CHAPTER 32 Spinal Disorders Associated with Skeletal Dysplasias and Metabolic Diseases
Spinal disorders associated with skeletal dysplasias and metabolic diseases may be called orthopaedic trivia by some. None of these conditions are found in a large number of patients, and the numbers of syndromes and subclassifications seem to be almost endless. More than 200 types of short stature syndromes have been described to date, and the mapping of the human genome has led to discovery and clarification of many new metabolic disorders. Despite the relative rarity of these conditions in any spine surgeon’s practice, it is important for physicians and surgeons to know enough features of these disorders to allow for their early recognition and diagnosis. Because the natural history of spinal disorders in each of these syndromes is often quite different, it is important initially to establish as accurate a diagnosis as possible, particularly in disorders in which spinal deformity or stenosis may lead to spinal cord compression.1
Skeletal Dysplasias
Achondroplasia
The cause of hypotonia remains unclear regarding whether it is constitutional or the result of a partial neurologic deficit. There was speculation that foramen magnum stenosis might be the cause of the hypotonia, but this has been shown not to be the case.2 Some hypotonia may result from spinal cord compression at the foramen magnum because there is a relative increased frequency of sleep apnea in infants with achondroplasia, sometimes leading to sudden death.3 Sleep apnea monitors are often used for the first several months in many of these infants, and if sleep apnea is clinically significant, evaluation of potential spinal cord compression at the foramen magnum is needed.4,5 This evaluation includes not only careful recording of sleep monitor findings, but also sleep laboratory studies for apnea and oxygen saturation levels and computed tomography (CT) and magnetic resonance imaging (MRI) of the brainstem and upper cervical spine.
Somatosensory evoked potential (SSEP) monitoring has been shown to be helpful in establishing the diagnosis of cervical myelopathy in these patients.6 If SSEP testing is done, subcortical recording of SSEPs from stimulation of the median nerve is more sensitive and specific in diagnosing high cervical myelopathy than stimulation of the posterior tibial nerve.7
Previous studies have divided this foramen magnum area compression into two major types—the first involving cervical cord compromise from direct impingement of the posterior rim of the foramen magnum and the second caused by the posterior foramen magnum rim invaginating into the ring of the atlas. In these studies, it is stressed that the neural compression is of the high cervical spinal cord, not of the brainstem itself.8 Autopsy studies have noted histologic changes in the upper cervical spinal cord similar to changes seen in the central cord syndrome, and some authors believe that if one avoids placing the head of a young child with achondroplasia in hyperextension, the risk of spinal compression is lessened.9 Persistent foramen magnum compression may play a role in the later finding of a syrinx in the cervical spinal cord.10
Some degree of foramen magnum stenosis is present in essentially all children with achondroplasia. By using CT to measure the size of the foramen magnum, it has been shown that 96% have a foramen magnum size smaller than 3 standard deviations below the mean.8 How and whom to treat remains unclear, however. One study of 32 children with achondroplasia showed 28% with a history of sleep apnea and 22% with abnormal sleep study results, both of which improved in the 6 children who had foramen magnum decompression.6 Foramen magnum decompression has been reported to be performed more safely and successfully when combined with external ventricular drainage to manage the abnormal cerebrospinal fluid dynamics in this compressive condition.11 Diverse symptoms and signs such as ataxia, incontinence, and respiratory problems have been reported to be successfully treated by foramen magnum decompression and atlas laminectomy in patients ranging in age from 7 months to 30 years.12 Some authors recommend that prophylactic cervicomedullary decompression be done, even in asymptomatic children, if T2-weighted MRI signal changes in the spinal cord are present.13
Although successful foramen magnum decompression has been reported in infants,14 other authors maintain that if appropriate sleep apnea monitoring is continued until the child is 2 or 3 years old, there is a natural relative increase in the foramen magnum size with growth, relieving enough of the spinal cord compression to avoid the need for surgical treatment.15 Reported mortality and morbidity rates from foramen magnum decompression are thought by these authors to be greater than if no treatment except sleep monitoring is done. It is rarely necessary to perform foramen magnum decompression in older children or in adults. If a child has required cervicomedullary decompression, there seems to be an increased risk for symptomatic thoracolumbar stenosis requiring laminectomies before adolescence.16
In the cervical spine below the foramen magnum in achondroplasia, the principal spinal disorder is diffuse spinal stenosis. Although a small spinal canal is present from birth, signs or symptoms of neural compression in the lower cervical spine are usually not noted until middle age or later.17 At those ages, neural compression results from osteophytes that develop with time from degenerative disc changes. The cumulative effect of previous small cervical spine dimensions and osteophyte compression leads to neurologic deficits that require treatment. If pain or sensory changes in the upper extremities are the only findings, conservative care with a cervical orthosis and anti-inflammatory medications is used initially.
If a motor deficit in the upper or lower extremities is present and pain is unrelieved by nonoperative treatment, laminectomy at multiple levels is needed.18,19 MRI defines the levels of compression, which are usually multiple. As a result, when laminectomy of the cervical spine is needed in achondroplasia, often most of the cervical spine below the axis needs to have the laminae removed to relieve the neural compression. In addition, these patients may need laminectomies in the thoracic and lumbar spine, and some patients with achondroplasia require laminectomy decompression from the skull to the sacrum.20 Foraminotomies are needed at levels shown on MRI to have neural foraminal stenosis from adjacent osteophytes. After multilevel laminectomy, cervical spine fusion is generally not needed in adults, but in the rare instance that cervical laminectomy in a child with achondroplasia should become needed, there is an increased chance of postlaminectomy kyphosis, and careful follow-up evaluation is needed. Although atlantoaxial instability is commonly seen in some other skeletal dysplasias, this instability is rarely seen in achondroplasia.21,22
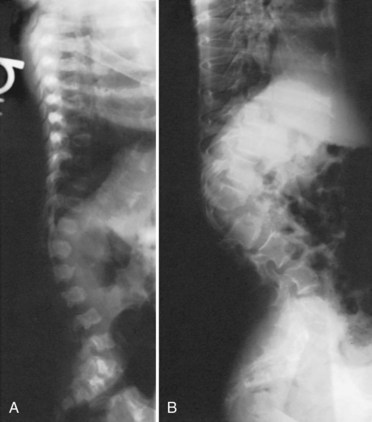
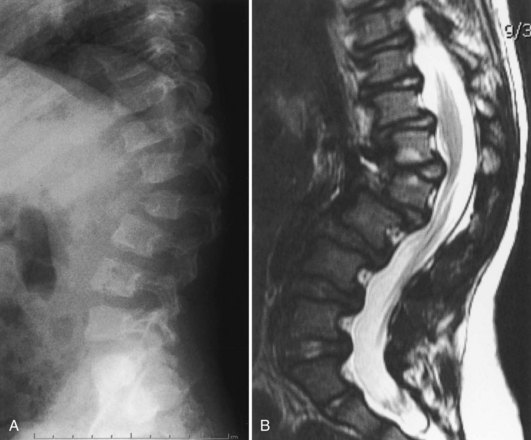
Of all the spinal segments, the middle and upper thoracic spine is the least involved in spinal deformity or cord compression in achondroplasia. The most common spinal deformities and stenosis, with subsequent neurologic problems, occur in the thoracolumbar and lumbar spine. Thoracolumbar kyphosis is usually present at birth to some degree but is more noticeable when the infant begins to sit (Fig. 32–1A). As sitting begins, the entire spine appears kyphotic, at least partly owing to the generalized hypotonia present at this age. Although some authors have advocated limiting infants to a reclined, rather than a fully upright, sitting position to avoid the development of thoracolumbar kyphosis,23 this does not seem to be necessary in clinical practice. In more than 90% of children with achondroplasia, thoracolumbar kyphosis improves without treatment as the standing position is assumed and lumbar lordosis develops. Because walking typically is achieved by 18 to 24 months of age, this is the time that the resolution of the thoracolumbar kyphosis begins, followed by the gradual continual improvement over the subsequent 2 to 3 years.
A lateral spinal radiograph shows initial relative anterior wedging of the thoracolumbar vertebrae, but this generally resolves as standing and walking occur, with the radiograph showing a gradual filling in of the anterior aspects of the vertebral bodies at the thoracolumbar apex. In the author’s experience, bracing to correct this kyphosis has limited value, and the use of a thoracolumbosacral orthosis (TLSO) is poorly tolerated by many children with achondroplasia, partly owing to the difficulty they have with TLSO wear in reaching their feet owing to their short extremities. An orthosis that uses a soft front while supporting the kyphosis has been reported to be successful,24–27 although proper patient selection for bracing remains problematic. The use of stretching exercises for the hip flexion contractures (always present in this condition), as a means to decrease lumbar lordosis and control the thoracolumbar kyphosis, has been advocated, but documentation of the effectiveness of this passive stretching program is difficult, particularly when most thoracolumbar kyphoses resolve with no treatment.28
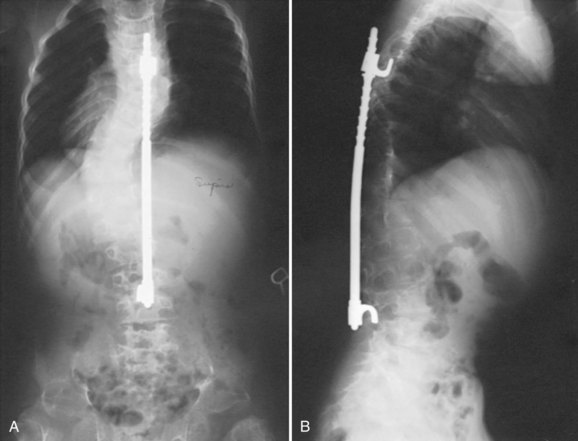
In a few young patients with achondroplasia, thoracolumbar kyphosis does not improve and becomes progressively worse with time. If there is significant wedging of one or more of the thoracolumbar vertebrae at 5 or 6 years of age, surgical treatment is needed to allow partial correction and prevent continued progression of the deformity (Fig. 32–1B).29 This approach is based on review of early childhood x-rays of teenagers later requiring treatment for severe thoracolumbar kyphosis and neurologic deficits. It is generally possible by age 5 or 6 years to determine whether or not the thoracolumbar wedging would improve without treatment or progress to a more severe deformity. Using this approach at this earlier age, the kyphotic deformity generally can be corrected better and more safely, leading to the prevention of localized increased kyphosis and early spinal cord compression.
As thoracolumbar kyphosis increases, so does the compensatory lumbar lordosis. Because the lumbar spine capacity decreases as lumbar lordosis increases, control of the thoracolumbar kyphosis seems to delay the onset of lumbar spinal stenosis symptoms. Whether this early kyphosis fusion will eventually lead to less of a need for decompressive lumbar laminectomy has not been determined. Circumferential fusion of the kyphosis thoracolumbar area in children younger than 2 years has been reported to lead to hypoplastic vertebral bodies and iatrogenic spinal stenosis, apparently as a result of inhibition of circumferential vertebral growth after the fusion.30 In an average-sized individual, it has been shown, however, that the spinal canal achieves its adult dimensions by age 6 years, so circumferential fusion after this age should not lead to iatrogenic stenosis.
In young children with persistent thoracolumbar kyphosis at age 6, the author’s favored surgical technique involves a combined anterior and posterior spinal fusion on the same operative day.29 The child is positioned with a beanbag and tape in a lateral decubitus position with the table tilted about 20 degrees, to allow for simultaneous exposures for the anterior and posterior spinal surgery. Through a thoracoretroperitoneal approach, anterior discectomies are done at three or four levels at the apex of the kyphotic deformity. Because the surgical approach at this level commonly involves an approach through the rib bed of either the 10th or the 11th rib, the portion of the rib removed is saved for an anterior strut graft.
In the author’s surgical series of more than 25 patients with achondroplasia with kyphotic deformity requiring surgical treatment, more than half have temporarily lost SSEPs at the time of the initial correction of the kyphosis. None of these patients have had permanent neurologic deficit postoperatively. If the SSEPs are lost, however, the rod must be removed and bent into more kyphosis before reinsertion of the rods. Postoperatively, a TLSO brace may be used for about 3 months during the day to allow for some protection for the instrumentation and fusion, but the child remains ambulatory at all times. After 3 months postoperatively, sports activity restrictions are stopped, and full activity is resumed (Fig. 32–2). In another series of 12 immature patients with achondroplasia treated with pedicle screw posterior instrumentation and fusion, no loss of evoked potentials was noted, and overall mean kyphosis correction was 50%.31 The exact cause of this neurologic compromise is unclear but may be related to buckling of the ligamentum flavum with kyphosis correction, leading to further spinal canal compromise.
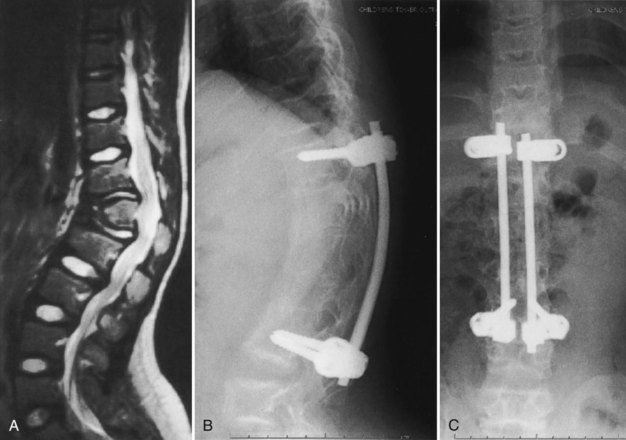
An alternative approach has been reported to treat persistent thoracolumbar kyphosis in which spinal instrumentation is placed in the anterior spine rather than in the posterior spine.32 An anterior strut bone graft is placed, and posterior fusion, without instrumentation, is done. This posterior fusion is repeated about 4 months after the initial surgery. A body jacket cast is used for 6 months, and a brace is used for another 3 months. Using this approach in four patients, Ain and Shirley32 obtained solid fusion, kyphosis correction of 23% to 31%, and no worsening of neurologic function.
Although clinically the principal thoracic and lumbar problem in preadolescent children with achondroplasia is persistent thoracolumbar kyphosis, neurologic compromise from spinal stenosis is the most common spinal disorder in teenagers and adults. Lutter and Langer33 separated these neurologic manifestations into four types: I, progressive, insidious onset; II, intermittent claudication; III, nerve root compression; and IV, acute onset of paraplegia. Types I and II are the most common. In type I, there is a slow but progressive onset of back pain, associated with lower extremity paresthesias and sensory loss.
Urologic function is often impaired, subclinically at first but later leading to incontinence.34 If urologic problems are suspected in the initial stages, voiding cystourethrogram can help to diagnose urinary control problems. If voiding cystourethrogram is abnormal, MRI of the thoracic and lumbar spine is indicated. If significant spinal stenosis is present, laminectomy decompression is needed to reverse the urologic problems. In one series of 22 pediatric patients with achondroplasia with symptoms and signs of spinal claudication and requiring laminectomy surgery, 77% had bladder incontinence. These patients requiring laminectomy at this young age had narrower lumbar interpediculate distances and greater thoracolumbar kyphosis than young patients with achondroplasia not requiring laminectomy.35
In achondroplasia with type II with neurologic manifestations, there is also a slow, progressive onset of symptoms, with the patient first noting a decreased ability to walk distances. Pain and weakness in the legs result when standing and walking occur, and these symptoms are relieved by the patient squatting, sitting, leaning forward, or lying down. These all are spinal positional changes that flex the lumbar spine to allow more room for the cauda equina and relieve the spinal claudication symptoms. It has been shown in a stillborn infant with achondroplasia that the capacity or space within the lumbar spine is nearly doubled by fully flexing the lumbar spine when compared with the extended or lordotic position.28
For any of these types, if neurologic deficits are suspected from the history or shown on physical examination, MRI is indicated to localize the site of abnormality. The problem most common in the interpretation of MRI is to determine which level is causing the neurologic signs or symptoms because there is diffuse stenosis usually present in the lower thoracic spine and the entire lumbar spine. Imaging studies show that the primary stenosis is from the narrowing of the interpediculate distances because the anteroposterior dimension of the spinal canal is relatively more normal until osteophytes from disc degeneration protrude into the spinal canal.36,37 Occasionally, CT myelography may be used to evaluate for levels of spinal stenosis, but if this is done, the myelographic dye should be placed via cisternal puncture in the upper cervical spine and not by lumbar puncture because lumbar puncture, with loss of cerebrospinal fluid, may lead to increased neurologic deficits in achondroplasia.38
The selection of the surgical procedures best suited to the individual patient with achondroplasia depends on the physical examination and the imaging findings. In a patient with type III neurologic manifestations, a limited laminectomy, disc excision, and foraminotomy generally suffice to relieve the symptoms. More commonly, in the other types of neurologic manifestations, multilevel decompressive laminectomy is the surgical treatment of choice.39,40 Laminoplasty was reported to be successful for complete relief of symptoms in 71% of 35 patients with achondroplasia and lumbar stenosis, but the use of this procedure has not been widespread.41 Even if multilevel laminectomy is done in these patients, fusion may not be needed unless there is a preexisting thoracolumbar kyphosis of about 30 degrees or more. If there is preexisting kyphosis at this level, pedicle screw fixation with dual-rod instrumentation is used with no instrumentation, no matter how small, within the spinal canal at the thoracic levels without laminectomy, although posterior pedicle screws can be used with laminectomy.
In situations with marked thoracolumbar kyphosis and multilevel stenosis on MRI, it is often difficult to determine the exact level of neurologic compromise. Generally, if depressed knee and ankle deep tendon reflexes and leg weakness are present, lumbar laminectomy seems to suffice for treatment. If there is MRI evidence of significant anterior spinal cord compression at the apex of a thoracolumbar kyphosis, and hyperreflexia is present together with leg weakness and sensory changes, anterior partial vertebrectomy or posterior pedicle subtraction osteotomy to decompress the anterior spinal cord at the apex of the kyphosis may be needed, in addition to multilevel lumbar and lower thoracic laminectomies. A report of four patients with achondroplasia and marked kyphosis who had decompression through a pedicle subtraction osteotomy noted that there was a mean kyphosis correction of 44%, but that, despite final improvement in neurologic status, transient postoperative weakness was noted in two of the four patients.42
In achondroplastic patients without coincident kyphosis but with neurologic deficits secondary to the diffuse stenosis, multilevel laminectomy is the treatment of choice (Fig. 32–3).39,40 Before surgery, it is essential to view the entire thoracic and lumbar spine on MRI to assess for all levels of stenosis that may be causing lower extremity weakness. It is important to decompress all levels that appear to have neural compression on MRI, and most often the levels for laminectomy extend from around T10 to S1. If only a single-level or double-level laminectomy is done at what appear to be the most involved areas, recurrence of new symptoms within months of the decompressive surgery is common as new levels of compression develop adjacent to the initial sites of decompression.
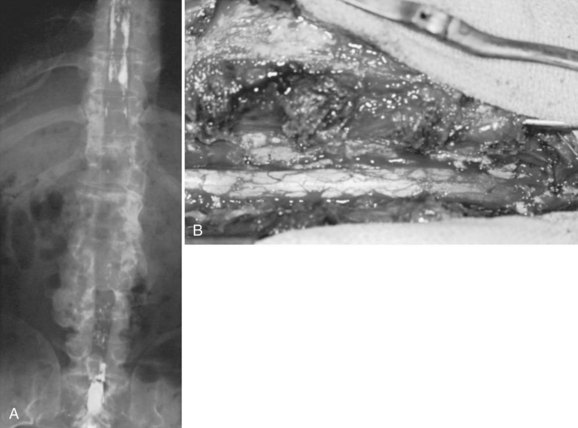
Because of the severe spinal stenosis present in achondroplasia, with the absence of epidural fat and concurrent loss of most of the subarachnoid space seen at the time of surgery, special precautions and surgical techniques are needed to complete these laminectomies more safely.34 The use of rongeurs within the spinal canal during laminectomy should be limited, owing to the severe stenosis. A postoperative increase in neurologic deficits is common, even if extreme care is taken during the laminectomy procedure. The use of a high-speed bur to transect the lamina on each side just medial to the facet or at the facet level is recommended. After the laminae have been transected, the posterior elements are lifted dorsally with a clamp, with the intent to avoid placing instruments within the stenotic spinal canal, which may injure the neural elements.
Foraminotomies can be done as needed after the laminae are removed, but MRI studies showed that although the foramina in achondroplasia lumbar spines were smaller than in control groups, the percentage of foraminal space occupied by the nerve root was similar between the two groups. The conclusion was that spinal stenosis symptoms arose from the central canal stenosis, not from foraminal stenosis. It would seem that foraminotomies in these patients are usually not needed.43 Unless there is increased kyphosis in the area of laminectomies, the author has noted that fusion may not be needed, even if laminectomies include removal of the facets. It has been reported more recently, however, that 10 skeletally immature patients with achondroplasia required spinal instrumentation and posterior fusion after laminectomies across the thoracolumbar region.44
After extensive laminectomy surgery, as described earlier, there may be areas of thinned dura that later form pseudomeningoceles and lead to later neurologic deterioration or pain or both. Using the paraspinal muscles to obliterate the dead space left by removal of the bony laminae seems to be helpful in decreasing the formation of these postlaminectomy pseudomeningoceles.34 Ain and colleagues45 reported that 61% of 98 achondroplasia patients undergoing laminectomy surgery had at least one perioperative complication, including 37% with dural tears, 23% with neurologic complications, 9% with infection, and 1 death.
Signs and symptoms of recurrent spinal stenosis may occur years after initial decompressive laminectomies. The most common cause of recurrent stenosis seems to be facet hypertrophy and disc disease, although scar tissue may form over the decompressed dural sac as well. MRI studies with gadolinium enhancement often help to visualize the scar tissue present and where the cauda equina compression has recurred. In one series of eight patients with restenosis, repeat decompression helped improve motor function in some, but three of these patients had significant complications.46
In patients with significant thoracolumbar kyphosis and lumbar spine stenosis, both of these conditions can be a possible cause of the underlying neurologic deficit.47 In this setting, MRI of the entire spine is used to ascertain all levels of compression. To treat lumbar spinal stenosis and anterior spinal cord compression at the apex of the kyphosis, anterior decompression and fusion at the apex of the kyphosis, together with a posterior multilevel laminectomy and instrumented fusion, is needed.29,47 Anterior decompression and fusion may be done through an anterior approach by vertebrectomy and strut bone graft fusion at the apex of the deformity or by an approach through the pedicles for anterior decompression with cage and bone graft stabilization. After posterior decompression at multiple levels is completed, pedicle screw and rod instrumentation is inserted, and bone graft is placed to complete the fusion. The pedicle anatomy of the thoracic and lumbar spine has been shown by CT to be markedly different from that of the normal spine. In addition to other findings reported, all pedicles are directed cranially, the pedicle starting points diverge from T9 to L5, and the maximal screw path length is at L2.48,49 Instead of stainless steel implants, titanium spinal implants are used to obtain better images at MRI if there is a subsequent need to evaluate the spinal cord further.
Diastrophic Dysplasia
Characteristic diagnostic features include micromelia with markedly short stature; “hitchhiker’s thumb”; stiff proximal interphalangeal joints of the fingers; severe equinovarus foot deformity; and, within a few weeks of birth, the formation of external ear cysts, which lead to scarring and the classic “cauliflower ear” appearance.50 Intelligence is normal. A cleft palate is present in about 25% of these children.
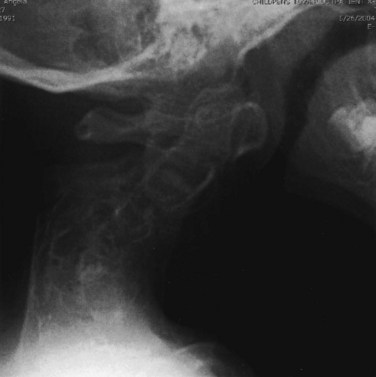
The spine has several areas of involvement in diastrophic dysplasia. All patients have spina bifida of the cervical spine, although symptoms directly related to this anatomic feature do not seem to occur.51,52 Upper cervical abnormalities seen in some of the other skeletal dysplasias, such as foramen magnum stenosis and atlantoaxial instability, are not present in diastrophic dysplasia.53 The primary cervical spine abnormality in this condition is mid-cervical kyphosis,54,55 although a review of 122 patients from Finland found only a 4% incidence of cervical kyphosis.56 At a young age, many patients with diastrophic dysplasia have mild cervical kyphosis, but most of these cases resolve with time and growth, by an average of 7 years of age.51,57
In patients with progressive cervical kyphosis, anterior and posterior fusion is recommended for optimal stabilization (Fig. 32–4). A halo brace is used to allow partial correction and postoperative immobilization until the fusion is solid. The kyphotic neck is usually relatively stiff, and aggressive attempts to correct the kyphosis are more likely to lead to iatrogenic neurologic injury. Laminectomy does not have a role here. If there is spinal cord compression that requires treatment, anterior decompression with vertebrectomies at the apex is the surgical approach of choice.
Kyphoscoliosis is the primary spinal disorder in the thoracic spine. In one study, 40% of children with diastrophic dysplasia developed mild to moderate scoliosis.58 In these patients, this deformity is not large enough to require treatment except for periodic follow-up, although occasionally a brace is used for a time. About 30% of patients with this condition develop a severe, rigid, progressive thoracic scoliosis associated with a sharply angular mid-thoracic kyphosis. In a more recent study from Finland, of 86 patients with diastrophic dysplasia and scoliosis, 11 cases were severe, 41 cases were “idiopathic-like,” and 33 cases were mild and nonprogressive.59
In a review of 43 patients with diastrophic dysplasia,60 if significant kyphoscoliosis was to develop, the onset of the spinal deformity was before age 4 years. In another review of 88 patients, 70 had scoliosis measuring an average of 42 degrees (range 11 to 188 degrees).61 Lung function generally has been shown to be relatively normal in diastrophic dysplasia, but pulmonary function declines with increasing thoracic kyphoscoliosis.62
Imaging studies, such as tomography or three-dimensional CT reconstructions of the spine, often show a wedge-shaped vertebra at the apex of the thoracic kyphoscoliosis, similar to what one would see with a congenital hemivertebra.60 Although orthotic treatment can be attempted early if there is flexibility proven on lateral bending radiographs, most of these early-onset deformities are very stiff. The treatment goal is to prevent progressive deformity rather than to wait and treat a severe deformity, an approach that is analogous in many ways to the treatment approach used for congenital scoliosis or kyphosis.
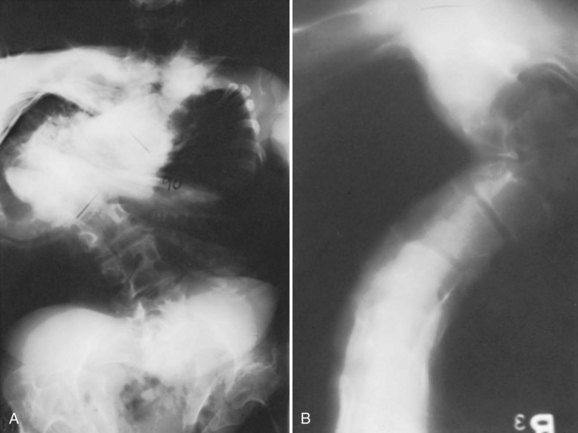
Submuscular spinal instrumentation without fusion to allow continued growth can be used in some cases, if there is a large curve in a young child (Fig. 32–5), but significant flexibility of the spine must be shown before this type of instrumentation treatment. If there is no real correction on lateral bending radiographs, anterior and posterior spinal fusion and posterior spinal instrumentation with modest correction is recommended at the stage at which progression has been proven greater than 50 degrees. Spinal growth in diastrophic dysplasia seems to be complete by about age 8 years, so definitive spinal fusion can be done in this condition at an earlier age than with most childhood spinal deformities with less fear of inhibiting later trunk growth.
Some of the most severe cases of kyphoscoliosis the author has seen have been in patients with diastrophic dysplasia (Fig. 32–6). These deformities may be severe enough to cause swallowing difficulties owing to the aberrant path taken by the esophagus. Despite the severity of these deformities, neurologic signs and symptoms related to spinal cord compression in the thoracic and lumbar regions are very rare, in contrast to what is expected from severe kyphotic deformities in general. In patients with diastrophic dysplasia, if neurologic defects or paraplegia is present in conjunction with severe thoracic kyphoscoliosis, the cause is usually iatrogenic, secondary to surgical attempts to instrument and correct the spinal deformity aggressively. There is minimal flexibility in these severe curves, so motor evoked potential and SSEP monitoring is required intraoperatively when spinal instrumentation and fusion is performed to detect early any neurologic changes from overstretching of the spinal cord in the face of a rigid spine. The vertebrae and the spinal canal in this condition are of sufficient size to accept appropriately sized pedicle screws, hooks, and wires or cables as part of spinal instrumentation. A goal in treating patients with scoliosis and diastrophic dysplasia is, however, to detect this deformity early, monitor this closely, and fuse the spinal deformity area before it progresses to a severe degree. Anterior and posterior fusion at an early age for progressive curves seems to be the best approach in treatment to prevent the severe deformity seen in the past in some adults with this condition.63
Most patients with diastrophic dysplasia also have a marked lumbosacral lordosis. The sacrum itself also develops a lordotic position with growth. This lordosis is exaggerated further by posterior vertebral body wedging that occurs in L5. In addition, owing to hip flexion contractures that are always present, lumbar lordosis is increased even more when the patient is standing. This standing hip and spine position is also partially compensatory for knee flexion contractures, which are common in these patients. In a study of walking difficulties in patients with diastrophic dysplasia, the walking difficulties were only rarely related to low lumbar spinal stenosis, however.64 In some patients with diastrophic dysplasia, interpediculate narrowing in the L5 and S1 vertebrae is seen on radiographs and MRI, but decompressive laminectomy is rarely required.60,61 There is no interpediculate narrowing in the upper lumbar spine.
Spondyloepiphyseal Dysplasia
Spondyloepiphyseal Dysplasia Tarda
Spondyloepiphyseal dysplasia tarda affects only males and is inherited as an X-linked recessive condition. This condition is caused by mutations in the SEDL gene in chromosomal location Xp22.2-p22.1.65 At birth, normal body proportions seem to be present, and the diagnosis is commonly not established until late childhood or early adolescence. The trunk is short but not dramatically so; radiographs show platyspondyly,66 a characteristic hump-shaped buildup of bone in the central and posterior aspects of the vertebral body, and a delay in ossification of the vertebral ring apophysis. Scoliosis and kyphosis are uncommon; although low back pain may result from a combination of disc degenerative disease and increased lumbar lordosis, the spine is not the most important feature of this condition. Multiple disc herniations have been reported.67
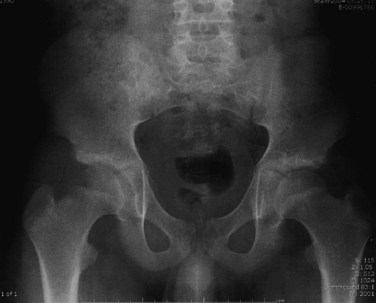
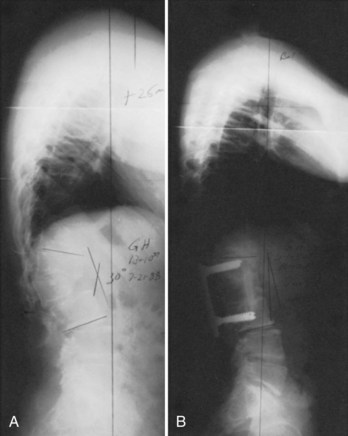
The most common reason that patients with spondyloepiphyseal dysplasia present to an orthopaedist is for treatment of hip pain and stiffness (Fig. 32–7).65 Premature hip osteoarthritis is a feature of this condition, often starting in the preadolescent years and progressing with age. Increased lumbar lordosis may be exaggerated by hip flexion contractures from hip arthritis. Total hip arthroplasty is commonly needed in early adult life. If this condition is suspected in a child, a family history of the need for hip replacement in early adult life may help make the diagnosis of spondyloepiphyseal dysplasia tarda.
Spondyloepiphyseal Dysplasia Congenita
Spondyloepiphyseal dysplasia congenita is recognizable at birth with imaging findings in conjunction with short-trunk dwarfism.68 There is delayed ossification in the vertebral bodies, and coxa vara is present. Hands and feet are of relatively normal size. As the child ages, there continues to be absent or delayed ossification of the femoral heads and irregularities in the epiphyseal and the metaphyseal areas of the long bones. From the medical standpoint, retinal detachment is common in this condition, and the parents need to be aware of this to arrange periodic eye evaluations.
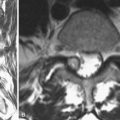
Atlantoaxial instability is the most commonly seen cervical spine problem in children with spondyloepiphyseal dysplasia congenita, with nearly half of children having this finding.50,69 In infancy, some hypotonia is present, but this should resolve with age. Failure to attain motor milestones progressively in the lower or upper extremities should direct attention to the cervical spine because atlantoaxial instability is often found early in childhood, sometimes at 1 year of age.
Cervical spine radiographs in a young child may be difficult to interpret. In a normal child, there is increased flexibility and motion on flexion and extension compared with an adult, so this has to be taken into consideration. In addition, in spondyloepiphyseal dysplasia congenita, there is a delay in posterior element ossification, so it is even more difficult to delineate clearly instability with flexion and extension radiographs in the upper cervical spine. Because odontoid hypoplasia is the underlying anatomic abnormality, the abnormal motion may be either excessive flexion motion or excessive extension motion (Fig. 32–8).
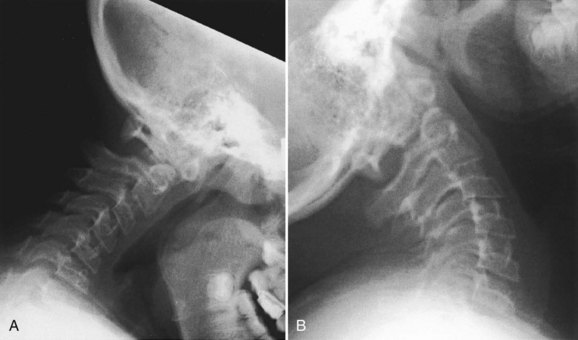
Although radiographs are the first step in the evaluation of upper cervical stability, flexion-extension sagittal plane MRI is extremely useful not only to view the anatomic abnormalities, but also to see if there is indentation into the spinal cord or narrowing of the spinal canal in either full flexion or full extension positions. If MRI findings are normal except for odontoid hypoplasia and there is 5 mm or less of motion on flexion and extension lateral cervical spine radiographs, ongoing follow-up is indicated periodically throughout childhood. If there is more than 5 mm of motion with neck extension and flexion or if there is indentation or signal change at the cervical spinal cord, posterior upper cervical spinal fusion is indicated. Laminectomy at the upper cervical spine is generally not needed in patients with spondyloepiphyseal dysplasia congenita and has no role as the only treatment for upper cervical cord compression caused by atlantoaxial instability. If the upper cervical spine sagittal canal diameter is narrowed and myelopathy is present, C1 laminectomy may be indicated in addition to occiput-C2 posterior fusion.70
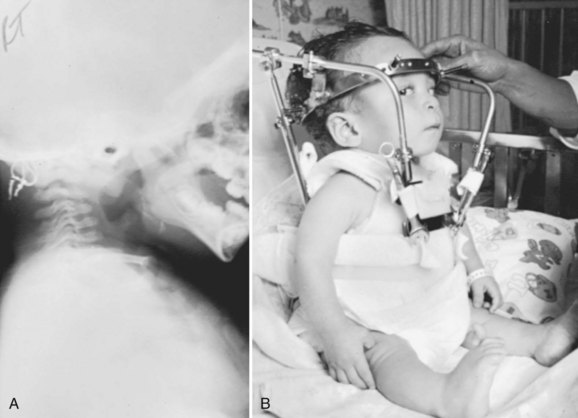
Upper cervical fusion may include only C1 and C2 but commonly extends from the occiput to C2 in these young children; partly owing to the lack of ossification of the posterior elements at this level at this age, this fusion usually is needed. It is more difficult to obtain secure wire fixation of the laminae, and if the instability is excessive, C1 extension movement and overreduction can occur with posterior wiring. To stabilize the upper cervical spine, the author prefers to use a halo brace, applied in the operating room as the first step in the fusion surgery. The number of halo pins used increases in younger children to obtain adequate stability, with six to eight pins usually placed for children younger than 5 years. In these young children, torque screwdrivers are used to tighten the pins to 3 or 4 psi rather than the 6 to 7 psi used in older individuals (Fig. 32–9).
Decortication of the laminae of C1 and C2 is completed with a high-speed bur, and cancellous bone graft is laid directly on the decorticated laminae. The corticocancellous strips are placed dorsal to the cancellous bone, and a two-layer or three-layer muscle closure is used to hold the corticocancellous bone strips firmly in place. The author has used this technique on many occasions. Employing halo brace immobilization for 3 months, the author has not noted slippage of the graft in children of this age, even without internal fixation. In a series of patients with skeletal dysplasia (including several with spondyloepiphyseal dysplasia congenita) treated for atlantoaxial instability, 92% achieved a solid fusion, and 88% had improvement in neurologic function.71
Pseudoachondroplasia
In pseudoachondroplasia, there is a normal facial appearance; epiphyseal and metaphyseal changes are evident in the long bones on radiographs; and spinal radiographs show flattened vertebral bodies with a central, anterior tonguelike projection and normal interpediculate distances throughout the spine (Fig. 32–10). The primary orthopaedic problems in this dysplasia that require treatment are the angular deformities of the lower extremities requiring corrective osteotomy (often more than once) and premature osteoarthritis of the hips requiring total hip arthroplasty at a relatively early age.
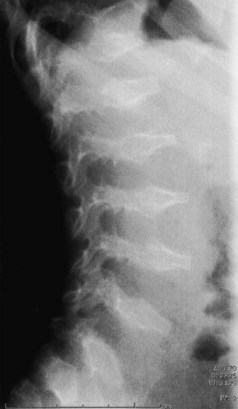
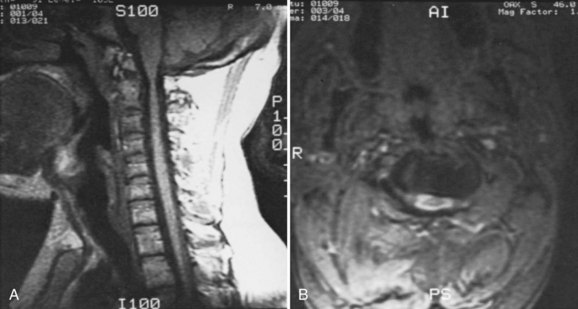
Atlantoaxial instability, a condition essentially never seen in achondroplasia, is not unusual in pseudoachondroplasia. This instability may be due partly to the generalized laxity present in all joints in this dysplasia and is not usually a result of odontoid hypoplasia seen in other skeletal dysplasias. Upper cervical instability caused by os odontoideum has been reported in 60% of 15 patients with pseudoachondroplasia, but no surgery was needed in this group.72 It is recommended that before any orthopaedic procedures that require anesthesia, flexion and extension lateral cervical radiographs should be obtained to evaluate these patients for instability. If atlantoaxial instability is diagnosed, posterior atlantoaxial instrumentation and fusion is needed (Fig. 32–11).
Mucopolysaccharidoses
Several syndromes have been described with abnormal metabolism of mucopolysaccharides. Sanfilippo syndrome (mucopolysaccharidosis [MPS] type III) and Scheie syndrome (MPS type V or type I-S) rarely have spinal manifestations, although Scheie syndrome may have dural thickening that can lead to neurologic deficits.73 In the remaining types, short-trunk dwarfism is usually seen, and thoracolumbar kyphosis is common. In addition, cervical spine abnormalities are often seen.
Significant anesthetic and other perioperative considerations need to be taken into account in this group of patients if surgery of any type is planned.74,75 At the time of surgery, intubation is often difficult and frequently requires use of fiberoptic intubation techniques. Care must be taken postoperatively to monitor breathing owing to airway compromise. An intensive care unit stay postoperatively is recommended because reintubation, if needed, may be very difficult.74,76,77
Hurler Syndrome (Mucopolysaccharidosis Type I)
Although an infant with Hurler syndrome appears normal at birth, short stature becomes apparent early along with delays in motor and mental development. Thoracolumbar kyphosis is present at birth but often is not noted initially, even though anterior beaking of the apical vertebral bodies is seen if radiographs were obtained. By 2 years of age, corneal clouding, coarse facial features with a large tongue and large lips, stiff joints, and hernias are obvious, and further motor and mental deterioration is noted. Many of these physical signs develop in the 1st year of life, and early diagnosis is key to an improved prognosis. Laboratory studies show excessive dermatan sulfate and heparan sulfate secretion. Atlantoaxial instability may be present.78,79
In the past, surgical treatment was not usually indicated for the spine or hip abnormalities seen in Hurler syndrome because death in early childhood was expected. More recently, bone marrow transplantation and enzyme replacement therapy have been used in these patients, however, with significant improvement in quality of life, survival, and life expectancy. Although these therapies can delay or prevent cardiac and neurologic deterioration in Hurler syndrome, numerous orthopaedic manifestations are present that usually require treatment, such as progressive thoracolumbar kyphosis or atlantoaxial instability.80,81 In one group of 10 patients followed for a mean of 8.7 years after bone marrow transplantation, all showed a decrease, however, in the amount of odontoid dysplasia.82 In another group of patients with Hurler syndrome who underwent bone marrow transplantation, high lumbar kyphosis was the most common spinal problem seen (Fig. 32–12).83
Hunter Syndrome (Mucopolysaccharidosis Type II)
Hunter syndrome is a lysosomal storage disease with a defect in iduronate-2-sulfatase that leads to a buildup of certain glycosaminoglycans and adverse neurologic effects. It is inherited in a X-linked recessive manner, affecting only males, and appearance at birth is normal. These children grow normally for about 2 years, at which time an abnormality is suspected. Life expectancy may extend well into adult life, although death in the 2nd decade of life may occur because of cardiopulmonary problems. Some noncharacteristic vertebral changes are present, but lumbar kyphosis may be marked and require surgical treatment.84 Evaluation of the upper cervical spine is needed because MRI studies may show anterior spinal cord compression secondary to a thickening of the soft tissue posterior to the odontoid, owing to deposition of the mucopolysaccharide.85
Morquio Syndrome (Mucopolysaccharidosis Type IV)
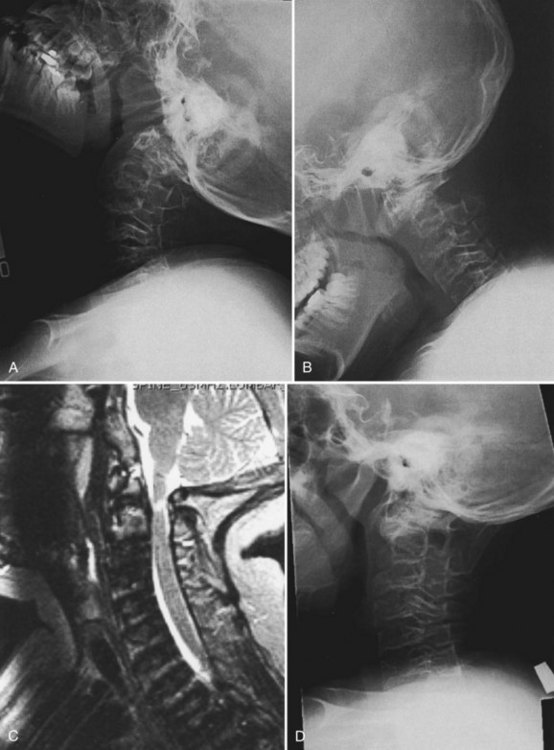
The most common spinal problem requiring treatment is odontoid hypoplasia, which leads to atlantoaxial instability.55,86–88 Compression at the upper cervical spine is compounded by the frequent presence of an anterior soft tissue mass from the deposition of mucopolysaccharide. Odontoid hypoplasia is present in most children with Morquio syndrome, and evaluation of the cervical spine is needed before orthopaedic surgery for the hips or legs.89 Instability is most often noted between 6 and 12 years of age. Lateral cervical spine radiographs are obtained in flexion and extension, and if there is more than 5 mm of motion, posterior upper cervical spinal fusion is needed.
In addition to instability, there is extradural soft tissue thickening, which is a contributing cause to the compression here that is often worse than is apparent from the radiographs alone.90 As with other dysplasias, if the radiographs are unclear regarding what instability may be present, a sagittal view MRI study with the neck in flexion and in extension is useful. When instability is present, posterior cervical fusion has been shown to be beneficial even with long-term follow-up.71,91 The author prefers occipitoaxial fusion without instrumentation, using iliac crest bone graft for fusion and a halo brace for immobilization for 3 months until fusion is complete (Fig. 32–13). In some children with Morquio syndrome, there may be mid-cervical or lower cervical spine stenosis or instability below the atlantoaxial level. The more levels of the cervical spine that require decompression and fusion, the harder airway access becomes, owing to a combination of limited neck motion and a progressive pectus carinatum deformity.
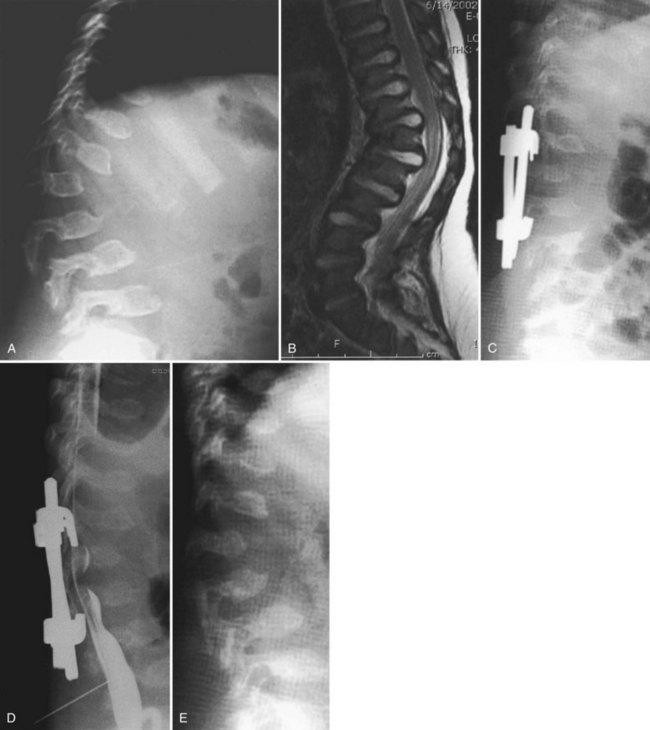
In a young child, use of instrumentation inside the spinal canal has been known to cause neurologic deficits in the lower extremities in some cases (Fig. 32–14). If it is determined that surgery is needed to treat thoracolumbar kyphosis, it may be safer to combine pedicle screw instrumentation and fusion with multilevel laminectomy (Fig. 32–15) or to use anterior spinal instrumentation.92
Maroteaux-Lamy Syndrome (Mucopolysaccharidosis Type VI)
Children with Maroteaux-Lamy syndrome appear normal at birth, and short-trunk dwarfism is noted by age 2 or 3 years. Many of the physical features resemble Hurler syndrome, but intelligence is normal. Diagnostic findings include increased urinary excretion of dermatan sulfate and arylsulfatase B deficiency in fibroblasts and white blood cells. This syndrome typically has hip involvement, which on radiographs resembles Legg-Perthes disease. Stenosis at the level of the foramen magnum and upper cervical spine, probably resulting from thickening of the posterior longitudinal ligament, can be improved by laminectomy when neurologic symptoms and signs are present.93 Thoracolumbar kyphosis is common, and the vertebrae are flattened on spinal radiographs. Laminectomy may be needed if MRI findings correlate with lower extremity loss of function. Spinal fusion should be considered for progressive kyphotic deformity. Spinal cord monitoring is essential, and pedicle screw instrumentation should be attempted because placing implants within the spinal canal has been known to lead to iatrogenic neurologic deficits.
Miscellaneous Syndromes
Kniest Syndrome
The clinical features of Kniest syndrome are closely related to the features found with spondyloepiphyseal dysplasia congenita. Life expectancy and intelligence are normal. Imaging findings outside of the spine include metaphyseal widening of the long bones, coxa vara, delay in epiphyseal ossification, and sometimes angular deformity of the lower extremities.94,95 These children walk with a marked external foot progression angle because of femoral external rotation. The presence of coronal and sagittal vertebral clefts has been reported in 63% of infants with Kniest syndrome and may be helpful in establishing a diagnosis.96
Odontoid hypoplasia with resultant atlantoaxial instability is the most common spinal problem requiring treatment.97 Use of flexion-extension lateral cervical spine radiographs or flexion-extension sagittal view MRI allows for serial evaluation of this instability to determine any need for posterior occipitoaxial fusion. The criteria for surgery and surgical technique are the same as described earlier for spondyloepiphyseal dysplasia congenita. Scoliosis is common but because of the limited trunk growth does not always require treatment. Periodic thoracolumbar spine radiographs are indicated, however, for evaluation and identification of patients who would benefit from either orthotic or surgical treatment. If surgical treatment is needed, small but standard spinal instrumentation can be used. Finally, excessive lumbar lordosis is present, in part from the spine and in part from the expected hip flexion contractures usually present. Because proximal femoral osteotomy is often used to treat coxa vara and external femoral rotation, it is recommended that this femoral osteotomy also include an extension component, which allows some correction of the flexible component of the excessive lumbar lordosis.
Metatropic Dysplasia
Metatropic dysplasia is rare; the name is derived from the apparent change in body proportions with increasing age. This condition seems to be not only a disorder of endochondral ossification, but also seems to be associated with defects in the longitudinal proliferation and maturation of chondrocytes and in the production of normal matrix. The uncoupling of endochondral and perichondral growth seen in this condition may explain why the long bones are dumbbell-shaped, as is seen on radiographs in this dysplasia.98 As growth occurs, the trunk becomes disproportionately short owing to flattened vertebrae and scoliosis or hyperkyphosis. Life expectancy usually extends into early adulthood. Scoliosis or kyphosis appears early in childhood and is difficult to manage. Orthotic management is attempted early, but growing rod instrumentation without fusion with or without apical fusion should be considered in young children with severe scoliosis. Definitive spinal instrumentation and fusion, commonly involving anterior and posterior fusion, is usually needed by preadolescence if not earlier.
Odontoid hypoplasia has also been reported to be commonly present in this condition and may require upper cervical fusion.99 More recently, cervical spinal stenosis has been reported to be a common feature of this condition, and decompressive laminectomy may be needed in addition to fusion in the upper cervical spine.100 An additional feature of metatropic dysplasia that should be considered is the presence of enlarged ventricles on head CT scans.
Chondrodysplasia Punctata
Also known as Conradi-Hünermann syndrome, chondrodysplasia punctata can be diagnosed at birth by short limbs; ichthyosis; flat facial features; and, in particular, radiographic findings of punctate calcification in the epiphyses at the ends of the long bones. These stippled epiphyses are present even in very young children. Coronal and sagittal vertebral clefts are present in 79% of infants with this dysplasia.96
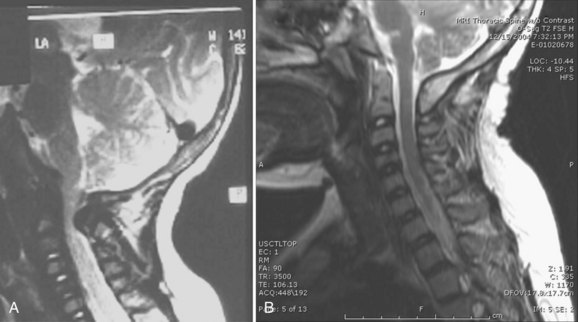
Atlantoaxial instability, in one reported case leading to death from cervical cord compression,101 and upper cervical stenosis can be seen, so cervical spine imaging is needed in chondrodysplasia punctata (Fig. 32–16).102–105 Scoliosis is common and often has its onset in early childhood. There are two main types of scoliosis: One slowly progresses with growth, and the other is a dysplastic type that is rapidly progressive. Orthotic treatment may suffice in curves with slower progression, but spinal instrumentation and anterior and posterior fusion is needed in larger dysplastic curves.106 Growth hormone has been successful in some patients in improving their final height. Life expectancy and intelligence are normal if the child survives the newborn period.
Camptomelic Dysplasia
The most prominent feature in camptomelic dysplasia at birth is bowing of the long bones of the lower extremities. Delayed ossification of the mid-thoracic pedicles is very useful in helping to establish this diagnosis. Cervical kyphosis is reported in 38%,107 and spinal cord injury as a result of this cervical kyphosis has been reported.108 Scoliosis and thoracic hyperkyphosis develop very early in childhood and often develop into severe deformity. Many of these children die in early childhood from pulmonary causes. Orthotic treatment is difficult because of the pulmonary compromise seen in these children. If patients survive past early childhood and have progressive kyphoscoliosis, spinal instrumentation and fusion has been reported to be successful.107
Spondyloepimetaphyseal Dysplasia with Joint Laxity
Early development of severe and progressive kyphoscoliosis during infancy is seen in spondyloepimetaphyseal dysplasia with joint laxity. Left untreated, this deformity leads to death in early childhood resulting from either spinal cord compression or cardiorespiratory failure in most patients.109
Spondylometaphyseal Dysplasia, Kozlowski Type
The Kozlowski type of spondylometaphyseal dysplasia is a short-trunk dwarfism that is usually not diagnosed until preschool age. Platyspondyly causes the short trunk, and kyphosis is common. Life expectancy is normal, and the kyphotic deformity needs to be followed and treated according to how severe it becomes.110
Summary
Pearls
Key Points
1 Sciubba DM, Noggle JC, Marupudi NI, et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007;106(5 Suppl):372-378.
2 Ain MC, Chang TL, Schkrohowsky JG, et al. Rates of perioperative complications associated with laminectomies in patients with achondroplasia. J Bone Joint Surg Am. 2008;90:295-298.
3 Remes V, Marttinen E, Poussa M, et al. Cervical kyphosis in diastrophic dysplasia. Spine (Phila Pa 1976). 1999;24:1990-1995.
4 Remes V, Tervahartiala P, Poussa M, et al. Thoracic and lumbar spine in diastrophic dysplasia: A clinical and magnetic resonance imaging analysis. Spine (Phila Pa 1976). 2001;26:187-195.
5 Ain MC, Chaichana KL, Schkrohowsky JG. Retrospective study of cervical arthrodesis in patients with various types of skeletal dysplasia. Spine (Phila Pa 1976). 2006;31:E169-E174.
6 Weisstein JS, Delgado E, Steinback LS, et al. Musculoskeletal manifestations of Hurler syndrome: Long term followup after bone marrow transplantation. J Pediatr Orthop. 2004;24:97-101.
1 Lachman RS. Neurologic abnormalities in the skeletal dysplasias: A clinical and radiological perspective. Am J Med Genet. 1997;69:33-43.
2 Reynolds KK, Modaff P, Pauli RM. Absence of correlation between infantile hypotonia and foramen magnum size in achondroplasia. Am J Med Genet. 2001;101:40-45.
3 Pauli RM, Scott CIJr, Wassman ERJr, et al. Apnea and sudden unexpected death in infants with achondroplasia. J Pediatr. 1984;104:342-348.
4 Fremion AS, Garg BP, Kalsbeck J. Apnea as the sole manifestation of cord compression in achondroplasia. J Pediatr. 1984;104:398-401.
5 Reid CS, Pyeritz RE, Kopits SE, et al. Cervicomedullary compression in young patients with achondroplasia: Value of comprehensive neurologic and respiratory evaluation. J Pediatr. 1987;110:522-530.
6 Nelson FW, Hecht JT, Horton WA, et al. Neurological basis of respiratory complications in achondroplasia. Ann Neurol. 1988;24:89-93.
7 Li L, Muller-Forell W, Oberman B, et al. Subcortical somatosensory evoked potentials after median nerve and posterior tibial nerve stimulation in high cervical cord compression of achondroplasia. Brain Dev. 2008;30:499-503.
8 Wang H, Rosenbaum AE, Reid CS, et al. Pediatric patients with achondroplasia: CT evaluation of the craniocervical junction. Radiology. 1987;164:515-519.
9 Yang SS, Corbett DP, Brough AJ, et al. Upper cervical myelopathy in achondroplasia. Am J Clin Pathol. 1977;68:68-72.
10 Hecht JT, Butler IJ, Scott CIJr. Long-term neurological sequelae in achondroplasia. Eur J Pediatr. 1984;143:58-60.
11 Carson B, Winfield J, Wang H, et al. Surgical management of cervicomedullary compression in achondroplastic patients. In: Nicoletto B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:207-214.
12 Ryken TC, Menezes AH. Cervicomedullary compression in achondroplasia. J Neurosurg. 1994;81:43-48.
13 Benglis DM, Sandberg DI. Acute neurological deficit after minor trauma in an infant with achondroplasia and cervicomedullary compression: Case report and review of the literature. J Neurosurg. 2007;107(2 Suppl):152-155.
14 Keiper GLJr, Koch B, Crone KR. Achondroplasia and cervicomedullary compression: Prospective evaluation and surgical treatment. Pediatr Neurosurg. 1999;31:78-83.
15 Wassman ERJr, Rimoin DL. Cervicomedullary compression with achondroplasia [letter]. J Pediatr. 1988;113:411.
16 Sciubba DM, Noggle JC, Marupudi NI, et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007;106(5 Suppl):372-378.
17 Kahonovitz N, Rimoin DL, Sillence DO. The clinical spectrum of lumbar spine disease in achondroplasia. Spine (Phila Pa 1976). 1982;7:137-140.
18 Morgan DF, Young RF. Spinal neurological complications of achondroplasia: Results of surgical treatment. J Neurosurg. 1980;52:463-472.
19 Pyeritz RE, Sack GH, Udvarhelyi GB. Cervical and lumbar laminectomy for spinal stenosis in achondroplasia. Johns Hopkins Med J. 1980;146:203-206.
20 Uematsu S, Wang H, Kopits SE, et al. Total craniospinal decompression in achondroplastic stenosis. Neurosurgery. 1994;35:250-257.
21 Gulati DR, Rout D. Atlantoaxial dislocation with quadriparesis in achondroplasia: Case report. J Neurosurg. 1974;40:394-396.
22 Hammerschlag W, Ziv I, Wald U, et al. Cervical instability in an achondroplastic infant: Case report. J Pediatr Orthop. 1988;8:481-484.
23 Hall JG. Kyphosis in achondroplasia: Probably preventable. J Pediatr. 1988;112:166-167.
24 Kopits SE. Thoracolumbar kyphosis and lumbosacral hyperlordosis in achondroplastic children. In: Nicoletti B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:241-255.
25 Siebens AA, Hungerford DS, Kirby NA. Achondroplasia: Effectiveness of an orthosis in reducing deformity of the spine. Arch Phys Med Rehabil. 1987;68:384-388.
26 Siebens AA, Kirby N, Hungerford DS. Orthotic correction of sitting abnormality in achondroplastic children. In: Nicoletti B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:313-320.
27 Winter RB, Herring JA. Kyphosis in an achondroplastic dwarf. J Pediatr Orthop. 1983;3:250-252.
28 Siebens AA, Hungerford DS, Kirby NA. Curves of the achondroplastic spine: A new hypothesis. Johns Hopkins Med J. 1978;142:205-210.
29 Tolo VT. Surgical treatment of kyphosis in achondroplasia. In: Nicoletti B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:257-259.
30 Sensenbrenner JA. Achondroplasia with hypoplastic vertebral bodies secondary to surgical fusion. Birth Defects. 1974;10:356-357.
31 Ain MC, Browne JA. Spinal arthrodesis with instrumentation for thoracolumbar kyphosis in pediatric achondroplasia. Spine (Phila Pa 1976). 2004;29:2075-2080.
32 Ain MC, Shirley ED. Spinal fusion for kyphosis in achondroplasia. J Pediatr Orthop. 2004;24:541-545.
33 Lutter LD, Langer LO. Neurologic symptoms in achondroplastic dwarfs—surgical treatment. J Bone Joint Surg Am. 1977;59:87-92.
34 Uematsu S, Wang H, Hurko O, et al. The subarachnoid fluid space in achondroplastic spinal stenosis: The surgical implications. In: Nicoletti B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:275-281.
35 Schkrohowsky JG, Hoernschemeyer DG, Carson BS, et al. Early presentation of spinal stenosis in achondroplasia. J Pediatr Orthop. 2007;27:119-122.
36 Lutter LD, Lonstein JE, Winter RE, et al. Anatomy of the achondroplastic lumbar canal. Clin Orthop Relat Res. 1977;126:139-142.
37 Lonstein JE. Anatomy of the lumbar spinal canal. Basic Life Sci. 1988;48:219-226.
38 Suss RA, Udvarhelyi GB, Wang H, et al. Myelography in achondroplasia: Value of a lateral C1-2 puncture and non-ionic, water-soluble contrast medium. Radiology. 1983;149:159-163.
39 Alexander EJr. Significance of the small lumbar spinal canal: Cauda equina compression syndromes due to spondylosis. Part 5: Achondroplasia. J Neurosurg. 1969;31:513-519.
40 Streeten E, Uematsu S, Hurko O, et al. Extended laminectomy for spinal stenosis in achondroplasia. In: Nicoletti B, Kopits SE, Ascani E, et al, editors. Human Achondroplasia: A Multidisciplinary Approach. New York: Plenum Press; 1988:261-273.
41 Thomeer RT, van Dijk JM. Surgical treatment of lumbar stenosis in achondroplasia. J Neurosurg. 2002;96(3 Suppl):292-297.
42 Qi X, Matsumoto M, Ishii K, et al. Posterior osteotomy and instrumentation for thoracolumbar kyphosis in patients with achondroplasia. Spine (Phila Pa 1976). 2006;31:E606-E610.
43 Modi HN, Suh SW, Song HR, et al. Lumber nerve root occupancy in the foramen in achondroplasia: A morphometric analysis. Clin Orthop Relat Res. 2008;466:907-913.
44 Ain MD, Shirley ED, Pirouzmanesh A, et al. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine (Phila Pa 1976). 2006;31:197-201.
45 Ain MC, Chang TL, Schkrohowsky JG, et al. Rates of perioperative complications associated with laminectomies in patients with achondroplasia. J Bone Joint Surg Am. 2008;90:295-298.
46 Ain MC, Elmaci I, Hurko O, et al. Reoperation for spinal restenosis in achondroplasia. J Spinal Disord. 2000;13:168-173.
47 Hancock DO, Phillips DG. Spinal compression in achondroplasia. Paraplegia. 1965;3:23-33.
48 Srikumaran U, Woodard EJ, Leet AI, et al. Pedicle and spinal canal parameters of the lower thoracic and lumbar vertebrae in the achondroplast population. Spine (Phila Pa 1976). 2007;32:2423-2431.
49 Kumar CP, Song HR, Lee SH, et al. Thoracic and lumbar pedicle morphometry in achondroplasia. Clin Orthop Relat Res. 2007;454:180-185.
50 Walker BA, Scott CI, Hall JG, et al. Diastrophic dwarfism. Medicine (Baltimore). 1972;51:41-59.
51 Bethem D, Winter RB, Lutter L. Disorders of the spine in diastrophic dwarfism: A discussion of nine patients and review of the literature. J Bone Joint Surg Am. 1980;62:529-536.
52 Herring JA. The spinal disorder in diastrophic dwarfism. J Bone Joint Surg Am. 1978;60:177-182.
53 Remes V, Tervahartiala P, Poussa M, et al. Cervical spine in diastrophic dysplasia: An MRI analysis. J Pediatr Orthop. 2000;20:48-53.
54 Hensinger RN. Kyphosis secondary to skeletal dysplasias and metabolic disease. Clin Orthop Relat Res. 1977;128:113-128.
55 Kopits SE. Orthopaedic complications of dwarfism. Clin Orthop Relat Res. 1976;114:153-179.
56 Remes VM, Marttinen EJ, Poussa MS, et al. Cervical spine in patients with diastrophic dysplasia—radiographic findings in 122 patients. Pediatr Radiol. 2002;32:621-628.
57 Remes V, Marttinen E, Poussa M, et al. Cervical kyphosis in diastrophic dysplasia. Spine (Phila Pa 1976). 1999;24:1990-1995.
58 Poussa M, Merikanto J, Ryoppy S, et al. The spine in diastrophic dysplasia. Spine (Phila Pa 1976). 1991;16:881-887.
59 Remes V, Poussa M, Peltonen J. Scoliosis in patients with diastrophic dysplasia: A new classification. Spine (Phila Pa 1976). 2001;26:1689-1697.
60 Tolo VT, Kopits SE. Spinal deformity in diastrophic dysplasia. Orthop Trans. 1983;7:31-32.
61 Remes V, Tervahartiala P, Poussa M, et al. Thoracic and lumbar spine in diastrophic dysplasia: A clinical and magnetic resonance imaging analysis. Spine (Phila Pa 1976). 2001;26:187-195.
62 Remes V, Helenius I, Peltonen J, et al. Lung function in diastrophic dysplasia. Pediatr Pulmonol. 2002;33:277-282.
63 Matsuyama Y, Winter RB, Lonstein JE. The spine in diastrophic dysplasia: The surgical arthrodesis of thoracic and lumbar deformities in 21 patients. Spine (Phila Pa 1976). 1999;15:2325-2331.
64 Reems V, Poussa M, Lonnqvist T, et al. Walking ability in patients with diastrophic dysplasia: A clinical, electroneurophysiological, treadmill and MRI analysis. J Pediatr Orthop. 2004;24:546-551.
65 Fiedler J, Bergmann C, Brenner RE. X-linked spondyloepiphyseal dysplasia tarda: Molecular cause of heritable disorder associated with early degenerative joint disease. Acta Orthop Scand. 2003;74:737-741.
66 Fiedler J, Frances AM, LeMerrer M, et al. X-linked spondyloepiphyseal dysplasia tarda: Molecular cause of heritable platyspondyly. Spine (Phila Pa 1976). 2003;28:478-482.
67 Nakamura I, Hoshino Y. Multiple disc herniations in spondyloepiphyseal dysplasia tarda: A case report. Int Orthop. 1998;22:404-406.
68 Spranger J, Wiedemann HR. Dysplasia spondyloepiphysaria congenita. Helv Paediatr Acta. 1966;21:598-611.
69 Takeda E, Hashimoto T, Tayama M, et al. Diagnosis of atlantoaxial subluxation in Morquio’s syndrome and spondyloepiphyseal dysplasia congenita. Acta Paediatr Jpn. 1991;33:633-638.
70 Miyoshi K, Nakamura K, Haga N, et al. Surgical treatment for atlantoaxial subluxation with myelopathy in spondyloepiphyseal dysplasia congenita. Spine (Phila Pa 1976). 2004;29:E488-E491.
71 Ain MC, Chaichana KL, Schkrohowsky JG. Retrospective study of cervical arthrodesis in patients with various types of skeletal dysplasia. Spine (Phila Pa 1976). 2006;31:E169-E174.
72 Shetty GM, Song HR, Unnikrishman R, et al. Upper cervical spine instability in pseudoachondroplasia. J Pediatr Orthop. 2007;27:782-787.
73 Sostrin RD, Hasso AN, Peterson DI, et al. Myelographic features of mucopolysaccharidoses: A new sign. Radiology. 1977;125:421-424.
74 Morgan KA, Rehman MA, Schwartz RE. Morquio’s syndrome and its anaesthetic considerations. Paediatr Anaesth. 2002;12:641-644.
75 Dullenkopf A, Holzmann D, Feurer R, et al. Tracheal intubation in children with Morquio syndrome using the angulated video-intubation laryngoscope. Can J Anaesth. 2002;49:198-202.
76 Belani KG, Krivit W, Carpenter BL, et al. Children with mucopolysaccharidosis: Perioperative care, morbidity, mortality, and new findings. J Pediatr Surg. 1993;28:403-408.
77 Tobias JD. Anesthetic care for the child with Morquio syndrome: General versus regional anesthesia. J Clin Anesth. 1999;11:242-246.
78 Brill CB, Rose JS, Godmilow L, et al. Spastic quadriparesis due to C1-C2 subluxation in Hurler syndrome. J Pediatr. 1978;92:441-443.
79 Thomas SL, Childress MH, Quinton B. Hypoplasia of the odontoid with atlanto-axial subluxation in Hurler’s syndrome. Pediatr Radiol. 1985;15:353-354.
80 Weisstein JS, Delgado E, Steinback LS, et al. Musculoskeletal manifestations of Hurler syndrome: Long term followup after bone marrow transplantation. J Pediatr Orthop. 2004;24:97-101.
81 Malm G, Gustafsson B, Berglund G, et al. Outcome in six children with mucopolysaccharidosis type III, Hurler syndrome, after haematopoietic stem cell transplantation (HSCT). Acta Paediatr. 2008;97:1108-1112.
82 Hite SH, Peters C, Krivit W. Correction of odontoid dysplasia following bone-marrow transplantation and engraftment (in Hurler syndrome MPAS 1H). Pediatr Radiol. 2000;30:464-470.
83 Tandon V, Williamson JB, Cowie RA, et al. Spinal problems in mucopolysaccharidosis I (Hurler syndrome). J Bone Joint Surg Br. 1996;78:938-944.
84 Benson PF, Button LR, Fensom AH, et al. Lumbar kyphosis in Hunter’s disease (MPS II). Clin Genet. 1979;16:317-322.
85 Parsons VJ, Hughes DG, Wraith JE. Magnetic resonance imaging of the brain, neck and cervical spine in mild Hunter’s syndrome (mucopolysaccharidoses type II). Clin Radiol. 1996;51:719-723.
86 Nelson J, Thomas PS. Clinical findings in 12 patients with MPS IV A (Morquio’s disease): Further evidence for heterogeneity. Part III: Odontoid dysplasia. Clin Genet. 1988;33:126-130.
87 Blaw MF, Langer LO. Spinal cord compression in Morquio-Brailsford disease. J Pediatr. 1969;74:593-600.
88 Lipson SJ. Dysplasia of the odontoid process in Morquio’s syndrome causing quadriparesis. J Bone Joint Surg Am. 1977;59:340-344.
89 Hughes DG, Chadderton RD, Cowie RA, et al. MRI of the brain and craniocervical junction in Morquio’s disease. Neuroradiology. 1997;39:381-385.
90 Stevens JM, Kendall BE, Crockard HA, et al. The odontoid process in Morquio-Brailford’s disease: The effects of occipitocervical fusion. J Bone Joint Surg Br. 1991;73:851-858.
91 White KK, Steinman S, Mubarak SJ. Cervical stenosis and spastic quadriplegia in Morquio disease (MPS IV): A case report with twenty-six year followup. J Bone Joint Surg Am. 2009;91:438-442.
92 Dalvie SS, Noordeen MH, Vellodi A. Anterior instrumented fusion for thoracolumbar kyphosis in mucopolysaccharidosis. Spine (Phila Pa 1976). 2001;26:E539-E541.
93 Thorne JA, Javadpour M, Hughes DG, et al. Craniovertebral abnormalities in type VI mucopolysaccharidosis (Maroteaux-Lamy syndrome). Neurosurgery. 2001;48:849-852.
94 Bethem D, Winter RB, Lutter L, et al. Spinal disorders of dwarfism: Review of the literature and report of eighty cases. J Bone Joint Surg Am. 1981;63:1412-1425.
95 Lachman RS, Rimoin DL, Hollister DW, et al. The Kniest syndrome. Am J Roentgenol Radium Ther Nucl Med. 1975;123:805-814.
96 Westvik J, Lachman RS. Coronal and sagittal clefts in skeletal dysplasias. Pediatr Radiol. 1998;28:764-770.
97 Merrill KD, Schmidt TL. Occipitoatlantal instability in a child with Kniest syndrome. J Pediatr Orthop. 1989;9:338-340.
98 Boden SD, Kaplan FS, Fallon MD, et al. Metatropic dwarfism: Uncoupling of endochondral and perichondral growth. J Bone Joint Surg Am. 1987;69:174-184.
99 Shohat M, Lachman R, Rimoin DL. Odontoid hypoplasia with vertebral cervical subluxation and ventriculomegaly in metatropic dysplasia. J Pediatr. 1989;114:239-243.
100 Leet AL, Sampath JS, Scott CIJr, et al. Cervical spinal stenosis in metatropic dysplasia. J Pediatr Orthop. 2006;26:347-352.
101 Afshani E, Girdany BR. Atlanto-axial dislocation in chondrodysplasia punctata: Report of the findings in two brothers. Radiology. 1972;102:399-401.
102 Khanna AJ, Braverman ME, Valle D, et al. Cervical stenosis secondary to rhizomelic chondrodysplasia punctata. Am J Med Genet. 2001;99:63-66.
103 Goodman P, Dominguez R. Cervicothoracic myelopathy in Conradi-Hunermann disease: MRI diagnosis. Magn Reson Imaging. 1990;8:647-650.
104 Violas P, Fraisse B, Chapuis M, et al. Cervical spine stenosis in chondrodysplasia punctata. J Pediatr Orthop B. 2007;16:443-445.
105 Garnier A, Dauger S, Eurin D, et al. Brachytelephalangic chondrodysplasia punctata with severe spinal cord compression: Report of four new cases. Eur J Pediatr. 2007;166:127-131.
106 Mason DE, Sanders JO, MacKenzie WG, et al. Spinal deformity in chondrodysplasia punctata. Spine (Phila Pa 1976). 2002;27:1995-2002.
107 Coscia MF, Bassett GS, Bowen JR, et al. Spinal abnormalities in camptomelic dysplasia. J Pediatr Orthop. 1989;9:6-14.
108 Lekovic GP, Rekate HL, Dickman CA, et al. Congenital cervical instability in a patient with camptomelic dysplasia. Childs Nerv Syst. 2006;22:1212-1214.
109 Kozlowski K, Beighton P. Radiographic features of spondylo-epimetaphyseal dysplasia with joint laxity and progressive kyphoscoliosis: Review of 19 cases. Fortschr Roentgenstr. 1984;141:337-341.
110 Kozlowski KS. Chondrodysplasia spondylometaphysealis. Birth Defects. 1975;11:183-185.