18 Support of Renal Function
After reading this chapter, you should be able to:
• summarise the physiology of urine production
• describe the most likely causes of renal failure in the critically ill adult
• differentiate between acute and chronic renal failure
• outline treatment approaches in managing renal failure in critical illness
• appreciate historical developments in dialysis
• describe the indications for renal replacement therapy in critical care
• understand the principles and challenges associated with nursing management of continuous renal replacement therapy in critical care.
Introduction
Sudden deterioration of kidney function, to the point where there is retention of nitrogenous wastes, or acute renal failure (ARF), is a common manifestation of critical illness and is often associated with failure of other organs. Acute renal failure is a syndrome with numerous causes, including glomerulonephritis, prerenal azotaemia, urinary tract obstruction and vasculitis. Acute tubular necrosis (ATN) is a collective term commonly used to describe acutely deteriorating renal function, reflecting pathological changes from various renal insults of a nephrotoxic or ischaemic origin. Factors that cause renal function to deteriorate are not, however, always ischaemic or necrotic in origin, and a syndrome with degrees of failure is often evident. Therefore a new consensus definition and classification system has been established.1 This approach describes staging of ARF severity and embraces the concept of acute kidney injury (AKI) where, like other organs of the body, a dynamic spectrum is found, from small indiscrete changes in function that are immediately reversible, through to gross signs and irreversible organ failure.2
Acute renal failure is defined by a rapid deterioration in renal function (hours to days), which is easily detected by commonly measured markers of kidney performance, including blood urea nitrogen, serum creatinine, and a failed ability to adequately regulate electrolytes, sodium and water balance.3 While generally reversible, ARF can be life-threatening in the critically ill patient if acid–base balance, electrolyte levels (particularly potassium) or fluid overloads are not effectively diagnosed and managed.
The preferred serum marker of renal function is the serum creatinine level. The exact level of serum creatinine that is considered excessive is disputed; however, a doubling of the baseline serum creatinine or levels in excess of 200 µmol/L is commonly agreed on as being indicative of ARF.3 Urine output is also a key factor in determining the severity of ARF. It is well established that oliguric renal failure, that is, a urine output of less than 0.5 mL/kg/h in adults and 1 mL/kg/h in infants, is associated with poorer patient outcome than the non-oliguric form.4
Acute renal failure is reported to occur in 20–25% of intensive care patient admissions, much higher than the broader hospital rate of 5%.5,6,7 In critical care, ARF often forms part of the multiple organ dysfunction syndrome, whose cause has often been associated with sepsis, trauma, pneumonia or cardiovascular dysfunctions (see Chapter 21). Mortality in intensive care ARF is high, with those patients requiring renal replacement therapy (RRT) having worse outcomes than those patients who can be managed without this intervention.8
Related Anatomy and Physiology
Anatomy of Kidneys, Nephron and Urinary Drainage System
The functional anatomy of the renal system includes the two kidneys, ureters, bladder and urethra (see Figure 18.1). The ureters, bladder and urethra collect, drain and temporarily store the urine produced from each kidney.9 While important in providing the conduit for the final excretion of urine, the kidney is the primary organ of interest in the renal system, particularly in critical care practice, and hence will be described in more detail from the anatomical and physiological perspectives.
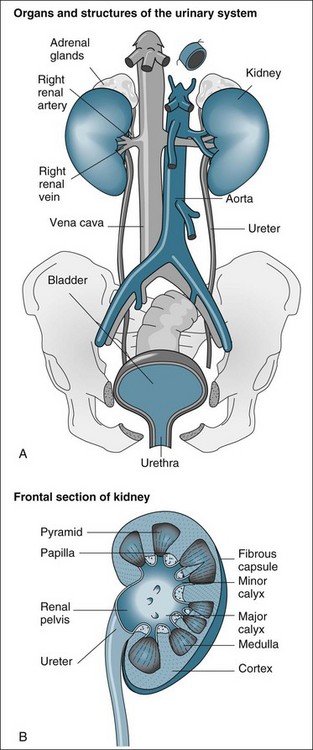
The kidneys are located in the retroperitoneal space on the posterior wall of the abdominal cavity, encased in a protective combination of the ribs, muscle, fat, tendon and the renal capsule. Each adult kidney weighs approximately 140 g. The kidneys may develop a different anatomical appearance, or vary in number and location from the classic description provided here. The functional unit of the kidney is the nephron, which consists of a filtrate-collecting device (the Bowman’s capsule), a convoluted tubule that varies in length and diameter, finally attaching to a common filtrate-collecting tubule and duct (see Figure 18.2). Within the Bowman’s capsule rests the glomerulus, a tuft of interlaced capillaries that arise from the afferent arteriole. The efferent arteriole then drains from the glomerulus via a closely entwined network called the peritubular capillaries, until these collect in the venous network of the kidney.
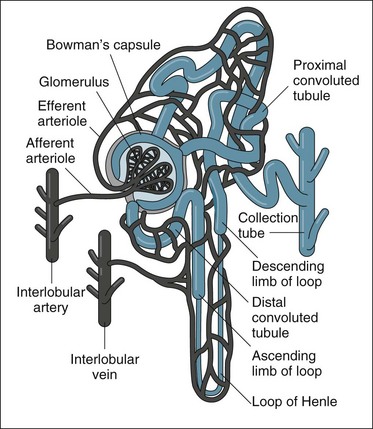
The glomeruli and nephrons lie in the cortical area of the kidney, while the collecting ducts gather together into the renal pyramids, which lie in the medulla of the kidney. The pyramids drain into the calyces of the kidney, which then drain into the renal pelvis where urine is gathered to drain into the ureter. The major blood vessels of the kidney, the renal artery and veins also enter the renal capsule through the pelvis of the kidney.9
Urine Production, Regulation of Gfr and Filtrate Reabsorption in the Nephron
Urine production consists of a three-stage process, which occurs in the nephron: glomerular filtration, tubular reabsorption and tubular secretion (see Figure 18.3). As previously noted, the production of urine is highly dependent on delivery of blood under pressure to the glomerulus. This results in the first step of the urine production process, glomerular filtration. The glomerular filtration rate (GFR) is about 125 mL/min under normal conditions. Changes in the diameter of the afferent and efferent arteriole help regulate glomerular blood flow, but this is unable to compensate for large variations of mean blood pressure; hence, filtration rates may rise or fall markedly over the course of a day.10
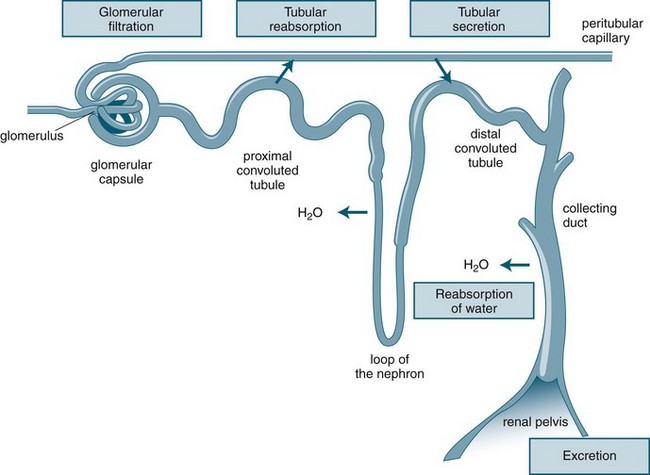
As the filtrate transgresses the glomerulus it is collected into the Bowman’s capsule and delivered into the proximal convoluted tubule, loop of Henle and then the distal convoluted tubule, where a number of processes result in the reabsorption of about 99% of the glomerular filtrate. The remaining fluid within the tubule drains into the collecting tubule to form urine. This fluid has substantially different properties from the original glomerular filtrate, as fluid and many electrolytes and glucose are reabsorbed by the peritubular capillaries.10
Along with blood pressure, the sodium content of the extracellular fluid is critical in maintaining fluid balance, as it constitutes the major electrolyte and osmotic agent of the glomerular filtrate. It is imperative that sodium intake and loss is equally balanced, as excessive losses will result in associated fluid loss and excessive intake will result in fluid retention. If sodium balance is not maintained, other compensations such as a rise in blood pressure may result to restore fluid balance. As blood pressure rises, the excretion of sodium also increases by way of the production of additional glomerular filtrate. In this way, water and sodium balance are inextricably linked.10
Hormonal and Neural Regulation of Renal Function
Various feedback mechanisms exist that assist in precisely adjusting the final amount of fluid and electrolyte to be excreted from the kidney. These include the sympathetic nervous system response, angiotension II, aldosterone, antidiuretic hormone and atrial natriuretic peptide. All these mechanisms work in synchrony with blood pressure and sodium balance in ensuring a highly regulated circulating and extracellular fluid volume.10
Renin–Angiotensin–Aldosterone System (RAAS)
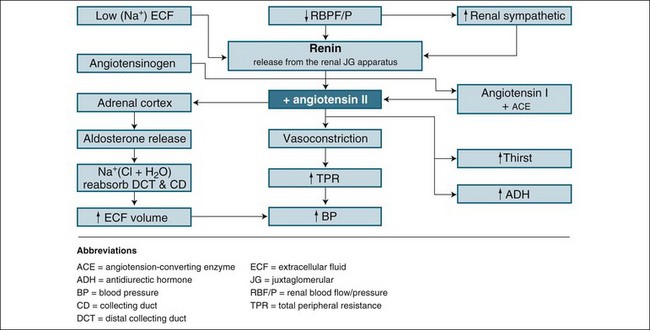
Renin is the chemical trigger to initiate a cascade system that results in two powerful hormones acting on the kidney to significantly influence sodium and water excretion (see Figure 18.4). Renin is produced and released from the juxtaglomerular apparatus, a collection of cells in the macula densa of the distal tubule, and the adjacent afferent arteriole next to the glomerulus, which monitors blood sodium concentration. When released, renin stimulates the activation of angiotensin I from angiotensinogen. Under the influence of coenzyme A, angiotensin I converts to angiotensin II, a potent vasoconstrictor and stimulus to reabsorb sodium and water. The vasoconstrictor effect raises blood pressure and flow to the glomerulus, inhibiting further renin release (a negative feedback mechanism) as perfusion pressure normalises. This allows the return of natriuresis (sodium excretion) and diuresis. This response is essential in assisting with retaining fluid in the event of a falling blood pressure, or boosting fluid excretion as blood pressure rises. It also responds effectively to a rise in sodium intake by reducing angiotensin II formation and allowing a larger natriuresis, resulting in maintenance of sodium balance, a key to tissue fluid distribution and balance.10
Atrial Natriuretic Peptide
Atrial natriuretic peptide (ANP) is a hormone released from the atria of the heart in response to atrial stretching during periods of increased circulating fluid volume. ANP is therefore often described as having an antagonising effect to the RAAS (which acts primarily to preserve sodium and water). These natriuretic, and hence diuretic, effects are mild and self-limiting, and occur in response to mild rises in GFR and reductions in sodium reabsorption. As blood pressure falls, the drop in GFR compensates for the effect of ANP, ensuring that excessive loss of sodium and water does not occur.10
Regulation of Acid–base and Electrolyte Balance
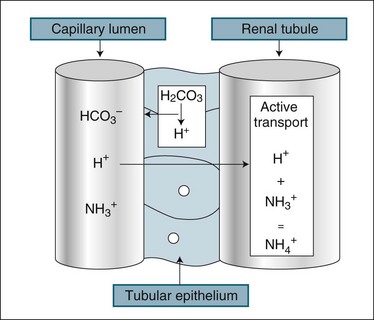
The kidney assists in the management of body pH by regulation of the excretion of H+ and HCO3− ions. While the renal response to alkalosis or acidosis is slow in comparison with plasma buffers and respiratory regulation (see Chapter 13), it does result in a net loss of H+ ions or recovery of HCO3− ions, which are the basis of human pH balance (see Figure 18.5). During acidosis the kidney raises H+ secretion by active transport to combine with ammonia (NH3+) in the renal tubule to form ammonium (NH4+), which is unable to be reabsorbed. Coincidentally, raised H+ excretion increases the reabsorption of sodium, which increases the alkalytic ion, bicarbonate (HCO3−). Conversely, during alkalosis the reabsorption of hydrogen ions is increased. These changes in secretion of hydrogen ion concentration in the renal filtrate alter the pH of the urine down to a maximum level of 4. The buffering of H+ with ammonia reduces the acidifying effect of the hydrogen ions, particularly as some ammonium combines with chloride to form ammonium chloride.10
Role as an Endocrine Organ
The kidney has two homeostatic roles as an endocrine organ. Although neither have effects relevant to acute illness, patients with chronic renal dysfunction often need supplementation to overcome the loss of renal endocrine support. Erythropoietin is important in stimulating the generation of new red blood cells and is released from the kidney in response to a sustained drop in arterial blood oxygen levels. Calcitriol helps regulate the absorption of calcium from the gut, which in turn promotes bone resorption of calcium and the reabsorption of calcium in the kidney. The kidney also acts to convert vitamin D to its active form, which is necessary for the maintenance of body calcium levels.10
Pathophysiology and Classification of Renal Failure
Diseases of the kidneys affect structure and therefore the function of the nephrons in some way. Pathology such as this, if untreated, may not cause complete loss of renal function (i.e. ARF), but is dependent on the amount of nephron damage or ‘injury’ occurring at the time of the illness, and whether the patient has had any previous illness that resulted in undetected kidney damage.11 By focusing on factors that resulted in kidney injury, both individually and collectively, then more serious damage that may result in failure can be averted. This concept is more clearly described in the later section on ATN and AKI which includes the RIFLE Criteria.1,2
The conventional classification of ARF is based on the perceived causative mechanisms, as outlined by numerous authors,12,13 however, irrespective of causative mechanism, the same renal replacement therapies are suitable to treat this:14
Prerenal Causes
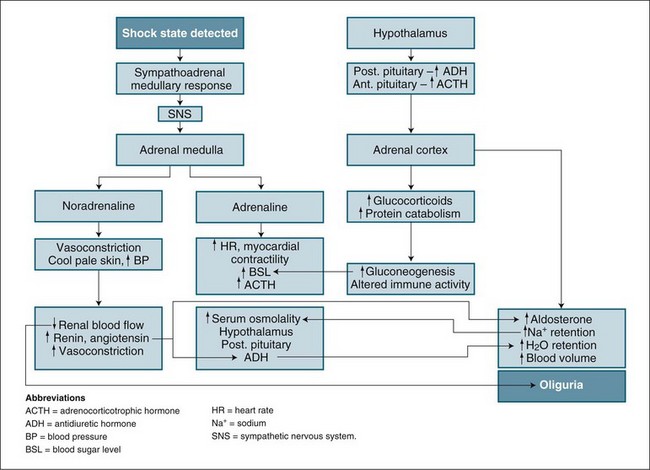
Prerenal factors affecting blood supply to the kidneys, such as hypovolaemia, cardiac failure or hypotension/shock, can cause ARF. The mechanism and outcome are easily related. As blood flow to the kidneys is reduced, less glomerulofiltration occurs, urine production diminishes and wastes accumulate. This state can be reversed by restoration of blood volume or blood pressure. In the short term (1–2 hours), nephrons remain structurally normal and respond by limiting fluid lost by urine production while concentrating the excretion of waste products. The physiological process combines the neuroendocrine control of the hypothalamus and the sympathomimetic response, which then regulates both antidiuretic hormone secretion and the stimulation of the renin–angiotensin–aldosterone system (see Figure 18.4). This process is highly influenced by any preexisting illness or concurrent factors such as diabetes and systemic infection.12
Intrarenal (Intrinsic) Causes
Intrinsic damage to the nephron structure and function can be due to infective or inflammatory illness, toxic drugs, toxic wastes from systemic inflammation in sepsis, vascular obstructive thrombus or emboli. In differentiating this type of ARF, a process of elimination has been suggested where failure of kidney function persists after the restoration of adequate perfusion (blood flow), or where no loss of perfusion has occurred, and there is no obstruction to urine flow.15 Diagnosis is made by exclusion of other causes. The common causes of this type of ARF, glomerulonephritis, nephrotoxicity and chronic vascular insufficiency, are discussed below.
Glomerulonephritis
This condition is caused by either an infective or a non-infective inflammatory process damaging the glomerular membrane or a systemic autoimmune illness attacking the membrane.14 Either cause results in a loss of glomerular membrane integrity, allowing larger blood components such as plasma proteins and white blood cells to cross the glomerular basement membrane. This causes a loss of blood protein, tubular congestion and failure of normal nephron activity. Resolution is based on treating the cause, such as an infection or autoimmune inflammatory illness.16
Nephrotoxicity
Nephrotoxicity occurs as a result of damage to nephron cells from a wide range of agents, including many drugs used in critical care (e.g. antibiotics, anti-inflammatory agents, cancer drugs, radio-opaque dyes).17 Toxic products of muscle breakdown in severe illness and trauma, commonly called Rhabdomyolysis (see Chapter 23 for trauma association),4,13,18 blood product administration reactions and blood cell damage associated with major surgery are also causative agents.19 As these agents may often be given concurrently, a cumulative effect, along with intermittent falls in renal perfusion, may result in the development of intrinsic ARF.
Vascular Insufficiency
One-third of patients who develop ARF in the ICU have chronic renal dysfunction.20 This chronic dysfunction may be undiagnosed prior to the critical illness, and may be related to diabetes, the ageing process and/or long-term hypertension. These factors create a reduction in both large and microvasculature blood flow into and within the kidney, therefore reducing glomerular filtration activity and affecting the reabsorption and diffusive process of the nephron. This reduction in blood flow is exacerbated by degenerative vessel obstruction with atheromatous plaque, particularly pronounced in diabetic patients due to ineffective glucose metabolism. Diabetic patients are more likely to develop ARF associated with medical care in hospital from what may otherwise seem to be a relatively trivial insult to the kidneys in a younger, healthy patient. The event may be enough to trigger ARF in these patients, as they lack any degree of ‘renal reserve’ or tolerance to events such as low blood pressure or administration of nephrotoxic drugs normally filtered by the kidneys.13
Postrenal Causes
Urinary tract obstruction is the primary postrenal cause of ARF, and is uncommon in the critical care setting as it is rarely associated with acute onset renal failure.13 Postrenal obstruction is more common in the community and is associated with urological disorders such as prostate gland enlargement in males, urinary tract tumours and renal calculi formation impairing urine outflow. It is essential that blockage of any urinary drainage device be excluded in the critically ill patient when undertaking an assessment of apparent oliguria.
Acute Tubular Necrosis and Acute Kidney Injury
Intrinsic ARF (described above) is often associated with typical microscopic changes on pathology examination of kidney tissue. This pathology is termed acute tubular necrosis (ATN), and possibly explains how and why, in the acute setting, kidneys can fail abruptly to minimal to no function (no urine output and therefore no waste clearance), and can then after a period of time, with artificial support, recover to normal function in many patients.21 This is an interesting area for current research into the mechanisms responsible for acute kidney failure that are yet to be fully understood.
Acute tubular necrosis describes damage to the tubular portion of the nephron and may range from subtle metabolic changes to total dissolution of cell structure, with tubular cells ‘defoliating’ or detaching from the tubule basement membrane.22 Most ARF is multifactorial in origin and may involve more than one causative mechanism and is not always an ischaemic or necrotic event.15 In critical illness, the most common combination causing ARF is the administration of nephrotoxic agents in association with prolonged hypoperfusion or ischaemia (oxygen deprivation).22 This type of tubular necrosis can be further mediated by infection, blood transfusion reactions, drugs, ingested toxins and poisons, or be a complication of heart failure or major cardiovascular surgery. The initial insult can also be compounded by metabolic disturbances and subsequent systemic infection.
ATN is the causative mechanism for up to 30% of acute kidney failure in the intensive care setting,23 with the precise causative illness not easily identifiable in critically ill patients with multiple co-morbidities, for example diabetes, advanced age, investigations requiring radio-opaque dye administration, potent and nephrotoxic drug administration or major surgery with an inflammatory state due to an underlying infection. This is the context of critical illness and ARF where, despite modulation of the cause and support with artificial renal replacement therapies, mortality ranges from 28–90% depending on diagnostic criteria or definition.3,24,25
This type of kidney damage is of particular importance, as ATN is abrupt in onset and causes a rapid cessation of normal nephron function, a picture typical of any critical illness and failure of other body organs. As this failure is commonly mediated by a loss in total or regional blood flow to the kidney,26 it is more pronounced in the kidney medulla or outer regions sensitive to reduced blood flow. The cause of this loss in blood flow may be multifactorial but is commonly associated with shock and consequent low blood pressure (see Figure 18.6). Tubular cells suffer an ischaemic insult, causing a shedding of the cells from the nephron basement membrane. This shedding of cells has an initial loss of cell polarity, and then cell death, with a ‘patchy’ occurrence along the tubule basement membrane.21 In addition, some cells detach themselves before death in a response known as apoptosis (cell self-death)21 (see Chapter 21). The response is aimed at organ survival, with some individual cells ‘sacrificing’ themselves during a period of crisis. This protective response reduces oxygen demand by initiating cell death in some tubules, while others differentiate and/or proliferate for repair, and allows continuation of some normal function. If the causative process abates, remaining cells regenerate by differentiation and proliferation, tissue repair occurs with restoration of normal epithelium in some tubules and nephron function returns.
During this period cellular ‘debris’ collects in the tubule loops, causing obstruction of tubular flow, with backleak of filtrate occurring through the ‘patchy’ exposed tubular membrane surface. An inflammatory process is also stimulated due to release of cell adhesion factors and leucocyte activation,27 which in turn causes further vasoconstriction and ischaemia28 in the acute stage. The backleakage and static tubular fluid creates a concentrate that, by diffusion, raises blood levels of wastes such as urea, creatinine and other toxins. Along with this cessation of urine flow, toxicity occurs with high serum levels of wastes such as urea, creatinine, potassium and undefined toxins.27 This is the clinical state of ARF associated with the pathology of ATN and now referred to as acute kidney injury (AKI) to better describe the total ‘picture’ of pre-illness status, immediate causative events and degree of injury determined by patient serum creatinine or urine output.1,2
Some authors prefer the term ‘ATN’ to describe ARF,11 using it as a surrogate for ARF in the acute setting, as it focuses on the pathophysiology of tubular damage, recognising this damage as a final outcome of all causative factors. However, more recently, with the development of a consensus definition for ARF describing stages of illness severity, the term acute kidney injury (AKI) is now used reflecting pathophysiology, the outcome of many causative factors, and the clinical context where small derangements may be evident with reversibility of dysfunction and recovery, through to irreversible damage with kidney failure.1,2
The kidneys are vital human body organs essential to sustaining life. An important interrelationship of the kidneys and other body organs exists, with the brain, heart, liver and lungs dependent on receiving ‘clean’ blood to function. As toxins accumulate in ARF these organs become dysfunctional,29,30 although many of these interrelationships (e.g. the liver) are poorly understood.31
Acute Renal Failure: Clinical and Diagnostic Criteria for Classification and Management
Clinical Assessment
Clinical assessment of the patient with impending renal failure can involve myriad tests and investigations; however, the majority of these are not used to assess the critically ill patient. The clinical history is important in differentiating preexisting renal disease and cataloguing the numerous factors already discussed that can contribute to renal dysfunction. As the majority of renal failure patients in the ICU will succumb to the combination of prerenal renal failure and ATN, the key assessments used in monitoring renal function are urine output, serum creatinine and urea levels, combined with more general haemodynamic measures including HR, CVP, BP, PCWP. These measures are essential for the critically ill patient, and alterations provide the diagnostic key. They also link into the wider assessment of fluid and electrolyte balance, as described in Chapters 9 and 19.
Diagnosis
The management of ARF begins with making the diagnosis, based on the patient’s presenting signs and symptoms linked to a patient history. A long-term history of renal disease involving urinary tract infections, diabetes, cardiac failure and systemic inflammatory illnesses are all highly relevant.11 Immediate history of presentation to a hospital involving surgery or any life-threatening illness with associated shock is also highly relevant in association with reduced urine output volumes over time.
Nurses in the critical care setting who measure urine output hourly, readily recognise a key sign of impending renal dysfunction. Oliguria in the absence of catheter obstruction should be responded to quickly, as this suggests inadequate kidney blood flow and to some extent is a delayed observation considering the continual moderation of kidney function producing urine output. Persisting oliguria or the onset of anuria with associated rises in blood creatinine defines renal failure. This sequence of events can be identified in a criteria pathway published following a consensus meeting of physicians. The ‘RIFLE’ criteria32 – risk, injury, failure, and outcome criteria of loss and end-stage disease – provides an increasingly widely-accepted approach to diagnosing and classifying ARF.
Consensus Definition: the Rifle Criteria
The RIFLE criteria are indicated in Figure 18.7, and use raising creatinine and lowering urine output as highly sensitive and specific indicators for a continuum of renal failure. This is a useful classification system to grade loss of kidney function, reflecting stages of injury to the kidney before failure occurs. Without this, the small but important losses of kidney function before the failure state are not adequately considered.1 This approach provides a consensus definition for loss of kidney function that is useful for clinical practice and research into this area, with clinicians all talking the same ‘language’ when comparing patients and/or results from clinical trials. The shape of the diagram indicates that more people will develop symptoms of ARF linked to kidney ‘injury’ and be considered ‘at risk’ (high sensitivity) than those at the bottom of the definition, who are fewer in number but need to fulfil strict criteria (high specificity).
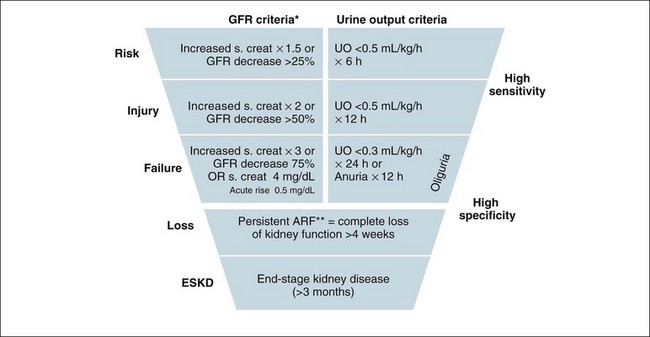
FIGURE 18.7 Criteria for diagnosis of acute renal failure: the risk, injury and failure criteria with outcomes of loss and end-stage renal disease (RIFLE).32
To better understand the RIFLE criteria in a clinical care context, the following discussion is useful to consider in association with a review of Figure 18.7. In a hospital setting, those patients with a risk of renal injury would be identified by a low urine output of less than 0.5 mL/kg/h for 6 hours. In this situation their creatinine would be expected to rise, indicating a concurrent reduction in glomerular filtration rate. Reducing renal risk requires basic measures such as increasing their fluid intake and continuing to closely monitor urine output, whilst reviewing the patient’s medications, haemodynamic state and possible other causes of injury in order to prevent further deterioration.
Clinical Management
In the critically ill patient, kidney function failure may be associated with an initial renal response to a fall in perfusion associated with systemic shock. As the majority of patients recover their renal function from ICU ARF,8 initial clinical management is aimed at reducing further renal damage. If kidney function becomes so compromised that blood pH, fluid and electrolyte balance cannot be sustained, then a replacement therapy will need to be introduced. This is continued until kidney function is marked by the return of urine production or patients are moved to a more chronic form of replacement therapy, such as intermittent haemodialysis.
Reducing Further Insults to the Kidneys
After diagnosis, the next management principle is to remove or modify any cause that may exacerbate the pathological process associated with ARF. Further interventions and investigations are performed in relation to the findings from the history and presentation. These may include:13
• further intravenous fluid resuscitation (despite an oligo-anuric state) and restoration of blood pressure
• physical or diagnostic assessment for renal outflow obstruction and alleviation if present
• ceasing or modifying the dose of any nephrotoxic drugs or agents and treating infection with alternative, less toxic antibiotics.
Initial management strategies for developing ARF remain conservative, with careful management of fluid (once adequate circulating volume is assured by initial fluid resuscitation) and haemodynamics, encouraging diuresis if present, monitoring blood profiles for changes in urea and electrolytes, and limiting or reformulating the administration of agents that may contribute to the accumulation of urea and electrolytes (e.g. enteral or IV nutritional supplements). The use of agents such as mannitol, dopamine and frusemide, while popular in practice, have not been shown to be of any value in improving outcome in patients at risk of ARF.13,20
Despite these efforts, life-threatening biochemistry may arise in ARF, such as severe acidosis and hyperkalaemia (a pH of <7.1 and a serum potassium >6.5 mmol/L) that requires immediate treatment and can be an indication for beginning RRT without elevation of serum creatinine and oliguria and fluid overload.33
Nutrition
When ARF is persistent, providing nutritional support is another important management strategy. A review of nutrition in ARF suggested that an intake of 30–35 kcal/kg/day and a protein intake of 1–2 g/kg/day is essential due to the combined increase in protein catabolism and caloric requirements of associated critical illness.34 This nutrition is provided by the enteral or parenteral route and constitutes an increase in body fluids and nitrogen load. Ironically, this aspect of managing ARF in critical illness may also be an indication for starting RRT35 (see Chapter 19 for further discussion on nutritional requirements in critical illness).
Renal Replacement Therapy
If conservative measures fail, then the ongoing management of the patient with ARF requires RRT. This enables control of blood biochemistry, prevents toxin accumulation, and allows removal of fluids so that adequate nutrition can be achieved. The criteria and indications for initiating RRT are listed in Box 18.1. One indication is sufficient to initiate RRT, while two or more make RRT urgent and mandatory. Combined derangements can create the necessity to commence therapy before individually-defined limits have been reached. Early initiation of treatment is widely advocated and may confer more rapid renal recovery.
Box 18.1
Proposed criteria for the initiation of renal replacement therapy in adult critically ill patients14
Renal Dialysis
History
Dialysis is a term describing RRT and refers to the purification of blood through a membrane by diffusion of waste substances.36 Table 18.1 outlines the historical events in the development of dialysis. The Kolff rotating drum kidney, one of the earliest attempts at RRT (illustrated in Figure 18.8), used cellulose tubing rolled around a wooden skeleton built as a large, drum-styled cage. Cellulose acetate (material similar to ‘sticky tape’) tubing was strong, did not burst under pressure and could be sterilised.37 The drum with the blood-filled cellulose tubing wound around it was immersed in a bath of weak salt solution, and as blood passed through, the rotating cellulose tubing allowed waste exchange to occur by diffusion. This method and the diagram included is useful information as it is essentially the key concepts for how dialysis is today with modern machines and dialysis membranes. A major impediment to the safe use of this system was, however, the large amount of patient blood required in the tubing and membrane.
| Time period and developer | Description |
|---|---|
| 1854: Thomas Graham, Scottish chemist | First used the term ‘dialysis’ to describe the transport of solutes through an ox bladder, which drew attention to the concept of a membrane for solute removal from fluid. |
| 1920s: George Haas, German physician | First human dialysis was carried out, performing six treatments on six patients. Haas failed to make further progress with the treatment but is recognised as an early pioneer of dialysis. |
| 1920–30s | Synthetic polymer chemistry allowed development of cellulose acetate, a membrane integral to the further development of dialysis treatments. |
| 1940s: Willem Kolff, Dutch physician | The discovery of heparin, an anticoagulant, enabled further development of dialysis during World War II, the Kolff rotating drum kidney. |
| 1940–50s: Kolff and Allis-Chalmers, USA | Further modification of Kolff dialyser and the development of improved machines. |
| 1950s: Fredrik Kiil, Norway | Developed the parallel plate dialyser made of a new cellulose, Cuprophane. This required a pump to push the blood through the membrane and return the blood to the patient. |
| 1950–60s | Dialysis began to be widely used to treat kidney failure. |
| 1960s: Richard Stewart and Dow Chemical, USA | The hollow-fibre membrane dialyser used a membrane design of a cellulose acetate bundle, with 11,000 fibres providing a surface area of 1 m2. |
| 1970s | Use of first CAVH circuits for diuretic resistant oedema by Kramer |
| 1980s | First continuous therapies using blood pump and IV pumps to control fluids removal and substitution: Australia and New Zealand led the way |
| 1990s | New purpose built machines used; Gambro Prisma, Baxter BM 11 + 14 to provide pump controlled therapies with integrated automated fluid balance using scales to measure fluids. Cassette circuits, automated priming; new membranes |
| 2000 | Further purpose built machines using direct measurement for waste and substitution fluids via Hygieia–Kimal machine. Introduction of high fluid exchange rates for sepsis treatment. introduction of dialysis based machines in ICU for daily ‘hybrid’ treatments: SLEDD and SLEDDf |
| 2010 | Multiple CRRT machines; more advanced graphics interface and smart alarms. Waste disposal systems. High flux, porous membranes |

This large extracorporeal blood volume became a focus for further development of the therapy. The goal was to develop a membrane for solute exchange with a greater surface area than the cellulose membrane used by Kolff but needing less blood volume. This led to the development of the hollow-fibre filter membrane structure in the 1960s, the same design concept that is used today. Since then significant developments have occurred, with new fibres using the polymer polysulfone or other artificial synthetic chemical structures that better imitate the nephron glomerulus and the ability to transfer wastes and plasma water38 for an effective ‘artificial kidney’.
This combination of extracorporeal circuit (EC), blood pump and filter membrane (or artificial kidney or dialyser), and the associated nursing management is now commonly known as haemodialysis. The major treatment components are essentially the same as those first developed in the 1960s, with the key component being the device membrane. Over the past 50 years, industrial and scientific developments such as plastics moulding and electronics have made current dialysis techniques safe, effective and a life-sustaining treatment for the millions of people who suffer acute and chronic kidney failure.39,40
Historical Perspectives of Dialysis Nursing
Key to the application of these technical and scientific developments has been the role of nurses, who have made a substantial contribution to the safety and efficiency of dialysis. Barbara Coleman is recognised as the first dialysis nurse to publish a treatment protocol for dialysis using the rotating drum machine in 1952.39,41 Nursing of dialysis patients has developed into a specialist field of knowledge and skill, with nurses combining their holistic view of patient management with the specialist needs of patients with renal failure, from the outpatient setting to the ICU,42 including a collaborative approach to further adaptations of dialysis best suited to the critically ill.43,44
Development of Renal Replacement Therapy in Critical Care
Historically, ARF was treated in the ICU with the use of peritoneal dialysis (PD), which did not require specialist nurses or physicians.45 This simple technique removes wastes by infusing a dialysis fluid into the abdomen, allowing diffusion and osmosis to occur between the peritoneum and fluid before draining out again in repeated cycles.46 This was performed by the ICU nurse and prescribed by ICU physicians, but was inadequate in its clearance of waste and fluid volume, and was associated with infection, limiting respiratory function and exacerbated glucose intolerance.14,47
In 1977 Peter Kramer, a German ICU physician, frustrated with the limitations of PD and the delays in gaining a dialysis nurse and machine to attend the ICU, developed a new dialytic technique by inserting a catheter into the femoral artery and allowing blood to flow to a membrane and back to the femoral vein. As the blood passed through the membrane, plasma water was filtered out.48 The technique was called continuous arteriovenous haemofiltration (CAVH). It was later renamed slow continuous ultrafiltration (SCUF), as it enabled the removal of plasma water in addition to dissolved wastes (convective clearance of solutes) at a flow rate of 200–600 mL/h by passive drainage from the membrane as blood flowed through it. Kramer reported ‘considerable therapeutic potential’ after treating 12 patients in the ICU, although suggesting that this level of ultrafiltration and waste clearance was inadequate for optimal support of ARF in many critically ill patients.6 This marked the beginning of continuous RRT in the ICU as an intervention prescribed and managed by ICU nurses and doctors for patients with ARF. A schematic diagram of Kramer’s original technique is illustrated in Figure 18.9.
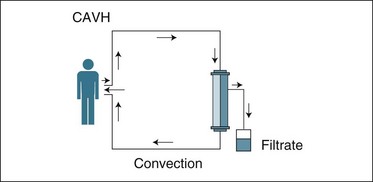
FIGURE 18.9 CAVH used in the ICU in the late 1970s. Patient arterial blood pressure provides flow to the membrane, and blood returns via a large vein. Plasma water removal occurs via filtration and is pressure-dependent.48
The increase in demand for chronic renal failure dialysis made it problematic to provide any service to patients in the ICU with ARF by dialysis nurses. Dialysis for patients in the ICU was often delayed or unavailable while using the same human and material resources from a dialysis unit.49 It was thus inevitable that in the ARF context ICU nurses would adopt the role of the dialysis nurse, in addition to their established role in ICU of caring for critically ill patients using mechanical ventilation and haemodynamic interventions. Physician-prescribing rights also favoured this role for nursing in Australia and New Zealand, as the ICU physician can prescribe renal dialysis without the referral of the patient to a nephrologist that is required in other countries.8,49,50 In the Australian context, this resulted in the provision of renal support for critically ill patients by ICU nurses alone, or in a shared role with renal dialysis nurses.50
Different approaches or models of care are created by many factors, including the presence of a dialysis service in a hospital, the number of patients requiring treatment in an ICU per annum, physician rights of prescribing as specified by health insurance scheme structures, and the number, professional competence and flexibility of the nursing workforce available for an ICU or dialysis service in a given hospital.44,50,51
Refinement of Renal Replacement Therapy
Although CAVH as developed by Kramer was useful in removing excessive body water and some wastes, a dialysis blood pump enabled a much more efficient technique for therapeutic benefit in the ICU patient with ARF. The use of roller blood pumps to generate pressure and a reliable flow of blood, thus eliminating the need for arterial puncture and access, was introduced by two German groups.49 This approach, termed continuous veno-venous haemofiltration (CVVH), could reliably pump blood at a constant rate and achieve ultrafiltration volumes of 1000 mL/h. This therapy was able to remove large volumes of plasma water and, if run continuously with similar amounts replaced by a balanced plasma water substitute, an effective clearance of wastes similar to a high-intensity dialysis treatment could be achieved without cardiovascular instability.
With further modifications to the circuit and filter set-up, a diffusive component was added to the therapy by running a dialysate volume through the haemofilter, flowing between the membrane fibres and countercurrent to blood flow. This was termed continuous veno-venous haemodiafiltration (CVVHDf). To deliver continuous forms of veno-venous RRT required the introduction of blood pumps from modified dialysis machines into the ICU, and created a major education and training need for critical care nursing.52 This was first met in the Australian setting by dialysis nurses until independent practice was achieved. This training often used old dialysis machines and equipment that had been superseded in the chronic dialysis setting by new, more efficient dialysis machines. The old machines were suitable for the ICU to use because of their technical simplicity, with low or no purchase cost.42 As CRRT became more widespread in the ICU, this encouraged manufacturers to design and market purpose-built CRRT machines with automation, intelligence software for alarm detection and practical solutions.53
As already noted, in the Australian and New Zealand setting CRRT is provided by ICU nursing staff with medical prescription by ICU physicians, with nephrologists focused on chronic disease and care of patients outside the ICU. In North America, CRRT is not always the preferred choice for supporting ARF in ICU, and intermittent dialysis techniques prevail.54 As a result of this mixed application of techniques, there have been a number of publications highlighting the success, utility and methods for CRRT in the ICU setting,47,55,56 with a smaller number of publications either making comparisons between approaches or continuing to support intermittent techniques using dialysis in the ICU.57–60
While this application of both CRRT and intermittent haemodialysis (IHD) has prevailed for the management of ARF, new ‘hybrid’ approaches have evolved that combine some benefits of both CRRT and IHD, but also do suffer from the compromise nature of the approach. These are daily, limited time frame treatments, with one variant of the approach termed EDDf (extended daily diafiltration), which provides a further treatment option for ARF in the ICU. While daily treatments are emerging as the only method of RRT for ICU patients in a limited number of hospitals59,61,62 evidence as to their utility in treating the most critically ill patients is lacking. This means that continuous forms of RRT prevail as the commonest method for treating ARF in the ICU in Australia and Europe.
Approaches to Renal Replacement Therapy
Both IHD and CRRT require a machine to pump blood and fluids; pressure and flow devices to monitor treatment; a tubing and filter membrane set that together create an extracorporeal circuit (EC) (outside the body blood pathway); and a catheter connecting the patient’s circulation to the circuit (see Figure 18.10). This catheter enables blood to be drawn from and returned to the patient (known as ‘access’). Access can be achieved by three different techniques:
• temporary catheters inserted via a skin puncture into an artery (A) for drawing blood and a vein (V) to return the blood (AV access)
• a surgical joining of an artery and vein (usually in the forearm), making a large vessel that is punctured with needles to both draw and return the blood (AV fistula)
• a catheter with two lumens to draw and return blood via a large central vein63 (veno-venous access catheter).
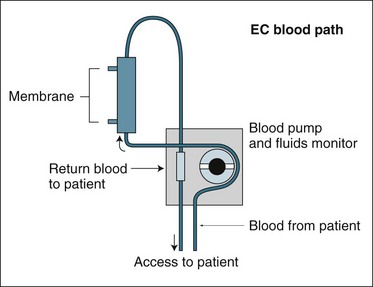
In the acute renal failure setting and where temporary treatment is anticipated, the two-lumen catheter is recommended.8,64
Haemodialysis, Haemofiltration and Haemodiafiltration
There are numerous approaches to the delivery of RRT. Haemodialysis, haemofiltration and haemodiafiltration are three common techniques used to achieve artificial kidney support in ARF. The basic blood path or circuit for these therapies is indicated in Figure 18.10 and is useful to review as a basis for understanding each of the three circuits for each therapy and where the RRT fluids are then applied to this circuit differentiating them as alternative techniques.
The extracorporeal component is a common factor in all these different circuit designs. The difference between treatments is how the solutes (urea, creatinine and other waste products) and solvent (blood plasma water) are removed from the blood as it passes through the filter membrane (artificial kidney), and the intermittent versus continuous prescription of the therapy. This is determined by the way in which the dialysis fluids are mixed with or exposed to the blood, the rate and direction of blood and fluid flows, and how fluid loss or a negative fluid balance is achieved. The three physical mechanisms of fluid and solute management are convection, diffusion, and ultrafiltration. Table 18.2 lists the commonly-used abbreviations to describe the timing of treatment, blood access for the therapy and mode of solute removal.
TABLE 18.2 Abbreviations describing modes of renal replacement therapy
| Timing of therapy | Route of access | Mechanism of solute removal |
|---|---|---|
Convection
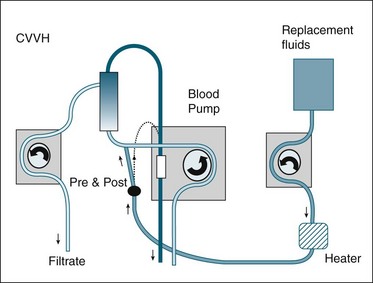
Convection is the process whereby dissolved solutes are removed with blood plasma water as it is filtered through the dialysis membrane. The word is derived from the Latin convehere, meaning ‘to remove or to carry along with’.65 This process is very similar to that occurring in the native kidney glomerulus, as plasma water is filtered across the nephron tubule via the Bowman’s capsule. In RRT, the plasma water with the dissolved wastes is discarded; the plasma water deficit is then replaced with manufactured artificial plasma water in equal or slightly lower amounts to achieve a desired fluid balance. This blood washing (purification) process is commonly known as haemofiltration. When applied on a continuous basis in the ICU, haemofiltration can adequately replace essential renal functions, and is particularly effective in managing fluid balance.66–70 Figure 18.11 illustrates the circuit and set-up for continuous veno-venous hemofiltration.
Diffusion
Diffusion refers to the physical movement of solutes across a semipermeable membrane from an area of high concentration to that of a relatively low concentration;66 that is, solutes move across a concentration gradient.71 A higher concentration gradient results in a greater rate of diffusive clearance. As blood passes through the dialysis membrane, dialysate fluid, reflecting normal blood chemistry, is exposed to the blood on the opposing side of the membrane fibre. Diffusive clearance is continuous as solute exchange occurs by diffusion with the dialysate fluid and the blood continually moving in and out of the membrane. As ‘dirty’ or waste-laden blood enters the membrane and ‘clean’, fresh dialysate is in continuous supply, this process performs an effective waste-removal process. The two mediums are usually established in a countercurrent or opposing flow to each other, making diffusion another process, mimicking the normal nephron function of the kidneys.71
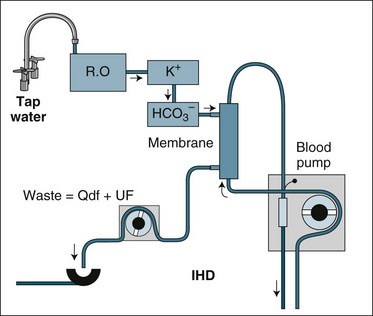
Diffusive clearance technique can be performed with increasing intensity and effect by making the blood and dialysate flow faster, with technical problems associated with delivering the high fluid and blood flow being the main limiting factor increasing clearance. The two flows need to be maintained in relation to each other; for the diffusive clearance to be efficient the dialysate flow must always equal or exceed the blood flow. A common setting for an intermittent dialysis treatment would be a blood flow and dialysate fluid flow of 300 mL/min each. A faster blood flow is not useful unless dialysate flow is also increased, as more waste solutes will not be cleared if the dialysate fluid and blood are in diffusive equilibrium. The technique of solute removal using diffusion alone is termed dialysis; when used with blood, the process is termed haemodialysis (HD). When applied on an intermittent basis, as is normal for patients receiving RRT for chronic renal failure, it is called intermittent haemodialysis (IHD).72 Figure 18.12 illustrates the circuit set-up for IHD.
Ultrafiltration
Ultrafiltration is the process that allows plasma water to leave the blood, achieving body fluid or water loss. Dialysis nurses measure a fluid loss by weighing the patient before and after a treatment. This process is primarily used to achieve fluid balance, an important function of the kidneys.33,66 The only difference between this process and the convective clearance of solutes is that this fluid is not replaced, and it is therefore not considered an adequate solute management method. Ultrafiltration cannot be undertaken in large amounts without fluid replacement, as it would cause hypovolaemia. It is therefore implemented during a dialysis period by removing small amounts each hour (e.g. 250 mL/h for 4 hours) of the intermittent treatment cycle.
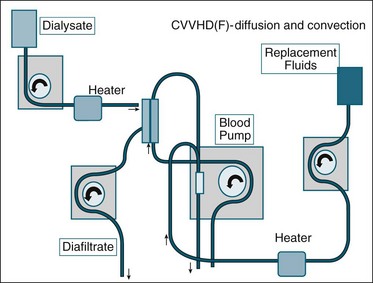
There are different therapeutic effects from each form of RRT and different operational prescriptions of blood and fluid flow. Combinations of convection and diffusion can be used, known as haemodiafiltration (CVVHDf).66 An increase in the diffusive component (i.e. raising the dialysate flow rate in CVVHDf) will increase the removal of small-molecular-weight substances such as potassium and assist with hydrogen ion exchange via buffer solution. This can also be achieved via increasing filtration fluid flow (convective clearance), which will also add an increase in clearance of larger molecules, for example those associated with severe infection and systemic inflammation or sepsis. Figure 18.13 illustrates the circuit and set-up for CVVHDf.
Major Circuit Components for Crrt
Membranes
The filter or haemofilter (blood filter) is the primary functional component of the RRT system, responsible for separating plasma water from the blood and/or allowing the exchange of solutes across the membrane by diffusion. The filter is made of a plastic casing, containing a synthetic polymer inner structure arranged in longitudinal fibres. A schematic diagram of a haemofilter is shown in Figure 18.14. The fibres are hollow and have pores along their length with a size of 15,000–30,000 daltons. This allows plasma water to pass through, carrying dissolved wastes out of the blood (most of which have a molecular weight <20,000 daltons), while larger plasma proteins and blood cells (at least 60–70,000 daltons) are retained.65 Plasma water separated from the blood in this way is carried away from the filter by a side exit port and a pump, where it is measured and directed into a collection bottle or bag as waste; this convective clearance of solutes is similar to urine produced by the normal kidneys. The plasma water loss is replaced in equal volume with a commercially-manufactured plasma water substitute, either after the ‘filtered’ blood exits the haemofilter (postdilution) or prior to the blood entering the haemofilter (predilution) or both at the same time. The plasma water replacement contains no metabolic wastes and achieves blood purification as it is continuously replaced.67–7073
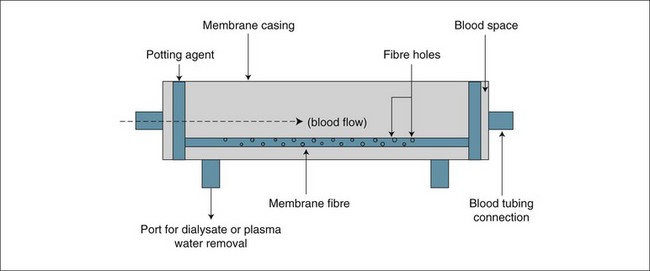
Filter membrane polymers are of different materials: AN69 (acrylonitrile/sodium methallyl sulfonate), PAN (polyacrylonitrile) or PA (polyamide), and polysulfone;74 however, all demonstrate similar artificial kidney effects and are generally applied according to the physician’s preference.42 The most important characteristics of filters used in continuous modes of therapy are: (a) a high plasma water clearance rate at low blood flow rates and circuit pressures; and (b) high permeability to middle-sized molecular weight substances (500–15,000 daltons, e.g. inflammatory cytokines), which are often encountered in critical illness.
Vascular Access
As previously noted, in order to establish CRRT it is necessary to create a blood flow outside the body using the EC. Blood is most commonly accessed from the venous circulation of the critical care patient via a catheter placed in a central vein (e.g. femoral). Blood is both withdrawn from the vein and returned to the same vein – that is, veno-venous (VV) access by means of a double (dual)-lumen catheter.70 When the same procedure is carried out by accessing the blood from the patient’s systemic circulation via an artery and returning it to a vein, the term arteriovenous (AV) is used.75,76 In this system there is no mechanical blood pump required, as the patient’s arterial blood pressure provides a flow of blood in the EC. Veno-venous haemofiltration (CVVH) has the advantages of requiring only a single venipuncture, a reliable blood flow delivered from a blood pump, and alternative venous access sites if site infection or access is difficult.77 While it is easy to establish flow within AV-driven circuits and no complex system of blood pumps and pressure sensors is necessary, this method is susceptible to flow problems associated with low patient arterial blood pressure and high venous pressures, a common occurrence in critically ill patients.
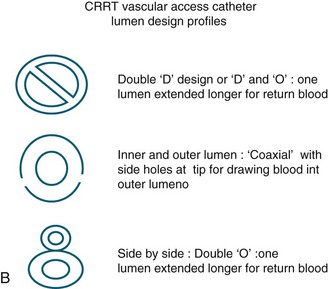
The dual-lumen catheter used for veno-venous access has an internal diameter of 1.5–3 mm and the ends of the catheter are sufficiently separated from each other in the patient’s vein to prevent filtered blood from mixing with unfiltered blood when used in the recommended sequence.77 This ensures that filtered blood does not simply pass back through the artificial kidney, where there would be minimal waste clearance compared with ‘fresh’ unfiltered blood; this design is illustrated in Figure 18.15a. The catheter must be small enough to place into a vein but large enough to provide blood flow of at least 200 mL/min for an adult CRRT circuit. Catheters are made with different arrangement of the lumens revealing variation in their cross section profile (see Figure 18.15b) There is no evidence to suggest which profile is better, but the larger the internal diameter, the less likely flow will be obstructed during patient care with CRRT. After a catheter is threaded into a vein, the blood flow may be adequate, but later during patient care it may obstruct due to different nursing interventions and patient movement, which may alter blood flows within the low pressure venous system.70

Insertion sites may be affected by nursing care interventions. Placement of the catheter is usually in the subclavian or femoral vein, and occasionally in the internal jugular vein.78 Anecdotally, the subclavian position is more easily managed for dressing and securing, continuous observation and patient comfort, but is more problematic in terms of flow reliability. Intrathoracic pressure changes associated with physiotherapy or spontaneous patient coughing and breathing, coupled with the upright position of patients, may hinder blood flow from the subclavian-sited access catheter. While these issues are not encountered with a femoral-placed catheter, flow problems can arise due to side lying and flexion at the groin or hip.42
The flow performance of any vascular-access catheter can be affected by the patient’s position in bed, spontaneous movement and repositioning activities as part of routine nursing care in the critically ill for pressure-area prevention. Catheter lumen outlet or inlet obstruction can be due to contact with the vessel wall, or to a sharp bend occurring due to the patient’s movement. These factors contribute to compromising blood flow in the EC,42,77 and have been identified by ultrasound Doppler flow probe attached to the circuit tubing.79
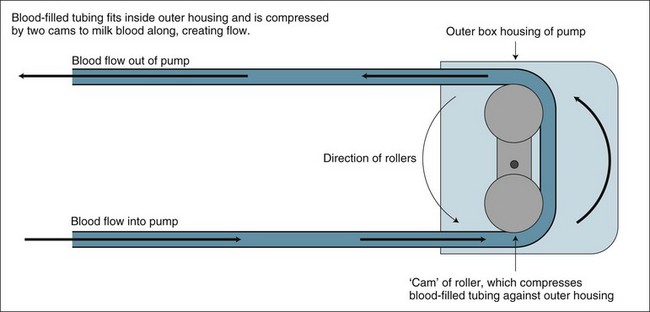
Blood Pump
In veno-venous modes, a pump component is essential as part of the patient’s blood volume flows externally to the body via the EC. Blood flow is maintained by a ‘roller pump’ (see Figure 18.16), that propels the blood along the tubing in a peristaltic fashion (milking along by compression of the tubing), compressing the blood-filled tubing but having no contact with the blood itself. This roller rotates at a rate providing a flow of fresh unfiltered blood to the haemofilter, enabling it to clear metabolic waste products.
Venous Return Line Bubble Trap Chamber
The purpose of this chamber is to prevent any gas bubbles in the EC from entering the patient’s circulation by allowing them to rise to the top of a small, vertically positioned collection reservoir (see Figure 18.17). Venous pressure is commonly measured via a tubing connection into the top of the venous chamber, and additional IV fluids can be administered into this chamber via a secondary tube connection. The level of the blood in the chamber must be below the top, to prevent spillage into the pressure monitoring line. It is advised that the blood level be adjusted to near full but allow for visual inspection of incoming blood flow and to ensure that any air bubbles are trapped here.69 As this creates a gas–blood interface within the venous chamber there is a potential source of venous chamber clotting and hence circuit failure.42,69,80 Addition of replacement fluids into this chamber when using post-dilution fluid administration can cause a plasma fluid layer to develop above the blood level and may reduce clotting by stopping blood foaming on its surface and eliminates air or gas contact with the blood.81
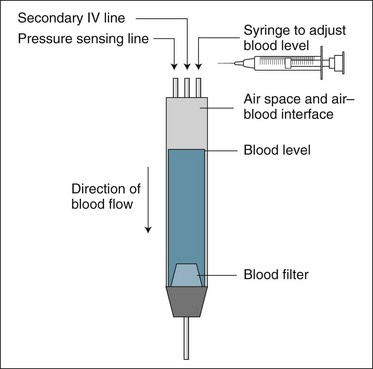
Anticoagulation
There are several different drugs utilised to prevent blood clotting in the EC; heparin, prostacyclin and sodium citrate have been used separately or in various combinations (see Table 18.3).82–84 As blood comes into contact with the plastic tubing and the polymer fibres of the filter, various clotting systems are activated. This is a normal action of blood when exposed to non-biological surfaces. The aim of anti-clotting drugs is to delay clot formation while the blood is outside the body, particularly when within the densely-packed fibres of the filter. As calcium, blood platelet cells or thrombin are vital in clot formation,80 these drugs are targeted to one of these elements. This targeting must not be too pronounced, as the patient may begin to bleed when the blood returns from the EC to the body.80
| Drug | Benefits | Precautions |
|---|---|---|
| Heparin | Inexpensive, wide experience, easily reversed, easily monitored, short half-life | Sensitivity reactions, heparin-induced thrombocytopenia, to be effective means increased risk of bleeding systemically |
| Low-molecular-weight heparin (LMWH) | Moderately inexpensive, increasing experience, less likely to result in sensitivity reactions | Difficult to monitor, not easily reversed, longer half-life, dosing varies between types of LMWH |
| Prostacyclin | Very short-acting, has a physiological role in inhibiting platelet activity, does not exacerbate other drug reactions | Expensive, no measure of effectiveness, narrow dose range with associated hypotension, individual patients sensitive to haemodynamic effects, unstable in solution |
| Citrate-based solutions | Limit anticlotting effect to EC – ‘Regional anticoagulation’; results suggest very effective in prolonging circuit life | Substantial metabolic effect if not adequately managed (serum ionised calcium must be monitored closely); requires additions to extracorporeal circuit to administer and reverse and use of specialised replacement/dialysate solutions. Not useful when liver failure present, citrate is converted to bicarbonate by the liver providing the necessary buffer for RRT. Acidosis may occur in liver failure |
| No anti-clotting agent (with saline flushes) | No side effects, no exacerbation of unstable haematological status, liver failure | May encounter very short circuit life that consumes remaining haematological components, risk of fluid overload if saline flushes not part of fluid balance. No evidence saline flushes have any benefit |
Heparin is the most commonly-used agent for the prevention of clotting, as it is inexpensive, widely available and easily reversed by another drug, protamine.82 Heparin is commonly administered into the EC before the blood enters the filter, although the optimal place to administer any anticoagulant drug during CRRT is not agreed upon.85,86 A bolus is often given prior to circuit connection, either in the circuit prime or via the venous access catheter. A maintenance dose (5–15 units/kg/h) is then adjusted against the relevant laboratory tests and a visual inspection for clotting in the EC is undertaken, particularly noting the venous bubble trap.
Citrate is another popular anticoagulant for CRRT as an alternative to heparin. Citrate buffers pH and chelates calcium inducing anticoagulation of blood by reducing serum ionised calcium level. For anticoagulation, the dose and dose rate of citrate is commonly set to achieve a reduction in the CRRT circuit blood ionised calcium level to <0.3 mmol/L.87–89 As ionised calcium is essential for the progression of the coagulation cascade to form a stable clot, an anticoagulant effect is achieved when the calcium is bound or chelated.88 A continuous infusion of citrate is administered into the CRRT circuit, as patient blood enters the circuit similar to heparin administration. A new approach in Australia includes citrate as an additive to commercially-prepared CRRT replacement fluids.90 When circuit blood returns to the patient circulation, it mixes with systemic blood and the calcium concentration is restored to normal; free citrate not binding to calcium is metabolised by the liver to provide carbon dioxide and bicarbonate as a necessary buffer.87–89 However, citrate-bound calcium is lost in the waste fluid removed and requires replacement by a separate calcium infusion to maintain serum calcium levels to normal; at 1.0–1.3 mmol/L.87 With this method, the circuit is anticoagulated, but the patient is not (also called a ‘regional’ method of anticoagulation) as the patient blood calcium level is restored to normal making this approach safer compared to heparin use and can be applied in auto-anticoagulated patients where premature circuit clotting continues to occur.91 Due to the complex nature of the citrate-based anticoagulation approach a number of different protocols have been proposed to aid in management.92 Not all methods will be applied in the one ICU, and local expertise development of one method and an alternative is common. Recent reviews provide a good synopsis of each method.93
Fluids and Fluid Balance
A key component of any CRRT is the administration of a replacement solution for the fluid removed during haemofiltration (see Table 18.4). This same physiologically balanced solution is also used as a dialysis fluid if a dialytic mode is included. The solution may have a low potassium level, so this electrolyte can be removed rapidly if hyperkalaemia is part of the original indication for CRRT. Potassium must therefore be added later to the solution to ensure that hypokalaemia does not occur. In Australia and New Zealand these fluids are commercially prepared, with the only major choice being between the type of acid buffer: lactate or bicarbonate. Some small studies have compared the use of lactate and bicarbonate fluid buffer because of a concern that the lactate solution reduced heart performance by causing cardiovascular depression and made accurate determination of patient lactic acid accumulation difficult to interpret.94–97 In most settings the use of lactate-buffered fluids in patients with cardiac and liver failure is avoided, as lactate accumulation (levels above 5 mmol/L) in these patients indicates inadequate metabolism of lactate to bicarbonate by the liver, with increased acidaemia.13 While bicarbonate solutions may appear to have an advantage over lactate-based solutions in critically ill patients, they are more expensive causing some physicians to only prescribe them when lactate accumulation is anticipated or after it occurs.
TABLE 18.4 Typical replacement/dialysate fluid constituents for CRRT
| Component | Bicarbonate-based solution (mmol/L) | Lactate-based solution (mmol/L) |
|---|---|---|
| Buffer | 25.00 | 45.00 |
| Potassium | 0.00 | 1.00 |
| Sodium | 140.00 | 140.00 |
| Glucose | 0.00 | 10.00 |
| Calcium | 1.63 | 1.63 |
| Magnesium | 0.75 | 0.75 |
| Chloride | 100.75 | 100.75 |
The higher cost, problems with reconstituting bicarbonate solution bags and manual handling of large (5 L) fluid bags has increased interest in ‘online’ fluid production from tapwater at the bedside. This approach can be cheaper, requires no bag changing or reconstituting by nurses,98 but involves installation of a complex and expensive reverse osmosis machine, the cost of which would be offset if large volumes of fluid were then consumed from this online manufacture. This approach may alter fluid selection and use in the future, and is an essential feature of chronic dialysis and use of ICU daily dialysis (EDDf) modes of therapy.59–61
Fluid balance maintenance is a key nursing responsibility in managing CRRT. Most machines now available to provide CRRT incorporate a mode selection program, including an automatic fluid balance system. This ensures delivery of a fluid prescription based on the input provided by the programming nurse. As many litres of fluid may be exchanged in an hour (25–35 mL/kg), the default setting is usually a net machine balance, where all fluids administered as either dialysate or replacement are recovered or balanced. The machine cannot, however, include fluids administered or expelled directly from the patient, so a fluid maintenance schedule must be established. This schedule is also based on input and output being equal. For example, if more fluid is being infused into the patient than being lost, as would be expected in ARF, then additional fluid would need to be recovered via the CRRT circuit: that is, less replacement fluid administered or more waste created. Consider the calculation steps in the example in Table 18.5.
TABLE 18.5 Example of CVVH fluid maintenance schedule to calculate replacement solution dose/hour
| Fluids in (mL) | Fluids out (mL) | Fluid balance (mL) |
|---|---|---|
| 1. Drugs, IV, NG = 120 mL/h (heparin, inotropes, intraflows, NG feeds) | 2. drains = 10 mL/h insensible losses = 25 mL/h | + 85 mL (+ve balance/h) |
| 3. CVVH fluid removal dose = 2500 mL/h (25–35 mL/kg/h) | ||
| 4. Total in = 120 mL/h | 5. Total out = 35 mL/h (from 2) + 2500 mL ultrafiltrate = 2535 mL | |
| 6. Patient overloaded on clinical examination; need to remove an additional 25 mL/h | 7. Replacement solution/h = 2535 (−120 mL fluid input; −25 mL to reduce fluid overload) = 2390 mL/h |
Irrespective of the accuracy of fluid replacement assessment prior to therapy, individual patient assessment for fluid status must occur at least twice a day. Subtle temperature changes in the patient, fluid boluses, diarrhoea and variable absorption of feed may all contribute fluid losses not included in routine fluid maintenance. As treatment progresses over time, this may exacerbate a general loss of body fluid and dehydrate the patient. Regular weighing of patients may assist in assessing this situation. Electrolyte disturbances may also occur despite use of balanced replacement solutions. Particular attention should focus on regular assessment of fluid and electrolytes, especially potassium, sodium, phosphate and magnesium levels (see Chapter 19).
Proposed Benefits of Crrt and Comparisons with Ihd
As a continuous therapy, CRRT is considered a better therapy than IHD for critically ill patients33 for a number of reasons (see Table 18.6). Critically ill patients are often haemodynamically unstable and tolerate large fluid shifts poorly (see Chapter 20). The CRRT approach may be better at improving blood pressure stability than IHD. It has also been proposed that CRRT may be beneficial to patients with severe sepsis through the continuous clearance of inflammatory mediators, which are linked to the systemic inflammatory response syndrome. Despite this, CRRT alone may not be of any benefit to this condition, as many mediators in sepsis are not cleared via convection and antiinflammatory mediators may also be removed, which may have a counter effect to the proinflammatory mediator removal.91
| Factors | CRRT | IHD |
|---|---|---|
| Blood pressure stability | Less instability because of lower blood and dialysate flow rates and the continuous nature of the treatment with slower fluid removal rate | Removal of large volumes of fluid in a short time frame, causing hypotension and blood pressure instability during treatment |
| Waste clearance | Control of urea and creatinine (key wastes) achieved slowly and steadily; critical in managing patients with intracranial pathologies | Peaks and troughs of urea and creatinine associated with daily or second daily treatments |
| Nutritional support | Larger volumes of nutritional fluid can be administered with concurrent clearance of products of protein digestion and fluid load | Continuous feeding difficult because of fluid volume requirement and accumulation while off treatment |
| Electrolyte, acid–base and body water homeostasis | Ability to adjust dose of treatment and replacement of electrolytes, acid–base balance and body water balance to meet patient needs as necessary | Potential acute changes in pH during treatment; acid and electrolyte accumulation while off treatment |
| Neurotrauma and surgery patients | Fare much better when managed by CRRT without acute changes in solute levels such as urea and sodium | Fluid shifts can aggravate cerebral oedema and be life-threatening; IHD contraindicated for patients with raised ICP |
| Sepsis or severe infections | Blood-borne mediators in sepsis and inflammatory illness (cytokines) better removed by the convective process in CRRT (CVVH) | Diffusive and intermittent process in IHD not as effective at removing the mediators of inflammation due to molecular size |
The efficiency in initiation and application, the nursing workload and costs of the two approaches are also important considerations. Delays in waiting for dialysis nurses to initiate treatment may result in patients developing a worsening acidosis, higher potassium and urea levels, reduced or no nutritional intake and oedema with heart failure.88 This suggests that prescribing IHD and using the limited resources of dialysis nurses for a service to the ICU is inefficient, resulting in patients deteriorating further before treatment. As the IHD treatment may also be poorly tolerated when started and then seen as less ‘well tolerated’ when delay has created less stable patients, this can be avoided by the prompt initiation of CRRT.
Depending on nursing organisational structures, CRRT can be cheaper in the ICU, where one nurse cares for the critically ill patient and the CRRT. This is a primary argument supporting CRRT in the Australian and New Zealand context,42 where an IHD treatment means two nursing salaries are apportioned to one patient for the period of treatment in the ICU. There are mixed models, where a dialysis nurse initiates and terminates a treatment, leaving the ICU nurse to manage the machine and treatment for the time in between.70 Comparison of approaches is, however, difficult because of variable staffing and system structures in different countries. What little evidence is available suggests that cost differences between CRRT and IHD are minimal,51 however this depends on how this is calculated and what the costing includes, and reports highlight a great deal of variation between centres and countries for items included.99
Continuous Renal Replacement Machines
There are many machines available for CRRT in the ICU. Identifying the machine that a particular ICU should use or purchase is a common question from nurses, and there is limited literature available to guide this decision, however recent literature provides some comparisons of their technical characteristics.100 Table 18.7 outlines the major differences in machines or system approaches. Two machines adopting common design features from Table 18.7 are shown in Figures 18.18 (Prismaflex; Hospal, Lyon, France) and 18.19 (Infomed HF 440; Infomed, Geneva, Switzerland), each highlighting the major technical differences in how CRRT machines are presented and used.


Nursing Practice
Key nursing management themes found in the literature useful for application of CRRT are: education and training;42,101,102 methods for fluids management and fluid balance;103,104 anticoagulation approaches;92 and machines used.99,100,105 Nursing protocols developed in the early 1980s focused on how to prepare and prime a CRRT system, as modified dialysis pumps with IV pumps were often used.42 This was an adaptive technology and required a very manual, idiosyncratic approach. These days, with automated technology and on-screen prompts and sequential step-by-step diagrams on machine screen displays, such policies are not required, and the key headings in Table 18.8 along with the key nursing themes above, are a roadmap for protocol development. Table 18.8 also provides a brief problem and intervention-based review of nursing management for CRRT. For each group of nursing interventions, a practice recommendation is included. These recommendations could be the framework for a nursing standard or policy for CRRT. A number of publications provide clinically-focused outlines of nursing management for the CRRT technique and patient care.54,106 Additional information for the specific equipment and products used by an individual ICU are also necessary. This would include components such as size and type of membranes used, access catheters, fluids (bicarbonate and lactate) and additives.
| Nursing area | Potential problem | Key nursing interventions required |
|---|---|---|
| Patient and machine/system preparation before use |
1. Maintain access catheter alignment, preventing kinks
2. Do not place extra connections or taps between access catheter and circuit lines
3. Blood pump speed >150 mL/min
4. Ensure venous chamber filled well above air sensor, bubbles removed
6. Use fluids chart or similar to account for all fluids used including anticoagulant
7. Potassium additive to CRRT fluid is often required after 24–48 hours of treatment; some patients are hypokalaemic despite acute renal failure
1. Monitor vital signs hourly, consider any link between changes and use of RRT; e.g. low CVP and inadvertent fluid loss occurring
.
1. If transmembrane pressure (TMP) or prefilter pressure (P-IN) >250 mmHg, consider electively returning blood by saline infusion into circuit and ceasing treatment
2. Assess patient’s urea and creatinine measures; they should be reducing or stable
1. Use concentrated heparin to fill deadspace of catheter when not in use >4 hours. Use 1000 IU/mL and follow manufacturer’s specifications for volume required
2. Always cease a circuit before it clots, return patient blood
3. Use asepsis for disconnection procedure
Recommendation: access catheter should not be used for other purposes/infusions when RRT is not connected.
1. Flush out any excess blood residue in circuit, keep blood pump operational with saline in circuit
2. Circuits in use for >24 hours before disconnection or not restarted after 6 hours following temporary disconnection, consider discarding
3. After restarting circuit, increase fluid loss to remove fluid used to re-establish RRT
Summary
Case study
After initial assessment the following therapy was prescribed:
• broad-spectrum antibiotic cover; already commenced but adjusted
• continuing ventilatory support, lightening sedation to better assess neurological status
• change of CVC line, insertion of PA catheter and arterial line, and assessment of cardiac output and haemodynamic profile
• administration of boluses of 0.9% saline to restore circulating fluid deficit
• commencing noradrenaline infusion at low dose until haemodynamic profile improved
Learning activities
Learning activities 1–4 relate to the Case study.
1. Construct a database for your ICU to collect the filter ‘life’ for CRRT using a common spreadsheet program (e.g. Microsoft Excel). Include in this database:
2. Compare data from two facilities that use a different method or machine (e.g. anticoagulation). Locate a friend or colleague from a different ICU, collect data and then compare.
3. Construct a simple dot-point checklist of the machine and RRT circuit before connecting to the patient. The list should be no longer than one page and should include machine power, tubing connections, fluid balance, anticoagulation and alarms. If such a list or check routine exists, does it need modifying or updating?
4. All staff enjoy case presentations. Review a patient throughout his/her illness when treated with RRT, and construct a case presentation. Even if the case is routine, it may be useful to compare and contrast with a problematic case. Include in the case: medical history, diagnosis and ARF presentation, biochemistry and diagnostic test results. Include initiation of RRT, solute levels, effective treatment time, problems, successes and outcomes. What could be improved for another patient treatment?
5. Create a learning resource for staff caring for a patient during RRT, for example, a ‘tips and tricks’ list to aid new users and also to ensure efficiency and safety. What are the most common problems or errors when using your RRT in the ICU? List these and devise a guide for their prevention and/or remedy.
Paediatric CRRT. This US-based website specialises in resources for paediatric CRRT. It hosts numerous weblinks from companies and has a list server of interested individuals in paediatric CRRT; the site also promotes and provides links to past and future meetings on paediatric CRRT www.pcrrt.com/
CRRT Information. This US-based site enjoys wide industry support and features reports and programs from an annual international meeting on CRRT. Mainly focused on adult therapies, the site has industry links, protocols for adaptation to local conditions, video lectures, and recent information on CRRT www.crrtonline.com
1 Kellum JA, Bellomo R, Ronco C. Definition and classification of acute kidney injury. Nephron Clin Prac. 2008;109:c182–c187.
2 Kellum JA, Bellomo R, Ronco C. The concept of acute kidney injury and the RIFLE criteria. In: Ronco C, Bellomo R, Kellum J, eds. Contributions to Nephrology, Vol. 156. Basel: Karger; 2007:10–16.
3 Esson ML, Schrier RW. Diagnosis and treatment of acute tubular necrosis. Ann Intern Med. 2002;137:744–752.
4 Myers BD, Moran SM. Haemodynamically mediated acute renal failure. New Engl J Med. 1986;314:97–105.
5 Brivet FG, Kleinknecht DJ, Lourat P, Landais PJ. Acute renal failure in intensive care units: causes, outcome, and prognostic factors of hospital mortality; a prospective multicentre study. French Study Group in Acute Renal Failure. Crit Care Med. 1996;24:192–198.
6 Bellomo R, Mehta R. Acute renal replacement in the intensive care unit: now and tomorrow. New Horizons. 2005;3(4):760–767.
7 Lameire N, Van Biesen W, Vanholder R, Colardijn F. The place of intermittent hemodialysis in the treatment of acute renal failure in the ICU patient. Kidney Int. 1998;53(Suppl 66):S110–S119.
8 Silvester W, Bellomo R, Cole L. Epidemiology, management, and outcome of severe acute renal failure of critical illness in Australia. Crit Care Med. 2001;29:1910–1915.
9 . Gray’s anatomy of the human body: the Bartleby.com edition. [Cited September 2005]. Available from http://education.yahoo.com/reference/gray/
10 Unit V. the kidneys and body fluids. Guyton AC, Hall JE. Textbook of medical physiology, 11th edn, Philadelphia: WB Saunders, 2006.
11 Endre ZH. Acute renal failure. Whitworth JA, Lawrence JR, Kincaid-Smith P. Textbook of renal disease, 2nd edn, Edinburgh: Churchill Livingstone, 1994.
12 Horl WH, Druml W, Stevens PE. Pathophysiology of ARF in the ICU. Int J Artificial Organs. 1996;19(2):84–86.
13 Bellomo R. Acute renal failure. Bersten A, Soni N. Oh’s intensive care manual, 6th edn, Elsevier: Butterworth-Heinemann, 2009.
14 Bellomo R. Renal replacement therapy. Bersten A, Soni N. Oh’s intensive care manual, 6th edn, Elsevier: Butterworth-Heinemann, 2009.
15 Cumming AD. Acute renal failure: definitions and diagnosis. In: Ronco C, Bellomo R. Critical care nephrology. Dordrecht: Kluwer Academic, 1998.
16 Glassock R, Brenner B. The major glomerulopathies. In: Petersdorf R, Adams R, Braunwald E, Isselbacher K, Martin J, Wilson J. Harrison’s principles of internal medicine. 10th edn. New York: McGraw Hill; 1983:1632–1642.
17 Bennett M. Drugs and the kidney. Whitworth JA, Lawrence JR. Textbook of renal diseases, 2nd edn, Edinburgh: Churchill Livingstone, 1994.
18 Iaina A, Peer G. Post surgery/polytrauma and acute renal failure. In: Ronco C, Bellomo R. Critical care nephrology. Dordrecht: Kluwer Academic, 1998.
19 Endre ZH. Post cardiac surgery acute renal failure in the 1990s. Aust J Med. 1997;25:278–279.
20 Cole L, Bellomo R, Silvester W, Reeves JH. A prospective, multicenter study of the epidemiology, management and outcome of severe acute renal failure in a ‘closed’ ICU system. Am J Respir Crit Care Med. 2000;162:191–196.
21 Sheridan A, Bonventre J. Pathophysiology of ischaemic acute renal failure. Contrib Nephrol. 2001;132:7–21.
22 Racusen LC. The histopathology of acute renal failure. In: Ronco C, Bellomo R. Critical care nephrology. Dordrecht: Kluwer Academic, 1998.
23 Groeneveld A, Tran D, van der Meulen J, Nauta J, Thijs L. Acute renal failure in the medical intensive care unit: predisposing, complicating factors and outcome. Nephron. 1991;59:602–610.
24 Consentino F, Chaff C, Piedmonte M. Risk factors influencing survival in ICU acute renal failure. Nephrol Dial Transplant. 1994;9:179–182.
25 Schiffle H, Lang SM, Fischer R. Daily hemodialysis and the outcome of acute renal failure. New Engl J Med. 2002;346:305–310.
26 Bonventre JV. Pathophysiology of ischemic acute renal failure. Inflammation, lung-kidney cross talk, and biomarkers. Contrib Nephrol. 2004;144:19–30.
27 Bonventre JV. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J Am Soc Nephrol. 2003;14(Suppl 1):S55–S61.
28 Sheridan AM, Bonventre JV. Cell biology and molecular mechanisms of injury in ischaemic acute renal failure. Curr Opin Nephrol. 2000;9(4):427–434.
29 Kellum JA, Hoste EA. Acute renal failure in the critically ill: impact on morbidity and mortality. Contrib Nephrol. 2004;144:1–11.
30 Chien-Chu Chun, Landon KS, Rabb H. Mechanisms underlying combined acute renal failure and acute lung injury in the intensive care unit. Contrib Nephrol. 2004;144:53–62.
31 Bataller R, Sort P, Gines P, Arroyo V. Hepatorenal syndrome: definition, pathophysiology, clinical features and management. Kidney Int. 1998;53(Suppl. 66):s47–s53.
32 Bellomo R, Ronco C, Kellum J, Mehta R, Palevsky P, ADQI working group. Acute renal failure: definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204–R212.
33 Bellomo R, Ronco C. Continuous versus intermittent renal replacement therapy in the intensive care unit. Kidney Int. 1998;53(Suppl 66):s125–s128.
34 Kierdorf H. The nutritional management of acute renal failure in the intensive care unit. New Horiz. 1995;3(4):699–707.
35 Kierdorf H. Continuous versus intermittent treatment: clinical results in acute renal failure. Contrib Nephrol. 1991;93:1–12.
36 Krebs WA, ed. Concise Oxford Dictionary. London: Oxford Press, 1987.
37 Cameron JS. Practical haemodialysis began with cellophane and heparin: the crucial role of William Thalhimer (1884–1961). Nephrol Dial Transplantat. 2000;15:1086–1091.
38 Vienken J, Diamantoglou M, Henne W, Nederlof B. Artificial dialysis membranes: from concept to large scale production. Am J Nephrol. 1999;19:355–362.
39 McBride P. Industry’s contributions to the development of renal care. ANNA J. 1989;16(3):217–226.
40 Ronco C, La Greca G. The role of technology in hemodialysis. Contrib Nephrol. 2002;137:1–12.
41 Coleman B, Merrill JP. The artificial kidney. Am J Nurs. 1952;52(3):327–329.
42 Baldwin I, Elderkin T. Continuous hemofiltration: nursing perspectives in critical care. New Horizons. 1995;3(4):738–747.
43 Martin R, Jurschak J. Nursing management of continuous renal replacement therapy. Semin Dial. 1996;9(2):192–199.
44 Mehta R, Martin R. Initiating and implementing a continuous renal replacement therapy program. Semin Dial. 1996;9(2):80–87.
45 Ash SR, Wimberly AL, Mertz SL. PD for acute and ESRD: an update. Hosp Pract. 1983;2:179–210.
46 Wild J. Peritoneal dialysis. Thomas N, ed. Renal nursing, 2nd edn, London: Baillière Tindall, 2002.
47 Mehta R, Letteri JM. Current status of renal replacement therapy for acute renal failure. Am J Nephrol. 1999;19:377–382.
48 Kramer P, Wigger W, Rieger J, Matthaei D, Scheler F. Arteriovenous haemofiltration: a new and simple method for treatment of overhydrated patients resistant to diuretics. Klin Wochenschr. 1977;55:1121–1122.
49 Burchardi H. History and development of continuous renal replacement techniques. Kidney Int. 1998;53(Suppl 66):S120–S124.
50 Bihari DJ. Acute renal failure in the intensive care unit: the role of the specialist in intensive care. Semin Dial. 1996;9(2):204–208.
51 Moreno L, Heyka R, Pagannini E. Continuous renal replacement therapy: cost considerations and reimbursement. Semin Dial. 1996;9(2):209–214.
52 Ronco C, Brendolan A, Bellomo R. Current technology for continuous renal replacement therapies. In: Ronco C, Bellomo R. Critical care nephrology. Dordrecht: Kluwer Academic, 1998.
53 Uchino S, Bellomo R, Morimatsu H, Morgera S, Schetz M, et al. Continuous renal replacement therapy: a worldwide practice survey. The beginning and ending supportive therapy for the kidney (B.E.S.T. kidney) investigators. Intens Care Med. 2007;33(9):1563–1570.
54 Baldwin I, Fealy N. Nursing for renal replacement therapies in the Intensive Care Unit: historical, educational, and protocol review. Blood Purification. 2009;27:174–181.
55 Kaplan A. Current assessment of CRRT: appropriate optimism or unwarranted enthusiasm? Advances in RRT. 9(4), 2002.
56 Ronco C, Bellomo R, Kellum J. Continuous renal replacement therapy: opinions and evidence. Adv Renal Replace Ther. 2002;9(4):229–244.
57 Kellum JA, Angus DC, Johnson JP, Leblanc M, Griffin M, et al. Continuous versus intermittent renal replacement therapy: a meta-analysis. Intens Care Med. 2002;28:29–37.
58 Lameire N, Van Biesen WV, Vanholder R, Colardijn F. The place of intermittent hemodialysis in the treatment of acute renal failure in the ICU patient. Kidney Int. 1998;53(Suppl 66):s110–s119.
59 Van Biesen W, Vanholder R, Lamiere N. Dialysis strategies in critically ill acute renal failure patients. Curr Opin Crit Care. 2003;9:491–495.
60 Palevsky PM, Zhang JH, O’Connor TZ, Chertow GM, et al. Intensity of renal support in critically ill patients with acute kidney injury. N Eng J Med. 2008;359(1):7–20.
61 Marshal M, Ma T, Galler D, Rankin A, Williams A. Sustained low-efficiency daily diafiltration (SLEDD-f) for critically ill patients requiring renal replacement therapy: towards an adequate therapy. Nephrol Dial Transplant. 2004;19:877–884.
62 Baldwin I, Bellomo R. Sustained low efficiency dialysis in the ICU. Int J Intens Care. 2002;9:177–187.
63 Harris DCH, Stewart JH. Dialysis. Whitworth JA, Lawrence JR. Textbook of renal diseases, 2nd edn, Edinburgh: Churchill Livingstone, 1994.
64 Davenport A, Mehta S. The acute dialysis quality initiative–Part VI: access and anticoagulation in CRRT. Adv Renal Replace Ther. 2002;9(4):273–281.
65 Ofsthun NJ, Colton CK, Lysaght MJ. Determinants of fluid and solute removal rates during hemofiltration. In: Henderson LW, Quellhorst G, Baldamus CA, Lysaght MJ. Hemofiltration. Berlin: Springer-Verlag, 1986.
66 Ronco C, Bellomo R. Basic mechanisms and definitions for continuous renal replacement therapies. Int J Artificial Organs. 1996;19:95–99.
67 Bellomo R. Hemofiltration. In: Ayres SM, Grenvik A, Holbrook PR, Shoemaker WC. Textbook of critical care. 3rd edn. Philadelphia: WB Saunders; 1995:1041–1053.
68 Winkleman C. Hemofiltration: a new technique in critical care nursing. Heart Lung. 1985;14(3):265–271.
69 Dirkes S. How to use the new CVVH renal replacement systems. Am J Nurs. 1994;94:67–73.
70 Golper TA, Price J. Continuous veno-venous hemofiltration for acute renal failure in the intensive care setting. ASAIO J. 1994;40:936–939.
71 Thomas N. Haemodialysis. Thomas N, ed. Renal nursing, 2nd edn, London: Baillière Tindall, 2002.
72 Bellomo R, Ronco C, Mehta R. Technique of continuous renal replacement therapy: nomenclature for continuous renal replacement therapies. Am J Kidney Dis. 1996;28(5 Supp. 3):s2–s7.
73 Macias W, Mueller B, Scarim S, Robinson M, Rudy D. Continuous venovenous hemofiltration: an alternative to continuous arteriovenous hemofiltration and hemodiafiltration in acute renal failure. Am J Kidney Dis. 1991;18:451–458.
74 Relton S, Greenberg A, Palevsky P. Dialysate and blood flow dependence of diffusive solute clearance during CVVHD. ASAIO J. 1992;38(3):M691–M696.
75 Ronco C, Bellomo R. Continuous renal replacement therapies: the need for a standard nomenclature. Contrib Nephrol. 1995;116:28–33.
76 Yohay DA, Butterly DW, Schwab SJ, Quarles LD. Continuous arteriovenous hemodialysis: effect of dialyzer geometry. Kidney Int. 1992;42(2):448–451.
77 Uldall R. Vascular access for continuous renal replacement therapy. Semin Dial. 1996;9(2):93–97.
78 Kox WJ, Rohr U, Wauer H. Practical aspects of renal replacement therapy. Int J Artificial Organs. 1996;19(2):100–105.
79 Baldwin I, Bellomo R, Koch B. A technique for the monitoring of blood flow during continuous hemofiltration. Intens Care Med. 2002;28:1361–1364.
80 Webb AR, Mythen MG, Jacobsen D, Mackie IJ. Maintaining blood flow in the extracorporeal circuit: haemostasis and anticoagulation. Intens Care Med. 1995;21:84–93.
81 Baldwin I. Factors affecting circuit patency and filter life. In: Ronco C, Bellomo R, Kellum J, eds. Contributions to Nephrology, Vol. 156. Basel: Karger; 2007:178–184.
82 Gretz N, Quintel M, Ragaller M, Odenwalder W, Bender HJ, Rohmeiss SM. Low-dose heparinization for anticoagulation in intensive care patients on continuous hemofiltration. Contrib Nephrol. 1995;116:130–135.
83 Langeneker SA, Felfernig M, Werba A, Meuller CM, Chiari A, Zinpfer M. Anticoagulation with prostacyclin and heparin during continuous venovenous hemofiltration. Crit Care Med. 1994;22(11):1774–1781.
84 Cassina T, Mauri R, Engeler A, Giannini O. Continuous veno-venous haemofiltration with regional citrate anticoagulation: A four year single center experience. Int. Journal of Artificial Organs. 2008;31(11):937–943.
85 Baldwin I, Tan HK, Bridge N, Bellomo R. Possible strategies to prolong circuit life during hemofiltration: three controlled studies. Renal Failure. 2002;24(6):839–848.
86 Leslie G, Jacobs I, Clarke G. Proximally delivered high volume heparin does not improve circuit life in continuous venovenous haemodiafiltration (CVVHD). Intens Care Med. 1996;22:1261–1264.
87 Monchi M, Berghmans D, Ledoux D, Canivet JL, Dubois B, Damas P. Citrate vs. heparin for anticoagulation in continuous venovenous hemofiltration: A prospective randomized study. Intens Care Med. 2004;30(7):260–265.
88 Tolwani A, Campbell R, Schenk M, Allon M, Warnock D. (2001) Simplified citrate anticoagulation for continuous renal replacement therapy. Kidney Int. 2001;60(1):370–374.
89 Palsson R, Niles JL. Regional citrate anticoagulation in continuous venovenous hemofiltration in critically ill patients with a high risk of bleeding. Kidney Int. 1999;55(5):1991–1997.
90 Naka T, Egi M, Bellomo R, Cole L, French C, et al. Commercial low citrate anticoagulation haemofiltration in high risk patients with frequent filter clotting. Anaesthes Intens Care. 2005;33(5):601–608.
91 Mehta R, McDonald B, Aguilar M, Ward D. Regional citrate anticoagulation in continuous arteriovenous hemodialysis in critically ill patients. Kidney Int. 1990;38:976–981.
92 Davies H, Morgan D, Leslie GD. A Regional Citrate Anticoagulation Protocol for Pre-dilutional CVVHDf: The ‘Alabama Concept’. Australian Critical Care. 2008;21(3):154–156.
93 Tolwani AJ, Wille K. Anticoagulation for continuous renal replacement therapy. Seminars in Dialysis. 2009;22(2):141–145.
94 Heering P, Ivens K, Thumer O, Brause M, Grabensee B. Acid–base balance and substitution fluid during continuous hemofiltration. Kidney Int. 1999;56(Suppl 72):s37–s40.
95 Barenbrock M, Hausberg M, Matzkies F, de la Motte S, Schaefer RM. Effects of bicarbonate and lactate buffered replacement fluids on cardiovascular outcome in CVVH patients. Kidney Int. 2000;58(4):1751–1757.
96 Kierdorf HP, Leue C, Arns S. Lactate or bicarbonate buffered solutions in continuous extracorporeal renal replacement therapies. Kidney Int. 1999;56(Suppl 72):s32–s36.
97 Thomas AN, Guy JM, Kishen R, Geraghty IF, Bowles BJM, Vadgama P. Comparison of lactate and bicarbonate buffered haemofiltration fluids: use in critically ill patients. Nephrol Dial Transplant. 1997;12(6):1212–1217.
98 Bellomo R, Baldwin I, Fealy N. Prolonged intermittent renal replacement therapy in the intensive care unit. Crit Care Resusc. 2002;4:281–290.
99 Nattachai S, Lawsin L, Uchino S, Bellomo R, Kellum JA, BEST kidney Investigators. Cost of acute renal replacement therapy in the intensive care unit: results from The Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Study. Critical Care Forum. 2010. Available at http://ccforum.com/content/14/2/R46
100 Cruz D, Bobek I, Lentini P, Soni S, Chionh CY, Ronco C. Machines for Continuous Renal Replacement Therapy. Seminars in Dialysis. 2009;22(2):123–132.
101 Baldwin I. Training management and credentialling for CRRT in critical care. Am J Kidney Dis. 1997;30(5):S112–S116.
102 Craig M. Continuous venous to venous hemofiltration – implementing and maintaining a program: examples and alternatives. Crit Care Nurs Clin N Am. 1998;10:219–233.
103 Schetz M, Leblanc M, Murray P. The acute dialysis quality initiative – part VII: fluid composition and management in CRRT. Adv Renal Replace Ther. 2002;9(4):282–289.
104 Mehta R. Fluid management in continuous renal replacement therapy. Semin Dial. 1996;9:140–144.
105 Ronco C. Machines used for continuous renal replacement therapy. In: Kellum J, Bellomo R, Ronco C. Continuous renal replacement therapy. New York: Oxford University Press, 2010.
106 Baldwin I, Fealy N. Clinical nursing for the application of renal replacement therapies in the Intensive Care Unit. Seminars in Dialysis. 2009;22(2):189–193.
107 Phipps W, Monahan F, Sands J, et al. Medical-surgical nursing, 7th edn. St Louis: Mosby; 2003.
108 Mader S. Inquiry into Life, 11th edn. New York: McGraw-Hill; 2006.