Structural and Molecular Bases of Cardiac Inward Rectifier Potassium Channel Function
Background
Inwardly rectifying potassium (Kir) channels are important for stabilizing the resting membrane potential, establishing the threshold of excitation, and modulating the repolarization phase of the cardiac action potential.1 Inward rectification is a process in which the conductance of the Kir channel increases with membrane hyperpolarization, but decreases with depolarization to potentials positive to the potassium equilibrium potential. In essence, Kir channels behave as “bio-diodes,” preferentially passing current in one direction. The molecular basis of rectification in Kir channels is a physical occlusion of the ion permeation pathway by depolarization-induced movement of intracellular cations, such as magnesium and polyamines.1 Only few Kir channels, however, display strong rectification.2 Strong rectification enables the Kir channels to stabilize the resting membrane potential, as well as to protect the cell from an excessive loss of K+ ions during the plateau phase of an action potential.1 The molecular correlates of these channels are primary subunits encoded by the KCNJ family of genes.3 Mutations in KCNJ genes have been associated with various channelopathies,2 demonstrating the importance of Kir channels in normal cardiac excitation. This chapter will focus on three well-studied Kir channels in the myocardial cells: the classical inward rectifier potassium channels (IK1), the acetylcholine-activated potassium channels (KACh), and the adenosine triphosphate (ATP)-sensitive potassium channels (KATP). Additional information on the topics covered in this chapter can be obtained from recent review articles.2,4
A Family of Genes Encode Inward Rectifier Potassium Channels
Channels belonging to the Kir family are structurally and functionally different from voltage-gated potassium channels.2,4 The genes that encode Kir channels are ascribed the KCNJ nomenclature and are categorized into seven subfamilies based on the gene products (Kir1-7; Table 4-1; Figure 4-1). In general, Kir channels consist of homomeric or heteromeric complexes of the respective Kir subunits, but as will be discussed, functionality of the resulting channels depends partially or solely on interactions with additional (accessory) proteins.2 From a functional perspective, there are three distinct classes of cardiac Kir channels (IK1, IKACh, and IKATP). As will be discussed later in this chapter, based on the degree of inward rectification the underlying channels can be considered as strong (IK1; KACh) or weak (KATP) inward rectifiers.2
Table 4-1
Diversity of α-Subunit Proteins in the Family of Inward Rectifier Potassium Channels
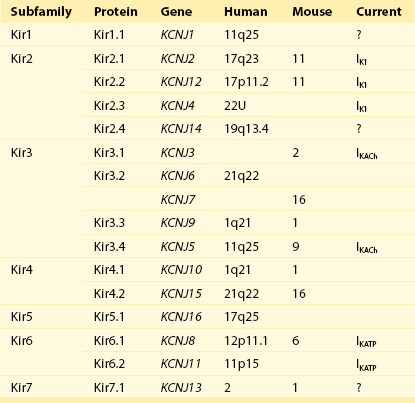
(Modified from Nerbonne JM, Nichols CG, Schwarz TL, et al: Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ Res 89:944–956, 2001).
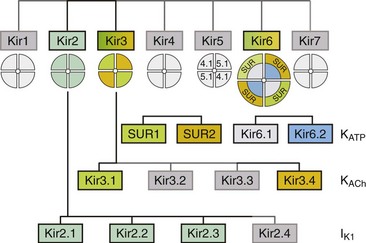
Figure 4-1 The family of inward rectifier potassium channels. All members of the Kir family share significant structural similarity, but only Kir2 and Kir3 subfamilies represent channels carrying classical strongly rectifying currents. Four members of each Kir2 and Kir3 subfamilies were cloned in mammals. Heteromeric assemblies of Kir2.1, Kir2.2, and Kir2.3 subunits underlie IK1 current, and heteromeric assembly of Kir3.1 and Kir3.4 subunits underlies IKACh current. Weakly rectifying KATP channels are composed of pore-forming Kir6.1 and Kir6.2 subunits and auxiliary SUR1and SUR2 subunits. (Modified from Anumonwo JM, Lopatin AN: Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol 48:45–54, 2010.)
Classical Cardiac Inward Rectifier Potassium Channels
Structure and Function
Kir2 Subfamily Underlies Cardiac IK1
The Kir2 subfamily consists of five members (Kir2.1-Kir2.5), of which only Kir2.1-Kir2.4 are expressed in the mammalian heart. Kir2.5 is expressed in the fish. As shown in Table 4-1, the following genes encode the mammalian cardiac Kir channels: KCNJ2, KCNJ12, KCNJ4, and KCNJ14, for Kir2.1 through Kir2.4, respectively. There is evidence that in the mammalian heart Kir2.1-Kir2.3 isoforms are expressed in cardiac myocytes, and that Kir2.4 is probably only expressed in neuronal cells. It is well established that members of the Kir2 subfamily underlie IK1, although the subunit composition varies among species and cell types, and channel complexes are likely formed as hetero-tetrameric structures.
Crystal Structure of Kir2 Channels
In recent years, x-ray crystallographic structures of both bacterial and mammalian homologs of several Kir channels have been obtained.5 Figure 4-2 highlights some important and common features of Kir channel structure based on the results of work with Kir2.1 and Kir2.2 mammalian channels.6 It is now firmly established that Kir channels are tetramers of distinct subunits, each having two transmembrane domains (M1 and M2), relatively small N-terminal, and large C-terminal cytoplasmic domains, and a pore-forming structure between M1 and M2 (see Figure 4-2). The pore structure contains pore helix directed toward the conduction pathway and the characteristic GYG (or GFG) motif, also known as K+ channel signature sequence, that contributes to the selectivity filter in all potassium channels. The M1 and M2 transmembrane domains in each subunit are arranged as an antiparallel coiled-coil and make contact with each other. Kir2 channels have a negatively charged amino acid (D172 in Kir2.1) located in approximately the middle of the pore, which has a critical role in the phenomenon of inward rectification discussed later.
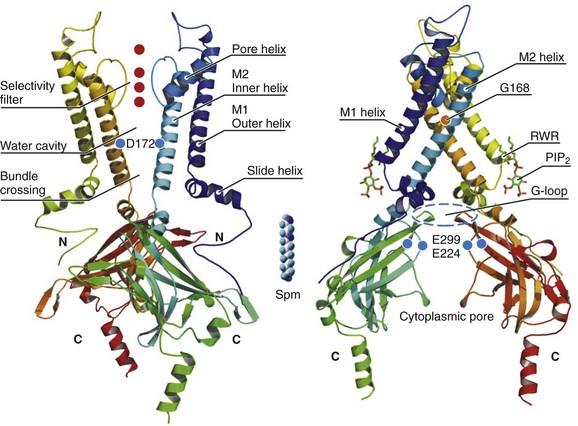
Figure 4-2 Architecture of a typical Kir channel. X-ray structure of chicken Kir2.2 crystallized in the presence of PIP2 (Accession code 3SPI, available in the Protein Data Bank; from Hansen et al.6). Only two opposing subunits are shown for clarity. Right, The structure is rotated approximately 90 degrees to better present the intracellular ion conduction pathway and bound molecules of PIP2. Numbering of the amino acids corresponds to that in Kir2.1. Red circles represent K+ ions in the selectivity filter. Blue circles show the location of three residues most critical for inward rectification. Spermine molecule (Spm) is shown in the middle at the same scale as the Kir2.2 structure. Analysis and visual presentation of the structures were performed using DeepView Swiss-PdbViewer and PyMol software.
The so-called slide helix formed by the N-terminus region just preceding the M1 helix is another unique and important regulatory feature of both bacterial and mammalian Kir channels.7 This helix intercalates between the inner leaflet of the plasma membrane and cytosol likely because of its amphiphilic nature. The important functional role of the slide helix is highlighted by the fact that many loss-of-function mutations associated with Andersen-Tawil syndrome (LQT7) are located in the slide helix.
A large C-terminal domain provided by four Kir2.x subunits consists primarily of β-strands and potentially strongly interacts with a smaller N-terminal domain. As shown in Figure 4-2, C- terminal domain forms a large intracellular vestibule, approximately 30 Å in length, for easy ion passage and likely provides binding sites for various intracellular agents. The cytoplasmic domain harbors a number of residues (e.g., E224 and E299 in Figure 4-2) known to contribute to inward rectification.
A membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) is an important structural and regulatory component of Kir channels.8 In the membrane, PIP2 likely interacts with hydrophobic amino acids on both M1 and M2 transmembrane helices as well as with a well-conserved (among Kir channels) RWR motif located just at the end of the M1 domain (see Figure 4-2). The interaction of PIP2 with a specific region in the CTD leads to an approximately 6 Å translation of the entire CTD toward the membrane associated with the movement of the M1 helix, which ultimately leads to the opening of the gate.6
Mechanism of Polyamine-Induced Rectification
The key experiments conducted in mid-1990s with the first cloned members of the Kir2 subfamily have clearly demonstrated that almost all essential properties of classical strong inward rectification could be explained by a voltage-dependent block of the channel by ubiquitous intracellular organic cations, the polyamines.9 Micromolar concentrations of free polyamines (primarily spermine and spermidine) are sufficient to reproduce the degree of rectification observed in native cells. The strength of rectification varies among the different members of the Kir subfamily, and although every Kir channel shows some degree of inward rectification, they can be broadly grouped as either strongly rectifying (Kir2 and Kir3) or weakly rectifying (Kir1 and Kir6) channels. It is well established that the time-dependent activation of strong, inward rectifiers during membrane hyperpolarization reflects the exit of polyamines (primarily sperimine) from the pore.
Rectification Properties Are Related to Electrostatic Interactions in the Cytoplasmic Pore of a Kir Channel
Early work with cloned Kir channels established that strong inward rectification by intracellular polyamines depends on three negatively charged residues located in the second transmembrane domain (D172; the rectification controller; see Figure 4-2) and in the C-terminal tail (E224 and E299; Kir2.1 amino acid [aa] numbering; see Figure 4-2). Positively charged polyamines enter the Kir channel pore to physically occlude it, a process aided by negative charges provided by aspartate and glutamate residues. Both electrophysiologic and structural data are consistent with the idea that channel block occurs in two sequential steps. A weakly voltage-dependent (shallow) blocking step involves entry of polyamine into the Kir2.1 pore at a site provided by a ring of negative charges (E224 and E299) in C-terminus. A more strongly voltage-dependent blocking step reflects the movement of polyamine to its deep binding site near D172 residue. It is also believed that the strong voltage dependence of the polyamine block arises not only from the high valency (z) of polyamines (z≈4 for spermine) but also from a displacement (push) of K+ ions through the pore.10 Among the polyamines, spermine is the most voltage-dependent blocker and also has the highest potency for blocking the channel.
A characteristic feature of Kir2.1 and Kir3 channels is a flexible cytoplasmic pore-facing G-loop (see Figure 4-2) that forms a girdle around the central axis of the Kir channel.11 It was estimated that this girdle constricts the ion permeation pathway to approximately 3 Å. Mutations in the G-loop were shown to disrupt inward rectification. In addition to the previously described E224 and E299 residues, it was shown that A255 and A259 located farther away from the pore axis are also involved in channel rectification. Electrophysiologic experiments using cysteine modifications in the pore region of a mutant Kir6.2 channel (N160D; equivalent to D172 in Kir2.1) showed that spermine binds at a deep site beyond the rectification controller residue D172, a site that is close to the extracellular mouth of the pore. In another study, refinement of the crystal structures of bacterial KirBac1.1 and KirBac3.1 allowed identification of the shallow polyamine binding sites at the cytoplasmic interface between the two subunits.12 These observations notwithstanding, the precise mechanism involved in rectification is still being worked out, and the exact location of polyamine binding sites in Kir channels is still a controversial issue.
Differential Properties of Kir2.x Subfamily
Consistent with their overall significant sequence homology, all Kir2 channels have the three mentioned residues (D172, E224, and E299) important for rectification at the equivalent positions. Recent studies, however, unexpectedly showed that rectification properties are rather different in the three Kir2 isoforms.13 Specifically, the data show that Kir2.2 channels display significantly stronger voltage dependence of rectification than that observed in Kir2.1 and Kir2.3 channels. The voltage dependence of steady-state rectification is different between Kir2.x channels, as are single-channel conductance and kinetics properties of the currents. In particular, polyamine unblock (activation) at negative membrane potentials in Kir2.3 channels is several-fold slower than in Kir2.1 and Kir2.2 channels. Single-channel conductance is smallest in Kir2.3 (approximately 10 pS), medium in Kir.2.1 (approximately 25 pS) and largest in Kir2.2 (approximately 35 pS).
Cellular and Membrane Localization
Molecular biologic studies are also consistent with location-dependent expression of specific Kir2 isoforms. In particular, real-time reverse-transcriptase polymerase chain reaction analysis of Kir2 transcripts in the human heart showed the following relative expression levels: in Purkinje fibers, Kir2.1 > Kir2.3 > Kir2.2; and in the right ventricle, Kir2.1 > Kir2.2 > Kir2.3; and the sequence was reversed in right atrium, Kir2.3 > Kir2.2 > Kir2.1.14 Information on expression patterns of Kir2 subunits can also be gleaned from functional data using the unique properties of corresponding channels. For example, in cardiac atrial and ventricular myocytes, unitary conductance values display a wide spectrum ranging from 10 to 15 pS (as in Kir2.3 channels) to as high as 40 to 45 pS (as in Kir2.2 channels).
Evidence shows that IK1 channels are located not only in the non–T-tubular component of sarcolemma of ventricular myocytes, but also in the intercalated discs and in the T-tubules. For example, accumulation and depletion of K+ only in T-tubules lead to changes in whole-cell IK1.15 IK1 accumulation/depletion phenomena are not observed in atrial cells, which essentially lack T-tubules and in ventricular myocytes, in which T-tubules are removed by osmotic shock.15
Pharmacology and Regulation
Kir2.x and IK1 channels can be regulated in a number of ways.2 Most studies on adrenergic stimulation show that inward IK1 currents are suppressed by activation of both α and β receptors, although opposite effects were also described. In addition, adrenergic regulation is clearly dependent on the type of receptors and subunit composition of the channel.
Both isoproterenol and forskolin inhibit IK1 in human ventricular myocytes, suggesting involvement of protein kinase A (PKA)-mediated phosphorylation of underlying Kir2.x subunits. Molecular details of the phenomenon, however, are contradictory. For example, it has been shown that the application of a catalytic subunit of cyclic adenosine monophosphate (cAMP) dependent PKA leads to activation of Kir2.1 channels expressed in Xenopus oocytes, but to Kir2.1 inhibition when the channels they are expressed in a mammalian cell line (COS-7). The data on PKA regulation of native IK1 channels is limited and somewhat controversial; however, most of the studies show that IK1 channels are inhibited by exposure of the cytosolic side of the membrane to purified catalytic subunit of PKA. Studies in exogenous-expressing systems also showed the involvement of PKC in negative regulation of Kir2.1 channels. Consistent with the latter, experiments using human atrial myocytes show that α1-adrenergic stimulation likely reduces IK1 via a PKC-dependent mechanism. Kir channels are also targets for phosphorylation by tyrosine kinases. In Kir2.1 channels, the site of downregulation was targeted to a single Y242 residue in the C-terminus.16
PIP2 is an important component in membrane-delimited second messenger signaling system and a powerful activator of Kir channels.17 There is significant evidence that PIP2 regulates the channel gating primarily through specific electrostatic interactions with the cytosolic part of the channel (see Figure 4-2).6 It is also clear that various properties of Kir2 channels are modulated by PIP2. For example, pH sensitivity of Kir2.3 channels is strongly dependent on the strength of channel-PIP2 interaction.
Various common cations, such as Ca2+ and H+, contribute to regulation of Kir2 channels as well.2 Intracellular Ca2+ blocks IK1 channels in a voltage-dependent manner. There is evidence that a transient increase in intracellular Ca2+ during action potential can lead to significant blockage of IK1.18 Regulation of IK1 by intracellular pH (pHi) is dependent on the species and the type of tissue. For example, rat and guinea pig ventricular IK1 is not sensitive to physiologically relevant changes in pHi. In contrast, IK1 in sheep ventricular myocytes is inhibited by intracellular H+ with pKa of approximately 7.4. The difference in pH sensitivity is likely due to differences in subunit composition of IK1. For example, channels composed of Kir2.3 subunits exhibit strong sensitivity to pHi within a physiologically relevant range, whereas Kir2.1 and Kir2.2 channels are relatively insensitive to H+.
Pharmacologic tools for modulation of Kir2.x and IK1 channels are limited. The most useful research tool for studying Kir2 channels is Ba2+ (inhibitory concentration of 50% [IC50] for inward currents, 0.5 to 10 µM). Ba2+ action is subunit dependent. For example, Ba2+ blocks Kir2.2 channels fivefold to sevenfold more efficiently than Kir2.1 channels. In the early 1990s, a compound RP 58866 and its active enantiomer, terikalant, were shown to be selective blockers of IK1 (IC50, 5 to 20 µM). However, later studies revealed that terikalant also inhibits many other K+ channels, some with even higher potency (IC50 in submicromolar range for IKr channels). Similarly, it was found that LY97119 compound (LY, a tertiary homolog of clofilium) blocks IK1 in the low micromolar range, but it also blocked Ito (transient outward current) at submicromolar concentrations. Perhaps chloroquine (an antimalarial drug) is the most potent blocker of IK1 with an IC50 of approximately 0.5 µM. However, chloroquine does not discriminate among IK1, IKACh, and IKATP, and it has been shown to affect other currents (e.g., INa) in the low micromolar range. Activators of Kir2 and IK1 channels were also described. In particular, flecainide, a widely used antiarrhythmic drug, increases Kir2.1 currents by approximately 50% at a concentration of 1 µM, but has no effect on current carried through Kir2.2 and Kir2.3 channels.19 Arachidonic acid and the antiinflammatory agent tenidap were shown to specifically activate Kir2.3 channels, with a greater than twofold maximum increase in inward current with IC50 of approximately 0.5 and 1.3 µM, respectively. Zacopride, a gastrointestinal prokinetic drug, was recently found to be a selective IK1 channel agonist. The activating effect, however, is modest, with a maximum increase in IK1 of approximately 34% at a concentration of 1 µM.
Channelopathies
There are at least four known channelopathies associated with IK1 channels, all originating from mutations in KCNJ2: ATS, short QT (SQT) syndrome, familial atrial fibrillation (FAF) and catecholaminergic polymorphic ventricular tachycardia (CPVT) (Figure 4-3).2
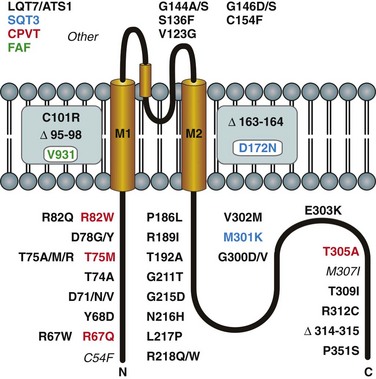
Figure 4-3 Channelopathies of the classical inward rectifier channel, IK1, associated with mutations in Kir2.1 subunit. Mutant residues are color coded to represent the long QT (LQT7/ATS1; black), catecholaminergic polymorphic ventricular tachycardia (CPVT; red), familial atrial fibrillation (FAF; green), and short QT3 (SQT3; blue). (Modified from Anumonwo JM, Lopatin AN: Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol 48:45–54, 2010.)
LQT7/ATS1 mutations are numerous (see Figure 4-3) and result in nonfunctional channels when exogenously expressed in heterologous system.20 Because affected patients are heterozygous for the mutant and wild type (WT) alleles, and the channel is composed of four subunits, a dominant-negative effect of mutated subunit can lead to reduced IK1 current in cardiac myocytes (and other cells). The dominant-negative effect of LQT7/ATS1 mutations has been demonstrated using cloned channels. For example, coexpression of WT and D71V mutants results in barely measurable inward Kir2 currents compared with those produced by expression of WT subunits alone. The magnitude of dominant negative effect, however, is variable in different mutants.
SQT syndrome is characterized by an abnormally short QT interval (less than 300 ms) and increased risk of having fibrillation and sudden death. Currently, three forms of SQT syndrome have been described. SQT1 and SQT2 syndromes result from mutations in genes underlying two voltage-gated potassium channels, HERG (KCNH2) and IKs (KCNQ1). A third variant of SQT syndrome (SQT3) originating from mutation in KCNJ2 has been described recently.21 Genetic analysis revealed a charge-neutralizing substitution (D172N) in the critical place of the channel responsible for strong inward rectification (see Figure 4-2). Accordingly, coexpression of WT and D172N mutant subunits showed decreased rectification of heteromeric channel. Computer simulations showed that reduced rectification of IK1 can explain some of the characteristic features of an electrocardiograph, such as tall and asymmetric T waves, observed in affected patients.
Recently, a second mutation in KCNJ2 associated with SQT syndrome has been identified (M301K).22 In contrast to D172N mutation, exogenous expression of M301K subunits alone did not produce measurable currents. However, coexpression of both WT and M301K subunits resulted in large K+ currents displaying significantly reduced inward rectification (relatively larger outward currents). Given that the M301K mutation resides on KCNJ2, a gene already associated with SQT3 syndrome, it is reasonable to classify M301K as another SQT3 mutation.
One study described association of single V93I mutation in KCNJ2 with familial atrial fibrillation.23 Electrophysiologic experiments with cloned channels showed, in particular, that V93I mutation leads to a relative increase in the magnitude of the outward current, or a decrease in the strength of inward rectification. Regarding the inward rectification, the effect of V91I mutation on Kir2.1 current resembles that found in D172N mutant channels. However, patients carrying the V93I mutation display a normal QT interval in contrast to patients affected by D172N mutation.
Mutations in KCNJ2 were also linked to another type of excitability disorder—catecholaminergic polymorphic ventricular tachycardia (CPVT).24 CPVT is a heritable disorder characterized by frequent ventricular arrhythmias and sudden cardiac death associated with physical activity or adrenergic stimulation. Four KCNJ2 mutations associated with CPVT have been identified (see Figure 4-3). Three of them are located in the N-terminus and one in the distal C-terminus of Kir2.1. The electrical phenotype of these mutations included prominent U waves, ventricular ectopy, and polymorphic ventricular tachycardia. In contrast to closely located mutations associated with LQT7/ATS1 syndrome, CPVT mutations were not associated with dysmorphic features or with skeletal muscle abnormalities. Electrophysiologic analysis of exogenously expressed mutants showed that they did not produce any measurable current. In addition, two of the mutations (T75M, R82W) displayed strong dominant negative effects when coexpressed with WT channels. T305A mutation did not exert a significant dominant-negative effect on inward currents, but strongly increased the strength of inward rectification, leading to a relative decrease in outward current. Finally, some mutations in KCNJ2 could not be easily classified into the four categories mentioned previously, and for this reason they are referred to as other in Figure 4-3. For example, C54F mutation was found in a patient who showed cardiac abnormalities (arrhythmia was not specified) upon corticosteroid intake.
Acetylcholine-Activated Potassium Channels
Structure and Function
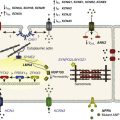
In the heart, Kir3.1/Kir3.4 channels are responsible for the effects of acetylcholine and adenosine, and they act through a coupling mechanism involving a receptor, a G protein (Go/Gi family), and the potassium channel.2,25 Because channel gating requires a G protein, Kir3.1/Kir3.4 channels are considered a type of KG channel.4 For channel activation, the G-protein–coupled receptor (Figure 4-4) can be a muscarinic (M2) or a purinergic (P1) receptor, which are activated by acetylcholine or adenosine, respectively. The ultimate result of channel activation is the opening of Kir3.1/Kir3.4 channels, which permits K+ efflux and consequently hyperpolarizes the cell membrane.
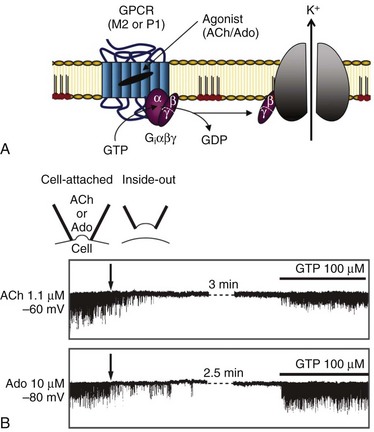
Figure 4-4 Activation of Kir3.1/Kir3.4 channels by muscarinic (M2) and purinergic (P1) G-protein coupled receptors (GPCRs). A, Membrane-delimited pathway for activation of acetylcholine (Ach) and adenosine (Ado) gated Kir channels. Ligand-GPCR interaction enhances GTP association with G-protein α-subunit, and results in the release of the β/γ-subunits for the activation of the Kir channel. Potassium efflux hyperpolarizes the cell membrane. B, Requirement of GTP for Kir3.1/Kir3.4 activation by ACh (top single channel traces) and Ado (bottom single channel traces). Holding potentials for experiments are annotated. Channel activity is present in the cell-attached patch mode, with ACh or Ado present in the pipette (top inset). Patch excision (at arrow; inside-out patch configuration) loss of GTP results in loss of channel activity. Subsequent application of GTP (100 µM; intracellular side) restored channel activity. (A, Modified from Breitwieser GE: GIRK channels: hierarchy of control. Am J Physiol Cell Physiol 289:C509–C511, 2005. B, Modified from Hibino H, Inanobe A, Furutani K et al: Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366, 2010.)
As in a typical potassium channel, ion selectivity in Kir3.x channels is conferred by the presence of the signature sequence (T-X-G-Y/F-G),4,26 and the mechanism of rectification involves an asparagine or an aspartate residue for interactions with the polyamines (details of rectification have been discussed previously). Thus, the mechanism of rectification is similar to that described for Kir2 channels, and Kir3.x channels belong to the class of strong inward rectifier channels.2 Membrane topology of Kir3 channels is similar to that described for Kir2 channels (see Figure 4-2).
Exactly how do Kir3.1/Kir3.4 channels and G proteins interact to cause channel opening? First, a brief discussion of G proteins is necessary. G proteins are complexes consisting of an α (molecular weight [MW], ~40,000), a β (MW, ~35,000) and a γ (MW, ~8000) subunit, which transduce signals from membrane receptors (e.g., muscarinic [M2]) to an effector, such as a KG channel.4,25 Four subfamily members of the guanosine diphosphate (GDP) Gα subunit dictate selectivity of signaling (activation of adenylate cyclase [Gαs], inhibition of adenylate cyclase [Gαi], and activation of phospholipase [Gq]).25 Normally, Gα is bound to GDP in the absence of an agonist, and the Gα/GDP complex is coupled to the receptor and has low GTPase activity. With vagal stimulation and ligand-receptor interaction, GDP is exchanged for GTP and results in the uncoupling of Gβγ from Gα (see Figure 4-4, A). The released Gβγ subunits in turn activate the Kir3.1/Kir3.4 channels. There is a critical requirement for GTP for channel activation (see Figure 4-4, B). A variety of experiments have determined the precise molecular interactions involved in the Gβγ-induced Kir3.1/Kir3.4 channel gating, and have shown that the cytoplasmic region of the channel is intimately involved with gating. For example, crystal structure analyses of the cytoplasmic region of Kir3.1 suggest that the C-terminus of two neighboring Kir3 channels subunits bind to each other, and that an N-terminus is positioned between the two C-terminal domains.27 The study also showed that for Kir3.1 channels, the cytoplasmic residues L262, L333, and E336 are important in gating, and the equivalent residues are H64 and L262 in Kir3.4 channels.
Cellular and Membrane Localization
There are tissue-dependent differences in expression of IKACh in the myocardium, with a very high density of expression in nodal tissues.2,4 Following the cloning of genes underlying Kir3 channels in the early to middle 1990s, investigators have probed myocardial tissues to examine localization of the gene products. Overall, these studies reported an abundance of Kir3.1 and Kir3.4 mRNA in nodal and atrial, but not in ventricular tissues, which would be consistent with tissue distribution of IKACh. In one such study,28 a comprehensive analysis was performed using Western blot and immunofluorescence to examine channel distribution in sinus-nodal, atrial, and ventricular tissues, and showed similar results of expression pattern across species (rat, ferret, and guinea pig hearts). It was reported that, whereas there was minimal expression of Kir3.1 in the ventricles of all species tested, Kir3.1 and Kir 3.4 were highly expressed in atrial tissue of all the species. It was noted that although there were relatively high levels of expression in the atria, significant quantitative differences in Kir3.1 and Kir3.4 protein levels were found in the different species. Furthermore, it was demonstrated that Kir3.1 and M2 receptor colocalized in the sinoatrial node. In nodal and atrial tissue, immunofluorescence showed localization of Kir3.x/M2 receptors more on the outer (lateral) membranes than in T-tubular membranes.28 Similar quantitative differences in expression have been reported for atrial versus ventricular tissues in the human myocardium.28,29
Pharmacology and Regulation
It is well established that Kir3.1/Kir3.4 are generally insensitive to classical potassium channel blockers such as or 4-AP or TEA.4 However, experiments in cardiac myocytes isolated from rabbit hearts show that the toxin, tertiapin, is a selective blocker of IKACh.30 Channel block is highly potent, with an affinity in the nanomolar range. Cardiac Kir3.1/Kir3.4 channels are also inhibited by quinine, quinidine, and verapamil; however, affinities of the these drugs are in the micromolar range.4 Based on the results of experiments in heterologous (oocyte) expression systems, current through Kir3.x channels is enhanced by “intoxicating” concentrations of ethanol, and an approximately 43-aa stretch on the C-terminus has been identified as important for the ethanol effect.31 Similarly, in a heterologous expression system, Kir3.x channels displayed sensitivity to intracellular acidification, an inhibitory effect that was dependent on histidine residues in the N- and C-terminal regions of the channels.4 A variety of other agents have also been shown to modulate Kir3.x channels. For example, similar to all Kir channels, Kir3.1/Kir3.4 require PIP2 for channel activity.4 Curiously, however, the PIP2 effect is enhanced by other factors such as the G-protein Gβγ complex, as well as by intracellular cations. In general, Kir3.x channel activity is also sensitive to mechanical stretch, can be modified by phosphorylating agents, and is sensitive (negatively) to regulators of G-protein signaling.4
Channelopathies
Over four decades ago, a mouse with a striking locomotor deficiency (weaving) was described, and the defect has subsequently been traced to a naturally occurring gain-in-function mutation in the Kir3.2 channel.32 Additional neurologic defects attendant to this mutation earned the weaver mouse the title of the “most cantankerous rodent.”33 There is relatively little information available on inherited cardiac channelopathies associated with Kir3.1/Kir3.4 channels. However, alterations in Kir3.1/Kir3.4 channel activity have been reported in certain cardiac rhythm abnormalities.34 Electrical remodeling in atrial fibrillation patients has been shown to increase the constitutively active component of IKACh, which could cause an abbreviation of action potential duration. More recently, a study was performed in a Long QT syndrome large Chinese family (49 individuals) with autosomal-dominant Long QT syndrome (LQTS).35 The locus of the LQTS-associated gene was mapped to chromosome 11q23.3-24.3. A combination of biochemistry and cell electrophysiology in heterologous expression systems was used to demonstrate that a heterozygous G387R mutation on the Kir3.4 (KCNJ5) identified in all affected family members was responsible for reduced channel expression on the sarcolemma.
ATP-Sensitive Potassium Channels
Structure and Function
Among all inward rectifiers, KATP channels are the most unique in their molecular architecture.36 As in other Kir channels, the ion conduction pathway is provided by a tetrameric arrangement of pore-forming subunits, but additional auxiliary regulatory subunits are necessary for the channel to be fully functional (Figure 4-5, B). The two known isoforms of the pore-forming subunits are encoded by Kir6.1 (KCNJ8) and Kir6.2 (KCNJ11) genes. The two isoforms of auxiliary subunits are encoded by SUR1 (ABCC8) and SUR2 (ABCC9) genes (the SUR name originates from “sulfonylureas,” a class of drugs known to inhibit KATP channels by acting on the auxiliary subunit). SUR2 gene can be alternatively spliced at the very C-terminus (last 42 aa) leading to SUR2A and SUR2B isoforms. Genomic arrangements of SUR and Kir6 genes are unique as well. Specifically, SUR1 is followed by Kir6.2 in a close proximity on chromosome 11pp15.1, whereas while SUR2 is followed by Kir6.1 on chromosome 12p12.1. The consequences of this arrangement regarding the regulation at a genomic level are not clear at this time. Although heteromeric assembly of Kir6.x subunits produces functional channels in vitro, it remains unclear whether heteromeric Kir6.x complexes exist in native tissues. In contrast, both SUR1/Kir6.2 and SUR2/Kir6.2 channels likely exist in native tissues. Membrane topology and general organization of Kir6.x subunits is highly similar to that in well-characterized Kir2.x channels (see Figure 4-2).
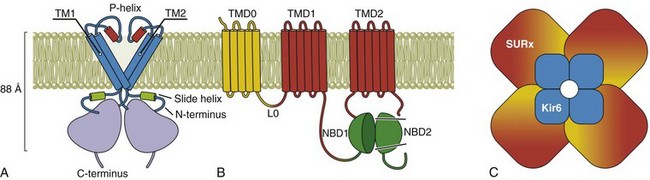
Figure 4-5 Molecular structure of KATP channel. A, A pore-forming subunit of KATP channel is encoded by Kir6.x genes and contains two transmembrane helical domains TM1 and TM2, a pore-forming region (P-helix), a large C-terminal domain, and characteristic N-terminal domain (slide helix) interfacing inner leaflet of the membrane and cytoplasmic C-terminus. B, An auxiliary subunit to the channel is encoded by SUR1 and SUR 2A/B genes and consists of the seventeen transmembrane helices organized in several distinct domains and several important cytoplasmic regions. TMD0 domain and L0 region are responsible for the interaction with Kir6.x subunits and regulation of gating of the channel. TMD1 and TMD2 domains are followed by nucleotide binding domains, NBD1 and NBD2, which form two nucleotide binding sites at their interface. C, Arrangement of Kir6.x and SUR.x subunits in an octameric KATP channel complex. Modeling studies predict one adenosine triphosphate binding site at each of the Kir6 interfaces and Mg2+-nucleotide binding sites in the NBD domains of SUR subunit. (Modified from Flagg TP, Enkvetchakul D, Koster JC, et al: Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev 90:799–829, 2010.)
KATP channels display weak rectification; however, it has been shown that just a single amino acid substitution in the so-called rectification controller region of inward rectifiers (see Figure 4-2, Kir2.1; N160D in Kir6.2) converts Kir6.2 into a strongly rectifying channel.37 A defining property of KATP channels is their characteristic sensitivity to intracellular ATP (hence the name ATP-dependent K+ channels). Under normal conditions, both exogenously expressed Kir6.2/SUR2 channels and native channels in cardiac myocytes are inhibited by ATP in the micromolar range (10 to 100 µM) by direct binding to the channel rather than through phosphorylation mechanisms. In the absence of intracellular Mg2+ ions, adenosine diphosphate (ADP) and other nucleotides also inhibit the channel. Modeling studies suggests that the ATP binding pocket is located at the interface of the N and C terminus of each Kir6.x subunit; therefore, the KATP channel possesses four ATP binding sites.
The overall structure of auxiliary SUR subunit is presented in Figure 4-5, B. It is believed that SUR interacts with Kir6.x subunits to modulate channel gating through TMD0 domain and L0 linker region. Experimental data are consistent with the idea that intracellular ATP induces dimerization of nucleotide-binding domains NBD1 and NBD2, converting them into a catalytically active site for Mg2+-dependent hydrolysis of ATP (leading to MgATP). Hydrolysis of ATP is followed by a conformational change that is then transduced to the Kir6.x subunit; however, MgADP is an even more potent activator of the KATP channel. The latter puts the hydrolysis hypothesis into question. Accordingly, it has been suggested that NBDs can be locked in a post-hydrolytic state by MgADP and other nucleotides to sustain the active state of the channel.
Cellular and Membrane Localization
KATP channels are found in virtually every kind of cardiac tissues, but they are most prominent in cardiac myocytes and smooth muscle cells. Detailed experimental analysis reveals significant differences in various properties of native KATP channels, suggesting different subunit organization in every case.38 A significant amount of evidence suggest that ventricular sarcolemmal KATP channels are likely composed of Kir6.2 and SUR2A subunits. In particular, functional sarcolemmal KATP channels are absent in Kir6.2 knockout mice, and various channel properties (including large single channel conductance, high sensitivity to pinacidil and cromakalim, and low sensitivity to diazoxide) are similar to those obtained from ventricular myocytes. The latter is also supported by a relatively low level of SUR1 (a pancreatic isoform) in the ventricles, and the finding that the activity of KATP channels is essentially unaffected in ventricular myocytes from SUR1 knockout mice. Recent experiments with mice showed, however, that SUR1 subunit of KATP channel may be a dominant isoform in atrial tissue. In particular, activity of KATP channels could not be detected in atrial myocytes isolated from SUR1 knockout mice, and the pharmacologic profile of atrial KATP channels is reminiscent of that conferred by SUR1 (higher sensitivity to diazoxide, lower to pinacidil) rather than SUR2 subunits. It remains unresolved whether this subunit composition exists in atrial tissue of other animals and humans.
KATP channels in smooth muscle display a number of properties distinct from that in cardiac myocytes, suggesting unique subunit composition.36 Significantly smaller channel densities (up to 100-fold per cell), generally low single channel conductance (~30 pS), and a lack of channel activity upon excision of the membrane patch are some of the common features of smooth muscle KATP channels. Studies with exogenously expressing systems showed that cloned KATP channels originating from coexpression of Kir6.1 and SUR2B subunits resemble native KATP channels in smooth muscle, for the most part. A strongest support for Kir6.1/SUR2B composition of smooth muscle KATP channels comes from experiments with genetically modified mice. Specifically, KATP currents cannot be recorded in aortic smooth muscle cells isolated from either Kir6.1 or SUR2 knockout mice, whereas the activity of KATP channels is preserved in these cells in Kir6.2 knockout mice.
Mitochondrial KATP channels received a lot of attention since they were first described at this location in early 1990s. In contrast to sarcolemmal KATP channels, however, their molecular identity remains highly controversial.36 Although exogenously coexpressed Kir6.1 and SUR1 subunits produce KATP channels with many properties resembling those found in mitochondria, the activity of mitochondrial KATP channels in Kir6.1 knockout mice was not affected. Recent promising developments in this area include the identification of mitochondria-specific short-form of SUR2 subunits generated by a nonconventional intraexonic splicing, which can underlie mitochondrial KATP channels.39 There is also strong evidence that the pore-forming subunit of mitochondrial KATP channels is encoded by KCNJ1 (Kir1.140).
Pharmacology and Regulation
Pharmacology of KATP channels is extensive, and regulation is complex relative to other members of Kir family, which is in part due to the structural complexity of the channel.4 Cardiac KATP channels are regulated by a variety of mechanisms, and important quantitative details of their modulation surely depend on the specific subunit composition. The most prominent and characteristic mechanism involves regulation by intracellular nucleotides such as ATP and ADP. Intracellular ATP is a potent blocker of the channel while ADP (in the presence of Mg2+ ions) has an activating effect. An overwhelming level of ATP under normal conditions keeps the channel essentially shut (native channels in cardiac myocytes are half blocked by 50 to 100 µM ATP), whereas metabolic disturbances leading to both a drop in ATP levels and a consequent rise in ADP levels result in channel opening (in the presence of intracellular Mg2+ ions). Experiments with isolated membrane patches show that phospholipids, especially PIP2, are potent activators of native (and cloned) KATP channels while their hydrolysis reduces channel activity. Regulation of KATP channels by PKA-dependent phosphorylation is well documented in smooth muscles, but the data are limited in cardiac myocytes. It has been shown that the activity of atrial KATP channels can be increased by stretch suggesting the involvement of channel-cytoskeleton interactions. Intracellular pH is another potent regulator of native KATP channels with acidification leading to increase in channel activity.
Channelopathies
Mutations in genes underlying KATP channels have been associated with cardiovascular disorders. Specifically, F1524S and A1513T substitutions in the SUR2A gene (missense and frameshift, respectively) were linked to dilated cardiomyopathy. Both mutations were mapped to the locus of SUR2A, which is responsible for catalytic activity of NBD2 domains, and likely exert their actions through reduced activation of KATP channel. The other reported mutation in the SUR2A gene was associated with adrenergic atrial fibrillation originating in the vein of Marshall, a well-known location for this type of fibrillation. As in the previous case, the underlying missense mutation in the SUR2A gene (T1547I substitution) likely affects the functioning of NBD2 leading to a compromised channel regulation by adenine nucleotides. Recently, the novel gain-of-function mutation (S422L substitution) in the pore-forming Kir6.1 subunit (when coexpressed with SUR2A) was associated with ventricular fibrillation and linked to J-wave syndrome susceptibility.41
References
1. Lopatin, AN, Nichols, CG. Inward rectifiers in the heart: an update on I(K1). J Mol Cell Cardiol. 2001; 33(4):625–638.
2. Anumonwo, JM, Lopatin, AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010; 48(1):45–54.
3. Nerbonne, JM, Nichols, CG, Schwarz, TL, et al. Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ Res. 2001; 89(11):944–956.
4. Hibino, H, Inanobe, A, Furutani, K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010; 90(1):291–366.
5. Doyle, DA, Morais Cabral, J, Pfuetzner, RA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998; 280(5360):69–77.
6. Hansen, SB, Tao, X, MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2. 2. Nature. 2011; 477(7365):495–498.
7. Enkvetchakul, D, Jeliazkova, I, Bhattacharyya, J, et al. Control of inward rectifier K channel activity by lipid tethering of cytoplasmic domains. J Gen Physiol. 2007; 130(3):329–334.
8. Logothetis, DE, Jin, T, Lupyan, D, et al. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007; 455(1):83–95.
9. Lopatin, AN, Makhina, EN, Nichols, CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. J Gen Physiol. 1995; 106(5):923–955.
10. Xu, Y, Shin, HG, Szep, S, et al. Physical determinants of strong voltage sensitivity of K(+) channel block. Nat Struct Mol Biol. 2009; 16:1252–1258.
11. Pegan, S, Arrabit, C, Zhou, W, et al. Cytoplasmic domain structures of Kir2. 1 and Kir3. 1 show sites for modulating gating and rectification. Nat Neurosci. 2005; 8(3):279–287.
12. Clarke, OB, Caputo, AT, Hill, AP, et al. Domain reorientation and rotation of an intracellular assembly regulate conduction in Kir potassium channels. Cell. 2010; 141(6):1018–1029.
13. Panama, BK, Lopatin, AN. Differential polyamine sensitivity in inwardly rectifying Kir2 potassium channels. J Physiol. 2006; 571(2):287–302.
14. Gaborit, N, Le Bouter, S, Szuts, V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007; 582(Pt 2):675–693.
15. Cheng, L, Wang, F, Lopatin, AN. Metabolic stress in isolated mouse ventricular myocytes leads to remodeling of t-tubules. Am J Physiol Heart Circ Physiol. 2011; 301:H1984–H1995.
16. Wischmeyer, E, Doring, F, Karschin, A. Acute suppression of inwardly rectifying Kir2. 1 channels by direct tyrosine kinase phosphorylation. J Biol Chem. 1998; 273(51):34063–34068.
17. Huang, CL, Feng, S, Hilgemann, DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998; 391(6669):803–806.
18. Zaza, A, Rocchetti, M, Brioschi, A, et al. Dynamic Ca2+-induced inward rectification of K+ current during the ventricular action potential. Circ Res. 1998; 82(9):947–956.
19. Caballero, R, Dolz-Gaiton, P, Gomez, R, et al. Flecainide increases Kir2. 1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc Natl Acad Sci U S A. 2010; 107(35):15631–15636.
20. Plaster, NM, Tawil, R, Tristani-Firouzi, M, et al. Mutations in Kir2. 1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001; 105(4):511–519.
21. Priori, SG, Pandit, SV, Rivolta, I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005; 96(7):800–807.
22. Hattori, T, Makiyama, T, Akao, M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2. 1 currents. Cardiovasc Res. 2012; 93:666–673.
23. Xia, M, Jin, Q, Bendahhou, S, et al. A Kir2. 1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005; 332(4):1012–1019.
24. Watanabe, H, Knollmann, BC. Mechanism underlying catecholaminergic polymorphic ventricular tachycardia and approaches to therapy. J Electrocardiol. 2011; 44(6):650–655.
25. Wettschureck, N, Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005; 85(4):1159–1204.
26. Bichet, D, Haass, FA, Jan, LY. Merging functional studies with structures of inward-rectifier K(+) channels. Nat Rev Neurosci. 2003; 4(12):957–967.
27. Nishida, M, MacKinnon, R. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1. 8 A resolution. Cell. 2002; 111(7):957–965.
28. Dobrzynski, H, Marples, DD, Musa, H, et al. Distribution of the muscarinic K+ channel proteins Kir3. 1 and Kir3. 4 in the ventricle, atrium, and sinoatrial node of heart. J Histochem Cytochem. 2001; 49(10):1221–1234.
29. Koumi, S, Wasserstrom, JA. Acetylcholine-sensitive muscarinic K+ channels in mammalian ventricular myocytes. Am J Physiol. 1994; 266(5 Pt 2):H1812–H1821.
30. Kitamura, H, Yokoyama, M, Akita, H, et al. Tertiapin potently and selectively blocks muscarinic K(+) channels in rabbit cardiac myocytes. J Pharmacol Exp Ther. 2000; 293(1):196–205.
31. Aryal, P, Dvir, H, Choe, S, et al. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009; 12(8):988–995.
32. Patil, N, Cox, DR, Bhat, D, et al. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995; 11(2):126–129.
33. Herrup, K. The weaver mouse: a most cantankerous rodent. Proc Natl Acad U S A. 1996; 93(20):10541–10542.
34. Voigt, N, Trausch, A, Knaut, M, et al. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol. 2010; 3(5):472–480.
35. Yang, Y, Liang, B, Liu, J, et al. Identification of a Kir3. 4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010; 86(6):872–880.
36. Flagg, TP, Enkvetchakul, D, Koster, JC, et al. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010; 90(3):799–829.
37. Shyng, S, Ferrigni, T, Nichols, CG. Control of rectification and gating of cloned KATP channels by the Kir6. 2 subunit. J Gen Physiol. 1997; 110(2):141–153.
38. Bao, L, Kefaloyianni, E, Lader, J, et al. Unique properties of the ATP-sensitive K(+) channel in the mouse ventricular cardiac conduction system. Circ Arrhythm Electrophysiol. 2011; 4(6):926–935.
39. Ye, B, Kroboth, SL, Pu, JL, et al. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ Res. 2009; 105(11):1083–1093.
40. Foster, DB, Ho, AS, Rucker, J, et al. Mitochondrial ROMK channel is a molecular component of MitoKATP. Circ Res. 2012; 111:446–454.
41. Medeiros-Domingo, A, Tan, BH, Crotti, L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6. 1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010; 7(10):1466–1471.