[level-membership-for-internal-medicine-category]
88 |
Stem Cell Biology |
Stem cell biology is a rapidly expanding field that explores the characteristics and possible clinical applications of a variety of stem cells that serve as the progenitors of more differentiated cell types. In addition to potential therapeutic applications (Chap. 90e), patient-derived stem cells can also be used as disease models and as a means of testing drug efficacy. Stem cells and their niche are a major focus of medical research because they play central roles in tissue and organ homeostasis and repair, which are important aspects of aging and disease.
IDENTIFICATION, ISOLATION, AND DERIVATION OF STEM CELLS
Resident Stem Cells The definition of stem cells remains elusive. Stem cells were originally postulated as unspecified or undifferentiated cells that provide a source of renewal of skin, intestine, and blood cells throughout life. These resident stem cells have been identified in a variety of organs (e.g., epithelia of the skin and digestive system, bone marrow, blood vessels, brain, skeletal muscle, liver, testis, and pancreas) based on their specific locations, morphology, and biochemical markers.
Isolated Stem Cells Unequivocal identification of stem cells requires their separation and purification, usually based on a combination of specific cell-surface markers. These isolated stem cells (e.g., hematopoietic stem [HS] cells) can be studied in detail and used in clinical applications, such as bone marrow transplantation (Chap. 89e). However, the lack of specific cell-surface markers for other types of stem cells has made it difficult to isolate them in large quantities. This challenge has been partially addressed in animal models by genetically marking different cell types with green-fluorescent protein driven by cell-specific promoters. Alternatively, putative stem cells have been isolated from a variety of tissues as side population (SP) cells using fluorescence-activated cell sorting after staining with the Hoechst 33342 dye.
Cultured Stem Cells It is desirable to culture and expand stem cells in vitro to obtain a sufficient quantity for analysis and potential therapeutic use. Although the derivation of stem cells in vitro has been a major obstacle in stem cell biology, the number and types of cultured stem cells have increased progressively (Table 88-1). Cultured stem cells derived from resident stem cells are often called adult stem cells or somatic stem cells to distinguish them from embryonic stem (ES) and embryonic germ (EG) cells. However, considering the existence of embryo-derived, tissue-specific stem cells (e.g., trophoblast stem [TS] cells) and the possible derivation of similar cells from an embryo/fetus (e.g., neural stem [NS] cells), it is more appropriate to use the term, tissue stem cells.
|
EXAMPLES OF CULTURED STEM CELLS |
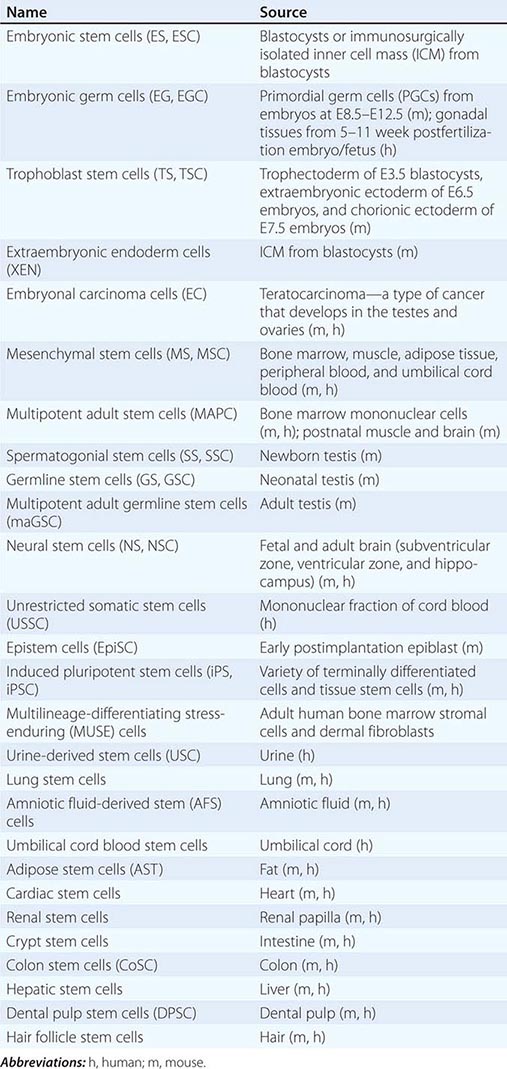
Successful derivation of cultured stem cells (both embryonic and tissue stem cells) often requires the identification of necessary growth factors and culture conditions, mimicking the microenvironment or niche of the resident stem cells. Recently, long-term maintenance of tissue stem cells in vitro is increasingly possible by growing them as three-dimensional (3D) organoids, which contain both stem cells and niche cells (Chap. 92e). For example, intestinal stem cells can now be cultured as “epithelial mini-guts” in the presence of R-spondin, epidermal growth factor (EGF), and noggin on Matrigel. Similarly, lung stem cells can be cultured as self-renewing “alveolospheres.” A growing list of cultured stem cells, although not comprehensive, is shown in Table 88-1. Please note that the establishment of cultured stem cells is often under dispute due to the difficulties in assessing the characteristics of these cells.
SELF-RENEWAL AND PROLIFERATION OF STEM CELLS
Symmetric and Asymmetric Cell Division The most widely accepted stem cell definition is a cell with a unique capacity to produce unaltered daughter cells (self-renewal) and to generate specialized cell types (potency). Self-renewal can be achieved in two ways. Asymmetric cell division produces one daughter cell that is identical to the parental cell and one daughter cell that is different from the parental cell and is a progenitor or differentiated cell. Asymmetric cell division does not increase the number of stem cells. Symmetric cell division produces two identical daughter cells. For stem cells to proliferate in vitro, they must divide symmetrically.
Unlimited Expansion In Vitro Resident stem cells are often quiescent and divide infrequently. However, once the stem cells are successfully cultured in vitro, they often acquire the capacity to divide continuously and the ability to proliferate beyond the normal passage limit typical of primary cultured cells (sometimes called immortality). These features are primarily seen in ES cells but have also been demonstrated for tissue stem cells, such as NS cells and mesenchymal stem (MS) cells, thereby enhancing the potential of these cells for therapeutic use (Table 88-1).
Stability of Genotype and Phenotype The capacity to actively proliferate is often associated with the accumulation of chromosomal abnormalities and mutations. Mouse ES cells appear to be an exception to this rule and tend to maintain their euploid karyotype and genome integrity. By contrast, human ES cells appear to be more susceptible to mutations after long-term culture. However, it is also important to note that even euploid mouse ES cells can form teratomas when injected into immunosuppressed animals, raising concerns about the possible formation of tumors after transplanting actively dividing stem cells.
POTENCY AND DIFFERENTIATION OF STEM CELLS
Developmental Potency The term potency is used to indicate a cell’s ability to differentiate into multiple specialized cell types. The current lack of knowledge about the molecular nature of potency requires the experimental manipulation of stem cells to demonstrate their potency. For example, in vivo testing can be done by injecting stem cells into mouse blastocysts or immunosuppressed adult mice and determining how many different cell types are formed from the injected cells. However, these in vivo assays are not applicable to human stem cells. In vitro testing can be performed by differentiating cells in various culture conditions to determine how many different cell types are formed from the cells. The formal test of self-renewal and potency is performed by demonstrating that a single cell possesses such abilities in vitro (clonality). Cultured stem cells are tentatively grouped according to their potency (Fig. 88-1). Only some examples are shown, because many cultured stem cells, especially human cells, lack definitive information about their developmental potency.
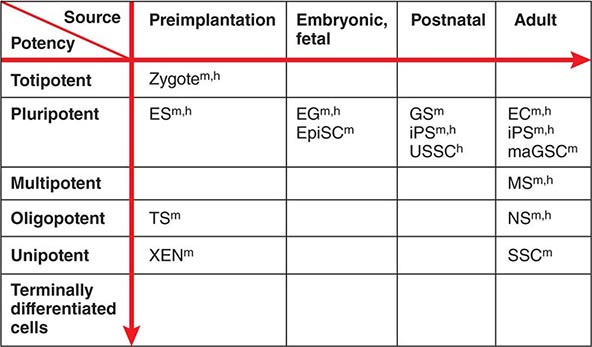
FIGURE 88-1 Potency and source developmental stage of cultured stem cells. For abbreviations of stem cells, see Table 88-1. Note that stem cells are often abbreviated with or without “cells,” e.g., ES cells or ESCs for embryonic stem cells. h, human; m, mouse.
From Totipotency to Unipotency Totipotent cells can form an entire organism autonomously. Only a fertilized egg (zygote) possesses this feature. Pluripotent cells (e.g., ES cells) can form almost all of the body’s cell lineages (endoderm, mesoderm, and ectoderm), including germ cells. Multipotent cells (e.g., HS cells) can form multiple cell lineages but cannot form all of the body’s cell lineages. Oligopotent cells (e.g., NS cells) can form more than one cell lineage but are more restricted than multipotent cells. Oligopotent cells are sometimes called progenitor cells or precursor cells; however, these terms are often more strictly used to define partially differentiated or lineage-committed cells (e.g., myeloid progenitor cells) that can divide into different cell types but lack self-renewing capacity. Unipotent cells or monopotent cells (e.g., spermatogonial stem [SS] cells) can form a single differentiated cell lineage.
Nuclear Reprogramming Development naturally progresses from totipotent fertilized eggs to pluripotent epiblast cells to multipotent cells and, finally, to terminally differentiated cells. According to Waddington’s epigenetic landscape, this is analogous to a ball moving down a slope. The reversal of the terminally differentiated cells to totipotent or pluripotent cells (called nuclear reprogramming) can thus be seen as an uphill gradient. Nuclear reprogramming has been achieved using nuclear transplantation, or nuclear transfer (NT), procedures (often called “cloning”), where the nucleus of a differentiated cell is transferred into an enucleated oocyte. Although this is an error-prone procedure with a very low success rate, live animals have been produced using adult somatic cells as donors in sheep, mice, and other mammals. In mice, it has been demonstrated that ES cells derived from blastocysts made by somatic cell NT are indistinguishable from normal ES cells. NT can potentially be used to produce patient-specific ES cells carrying a genome identical to that of the patient, although such strategies have not been pursued due to ethical issues and technical challenges. Recent success in generating human ES cells by NT has rekindled an interest in this area; however, the limited supply of human oocytes will still be a major problem for clinical applications of NT.
An alternative approach that has become a method of choice is the direct conversion of terminally differentiated cells into ES-like cells (called induced pluripotent stem [iPS] cells) by overexpressing a combination of key transcription factors (TFs). The original method was to infect mouse embryonic fibroblast cells with retrovirus vectors carrying four TFs [Pou5f1 (Oct4), Sox2, Klf4, and Myc] and to identify rare ES-like cells in culture. This approach was soon adapted to human cells, followed by a more refined procedure (e.g., the use of fewer TFs, different cell types, and different gene-delivery methods). Because a clinical trial using iPS cells is imminent, the safety of iPS-based therapy is a major concern and a variety of measures are being taken to ensure the safety. For example, it has now become a standard to use footprint-free methods such as an episomal vector, Sendai virus vector, and synthetic mRNAs to deliver reprogramming factors into cells, resulting in the production of patient-specific iPS cells with minimal alteration of their genetic makeup. In addition to cell replacement therapy, disease-specific iPS cells are expected to play a role in modeling human disease in vitro and in screening drugs for personalized medicine.
It has also become possible to convert one type of terminally differentiated cell (e.g., fibroblast cell) into another type of terminally differentiated cell (e.g., cardiac muscle, neuron, or hepatocyte) by overexpressing specific sets of TFs (called direct reprogramming). Direct reprogramming can bypass the step of making iPS cells, possibly providing the safer route to desired cell types for therapy; however, the technology is currently limited by its low efficiency.
Stem Cell Plasticity, Transdifferentiation, and Facultative Stem Cells The prevailing paradigm in developmental biology is that once cells are differentiated, their phenotypes are stable. However, more recent studies show that tissue stem cells, which have traditionally been thought to be lineage-committed multipotent cells, possess the capacity to differentiate into cell types outside their lineage restrictions (called transdifferentiation). For example, HS cells may be converted into neurons as well as germ cells. This feature may provide a means to use tissue stem cells derived directly from a patient for therapeutic purposes, thereby eliminating the need to use embryonic stem cells or elaborate procedures such as nuclear reprogramming of a patient’s somatic cells. However, more strict criteria and rigorous validation are required to establish tissue stem cell plasticity. For example, observations of transdifferentiation may reflect cell fusion, contamination with progenitor cells from other cell lineages, or persistence of pluripotent embryonic cells in adult organs. Therefore, the assignment of potency to each cultured stem cell in Fig. 88-1 should be considered with caution. Whether transdifferentiation exists and can be used for therapeutic purposes remains to be determined conclusively. A similar, but distinct, concept is the facultative stem cell, which is defined as a unipotent cell or a terminally differentiated cell that can function as a stem cell upon tissue injury. The presence of such cells has been proposed for some organs such as liver, intestine, pancreas, and testis, but is still debated.
Directed Differentiation of Stem Cells Pluripotent stem cells (e.g., ES and iPS cells) can differentiate into multiple cell types, but in culture, they normally differentiate into heterogeneous cell populations in a stochastic manner. However, for therapeutic uses, it is desirable to direct stem cells into specific cell types (e.g., insulin-secreting beta cells). This is an active area of stem cell research, and protocols are being developed to achieve this goal. In any of these directed cell differentiation systems, the cell phenotype must be evaluated critically. Alternatively, the heterogeneity of the cell population derived from pluripotent stem cells can be actively exploited, as different types of cells interact with each other in culture and further enhance their own differentiation. In some instances, e.g., optic cup, self-organizing tissue morphogenesis has been demonstrated in 3D culture.
MOLECULAR CHARACTERIZATION OF STEM CELLS
Genomics and Proteomics In addition to standard molecular biological approaches, high-throughput genomics and proteomics have been extensively applied to the analysis of stem cells. For example, DNA microarray analyses have revealed the expression levels of essentially all genes and identified specific markers for some stem cells. Chromatin immunoprecipitation coupled with next-generation sequencing technologies, capable of producing billions of sequence reads in a single run, has revealed chromatin modifications (“epigenetic marks”) relevant to stem cell properties. Similarly, the protein profiles of stem cells have been assessed by using mass spectrometry. These methods are beginning to provide a novel means to characterize and classify various stem cells and the molecular mechanisms that give them their unique characteristics.
ES Cell Regulation It is important to identify genes involved in the regulation of stem cell function and to examine the effects of altered gene expression on ES and other stem cells. For example, core networks of TFs such as Pou5f1 (Oct4), Nanog, and Sox2, govern key gene regulatory pathways/networks for the maintenance of self-renewal and pluripotency of mouse and human ES cells. These TF networks are modulated by specific external factors through signal transduction pathways, such as leukemia inhibitory factor (Lif)/Stat3, mitogen-activated protein kinase 1/3 (Mapk1/3), the transforming growth factor β (TGFβ) superfamily, and Wnt/glycogen synthase kinase 3 beta (Gsk3b). Inhibitors of Mapk1/3 and Gsk3b signaling enhance the derivation of ES cells and help maintain ES cells in full pluripotency (“ground” or “naive state”). Recent data also indicate that 20–25 nucleotide RNAs, called microRNAs (miRNAs), play an important role in regulating stem cell function by repressing the translation of their target genes. For example, it has been shown that miR-21 regulates cell cycle progression in ES cells and miR-128 prevents the differentiation of hematopoietic progenitor cells. These types of analyses should provide molecular clues about the function of stem cells and lead to a more effective means to manipulate stem cells for future therapeutic use.
89e |
Hematopoietic Stem Cells |
All of the cell types in the peripheral blood and some cells in every tissue of the body are derived from hematopoietic (hemo: blood; poiesis: creation) stem cells. If the hematopoietic stem cell is damaged and can no longer function (e.g., due to a nuclear accident), a person would survive 2–4 weeks in the absence of extraordinary support measures. With the clinical use of hematopoietic stem cells, tens of thousands of lives are saved each year (Chap. 139e). Stem cells produce hundreds of billions of blood cells daily from a stem cell pool that is estimated to be only in the tens of thousands. How stem cells do this, how they persist for many decades despite the production demands, and how they may be better used in clinical care are important issues in medicine.
The study of blood cell production has become a paradigm for how other tissues may be organized and regulated. Basic research in hematopoiesis includes defining stepwise molecular changes accompanying functional changes in maturing cells, aggregating cells into functional subgroups, and demonstrating hematopoietic stem cell regulation by a specialized microenvironment; these concepts are worked out in hematology, but they offer models for other tissues. Moreover, these concepts may not be restricted to normal tissue function but extend to malignancy. Stem cells are rare cells among a heterogeneous population of cell types, and their behavior is assessed mainly in experimental animal models involving reconstitution of hematopoiesis. Thus, much of what we know about stem cells is imprecise and based on inferences from genetically manipulated animals.
CARDINAL FUNCTIONS OF HEMATOPOIETIC STEM CELLS
All stem cell types have two cardinal functions: self-renewal and differentiation (Fig. 89e-1). Stem cells exist to generate, maintain, and repair tissues. They function successfully if they can replace a wide variety of shorter-lived mature cells over prolonged periods. The process of self-renewal (see below) assures that a stem cell population can be sustained over time. Without self-renewal, the stem cell pool would become exhausted and tissue maintenance would not be possible. The process of differentiation leads to production of the effectors of tissue function: mature cells. Without proper differentiation, the integrity of tissue function would be compromised and organ failure or neoplasia would ensue.
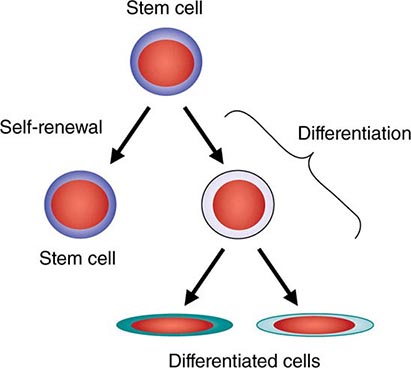
FIGURE 89e-1 Signature characteristics of the stem cell. Stem cells have two essential features: the capacity to differentiate into a variety of mature cell types and the capacity for self-renewal. Intrinsic factors associated with self-renewal include expression of Bmi-1, Gfi-1, PTEN, STAT5, Tel/Atv6, p21, p18, MCL-1, Mel-18, RAE28, and HoxB4. Extrinsic signals for self-renewal include Notch, Wnt, SHH, and Tie2/Ang-1. Based mainly on murine studies, hematopoietic stem cells express the following cell surface molecules: CD34, Thy-1 (CD90), c-Kit receptor (CD117), CD133, CD164, and c-Mpl (CD110, also known as the thrombopoietin receptor).
In the blood, mature cells have variable average life spans, ranging from 7 h for mature neutrophils to a few months for red blood cells to many years for memory lymphocytes. However, the stem cell pool is the central, durable source of all blood and immune cells, maintaining a capacity to produce a broad range of cells from a single cell source, yet keeping itself vigorous over decades of life. As an individual stem cell divides, it has the capacity to accomplish one of three division outcomes: two stem cells, two cells destined for differentiation, or one stem cell and one differentiating cell. The former two outcomes are the result of symmetric cell division, whereas the latter indicates a different outcome for the two daughter cells—an event termed asymmetric cell division. The relative balance for these types of outcomes may change during development and under particular kinds of demands on the stem cell pool.
DEVELOPMENTAL BIOLOGY OF HEMATOPOIETIC STEM CELLS
During development, blood cells are produced at different sites. Initially, the yolk sac provides oxygen-carrying red blood cells, and then the placenta and several sites of intraembryonic blood cell production become involved. These intraembryonic sites engage in sequential order, moving from the genital ridge at a site where the aorta, gonadal tissue, and mesonephros are emerging to the fetal liver and then, in the second trimester, to the bone marrow and spleen. As the location of stem cells changes, the cells they produce also change. The yolk sac provides red cells expressing embryonic hemoglobins while intraembryonic sites of hematopoiesis generate red cells, platelets, and the cells of innate immunity. The production of the cells of adaptive immunity occurs when the bone marrow is colonized and the thymus forms. Stem cell proliferation remains high, even in the bone marrow, until shortly after birth, when it appears to dramatically decline. The cells in the bone marrow are thought to arrive by the bloodborne transit of cells from the fetal liver after calcification of the long bones has begun. The presence of stem cells in the circulation is not unique to a time window in development; however, hematopoietic stem cells appear to circulate throughout life. The time that cells spend freely circulating appears to be brief (measured in minutes in the mouse), but the cells that do circulate are functional and can be used for transplantation. The number of stem cells that circulate can be increased in a number of ways to facilitate harvest and transfer to the same or a different host.
MOBILITY OF HEMATOPOIETIC STEM CELLS
Cells entering and exiting the bone marrow do so through a series of molecular interactions. Circulating stem cells (through CD162 and CD44) engage the lectins (carbohydrate binding proteins) P- and E-selectin on the endothelial surface to slow the movement of the cells to a rolling phenotype. Stem cell integrins are then activated and accomplish firm adhesion between the stem cell and vessel wall, with a particularly important role for stem cell VCAM-1 engaging endothelial VLA-4. The chemokine CXCL12 (SDF1) interacting with stem cell CXCR4 receptors and ionic calcium interacting with the calcium sensing receptor appear to be important in the process of stem cells getting from the circulation to where they engraft in the bone marrow. This is particularly true in the developmental move from fetal liver to bone marrow.
However, the role for CXCR4 in adults appears to be more related to retention of stem cells in the bone marrow rather than the process of getting them there. Interrupting that retention process through either specific molecular blockers of the CXCR4/CXCL12 interaction, cleavage of CXCL12, or downregulation of the CXCR4 receptor can all result in the release of stem cells into the circulation. This process is an increasingly important aspect of recovering stem cells for therapeutic use as it has permitted the harvesting process to be done by leukapheresis rather than bone marrow punctures in the operating room. Granulocyte colony-stimulating factor and plerixafor, a macrocyclic compound that can block CXCR4, are both used clinically to mobilize marrow hematopoietic stem cells for transplant. Refining our knowledge of how stem cells get into and out of the bone marrow may improve our ability to obtain stem cells and make them more efficient at finding their way to the specific sites for blood cell production, the so-called stem cell niche.
HEMATOPOIETIC STEM CELL MICROENVIRONMENT
The concept of a specialized microenvironment, or stem cell niche, was first proposed to explain why cells derived from the bone marrow of one animal could be used in transplantation and again be found in the bone marrow of the recipient. This niche is more than just a housing site for stem cells, however. It is an anatomic location where regulatory signals are provided that allow the stem cells to thrive, to expand if needed, and to provide varying amounts of descendant daughter cells. In addition, unregulated growth of stem cells may be problematic based on their undifferentiated state and self-renewal capacity. Thus, the niche must also regulate the number of stem cells produced. In this manner, the niche has the dual function of serving as a site of nurture but imposing limits for stem cells: in effect, acting as both a nutritive and constraining home.
The niche for blood stem cells changes with each of the sites of blood production during development, but for most of human life it is located in the bone marrow. Within the bone marrow, the perivascular space particularly in regions of trabecular bone serves as a niche. The mesenchymal and endothelial cells of the marrow microvessels produce kit ligand and CXCL12, both known to be important for hematopoietic stem cells. Other cell types, such as sympathetic neurons, nonmyelinating Schwann cells, macrophages, osteoclasts, and osteoblasts, have been shown to regulate stem cells, but it is unclear whether their effects are direct or indirect. Extracellular matrix proteins like osteopontin also affect stem cell function. The endosteal region is particularly important for transplanted cells, suggesting that there may be distinctive features of that region that are yet to be defined that are important mediators of stem cell engraftment. The functioning of the niche as a supportive context for stem cells is of obvious importance for maintaining hematopoiesis and in transplantation. An active area of study involves determining whether the niche is altered in disease and whether drugs can modify niche function to improve transplantation or normal stem cell function in hematologic disease.
EXCESS CAPACITY OF HEMATOPOIETIC STEM CELLS
In the absence of disease, one never runs out of hematopoietic stem cells. Indeed, serial transplantation studies in mice suggest that sufficient stem cells are present to reconstitute several animals in succession, with each animal having normal blood cell production. The fact that allogeneic stem cell transplant recipients also never run out of blood cells in their life span, which can extend for decades, argues that even the limiting numbers of stem cells provided to them are sufficient. How stem cells respond to different conditions to increase or decrease their mature cell production remains poorly understood. Clearly, negative feedback mechanisms affect the level of production of most of the cells, leading to the normal tightly regulated blood cell counts. However, many of the regulatory mechanisms that govern production of more mature progenitor cells do not apply or apply differently to stem cells. Similarly, most of the molecules shown to be able to change the size of the stem cell pool have little effect on more mature blood cells. For example, the growth factor erythropoietin, which stimulates red blood cell production from more mature precursor cells, has no effect on stem cells. Similarly, granulocyte colony-stimulating factor drives the rapid proliferation of granulocyte precursors but has little or no effect on the cell cycling of stem cells. Rather, it changes the location of stem cells by indirect means, altering molecules such as CXCL12 that tether stem cells to their niche. Molecules shown to be important for altering the proliferation, self-renewal, or survival of stem cells, such as cyclin-dependent kinase inhibitors, transcription factors like Bmi-1, or microRNA-processing enzymes like Dicer, have little or different effects on progenitor cells. Hematopoietic stem cells have governing mechanisms that are distinct from the cells they generate.
HEMATOPOIETIC STEM CELL DIFFERENTIATION
Hematopoietic stem cells sit at the base of a branching hierarchy of cells culminating in the many mature cell types that compose the blood and immune system (Fig. 89e-2). The maturation steps leading to terminally differentiated and functional blood cells take place both as a consequence of intrinsic changes in gene expression and niche-directed and cytokine-directed changes in the cells. Our knowledge of the details remains incomplete. As stem cells mature to progenitors, precursors, and, finally, mature effector cells, they undergo a series of functional changes. These include the obvious acquisition of functions defining mature blood cells, such as phagocytic capacity or hemoglobin synthesis. They also include the progressive loss of plasticity (i.e., the ability to become other cell types). For example, the myeloid progenitor can make all cells in the myeloid series but none in the lymphoid series. As common myeloid progenitors mature, they become precursors for either monocytes and granulocytes or erythrocytes and megakaryocytes, but not both. Some amount of reversibility of this process may exist early in the differentiation cascade, but that is lost beyond a distinct stage in normal physiologic conditions. With genetic interventions, however, blood cells, like other somatic cells, can be reprogrammed to become a variety of cell types.
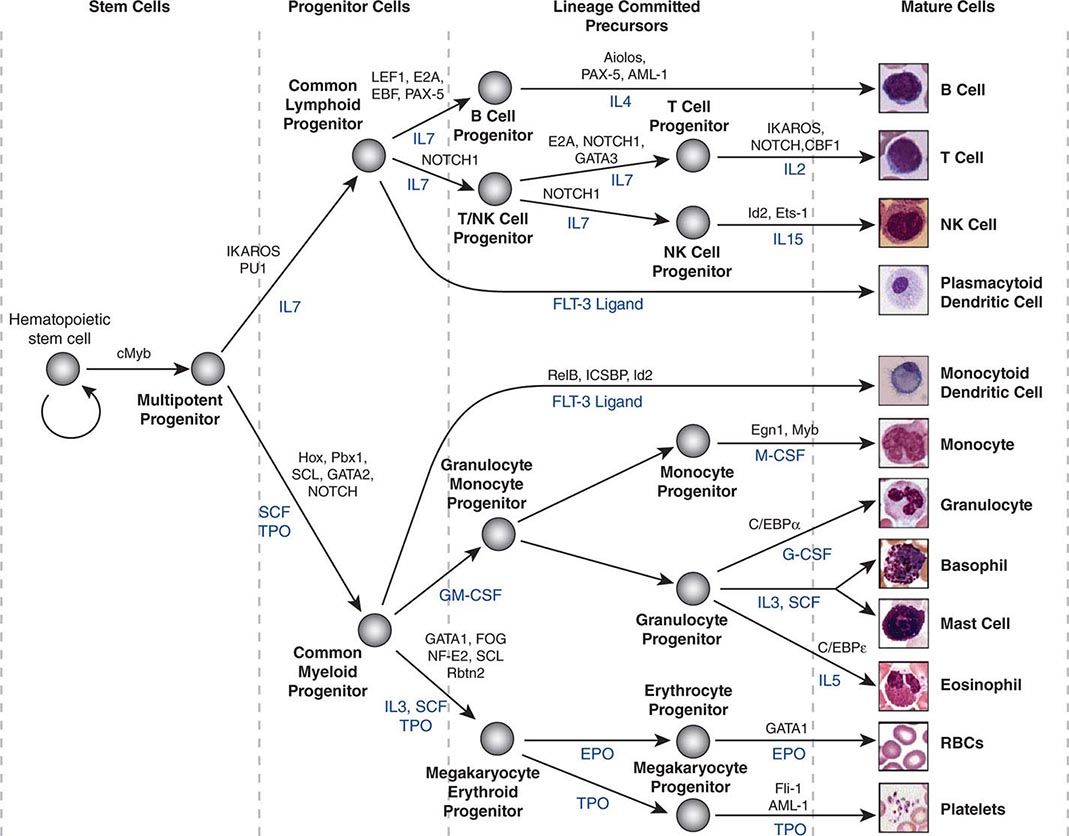
FIGURE 89e-2 Hierarchy of hematopoietic differentiation. Stem cells are multipotent cells that are the source of all descendant cells and have the capacity to provide either long-term (measured in years) or short-term (measured in months) cell production. Progenitor cells have a more limited spectrum of cells they can produce and are generally a short-lived, highly proliferative population also known as transient amplifying cells. Precursor cells are cells committed to a single blood cell lineage but with a continued ability to proliferate; they do not have all the features of a fully mature cell. Mature cells are the terminally differentiated product of the differentiation process and are the effector cells of specific activities of the blood and immune system. Progress through the pathways is mediated by alterations in gene expression. The regulation of the differentiation by soluble factors and cell-cell communications within the bone marrow niche are still being defined. The transcription factors that characterize particular cell transitions are illustrated on the arrows; the soluble factors that contribute to the differentiation process are in blue. This picture is a simplification of the process. Active research is revealing multiple discrete cell types in the maturation of B cells and T cells and has identified cells that are biased toward one lineage or another (rather than uncommitted) in their differentiation. EPO, erythropoietin; RBC, red blood cell; SCF, stem cell factor; TPO, thrombopoietin.
As cells differentiate, they may also lose proliferative capacity (Fig. 89e-3). Mature granulocytes are incapable of proliferation and only increase in number by increased production from precursors. The exceptions to the rule are some resident macrophages, which appear capable of proliferation, and lymphoid cells. Lymphoid cells retain the capacity to proliferate but have linked their proliferation to the recognition of particular proteins or peptides by specific antigen receptors on their surface. Like many tissues with short-lived mature cells such as the skin and intestine, blood cell proliferation is largely accomplished by a more immature progenitor population. In general, cells within the highly proliferative progenitor cell compartment are also relatively short-lived, making their way through the differentiation process in a defined molecular program involving the sequential activation of particular sets of genes. For any particular cell type, the differentiation program is difficult to speed up. The time it takes for hematopoietic progenitors to become mature cells is ~10–14 days in humans, evident clinically by the interval between cytotoxic chemotherapy and blood count recovery in patients.
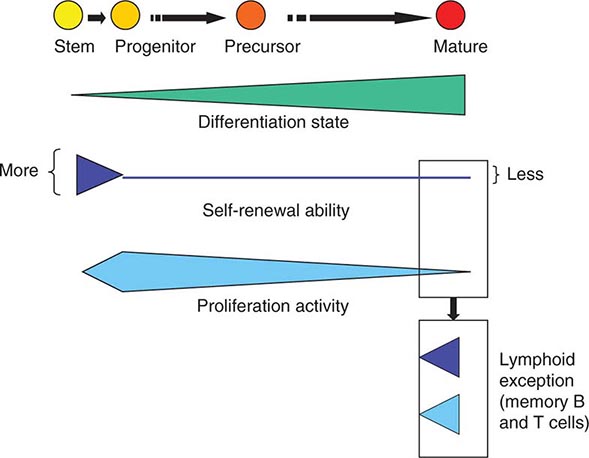
FIGURE 89e-3 Relative function of cells in the hematopoietic hierarchy. The boxes represent distinct functional features of cells in the myeloid (upper box) versus lymphoid (lower box) lineages.
Although hematopoietic stem cells are generally thought to have the capacity to form all cells of the blood, it is becoming clear that individual stem cells may not be equal in their differentiation potential. That is, some stem cells are “biased” to become mature cells of a particular type. In addition, the general concept of cells having a binary choice of lymphoid or myeloid differentiation is not entirely accurate. A cell population with limited myeloid (monocyte and granulocyte) and lymphoid potential is now added to the commitment steps stem cells may undergo.
SELF-RENEWAL
The hematopoietic stem cell must balance its three potential fates: apoptosis, self-renewal, and differentiation. The proliferation of cells is generally not associated with the ability to undergo a self-renewing division except among memory T and B cells and among stem cells. Self-renewal capacity gives way to differentiation as the only option after cell division when cells leave the stem cell compartment, until they have the opportunity to become memory lymphocytes. In addition to this self-renewing capacity, stem cells have an additional feature characterizing their proliferation machinery. Stem cells in many mature adult tissues may be heterogeneous with some being deeply quiescent, serving as a deep reserve, whereas others are more proliferative and replenish the short-lived progenitor population. In the hematopoietic system, stem cells are generally cytokine-resistant, remaining dormant even when cytokines drive bone marrow progenitors to proliferation rates measured in hours. Stem cells, in contrast, are thought to divide at far longer intervals, measured in months to years, for the most quiescent cells. This quiescence is difficult to overcome in vitro, limiting the ability to effectively expand human hematopoietic stem cells. The process may be controlled by particularly high levels of cyclin-dependent kinase inhibitors like p57 or CDKN1c that restrict entry of stem cells into the cell cycle, blocking the G1-S transition. Exogenous signals from the niche also appear to enforce quiescence, including the activation of the tyrosine kinase receptor Tie2 on stem cells by angiopoietin 1 on niche cells.
The regulation of stem cell proliferation also appears to change with age. In mice, the cyclin-dependent kinase inhibitor p16INK4a accumulates in stem cells in older animals and is associated with a change in five different stem cell functions, including cell cycling. Lowering expression of p16INK4a in older animals improves stem cell cycling and capacity to reconstitute hematopoiesis in adoptive hosts, making them similar to younger animals. Mature cell numbers are unaffected. Therefore, molecular events governing the specific functions of stem cells are being gradually made clear and offer the potential of new approaches to changing stem cell function for therapy. One critical stem cell function that remains poorly defined is the molecular regulation of self-renewal.
For medicine, self-renewal is perhaps the most important function of stem cells because it is critical in regulating the number of stem cells. Stem cell number is a key limiting parameter for both autologous and allogeneic stem cell transplantation. Were we to have the ability to use fewer stem cells or expand limited numbers of stem cells ex vivo, it might be possible to reduce the morbidity and expense of stem cell harvests and enable use of other stem cell sources. Specifically, umbilical cord blood is a rich source of stem cells. However, the volume of cord blood units is extremely small, and therefore, the total number of hematopoietic stem cells that can be obtained in any single cord blood unit is generally only sufficient to transplant an individual of <40 kg. This limitation restricts what would otherwise be an extremely promising source of stem cells. Two features of cord blood stem cells are particularly important. (1) They are derived from a diversity of individuals that far exceeds the adult donor pool and therefore can overcome the majority of immunologic cross-matching obstacles. (2) Cord blood stem cells have a large number of T cells associated with them, but (paradoxically) they appear to be associated with a lower incidence of graft-versus-host disease when compared with similarly mismatched stem cells from other sources. If stem cell expansion by self-renewal could be achieved, the number of cells available might be sufficient for use in larger adults. An alternative approach to this problem is to improve the efficiency of engraftment of donor stem cells. Graft engineering is exploring methods of adding cell components that may enhance engraftment. Furthermore, at least some data suggest that depletion of host NK (natural killer) cells may lower the number of stem cells necessary to reconstitute hematopoiesis.
Some limited understanding of self-renewal exists and, intriguingly, implicates gene products that are associated with the chromatin state, a high-order organization of chromosomal DNA that influences transcription. These include members of the polycomb family, a group of zinc finger–containing transcriptional regulators that interact with the chromatin structure, contributing to the accessibility of groups of genes for transcription. One member, Bmi-1, is important in enabling hematopoietic stem cell self-renewal through modification of cell cycle regulators such as the cyclin-dependent kinase inhibitors. In the absence of Bmi-1 or of the transcriptional regulator, Gfi-1, hematopoietic stem cells decline in number and function. In contrast, dysregulation of Bmi-1 has been associated with leukemia; it may promote leukemic stem cell self-renewal when it is overexpressed. Other transcription regulators have also been associated with self-renewal, particularly homeobox, or “hox,” genes. These transcription factors are named for their ability to govern large numbers of genes, including those determining body patterning in invertebrates. HoxB4 is capable of inducing extensive self-renewal of stem cells through its DNA-binding motif. Other members of the hox family of genes have been noted to affect normal stem cells, but they are also associated with leukemia. External signals that may influence the relative self-renewal versus differentiation outcomes of stem cell cycling include specific Wnt ligands. Intracellular signal transducing intermediates are also implicated in regulating self-renewal. They include PTEN, an inhibitor of the AKT pathway, and STAT5, both of which are downstream of activated growth factor receptors and necessary for normal stem cell functions including self-renewal, at least in mouse models. The connections between these molecules remain to be defined, and their role in physiologic regulation of stem cell self-renewal is still poorly understood.
CANCER IS SIMILAR TO AN ORGAN WITH SELF-RENEWING CAPACITY
The relationship of stem cells to cancer is an important evolving dimension of adult stem cell biology. Cancer may share principles of organization with normal tissues. Cancer cells are heterogeneous even within a given patient and may have a hierarchical organization of cells with a base of stem-like cells capable of the signature stem cell features: self-renewal and differentiation. These stem-like cells might be the basis for perpetuation of the tumor and represent a slowly dividing, rare population with distinct regulatory mechanisms, including a relationship with a specialized microenvironment. A subpopulation of self-renewing cells has been defined for some, but not all, cancers. A more sophisticated understanding of the stem cell organization of cancers may lead to improved strategies for developing new therapies for the many common and difficult-to-treat types of malignancies that have been relatively refractory to interventions aimed at dividing cells.
Does the concept of cancer stem cells provide insight into the cellular origin of cancer? The fact that some cells within a cancer have stem cell–like properties does not necessarily mean that the cancer arose in the stem cell itself. Rather, more mature cells could have acquired the self-renewal characteristics of stem cells. Any single genetic event is unlikely to be sufficient to enable full transformation of a normal cell to a frankly malignant one. Rather, cancer is a multistep process, and for the multiple steps to accumulate, the cell of origin must be able to persist for prolonged periods. It must also be able to generate large numbers of daughter cells. The normal stem cell has these properties and, by virtue of its having intrinsic self-renewal capability, may be more readily converted to a malignant phenotype. This hypothesis has been tested experimentally in the hematopoietic system. Taking advantage of the cell-surface markers that distinguish hematopoietic cells of varying maturity, stem cells, progenitors, precursors, and mature cells can be isolated. Powerful transforming gene constructs were placed in these cells, and it was found that the cell with the greatest potential to produce a malignancy was dependent on the transforming gene. In some cases, it was the stem cell, but in others, the progenitor cell functioned to initiate and perpetuate the cancer. This shows that cells can acquire stem cell–like properties in malignancy.
WHAT ELSE CAN HEMATOPOIETIC STEM CELLS DO?
Some experimental data have suggested that hematopoietic stem cells or other cells mobilized into the circulation by the same factors that mobilize hematopoietic stem cells are capable of playing a role in healing the vascular and tissue damage associated with stroke and myocardial infarction. These data are controversial, and the applicability of a stem cell approach to nonhematopoietic conditions remains experimental. However, reprogramming technology offers the potential for using the readily obtained hematopoietic stem cell as a source for cells with other capabilities.
The stem cell, therefore, represents a true dual-edged sword. It has tremendous healing capacity and is essential for life. Uncontrolled, it can threaten the life it maintains. Understanding how stem cells function, the signals that modify their behavior, and the tissue niches that modulate stem cell responses to injury and disease are critical for more effectively developing stem cell–based medicine. That aspect of medicine will include the use of the stem cells and the use of drugs to target stem cells to enhance repair of damaged tissues. It will also include the careful balance of interventions to control stem cells where they may be dysfunctional or malignant.
90e |
Applications of Stem Cell Biology in Clinical Medicine |
Damage to an organ initiates a series of events that lead to the reconstruction of the damaged tissue, including proliferation, differentiation, and migration of various cell types; release of cytokines and chemokines; and remodeling of the extracellular matrix. Endogenous stem and progenitor cells are among the cell populations that are involved in these injury responses. In normal steady-state conditions, an equilibrium is maintained in which endogenous stem cells intrinsic to the tissue replenish dying cells. After tissue injury, stem cells in organs such as the liver and skin have a remarkable ability to regenerate the organ, whereas other stem cell populations, such as those in the heart and brain, have a much more limited capability for self-repair. In rare circumstances, circulating stem cells may contribute to regenerative responses by migrating into a tissue and differentiating into organ-specific cell types. The goal of stem cell therapies is to promote cell replacement in organs that are damaged beyond their ability to self-repair.
GENERAL STRATEGIES FOR STEM CELL REPLACEMENT
At least three different therapeutic concepts for cell replacement can be envisaged (Fig. 90e-1). One therapeutic approach involves direct administration of stem cells. The cells may be injected directly into the damaged organ, where they can differentiate into the desired cell type. Alternatively, stem cells may be injected systemically since they have the capacity to home in on damaged tissues by following gradients of cytokines and chemokines released by the diseased organ. A second approach involves transplantation of differentiated cells derived from stem cells. For example, pancreatic islet cells can be generated from stem cells before transplantation into diabetic patients, and cardiomyocytes can be generated to treat ischemic heart disease. A third approach involves stimulation of endogenous stem cells to facilitate repair. This goal might be accomplished by administration of appropriate growth factors and drugs that amplify the number of endogenous stem/progenitor cells and/or direct them to differentiate into the desired cell types. Therapeutic stimulation of precursor cells is already a clinical reality in the hematopoietic system, where factors such as erythropoietin, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor are used to increase production of specific blood elements. In addition to these strategies for cell replacement, a number of other approaches could involve stem cells for ex vivo or in situ generation of tissues, a process termed tissue engineering (Chap. 92e). Stem cells are also excellent candidates as vehicles for cellular gene therapy (Chap. 91e). Finally, transplanted stem cells may exert paracrine effects on damaged tissues without the differentiation and replacement of lost cells.
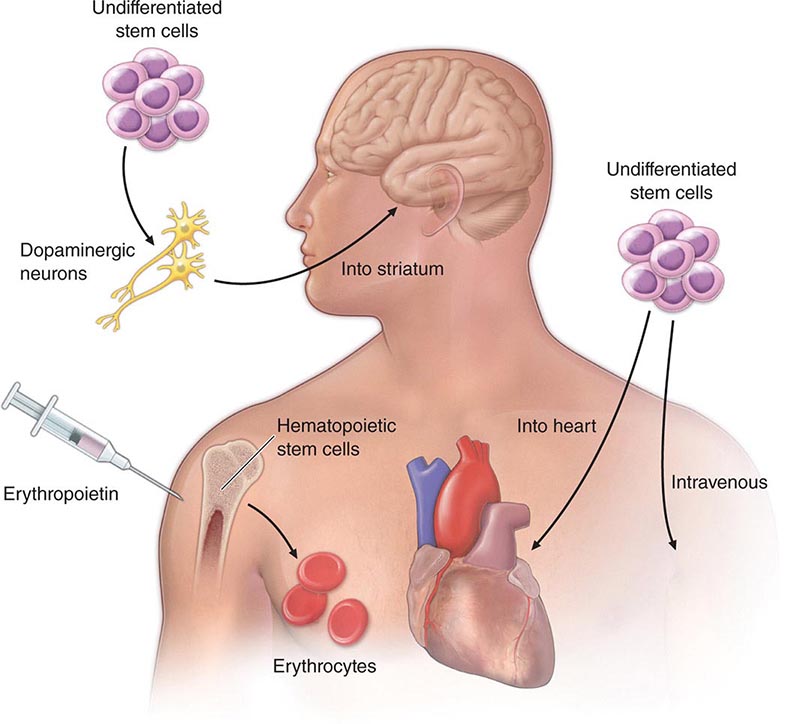
FIGURE 90e-1 Strategies for transplantation of stem cells. 1. Undifferentiated or partially differentiated stem cells may be injected directly into the target organ or intravenously. 2. Stem cells may be differentiated ex vivo before injection into the target organ. 3. Growth factors or other drugs may be injected to stimulate endogenous stem cell populations.
Stem cell transplantation is not a new concept but rather is already part of established medical practice. Hematopoietic stem cells (Chap. 89e) are responsible for the long-term repopulation of all blood elements in recipients of bone marrow transplants, and hematopoietic stem cell transplantation is the gold standard against which other stem cell transplantation therapies will be measured. Transplantation of differentiated cells is also a clinical reality, and donated organs and tissues are often used to replace damaged tissues. However, the need for transplantable tissues and organs far outweighs the available supply, and organ transplantation has limited potential for some tissues, such as the brain. Stem cells offer the possibility of a renewable source of replacement cells for virtually all organs.
SOURCES OF STEM CELLS FOR TISSUE REPAIR
A variety of different types of stem cells (Chap. 88) could be used in regenerative strategies, including embryonic stem (ES) cells, induced pluripotent stem (iPS) cells, umbilical-cord blood stem cells (USCs), organ-specific somatic stem cells (e.g., neural stem cells for treatment of the brain), and somatic stem cells that generate cell types specific for the target organ rather than the donor organ (e.g., bone marrow mesenchymal stem cells or CD34+ hematopoietic stem cells for cardiac repair). Although each cell type has potential advantages and disadvantages, there are a number of generic problems in developing any of these cell types into a useful and reliable clinical tool.
Embryonic Stem Cells Embryonic stem cells have the potential to generate all the cell types in the body; thus, in theory, there are no restrictions on the organs that could be regenerated. ES cells can self-renew endlessly, so that a single cell line with carefully characterized traits potentially could generate almost limitless numbers of cells. In the absence of moral or ethical constraints (see “Ethical Issues,” below), unused human blastocysts from fertility clinics could be used to derive new ES cell lines that are matched immunologically with potential transplant recipients. Alternatively, somatic cell nuclear transfer (“therapeutic cloning”) could be used to create ES cell lines that are genetically identical to those of the patient, although this endeavor has been technically refractory for human cells. However, human ES cells are difficult to culture and grow slowly. Techniques for differentiating them into specific cell types are just beginning to be developed. Cells tend to develop abnormal karyotypes and other abnormalities with increased time in culture, and ES cells have the potential to form teratomas if all cells are not committed to the desired cell types before transplantation. Further, human ES cells are ethically controversial and, on these grounds, would be unacceptable to some patients and physicians despite their therapeutic potential. Nevertheless, there have been limited clinical trials of ES-derived cells in a number of disorders, including macular degeneration, myopia, and spinal cord injury.
Induced Pluripotent Stem Cells The field of stem cell biology was transformed by the discovery that adult somatic cells can be converted (“reprogrammed”) into pluripotent cells through the overexpression of four transcription factors normally expressed in pluripotent cells (Chap. 88). These iPS cells share most properties with ES cells, although there are distinct differences in gene expression between ES and iPS cells. The initial use of viruses to insert the transcription factors into somatic cells made the resulting cells unsuitable for clinical use. However, a number of strategies have since been developed to circumvent this problem, including the insertion of modified mRNAs, proteins, or microRNAs rather than cDNAs; the use of non-integrating viruses such as Sendai virus; the insertion of transposons with the programming factors, followed by their subsequent removal; and the use of floxed viral constructs, followed by treatment with Cre recombinase to excise those constructs. The safety of iPS cells in humans remains to be demonstrated, but clinical trials in macular degeneration and other disorders are planned. Potential advantages of iPS cells are that somatic cells from patients would generate pluripotent cells genetically identical to those of the patient and that these cells are not subject to the same ethical constraints as ES cells. It is not clear whether the differences in gene expression between ES and iPS cells will have any impact on their potential clinical utility, and studies of both cell types will be essential to resolve this issue.
Umbilical-Cord Stem Cells Umbilical-cord blood stem/progenitor cells (USCs) are widely and readily available. These cells appear to be associated with less graft-versus-host disease than are some other cell types, such as marrow stem cells. They have less human leukocyte antigen restriction than adult marrow stem cells and are less likely to be contaminated with herpesvirus. However, it is unclear how many different cell types can be generated from USCs, and methods for differentiating these cells into nonhematopoietic phenotypes are largely lacking. Nevertheless, there are ongoing clinical trials of these cells in dozens of disorders, including cirrhosis, cardiopathies, multiple sclerosis, burns, stroke, autism, and critical limb ischemia.
Organ-Specific Multipotent Stem Cells Organ-specific multipotent stem cells have the advantage of already being somewhat specialized so that the inducement of desired cell types may be easier. Cells potentially could be obtained from the patient and amplified in cell culture, circumventing the problems associated with immune rejection. Stem cells are relatively easy to harvest from some tissues, such as bone marrow and blood, but are difficult to harvest from other tissues, such as heart and brain. Moreover, these populations of cells are more limited in potentiality than are pluripotent ES or iPS cells, and they may be difficult to obtain in large quantities from many organs. Therefore, substantial efforts have been devoted to developing techniques for using more easily obtainable stem cell populations, such as bone marrow mesenchymal stem cells (MSCs), CD34+ hematopoietic stem cells (HSCs), cardiac mesenchymal cells, and adipose-derived stem cells (ASCs), for use in regenerative strategies. Tissue culture evidence suggests that these stem cell populations may be able to generate differentiated cell types unrelated to their organ source (including myocytes, chondrocytes, tendon cells, osteoblasts, cardiomyocytes, adipocytes, hepatocytes, and neurons) in a process known as transdifferentiation. However, it is still unclear whether these stem cells are capable of generating differentiated cell types that integrate into organs, survive, and function after transplantation in vivo. A number of early studies of MSCs transplanted into heart, liver, and other organs suggested that the cells had differentiated into organ-specific cell types with beneficial effects in animal models of disease. Unfortunately, subsequent studies revealed that the stem cells had simply fused with cells resident in the organs and that the observed beneficial effects were due to paracrine release of trophic and anti-inflammatory cytokines. Further studies will be necessary to determine whether transdifferentiation of MSCs, ASCs, or other stem cell populations occurs at a high enough frequency to make these cells useful for stem cell replacement therapy. Despite the remaining issues, clinical trials of MSCs, autologous HSCs, USCs, and ASCs are being performed in many disorders, including ischemic cardiac disease, cardiomyopathy, diabetes, stroke, cirrhosis, and muscular dystrophy.
Regardless of the source of the stem cells used in regenerative strategies, a number of generic problems must be overcome for the development of successful clinical applications. These problems include the devising of methods to reliably generate large numbers of specific cell types, to minimize the risk of tumor formation or proliferation of inappropriate cell types, to ensure the viability and function of the engrafted cells, to overcome immune rejection when autografts are not used, and to facilitate revascularization of regenerated tissue. Each organ system will also pose tissue-specific problems for stem cell therapies.
DISEASE-SPECIFIC APPLICATIONS OF STEM CELLS
Ischemic Heart Disease and Cardiomyocyte Regeneration Because of the high prevalence of ischemic heart disease, extensive efforts have been devoted to the development of strategies for stem cell replacement of cardiomyocytes. Historically, the adult heart has been viewed as a terminally differentiated organ without the capacity for regeneration. However, recent studies have demonstrated that the heart has the capacity for low levels of cardiomyocyte regeneration (Chap. 265e). This regeneration appears to be accomplished by cardiac stem cells resident in the heart and possibly also by cells originating in the bone marrow. The heart might be an ideal source of stem cells for therapeutic use, but techniques for isolating, characterizing, and amplifying large numbers of these cells have not yet been perfected. For successful myocardial repair, stem cell therapy must deliver cells either systemically or locally, and the cells must survive, engraft, and differentiate into functional cardiomyocytes that couple mechanically and electrically with the recipient myocardium. The optimal method for cell delivery is not clear, and various experimental and clinical studies have successfully employed intramyocardial, transendocardial, intravenous, intracoronary, and retrograde coronary venous injections. In experimental myocardial infarction, functional improvements have been achieved after transplantation of a variety of different cell types, including ES cells, HSCs, MSCs, USCs, and ASCs. Early studies suggested that each of these cell types might have the potential to engraft and generate cardiomyocytes. However, most investigators have found that the generation of new cardiomyocytes by these cells is at best a rare event and that graft survival over long periods is poor. The preponderance of evidence suggests that the observed beneficial effects of most experimental therapies were not derived from direct stem cell generation of cardiomyocytes but rather from indirect effects of the stem cells on resident cells. It is not clear whether these effects reflect the release of soluble trophic factors, the induction of angiogenesis, the release of anti-inflammatory cytokines, or another mechanism. A wide variety of cell delivery methods, cell types, and cell doses have been used in a progressively enlarging series of clinical trials, but the fate of the cells and the mechanisms by which they alter cardiac function are still open questions. In aggregate, however, these studies have shown a small but measurable improvement in cardiac function and, in some cases, reduction in infarct size. In short, the available evidence suggests that the beneficial clinical impact reflects an indirect effect of the transplanted cells rather than genuine cell replacement.
Diabetes Successes with islet cell and pancreas transplantation have provided proof of concept for cell-based therapies for type 1 diabetes. However, the demand for donor pancreases far exceeds the number available, and maintenance of long-term graft survival is a problem. The search for a renewable source of stem cells capable of regenerating pancreatic islets has therefore been intensive. Pancreatic beta cell turnover occurs even in the normal pancreas, although the source of the new beta cells remains controversial. This persistent turnover suggests that, in principle, it should be possible to develop strategies for reconstituting the beta cell population in diabetics. Attempts to devise techniques for promoting endogenous regenerative processes by using combinations of growth factors, drugs, and gene therapy have failed thus far, but this remains a potentially viable approach. A number of different cell types are candidates for use in stem cell replacement strategies, including iPS cells, ES cells, hepatic progenitor cells, pancreatic ductal progenitor cells, and MSCs. Successful therapy will depend on the development of a source of cells that can be amplified to produce large numbers of progeny with the ability to synthesize, store, and release insulin when it is required, primarily in response to changes in the ambient level of glucose. The proliferative capacity of the replacement cells must be tightly regulated to avoid excessive expansion of beta cell numbers and the consequent development of hyperinsulinemia/hypoglycemia; moreover, the cells must withstand immune rejection. Although it has been reported that ES and iPS cells can be differentiated into cells that produce insulin, these cells have a low content of insulin and a high rate of apoptosis and generally lack the capacity to normalize blood glucose levels in diabetic animals. Thus, ES and iPS cells have not yet been useful for the large-scale production of differentiated islet cells. During embryogenesis, the pancreas, liver, and gastrointestinal tract are all derived from the anterior endoderm, and transdifferentiation of pancreas to liver and vice versa has been observed in a number of pathologic conditions. There is also substantial evidence that multipotential stem cells reside within gastric glands and intestinal crypts. These observations suggest that hepatic, pancreatic, and/or gastrointestinal precursor cells may be reasonable candidates for cell-based therapy for diabetes, although it is unclear whether insulin-producing cells derived from pancreatic stem cells or liver progenitors can be expanded in vitro to clinically useful numbers. MSCs and neural stem cells both reportedly have the capacity to generate insulin-producing cells, but there is no convincing evidence that either cell type will be clinically useful. Clinical trials of MSCs, USCs, HSCs, and ASCs in both type 1 and type 2 diabetes are ongoing.
Nervous System Substantial progress has been made in the development of methodologies for generating neural cells from different stem cell populations. Human ES or iPS cells can be induced to generate cells with the properties of neural stem cells, and these cells in turn give rise to neurons, oligodendroglia, and astrocytes. Reasonably large numbers of these cells can be transplanted into the rodent brain with formation of appropriate cell types and no tumor formation. Multipotent stem cells present in the adult brain also can be easily amplified in number and used to generate all the major neural cell types, but the need for invasive procedures to obtain autologous cells is a major limitation. Fetal neural stem cells derived from miscarriages or abortions are an alternative but raise ethical concerns. Nevertheless, clinical trials of fetal neural stem cells have commenced in amyotrophic lateral sclerosis (ALS), stroke, and several other disorders. Transdifferentiation of MSCs and ASCs into neural stem cells, and vice versa, has been reported by numerous investigators, and clinical trials of such cells have begun for a number of neurologic diseases. Clinical trials of a conditionally immortalized human cell line and of USCs in stroke are also in progress. Because of the incapacitating nature of neural disorders and the limited endogenous repair capacity of the nervous system, clinical trials of stem cells in neurologic disorders have been particularly numerous, including trials in spinal cord injury, multiple sclerosis, epilepsy, Alzheimer’s disease, ALS, acute and chronic stroke, numerous genetic disorders, traumatic brain injury, Parkinson’s disease, and others. In diseases such as ALS, possible benefits are more likely to be due to indirect trophic effects than to neuron replacement. In Parkinson’s disease, the major motor features of the disorder result from the loss of a single cell population: dopaminergic neurons within the substantia nigra; this circumstance suggests that cell replacement should be relatively straightforward. However, two clinical trials of fetal nigral transplantation failed to meet their primary endpoint and were complicated by the development of dyskinesia. Transplantation of stem cell–derived dopamine-producing cells offers a number of potential advantages over the fetal transplants, including the ability of stem cells to migrate and disperse within tissue, the potential for engineering regulatable release of dopamine, and the ability to engineer cells to produce factors that will enhance cell survival. Nevertheless, the experience with fetal transplants points out the difficulties that may be encountered.
At least some of the neurologic dysfunction after spinal cord injury reflects demyelination, and both ES cells and MSCs can facilitate remyelination after experimental spinal cord injury (SCI). Clinical trials of MSCs in this disorder have commenced in a number of countries, and SCI was the first disorder targeted for the clinical use of ES cells. However, the ES cell trial in SCI was terminated early for nonmedical reasons. At present, no population of transplanted stem cells has been shown to have the capacity to generate neurons that extend axons over long distances to form synaptic connections (as would be necessary for replacement of upper motor neurons in ALS, stroke, or other disorders). For many injuries, including SCI, the balance between scar formation and tissue repair/regeneration may prove to be an important consideration. For example, it may ultimately prove necessary to limit scar formation so that axons can reestablish connections.
Liver Liver transplantation is currently the only successful treatment for end-stage liver diseases, but the shortage of liver grafts limits its application. Clinical trials of hepatocyte transplantation demonstrate its potential as a substitute for organ transplantation, but this approach is limited by the paucity of available cells. Potential sources of stem cells for regenerative strategies include endogenous liver stem cells (such as oval cells), ES cells, MSCs, and USCs. Although a series of studies in humans as well as animals suggested that transplanted MSCs and HSCs can generate hepatocytes, fusion of the transplanted cells with endogenous liver cells, giving the erroneous appearance of new hepatocytes, appears to be the underlying event in most circumstances. The available evidence suggests that transplanted HSCs and MSCs can generate hepatocyte-like cells in the liver only at a very low frequency, but there are beneficial consequences presumably related to indirect paracrine effects. ES cells can be differentiated into hepatocytes and transplanted in animal models of liver failure without the formation of teratomas. Clinical trials are in progress in cirrhosis with numerous cell types, including MSCs, USCs, HSCs, and ASCs.
Other Organ Systems and the Future The use of stem cells in regenerative strategies has been studied for many other organ systems and cell types, including skin, eye, cartilage, bone, kidney, lung, endometrium, vascular endothelium, smooth muscle, and striated muscle, and clinical trials in these and other organs are ongoing. In fact, the potential for stem cell regeneration of damaged organs and tissues is virtually limitless. However, numerous obstacles must be overcome before stem cell therapies can become a widespread clinical reality. Only HSCs have been adequately characterized by surface markers so that they can be unambiguously identified, a prerequisite for reliable clinical applications. The pathways for differentiating stem cells into specific cellular phenotypes are largely unknown, and the ability to control the migration of transplanted cells or predict the response of the cells to the environment of diseased organs is presently limited. Some strategies may employ the coadministration of scaffolding, artificial extracellular matrix, and/or growth factors to orchestrate differentiation of stem cells and their organization into appropriate constituents of the organ. There is currently no way to image stem cells in vivo after transplantation into humans, and it will be necessary to develop techniques to do so. Fortunately, stem cells can be engineered before transplantation to contain a contrast agent that may make their in vivo imaging feasible. The potential for tumor formation and the problems associated with immune rejection are impediments, and it will also be necessary to develop techniques for ensuring vascularization of regenerated tissues. There already are many strategies for supporting cell replacement, including coadministration of vasoactive endothelial growth factor to foster vascularization of the transplant. Some strategies also include the genetic engineering of stem cells with an inducible suicide gene so that the cells can be easily eradicated in the event of tumor formation or another complication. The potential for stem cell therapies to revolutionize medical care is extraordinary, and disorders such as myocardial infarction, diabetes, and Parkinson’s disease, among many others, are potentially curable by such therapies. However, stem cell–based therapies are still at a very early stage of development, and perfection of techniques for clinical transplantation of predictable, well-characterized cells is going to be a difficult and lengthy undertaking.
ETHICAL ISSUES
Stem cell therapies raise ethical and socially contentious issues that must be addressed in parallel with the scientific and medical opportunities. Society has great diversity with respect to religious beliefs, concepts of individual rights, tolerance for uncertainty and risk, and boundaries for how scientific interventions should be used to alter the outcome of disease. In the United States, the federal government has authorized research using existing human ES cell lines but still restricts the use of federal funds for developing new human ES cell lines. Ongoing studies of existing lines have indicated that they develop abnormalities with time in culture and that they may be contaminated with mouse proteins. These findings highlight the need to develop new human ES cell lines. The development of iPS cell technology may lessen the need for deriving new ES cell lines, but it is still not clear whether the differences in gene expression by ES and iPS cells are important for potential clinical use.
In considering ethical issues associated with the use of stem cells, it is helpful to draw from experience with other scientific advances, such as organ transplantation, recombinant DNA technology, implantation of mechanical devices, neuroscience and cognitive research, in vitro fertilization, and prenatal genetic testing. These and other precedents have pointed to the importance of understanding and testing fundamental biology in the laboratory setting and in animal models before applying new techniques in carefully controlled clinical trials. When these trials occur, they must include full informed consent and careful oversight by external review groups.
Ultimately, there will be medical interventions that are scientifically feasible but ethically or socially unacceptable to some members of a society. Stem cell research raises fundamentally difficult questions about the definition of human life, and it has raised deep fears about the ability to balance issues of justice and safety with the needs of critically ill patients. Health care providers and experts with backgrounds in ethics, law, and sociology must help guard against the premature or inappropriate application of stem cell therapies and the inappropriate involvement of vulnerable population groups. However, these therapies offer important new strategies for the treatment of otherwise irreversible disorders. An open dialogue among the scientific community, physicians, patients and their advocates, lawmakers, and the lay population is critically important to raise and address important ethical issues and balance the benefits and risks associated with stem cell transfer.
91e |
Gene Therapy in Clinical Medicine |
Gene transfer is a novel area of therapeutics in which the active agent is a nucleic acid sequence rather than a protein or small molecule. Because delivery of naked DNA or RNA to a cell is an inefficient process, most gene transfer is carried out using a vector, or gene delivery vehicle. These vehicles have generally been engineered from viruses by deleting some or all of the viral genome and replacing it with the therapeutic gene of interest under the control of a suitable promoter (Table 91e-1). Gene transfer strategies can thus be described in terms of three essential elements: (1) a vector; (2) a gene to be delivered, sometimes called the transgene; and (3) a physiologically relevant target cell to which the DNA or RNA is delivered. The series of steps in which the vector and donated DNA enter the target cell and express the transgene is referred to as transduction. Gene delivery can take place in vivo, in which the vector is directly injected into the patient, or, in the case of hematopoietic and some other target cells, ex vivo, with removal of the target cells from the patient, followed by return of the gene-modified autologous cells to the patient after manipulation in the laboratory. The latter approach effectively combines gene transfer techniques with cellular therapies (Chap. 90e).
|
CHARACTERISTICS OF GENE DELIVERY VEHICLES |
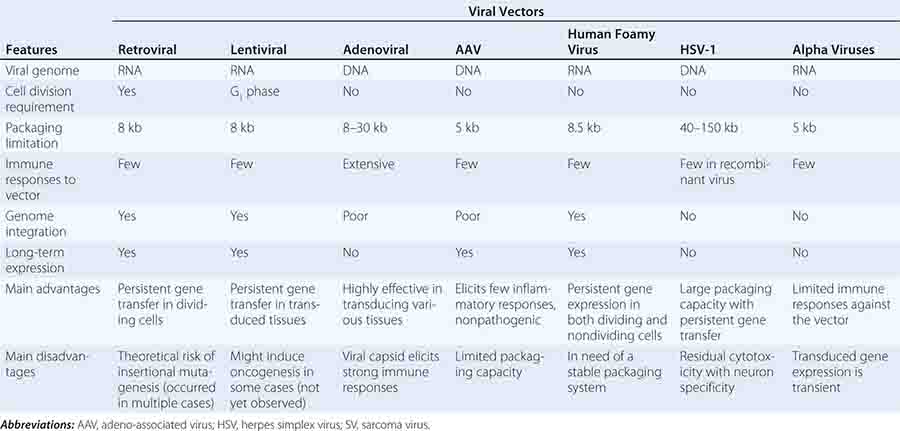
Gene transfer is one of the most powerful concepts in modern molecular medicine and has the potential to address a host of diseases for which there are currently no available treatments. Clinical trials of gene therapy have been under way since 1990; a recent landmark in the field was the licensing, in 2012, of the first gene therapy product approved in Europe or the United States (see below). Given that vector-mediated gene therapy is arguably one of the most complex therapeutics yet developed, consisting of both a nucleic acid and a protein component, this time course from first clinical trial to licensed product is noteworthy for being similar to that seen with other novel classes of therapeutics, including monoclonal antibodies or bone marrow transplantation. Over 5000 subjects have been enrolled in gene transfer studies, and serious adverse events have been rare. Some of the initial trials were characterized by an overabundance of optimism and a failure to be appropriately critical of preclinical studies in animals; in addition, it was in some contexts not fully appreciated that animal studies are only a partial guide to safety profiles of products in humans (e.g., insertional mutagenesis). Initial exuberance was driven by many factors, including an intense desire to develop therapies for hitherto untreatable diseases, lack of understanding of risks, and, in some cases, undisclosed financial conflicts of interest. After a teenager died of complications related to vector infusion, the field underwent a retrenchment; continued efforts led to a more nuanced understanding of the risks and benefits of these new therapies and more sophisticated selection of disease targets. Currently, gene therapies are being developed for a wide variety of disease entities (Fig. 91e-1).
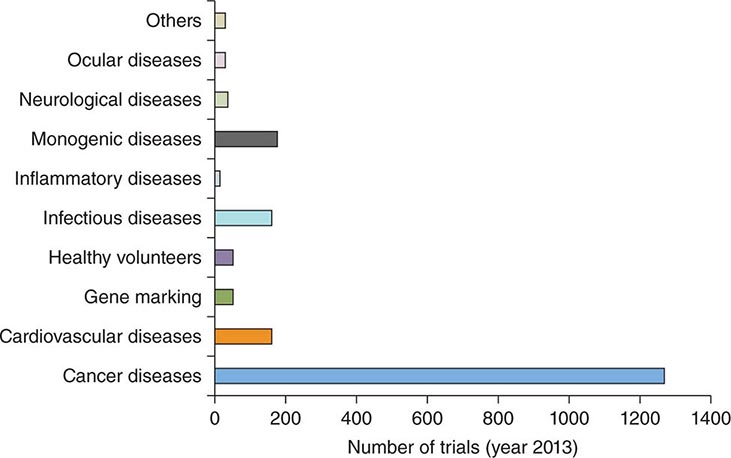
FIGURE 91e-1 Indications in gene therapy clinical trials. The bar graph classifies clinical gene transfer studies by disease. A majority of trials have addressed cancer, with monogenic disorders, infectious diseases, and cardiovascular diseases the next largest categories. (Adapted from SL Ginn et al: J Gene Med 15:65-77, 2013. Published online in Wiley Online.)
GENE TRANSFER FOR GENETIC DISEASE
Gene transfer strategies for genetic disease generally involve gene addition therapy, an approach characterized by transfer of the missing gene to a physiologically relevant target cell. However, other strategies are possible, including supplying a gene that achieves a similar biologic effect through an alternative pathway (e.g., factor VIIa for hemophilia A); supplying an antisense oligonucleotide to splice out a mutant exon if the sequence is not critical to the function of the protein (as has been done with the dystrophin gene in Duchenne’s muscular dystrophy); or downregulating a harmful effect through a small interfering RNA (siRNA). Two distinct strategies are used to achieve long-term gene expression: one is to transduce stem cells with an integrating vector, so that all progeny cells will carry the donated gene; and the other is to transduce long-lived cells, such as skeletal muscle or neurons. In the case of long-lived cells, integration into the target cell genome is unnecessary. Instead, because the cells are nondividing, the donated DNA, if stabilized in an episomal form, will give rise to expression for the life of the cell. This approach thus avoids problems related to integration and insertional mutagenesis.
IMMUNODEFICIENCY DISORDERS: PROOF OF PRINCIPLE
Early attempts to effect gene replacement into hematopoietic stem cells (HSCs) were stymied by the relatively low transduction efficiency of retroviral vectors, which require dividing target cells for integration. Because HSCs are normally quiescent, they are a formidable transduction target. However, identification of cytokines that induced cell division without promoting differentiation of stem cells, along with technical improvements in the isolation and transduction of HSCs, led to modest but real gains in transduction efficiency.
The first convincing therapeutic effect from gene transfer occurred with X-linked severe combined immunodeficiency disease (SCID), which results from mutations in the gene (IL2RG) encoding the γc subunit of cytokine receptors required for normal development of t and natural killer (NK) cells (Chap. 374). Affected infants present in the first few months of life with overwhelming infections and/or failure to thrive. In this disorder, it was recognized that the transduced cells, even if few in number, would have a proliferative advantage compared to the nontransduced cells, which lack receptors for the cytokines required for lymphocyte development and maturation. Complete reconstitution of the immune system, including documented responses to standard childhood vaccinations, clearing of infections, and remarkable gains in growth occurred in most of the treated children. However, among 20 children treated in two separate trials, five eventually developed a syndrome similar to T cell acute lymphocytic leukemia, with splenomegaly, rising white counts, and the emergence of a single clone of T cells. Molecular studies revealed that, in most of these children, the retroviral vector had integrated within a gene, LMO-2 (LIM only-2), which encodes a component of a transcription factor complex involved in hematopoietic development. The retroviral long terminal repeat increases the expression of LMO-2, resulting in T cell leukemia.
The X-linked SCID studies were a watershed event in the evolution of gene therapy. They demonstrated conclusively that gene therapy could cure disease; of the 20 children eventually treated in these trials, 18 achieved correction of the immunodeficiency disorder. Unfortunately, 5 of the 20 patients later developed a leukemia-like disorder, and one died of this complication; the rest are alive and free of complications at time periods ranging up to 14 years after initial treatment. These studies demonstrated that insertional mutagenesis leading to cancer was more than a theoretical possibility (Table 91e-2). As a result of the experience in these trials, all protocols using integrating vectors in hematopoietic cells must include a plan for monitoring sites of insertion and clonal proliferation. Strategies to overcome this complication have included using a “suicide” gene cassette in the vector, so that errant clones can be quickly ablated, or using “insulator” elements in the cassette, which can limit the activation of genes surrounding the insertion site. Lentiviral vectors, which can efficiently transduce nondividing target cells, are also likely to be safer than retroviral vectors, based on patterns of integration; the field is thus gradually moving toward these to replace retroviral vectors.
|
POTENTIAL COMPLICATIONS OF GENE THERAPY |
More clear-cut success has been achieved in a gene therapy trial for another form of SCID, adenosine deaminase (ADA) deficiency (Chap. 374). ADA-SCID is clinically similar to X-linked SCID, although it can be treated by enzyme replacement therapy with a pegylated form of the enzyme (PEG-ADA), which leads to immune reconstitution but not always to normal T cell counts. Enzyme replacement therapy is expensive (annual costs: $200,000–$300,000 in U.S. dollars). The initial trials of gene therapy for ADA-SCID were unsuccessful, but modifications of this protocol to include the use of HSCs rather than T cells as the target for transduction; discontinuation of PEG-ADA at the time of vector infusion, so that the transduced cells have a proliferative advantage over the nontransduced; and the use of a mild conditioning regimen to facilitate engraftment of the transduced cells have led to success without the complications seen in the X-linked SCID trials. There have been no complications in the 10 children treated on the Milan protocol, with a median follow-up of >8 years. ADA-SCID, then, is an example where gene therapy has changed therapeutic options for patients. For those with a human leukocyte antigen (HLA)-identical sibling, bone marrow transplantation is still the best treatment option, but this includes only a minority of those affected. For those without an HLA-identical match, gene therapy has comparable efficacy to PEG-ADA, does not require repetitive injections, and does not run the risk of neutralizing antibodies to the bovine enzyme.
NEURODEGENERATIVE DISEASES: EXTENSION OF PRINCIPLE
The SCID trials gave support to the hypothesis that gene transfer into HSCs could be used to treat any disease for which allogeneic bone marrow transplantation was therapeutic. Moreover, the use of genetically modified autologous cells carried several advantages including no risk of graft-versus-host disease, guaranteed availability of a “donor” (unless the disease itself damages the stem cell population of the patient), and low likelihood of failure of engraftment. Cartier and Aubourg capitalized on this realization to conduct the first trial of lentiviral vector transduction of HSCs for a neurodegenerative disorder, X-linked adrenoleukodystrophy (ALD). X-linked ALD is a fatal demyelinating disease of the central nervous system caused by mutations in the gene encoding an adenosine triphosphate–binding cassette transporter. Deficiency of this protein leads to accumulation of very-long-chain fatty acids in oligodendrocytes and microglia, disrupting myelin maintenance by these cells. Affected boys present with clinical and neuroradiographic evidence of disease at age 6–8 and usually die before adolescence. Following lentiviral transduction of autologous HSCs in young boys with the disease, dramatic stabilization of disease occurred, demonstrating that stem cell transduction could work for neurodegenerative as well as immunologic disorders. Investigators in Milan carried this observation one step further to develop a treatment for another neurodegenerative disorder that has previously responded poorly to bone marrow transplantation. Metachromatic leukodystrophy is a lysosomal storage disorder caused by mutations in the gene encoding arylsulfatase A (ARSA). The late infantile form of the disease is characterized by progressive motor and cognitive impairment, and death within a few years of onset, due to accumulation of the ARSA substrate sulfatide in oligodendrocytes, microglia, and some neurons. Recognizing that endogenous levels of production of ARSA were too low to provide cross-correction by allogeneic transplant, Naldini and colleagues engineered a lentiviral vector that directed supraphysiologic levels of ARSA expression in transduced cells. Transduction of autologous HSCs from children born with the disease, at a point when they were still presymptomatic, led to preservation and continued acquisition of motor and cognitive milestones at time periods as long as 32 months after affected siblings had begun to lose milestones. These results illustrate that the ability to engineer levels of expression can allow gene therapy approaches to succeed where allogeneic bone marrow transplantation cannot. It is likely that a similar approach will be used in other neurodegenerative conditions.
Transduction of HSCs to treat the hemoglobinopathies is an obvious extension of studies already conducted but represents a higher hurdle in terms of the extent of transduction required to achieve a therapeutic effect. Trials are now under way for thalassemia and for a number of other hematologic disorders, including Wiskott-Aldrich syndrome, and chronic granulomatous disease.
LONG-TERM EXPRESSION IN GENETIC DISEASE: IN VIVO GENE TRANSFER WITH RECOMBINANT ADENO-ASSOCIATED VIRAL VECTORS
Recombinant adeno-associated viral (AAV) vectors have emerged as attractive gene delivery vehicles for genetic disease. Engineered from a small replication-defective DNA virus, they are devoid of viral coding sequences and trigger very little immune response in experimental animals. They are capable of transducing nondividing target cells, and the donated DNA is stabilized primarily in an episomal form, thus minimizing risks arising from insertional mutagenesis. Because the vector has a tropism for certain long-lived cell types, such as skeletal muscle, the central nervous system (CNS), and hepatocytes, long-term expression can be achieved even in the absence of integration.
FIRST LICENSED PRODUCT
These features of AAV were used to develop the first licensed gene therapy product in Europe, an AAV vector for treatment of the autosomal recessive disorder lipoprotein lipase (LPL) deficiency. This rare disorder (1–2/million) is due to loss-of-function mutations in the gene encoding LPL, an enzyme normally produced in skeletal muscle and required for the catabolism of triglyceride-rich lipoproteins and chylomicrons. Affected individuals have lipemic serum and may have eruptive xanthomas, hepatosplenomegaly, and in some cases, recurrent bouts of acute pancreatitis. Clinical trials demonstrated the safety of intramuscular injection of AAV-LPL and its efficacy in reducing frequency of pancreatitis episodes in affected individuals, leading to drug approval in Europe. Additional clinical trials currently under way that use AAV vectors in the setting of genetic disease include those for muscular dystrophies, α1 antitrypsin deficiency, Parkinson’s disease, Batten’s disease, hemophilia B, and several forms of congenital blindness.
HEMOPHILIA
Hemophilia (Chap. 78) has long been considered a promising disease model for gene transfer, because the gene product does not require precise regulation of expression and biologically active clotting factors can be synthesized in a variety of tissue types, permitting latitude in the choice of target tissue. Moreover, raising circulating factor levels from <1% (levels seen in those severely affected) into the range of 5% greatly improves the phenotype of the disease. Preclinical studies with recombinant AAV vectors infused into skeletal muscle or liver have resulted in long-term (>5 years) expression of factor VIII or factor IX in the hemophilic dog model. Administration to skeletal muscle of an AAV vector expressing factor IX in patients with hemophilia B was safe and resulted in long-term expression as measured on muscle biopsy, but circulating levels never rose to >1% for sustained periods, and a large number of IM injections (>80–100) was required to access a large muscle mass. Intravascular vector delivery has been used to access large areas of skeletal muscle in animal models of hemophilia and will likely be tested for this and other disorders in upcoming trials.
The first trial of an AAV vector expressing factor IX delivered to the liver in humans with hemophilia B resulted in therapeutic circulating levels at the highest dose tested, but expression at these levels (>5%) lasted for only 6–10 weeks before declining to baseline (<1%). A memory T cell response to viral capsid, present in humans but not in other animal species (which are not natural hosts for the virus), likely led to the loss of expression (Table 91e-2). In response to these findings, a second trial included a short course of prednisolone, to be administered if factor IX levels began to decline. This approach resulted in long-term expression of factor IX, in the range of 2–5%, in men with severe hemophilia B. Current efforts are focused on expanding these trials, and extending the approach to hemophilia A.
RETINAL GENE THERAPY
A logical conclusion from the early experience with AAV in liver in the hemophilia trial was that avoidance of immune responses was key to long-term expression. Thus immunoprivileged sites such as the retina began to attract substantial interest as therapeutic targets. This inference has been elegantly confirmed in the setting of the retinal degenerative disease Leber’s congenital amaurosis (LCA). Characterized by early-onset blindness, LCA is not currently treatable and is caused by mutations in several different genes; ~15% of cases of LCA are due to a mutation in a gene, RPE65, encoding a retinal pigment epithelial-associated 65-kDa protein. In dogs with a null mutation in RPE65, sight was restored after subretinal injection of an AAV vector expressing RPE65. Transgene expression appears to be stable, with the first animals treated >10 years ago continuing to manifest electroretinal and behavioral evidence of visual function. As is the case for X-linked SCID, gene transfer must occur relatively early in life to achieve optimal correction of the genetic disease, although the exact limitations imposed by age have not yet been defined. AAV-RPE65 trials carried out in both the United States and the United Kingdom have shown restoration of visual and retinal function in over 30 subjects, with the most marked improvement occurring in the younger subjects. Trials for other inherited retinal degenerative disorders such as choroideremia are under way, as are studies for certain complex acquired disorders such as age-related macular degeneration, which affects several million people worldwide. The neovascularization that occurs in age-related macular degeneration can be inhibited by expression of vascular endothelial growth factor (VEGF) inhibitors such as angiostatin or through the use of RNA interference (RNAi)-mediated knockdown of VEGF. Early-phase trials of siRNAs that target VEGF RNA are under way, but these require repeated intravitreal injection of the siRNAs; an AAV vector–mediated approach, which would allow long-term inhibition of the biological effects of VEGF through a soluble VEGF receptor, is now in clinical testing.
GENE THERAPY FOR CANCER
The majority of clinical gene transfer experience has been in subjects with cancer (Fig. 91e-1). As a general rule, a feature that distinguishes gene therapies from conventional cancer therapeutics is that the former are less toxic, in some cases because they are delivered locally (e.g., intratumoral injections), and in other cases because they are targeted specifically to elements of the tumor (immunotherapies, antiangiogenic approaches). Because cancer is a disease of aging, and many elderly are frail, the development of therapeutics with milder side effects is an important goal.
LOCAL APPROACHES
Cancer gene therapies can be divided into local and systemic approaches (Table 91e-3). Some of the earliest cancer gene therapy trials focused on local delivery of a prodrug or a suicide gene that would increase sensitivity of tumor cells to cytotoxic drugs. A frequently used strategy has been intratumoral injection of an adenoviral vector expressing the thymidine kinase (TK) gene. Cells that take up and express the TK gene can be killed after the administration of ganciclovir, which is phosphorylated to a toxic nucleoside by TK. Because cell division is required for the toxic nucleoside to affect cell viability, this strategy was initially used in aggressive brain tumors (glioblastoma multiforme) where the cycling tumor cells were affected but the nondividing normal neurons were not. More recently, this approach has been explored for locally recurrent prostate, breast, and colon tumors, among others.
|
GENE THERAPY STRATEGIES IN CANCER |
Another local approach uses adenoviral-mediated expression of the tumor suppressor p53, which is mutated in a wide variety of cancers. This strategy has resulted in complete and partial responses in squamous cell carcinoma of the head and neck, esophageal cancer, and non-small-cell lung cancer after direct intratumoral injection of the vector. Response rates (~15%) are comparable to those of other single agents. The use of oncolytic viruses that selectively replicate in tumor cells but not in normal cells has also shown promise in squamous cell carcinoma of the head and neck and in other solid tumors. This approach is based on the observation that deletion of certain viral genes abolishes their ability to replicate in normal cells but not in tumor cells. An advantage of this strategy is that the replicating vector can proliferate and spread within the tumor, facilitating eventual tumor clearance. However, physical limitations to viral spread, including fibrosis, intermixed normal cells, basement membranes, and necrotic areas within the tumor, may limit clinical efficacy. Oncolytic viruses are licensed and available in some countries but not in the United States.
SYSTEMIC APPROACHES
Because metastatic disease rather than uncontrolled growth of the primary tumor is the source of mortality for most cancers, there has been considerable interest in developing systemic gene therapy approaches. One strategy has been to promote more efficient recognition of tumor cells by the immune system. Approaches have included transduction of tumor cells with immune-enhancing genes encoding cytokines, chemokines, or co-stimulatory molecules; and ex vivo manipulation of dendritic cells to enhance the presentation of tumor antigens. Recently, considerable success has been achieved using lentiviral transduction of autologous lymphocytes with a cDNA encoding a chimeric antigen receptor (CAR). The CAR moiety consists of a tumor antigen-binding domain (e.g., an antibody to the B cell antigen CD19) fused to an intracellular signaling domain that allows T cell activation. The transduced lymphocytes can then recognize and destroy cells bearing the antigen. This CAR–T cell approach has proven extraordinarily successful in the setting of refractory chronic lymphocytic leukemia and pre-B-cell acute lymphoblastic leukemia. Infusion of gene-modified T cells engineered to recognize the B cell antigen CD19 has resulted in >1000-fold expansion in vivo, trafficking of the T cells to the bone marrow, and complete remission in a subset of patients who had failed multiple chemotherapy regimens. The cells persist as memory CAR+ T cells, providing ongoing antitumor functionality. Some patients experience a delayed tumor lysis syndrome requiring intensive medical management. This approach also causes an on-target toxicity, leading to B cell aplasia that necessitates lifelong IgG infusions. Current results indicate that long-lasting remissions can be achieved and the strategy can theoretically be extended to other tumor types if a tumor antigen can be identified.
Gene transfer strategies have also been developed for inhibiting tumor angiogenesis. These have included constitutive expression of angiogenesis inhibitors such as angiostatin and endostatin; use of siRNA to reduce levels of VEGF or VEGF receptor; and combined approaches in which autologous T cells are genetically modified to recognize antigens specific to tumor vasculature. These studies are still in early-phase testing.
Another novel systemic approach is the use of gene transfer to protect normal cells from the toxicities of chemotherapy. The most extensively studied of these approaches has been transduction of hematopoietic cells with genes encoding resistance to chemotherapeutic agents, including the multidrug resistance gene MDRI or the gene encoding O6-methylguanine DNA methyltransferase (MGMT). Ex vivo transduction of hematopoietic cells, followed by autologous transplantation, is being investigated as a strategy for allowing administration of higher doses of chemotherapy than would otherwise be tolerated.
GENE THERAPY FOR VASCULAR DISEASE
The third major category addressed by gene transfer studies is cardiovascular disease. Initial experience was in trials designed to increase blood flow to either skeletal (critical limb ischemia) or cardiac muscle (angina/myocardial ischemia). First-line treatment for both of these groups includes mechanical revascularization or medical management, but a subset of patients are not candidates for or fail these approaches. These patients formed the first cohorts for evaluation of gene transfer to achieve therapeutic angiogenesis. The major transgene used has been VEGF, attractive because of its specificity for endothelial cells; other transgenes have included fibroblast growth factor (FGF) and hypoxia-inducible factor 1, α subunit (HIF-1α). The design of most of the trials has included direct IM (or myocardial) injection of either a plasmid or an adenoviral vector expressing the transgene. Both of these vectors are likely to result in only short-term expression of VEGF, which may be adequate because there is no need for continued transgene expression once the new vessels have formed. Direct injection favors local expression, which should help to avoid systemic effects such as retinal neovascularization or new vessel formation in a nascent tumor. Initial trials of adeno-VEGF or plasmid-VEGF injection resulted in improvement over baseline in angiographically detectable vasculature, but no change in amputation frequency or cardiovascular mortality. Studies using different routes of administration or different transgenes are currently under way.
More recent studies have used AAV vectors to develop a therapeutic approach for individuals with refractory congestive heart failure. In preclinical studies, a vector encoding sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) demonstrated positive left ventricular inotropic effects in a swine model of volume-overloaded heart failure. Results of a phase II study in which vector was infused via the coronary arteries in patients with congestive heart failure demonstrated safety and some indications of efficacy; larger studies are now planned.
OTHER APPROACHES
This chapter has focused on gene addition therapy, in which a normal gene is transferred to a target tissue to drive expression of a gene product with therapeutic effects. Another powerful technique under development is genome editing, in which a mutation is corrected in situ, generating a wild-type copy under the control of the endogenous regulatory signals. This approach makes use of novel reagents including zinc finger nucleases, TALENs and CRISPR, which introduce double-stranded breaks into the DNA near the site of the mutation and then rely on a donated repair sequence and cellular mechanisms for repair of double-strand breaks to reconstitute a functioning gene. Another strategy recently introduced into clinical trials is the use of siRNAs or short hairpin RNAs as transgenes to knock down expression of deleterious genes (e.g., mutant huntingtin in Huntington’s disease or genes of the hepatitis C genome in infected individuals).
SUMMARY
The power and versatility of gene transfer approaches are such that there are few serious disease entities for which gene transfer therapies are not under development. The development of new classes of therapeutics typically takes two to three decades; monoclonal antibodies and recombinant proteins are recent examples. Gene therapeutics, which entered clinical testing in the early 1990s, traversed the same time course. Examples of clinical success are now abundant, and gene therapy approaches are likely to become increasingly important as a therapeutic modality in the twenty-first century. A central question to be addressed is the long-term safety of gene transfer, and regulatory agencies have mandated a 15-year follow-up for subjects enrolled in gene therapy trials (Table 91e-4). Realization of the therapeutic benefits of modern molecular medicine will depend on continued progress in gene transfer technology.
|
TAKING HISTORY FROM SUBJECTS ENROLLED IN GENE TRANSFER STUDIES |
aFactors influencing long-term risk include: integration of the vector into the genome, vector persistence without integration, and transgene-specific effects.
92e |
Tissue Engineering |
Tissue engineering is a field that applies principles of regenerative medicine to restore the function of various organs by combining cells with biomaterials. It is multidisciplinary, often combining the skills of physicians, cell biologists, bioengineers, and material scientists, to recapitulate the native three-dimensional architecture of an organ, the appropriate cell types, and the supportive nutrients and growth factors that allow normal cell growth, differentiation, and function. Tissue engineering is a relatively new field, originating in the late 1970s. Early studies focused on efforts to create skin substitutes using biomaterials and epithelial skin cells with a goal of providing barrier protection for patients with burns. The early strategies employed a tissue biopsy, followed by ex vivo expansion of cells seeded on scaffolds. The cell–scaffold composite was later implanted back into the same patient, where the new tissue would mature. However, there were many hurdles to overcome. The three major challenges in the field of tissue engineering involved: (1) the ability to grow and expand normal primary human cells in large quantities; (2) the identification of appropriate biomaterials; and (3) the requirement for adequate vascularization and innervation of the engineered constructs.
ISOLATION AND GROWTH OF CELLS
The original model for tissue engineering focused largely on the isolation of tissue from the organ of interest, the growth and expansion of the tissue-specific cells, and the seeding of these cells onto three-dimensional scaffolds. Just a few decades ago, most primary cultures of human cells could not be grown and expanded in large quantities, representing a major impediment to the engineering of human tissues. However, the identification of specific tissue progenitor cells in the 1990s allowed expansion of multiple cell types, and progress has occurred steadily since then. Some cell types are more amenable to expansion than others, reflecting in part their native regenerative capacity but also varying requirements for nutrients, growth factors, and cell–cell contacts. As an example of progress, after years of effort, protocols for the growth and expansion of human cardiomyocytes are now available. However, there are still many tissue-specific cell types that cannot be expanded from tissue sources, including the pancreas, liver, and nerves. The discovery of pluripotent or highly multipotent stem cells (Chap. 88) may ultimately allow most human cell types to be used for tissue engineering. The stem cell characteristics depend on their origin and their degree of plasticity, with cells from the earliest developmental stages, such as embryonic stem cells, having the greatest plasticity. Induced pluripotent stem cells have the advantage that they can be derived from individual patients, allowing autologous transplants. They can also be differentiated, in vitro, along cell-specific lineages, although these protocols are still at an early stage of development. Human embryonic and induced pluripotent stem cells have a very high replicative potential, but they also have the potential for rejection and tumor formation (e.g., teratomas). The more recently described amniotic fluid and placental stem cells have a high replicative potential but without an apparent propensity for tumor formation. Moreover, they have the potential to be used in an autologous manner without rejection. Adult stem cells, such as those derived from bone marrow, also have less propensity for tumor formation and, if used in an autologous manner, will not be rejected, but their replicative potential is limited, especially for endoderm and ectoderm cells.
Stem cells can be derived from autologous or heterologous sources. Heterologous cells can be used when only temporary coverage is needed, such as replacing skin after a burn or wound. However, if a more permanent construct is required, autologous cells are preferred to avoid rejection. There are also practical issues related to tissue sources. For example, if a patient presents with end-stage heart disease, obtaining a cardiac tissue biopsy for cell expansion is unlikely to be feasible, and bone marrow–derived mesenchymal cells may provide an alternative.
BIOMATERIALS AS SCAFFOLDS FOR TISSUE ENGINEERING
The biomaterials used to create the scaffolds for tissue engineering require specific properties to enhance the long-term success of the implanted constructs. Ideally, the biomaterials should be biocompatible; elicit minimal inflammatory responses; have appropriate biomechanical properties; and promote cell attachment, viability, proliferation, and differentiated function. Ideally, the scaffolds should replicate the biomechanical and structural properties of the tissue being replaced. In addition, biodegradation should be controlled such that the scaffold retains its structural integrity until the cells deposit their own matrix. If the scaffolds degrade too quickly, the constructs may collapse. If the scaffolds degrade too slowly, fibrotic tissue may form. Also, the degradation of the scaffolds should not alter the local environment unfavorably, because this can impair the function of cells or newly formed tissue.
The first scaffolds designed for tissue regeneration were naturally derived materials, such as collagen. The first artificially derived material for tissue engineering used a biodegradable scaffold made of polyglycolic acid. Naturally derived scaffolds have properties very similar to the native matrix, but there is an inherent batch-to-batch variability, whereas the production of artificially derived biomaterials can be better controlled, allowing for more uniform results. More recently, combination scaffolds, made of both naturally and artificially derived biomaterials, have been used for tissue engineering.
An emerging area is the use of peptide nanostructures to facilitate tissue engineering. Some of these are self-assembling peptide amphiphiles that allow scaffolds to form in vivo, for example at sites of spinal cord injury where they have been used experimentally to prevent scar formation and facilitate nerve and blood vessel regeneration. Peptide nanostructures can be combined with other biomaterials, and they can be linked to growth factors, antibodies, and various signaling molecules that can modulate cell behavior during organ regeneration.
VASCULARIZATION AND INNERVATION
Implanted tissue-engineered constructs require adequate vascularity and innervation. Judah Folkman, a pioneer in the field of angiogenesis, made the observation that cells could survive in volumes up to 3 mm3 via nutrient diffusion alone, but larger cell volumes required vascularization for survival. Adequate vascularity was also essential for normal innervation to occur. This was a major challenge in the field of tissue engineering, which largely depended on the patient’s native angiogenesis and innervation. Even if sufficient cell quantities are available, there is a theoretical limit on the types of tissue constructs that could be created. In response to this challenge, material scientists designed scaffolds with much greater porosity and architecture. Scaffold designs included the creation of thin, porous sponges comprised of 95% air, markedly increasing the surface area for the resident cells. These properties promoted increased vascularity and innervation. The addition of growth factors, such as vascular endothelial growth factor and nerve growth factor, has been used to enhance angiogenesis and innervation.
LEVEL OF COMPLEXITY FOR THE ENGINEERING OF TISSUES AND ORGANS
All human tissues are complex. However, from an architectural aspect, tissues can be categorized under four levels. Flat tissue structures, such as skin, are the least complex (level 1), comprised predominantly of a single epithelial cell type. Tubular structures, such as blood vessels and the trachea, are more complex architecturally (level 2) and must be constructed to ensure that the structure does not collapse over time. These tissues typically have two major cell types. They are designed to act as a conduit for air or fluid at a steady state within a defined physiologic range. Hollow nontubular organs, such as the stomach, bladder, or uterus, are more complex architecturally (level 3). The cells are functionally more complex, and these cell types often have a functional interdependence. By far, the most complex are the solid organs (level 4), because the amount of cells per cm2 are exponentially greater than any of the other tissue types.
For the first three tissue levels (1–3), when the constructs are initially implanted, the cell layering on the scaffolds is thin, not unlike that seen in tissue culture matrices. The cell layering continues to mature, in concert with the recipient’s native angiogenesis and neo-innervation. For level 4 (solid) organs, the vascularity requirements are substantial, and native tissue angiogenesis is not sufficient. The engineering strategies for tissues vary according to their complexity level.
STRATEGIES FOR TISSUE ENGINEERING
The basic principles of tissue engineering involve the use of the relevant cell populations, where the cell biology is well understood and the cells can be reproducibly retrieved and expanded, and the use of optimized biomaterials and scaffold designs. Cell seeding can be performed using various techniques, including static or flow-based systems that use bioreactors.
Most techniques for the engineering of tissues fall under one of five strategies (Fig. 92e-1):
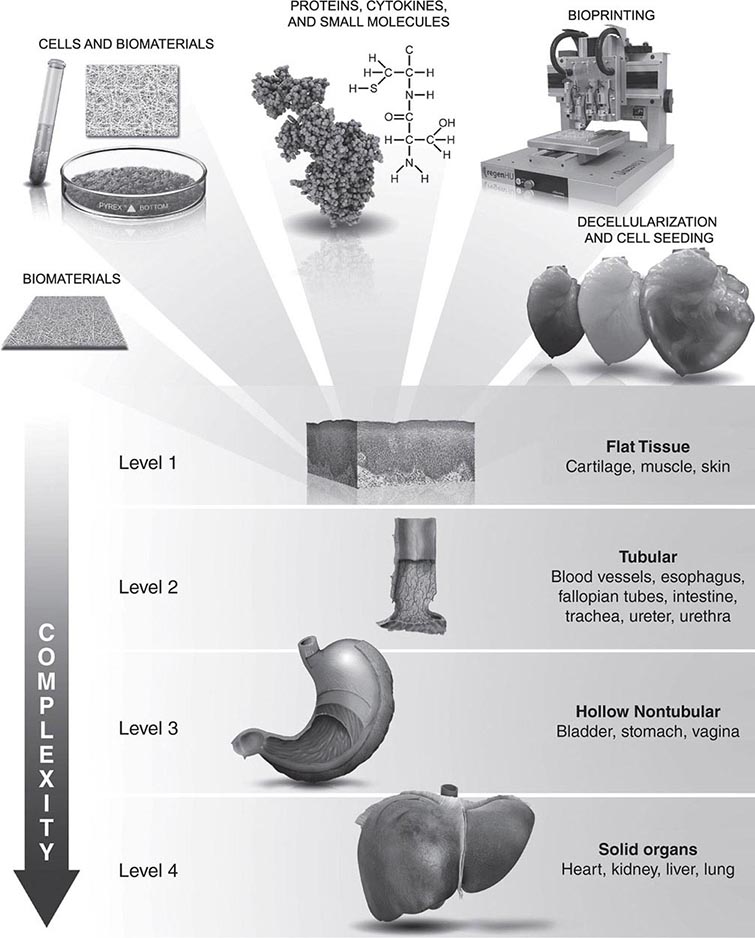
FIGURE 92e-1 Strategies for tissue and organ engineering.
1. Scaffolds can be used alone, without cells, and implanted, where they depend on native cell migration onto the scaffold from the adjacent tissue for regeneration. The first use of decellularized scaffolds for tissue regeneration was for urethral reconstruction. These techniques are most optimal when the size of the defect is relatively small, usually <0.5 cm from each tissue edge. Larger defects tend to heal by scarring, due to the deposition of fibroblasts, and eventual fibrosis. Scaffolds alone have also been used for other applications, including for wound coverage, soft tissue coverage after joint surgery, urogynecologic applications for sling surgery, and as materials for hernia repair.
2. A more recent strategy in tissue engineering involves the use of proteins, cytokines, genes, or small molecules that induce in situ tissue regeneration, either alone or with the use of scaffolds. For example, gene transcription factors used in the mouse pancreas led to tissue regeneration. Surgically implanted decellularized heart valve scaffolds, coated with proteins that attract vascular stem cells, led to the creation of in situ cell-seeded functional heart valves in sheep. Drugs that induce muscle regeneration are being tested clinically. Small molecules that induce tissue regeneration are currently under investigation for multiple applications, including growth of skin and hair and for musculoskeletal applications.
3. The most common strategy for the engineering of tissues uses scaffolds seeded with cells. The most direct and established type of tissue engineering uses flat scaffolds, either artificial or naturally derived, that are seeded with cells and used for the replacement or repair of flat tissue structures. The flat scaffolds can also be sized and molded at the time of surgical implantation, or they can be shaped prior to cell seeding, for example, for tubular organs such as blood vessels or nontubular hollow tissues such as bladders. Bioreactors are often used to expose the cell–scaffold construct to mechanical forces, such as, stress, strain, and pulsatile flow that aid in the normal development of the cells into tissues (Video 92e-1, engineered heart valve in a pulsatile bioreactor showing the valves opening and closing). This strategy is the most common method used for tissue regeneration to date, and tissues and organs, such as skin, blood vessels, urethras, tracheas, vaginas, and bladders, have been engineered and implanted in patients using these techniques.
4. The fourth strategy in tissue engineering is applicable for solid organs, where discarded organs are exposed to mild detergents and are decellularized, leaving behind a three-dimensional scaffold that preserves its vascular tree. The scaffold can then be reseeded with the patient’s own expanded vascular and tissue-specific cells. This strategy was used initially to create solid phallic structures in rabbits that were functional and able to produce offspring. Similar strategies were also used to recellularize miniature heart, liver, and kidney structures, with limited functionality to date, but with an established proof of concept (Video 92e-2, a dye is injected through the portal artery of a decellularized liver showing an intact vascular tree). These techniques are currently under investigation and have not been used clinically to date.
5. The fifth strategy for tissue engineering involves the use of bioprinting. These technologies arose through the use of modified desktop inkjet printers over a decade ago. The inkjet cartridges were filled with a cell–hydrogel combination instead of ink. A rudimentary three-dimensional elevator was lowered each time the cartridge deposited the cells and hydrogel, thus building miniature solid structures, such as two-chambered heart organoids, one layer at a time. More sophisticated bioprinters have now been built that have additional computer-aided design (CAD) and three-dimensional printing technologies. The information to print the organ can be personalized using the patient’s own imaging studies that help to define the size and shape of the particular tissue (Video 92e-3, a modified inkjet printer shows the three-dimensional construction of a two-chambered heart and how the structure beats with the cardiomyocytes in synchrony). Bioprinting is a tool that allows a scale-up option for the production of engineered tissues. Its use is still experimental and has not been applied clinically to date.
FUTURE DIRECTIONS
A number of engineered tissues, including architecturally flat, tubular, and hollow nontubular organs, have been implanted in patients dating back to the 1990s. These include bladders, blood vessels, urethras, vaginal organs, tracheas, and skin for permanent replacement (Table 92e-1). Various types of skin substitutes, which were used as temporary “living wound dressings” to cover burn areas until skin grafts could be obtained from the same patient, were implanted starting in the 1990s. However, the use of engineered skin as a permanent replacement occurred only recently. Many engineered tissues are still being used in patients under regulatory guidelines for clinical trials. To date, solid organs have not yet been engineered for clinical use.
|
ENGINEERED TISSUES IN PATIENTS |
Tissue engineering is a rapidly evolving field where new technologies are continuously being applied to achieve success. The field still has many challenges ahead, including the long regulatory timelines required for the approval of widespread use, the need for improved scale-up production technologies, and the cost of the technologies, which include multiple processes involving biologics. Nonetheless, the list of tissues and organs being implanted in patients keeps growing, and the ability of these technologies to improve health has been demonstrated. More patients should be able to benefit from these technologies in the coming years.
VIDEO 92e-1 Engineered heart valve in a pulsatile bioreactor showing the valves opening and closing.
VIDEO 92e-2 A dye is injected through the portal artery of a decellularized liver showing an intact vascular tree.
VIDEO 92e-3 A modified inkjet printer shows the three-dimensional construction of a two-chambered heart and how the structure beats with the cardiomyocytes in synchrony.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
88 |
Stem Cell Biology |
Stem cell biology is a rapidly expanding field that explores the characteristics and possible clinical applications of a variety of stem cells that serve as the progenitors of more differentiated cell types. In addition to potential therapeutic applications (Chap. 90e), patient-derived stem cells can also be used as disease models and as a means of testing drug efficacy. Stem cells and their niche are a major focus of medical research because they play central roles in tissue and organ homeostasis and repair, which are important aspects of aging and disease.
IDENTIFICATION, ISOLATION, AND DERIVATION OF STEM CELLS
Resident Stem Cells The definition of stem cells remains elusive. Stem cells were originally postulated as unspecified or undifferentiated cells that provide a source of renewal of skin, intestine, and blood cells throughout life. These resident stem cells have been identified in a variety of organs (e.g., epithelia of the skin and digestive system, bone marrow, blood vessels, brain, skeletal muscle, liver, testis, and pancreas) based on their specific locations, morphology, and biochemical markers.
Isolated Stem Cells Unequivocal identification of stem cells requires their separation and purification, usually based on a combination of specific cell-surface markers. These isolated stem cells (e.g., hematopoietic stem [HS] cells) can be studied in detail and used in clinical applications, such as bone marrow transplantation (Chap. 89e). However, the lack of specific cell-surface markers for other types of stem cells has made it difficult to isolate them in large quantities. This challenge has been partially addressed in animal models by genetically marking different cell types with green-fluorescent protein driven by cell-specific promoters. Alternatively, putative stem cells have been isolated from a variety of tissues as side population (SP) cells using fluorescence-activated cell sorting after staining with the Hoechst 33342 dye.
Cultured Stem Cells It is desirable to culture and expand stem cells in vitro to obtain a sufficient quantity for analysis and potential therapeutic use. Although the derivation of stem cells in vitro has been a major obstacle in stem cell biology, the number and types of cultured stem cells have increased progressively (Table 88-1). Cultured stem cells derived from resident stem cells are often called adult stem cells or somatic stem cells to distinguish them from embryonic stem (ES) and embryonic germ (EG) cells. However, considering the existence of embryo-derived, tissue-specific stem cells (e.g., trophoblast stem [TS] cells) and the possible derivation of similar cells from an embryo/fetus (e.g., neural stem [NS] cells), it is more appropriate to use the term, tissue stem cells.
|
EXAMPLES OF CULTURED STEM CELLS |
Successful derivation of cultured stem cells (both embryonic and tissue stem cells) often requires the identification of necessary growth factors and culture conditions, mimicking the microenvironment or niche of the resident stem cells. Recently, long-term maintenance of tissue stem cells in vitro is increasingly possible by growing them as three-dimensional (3D) organoids, which contain both stem cells and niche cells (Chap. 92e). For example, intestinal stem cells can now be cultured as “epithelial mini-guts” in the presence of R-spondin, epidermal growth factor (EGF), and noggin on Matrigel. Similarly, lung stem cells can be cultured as self-renewing “alveolospheres.” A growing list of cultured stem cells, although not comprehensive, is shown in Table 88-1. Please note that the establishment of cultured stem cells is often under dispute due to the difficulties in assessing the characteristics of these cells.
SELF-RENEWAL AND PROLIFERATION OF STEM CELLS
Symmetric and Asymmetric Cell Division The most widely accepted stem cell definition is a cell with a unique capacity to produce unaltered daughter cells (self-renewal) and to generate specialized cell types (potency). Self-renewal can be achieved in two ways. Asymmetric cell division produces one daughter cell that is identical to the parental cell and one daughter cell that is different from the parental cell and is a progenitor or differentiated cell. Asymmetric cell division does not increase the number of stem cells. Symmetric cell division produces two identical daughter cells. For stem cells to proliferate in vitro, they must divide symmetrically.
Unlimited Expansion In Vitro Resident stem cells are often quiescent and divide infrequently. However, once the stem cells are successfully cultured in vitro, they often acquire the capacity to divide continuously and the ability to proliferate beyond the normal passage limit typical of primary cultured cells (sometimes called immortality). These features are primarily seen in ES cells but have also been demonstrated for tissue stem cells, such as NS cells and mesenchymal stem (MS) cells, thereby enhancing the potential of these cells for therapeutic use (Table 88-1).
Stability of Genotype and Phenotype The capacity to actively proliferate is often associated with the accumulation of chromosomal abnormalities and mutations. Mouse ES cells appear to be an exception to this rule and tend to maintain their euploid karyotype and genome integrity. By contrast, human ES cells appear to be more susceptible to mutations after long-term culture. However, it is also important to note that even euploid mouse ES cells can form teratomas when injected into immunosuppressed animals, raising concerns about the possible formation of tumors after transplanting actively dividing stem cells.
POTENCY AND DIFFERENTIATION OF STEM CELLS
Developmental Potency The term potency is used to indicate a cell’s ability to differentiate into multiple specialized cell types. The current lack of knowledge about the molecular nature of potency requires the experimental manipulation of stem cells to demonstrate their potency. For example, in vivo testing can be done by injecting stem cells into mouse blastocysts or immunosuppressed adult mice and determining how many different cell types are formed from the injected cells. However, these in vivo assays are not applicable to human stem cells. In vitro testing can be performed by differentiating cells in various culture conditions to determine how many different cell types are formed from the cells. The formal test of self-renewal and potency is performed by demonstrating that a single cell possesses such abilities in vitro (clonality). Cultured stem cells are tentatively grouped according to their potency (Fig. 88-1). Only some examples are shown, because many cultured stem cells, especially human cells, lack definitive information about their developmental potency.
FIGURE 88-1 Potency and source developmental stage of cultured stem cells. For abbreviations of stem cells, see Table 88-1. Note that stem cells are often abbreviated with or without “cells,” e.g., ES cells or ESCs for embryonic stem cells. h, human; m, mouse.
From Totipotency to Unipotency Totipotent cells can form an entire organism autonomously. Only a fertilized egg (zygote) possesses this feature. Pluripotent cells (e.g., ES cells) can form almost all of the body’s cell lineages (endoderm, mesoderm, and ectoderm), including germ cells. Multipotent cells (e.g., HS cells) can form multiple cell lineages but cannot form all of the body’s cell lineages. Oligopotent cells (e.g., NS cells) can form more than one cell lineage but are more restricted than multipotent cells. Oligopotent cells are sometimes called progenitor cells or precursor cells; however, these terms are often more strictly used to define partially differentiated or lineage-committed cells (e.g., myeloid progenitor cells) that can divide into different cell types but lack self-renewing capacity. Unipotent cells or monopotent cells (e.g., spermatogonial stem [SS] cells) can form a single differentiated cell lineage.
Nuclear Reprogramming Development naturally progresses from totipotent fertilized eggs to pluripotent epiblast cells to multipotent cells and, finally, to terminally differentiated cells. According to Waddington’s epigenetic landscape, this is analogous to a ball moving down a slope. The reversal of the terminally differentiated cells to totipotent or pluripotent cells (called nuclear reprogramming) can thus be seen as an uphill gradient. Nuclear reprogramming has been achieved using nuclear transplantation, or nuclear transfer (NT), procedures (often called “cloning”), where the nucleus of a differentiated cell is transferred into an enucleated oocyte. Although this is an error-prone procedure with a very low success rate, live animals have been produced using adult somatic cells as donors in sheep, mice, and other mammals. In mice, it has been demonstrated that ES cells derived from blastocysts made by somatic cell NT are indistinguishable from normal ES cells. NT can potentially be used to produce patient-specific ES cells carrying a genome identical to that of the patient, although such strategies have not been pursued due to ethical issues and technical challenges. Recent success in generating human ES cells by NT has rekindled an interest in this area; however, the limited supply of human oocytes will still be a major problem for clinical applications of NT.
An alternative approach that has become a method of choice is the direct conversion of terminally differentiated cells into ES-like cells (called induced pluripotent stem [iPS] cells) by overexpressing a combination of key transcription factors (TFs). The original method was to infect mouse embryonic fibroblast cells with retrovirus vectors carrying four TFs [Pou5f1 (Oct4), Sox2, Klf4, and Myc] and to identify rare ES-like cells in culture. This approach was soon adapted to human cells, followed by a more refined procedure (e.g., the use of fewer TFs, different cell types, and different gene-delivery methods). Because a clinical trial using iPS cells is imminent, the safety of iPS-based therapy is a major concern and a variety of measures are being taken to ensure the safety. For example, it has now become a standard to use footprint-free methods such as an episomal vector, Sendai virus vector, and synthetic mRNAs to deliver reprogramming factors into cells, resulting in the production of patient-specific iPS cells with minimal alteration of their genetic makeup. In addition to cell replacement therapy, disease-specific iPS cells are expected to play a role in modeling human disease in vitro and in screening drugs for personalized medicine.
It has also become possible to convert one type of terminally differentiated cell (e.g., fibroblast cell) into another type of terminally differentiated cell (e.g., cardiac muscle, neuron, or hepatocyte) by overexpressing specific sets of TFs (called direct reprogramming). Direct reprogramming can bypass the step of making iPS cells, possibly providing the safer route to desired cell types for therapy; however, the technology is currently limited by its low efficiency.
Stem Cell Plasticity, Transdifferentiation, and Facultative Stem Cells The prevailing paradigm in developmental biology is that once cells are differentiated, their phenotypes are stable. However, more recent studies show that tissue stem cells, which have traditionally been thought to be lineage-committed multipotent cells, possess the capacity to differentiate into cell types outside their lineage restrictions (called transdifferentiation). For example, HS cells may be converted into neurons as well as germ cells. This feature may provide a means to use tissue stem cells derived directly from a patient for therapeutic purposes, thereby eliminating the need to use embryonic stem cells or elaborate procedures such as nuclear reprogramming of a patient’s somatic cells. However, more strict criteria and rigorous validation are required to establish tissue stem cell plasticity. For example, observations of transdifferentiation may reflect cell fusion, contamination with progenitor cells from other cell lineages, or persistence of pluripotent embryonic cells in adult organs. Therefore, the assignment of potency to each cultured stem cell in Fig. 88-1 should be considered with caution. Whether transdifferentiation exists and can be used for therapeutic purposes remains to be determined conclusively. A similar, but distinct, concept is the facultative stem cell, which is defined as a unipotent cell or a terminally differentiated cell that can function as a stem cell upon tissue injury. The presence of such cells has been proposed for some organs such as liver, intestine, pancreas, and testis, but is still debated.
Directed Differentiation of Stem Cells Pluripotent stem cells (e.g., ES and iPS cells) can differentiate into multiple cell types, but in culture, they normally differentiate into heterogeneous cell populations in a stochastic manner. However, for therapeutic uses, it is desirable to direct stem cells into specific cell types (e.g., insulin-secreting beta cells). This is an active area of stem cell research, and protocols are being developed to achieve this goal. In any of these directed cell differentiation systems, the cell phenotype must be evaluated critically. Alternatively, the heterogeneity of the cell population derived from pluripotent stem cells can be actively exploited, as different types of cells interact with each other in culture and further enhance their own differentiation. In some instances, e.g., optic cup, self-organizing tissue morphogenesis has been demonstrated in 3D culture.
MOLECULAR CHARACTERIZATION OF STEM CELLS
Genomics and Proteomics In addition to standard molecular biological approaches, high-throughput genomics and proteomics have been extensively applied to the analysis of stem cells. For example, DNA microarray analyses have revealed the expression levels of essentially all genes and identified specific markers for some stem cells. Chromatin immunoprecipitation coupled with next-generation sequencing technologies, capable of producing billions of sequence reads in a single run, has revealed chromatin modifications (“epigenetic marks”) relevant to stem cell properties. Similarly, the protein profiles of stem cells have been assessed by using mass spectrometry. These methods are beginning to provide a novel means to characterize and classify various stem cells and the molecular mechanisms that give them their unique characteristics.
ES Cell Regulation It is important to identify genes involved in the regulation of stem cell function and to examine the effects of altered gene expression on ES and other stem cells. For example, core networks of TFs such as Pou5f1 (Oct4), Nanog, and Sox2, govern key gene regulatory pathways/networks for the maintenance of self-renewal and pluripotency of mouse and human ES cells. These TF networks are modulated by specific external factors through signal transduction pathways, such as leukemia inhibitory factor (Lif)/Stat3, mitogen-activated protein kinase 1/3 (Mapk1/3), the transforming growth factor β (TGFβ) superfamily, and Wnt/glycogen synthase kinase 3 beta (Gsk3b). Inhibitors of Mapk1/3 and Gsk3b signaling enhance the derivation of ES cells and help maintain ES cells in full pluripotency (“ground” or “naive state”). Recent data also indicate that 20–25 nucleotide RNAs, called microRNAs (miRNAs), play an important role in regulating stem cell function by repressing the translation of their target genes. For example, it has been shown that miR-21 regulates cell cycle progression in ES cells and miR-128 prevents the differentiation of hematopoietic progenitor cells. These types of analyses should provide molecular clues about the function of stem cells and lead to a more effective means to manipulate stem cells for future therapeutic use.
89e |
Hematopoietic Stem Cells |
All of the cell types in the peripheral blood and some cells in every tissue of the body are derived from hematopoietic (hemo: blood; poiesis: creation) stem cells. If the hematopoietic stem cell is damaged and can no longer function (e.g., due to a nuclear accident), a person would survive 2–4 weeks in the absence of extraordinary support measures. With the clinical use of hematopoietic stem cells, tens of thousands of lives are saved each year (Chap. 139e). Stem cells produce hundreds of billions of blood cells daily from a stem cell pool that is estimated to be only in the tens of thousands. How stem cells do this, how they persist for many decades despite the production demands, and how they may be better used in clinical care are important issues in medicine.
The study of blood cell production has become a paradigm for how other tissues may be organized and regulated. Basic research in hematopoiesis includes defining stepwise molecular changes accompanying functional changes in maturing cells, aggregating cells into functional subgroups, and demonstrating hematopoietic stem cell regulation by a specialized microenvironment; these concepts are worked out in hematology, but they offer models for other tissues. Moreover, these concepts may not be restricted to normal tissue function but extend to malignancy. Stem cells are rare cells among a heterogeneous population of cell types, and their behavior is assessed mainly in experimental animal models involving reconstitution of hematopoiesis. Thus, much of what we know about stem cells is imprecise and based on inferences from genetically manipulated animals.
CARDINAL FUNCTIONS OF HEMATOPOIETIC STEM CELLS
All stem cell types have two cardinal functions: self-renewal and differentiation (Fig. 89e-1). Stem cells exist to generate, maintain, and repair tissues. They function successfully if they can replace a wide variety of shorter-lived mature cells over prolonged periods. The process of self-renewal (see below) assures that a stem cell population can be sustained over time. Without self-renewal, the stem cell pool would become exhausted and tissue maintenance would not be possible. The process of differentiation leads to production of the effectors of tissue function: mature cells. Without proper differentiation, the integrity of tissue function would be compromised and organ failure or neoplasia would ensue.
FIGURE 89e-1 Signature characteristics of the stem cell. Stem cells have two essential features: the capacity to differentiate into a variety of mature cell types and the capacity for self-renewal. Intrinsic factors associated with self-renewal include expression of Bmi-1, Gfi-1, PTEN, STAT5, Tel/Atv6, p21, p18, MCL-1, Mel-18, RAE28, and HoxB4. Extrinsic signals for self-renewal include Notch, Wnt, SHH, and Tie2/Ang-1. Based mainly on murine studies, hematopoietic stem cells express the following cell surface molecules: CD34, Thy-1 (CD90), c-Kit receptor (CD117), CD133, CD164, and c-Mpl (CD110, also known as the thrombopoietin receptor).
In the blood, mature cells have variable average life spans, ranging from 7 h for mature neutrophils to a few months for red blood cells to many years for memory lymphocytes. However, the stem cell pool is the central, durable source of all blood and immune cells, maintaining a capacity to produce a broad range of cells from a single cell source, yet keeping itself vigorous over decades of life. As an individual stem cell divides, it has the capacity to accomplish one of three division outcomes: two stem cells, two cells destined for differentiation, or one stem cell and one differentiating cell. The former two outcomes are the result of symmetric cell division, whereas the latter indicates a different outcome for the two daughter cells—an event termed asymmetric cell division. The relative balance for these types of outcomes may change during development and under particular kinds of demands on the stem cell pool.
DEVELOPMENTAL BIOLOGY OF HEMATOPOIETIC STEM CELLS
During development, blood cells are produced at different sites. Initially, the yolk sac provides oxygen-carrying red blood cells, and then the placenta and several sites of intraembryonic blood cell production become involved. These intraembryonic sites engage in sequential order, moving from the genital ridge at a site where the aorta, gonadal tissue, and mesonephros are emerging to the fetal liver and then, in the second trimester, to the bone marrow and spleen. As the location of stem cells changes, the cells they produce also change. The yolk sac provides red cells expressing embryonic hemoglobins while intraembryonic sites of hematopoiesis generate red cells, platelets, and the cells of innate immunity. The production of the cells of adaptive immunity occurs when the bone marrow is colonized and the thymus forms. Stem cell proliferation remains high, even in the bone marrow, until shortly after birth, when it appears to dramatically decline. The cells in the bone marrow are thought to arrive by the bloodborne transit of cells from the fetal liver after calcification of the long bones has begun. The presence of stem cells in the circulation is not unique to a time window in development; however, hematopoietic stem cells appear to circulate throughout life. The time that cells spend freely circulating appears to be brief (measured in minutes in the mouse), but the cells that do circulate are functional and can be used for transplantation. The number of stem cells that circulate can be increased in a number of ways to facilitate harvest and transfer to the same or a different host.
MOBILITY OF HEMATOPOIETIC STEM CELLS
Cells entering and exiting the bone marrow do so through a series of molecular interactions. Circulating stem cells (through CD162 and CD44) engage the lectins (carbohydrate binding proteins) P- and E-selectin on the endothelial surface to slow the movement of the cells to a rolling phenotype. Stem cell integrins are then activated and accomplish firm adhesion between the stem cell and vessel wall, with a particularly important role for stem cell VCAM-1 engaging endothelial VLA-4. The chemokine CXCL12 (SDF1) interacting with stem cell CXCR4 receptors and ionic calcium interacting with the calcium sensing receptor appear to be important in the process of stem cells getting from the circulation to where they engraft in the bone marrow. This is particularly true in the developmental move from fetal liver to bone marrow.
However, the role for CXCR4 in adults appears to be more related to retention of stem cells in the bone marrow rather than the process of getting them there. Interrupting that retention process through either specific molecular blockers of the CXCR4/CXCL12 interaction, cleavage of CXCL12, or downregulation of the CXCR4 receptor can all result in the release of stem cells into the circulation. This process is an increasingly important aspect of recovering stem cells for therapeutic use as it has permitted the harvesting process to be done by leukapheresis rather than bone marrow punctures in the operating room. Granulocyte colony-stimulating factor and plerixafor, a macrocyclic compound that can block CXCR4, are both used clinically to mobilize marrow hematopoietic stem cells for transplant. Refining our knowledge of how stem cells get into and out of the bone marrow may improve our ability to obtain stem cells and make them more efficient at finding their way to the specific sites for blood cell production, the so-called stem cell niche.
HEMATOPOIETIC STEM CELL MICROENVIRONMENT
The concept of a specialized microenvironment, or stem cell niche, was first proposed to explain why cells derived from the bone marrow of one animal could be used in transplantation and again be found in the bone marrow of the recipient. This niche is more than just a housing site for stem cells, however. It is an anatomic location where regulatory signals are provided that allow the stem cells to thrive, to expand if needed, and to provide varying amounts of descendant daughter cells. In addition, unregulated growth of stem cells may be problematic based on their undifferentiated state and self-renewal capacity. Thus, the niche must also regulate the number of stem cells produced. In this manner, the niche has the dual function of serving as a site of nurture but imposing limits for stem cells: in effect, acting as both a nutritive and constraining home.
The niche for blood stem cells changes with each of the sites of blood production during development, but for most of human life it is located in the bone marrow. Within the bone marrow, the perivascular space particularly in regions of trabecular bone serves as a niche. The mesenchymal and endothelial cells of the marrow microvessels produce kit ligand and CXCL12, both known to be important for hematopoietic stem cells. Other cell types, such as sympathetic neurons, nonmyelinating Schwann cells, macrophages, osteoclasts, and osteoblasts, have been shown to regulate stem cells, but it is unclear whether their effects are direct or indirect. Extracellular matrix proteins like osteopontin also affect stem cell function. The endosteal region is particularly important for transplanted cells, suggesting that there may be distinctive features of that region that are yet to be defined that are important mediators of stem cell engraftment. The functioning of the niche as a supportive context for stem cells is of obvious importance for maintaining hematopoiesis and in transplantation. An active area of study involves determining whether the niche is altered in disease and whether drugs can modify niche function to improve transplantation or normal stem cell function in hematologic disease.
EXCESS CAPACITY OF HEMATOPOIETIC STEM CELLS
In the absence of disease, one never runs out of hematopoietic stem cells. Indeed, serial transplantation studies in mice suggest that sufficient stem cells are present to reconstitute several animals in succession, with each animal having normal blood cell production. The fact that allogeneic stem cell transplant recipients also never run out of blood cells in their life span, which can extend for decades, argues that even the limiting numbers of stem cells provided to them are sufficient. How stem cells respond to different conditions to increase or decrease their mature cell production remains poorly understood. Clearly, negative feedback mechanisms affect the level of production of most of the cells, leading to the normal tightly regulated blood cell counts. However, many of the regulatory mechanisms that govern production of more mature progenitor cells do not apply or apply differently to stem cells. Similarly, most of the molecules shown to be able to change the size of the stem cell pool have little effect on more mature blood cells. For example, the growth factor erythropoietin, which stimulates red blood cell production from more mature precursor cells, has no effect on stem cells. Similarly, granulocyte colony-stimulating factor drives the rapid proliferation of granulocyte precursors but has little or no effect on the cell cycling of stem cells. Rather, it changes the location of stem cells by indirect means, altering molecules such as CXCL12 that tether stem cells to their niche. Molecules shown to be important for altering the proliferation, self-renewal, or survival of stem cells, such as cyclin-dependent kinase inhibitors, transcription factors like Bmi-1, or microRNA-processing enzymes like Dicer, have little or different effects on progenitor cells. Hematopoietic stem cells have governing mechanisms that are distinct from the cells they generate.
HEMATOPOIETIC STEM CELL DIFFERENTIATION
Hematopoietic stem cells sit at the base of a branching hierarchy of cells culminating in the many mature cell types that compose the blood and immune system (Fig. 89e-2). The maturation steps leading to terminally differentiated and functional blood cells take place both as a consequence of intrinsic changes in gene expression and niche-directed and cytokine-directed changes in the cells. Our knowledge of the details remains incomplete. As stem cells mature to progenitors, precursors, and, finally, mature effector cells, they undergo a series of functional changes. These include the obvious acquisition of functions defining mature blood cells, such as phagocytic capacity or hemoglobin synthesis. They also include the progressive loss of plasticity (i.e., the ability to become other cell types). For example, the myeloid progenitor can make all cells in the myeloid series but none in the lymphoid series. As common myeloid progenitors mature, they become precursors for either monocytes and granulocytes or erythrocytes and megakaryocytes, but not both. Some amount of reversibility of this process may exist early in the differentiation cascade, but that is lost beyond a distinct stage in normal physiologic conditions. With genetic interventions, however, blood cells, like other somatic cells, can be reprogrammed to become a variety of cell types.
FIGURE 89e-2 Hierarchy of hematopoietic differentiation. Stem cells are multipotent cells that are the source of all descendant cells and have the capacity to provide either long-term (measured in years) or short-term (measured in months) cell production. Progenitor cells have a more limited spectrum of cells they can produce and are generally a short-lived, highly proliferative population also known as transient amplifying cells. Precursor cells are cells committed to a single blood cell lineage but with a continued ability to proliferate; they do not have all the features of a fully mature cell. Mature cells are the terminally differentiated product of the differentiation process and are the effector cells of specific activities of the blood and immune system. Progress through the pathways is mediated by alterations in gene expression. The regulation of the differentiation by soluble factors and cell-cell communications within the bone marrow niche are still being defined. The transcription factors that characterize particular cell transitions are illustrated on the arrows; the soluble factors that contribute to the differentiation process are in blue. This picture is a simplification of the process. Active research is revealing multiple discrete cell types in the maturation of B cells and T cells and has identified cells that are biased toward one lineage or another (rather than uncommitted) in their differentiation. EPO, erythropoietin; RBC, red blood cell; SCF, stem cell factor; TPO, thrombopoietin.
As cells differentiate, they may also lose proliferative capacity (Fig. 89e-3). Mature granulocytes are incapable of proliferation and only increase in number by increased production from precursors. The exceptions to the rule are some resident macrophages, which appear capable of proliferation, and lymphoid cells. Lymphoid cells retain the capacity to proliferate but have linked their proliferation to the recognition of particular proteins or peptides by specific antigen receptors on their surface. Like many tissues with short-lived mature cells such as the skin and intestine, blood cell proliferation is largely accomplished by a more immature progenitor population. In general, cells within the highly proliferative progenitor cell compartment are also relatively short-lived, making their way through the differentiation process in a defined molecular program involving the sequential activation of particular sets of genes. For any particular cell type, the differentiation program is difficult to speed up. The time it takes for hematopoietic progenitors to become mature cells is ~10–14 days in humans, evident clinically by the interval between cytotoxic chemotherapy and blood count recovery in patients.
FIGURE 89e-3 Relative function of cells in the hematopoietic hierarchy. The boxes represent distinct functional features of cells in the myeloid (upper box) versus lymphoid (lower box) lineages.
Although hematopoietic stem cells are generally thought to have the capacity to form all cells of the blood, it is becoming clear that individual stem cells may not be equal in their differentiation potential. That is, some stem cells are “biased” to become mature cells of a particular type. In addition, the general concept of cells having a binary choice of lymphoid or myeloid differentiation is not entirely accurate. A cell population with limited myeloid (monocyte and granulocyte) and lymphoid potential is now added to the commitment steps stem cells may undergo.
SELF-RENEWAL
The hematopoietic stem cell must balance its three potential fates: apoptosis, self-renewal, and differentiation. The proliferation of cells is generally not associated with the ability to undergo a self-renewing division except among memory T and B cells and among stem cells. Self-renewal capacity gives way to differentiation as the only option after cell division when cells leave the stem cell compartment, until they have the opportunity to become memory lymphocytes. In addition to this self-renewing capacity, stem cells have an additional feature characterizing their proliferation machinery. Stem cells in many mature adult tissues may be heterogeneous with some being deeply quiescent, serving as a deep reserve, whereas others are more proliferative and replenish the short-lived progenitor population. In the hematopoietic system, stem cells are generally cytokine-resistant, remaining dormant even when cytokines drive bone marrow progenitors to proliferation rates measured in hours. Stem cells, in contrast, are thought to divide at far longer intervals, measured in months to years, for the most quiescent cells. This quiescence is difficult to overcome in vitro, limiting the ability to effectively expand human hematopoietic stem cells. The process may be controlled by particularly high levels of cyclin-dependent kinase inhibitors like p57 or CDKN1c that restrict entry of stem cells into the cell cycle, blocking the G1-S transition. Exogenous signals from the niche also appear to enforce quiescence, including the activation of the tyrosine kinase receptor Tie2 on stem cells by angiopoietin 1 on niche cells.
The regulation of stem cell proliferation also appears to change with age. In mice, the cyclin-dependent kinase inhibitor p16INK4a accumulates in stem cells in older animals and is associated with a change in five different stem cell functions, including cell cycling. Lowering expression of p16INK4a in older animals improves stem cell cycling and capacity to reconstitute hematopoiesis in adoptive hosts, making them similar to younger animals. Mature cell numbers are unaffected. Therefore, molecular events governing the specific functions of stem cells are being gradually made clear and offer the potential of new approaches to changing stem cell function for therapy. One critical stem cell function that remains poorly defined is the molecular regulation of self-renewal.
For medicine, self-renewal is perhaps the most important function of stem cells because it is critical in regulating the number of stem cells. Stem cell number is a key limiting parameter for both autologous and allogeneic stem cell transplantation. Were we to have the ability to use fewer stem cells or expand limited numbers of stem cells ex vivo, it might be possible to reduce the morbidity and expense of stem cell harvests and enable use of other stem cell sources. Specifically, umbilical cord blood is a rich source of stem cells. However, the volume of cord blood units is extremely small, and therefore, the total number of hematopoietic stem cells that can be obtained in any single cord blood unit is generally only sufficient to transplant an individual of <40 kg. This limitation restricts what would otherwise be an extremely promising source of stem cells. Two features of cord blood stem cells are particularly important. (1) They are derived from a diversity of individuals that far exceeds the adult donor pool and therefore can overcome the majority of immunologic cross-matching obstacles. (2) Cord blood stem cells have a large number of T cells associated with them, but (paradoxically) they appear to be associated with a lower incidence of graft-versus-host disease when compared with similarly mismatched stem cells from other sources. If stem cell expansion by self-renewal could be achieved, the number of cells available might be sufficient for use in larger adults. An alternative approach to this problem is to improve the efficiency of engraftment of donor stem cells. Graft engineering is exploring methods of adding cell components that may enhance engraftment. Furthermore, at least some data suggest that depletion of host NK (natural killer) cells may lower the number of stem cells necessary to reconstitute hematopoiesis.
Some limited understanding of self-renewal exists and, intriguingly, implicates gene products that are associated with the chromatin state, a high-order organization of chromosomal DNA that influences transcription. These include members of the polycomb family, a group of zinc finger–containing transcriptional regulators that interact with the chromatin structure, contributing to the accessibility of groups of genes for transcription. One member, Bmi-1, is important in enabling hematopoietic stem cell self-renewal through modification of cell cycle regulators such as the cyclin-dependent kinase inhibitors. In the absence of Bmi-1 or of the transcriptional regulator, Gfi-1, hematopoietic stem cells decline in number and function. In contrast, dysregulation of Bmi-1 has been associated with leukemia; it may promote leukemic stem cell self-renewal when it is overexpressed. Other transcription regulators have also been associated with self-renewal, particularly homeobox, or “hox,” genes. These transcription factors are named for their ability to govern large numbers of genes, including those determining body patterning in invertebrates. HoxB4 is capable of inducing extensive self-renewal of stem cells through its DNA-binding motif. Other members of the hox family of genes have been noted to affect normal stem cells, but they are also associated with leukemia. External signals that may influence the relative self-renewal versus differentiation outcomes of stem cell cycling include specific Wnt ligands. Intracellular signal transducing intermediates are also implicated in regulating self-renewal. They include PTEN, an inhibitor of the AKT pathway, and STAT5, both of which are downstream of activated growth factor receptors and necessary for normal stem cell functions including self-renewal, at least in mouse models. The connections between these molecules remain to be defined, and their role in physiologic regulation of stem cell self-renewal is still poorly understood.
CANCER IS SIMILAR TO AN ORGAN WITH SELF-RENEWING CAPACITY
[/not-level-membership-for-internal-medicine-category]
