Chapter 56 Supratentorial Tumors in the Pediatric Population
Multidisciplinary Management
Epidemiology
In the pediatric population, primary central nervous system (CNS) malignancies are the most common solid organ tumor, totaling approximately 20% of cancers in children ages 1 to 4 years and approximately 30% in children ages 5 to 9 years. In the United States, there are more than 3700 newly diagnosed cases of CNS tumors per year in children below 19 years of age. With a 66% 5-year survival rate for this age group, it is estimated that 26,000 children in the United States have a CNS tumor. The incidence rate for CNS tumors is higher in males than in females (29.9 vs. 25.1 per million) and is greatest in Caucasian individuals (29.4 per million). Based on the World Health Organization (WHO) classification system, 75% of all primary childhood CNS tumors are of neuroepithelial origin (gliomas). These include astrocytic tumors, embryonal CNS tumors, neuronal and mixed neuronal–glial tumors, and ependymomas. Ependymomas account for 15% of nonastrocytic neoplasms in the pediatric supratentorial space. Supratentorial tumors constitute 31% of pediatric CNS tumors (Table 56-1).1,2
Table 56-1 Breakdown of the Anatomic Location of Pediatric Supratentorial Tumors
| Supratentorial Brain Lesions | Patients |
|---|---|
| Temporal lobe | 7% |
| Frontal lobe | 6% |
| Multilobar | 6% |
| Ventricle | 6% |
| Parietal lobe | 4% |
| Occipital lobe | 2% |
| Total patients | 31% |
Matula C. Tumors of the pineal region. In Rengachary SS and Ellenbogen RG (eds), Principles of Neurosurgery, 2nd ed. Philadelphia, Elsevier, 2005.
Clinical Presentation
The clinical presentation is often determined by the tumor type and location, because symptoms are produced by local invasion, compression of adjacent structures, and increased intracranial pressure. More than 50% of patients have had symptoms for 6 months or longer at the time of diagnosis.3 They can be broadly categorized into generalizing and localizing symptoms. Generalizing symptoms are due to increased intracranial pressure, often a result of blockage of normal cerebrospinal fluid (CSF) flow. The most common generalizing symptoms for childhood CNS tumors include headache, nausea and vomiting, and lethargy.4 Localizing symptoms result from direct irritation, destruction, or impairment of neuronal function caused by the tumor. These include seizures, endocrinopathies, hemiparesis, cranial nerve deficits, and visual field defects5 (Table 56-2).

When considering the youngest pediatric patients, the constellation of symptoms differs because children are often unable to articulate their complaints. In this case, a thorough history with the caregiver and a physical exam are essential to uncover subtle and nonspecific signs. For children under the age of 4, the most commonly found symptoms include macrocephaly, nausea, vomiting, irritability, and lethargy.4
In the case of supratentorial tumors, symptoms are generally nonspecific and children typically have a delayed diagnosis when compared to those with infratentorial tumors. However, seizures are often seen in low-grade supratentorial lesions, either as an isolated finding (30% of patients) or with other symptoms, such as changes in personality, cognition, and development.2
Though headache is the most common manifestation of CNS tumors in children, its character is nonspecific and can vary from focal to diffuse. Thus, headaches in pediatric and adolescent patients are often attributed to benign causes, including migraines or tension headaches. However, on closer examination, these patients often present with other neurologic symptoms. Therefore, children with headaches in the setting of another neurologic deficit require further evaluation with neuroimaging. The options for imaging children include ultrasound if the fontanelle is open; x-ray, which can be of limited use in select cases; computed tomography (CT); magnetic resonance imaging (MRI); and if vascular abnormalities are suspected, angiography. The use of CT scans should be limited in the pediatric population, given the increasing evidence for radiation-induced malignancies that occur many years after image acquisition.6 If neuroimaging does confirm the presence of a mass, tissue diagnosis is often necessary to determine the appropriate treatment regimen.4
Low-Grade Glioma
Background
Pediatric low-grade gliomas, consisting of WHO grade I to II tumors, are a heterogeneous group of lesions that include pilocytic astrocytomas, oligodendrogliomas, and mixed neuroglial tumors, with pilocytic astrocytomas predominating. The overall prognosis is favorable with an indolent course and a remote risk of malignant transformation.5 While rare, there have even been case reports of spontaneous regression of low-grade astrocytomas.7,8 Low-grade astrocytomas account for 30% to 50% of all pediatric CNS tumors,9 with hemispheric (10%-15%), deep midline (10%-15%), and optic pathways (5%) being the most common locations.
The two most common pediatric low-grade gliomas are pilocytic astrocytomas (WHO grade I) and diffuse fibrillary astrocytoma (FA) (WHO grade II). Pilocytic astrocytomas typically occur in children between the ages of 5 to 19, whereas only 10% of FAs occur before the age of 20.5 Other less common pathologies include pilomyxoid astrocytoma, pleomorphic xanthoastrocytoma (PXA), ganglioglioma, subependymal giant cell astrocytoma, and oligodendroglioma. Pilomyxoid astrocytomas (WHO grade II) were first introduced in 1999; tend to occur early in life, with a median age of 18 months; and typically occur in the hypothalamus.10 Gangliogliomas and PXAs, both WHO grade II, typically occur in the temporal lobe and can present with seizures. Subependymal giant cell astrocytomas are WHO grade I lesions that occur almost exclusively in patients with tuberous sclerosis. Oligodendrogliomas (WHO grade II) are rare and account for less than 5% of brain tumors in children. When they do occur, they typically are found in the frontal lobe.5
The molecular triggers that lead to formation of low-grade gliomas are poorly understood. Few recurrent genetic changes have been described. Karyotype analysis has only revealed gain of chromosome 7 in a minority of tumors.11,12 Recently, groups have identified recurrent duplications in 7q34 in a large fraction of pilocytic astrocytomas, implicating BRAF and the RAS/RAF/MEK pathway in the pathogenesis.13,14
Pilocytic astrocytomas are the most common glial tumor in the pediatric patient and can occur anywhere in the CNS. The most common supratentorial location is the optic pathway, contributing to approximately 60% of optic pathway gliomas.5 These lesions are typically diagnosed early, with 75% of optic pathway gliomas being diagnosed before 10 years of age. More than 70% of children with optic pathway gliomas have neurofibromatosis type 1 (NF1), and many of those with NF1 develop bilateral optic nerve lesions.15
Optic Pathway Gliomas
Epidemiology
Optic nerve gliomas comprise approximately 1% of all intracranial tumors, are most commonly unilateral, and occur more frequently in females.16,17 While they can present at any age, 75% become symptomatic in the first decade of life and 90% become symptomatic before age 20.18 Rush et al. reported that in their series of 33 patients that the median age at diagnosis was 6.5 years and the mean age was 10.9 years, with a range of 2 to 46 years.19 The signs and symptoms of optic nerve gliomas typically consist of decreased visual function, proptosis, optic disc swelling, and strabismus.17 Patients can have vascular compromise of the optic apparatus from chronic compression of the central retinal vein, leading to occlusion, venous stasis retinopathy, optociliary shunt vessels, or neovascular glaucoma.17 In rare cases, acute loss of vision can occur in the setting of tumor hemorrhage. Patients typically do not present with orbital or ocular pain.17
A firm link has been established between NF1 and optic pathway gliomas. The incidence of NF1 among patients with optic nerve or chiasmal gliomas has ranged from 10% to 70% based on the series, while the incidence of optic nerve glioma in patients with NF1 varies from 8% to 31%.17,20
Diagnosis
The diagnosis of an optic nerve glioma can largely be made by either CT or MRI. The radiographic appearance varies depending on whether the person has NF1. In patients without NF1, the optic nerve is almost always enlarged in a fusiform manner with a clear-cut margin produced by the intact dural sheath. In patients with NF1, the nerve is irregular, with kinking and buckling secondary to the mass.17 On MRI, the tumor is typically hypo- to isointense on T1-weighted imaging and hyperintense on T2-weighted imaging. After intravenous administration of gadolinium-based contrast agents, the glioma may not enhance, have patchy enhancement, or demonstrate avid enhancement (Fig. 56-1).21 Additionally, MRI imaging may demonstrate abnormalities that extend beyond the optic nerve into the chiasm.17 Patients may have an enlarged optic canal ipsilateral to the side of the lesion, but this does not always indicate extension of the tumor intracranially. Arachnoid hyperplasia may be sufficient by itself to cause canal enlargement. Conversely, a normal caliber optic canal does not rule out the possibility of tumor extension beyond the orbit.17
The natural history of optic nerve gliomas is usually benign, with most growing slowly in a self-limited manner or, in extremely rare cases, spontaneously regressing.8 As a result, many patients retain excellent or maintain existing visual function without treatment. Rarely, WHO grade III and IV lesions arise from the optic nerve and result in rapid visual loss, neurologic deficit, and eventual death. These high-grade lesions almost always occur in adults.22
Pathology
The gross appearance of an optic nerve glioma often causes diffuse expansion of the nerve that may extend the entire length of the nerve or affect only a portion. The expanded portion can be solid or gelatinous, with hemorrhagic and necrotic regions. In children with NF1, the tumor not only expands the nerve but often breaks through pia mater and enters the subdural space.17,23 However, as long as the glioma lies within the confines of the orbit or optic canal, the tumors are typically limited to within the optic dural sheath. If the glioma extends intracranially, it can remain intraneural or become an expansile mass that can compress adjacent structures, including the chiasm or contralateral optic nerve.17
The most common type of optic nerve tumor is the pilocytic astrocytoma, which has three predominant histologic patterns: (1) coarsely reticulated, (2) finely reticulated, and (3) coarsely fibrillated or spindle cell.21 The most common pattern is coarsely reticulated, with a biphasic pattern histologically of coarse bipolar astrocytes that are either tightly compacted around blood vessels or loosely associated around microcystic spaces. These tumors frequently have Rosenthal fibers and eosinophilic granular bodies. Finely reticulated pilocytic astrocytomas can be confused with WHO grade II diffuse low-grade astrocytomas and demonstrate an expansion of the indigenous neuroglia of the optic nerve. Within the finely reticulated tumors, a delicate reticulated syncytium of neuroglia fibers is embedded among multiple, small, round or ovoid nuclei. The coarsely fibrillated variant consists of coarse neuroglial fibrils and spindle cells arranged in bundles and is more often seen in adults.21
Management
The three major treatment modalities are surgery, chemotherapy, and radiation therapy. Surgery still plays a role in the management of optic pathway gliomas, especially in individuals without NF1 for whom MRI findings are not compelling and tissue diagnosis is required.24–26 Often in those cases, stereotactic or open biopsies can be safely performed. However, in cases where a large tumor creates mass effect or hydrocephalus, recurrent tumors refractory to chemo- or radiotherapy or cystic lesions with compression of the optic pathway can benefit from more radical surgery.25,26 Advocates of aggressive surgical resection argue that radical tumor excision allows long-term remission, given the slow-growing nature of pilocytic astrocytomas. Additionally, it could delay the initiation of radiation therapy, which has significant long-term sequelae in children. While some studies have demonstrated that aggressive resection can be safely performed if done carefully,27,28 the outcomes after surgery remain variable. For example, Sawamura et al. reported on the visual outcomes after 26 cases treated surgically with the intent to debulk as much tumor as possible.25 Of this group, 20 patients underwent radical resection with greater than 90% tumor removal, and the remaining 6 patients had partial resection. Of the 26 children, only 2 had visual improvement, 2 remained static, 10 remained blind, and 12 worsened.
Thalamic Glioma
Pediatric thalamic gliomas arise as primary gliomas or secondary gliomas from adjacent structures, including the cerebral hemispheres, caudate nuclei, brain stem, or pineal gland. The deep, central location of the thalamus makes surgical treatment of these tumors difficult without significant patient morbidity and mortality. Even though thalamic gliomas are rare and account for 1% to 1.5% of all CNS tumors, both low-grade, well-circumscribed lesions and high-grade, diffuse, infiltrating gliomas have been described.29 Although pilocytic astrocytomas classically occur in the hypothalamic/optic pathway, they can also occur as unilateral thalamic masses. Higher-grade FAs also occur in this region and are typically unilateral, with little involvement of the visual pathways. Finally, bilateral infiltrating astrocytomas, a rare variant of FAs, are lesions in bilateral thalami that have a poor outcome due to their intrinsic aggressive biology and the difficulty in attaining adequate surgical debulking of the tumor, resulting in mass effect on the thalamus.30,31 Treatment considerations concerning the necessity, feasibility, and safety of surgery in children with these lesions are varied.
Epidemiology
Thalamic gliomas are rare and account for 0.84% to 5.2% of pediatric intracranial tumors.29 This discrepancy in incidence is due to difficulty in differentiating primary thalamic tumors from secondary lesions that grow to involve thalamic structures. In a retrospective analysis of 69 children with thalamic tumors, 32 had low-grade thalamic tumors and 22 had high-grade tumors; the remaining 15 had 9 bilateral thalamic and 6 thalamopeduncular tumors. It was found that low-grade lesions had statistically improved survival rates, particularly when patients had symptom duration longer than 2 months and tumor excision greater than 90%.32
Pathology
Primary thalamic gliomas include pilocytic astrocytomas, FAs, and bilateral infiltrating astrocytomas. Pilocytic astrocytomas are low-grade gliomas, with histologic grade I. FAs are a heterogeneous group of astrocytic cells and are graded by the WHO from grade II to grade IV. They are prone to malignant progression. Bilateral infiltrating astrocytomas are a rare variant of FAs histologically that are highly infiltrating but well-differentiated grade II tumors.30 They are extremely rare, and though benign and well differentiated, patients have a poor prognosis due to thalamic nuclei involvement and inadequate surgical resection.31 It is unknown whether these bilateral tumors arise from one side of thalamic nuclei and cross the midline to the other or arise independently from tumors in the subependymal region of the third ventricle.
Diagnosis
Clinical signs and symptoms include weakness, increased intracranial pressure, motor deficits, seizures, ophthalmologic abnormalities, and in some cases, mental deterioration and personality change. It has been reported that the majority of patients have symptoms of increased intracranial pressure, headache, and vomiting.33 Though the thalamus plays an important functional role in movement, only 12% of patients have movement disorders such as tremors and dystonia. More commonly, 30% of patients present with epilepsy, despite the tumor’s deep-seated location. Another common finding in thalamic lesions includes contralateral paresis that affects one or both extremities.34 In addition, clinical presentation is correlated with tumor location. Specifically, tumors of the anterolateral thalamus are associated with sensation deficits and paresis, while those of the posteromedial thalamus are associated with early hydrocephalus.35 The presentation can also include deficits in vision such as hemianopia, a mitotic poorly reactive pupil, and ipsilateral oculomotor palsy.34
Patients typically undergo MRI prior to treatment and during follow-up. Thalamic lesions are heterogeneous and often cystic, with calcification, edema, and contrast enhancement on T1- and T2-weighted MRI32 (Fig. 56-2). The most common neuroimaging finding is hydrocephalus. Imaging is helpful in distinguishing between pilocytic astrocytomas and more infiltrative FAs. Pilocytic astrocytomas are characteristically well circumscribed, solid, and contrast enhancing. Grade II FAs are diffuse and hyperintense on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences, and grade IV FAs enhance with contrast. Bithalamic infiltrating astrocytomas present as symmetrical T2 signal hyperintensity in both thalami.36
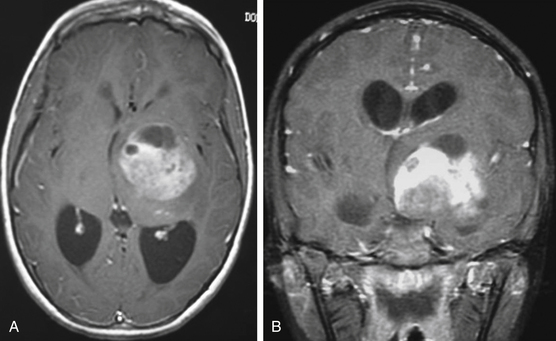
Management
The correlation between extent of resection and prognosis of a particular tumor type is of considerable importance. Historically, biopsy with irradiation was the standard of treatment due to patient morbidity and mortality with surgery. However, in low-grade thalamic tumors, radical resection can be curative. Consensus within the literature for low-grade astrocytomas includes stereotactic biopsy to verify histology, followed by surgery for curative intent. In a study examining the mortality rate of complete resection of benign thalamic astrocytomas in 26 children, the procedure was found to be associated with improved prognosis when postoperative MRI showed no residual tumor but was associated with a 7.7% mortality rate.37 Surgical resection of bilateral infiltrating astrocytomas of the thalamus is difficult to achieve, and biopsy for histologic diagnosis is preferred. These patients have a poor outcome despite adjuvant radiation and chemotherapy.
Anatomic considerations are important because the thalamus is adjacent to critical structures. The thalamus is bound anteriorly by the foramen of Monro and posteriorly by the posterior commissure. It also is bound superiorly by the stria medullaris and inferiorly by the hypothalamic sulcus. Finally, the thalamus is medially bound by the third ventricle and laterally bound by the internal capsule. The terminal vein lies in the groove between the thalamus and the caudate and serves as a landmark for the position of the internal capsule.34 Factors that must be considered by neurosurgeons include MRI appearance of the tumor, extent of tumor involvement, regions of enhancement, evidence of hydrocephalus, and relationship of the tumor to vital vascular structures.34 Surgical approaches that have been used with endoscopic or stereotactic guidance include transcallosal–interhemispheric, infratentorial–supracerebellar, and trans-sylvian transinsular approaches.34 Surgical resections can be performed while minimizing morbidity using concomitant neuronavigation and intraoperative neuromonitoring of the corticospinal tracts.
If the patient is in extremis because of elevated intracranial pressure secondary to hydrocephalus, the surgeon should perform emergent CSF drainage. The most rapid method is to place an extraventricular drain (EVD). If surgery for gross total resection is planned, the surgeon can attempt to wean the EVD postoperatively because the normal CSF flow is sometimes restored. If only biopsy is planned or a CSF blockade persists, the surgeon can perform an endoscopic third ventriculostomy or place a ventriculoperitoneal shunt.38
Postoperative imaging is obtained to evaluate the extent of resection and residual tumor. Adjuvant radiation and chemotherapy for high-grade gliomas are required. In a study of 57 patients, despite surgical resection and multimodal therapy, the median survival length remained 73 weeks.33 In contrast, children who have low-grade gliomas that have been resected in their entirety can be observed serially for tumor recurrence. Upward of 90% 10-year survival rates have been reported for low-grade thalamic gliomas. For partially resected or unresectable low-grade thalamic tumors, the timing of additional therapy is controversial. Most surgeons advocate a “wait and see” approach. Often adjuvant therapy can be delayed until signs of clinical worsening or radiographic growth. In one series looking at 128 children with subtotal resection of low-grade gliomas, 58% had no evidence of tumor progression 7 years after diagnosis despite undergoing no additional therapy. Furthermore, there was no difference in survival in those children that did receive immediate postoperative radiotherapy and those that not.39
Given the increasing data regarding the detrimental effects of radiation on children, many centers recommend chemotherapy as frontline adjuvant therapy in children with progressive low-grade gliomas. One combination that has shown promise is a combination of carboplatin and vincristine, which has been shown to generate a 68% 3-year progression-free survival rate.40 Unfortunately, 40% of children experience hypersensitivity reactions with carboplatin. These patients are often started on a regimen of 6-thioguanine, procarbazine, lomustine, dibromodulcitol, and vincristine, which has shown a 45% 3-year progression-free survival rate.40
Pediatric Glioblastoma
Glioblastoma multiforme (GBM) are WHO grade IV gliomas, which are rarely diagnosed in the pediatric population but cause significant morbidity and mortality. While GBMs are primarily an adult-age tumor, pediatric GBMs are also classified as high-grade gliomas, along with anaplastic astrocytomas and intrinsic pontine gliomas.41,42 Anatomically, the majority of these tumors are located supratentorially, but GBMs also occur in the brain stem.43 The current treatment recommendations include aggressive surgery, radiotherapy, and chemotherapy.
Epidemiology and Prognosis
Pediatric GBMs compose 3% of childhood primary brain tumors and are rarely diagnosed among children and adolescents. Overall, the prognosis of glioblastoma is better in children than in adults.44 The majority of patients experience recurrence after surgical resection within 2 years of diagnosis, with the overall survival rate at 67% 1 year, 52% 2 years, and 40% 5 years after diagnosis.44,45 The median overall survival length is 43 months, with a median progression-free survival length of 12 months.44 Survival is correlated with tumor location, and patients with superficially located GBM had a median survival length of 52 months, while those with deeply located tumors had a median survival length of 7 months.44 Furthermore, there is no significant relationship between patient age and survival.45 Histologic features, while prognostically significant in adults, have been found to have no association with survival in pediatric patients.45 Extent of resection was found to be a significant predictor of outcome. Radical tumor resection, defined as resection of greater than 90% of the tumor by postoperative imaging, was found to have a survival advantage when compared to partial resection in pediatric patients.42 Specifically, the median survival length for patients with completely resected tumors was 106 months, while those with incompletely resected tumors had a median survival length of 11 months.44
Pathology
GBMs are classified as WHO grade IV infiltrating astrocytomas. Pathologic diagnosis is based on histologic features that include pleomorphism, mitotic figures, endothelial proliferation, and tumor necrosis.46 Unlike adults, histologic grading has not been found to have a strong prognostic significance for children, as the difference in overall progression-free survival of pediatric glioblastoma and anaplastic astrocytoma is not statistically significant.47 Individual genetic alterations such as epithelial growth factor receptor (EGFR) amplification, p53 mutations, retinoblastoma loss, IDH1 mutations, and deletion of phosphatase and tensin homology (PTEN) are common in adults; however, such alterations are distinctly different in pediatric GBMs.46 In pediatric GBMs examined in the Children’s Cancer Group cohort of 945 patients, EGFR, PTEN, and IDH1 gene expression aberrance were rare in pediatrics, while p53 alterations were more frequent.47,48 In addition, overexpression of O-6-methylguanine-DNA methyltransferase, which is an important mechanism of resistance to temozolomide, is limited in pediatric glioblastomas and has driven investigation of the use of temozolomide in children.49 Pediatric GBMs are molecularly distinct from their adult counterparts and warrant therapies specifically directed toward these molecular targets.
Diagnosis
Presenting signs and symptoms vary by location and age. The history may include a short evolution of symptoms on the order of days and weeks to months.42 Cortical lesions may be manifested through seizures, hemiparesis, visual deficits, and headaches. Indications of obstructed CSF leading to hydrocephalus include morning headaches, vomiting, and papilledema.42 Infants commonly present with nonspecific symptoms, including irritability, lethargy, vomiting, failure to thrive, and macrocephaly. Nonspecific symptoms coupled with a neurologic deficit are particularly suspicious for a brain lesion in young children.
MRI is the preferred diagnostic imaging modality to determine whether symptoms are consistent with high-grade glioma. Imaging including axial, sagittal, and coronal planes pre- and postgadolinium should be obtained. T1 sequence characteristics include irregular margins, enhancement with contrast, invasion of surrounding brain parenchyma, heterogeneous enhancement patterns, and areas of cystic necrosis with a rim of enhancement (Fig. 56-3). Edema is commonly evidenced on FLAIR or T2. When imaging is consistent with high-grade glioma, biopsy—at a minimum—is indicated for tissue diagnosis. Stereotactic biopsy is appropriate for deep-seated tumors in functionally important areas.42 Further surgical management, including open resection, is dictated by tumor location.
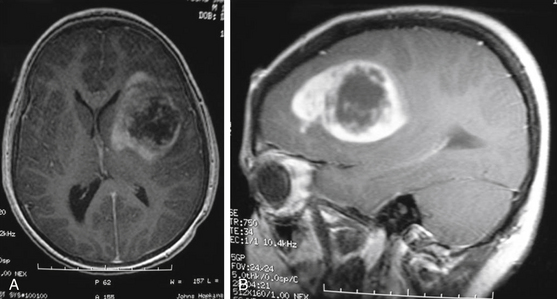
Management
Therapy for pediatric GBMs consists of surgery, radiotherapy, and chemotherapy. Complete or near-complete surgical resection is recommended when operative risks are acceptable, because gross total resection of the tumor significantly improves prognosis compared to subtotal resection.42 In 131 children with GBM treated with surgery, as well as chemotherapy and radiation, patients undergoing gross total resection had a 5-year progression-free survival rate of 26% compared to a rate of 4% in patients with subtotal resection.50 Because of the survival advantage for radical tumor resection, surgery with the intent of providing maximal safe resection while preserving normal or near-normal neurologic function should be attempted. However, in diencephalic tumors, more conservative management is warranted.
Focal radiotherapy is commonly utilized, because progression in the majority of patients occurs locally.45 Specifically, doses between 54 and 60 gray is delivered to a target volume of the tumor bed and additional margin for tumor infiltration.51 Use of fractionated radiotherapy, brachytherapy, stereotactic radiosurgery, and gamma knife radiosurgery allows for sparing of uninvolved surrounding tissues. Irradiation of the entire neuraxis is reserved for leptomeningeal spread, which occurs in 10% to 31% of patients at the time of progression.45 Radiation therapy is primarily indicated for pediatric brain tumors in patients older than 3 years. For children younger than 3 years, multiagent chemotherapy is recommended to delay radiation therapy.52 In the Children’s Cancer Group study, 299 enrolled infants with malignant brain tumors were found to have a high response rate (42%) to intensified induction therapy, but unfortunately the 5-year event-free survival rate was only 27%.52
The role of multimodal therapy for pediatric GBM with chemotherapy is less clear, with one study demonstrating a long-term survival rate of less than 20% in a study of 58 pediatric patients, despite the addition of chemotherapy.51 The improvement in outcome with temozolomide in adult GBMs has spurred interest in its use in the pediatric population. The efficacy of temozolomide in children with high-grade gliomas has been found to vary, and in a phase II trial with temozolomide the median progression-free survival was found to be 3 months in 24 patients.53,54 New therapies, as well as clear data on chemotherapy, are needed to achieve improved outcome in children.
Pineal Region Tumors
Tumors of the pineal region make up 0.4% to 2% of all primary CNS tumors in children.55 Germ cell tumors, pineal parenchymal tumors, and astrocytomas make up the vast majority of lesions in this area. When considering all CNS germ cell tumors, two thirds occur in the pineal region.56,57
Presenting Signs and Symptoms
The most common symptoms of pineal region tumors are those related to hydrocephalus due to obstruction of the outflow of the third ventricle at the sylvian aqueduct.58,59 The symptoms of headache, lethargy, increasing head circumference, seizures, nausea, and vomiting are common in the early symptomatic period, but papilledema, subacute cognitive deficits, and obtundation can develop with worsening hydrocephalus. The pathognomonic constellation of signs known as Parinaud’s syndrome occurs in 50% to 75% of patients and refers to the triad of paralysis of upward gaze, retraction nystagmus, and near-light papillary dissociation occurring as a result of compression of the superior colliculus in the pretectal region.60,61 As the tumor involves greater ventral midbrain, there is a greater impairment of downward gaze.
Less common presentations occur in patients presenting late in their disease with large tumors and relate to infiltration or compression of adjacent structures. Compression of the periaqueductal gray region can cause mydriasis, convergence spasm, papillary inequality, and convergence of refractory nystagmus. Compression or infiltration of the cerebellar efferent fibers within the superior cerebellar peduncle can cause motor impairments such as ataxia and dysmetria. Infiltration of the thalamus or internal capsule can occur with large, invasive tumors and cause contralateral hemihypesthesia, paresthesia, and typical thalamic pain syndromes.62 Exceedingly rare signs include compression of the inferior colliculus, causing hearing deficits; compression of elevatory inhibitory fibers in the posterior commissure, causing lid retraction (Collier’s sign); and fourth nerve palsy, causing double vision.63,64 While rare in children, drop metastasis from CSF or extracranial metastasis can occur and present with radiculopathy; myelopathy, which is more commonly observed in pineoblastomas; and germ cell tumors.65,66
Children with tumors of the pineal gland can present with symptoms of endocrine dysfunction, including diabetes insipidus and pseudoprecocious puberty either primarily due to tumor involvement of the hypothalamus or secondarily as a consequence of hydrocephalus.67 Precocious puberty has historically been reported to occur in approximately 10% of males, but reexamination suggests true precocious puberty is rare and may be a uncommon finding. This occurs due to ectopic secretion of beta-human chorionic gonadotropin (β-hCG) by choriocarcinomas and germinomas. Alternatively, mass effect on the posterior diencephalon augments secretion of gonadotropins by blocking the inhibitory effect on the median eminence of the hypothalamus or by decreasing secretion of a substance in the pineal gland with antigonadotropic effect.62 This is termed pseudopuberty because the mechanism is secondary stimulation of the Leydig cell to secrete androgen, resulting in the development of secondary sexual traits prematurely.
Diagnosis
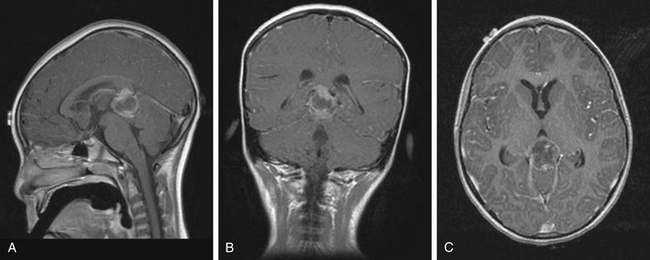
High-resolution MRI with and without gadolinium is important to appreciate tumor size, lateral and superior extension, contrast enhancement, marginal irregularities, and relationships with surrounding structures. Particularly important are the degree of involvement and position of the tumor within the third ventricle, extension into or above the corpus callosum, superolateral extension into the region of the ventricular trigone, involvement or compression of the quadrigeminal region, relation to the anterior cerebellar vermis, and location of the deep venous system. Angiography is not routinely recommended unless the MRI suggests potential vascular involvement with the Galen vein or an arteriovenous malformation. While imaging is not predictably diagnostic, radiologic patterns that are associated with specific tumor histology types can provide valuable information for preoperative planning and postoperative adjuvant management (Table 56-3).
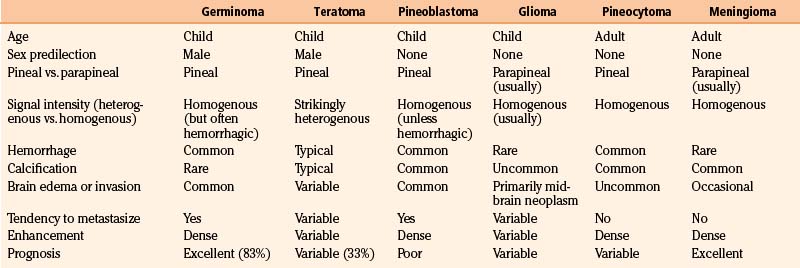
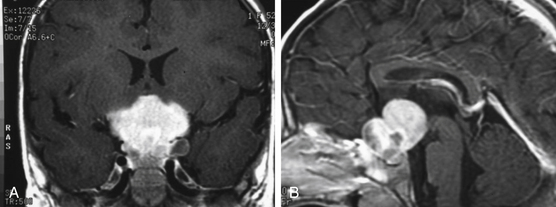
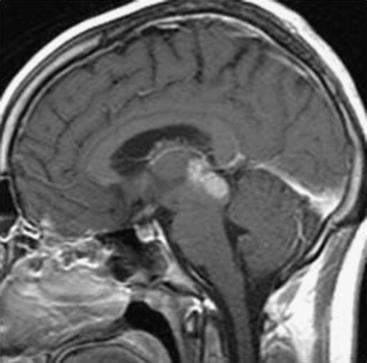
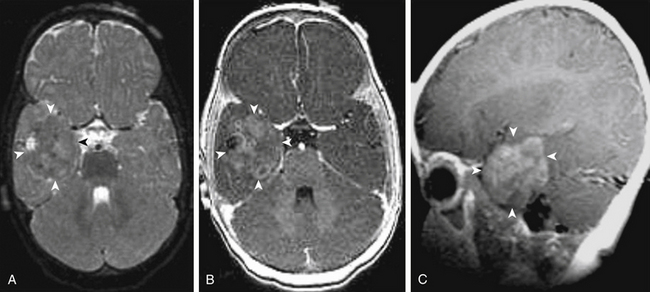
As a rule, malignant gliomas and germ cell tumors invade through the wall of the third ventricle, while pineal parenchymal tumors, meningiomas, and low-grade astrocytomas are more likely to cause expansive compression.68 Pineocytomas and pineoblastomas are typically hypointense to isointense on T1-weighted MRI, display increased T2 signal, and enhance homogenously after gadolinium administration. Pineoblastomas (Fig. 56-4) are larger (>4 cm) and have an irregular shape compared to pineocytomas. Astrocytomas are hypointense on T1-weight MRI and hyperintense on T2-weighted MRI. These lesions enhance in variable patterns because they can arise from either the glial stroma of the pineal gland or the surrounding tissue. Calcium may be present in either pineal cell tumors or astrocytomas. Meningiomas display smooth, distinct borders and often have a homogenously enhancing dural tail. A lesion fitting this pattern located dorsal relative to the deep venous system should raise suspicion for a tentorial meningioma. Germinomas are isointense on T1 and slightly hyperintense on T2 MRI. They enhance strongly in a homogenous pattern (Fig. 56-5) and can display a peripheral calcification that surrounds the pineal gland. By contrast, the calcification in pineocytomas can be differentiated from germinomas in that there is primarily intratumoral calcium; they may also have intratumoral cysts. Teratomas have a heterogeneous appearance on MRI, are multilocular, and enhance in an irregular pattern. These lesions are often well circumscribed and can be distinguished from other pineal region tumors by the low attenuation from the adipose tissue. Other malignant nongerminomatous germ cell tumors have a mixture of benign and malignant germ cell components and a heterogeneous appearance. Choriocarcinoma, a specific subtype, is characterized by areas of intratumoral hemorrhage. Incidental findings of a mostly cystic pineal gland, known as pineal cysts, are becoming more common with the increasing use of MRI scans. These lesions do not require surgery unless they are progressing in size or cause symptomatic obstruction of the aqueduct and may be followed with serial MRI scans.
Laboratory Data
The measurements of markers specific for tumors of the pineal gland are important to establish a diagnosis and to follow post-treatment response and recurrence (Table 56-4). Alpha-fetoprotein (AFP) and β-hCG are the most specific makers for malignant germ cell tumors, and these levels are important to ascertain as the treatment of these lesions is nonoperative.58 AFP is greatly elevated with endodermal sinus tumors, but smaller elevations occur with immature teratomas and embryonal cell carcinomas.58 β-hCG is elevated in choriocarcinomas and germinomas containing a syncytiotrophoblastic giant cell, which is associated with a worse prognosis.69 Lactate dehydrogenase and placental alkaline phosphate are less specific than AFP and β-hCG but are also often used to follow response to therapy. The candidate tumor makers for pineal parenchymal cell tumors are melatonin and the S antigen, but neither has proved valuable for establishing diagnosis or for following recurrence. Theoretically, undetectable levels of melatonin in the serum following surgery suggest a gross-total resection. A combination of tumor-maker evaluation and neuroimaging should be a routine part of the preoperative evaluation; it provides the greatest diagnostic accuracy and plays an important in optimal surgical and medical management.

Surgical Indication
Any patient presenting acutely with hydrocephalus should have an immediate CSF diversion procedure, either by external ventricular drainage or by endoscopic third ventriculostomy. The decision for more definitive CSF diversion can be made at the time of surgical resection. If surgical resection is not likely to occur soon and there is a concern for worsening hydrocephalus, the primary concern should be interval CSF diversion. Often, surgical or nonsurgical treatment of the tumor results in decreased tumor size with restoration of normal CSF flow.70 In cases of nonacute hydrocephalus, definitive CSF diversion can take place at the time of biopsy with endoscopic third ventriculostomy or with a ventriculoperitoneal shunt or temporary ventricular drain. Ultimately, the decision of CSF diversion must be based on the degree of normalization of CSF flow at the end of treatment. While extremely rare, metastasis of pineal cell tumors from CSF diversion has been reported.71,72
Biopsy
In cases of nondiagnostic CSF findings, the decision must be made to attempt stereotactic biopsy or open biopsy with planned resection. The advancement of neuroendoscopic and image-guided stereotactic techniques has played an important role in the diagnosis of these lesions. Large retrospective series of both adults and children report mortality rates of less than 1.5%, and morbidity rates ranging from 2% to 10%, with diagnosis made in greater than 90% of cases.73 To ensure safety, consider variation in anatomy due to the tumor, including displacement of vasculature. Some tumors, such as pineoblastoma or choriocarcinoma, are also known to be highly vascular. In cases of a clearly nonresectable lesion, evidence of metastatic disease, or a patient who is a poor surgical candidate, histologic diagnosis is still crucial for adjuvant management with chemotherapy and radiation therapy. The primary drawback of endoscopic or sterotactic biopsy is the inability to obtain sufficient tissue for definitive diagnosis, particularly in tumors with mixed components, such as teratomas. In cases of clearly resectable tumors, the risks of biopsy should be avoided and the preferred approach is open biopsy with resection at the same time.
Surgical Approaches
Anatomic Considerations
The anatomic considerations of surgical resection of pineal region tumors have been well described by prior authors, notably Bruce et al.59 In their large experience, among the most important considerations are the location and degree of attachment of these lesions to the undersurface of the velum interpositium, including the choroid plexus, deep venous system, and choroidal arteries. Because tumors rarely extend above the velum interpositium, the major vascular supply occurs from within the velum interpositium, mainly through the posterior medial and lateral choroidal arteries and with anastomoses to the quadrigeminal and pericallosal arteries.59 The majority of these lesions are centered on the pineal gland, and they may extend to the midportion of the third ventricle and compress the anterior portion of the cerebellum with posterior extension. Important vascular structures, including the Galen vein, internal cerebral veins, basal Rosenthal veins, and precentral cerebellar veins, commonly encapsulate these tumors. Rarely, the internal cerebral veins can be found ventral to the tumor. Essential information that must be taken into account includes knowing the tumor’s position within the third ventricle, its lateral and supratentorial extension, its position relative do the deep venous system, and the degree of brain stem involvement.
Approaches
At least five distinct approaches have been developed for lesions of the pineal gland because of its location in the geometric center of the intracranial cavity.62 The two most common approaches are the infratentorial–supracerebellar and the occipital–transtentorial approaches; in certain cases, the parietal/transcallosal–interhemispheric approach is also used. The midline sagittal MRI with gadolinium is often the best sequence with which to make a decision regarding the approach.
The primary advantage of the occipital–transtentorial approach is the excellent view provided above and below the tentorium. The major disadvantages of the transcallosal–interhemispheric approach are the extensive reaction required on the parietal lobe, the potential risk of venous infarction if disruption of the bridging veins between the sagittal sinus and the parietal lobe occurs, and the need to work around the veins of the deep venous system that overlie the tumor.59,62 The major disadvantages of the occipital–transtentorial approach also include the need to work around the deep venous system around the tumor and the high frequency of visual deficits due to retraction of the occipital lobe and damage to the calcarine cortex.59
Positions
Patient positioning is an important part of the preoperative plan and is largely dictated by the approach selected. In the transcallosal–interhemispheric and infratentorial–supracerebellar approaches, the sitting slouch position can be used. The major advantages of this position include minimal blood pooling in the operative field and the cerebellum falling away because of gravity to improve exposure. The major disadvantages include air embolus, pneumocephalus, and epidural or subdural hematoma from ventricular and cortical collapse, though these are rare occurrences.59 This position is contraindicated in children younger than 2 years with severe hydrocephalus, in which case the three quarter prone, lateral decubitus, and Concorde position are appropriate alternatives. In the occipital–transtentorial approach, the three quarter prone position or lateral decubitus position can be used. This position reduces the risk of complications of the sitting slouch position but does not allow gravity to work in the surgeon’s favor.59 The Concorde position was designed specifically for tumors of the pineal region and combines aspects of both the prone and the semisitting positions. While this position is more comfortable for the surgeon and reduces the risk of air embolism, a major disadvantage is the pooling of blood in the operative field.59 Ultimately, the approach and position selected depend on the presence and degree of hydrocephalus, whether the tumor is more anterior or posterior in location, and surgeon experience and preference.
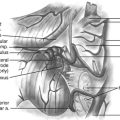
The infratentorial–supracerebellar approach was first described by Karuse in 1913 and popularized by Stein in 1979.74 This is the preferred approach for tumors of the pineal region because of the wide lateral exposure obtained and because it provides an intuitive orientation for the surgeon.59 The primary anatomic structures exposed are the cerebellum, cerebellar veins, cerebellar vermian veins, internal cerebellar veins, basal Rosenthal veins, Galen vein, pineal body, posterior commissure, quadrigeminal plate, splecinum of corpus callosum, and posterior third ventricle. The skin incision is made along the midline from the occipital region down to the spinous process of the third vertebral body. A wide craniotomy is performed to include the transverse sinuses and torcula. After the dura is opened, the arachnoid is dissected and the superomedial cerebellar bridging veins are divided to allow for inferior retraction on the cerebellar hemispheres. Alternatively, some degree of upward retraction on the sinus complex decreases the degree of downward retraction on the cerebellum. An operating microscope is then brought in and the arachnoid spanning the interval between the cerebellar vermis and the central posterior incisura is opened with microdissection to exposure the pineal gland. Several vascular structures are important to consider. The precentral cerebellar vein, which can be seen extending from the edge of the cerebellar vermis to the Galen vein, can be cauterized and divided. In the lateral aspects of the exposure, the Rosenthal veins can be seen coursing upward toward the confluence of veins. Branches of the choroidal arteries often supply these tumors. The tumor may be removed with a combination of tumor forceps, curets, suction, cautery, or ultrasonic aspiration based on its consistency.59
Occipital–Transtentorial Approach
Compared to the infratentorial–supracerebellar approach, the occipital–transtentorial approach is more laterally directed and the primary anatomic structures exposed include the splenium of the corpus callosum, internal cerebral veins, basal Rosenthal veins, Galen vein, precentral cerebellar vermian vein, pineal body, posterior commissure, and quadrigeminal plate. An inverted L-shaped skin incision is made to clearly expose the inion. The superior margin of the bone flap is 1 to 2 cm below the lambdoid suture, and the inferior margin is below the inion. The dura is opened in an H shape. Retraction of the occipital lobe laterally exposures the posterior edge of the incisura, where it is attached to the falx cerebri. The tentorium is incised, and sharp dissection of the arachnoid brings the Galen vein and the ipsilateral basal Rosenthal vein into view. The pineal gland is visible inferior to this venous complex.59
Postoperative Management and Adjuvant Therapy
Immediate Postoperative Management
The most common postoperative complications following pineal region surgery are extraocular movement dysfunction, ataxia, and altered mental status. These findings are particularly pronounced in patients with invasive and malignant tumors, prior radiation therapy, and severe preoperative neurologic deficits.59 A potential lethal complication is postoperative hemorrhage in a subtotally resected lesion, particularly vascular lesions such as primary pineal cell tumors. Acute worsening of symptoms up to 1 week following surgery or hydrocephalus should raise concern for this phenomenon. Less common postoperative complications include venous infarction, hemorrhage related to third ventriculostomy if performed, aseptic meningitis, and in the case of supratentorial approaches, seizures, hemianopsia, and hemiparesis. An MRI should be obtained 24 to 48 hours following surgery to assess the extent of resection and as a baseline for future studies.59
Chemotherapy
Primary chemotherapy or adjuvant chemotherapy plays an important role in the management of children with tumors of the pineal region. Germinomas and nongerminomatous germ cell tumors are primarily treated with radiation, but their chemosensitivity has been demonstrated and chemotherapy is now routinely used to improve the efficacy and reduce the dose of subsequent radiation therapy.75 Common agents used with germinomas include cyclophosphamide, ifosfamide, etoposide, cisplatin, carboplatin, and bleomycin. Common agents used in nongerminomatous germ cell tumors include platinum-based regimens, often used in combination with vinblastine, etoposide, and/or bleomycin.76 Children with pineoblastomas receive the same chemotherapy regimen for medulloblastomas, particularly infants to delay the age at first radiation treatment.76 There are limited data regarding the use of chemotherapy in children with pineocytoma because they are treated primarily with surgical resection and radiation therapy.
Radiation Therapy
While radiation therapy also plays an important role in the management of children with pineal region tumors, there is greater concern given the effects of radiation on cognitive development, particularly because these children are often long-term survivors. Germinomas are among the most radiosensitive tumors, with greater than 90% disease control with radiation therapy alone. The addition of chemotherapy reduces the dose of radiation required and is particularly effective in children presenting prior to puberty.76 There is great ongoing debate as to the type, dose, volume, and regions that should be irradiated. Though the data are limited, it appears that postoperative radiation therapy improves the outcome in children with pineoblastoma and pineocytoma, particularly in those with subtotal resection or with progressive disease.77 For pineoblastoma, the use of craniospinal irradiation is frequently recommended depending on patient age.
Long-Time Follow-Up
Second-look surgery is considered in relapse germ cell tumors, particularly when nongerminomatous germ cell tumors do not have complete radiographic response to adjuvant chemotherapy. The differential for residual on neuroimaging may be necrosis and fibrosis or residual malignant elements. In some rare instances, growing teratoma syndrome is a pathophysiologic phenomena in mature teratoma, where tumor masses enlarge during chemotherapy in the presence of normal or declining tumor markers.78 Second-look surgery is also considered when patients have persistently positive tumor markers in CSF and/or serum, which require histologic evaluation and confirmation.79
Intraventricular Masses
Intraventricular tumors are rare masses that are found more commonly in the pediatric population. These tumors are a heterogeneous group of histologically varying tumors that can be divided into primary and secondary intraventricular tumors. Primary tumors are considered those that develop from the ependyma and subependymal glia of the ventricular lining, the epithelium of the choroid plexus, and the arachnoid supporting tissue.80 Secondary, paraventricular tumors are masses that develop within the brain parenchyma, rather than the ventricular wall. They are defined as secondary intraventricular masses when more than two thirds of the tumor’s surface bulges into the ventricle.80 For example, third ventricular tumors tend to be germ cell tumors, pineal cell tumors, and glial cell tumors from the pineal gland that occupy this ventricular space.
Though extremely heterogenous, intraventricular tumors are grouped together due to their commonalities in clinical development, management, and difficulty in surgical resection due to their deep-seated location. The most common pediatric intraventricular tumors include ependymomas, choroid plexus papillomas (CPPs), and astrocytomas that frequently occur in the lateral and frontal horn.81–83 Less common intraventricular tumors include subependymal giant cell astrocytomas, oligodendrogliomas, subependymomas, pilocytic astrocytomas, neurocytoma, and choroid plexus carcinomas.82 The vast majority of intraventricular tumors are benign, and these rare intraventricular tumors make up 0.8% to 1.6% of all CNS tumors. These tumors occupy a nonfunctional space and are often able to grow into a considerable size before clinical manifestations. Given their benign nature, these masses are treated surgically but pose a significant challenge to neurosurgeons.
Epidemiology
Intraventricular tumors are more common in children than adults and comprise 16% of childhood and adolescent intracranial tumors. In a retrospective study of 112 pediatric patients with intraventricular tumors within the lateral ventricles, it was found that 42.8% of tumors occurred in the frontal horn and foramen of Monro, 22.3% in the body of the ventricle and septum pellucidum, 19.7% in the atrium or trigone, 8.9% in the temporal horn, and 6.3% in the occipital horn.81,84 Tumor type varies with age. Younger children with lateral ventricular neoplasms most commonly have CPPs or choroid plexus carcinomas, while in older children low-grade gliomas such as subependymal giant cell astrocytomas, pilocytic astrocytomas, and ependymomas are more frequent.83,85
Ependymomas
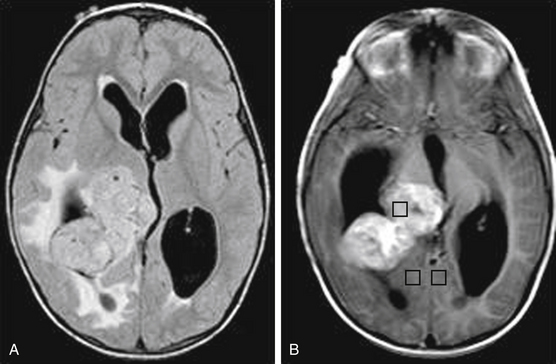
Ependymomas are uncommon, slow-growing, glial tumors that arise either within or adjacent to the ependymal lining of the ventricular system. Within pediatric patients, the median age of diagnosis is 5 years. These patients commonly present with seizures and focal neurologic defects when the tumors are in the supratentorial space.86 On pathologic exam, they are commonly characterized by calcification, hemorrhage, and cysts. Upon neuroimaging, ependymomas are hypointense on T1-weighted and hyperintense on T2-weighted MRI, and they enhance prominently with gadolinium contrast (Fig. 56-6). Their diagnosis requires biopsy and histologic confirmation. Due to the favorable outcome of gross resection of these benign tumors, open resection is recommended over stereotactic biopsy.87 Gross total resection followed by adjuvant radiation is recommended for children older than 3 years, and adjuvant chemotherapy is advisable for those younger than 3 years old to avoid radiation. Management also includes MRI of the entire neuraxes, as well as CSF cytology to exclude metastatic disease.88
Choroid Plexus Papillomas
CPPs arise from the endogenous neuroepithelial tissue of the choroid plexus.89 CPPs are distributed according to ventricular surface area, where 50% of these tumors occur in the lateral ventricle, 40% in the fourth ventricle, and 5% in the third ventricle. These tumors make up 10% to 20% of intracranial tumors in pediatric patients younger than 1 year. CPPs are benign WHO grade I tumors that histologically resemble normal neuroepithelium of the choroid plexus.90 CT and MRI are recommended imaging modalities. CPPs are often calcified and can be difficult to distinguish from ependymomas. On MRI, CPPs are hypo- to isointense on T1, are hyperintense on T2, and enhance with contrast (Fig. 56-7). The vascular nature of these tumors, which commonly obtain blood supply from the choroidal arteries, may lead to flow voids visible on MRI. Management includes complete surgical resection, with adjuvant radiotherapy reserved for recurrent tumors.91 Malignant transformation into a choroid plexus carcinoma rarely occurs, and the majority of incompletely resected CPPs remain benign.
Clinical Presentation
The range of pathologies that arise within the ventricles, as well as the diversity in location of intraventricular tumors, does not lend to a stereotypic neurologic or behavioral pattern of signs and symptoms. However, the majority of clinical manifestations result from either obstructed CSF flow or hydrocephalus secondary to an overproduction of CSF.83 Common symptoms include papilledema, headache, motor disturbance, sensory disturbance, nausea and vomiting, visual field defects, low vision, and mental status changes.82 Memory loss secondary to mass effect on the fornix from direct compression or hydrocephalus is also known to occur. Finally, in rare instances, spontaneous bleeding and acute clinical deterioration have been reported.80
Surgical Management
Treatment of intraventricular tumors is controversial due to the challenges presented by their location, size, association with hydrocephalus, and surrounding functional anatomy. Imaging with MRI is the preferred modality for preoperative evaluation. Important considerations include tumor size, tumor vascular supply and drainage, hydrocephalus, and surrounding neural structures.80 Finally, in instances where intraventricular tumors are inaccessible, intraventricular endoscopy can be utilized to achieve diagnosis. Such minimally invasive procedures are attractive and successful for smaller lesions or radiosensitive tumors that require biopsy for pathologic diagnosis, followed by definitive treatment with radiation or chemotherapy.92
Relevant Anatomy
During surgical resection, visible landmarks within the lateral ventricle should be identified to orient the surgeon. In particular, the veins that line the white ependymal surface are significant. The thalamostriate vein lies in the groove separating the caudate nucleus from the thalamus. This vein joins the septal vein, and together the two form the internal cerebral vein that lies proximal to the foramen of Monro, beneath the choroid plexus. The choroidal fissure serves as another important intraventricular landmark, because the choroid plexus lies within it, and the fissure divides the fornix and thalamus. The choroid plexus itself is within the medial aspect of the body, atrium, and temporal horns of the lateral ventricles. It courses through the foramen of Monro to the roof of the third ventricle.80 Additional important functional structures adjacent to the ventricles include the Rolandic area, language areas on the dominant hemisphere, fornices, internal cerebral veins, and the Galen vein.82
Surgical Approach
The ideal surgical exposure allows sufficient exposure for removal of the tumor, rapid identification of supplying vessels, and minimal brain retraction and damage to functional cortex.80 The most direct trajectory between the surgeon and the lesion is often the most appropriate, but the choice is variable and depends on experience (Table 56-5). The tumor location within the ventricle, hydrocephalus, hemispherical dominance, tumor size, tumor blood vessel supply, and histopathologic features are all significant factors. Stereotactic guidance systems are helpful in choosing tailored approaches that allow for safe and aggressive tumor removal. Historically, neurosurgeons have used a transcortical approach to the lateral ventricle. More recently, transcallosal–interhemispheric approaches are more frequently used due to the provision of direct access to the ventricles without damaging cortical brain tissue.93–95 For third and posterior intraventricular tumors, transcortical temporal, superior parietal lobule, and transcallosal–interhemispheric approaches are the most common.96
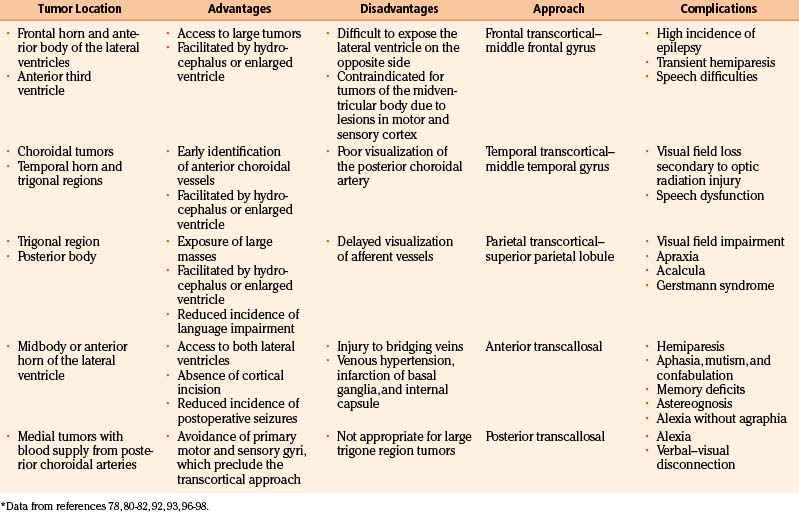
The most common surgical approaches to lateral ventricle tumors are anterior, temporal, and parietal transcortical approaches, followed by anterior and posterior interhemispheric approaches.80 Anterior approaches include anterior transcallosal, transcortical, and frontal. Posterior approaches include posterior transcallosal, transcortical, and occipital. Inferior approaches include temporal and posterior frontotemporal.
Intraoperative Considerations
Highly vascular intraventricular tumors necessitate division and coagulation of arterial supply prior to resection of the tumor. Once hemostasis is achieved, it is recommended that the ventricle be filled with saline and emptied to identify leaking vessels. The tumor resection cavity should also be filled with saline to prevent collapse of brain parenchyma once retractors are removed. Lining the cavity with hemostatic agents should be done cautiously, given the possibility of dislodging and obstructing CSF flow. Septal fenestration and placement of a ventricular drain is also recommended to avoid postoperative hydrocephalus by diversion of blood, debris, and CSF.80
Postoperative Management
Patients require intensive care unit monitoring postoperatively. Medications include 24 hours of antibiotics and a 10- to 14-day corticosteroid taper. If a transcortical approach was utilized, antiepileptic prophylaxis should be considered. Postoperative MRI is also needed to confirm the extent of tumor resection. If an external ventricular drain was placed, this can be weaned over 3 to 5 days. Patients need close monitoring because complication rates of intraventricular tumors can approach 20%.82 The most common complications include severe brain edema, intraventricular hemorrhage, subdural hematoma, and epidural hematoma. Severe memory difficulties can also occur because tumors of the lateral ventricles can lead to forniceal injury.80,95,97
Ahn Y., et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst. 2006;22(9):1136-1142.
Albright A.L. Feasibility and advisability of resections of thalamic tumors in pediatric patients. J Neurosurg. 2004;100(5 suppl Pediatrics):468-472.
Apuzzo M.L., et al. Transcallosal, interfornicial approaches for lesions affecting the third ventricle: surgical considerations and consequences. Neurosurgery. 1982;10(5):547-554.
Bhattacharjee M.B., et al. Cytogenetic analysis of 120 primary pediatric brain tumors and literature review. Cancer Genet Cytogenet. 1997;97(1):39-53.
Bruce J., editor. Pineal region masses: clinical features and management. In: R.D. Schmidek H. ed. Schmidek and Sweet’s Operative Neurosurgical Techniques: Indications, Methods, and Results, 5th ed. Vol. 2, Saunders, 2005. 786-797
Bruce J.N. Pineal tumors. In: Winn H.R., editor. Youman’s Neurological Surgery. WB Saunders; 2004:1011-1029.
Cuccia V., Monges J. Thalamic tumors in children. Childs Nerv Syst. 1997;13(10):514-520. discussion 521
Dutton J.J. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38(5):427-452.
Fisher P.G., et al. Outcome analysis of childhood low-grade astrocytomas. Pediatr Blood Cancer. 2008;51(2):245-250.
Heideman R.L., et al. Supratentorial malignant gliomas in childhood: a single institution perspective. Cancer. 1997;80(3):497-504.
Jennings M.T., Gelman R., Hochberg F. Intracranial germ-cell tumors: natural history and pathogenesis. J Neurosurg. 1985;63(2):155-167.
Lau C., Teo W.-Y. Clinical manifestations and diagnosis of central nervous system tumors in children. UpToDate Online. 17.2, 2009. May 26, 2009 [cited 2009 August 18]
Listernick R., et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149.
Lozier A.P., Bruce J.N. Surgical approaches to posterior third ventricular tumors. Neurosurg Clin N Am. 2003;14(4):527-545.
Miller N.R. Primary tumours of the optic nerve and its sheath. Eye (Lond). 2004;18(11):1026-1037.
Perkins SM, et al. Glioblastoma in children: a single-institution experience. Int J Radiat Oncol Biol Phys.
Pollack I.F., et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg. 2006;105(suppl 5):418-424.
Pytel P. Spectrum of pediatric gliomas: implications for the development of future therapies. Expert Rev Anticancer Ther. 2007;7(suppl 12):S51-S60.
Ruggiero A., et al. Phase II trial of temozolomide in children with recurrent high-grade glioma. J Neurooncol. 2006;77(1):89-94.
Sawamura Y., et al. Role of surgery for optic pathway/hypothalamic astrocytomas in children. Neuro Oncol. 2008;10(5):725-733.
Sievert A.J., Fisher M.J. Pediatric low-grade gliomas. J Child Neurol. 2009;24(11):1397-1408.
Song KS, et al. Long-term outcomes in children with glioblastoma. J Neurosurg Pediatr. 6(2):145–9.
Souweidane M.M., Hoffman H.J. Current treatment of thalamic gliomas in children. J Neurooncol. 1996;28(2-3):157-166.
Stein B.M. Supracerebellar–infratentorial approach to pineal tumors. Surg Neurol. 1979;11(5):331-337.
Stoiber EM, et al. Long term outcome of adolescent and adult patients with pineal parenchymal tumors treated with fractionated radiotherapy between 1982 and 2003—a single institution’s experience. Radiat Oncol. 5:122.
Winkler P.A., et al. The transcallosal interforniceal approach to the third ventricle: anatomic and microsurgical aspects. Neurosurgery. 1997;40(5):973-981. discussion 981-2
1. Lau C., Teo W.-Y. Epidemiology of central nervous system tumors in children. UpToDate Online. 17.2, 2009. June 18, 2009 [cited 2009 August 18]
2. Winn H.R., Youmans J.R. Section 7, Pediatric, in Youmans neurological surgery. Philadelphia, Pa: W.B. Saunders; 2004. : 4 v. (lxiv, 5296, cviii)
3. Fisher P.G., et al. Outcome analysis of childhood low-grade astrocytomas. Pediatr Blood Cancer. 2008;51(2):245-250.
4. Lau C., Teo W.-Y. Clinical manifestations and diagnosis of central nervous system tumors in children. UpToDate Online. 17.2, 2009. May 26, 2009 [cited 2009 August 18]
5. Sievert A.J., Fisher M.J. Pediatric low-grade gliomas. J Child Neurol. 2009;24(11):1397-1408.
6. Brenner D.J., Hall E.J. Computed tomography—an increasing source of radiation exposure. N Engl J Med. 2007;357(22):2277-2284.
7. Rozen W.M., Joseph S., Lo P.A. Spontaneous regression of low-grade gliomas in pediatric patients without neurofibromatosis. Pediatr Neurosurg. 2008;44(4):324-328.
8. Parsa C.F., et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119(4):516-529.
9. Freeman C.R., Farmer J.P., Montes J. Low-grade astrocytomas in children: evolving management strategies. Int J Radiat Oncol Biol Phys. 1998;41(5):979-987.
10. Komotar R.J., et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004;54(1):72-79. discussion 79-80
11. White F.V., et al. Nonrandom chromosomal gains in pilocytic astrocytomas of childhood. Hum Pathol. 1995;26(9):979-986.
12. Bhattacharjee M.B., et al. Cytogenetic analysis of 120 primary pediatric brain tumors and literature review. Cancer Genet Cytogenet. 1997;97(1):39-53.
13. Jones D.T., et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673-8677.
14. Pfister S., et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118(5):1739-1749.
15. Schmidek H.H., Roberts D.W. Schmidek & Sweet operative neurosurgical techniques: indications, methods, and results, 5th ed. Philadelphia: Saunders Elsevier; 2006:. 2 v. (xxxix, 2337, 67 p.)
16. Dutton J.J. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38(5):427-452.
17. Miller N.R. Primary tumours of the optic nerve and its sheath. Eye (Lond). 2004;18(11):1026-1037.
18. Chutorian A.M., et al. Optic gliomas in children. Neurology. 1964;14:83-95.
19. Rush J.A., et al. Optic glioma. Long-term follow-up of 85 histopathologically verified cases. Ophthalmology. 1982;89(11):1213-1219.
20. Listernick R., et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143-149.
21. Cummings T.J., et al. Gliomas of the optic nerve: histological, immunohistochemical (MIB-1 and p53), and MRI analysis. Acta Neuropathol. 2000;99(5):563-570.
22. Spoor T.C., et al. Malignant gliomas of the optic nerve pathways. Am J Ophthalmol. 1980;89(2):284-292.
23. Stern J., Jakobiec F.A., Housepian E.M. The architecture of optic nerve gliomas with and without neurofibromatosis. Arch Ophthalmol. 1980;98(3):505-511.
24. Campagna M, et al. Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer. 55(6): 1083–1088.
25. Sawamura Y., et al. Role of surgery for optic pathway/hypothalamic astrocytomas in children. Neuro Oncol. 2008;10(5):725-733.
26. Ahn Y., et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst. 2006;22(9):1136-1142.
27. Gillett G.R., Symon L. Hypothalamic glioma. Surg Neurol. 1987;28(4):291-300.
28. Konovalov A., Gorelyshev S., Serova N. Surgery of giant gliomas of chiasma and IIIrd ventricle. Acta Neurochir (Wien). 1994;130(1-4):71-79.
29. Di Rocco C., Iannelli A. Bilateral thalamic tumors in children. Childs Nerv Syst. 2002;18(8):440-444.
30. Burger P.C., et al. Pathology of diencephalic astrocytomas. Pediatr Neurosurg. 2000;32(4):214-219.
31. Rajput DK. et al. Bilateral thalamic glioma in a 6-year-old child. J Pediatr Neurosci. 5(1):45-48.
32. Puget S., et al. Thalamic tumors in children: a reappraisal. J Neurosurg. 2007;106(suppl 5):354-362.
33. Krouwer H.G., Prados M.D. Infiltrative astrocytomas of the thalamus. J Neurosurg. 1995;82(4):548-557.
34. Albright A.L. Feasibility and advisability of resections of thalamic tumors in pediatric patients. J Neurosurg. 2004;100(5 suppl Pediatrics):468-472.
35. Souweidane M.M., Hoffman H.J. Current treatment of thalamic gliomas in children. J Neurooncol. 1996;28(2-3):157-166.
36. Lefton D.R., et al. Radiologic features of pediatric thalamic and hypothalamic tumors. Crit Rev Diagn Imaging. 2000;41(4):237-278.
37. Cuccia V., Monges J. Thalamic tumors in children. Childs Nerv Syst. 1997;13(10):514-520. discussion 521
38. Selvapandian S. Endoscopic management of thalamic gliomas. Minim Invasive Neurosurg. 2006;49(4):194-196.
39. Fisher B.J., et al. Results of a policy of surveillance alone after surgical management of pediatric low grade gliomas. Int J Radiat Oncol Biol Phys. 2001;51(3):704-710.
40. Packer R.J., et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86(5):747-754.
41. Kleihues P., Sobin L.H. World Health Organization classification of tumors. Cancer. 2000;88(12):2887.
42. Reddy A.T., Wellons J.C.3rd. Pediatric high-grade gliomas. Cancer J. 2003;9(2):107-112.
43. Perkins SM, et al. Glioblastoma in Children: a single-institution experience. Int J Radiat Oncol Biol Phys.
44. Song KS, et al. Long-term outcomes in children with glioblastoma. J Neurosurg Pediatr. 6(2): 145–149.
45. Heideman R.L., et al. Supratentorial malignant gliomas in childhood: a single institution perspective. Cancer. 1997;80(3):497-504.
46. Pytel P. Spectrum of pediatric gliomas: implications for the development of future therapies. Expert Rev Anticancer Ther. 2007;7(suppl 12):S51-S60.
47. Antonelli M, et al. Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J Neurooncol. 99(2): 209–215.
48. Pollack I.F., et al. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: results from the Children’s Cancer Group 945 cohort. J Neurosurg. 2006;105(suppl 5):418-424.
49. Pollack I.F., et al. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: results from the CCG-945 Cohort. J Clin Oncol. 2006;24(21):3431-3437.
50. Kubo O. [Histological diagnosis of brain tumors: (1) Ependymoma]. No Shinkei Geka. 1989;17(1):7-14.
51. Broniscer A. Past, present, and future strategies in the treatment of high-grade glioma in children. Cancer Invest. 2006;24(1):77-81.
52. Geyer J.R., et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol. 2005;23(30):7621-7631.
53. Kumagai Y. et al. Induction of corneal epithelium-like cells from cynomolgus monkey embryonic stem cells and their experimental transplantation to damaged cornea. Cornea. 29(4): 432–438.
54. Ruggiero A., et al. Phase II trial of temozolomide in children with recurrent high-grade glioma. J Neurooncol. 2006;77(1):89-94.
55. Burger P.C., Scheithauer B.W. Tumors of the central nervous system. In: AFIP atlas of tumor pathology. Washington, DC: American Registry of Pathology; 2007:321-327.
56. Jennings M.T., Gelman R., Hochberg F. Intracranial germ-cell tumors: natural history and pathogenesis. J Neurosurg. 1985;63(2):155-167.
57. Rickert C.H., Hasselblatt M. Cytogenetic features of ependymoblastomas. Acta Neuropathol. 2006;111(6):559-562.
58. Bruce J.N. Pineal tumors. In: Winn H.R., editor. Youman’s Neurological Surgery. WB Saunders; 2004:1011-1029.
59. Bruce J., editor, Pineal region masses: clinical features and management. In: R.D. Schmidek H, ed. Schmidek and Sweet’s Operative Neurosurgical Techniques: Indications, Methods, and Results, 5th ed. vol. 2. Saunders, 2005:786-797
60. Parinaud H. Paralysis of the movement of convergence of the eyes. Brain. 1886;9(3):330-341.
61. Posner M., Horrax G. Eye signs in pineal tumors. J Neurosurg. 1946;3:15-24.
62. Matula C.. Tumors of the pineal region, Rengachary S.S., Ellenbogen R.G. Principles of Neurosurgery, 2nd ed., Philadelphia: Elsevier, 2005. 501-520
63. Missori P., Delfini R., Cantore G. Tinnitus and hearing loss in pineal region tumours. Acta Neurochir (Wien). 1995;135(3-4):154-158.
64. DeMonte F., Zelby A.S., Al-Mefty O. Hearing impairment resulting from a pineal region meningioma. Neurosurgery. 1993;32(4):665-668.
65. Balmaceda C. Pineal gland masses. UpToDate Online. 17.2, 2009. December 11, 2006 [cited 2009 August 21]
66. Luther N, et al. Subarachnoid dissemination of intraventricular tumors following simultaneous endoscopic biopsy and third ventriculostomy. J Neurosurg Pediatr. 5(1): p. 61–67.
67. Fetell M.R., Stein B.M. Neuroendocrine aspects of pineal tumors. Neurol Clin. 1986;4(4):877-905.
68. Moschovi MC, G, Pineal gland masses, in UpToDate. 2011, UpToDate.
69. Yoshida J., et al. Prognosis of intracranial germ cell tumours: effectiveness of chemotherapy with cisplatin and etoposide (CDDP and VP-16). Acta Neurochir (Wien). 1993;120(3-4):111-117.
70. Edwards M.S., et al. Pineal region tumors in children. J Neurosurg. 1988;68(5):689-697.
71. Altundag O.O., Celik I., Kars A. Pineal germ cell tumor metastasis via ventriculoperitoneal shunt. Am J Clin Oncol. 2002;25(1):104-105.
72. Saibara T., et al. [Abdominal metastasis of a pineal region tumor through ventriculoperitoneal shunt. Case report]. Neurol Med Chir (Tokyo). 1991;31(13):1012-1017.
73. Regis J., et al. Pineal region tumors and the role of stereotactic biopsy: review of the mortality, morbidity, and diagnostic rates in 370 cases. Neurosurgery. 1996;39(5):907-912. discussion 912-4
74. Stein B.M. Supracerebellar–infratentorial approach to pineal tumors. Surg Neurol. 1979;11(5):331-337.
75. Balmaceda C., et al. Chemotherapy without irradiation—a novel approach for newly diagnosed CNS germ cell tumors: results of an international cooperative trial. The First International Central Nervous System Germ Cell Tumor Study. J Clin Oncol. 1996;14(11):2908-2915.
76. Hinkes B.G., et al. Childhood pineoblastoma: experiences from the prospective multicenter trials HIT-SKK87, HIT-SKK92 and HIT91. J Neurooncol. 2007;81(2):217-223.
77. Stoiber EM, et al. Long term outcome of adolescent and adult patients with pineal parenchymal tumors treated with fractionated radiotherapy between 1982 and 2003—a single institution’s experience. Radiat Oncol. 5: 122
78. O’Callaghan A.M., et al. The growing teratoma syndrome in a nongerminomatous germ cell tumor of the pineal gland: a case report and review. Cancer. 1997;80(5):942-947.
79. Echevarria M.E., Fangusaro J., Goldman S. Pediatric central nervous system germ cell tumors: a review. Oncologist. 2008;13(6):690-699.
80. Anderson R.C., Ghatan S., Feldstein N.A. Surgical approaches to tumors of the lateral ventricle. Neurosurg Clin N Am. 2003;14(4):509-525.
81. Delfini R., et al. Tumors of the lateral ventricles. Neurosurg Rev. 1991;14(2):127-133.
82. Gokalp H.Z., et al. Tumours of the lateral ventricle. A retrospective review of 112 cases operated upon 1970-1997. Neurosurg Rev. 1998;21(2-3):126-137.
83. Pendl G., Ozturk E., Haselsberger K. Surgery of tumours of the lateral ventricle. Acta Neurochir (Wien). 1992;116(2-4):128-136.
84. Santoro A., et al. Surgical approaches to tumours of the lateral ventricles in the dominant hemisphere. J Neurosurg Sci. 2002;46(2):60-65. discussion 65
85. Jelinek J., et al. Lateral ventricular neoplasms of the brain: differential diagnosis based on clinical, CT, and MR findings. AJNR Am J Neuroradiol. 1990;11(3):567-574.
86. Shuangshoti S., et al. Supratentorial extraventricular ependymal neoplasms: a clinicopathologic study of 32 patients. Cancer. 2005;103(12):2598-2605.
87. Hukin J., et al. Treatment of intracranial ependymoma by surgery alone. Pediatr Neurosurg. 1998;29(1):40-45.
88. Grill J., Pascal C., Chantal K. Childhood ependymoma: a systematic review of treatment options and strategies. Paediatr Drugs. 2003;5(8):533-543.
89. Louis D.N., et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97-109.
90. Koeller K.K., Sandberg G.D. From the archives of the AFIP. Cerebral intraventricular neoplasms: radiologic–pathologic correlation. Radiographics. 2002;22(6):1473-1505.
91. Wolff J.E., et al. Choroid plexus tumours. Br J Cancer. 2002;87(10):1086-1091.
92. Najjar MW, et al. Endoscopy in the management of intra-ventricular lesions: preliminary experience in the Middle East. Clin Neurol Neurosurg. 112(1): 17–22.
93. Woiciechowsky C., et al. Transcallosal removal of lesions affecting the third ventricle: an anatomic and clinical study. Neurosurgery. 1995;36(1):117-122. discussion 122-3
94. Shucart W.A., Stein B.M. Transcallosal approach to the anterior ventricular system. Neurosurgery. 1978;3(3):339-343.
95. Apuzzo M.L., et al. Transcallosal, interfornicial approaches for lesions affecting the third ventricle: surgical considerations and consequences. Neurosurgery. 1982;10(5):547-554.
96. Lozier A.P., Bruce J.N. Surgical approaches to posterior third ventricular tumors. Neurosurg Clin N Am. 2003;14(4):527-545.
97. Winkler P.A., et al. The transcallosal interforniceal approach to the third ventricle: anatomic and microsurgical aspects. Neurosurgery. 1997;40(5):973-981. discussion 981-2