine was associated with long-term alterations in spinal cord function and gait disturbances,157 whereas, in a separate study, even high-dose intrathecal morphine produced no signs of spinal cord toxicity.215
Putative Mechanisms for Neurotoxicity
The exact mechanisms that trigger the above responses to anesthetics and sedatives in the immature brain remain unresolved. Elucidating these mechanisms is critical in order to establish the relevance of these findings for pediatric anesthesia and neonatal critical care medicine, as well as to develop mitigating interventions, if necessary. The current, overarching hypothesis is that anesthetics and sedatives interfere with normal GABAA and NMDA receptor–mediated activity, which are the putative targets for unconsciousness, amnesia, and immobility,216 but are also essential for mammalian central nervous system development.9,217 Some have suggested that administering GABAA-receptor agonists and/or NMDA-receptor antagonists may cause abnormal neuronal inhibition during a vulnerable period in brain development, triggering apoptosis in susceptible neurons, which in turn leads to neurocognitive impairment and decreased neuronal density in adults.22,60,85,97 Other lines of evidence suggest that the NMDA receptor–blocking properties of ketamine may upregulate NMDA receptors, rendering the neurons more susceptible to excitotoxic injury caused by endogenous glutamate immediately after ketamine withdrawal.135,139 However, several observations violate both hypotheses; neuronal cell death has been reported during exposure to anesthetics and not only after their discontinuation. Moreover, several anesthetics with minimal NMDA-receptor interaction, such as propofol and barbiturates, have demonstrated robust neurotoxic properties, whereas the neurotoxic potency of the NMDA-antagonist xenon is limited, therefore casting doubt on receptor upregulation being the sole mechanism for anesthetic neurotoxicity. In terms of abnormal neuronal inhibition being the main trigger for apoptosis in developing neurons, GABAA-receptor stimulation indeed decreases neuronal activity in the mature brain; however, it also causes excitation in developing neurons,218 thereby contradicting the inhibition hypothesis. In immature neurons, intracellular chloride (Cl−) concentration is high, thus GABA-induced opening of Cl− channels allows this anion to exit the cell, leading to membrane depolarization. On the other hand, the intracellular Cl− concentration is low in mature neurons. When anesthetics open Cl− channels in mature neurons, ions enter the cell, thereby hyperpolarizing the membrane. This reversal of the cellular Cl− gradient occurs as a result of a switch from the immature Na+-K+-2Cl− cotransporter 1 (NKCC1) to the mature brain form, K+-Cl− cotransporter 2 (KCC2).219 Along these lines, studies in neonatal rats demonstrated excitatory properties in the brain and episodes of epileptic seizures during sevoflurane anesthesia in neonatal rats.204 Isoflurane has also been shown to cause an excessive release of Ca2+ from the endoplasmic reticulum via over-activation of inositol 1,4,5-trisphosphate receptors (InsP3Rs) in neonatal rats in vivo and in vitro.220 A similar mechanism may be linked to the production of Alzheimer-associated increases in β-amyloid protein levels after anesthesia.221 Moreover, whereas xenon and hypothermia cause neuronal inhibition, they do not appear to exacerbate isoflurane-induced neuronal cell death, as expected by cumulative neuronal inhibition, but rather significantly reduce it.94,101,207
Importantly, evidence indicates that equianesthetic concentrations of the three contemporary inhalational anesthetics cause similar degrees of neuroapoptosis, suggesting that it is the anesthetic depth and not the doses of the anesthetics that determines the cytotoxic potency.71 However, other studies have failed to link the anesthetic and the apoptotic mechanisms. Specifically, although racemic ketamine and (S)-ketamine both elicit their anesthetic effects via NMDA-receptor blockade, (S)-ketamine induces up to 80% less cell death in vitro when compared with equipotent doses of racemic ketamine.154 Moreover, concomitant administration of the GABAA-receptor antagonist gabazine did not attenuate neuroapoptosis induced by the GABA agonist isoflurane, whereas the α2-agonist dexmedetomidine did.73 Decreases in anesthetic-induced neuronal activity may therefore be less important than the disruption of the neuronal balance of excitation and inhibition, as demonstrated by a series of studies that examined anesthesia-induced dendritic morphologic changes in mice.70,152 Whereas simultaneous blockade of excitatory and inhibitory activity with tetrodotoxin did not lead to structural changes during synaptogenesis as would have been expected from a causative relationship between neuronal inhibition and structural damage, the administration of either GABAA-agonistic or NMDA-antagonistic compounds alone altered synaptogenesis.152
It is not entirely clear at this time whether cytotoxicity is a direct effect of the anesthetic itself, or of anesthetic byproducts, or by the metabolic acidosis and respiratory derangements that have been observed during anesthesia in small rodents.105,107,222 Hypercarbia can trigger widespread neuroapoptosis, even in unanesthetized neonatal rats exposed to increased partial pressures of carbon dioxide. Whereas apoptotic cell death was quantitatively indistinguishable from neurodegeneration in isoflurane-treated litter mates, which were also hypercarbic, adult neurocognitive impairment was only observed in the isoflurane-treated animals.107
Lastly, experimental models of neurodegeneration have implicated reentry of postmitotic neurons into the cell cycle, leading to cell death. Ketamine exposure has been found to induce aberrant cell cycle reentry, leading to apoptotic cell death in the developing rat brain.155
Specific Anesthetic and Sedative Agents
In order to provide a succinct overview of the available laboratory data, we briefly review the effects of each class of anesthetics separately. Although the effects of some anesthetics, such as ketamine and isoflurane, have been extensively studied on the developing brain, the effects of others, such as xenon and desflurane, have not. However, the current data suggest that all anesthetics exert deleterious effects to some degree, on the developing animal brain (see E-Table 23-1). It is important to appreciate that although the MAC values for inhalational anesthetics are fairly constant across species, equipotent dosing of intravenous (IV) medications is not. That is, the dose of most IV medications to effect sedation or anesthesia in animals is approximately 6- to 10-fold greater than that in humans. The dosing is further complicated by the different routes by which drugs are administered in neonatal animals, which include the subcutaneous and intraperitoneal (IP) routes, as opposed to the oral or IV routes in humans. The possible importance of the interspecies differences in the pharmacology of IV medications on neuroapoptosis and cognitive dysfunction has not been addressed.
Ketamine
Possibly the most frequently studied anesthetic is ketamine, an antagonist of the NMDA and glutamate receptor that also interacts with other cell membrane proteins, such as muscarinic and opioid receptors, as well as voltage-gated calcium channels. Ketamine’s properties, which include potent analgesia, dissociative anesthesia, and relative hemodynamic stability, have made it a popular choice for procedural sedation in children, as well as for induction of anesthesia in children with critical congenital heart disease or pulmonary hypertension.223–225 However, about 10 years ago, a seminal study that examined the effects of repeated IP injections of ketamine on the brain of neonatal rats observed widespread apoptosis.17 Seven injections of 20 mg/kg of ketamine, administered over a 9 hour period in evenly divided intervals, to 7-day-old rat pups caused a 3- to 31-fold increase in degenerating neurons, depending on brain region. This led to speculation that these changes might contribute to neuropsychiatric disorders.17 These initial findings for ketamine have been confirmed in more than fifty studies in small rodents, as well as nonhuman primates, both in vitro and in vivo (see E-Table 23-1). Several of these studies have identified relationships between neurodegeneration and dose, duration of treatment, as well as animal species and age during exposure. Single doses of up to 75 mg/kg or multiple IP injections up to 17 mg/kg/hr for 6 hours were not neurotoxic to neonatal rat brains,128 whereas single doses between 20 and 50 mg/kg were neurotoxic to neonatal mice brains.74,134 Six or seven repeated injections of 20 to 25 mg/kg consistently cause apoptosis in neonatal rat brains.17,128,131,149,150 Although the doses of ketamine in these small-animal models appeared to be excessive and plasma concentrations in the rodents were up to 7 times greater than those in humans,131 these increased doses are consistent with the requirements for IV anesthesia in small animals (refer to later section on interspecies comparison). Coadministration of midazolam, diazepam, propofol, or thiopental compounded the neuronal injury caused by ketamine.74,134,142 Studies in rats, mice, and nonhuman primates suggest that the susceptibility to ketamine-induced neurotoxicity is limited to a brief period after birth, with a maximum impact between 3 and 7 days of age in small rodents and less than 35 days of age in monkeys.17,139 Beyond these ages, ketamine-induced neuroapoptosis subsides. In addition to apoptosis, both small rodents and nonhuman primates that were anesthetized with ketamine have exhibited impaired learning tasks later in life.129,130,158
In summary, ketamine is the most frequently studied anesthetic, in terms to its neurotoxic effects, and has repeatedly been shown to cause widespread apoptosis (an effect that is exacerbated by the coadministration of other anesthetics), as well as neurologic impairment in adult animals exposed early in life. Importantly, long-lasting learning impairment has been demonstrated in nonhuman primates, the closest animal model to humans.158 However, these animals were anesthetized with ketamine for 24 hours to produce this effect, a duration that exceeds the usual clinical scenario. Furthermore, it is unclear whether the observed learning abnormalities could not be explained by a reduction in motivation to perform the learning tasks.158
Inhalational Anesthetics
The second most commonly studied class of drugs is the inhalational anesthetics (see E-Table 23-1). The anesthetics desflurane, sevoflurane, isoflurane, enflurane, and halothane exert their anesthetic properties predominantly by their agonistic effects on the GABAA receptor, but also by differing degrees on glycine, NMDA, acetylcholine, serotonin (5-HT3), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors. Whereas GABA represents the main inhibitory neurotransmitter in the adult central nervous system, it has excitatory properties in the developing brain,218 which may have implications for neurotoxicity, as discussed previously. Most studies of inhalational anesthetics examined either isoflurane alone or a combination of midazolam, isoflurane, and nitrous oxide. This combination of GABA agonists and NMDA antagonists has been repeatedly found to cause widespread increases in brain cell degeneration in neonatal animals.85,87,91,92 In addition to the immediate deleterious effects on brain structure, long-term abnormalities in spatial learning tasks and decreased neuronal cell density in adult rats have been observed after exposure to this anesthetic combination early in life.85,97 One MAC of isoflurane administered as the sole anesthetic for 4 hours to neonatal rats led to neurocognitive deficits in rats when they matured to adults,106,107 whereas up to 0.6 MAC for 6 hours in neonatal mice, while causing widespread neuronal degeneration immediately after exposure, failed to cause neurocognitive deficits or decreases in neuronal density in adulthood.105,115 These inconsistent findings raise the question of whether neonatal neuronal apoptotic cell death is linked directly to behavioral and learning abnormalities in adulthood. Neuronal cell death has also been observed in neonatal rhesus monkeys after 5 hours of 0.75% to 1.5% isoflurane,114 although long-term neurologic studies in this species have not yet been published.
Similar to isoflurane, sevoflurane has also been shown to induce neuroapoptosis in neonatal mice,71,115,202 although the long-term effects on learning and behavior are conflicting.72,115,202 To date, few studies have examined desflurane in this context. Desflurane causes age- and species-dependent neuronal cell death in 7-day-old mice, but not 16-day-old rats.70,71
Few studies have compared the neurotoxicity of contemporary anesthetics. At equianesthetic concentrations of desflurane, isoflurane, and sevoflurane in neonatal mice, the degree of neuronal degeneration in the superficial neocortex, a brain region significantly affected in this model, is similar.71 These results contrast with those from a study of lower concentrations of sevoflurane and isoflurane, in which cell death was less after the former than the latter, but without differences in neurocognitive performance in adult rats.115 In another comparative mouse study, desflurane caused a greater degree of neurodegeneration than isoflurane and sevoflurane, and long-term neurologic impairment only occurred after desflurane.72 The significance of these differential findings remains unknown, but could be related to methodologic differences.
Although largely phased out from clinical pediatric anesthesia practice, halothane and enflurane have been shown to induce brain abnormalities. These anesthetics were studied in models of intermittent or chronic occupational exposure during pregnancy, and caused subsequent delayed synaptogenesis and behavioral and learning abnormalities in rats after fetal exposure.13–1676
Nitrous Oxide
Nitrous oxide, an NMDA antagonist, is the oldest anesthetic in clinical use, although its low potency (MAC of 115% in adult humans) necessitates the coadministration of other anesthetics to provide surgical anesthesia. Anesthetic combinations with nitrous oxide often include the GABA agonist midazolam, and the mixed GABA-agonist/NMDA-antagonist isoflurane.* In rats, nitrous oxide alone did not induce neuronal apoptosis,85,87,94 whereas in an in vitro study, it did cause neuronal cell death in mouse hippocampal slices.94 When administered in combination, however, nitrous oxide has been shown to exacerbate neuronal cell death induced by isoflurane, and also to contribute to long-term neurologic abnormalities in rats when combined with isoflurane and midazolam.85
Xenon
Due to the cost differential between other inhalational anesthetics and this rare, colorless, and odorless noble gas, xenon has not reached widespread clinical anesthesia practice, despite its NMDA-antagonistic, anesthetic properties.226 Xenon has a relatively low anesthetic potency, with a MAC measuring between 65% and 70% in adults227,228; its low blood-gas solubility speeds emergence from anesthesia.229 Xenon’s effects on neuronal apoptosis have been examined by two groups, with slightly differing results. Although 75% xenon for 6 hours did not cause neuronal apoptosis in 7-day-old rat pups,94 70% xenon for 4 hours did increase neuroapoptosis in 7-day-old mice.207 Interestingly, both studies demonstrated that xenon decreased the neurodegeneration induced by isoflurane anesthesia,94,207 which may have relevance to the phenomenon’s putative mechanism, as discussed.
Benzodiazepines
Benzodiazepines, such as clonazepam, diazepam, and midazolam, have been investigated regarding their effects on the immature brain, either alone or in combination with other drugs. These GABA agonists are frequently used in toddlers and older children for preoperative anxiolysis, but infrequently in neonates and infants. Studies have shown that they increase neuronal degeneration in small-animal models, depending on the dose, region of the brain, species, and age of the animal. Although single doses of 5 mg/kg of diazepam or 9 mg/kg of midazolam IP did not increase neuronal cell death in neonatal rats,68,85 5 mg/kg subcutaneous diazepam did cause neuronal cell death in some brain regions in mice, though this was not associated with learning deficits in adults.74 In these studies, the neuroapoptosis associated with diazepam was significantly augmented by the coadministration of other sedatives, such as ketamine.74 Studies have consistently reported increased neuroapoptosis in neonatal rats after diazepam in doses of 10 mg/kg or greater,68,69 an effect that was prevented by the coadministration of the benzodiazepine-antagonist flumazenil in one study.68 Two studies reported no neurocognitive learning disabilities in adult mice after they were sedated with diazepam or midazolam as neonates.74,166
Chloral Hydrate
The sedative chloral hydrate, a chlorination product of ethanol that is a GABA agonist as well as NMDA antagonist, has been largely supplanted by barbiturates and benzodiazepines in pediatric clinical practice. However, it is still being used in doses of up to 120 mg/kg for sedation for radiology studies,230 and its neurotoxic properties have been investigated. Preliminary results indicate that it causes neuroapoptosis in the cerebral cortex and the caudate-putamen complex in immature mouse pups in doses of 100 mg/kg or greater.67 The neurofunctional outcome in adult mice, however, has not as yet been investigated.
Barbiturates
Barbiturates act largely via the GABAA receptor, but also exert effects via nicotinic acetylcholine, AMPA, and kainate receptors. Thiopental alone, in doses of 25 mg/kg subcutaneously in neonatal mice, did not induce apoptosis, although when doses of 5 mg/kg were administered in conjunction with 25 mg/kg of ketamine subcutaneously, neuronal degeneration occurred and was associated with long-term impairment of learning and memory.142 Pentobarbital and phenobarbital have been shown to induce neurodegeneration in mouse and rat pups. Furthermore, after receiving these sedatives as neonates, long-term alterations in brain protein expression and learning and memory have been observed,103,184,186 although in one study these long-term alternations may be attributed in part to hypoxia and hypercarbia during the neonatal sedation.184 Interestingly, estradiol has been shown to attenuate phenobarbital-induced neuroapoptosis.68,185
Propofol
In recent years, propofol has supplanted the barbiturates as the IV induction agent of choice in children. Propofol predominantly acts via GABA and glycine receptor–agonistic properties, but also weakly on nicotinic, AMPA, and NMDA receptors, and has been repeatedly studied regarding its neurotoxic profile, both in vitro and in vivo. The overall consensus of this body of literature is that propofol, in a dose- and exposure time–dependent fashion, has dramatic effects on the developing brain in animals. Propofol has consistently caused neuroapoptosis after doses exceeding 50 mg/kg (subcutaneous or IP) or repeated doses exceeding 20 mg/kg/hr for 4 to 5 hours in neonatal small rodents.142,148,191,196 Interestingly, lithium protected from propofol-induced neuroapoptosis in neonatal mice.148 However, after 24 hours of IV anesthesia with propofol (6 mg/kg/hr) and fentanyl (10 µg/kg/hr), there was no evidence of apoptosis.192 Apart from overt neuronal cell death, propofol decreases the GABAergic enzyme glutamic acid decarboxylase,188 decreases nerve growth factors,194,198 and causes neurite growth cone collapse in tissue culture.190 In addition, propofol alters dendritic spine architecture in developing rats, depending on the age at the time of anesthetic exposure.197 For example, dendritic spine density decreased if the rat was anesthetized during week 1 of life, but it increased spine development if exposure took place during week 3 of life; the mechanism of these differing responses remains elusive.197
Dexmedetomidine
Unlike other anesthetics and sedatives, dexmedetomidine is a sedative and analgesic that does not interact with GABA, NMDA, or opioid receptors, but rather acts by stimulating presynaptic α2-adrenergic receptors. Given this different mechanism of action, it is interesting that dexmedetomidine, administered in three IP injections of 1 to 25 µg/kg at 0, 2, and 4 hours to 7-day-old rat pups, did not cause neuronal apoptosis, but attenuated isoflurane-induced neuroapoptosis. Even a dose of 75 µg/kg dexmedetomidine (75-fold greater than the median effective dose needed for sedation), did not cause apoptosis in this model. Furthermore, dexmedetomidine prevented the long-term cognitive impairment from isoflurane.73
Opioid Analgesics
To date, only one study investigated neurotoxic properties of opioids in relation to an inhalational anesthetic regimen. Mechanically ventilated, neonatal pigs, sedated with an IV bolus of 30 µg/kg fentanyl followed by 15 µg/kg/hr for 4 hours, exhibited significantly less neuroapoptosis in several regions of the brain, compared with an intramuscular injection of 1 mg/kg of midazolam, followed by 4 hours of 0.55% isoflurane and 75% nitrous oxide.79 These initial findings are interesting, although future neurotoxicity studies that include opioid infusions need to also include adjuvants that produce amnesia, as is customary in pediatric anesthesia practice. Moreover, when comparing different anesthetic regimens regarding their neurotoxic potential, equianesthetic potency should be established during the study.
Importantly, however, the long-term consequences of opioid administration to the immature brain needs to be further elucidated before recommending such a regimen as an alternative strategy. Similar to GABA and NMDA receptors, opioid receptors are also intimately involved in early brain development and synaptogenesis,231,232 which would make it plausible that opioids could similarly affect brain development during the critical period of synaptogenesis. Increased neuronal cell death and decreased neuronal density after perinatal exposure to opioid receptor agonists, such as morphine and heroin, have been observed in developing animals.82,83,233 Chronic buprenorphine and methadone treatment early in life have been found to diminish concentrations of nerve growth factors in the immature brain.66,164 Moreover, immediate and permanent reductions in µ-opioid receptor density have been observed after perinatal exposure to morphine,168,169 and may be associated with long-term impairment of memory and cognitive function in small animals,171,174–176,179–181 as well as exaggerated nociceptive responses to a pain challenge later in life.177,182 Stimulation of the κ-opioid receptor may amplify neuronal cell death induced by proapoptotic agents.234 High-dose fentanyl has been shown to significantly exacerbate white-matter brain lesions induced by glutamatergic overstimulation in mice.78
[/not-level-membership-for-anesthesiology-category]
