[level-membership-for-neurology-category]
CHAPTER 40 SPINAL DISEASE: NEOPLASTIC, DEGENERATIVE, AND INFECTIVE SPINAL CORD DISEASES AND SPINAL CORD COMPRESSION
The long, cylindrically shaped spinal cord sits within the bony vertebral canal, tightly enveloped by the meninges. It extends from the foramen magnum to the first lumbar vertebra, where it terminates in the conus medullaris. Below this, the lumbosacral nerve roots form the cauda equina. Anatomically, the spinal cord has a distinctive structural arrangement, illustrated in Figure 40-1A. Functionally, the spinal cord plays a key role in sensorimotor conduction. Because of its special features and relatively primitive reflexes, diseases of the spinal cord give rise to a number of distinctive, sometimes overlapping clinical syndromes (Tables 40-1 and 40-2; see Fig. 40-1B).
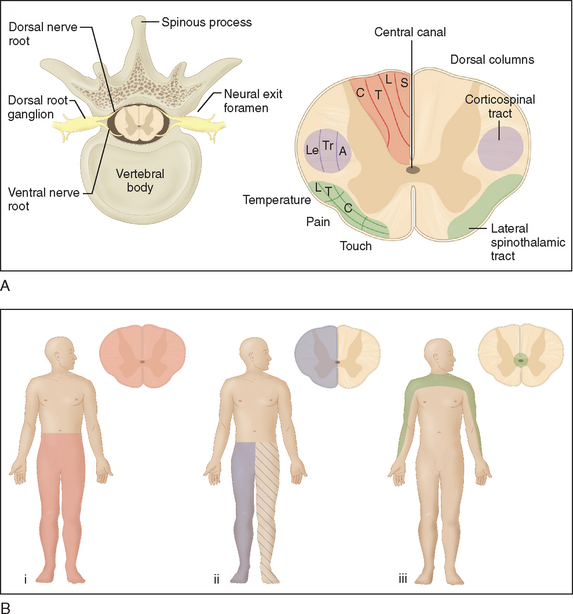
TABLE 40-1 Red Flag Features Indicative of Cord Disease
TABLE 40-2 Spinal Cord Syndromes
| Syndrome | Features |
|---|---|
| Complete transverse spinal cord syndrome (see Fig. 40-1Bi) | |
| Brown-Séquard (hemicord) syndrome (see Fig. 40-1Bii) | |
| Sensorimotor spinal tract syndrome |
Corticospinal tract involvement: upper motor neuron limb weakness (arm and leg weakness caused by cervical cord lesion; leg weakness caused by a lesion rostral to the upper lumbar spine):
|
| Radicular syndrome | |
| Syringomyelic or central cord syndrome (see Fig. 40-1Biii) | |
| Anterior horn syndrome | Caused by selective involvement of the motor neurons; segmental weakness, atrophy, fasciculations, and absent or diminished reflexes |
| Anterior spinal cord syndrome | Usually associated with vascular lesions of the anterior spinal artery, resulting in ischemia and neurological deficits within its area of supply (see Fig. 40-2); bilateral upper motor neuron weakness and spinothalamic sensory deficit below the lesion; dorsal column function is spared |
| Cauda equina syndrome | Pain usually an early feature; asymmetrical lower motor neuron (areflexic, flaccid, atrophic) paralysis of lower limbs; sphincter dysfunction (urinary or fecal incontinence, diminished anal tone) and radicular sensory loss; may difficult to differentiate clinically from lesions of the lumbosacral plexus |
| Conus medullaris lesions | Early and prominent sphincter dysfunction; back pain; sensory disturbance in sacral dermatomes (perineal: S3-S5; leg: L5-S2); loss of anal tone and reflex; impotence; leg weakness inconsistent, usually mild |
Segmental (myotomal) lower motor neuron weakness, with depressed or absent deep tendon reflexes, muscle atrophy, and flaccidity/decreased tone, may be present at the level of the lesion, as a result of involvement of anterior horn motor neurons or anterior (motor) nerve roots; this helps clinicians localize the lesion (Table 40-3). Segmental (dermatomal) sensory loss may also be present (Fig. 40-2).
TABLE 40-3 Segmental Innervation of Muscles and Tendon and Cutaneous Reflexes
| Segment/Nerve Root | Muscle | Tendon or Cutaneous Reflex |
|---|---|---|
| C3, C4 | Trapezius | — |
| C4, C5 | Rhomboids | — |
| C5 | Deltoid | — |
| C5, C6 | Supraspinatus, infraspinatus, biceps | Biceps reflex |
| C6 | Brachioradialis | Brachioradialis reflex |
| C7 | Triceps, extensor digitorum | Triceps reflex |
| C8 | Flexor digitorum superficialis and profundus | Finger-jerk reflex |
| T1 | Intrinsic muscles of hand | — |
| T7-10 | Upper rectus abdominis | Upper abdominal reflex |
| T10-12 | Lower rectus abdominis | Lower abdominal reflex |
| L1 | Iliopsoas | Cremasteric reflex |
| L2 | Adductor magnus | Adductor and cremasteric reflexes |
| L3, L4 | Quadriceps femoris | Knee-jerk (patellar) reflex |
| L4 | Tibialis anterior | — |
| L5 | Extensor hallucis longus | Plantar reflex |
| L5, S1 | Hamstrings | Ankle-jerk reflex |
| S1 | Extensor digitorum brevis | — |
| S1, S2 | Soleus, gastrocnemius | — |
| S4, S5 | Anal reflex |
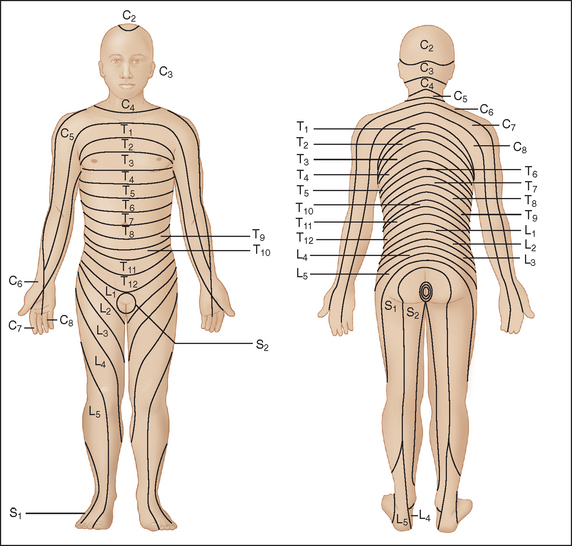
Figure 40-2 Distribution of dermatomes on the anterior surface of the body.
(From Merck and Company: Merck Manual of Diagnosis and Therapy, Section 14: Neurologic Disorders; redrawn from Keegan JJ, Garrett FD: The segmental distribution of the cutaneous nerves in the limbs of man. Anat Rec 102:409-437, 1948.)
Radicular symptoms and signs (see Table 40-2) result from irritation and compression of spinal nerve roots by extramedullary spinal cord lesions, usually before the involvement of long tracts (radiculomyelopathy). Radicular features are unusual with intramedullary lesions; when they do occur, they are most often present in the context of demyelinating disease.
Sensorimotor long tract signs usually arise as a result of compression, rather than invasion or destruction, of spinal white matter fasciculi and occur early with intramedullary lesions. Either sensory or motor function may be affected first; the distribution and extent of long tract symptoms and signs are dependent on the site and size of the lesion. A sensory level, below which perception of pain and temperature is altered or lost, is pathognomonic of a spinal cord lesion and should prompt imaging of the spinal cord. Often the signs are subtle in the early stages, and the description of a sensory level should alert the clinician. The highest level of sensory deficit indicates the lower possible level of the lesion, and the pathology may be present anywhere above this level; the sensory level may in fact be well below the actual lesion, and this must be kept in mind when imaging is ordered. Lamination of sensory fibers (see Fig. 40-1A) can give rise to suspended sensory deficits (i.e., isolated sensory deficit) at the level of the lesion or several segments caudally. Sacral sensation may be spared with deeper intramedullary lesions caused by spinothalamic tract lamination. Progressive spinal cord lesions, such as intramedullary neoplasms, may engender sequential deficits.
IMAGING
Magnetic resonance imaging (MRI) has enabled characterization of spinal cord diseases, including their location and internal structure, with the use of different magnetic resonance scanning sequences. The basic spinal MRI study includes T1- and T2-weighted sequences in the sagittal plane and contrast material (gadolinium)–enhanced T1-weighted images in the sagittal and axial planes. Myelography involves the introduction of radiopaque dye to the spinal fluid, through lumbar or sometimes cervical roots, but is limited in resolution and the length of cord that can be imaged in one episode. Obstruction to the cerebrospinal column and distortion and enlargement of the cord may all be directly visualized. Combining myelography with computed tomographic scanning of the spine is useful, allowing a greater extent of the cord to be accurately imaged in the one sitting. Plane films of the spine may reveal bony lesions and other local pathological processes, but their utility is quite limited.
SPINAL CORD INFECTIONS
Potentially any structure of the spinal cord and its surrounding meninges and vertebral column may be involved in infectious processes. The causative agents include bacteria, viruses, parasites, and fungi (Table 40-4).
AIDS, acquired immunodeficiency syndrome; CMV, cytomegalovirus; EBV, Epstein-Barr virus; HSV, herpes simplex virus; HTLV-1, human T cell leukemia/lymphoma virus type 1; VZV, varicellazoster virus.
Infectious and Parainfectious Myelopathies
Myelitis is a process intrinsic to the spinal cord; this definition excludes expansive lesions.1 Transverse myelitis is characterized by a focal inflammatory process within the spinal cord, leading to varying degrees of neural injury and dysfunction of neural pathways passing through the inflamed segment.2 This in turn leads to the abrupt onset of varying degrees of weakness, sensory alteration, and autonomic dysfunction.
The diagnostic criteria for transverse myelitis include the following:1,3
Transverse myelitis forms a part of a spectrum of neuroinflammatory conditions and may occur as part of a multifocal central nervous system (CNS) disease, such as multiple sclerosis; as part of a multisystem disease, such as systemic lupus erythematosus; or as an idiopathic, isolated entity.2 Of all cases of idiopathic transverse myelitis, 30% to 60% were preceded by a respiratory, gastrointestinal, or systemic infectious illness.2–5
The underlying pathogenesis of transverse myelitis is varied, depending on the specific underlying disease process. For instance, connective tissue disease–associated transverse myelitis may be secondary to a CNS vasculitis or thrombotic infarction of the cord.6 Most patients have cerebrospinal fluid (CSF) pleocytosis and foci of blood-brain barrier breakdown within the spinal cord.2
Two large etiological groups of inflammatory transverse myelitis are identifiable: the inflammatory demyelinating autoimmune myelopathies, such as multiple sclerosis, and the usually monophasic infectious encephalomyelitides.1 More than 40 such infectious encephalomyelitides have been identified, of which approximately 10 have been established as “pure infectious” myelitides by CSF virus isolation or polymerase chain reaction (PCR) techniques, which indicate that neurological injury resulted directly from microbial infection.1,2,8 The remainder are believed to be parainfectious, caused namely by a microbial infection, which in turn incites an immune response against neural tissue. In the latter, the infection may be remote from the immunological response.2 The latency between myelitis and preceding infection does not significantly differ between infectious and postinfectious myelitis. An interval of 9 ± 6 days has been reported for parainfectious cases, 5 days in mumps-related myelitis, 12 days in zoster-related myelitis, and 10 days in Mycoplasma-related myelitis.3,9–11
Although diagnosis of infectious and parainfectious transverse myelitis requires identification of the infectious agent within the CNS, some autoimmune mechanisms, such as molecular mimicry and superantigen-mediated disease, require only peripheral immune activation.2 In molecular mimicry, the putative mechanism of neural injury is antibody-mediated damage, secondary to cross-reaction of antibodies against an infectious agent with molecularly similar antigens in host neural tissue.2
Transverse myelitis is associated pathologically on tissue sampling (biopsy or autopsy) with inflammation.2 Both gray and white matter compartments are affected, although in postinfectious myelitis, white matter changes, demyelination, and axonal injury are prominent. Focal infiltration by lymphocytes and monocytes into segments of the spinal cord and perivascular spaces and activation of astroglial and microglial cells are observed.2 In some biopsies performed during the acute phase of myelitis, infiltration of CD4+ and CD8+ lymphocytes is prominent. Subsequently, during the subacute phase, monocyte and macrophage infiltration may become more prominent. In cases in which transverse myelitis is associated with autoimmune disease such as systemic lupus erythematosus, focal areas of ischemia or infarction secondary to vasculitis may be present, without prominent inflammation, a finding not usually seen in most cases of infectious transverse myelitis.12
PCR techniques, performed on serum and CSF, have facilitated the rapid, sensitive, and noninvasive diagnosis of many infectious forms, particularly viral, of myelitis.1,13,14 Diagnosis is often based on the detection of viral DNA in CSF by PCR.13 Further supportive evidence for a particular viral agent as the cause of myelitis, of value when viral PCR results are negative, is the presence of specific immunoglobulins M (IgM) and G (IgG) antibodies in the CSF, suggestive of intrathecal synthesis.1,13 This is especially the case when the serum: CSF ratio of these antibodies is depressed in relation to total IgG and albumin levels.
Infectious and parainfectious transverse myelitis are the focus of discussion in this chapter. The list of antecedent infections includes herpesviruses and Listeria monocytogenes, although in most of these cases, causality has not been established. Infectious and parainfectious transverse myelitis can be further subdivided, on the basis of the components of the neural axis involved, into meningomyelitis, transverse myelitis without meningitis, encephalomyelitis, and anterior horn syndrome with meningitis, among other entities.1 Meningitis is defined here as the clinical syndrome of fever and neck stiffness.1,2
Clinical Features
The clinical picture of acute transverse myelitis is one of rapid evolution of neurological deficit; however, the distribution of dysfunction, including the modalities affected, depends on the extent and distribution of inflammation within the spinal cord.2 On examination, there is often a clearly defined level of sensory dysfunction.2 When the maximum level of neurological deficit is reached, approximately 50% of affected patients are paraplegic, and 80% to 94% of patients experience sensory disturbances, including numbness, paresthesias, and bandlike dysesthesias.2–5,15,16 Manifestations of autonomic dysfunction include urinary urgency, urinary or fecal incontinence, difficulty or inability opening bladder or bowels, and sexual dysfunction.1
Imaging
Acute inflammation is evident on spinal MRI.1,2 This includes swelling of the spinal cord, in inflamed segments, as well as increased signal intensity on T2-weighted imaging. In cases in which nerve roots are involved in the inflammatory process (radiculomyelitis), enhancement of the affected nerve roots with administration of intravenous gadolinium contrast material may be seen.2,13 However, there are several reports in the literature of clinical cases of myelitis in which there is no demonstrable abnormality of the spinal cord on imaging.1,2,13
Differentiating between Infectious/Parainfectious Transverse Myelitis and Multiple Sclerosis
In multiple sclerosis, it is unusual to have complete transverse myelitis; there was only one such case of 308 in the Göteborg series.18 The weakness and numbness tend to be less severe in multiple sclerosis.19 Pathologically, CSF oligoclonal bands present in multiple sclerosis are absent in infectious transverse myelitis.1,19 Differences in MRI of the spine are also reported.1,18–20 In multiple sclerosis, the areas of T2 hyperintensity tend to be multifocal, smaller, and limited to one segment of the cord in transverse segments; in infectious and parainfectious transverse myelitis, in contrast, T2 hyperintensities are confluent and elongated, often extending over several spinal segments. Furthermore, plaques of demyelination are usually seen on cerebral MRI in multiple sclerosis but not in infectious transverse myelitis.
Specific Entities
Echovirus meningomyelitis
There have been case reports of echovirus (types 11 and 18) isolated from either pharyngeal exudate or CSF of children with clinical meningomyelitis, with transient T2 hyperintensity of the spinal cord.21,22
Coxsackie virus–related myelitis and meningomyelitis
There exist several case reports of patients with clinical and MRI evidence of transverse myelitis with or without meningitis, from whom Coxsackie virus (B4, B3, B5, A5, and A9) had been isolated or in whom a high titer of specific antibody to this virus had been demonstrated in the CSF or serum.1,23,24 For instance, in one case report of a 6-year-old with rapidly progressive paraplegia, with associated bladder and bowel dysfunction, a rising titer of serum anti-Coxsackie virus B5 antibody was demonstrated.25 The virus was also isolated from the stool. MRI in this case demonstrated diffuse swelling of the spinal cord on T1-weighted imaging.
Mumps-related meningomyelitis
The commonest neurological manifestation of mumps virus infection is meningitis; encephalitis occurs in fewer than 0.1% of cases.9 However, there are case reports of myelitis associated with mumps viremia. For instance, a 10-year-old boy developed flaccid paraparesis and neck stiffness in the setting of parotitis and orchitis that are clinically indicative of mumps infection.26 MRI demonstrated T2 hyperintensity from C2 to T12 spinal cord segments, in keeping with myelitis. Mumps infection was confirmed by the presence of IgM class antibodies against mumps in the serum. In another report, a young woman was diagnosed with mumps-associated transverse myelitis on the basis of clinical findings, MRI demonstration of a swollen spinal cord with uniform high-signal change on T2 weighting, and acute mumps viremia.27 No reports of mumps virus isolation from the CSF, in the setting of transverse myelitis, have been found.
Herpes simplex virus–related myelitis
Although a well-known cause of viral encephalitis, herpes simplex virus (HSV) is a rare cause of myelitis.13,28–30 This entity was first described by Klatersky and associates and has since been reported in both immunocompromised and immunocompetent patients.13,30 Subtle spinal cord involvement by HSV infection may be underrecognized.
HSV type 2 infection and, less frequently, HSV type 1 infection cause genital infection, with vesicles and ulceration.9,32 After primary genital infection, the virus persists latently within the sensory neurons of the dorsal root ganglia and produces recurrent cutaneous disease with intermittent reactivation and retrograde spread along the sensory nerve.9,13,32
On occasion, HSV reactivation may be followed by radiculomyelitis, variably affecting the cauda equina, conus medullaris, and thoracic cord.32 The pathogenesis of HSV-associated myelitis is not well understood, although intra-axonal spread of virus from the sensory ganglion into the spinal cord through the dorsal roots is thought to be the likely mechanism.28 Most cases of HSV-related myelitis described in the literature have been associated with type 2 infection, probably reflective of the recurrence of latent infection mainly with the more commonly HSV type 2 venereal infections.13
In six of a series of nine patients with HSV-related myelitis reported by Nakajima and colleagues, disease onset was marked by bilateral lower limb sensorimotor disturbance and urinary symptoms, with ascending progression of the myelopathy to thoracic or cervical level within the course of a few weeks.13 Most cases of HSV-related myelitis described in the literature followed a similar clinical pattern of an acute, monophasic, usually fatal ascending necrotizing myelitis, with death resulting from respiratory paralysis or meningoencephalitis.13 However, other clinical patterns, including a self-limited, resolving transverse myelitis, a relapsing-remitting form, and chronic myelitis, have been described since the development of improved diagnostic techniques such as PCR which have enabled noninvasive diagnosis.13,28–30,33 In three of the nine patients studied by Nakajima and colleagues, transverse myelitis started in the cervicothoracic cord, with a nonascending pattern and milder sequelae than in the cases of ascending myelitis. There have also been other case reports of transverse myelitis of the cervical and thoracic cord, caused by HSV, that resolved.34 In these cases, demyelination without necrosis was postulated to be the underlying pathological process responsible for myelitis.13
In early reports, HSV-related myelitis was described as being frequently associated, in close temporal relation, with cutaneous herpetic eruptions, which provided a clue as to the diagnosis.13,30 However, it has since then been suggested that this association is seen in very few cases of HSV-related myelitis, and therefore this feature is not useful for early diagnosis. HSV per se is rarely isolated from the CSF.13 Subtle clinical involvement of the spinal cord in patients with genital HSV infections are probably much more common than appreciated, however, and symptoms related to cord involvement are often a prominent feature of the prodromal phase of the illness.
The diagnosis of HSV meningoencephalitis is usually based on the detection of HSV DNA in CSF by PCR, with reported sensitivity and specificity of 98% and 94%, respectively.35 In most cases reported since 2000, PCR has been used in the diagnosis of HSV-related myelitis, although numbers have been too small to obtain data on specificity and sensitivity.28 Further evidence of herpetic infection as the cause of myelitis is the presence of anti-HSV IgM and IgG antibodies in the CSF, suggestive of intrathecal synthesis.28 Although demonstration of seroconversion against HSV concurrent with the episode of myelitis enables diagnosis, it should be kept in mind that anti-HSV antibody titers are not always elevated during the early stage of disease.13 CSF pleocytosis, although usually present, is also not a constant feature.29
MRI findings include enlargement of the conus medullaris; intramedullary T2 hyperintensity, particularly of the posterior funiculi; and gadolinium enhancement of the affected dorsal nerve roots, with no enhancement of the ventral roots.13,28 This is in keeping with the expected pattern of involvement of spinal cord structures, if infection is indeed caused by retrograde spread of virus into the cord from dorsal root ganglia. In one case, both T1 and T2 hyperintensities were observed, suggestive of hemorrhagic necrosis of the affected parts of the cord.13 Follow-up imaging may demonstrate atrophy of the cord in the previously inflamed regions.33
Treatment of HSV-related encephalitis with acyclovir is established; however, there are case reports of the value of this antiviral agent only in the treatment of HSV-related myelitis.28 Corticosteroids, in conjunction with antiviral therapy, may prevent ascending necrotizing myelopathy and improve survival.28 With regard to long-term prognosis, in the series by Nakajima and colleagues, three patients recovered, whereas the remaining six had severe persistent neurological deficits, such as paraplegia, despite antiviral therapy.13
Zoster-related transverse myelitis
Varicellazoster virus (VZV) infection may be associated with a wide spectrum of neurological complications, myelitis being the most poorly characterized of these.36 Several reports describe a postinfectious VZV-related myelitis, which is usually an acute, relatively benign transverse myelopathy, associated with a self-limiting paraparesis and sometimes sensory symptoms and sphincter disturbance.37 Chronic and remitting-relapsing temporal profiles of myelopathy have been described in association with VZV.36
Less commonly, a progressive ascending myelopathy, usually fatal in outcome, is seen in immunocompromised individuals and is thought to be caused by direct VZV invasion of the spinal cord.10,38 Ten cases of necrotizing vasculitis associated with VZV meningoencephalomyelitis have been reported in the literature, all with underlying immunodeficiency, secondary to either malignancy or human immunodeficiency virus (HIV) infection, and all cases fatal within 4 to 32 days.38 The patients typically developed radicular or central back pain within 2 weeks of zoster infection, in association with bilateral motor and sensory signs.
The diagnosis of VZV-related myelitis is historically based on the onset of myelopathy within days to weeks of a typical rash.37 The symptoms may be most pronounced ipsilateral to and at the level of the rash. However, there are reports of VZV-related myelitis, confirmed by positive VZV PCR or culture from the CSF, with no history of cutaneous rash.37,39
VZV per se is rarely isolated from blood or CSF, although the latter usually shows an often profound pleocytosis.37 PCR techniques have enabled the detection of VZV DNA in the CSF of patients suspected of having VZV-related myelopathy, even months later, which enables confirmation of the diagnosis and suggests a role of virus persistence in the pathogenesis of disease.37,38 VZV as the etiological agent in myelopathy is also supported by the presence of antibody (IgG and IgM) to VZV in the CSF, indicative of presence of viral antigen in the nervous system and intrathecal antibody synthesis.37,40
The underlying pathological process in VZV-related myelitis is thought to be a small- or large-vessel vasculopathy, depending on the immune status of the patient.38 Small-vessel disease is seen almost exclusively in immunocompromised patients and includes a necrotizing vasculitis of leptomeningeal vessels, especially around the spinal cord and brainstem.38 VZV has been detected in large and small vessels of the nervous system by PCR, in situ hybridization for VZV antigen, and immunohistochemistry in these cases.38,41,42 Pathological examination may demonstrate intranuclear viral inclusions.38
VZV-related vasculopathy may occur as late as 5 to 6 months after the zoster rash.39 In these patients, the detection of antibody to VZV in CSF, even in the absence of amplifiable VZV DNA, is diagnostic.39,40 There are other reports of zoster-related myelitis in which PCR results for VZV were negative in the presence of anti-VZV antibodies in the CSF or became positive only after several days of clinical symptoms.38,40 It is presumed that the reason why patients with VZV-related vasculopathy may not contain VZV DNA is that active viral replication is confined to CNS arteries.39 Hence CSF PCR for VZV DNA is not as sensitive a test as CSF PCR for HSV.40
MRI demonstrates hyperintensity and swelling, consistent with edema, on T2-weighted sequences.36,38 On T1-weighted images, with gadolinium contrast material, leptomeningeal enhancement over the cerebellum and brainstem is seen in cases of ascending myelitis.38
The treatment of VZV-related myelitis is aggressive antiviral therapy, usually with intravenous acyclovir; however, response is variable.36 VZV strains that are resistant to acyclovir, as well as to related newer compounds such as famciclovir and valacyclovir, have been reported, in both immunodeficient and immunocompetent patients.38,43,44 Resistance results from deficiency or absence of viral thymidine kinase, the enzyme on which these drugs are dependent for intracellular activation via phosphorylation.38,44 These thymidine kinase–deficient VZV mutant strains are, however, susceptible to foscarnet and acyclic nucleoside phosphonates, which are active independently of thymidine kinase.44 Because diagnostic tests for VZV-mediated myelitis are potentially insensitive, management of patients suspected of having this condition should include broadening of antiviral therapy to overcome possible resistance if there is clinical progression or inadequate response to therapy.38
Cytomegalovirus-related myelitis
In the setting of cellular immunodeficiency, such as the acquired immunodeficiency syndrome (AIDS), cytomegalovirus infection of the cord can produce a necrotizing transverse myelitis, typically when CD4 counts are less than 50.45 It is exceedingly rare in immunocompetent persons, although five such cases of cytomegalovirus-associated transverse myelitis were reported by Giobbia and coworkers.46 In all these cases, CSF PCR for cytomegalovirus yielded negative results. The diagnosis was instead obtained on the basis of serological data: namely, cytomegalovirus antigenemia (positivity for cytomegalovirus pp65 antigen) and high serum titers of specific cytomegalovirus IgM and IgG antibodies, positive blood PCR results for cytomegalovirus, and blood or urine cultures. Of note, however, PCR performed on CSF of patients with AIDS demonstrated this test to predict histopathologically confirmed cytomegalovirus-associated CNS disease, including myelitis.47 It is unclear as to whether neuronal injury is immune mediated or caused by a cytotoxic effect of viral infection.
Epstein-Barr virus–related myelitis
Epstein-Barr virus, like VZV, can infect multiples sites of the neuraxis and can therefore produce protean clinical manifestations.48 Both CNS and peripheral nervous system disease can occur, with reports of meningitis, encephalitis, transverse myelitis, neuritis, and overlapping syndromes.48–50 The incidence of Epstein-Barr virus–related neurological disease is not known; however, it has been estimated to occur in 1% to 5% of individuals with infectious mononucleosis.48 The mechanism by which Epstein-Barr virus produces neurological disease is unknown; however, the development of neurological disease over weeks is suggestive of either a subacute to chronic viral infection or a postinfectious, immune-mediated process.48
CSF typically shows pleocytosis, with elevated protein levels.48 The diagnosis is made by the isolation of Epstein-Barr virus DNA from CSF, as well as from peripheral blood monocytes, and the exclusion of other herpesvirus infections through PCR techniques.48,51 Serological evidence is supportive.48 Findings on MRI are variable and may demonstrate increased T2 signal intensity; however, normal findings are also reported in the presence of neurological deficits.48
Unfortunately, no definitive treatment for Epstein-Barr virus nervous system infections exists. Although corticosteroids and immunoglobulins have been used, their effect on disease progression is unknown.48 Antiviral agents have not been shown to be clinically useful. Significant neurological deficits may persist after resolution of the acute phase of myelitis.48,51
Mycoplasma-related myelitis
It is estimated that CNS manifestations, most commonly encephalitis, occur in 1 per 1000 patients with Mycoplasma pneumoniae infections.52 Myelitis, albeit atypical, and radiculitis have been reported in association with meningitis. In 19 cases of Mycoplasma-associated transverse myelitis, patients often developed a flaccid paraparesis, with bladder paresis.11 A definite sensory level was described in only a few cases. There is usually slight CSF pleocytosis, and M. pneumoniae was isolated in at least seven reported cases. In one case, in a patient who had no antecedent respiratory infection, M. pneumoniae was cultivated from a nasopharyngeal aspirate and detected in the CSF by PCR.11,53 Treatment is with doxycycline for 14 days, although the effects of therapy are debated.1 The overall prognosis is generally good, with complete recovery in most cases, although there is one case report of persistent paraplegia after Mycoplasma-associated transverse myelitis.54
Other Rare Infectious Causes of Myelitis
Cases of transverse myelitis associated with Burkholderia pseudomallei (melioidosis) have been described.55 Borrelia recurrentis may also produce myelitis, usually radiculomyelitis rather than isolated myelitis.56 Even so, only 5% of 330 cases of neuroborreliosis were classified as acute meningomyelitis or meningomyeloradiculitis. Transverse myelitis may occur in tuberculous meningitis, usually with severe leptomeningitis.1
Other Myelopathies
Subacute combined degeneration of the cord, classically described in association with vitamin B12 deficiency, affects the posterior and lateral columns of the spinal cord.57 A similar syndrome of posterior column degeneration is also seen in tertiary syphilis (Treponema pallidum infection), and is referred to as tabes dorsalis.57 This manifests clinically with a spastic weakness and ataxia of the lower limbs. Chronic meningeal inflammation may be present and may involve the ventral nerve roots. Other clinical spinal syndromes described in association with syphilitic infection are syphilitic meningomyelitis (a chronic meningitis resulting in subpial myelinated fiber loss, with predominantly bilateral corticospinal tract involvement) and spinal meningovascular syphilis, which may result in an anterior spinal artery syndrome.57
The myelopathy associated with the late stages of AIDS also affects the posterior and lateral columns, as demonstrated in histological studies.1,57 Clinical features of spinal cord disease are often obscured by other neurological complications of AIDS, such as neuropathy and encephalopathy, which result from HIV per se or from opportunistic infections. Vacuolation of myelin and relative sparing of the axons occur, and lipid-laden macrophages are typically abundant.57,58 This vacuolar myopathy was seen in 48% of autopsy examinations of the cord of 90 patients with AIDS, although the frequency of encephalitis was higher in this population.58 There was no correlation between the proviral HIV type I load and the degree of myelopathy.58 Likewise, there was no correlation between opportunistic infections, particularly cytomegalovirus, and the presence and severity of myelopathy. In a separate series of 21 patients with AIDS-associated myelopathy, MRI demonstrated spinal cord atrophy in 15 cases and diffuse intrinsic signal abnormality in six cases.59
Tropical spastic paraparesis is a syndrome of slowly progressive central paraparesis of proximal lower limb dominance, caused by infection with the human T cell leukemia/lymphoma virus type 1 (HTLV-1). Sensory signs are characteristically minimal and usually only in the lower limbs; paresthesias and ataxia, with diminished proprioceptive function and vibration sense, have been described.57,60 Sphincter dysfunction usually occurs early. An associated polyneuropathy has been reported. Levels of CSF antibodies to HTLV-1 are elevated.57 On pathological examination, the corticospinal tracts and posterior columns are affected by an inflammatory myelitis, with focal spongiform demyelination and necrosis.57,60 Foci of gray matter destruction are also seen.
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis occurs typically after childhood viral exanthemata, upper respiratory tract infection, or vaccination, with a short latency.1 Although the majority of postinfectious cases follow viral infections, such as measles, mumps, and Epstein-Barr virus infection, acute disseminated encephalomyelitis can occur after bacterial or parasitic infections.1,2 Most cases are disseminated, as the name implies, with alteration of sensorium; however, in some cases, spinal disease dominates the clinical picture.1
Anterior Horn Cell Syndromes
The hallmark of damage to lower motor neurons is flaccid paralysis. Viral infection and the resultant inflammation cause direct damage to the motor neurons residing in the anterior horns of the spinal cord, producing an anterior myelitis.61 Viral invasion of anterior horn cells usually occurs as part of an acute viral meningitic illness, with fevers, headache, and meningism.61 This is followed by the rapid onset of asymmetrical weakness without any sensory deficits.61 Bulbar and respiratory musculature may be affected, and ventilatory support may be needed. Examination of CSF demonstrates moderate pleocytosis.61 Neurophysiological testing demonstrates low compound muscle action potential, normal sensory nerve action potentials, and sharp waves and fibrillations on electromyography.61
Poliomyelitis
Acute poliomyelitis is now rarely encountered in the industrialized world, because of the World Health Organization’s global eradication program. Nonetheless, pockets of endemic disease persist in sub-Saharan Africa and the Indian subcontinent, and it continues to occur sporadically elsewhere.62 There is also a small incidence of live vaccine–related poliomyelitis.62
Poliomyelitis is caused by an enterovirus whose main route of infection is via the gastrointestinal tract.62 In approximately 5% of infections, after nonspecific flulike symptoms, nervous system infection occurs and is initially manifested as meningitis, with high fevers, neck stiffness, and headache.62 The onset of spinal poliomyelitis is heralded by myalgias, with the subsequent development of asymmetrical, lower limb–predominant flaccid weakness or paralysis, reaching a maximum within 48 hours of onset.62
Diagnosis is based on virus isolation of nasopharyngeal secretions and/or the stool. CSF examination demonstrates elevated protein levels and pleocytosis. Serological diagnosis, with demonstration of antipoliovirus IgG and IgM antibodies, can establish the diagnosis in the absence of viral isolate. PCR techniques now enable rapid diagnosis, identification of serotype, and differentiation of wild-type from vaccine-related disease.63 MRI may demonstrate high signal intensity on T2-weighted imaging in the region of the anterior horn cells.64
There is, unfortunately, no curative treatment.62 Management is supportive, aimed at symptom control with analgesia and prevention of complications. The aim of strict bed rest is to prevent progression of paralysis. Passive exercise during the acute phase, followed by more intensive physiotherapy, can prevent or minimize contractures.65
Poliomyelitis-like Syndromes
Acute flaccid paralysis secondary to anterior horn cell disease may be associated with other enteroviruses (e.g., enterovirus 71, Coxsackie virus A7, echoviruses), tickborne encephalitides, flaviviruses (e.g., Japanese encephalitis, Murray Valley encephalitis virus, and West Nile virus), herpesviruses (cytomegalovirus, Epstein-Barr virus, VZV), and HIV-related opportunistic infections.57,61–68
West Nile virus, a flavivirus, usually causes a mild febrile illness.61 Neurological disease is reported in 1 per 150 infected persons and usually consists of meningitis and encephalitis; however, there are numerous reports of patients with poliomyelitis-like acute flaccid paralysis, occurring during the acute febrile illness.61,68 Diagnosis is based on identification of anti–West Nile virus antibodies in serum or CSF.61 Confirming an anterior horn cell process, high signal intensity in the anterior horns on T2-weighted MRI has been demonstrated.69 Histopathological examination of autopsy specimens has demonstrated anterior myelitis, with perivascular lymphocytic infiltrate, monocytic infiltration, and gliosis.70
Enterovirus 71, which causes outbreaks of hand-foot-and-mouth disease, a common exanthema of childhood characterized by fevers, palmar and plantar rash, and oromucosal ulceration, may be associated with neurological complications, including an acute flaccid paralysis.66
Spinal Epidural Abscess
Spinal epidural abscess is an uncommon condition, occurring with an estimated incidence of 0.2 to 2.8 cases per 10,000 per year.71–74 The incidence peaks in the sixth and seventh decades of life.71 Although this condition is potentially fatal, with devastating neurological sequelae if left untreated, early recognition and prompt institution of appropriate therapy can avert complications.
Risk factors for spinal epidural abscess include immunocompromised states and are identified in Table 40-5.72 In 20% of cases, there is no identifiable risk factor.71,76
AIDS, acquired immunodeficiency syndrome.
The most common causative agent is Staphylococcus aureus, accounting for 57% to 75% of reported cases.72,73 Mycobacterium tuberculosis is the next most frequent cause, accounting for 25% of spinal epidural abscess in one series.77 Tuberculous osteomyelitis of the vertebra (Pott’s disease) can give rise to epidural extension, with abscess formation.57 Tuberculous meningitis may also occur. Gram-positive cocci other than S. aureus were cultured in 10% of patients, gram-negative organisms in 18%, and anaerobes in 2% in one series.75
Fungal infections of the spinal cord are rare, and usually occur in immunocompromised persons.57,76 Blastomyces, Coccidioides, and Aspergillus organisms may invade the epidural space via the intervertebral foramina or as a result of direct extension from a focus of vertebral osteomyelitis.57 Both Coccidioides and Blastomyces organisms may hematogenously seed the spinal cord or surrounding meninges. Cryptococcus infection, which can cause meningoencephalitis, is a rare cause of spinal infection.57
In the majority of cases, spinal epidural abscess is thought to arise from hematogenous spread of organisms from remote mucocutaneous sources, such as the site of pharyngitis or dental sepsis.72 These abscesses are usually located posteriorly in the epidural space, the thoracic spine, or the lumbar spine. Contiguous spread of infection into the epidural space from a source adjacent to the spine, such as that of diskitis or vertebral osteomyelitis, is also well described and usually results in an anterior epidural abscess.72,76 Direct extension from retropharyngeal or retroperitoneal abscesses via the vertebral foramina may occur.72 Blunt trauma precedes spinal epidural abscess in 15% to 35% of cases.78 The postulated mechanism in such cases is hematogenous seeding of the nutrient-rich environment of a vertebral hematoma.
The mechanism leading to neurological dysfunction in spinal epidural abscess is incompletely understood. Neurological deficits are disproportionate to the degree of direct compression of neural structures by the extradural mass within the rigid bony spinal canal.71 Epidural edema and inflammation with involvement of the epidural venous plexus, resulting in venous ischemia and infarction of the spinal cord, are postulated to potentiate the effects of compression.74,75
Clinical Features
Clinical manifestation is usually nonspecific, and symptoms can evolve over a few hours to several months.71 The most common presenting symptoms are fever, malaise, and back pain.71 The presence of overt neurological symptoms, including radicular pain, weakness, and sphincter dysfunction, is suggestive of disease progression. Systemic sepsis may occur. In patients with chronic infection, constitutional symptoms of fever, anorexia, and weight loss may dominate the clinical picture. There may be focal tenderness of the spine, and neurological deficits may be evident.71
Laboratory investigation usually reveals leukocytosis, with elevated levels of inflammatory markers (C-reactive protein and, if the process is more chronic, erythrocyte sedimentation rate). Blood cultures are positive in more than 60% of cases.75
The imaging modality of choice for diagnosis is MRI with gadolinium contrast material, because it enables the abscess to be distinguished from adjacent compressed thecal sac and differential diagnoses such as herniated intervertebral disk (Fig. 40-3).71 During the early phlegmonous stage of infection, homogenous enhancement is seen. Once the necrotic center liquefies, with surrounding inflammatory tissue, peripheral enhancement is seen with gadolinium. Open biopsy or computed tomography–guided aspiration provides tissue for culture, to isolate the causative organism and facilitate directed therapy.71,72
Treatment
The treatment of choice in most patients is surgical evacuation and decompression, followed by intravenous broad-spectrum bactericidal antibiotic therapy for at least 4 to 6 weeks.72 Less invasive endoscopic and percutaneous drainage procedures have been described.79 In patients who present with minimal neurological dysfunction or who are poor surgical candidates, conservative treatment in the form of long-term antibiotic therapy may be undertaken.72 Serial MRI and close neurological monitoring are imperative in these cases; indications for surgical intervention are neurological progression and increasing size of the abscess. Neither conservative management nor surgical treatment has been shown to be superior; evidence indicates that both modalities have good outcomes if treatment is initiated promptly.81,82
Prognosis is dependent on the clinical, particularly neurological, condition of the patient at presentation. Poorer outcomes have been reported when treatment has been delayed.72 Good outcomes are reported when the duration of neurological deficit is less than 72 hours, when the degree of thecal sac compression is less than 50%, and in patients younger than 60 years.82 The rate of mortality from epidural abscess is approximately 14%.72
Intramedullary Spinal Cord Abscess
This rare entity, first described by Hans Chiari in 1900, is indistinguishable on the basis of symptoms from epidural abscess, producing fever, pain, and motor and sensory deficits with progression.57 It may occur in the setting of systemic sepsis, either septicemia or infective endocarditis, with hematogenous seeding of the spinal cord. Contiguous spread from a local source of sepsis, via the intervertebral foramina, is also possible. CSF examination demonstrates pleocytosis, with elevated protein levels. MRI is the imaging modality of choice.57 Causative agents reported in the literature include Listeria monocytogenes and M. pneumoniae. Solitary tuberculoma of the spinal cord is extremely rare.57
Parasitic Spinal Cord Disease
Parasitic infections of the spinal cord are rare; however, schistosomiasis (Schistosoma haematobium, Schistosoma japonicum and Schistosoma mansoni) is a cause of myelitis encountered in eastern Asia, Africa, and South America.57 The ova of the schistosomes evoke a granulomatous myelomeningoradiculitis, with destruction of both gray and white matter. Less commonly, an acute transverse myelitis and spinal cord compression caused by a localized granuloma may be present.57
Vertebral Osteomyelitis
Accounting for approximately 2% to 7% of all cases of osteomyelitis, spinal osteomyelitis occurs more often in elderly persons.83 As in epidural abscess, the most common causative agent, reported to account for more than 50% of cases, is S. aureus.83,84 Hematogenous seeding in association with conditions producing bacteremia, such as infective endocarditis, and, less frequently, with direct inoculation of bacteria result in pyogenic infection.83 Infection is characteristically confined to the vertebral body and intervertebral disk; however, in as many as 20% of cases, the posterior osseous elements are involved.83,84
Clinical features are usually insidious. Back and neck pain is the commonest manifestation, occurring in 90% of affected patients, and neurological deficits secondary to nerve root or spinal cord impingement are reported in 20%.83,84 Complications include spinal deformity secondary to vertebral destruction and epidural abscess formation, both of which may lead to compression of spinal cord and nerve roots.83 Paraspinal or retrospinal abscess formation may occur in association with spinal osteomyelitis.
Laboratory investigations are of limited value. MRI is the preferred diagnostic imaging modality, with an accuracy of approximately 90%, enabling visualization of spinal cord, nerve roots, extradural space, and bony elements.83 Single photon emission computed tomography with gallium 67, a functional imaging modality, has also been shown to be of value in diagnosis and may be a valuable adjunct in patients in whom the diagnosis remains uncertain.83
Early surgical decompression, with internal fixation to provide spinal stability, is the contemporary approach to vertebral osteomyelitis with neurological deficit and focal spinal deformity.85 Nonsurgical management—namely, broad-spectrum intravenous bactericidal antibiotic therapy—is indicated in patients with minimal or no neurological deficits and localized kyphotic deformities, although it may fail, necessitating surgical intervention.85
Spinal Meningitis and Meningomyelitis
Infections of the spinal meninges may involve primarily the pia mater and arachnoid mater, to produce a leptomeningitis, or the dura mater, causing a pachymeningitis.57 These processes may be either acute or chronic. When the spinal cord is also involved, the process is referred to as meningomyelitis. Viral agents reported to cause myelomeningitis and meningitis include Coxsackie viruses, enteroviruses, and herpesviruses.57 Bacteria, including M. tuberculosis and T. pallidum (syphilis), may cause meningomyelitis, as may some fungal and parasitic infections. Involvement of pial blood vessels can lead to thrombosis, with infarction of the spinal cord (myelomalacia).57 Progressive pial constrictive fibrosis (spinal arachnoiditis) can result from chronic meningeal inflammation, resulting in compression of neural structures and their vascular supply.57 Damage to spinal nerve roots, particularly in the dorsal roots in the lumbosacral region, may occur with protracted meningitis, as seen in syphilitic myelomeningitis (tabes dorsalis).57 Clinical features include those of meningeal irritation (fever, neck or back pain) and symptoms and signs attributable to the particular pattern of involvement of spinal structures. Examination of CSF demonstrates pleocytosis, with elevated protein levels.57 Gram and other specialized stains may reveal causative agents in cases of pyogenic meningitis, and microbial culture is an imperative diagnostic investigation. MRI with gadolinium contrast material demonstrates leptomeningeal enhancement.57
DEGENERATIVE DISEASES OF THE SPINAL CORD
Syringomyelia
Primary syringomyelia is a chronic, progressive degenerative disease of the spinal cord, characterized by brachial amyotrophy and dissociated segmental sensory deficit. Cavitation of the central spinal cord is seen, usually in the cervical region, although extension of the process rostrally into the medulla and pons of the brainstem (syringobulbia) or caudally into the thoracic and even the lumbar spine may be seen.57 Syringomyelia may also occur in association with intramedullary spinal cord tumors, mostly gliomas, in 25% to 57% of cases in some series.86 Traumatic myelopathy, radiation myelopathy, myelomalacia (spinal cord infarction), hematomyelia, and spinal arachnoiditis are other associated conditions.57
Bony abnormalities of the vertebral column (e.g., thoracic scoliosis and fusion of vertebrae [Klippel-Feil anomaly]) and the base of the skull (e.g., platybasia or basilar invagination) are frequently associated conditions.57,87 Caudal displacement of the posterior fossa structures—namely, the brainstem and cerebellum—producing a Chiari type I malformation is seen in approximately 90% of cases of syringomyelia.57 A suggested scheme of classification is found in Table 40-6.88
Pathogenesis
A number of hypotheses regarding the mechanism of formation of syrinx have been put forward. Many of these suggest that disturbances in CSF hydrodynamics result in interruption of CSF flow between the spinal and intracranial subarachnoid compartments.57,87,89
An early theory was that syringomyelia arose from the prevention of normal flow of CSF by congenital failure of opening of the outlets of the fourth ventricle (foramina of Luschka and Magendie).90 A pulse wave of CSF pressure, generated by systolic pulsation of the choroid plexuses, is transmitted into the spinal cord via the central canal, resulting in dilatation of the central canal, with a diverticulum that ramifies from the central canal and dissects along gray matter and white matter tracts. Indeed, the frequent association of syrinx with craniocervical junction malformations, which could potentially block the flow of CSF, supports this theory. In a more recent report, it was postulated that the primary event in the formation of syrinx in many cases is reduced volume of the posterior cranial fossa, such as that caused by basilar invagination, with resultant Chiari malformation caused by caudal displacement of structures. Long-standing pulsatile pressure of the herniated structures on the spinal cord leads to syrinx formation as a tertiary event. It was suggested that this was a mechanism serving to protect vital neural structures at the level of the craniovertebral junction at the cost of spinal cord. One report questioned prevailing hydrodynamic theories, on the basis of the facts that syrinx pressure exceeds CSF pressure and the composition of syrinx fluid is not identical to that of CSF.89 An alternative theory is abnormal spinal cord structure, with a tendency to cavitate.57,87
The syrinx initially occupies the central gray matter of the cervical spinal cord, interrupting the crossing spinothalamic tract fibers in the anterior commissure over several cord segments. Progressive symmetrical or asymmetrical enlargement leads to interruption of the posterior and anterior horns of the spinal cord and, finally, the white matter funiculi (lateral and posterior).57
Clinical Features
Types I and II syringomyelia are sporadic and usually begin in middle age; in some cases, an abnormality may be evident at birth. Symptoms develop insidiously, although there are reports of sudden and dramatic worsening after violent strain or a paroxysm of coughing.57 Intermittent progression is the usual course, with patients being bedridden within 5 to 10 years; however, the course is extremely variable, and some patients remain stable for many years.57
The clinical picture varies according to subtype of syringomyelia, particularly the presence or absence of a Chiari malformation. The cross-sectional and longitudinal extent of syrinx determines the clinical profile at any given time in the evolution of the disease. Pain, which is usually unilateral or asymmetrical, is experienced by approximately 50% of patients with types I and II syringomyelia.57
Segmental weakness and atrophy of the upper limbs, which may be asymmetrical, with loss of deep tendon reflexes, are also characteristic. However, there are exceptions in which motor function is preserved. Eventually, spastic lower limb weakness and ataxia develop, with impairment of lower limb proprioception and vibration sense, as a result of corticospinal and posterior column involvement in the inferior medulla and cervical spinal cord, respectively. In some instances, especially when there is a coexistent Chiari malformation, an upper motor neuron pattern of weakness may also be seen in the upper limbs. The presence of a Chiari malformation or a foramen magnum lesion is also indicated by nystagmus, cerebellar ataxia, and occipitonuchal pain with maneuvers that increase intracranial pressure.57 Ipsilateral Horner’s syndrome results with involvement of the intermediolateral cell column at C8, T1, and T2 levels.57
MRI is the investigation of choice, demonstrating syrinx and associated abnormalities, including Chiari malformation and foramen magnum lesions (Fig. 40-4).57
Treatment
Several surgical procedures have been reported for the treatment of syringomyelia, with the approach tailored to the individual clinical and imaging characteristics.91 There have been no controlled, prospective, or multi-institutional studies on which to base management decisions, reflective of the complexity of treatment selection. The two major approaches to types I and II are syringosubarachnoid shunting and decompression of the foramen magnum and upper cervical spinal canal.91 Decompression has been reported to ameliorate lower limb signs and symptoms; however, upper limb sensorimotor deficits persist.92 Conversely, shunting has produced unpredictable results, although there is some evidence that it may be superior to decompression as a first-line treatment for syringomyelia with Chiari I malformation.57
Cervical Spondylotic Myelopathy
Cervical spondylotic myelopathy (CSM) results from narrowing of the spinal canal, secondary to degenerative or congenital vertebral and disk disease. It is the commonest spinal cord disease in patients older than 55 years.93–95 Clinical manifestations are diverse, and it is often asymptomatic.
Pathophysiology
The primary pathophysiological abnormality is reduced sagittal diameter of the spinal canal, and a dimension of less than 13 mm is thought to be critical in the later development in relation to superimposed degenerative change.93 The mechanical factors involved in the process of CSM fall into two groups: static factors (Table 40-7) and dynamic changes in the spinal column and spinal cord during flexion and extension of the cervical spine under normal physiological loads.93,96 Direct compression of neural structures, as a result of these mechanical factors, is compounded by ischemia of the spinal cord, secondary to compression of the arterial supply (either larger vessels, such as the anterior spinal artery, or the pial plexuses and small penetrating arteries) or draining veins of the spinal cord and nerve roots by degenerative elements.93 Indeed, the highest frequency of myelopathy occurs at the C5-C7 level, which has the most tenuous vascular supply.93
TABLE 40-7 Static Factors Contributing to Cervical Spondylotic Myelopathy
Clinical Features
Symptoms and signs are varied, and there are no pathognomonic clinical features. Onset tends to be insidious. Neck, subscapular, or shoulder pain may occur. Loss of hand dexterity may be a marked early feature. Gait disturbance, the commonest presenting symptom of CSM, is a manifestation of long tract dysfunction, with upper motor neuron weakness resulting from corticospinal tract involvement and compounded by proprioceptive (dorsal column) dysfunction.93 Bowel and bladder dysfunction is also common, reported to occur in 15% to 50% of affected patients.93–98 Segmental lower motor neuron deficits are seen at the compressed level of the spinal cord, most commonly at the C5-C6 level.
Diagnosis
MRI is the imaging modality of choice for evaluating cervical degenerative disease, enabling evaluation of the spinal cord, vertebral bodies, disks, osteophytes, and ligaments.93 In addition to indentation, T2 hyperintensity of the spinal cord may be seen at the level of compression, caused by edema, inflammatory changes, ischemia, myelomalacia, or gliosis (Fig. 40-5).93
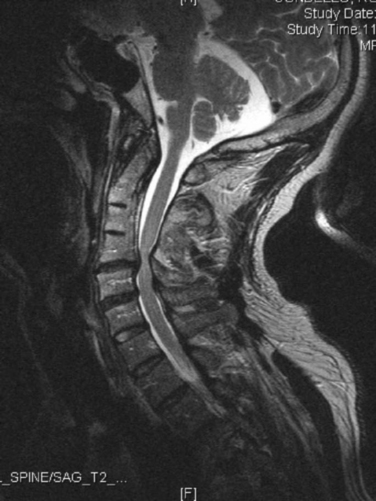
The responsible lesion may be difficult to distinguish from clinically silent cord compression on MRI, particularly in elderly patients. In order to avert the high rate of morbidity associated with extensive spinal surgery, the use of electrophysiological measurement of spinal cord evoked potentials may be a valuable adjunctive tool for localizing the spinal level at which clinically significant compression occurs in cervical myelopathy.99 Most often, however, careful clinical judgment is necessary to distinguish clinically relevant CSM from the sometimes striking incidental changes seen in older patients. Some clinicians believe that flexion-extension views of the spine are a useful discriminatory test.
Treatment
Traditionally, first-line management is conservative, because of the significant morbidity associated with spinal surgery.93 However, there are a number of arguments in favor of surgery over medical management. Up to 70% of patients treated medically show no improvement or deteriorate neurologically, as measured by the degree of motor disability and the ability to perform activities of daily living.93 Furthermore, patients with CSM are at increased risk of spinal cord injury from relatively mild traumatic events, because the spinal cord has limited room to move.93 Finally, it has been shown that early surgical intervention can improve prognosis, particularly if symptoms are of less than 1 year’s duration.95,100,101
The two traditional surgical approaches to CSM are a dorsal approach—namely, cervical laminectomy—and a ventral approach, either diskectomy or corpectomy at one or more levels, with interbody fusion.93 After corpectomy, cervical plating is typically necessary to provide stability until fusion occurs.93 No significant difference in outcome of one approach over the other has been demonstrated.93,98,101,102 The choice is therefore guided by the surgeon’s preference, the relative location of the stenosis (dorsal versus ventral), and the alignment of the spinal cord (kyphosis versus lordosis), as well as by individual patient factors.93 Cervical total disk replacement is an evolving modality for the treatment of disk degeneration and herniation in the cervical spine.103
Spondylotic Lumbar Stenosis
Osteoarthritic or spondylitic changes in the lumbar region, compounded by an unusually small spinal canal, may lead to compression of the cauda equina, with the nerve roots compressed between the posterior surface of the vertebral body and the ligamentum flavum posterolaterally.57 Symptoms of gradually ascending numbness and leg pain, associated with weakness, occur in the upright position or with ambulation (especially downhill) and are relieved by sitting down or lying down with hips and knees flexed.57 The intermittent nature of these symptoms and their relation to ambulation (hence the term neurogenic claudication) may lead to misdiagnosis of lower limb vascular insufficiency. Back pain occurs in varying severity, and sphincter disturbance is rare. Diagnosis is based on MRI demonstration of narrowing of the lumbar spinal canal, and treatment is decompressive surgery.57
Degenerative Lumbar Intervertebral Disk Disease
Herniation of lumbar intervertebral disks is a major cause of morbidity, resulting in severe, chronic, or recurrent low back pain. The incidence peaks in the third to fourth decades of life.57 The disk between the L5 and S1 vertebrae is most commonly involved, with the more cranial intervertebral disks affected with diminishing frequency.57,104 Thoracic disks are rarely affected, accounting for only 0.5% of all surgically confirmed disk prolapses.57
Protrusion of the nucleus pulposus posteriorly (through the annulus) occurs with a flexion injury, with a maneuver that increases intraspinal pressure (e.g., sneezing), or even with a trivial movement, in the setting of degenerative changes within the nucleus pulposus, annulus fibrosus, and posterior longitudinal ligaments. In severe cases, the nucleus pulposus may protrude entirely through the annulus. More often, a fragment extrudes or protrudes through tears in the annulus, usually to a side, to impinge on a nerve root. A large fragment may compress the root or roots against the articular process or lamina, giving rise to radicular symptoms. Because this extruded material is often not resorbed, chronic irritation of neural structures may result. Herniation of the disk into the adjacent vertebral body gives rise to a Schmorl nodule, which may be associated with back pain but no signs or symptoms of nerve root compression.
Clinical Features
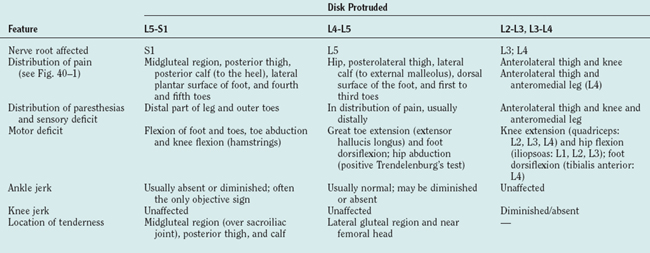
The full complement of features of the syndrome of prolapsed lumbar intervertebral disk is detailed in Table 40-8.57,104 Disk degeneration without herniation may lead to low back pain. The pain of disk herniation can be severe, necessitating bed rest and avoidance of any movement. Signs of nerve root involvement may be elicited on the affected side. The clinical features associated with compression of particular nerve roots are detailed in Table 40-9. Frequently, more than one nerve root is compressed by a single herniated disk, giving rise to multilevel signs and symptoms.57 Large central protrusions can produce bilateral features, as well as cauda equina syndrome; however, this is rare.
|
Position of greatest comfort: dorsal decubitus position (supine, with legs flexed at knees and hips; shoulders elevated on pillows in order to obliterate the lumbar lordosis
|
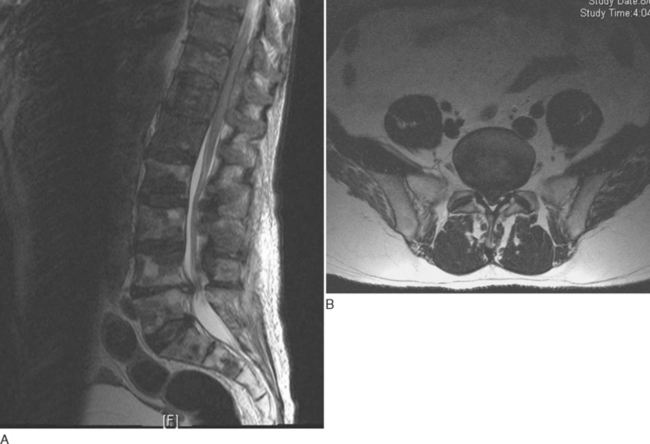
Diagnosis
MRI of the lumbosacral spine is the “gold standard” investigation for diagnosis, demonstrating the protruded disk and annular tears and excluding differential diagnoses such as extradural tumor (Fig. 40-6).104,105 Oblique views, in addition to the traditional protocol of axial and sagittal views, enable visualization of the neural exit foramina, facilitating detection of foraminal nerve root impingement.106 MRI also allows other sites of degenerative disease, including clinically silent lesions, to be identified. Electromyography and nerve conduction studies are valuable adjuncts, particularly when the clinical significance of a lesion is unclear and other differential diagnoses such as a peripheral neuropathy are being entertained. Prolonged F wave latencies and denervation potentials in the paraspinal muscles and in other muscles in a myotomal distribution indicate a radicular rather than a peripheral nerve lesion.57
Treatment
Most patients with herniated lumbar intervertebral disk may be treated conservatively, with analgesia and bed rest for the first 48 hours, followed by gradual mobilization and physiotherapy.107 Indications for surgical management include the presence of cauda equina compression, progressive or profound neurological deficit, and disabling pain after 4 to 6 weeks of conservative management.107 Spinal fusion is the traditional surgical approach.107
TUMORS OF THE SPINAL CORD
Most of these tumors are potentially surgically resectable; therefore, early detection, before the occurrence of irreversible neurological damage, is imperative. Tumors of the spinal cord can be divided into primary neoplasms, arising from the cord itself or the surrounding meninges, and secondary neoplasms, which have metastasized to the spine from elsewhere in the body. Further subdivision, into intramedullary and extramedullary lesions, is based on the anatomical distribution within the spinal canal (Table 40-10). Primary spinal tumors are usually intradural, in contrast to the majority of secondary tumors, which are extradurally based.
|
Arising outside the spinal cord; extradural: arising within the epidural tissues or the vertebral bodies; intradural: arising within the spinal roots or the leptomeninges
|
Epidemiology
Spinal cord tumors are rarer than brain tumors, and in contrast to the latter, the majority are benign.57 A Mayo Clinic series of 8784 primary tumors of the CNS revealed that 15% arose from the spinal cord.57 Most seen in the general hospital population are extramedullary; approximately 40% are intradural-extramedullary, 55% are extradural-extramedullary, and only 5% are intramedullary.57
Clinical Features
The clinical sequelae of most spinal cord tumors result from local compressive effects rather than direct invasion. The onset of symptoms is usually gradual, progressing over weeks to months, and asymmetrical.57 Patients often complain of pain in the spine; about one half experience tenderness of spinous processes over the tumor. The pain is characteristically central or dermatomal and is exacerbated by maneuvers increasing intraspinal pressure, such as sneezing, coughing, and recumbency.
There are three major, sometimes overlapping clinical neurological syndromes that spinal cord tumors can engender, addressed in the overview:57 (1) a sensorimotor spinal tract syndrome, (2) a painful radicular-spinal cord syndrome, and (3) a syringomyelic syndrome. Spinal cord compression, manifested by clinical features of segmental and long tract involvement, is covered in greater detail later in this chapter. It is often preceded by radicular symptoms, which are common.
Tumors in the region of the foramen magnum may produce quadriparesis, with pain in the occipitonuchal region, neck stiffness, weakness, and atrophy of hand and dorsal neck muscles, as well as varying sensory signs.57 In rare cases, extramedullary tumors of the thoracolumbar spine may be associated with dementia and communicating hydrocephalus, ameliorated by shunting and tumor excision.57
Imaging
MRI has enabled characterization of spinal cord tumors, including their location within the spinal canal and internal structure, through the use of different magnetic resonance scanning sequences.108 The basic spinal magnetic resonance study includes T1- and T2-weighted sequences in the sagittal plane, and contrast material (gadolinium)–enhanced T1-weighted images in the sagittal and axial planes.108,109 In comparison with intracranial neoplasms, most spinal cord tumors, even low-grade forms, are enhanced after the administration of intravenous contrast material, which enables differentiation of solid from cystic components of the tumor. Identification of the extent of the solid enhancing component of tumors is vital, because current neurosurgical practice dictates that laminotomy and laminectomy be limited to this area, in order to minimize the surgical morbidity associated with spinal surgery.
Intramedullary Spinal Cord Neoplasms
Intramedullary spinal cord neoplasms are rare, accounting for 4% to 10% of all CNS tumors and 20% of all intraspinal tumors in the adult population.108 They arise within the substance of the spinal cord itself, distorting, invading and destroying white matter tracts and the central gray matter structures.
Primary intramedullary tumors have the same cellular origins as primary tumors of the brain.108 Ninety percent to 95% are gliomas, the majority of these being either ependymomas or astrocytomas, although rarer types such subependymomas do occur in the spinal cord.108 Metastases are far less common in the intramedullary than the extramedullary compartment.57,108
There is usually a long antecedent history before the diagnosis.108 Pain is the most common symptom, with mixed sensorimotor tract symptoms and signs.108 Involvement of the central gray matter leads to a syringomyelic central cord syndrome.57 Sacral sparing has been reported to help distinguish between intramedullary and extramedullary tumors; however, no clinical feature is unique to intramedullary tumors.108 Therefore, distinction between the clinical profiles of intramedullary and extramedullary lesions is seldom dependable, and imaging is vital for diagnosis.
The key findings in intramedullary spinal cord tumors on MRI are as follows:108,110,111
Polar cysts, located at the rostral or caudal extent of solid lesions, represent reactive dilatation of the central canal (syringomyelia). They are seen in approximately 60% of intramedullary spinal tumors and are not enhanced with contrast material.108 Conversely, tumoral cysts occur within the tumor itself and usually demonstrate peripheral enhancement. They occur more commonly in astrocytomas than in ependymomas.108
There is an increased incidence of intramedullary spinal cord tumors in patients with neurofibromatosis.112 Ependymomas are associated with type 2 disease, whereas astrocytomas are seen more frequently in type 1 disease. Interestingly, cytogenetic analysis has revealed the mutations of type 2 neurofibromatosis in some cases of sporadic spinal cord ependymoma.
Glial Neoplasms
Ependymoma
Ependymoma is the most common intramedullary spinal neoplasm in adults, reported to account for 60% of all glial cord tumors.113 The mean age at presentation is 38.8 years, and there is a male preponderance.108,113–115 They are believed to arise from the ependymal cells that line the central canal and are usually slow-growing tumors, which displace rather than infiltrate adjacent neural tissue; the mean duration of symptoms before diagnosis is 36.5 months.108,113–114 They tend to produce symmetrical expansion of the cord, as a result of their central location. Syringomyelia occurs in 9% to 50% of cases.108 Polar cysts are common. The cervical region is most often affected (44% of cases), with 23.5% of cases occurring at the cervicothoracic junction, and 26% occur in the thoracic cord alone.108 Metastasis to the retroperitoneum, lungs, and lymph nodes, may occur.115
Six histological subtypes are recognized: cellular (the most common type), papillary, clear cell, tanycytic, myxopapillary, and melanotic (the rarest).116 Almost all ependymomas are, however, low grade (World Health Organization classification grade I or grade II).108 Myxopapillary ependymomas, constituting 13% of all spinal ependymomas, tend to occur in the conus medullaris and filum terminale, in contrast to other subtypes, and consequently are the most common neoplasm in this region of the cord.108
Most affected patients have relatively mild symptoms and are ambulatory at presentation.108 Back or neck pain is reported in 67%.114,117,118 There are often no objective neurological signs. Sensory deficits predominate and are reported in 52%, probably caused by compression or interruption of spinothalamic tracts.108 Motor weakness is reported in 46% but may predominate in very large ependymomas, and sphincter dysfunction occurs in 15%.113,114,117,118 In rare cases, ependymomas may cause subarachnoid hemorrhage.119
On computed tomographic imaging, ependymomas appear either isoattenuated or slightly hyperattenuated in relation to the adjacent normal spinal cord tissue, but they are enhanced intensely after the administration of iodinated intravenous contrast material.113 On T1-weighted MRI (Fig. 40-7), most are isointense or hypointense in relation to normal spinal cord, but they are hyperintense on T2-weighted sequences.120,121 Approximately 20% to 33% of these tumors demonstrated a rim of hypointensity at the poles, the “cap sign,” on T2-weighted images, corresponding to hemosiderin deposition as a result of hemorrhage.117,120,121 Edema surrounding the tumor mass is seen in 60% of cases.117 At least some enhancement is seen with gadolinium in 84% of cases, with 89% of those displaying well-defined margins on contrast material–enhanced images.120,121
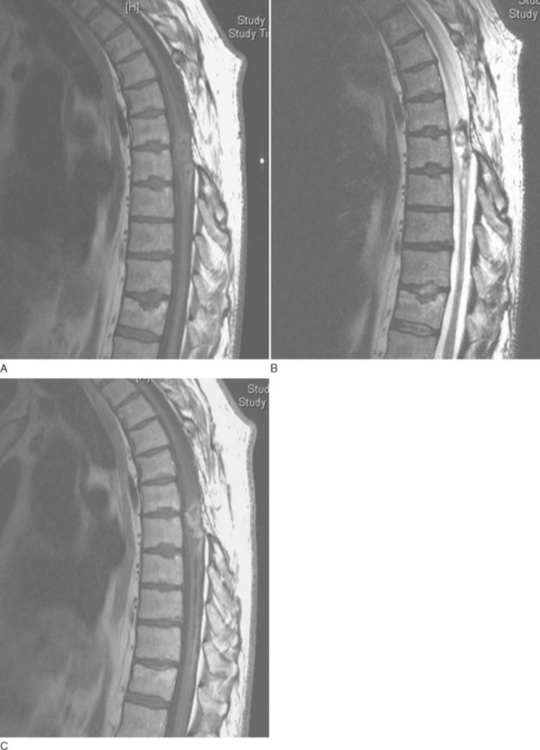
The treatment of choice is laminectomy and microsurgical resection.113,117,118 Adjuvant radiotherapy is reserved for cases of recurrent disease.113,117 In the immediate postoperative period, patients often deteriorate neurologically; this is secondary to edema and possibly transient interference with spinal cord blood flow. Preoperative neurological status is the key predictor of ultimate outcome.114 Milder neurological deficits at presentation, smaller lesions, and shorter symptom duration are factors associated with better postoperative outcome. The 5-year survival rate is 82%, regardless of neurological status at presentation.108,113
Astrocytoma
Astrocytomas constitute approximately one third of all spinal cord gliomas and are the most common intramedullary tumor in children. As with ependymoma, there is a male preponderance (58% of cases); however, the mean age at presentation is younger (29 years) (Table 40-11).117 The thoracic cord is most commonly involved (67% of cases), followed by the cervical cord (49%).122 Holocord (involvement of the entire spinal cord) occurs in up to 60% of children with this condition.117
| Feature | Ependymoma | Astrocytoma |
|---|---|---|
| Characteristic population (mean age at presentation) | Adult (38.8 years) | Pediatric (29 years) |
| Location within spinal cord | Central | Eccentric |
| Gross morphology | Circumscribed | Poorly defined |
| Presence of hemorrhage | Common | Uncommon |
| Contrast enhancement | Intense, focal, homogenous | Patchy, irregular |
| Conus medullaris/filum terminale involvement | Yes | No |
Adapted from Koeller KK, Rosenblum S, Morrison AL: Neoplasms of the spinal cord and filum terminale: radiologic-pathologic correlation. Radiographics 2000; 20:1721-1749.
Astrocytomas are characteristically ill-defined, fusiform intramedullary enlargements.117 Small, eccentric tumor cysts are common, and syringes may be present. Histologically, astrocytomas are highly cellular and have no surrounding capsule, as a result of which malignant cells infiltrate along the network of normal astrocytes, oligodendrocytes, and axons.115 The degree of pleomorphism correlates with the biological behavior of these tumors. The World Health Organization’s system of classification subdivides astrocytomas into four grades.107,115 Grade I tumors are the most benign, accounting for 75% of spinal cord astrocytomas. Grade II lesions are the classic low-grade astrocytomas, whereas grade III lesions, known as anaplastic astrocytomas, are less differentiated. Grade IV astrocytomas, known as glioblastoma multiforme and displaying the most aggressive biological behavior, are rare in the spinal cord, constituting only 0.2% to 1.5% of spinal cord astrocytomas.
The most common manifestations are pain and sensory deficit, occurring in approximately 53% of cases.117 In contrast to ependymomas, in which dysesthesias dominate the sensory symptoms as a result of spinothalamic tract involvement, astrocytomas tend to manifest with paresthesias.118 Motor dysfunction is less common (41.4%), and sphincter dysfunction is infrequent.117 On examination, objective neurological deficits are often absent in lower grade (grades I and II) astrocytomas, which results in diagnostic delay.117 In young children, symptoms occur sooner, with a median duration of 5 months before diagnosis; however, the lesions tend to be low grade, with slow growth and low recurrence rates.123
MRI features of astrocytomas include poor margination and isointensity or hypointensity in relation to normal spinal cord on T1-weighted sequences, with contrast material enhancement.108,123 On T2-weighted imaging, these lesions are hyperintense. In contrast to ependymomas, astrocytomas tend to occur eccentrically (57%) in the cord, reflective of their parenchymal origin, and the “cap sign” is not seen.124 Leptomeningeal spread is reported for 60% of intramedullary glioblastoma multiforme tumors.125
The prognosis for adult patients with astrocytoma is worse than that for ependymoma; mortality rates are much higher than in patients with comparable ependymomas, even in cases of gross total resection.126 This is probably reflective of the infiltrative nature of astrocytomas, with extension of the neoplastic cells well beyond the gross margin of the tumor, which therefore necessitates more extensive resection of spinal cord, with increased risk of postsurgical neurological deficits.126
The treatment of these tumors is therefore somewhat controversial.108 Some neurosurgeons advocate gross total resection, whereas others prefer initial intraoperative biopsy and frozen section examination, proceeding to debulking in high-grade lesions.117,126 As malignant cells invariably remain despite apparent total macroscopic debulking, gross total resection may not be carried out, with the aim of averting significant neurological morbidity.126
Radiotherapy may be administered, with the aim of eradicating residual disease. Annual surveillance MRI is recommended after treatment.126
Nonglial Neoplasms
Hemangioblastoma
Constituting 1.0% to 7.2% of all spinal cord neoplasms, these tumors usually occur in patients younger than 40 years and may be extramedullary in 25%.108,117,127 They grow slowly and usually involve either the thoracic (50%) or cervical (40%) cord.108 Most cord hemangioblastomas are solitary; however, multiple lesions are seen in the von Hippel–Lindau syndrome, which is reported to occur in one third of patients with hemangioblastoma.128,129
Spinal hemangioblastomas are highly vascular, circumscribed lesions, which characteristically display prominent dilated, tortuous surface vessels.117,127 Histopathologically, the tumors are composed of sheets of large, pale stromal cells with hyperchromatic nuclei, interspersed with blood vessels of varying sizes.108 Foci of hemorrhage are usually prominent.108
Clinical manifestations, usually slowly progressive, include sensory symptoms (39%), usually impaired proprioception; motor dysfunction (31%); and pain (31%).108 Subarachnoid hemorrhage and hematomyelia are rare complications.128
Spinal angiography demonstrates a highly vascular mass, with a dense, prolonged blush and prominent draining veins.108 On T1-weighted MRI, hemangioblastomas display varying signal intensity, usually isointensity (50%) or hyperintensity (25%) in relation to normal cord, with intense and homogenous contrast material enhancement.117,124 High signal intensity, with flow voids, is seen on T2-weighted images; however, there are reports of hemangioblastomas that are not visible on this sequence.108,117 Three-dimensional magnetic resonance angiography can provide valuable noninvasive preoperative information regarding the architecture of feeding vessels.130
Metastases
Intramedullary spinal metastases are rare, reported to be found in 0.9% to 2.1% of patients with cancer at autopsy.131 The most common primary cancer to metastasize to the spinal cord is lung carcinoma, followed by breast carcinoma (11%) (Table 40-12).132 The cervical cord is most commonly affected (45%) and then the thoracic cord (35%); the lumbar spinal cord is infrequently involved (8%).132 Most metastases are solitary.132
| Primary Tumor | |
|---|---|
| Lung carcinoma | 40%-85% |
| Breast | 11% |
| Melanoma | 5% |
| Renal cell carcinoma | 4% |
| Colorectal carcinoma | 3% |
| Unknown primary tumor | 5% |
Symptoms progress rapidly, in contrast to the gradual onset of symptoms characterizing primary intramedullary neoplasms.108 Weakness is almost universal, and pain (70% of cases), sphincter dysfunction (60%), and paresthesias (50%) are other common manifestations.108,132
MRI typically reveals mild cord expansion over several segments, with a central area of low signal intensity on T1-weighted imaging and high signal intensity, reflecting edema and tumor infiltration, on T2-weighted sequences.133 Intense, homogenous enhancement is seen with administration of contrast material.108 Peritumoral edema is often florid, and cysts are rare, in contrast to primary intramedullary neoplasms.108
Radiotherapy and corticosteroids are the mainstays of therapy in the setting of widely metastatic disease, although microsurgical resection of discrete lesions has been advocated, to improve the quality of remaining life.131,132 Prognosis is almost universally poor, with two thirds of these patients dying within 6 months of diagnosis.131
Lymphoma
CNS lymphoma is a rare condition, accounting for only 1% of all lymphomas.134–136 Most cases of spinal cord lymphoma involve the vertebra and the epidural compartment, although isolated intramedullary lymphoma occurs in 3.3% of cases of CNS lymphoma, and is usually a solitary lesion. The mean age at presentation is 47. The cervical cord is most often affected.108,134–136 Clinical features include weakness, progressive gait dysfunction, and sensory disturbance.135
Spinal cord lymphoma is usually of B cell phenotype, as demonstrated on immunohistochemistry study, although primary spinal cord T cell lymphomas have been reported, and this may be related to previous human T cell lymphoma virus–related myelopathy.134,135 On MRI, spinal cord lymphomas demonstrate high T2-weighted signal intensity, with intense enhancement after administration of contrast material. Management consists of radiotherapy to the lymphomatous mass, with systemic and intrathecal chemotherapy.134,135
Extramedullary Spinal Cord Neoplasms
Extramedullary tumors arise outside the spinal cord and are subdivided on the basis of their relationship to the dura mater: extradural if they arise within the epidural tissues or the vertebral bodies and intradural if they arise within the spinal roots or the leptomeninges. Intradural tumor growth, accounting for approximately 10% of CNS tumors, usually takes the form of a leptomeningeal carcinomatosis or lymphomatosis.57,137,138
The commonest extramedullary tumors are meningiomas and neurilemmomas (neurofibroma and schwannoma), together constituting 55% of all spinal neoplasms, and these are more often intradural than extradural.57 Metastatic tumors and lymphoma usually occur in the extramedullary extradural compartment and are far commoner than intramedullary disease.57 In fact, extradural myeloma, lymphoma, and carcinoma together form the largest group of spinal tumors. They either seed the extradural space hematogenously or extend directly from vertebral deposits or extraspinal sites via the intervertebral foramina.137
Meningioma
Meningiomas represent 16% to 25% of all spinal tumors and 25% to 46% of primary spinal cord tumors.139–141 They are usually encapsulated and extradural and tend to develop on the lateral or posterolateral surface of the spinal cord, which renders them readily surgically resectable.142 However, an “en plaque” form, which usually occurs intradurally, characterized by growth in a sheetlike or collar-like manner around the spinal cord with infiltration of the pia mater, was reported to occur in 3.1% of cases.143 These rare variants also have a tendency to infiltrate the arachnoid plane and the spinal cord parenchyma and are difficult to resect.142
Meningiomas tend to be slow growing, although the clinical history is shorter with the en plaque variant, and produce symptoms through compression of neural structures.139–141
MRI is the imaging modality of choice for the demonstration of meningiomas, with either isointensity or hypointensity on T1-weighted imaging and hyperintensity on T2-weighted sequences, and intense enhancement with intravenous administration of gadolinium (Fig. 40-8).143 The extent of extradural extension and compression of neural structures can be delineated on MRI. Computed tomography is useful for detecting the presence of calcification, an adverse factor in surgical outcome.141
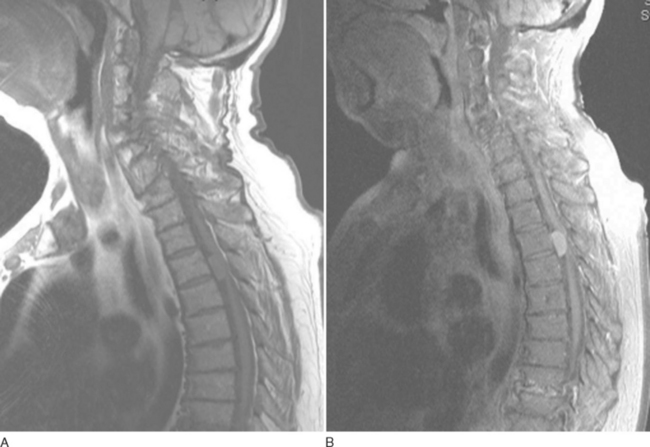
Surgical resection is the mainstay of therapy, with radiotherapy used as an adjuvant treatment or as alternative therapy in patients who are poor surgical candidates.141
Schwannoma
Schwannomas are benign, solitary, slow-growing neoplasms, composed of differentiated neoplastic Schwann cells derived from nerve sheaths.138,144,145 They account for approximately 20% of all primary spinal neoplasms.138,144 They usually occur in the intradural extramedullary compartment, and approximately 40% of intradural extramedullary tumors are either schwannomas or neurofibromas.57,138 Spinal cord schwannomas arise predominantly in association with dorsal sensory nerve roots.144 Treatment is surgical excision, usually via a posterior or posterolateral approach, with good outcomes reported.145
SPINAL CORD COMPRESSION
Metastatic epidural spinal cord compression (MESCC) is defined as compression of the dural sac and its contents (spinal cord and/or cauda equina) by an extradural tumor mass, with lower motor neuron features at this level and upper motor neuron features below this level of the spinal cord.57,146 Radiological evidence for this is indentation of the theca at the level of clinical features.146 Subclinical MESCC is defined as the presence of radiographic features in the absence of clinical features.
It is imperative that symptoms and signs of MESCC be detected early to prevent profound neurological deficits by instituting therapy early. Delay in recognition and inappropriate referral for treatment can result in deterioration and irreversible motor deficit. Education of patients (and clinicians) about the symptoms of MESCC is therefore important, especially for patients known to have vertebral disease or asymptomatic MESCC.146
Epidemiology
A population-based study of 3400 patients from 1990 to 1995 demonstrated that MESCC is relatively common; 2.5% who died from their disease had had at least one admission with this complication in the 5 years preceding death.147 The incidence of MESCC varies widely with the type of primary tumor; 7% of patients with myeloma experienced MESCC, in contrast to 0.2% of patients with pancreatic cancer.147 In one retrospective analysis, breast, lung, and prostate cancers, which have a particular predilection for bony metastasis, accounted for 21%, 24%, and 20%, respectively, of episodes of MESCC; 15% of patients experienced multiple episodes.148
A multivariate analysis of patient, imaging, and neurological factors in 258 patients revealed six major predictive risk factors for MESCC (Table 40-13).146,149 Patients with none of these six risk factors had a 4% risk of developing MESCC, in contrast to a 87% risk of MESCC in those with all six risk factors. The estimated lifetime risk of MESCC was 19.3% for asymptomatic patients with all risk factors identified in Table 40-13, whose primary malignancies were prostate cancer, breast cancer, and renal carcinoma and myeloma. Conversely, the estimated lifetime incidence of MESCC for neurologically asymptomatic patients with leukemia, ovarian, gastric, and pancreatic primary cancers was 0.048%.
TABLE 40-13 Factors Predictive of Risk of Metastatic Epidural Spinal Cord Compression
From Talcott JA, Stomper PC, Drislane FW, et al: Assessing suspected spinal cord compression: a multidisciplinary outcomes analysis of 342 episodes. Support Care Cancer 1999; 7:31-38.
The natural history of untreated MESCC is usually of progressive and relentless pain, paralysis, sensory deficit, and sphincter dysfunction.146 Approximately 70% of patients are reported to suffer loss of neurological function between the onset of symptoms and the start of treatment.150 The majority of delays were attributable to lack of symptom recognition by the patient and diagnostic delay at the primary care level. In a study of 98 patients, Rades and colleagues found that those with slower development of motor deficits before treatment (>14 days) had the best functional outcome, in comparison with patients with more rapid development (<14 days) of motor deficits.151
Clinical Features
Again, a high degree of suspicion for cord compression in patients who present with vague sensorimotor or sphincteric dysfunction is crucial in this clinical emergency. Any suggestion of a sensory or motor level in a neurological syndrome, with or without signs, particularly in patients known to be at risk for cord compression cannot be ignored, and the most significant delays in treatment occur at the primary care level through failure to recognize the syndrome and its importance. Minimizing disability through early therapy is the key to successful treatment. Approximately 90% of patients with MESCC have pain (local back pain or radicular pain), and up to 50% are paretic (nonambulatory and paraparetic) or paraplegic.152–154 Other frequent symptoms of MESCC are weakness, sensory alteration, and bladder dysfunction154 (see Table 40-2 for features of spinal cord compression).
Investigations
Studies evaluating the utility of MRI in identifying MESCC support the use of whole-spine MRI for patients with known malignancy and suspected MESCC.146,155 Findings on MRI are of an epidural mass with indentation of the thecal sac (Fig. 40-9).
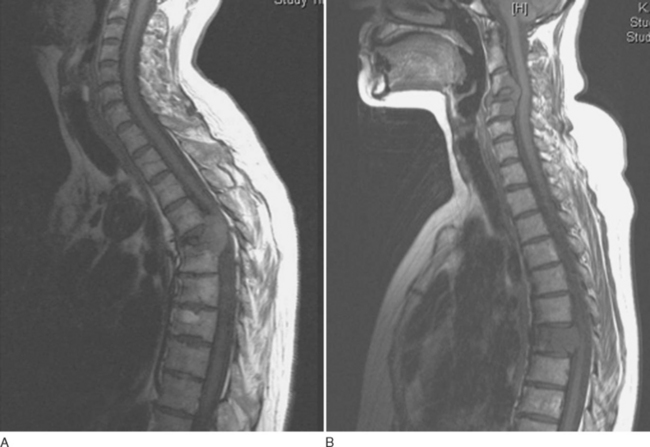
Role of Systemic Corticosteroids
The role of corticosteroids in the management of MESCC is somewhat controversial, and there is a paucity of evidence supporting clinical practice.146 High-dose maintenance dexamethasone has been shown to significantly improve ambulation in comparison with no corticosteroids—however, with an increased incidence of adverse effects, including psychosis and gastric ulcers that necessitate surgery.156 In a small randomized controlled trial (n = 37), high-dose (100-mg) bolus dexamethasone was compared with moderate-dose (10-mg) bolus dexamethasone, before maintenance dexamethasone (16 mg daily for the duration of radiotherapy). Eight percent of patients administered the moderate dose and 20% of those administered high-dose dexamethasone demonstrated improved neurological status, although this difference was not statistically significant.157
Treatment
Radiotherapy
Radiotherapy is the mainstay of treatment of MESCC for both radiosensitive and radioresistant tumors, and many studies have addressed its role. The evidence suggests that patients who have no bony compression stand to benefit most, whereas those with bony compression may require surgical decompression in addition.146 Patients with bony compression, particularly those with mild to moderate motor deficit, were less likely to recover or retain the ability to ambulate after radiotherapy than were patients without bony compression.146
Results of an analysis of ambulatory outcomes in patients with MESCC with and without bony compression or spinal instability suggested that the presence of bony compression is a negative predictive factor for the ability to ambulate after radiotherapy.146 For instance, in one retrospective review of 46 patients treated with radiotherapy for MESCC, 66% (23 of 35) of patients with no bony compression were ambulant after radiotherapy, whereas only 27% (3 of 11) of those with bony compression were able to walk.158
In one of the most publicized trials in the area of management of MESCC, Patchell and colleagues compared maximum decompressive surgical resection in addition to radiotherapy (30 Gy in 10 fractions) with radiotherapy alone, in a randomized, nonblinded multicenter trial.159 The primary endpoints were the ability to walk and ambulatory time after treatment. Patients who underwent combined-modality treatment had better outcomes than did those treated with radiotherapy alone, in terms of more time ambulatory (median ambulation 126 versus 35 days, respectively, P = 0.006) and better analgesic control, in addition to a trend toward improved survival (P = 0.08). Only 3 (19%) of patients undergoing radiotherapy alone regained ambulation, in contrast to 9 (58%) of 16 patients treated with the combined-modality approach (P < 0.03). Criticisms of Patchell and colleagues’ study include its relatively small size and its restrictive inclusion criteria, particularly a single focus of MESCC, which may be unrealistic in the cancer population affected by MESCC and may limit the applicability of this study.
The prescription of radiotherapy used to treat MESCC varies both within and between centers, as reflected in the various study protocols published. Dosages studied include 30 Gy in 10 fractions, 37.5 Gy in 15 fractions, 40 Gy in 20 fractions, 28 Gy in 7 fractions and split doses of 15 Gy in 3 fractions followed by 15 Gy in 5 fractions.146 No one regimen has been demonstrated to be superior to the others for any cohort of patients.146
Patients who develop recompression in-field may be candidates for repeated irradiation, particularly if the latency between the completion of the last course of radiotherapy and recurrence of MESCC is more than 6 weeks. Repeated irradiation may lead to radiation myelopathy in a relatively short interval; however, one retrospective review reported no occurrence of radiation myelopathy when the cumulative dose to the cord was less than 100 Gy.160
Surgery
Before the availability of radiotherapy for MESCC, surgical decompression, via laminectomy, was the mainstay of therapy.146 However, since the introduction of radiotherapy, and in the context of several large trials demonstrating no benefit from surgery alone or in combination with radiotherapy, there is little consensus regarding the operative indications in patients with MESCC. It is generally accepted by those who manage MESCC that spinal instability is an indication for surgical intervention.146,159
Patchell and colleagues found that MESCC patients undergoing surgery in addition to radiotherapy were more likely to retain or regain their ambulatory status than were patients receiving radiotherapy alone (P = 0.006).159 Surgery is, however, associated with significant morbidity in this population of cancer patients, even in the preradiotherapy setting, and may be an essentially palliative procedure.146 Postoperative complication rates of up to 54% have been reported in the literature, as have mortality rates of 0% to 13%, which must be taken into consideration in the decision between surgery and radiotherapy.146
A posterior surgical approach, with laminectomy, is traditionally adopted. However, the compressive mass in most cases of MESCC originates anteriorly, from the vertebral body.161,162 Laminectomy per se does not remove this compressive mass, which may be difficult to access posteriorly.146,162 Furthermore, this approach may contribute to mechanical instability of the spinal column, by removing the intact posterior osseous elements, thereby potentiating spinal cord compression.162 With anterior and lateral surgical approaches, several uncontrolled studies have demonstrated good outcomes, with improved neurological status and analgesia, even in patients with severe motor deficits.163 However, there is an absence of randomized trials supporting these alternative approaches.
Prognosis
The strongest prognostic factor for overall survival and ability to ambulate after treatment is pretreatment neurological status, particularly motor function.146 In one series of patients with MESCC without bony compression, 100% of ambulatory patients retained ambulation after radiotherapy, whereas patients who were ambulatory with assistance, were paraparetic, or were paraplegic had ambulatory rates of 94%, 60%, and 11%, respectively.164 Patients with paralysis either at presentation or after treatment have a much shorter life expectancy than do ambulatory patients.146,153,164
Nonneoplastic Spinal Cord Compression
Various nonneoplastic causes of spinal cord compression have been described. Causes addressed elsewhere in this chapter include benign spinal tumors, spinal epidural abscess, degenerative disk disease, and spondylotic disease. The last is easily the most common cause of cord compression but is often asymptomatic. Paget’s disease causes enlargement of the vertebral bodies, laminae, and pedicles, producing narrowing of the spinal canal, which may lead to a clinical picture of spinal cord compression.57 Other causes include traumatic or osteopenic vertebral fracture, tuberculous granulomata and hemangiomas, and intraspinal (either extradural or intradural) lipoma.57 Rare causes of spinal cord compression include solitary osteochondromas of vertebral bodies, vertebral extramedullary hematopoiesis, and arachnoid diverticula.57
Spinal cord sarcoidosis is another rare cause of spinal cord compression.165 Neurological manifestations occur in 5% of patients with sarcoidosis, but spinal cord lesions occur in fewer than 10% of these. Intramedullary, intradural extramedullary, and extradural disease can occur, with the midcervical and thoracic cord being most often affected. MRI demonstrates isointensity on T1-weighted imaging and typically hyperintensity on T2-weighted imaging.165
Andersen O. Myelitis. Current Opin Neurol. 2000;13:311-316.
Kaplin AI, Krishnan C, Deshpande DM, et al. Diagnosis and management of acute myelopathies. Neurologist. 2005;11:2-18.
McCormick WE, Steinmetz MP, Benzel EC. Cervical spondylotic myelopathy: make the difficult diagnosis, then refer for surgery. Cleve Clinic J Med. 2003;70:899-904.
Nakajima H, Furutama D, Fimura F, et al. Herpes simplex myelitis: clinical manifestations and diagnosis by the polymerase chain reaction. Eur Neurol. 1998;38:163-168.
Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366:643-648.
1 Andersen O. Myelitis. Current Opin Neurol. 2000;13:311-316.
2 Kaplin AI, Krishnan C, Deshpande DM, et al. Diagnosis and management of acute myelopathies. Neurologist. 2005;11:2-18.
3 Jeffery D, Mandler R, Dacis L. Transverse myelitis: retrospective analysis of 33 cases, with differentiation of cases associated with multiple sclerosis and parainfectious events. Arch Neurol. 1993;50:532-535.
4 Berman M, Feldman S, Alter M, et al. Acute transverse myelitis: incidence and aetiologic considerations. Neurology. 1981;31:966-971.
5 Christensen PB, Wermuth L, Hinge HH, et al. Clinical course and long-term prognosis of acute transverse myelopathy. Acta Neurol Scand. 1990;81:431-435.
6 Adrianakos AA, Duffy J, Suzuki, et al. Transverse myelitis in systemic lupus erythematosus: report of three cases and review of the literature. Ann Intern Med. 1975;83:616-624.
7 Nakano I, Mannen T, Mizutani T, et al. Peripheral white matter lesions of the spinal cord with changes in small arachnoid arteries in systemic lupus erythematosus. Clin Neuropathol. 1989;8:102-108.
8 Griffin D. Encephalitis, myelitis and neuritis. In: Mandell G, Bennett J, Dolin R, editors. Mandell, Douglas and Bennett’s Principles and Practice of Infectious Diseases. Philadelphia: Churchill Livingstone; 2000:1009-1016.
9 Gnann J. Mumps virus diseases. In: McKendall R, Stroop W, editors. Handbook of Neurology. New York: Dekker; 1994:563-573.
10 Devinsky O, Cho E, Petito C, et al. Herpes zoster myelitis. Brain. 1991;114:1181-1196.
11 Albucher J, Lauque D, Geyer I, et al. Transverse myelitis caused by Mycoplasma pneumoniae infection. Rev Neurol (Paris). 1995;151:350-353.
12 de Macedo DD, de Mattos JP, Borges TM. [Transverse myelopathy and systemic lupus erythematosus: report of a case and review of the literature]. Arq Neuropsiquiatr. 1979;37:76-84.
13 Nakajima H, Furutama D, Fimura F, et al. Herpes simplex myelitis: clinical manifestations and diagnosis by the polymerase chain reaction. Eur Neurol. 1998;38:163-168.
14 Aurelius E, Johansson B, Skoldenberg B, et al. Rapid diagnosis of herpes simplex encephalitis by nested polymerase chain reaction assay of cerebrospinal fluid. Lancet. 1991;337:189-192.
15 Misra UK, Kalita J, Kumar S. A clinical, MRI and neurophysiological study of acute transverse myelitis. J Neurol Sci. 1996;138:150-156.
16 Lipton HL, Teasdall RD. Acute transverse myelopathy in adults: a follow up study. Arch Neurol. 1973;28:252-257.
17 Sakakibara R, Hattori T, Yasuda K, et al. Micturition disturbance in acute transverse myelitis. Spinal Cord. 1996;34:481-485.
18 Runmarker B, Andersen O. Prognostic factors in a multiple sclerosis incidence cohort with twenty-five years of follow up. Brain. 1993;116:117-134.
19 Scott T, Bhagavatula K, Snyder R, et al. Transverse myelitis: comparison with spinal cord presentations of multiple sclerosis. Neurology. 1998;50:429-433.
20 Bakshi R, Kinkel RR, Mechtler LL, et al. Magnetic resonance imaging findings in 22 cases of myelitis: comparison between patients with and without multiple sclerosis. Eur J Neurol. 1998;5:35-48.
21 Kibe T, Fujimoto S, Ishikawa T, et al. Serial MRI findings of benign poliomyelitis. Brain Dev. 1996;18:147-149.
22 Takahashi S, Miyamoto A, Oki J, et al. Acute transverse myelitis caused by ECHO virus type 18 infection. Eur J Paediatr. 1995;154:378-380.
23 Ku B, Lee K. Acute transverse myelitis caused by Coxsackie virus B4 infection: a case report. J Korean Med Sci. 1998;13:449-453.
24 Graber D, Fossoud C, Grouteau E, et al. Acute transverse myelitis and Coxsackie A9 virus infection. Paediatr Infect Dis J. 1994;13:77.
25 Minami K, Tsuda Y, Maeda H, et al. Acute transverse myelitis caused by Coxsackie virus B5 infection. J Paediatr Child Health. 2004;40:66-68.
26 Bansal R, Kalita J, Misra U, et al. Myelitis: a rare presentation of mumps. Paediatr Neurosurg. 1998;28:204-206.
27 Bajaj NP, Rose P, Clifford-Jones R, et al. Acute transverse myelitis and Guillain-Barré syndrome overlap with serological evidence for mumps viraemia. Acta Neurol Scand. 2001;104:239-242.
28 Gobbi C, Tosi C, Stadler C, et al. Recurrent myelitis associated with herpes simplex virus type 2. Eur Neurol. 2001;46:215-219.
29 Petereit HF, Bamborschke S, Lanfermann H. Acute transverse myelitis caused by herpes simplex virus. Eur Neurol. 1996;36:52-53.
30 Klatersky J, Cappel R, Snoeck JM, et al. Ascending myelitis in association with herpes simplex virus. N Engl J Med. 1972;287:182-184.
32 Kueker W, Schaade L, Ritter K, et al. MRI follow up of herpes simplex virus (type 1) radiculomyelitis. Neurology. 1995;52:1102-1103.
33 Shyu WC, Lin JC, Chang BC, et al. Recurrent ascending myelitis: an unusual presentation of herpes simplex virus type 1 infection. Ann Neurol. 1993;34:625-627.
34 Hirai T, Korogi Y, Hamatake S, et al. Case report: varicellazoster virus myelitis—serial MR findings. Br J Radiol. 1996;69:1187-1190.
35 Lakeman FD, Whitley RJ. Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis. 1995;171:857-863.
36 Gilden DH, Beinlich B, Rubinstein EM, et al. Varicellazoster virus: an expanding spectrum. Neurology. 1994;44:1818-1823.
37 Gilden DH, Mahalingam R, Dueland AN, et al. Herpes zoster: pathogenesis and latency. Melinck J, editor. Progress in Medical Virology. vol 39. Basel, Switzerland: S. Karger; 1992:19-75.
38 McKelvie PA, Collins S, Thyagarajan D, et al. Meningoencephalomyelitis with vasculitis due to varicella zoster virus: a case report and review of the literature. Pathology. 2002;34:88-93.
39 Morita Y, Osaki Y, Doi Y, et al. Chronic active VZV infection manifesting as zoster sine herpete, zoster paresis and myelopathy. J Neurol Sci. 2003;212:7-9.
40 Gilden DH, Bennett JL, Kleinschmidt-deMasters BK, et al. The value of cerebrospinal fluid antiviral antibody in the diagnosis of neurological disease produced by varicella zoster virus. J Neurol Sci. 1998;159:140-144.
41 Chretien F, Gray F, Lescs MC, et al. Acute varicellazoster virus ventriculitis and meningomyeloradiculitis in acquired immunodeficiency syndrome. Acta Neuropathol (Berlin). 1993;86:659-665.
42 Gray F, Belec L, Lescs MC, et al. Varicellazoster virus infection of the central nervous system in the acquired immune deficiency syndrome. Brain. 1994;117:987-999.
43 Fillet AM, Dumont B, Caumes E, et al. Acyclovir-resistant varicellazoster virus: phenotypic and genetic characteristics. J Med Virol. 1998;55:250-254.
44 Snoek R, Andrei G, De Clercq E. Current pharmacological approaches to the therapy of varicella zoster infections: a guide to treatment. Drugs. 1999;57:187-206.
45 Corti M, Soto I, Villafane NF, et al. [Acute necrotising myelitis in an AIDS patient]. Medicina. 2003;63:143-146.
46 Giobbia M, Carniato A, Scotton PG, et al. Cytomegalovirus-associated transverse myelitis in a non-immunocompromised patient. Infection. 1999;27:228-230.
47 Burke D, Leonard D, Imperiale T, et al. The utility of clinical and radiographic features in the diagnosis of cytomegalovirus central nervous system disease in AIDS patients. Mol Diag. 1999;4:37-43.
48 Majid A, Galetta SL, Sweeney CJ, et al. Epstein-Barr virus myeloradiculitis and encephalomyeloradiculitis. Brain. 2002;125:159-165.
49 Rubin DI, Daube JR. Subacute sensory neuropathy associated with Epstein-Barr virus. Muscle Nerve. 1999;22:1607-1610.
50 Feinberg WM, Zonis J, Minnich LL. Epstein-Barr virus–associated myelopathy in an adult. Arch Neurol. 1984;41:454-455.
51 Merelli E, Bedin R, Sola P, et al. Encephalomyeloradiculopathy associated with Epstein-Barr virus: primary infection or reactivation? Acta Neurol Scand. 1997;96:416-420.
52 Koskiniemi M. CNS manifestations associated with Mycoplasma pneumoniae infections: summary of cases at the University of Helsinki and review. Clin Infect Dis. 1993;17:S52-S57.
53 Abele-Horn M, Franck W, Busch U, et al. Transverse myelitis associated with Mycoplasma pneumoniae infection. Clin Infect Dis. 1997;26:909-912.
54 Heller L, Keren O, Mendelson L, et al. Transverse myelitis associated with Mycoplasma pneumoniae: case report. Paraplegia. 1990;28:522-525.
55 Haran MJ, Jenney AW, Keenan RJ, et al. Paraplegia secondary to Burkholderia pseudomallei myelitis: a case report. Arch Phys Med Rehab. 2001;82:1630-1632.
56 Oschmann P, Dordorf W, Hornig C, et al. Stages and syndromes of neuroborreliosis. J Neurol. 1998;245:262-272.
57 Adams RD, Victor M. Diseases of the spinal cord. In: Adams RD, Victor M, editors. Principles of Neurology. 5th ed. New York: McGraw-Hill; 1993:1078-1116.
58 Shepherd E, Brettle R, Liberski P, et al. Spinal cord pathology and viral burden in homosexuals and drug users with AIDS. Neuropathol Appl Neurobiol. 1999;25:2-10.
59 Chong J, Di Rocco A, Tagliati M, et al. MR findings in AIDS-associated myelopathy. Am J Neuroradiol. 1999;20:1412-1416.
60 Manns A, Hisada M, La Grenado L. Human T-lymphotropic virus type 1 infection. Lancet. 1999;353:1951-1958.
61 Solomon T, Willison H. Infectious causes of acute flaccid paralysis. Curr Opin Infect Dis. 2003;16:375-381.
62 Howard RS. Poliomyelitis and the postpolio syndrome. BMJ. 2005;330:1314-1318.
63 Kilpatrick DR, Nottay B, Yang CF, et al. Group specific identification of poliovirus by PCR using primers containing mixed-base or deoxyinosine residues at positions of codon degeneracy. J Clin Microbiol. 1996;34:2990-2996.
64 Rao DG, Bateman DE. Hyperintensities of the anterior horn cells on MRI due to poliomyelitis. J Neurol Neurosurg Psychiatry. 1997;63:720.
65 Jubelt B, Agre JC. Characteristics and management of postpolio syndrome. JAMA. 2000;284:412-414.
66 Chen CY, Chang YC, Huang C, et al. Acute flaccid paralysis in infants and young children with enterovirus 71 infection: MR imaging findings and clinical correlates. Am J Neuroradiol. 2001;22:200-205.
67 Leis AA, Stokic DS, Polk JL, et al. A poliomyelitis-like syndrome from West Nile virus infection. N Engl J Med. 2002;347:1279-1280.
68 Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358:261-264.
69 Li J, Loeb JA, Shy ME, et al. Asymmetric flaccid paralysis: a neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003;53:703-710.
70 Leis AA, Fratkin J, Stokic DS, et al. West Nile poliomyelitis. Lancet Infect Dis. 2003;3:9-10.
71 Chao D, Nanda A. Spinal epidural abscess: a diagnostic challenge. Am Fam Physician. 2002;65:1341-1346.
72 Mackenzie AR, Laing RBS, Smith CC, et al. Spinal epidural abscess: the importance of early diagnosis and treatment. J Neurol Neurosurg Psychiatry. 1998;65:209-212.
73 Baker AS, Ojemann RG, Swartz MN, et al. Spinal epidural abscess. N Engl J Med. 1975;293:463-468.
74 Hlavin ML, Kaminski KJ, Ross JS, et al. Spinal epidural abscess: a 10 year perspective. Neurosurgery. 1990;27:177-184.
75 Danner RL, Hartman BJ. Update of spinal epidural abscess: 35 cases and review of the literature. Rev Infect Dis. 1987;9:265-274.
76 Vilke GM, Honingford EA. Cervical spine epidural abscess in a patient with no predisposing risk factors. Ann Emerg Med. 1996;27:777-780.
77 Kaufman DM, Kaplan JG, Litman N. Infectious agents in spinal epidural abscess. Neurology. 1980;30:844-850.
78 Verner EF, Musher DM. Spinal epidural abscess. Med Clin North Am. 1985;69:375-384.
79 Hori K, Kano T, Fukushige T, et al. Successful treatment of epidural abscess with percutaneously introduced 4-French catheter for drainage. Anesth Analg. 1997;84:1384-1386.
80 Wang JS, Fellows DG, Vakharia S, et al. Epidural abscess: early magnetic resonance imaging detection and conservative therapy. Anesth Analg. 1996;82:1069-1071.
81 Manfredi PL, Herskovitz S, Folli F, et al. Spinal epidural abscess: treatment options. Eur Neurol. 1998;40:58-60.
82 Khanna RK, Malik GM, Rock JP, et al. Spinal epidural abscess: evaluation of factors influencing outcome. Neurosurgery. 1996;39:958-964.
83 Love C, Patel M, Lonner BS, et al. Diagnosing spinal osteomyelitis: a comparison of bone and Ga-67 scintigraphy and magnetic resonance imaging. Clin Nucl Med. 2000;25:963-977.
84 Sapico FL, Montgomerie JZ. Pyogenic vertebral osteomyelitis: report of nine cases and review of the literature. Rev Infect Dis. 1979;1:754.
85 Rezai AR, Woo HH, Errico TJ, et al. Contemporary management of spinal osteomyelitis. Neurosurgery. 1999;44:1018-1025.
86 Keung YK, Cobos E, Whitehead RP, et al. Secondary syringomyelia due to intramedullary spinal cord metastasis: case report and review of the literature. Am J Clin Oncol. 1997;20:577-579.
87 Goel A. Is syringomyelia pathology or a natural protective mechanism? J Postgrad Med. 2001;47:87-88.
88 Barnett HJM, Foster JB, Hodgson P. Syringomyelia. Philadelphia: WB Saunders, 1973.
89 Levine DN. The pathogenesis of syringomyelia associated with lesions at the foramen magnum: a critical review of existing theories and proposal of a new hypothesis. J Neurol Sci. 2004;220:3-21.
90 Gardner WJ. Hydrodynamic mechanism of syringomyelia: its relationship to myelocele. J Neurol Neurosurg Psychiatry. 1965;28:247.
91 Hida K, Iwasaki Y, Koyanagi I, et al. Surgical indication and results of foramen magnum decompression versus syringosubarachnoid shunting for syringomyelia associated with Chiari I malformation. Neurosurgery. 1995;37:673-678.
92 Logue V, Edwards MR. Syringomyelia and its surgical treatment: an analysis of 75 cases. J Neurol Neurosurg Psychiatry. 1981;44:273.
93 McCormick WE, Steinmetz MP, Benzel EC. Cervical spondylotic myelopathy: make the difficult diagnosis, then refer for surgery. Cleve Clin J Med. 2003;70:899-904.
94 Small JM, Dillin WH, Watkins RG. Clinical syndromes in cervical myelopathy. In: Herkowitz H, Garfin SR, Balderston RA, et al, editors. The Spine. 4th ed. Philadelphia: WB Saunders; 1999:465-474.
95 Montgomery DM, Brower RS. Cervical spondylotic myelopathy: clinical syndrome and natural history. Orthop Clin North Am. 1992;23:487-493.
96 White AA, Panjabi MM. Biomechanical considerations in the surgical management of cervical spondylitic myelopathy. Spine. 1988;13:856-860.
97 Lundsford LD, Bissonette D, Dorub D. Anterior surgery for cervical disc disease, part 2. J Neurosurg. 1980;53:12-19.
98 Hukuda S, Mochizuki T, Ogata M, et al. Operations for cervical spondylotic myelopathy: a comparison of the results of anterior and posterior procedures. J Bone Joint Surg Br. 1985;67:609-615.
99 Tanaka N, Fujimoto Y, Yasunaga Y, et al. Functional diagnosis using multimodal spinal cord evoked potentials in cervical myelopathy. J Orthopaed Sci. 2005;10:3-7.
100 Phillips DG. Surgical treatment of myelopathy with cervical spondylosis. J Neurol Neurosurg Psychiatry. 1973;36:879-884.
101 Ebersold MJ, Pare MC, Quast LM. Surgical treatment for cervical spondylotic myelopathy. J Neurosurg. 1995;82:745-751.
102 Iwasaki M, Ebara S, Miyamoto S, et al. Expansive lamino-plasty for cervical radiculomyelopathy due to soft disc herniation. Spine. 1996;21:32-38.
103 Durbukhula MM, Ghiselli G. Cervical total disc replacement part I: rationale, biomechanics and implant types. Orthop Clin North Am. 2005;36:349-354.
104 Boden SD, Wiesel SW. Lumbar spine imaging: role in clinical decision making. J Am Acad Orthop Surg. 1996;4:238-248.
105 Haughton VM. MR imaging of the spine. Radiology. 1988;166:297-301.
106 Humphreys SC, An HS, Eck JC, et al. Oblique MRI as a useful adjunct in evaluation of cervical foraminal impingement. Loyola Univ Orthop J. 1997;6:4-7.
107 Humphreys SC, Eck JC. Clinical evaluation and treatment options for herniated lumbar disc. Am Fam Physician. 1999;59:575-582. 587–588.
108 Koeller KK, Rosenblum S, Morrison AL. Neoplasms of the spinal cord and filum terminale: radiologic-pathologic correlation. Radiographics. 2000;20:1721-1749.
109 Sze G, Stimac GK, Bartlett C, et al. Multicenter study of gadopentetate dimeglumine as an MR contrast agent: evaluation in patients with spinal tumors. Am J Neuroradiol. 1990;11:967-974.
110 Takemoto K, Matsumura Y, Hashimoto H, et al. MR imaging of intraspinal tumors: capability in histological differentiation and compartmentalisation of extramedullary tumors. Neuroradiology. 1988;30:303-309.
111 Lee M, Epstein FJ, Rezai AR, et al. Nonneoplastic intramedullary spinal cord lesions mimicking tumors. Neurosurgery. 1998;43:788-795.
112 Lee M, Rezai AR, Freed D, et al: Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery 196; 38:32–37.
113 Ferrante L, Mastronardi L, Celli P, et al. Intramedullary spinal cord ependymomas: a study of 45 cases with long term follow up. Acta Neurochir. 1992;119:74-79.
114 Hoshimaru M, Koyama T, Hashimoto N, et al. Results of microsurgical treatment for intramedullary spinal cord ependymomas: analysis of 36 cases. Neurosurgery. 1999;44:264-269.
115 Wolff M, Santiago H, Duby MM. Delayed distant metastasis from a subcutaneous sacrococcygeal ependymoma: case report with tissue culture, ultrasound observation and review of the literature. Cancer. 1972;30:1046-1067.
116 Burger PC, Scheithauer BW. Tumors of neuroglia and choroid plexus epithelium. In: Burger PC, Scheithauer BW, editors. Tumors of the Central Nervous System. Washington, DC: Armed Forces Institute of Pathology; 1994:25-161.
117 Brotchi J, Fischer G. Treatment. In: Fisher G, Brotchi J, editors. Intramedullary Spinal Cord Tumors. Stuttgart, Germany: Thieme; 1996:60-84.
118 Epstein FJ, Fermer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
119 Djindjian M, Djindjian R, Houdart R, et al. Subarachnoid haemorrhage due to intraspinal tumors. Surg Neurol. 1978;9:223-229.
120 Fine MJ, Kricheff II, Freed D, et al. Spinal cord ependymomas: MR imaging features. Radiology. 1995;197:655-658.
121 Kahan H, Sklar EML, Post MJD, et al. MR characteristics of histopathologic subtypes of spinal ependymoma. Am J Neuroradiol. 1996;17:143-150.
122 Epstein F, Epstein N. Surgical treatment of spinal cord astrocytomas of childhood. J Neurosurg. 1982;57:685-689.
123 Constatini S, Houten J, Miller D, et al. Intramedullary spinal cord tumors in children under the age of 3 years. J Neurosurg. 1996;85:1036-1043.
124 Froment JC, Baleriaux D, Turjman F, et al. Fisher G, Brotchi J, editors. Intramedullary Spinal Cord Tumors. Stuttgart, Germany: Thieme, 1996.
125 Ciapetta P, Salvati M, Capoccia G, et al. Spinal glioblastomas: report of seven cases and review of the literature. Neurosurgery. 1991;28:302-306.
126 Cooper P. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long term results in 51 patients. Neurosurgery. 1989;25:855-859.
127 Murota T, Symon L. Surgical management of hemangioblastoma of the spinal cord: report of 18 cases. Neurosurgery. 1989;25:699-708.
128 Neumann HPH, Eggert HR, Weigel K, et al. Hemangioblastomas of the central nervous system: a 10 year study with special reference to von Hippel–Lindau syndrome. J Neurosurg. 1989;70:24-30.
129 Browne TR, Adams RD, Robertson GH. Hemangioblastoma of the spinal cord. Arch Neurol. 1976;33:435-441.
130 Mascalchi M, Quilici N, Ferrito G, et al. Identification of the feeding arteries of spinal vascular lesions via phase-contrast MR angiography with three-dimensional acquisition and phase display. Am J Neuroradiol. 1997;18:351-358.
131 Costigan DA, Winkelman MD. Intramedullary spinal cord metastasis: a clinicopathological study of 13 cases. J Neurosurg. 1985;62:227-233.
132 Findlay JM, Bernstein M, Vanderlinden RG, et al. Microsurgical resection of solitary intramedullary spinal cord metastases. Neurosurgery. 1987;21:911-915.
133 Post MJD, Quencer RM, Green BA, et al. Intramedullary spinal cord metastasis, mainly of nonneurogenic origin. Am J Neuroradiol. 1987;8:339-346.
134 Koeller KK, Smirniotopoulos JG, Jones RV. Primary central nervous system lymphoma: radiologic-pathologic correlation. Radiographics. 1997;17:1497-1526.
135 Schild SE, Wharen REJr, Menke DM, et al. Primary lymphoma of the spinal cord. Mayo Clin Proc. 1995;70:256-260.
136 Bluemke DA, Wang H. Primary spinal cord lymphoma: MR appearance. J Comput Assist Tomogr. 1990;14:812-814.
137 McCormick PC, Stein BM. Spinal cord tumors in adults. In: Youmans JR, editor. Neurological Surgery. Philadelphia: WB Saunders; 1996:3102-3122.
138 McCormick PC, Post KD, Stein BM. Intradural extramedullary tumors in adults. Neurosurg Clin North Am. 1990;1:591-608.
139 Gezen F, Kahraman S, Çanakci Z, et al. Review of 36 cases of spinal cord meningioma. Spine. 2000;25:727-731.
140 Levy WJ, Bay J, Dohn D. Spinal cord meningioma. J Neurosurg. 1982;57:804-812.
141 Roux FX, Nataf F, Pinaudeau M, et al. Intraspinal meningiomas: review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46:458-464.
142 Nicholas DS, Weller RO. The fine anatomy of the human spinal meninges: light and scanning electron microscopy study. J Neurosurg. 1988;69:276-282.
143 Caroli E, Acqui M, Roperto R, et al. Spinal en plaque meningiomas: a contemporary experience. Neurosurgery. 2004;55:1275-1279.
144 O’Toole JE, McCormick PC. Midline ventral intradural schwannoma of the cervical spinal cord resected via anterior corpectomy and reconstruction: technical case report and review of the literature. Neurosurgery. 2003;52:1482-1486.
145 Conti P, Pansini G, Mouchaty H, et al. Spinal neurinomas: retrospective analysis and long-term outcome of 179 consecutive operated cases and review of the literature. Surg Neurol. 2004;61:34-43.
146 Loblaw DA, Perry J, Chambers A, et al. Systematic review of the diagnosis and management of malignant extradural spinal cord compression: The Cancer Care Ontario Practice Guidelines Initiative’s Neuro-Oncology Disease Site Group. J Clin Oncol. 2005;23:2028-2037.
147 Loblaw DA, Laperriere NJ, Mackillop WJ. A population-based study of malignant spinal cord compression in Ontario cancer patients. Clin Oncol (R Coll Radiol). 2003;15:211-217.
148 Loblaw DA, Smith K, Lockwood G, et al. The Princess Margaret Hospital experience of malignant spinal cord compression. Proc Am Soc Clin Oncol. 2003;22:19.
149 Talcott JA, Stomper PC, Drislane FW, et al. Assessing suspected spinal cord compression: a multidisciplinary outcomes analysis of 342 episodes. Support Care Cancer. 1999;7:31-38.
150 Husband DJ. Malignant spinal cord compression: prospective study of delays in referral and treatment. BMJ. 1998;317:18-21.
151 Rades D, Heidenreich F, Karstens JH. Final results of a prospective study of the prognostic value of the time to develop motor deficits before irradiation in metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2002;53:975-979.
152 Perrin RG. Metastatic tumors of the axial spine. Curr Opin Oncol. 1992;4:525-532.
153 Sundaresan N, Galicich JH. Treatment of spinal metastases by vertebral body resection. Cancer Invest. 1984;2:383-397.
154 Helweg-Larsen S, Sorensen PS. Symptoms and signs in metastatic spinal cord compression: a study of progression from first symptom until diagnosis in 153 patients. Eur J Cancer. 1994;30A:396-398.
155 Cook AM, Lau TN, Tomlinson MJ, et al. Magnetic resonance imaging of the whole spine in suspected malignant spinal cord compression: impact on management. Clin Oncol (R Coll Radiol). 1998;10:39-43.
156 Sorensen S, Helweg-Larsen S, Mouridsen H, et al: Effect of high dose dexamethasone in carcinomatous metastatic spinal cord compression treated with radiotherapy: a randomised trial. Eur J Cancer 194; 30A:22–27.
157 Vecht CJ, Haaxma-Reiche H, van Putten WL, et al. Initial bolus of conventional versus high dose dexamethasone in metastatic spinal cord compression. Neurology. 1989;39:1255-1257.
158 Pigott KH, Baddeley H, Maher EJ. Pattern of disease in spinal cord compression on MRI scan and implications for treatment. Clin Oncol (R Coll Radiol). 1994;6:7-10.
159 Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366:643-648.
160 Wong CS, van Dyk J, Milosevic M, et al. Radiation myelopathy following single courses of radiotherapy and retreatment. Int J Radiat Oncol Biol Phys. 1994;30:575-581.
161 Young RF, Post EM, King GA. Treatment of spinal epidural metastases: randomised prospective comparison of laminectomy and radiotherapy. J Neurosurg. 1980;53:741-748.
162 van den Bent MJ. Surgical resection improves outcome in metastatic epidural spinal cord compression [Comment]. Lancet. 2005;366:609-610.
163 Sundaresan N, Rothman A, Manhart K, et al. Surgery for solitary metastases of the spine: rationale and results of treatment. Spine. 2002;27:1802-1806.
164 Maranzano E, Latini P, Beneventi S, et al. Radiotherapy without steroids in selected metastatic spinal cord compression patients: a phase II trial. Am J Clin Oncol. 1996;19:179-183.
165 Hamasaki T, Noda M, Kamei N, et al. Intradural extramedullary mass formation in spinal cord sarcoidosis: case report and literature review. Spine. 2003;28:E420-E423.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 40 SPINAL DISEASE: NEOPLASTIC, DEGENERATIVE, AND INFECTIVE SPINAL CORD DISEASES AND SPINAL CORD COMPRESSION
The long, cylindrically shaped spinal cord sits within the bony vertebral canal, tightly enveloped by the meninges. It extends from the foramen magnum to the first lumbar vertebra, where it terminates in the conus medullaris. Below this, the lumbosacral nerve roots form the cauda equina. Anatomically, the spinal cord has a distinctive structural arrangement, illustrated in Figure 40-1A. Functionally, the spinal cord plays a key role in sensorimotor conduction. Because of its special features and relatively primitive reflexes, diseases of the spinal cord give rise to a number of distinctive, sometimes overlapping clinical syndromes (Tables 40-1 and 40-2; see Fig. 40-1B).
TABLE 40-1 Red Flag Features Indicative of Cord Disease
TABLE 40-2 Spinal Cord Syndromes
| Syndrome | Features |
|---|---|
| Complete transverse spinal cord syndrome (see Fig. 40-1Bi) | |
| Brown-Séquard (hemicord) syndrome (see Fig. 40-1Bii) | |
| Sensorimotor spinal tract syndrome |
Corticospinal tract involvement: upper motor neuron limb weakness (arm and leg weakness caused by cervical cord lesion; leg weakness caused by a lesion rostral to the upper lumbar spine):
|
| Radicular syndrome | |
| Syringomyelic or central cord syndrome (see Fig. 40-1Biii) | |
| Anterior horn syndrome | Caused by selective involvement of the motor neurons; segmental weakness, atrophy, fasciculations, and absent or diminished reflexes |
| Anterior spinal cord syndrome | Usually associated with vascular lesions of the anterior spinal artery, resulting in ischemia and neurological deficits within its area of supply (see Fig. 40-2); bilateral upper motor neuron weakness and spinothalamic sensory deficit below the lesion; dorsal column function is spared |
| Cauda equina syndrome | Pain usually an early feature; asymmetrical lower motor neuron (areflexic, flaccid, atrophic) paralysis of lower limbs; sphincter dysfunction (urinary or fecal incontinence, diminished anal tone) and radicular sensory loss; may difficult to differentiate clinically from lesions of the lumbosacral plexus |
| Conus medullaris lesions | Early and prominent sphincter dysfunction; back pain; sensory disturbance in sacral dermatomes (perineal: S3-S5; leg: L5-S2); loss of anal tone and reflex; impotence; leg weakness inconsistent, usually mild |
Segmental (myotomal) lower motor neuron weakness, with depressed or absent deep tendon reflexes, muscle atrophy, and flaccidity/decreased tone, may be present at the level of the lesion, as a result of involvement of anterior horn motor neurons or anterior (motor) nerve roots; this helps clinicians localize the lesion (Table 40-3). Segmental (dermatomal) sensory loss may also be present (Fig. 40-2).
TABLE 40-3 Segmental Innervation of Muscles and Tendon and Cutaneous Reflexes
| Segment/Nerve Root | Muscle | Tendon or Cutaneous Reflex |
|---|---|---|
| C3, C4 | Trapezius | — |
| C4, C5 | Rhomboids | — |
| C5 | Deltoid | — |
| C5, C6 | Supraspinatus, infraspinatus, biceps | Biceps reflex |
| C6 | Brachioradialis | Brachioradialis reflex |
| C7 | Triceps, extensor digitorum | Triceps reflex |
| C8 | Flexor digitorum superficialis and profundus | Finger-jerk reflex |
| T1 | Intrinsic muscles of hand | — |
| T7-10 | Upper rectus abdominis | Upper abdominal reflex |
| T10-12 | Lower rectus abdominis | Lower abdominal reflex |
| L1 | Iliopsoas | Cremasteric reflex |
| L2 | Adductor magnus | Adductor and cremasteric reflexes |
| L3, L4 | Quadriceps femoris | Knee-jerk (patellar) reflex |
| L4 | Tibialis anterior | — |
| L5 | Extensor hallucis longus | Plantar reflex |
| L5, S1 | Hamstrings | Ankle-jerk reflex |
| S1 | Extensor digitorum brevis | — |
| S1, S2 | Soleus, gastrocnemius | — |
| S4, S5 | Anal reflex |
Figure 40-2 Distribution of dermatomes on the anterior surface of the body.
(From Merck and Company: Merck Manual of Diagnosis and Therapy, Section 14: Neurologic Disorders; redrawn from Keegan JJ, Garrett FD: The segmental distribution of the cutaneous nerves in the limbs of man. Anat Rec 102:409-437, 1948.)
Radicular symptoms and signs (see Table 40-2) result from irritation and compression of spinal nerve roots by extramedullary spinal cord lesions, usually before the involvement of long tracts (radiculomyelopathy). Radicular features are unusual with intramedullary lesions; when they do occur, they are most often present in the context of demyelinating disease.
Sensorimotor long tract signs usually arise as a result of compression, rather than invasion or destruction, of spinal white matter fasciculi and occur early with intramedullary lesions. Either sensory or motor function may be affected first; the distribution and extent of long tract symptoms and signs are dependent on the site and size of the lesion. A sensory level, below which perception of pain and temperature is altered or lost, is pathognomonic of a spinal cord lesion and should prompt imaging of the spinal cord. Often the signs are subtle in the early stages, and the description of a sensory level should alert the clinician. The highest level of sensory deficit indicates the lower possible level of the lesion, and the pathology may be present anywhere above this level; the sensory level may in fact be well below the actual lesion, and this must be kept in mind when imaging is ordered. Lamination of sensory fibers (see Fig. 40-1A) can give rise to suspended sensory deficits (i.e., isolated sensory deficit) at the level of the lesion or several segments caudally. Sacral sensation may be spared with deeper intramedullary lesions caused by spinothalamic tract lamination. Progressive spinal cord lesions, such as intramedullary neoplasms, may engender sequential deficits.
IMAGING
Magnetic resonance imaging (MRI) has enabled characterization of spinal cord diseases, including their location and internal structure, with the use of different magnetic resonance scanning sequences. The basic spinal MRI study includes T1- and T2-weighted sequences in the sagittal plane and contrast material (gadolinium)–enhanced T1-weighted images in the sagittal and axial planes. Myelography involves the introduction of radiopaque dye to the spinal fluid, through lumbar or sometimes cervical roots, but is limited in resolution and the length of cord that can be imaged in one episode. Obstruction to the cerebrospinal column and distortion and enlargement of the cord may all be directly visualized. Combining myelography with computed tomographic scanning of the spine is useful, allowing a greater extent of the cord to be accurately imaged in the one sitting. Plane films of the spine may reveal bony lesions and other local pathological processes, but their utility is quite limited.
SPINAL CORD INFECTIONS
Potentially any structure of the spinal cord and its surrounding meninges and vertebral column may be involved in infectious processes. The causative agents include bacteria, viruses, parasites, and fungi (Table 40-4).
AIDS, acquired immunodeficiency syndrome; CMV, cytomegalovirus; EBV, Epstein-Barr virus; HSV, herpes simplex virus; HTLV-1, human T cell leukemia/lymphoma virus type 1; VZV, varicellazoster virus.
Infectious and Parainfectious Myelopathies
Myelitis is a process intrinsic to the spinal cord; this definition excludes expansive lesions.1 Transverse myelitis is characterized by a focal inflammatory process within the spinal cord, leading to varying degrees of neural injury and dysfunction of neural pathways passing through the inflamed segment.2 This in turn leads to the abrupt onset of varying degrees of weakness, sensory alteration, and autonomic dysfunction.
The diagnostic criteria for transverse myelitis include the following:1,3
Transverse myelitis forms a part of a spectrum of neuroinflammatory conditions and may occur as part of a multifocal central nervous system (CNS) disease, such as multiple sclerosis; as part of a multisystem disease, such as systemic lupus erythematosus; or as an idiopathic, isolated entity.2 Of all cases of idiopathic transverse myelitis, 30% to 60% were preceded by a respiratory, gastrointestinal, or systemic infectious illness.2–5
The underlying pathogenesis of transverse myelitis is varied, depending on the specific underlying disease process. For instance, connective tissue disease–associated transverse myelitis may be secondary to a CNS vasculitis or thrombotic infarction of the cord.6 Most patients have cerebrospinal fluid (CSF) pleocytosis and foci of blood-brain barrier breakdown within the spinal cord.2
Two large etiological groups of inflammatory transverse myelitis are identifiable: the inflammatory demyelinating autoimmune myelopathies, such as multiple sclerosis, and the usually monophasic infectious encephalomyelitides.1 More than 40 such infectious encephalomyelitides have been identified, of which approximately 10 have been established as “pure infectious” myelitides by CSF virus isolation or polymerase chain reaction (PCR) techniques, which indicate that neurological injury resulted directly from microbial infection.1,2,8 The remainder are believed to be parainfectious, caused namely by a microbial infection, which in turn incites an immune response against neural tissue. In the latter, the infection may be remote from the immunological response.2 The latency between myelitis and preceding infection does not significantly differ between infectious and postinfectious myelitis. An interval of 9 ± 6 days has been reported for parainfectious cases, 5 days in mumps-related myelitis, 12 days in zoster-related myelitis, and 10 days in Mycoplasma-related myelitis.3,9–11
Although diagnosis of infectious and parainfectious transverse myelitis requires identification of the infectious agent within the CNS, some autoimmune mechanisms, such as molecular mimicry and superantigen-mediated disease, require only peripheral immune activation.2 In molecular mimicry, the putative mechanism of neural injury is antibody-mediated damage, secondary to cross-reaction of antibodies against an infectious agent with molecularly similar antigens in host neural tissue.2
Transverse myelitis is associated pathologically on tissue sampling (biopsy or autopsy) with inflammation.2 Both gray and white matter compartments are affected, although in postinfectious myelitis, white matter changes, demyelination, and axonal injury are prominent. Focal infiltration by lymphocytes and monocytes into segments of the spinal cord and perivascular spaces and activation of astroglial and microglial cells are observed.2 In some biopsies performed during the acute phase of myelitis, infiltration of CD4+ and CD8+ lymphocytes is prominent. Subsequently, during the subacute phase, monocyte and macrophage infiltration may become more prominent. In cases in which transverse myelitis is associated with autoimmune disease such as systemic lupus erythematosus, focal areas of ischemia or infarction secondary to vasculitis may be present, without prominent inflammation, a finding not usually seen in most cases of infectious transverse myelitis.12
PCR techniques, performed on serum and CSF, have facilitated the rapid, sensitive, and noninvasive diagnosis of many infectious forms, particularly viral, of myelitis.1,13,14 Diagnosis is often based on the detection of viral DNA in CSF by PCR.13 Further supportive evidence for a particular viral agent as the cause of myelitis, of value when viral PCR results are negative, is the presence of specific immunoglobulins M (IgM) and G (IgG) antibodies in the CSF, suggestive of intrathecal synthesis.1,13 This is especially the case when the serum: CSF ratio of these antibodies is depressed in relation to total IgG and albumin levels.
Infectious and parainfectious transverse myelitis are the focus of discussion in this chapter. The list of antecedent infections includes herpesviruses and Listeria monocytogenes, although in most of these cases, causality has not been established. Infectious and parainfectious transverse myelitis can be further subdivided, on the basis of the components of the neural axis involved, into meningomyelitis, transverse myelitis without meningitis, encephalomyelitis, and anterior horn syndrome with meningitis, among other entities.1 Meningitis is defined here as the clinical syndrome of fever and neck stiffness.1,2
Clinical Features
The clinical picture of acute transverse myelitis is one of rapid evolution of neurological deficit; however, the distribution of dysfunction, including the modalities affected, depends on the extent and distribution of inflammation within the spinal cord.2 On examination, there is often a clearly defined level of sensory dysfunction.2 When the maximum level of neurological deficit is reached, approximately 50% of affected patients are paraplegic, and 80% to 94% of patients experience sensory disturbances, including numbness, paresthesias, and bandlike dysesthesias.2–5,15,16 Manifestations of autonomic dysfunction include urinary urgency, urinary or fecal incontinence, difficulty or inability opening bladder or bowels, and sexual dysfunction.1
Imaging
Acute inflammation is evident on spinal MRI.1,2 This includes swelling of the spinal cord, in inflamed segments, as well as increased signal intensity on T2-weighted imaging. In cases in which nerve roots are involved in the inflammatory process (radiculomyelitis), enhancement of the affected nerve roots with administration of intravenous gadolinium contrast material may be seen.2,13 However, there are several reports in the literature of clinical cases of myelitis in which there is no demonstrable abnormality of the spinal cord on imaging.1,2,13
Differentiating between Infectious/Parainfectious Transverse Myelitis and Multiple Sclerosis
In multiple sclerosis, it is unusual to have complete transverse myelitis; there was only one such case of 308 in the Göteborg series.18 The weakness and numbness tend to be less severe in multiple sclerosis.19 Pathologically, CSF oligoclonal bands present in multiple sclerosis are absent in infectious transverse myelitis.1,19 Differences in MRI of the spine are also reported.1,18–20 In multiple sclerosis, the areas of T2 hyperintensity tend to be multifocal, smaller, and limited to one segment of the cord in transverse segments; in infectious and parainfectious transverse myelitis, in contrast, T2 hyperintensities are confluent and elongated, often extending over several spinal segments. Furthermore, plaques of demyelination are usually seen on cerebral MRI in multiple sclerosis but not in infectious transverse myelitis.
Specific Entities
Echovirus meningomyelitis
There have been case reports of echovirus (types 11 and 18) isolated from either pharyngeal exudate or CSF of children with clinical meningomyelitis, with transient T2 hyperintensity of the spinal cord.21,22
Coxsackie virus–related myelitis and meningomyelitis
There exist several case reports of patients with clinical and MRI evidence of transverse myelitis with or without meningitis, from whom Coxsackie virus (B4, B3, B5, A5, and A9) had been isolated or in whom a high titer of specific antibody to this virus had been demonstrated in the CSF or serum.1,23,24 For instance, in one case report of a 6-year-old with rapidly progressive paraplegia, with associated bladder and bowel dysfunction, a rising titer of serum anti-Coxsackie virus B5 antibody was demonstrated.25 The virus was also isolated from the stool. MRI in this case demonstrated diffuse swelling of the spinal cord on T1-weighted imaging.
Mumps-related meningomyelitis
The commonest neurological manifestation of mumps virus infection is meningitis; encephalitis occurs in fewer than 0.1% of cases.9 However, there are case reports of myelitis associated with mumps viremia. For instance, a 10-year-old boy developed flaccid paraparesis and neck stiffness in the setting of parotitis and orchitis that are clinically indicative of mumps infection.26 MRI demonstrated T2 hyperintensity from C2 to T12 spinal cord segments, in keeping with myelitis. Mumps infection was confirmed by the presence of IgM class antibodies against mumps in the serum. In another report, a young woman was diagnosed with mumps-associated transverse myelitis on the basis of clinical findings, MRI demonstration of a swollen spinal cord with uniform high-signal change on T2 weighting, and acute mumps viremia.27 No reports of mumps virus isolation from the CSF, in the setting of transverse myelitis, have been found.
Herpes simplex virus–related myelitis
Although a well-known cause of viral encephalitis, herpes simplex virus (HSV) is a rare cause of myelitis.13,28–30 This entity was first described by Klatersky and associates and has since been reported in both immunocompromised and immunocompetent patients.13,30 Subtle spinal cord involvement by HSV infection may be underrecognized.
HSV type 2 infection and, less frequently, HSV type 1 infection cause genital infection, with vesicles and ulceration.9,32 After primary genital infection, the virus persists latently within the sensory neurons of the dorsal root ganglia and produces recurrent cutaneous disease with intermittent reactivation and retrograde spread along the sensory nerve.9,13,32
On occasion, HSV reactivation may be followed by radiculomyelitis, variably affecting the cauda equina, conus medullaris, and thoracic cord.32 The pathogenesis of HSV-associated myelitis is not well understood, although intra-axonal spread of virus from the sensory ganglion into the spinal cord through the dorsal roots is thought to be the likely mechanism.28 Most cases of HSV-related myelitis described in the literature have been associated with type 2 infection, probably reflective of the recurrence of latent infection mainly with the more commonly HSV type 2 venereal infections.13
In six of a series of nine patients with HSV-related myelitis reported by Nakajima and colleagues, disease onset was marked by bilateral lower limb sensorimotor disturbance and urinary symptoms, with ascending progression of the myelopathy to thoracic or cervical level within the course of a few weeks.13 Most cases of HSV-related myelitis described in the literature followed a similar clinical pattern of an acute, monophasic, usually fatal ascending necrotizing myelitis, with death resulting from respiratory paralysis or meningoencephalitis.13 However, other clinical patterns, including a self-limited, resolving transverse myelitis, a relapsing-remitting form, and chronic myelitis, have been described since the development of improved diagnostic techniques such as PCR which have enabled noninvasive diagnosis.13,28–30,33 In three of the nine patients studied by Nakajima and colleagues, transverse myelitis started in the cervicothoracic cord, with a nonascending pattern and milder sequelae than in the cases of ascending myelitis. There have also been other case reports of transverse myelitis of the cervical and thoracic cord, caused by HSV, that resolved.34 In these cases, demyelination without necrosis was postulated to be the underlying pathological process responsible for myelitis.13
In early reports, HSV-related myelitis was described as being frequently associated, in close temporal relation, with cutaneous herpetic eruptions, which provided a clue as to the diagnosis.13,30 However, it has since then been suggested that this association is seen in very few cases of HSV-related myelitis, and therefore this feature is not useful for early diagnosis. HSV per se is rarely isolated from the CSF.13 Subtle clinical involvement of the spinal cord in patients with genital HSV infections are probably much more common than appreciated, however, and symptoms related to cord involvement are often a prominent feature of the prodromal phase of the illness.
The diagnosis of HSV meningoencephalitis is usually based on the detection of HSV DNA in CSF by PCR, with reported sensitivity and specificity of 98% and 94%, respectively.35 In most cases reported since 2000, PCR has been used in the diagnosis of HSV-related myelitis, although numbers have been too small to obtain data on specificity and sensitivity.28 Further evidence of herpetic infection as the cause of myelitis is the presence of anti-HSV IgM and IgG antibodies in the CSF, suggestive of intrathecal synthesis.28 Although demonstration of seroconversion against HSV concurrent with the episode of myelitis enables diagnosis, it should be kept in mind that anti-HSV antibody titers are not always elevated during the early stage of disease.13 CSF pleocytosis, although usually present, is also not a constant feature.29
MRI findings include enlargement of the conus medullaris; intramedullary T2 hyperintensity, particularly of the posterior funiculi; and gadolinium enhancement of the affected dorsal nerve roots, with no enhancement of the ventral roots.13,28 This is in keeping with the expected pattern of involvement of spinal cord structures, if infection is indeed caused by retrograde spread of virus into the cord from dorsal root ganglia. In one case, both T1 and T2 hyperintensities were observed, suggestive of hemorrhagic necrosis of the affected parts of the cord.13 Follow-up imaging may demonstrate atrophy of the cord in the previously inflamed regions.33
Treatment of HSV-related encephalitis with acyclovir is established; however, there are case reports of the value of this antiviral agent only in the treatment of HSV-related myelitis.28 Corticosteroids, in conjunction with antiviral therapy, may prevent ascending necrotizing myelopathy and improve survival.28 With regard to long-term prognosis, in the series by Nakajima and colleagues, three patients recovered, whereas the remaining six had severe persistent neurological deficits, such as paraplegia, despite antiviral therapy.13
Zoster-related transverse myelitis
Varicellazoster virus (VZV) infection may be associated with a wide spectrum of neurological complications, myelitis being the most poorly characterized of these.36 Several reports describe a postinfectious VZV-related myelitis, which is usually an acute, relatively benign transverse myelopathy, associated with a self-limiting paraparesis and sometimes sensory symptoms and sphincter disturbance.37 Chronic and remitting-relapsing temporal profiles of myelopathy have been described in association with VZV.36
Less commonly, a progressive ascending myelopathy, usually fatal in outcome, is seen in immunocompromised individuals and is thought to be caused by direct VZV invasion of the spinal cord.10,38 Ten cases of necrotizing vasculitis associated with VZV meningoencephalomyelitis have been reported in the literature, all with underlying immunodeficiency, secondary to either malignancy or human immunodeficiency virus (HIV) infection, and all cases fatal within 4 to 32 days.38 The patients typically developed radicular or central back pain within 2 weeks of zoster infection, in association with bilateral motor and sensory signs.
The diagnosis of VZV-related myelitis is historically based on the onset of myelopathy within days to weeks of a typical rash.37 The symptoms may be most pronounced ipsilateral to and at the level of the rash. However, there are reports of VZV-related myelitis, confirmed by positive VZV PCR or culture from the CSF, with no history of cutaneous rash.37,39
VZV per se is rarely isolated from blood or CSF, although the latter usually shows an often profound pleocytosis.37 PCR techniques have enabled the detection of VZV DNA in the CSF of patients suspected of having VZV-related myelopathy, even months later, which enables confirmation of the diagnosis and suggests a role of virus persistence in the pathogenesis of disease.37,38 VZV as the etiological agent in myelopathy is also supported by the presence of antibody (IgG and IgM) to VZV in the CSF, indicative of presence of viral antigen in the nervous system and intrathecal antibody synthesis.37,40
The underlying pathological process in VZV-related myelitis is thought to be a small- or large-vessel vasculopathy, depending on the immune status of the patient.38 Small-vessel disease is seen almost exclusively in immunocompromised patients and includes a necrotizing vasculitis of leptomeningeal vessels, especially around the spinal cord and brainstem.38 VZV has been detected in large and small vessels of the nervous system by PCR, in situ hybridization for VZV antigen, and immunohistochemistry in these cases.38,41,42 Pathological examination may demonstrate intranuclear viral inclusions.38
VZV-related vasculopathy may occur as late as 5 to 6 months after the zoster rash.39 In these patients, the detection of antibody to VZV in CSF, even in the absence of amplifiable VZV DNA, is diagnostic.39,40 There are other reports of zoster-related myelitis in which PCR results for VZV were negative in the presence of anti-VZV antibodies in the CSF or became positive only after several days of clinical symptoms.38,40 It is presumed that the reason why patients with VZV-related vasculopathy may not contain VZV DNA is that active viral replication is confined to CNS arteries.39 Hence CSF PCR for VZV DNA is not as sensitive a test as CSF PCR for HSV.40
MRI demonstrates hyperintensity and swelling, consistent with edema, on T2-weighted sequences.36,38 On T1-weighted images, with gadolinium contrast material, leptomeningeal enhancement over the cerebellum and brainstem is seen in cases of ascending myelitis.38
The treatment of VZV-related myelitis is aggressive antiviral therapy, usually with intravenous acyclovir; however, response is variable.36 VZV strains that are resistant to acyclovir, as well as to related newer compounds such as famciclovir and valacyclovir, have been reported, in both immunodeficient and immunocompetent patients.38,43,44 Resistance results from deficiency or absence of viral thymidine kinase, the enzyme on which these drugs are dependent for intracellular activation via phosphorylation.38,44 These thymidine kinase–deficient VZV mutant strains are, however, susceptible to foscarnet and acyclic nucleoside phosphonates, which are active independently of thymidine kinase.44 Because diagnostic tests for VZV-mediated myelitis are potentially insensitive, management of patients suspected of having this condition should include broadening of antiviral therapy to overcome possible resistance if there is clinical progression or inadequate response to therapy.38
Cytomegalovirus-related myelitis
In the setting of cellular immunodeficiency, such as the acquired immunodeficiency syndrome (AIDS), cytomegalovirus infection of the cord can produce a necrotizing transverse myelitis, typically when CD4 counts are less than 50.45 It is exceedingly rare in immunocompetent persons, although five such cases of cytomegalovirus-associated transverse myelitis were reported by Giobbia and coworkers.46 In all these cases, CSF PCR for cytomegalovirus yielded negative results. The diagnosis was instead obtained on the basis of serological data: namely, cytomegalovirus antigenemia (positivity for cytomegalovirus pp65 antigen) and high serum titers of specific cytomegalovirus IgM and IgG antibodies, positive blood PCR results for cytomegalovirus, and blood or urine cultures. Of note, however, PCR performed on CSF of patients with AIDS demonstrated this test to predict histopathologically confirmed cytomegalovirus-associated CNS disease, including myelitis.47 It is unclear as to whether neuronal injury is immune mediated or caused by a cytotoxic effect of viral infection.
Epstein-Barr virus–related myelitis
Epstein-Barr virus, like VZV, can infect multiples sites of the neuraxis and can therefore produce protean clinical manifestations.48 Both CNS and peripheral nervous system disease can occur, with reports of meningitis, encephalitis, transverse myelitis, neuritis, and overlapping syndromes.48–50 The incidence of Epstein-Barr virus–related neurological disease is not known; however, it has been estimated to occur in 1% to 5% of individuals with infectious mononucleosis.48 The mechanism by which Epstein-Barr virus produces neurological disease is unknown; however, the development of neurological disease over weeks is suggestive of either a subacute to chronic viral infection or a postinfectious, immune-mediated process.48
CSF typically shows pleocytosis, with elevated protein levels.48 The diagnosis is made by the isolation of Epstein-Barr virus DNA from CSF, as well as from peripheral blood monocytes, and the exclusion of other herpesvirus infections through PCR techniques.48,51 Serological evidence is supportive.48 Findings on MRI are variable and may demonstrate increased T2 signal intensity; however, normal findings are also reported in the presence of neurological deficits.48
Unfortunately, no definitive treatment for Epstein-Barr virus nervous system infections exists. Although corticosteroids and immunoglobulins have been used, their effect on disease progression is unknown.48 Antiviral agents have not been shown to be clinically useful. Significant neurological deficits may persist after resolution of the acute phase of myelitis.48,51
Mycoplasma-related myelitis
It is estimated that CNS manifestations, most commonly encephalitis, occur in 1 per 1000 patients with Mycoplasma pneumoniae infections.52 Myelitis, albeit atypical, and radiculitis have been reported in association with meningitis. In 19 cases of Mycoplasma-associated transverse myelitis, patients often developed a flaccid paraparesis, with bladder paresis.11 A definite sensory level was described in only a few cases. There is usually slight CSF pleocytosis, and M. pneumoniae was isolated in at least seven reported cases. In one case, in a patient who had no antecedent respiratory infection, M. pneumoniae was cultivated from a nasopharyngeal aspirate and detected in the CSF by PCR.11,53 Treatment is with doxycycline for 14 days, although the effects of therapy are debated.1 The overall prognosis is generally good, with complete recovery in most cases, although there is one case report of persistent paraplegia after Mycoplasma-associated transverse myelitis.54
Other Rare Infectious Causes of Myelitis
Cases of transverse myelitis associated with Burkholderia pseudomallei (melioidosis) have been described.55 Borrelia recurrentis may also produce myelitis, usually radiculomyelitis rather than isolated myelitis.56 Even so, only 5% of 330 cases of neuroborreliosis were classified as acute meningomyelitis or meningomyeloradiculitis. Transverse myelitis may occur in tuberculous meningitis, usually with severe leptomeningitis.1
Other Myelopathies
Subacute combined degeneration of the cord, classically described in association with vitamin B12 deficiency, affects the posterior and lateral columns of the spinal cord.57 A similar syndrome of posterior column degeneration is also seen in tertiary syphilis (Treponema pallidum infection), and is referred to as tabes dorsalis.57 This manifests clinically with a spastic weakness and ataxia of the lower limbs. Chronic meningeal inflammation may be present and may involve the ventral nerve roots. Other clinical spinal syndromes described in association with syphilitic infection are syphilitic meningomyelitis (a chronic meningitis resulting in subpial myelinated fiber loss, with predominantly bilateral corticospinal tract involvement) and spinal meningovascular syphilis, which may result in an anterior spinal artery syndrome.57
The myelopathy associated with the late stages of AIDS also affects the posterior and lateral columns, as demonstrated in histological studies.1,57 Clinical features of spinal cord disease are often obscured by other neurological complications of AIDS, such as neuropathy and encephalopathy, which result from HIV per se or from opportunistic infections. Vacuolation of myelin and relative sparing of the axons occur, and lipid-laden macrophages are typically abundant.57,58 This vacuolar myopathy was seen in 48% of autopsy examinations of the cord of 90 patients with AIDS, although the frequency of encephalitis was higher in this population.58 There was no correlation between the proviral HIV type I load and the degree of myelopathy.58 Likewise, there was no correlation between opportunistic infections, particularly cytomegalovirus, and the presence and severity of myelopathy. In a separate series of 21 patients with AIDS-associated myelopathy, MRI demonstrated spinal cord atrophy in 15 cases and diffuse intrinsic signal abnormality in six cases.59
Tropical spastic paraparesis is a syndrome of slowly progressive central paraparesis of proximal lower limb dominance, caused by infection with the human T cell leukemia/lymphoma virus type 1 (HTLV-1). Sensory signs are characteristically minimal and usually only in the lower limbs; paresthesias and ataxia, with diminished proprioceptive function and vibration sense, have been described.57,60 Sphincter dysfunction usually occurs early. An associated polyneuropathy has been reported. Levels of CSF antibodies to HTLV-1 are elevated.57 On pathological examination, the corticospinal tracts and posterior columns are affected by an inflammatory myelitis, with focal spongiform demyelination and necrosis.57,60 Foci of gray matter destruction are also seen.
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis occurs typically after childhood viral exanthemata, upper respiratory tract infection, or vaccination, with a short latency.1 Although the majority of postinfectious cases follow viral infections, such as measles, mumps, and Epstein-Barr virus infection, acute disseminated encephalomyelitis can occur after bacterial or parasitic infections.1,2 Most cases are disseminated, as the name implies, with alteration of sensorium; however, in some cases, spinal disease dominates the clinical picture.1
Anterior Horn Cell Syndromes
The hallmark of damage to lower motor neurons is flaccid paralysis. Viral infection and the resultant inflammation cause direct damage to the motor neurons residing in the anterior horns of the spinal cord, producing an anterior myelitis.61 Viral invasion of anterior horn cells usually occurs as part of an acute viral meningitic illness, with fevers, headache, and meningism.61 This is followed by the rapid onset of asymmetrical weakness without any sensory deficits.61 Bulbar and respiratory musculature may be affected, and ventilatory support may be needed. Examination of CSF demonstrates moderate pleocytosis.61 Neurophysiological testing demonstrates low compound muscle action potential, normal sensory nerve action potentials, and sharp waves and fibrillations on electromyography.61
Poliomyelitis
Acute poliomyelitis is now rarely encountered in the industrialized world, because of the World Health Organization’s global eradication program. Nonetheless, pockets of endemic disease persist in sub-Saharan Africa and the Indian subcontinent, and it continues to occur sporadically elsewhere.62 There is also a small incidence of live vaccine–related poliomyelitis.62
Poliomyelitis is caused by an enterovirus whose main route of infection is via the gastrointestinal tract.62 In approximately 5% of infections, after nonspecific flulike symptoms, nervous system infection occurs and is initially manifested as meningitis, with high fevers, neck stiffness, and headache.62 The onset of spinal poliomyelitis is heralded by myalgias, with the subsequent development of asymmetrical, lower limb–predominant flaccid weakness or paralysis, reaching a maximum within 48 hours of onset.62
Diagnosis is based on virus isolation of nasopharyngeal secretions and/or the stool. CSF examination demonstrates elevated protein levels and pleocytosis. Serological diagnosis, with demonstration of antipoliovirus IgG and IgM antibodies, can establish the diagnosis in the absence of viral isolate. PCR techniques now enable rapid diagnosis, identification of serotype, and differentiation of wild-type from vaccine-related disease.63 MRI may demonstrate high signal intensity on T2-weighted imaging in the region of the anterior horn cells.64
There is, unfortunately, no curative treatment.62 Management is supportive, aimed at symptom control with analgesia and prevention of complications. The aim of strict bed rest is to prevent progression of paralysis. Passive exercise during the acute phase, followed by more intensive physiotherapy, can prevent or minimize contractures.65
Poliomyelitis-like Syndromes
Acute flaccid paralysis secondary to anterior horn cell disease may be associated with other enteroviruses (e.g., enterovirus 71, Coxsackie virus A7, echoviruses), tickborne encephalitides, flaviviruses (e.g., Japanese encephalitis, Murray Valley encephalitis virus, and West Nile virus), herpesviruses (cytomegalovirus, Epstein-Barr virus, VZV), and HIV-related opportunistic infections.57,61–68
West Nile virus, a flavivirus, usually causes a mild febrile illness.61 Neurological disease is reported in 1 per 150 infected persons and usually consists of meningitis and encephalitis; however, there are numerous reports of patients with poliomyelitis-like acute flaccid paralysis, occurring during the acute febrile illness.61,68 Diagnosis is based on identification of anti–West Nile virus antibodies in serum or CSF.61 Confirming an anterior horn cell process, high signal intensity in the anterior horns on T2-weighted MRI has been demonstrated.69 Histopathological examination of autopsy specimens has demonstrated anterior myelitis, with perivascular lymphocytic infiltrate, monocytic infiltration, and gliosis.70
Enterovirus 71, which causes outbreaks of hand-foot-and-mouth disease, a common exanthema of childhood characterized by fevers, palmar and plantar rash, and oromucosal ulceration, may be associated with neurological complications, including an acute flaccid paralysis.66
Spinal Epidural Abscess
Spinal epidural abscess is an uncommon condition, occurring with an estimated incidence of 0.2 to 2.8 cases per 10,000 per year.71–74 The incidence peaks in the sixth and seventh decades of life.71 Although this condition is potentially fatal, with devastating neurological sequelae if left untreated, early recognition and prompt institution of appropriate therapy can avert complications.
Risk factors for spinal epidural abscess include immunocompromised states and are identified in Table 40-5.72 In 20% of cases, there is no identifiable risk factor.71,76
TABLE 40-5 Risk Factors for Spinal Epidural Abscess
[/not-level-membership-for-neurology-category]
