CHAPTER 38 SPINE AND SPINAL CORD: DEVELOPMENTAL DISORDERS
Developmental disorders of the spine and spinal cord occur because of defects in the development and maturation of the nervous system. A basic understanding of embryology is therefore important in appreciating the nature and scope of these disorders. There are various ways of approaching and thinking about the developmental disorders:
As knowledge and understanding of these disorders continue to expand, the most reasonable approach is to integrate advances in genetic and etiological models within the traditional phenotypic clinicoanatomical framework. Developmental disorders can be classified anatomically, depending on whether they affect primarily the spinal cord and neural tissues or the spine and supporting structures (Table 38-1).
TABLE 38-1 Developmental Disorders That Can Affect Vertebrospinal Structures
| Disorders Affecting the Spine | Disorders Affecting the Spinal Cord and Neural Tissues | Vascular Malformations |
|---|---|---|
| A variety of vascular malformations can affect the spine and spinal cord | ||
| Scoliosis | Developmental cysts |
NORMAL DEVELOPMENT OF THE NERVOUS SYSTEM
The human embryonic period spans the first 8 postfertilization weeks and is classified on the basis of a morphological system. There are 23 distinct Carnegie stages, each stage covering a period of 2 to 3 days. This staging system facilitates an understanding of the timing and sequence of embryonic development. The fetal period spans the 9th week after fertilization to birth. Fetal age is thought of mainly as measurements, inasmuch as a similar morphological staging system is not available. The great majority of congenital malformations begin during the embryonic period.1–3
The neural tube is the embryonic structure that develops into the brain and spinal cord. Primary neurulation refers to the development of the neural tube from the neural plate. Complete development and closure of the neural tube occurs from days 17 to 30 after conception and spans Carnegie stages 8 to 12 in the embryonic period (Table 38-2). The caudal eminence is the continuation of the primitive streak and develops into the terminal portions of the notochord, somites, vertebrae, and hindgut. The neural cord arises from the caudal eminence and forms the caudal portion of the spinal cord. This process is secondary neurulation. The level of the junction between primary and secondary neurulation is at the level of the future second sacral vertebra. There is elongation of the previous neural tube together with formation of the lower sacral and coccygeal segments. This part of the development of the nervous system commences in stage 12 at 4 weeks and ends approximately at stage 20.2–6
TABLE 38-2 Important Stages in Primary Neurulation and the Formation and Closure of the Neural Tube
| Development Stage No. | Days in Embryonic Life | Nervous System Development |
|---|---|---|
| 8 | 17-19 | Development of the neural plate |
| 9 | 19-21 | Development of the neural folds |
| 10 | 22-23 | First steps in the fusion of the neural folds: There are two initial sites of fusion: fusion proceeds bidirectionally from the lower medullary rhombencephalic site and in a caudal direction from the higher prosencephalic site. The fusions terminate in two neuropores: the rostral and caudal neuropores. |
| 11 | 23-26 | Closure of the rostral neuropore |
| 12 | 26-30 | Closure of the caudal neuropore |
Based on a table from Lemire RJ: Neural tube defects. JAMA 1988; 259:558-562 Copyright © 1988 American Medical Association. All rights reserved; and on information from O’Rahilly R, Muller F: The two sites of fusion of the neural folds and the two neuropores in the human embryo. Teratology 2002; 65:162-170.
The vertebral bodies of the spine are generated from the mesenchymal cells. The process of gastrulation occurs at day 14 in embryonic life to generate the mesenchymal cells that will in turn develop into the head, cardiac, and the paraxial and lateral mesoderm. At 20 to 30 days in embryonic life, the paraxial mesoderm, in a process of somatogenesis, subdivides into spherical somite segments on each side of the spinal cord in a rostral-to-caudal direction. The somites first appear in stage 9; differentiation commences at stage 10. The somites mature and further subdivide into the sclerotome to form the vertebral bodies, the myotome to form the musculature, and the dermatome to form the dermis. The sclerotome undergoes a resegmentation process in which the caudal end of one somite links with the rostral end of the next somite to form a vertebral body.7–9
DEVELOPMENTAL DISORDERS AFFECTING THE SPINE
Klippel-Feil Anomaly
Epidemiology
The Klippel-Feil anomaly occurs in 1 per 40,000 to 1 per 42,000 births, with a female-to-male ratio of 3:2.8
Clinical Features
Individuals with Klippel-Feil anomaly may be asymptomatic. Clinical manifestations can range from pain, through orthopedic or neurological manifestations, to a more widespread malformation disorder. The initial description was of a classical triad of shortening of the neck, limited range of neck movement, and a low posterior hairline. Affected individuals may complain of cervical pain and present with cosmetic deformities. The Klippel-Feil anomaly may be associated with spinal instability. This occurs if there are unstable fusion patterns or if stenosis and arthritis develop at the interspaces between the fused joints. The associated axial and spine anomalies include cervical or fused ribs, cleft vertebrae or hemivertebrae, and kyphoscoliosis.8
The Klippel-Feil anomaly may be associated with other spinal developmental disorders, including syrinx, tethered cord, and split cord malformations (SCMs). Alternatively, the Klippel-Feil anomaly could be part of a widespread multisystem developmental disorder as well, in which other features include hearing deficits, congenital heart disease, and genitourinary manifestations.8
Etiology and Pathophysiology
The Klippel-Feil anomaly results from failure of normal segmentation of the vertebral column in early embryonic life. The exact cause and mechanism behind this syndrome are unclear; the Klippel-Feil anomaly may result from disruptions in isolated genes that govern segmentation, or it may result from complex gene-gene or gene-environment interactions.8
Management
Diagnostic investigations center on imaging studies. Good-quality radiographs and computed tomographic (CT) scans of the cervical spine are necessary to show the extent of cervical fusion and to evaluate potential instability (Fig. 38-1). Magnetic resonance imaging (MRI) studies, including flexion and extension views, are useful for evaluating instability or stenosis in the cervical spine and for assessing neurocompression of the spinal cord. MRI should be performed on the whole neuraxis in order to look for the associated developmental anomalies. Further evaluation is targeted at the associated conditions, including audiology assessments and cardiac and renal investigations.8
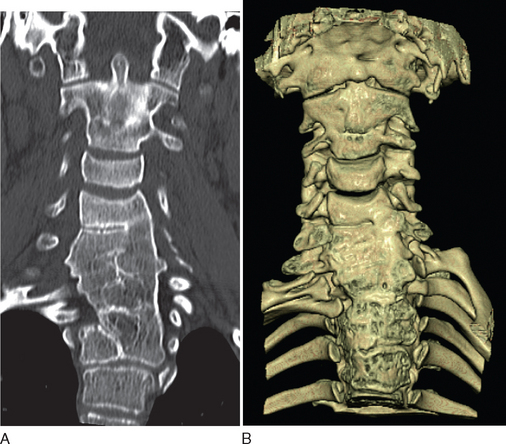
Management depends on the symptoms and the presence of other malformations. Treatment modalities may include simple measures of modifying activities and the use of traction and bracing to alleviate deformities. Surgical intervention to stabilize the spine is indicated if there is neurological impairment or symptomatic cervical instability. Surgical options include occipitocervical arthrodesis and combined atlantoaxial and subaxial arthrodesis.8
Basilar Impression
Definition
Basilar impression (or basilar invagination) occurs when there is an abnormal upward displacement of the basilar and condylar portions of the occipital bone. This leads to invagination of the foramen magnum into the posterior cranial fossa with associated translocation of the upper cervical vertebrae into this depression. We believe the terms basilar impression and basilar invagination are interchangeable, but some authors differentiate basilar impression from basilar invagination on the basis of differences in anatomical definitions and causation (e.g., acquired or primary).10,11 Some texts include platybasia in the spectrum of basilar impression. Platybasia is an abnormal flattening of the base of the skull with an abnormally obtuse angle between the plane of the clivus and the plane of the anterior fossa. This may occur together with basilar impression but is of no real clinical significance.
For instance, if Chamberlain’s line is the defining parameter, basilar impression is present when more than one third of the odontoid process lies above this line.10,12
Epidemiology
Basilar impression is the most common of the craniocervical malformations; clinical manifestations usually occur from the second decade onward.11 The true incidence is unclear.
Clinical Features
Basilar impression may be asymptomatic or may manifest with deformities, including a shortened neck. Movement or exercise-induced occipital headache is a common feature. Oculomotor features include horizontal and upward-beat or downward-beat nystagmus. Other cranial nerve symptoms include facial nerve spasm, trigeminal sensory symptoms, and trigeminal neuralgia. Pontomedullary compression can lead to pyramidal tract signs, with the lower limbs typically more affected than the upper limbs. Sleep apnea and respiratory depression have been described.11,12
Primary basilar impression is often associated with other developmental anomalies, including occipitalization of the atlas, the Klippel-Feil anomaly, the Arnold-Chiari malformation, and syringomyelia or syringobulbia.12
Etiology and Pathophysiology
Management
The treatment goal is to relieve current compression, and to prevent future compression, of the neuraxis. The treatment modalities for symptomatic basilar impression include cervical traction and surgical intervention. Cervical traction has been reported to cause vertebral artery dissection, as well as potentially causing infection, cranial fractures, and hemorrhage. Anterior compression is managed with an anterior surgical approach; posterior or circumferential compression is managed with a posterior approach. The role of prophylactic surgery or prolonged immobilization with asymptomatic lesions is unclear.10,12
Occipitalization of the Atlas
Clinical Features
This condition may be asymptomatic. However, atlantoaxial dislocation can occur in up to 60% of affected patients. In this condition, there is excessive compensatory stress on the atlantoaxial joint as a result of the decreased movement of the atlantooccipital joint. This situation increases the possibility of developing traumatic dislocation or cervical degenerative joint disease. Occipitalization of the atlas is associated with other developmental disorders as well, including basilar impression, Klippel-Feil anomaly, and Chiari malformations.13
Atlantoaxial Dislocation
Definition
Epidemiology
The various degrees of C1 malformations have a quoted prevalence rate of 4%, which is based on autopsy studies.14
Clinical Features
Abnormalities in this region may be asymptomatic and found incidentally, or they may manifest with pain. The vertebral anomalies may lead to atlantoaxial instability and dislocation. This may result in cervicomedullary compression, which may manifest either spontaneously or after trauma. Affected individuals are especially prone to cord compression during extension injuries: for instance, with intubation or after neck injuries. These anomalies should be considered when a child presents with a neurological deficit or neck pain after minor trauma.14,15
Congenital atlantoaxial instability may be seen in association with Down’s syndrome. Other congenital craniovertebral junction and spinal disorders may occur in tandem as well, including Chiari type I and SCMs. It is important to be aware that atlantoaxial instability may not be congenital in nature but may be caused by an underlying condition, such as rheumatoid arthritis.14,15
Etiology and Pathophysiology
The craniovertebral junction malformations arise from defects during the ossification process. The malformations lead to instability in the atlantoaxial joint with subsequent dislocation. Subluxation usually occurs in the horizontal plane, with C1 moving anteriorly to C2. In other settings, progressive cervical myelopathy rarely occurs above C2; the usual scenario of a myelopathy is in the context of acquired degenerative change and usually occurs below C3. The risk of neurocompression depends on the underlying malformation and the joint stability. The actual degree of neurocompression depends on the extent of the subluxation and the width of the spinal canal. The normal sagittal diameter of the spinal canal is 16 to 25 mm at the level of the atlas; the spinal cord is at risk when the canal diameter is less than 14 mm.14
Management
The use of high-quality imaging with plain radiographs, CT scans (including fine cuts), and MRI is necessary in the evaluation of atlantoaxial dislocation and craniocervical anomalies. Flexion and extension views are especially useful for assessing joint stability. MRI is crucial in the assessment of the neural structures, especially with regard to cord compression. Surgery is indicated in order to decrease the possibility that significant neurological deficits may develop and should be considered when there is neurocompression or when the individual is symptomatic. Surgery for atlantoaxial and craniocervical instability is a challenging field, especially because surgeons have to work with abnormal anatomy while taking into account the ongoing growth potential for the child. Depending on the underlying malformation, surgical management consists of decompression procedures and joint fusion and stabilization.14–17
Scoliosis
Epidemiology
Idiopathic scoliosis affects 2% to 4% of children aged 10 to 16 years. The incidence is equal in boys and girls for curves of less than 20 degrees. Girls, however, significantly outnumber boys, at 5:1, for curves of more than 20 degrees. Secondary neurogenic scoliosis is more common in the younger population. For instance, of patients with the Chiari malformation and syringomyelia, 82% younger than 20 years have scoliosis, as opposed to only 16% of those older than 20 years.18,19
Clinical Features
Individuals with scoliosis may present with back pain or, more obviously, with the spinal curvature deformity. In evaluating scoliosis, it is imperative to be aware that the spinal deformity may be the presenting feature of an underlying spinal cord or brainstem anomaly in 4% to 58% of affected patients. Individuals with scoliosis may present with the neurological or orthopedic features of the underlying developmental disorder.18,19
Etiology and Pathophysiology
Secondary scoliosis is thought to arise from a generalized paresis of truncal musculature or congenital changes in the vertebrae. In addition, scoliosis in the Chiari-syringomyelia complex may be caused by the abnormal intramedullary pressures within the spinal cord that interfere with the postural tonic reflexes.18,19
Management
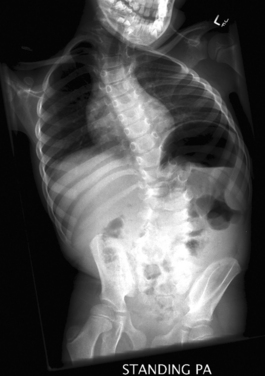
It is important to treat or rule out an underlying spinal cord or brainstem disorder in a young patient with scoliosis. Evaluation includes the use of radiographs (Fig. 38-2) to chart the pattern of scoliosis and MRI to look for associated anomalies in the neural elements. Posterior fossa decompression for the Chiari-syringomyelia malformation can lead to an improvement in the scoliosis, especially if done early in the patient’s life. Initial treatment may involve use of a spinal orthotic brace as well. Spinal fusion surgery may ultimately be required for surgical correction of the scoliosis.18,19
DEVELOPMENTAL DISORDERS AFFECTING THE SPINAL CORD AND NEURAL TISSUES
Chiari Malformation
Definition
In 1891, Hans Chiari first described the disorder that now bears his name as cerebellar tonsillar herniation below the plane of the foramen magnum into the spinal canal. Further contributions and observations were made by Julius Arnold and John Cleland. Chiari malformations are also known as cerebellar ectopy and are classified into four types:
Epidemiology
Chiari type I malformations have a prevalence of 0.6% to 0.9% in studies based on MRI. Symptomatic Chiari type I malformations are slightly more common in girls and women, with a female-to-male ratio of 1.3:1 to 1.7:1.22,23
Clinical Features
As with the other developmental disorders, the Chiari malformation may be asymptomatic or, alternatively, may be symptomatic with a wide spectrum of clinical manifestations. A very common manifestation is headaches with or without cervical pain. The headache is typically a protracted occipital-suboccipital headache and is exacerbated by the Valsalva maneuver, postural changes, and coughing or straining. Another common complaint is of weakness and altered sensation, including paresthesia and dysesthesias. A myelopathy may manifest with the sensorimotor or sphincter disturbances. Individuals with Chiari malformations may present with ataxia and other cerebellar signs. Downward-beating nystagmus and other oculomotor disturbances may occur. Other features of brainstem dysfunction may result, including cranial neuropathies, neuro-otological symptoms, sleep apnea, and dysphagia.21–24
Syringomyelia occurs in 32% to 74% of individuals with Chiari type I malformations. A cervical syrinx may manifest with upper limb neurological deficits, whereas a thoracic syrinx may lead to scoliosis. In addition, osseous anomalies of the base of skull, including basilar impression, may be seen with Chiari type I malformations. Chiari type II malformations are often associated with an NTD, other brainstem deformities, and hydrocephalus. Features of raised intracranial pressure are commonly the clinical manifestations of the associated hydrocephalus. The Chiari type I malformation is usually asymptomatic in childhood and tends to manifest in the second to third decade of adulthood. The Chiari type II malformation, however, is usually evident in childhood.21,23,24
Etiology and Pathophysiology
The Chiari malformation probably arises from underdevelopment of the occipital enchondrium, which leads to an undeveloped occipital bone with a small and shallow posterior fossa. This in turn leads to overcrowding of the posterior fossa and a downward herniation of the brain. The occipital enchondrium originates from the occipital somite, which in turn is derived from paraxial mesoderm. If the occipital enchondrium is more severely affected, basilar impression may result as well. The degree of tonsillar herniation is thought to be correlated to some extent with the severity of symptoms.21,24
Management
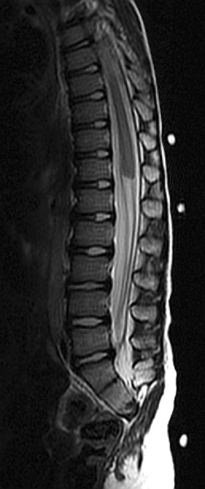
The main investigation for a suspected Chiari malformation is MRI of the neuraxis. Sagittal views are particularly important in confirming the diagnosis and demonstrating the degree of cerebellar and brainstem herniation and compression (Fig. 38-3). MRI is also useful for identifying the associated hydrocephalus or syrinx and identifying other developmental disorders, including NTDs, although these are usually obvious clinically.
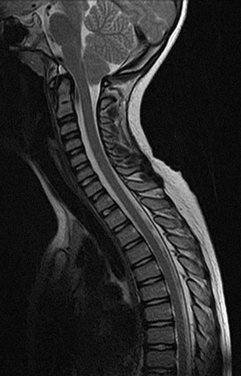
If there are neurological symptoms or signs, surgical intervention is usually indicated, to stabilize or ameliorate symptoms. The goal of surgery is to relieve brainstem and cerebellar compression. Ventral compression can be surgically treated with a transoral clivus odontoid resection. If there is no ventral compression, posterior fossa decompression is usually undertaken. A ventriculoperitoneal shunt may be inserted for associated hydrocephalus. The syrinx usually improves after recovery of cerebrospinal fluid (CSF) flow dynamics. If indicated, especially if the syrinx persists despite decompression, a shunt procedure for the syrinx can be undertaken as well.18,19
Syrinx
Clinical Features
A syrinx may occur in isolation or may be associated with an abnormality in the craniocervical junction. A common association is with the Chiari type I malformation. The level of the syrinx is variable and may in fact be several levels below the foramen magnum. Syringes may also be secondary to either trauma or focal spinal cord pathology, including tumors.25
The clinical picture in syringomyelia can be variable. The typical description is of a dissociated suspended sensory loss of compromised pain and temperature sensation with intact vibration and joint position sense, in a capelike distribution over the arms and trunk. Associated motor features include myelopathy, leading to spasticity and weakness with lower motor neurone signs at the level of the lesion and upper motor neuron signs below the level of the syrinx. A syrinx can manifest with pain, including radicular pain, neck pain, and headaches. Involuntary movements were characterized in 22% of patients with syringomyelia and syringobulbia, including torticollis, myoclonus, tremors, dystonia, myokymia, respiratory synkinesis, and inverse masticatory muscle activity.26,27
Etiology and Pathophysiology
The source of fluid in syringomyelia has been postulated to be CSF itself. The exact mechanism for the formation of syringomyelia in a Chiari malformation is unclear. There are two initial principal theories that rely on a patent communication between the fourth ventricle and the syrinx: According to the “water-hammer” theory, the pulsatile transmission of CSF from the choroid plexus is transmitted via an abnormal fourth ventricle to hammer out a dilatation within the spinal cord. The abnormal communication is considered to result from a delayed and incomplete embryonic opening of the outlet of the fourth ventricle. According to the “one-way-valve” theory, there are intermittent high pressures generated in the spinal cord, as a result of an uneven pressure gradient during the Valsalva maneuver. During the Valsalva maneuver, high intrathoracic pressures are transmitted to the epidural spinal veins, leading to an ascending pressure wave pushing CSF from the spinal column to the cranial subarachnoid space. Because there is a downward obstruction to the flow of CSF, this pressure is then translated into CSF movement from the fourth ventricle through the patent central canal into the syrinx. Investigators have reexamined the mechanism of syringomyelia formation, in which magnetic resonance images did not demonstrate a patent communication between the fourth ventricle and syrinx. Instead, it was postulated that during systole, the brain expands with blood. This imparts a systolic pressure wave that moves the cerebellar tonsils downward and acts on the spinal CSF along the surface of the cord to compress the cord and propel fluid in the syrinx longitudinally with each pulse.18,19,25,28
Management
MRI is crucial in the evaluation of a syrinx because it shows the syrinx itself and the associated neurological disorders, including malformations at the craniocervical junction (Fig. 38-4).27
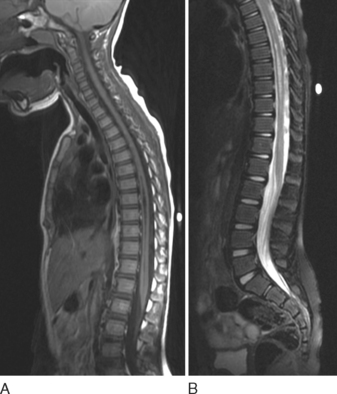
Surgical management consists mainly of either drainage or posterior fossa decompression. Traditionally, surgical intervention involved drainage with syringostomy and aspiration of the intramedullary fluid. There was later a paradigm shift with foramen magnum decompression, with or without plugging of the obex. More recently, there has been increasing interest in drainage procedures again, mainly because of the advent of microsurgical techniques and better shunt materials. Currently, surgery is indicated for a symptomatic syrinx and can be done either with posterior fossa decompression or as a shunt procedure. Shunts inserted in a syringosubarachnoid, syringopleural, or syringoperitoneal position are potentially associated with many complications, including shunt obstruction, dislocation or infection, spinal cord tethering, and septation within the syrinx cavity.27,29
Neural Tube Defects
Definition
NTDs result either from a failure of fusion of the neural tube in early postconceptional life or from a reopening of a previously closed neural tube.3,6 NTDs can be classified according to the following:


 Anencephaly consists of partial or total absence of the brain, with the undeveloped brain exposed to the surface through a defect in the skull and scalp.
Anencephaly consists of partial or total absence of the brain, with the undeveloped brain exposed to the surface through a defect in the skull and scalp. Encephalocele or cranial meningocele is an unfused midline defect in the skull that results in a closed lesion with eventration of the brain tissue and its coverings (encephalocele) or filled with only cerebrospinal fluid (cranial meningocele). These defects are classified by location into occipital, parietal, and anterior lesions.3,6
Encephalocele or cranial meningocele is an unfused midline defect in the skull that results in a closed lesion with eventration of the brain tissue and its coverings (encephalocele) or filled with only cerebrospinal fluid (cranial meningocele). These defects are classified by location into occipital, parietal, and anterior lesions.3,6Within the spine and spinal cord itself, spina bifida can span a wide range of anomalies, from mild defects with absent spinous processes to the huge defects seen with a myelomeningocele. There are two major working classification systems for spina bifida. Spina bifida can be classified depending on the pattern of the defect (Fig. 38-5):
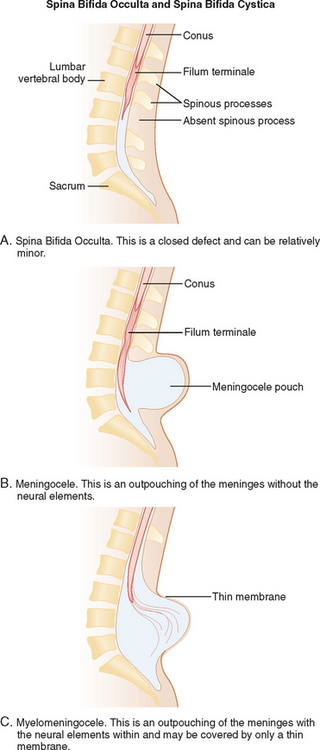
The other classification system categorizes spina bifida as follows:


Epidemiology
In the United States, NTDs occur in 1 per 1000 pregnancies. Rates of NTDs are higher in Mexico and northern China. NTDs are more common in girls than in boys.5,6
Clinical Features
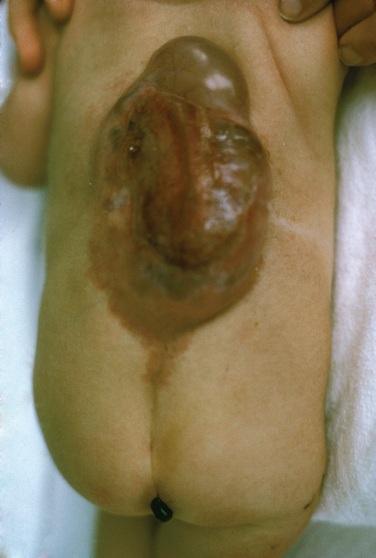
It is important to recognize the cutaneous manifestations of a closed NTD in the caudal regions. These can include lipomas, epidermoid sinus, dermoid cysts, hemangiomas, cutaneous nevi, and a deep sacral dimple. Clinicians must maintain a high index of suspicion when they see these cutaneous lesions and not dismiss them merely as isolated benign lesions.3
Urological abnormalities are common; most affected individuals lack sphincter control, and 80% have a neurogenic bladder. Although mainly in the lumbosacral region, spina bifida can also affect the thoracic region and, in rare cases, the cervical region. A lesion affecting the thoracic level significantly compromises lower limb strength.5,31
Orthopedic abnormalities, including clubfoot, contractures, and dislocated hips, can occur as well. Of all children with spina bifida, 90% have spinal curvature problems as well, including kyphosis, scoliosis, and kyphoscoliosis. Community ambulation is defined as the ability to move about the community without using a wheelchair. This can be achieved by 100% of patients with sacral and lower lumbar lesions and by 63% of patients with higher-level lesions. Other than the neurological level of the lesion and musculoskeletal deformities, other important factors influencing mobility and ambulation include balance disturbances, spasticity in the knees and hips, and the number of shunt revisions made.32,33
A host of late complications can occur in spina bifida, either as a consequence of the disorder and the deficits or as a consequence of the surgical intervention. The extent and severity of the neurological deficit itself can lead to severe medical complications as well. For instance, a neurogenic bladder can lead to recurrent urinary tract infections and chronic renal dysfunction. Severe sensorimotor deficits can lead to decubitus pressure sores and ulcers and other consequences of reduced mobility. Other late neurological complications include progressive hydrocephalus and the Chiari type II malformation, spinal cord tethering, and hydromyelia. Of individuals with myelomeningocele, 75% have an associated Chiari type II malformation that can be identified radiologically. Up to a third have clinically symptomatic lesions with manifestations, including sleep apnea, swallowing difficulties, stridor, headache, quadriparesis, scoliosis, and balance and coordination difficulties. Hydromyelia occurs in approximately 40% of individuals with myelomeningocele and manifests as progressive scoliosis, weakness of the upper limbs, spasticity, and an ascending motor loss in the lower limbs. Meningitis and serious nervous system infections cause significant morbidity as well.3,5,32
Individuals with spina bifida have a normal overall IQ score. Despite this, they have selective cognitive disabilities, including distractibility, a short attention span, and difficulties with perceptual organization and visual-motor integration. Practitioners involved in the care of individuals with spina bifida must monitor for the psychosocial complications of having a chronic disorder that is potentially disabling and deforming. All these factors have a significant effect on an individual’s ability to participate in education and employment opportunities and to engage in independent living.31
Prognosis
Spina bifida is a complex developmental disorder that is associated with relatively longer survival. Approximately 78% of all individuals with spina bifida survive to 17 years, and approximately 52% to 68% with spina bifida survive to the third decade. Survival depends significantly on the availability and delivery of optimal medical and surgical care. For instance, the early infant mortality rate in spina bifida is 10% in the United States and nearly 100% in northern China. The prognosis in spina bifida is relatively good, in contrast to anencephaly (affected children either are stillborn or die shortly after birth) and craniorachischisis (most affected fetuses are lost in early spontaneous abortion, or infants die after birth).3,5,6
The overall long-term prognosis in spina bifida, however, is not good in that there is significant morbidity associated with the condition. Survivors of spina bifida can have a multitude of problems ranging from the neurological deficits to the medical complications to the emotional and psychological scars. There is a significant burden of care on the community as well. The average monetary cost of medical care over a lifetime for an infant born in 1988 with spina bifida was $294,000 (in 1992 monetary terms).6
Etiology and Pathophysiology
The causes of spina bifida are heterogenous and consist of genetic and environmental factors. Spina bifida is likely to be the culmination of an interplay of genetic susceptibility and environmental factors. The combination of environmental and genetic triggers for spina bifida is supported by the geographical variation in the incidence rates.5,6
A genetic basis for NTDs is suggested by the racial differences in incidence and by the familial recurrence patterns. For instance, NTDs are more common in Hispanic and non-Hispanic white persons than in black persons in the United States. In addition, there is a degree of disease aggregation in families. The siblings of affected individuals have a 3% to 8% chance of having spina bifida, anencephaly, or both. There is also an increased risk of spina bifida in second- and third-degree relatives of affected individuals.5,6
The exact etiological factors remain unknown for the majority of cases of spina bifida. In a minority, there are recognized syndromes causing the spina bifida. Of all children with NTDs, 20% have additional congenital malformations. Genetic abnormalities, including chromosomal abnormalities and single gene mutations, and teratogenic causes are identified in 10% of affected children.5,6
Of all the environmental factors studied, inadequate maternal intake of either natural folate or synthetic folic acid before conception and during the early stages of pregnancy has the most robust association with the development of NTDs. The exact mechanism of protection from folic acid is unclear but is likely to be mediated by genes that regulate folate transport and metabolism. Various genetic defects in the folate-homocysteine metabolism and folate transport have been identified as being associated with NTDs.5
Only a few specific environmental factors, including maternal diabetes and maternal use of anticonvulsants, have been identified in the causation of NTDs. A history of pregestational diabetes in women is associated with a twofold to tenfold increase in the risk of malformations of the central nervous system, including spina bifida, in their children. The risk is related to the level of maternal metabolic control. Use of valproic acid or carbamazepine by women is associated with a 1% to 2% risk of spina bifida in their children. Other factors shown to lead to an increased risk of NTDs include maternal obesity and maternal hyperthermia.5,6
Prevention
Maternal folic acid supplementation before conception and during early pregnancy has been shown to prevent at least half the cases of NTDs and up to 70% of the cases of spina bifida. In 1991, a British study demonstrated that recurrent NTDs could be reduced by taking 4000 μg/day of folic acid. This was followed in 1992 by a Hungarian study that demonstrated that 800 μg/day of folic acid reduced the first occurrence of a neural tube defect. In a later (1999) study, a daily dose of 400 μg/day of folic acid was sufficient to prevent NTDs in an area of China with a high incidence of NTDs.5,6
The recommended dose is 400 μg/day of folic acid in women of childbearing age, especially if they are contemplating or planning a pregnancy. This is most consistently achieved through nutritional supplements. Other sources of folic acid include foods fortified with folic acid, including cereals and grains, and foods naturally rich in folate, including fruits and vegetables. There has been some concern that universal fortification of common foods may mask vitamin B12 deficiency.5,6
Management
There are four phases in the management of spina bifida, described as follows.
Prenatal diagnosis and management considerations
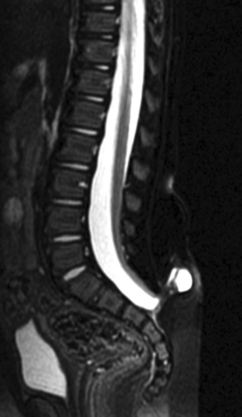
Prenatal management centers on diagnosis of the NTD and discussions about subsequent management. NTDs can be screened with maternal serum α-fetoprotein measurements and ultrasonography. The maternal serum α-fetoprotein level can be tested at the 16th week of pregnancy. False-positive results can arise from other malformations, the presence of multiple fetuses, and incorrect calculation of gestation dates. Ultrasonography is useful in diagnosing the NTD and in providing useful information about the overall growth of the fetus. It may, however, miss small myelomeningoceles. Once an NTD is diagnosed, ultrasonography is used to assess for other associated malformations and general limb movement, deformity, and paralysis. The use of fetal MRI is increasingly studied in this area as well (Fig. 38-8). Such prenatal imaging may have a bearing in predicting neurological deficit and functional outcome.3,5
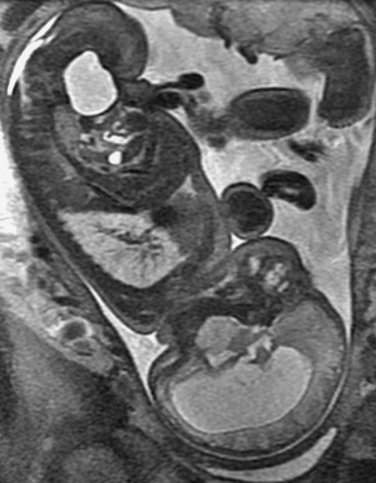
Amniocentesis is a more invasive investigation that can be used to follow up positive screening test results. The α-fetoprotein levels in the amniotic fluid at the 15th or 16th week of gestation are elevated in cases of an open NTD. Similarly, acetylcholinesterase levels are also elevated in cases of an open NTD. Fetal karyotype studies can be undertaken as well to investigate for chromosomal anomalies.3,5
Damage to the neural tissue can occur either in utero, as a result of exposure to toxic substances in the amniotic fluid or mechanical damage from contact with the uterine wall, or during delivery, as a result of contact with the birth canal. Despite this, there is currently insufficient medical evidence to support any particular mode of delivery in minimizing the neurological impairments from spina bifida. The elective use of cesarean section, however, may be warranted to minimize trauma if there is a large lesion and to reduce the risk of dehiscence after in utero surgical treatment.5,6
Perinatal surgical considerations
The next stage in management involves exploring the various surgical and therapeutic treatment options available. There is the choice of either in utero fetal surgery or the more traditional postnatal surgery. Postnatal surgical closure of a spinal lesion is usually undertaken within the first 48 hours after birth. If there is associated hydrocephalus, a ventricular shunt is usually inserted as well.5
There have been various studies exploring the role of in utero surgical repair of NTDs in fetuses as young as 22 weeks of gestational age. Studies have shown complete reversal of hindbrain herniation and a lower proportion of cases of hydrocephalus necessitating shunting (43% versus 85%). Initial reports have shown resolution of the associated Chiari type II malformations and a lower incidence of moderate to severe hindbrain herniation and hydrocephalus necessitating shunting. The urodynamic and leg function, however, remain similar in comparison with that in infants who are treated postnatally. The exact role of in utero surgery is unclear, especially with regard to the overall fetal and maternal risk-benefit ratio.5,6
Early childhood management
Assessing and optimizing ambulation and mobility are of prime importance in the management of spina bifida. The orthopedic deformities can be treated with early surgical intervention. If left untreated, orthopedic deformities, including scoliosis, kyphosis, and hip dysplasias, can compromise mobility and functional outcomes. Supportive aids, including braces, canes, and walkers, can be used to maintain erect posture to achieve effective community ambulation. Wheelchair assessment is also important, especially if the combination of deficits weigh against successful independent ambulation.5,6
The potential complications must be considered and sought. For instance, the medical team should watch for progressive hydrocephalus with baseline imaging studies and serial head circumference measurements. Children who have had shunt surgery need to be evaluated on a regular basis for potential markers of shunt malfunction, including the development of seizures. Urodynamic studies and renal tract ultrasonography are important in assessing urological function.5
Growth is affected in spina bifida. Growth hormone has been successfully used in improving the growth of children with spina bifida. The overall long-term outcomes, however, are unknown at this stage.6
Lifelong management and considerations
Approximately 47% of hospital admissions for spina bifida result from potentially preventable secondary complications, including recurrent urinary tract infections and skin ulceration.6 Careful urological monitoring and advice are important. This is especially so in providing techniques for complete bladder emptying and in ensuring adequate catheter management, balanced with judicious use of pharmacological agents and surgical intervention. Skin care is of prime importance, with strategies to minimize skin friction and to ensure early attention to skin irritation and ulcers. Approximately 43% of people with spina bifida have a latex allergy. Latex-containing products should be avoided in the care of patients with spina bifida.6
Tethered Cord Syndrome
Definition
All these causative conditions are sometimes grouped under the umbrella term spinal dysraphism, despite the clear structural and anatomical differences. The term refers broadly to this assorted group of congenital anomalies affecting the spine and spinal cord that have similarities in clinical manifestation and the consequent neurological deficits and deformities.34–36
Tethered cord syndrome (TCS) may also occur as a secondary condition as a result of surgical treatment for these disorders. For instance, the syndrome has been reported in 15% of patients with a previously repaired myelomeningocele, in whom surgical closure has led to posterior attachment of the neural placode at the site of repair.34,36
Clinical Features
There are four modes of presentation of TCS:
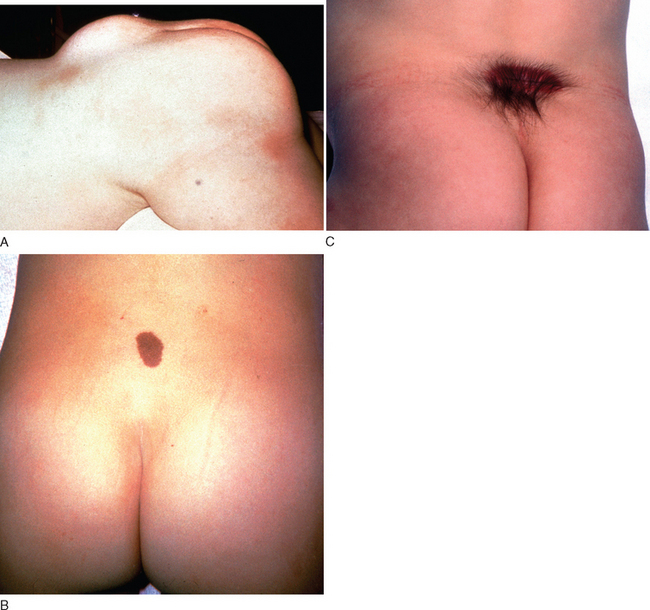
Spinal cord tethering may occur after surgery for spinal dysraphism; hence, neurological deterioration or progression must be carefully evaluated after surgery. A history of myelomeningocele repair in childhood is an important clue to underlying TCS if an adult presents with clinical features suggestive of the syndrome.34,36
Etiology and Pathophysiology
The spinal cord reaches the sacral-coccygeal region in early embryonic life. After 16 weeks of gestation, there is then a growth differential in which the bony vertebral column lengthens and grows beyond the spinal cord itself. This leads to ascension of the spinal cord to the level of L3 at 30 weeks of gestation and to the adult level of L1 or L2 by 2 months after birth.34,37
In embryonic life, the normal pattern of ascent is compromised if the conus is hinged to an abnormally low level by any of the underlying causative pathological entities responsible for TCS. In addition to the low-lying position, the neural tissues are abnormally stretched vertically. Studies have shown that, when placed under traction, the lumbosacral cord is vulnerable to hypoxic stress, with neuronal dysfunction resulting from mitochondrial oxidative derangement. Over time, neuronal dysfunction leads to neuronal damage. Other contributory factors include impairment of blood supply and problems with glucose metabolism. In essence, the TCS results from abnormal longitudinal traction that causes cord injury. There may be an additional factor with a compressive element from the underlying pathology:for instance, with a lipoma.34–36,38
With normal growth and development—for instance, during the growth spurts—there is further stretching of the spinal cord, causing increasing neuronal damage. This leads to the onset of neurological features in early childhood in individuals with more severe cord tethering. Alternatively, symptoms may emerge later in milder cases, either in the setting of aggravating features as discussed previously or over time after repeated normal day-to-day stretching of the spinal cord from head and neck flexion. The sudden tension may lead to irreversible neuronal damage in an already chronically tethered cord. The tension from the tethered spinal cord can be transmitted up the cord, with neural elements stretched at levels away from the site of the tethering. This is one of the factors explaining the combination of upper and lower motor neuron signs that may be seen.34,35,38
Management
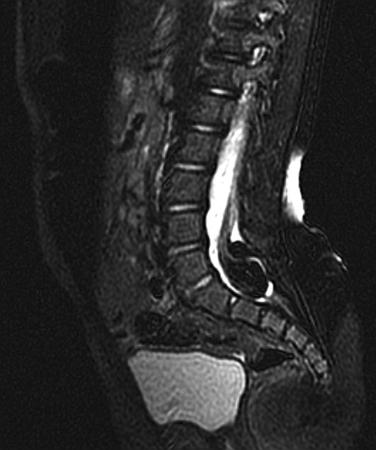
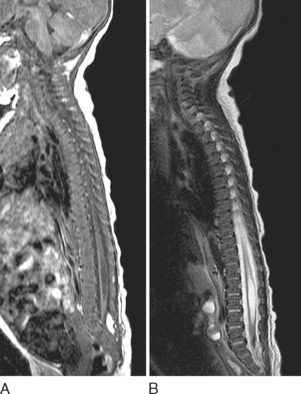
MRI is of critical importance in the evaluation of TCS. MRI is useful for assessing conus position and thickness of the filum terminale and in looking for associated vertebrospinal anomalies. The conus is low-lying when the tip is abnormally located below the lower border of the L2 vertebral body, and the filum is thickened if it appears wider than 1.5 to 2.0 mm on MRI. The other clues of TCS include posterior placement of the spinal cord, thickened filum terminale, and the sacral roots’ running in a lateral or cephalic course, as opposed to the usual cephalocaudal orientation. Plain radiographs and CT scans are useful in delineating the associated bony vertebral abnormalities. Although the advent of MRI has rendered most of the older procedures obsolete, including the use of hyperbaric and gas myelography, there is occasionally a role for myelography in further defining the nerve root anatomy and pathways.34,36
Evaluation of TCS is incomplete without urodynamic studies, because neurogenic bladder dysfunction may occur even in the absence of symptoms. It is therefore important to refer individuals with TCS for urodynamic assessment. It is especially useful to perform assessments both preoperatively and postoperatively.37
Surgical management is aimed at untethering the spinal cord and neural elements and leads to good neurological outcomes and improvement in symptoms. Compressive lesions, if present, should be removed as well. Improvement after untethering surgery is especially convincing if the deficits are mild or treated early. In an analysis of TCS in adults, 83% experienced relief of significant pain and 67% experienced an improvement in sensorimotor deficits. Improvement in sphincter disturbance, however, is more variable. In one study, for instance, only 39% of patients with sphincter disturbance experienced an improvement. There have been occasional reports as well of bladder dysfunction occurring after surgery. Although there are no differences in the clinical presentation for the different pathologies, there are differences in ease of surgical management, depending on causative etiology. Complex lipomas and adhesions from previous operations prove to be the most challenging. Despite this, the overall rate of complications from surgery is low, with reported immediate postoperative neurological morbidity rates of 0% to 5%. Postoperative CSF fistula is a possible complication that should be treated early with further surgery and dural repair.34,36,37
Together, all these factors positively support the role of surgical intervention. Early surgery is particularly crucial for preventing further neuronal damage. Surgery is recommended in infants upon diagnosis of a tethered cord, to prevent neurological deficits. Surgery is also advocated in later presentations with progressive neurological deterioration. Retethering may occur after surgery and must be differentiated from incomplete untethering during the initial operation. There is a range of operative options for retethered cords.34,36–38
Split Cord Malformations
Definition
The traditional terms used for double spinal cord malformations were diplomyelia, for a twinning and doubling of the cord within a single dural sac, and diastematomyelia, for two hemicords in separate dural sacs separated by a midline spur. These terms were derived from Greek words:myelos, meaning marrow or cord; diplous, meaning double; and diastema, meaning cleft. These malformations have been categorized as SCMs. They are rare disorders with complete or incomplete division of the spinal cord by an osseocartilaginous or fibrous septum, leading to double spinal cords. In type I SCM, there are two hemicords separated by a rigid dura-sheathed osseocartilaginous septum, each hemicord housed within its own dural tube. In type II SCM, the spinal cord sits within a single dural tube and is split into two independent segments for a variable vertical extent by a nonrigid fibrous median septum.39,40
Clinical Features
SCMs can be either asymptomatic or symptomatic. Affected children are more likely than affected adults to be asymptomatic. Both affected adults and children may present with cutaneous manifestations, the most common being hypertrichosis. Other cutaneous signs include capillary hemangiomas, a lipoma, and dermal sinus openings. SCMs may manifest with pain. This is a common feature in adulthood, and the pain may be localized to the spine at the site of the split cord, or it may be diffuse with a dysesthetic component. Neurological deficits, including progressive gait disturbance, worsening sensorimotor disturbance, and bladder or bowel dysfunction, may occur. SCMs most commonly affect the lumbar region; they also can occur in the cervical and thoracic regions. A cervical region SCM involves the upper limbs as well. Neurological decline is independent of SCM pattern and location type; neurological features occur equally in type I and type II SCMs, and progression rates are similar for cervical and thoracolumbar lesions. Trophic changes and nonhealing ulcers may eventuate in SCM. Urinary tract manifestations may include features of a neurogenic bladder, renal impairment, recurrent urinary tract infections, or incontinence. Urinary tract dysfunction, however, may be asymptomatic and must be carefully evaluated with urodynamic studies. Children can also present with orthopedic deformities, including scoliosis and foot deformities.30,40
SCMs may occur in conjunction with other vertebrospinal anomalies. The incidence of an coexisting myelomeningocele is 26% to 39% in individuals with SCM and is more common with type I SCM. The neural placode may involve both hemicords or only one of the hemicords as a hemimyelocele. Alternatively, involvement may be more limited with strands of neural tissue flowing from the hemicords to enter the myelomeningocele sac. SCMs may be associated with a low-lying conus and other tethering lesions, including tight filum terminale and lipomas. Abnormalities in the vertebral bodies adjacent to the SCM, including a bifid body, a widened body, and vertebral fusion, may be seen. Individuals with a cervical SCM may have an associated Klippel-Feil anomaly, with torticollis, a short neck, and webbed trapezii.30,40
Overall, the clinical manifestations are similar to those of TCS. There is one notable difference: Individuals with SCM may have a significant left-right differential with regard to lower limb neurology. This occurs either in the setting of a hemimyelocele, in which the side with the hemicord involved with the myelocele has much reduced function, or in the setting of an asymmetrical split within the spinal cord. In the latter situation, there is a large, major hemicord and a smaller, minor hemicord, with reduced function on the minor side.40
Epidemiology
There may be a slight female preponderance for SCM. The average age at diagnosis ranges from 3.9 to 5.7 years.30
Etiology and Pathophysiology
The exact cause of SCMs is unknown. Initially, diastematomyelia was thought to be a developmental disorder resulting from abnormal tissue protrusions splitting the spinal cord. Diplomyelia, on the other hand, was postulated to result from an abnormal twinning response in early embryonic development. Later work, however, has shown the two entities to be the same and suggests that SCMs arise from a common embryogenetic mechanism. In the early embryonic period, adhesions are formed between the ectoderm and endoderm. These adhesions develop into an accessory neurenteric canal, which subsequently condenses into the endomesenchymal tract. This abnormal tract splits the notochord and neural plate into two hemineural plates.39
Management
Investigations in SCMs are twofold. The first is to characterize the malformation, especially as part of an extensive presurgical workup. Plain radiographs are often unhelpful, although they occasionally show the bone abnormalities involved. CT scans and MRI have complementary roles. MRI is particularly valuable in demonstrating spinal cord anatomy and in defining the level and extent of the hemicord (Fig. 38-13). The number of vertebral bodies crossed is used to define the length of the split segment. In addition, it is important to perform MRI of the whole neuraxis to look for associated anomalies, including NTDs and lipomas. The high-resolution CT scan with fine-cut slices is especially useful in defining the spur characteristics and the bony vertebral body and posterior arches. The second role of investigations is to map out complications that may arise. Urodynamic studies are particularly important in this regard, especially if there are no overt renal tract features.40
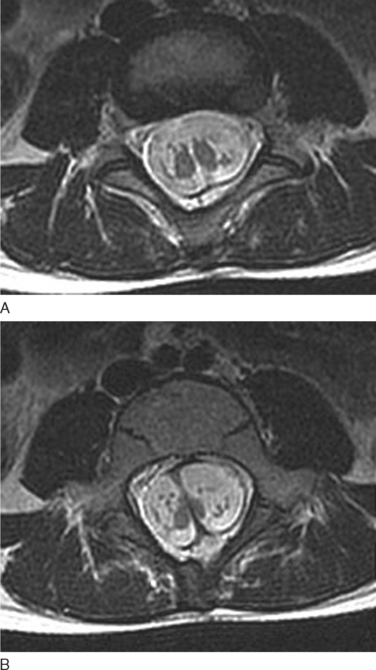
Most surgeons advocate prompt identification and early surgical intervention for SCMs to either improve outcomes or prevent further functional deterioration. The pain component responds the best to surgical management; improvement occurs within a month. With neurological deficits, surgery may improve symptoms or may prevent further progression. Neurological recovery is inversely proportional to the duration of symptoms before surgical intervention. Results with sphincter dysfunction, however, are generally less encouraging, with an overall 40% improvement rate. Surgery involves excision of the midline spur, together with the dural sheath encasing the spur and any associated soft tissue bands that may be attached to the hemicord. The surgical morbidity rate is slightly higher in type I SCM. Complications may include wound complications and CSF leakage. Coexisting anomalies can be surgically treated as well. Orthopedic deformities can occasionally be managed with appropriate operations, including tendon transfer and joint arthrodesis. Management of bladder dysfunction with monitoring of renal function and judicious use of catheterization is also important. There have been reports in the literature of SCMs’ being diagnosed prenatally with ultrasonography.40
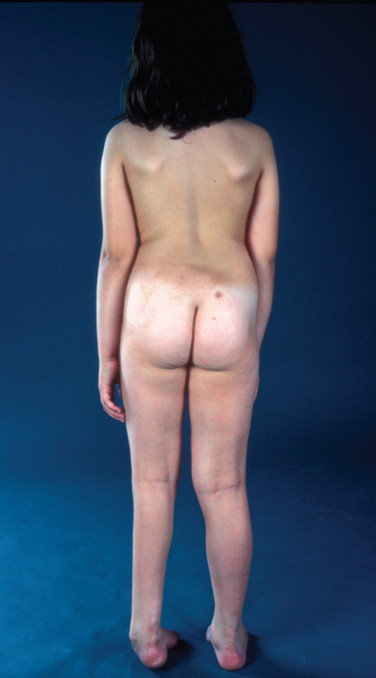
Sacral Agenesis
Definition
Some authors make a further distinction, with sacral agenesis referring to a symmetrical absence and sacral dysgenesis referring to an asymmetrical deformity. The syndrome of caudal regression encompasses a constellation of malformations in which multiple embryological defects of the caudal region occur in addition to sacral agenesis. The term caudal dysplasia refers to the anomalies of the spine and lower extremities that may be seen in infants of diabetic mothers.41–43
Epidemiology
Sacral agenesis is a rare congenital malformation. A maternal history of diabetes mellitus is strongly associated with sacral agenesis; 16% to 64% of individuals with caudal vertebral deformity or sacral agenesis have a diabetic mother.42,43
Clinical Features
The features of sacral agenesis depend on the severity and extent of the lesion. Minor defects in the sacrum and coccyx may be asymptomatic, or typical cutaneous stigmata (e.g., lipoma, nevus) may be exhibited. Affected individuals may complain of back pain or may present with orthopedic and musculoskeletal features, including lower limb deformities and contractures, scoliosis, spine instability, and hip dislocations. Complete sacral agenesis, especially if affecting the lumbar region as well, leads to completely thin and flaccid lower limbs. The neurological deficit tends to correlate with the level of the vertebral abnormality and is often associated with bladder or bowel dysfunction. Ambulatory capacity is correlated with the type of sacral agenesis.41–43
Two distinct subgroups have been identified. Individuals with high agenesis have high blunt conus termination and tend to present with the typical features of flat buttock cheeks and prominent iliac crests. Such morphological changes are less pronounced with low agenesis and a low-lying conus. The recurring theme of multiple developmental defects applies to sacral agenesis as well; other congenital vertebrospinal deformities, including syrinx, myelomeningocele, and lumbosacral lipoma, may be present.41–43
The features of the syndrome of caudal regression may be more variable and span a wider range of potential problems, including cleft lip or palate and gastrointestinal and genitourinary malformations. Gastrointestinal tract abnormalities include imperforate anus and malrotation of the bowel. Genitourinary malformations include a pelvic or solitary kidney, a urogenital sinus, a perineal urethra, labial abnormalities, and penile atrophy. A combination of orthopedic and neurological problems may lead to a “Buddha” position, with marked flexion and overlapping of the lower limbs and equinovarus. The most severe end of the spectrum includes the dramatic sirenomelia malformation, in which the “mermaid” fusion of the lower limbs is accompanied by extreme malformations of lower gastrointestinal and genitourinary tracts, including total agenesis of the kidneys, renal tract, anus, and rectum.41,43
Etiology and Pathophysiology
The exact cause of sacral agenesis is unclear but appears to be a defect either late in or after the neurulation stage of embryogenesis. This view is derived from the observation that sacral agenesis is a closed defect. An insult to the caudal eminence may be the cause of high agenesis, whereas a milder or later insult affecting differentiation may be the cause of the low agenesis.43
Management
As with the other vertebrospinal conditions, evaluation with MRI is necessary to delineate the neural structures and associated anomalies (Fig. 38-14). Bony definition may be improved on CT scanning. The primary aim of orthopedic intervention is to improve mobility or sitting and standing balance. This may be achieved through the use of orthotics and a range of surgical measures, including soft tissue releases, osteotomies, and open reduction of hip dislocations. Limb amputation has been undertaken for severe deformities that cannot be corrected or ameliorated with simpler operations. The role of neurosurgery is not as well defined, inasmuch as there are conflicting accounts of benefit in sacral agenesis. There have been reports favoring early preemptive neurosurgery for the low-lying variant.41,43
CONCLUSION
Botto LD, Moore CA, Khoury MJ, et al. Neural-tube defects. N Engl J Med. 1999;341:1509-1519.
Lemire RJ. Neural tube defects. JAMA. 1988;259:558-562.
Mitchell LE, Adzick NS, Melchionne J, et al. Spina bifida. Lancet. 2004;364:1885-1895.
Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
Pang D, Wilberger JEJr. Tethered cord syndrome in adults. J Neurosurg. 1982;57:32-47.
1 O’Rahilly R, Muller F. Prenatal ages and stages—measures and errors. Teratology. 2000;61:382-384.
2 O’Rahilly R, Muller F. Minireview: summary of the initial development of the human nervous system. Teratology. 1999;60:39-41.
3 Lemire RJ. Neural tube defects. JAMA. 1988;259:558-562.
4 O’Rahilly R, Muller F. The two sites of fusion of the neural folds and the two neuropores in the human embryo. Teratology. 2002;65:162-170.
5 Mitchell LE, Adzick NS, Melchionne J, et al. Spina bifida. Lancet. 2004;364:1885-1895.
6 Botto LD, Moore CA, Khoury MJ, et al. Neural-tube defects. N Engl J Med. 1999;341:1509-1519.
7 O’Rahilly R, Muller F. Somites, spinal ganglia, and centra. Enumeration and interrelationships in staged human embryos, and implications for neural tube defects. Cells Tissues Organs. 2003;173:75-92.
8 Tracy MR, Dormans JP, Kusumi K. Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res. 2004;424:183-190.
9 Muller F, O’Rahilly R. Segmentation in staged human embryos: the occipitocervical region revisited. J Anat. 2003;203:297-315.
10 Dickinson LD, Tuite GF, Colon GP, et al. Vertebral artery dissection related to basilar impression: case report. Neurosurgery. 1995;36:835-838.
11 Hayes M, Parker G, Ell J, et al. Basilar impression complicating osteogenesis imperfecta type IV: the clinical and neuroradiological findings in four cases. J Neurol Neurosurg Psychiatry. 1999;66:357-364.
12 Bhangoo RS, Crockard HA. Transmaxillary anterior decompressions in patients with severe basilar impression. Clin Orthop Relat Res. 1999;359:115-125.
13 Jain VK, Takayasu M, Singh S, et al. Occipital-axis posterior wiring and fusion for atlantoaxial dislocation associated with occipitalization of the atlas. Technical note. J Neurosurg. 1993;79:142-144.
14 May D, Jenny B, Faundez A. Cervical cord compression due to a hypoplastic atlas. Case report. J Neurosurg Spine. 2001;94:133-136.
15 Lowry DW, Pollack IF, Clyde B, et al. Upper cervical spine fusion in the pediatric population. J Neurosurg. 1997;87:671-676.
16 Dickman CA, Sonntag VK. Surgical management of atlantoaxial nonunions. J Neurosurg. 1995;83:248-253.
17 Gluf WM, Brockmeyer DL. Atlantoaxial transarticular screw fixation: a review of surgical indications, fusion rate, complications, and lessons learned in 67 pediatric patients. J Neurosurg Spine. 2005;2:164-169.
18 Eule JM, Erickson MA, O’Brien MF, et al. Chiari I malformation associated with syringomyelia and scoliosis: a twenty-year review of surgical and nonsurgical treatment in a pediatric population. Spine. 2002;27:1451-1455.
19 Muhonen MG, Menezes AH, Sawin PD, et al. Scoliosis in pediatric Chiari malformations without myelodysplasia. J Neurosurg. 1992;77:69-77.
20 Koehler PJ. Chiari’s description of cerebellar ectopy (1891). With a summary of Cleland’s and Arnold’s contributions and some early observations on neural-tube defects. J Neurosurg. 1991;75:823-826.
21 Nishikawa M, Sakamoto H, Hakuba A, et al. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40-47.
22 Meadows J, Kraut M, Guarnieri M, et al. Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000;92:920-926.
23 Wu YW, Chin CT, Chan KM, et al. Pediatric Chiari I malformations: do clinical and radiologic features correlate? Neurology. 1999;53:1271-1276.
24 Pascual J, Oterino A, Berciano J. Headache in type I Chiari malformation. Neurology. 1992;42:1519-1521.
25 Oldfield EH. Syringomyelia. J Neurosurg Spine. 2001;95:153-155.
26 Nogues MA, Leiguarda RC, Rivero AD, et al. Involuntary movements and abnormal spontaneous EMG activity in syringomyelia and syringobulbia. Neurology. 1999;52:823-834.
27 Vaquero J, Martinez R, Arias A. Syringomyelia-Chiari complex: magnetic resonance imaging and clinical evaluation of surgical treatment. J Neurosurg. 1990;73:64-68.
28 Oldfield EH, Muraszko K, Shawker TH, et al. Pathophysiology of syringomyelia associated with Chiari I malformation of the cerebellar tonsils. Implications for diagnosis and treatment. J Neurosurg. 1994;80:3-15.
29 Batzdorf U, Klekamp J, Johnson JP. A critical appraisal of syrinx cavity shunting procedures. J Neurosurg. 1998;89:382-388.
30 Kumar R, Bansal KK, Chhabra DK. Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg. 2002;36:119-127.
31 Simeonsson RJ, McMillen JS, Huntington GS. Secondary conditions in children with disabilities: spina bifida as a case example. Ment Retard Dev Disabil Res Rev. 2002;8:198-205.
32 Bartonek A, Saraste H. Factors influencing ambulation in myelomeningocele: a cross-sectional study. Dev Med Child Neurol. 2001;43:253-260.
33 McLone DG. Spina bifida today: problems adults face. Semin Neurol. 1989;9:169-175.
34 Pang D, Wilberger JEJr. Tethered cord syndrome in adults. J Neurosurg. 1982;57:32-47.
35 Yamada S, Zinke DE, Sanders D. Pathophysiology of “tethered cord syndrome.”. J Neurosurg. 1981;54:494-503.
36 Kirollos RW, Van Hille PT. Evaluation of surgery for the tethered cord syndrome using a new grading system. Br J Neurosurg. 1996;10:253-260.
37 Zoller G, Schoner W, Ringert RH. Pre- and postoperative urodynamic findings in children with tethered spinal cord syndrome. Eur Urol. 1991;19:139-141.
38 Yamada S, Iacono RP, Andrade T, et al. Pathophysiology of tethered cord syndrome. Neurosurg Clin North Am. 1995;6:311-323.
39 Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: part I: a unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-480.
40 Pang D. Split cord malformation: part II: clinical syndrome. Neurosurgery. 1992;31:481-500.
41 Guidera KJ, Raney E, Ogden JA, et al. Caudal regression: a review of seven cases, including the mermaid syndrome. J Pediatr Orthop. 1991;11:743-747.
42 Van Buskirk CS, Ritterbusch JF. Natural history of distal spinal agenesis. J Pediatr Orthop B. 1997;6:146-152.
43 O’Neill OR, Piatt JHJr, Mitchell P, et al. Agenesis and dysgenesis of the sacrum: neurosurgical implications. Pediatr Neurosurg. 1995;22:20-28.