[level-membership-for-anesthesiology-category]
10 Strategies for Blood Product Management and Reducing Transfusions
DESPITE ADVANCES IN PEDIATRIC SURGERY, the number of infants and children who sustain major operative blood loss remains high. Little information is available about when to expect coagulation defects in the pediatric age group,1,2 and most studies of massive blood transfusion have involved adult patients.3
Nothing changed the use of blood products more than the threat of the acquired immunodeficiency syndrome (AIDS).4–6 Although infection with human immunodeficiency virus (HIV) by blood transfusion has become rare, it was widely publicized in the lay press and is still feared by the public. Implementation of donor education programs, improved health history screening, new tests, and new test technologies (Table 10-1) have markedly altered the spectrum of transfusion-transmitted infectious agents in the developed world. The risks of some of the infectious and noninfectious hazards of transfusion are summarized in Table 10-2.
TABLE 10-1 Current Blood Screening Tests Used on Donated Blood in the United States
Hepatitis B surface antigen (HBsAg)
Hepatitis B core antibody (anti-HBc)
Hepatitis C virus antibody (anti-HCV)
Nucleic acid amplification testing for HCV RNA
Human immunodeficiency virus type 1 (HIV-1) antibody (anti-HIV-1)
Nucleic acid amplification testing for HIV-1 RNA
Human T-lymphotropic virus type 1 (HTLV-I) antibody (anti-HTLV-I)
HTLV-II antibody (anti-HTLV-II)
Serologic test for syphilis (Treponema pallidum)
*This test depends on the incidence in the geographic area.
†As of December 2011, at first donation or after residence in endemic area.
From American Association of Blood Banks. Facts about blood and blood banking, 2006. 509 Available at http://www.aabb.org/resources/bct/Pages/bloodfaq.aspx (accessed May 2012).
TABLE 10-2 Estimated Frequency of Complications per Number of Units Transfused
| Category | Complication | Frequency |
|---|---|---|
| Noninfectious | Allergic (urticarial) | 1 : 100 |
| Febrile, nonhemolytic | 1 : 100 | |
| Transfusion-associated circulatory overload | 1 : 1000 | |
| Delayed hemolytic | 1 : 1600 | |
| Transfusion-related acute lung injury | 1 : 10,000 | |
| Acute hemolytic | 1 : 50,000 | |
| Fatal acute hemolytic | 1 : 500,000 | |
| Infectious | Hepatitis B virus | 1 : 250,000 |
| Hepatitis C virus | 1 : 1,800,000 | |
| Human T-lymphotropic virus type I | 1 : 3,000,000 | |
| Human immunodeficiency virus type 1 | 1 : 2,300,000 | |
| Bacterial contamination of red blood cells | 1 : 50,000 | |
| Bacterial sepsis of red blood cells | 1 : 500,000 | |
| Bacterial contamination of platelets | 1 : 2,000 | |
| Bacterial sepsis of platelets | 1 : 75,000 |
Data from Davenport RD. Management of transfusion reactions. In: Mintz PD, editor. Transfusion therapy: clinical principles and practice. 3rd ed. Bethesda, Md.: AABB Press; 2011, p. 757-84; Alter HJ, Esteban-Mur JI. Transfusion transmitted hepatitis. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 718-45; Barbara JA, Dow BC. Retroviruses and other viruses. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 746-59; Park YA, Brecher ME. Bacterial contamination of blood products. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 773-90.
Despite marked reductions in the transmission of HIV, hepatitis C virus, and hepatitis B virus, transfusions can produce other deleterious effects.7,8 Every transfusion must be medically justified; benefits must be weighed against the potential infectious, immunologic, and metabolic risks.9 It is in the child’s best interest to transfuse with a clear clinical goal and in the anesthesiologist’s best interest to document the reason for each transfusion. It is not acceptable medical practice to administer a transfusion when it is of questionable benefit.
Blood Volume
The minimum acceptable hematocrit varies according to an individual child’s need. The balance between oxygen supply and demand depends on a number of factors, including the oxygen content of blood, cardiac output and its regional distribution, and metabolic needs. A child with severe pulmonary disease or cyanotic congenital heart disease probably requires a greater hematocrit than a healthy child to satisfy the metabolic oxygen demands. Preterm infants may require a greater hematocrit to prevent apnea, reduce cardiac and respiratory work, and possibly improve neurologic outcomes,10 although a Cochrane review suggests that this may not be the case.11 If there is uncertainty about the need to transfuse these infants, the neonatologist should be consulted.10,12,13 A healthy child readily tolerates a hematocrit well below 30%. It is our practice not to transfuse otherwise healthy infants up to about 3 months old until their hematocrits have decreased to 20% to 25% and hematocrits of older children have decreased to 20% if there is little potential for postoperative bleeding. The circulating blood volume must be maintained in every case. Observing the operative field to estimate blood loss and monitoring the vital signs, hematocrit, urine output, and the central venous pressure (CVP) help to assess the adequacy of volume replacement. If a procedure is expected to result in significant blood loss or fluid shifts, the anesthesiologist should strongly consider the use of a urine catheter, a central venous line, and invasive arterial monitoring. The child’s size or age should not be a deterrent to the use of a central venous catheter (Table 10-3).
TABLE 10-3 Estimated Predicted Blood Loss and Recommended Monitoring and Equipment
| Predicted Blood Loss | Recommended Monitors or Equipment |
|---|---|
| Less than 0.5 blood volume | Routine monitoring |
| 0.5-1.0 blood volume | Routing monitoring + urine catheter |
| 1.0 blood volume or more | Routine monitoring + urine catheter + CVP + arterial line |
| 1.0 blood volume or more with potential for rapid blood loss | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line + rapid-infusion device |
| Severe head injury | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line |
| Major trauma with unknown severity | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line (preferably in upper extremity or central) + rapid-infusion device |
CVP, Central venous pressure; IV, intravenous.
There are three approaches for estimating the MABL: an approximation of circulating RBC mass, a modified logarithmic equation, and a simple proportion.14,15 All three approaches yield clinically similar estimates of the MABL. The most straightforward method is to estimate the MABL by simple proportion.14 For purposes of discussion, we use a hematocrit of 25% as the minimum acceptable hematocrit.
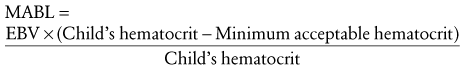
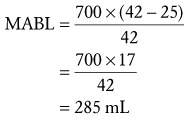
Initial therapy is directed at replacing fluid deficits and providing maintenance requirements (see Chapter 8). Additional fluid administration is directed at replacing blood loss and third space fluid losses. Although former recommendations for crystalloid replacement of blood loss were 2 to 3 mL per milliliter of shed blood, later studies suggest a smaller volume of replacement, and 1 to 2 mL of isotonic crystalloid or 1 mL of 5% albumin may be adequate.16–18 The latter type of replacement is expensive, and there is no clear evidence that colloid is superior to crystalloid.19 New starch volume expanders have been introduced that may hold promise in the future for use in children.

Blood Components and Alternatives
Red Blood Cell–Containing Components
Blood components containing RBCs are indicated for the treatment of symptomatic deficits of oxygen-carrying capacity.20,21 PRBCs are the most widely available RBC-containing blood component, although in settings where the collection facilities do not have the capability of making components, whole blood may be the only component available. Donor whole blood is collected in a preservative-anticoagulant solution that contains citrate, phosphate, dextrose (glucose), and adenine (CPDA) or just citrate, phosphate, and dextrose. In the latter case, the platelet-rich plasma is removed after centrifugation of the whole blood unit, and a solution containing adenine, dextrose, and occasionally mannitol is added to the PRBCs. The additive-solution systems permit storage for 42 days (compared with 35 days for CPDA) and better preservation of 2,3-diphosphoglycerate (DPG) levels. The characteristics of the CPDA and additive-solution PRBCs and of whole blood are shown in Table 10-4. Although the hematocrit is reduced in the additive-solution PRBCs, the red cell mass is the same.

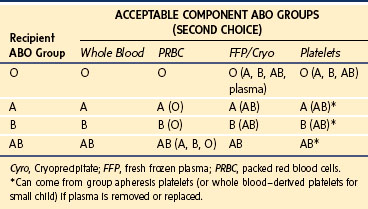
RBCs carry glycoconjugate antigens of the ABH histo-blood group system on the cell surface that are determined by six common alleles on chromosome 9.19a During the first year of life, infants begin to elaborate alloantibodies to whichever A or B antigens they lack. These isoagglutinins are invariably present after a few months and constitute a formidable immunologic obstacle to transfusion or transplantation across this ABO barrier. The RBCs for transfusion must be compatible with the ABO isoagglutinins of the intended transfusion recipient. Similarly, components with a large volume of plasma (e.g., whole blood, FFP, apheresis platelets) must be compatible with the A or B surface antigens expressed on the recipient’s RBCs. PRBCs must be ABO compatible with the recipient, whereas whole blood must be ABO identical. Table 10-5 summarizes the permissible combinations.
Only RBCs express the Rh(D) antigen. Rh(D)-positive patients may receive Rh(D)-positive or Rh(D)-negative RBCs. Rh(D)-negative patients are routinely given Rh(D)-negative RBCs for any elective transfusions, but in the setting of massive transfusion it may be necessary to switch to Rh(D)-positive RBCs to preserve the supply of Rh(D)-negative RBCs. The blood bank usually determines when to make this substitution based on the inventory and does so more quickly for a patient who is a male or a postmenopausal female (Table 10-6). The objective is to avoid exposing a female with childbearing potential to Rh(D)-positive RBCs and possibly triggering the production of the anti-D alloantibody, which is responsible for the most severe forms of hemolytic disease of the newborn. Table 10-7 shows the common initial volume of PRBCs needed to increase the hemoglobin level by 2 to 3 g/dL.

TABLE 10-7 Common Initial Doses of Blood Components and Expected Effects in Children
| Component | Dose | Effect |
|---|---|---|
| Packed red blood cells | 10-15 mL/kg | Increase hemoglobin by 2-3 g/dL |
| Platelets* | 5-10 mL/kg | Increase platelet count by 50,000-100,000/mm3 |
| Fresh frozen plasma | 10-15 mL/kg | Factor levels increase by 15%-20% |
| Cryoprecipitate | 1-2 units/kg | Increase fibrinogen by 60-100 mg/dL |
*This recommendation may be reduced pending the impact of the prophylactic platelet dose (PLADO) trial, as published for all age groups40 and for the pediatric age range.40a
The changes that occur to RBC during storage under conventional blood bank conditions has been well described.22 These observations have generated physiologically plausible hypotheses about how such changes may impair the function of the banked RBCs in vivo. The loss of intra-erythrocytic levels of 2,3-DPG and its corresponding decrease in the P50 value may reduce the ability of stored RBCs to relinquish bound O2 compared with 2,3-DPG–replete RBCs. The depletion of nitric oxide (NO) may reduce the vasodilatory properties of the RBCs, hence impairing their ability to maintain the patency of the small vessels in the microcirculation and blood flow to the tissues.23 Numerous changes in the composition and behavior of the RBC plasma membrane, including the loss and oxidation of membrane lipids and proteins and the rearrangement of some membrane constituents,24 correlate with changes in the shape and elasticity of the RBC membrane.25,26 The loss of elasticity in particular can impede the rapid movement of the RBCs through the microcirculation.
These hypotheses and some supportive data from animal models27 have led to a number of clinical studies (mostly in trauma, critical care, colorectal surgery, and cardiac surgery) of outcomes after using stored RBCs, but the results have been inconclusive.28–30 About half of the studies found a statistical association between at an unfavorable clinical outcome measure and the transfusion of RBCs, which had been stored for a longer time. However, no association was seen in one half of the studies, including two that were extensions of previous studies with positive findings. A small number of randomized, controlled trials addressed this issue without statistically significant differences in outcomes between patients receiving RBCs stored for different amounts of time,31 although two of them were underpowered.32,33 Knowing that RBCs change during storage raises the question of whether these changes affect the patients in a clinically meaningful way, a question that remains unanswered. Currently, there are four randomized, clinical trials being conducted in North America in populations of especially vulnerable patients: neonates in the intensive care unit, adults in the intensive care unit, and adults undergoing cardiac surgery.34–37 The results of these trials should help to guide future transfusion practice.
Platelets
Platelets may be obtained from a whole blood donation or collected by apheresis. Whole blood–derived platelets are separated by centrifugation and suspended in 40 to 60 mL of plasma at a concentration that is two to four times greater than in the circulation. Each unit contains a minimum of 5.5 × 1010 platelets and is stored at 20° to 24° C with gentle continuous agitation for a maximum of 5 days. One unit of whole blood–derived platelets can be expected to increase the platelet count of a 70-kg adult by 5000 to 10,000/mm3 and increase the count in an 18-kg child by 15,000/mm3.38,39 A unit of platelets obtained by apheresis contains at least 3 × 1011 platelets in 200 to 400 mL of plasma, or the equivalent of approximately 6 units of whole blood–derived platelets. A common dose for pediatric patients is 0.1 to 0.3 unit/kg of body weight, or 10 to 15 mL/kg (see Table 10-7); this dose usually produces an increment of 30,000 to 90,000/mm3. However, a rigorous reassessment of platelet dosing was just carried out in the prophylactic platelet dose trial.40,41 Doses equivalent to the standard dose of one pheresis pack (or 6 units) per m2, half of this dose, and double this dose were compared in 1272 adult and pediatric patients who received at least one platelet transfusion. Blood losses were determined with the World Health Organization (WHO) bleeding scale: grade 0 = no bleeding, grade 1 = petechiae, grade 2 = mild blood loss, grade 3 = gross blood loss, and grade 4 = debilitating blood loss. No differences were observed in bleeding outcomes (WHO grade 2 or greater) among the three doses; this was also true for the subset of 200 pediatric patients. The recommended platelet dose may be reduced in the near future, but this change awaits further discussion by the blood transfusion and hemostasis community.42–44 This trial did not involve patients undergoing surgical procedures with ongoing blood loss.
In the setting of dilutional thrombocytopenia with ongoing losses or a consumptive coagulopathy (e.g., disseminated intravascular coagulation), larger doses (≥0.3 unit/kg) may be required to boost the platelet count above 50,000/mm3. Because platelets are suspended in plasma that contains the anti-A and anti-B isoagglutinins, they should be ABO compatible with the recipient’s RBCs. Some blood donors have high-titer isoagglutinins that can produce hemolysis in transfusion recipients if a large enough volume of plasma is given.45 The transfusion of plasma-incompatible, whole blood–derived platelets to adult recipients does not produce clinically significant hemolysis because the volume of plasma given is so small relative to the plasma volume of an adult. However, apheresis platelets (and whole blood–derived platelets for small children) should be ABO compatible with the recipient’s RBCs. Because platelets do not express Rh antigens, matching for Rh(D) antigen is not necessary for apheresis platelets because they contain virtually no RBCs. However, whole blood–derived platelets may contain enough RBCs to provoke Rh alloimmunization so platelets from Rh(D)-negative donors are given preferentially to Rh(D)-negative recipients with childbearing potential. If a premenopausal female receives whole blood–derived platelets from an Rh(D)-positive donor, Rh immune globulin can be administered within 72 hours to prevent alloimmunization. Platelets should never be withheld in an emergency situation because of Rh(D) incompatibility.
Platelets are essential to hemostasis associated with the vascular injury of surgery and are necessary for the control of surgical bleeding. Platelets are also required for the maintenance of an intact endothelial barrier to spontaneous blood loss. The number of platelets required to provide adequate hemostasis in the surgical setting is much greater than the level needed to provide prophylaxis against spontaneous hemorrhage. A platelet count of 10,000/mm3 is considered adequate to prevent spontaneous bleeding or bleeding from minor invasive procedures (e.g., lumbar puncture, line placement) in an otherwise stable child. If overt signs of bleeding are present or a more significant hemostatic challenge in the form of a surgical procedure is imminent, a level of 30,000 to 50,000/mm3 may be required.46–50 A target level of 50,000/mm3 is appropriate in the setting of massive transfusion.47,51–53 Platelets may also be required for children with adequate counts but in whom platelet function is impaired. Many medications (e.g., aspirin; nonsteroidal antiinflammatory agents; dipyridamole; platelet P2Y12 receptor blockers such as clopidogrel or prasugrel; or glycoprotein IIa/IIIb receptor inhibitors such as abciximab, eptifibatide, or tirofiban; serotonin uptake antagonists such as Zoloft) and some conditions (e.g., renal failure with blood urea nitrogen levels above 60 mg/dL) cause abnormal platelet function, which may interfere with surgical hemostasis, in which case it may be necessary to maintain the platelet count at a somewhat greater level, at least until the effect of the medication dissipates or the child’s platelets have largely been replaced by banked platelets.54,55 In a few settings, such as intracranial, ophthalmic, and otologic surgery, even greater levels (100,000/mm3) are sought.
There is no clear-cut threshold below which the platelet count predicts clinical bleeding in the perioperative period. Each child must be individually assessed by constantly observing the surgical field for evidence of abnormal bleeding.56 Unfortunately, we lack a well-validated bedside tool to assess platelet function. The utility of the thromboelastogram and other devices to measure platelet function under controlled flow conditions, such as the platelet function analyzer (PFA-100), are being investigated.57–59 The standard technique for diagnosis and evaluation of thrombocytopathies remains Born-O’Brien platelet aggregometry, but it is not useful in the intraoperative setting.60 Most commonly, thrombocytopenia rather than a newly acquired platelet function defect is the problem in the operative setting and massive transfusion.
Several additional points should be considered61:
1. Not all hospitals have platelets in the inventory. Unless the need is anticipated before surgery, platelets may not be available when required.
2. For children who are thrombocytopenic before surgery, platelets should be infused just before the surgical procedure to ensure the greatest levels during the time of peak demand.
3. Platelets should be filtered only by large-pore filters (≥150 µm) or leukocyte-reduction filters (if indicated). Micropore filters may adsorb large numbers of platelets and therefore diminish the effectiveness of a platelet transfusion.
4. Platelets are suspended in plasma, which may help to replenish coagulation factors other than factors V and VIII, which are labile.
5. Platelets should not be refrigerated or placed in a cooler with ice before administration, because after they are transfused, cold platelets are rapidly cleared from the circulation.
Special Processing of Cellular Blood Components
Leukocytes collected with whole blood donations partition into platelet and PRBC components, and few intact leukocytes are present in FFP. Passenger leukocytes are responsible for most febrile, nonhemolytic transfusion reactions, HLA alloimmunization, and transmission of cytomegalovirus (CMV). To prevent the complications from these leukocytes, they can be very effectively removed (2 to 3 log reduction) by passage through leukocyte-reduction filters that may be done shortly after collection (prestorage leukoreduction) or at the bedside. Leukocyte reduction of RBCs by filtration is a superior technique to washing or freezing-deglycerolizing, which was used in the past. Table 10-8 provides indications for who may benefit from receiving leukocyte-reduced cellular components (i.e., RBCs or platelets).
TABLE 10-8 Indications for Leukocyte-Reduced Cellular Blood Components
• To prevent further febrile, nonhemolytic transfusion reactions in a child with a history of such reactions
• To prevent human leukocyte antigen alloimmunization in a child who may require long-term platelet transfusion support (e.g., leukemia, lymphoma)
• To prevent human leukocyte antigen alloimmunization in a transplant recipient
• To prevent cytomegalovirus infection and disease in a susceptible child
CMV transmission can also be reduced by screening donors for CMV exposure (testing for antibody to CMV), although leukocyte reduction is the more widely used approach. Even though primary CMV infection is benign in children with intact immune systems, some pediatric populations are at risk for developing systemic disease and should be protected from CMV transmission by blood. Only patients who have not previously been infected with CMV (i.e., CMV seronegative) are at risk. Children particularly susceptible to systemic CMV infections are listed in E-Table 10-1.

E-TABLE 10-1 Children at Risk for Cytomegalovirus Disease*
AIDS, Acquired immunodeficiency syndrome; CMV, cytomegalovirus; HIV, human immunodeficiency virus.
*Applies to CMV-seronegative patients only.
Transfused lymphocytes may mediate a graft-versus-host process in some recipients with impaired cellular immunity. Because this process involves the bone marrow and the usual targets (i.e., skin and gastrointestinal tract), the fatality rate is substantial. Transfusion-associated graft-versus-host disease (GVHD) can be prevented by exposing cellular blood components to gamma irradiation that disables the donor lymphocytes. Children who are considered to be at risk for transfusion-associated GVHD and who should receive irradiated cellular components are listed in E-Table 10-2. This complication can also occur in children with intact immune systems in the unusual circumstance when the transfusion donor is homozygous for an HLA haplotype that is shared with the recipient. In this case, the recipient’s immune system, although fully functional, cannot recognize the donor lymphocytes as foreign. The donor lymphocytes mount a GVHD attack on the recipient’s tissues, recognizing the mismatched haplotype. This situation is more likely to occur when the donor is a blood relative of the recipient. It is for this reason that blood and HLA-matched platelets donated by family members are routinely irradiated.

E-TABLE 10-2 Children at Risk for Transfusion-Associated Graft-versus-Host Disease
• Children with T-cell immune defects (e.g., severe combined immunodeficiency syndrome, Wiskott-Aldrich syndrome, DiGeorge syndrome)
• Children undergoing chemotherapy for leukemia, lymphoma, or neuroblastoma
• Children undergoing hematopoietic stem cell transplantation
• Children receiving fludarabine
• Fetus: intrauterine transfusion
• Neonate: exchange transfusion
• Children receiving platelets from HLA-matched donors
• Children receiving red blood cells or platelets from blood relatives
HLA, Human leukocyte antigen system.
Fresh Frozen Plasma
Fresh frozen plasma (FFP) represents the fluid portion of whole blood that is separated and frozen within 8 hours of collection. After thawing at 37° C, which usually requires 30 minutes, it may be administered within 24 hours if stored at 1° to 6° C. The volume of 1 unit varies from 180 to 300 mL and represents 7% to 10% of the coagulation factor activity in a 70-kg patient. It contains all of the clotting factors and regulatory proteins at approximately the native concentration, but after 6 hours at 1° to 6° C, the levels of the labile factors V and VIII begin to diminish.62 FFP does not provide functional platelets, nor does it contain leukocytes or RBCs.
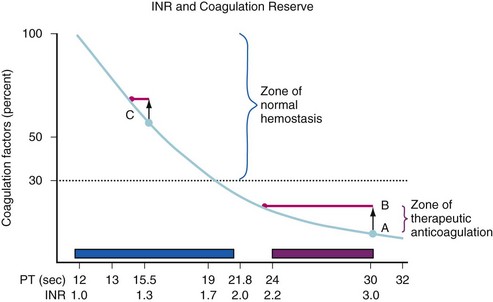
FFP is frequently administered without justification by evidence-based medicine.63 One major surgical indication for FFP is to correct the coagulopathy associated with massive blood transfusion (see Table 10-7). Other indications include a prolongation in the prothrombin time (PT) before surgery or in the setting of bleeding, the emergency reversal of warfarin, or the presence of a specific congenital or acquired coagulation protein deficiency for which a factor concentrate or a recombinant factor is not available (e.g., for factor XI deficiency).64 Administration of vitamin K should not be overlooked in children who are taking warfarin, have hepatic insufficiency, have been exclusively breastfed,65–67 who have been on broad-spectrum antibiotics (which often eliminate normal vitamin K–producing gastrointestinal flora), who have been on total parenteral nutrition for inadequate oral caloric intake, or who may have prolonged hospitalizations. Correction of a mild increase in the PT (e.g., international normalized ratio [INR] <1.5) is rarely necessary. Figure 10-1 shows the relationship between the level of coagulation factors and the in vitro clotting times, in this case the PT. Relatively modest levels of coagulation factors can support normal hemostasis, even though the PT is prolonged. When the PT is very prolonged (see Fig. 10-1, point A), the transfusion of 1 unit of FFP, which increases the coagulation factor levels by 7% to 10% in an adult, has a dramatic effect on shortening the PT. When the PT is only mildly prolonged, as at point C, where factor levels are already adequate for hemostasis, the increase in the factor levels by an additional 7% to 10% with 1 unit of FFP (in an adult) has a much smaller effect on the PT. This small effect does not achieve any improvement in hemostasis.
Cryoprecipitate
Cryoprecipitate is prepared by thawing FFP at 4° to 10° C and expressing most of the plasma, leaving behind precipitated protein that is then resuspended in a small volume of residual plasma (15 to 25 mL) and refrozen. This component contains 20% to 50% of the factor VIII from the original unit of plasma. It also contains von Willebrand factor (vWF), fibrinogen (approximately 250 mg), and factor XIII. It is indicated for the treatment of factor XIII deficiency, dysfibrinogenemia, and hypofibrinogenemia (see Table 10-7).68–77 It is no longer used for the treatment of von Willebrand disease or hemophilia A. Plasma concentrates of factor XIII and fibrinogen have been available in Europe for several years and recently have been licensed in the United States for use in patients with these congenital deficiencies.
Plasma-Derived and Recombinant Factor Concentrates
The most commonly administered factor concentrate is factor VIII, used in the treatment of hemophilia A. Children with hemophilia can have many problems related to their disease, including splenomegaly, abnormal liver function, and joint disease related to hemarthrosis. In the past, the use of pooled plasma products was associated with very high rates of transmission of viral hepatitis (especially hepatitis C virus) and HIV.78–80 The use of more rigorous viral removal and inactivation processes and the introduction of recombinant factor VIII and IX products81–85 have greatly reduced these problems.68–7586 Initial concerns that there may be an increased incidence of inhibitors in children who receive recombinant therapy compared with plasma-derived factor therapy have not been borne out.87 Mild hemophilia usually responds well to desmopressin (1-deamino-8-d-arginine vasopressin [DDAVP]) therapy.88,89
Children with hemophilia B (i.e., Christmas disease or factor IX deficiency) are managed with recombinant human factor IX and highly purified factor IX (preparations with various amounts of factors VII, X, and prothrombin) that are treated to inactivate or remove viruses.70,71,73,90–103 Careful planning of any surgical procedure for these children includes close communication with the child’s hematologist to ensure optimal therapy while reducing unnecessary transfusions (see Chapter 9).
Desmopressin
DDAVP, a synthetic analogue of vasopressin, can increase the levels of factor VIII : C (i.e., coagulant activity) and factor VIII : vWF in children with mild hemophilia A or von Willebrand disease.89,104–108 An IV dose of 0.3 µg/kg (maximum 20 µg; a subcutaneous preparation is available in Europe) increases the levels of both factors twofold to threefold within 30 to 60 minutes, with a half-life of 3 to 6 hours.105 Intranasal DDAVP is also effective, but onset is less rapid. Between 80% and 90% of children with von Willebrand disease are responders,109,110 and affected children should be tested for their responsiveness to IV DDAVP. This treatment is best suited to bleeding from surgical procedures, which ceases within 2 to 3 days. When bleeding continues beyond this period, as can occur with some orthopedic procedures, daily IV Humate P (or Alphanate or Koate DVI) can obviate possible tachyphylaxis with DDAVP. Products rich in the vWF allow better control over peak levels of factor VIII. When in excess of 200%, factor VIII predisposes to postoperative deep venous thrombosis and pulmonary embolism.
DDAVP has been used to treat the coagulopathy associated with uremia and cirrhosis.111,112 It may reduce elective surgical bleeding when the potential for blood loss is substantial, such as in cardiac surgery and spinal fusion.89,113–118 Although initial reports apparently demonstrated a benefit in patients who did not have a preexisting coagulopathy, other controlled studies failed to show an effect despite increases in factor VIII:C and vWF, and its use for these indications has largely been abandoned.119–121 Because of the potential for hyponatremia from water retention, use of DDAVP is avoided in children younger than 2 years, in children with CNS lesions, including a brain tumor, history of CNS irradiation, or recent neurosurgery or CNS trauma, and in elderly adults.
Albumin, Dextrans, Starches, and Gelatins
Albumin has the longest track record and the fewest adverse side effects.122,123 In the past, dextrans (i.e., high- and low-molecular-weight glucose polymers) were administered for volume expansion and hemodilution in children,124,125 but currently, their primary use is for antithrombosis, although their value for even this indication is questionable.126
Starches are branched polysaccharide polymers available in high-, medium-, and low-molecular-weight ranges (480,000 to 70,000 daltons). However, these compounds alter hemostasis by diluting clotting factors and impairing platelet function and the coagulation cascade.127,128 An additional concern, especially in children, is their accumulation in the reticuloendothelial system and the potential for unknown long-term adverse effects.129 Several studies have been carried out in children with minor changes in coagulation parameters that occurred when transfusions exceed 20 mL/kg.130–134 A 6% hydroxyethyl starch (HES 130/0.4) yielded clinical and physiologic profiles similar to those for 5% albumin in volumes up to 16 mL/kg in noncardiac surgery and in volumes up to 50 mL/kg in cardiac surgery, although at smaller cost.135,136 Several major reviews regarding the use of starches and gels in adult patients who are critically ill have raised significant concerns regarding adverse effects on coagulation137,138 and renal function,139,140 and they found inadequate overall safety data, even for the third-generation products.18,141 If these concerns have been raised in adult populations, we should have even greater concern about their use in children.
Gelatins are polypeptides derived from bovine collagen that seem to have a minimal effect on coagulation and provide reasonable plasma volume expansion. However, a significant number of life-threatening anaphylactic or anaphylactoid reactions have been reported, and their use in children remains somewhat limited.122,142–146
Red Blood Cell Substitutes
Blood substitutes offer the promise of agents with universal compatibility, minimal infectious risks, and prolonged shelf life (years rather than days) to carry oxygen to vital organs.147 Early efforts to develop these products involved a variety of human, bovine, and genetically engineered hemoglobin polymer solutions, perfluorocarbons, and lipid-encapsulated hemoglobin. Most failed in clinical trials because of severe complications such as renal failure, stroke, and vasoconstriction.148,149 One formulation, Hemospan (Sangart, Inc., San Diego), is still in clinical trials.150–152 It is designed to maximize oxygen delivery at the tissue level (i.e., capillary beds with impaired perfusion) rather than maximize oxygen-carrying capacity and have equivalency to hemoglobin.148,153–157 Further research is needed before these blood substitutes become available for use in children.
Massive Blood Transfusion
Massive blood transfusion may be defined as replacement of a patient’s entire blood volume one or more times. In children, the anesthesiologist must think in terms of percent of blood volumes lost rather than units of blood transfused. The composition of each blood component must be considered to anticipate problems and determine at what stage of a massive transfusion these problems may occur (see Table 10-3). Transfusion of large quantities of blood components may seriously affect coagulation, potassium and calcium concentrations, acid–base balance, body temperature, oxygen-hemoglobin dissociation, and hematocrit (i.e., oxygen-carrying capacity).
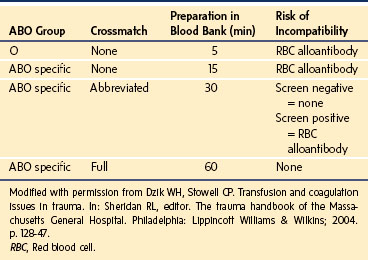
Most blood banks should have a system for the expedited release of blood products, often referred to as a massive transfusion protocol when there is inadequate time to perform complete serologic testing, including the crossmatch. Type O Rh-negative blood can be transfused into any child without the need for a crossmatch; type O Rh-positive blood may be transfused into male patients. After the blood bank has a sample of the child’s blood, it can be switched to type-specific blood and then to blood that has completed standard compatibility testing. Table 10-9 illustrates the process and risks associated with expedited release of RBCs.158 With massive blood loss, infusing crystalloid solutions alone can only worsen the underlying coagulopathy, such as from trauma-induced bleeding.
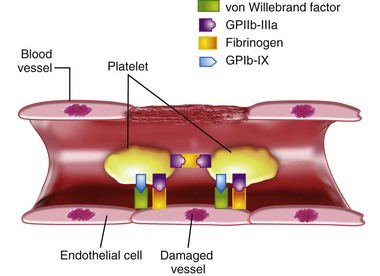
Coagulopathy
The coagulation system in children and adults involves platelets, coagulation proteins, and localized tissue factor, which initiate all steps of hemostasis. Figure 10-2 shows that an initial step is platelet adhesion to a wound or site of vessel wall injury, with adhesion being mediated by the vWF through its receptor on the platelet, the glycoprotein Ib–glycoprotein IX complex (GPIb-IX), and fibrinogen through the fibrinogen receptor on the platelet, the glycoprotein IIb–glycoprotein IIIa complex (GPIIb-IIIA). In flowing blood, initial platelet attachment is facilitated by vWF, whereas platelet spreading and more secure (shear stress–resistant) platelet-platelet aggregation is driven by fibrinogen and by the GPIIa-GPIIIa complex. However, there is also evidence that platelets attach even to intact endothelium, which has an activated phenotype, as after inflammatory cytokine exposure or sepsis. Platelets attach to the endothelium through high-molecular-weight von Willebrand multimers. Fibrinogen is attached to endothelium through upregulated integrins and selectins.
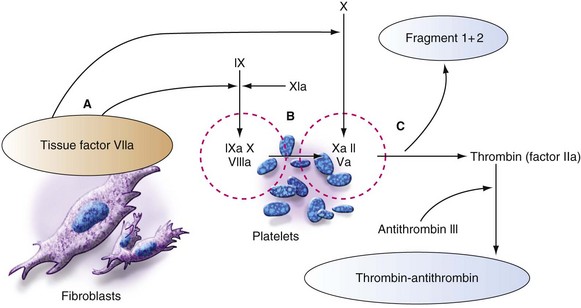
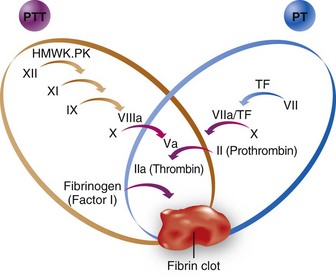
The surface-active enzyme complexes (Fig. 10-3) are active on the phospholipid surfaces provided by platelets, leukocytes, and endothelial cells but not in the bulk of the blood. In this regard, the conventional coagulation cascade shown in Figure 10-4 is oversimplified, although it is a convenient approach to understanding the PT and PTT. All of the steps must be considered in the milieu of flowing blood, such that the high-velocity gradients of arterioles (i.e., mucous membranes of the uterus, gastrointestinal tract, upper respiratory tract, oral cavity, and gums) and arteries favor thrombi with a greater proportion of platelets (i.e., white thrombi), and low-velocity gradient states such as those found in stasis or blood accumulation within a body cavity favor a greater proportion of red cells (i.e., red thrombi). This explains why a patient with von Willebrand disease, characterized by a defect in the protein that allows blood platelets to adhere to a wound, tends to bleed from mucous membranes, sites of high-velocity (arteriolar) gradients, whereas hemophiliacs tend to bleed into joint spaces and muscle planes, sites of low- or near-zero–velocity gradients.
The coagulopathy associated with massive blood transfusions is usually attributable to the dilution of clotting factors or platelets, or both. The point at which the deficiency in clotting factors is sufficient to produce a coagulopathy depends on the volume of blood lost and the type of blood component transfused (i.e., PRBCs or whole blood). Dilutional thrombocytopenia sufficient to cause clinical bleeding depends on the starting platelet count and the volume of blood replaced (Fig. 10-5). In some cases, the cause of bleeding is a consumptive coagulopathy such as fibrinolysis or disseminated intravascular coagulation (DIC).51,159–179 In other scenarios, bleeding is caused by hypothermia, severe metabolic acidosis, poor tissue perfusion, and the release of tissue factors. Body temperature should be maintained by using efficient blood-warming devices, acidosis should be treated, and normovolemia and cardiac output should be restored to prevent a coagulopathy from developing.166,180–183
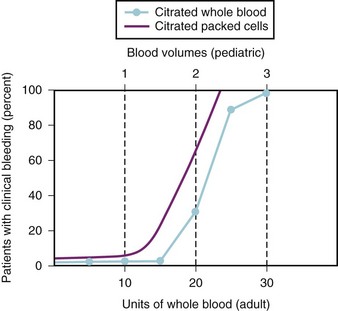
Dilutional Thrombocytopenia
Because of the dearth of published data on the effects of massive blood transfusion in children, we must rely on our clinical experience and data extrapolated from adults to anticipate the effects in children. A study of trauma patients during the Vietnam War reported that the onset of clinical bleeding occurred after about 15 units of whole blood had been transfused. The incidence of coagulopathy was unrelated to an abnormal PT or partial thromboplastin time (PTT) but correlated closely with a platelet count of less than 65,000/mm3.51 In a 70-kg adult, the estimated blood volume is approximately 5 L (70 mL/kg × 70 kg = 4900 mL), or approximately 10 units of whole blood. To relate the data in Figure 10-6 to children, assume the 70-kg adult has 5 L of blood, or 10 units of whole blood. If each 10 units of whole blood is one blood volume, some children are expected to develop a coagulopathy after losing about 1.5 blood volumes, but most develop clinical bleeding after losing 2.0 to 2.5 blood volumes. Studies of massive blood loss with whole blood replacement appear to support the conclusion that the coagulopathy at these levels of blood loss results from thrombocytopenia rather than a clotting factor deficiency.51,166–175 Consequently, children should be monitored for thrombocytopenia and possible transfusion of platelets or clotting factor deficiency (see further) after the loss of the first 1.0 to 1.5 blood volumes. After the platelet count has decreased to 50,000/mm3, it is likely that approximately one platelet dose (i.e., 6 units for an adult or 10 to 15 mL/kg for a child) will be required for each blood volume replaced. If a coagulopathy develops earlier than expected (i.e., before a 1.0-blood volume loss), a search should be initiated for other causes of bleeding, such as increased arterial or venous pressure in the surgical field or DIC.
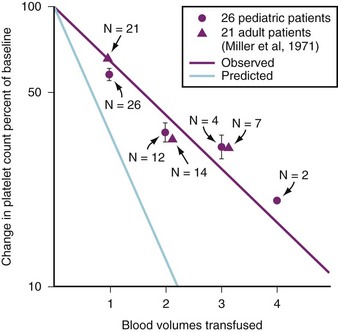
FIGURE 10-6 Percent of change in platelet count versus blood volumes transfused in adults and children.51,52 The magenta line represents observed values, whereas the blue line represents calculated values. This difference suggests increased bone marrow production and/or splenic recruitment of platelets during massive transfusion.
(From Miller RD, Robbins TO, Tong MJ, Barton SL. Coagulation defects associated with massive blood transfusions. Ann Surg 1971;174:794-801; Coté CJ, Liu LM, Szyfelbein SK, Goudsouzian NG, Daniels AL. Changes in serial platelet counts following massive blood transfusions in pediatric patients. Anesthesiology 1985;62:197-201.)
Studies of adult and pediatric patients with acute dilutional thrombocytopenia found that clinical bleeding correlated closely with a platelet count of 65,000/mm3 or less.51,166–175 Figure 10-6 compares the calculated reduction in platelet count with the observed decline in platelet count in adults and children. The observed decrement differs from the calculated reduction because platelets are mobilized from the bone marrow, lungs, and lymphatic tissues. The platelet count usually does not decrease to concentrations that cause bleeding until 2.0 to 2.5 blood volumes are lost in children, which is consistent with our observations, or until 20 to 25 units of whole blood are transfused in adults.52,173,184 Clinical bleeding does not usually occur in children whose platelet counts remain above 50,000/mm3, despite blood losses as great as 5.0 blood volumes (Fig. 10-7, A).52
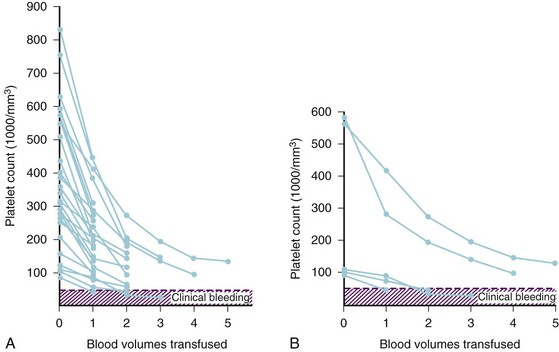
The starting platelet count is important for estimating how much blood loss can be tolerated before critical thrombocytopenia occurs. For example, with a starting platelet count of 600,000/mm3, dilutional thrombocytopenia is unlikely to occur until 4.0 or more blood volumes are lost, whereas with a starting count of 100,000/mm3, dilutional thrombocytopenia may be expected after 1.0 blood volume has been lost (see Fig. 10-7, B). Prophylactic transfusion of platelets typically is not indicated without documented evidence of dilutional thrombocytopenia, visible microvascular bleeding, and ongoing blood loss, although it should be anticipated based on the starting platelet count and the volume of blood lost.52,56,162,166,185
Although the primary platelet defect in massive transfusion is thrombocytopenia, some data suggest that platelets may not function normally (i.e., thrombocytopathy) after massive trauma or in the presence of hypothermia.182,186,187 This has not been our experience in the children we studied whose temperature remained within the normal range.52,167 The only simple test to assess platelet function is the bleeding time. However, this test is also sensitive to thrombocytopenia and its predictive value is of equivocal utility.162,186,188 The PFA-100 test shows less potential as a rapid screening tool than it once did, because the device uses citrated blood warmed to 37° C and is relatively insensitive to milder defects in platelet-vessel wall interaction, such as that in mild von Willebrand disease. There remains a critical need for a point-of-care device or simple test to assess platelet function. Currently, the platelet count is our best indication for the need for platelet transfusions in situations involving rapid blood loss.189 Other devices to measure whole blood clotting have been used to guide transfusion therapy, but their efficacy in improving outcomes has not been established.190
In several in vitro and animal model systems, recombinant factor VII (rFVIIa) activates factors IX and X on the surface of activated platelets, probably through the binding of rFVIIa to the platelet membrane (from which rFVIIa can also be taken up into storage sites within the platelet) and subsequent recruitment of circulating tissue factor.191–196 Although this has significantly improved hemostasis for hemophiliacs with inhibitors to factor VIII, there is limited evidence that rFVIIa reduces mortality for off-label use, as in cardiovascular surgery, trauma, and intracerebral hemorrhage. This agent increases the risk of thromboembolism.197
Factor Deficiency
Laboratory assessment of developing deficiencies in clotting factors plays an important part in transfusion decisions in managing massive transfusion. The PT (for the extrinsic system) measures the adequacy of factors VII, X, and V; prothrombin; and fibrinogen,51 whereas the PTT (for the intrinsic system) measures the adequacy of factors XII, XI, IX, VIII, X, and V; prothrombin; and fibrinogen (see Fig. 10-4). Banked whole blood contains normal plasma concentrations of all of the clotting factors and regulatory proteins, but it has a reduced concentration of factors V and VIII (20% to 50% of normal at the time of outdate). For coagulopathy due to a clotting factor deficiency to develop, factor VIII must be less than 30% of the normal concentration and factor V less than 20% of normal.167 For these to occur, at least 3.0 blood volumes must be exchanged with whole blood. In this scenario, the first coagulation test that is abnormal is the PTT because factor VIII is diluted to less than 30%.163
If blood loss is replaced with PRBCs, as is the current practice with blood banking techniques, the amount of plasma that is transfused is minimal because most of it was sequestered in the FFP fraction when it was separated. Massive replacement of blood loss with PRBCs and no other blood products quickly dilutes all of the clotting factors, including fibrinogen (see Fig. 10-4).* Data have confirmed that the PT and PTT are prolonged in children with multiple clotting factor deficiencies (e.g., during massive transfusion) at concentrations of clotting factors that are greater than in children with single clotting factor deficiencies (e.g., congenital coagulopathies).174,175 This was documented in adult patients who were transfused exclusively with PRBCs and crystalloid. The dilution of multiple clotting factors correlated with the volume of blood and crystalloid transfused.148 Diluting clotting factors to approximately 30% of normal may be expected replacing 1.0 to 1.5 blood volumes with PRBCs and crystalloid exclusively. Because moderate prolongations of the PT and PTT exist without overt signs of clinical bleeding,174 the indication for FFP should be onset of clinical coagulopathy. However, to avoid falling behind, the anesthesiologist should anticipate that the clotting factors will be diluted and that FFP should be infused after 1.0 blood volume of blood loss has been replaced with a combination of PRBCs and crystalloid. Documented deficiency of fibrinogen (<80 mg/dL) may also be corrected by transfusing FFP, but marked deficiency, particularly in the presence of a consumptive coagulopathy (e.g., DIC, fibrinolysis), may require the addition of cryoprecipitate (0.2 to 0.4 unit/kg).3,199,201–203 No published studies have examined massive blood replacement in infants and children using exclusively component therapy.
Our experience with 26 children (12 ± 4 years old, weight of 41.9 ± 15.8 kg; 22 Harrington rod procedures, three tumor excisions, and one Whipple procedure) who did not receive FFP or whole blood during surgery that involved blood losses between 0.5 and 1.0 blood volumes showed that none of the children demonstrated clinical signs of coagulopathy. Slight prolongations of the PT or PTT occurred when blood loss was 1.0 blood volume or less (Table 10-10). Two children who lost 1.5 blood volumes, and one who lost 2.0 blood volumes had prolonged PT and PTT values. The only child who had clinical coagulopathy was the one who lost 2.0 blood volumes, and the coagulopathy could have been ascribed to simultaneous dilutional thrombocytopenia.204,205
TABLE 10-10 Changes in Prothrombin and Partial Thromboplastin Times during Massive Blood Transfusions in Children
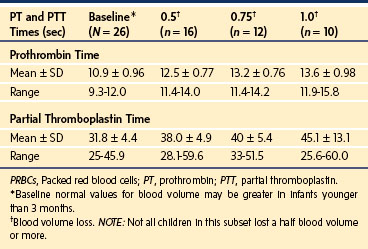
The magnitude of the change in PT or PTT that predicts a clinical coagulopathy is not well defined. However, the consensus panel of the National Institutes of Health and others suggest that greater than 1.5 times normal (or INR >2.0) should be considered pathologic.56,169,173,206–209 Our studies suggest that the PT and PTT are prolonged to more than 1.5 times normal when the blood loss is 1.5 or more blood volumes and when the blood loss has been replaced with only PRBCs and crystalloid or PRBCs and 5% albumin.204,205 Our clinical practice is to initiate FFP after loss of more than 1.0 blood volume has been replaced with PRBCs (Table 10-11). At that point, FFP is administered in a ratio of 1 unit for every 2 units of PRBCs transfused. The indications for and timing of FFP depend on which blood product has been transfused, the volume of that transfusion as it relates to the child’s blood volume, and whether the blood loss will continue perioperatively. The PT, PTT, fibrinogen concentration, and platelet count should be determined after each blood volume has been replaced and used to guide the need for additional FFP and platelets.
TABLE 10-11 Minimal Fresh Frozen Plasma Recommendations according to the Type of Blood Product Transfused and the Volume of Blood Lost
| Type of Blood Replaced | FFP Indicated | Volume FFP To Be Transfused |
|---|---|---|
| Whole blood | After 2.0-3.0 blood volumes lost and each blood volume thereafter | 25%-33% of each blood volume lost |
| PRBCs | After 1.0 blood volume lost and each blood volume thereafter | 1 U FFP / 2 U PRBCs |
FFP, Fresh frozen plasma; PRBCs, packed red blood cells; U, unit.
Recombinant factor VIIa (rFVIIa; NOVO Seven) is approved for use in the United States for hemophiliacs with high-titer inhibitors and for congenital factor VII deficiency. Several anecdotal reports describe its efficacy in controlling hemorrhage in a variety of other settings. However, in randomized clinical trials enrolling patients with partial hepatectomy, liver transplantation, prostate surgery, pelvic (orthopedic) surgery, trauma, or upper gastrointestinal tract bleeding, it conferred little or no benefit.210–215 In a large randomized clinical trial of intracranial hemorrhage, the patients in the largest-dose group showed a small improvement in hematoma expansion, but 10% also experienced thromboembolic complications. In adults receiving rFVIIa, there is a 1.4% to 10% incidence of major thromboembolic complications, including acute myocardial infarction and stroke.216 There are anecdotal reports of its successful use in children, but they provide little guidance because of the limited data.1,2,217–219,219a Until the results of controlled trials demonstrate a clear benefit for its use, rFVIIa should be used with great caution for off-label indications.210–215,218,220,221
Several studies215,222 have compared different transfusion strategies for treating trauma incurred during combat in the Middle East A consensus conference on massive transfusion concluded that the evidence did not support the up-front use of a 1 : 1 : 1 ratio of units of PRBC, FFP, and platelets, but it did support the early use of an antifibrinolytic medication (i.e., tranexamic acid).222 The consensus conference also recommended an integrated approach to managing massive transfusion that included rapid provision of PRBCs, the use of antifibrinolytics, and a foundation ratio of blood components directed by the results of standard coagulation testing (e.g., PT, PTT, platelet count, fibrinogen) or clot viscoelasticity, or both.222
Disseminated Intravascular Coagulation and Fibrinolysis
DIC and fibrinolysis are frequently associated with shock, trauma, and other forms of tissue damage, with release of procoagulants (e.g., tissue factor) and fibrinolytics (e.g., tissue plasminogen activator). In the presence of massive blood loss, these processes must be differentiated from dilutional coagulopathy. Differentiation may be difficult, because both are associated with pathologic oozing of blood in the surgical field and each may result in prolongation of the PT and PTT, as well as thrombocytopenia.223,224 With massive replacement using whole blood or PRBCs and adequate FFP, the fibrinogen level should remain normal; with uncompensated (acute) DIC, it may be decreased. However, replacing the blood loss with PRBCs, albumin, and crystalloid also leads to a reduction in fibrinogen.
The most helpful test for DIC and fibrinolysis is documentation of a significant increase in the level of D-dimer, a small peptide fragment generated during the digestion of fibrin by ongoing thrombolysis (i.e., through plasmin), along with evidence on the peripheral blood smear of schistocytes and helmet cells (i.e., microangiopathic hemolytic anemia).195,225,226 Abnormal RBCs and RBC fragments are thought to arise from the slicing action of immobilized fibrin strands in the microcirculation, although the precise mechanism remains unknown. A scoring system used for screening for potential DIC has been devised but not evaluated in the operating room setting.227 If pathologic oozing in the surgical field is observed and 1.0 blood volume or less has been lost in a child who had a normal platelet count and normal PT and PTT values preoperatively, it should be suspected that the child has developed a consumptive coagulopathy.
The most effective treatment for DIC is to eliminate the cause, such as correcting shock, acidosis, or sepsis. Heparin therapy remains controversial even in children with thrombotic manifestations of DIC and is not advisable in children with active bleeding, especially in the operative setting.224–226228
Hyperkalemia
RBCs leak potassium into the extracellular fluid during storage, particularly as the units age. The concentration of adenosine triphosphate (ATP) decreases, and the ATPase-driven Na+/K+ pump activity decreases. When the blood is outdated, 1 unit of whole blood or PRBCs has approximately 6 mEq of K+ in the plasma at a concentration that varies considerably (see Table 10-4) because this 6 mEq of K+ is distributed in the plasma and the anticoagulant solution of the stored whole blood and PRBC units. The K+ leak is more rapid from RBCs that have been irradiated.229–231 To avoid the accumulation of large amounts of extracellular K+, irradiated RBCs are stored for a maximum of 28 days (versus 35 or 42 days for whole blood or PRBCs).
Although extracellular K+ is present in banked RBCs, clinically important hyperkalemia after routine transfusion has not been reported.232–238 A study of serum potassium in neonates after exchange transfusion with PRBCs documented a decrease in serum potassium concentrations.239 A retrospective study of children undergoing massive intraoperative transfusion with PRBCs documented transient, but not life-threatening, hyperkalemia.240 It appears that clinically important hyperkalemia does not usually occur when PRBCs are administered at normal, slow infusion rates through peripheral IV access.241 This may be explained by of the combination of the small absolute amount of K+ (about 6 mEq per unit); its rapid reabsorption into the potassium-depleted, transfused RBCs; the large volume of distribution; and dilution with crystalloid or albumin during administration. Hyperkalemia in the setting of massive transfusion is usually the consequence of extensive tissue injury or acidemia resulting from inadequate tissue perfusion.
The need to relate the size of the patient to the rate of blood replacement is infrequently a problem in adults, but it is vitally important to consider in an infant or small child. An alert from the Society for Pediatric Anesthesia242 described 4 deaths among 11 children who developed hyperkalemia during transfusion; 8 were younger than 1 year, and 6 were younger than 6 months old. The Perioperative Cardiac Arrest Registry reported 8 hyperkalemic cardiac arrests related to blood transfusion.242,243 Hyperkalemia may become a problem when large volumes of whole blood or PRBCs are administered very rapidly in adults (rates ≥120 mL/min) and in infants and children undergoing rapid blood transfusion, particularly through a central venous catheter.240,244,245 A rapid transfusion rate of 120 mL/min in a 70-kg adult is equivalent to 1.5 to 2 mL/kg/min of blood, which is relatively easy to infuse in an infant or small child using a pressure bag or a rapid-transfusion device. An adult sustained a cardiac arrest and died after receiving Adsol-preserved PRBCs with a supernatant potassium concentration of 24 to 34 mEq/L at a rate of 6.4 mL/kg/min through a rapid infusion device (see Chapter 51).246 Similar transfusion rates and greater are possible in infants and children without such devices.240,247,248
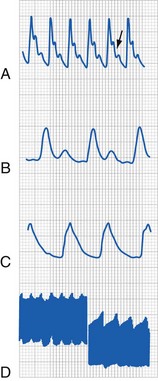
The principle is to avoid falling behind in replacing the blood loss and to avoid a situation in which a rapid and massive infusion of blood is required. Warming the blood and administering it through a peripheral IV line (rather than a central venous catheter) reduce the severe effects of hyperkalemia on the cardiac conduction system. When the rate of infusion of whole blood or PRBCs exceeds 1.5 to 2.0 mL/kg/min, the electrocardiogram must be closely monitored. If ventricular arrhythmias occur with peaked T waves in the setting of hyperkalemia (see Fig. 8-7), appropriate treatment should be instituted (e.g., calcium chloride or calcium gluconate, hyperventilation, sodium bicarbonate, albuterol, glucose and insulin); Kayexalate is the slowest and least effective in this situation and not recommended as an acute intervention.
These practices are consistent with the recommendations from the Society for Pediatric Anesthesia Wake Up Safe quality improvement initiative, which directs the physician to anticipate the blood loss and transfuse early and cautions that transfusing the hypovolemic child should be done slowly and through a peripheral IV catheter rather than rapidly through a central venous catheter.242 If the infant requires irradiated RBC blood components, it is preferable to transfuse them soon after irradiating the blood.
Hypocalcemia and Citrate Toxicity
Citrate works as an anticoagulant for stored blood components by chelating ionized calcium (iCa2+). As citrate in the blood component is transfused, it is rapidly taken up and metabolized by every nucleated cell in the body, although the primary site of clearance is the liver. During massive transfusion, particularly of whole blood or FFP, the influx of citrate may temporarily overwhelm the capacity to clear it, resulting in its accumulation, which causes the plasma concentration of iCa2+ to decrease.249–253 The plasma fraction in a unit of PRBCs, which contains the citrate, is a much smaller volume than in a unit of whole blood or FFP. Clinically, it is rare for the iCa2+ level to decrease unless the transfusion rate is very rapid; in adults, this rate must be 1.0 or more units of whole blood or FFP in 3 to 4 minutes.249 This effect on the iCa2+ concentration has been reported in neonates undergoing exchange transfusion and is more likely to occur in more-premature and lower-weight infants.253 In adult cardiac surgical patients who received whole blood at 1.5 mL/kg/min, the ventricular function curve did not improve (i.e., cardiac output did not increase although the iCa2+ decreased). When a similar volume of heparinized blood was administered at the same rate, the Frank-Starling response to volume loading was normal (i.e., cardiac output increased, but iCa2+ level did not change).254 These findings correlate well with earlier studies in that a decrease in iCa2+ concentration of clinical importance (i.e., decreased cardiac contractility) is expected to occur when the rate of infusion of citrated whole blood exceeds 1.5 to 2.0 mL/kg/min.249,254
The change in the iCa2+ level after transfusion of large volumes of PRBCs is less than that observed with whole blood or FFP. Although measurable decreases in the iCa2+ concentration have been observed during rapid transfusion, they have only rarely been associated with cardiac toxicity in adults. The rates of infusion of citrated whole blood that produce hypocalcemia and hyperkalemia are almost identical, whereas the cardiac electrophysiologic effects produced by hypocalcemia and hyperkalemia are opposite. It is important to observe the electrocardiogram for abnormalities, especially widening of the QRS complex, prolonged QT interval, and peaking of the T wave.167,255
Treatment for hypocalcemia and hyperkalemia is administration of exogenous calcium. Evidence from normal animals and children with extensive thermal injuries demonstrated that calcium chloride and calcium gluconate dissociate at a similar rate; hepatic metabolism of the gluconate moiety is not necessary (Fig. 10-8).256 Other studies during the anhepatic phase of liver transplantation also found equal ionization of calcium chloride and calcium gluconate, establishing that hepatic metabolism is not required to release calcium from gluconate.257 We recommend using calcium chloride or calcium gluconate to treat acute ionized hypocalcemia, with the caveat that calcium gluconate, which contains one third of the ionizable calcium of calcium chloride (by weight), be administered at a threefold greater dose (milligrams per kilogram) than the latter. Frequent, small boluses are as effective as single large boluses and result in smaller fluctuations in plasma iCa2+ values.256 Ideally, both forms of calcium should be slowly administered through a large peripheral or central vein, because both are sclerosing medications.
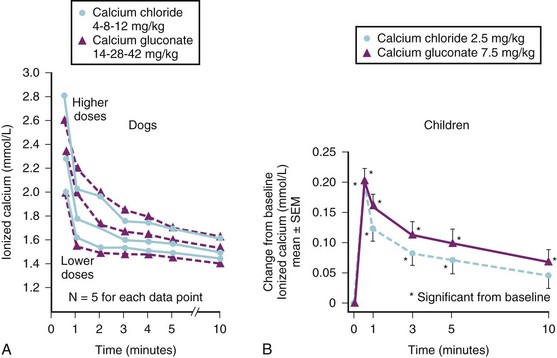
The volume of plasma and therefore the absolute amount of citrate in a unit of FFP are slightly less than the amounts in a unit of whole blood. However, the citrate load can be more rapidly delivered in FFP because it can be infused more rapidly owing to its low viscosity. It is easy to give a large citrate load in a brief period. Caution is urged when FFP or whole blood is rapidly infused, especially if the child already has a low iCa2+ concentration or impaired hepatic function (e.g., neonates and children undergoing liver transplantation). Figure 10-9, A, shows the changes in iCa2+ concentrations that occurred in children who had extensive thermal injuries and who received rapid FFP infusions at a rate between 1.0 to 2.5 mL/kg/min for 5 minutes. The maximum decrease in iCa2+ levels occurred between the fourth and fifth minute. There was no difference in the nadir in the iCa2+ concentration among the three largest rates of FFP infusion.258
If exogenous calcium is administered during rapid FFP transfusion, large decreases in the iCa2+ level can be avoided (see Fig. 10-9, B). We transfused FFP at a rate of 2 mL/kg/min for 10 minutes (equivalent to an average adult receiving 1400 mL FFP over 10 minutes) in six children with extensive burn injuries and measured very significant decreases in iCa2+ levels but observed no consistent adverse cardiovascular events. However, because these children had extensive burn injuries, we presumed they were hypermetabolic and therefore able to rapidly metabolize the excess citrate, which limits our ability to extrapolate these data to children without burn injuries (see Chapter 34).258
In dogs anesthetized with halothane, we determined that citrate-induced ionized hypocalcemia caused significantly greater cardiovascular depression as the concentration of expired halothane increased.259 These findings are consistent with the combined myocardial depression caused by ionized hypocalcemia and the myocardial depressant effects of calcium channel blockade caused by the halothane.260,261 Although halothane seems to exert the greatest calcium channel blocking activity of the inhalational anesthetics, all inhalational anesthetics depress the myocardium through this mechanism and therefore should augment the myocardial dysfunction associated with citrate-induced hypocalcemia, although similar data with sevoflurane and desflurane have not been forthcoming.262,263
The adverse cardiac effects of citrate-induced hypocalcemia may be increased if FFP is rapidly administered through a central venous catheter because there is less time to dilute the FFP and metabolize the citrate before it enters the heart and coronary vessels. FFP may be more safely administered through a peripheral IV site. Calcium should be administered during rapid transfusion of FFP (>1 mL/kg/min) to attenuate this transient but dangerous citrate toxicity, especially in the presence of potent inhalational anesthetics.259–263 Our clinical impression is that neonates and small infants are particularly vulnerable to the developing citrate toxicity because it is easier to administer a relatively large volume of FFP over a brief period to them and because citrate may not be eliminated as rapidly (i.e., first-pass effect through the liver) in infants. In addition to thermally injured patients, children undergoing liver transplantation and cardiac surgery are likely to require FFP and may develop hypocalcemia.264,265 Liver transplantation recipients are particularly susceptible to decreased iCa2+ levels during the anhepatic phase and during the pre-anhepatic phase of surgery because of impaired hepatic metabolism, impaired hepatic blood flow, and the reduced ability to metabolize citrate.266–270 An IV preparation of calcium should always be available when a major transfusion with FFP is anticipated.
Acid–base Balance
Massive transfusions usually occur in one of two situations: severe trauma with shock or major surgery with massive blood loss. In the first situation, severe metabolic acidosis may occur because of low cardiac output and diminished oxygen delivery. Correction of the acidosis with sodium bicarbonate may be a necessary part of the resuscitation, along with blood volume replacement. In this situation, impaired coagulation may occur because of the acidosis.164,180,271–273 In the operating room, intravascular volume is usually maintained, and because most instances of massive blood loss are anticipated, replacement of acute blood loss is more controlled. Even with repeated massive blood loss, metabolic acidosis is not usually a problem provided severe hypovolemia is avoided.274–276 Sodium bicarbonate therapy must be governed by the child’s acid–base status because metabolic acidosis does not usually occur with massive transfusion unless accompanied by severe hypovolemia, low cardiac output, or hypoxemia.
With massive blood transfusion, the child may develop a moderate to severe metabolic alkalosis due to the large volume of transfused citrate and its conversion to bicarbonate, which causes a metabolic alkalosis within hours of a massive transfusion.250,265,277–279 If exogenous bicarbonate therapy is superimposed on this endogenous metabolic alkalosis, a leftward shift of the oxyhemoglobin dissociation curve may result. It is important to determine the acid–base status before administering sodium bicarbonate to avoid overcorrecting the pH and shifting the oxyhemoglobin dissociation curve further.
Hypothermia
Hypothermia may be a significant problem associated with major blood loss and its replacement. Hypothermia decreases oxygen consumption and reduces oxygen demand. It may also increase oxygen consumption if the child shivers and decrease tissue delivery of oxygen by a leftward shift of the oxygen-hemoglobin dissociation curve; severe hypothermia (about 32° C) may induce a refractory ventricular tachycardia.167,280,281 Hypothermia may also profoundly compromise platelet function and impair the coagulation cascade.164,180–182,271,272,282,283
Prevention of hypothermia by all available means is considered an essential part of damage-control resuscitation of trauma patients.284–287 Banked blood products are stored between room temperature and 4° C, depending on the blood component. In the setting of high-volume transfusions, all blood products should be infused through a blood warmer. No other method should be considered (e.g., storing blood in a warming cupboard, immersing in hot water) because RBCs hemolyze readily with prolonged warming or overheating (>42° C). Warming blood and all other IV infusions with a high-capacity blood warmer, using hot air warming blankets and radiant warmers, placing plastic wrap around extremities, inserting a heated humidifier in the anesthesia circuit, covering the child’s head, and maintaining a warm to hot operating room contribute to maintaining thermal neutrality. Rapid-transfusion devices markedly improve the rapidity of transfusion and the thermokinetics involved.288–294 In one case, one author (CJC) and two nurses transfused more than 50,000 mL of blood products and crystalloid in less than 1 hour, and the child’s temperature remained at or exceeded 34.5° C.288
Monitoring during Massive Blood Transfusion
If massive blood loss can be anticipated, adequate monitoring should be instituted before surgery begins so that baseline information can be recorded. Large-bore peripheral IV access is preferable (E-Fig. 10-1) because these catheters have reduced resistance and they avoid giving blood products and drugs directly into the heart, as with a CVP line. If a child arrives in the operating room in shock (e.g., trauma patient), the physician must be careful to differentiate hypovolemia from other causes of shock (e.g., tension pneumothorax, cardiac tamponade) (see Chapters 38 and 39). Invasive monitoring inserted during resuscitation helps with the differentiation. Our philosophy is one of aggressive invasive monitoring to provide maximum data for evaluation and management of a critically hypovolemic child.


E-FIGURE 10-1 Rapid infusion intravenous catheters are available. A 20-gauge catheter is inserted into a peripheral vein, and a guidewire is advanced. A moderate skin nick is made with the provided scalpel blade, and the dilator (blue catheter) is advanced into the vein. The Rapid Infusion Catheter (white catheter; Rapid Infusion Catheter Exchange Set, Arrow International, Erding Germany) is then advanced into the vein, and the dilator and guidewire are removed. The Rapid Infusion Catheter is sutured in place (side hole on white catheter). In this case, an 8.5-Fr catheter that is only 6.4 cm long is in place, providing a large-bore, low-resistance catheter that is able to adequately function with rapid transfusion devices (see Figs. 51-1 and 51-2). These catheters are available in smaller sizes and from various manufacturers, and the devices are best combined with large-bore extension catheters.
1. Routine monitoring includes an electrocardiogram, blood pressure cuff, stethoscope, temperature, pulse oximetry, and expired carbon dioxide monitoring. The use of a pulse oximeter placed on the tongue may be particularly valuable in special circumstances when a child is vasoconstricted, hypothermic, or without peripheral pulses.295,296 Hypovolemia may occasionally manifest as pulsus paradoxus, identified by the peripheral pulse oximeter.297
2. A urinary catheter allows accurate quantitation of urine output and assessment of organ perfusion and intravascular volume status.
3. An arterial catheter enables continuous blood pressure monitoring, arterial blood gas measurements, and determinations of hematocrit, glucose, calcium, potassium, and clotting parameters. The adequacy of the circulating blood volume may be inferred from the shape of the arterial waveform, presence of the dicrotic notch, and absence of exaggerated respiratory variation (Fig. 10-10).
4. A CVP line may provide useful information, and its ease and safety of insertion have been demonstrated for children of all sizes.298 The use of ultrasound may improve the success and safety of insertion.299,300 CVP readings may vary depending on the location of the catheter tip and whether there is rapidly running fluid in the same catheter.301,302 In the latter case, the infusions should be interrupted intermittently to obtain readings. It is our clinical impression that in healthy, anesthetized, supine children, a very small change in CVP (2 to 3 mm Hg) may represent a change of as much as 10% to 15% of a child’s blood volume. In most children, right-sided pressures correlate well with left-sided pressures; the right atrial CVP usually is an accurate indicator of cardiac filling pressures of both ventricles. A CVP line provides access for blood sampling and a reliable site for IV administration of medications, fluid, and blood. A CVP line cannot always be relied on as a volume administration line because resistance is large through the long, narrow lumen; a centrally placed introducer is a reliable volume line.
5. Continuous noninvasive cardiac output devices may in the future provide further clinical guidance.
Thromboelastography provides a standardized means of quantitating the rapidity and quality of clot formation and a means for identifying fibrinolysis.303,304 This device was first used primarily during massive blood transfusion situations related to liver transplantation and now is used in several areas, particularly cardiac surgery.303–312 Some studies have found that thromboelastographic screening was not useful in predicting bleeding after cardiac surgery and was associated with a large percentage of false-positive results.313,314 Use of heparinase helps to improve the accuracy of the results by eliminating the effects of heparin, but this requires two machines to simultaneously sample blood (with and without heparinase) to have results within a useful period.315 This monitor may also be used to guide the effectiveness of antifibrinolytic therapy.310,311,316 The exact role of thromboelastography in the routine care of pediatric cardiac surgical patients, liver transplant recipients, and children who have had massive blood loss with ongoing coagulopathy has not been established. Tables 10-10 and 10-11 summarize the expected changes in various blood components when administered on a per kilogram basis and the estimated FFP requirement.
Infectious Disease Considerations
It is important for anesthesiologists to use basic precautions to minimize their risk when administering blood products and contacting body fluids. Blood and body fluids, even in infants, are capable of transmitting hepatitis B, hepatitis C, and HIV through parenteral exposure (e.g., cuts, needlestick), mucous membrane contact, or exposure to nonintact skin.4,317–326 Accidental needlesticks were the most common means of exposure in the operating room for anesthesiologists. The introduction of safe IV needles, a needleless IV system, the use of stopcocks, and never recapping used needles has reduced the incidence of this problem.191–193327 The incidence of HIV seroconversion after needle puncture is estimated to be 0.2% to 0.5% (∼1 in 300), although the conversion rate is much greater after a needlestick injury from individuals with hepatitis.4,194,317,323–325,328
Anesthesiologists must practice universal blood and body fluid precautions (e.g., gloves, goggles) and should minimize the use of needles and especially the practice of recapping needles. The management of infants may be less than optimal with the use of three-way stopcocks because of the fluid required to flush the system and the ease of introducing air into the IV line. In these infants, single-use needles without recapping or needleless systems are recommended. If an exposure occurs from a known HIV-positive patient or puncture from a needle of unknown origin, consult a specialist immediately to determine the need for immediate institution of prophylactic medical therapy as soon as possible.327,328 Early institution of drug therapy is recommended to reduce the potential for seroconversion (see Tables 49-6 and 49-7).
Methods to Reduce Exposure to Allogeneic Blood Components
Erythropoietin
Use of recombinant erythropoietin to promote endogenous RBC production can reduce the need for allogeneic RBC transfusions. This therapy has proved useful in many populations, including preterm infants, children on chemotherapy, children with renal failure, children of Jehovah’s Witnesses, and children undergoing elective major reconstructive surgery, spinal surgery, liver transplantation, or cardiac surgery. Coordination with the hematology department, blood banking, and the primary patient care team is required to take full advantage of this form of therapy.329–347 Although usually well tolerated, erythropoietin should be used with careful monitoring in patients with hypertension. Erythropoietin is available in a lyophilized (freeze-dried form for reconstitution) or in a dilute albumin solution. Some members of the Jehovah’s Witness faith who refuse albumin should receive the lyophilized formulation, which is albumin free.
Preoperative Autologous Blood Donation
Donation and storage of blood before elective surgery have reduced the use of allogeneic RBCs.230,330,343,348–361 Banked units of PRBCs may be stored for 35 to 42 days in the liquid state,362,363 permitting the donation of several units and the time required for the patient (usually teenagers) to regenerate the RBC mass before surgery. Children unable to mount an erythropoietic response to phlebotomy may only succeed in making themselves anemic, so administration of iron, vitamin C, and folate is important, as is monitoring for reticulocytosis to ensure the bone marrow is replenishing the donated RBCs. Autologous donation should not be attempted in children with significant cardiac ischemic disease or those with an active infection because bacteria can seed the collected unit and overgrow during storage. Autologous donation should be discouraged before procedures for which RBC transfusion is unlikely. Donated blood that is not used by the donor must be discarded rather than enter the general blood bank pool.
Patients or family of pediatric patients may wish to obtain blood from family members or friends (i.e., directed donation). Despite the perception that this donor pool may be safer than the pool of volunteer, allogeneic donors, there is no evidence that this is true. Directed donors are considered to be allogeneic donors and are screened and tested in the same manner as any volunteer donor. Because there is a greater risk of transfusion-associated GVHD with cellular components from a donor who is a blood relative, these units are irradiated to eliminate the possibility of this usually fatal complication of transfusion.364–366
Intraoperative Blood Recovery and Reinfusion: Autotransfusion
Recovery of blood from an operative site and reinfusion after some form of processing has been applied to major vascular, cardiac, and multiple trauma situations for many years.353,358–360,367–377 The common techniques used wash the recovered blood in a centrifuge so the product consists of the child’s RBCs suspended in saline at a hematocrit of 50% to 60%.368,375 Cellular debris, excess citrate or heparin, free hemoglobin, activated clotting factors, and clotted blood are almost completely removed. Autotransfusion avoids the infectious and immunologic risks of allogeneic transfusion and, if reinfusion is carried out in the operating room, minimizes the opportunity for mistransfusion.367,375,376,378
Intraoperative blood recovery is not widely used in infants and children.* The equipment is designed for adults, although some manufacturers have adapted standard devices for pediatric use. We have found blood recovery to be a useful adjunct to minimize allogeneic blood transfusion during scoliosis surgery. This technique may also be used in conjunction with preoperative autologous blood donation, further reducing the need for allogeneic RBC transfusions.358–360,371,372 The capital investment for the devices and the costs for the disposables and a trained operator are significant. However, these expenses can be offset if three fewer units of allogeneic RBCs are transfused. Development of pediatric-sized equipment should make this technique more widely used and more cost-effective even in smaller children.383
Contraindications
Major contraindications to blood-recovery devices include contamination of the operative field by bacteria (e.g., bowel trauma, abscess), cancer, and sickle cell disease (e.g., sickling in the device). Recovered blood should not be processed for reinfusion if the surgical field contains topical clotting agents, some topical antibiotics (e.g., Polymyxin, Neomycin), or other foreign materials (e.g., methylmethacrylate). Surgery for a malignancy is considered to be a relative contraindication because of the (theoretical) concern that malignant cells may be recovered and reinfused; this can be avoided by discarding blood recovered from the operative field while the tumor is being manipulated.384–386
Controlled Hypotension
Controlled hypotension, the intentional reduction of systemic perfusion pressure, has been used to reduce intraoperative blood loss or to provide a relatively bloodless operating field.329,387–398 Hypotensive anesthesia may be accomplished with several techniques, including continuous infusion of vasodilators, β-adrenergic blockade, deep inhalational anesthesia, and large-dose opioid infusions (e.g., remifentanil).388,399–401
Controlled hypotension is reserved for older children and teenagers undergoing major reconstructive or orthopedic surgery. The choice of technique and the degree of induced hypotension depend on the surgical procedure. For procedures in which a dry surgical field is the end point with little potential for rapid blood loss, a technique that may take some time for recovery is acceptable (e.g., deep inhalational agent with or without β-blockade). If surgery carries the possibility of rapid or massive blood loss, a technique that is rapidly reversed (e.g., nitroprusside, nitroglycerin, remifentanil) is probably safer. Controlled hypotension with mean arterial pressure (MAP) of 55 to 60 mm Hg is used less in pediatric anesthesia in recent years, although there are insufficient data to categorically state that the risks of controlled hypotension outweigh the purported benefits. Moderate hypotensive techniques with a MAP of 65 to 70 mm Hg are a more common practice and are likely associated with less risk, although no studies have been published in this regard.347
Physiology
All potent inhalational anesthetics decrease the cerebral metabolic rate for oxygen consumption (CMRo2) and increase cerebral blood flow. Isoflurane appears to offer the greatest advantage because it induces the greatest depression of CMRo2, and it has been used as the sole hypotensive agent.402–408 One of the most important considerations of any hypotensive technique is the effect that it has on cerebral blood flow. Studies of adults demonstrate that little change occurs in cerebral metabolism when the MAP is maintained above 55 mm Hg.402 There are no comparable data for children. However, experience with children during cardiac bypass suggests that children tolerate cerebral perfusion pressures below this level on an age-related basis. Brain ischemia has been documented when an MAP of 55 mm Hg is combined with hypocarbia; however, no similar pediatric studies have been carried out.407,408
Maintenance of normal arterial carbon dioxide tension (Paco2) is vitally important to ensure adequate cerebral blood flow during induced hypotension. The relationship of cerebral blood flow to Paco2 is described in greater detail in Chapter 24. To optimize cerebral blood flow, we typically maintain the MAP at 55 mm Hg or greater and the Paco2 at 35 to 45 mm Hg.
Hypotensive anesthesia may be induced by one of several techniques: direct myocardial depression with inhalational anesthetics alone or in combination with β-adrenergic blockade,404,409,410 direct vasodilators, and other medications (e.g., remifentanil). Because hypotensive anesthesia with inhalational anesthetics depresses myocardial function and requires time to wash out, a rapid offset is often difficult to achieve. We advocate alternative strategies (e.g., vasodilating agents, remifentanil) that provide more precise control of blood pressure without depressing the heart. The rapid offset of action of these latter agents may hold a particular advantage in children who incur rapid blood loss because the systolic blood pressure is quickly restored on termination while fluids are infused to reestablish euvolemia. Remifentanil offers a similar speed of reversal of cardiovascular depression while avoiding the need for high doses of an inhalational agent. Although β-adrenergic blockade decreases the requirements of vasodilators,411 cardiac arrest can occur with administration to children.412
If β-blockade is to be used safely, the clinician must understand the differences in half-lives of anesthetics and the best means for reversing their effects. Esmolol is very short acting, with a half-life in children of approximately 3 minutes.413 Nonanesthetized children have a greater requirement (µg/kg) than adults.414 In nonanesthetized children, a loading dose of 500 µg/kg/min is followed by a maintenance infusion at a rate of 25 to 200 µg/kg/min. Because there is limited published experience esmolol in children under anesthesia, a smaller starting dose (25 to 50 µg/kg/min) and titration of dose every 3 to 5 minutes (increase by 12.5 to 25 µg/kg/min) are indicated. Labetalol410,415–417 and propranolol418,419 have been used to help induce hypotensive anesthesia; the half-lives, onset of effect, and time to peak effect of these medications are much greater, and the effects are therefore less controllable and not recommended. Acute β-blocker toxicity can be reversed with high-dose IV glucagon (50 µg/kg followed by an infusion of 0.3 to 3.0 µg/kg/min [extrapolated from adult data])420–422 and possibly with vasopressin.423
Renal System
Renal blood flow autoregulates between an MAP of 80 and 180 mm Hg. However, general anesthesia with inhalational anesthetics profoundly affects autoregulation. Urinary output is a simple method to monitor the adequacy of intraoperative renal perfusion and function. Reducing the MAP decreases renal blood flow, although the effect on renal function, at least in the case of β-blockers, is insignificant. Renal function returns to normal after the period of controlled hypotension.415,424–427
Pulmonary System
An increase in physiologic dead space and intrapulmonary shunting during induced hypotension has been reported in adults and children.393,395,428–430 This does not seem to be a clinically important problem in children. Their smaller size compared with adults may reduce the gravitational pooling of blood in the lungs. Nonetheless, it is important to measure the arterial blood gases, expired carbon dioxide, and oxygen saturation to assess for this effect.393,395 An increasing difference between end-expired carbon dioxide values and measured arterial blood gas values suggest the development of a shunt and increased physiologic dead space. Children with severe scoliosis may have significant arterial-to-alveolar gradients for carbon dioxide even before surgery.
Hepatic System
Catecholamines, Paco2, circulating blood volume, and anesthetic agents influence the portal circulation.431 The liver is oxygenated by the arterial circulation but receives most of its blood flow through the portal circulation, and changes in portal blood flow may have profound effects on total hepatic blood flow. Hepatic oxygenation is maintained during periods of induced hypotension during anesthesia if the Paco2 is within the normal range and an adequate circulating blood volume is maintained.432
Pharmacology
Sodium Nitroprusside
Sodium nitroprusside has a very rapid onset of action (seconds), brief duration of action (minutes), and minimal side effects when used in the recommended dose range.433 This agent is extremely potent and is most safely administered by an infusion pump through a separate IV site and with a second pump to provide a continuous, uninterrupted, and stable infusion rate. Its principal mechanism of action is direct vascular smooth muscle relaxation, primarily causing arteriolar dilation and some venodilation.
Dosage.
The initial infusion rate for sodium nitroprusside is 0.5 to 1.0 µg/kg/min. The rate can be increased as needed to achieve the desired MAP.434,435 A satisfactory reduction in systemic perfusion pressure can usually be obtained well below the recommended maximum rate of 10 µg/kg/min.
Toxicity.
Cyanide toxicity is characterized by an unexplained metabolic acidosis, increased blood lactate concentrations, and an increased mixed venous oxygen content.435,436 The nitroprusside radical interacts with the sulfhydryl groups of erythrocytes, releasing cyanide. Nitroprusside contains five cyanide molecules; and as the cyanide is released, it is converted to nontoxic thiocyanate by the rhodanese enzyme system in the liver and is then excreted by the kidneys.436 If the amount of cyanide released overwhelms the capacity of the rhodanese system, cyanide toxicity (i.e., binding to the cytochrome electron transport system) results. This produces a change to anaerobic metabolism, metabolic acidosis, an increase in mixed venous oxygen content, and eventually death.435,437–441 Several pediatric anesthetic-related deaths have resulted from cyanide toxicity and its treatment.438–440
Three responses to sodium nitroprusside infusion may herald impending cyanide toxicity: more than 10 µg/kg/min required for a response, tachyphylaxis developing within 30 to 60 minutes, and immediate resistance to the drug.436 If any of these occur, sodium nitroprusside should be discontinued and the child investigated for possible cyanide toxicity. Treatment of cyanide poisoning is directed at reversal of the binding of cyanide to the cytochrome enzymes. This can be accomplished by producing methemoglobinemia with amyl nitrite. Methemoglobin has a greater affinity for cyanide than it does for the cytochrome system, forcing the reaction in the direction of forming cyanmethemoglobin. The breakdown of cyanmethemoglobin is promoted by administering thiosulfate, which reacts with the cyanide to form nontoxic thiocyanate, which is then excreted by the kidneys. Hydroxocobalamin may prevent toxicity by formation of cyanocobalamin.442
This treatment is not without hazard. As Posner states, “Overzealous treatment may merely convert a cytotoxic hypoxia to an anemic hypoxia.”442
Sodium nitroprusside is a safe medication if doses remain well within the guidelines that have been established by various investigators.443–445 For children, this is a maximum of 50 µg/kg/min for 30 minutes and 8 to 10 µg/kg/min for 3 hours, with frequent blood gas analyses.435,445 Because of the potential for toxicity and the availability of newer, less toxic vasodilators, this drug has had decreasing popularity for use for controlled hypotension and is now most commonly used for short-term control of blood pressure in special situations.
Nitroglycerin
Toxicity.
Nitroglycerin is relatively free of toxic side effects in the usual doses applied during hypotensive anesthesia.395,446,447 No toxicities or deaths have been reported with nitroglycerin when it is used for hypotensive anesthesia.395,446–448 However, several reports have described nitroglycerin-induced methemoglobinemia, and a study of patients undergoing anesthetic procedures using controlled hypotension did not find a relationship between the development of methemoglobin and the dose of nitroglycerin.449,450 Pulse oximetry may be of value in making the initial diagnosis (i.e., decreased saturation). However, if this occurs, accurate saturation determinations are not possible because of the interference in light absorbance caused by methemoglobin at both ends of the absorbance spectrum used by pulse oximeters.451,452 The use of other adjuncts (e.g., potent inhalation agents, other vasodilators, β-adrenergic blockade, potent opioid) reduces the total dose of nitroglycerin administered.
General Concepts of Hypotensive Anesthesia
Before using controlled hypotension, it is important to understand the rationale for using this technique.453 If it is used to reduce surgical blood loss, the preparation and monitoring of a child are different from the approach in a procedure in which the main objective for reducing the perfusion pressure is to improve operating conditions (e.g., microsurgical techniques). In the former case, direct assessment of circulating blood pressure and volume with an arterial line and central venous catheter is important, whereas in the latter case, only a direct means of measuring blood pressure (arterial line) is needed.
Anesthetic Management
All inhalational agents, by directly depressing cardiac output, have been used with various degrees of success as a single drug to produce controlled hypotension, but profound cardiovascular depression may be difficult to control and certainly is not readily reversible.404–406,409,454 We do not advocate hypotensive anesthesia using potent inhalational agents as the sole hypotensive agent because the cardiovascular depression is not rapidly reversed if a problem arises with a relative anesthetic overdose. However, small to moderate concentrations of inhalational anesthetic reduces the amount of vasodilator, β-blocker, or opioid necessary to reduce blood pressure.455
The availability of short-acting β-adrenergic blockers offers an alternative method to decrease MAP by directly depressing cardiac output. However, β-adrenergic blockade removes a valuable guide to the depth of anesthesia and volume status. Because the cardiac output in children approximately 2 years old or younger depends on heart rate (see Chapter 16), β-adrenergic blockade is not recommended in this age group. Low-dose, short-acting β-adrenergic blockade may be a reasonable adjunct to hypotensive anesthesia with inhalational anesthetics as a means of reducing the concentration of the anesthetic or as a supplement to reduce the vasodilator requirements.456 A rapid-acting β-blocker such as esmolol may be the best compromise because its half-life is brief (3 minutes), and it is administered as an infusion. Even with this type of control, serious adverse events have been reported with the use of β-blockers in children.412
Monitoring and Management Principles
Position.
Make the operative field the highest point of the child’s body to take advantage of gravitational forces to help reduce blood pressure. When positioning a child, care must also be taken to minimize any possible impedance to venous drainage that may contribute to blood loss. For example, abdominal pressure from a misplaced roll of sheets can markedly increase venous pressure during spinal instrumentation and completely offset any beneficial effects from reduced arterial pressure.397,453,457,458 If the head is the surgical site, the arterial transducer must be calibrated at head level rather than heart level to ensure adequate cerebral perfusion pressure.447,459
Laboratory Parameters.
Adequate levels of hemoglobin must be maintained to have sufficient oxygen-carrying capacity. Studies have indicated that at normal blood pressures, a hemoglobin value of 5 g/dL is well tolerated in laboratory animals, but ischemia may occur with a reduced hemoglobin concentration.460 There is some evidence that the combination of hemodilution and hypotensive anesthesia may fail to deliver adequate oxygenation to some vascular beds (e.g., renal, enteric mucosa).461,462 Although no similar studies have been conducted in children, for additional safety, we maintain the hemoglobin level at 9 to 10 g/dL during controlled hypotensive anesthesia. This is important for children undergoing spinal instrumentation, in which traction on the spinal cord may alter spinal cord blood flow.
Arterial blood gases must be carefully evaluated on a 30- to 60-minute basis to diagnose changes in oxygenation, ventilation, or perfusion or the development of drug toxicity (e.g., nitroprusside) or adverse anesthesia events.463–465 A large difference between arterial and expired carbon dioxide values may indicate a pulmonary shunt or air embolization. An increase in mixed venous oxygen content may signal cyanide toxicity. Adequate Pao2 must be maintained at all times. Continuous examination of the Pao2 is important during any hypotensive anesthetic technique because cerebral perfusion is directly related to Paco2.466,467 Normocarbia should be maintained. The combination of hypocarbia and hypotension should be avoided. The metabolic components of acid–base equilibrium must be monitored; development of acidosis reflects inadequate oxygen delivery or toxicity from the hypotensive agent (e.g., sodium nitroprusside).433
Although we do not advocate the routine use of β-adrenergic blockade, blood glucose values should be measured serially if these drugs are part of the chosen hypotensive technique. β-Adrenergic blockade inhibits glycogenolysis and may cause severe, unsuspected hypoglycemia in children.393,468,469
Contraindications
The risks of hypotensive anesthesia are significant.470 The risk-benefit ratio must always be considered on an individual basis, particularly with neurosurgical patients and those undergoing spinal instrumentation. Any systemic disease compromising the function of a major organ is a relative contraindication to the use of controlled hypotension. Most reported complications are related to the inexperience of the practitioner, inappropriate patient selection, unfamiliarity with the drugs involved, or inattention to details such as blood volume status, pH, Paco2, or blood glucose or not using infusion pumps to carefully titrate medications.* If a child is healthy and meticulous attention is paid to all the parameters previously detailed, the benefits of improved surgical technique, reduced surgical time, and decreased need for blood transfusion may outweigh the potential risks.
Normovolemic Hemodilution
Intentional isovolemic hemodilution is a useful adjunct to an anesthesiologist’s strategy for reducing allogeneic blood transfusions.329,330,353,460,473–480 Two basic methods can be applied:
1. Allow the surgical blood loss to continue until the child’s hematocrit value is in the high teens and maintain that hematocrit value until near the end of the procedure. At that time, the hematocrit can be increased to the desired value by transfusing PRBCs. This technique allows surgical bleeding to occur at a reduced hematocrit value, resulting in reduced loss of RBC mass.
2. Blood can be removed from a child at the beginning of the operation while replacing the volume with crystalloid solution (usually) and then returning the blood at the end of the procedure or when significant bleeding occurs.
The latter technique is preferable because it reserves a quantity of the child’s own blood, which can be returned at the end of the surgical procedure. For a Jehovah’s Witness, this technique often conforms to religious guidelines if direct continuity is maintained with the child’s circulation.460,481–488
During acute normovolemic hemodilution under anesthesia, the distribution of blood flow improves with the reduced hematocrit. Improved blood rheology is the major compensatory mechanism for maintaining oxygen delivery despite a reduced hematocrit. Oxygen extraction increases in the presence of an inadequate circulating blood volume or when the hematocrit decreases to less than 20%. If the hematocrit decreases to less than 15%, subendocardial myocardial ischemia may develop.489–492 At this extreme level of anemia, dissolved oxygen begins to assume a more important role in oxygen delivery.493 In several reports, extreme acute normovolemic hemodilution (hemoglobin as low as 2 g/dL) were well-tolerated,489,494–497 although we cannot endorse the use of this technique in children because it is impossible to assess the effects of such an extreme hemoglobin concentration on the long-term cognitive ability. Nonetheless, these reports489,497 indicate that healthy children can tolerate these extreme hematocrit concentrations provided they are anesthetized, normovolemic, slightly hypothermic, and ventilated with 100% oxygen. We limit the absolute minimum hematocrit to 15%, but we prefer to maintain the hematocrit closer to 20% at all times.
Technique and Key Concepts
When blood is collected for normovolemic hemodilution, blood flows directly from the arterial line into sterile blood bags that contain the appropriate anticoagulant. Each bag is weighed before any blood is transferred and then continuously during filling by placing it on a scale. The bag is frequently but gently agitated to ensure an even distribution of the anticoagulant. The total volume of blood to be withdrawn should be calculated preoperatively to reduce the hematocrit to the 20% to 25%. Care must be taken to replace the blood removed with 5% albumin milliliter for milliliter or 1.5 to 2 mL of lactated Ringer’s solution for each milliliter of blood removed. Sometimes, an even greater volume of replacement fluid is needed.479 A reasonable estimate of the adequacy of replacement is to obtain a baseline CVP and then maintain the same CVP as blood is withdrawn and replaced. It is preferable to hemodilute before the surgical incision to monitor changes in hemodynamic indices, although it can be performed during the initial phases of surgery. The major concern is to maintain a normal circulating blood volume and provide adequate oxygen-carrying capacity. It is important to make an educated guess about how much blood loss is anticipated during the surgery so that autologous blood can be reinfused in place of homologous blood. Because a small-pore filter (20 µm) traps many of the platelets that a large-pore filter (≥150 µm) allows to pass, the former is best avoided at this juncture. In many hospitals, use of this small pore filter has been discontinued.
The Jehovah’s Witness Patient
The children of Jehovah’s Witnesses present a particular medical and legal dilemma.498,499 Transfusion management of anyone with a religious objection to transfusion depends in part on the urgency of the surgical procedure and underlying medical condition of the child. If not emergent, a meeting with the patient (or the parents or guardian if a minor), the patient’s spiritual advisor (if the child so chooses), and representatives of the team that will be caring for the child should be held to allow the child to clearly articulate his or her wishes with respect to the refusal of transfusion and its consequences and to discuss possible alternatives, including the use of erythropoietin, iron therapy, acute normovolemic hemodilution, intraoperative cell recovery and reinfusion, and the use of antifibrinolytic medications.495,496,500–503 Specific inquiries should be made about each child’s beliefs regarding the use of albumin, plasma, cryoprecipitate, platelets, and intraoperative cell recovery and reinfusion if the circuit is not continuously connected to the child and, in particular, what the child’s response would be if a life-threatening event occurred while he or she was under anesthesia.481,504 These discussions should be carefully documented in the child’s record and informed consent signed beforehand. Not all hospitals or physicians are willing to participate in these cases, in which case arrangements should be made to transfer the child to the care of institutions or physicians who are willing to work within these constraints.
The courts have consistently ruled that the adult patient or emancipated minor has a right to refuse transfusion.482,505 Physicians have the moral and legal obligations to respect those beliefs if an adult or teenager has made an informed decision and understands that he or she may die or suffer permanent injury without a transfusion if a life-threatening situation occurs. However, in the case of a minor child, the Supreme Courts in the United States and Canada have ruled that the fate of the minor child cannot be determined by the parents’ religious convictions. The most important issue is full and open discussion of the effort that will be made to respect the person’s religious beliefs and avoid blood transfusions. In the case of minor children about whom a mutual understanding cannot be reached on avoiding blood transfusions, a court order can be obtained to save the child’s life. The parents should be informed about this possibility beforehand.506 The ethics of this issue are discussed in Chapter 5.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
10 Strategies for Blood Product Management and Reducing Transfusions
DESPITE ADVANCES IN PEDIATRIC SURGERY, the number of infants and children who sustain major operative blood loss remains high. Little information is available about when to expect coagulation defects in the pediatric age group,1,2 and most studies of massive blood transfusion have involved adult patients.3
Nothing changed the use of blood products more than the threat of the acquired immunodeficiency syndrome (AIDS).4–6 Although infection with human immunodeficiency virus (HIV) by blood transfusion has become rare, it was widely publicized in the lay press and is still feared by the public. Implementation of donor education programs, improved health history screening, new tests, and new test technologies (Table 10-1) have markedly altered the spectrum of transfusion-transmitted infectious agents in the developed world. The risks of some of the infectious and noninfectious hazards of transfusion are summarized in Table 10-2.
TABLE 10-1 Current Blood Screening Tests Used on Donated Blood in the United States
Hepatitis B surface antigen (HBsAg)
Hepatitis B core antibody (anti-HBc)
Hepatitis C virus antibody (anti-HCV)
Nucleic acid amplification testing for HCV RNA
Human immunodeficiency virus type 1 (HIV-1) antibody (anti-HIV-1)
Nucleic acid amplification testing for HIV-1 RNA
Human T-lymphotropic virus type 1 (HTLV-I) antibody (anti-HTLV-I)
HTLV-II antibody (anti-HTLV-II)
Serologic test for syphilis (Treponema pallidum)
*This test depends on the incidence in the geographic area.
†As of December 2011, at first donation or after residence in endemic area.
From American Association of Blood Banks. Facts about blood and blood banking, 2006. 509 Available at http://www.aabb.org/resources/bct/Pages/bloodfaq.aspx (accessed May 2012).
TABLE 10-2 Estimated Frequency of Complications per Number of Units Transfused
| Category | Complication | Frequency |
|---|---|---|
| Noninfectious | Allergic (urticarial) | 1 : 100 |
| Febrile, nonhemolytic | 1 : 100 | |
| Transfusion-associated circulatory overload | 1 : 1000 | |
| Delayed hemolytic | 1 : 1600 | |
| Transfusion-related acute lung injury | 1 : 10,000 | |
| Acute hemolytic | 1 : 50,000 | |
| Fatal acute hemolytic | 1 : 500,000 | |
| Infectious | Hepatitis B virus | 1 : 250,000 |
| Hepatitis C virus | 1 : 1,800,000 | |
| Human T-lymphotropic virus type I | 1 : 3,000,000 | |
| Human immunodeficiency virus type 1 | 1 : 2,300,000 | |
| Bacterial contamination of red blood cells | 1 : 50,000 | |
| Bacterial sepsis of red blood cells | 1 : 500,000 | |
| Bacterial contamination of platelets | 1 : 2,000 | |
| Bacterial sepsis of platelets | 1 : 75,000 |
Data from Davenport RD. Management of transfusion reactions. In: Mintz PD, editor. Transfusion therapy: clinical principles and practice. 3rd ed. Bethesda, Md.: AABB Press; 2011, p. 757-84; Alter HJ, Esteban-Mur JI. Transfusion transmitted hepatitis. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 718-45; Barbara JA, Dow BC. Retroviruses and other viruses. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 746-59; Park YA, Brecher ME. Bacterial contamination of blood products. In: Simon TL, Snyder EL, Solheim BG, et al., editors. Rossi’s principles of transfusion medicine. 4th ed. Oxford: AABB Press, Wiley-Blackwell; 2009, p. 773-90.
Despite marked reductions in the transmission of HIV, hepatitis C virus, and hepatitis B virus, transfusions can produce other deleterious effects.7,8 Every transfusion must be medically justified; benefits must be weighed against the potential infectious, immunologic, and metabolic risks.9 It is in the child’s best interest to transfuse with a clear clinical goal and in the anesthesiologist’s best interest to document the reason for each transfusion. It is not acceptable medical practice to administer a transfusion when it is of questionable benefit.
Blood Volume
The minimum acceptable hematocrit varies according to an individual child’s need. The balance between oxygen supply and demand depends on a number of factors, including the oxygen content of blood, cardiac output and its regional distribution, and metabolic needs. A child with severe pulmonary disease or cyanotic congenital heart disease probably requires a greater hematocrit than a healthy child to satisfy the metabolic oxygen demands. Preterm infants may require a greater hematocrit to prevent apnea, reduce cardiac and respiratory work, and possibly improve neurologic outcomes,10 although a Cochrane review suggests that this may not be the case.11 If there is uncertainty about the need to transfuse these infants, the neonatologist should be consulted.10,12,13 A healthy child readily tolerates a hematocrit well below 30%. It is our practice not to transfuse otherwise healthy infants up to about 3 months old until their hematocrits have decreased to 20% to 25% and hematocrits of older children have decreased to 20% if there is little potential for postoperative bleeding. The circulating blood volume must be maintained in every case. Observing the operative field to estimate blood loss and monitoring the vital signs, hematocrit, urine output, and the central venous pressure (CVP) help to assess the adequacy of volume replacement. If a procedure is expected to result in significant blood loss or fluid shifts, the anesthesiologist should strongly consider the use of a urine catheter, a central venous line, and invasive arterial monitoring. The child’s size or age should not be a deterrent to the use of a central venous catheter (Table 10-3).
TABLE 10-3 Estimated Predicted Blood Loss and Recommended Monitoring and Equipment
| Predicted Blood Loss | Recommended Monitors or Equipment |
|---|---|
| Less than 0.5 blood volume | Routine monitoring |
| 0.5-1.0 blood volume | Routing monitoring + urine catheter |
| 1.0 blood volume or more | Routine monitoring + urine catheter + CVP + arterial line |
| 1.0 blood volume or more with potential for rapid blood loss | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line + rapid-infusion device |
| Severe head injury | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line |
| Major trauma with unknown severity | Routine monitoring + urine catheter + CVP + arterial line + large-bore IV line (preferably in upper extremity or central) + rapid-infusion device |
CVP, Central venous pressure; IV, intravenous.
There are three approaches for estimating the MABL: an approximation of circulating RBC mass, a modified logarithmic equation, and a simple proportion.14,15 All three approaches yield clinically similar estimates of the MABL. The most straightforward method is to estimate the MABL by simple proportion.14 For purposes of discussion, we use a hematocrit of 25% as the minimum acceptable hematocrit.
Initial therapy is directed at replacing fluid deficits and providing maintenance requirements (see Chapter 8). Additional fluid administration is directed at replacing blood loss and third space fluid losses. Although former recommendations for crystalloid replacement of blood loss were 2 to 3 mL per milliliter of shed blood, later studies suggest a smaller volume of replacement, and 1 to 2 mL of isotonic crystalloid or 1 mL of 5% albumin may be adequate.16–18 The latter type of replacement is expensive, and there is no clear evidence that colloid is superior to crystalloid.19 New starch volume expanders have been introduced that may hold promise in the future for use in children.
Blood Components and Alternatives
Red Blood Cell–Containing Components
Blood components containing RBCs are indicated for the treatment of symptomatic deficits of oxygen-carrying capacity.20,21 PRBCs are the most widely available RBC-containing blood component, although in settings where the collection facilities do not have the capability of making components, whole blood may be the only component available. Donor whole blood is collected in a preservative-anticoagulant solution that contains citrate, phosphate, dextrose (glucose), and adenine (CPDA) or just citrate, phosphate, and dextrose. In the latter case, the platelet-rich plasma is removed after centrifugation of the whole blood unit, and a solution containing adenine, dextrose, and occasionally mannitol is added to the PRBCs. The additive-solution systems permit storage for 42 days (compared with 35 days for CPDA) and better preservation of 2,3-diphosphoglycerate (DPG) levels. The characteristics of the CPDA and additive-solution PRBCs and of whole blood are shown in Table 10-4. Although the hematocrit is reduced in the additive-solution PRBCs, the red cell mass is the same.
RBCs carry glycoconjugate antigens of the ABH histo-blood group system on the cell surface that are determined by six common alleles on chromosome 9.19a During the first year of life, infants begin to elaborate alloantibodies to whichever A or B antigens they lack. These isoagglutinins are invariably present after a few months and constitute a formidable immunologic obstacle to transfusion or transplantation across this ABO barrier. The RBCs for transfusion must be compatible with the ABO isoagglutinins of the intended transfusion recipient. Similarly, components with a large volume of plasma (e.g., whole blood, FFP, apheresis platelets) must be compatible with the A or B surface antigens expressed on the recipient’s RBCs. PRBCs must be ABO compatible with the recipient, whereas whole blood must be ABO identical. Table 10-5 summarizes the permissible combinations.
Only RBCs express the Rh(D) antigen. Rh(D)-positive patients may receive Rh(D)-positive or Rh(D)-negative RBCs. Rh(D)-negative patients are routinely given Rh(D)-negative RBCs for any elective transfusions, but in the setting of massive transfusion it may be necessary to switch to Rh(D)-positive RBCs to preserve the supply of Rh(D)-negative RBCs. The blood bank usually determines when to make this substitution based on the inventory and does so more quickly for a patient who is a male or a postmenopausal female (Table 10-6). The objective is to avoid exposing a female with childbearing potential to Rh(D)-positive RBCs and possibly triggering the production of the anti-D alloantibody, which is responsible for the most severe forms of hemolytic disease of the newborn. Table 10-7 shows the common initial volume of PRBCs needed to increase the hemoglobin level by 2 to 3 g/dL.
TABLE 10-7 Common Initial Doses of Blood Components and Expected Effects in Children
| Component | Dose | Effect |
|---|---|---|
| Packed red blood cells | 10-15 mL/kg | Increase hemoglobin by 2-3 g/dL |
| Platelets* | 5-10 mL/kg | Increase platelet count by 50,000-100,000/mm3 |
| Fresh frozen plasma | 10-15 mL/kg | Factor levels increase by 15%-20% |
| Cryoprecipitate | 1-2 units/kg | Increase fibrinogen by 60-100 mg/dL |
*This recommendation may be reduced pending the impact of the prophylactic platelet dose (PLADO) trial, as published for all age groups40 and for the pediatric age range.40a
The changes that occur to RBC during storage under conventional blood bank conditions has been well described.22 These observations have generated physiologically plausible hypotheses about how such changes may impair the function of the banked RBCs in vivo. The loss of intra-erythrocytic levels of 2,3-DPG and its corresponding decrease in the P50 value may reduce the ability of stored RBCs to relinquish bound O2 compared with 2,3-DPG–replete RBCs. The depletion of nitric oxide (NO) may reduce the vasodilatory properties of the RBCs, hence impairing their ability to maintain the patency of the small vessels in the microcirculation and blood flow to the tissues.23 Numerous changes in the composition and behavior of the RBC plasma membrane, including the loss and oxidation of membrane lipids and proteins and the rearrangement of some membrane constituents,24 correlate with changes in the shape and elasticity of the RBC membrane.25,26 The loss of elasticity in particular can impede the rapid movement of the RBCs through the microcirculation.
These hypotheses and some supportive data from animal models27 have led to a number of clinical studies (mostly in trauma, critical care, colorectal surgery, and cardiac surgery) of outcomes after using stored RBCs, but the results have been inconclusive.28–30 About half of the studies found a statistical association between at an unfavorable clinical outcome measure and the transfusion of RBCs, which had been stored for a longer time. However, no association was seen in one half of the studies, including two that were extensions of previous studies with positive findings. A small number of randomized, controlled trials addressed this issue without statistically significant differences in outcomes between patients receiving RBCs stored for different amounts of time,31 although two of them were underpowered.32,33 Knowing that RBCs change during storage raises the question of whether these changes affect the patients in a clinically meaningful way, a question that remains unanswered. Currently, there are four randomized, clinical trials being conducted in North America in populations of especially vulnerable patients: neonates in the intensive care unit, adults in the intensive care unit, and adults undergoing cardiac surgery.34–37 The results of these trials should help to guide future transfusion practice.
Platelets
Platelets may be obtained from a whole blood donation or collected by apheresis. Whole blood–derived platelets are separated by centrifugation and suspended in 40 to 60 mL of plasma at a concentration that is two to four times greater than in the circulation. Each unit contains a minimum of 5.5 × 1010 platelets and is stored at 20° to 24° C with gentle continuous agitation for a maximum of 5 days. One unit of whole blood–derived platelets can be expected to increase the platelet count of a 70-kg adult by 5000 to 10,000/mm3 and increase the count in an 18-kg child by 15,000/mm3.38,39 A unit of platelets obtained by apheresis contains at least 3 × 1011 platelets in 200 to 400 mL of plasma, or the equivalent of approximately 6 units of whole blood–derived platelets. A common dose for pediatric patients is 0.1 to 0.3 unit/kg of body weight, or 10 to 15 mL/kg (see Table 10-7); this dose usually produces an increment of 30,000 to 90,000/mm3. However, a rigorous reassessment of platelet dosing was just carried out in the prophylactic platelet dose trial.40,41 Doses equivalent to the standard dose of one pheresis pack (or 6 units) per m2, half of this dose, and double this dose were compared in 1272 adult and pediatric patients who received at least one platelet transfusion. Blood losses were determined with the World Health Organization (WHO) bleeding scale: grade 0 = no bleeding, grade 1 = petechiae, grade 2 = mild blood loss, grade 3 = gross blood loss, and grade 4 = debilitating blood loss. No differences were observed in bleeding outcomes (WHO grade 2 or greater) among the three doses; this was also true for the subset of 200 pediatric patients. The recommended platelet dose may be reduced in the near future, but this change awaits further discussion by the blood transfusion and hemostasis community.42–44 This trial did not involve patients undergoing surgical procedures with ongoing blood loss.
In the setting of dilutional thrombocytopenia with ongoing losses or a consumptive coagulopathy (e.g., disseminated intravascular coagulation), larger doses (≥0.3 unit/kg) may be required to boost the platelet count above 50,000/mm3. Because platelets are suspended in plasma that contains the anti-A and anti-B isoagglutinins, they should be ABO compatible with the recipient’s RBCs. Some blood donors have high-titer isoagglutinins that can produce hemolysis in transfusion recipients if a large enough volume of plasma is given.45 The transfusion of plasma-incompatible, whole blood–derived platelets to adult recipients does not produce clinically significant hemolysis because the volume of plasma given is so small relative to the plasma volume of an adult. However, apheresis platelets (and whole blood–derived platelets for small children) should be ABO compatible with the recipient’s RBCs. Because platelets do not express Rh antigens, matching for Rh(D) antigen is not necessary for apheresis platelets because they contain virtually no RBCs. However, whole blood–derived platelets may contain enough RBCs to provoke Rh alloimmunization so platelets from Rh(D)-negative donors are given preferentially to Rh(D)-negative recipients with childbearing potential. If a premenopausal female receives whole blood–derived platelets from an Rh(D)-positive donor, Rh immune globulin can be administered within 72 hours to prevent alloimmunization. Platelets should never be withheld in an emergency situation because of Rh(D) incompatibility.
Platelets are essential to hemostasis associated with the vascular injury of surgery and are necessary for the control of surgical bleeding. Platelets are also required for the maintenance of an intact endothelial barrier to spontaneous blood loss. The number of platelets required to provide adequate hemostasis in the surgical setting is much greater than the level needed to provide prophylaxis against spontaneous hemorrhage. A platelet count of 10,000/mm3 is considered adequate to prevent spontaneous bleeding or bleeding from minor invasive procedures (e.g., lumbar puncture, line placement) in an otherwise stable child. If overt signs of bleeding are present or a more significant hemostatic challenge in the form of a surgical procedure is imminent, a level of 30,000 to 50,000/mm3 may be required.46–50 A target level of 50,000/mm3 is appropriate in the setting of massive transfusion.47,51–53 Platelets may also be required for children with adequate counts but in whom platelet function is impaired. Many medications (e.g., aspirin; nonsteroidal antiinflammatory agents; dipyridamole; platelet P2Y12 receptor blockers such as clopidogrel or prasugrel; or glycoprotein IIa/IIIb receptor inhibitors such as abciximab, eptifibatide, or tirofiban; serotonin uptake antagonists such as Zoloft) and some conditions (e.g., renal failure with blood urea nitrogen levels above 60 mg/dL) cause abnormal platelet function, which may interfere with surgical hemostasis, in which case it may be necessary to maintain the platelet count at a somewhat greater level, at least until the effect of the medication dissipates or the child’s platelets have largely been replaced by banked platelets.54,55 In a few settings, such as intracranial, ophthalmic, and otologic surgery, even greater levels (100,000/mm3) are sought.
There is no clear-cut threshold below which the platelet count predicts clinical bleeding in the perioperative period. Each child must be individually assessed by constantly observing the surgical field for evidence of abnormal bleeding.56 Unfortunately, we lack a well-validated bedside tool to assess platelet function. The utility of the thromboelastogram and other devices to measure platelet function under controlled flow conditions, such as the platelet function analyzer (PFA-100), are being investigated.57–59 The standard technique for diagnosis and evaluation of thrombocytopathies remains Born-O’Brien platelet aggregometry, but it is not useful in the intraoperative setting.60 Most commonly, thrombocytopenia rather than a newly acquired platelet function defect is the problem in the operative setting and massive transfusion.
Several additional points should be considered61:
1. Not all hospitals have platelets in the inventory. Unless the need is anticipated before surgery, platelets may not be available when required.
2. For children who are thrombocytopenic before surgery, platelets should be infused just before the surgical procedure to ensure the greatest levels during the time of peak demand.
3. Platelets should be filtered only by large-pore filters (≥150 µm) or leukocyte-reduction filters (if indicated). Micropore filters may adsorb large numbers of platelets and therefore diminish the effectiveness of a platelet transfusion.
4. Platelets are suspended in plasma, which may help to replenish coagulation factors other than factors V and VIII, which are labile.
5. Platelets should not be refrigerated or placed in a cooler with ice before administration, because after they are transfused, cold platelets are rapidly cleared from the circulation.
Special Processing of Cellular Blood Components
Leukocytes collected with whole blood donations partition into platelet and PRBC components, and few intact leukocytes are present in FFP. Passenger leukocytes are responsible for most febrile, nonhemolytic transfusion reactions, HLA alloimmunization, and transmission of cytomegalovirus (CMV). To prevent the complications from these leukocytes, they can be very effectively removed (2 to 3 log reduction) by passage through leukocyte-reduction filters that may be done shortly after collection (prestorage leukoreduction) or at the bedside. Leukocyte reduction of RBCs by filtration is a superior technique to washing or freezing-deglycerolizing, which was used in the past. Table 10-8 provides indications for who may benefit from receiving leukocyte-reduced cellular components (i.e., RBCs or platelets).
TABLE 10-8 Indications for Leukocyte-Reduced Cellular Blood Components
• To prevent further febrile, nonhemolytic transfusion reactions in a child with a history of such reactions
• To prevent human leukocyte antigen alloimmunization in a child who may require long-term platelet transfusion support (e.g., leukemia, lymphoma)
• To prevent human leukocyte antigen alloimmunization in a transplant recipient
• To prevent cytomegalovirus infection and disease in a susceptible child
CMV transmission can also be reduced by screening donors for CMV exposure (testing for antibody to CMV), although leukocyte reduction is the more widely used approach. Even though primary CMV infection is benign in children with intact immune systems, some pediatric populations are at risk for developing systemic disease and should be protected from CMV transmission by blood. Only patients who have not previously been infected with CMV (i.e., CMV seronegative) are at risk. Children particularly susceptible to systemic CMV infections are listed in E-Table 10-1.
E-TABLE 10-1 Children at Risk for Cytomegalovirus Disease*
AIDS, Acquired immunodeficiency syndrome; CMV, cytomegalovirus; HIV, human immunodeficiency virus.
*Applies to CMV-seronegative patients only.
Transfused lymphocytes may mediate a graft-versus-host process in some recipients with impaired cellular immunity. Because this process involves the bone marrow and the usual targets (i.e., skin and gastrointestinal tract), the fatality rate is substantial. Transfusion-associated graft-versus-host disease (GVHD) can be prevented by exposing cellular blood components to gamma irradiation that disables the donor lymphocytes. Children who are considered to be at risk for transfusion-associated GVHD and who should receive irradiated cellular components are listed in E-Table 10-2. This complication can also occur in children with intact immune systems in the unusual circumstance when the transfusion donor is homozygous for an HLA haplotype that is shared with the recipient. In this case, the recipient’s immune system, although fully functional, cannot recognize the donor lymphocytes as foreign. The donor lymphocytes mount a GVHD attack on the recipient’s tissues, recognizing the mismatched haplotype. This situation is more likely to occur when the donor is a blood relative of the recipient. It is for this reason that blood and HLA-matched platelets donated by family members are routinely irradiated.
E-TABLE 10-2 Children at Risk for Transfusion-Associated Graft-versus-Host Disease
• Children with T-cell immune defects (e.g., severe combined immunodeficiency syndrome, Wiskott-Aldrich syndrome, DiGeorge syndrome)
• Children undergoing chemotherapy for leukemia, lymphoma, or neuroblastoma
• Children undergoing hematopoietic stem cell transplantation
• Children receiving fludarabine
• Fetus: intrauterine transfusion
• Neonate: exchange transfusion
• Children receiving platelets from HLA-matched donors
• Children receiving red blood cells or platelets from blood relatives
HLA, Human leukocyte antigen system.
Fresh Frozen Plasma
Fresh frozen plasma (FFP) represents the fluid portion of whole blood that is separated and frozen within 8 hours of collection. After thawing at 37° C, which usually requires 30 minutes, it may be administered within 24 hours if stored at 1° to 6° C. The volume of 1 unit varies from 180 to 300 mL and represents 7% to 10% of the coagulation factor activity in a 70-kg patient. It contains all of the clotting factors and regulatory proteins at approximately the native concentration, but after 6 hours at 1° to 6° C, the levels of the labile factors V and VIII begin to diminish.62 FFP does not provide functional platelets, nor does it contain leukocytes or RBCs.
FFP is frequently administered without justification by evidence-based medicine.63 One major surgical indication for FFP is to correct the coagulopathy associated with massive blood transfusion (see Table 10-7). Other indications include a prolongation in the prothrombin time (PT) before surgery or in the setting of bleeding, the emergency reversal of warfarin, or the presence of a specific congenital or acquired coagulation protein deficiency for which a factor concentrate or a recombinant factor is not available (e.g., for factor XI deficiency).64 Administration of vitamin K should not be overlooked in children who are taking warfarin, have hepatic insufficiency, have been exclusively breastfed,65–67 who have been on broad-spectrum antibiotics (which often eliminate normal vitamin K–producing gastrointestinal flora), who have been on total parenteral nutrition for inadequate oral caloric intake, or who may have prolonged hospitalizations. Correction of a mild increase in the PT (e.g., international normalized ratio [INR] <1.5) is rarely necessary. Figure 10-1 shows the relationship between the level of coagulation factors and the in vitro clotting times, in this case the PT. Relatively modest levels of coagulation factors can support normal hemostasis, even though the PT is prolonged. When the PT is very prolonged (see Fig. 10-1, point A), the transfusion of 1 unit of FFP, which increases the coagulation factor levels by 7% to 10% in an adult, has a dramatic effect on shortening the PT. When the PT is only mildly prolonged, as at point C, where factor levels are already adequate for hemostasis, the increase in the factor levels by an additional 7% to 10% with 1 unit of FFP (in an adult) has a much smaller effect on the PT. This small effect does not achieve any improvement in hemostasis.
Cryoprecipitate
Cryoprecipitate is prepared by thawing FFP at 4° to 10° C and expressing most of the plasma, leaving behind precipitated protein that is then resuspended in a small volume of residual plasma (15 to 25 mL) and refrozen. This component contains 20% to 50% of the factor VIII from the original unit of plasma. It also contains von Willebrand factor (vWF), fibrinogen (approximately 250 mg), and factor XIII. It is indicated for the treatment of factor XIII deficiency, dysfibrinogenemia, and hypofibrinogenemia (see Table 10-7).68–77 It is no longer used for the treatment of von Willebrand disease or hemophilia A. Plasma concentrates of factor XIII and fibrinogen have been available in Europe for several years and recently have been licensed in the United States for use in patients with these congenital deficiencies.
Plasma-Derived and Recombinant Factor Concentrates
The most commonly administered factor concentrate is factor VIII, used in the treatment of hemophilia A. Children with hemophilia can have many problems related to their disease, including splenomegaly, abnormal liver function, and joint disease related to hemarthrosis. In the past, the use of pooled plasma products was associated with very high rates of transmission of viral hepatitis (especially hepatitis C virus) and HIV.78–80 The use of more rigorous viral removal and inactivation processes and the introduction of recombinant factor VIII and IX products81–85 have greatly reduced these problems.68–7586 Initial concerns that there may be an increased incidence of inhibitors in children who receive recombinant therapy compared with plasma-derived factor therapy have not been borne out.87 Mild hemophilia usually responds well to desmopressin (1-deamino-8-d-arginine vasopressin [DDAVP]) therapy.88,89
Children with hemophilia B (i.e., Christmas disease or factor IX deficiency) are managed with recombinant human factor IX and highly purified factor IX (preparations with various amounts of factors VII, X, and prothrombin) that are treated to inactivate or remove viruses.70,71,73,90–103 Careful planning of any surgical procedure for these children includes close communication with the child’s hematologist to ensure optimal therapy while reducing unnecessary transfusions (see Chapter 9).
Desmopressin
DDAVP, a synthetic analogue of vasopressin, can increase the levels of factor VIII : C (i.e., coagulant activity) and factor VIII : vWF in children with mild hemophilia A or von Willebrand disease.89,104–108 An IV dose of 0.3 µg/kg (maximum 20 µg; a subcutaneous preparation is available in Europe) increases the levels of both factors twofold to threefold within 30 to 60 minutes, with a half-life of 3 to 6 hours.105 Intranasal DDAVP is also effective, but onset is less rapid. Between 80% and 90% of children with von Willebrand disease are responders,109,110 and affected children should be tested for their responsiveness to IV DDAVP. This treatment is best suited to bleeding from surgical procedures, which ceases within 2 to 3 days. When bleeding continues beyond this period, as can occur with some orthopedic procedures, daily IV Humate P (or Alphanate or Koate DVI) can obviate possible tachyphylaxis with DDAVP. Products rich in the vWF allow better control over peak levels of factor VIII. When in excess of 200%, factor VIII predisposes to postoperative deep venous thrombosis and pulmonary embolism.
DDAVP has been used to treat the coagulopathy associated with uremia and cirrhosis.111,112 It may reduce elective surgical bleeding when the potential for blood loss is substantial, such as in cardiac surgery and spinal fusion.89,113–118 Although initial reports apparently demonstrated a benefit in patients who did not have a preexisting coagulopathy, other controlled studies failed to show an effect despite increases in factor VIII:C and vWF, and its use for these indications has largely been abandoned.119–121 Because of the potential for hyponatremia from water retention, use of DDAVP is avoided in children younger than 2 years, in children with CNS lesions, including a brain tumor, history of CNS irradiation, or recent neurosurgery or CNS trauma, and in elderly adults.
Albumin, Dextrans, Starches, and Gelatins
Albumin has the longest track record and the fewest adverse side effects.122,123 In the past, dextrans (i.e., high- and low-molecular-weight glucose polymers) were administered for volume expansion and hemodilution in children,124,125 but currently, their primary use is for antithrombosis, although their value for even this indication is questionable.126
Starches are branched polysaccharide polymers available in high-, medium-, and low-molecular-weight ranges (480,000 to 70,000 daltons). However, these compounds alter hemostasis by diluting clotting factors and impairing platelet function and the coagulation cascade.127,128 An additional concern, especially in children, is their accumulation in the reticuloendothelial system and the potential for unknown long-term adverse effects.129 Several studies have been carried out in children with minor changes in coagulation parameters that occurred when transfusions exceed 20 mL/kg.130–134 A 6% hydroxyethyl starch (HES 130/0.4) yielded clinical and physiologic profiles similar to those for 5% albumin in volumes up to 16 mL/kg in noncardiac surgery and in volumes up to 50 mL/kg in cardiac surgery, although at smaller cost.135,136 Several major reviews regarding the use of starches and gels in adult patients who are critically ill have raised significant concerns regarding adverse effects on coagulation137,138 and renal function,139,140 and they found inadequate overall safety data, even for the third-generation products.18,141 If these concerns have been raised in adult populations, we should have even greater concern about their use in children.
[/not-level-membership-for-anesthesiology-category]
