Chapter 30 Spasticity Management
The definition of spasticity is most commonly cited as follows135:
From a clinical standpoint, it is important to distinguish between the positive symptoms related to the UMN syndrome (e.g., spastic dystonia, flexor spasms, exaggerated cutaneous reflexes, and autonomic hyperreflexia) and the negative symptoms (e.g., paresis, loss of fine dexterity, and fatigability).245 This difference relates directly to the expectations of outcome following treatment for a positive symptom of spasticity, without necessarily worsening the negative symptom of weakness.246
Causes of Spasticity
Like the UMN syndrome, spasticity can accompany diffuse or localized cerebral or spinal pathology. Anoxic, toxic, or metabolic encephalopathies can cause diffuse cerebral abnormalities,2 whereas localized cerebral injury can occur with tumor, abscess, cyst, vascular malformations, infarction, hemorrhage, or trauma. Trauma, inflammation, demyelinating disease, and degenerative and familial disorders, as well as compression by a mass (e.g., neoplasm, infection, or cyst) are examples of spinal cord disorders. An example of a combination of UMN and lower motor neuron pathology is amyotrophic lateral sclerosis, where spasticity can be the dominant feature in some patients. Spasticity is often cited as a significant problem in multiple sclerosis (see Chapter 52), traumatic brain injury (see Chapter 49), cerebral palsy (see Chapter 53), SCI (see Chapter 55), and stroke (see Chapter 50). Problematic spasticity occurs in 40% to 60% of patients with SCI and multiple sclerosis, which results in a significant impact on activities of daily living and patient independence. Almost two thirds of patients with cerebral palsy present with “spastic diplegia.”∗
Changes in the characteristics of a person’s regular spasticity can help to alert those affected and caregivers of problems in parts of the body where the patient no longer has voluntary movement or sensory appreciation; for example, an increase in spasm frequency in a person with complete tetraplegia who has developed an otherwise asymptomatic urinary tract infection. In neurolathyrism, a rare disorder, spasticity can be the main presenting symptom.166 Physiatrists are often asked to evaluate the patient who presents with the sudden worsening of spasticity, possibly a result of the onset of a new pathologic process, such as a urinary tract infection, urolithiasis, stool implication, pressure sore, fracture, dislocation, ingrown toe nail, excessively restrictive clothing, irritating condom drainage appliance, or even thyrotoxicosis.208 If there is a remediable cause of spasticity, it must be discovered and treated. If problematic spasticity persists in the absence of a remediable cause, then it is appropriate to pursue treatment until a therapeutic response is obtained. Inevitable complications are the natural history of suboptimal treatment of severe spasticity. These complications include skin breakdown, infection, bone fracture or dislocation, and more frequent inpatient hospitalization.17,180
Evaluation and Measurement
Of the many clinical monitoring tools described in the literature to assess the severity of spasticity, most researchers agree that assessment tools should be tailored to meet the individual characteristics of a given patient. A number of spasticity-measuring tools are used, which range from simple questionnaires and goniometry evaluations to more technologically complicated electromyographic and biomechanical analysis of limb resistance to mechanical displacement, and even video monitoring assessments of joint mobility.32,43,98,186
The Ashworth Scale,10,73,139 Oswestry Scale of Grading,91 and Degree of Adductor Muscle Tone224 are some of the tone intensity scales used to assess spasticity in SCI. As proposed originally, the Ashworth Scale is a simple five-point Likert scale in which the observer’s subjective opinion of the subject’s resting muscle tone ranges from “normal” at the lowest grade to “rigid” at the highest10 (Table 30-1). The original scale was modified by adjusting the lowest number from 0 to 1 and the highest scale from 4 to 5. Another modification from the original scoring scheme was the addition of a point between 1 and 2, where 1 was a “catch” at the end of joint motion range and 1+ was a catch earlier in the joint motion range nearer to midpoint.24 The Ashworth Scale has the advantage of ease of use in the clinical setting. This asset has been used in a number of pharmaceutical trials of antispasticity medications in which a simple measurement tool can be used easily by the participating clinicians to assess the efficacy of the intervention. A recent comprehensive review of engineering and medical literature concluded that the Ashworth Scale is in common use and has significant interrater agreement and good reliability, but it is not a functional outcome measure and can be biased by evaluator subjectivity.73 A monitoring test should be able not only to assess the change in spasticity during therapy but also to assess the functional effects of interventions. Such a test should have a well-defined scoring system, be reliable and sensitive to change, and have standard instructions.186
| Score | Definition |
|---|---|
| 0 | No increase in muscle tone |
| 1 | Slight increase in muscle tone, manifested by a catch and release |
| 2 | More marked increase in muscle tone through most of the range of motion, but affected limb is easily moved |
| 3 | Considerable increase in muscle tone—passive movement difficult |
| 4 | Limb rigid in flexion or extension |
Another method of observing the spasticity phenomenon is to assess the number of episodic spasms as reported by the patient. The Penn Spasm Frequency Score195 is an ordinal ranking of the frequency of leg spasms per day and per hour. One problem with this scale is that patients usually report that the number of spasms occurring per hour is often affected by their activity at the time. For example, they tend to report few spasms if resting comfortably or more if physically active. The duration of each spasm is also not considered.
The casual observation of the free swing of the knee in the “pendulum test” was formalized and provided objective data by the use of videomotion analysis. The advantages of videomotion analysis of the pendulum test include the ability to do the analysis anywhere a video recorder is available, freedom from the attachment of cumbersome recording devices to the patient, and processing by a nonbiased “blinded” observer who has had no contact with the patient.105,179
Pain can be assessed, whether or not it is associated with spasticity, by a self-administered test such as the Pain Intensity Descriptor Scale95 or by using a 10-cm visual analog scale.40,117 It is important to decipher whether the pain is from the spasticity itself or caused by other factors such as with neuropathic pain in SCI or multiple sclerosis, thalamic pain syndromes, or frozen shoulder in stroke. Standardized assessments of functional ability or caregiver burden might or might not be sensitive to changes in relative levels of spasticity. These include the Sickness Impact Profile,22 the 36-Item Short-Form Health Survey,6 the functional independence measure (FIM),5 and the Caregiver Dependency Scale.4 The Canadian Occupational Performance Measure (COPM)200 and the Goal Attainment Scale (GAS) have been shown to be sensitive to detect changes following intervention in cerebral palsy.49 The GAS has been shown to have potential of detecting functional changes.9 A multicenter trial of intrathecal baclofen with 138 patients reported improvement in both the performance and satisfaction scores on the COPM.101
Physiologic Mechanisms
No single pathophysiologic mechanism accounts for all the observable aspects of spasticity. Dysfunction within the central nervous system of descending pathways to and within the spinal cord causes a UMN syndrome that is often associated with exaggerated reflexes and spasticity, which includes velocity-dependent increased muscle tone.135 Although enhanced reflexes are sufficiently common and associated with the spasticity phenomenon to be part of its definition, measurement of the reflex amplitude in some patients (such as patients with stroke or neurologically complete SCI) has shown reductions compared with that in able-bodied subjects.176,178,207
Although the spinal α-motor neuron is considered to be the final common pathway for expression of spasticity, one should consider the more complex motor pathways involved in the disordered movements of spastic brain-injured patients. Spastic hypertonia encompasses a variety of conditions, including dystonia, rigidity, myoclonus, muscle spasm, clonus, cocontraction, posturing, and spasticity.109,158,174 The following sections briefly review the physiology of segmental reflexes.
The Monosynaptic Reflex
The physiologic components involved in the spinal stretch reflex response include the muscle spindle stretch receptor, the myelinated sensory neuron, the synapse, the homonymous α-motor neuron, and the muscle it innervates. As originally described in the decerebrate cat model, the stretch reflex shows a dramatic increase in extensor muscle tone on passive flexion of the extended hindlimbs. This stretch reflex has two components: a brisk, short-acting phasic component that responds to the initial dynamic change in length, and a weaker, longer-acting tonic component that responds to the steady stretch of the muscle at a new length.144
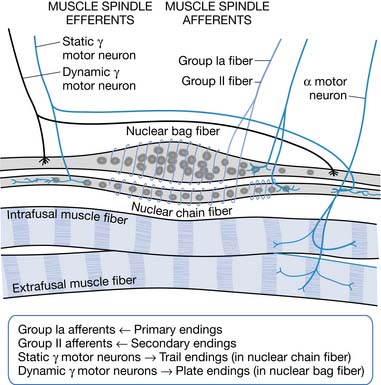
A change in muscle length can evoke a stretch reflex. Modified muscle fibers (intrafusal receptor organs) that detect changes in muscle length are called muscle spindles. Nuclear bag fibers and nuclear chain fibers are two types of specialized muscle spindle fibers (Figure 30-1). Nuclear bag fibers are further subdivided into dynamic and static nuclear bag fibers. Dynamic nuclear bag fibers are highly sensitive to the rate of change in muscle length, providing velocity sensitivity to muscle stretch.151 Static nuclear bag fibers and nuclear chain fibers are more sensitive to the steady-state, static or tonic, muscle length. The structural differences between these fibers are responsible for the physiologic differences in their sensitivities and for the two different components, phasic and tonic, of the stretch reflex. Intracellular muscle fibers are observed to undergo changes as a result of spasticity, as does the extracellular matrix.145
The Inverse Stretch Reflex
The Golgi tendon organ is sensitive to intramuscular tension and is innervated by myelinated Ib sensory afferents. The Golgi tendon organ is particularly sensitive to muscle tension created by active muscle contraction but has a high threshold for detecting passive stretch. Stimulation of Ib afferents leads to inhibition of the homonymous α-motor neuron and of its synergists. The excitation of its antagonistic motor neurons also stimulates Ib afferents. This behavior has been called the inverse myotactic reflex because its actions oppose those of the stretch (myotactic) reflex. It is also called Ib nonreciprocal inhibition. It should be noted that this reflex is stimulated by muscle tension, whereas the stretch reflex is stimulated by a change in muscle length. The Golgi tendon organ has been hypothesized to function as part of a muscle tension feedback system.137
Elevated Reflex Activity
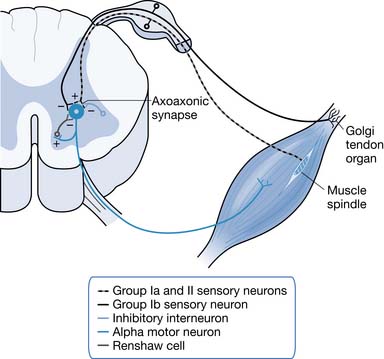
Long-term reductions in inhibition can contribute to hyperreflexia. Examples of inhibition types are as follows: recurrent Renshaw inhibition, reciprocal Ia inhibition, presynaptic inhibition, nonreciprocal Ib inhibition, and inhibition from group II afferents. Various lines of research have supported deficient presynaptic and nonreciprocal inhibition as significant contributors to spasticity. The supportive evidence for it being caused by deficient group II afferent-related and Renshaw inhibition is lacking.153 Presynaptic inhibition is mediated via a γ-aminobutyric acid (GABA)ergic mechanism that decreases the efficacy of Ia afferent transmitter release. Inhibitory interneurons synapse with the presynaptic terminal of the Ia afferent via an axoaxonic synapse where GABA is the neurotransmitter. Inhibitory interneurons involved in presynaptic inhibition are modulated by descending pathways. The loss or reduction of rostral control can reduce tonic levels of descending facilitation on inhibitory interneurons, leading to increased α-motor response to normal Ia afferent input.245
The Ia afferent presynaptic inhibitory interneurons are normally controlled by descending excitatory pathways. Reciprocal Ia inhibition decreases the chance for cocontraction of antagonistic and agonistic muscles during the stretch reflex or during voluntary movement. Evidence exists for decreased excitability of the inhibitory neurons after rostral lesions of the central nervous system. This dysfunction could lead to an increased cocontraction and weakness of voluntary movement.44 Nonreciprocal Ib inhibition has been found to be decreased or even replaced by facilitation in patients with spastic paresis and spastic dystonia, in this case both stroke and SCI subjects, but not in subjects without spastic dytonia.59
Patients with spastic paresis from SCI show increased rather than decreased levels of recurrent Renshaw inhibition. Renshaw cells are inhibitory neurons that are stimulated by collateral axons from α-motor neurons. When an α-motor neuron fires, it stimulates a Renshaw cell that in turn inhibits the initiating motor neuron and its synergists. The Renshaw cell also inhibits the IIa afferents presynaptic inhibitory interneuron associated with the initiating motor neuron. Because the Renshaw cell inhibits the inhibitory interneurons as well as agonist α-motor neurons, increased Renshaw cell activity might contribute to spasticity by decreasing reciprocal Ia inhibition.211 Hyperexcitability of α-motor neurons might contribute to spasticity. Examples of primary changes in membrane properties that would be expected to produce increased α-motor neuron discharge include a reduction in the area of dendritic membranes, deafferentation dendritic hyperexcitability, and an increase in the number of excitatory synaptic inputs as a result of sprouting (Figure 30-2).245
Multisynaptic Segmental Connections
The majority of spinal segmental connections are polysynaptic. In addition to the muscle spindle afferents making direct contact with the α-motor neuron of the agonist muscle, interposed interneurons connect these afferents and antagonistic α-motor neurons to opposing muscle groups, resulting in a polysynaptic connection. As mentioned above, the Golgi tendon organ contributes to this via Ib nonreciprocal inhibition. These reflex pathways coordinate muscle action around the joint. Interneurons also receive excitatory and inhibitory signals from descending pathways. Supraspinal centers can control joint stiffness through the modulation of excitatory and inhibitory input to segmental interneurons and interneuronal networks.41,108
The afferent fibers that produce generalized reflexive flexor movements have become known collectively as flexor reflex afferents. Interestingly, the response to cutaneous stimuli is not always one of generalized flexion. The vestibulocollic and cervicocollic reflexes produce patterns of coordinated ipsilateral limb flexion accompanied by contralateral limb extension with activation of group II and III fibers to either keep the head level during body tilt or to oppose a fall. Different modalities of stimuli can have differential effects, particularly evident after a neurologic injury. For example, after certain neurologic lesions, pressure applied to the plantar surface of the foot produces a marked extension of the leg, known as extensor thrust. In contrast, a pinprick in the same area leads to flexion withdrawal of the limb. The spinal circuits responsible for ipsilateral flexion and crossed limb extension also receive descending inputs and coordinate voluntary limb movements.
A cutaneous stimulus can modulate the activity of particular motor neurons. Touching an area of skin can cause a reflex contraction of specific muscles, usually those beneath the area of stimulation. This is an example of an exteroceptive response. Cutaneous stimuli might not always produce observable contractions. They can have subthreshold or facilitative effects. Proprioceptive information is transmitted from muscle spindles and Golgi tendon organs via group Ia, II, and Ib afferents. Finally, there are indications that pathway connectivity and neurotransmitter distribution might account for differential responses comparing the upper limbs with the lower limbs.159
Goal Setting
Because spasticity results from neurologic dysfunction within several regions in the central nervous system, the associated loss of voluntary motor function can be highly variable among patients with symptomatic spasticity. Prediction of the functional impact resulting from the presence of spasticity consequently can be challenging. Compare an individual with C4 tetraplegia who uses a mouth stick or suck and puff actuator to operate a computer, telephone, and numerous adapted electronic devices, as well as a head controller to operate an electric wheelchair, for example, with a person with T10 paraplegia. The presence of mild to moderate spasticity can alter the sitting position so that the control over the adaptive devices is lost for the person with tetraplegia, whereas the paraplegic person does not experience the same functional impact to that level of spasticity intensity. The functional goal for spasticity treatment should be one within the ability of the patient when the performance of the function is limited mainly by spasticity. Common examples of spasticity-limited functional goals are to improve speed and safety of wheelchair transfers, to improve the performance of activities of daily living such as dressing, and to facilitate perineal hygiene by reducing thigh adductor or pectoral muscle spasticity, thus facilitating ease of caregiver assistance. One must consider also the extent to which the spasticity helps the individual functionally; therefore the goal of functional improvement must consider the balance of treatment effects. Spasticity can be protective against skeletal muscle atrophy and indirectly affect functional independence, ambulation, and incidence of fracture.93 Spasticity has been reported to increase glucose uptake and thereby reduce the risk for diabetes in those with SCI.20
One potential functional goal might be the improvement of gait. Although patients with spasticity are reported to have disturbances of gait speed, timing, kinematics, and electromyographic patterns, the relative impact of spasticity on gait remains controversial.76,78,133,196 Although it seems logical that knee extensor muscle torque should correlate with the speed of “comfortable” walking, by experimental measurement it accounts for only 30% of the variance in gait speed in spastic stroke patients.23 Young244 concluded that not all abnormalities underlying “spastic gait” are caused by spasticity, and consequently are not affected by antispasticity drug treatment.
Nonpharmacologic Treatments
A regular exercise routine that includes daily range of motion exercise must be done, with a focus on muscle stretching. This can be accomplished with assistance from a therapist in the short term, but in the long term the exercise should be taught to be done by self or caregivers.31,102,197 Immediate reduction in spasticity can be seen objectively from passive movements or from stretch.187,220 Any spasticity-aggravating factors (e.g., urinary tract infection, constipation, skin ulceration, ingrown nails, and fractures) must be identified and treated.
A number of other helpful physical treatments can be used. Casting a joint with the muscle in a lengthened position can help maintain muscle length, with serial casting allowing for progressive improvement in joint range. With this treatment, however, one must be extremely vigilant not to cause skin breakdown from pressure points in the insensate limb. Externally applied repetitive cycling movements to the lower limbs using a motorized exercise bicycle has allowed some subjective improvements but no objective changes in torque resistance response to movement.121 Another approach to spasticity reduction is hippotherapy, which involves the rhythmic movements associated with riding a horse to regulate muscle tone. The short-term effect of hippotherapy has been shown in decreasing spasticity of the lower limb as noted by the Ashworth Scale and self-reported spasticity with a small crossover randomized clinical trial.138 In a recent review of the literature pertaining to randomized trials of antispasticity treatments for amyotrophic lateral sclerosis, the recommended treatment included individualized, moderate-intensity, endurance-type exercises for the trunk and limbs.11 Electrical stimulation of the spinal cord has been reported to result in reduction of spasticity,15,103 although the measurement of spasticity in these studies has been questioned.61 Several investigators have shown that electrical stimulation of the peripheral nerves can decrease spasticity in patients with SCI, stroke, or traumatic brain injury.∗ Other physical modalities that have been reported to ameliorate spasticity include application of tendon pressure,140 cold, warmth, vibration, splinting, bandaging, massage, low-power laser, and acupuncture.90,96 Some success has also been reported with magnetic stimulation over the thoracic spinal cord185 and topical application of 20% benzocaine,206 although these are not mainstays of therapy at this point.
Pharmacologic Treatments
Enhancement of Segmental Inhibition via GABA
Medications With GABA-Mimetic and GABA-Like Actions
Baclofen (Lioresal)
Baclofen is β-4-chlorophenyl GABA, which binds to and activates the bicuculline-insensitive GABAB receptors. Bicuculline is a toxin that antagonizes the inhibitory effects of endogenous GABA at GABAA receptors, which cause treated animals to convulse. Once a presynaptic GABAB receptor is activated, potassium conductance is altered, resulting in a net membrane hyperpolarization and a reduction in endogenous transmitter release.53,112 For example, in a presynaptic sensory neuron, release of GABA by a local interneuron and binding at the receptor on the sensory neuron produce inhibition of the primary afferent terminal, and result in a decrease in excitatory neurotransmitter release. Baclofen activation of receptors postsynaptically inhibits calcium conductance and causes inhibition of γ-motor neuron activity, reduced drive to intrafusal muscle fibers, and reduced muscle spindle sensitivity.234 The overall inhibitory effect of baclofen administration at the spinal cord level reduces sensory and motor neuron activation. It also reduces the activation of monosynaptic spinal reflexes and, to a lesser extent, polysynaptic spinal reflexes. Numerous clinical reports note the antispasticity effects of oral baclofen for patients with multiple sclerosis or SCI.∗ Orally delivered baclofen has recently been studied in patients with cerebral disorders and was found to have selective efficacy on lower limb spasticity but not on spasticity in the upper limbs.159 Short-term studies with multiple sclerosis patients suggest that gait enhancement is observable with effective spasticity treatment in selected patients.189,222
Baclofen absorption after oral administration occurs mainly in the proximal small intestine. This probably involves two different amino acid transporter systems as a result of competitive inhibition of absorption by the neutral and β amino acids. The kidney normally excretes the baclofen essentially unchanged, but the liver can metabolize as much as 15% of a given dose. This is why periodic liver function testing is advisable during baclofen treatment, and the dosage should be reduced in patients with impaired renal function. The average therapeutic half-life of baclofen is 3.5 hours but ranges from 2 to 6 hours. Baclofen dosing is usually initiated as 5 mg three times per day and increased gradually to a therapeutic level. The recommended maximum dosage is 80 mg/day in four divided doses.127 Reports of improved therapeutic effects with higher dosages have been published.1,128,222 Because baclofen treatment can produce sedation, patients should be cautioned regarding the operation of automobiles or other dangerous machinery and activities made hazardous by decreased alertness. Baclofen is excreted by the kidneys, so patients with renal impairment will likely require a lower dosage. The effects of chronic baclofen treatment during human pregnancy are largely unknown. In some patients, seizure control has been lost during treatment with baclofen.131 Abrupt discontinuation of baclofen can produce seizures, confusion, hallucinations, and rebound muscle spasticity with fever.231
Oral baclofen is a widely prescribed pharmaceutical in North America, and there are few reports of major toxicity. Massive overdose with oral baclofen has been reported, however, including a case report of a 57-year-old woman who ingested 2 g of baclofen, causing coma and hypoventilation. She was given naloxone, 50% dextrose, and activated charcoal. Initially her blood pressure was low, and later systolic hypertension was noted, followed 16 hours later by bradycardia and hypotension. Her pupils were small and unresponsive, and muscle stretch reflexes were absent. Plasma baclofen concentrations over time showed first-order elimination kinetics and a half-life of 8 hours.89
Modulating the Monoamines
Tizanidine (Zanaflex)
Tizanidine is an imidazoline derivative and agonist that binds to α2-receptor sites both spinally and supraspinally,46,210 similar to the α2-adrenergic agonist clonidine (see description following). The medical literature supports the notion that the pharmacologic effects include the restoration or enhancement of presynaptic inhibitory modulation of spinal reflexes in patients with spasticity.57,175,182,227 Tizanidine has been shown to decrease reflex activity, especially polysynaptic reflex activity.54,55,227 Tizanidine also has an antinociceptive effect as shown in animal models.54–56122 Several European and American studies have shown that tizanidine is equal in effectiveness to baclofen and diazepam but with a more favorable tolerability profile. The main advantage appears to be fewer complaints of treatment-related weakness. Furthermore, two clinical trials demonstrated that patients with spasticity improved muscle strength during tizanidine treatment.129,162 Gelber et al.87 describe a statistically significant improvement in upper limb spasticity, pain intensity, and quality of life in stroke patients by titrating dosages in 2-mg intervals, mindful of withdrawal rebound effects.
Tizanidine has been tested in a number of clinical trials in Europe and has been found to be safe, well tolerated, and beneficial in treating spasticity of various etiologies.74 Tizanidine is an α2 agonist like clonidine but has a much reduced potency and does not consistently induce a reduction in blood pressure or pulse, as clonidine does.33 Symptomatic hypotension has been reported when tizanidine is taken with an antihypertensive drug; thus the concomitant administration of tizanidine and antihypertensive drugs should be avoided. An important drug interaction between ciprofloxacin, an antibiotic, and tizanidine has prompted the USA FDA to approve safety labeling. As a result of ciprofloxacin-induced inhibition of cytochrome P450 1A2, hepatic metabolism of tizanidine is decreased. The resulting increase in tizanidine plasma concentration and clinically significant adverse events is a contraindication to the coadministration of tizanidine, taken orally, and ciprofloxacin, given intravenously.
Tizanidine is well absorbed after an oral dose, with extensive first-pass hepatic metabolism to inactive compounds that are subsequently eliminated in the urine. Therefore tizanidine should be used with caution in patients with known liver abnormality. Because the most common side effects reported during the clinical trials with tizanidine include dizziness and drowsiness, it is recommended that tizanidine therapy begin with a single dose of 2 to 4 mg at bedtime. The titration of tizanidine should be tailored to the patient. The maintenance dosage is the one at which the therapeutic goals have been met with the fewest side effects. The scored tizanidine tablets contain 4 mg. Dosage increases of 2 to 4 mg every 2 to 4 days are recommended; most clinicians experienced with tizanidine, however, recommend a slower and more gradual upward titration. This is particularly the case for patients with multiple sclerosis, who tend to experience side effects at lower dosages. The maximum recommended dosage is 36 mg/day. All trials, including those in people with SCI, multiple sclerosis, or cerebral disorders, have reported somnolence consistently in 42% to 46% of the patients.162,179,221
Alteration of Ion Channels
Dantrolene Sodium (Dantrium)
Dantrolene sodium is a hydantoin derivative whose primary pharmacologic effect is to reduce calcium flux across the sarcoplasmic reticulum of skeletal muscle. This action uncouples motor nerve excitation and skeletal muscle contraction.71,238 It is indicated for use in chronic disorders characterized by skeletal muscle spasticity, such as SCI, stroke, cerebral palsy, and multiple sclerosis. The oral formulation is prepared as a hydrated sodium salt to enhance absorption (approximately 70%), which occurs primarily in the small intestine. After a dose of 100 mg, the peak blood concentration of the free acid, dantrolene, occurs in 3 to 6 hours. The compound is hydroxylated, and the active metabolite, 5-hydroxydantrolene, peaks in 4 to 8 hours. Dantrolene sodium has been shown to produce a dose-dependent decrease in the stretch reflex109 and a percentage decrease of grip strength.79 Dantrolene is lipophilic and crosses cell membranes well, achieving wide distribution and significant placental concentration in the pregnant patient. Liver metabolism by mixed function oxidase and cytochrome P450 produces a 5-hydroxylation of the hydantoin ring and reduction of the nitro group to an amine, which is then acetylated. Urinary elimination of 15% to 25% of the unmetabolized drug is followed by urinary excretion of the metabolites after oral administration of the drug. The median elimination half-life is 15.5 hours after an oral dose and 12.1 hours after an intravenous dose.
The majority of placebo-controlled clinical trials of dantrolene have shown a reduction of muscle tone, stretch reflexes, and increased passive motion. The most consistent finding has been a reduction of clonus in patients with clonus.198 Mixed conclusions have been drawn regarding the effects of dantrolene sodium on gross motor performance and strength. In comparative trials with spasticity of different etiologies, some have suggested that the best responders to dantrolene sodium are those with stroke and cerebral palsy, and that patients with SCI improve the least, if at all. Most investigators agree, however, that patients with multiple sclerosis do not generally benefit from dantrolene treatment.148 In four trials of children with cerebral palsy, dantrolene sodium was found to be superior to placebo. The degree of improvement appeared greater in children than in adults. One study found dantrolene to be superior to baclofen, and another suggested equal efficacy to diazepam. In addition to its antispasticity effects, dantrolene has been used in the treatment of malignant hyperthermia and the neuroleptic malignant syndrome.231 Dantrolene has also been reported to be useful in the treatment of hyperthermia following abrupt baclofen withdrawal.126,148
Benzodiazepines
The pharmacologic and antispasticity effects of benzodiazepines are generally mediated by a functionally coupled benzodiazepine–GABA receptor chloride ionophore complex.45,188,212 As described above, the GABAA receptor supramolecular structure is envisioned as a heteropentameric glycoprotein of about 275 kDa, whose subunits react with GABA, benzodiazepines, steroids, barbiturates, and picrotoxin-like convulsants.146 More specifically, the duration of action is related to the receptor and pharmacodynamics; certain benzodiazepine subunits are either high-affinity or low-affinity receptors, as well as long-acting and short-acting benzodiazepines. Benzodiazepines enhance GABAA receptor current, which increases the opening frequency of the chloride ionophore without altering channel conductance or open duration.228 The relative length of action is also related to the duration of activity and rate of metabolism of the pharmacologically active metabolites. Examples of long-acting benzodiazepines are diazepam, chlordiazepoxide, and clonazepam. Oxazepam, alprazolam, and lorazepam are considered to be short acting without significant production of active metabolites. Benzodiazepines cross the placental barrier and are secreted into breast milk. Microsomal enzymes of the liver metabolize the benzodiazepines extensively.
Diazepam (Valium)
Diazepam is a benzodiazepine that is sedating, reducing agitation and anxiety. It decreases polysynaptic reflexes and has muscle relaxant, sedation, and antispasticity effects.58,148,193 Long used as a treatment for stiff person syndrome, its effects diminish over time, requiring alternative treatment such as intravenous propafol.107 Diazepam is usually started with a bedtime dose of 5 mg, increased to 10 mg as needed for adults, but is found to be effective when given 0.1 to 0.05 mg/kg and excessively sedating in children when given in excess of 1 mg/kg.150 Diazepam is well absorbed after an oral dose, with the peak blood level occurring typically in 1 hour. Diazepam is metabolized to the active compound N-desmethyldiazepam (nordiazepam) and then to oxazepam. The half-life of diazepam and its active metabolites is 20 to 80 hours, and it is 98% to 99% protein-bound. In patients with low serum albumin and lower protein-binding capacity, such as is often the case in patients with SCI or stroke, the incidence of undesirable sedation is increased. Daytime therapy is initiated with 2 mg and increased as needed; a clinical trial of children with cerebral palsy, however, suggests that a single bedtime dose was sufficient to improve passive stretching exercises throughout the day with the additional benefit of being a relatively low-cost pharmaceutical.150 Intravenous diazepam is reported to have been effective in the attenuation of intrathecal baclofen withdrawal.48
Diazepam intoxication causes a range of symptoms from somnolence to coma. Although it is generally regarded as having a wide margin of safety, treatment of patients with myelopathy has been linked to increased body weight.84 Benzodiazepine poisoning also has been reported.214 Near-term infants born with benzodiazepine intoxication are at risk. A case report notes a young mother at term who consumed a diazepam overdose of 250 to 300 mg and became drowsy but responsive. The fetal heart rate showed decreased variability and absence of accelerations. The benzodiazepine antagonist flumazenil, 0.3 mg, was given to the mother intravenously. Within 5 minutes, behavioral arousal in the mother and improved fetal heart rate variability were observed.225
Typical symptoms of patients in withdrawal from high-dose diazepam (>40 mg/day) are anxiety and agitation; restlessness; irritability; tremor; muscle fasciculation; twitching; nausea; hypersensitivity to touch, taste, smell, light, and sound; insomnia; nightmares; seizures; hyperpyrexia; and psychosis. The intensity of the symptoms and risk for death are related to the prewithdrawal dose. Symptoms of withdrawal from low-dose benzodiazepine (<40 mg/day) are more likely if the patient has taken the drug consistently for more than 8 months. Long-term usage of diazepam is common. In a study of 23 SCI treatment facilities with the Department of Veterans Affairs, 70% routinely used diazepam as an antispasticity treatment, and 67% of patients had been using it for more than 6 years.29 Onset of withdrawal symptoms occurs 1 to 2 days after a short-acting benzodiazepine is stopped or 2 to 4 days for a long-acting benzodiazepine. Even when the benzodiazepines are withdrawn slowly over 4 to 6 weeks, withdrawal symptoms can persist for 6 months.88
Clonazepam (Klonopin, Rivotril)
Clonazepam is a benzodiazepine that is indicated for the suppression of myoclonic, akinetic, or petit mal seizure activity. It can be used alone or as an adjunct for the treatment of spasticity and torsion dystonia.114 Well absorbed after an oral dose, the maximum blood concentrations of clonazepam occur in 1 to 2 hours. It is highly protein-bound and is metabolized in the liver. The half-life of clonazepam and its active 7-amino metabolite is 18 to 28 hours. Clonazepam, if used to treat spasticity, can be useful in suppressing spasms that disturb sleep at night. It is most commonly prescribed as 0.5 to 1 mg at night. If morning sedation is excessive, the tablet can be broken in half and 0.25 mg taken at night.
Gabapentin (Neurontin)
Gabapentin is an approved adjunctive treatment for epileptic seizure disorder. It is a structural derivative of GABA, but the mechanisms of gabapentin action are not fully elucidated. The off-label use of gabapentin for the treatment of spasticity, spastic hypertonia, and central pain syndromes in patients with SCI and multiple sclerosis has been cited in the literature.∗ Formica et al.80 conducted a randomized, double-blind, placebo-controlled trial regarding the efficacy and safety of gabapentin for treatment of spasticity in UMN injury. In this study, gabapentin was found to improve Ashworth Scale scores in the experimental versus control groups. These effects were found to occur with doses between 2700 and 3600 mg/day.80 The plasma level of gabapentin after oral administration peaks at 2 to 3 hours and has a half-life of 5 to 7 hours. Typically the required antispasticity dosage is 400 mg three times daily or higher to reduce tone in the multiple sclerosis and SCI population.67,99,168,201 Gabapentin can be associated with mild cognitive or behavioral effects and central nervous system depression, including somnolence, dizziness, ataxia, and fatigue. These adverse effects have been reported regardless of whether the medication is rapidly or slowly titrated. Gabapentin might also cause agitation in cognitively impaired patients.37 Abrupt cessation must be avoided because it can cause seizures in patients with brain lesions. Gabapentin awaits a demonstration of efficacy in the pediatric population but is suggested to be beneficial in treating elderly patients.14,183
Vigabatrin (Sabril)
Vigabatrin is approved in Canada for the treatment of drug-resistant seizures. It is γ-vinyl GABA (4-amino-hex-5-enoic acid) that irreversibly inhibits the activity of GABA transaminase. GABA transaminase exists in neurons and glia to regulate the intracellular balance of GABA, glutamate, and intermediate substances in energy metabolism such as α-ketoglutarate. Inhibition of GABA transaminase results in an elevation of GABA levels in the brain.72,97,100 Vigabatrin is well absorbed after an oral dose, and the peak plasma concentrations occur within 2 hours. It is widely distributed in the body and eliminated mainly by the kidney. The plasma half-life is 5 to 8 hours in young adults and 12 to 13 hours in elderly persons. The initial starting dose of vigabatrin is 500 mg twice per day and is often sedating. It is well absorbed orally, with peak plasma concentration within 2 hours. Like most antiepileptics, somnolence and drowsiness (28%) are unwanted side effects. Other adverse effects associated with vigabatrin are fatigue (28%), dizziness (21%), nystagmus (15%), abnormal vision (11%), agitation (11%), amnesia (10%), depression (10%), and paresthesia (9%).19,237
Pregabalin (Lyrica)
Pregabalin is approved in the United States and Europe for adjunctive therapy of partial seizures in adults and treatment of pain from diabetic neuropathy, postherpetic neuralgia, and fibromyalgia in adults. Pregabalin is structurally related to gabapentin, and the likely site of action of both drugs is similar, the α2-δ protein, an auxiliary subunit of voltage-gated calcium channels that reduces neuronal excitability.229 Bradley et al.26 highlight the difficulty in evaluating the efficacy of antispastic drugs when they conducted a retrospective case series reviewing a GABA analogue, pregabalin. In reviewing 22 patients who received pregabalin for spasticity, it was found the drug reduced symptoms, but there was difficulty in assessing the actual impact on the rehabilitation of patients because there is a bridge between clinical symptoms and functional recovery.26
Topiramate (Topamax)
Topiramate, another antiepileptic treatment, is a sulfamate-substituted monosaccharide (2,3:4,5-di-O-isopropylidene-β-D-fructopyranose sulfamate). It has been reported to be useful in the treatment of seizures and spasticity in children with Canavan disease, a rare autosomal recessive disease causing early-onset leukoencephalopathy and megalencephaly, as well as the infantile spasms of West syndrome when combined with vigabatrin.30,232 It has also been reported to be of therapeutic benefit in an elderly patient with vascular hemichorea–hemiballism.86 Topiramate is thought to have three possible mechanisms: blockade of state-dependent sodium channels, enhancement of GABAA receptor activation, and reduction of non-N-methyl-d-aspartic acid (NMDA) excitatory amino acid action. It is rapidly absorbed following oral administration, with approximately 80% bioavailability. Peak plasma concentration following a 400-mg dose occurs at approximately 2 hours. The half-life is 21 hours, and steady state is achieved after approximately 4 days. Topiramate is eliminated unchanged, primarily in the urine. Renal impairment can significantly prolong the elimination half-life of topiramate. Somnolence, dizziness, ataxia, and fatigue were the most frequently reported adverse reactions during clinical trials. The recommended initial dose of topiramate is 50 mg, usually in the evening, with dose increases by 50 mg/day each week to a maximum of 200 mg twice per day. Topiramate is available as 15- or 25-mg capsules or as sprinkle capsules.
Lamotrigine (Lamictal)
Lamotrigine is thought to act at voltage-sensitive sodium channels to stabilize neuronal membranes and inhibit the release of excitatory amino acid transmitters; it consequently has anticonvulsant effects. Lamotrigine has shown analgesic action in animal models of acute and chronic pain.172 Recent clinical trials with lamotrigine have shown promise in the treatment of chronic pain states and spasticity and concomitant chronic central pain.25 Skin-related adverse events have been reported during lamotrigine treatment, particularly in patients taking valproic acid.
Riluzole (Rilutec)
Riluzole is approved for symptomatic treatment of amyotrophic lateral sclerosis. It reportedly blocks the action of voltage-sensitive sodium channels, thereby preventing release of excitatory amino acids and producing a reduction in the stiffness associated with amyotrophic lateral sclerosis.21
Clonidine (Catapres, Dixarit)
Clonidine is best known as a treatment for hypertension. Because of its effects on the autonomic nervous system, however, clonidine has a number of effects on homeostatic processes. The most commonly described mechanism by which clonidine lowers blood pressure and heart rate appears to be an α2-mediated inhibition of locus coeruleus neurons and the intermediolateral cell column in the spinal cord. Clonidine has been termed a partial agonist because, when injected intravenously, the blood pressure increases as the α-receptors on blood vessels cause vasoconstriction and a brief increase in systemic blood pressure. Once the discharge rate of sympathetic preganglionic fibers is decreased via the central mechanism, the blood pressure decreases and the heart contraction rate slows.130,219,226,233
Clonidine’s effect on spinal reflex circuitry is to inhibit short-latency α-motor neuron response to group II muscle afferents, probably by augmenting presynaptic inhibition.211 It is likely that the antispasticity effect of clonidine is related to its α2-mediated presynaptic inhibition of sensory afferents.64,181,182 Clonidine is absorbed well from the gut, as well as through the skin, and has high bioavailability after an oral dose. The half-life is 5 to 19 hours, and 62% is excreted in the urine. Some patients with postural hypotension can develop symptomatic hypotension during clonidine treatment, and some patients can have an improvement of hypotension; therefore initial dosage should be low to evaluate the patient’s blood pressure response. A 25-mcg formulation is available in Canada (Dixarit) that can be taken orally twice per day to begin treatment. Alternatively, a 0.1-mg tablet can be cut into fourths (0.025 mg). The dose can be increased every 3 days by 0.05 mg/day as a three or four times per day dosage regimen. Clonidine is also available as a transdermal patch with two dosage formulations: 0.1 or 0.2 mg/day. The clonidine patch has been reported to be a useful treatment for spasticity.239,242 The patch is designed to deliver the indicated amount of clonidine daily for 7 days. Precaution should be used with individuals who develop an allergic skin reaction to the patch because they might have become sensitized to clonidine. A recent FDA alert warns physicians and patients to be aware of the potential burn hazard posed by wearing certain medication patches while being scanned in a magnetic resonance imager (MRI) because of the presence of metallic fibers in the patch. It is currently a recommendation for patients using transdermal clonidine to remove their patch during MRI examination to avoid a potential burn. A cautionary note for insulin-dependent diabetic patients is that clonidine can retard a tachycardia induced by a hypoglycemic reaction to insulin, which can delay the diagnosis. Additional side effects are dry mouth, ankle edema, and depression.
Cyproheptadine (Periactin)
Cyproheptadine has been a prescription pharmaceutical for many years, approved for the treatment of vascular headache, anorexia, and itching associated with hives. It is a nonselective compound with histamine and serotonin antagonist effects. It is deemed safe to use in pregnant women; in fact, it has been used in the treatment of repetitive spontaneous abortion. Cyproheptadine can decrease the duration of clonus in people with spasticity caused by SCI or multiple sclerosis.11 Cyproheptadine is reported to improve muscle firing patterns and increase walking speed in patients whose gait was limited by clonus.236 In a comparative clinical trial, cyproheptadine had similar antispasticity efficacy to that of clonidine and baclofen in spinal cord–injured patients.173 A recent report described a patient who was symptomatic from intrathecal baclofen withdrawal and who showed significant amelioration of symptoms by the use of cyproheptadine.165
Chlorpromazine (Thorazine, Largactil)
Chlorpromazine, a phenothiazine derivative developed in 1949, is a well-known sedative–antipsychotic drug that can depress reflex activity.27 Chlorpromazine is rarely used as an antispasticity treatment, but in an open clinical trial, resting muscle tone decreased with chlorpromazine and phenytoin (Dilantin) treatment.42 Adverse effects of chlorpromazine include sedation and the potential risk for tardive dyskinesia.
Inhibition of Excitatory Amino Acids
Orphenadrine Citrate (Norflex) and Memantine (Namenda)
Orphenadrine citrate is most commonly used as a “muscle relaxant,” but it has shown some efficacy as an antispasticity treatment in patients with SCI when given as an intravenous infusion.35 A related compound, orphenadrine hydrochloride (Disipal), is a treatment for Parkinson disease. Patch-clamp and binding studies have revealed that orphenadrine is an uncompetitive NMDA-type glutamate antagonist. Memantine (1-amino-3,5-dimethyladamantane) has anticonvulsant properties and has been used to treat Parkinson disease and dementia.7 Similar to orphenadrine, memantine inhibits binding of the NMDA antagonist MK-801.
Carisoprodol (Soma)
Similar to orphenadrine, metaxalone (Skelaxin), and cyclobenzaprine (Flexeril), carisoprodol is a “centrally acting” muscle relaxant. The antispasticity effects of carisoprodol, an isopropyl derivative of meprobamate, were tested by the then newly described Ashworth Scale in 1964.10 In that trial of 24 patients with multiple sclerosis, 1 patient was much improved, 16 improved, 2 slightly improved, and 5 showed no change. With an average dose of 350 mg four times per day, drowsiness is the main side effect.
Cannabinoids
Cannabis (Cesamet, Marinol, Sativex)
Great controversy exists in the United States concerning the medicinal use of marijuana, despite the long history of the use the leaves of the Cannabis sativa plant.170 The medicinal uses of this plant were discovered by China’s emperor around 2800 bc, Shen Nung, who wrote about its numerous therapeutic uses. O’Shaughnessy190 reported the use of hemp extract to treat muscle spasms associated with tetanus in 1842. Reynolds subsequently described the toxic effects and therapeutic use of cannabis in the treatment of epilepsy, chorea, and nocturnal spasms in 1890.203 The most studied of the various active alkaloids is Δ-9-tetrahydrocannabinol (THC), available as a prescription pharmaceutical dronabinol (Marinol), as the synthetic cannabinoid nabilone (Cesamet), or in Canada as a sublingual spray Sativex (THC and cannabidiol). The clinical indication for dronabinol and nabilone is nausea related to chemotherapy treatment and for Sativex is neuropathic pain in multiple sclerosis, although these compounds have been used clinically for spasticity.
Anecdotal reports suggest that smoking marijuana has a muscle-relaxing effect in people with SCI or multiple sclerosis, but users might experience significant rebound spasticity between doses.34,68 Numerous survey-type reports indicate improvement of spasticity with the use of cannabis in SCI, multiple sclerosis, and amyotrophic lateral sclerosis.3,34,39,147 An on-again, off-again study of one subject with SCI consistently showed reduced spasticity while on THC.154 Some placebo-controlled clinical trials of orally administered compounds might cast doubt on the efficacy of THC in objectively reducing spasticity in patients with multiple sclerosis.223 It might be, however, that the detection of the subjective relaxation was not measured by the Ashworth Scale but rather by the secondary outcome measure of the patient-reported treatment effect that was significantly improved compared with placebo.247 A small, essentially open-label study of people with SCI suggests that spasticity is reduced with THC.104
Bioavailability of dronabinol after an oral dose is 4% to 12% with less than 1% excreted via the urine, and 95% of the drug is bound to plasma proteins. The half-life is 20 to 44 hours. It is formulated in 2.5-, 5-, or 10-mg capsules. Nabilone is formulated as a pulvule containing 1 mg, with a recommended dose of 1 to 2 mg twice per day. With the discovery of the endocannabinoid system and the potential development of selective agonists and endocannabinoid degradation inhibitors, there might be greater future use of cannabinoids medicinally than that presently.69 Further studies on the use of cannabinoids in SCI and other people with spasticity are warranted.
Injectable Pharmacologic Therapy
The advantage of an injected, locally administered agent is to limit systemic effects while targeting a specific nerve or muscle. A good rationale exists for local therapy in the treatment of spasticity. Local treatment provides the opportunity to target specific muscles and body regions. For example, in a patient with severely spastic distal hand muscles, one can perform intramuscular injections of a given therapeutic agent to achieve a finely graded result. Additional benefits of local therapy include the prevention or amelioration of contracture and reduction of pain associated with spasticity. Specific targeted alteration of spastic muscle tone can improve sitting posture and reduce the need for splinting or bracing. For example, reducing spastic thigh adductor tone can improve perineal hygiene and ease of nursing care.224 Local therapies and oral or intrathecal antispasticity treatment can be used in conjunction.
Phenol and Alcohol
Before the introduction of botulinum toxin and intrathecal baclofen, phenol was the most widely used injectable agent in the treatment of spasticity.241 Phenol denatures the protein of nerve fibers and in low concentrations has the property of a reversible local anesthetic. By contrast, alcohol is thought to have a dehydration effect on nerve tissue, resulting in sclerosis of nerve fibers and the myelin sheath. These agents can be injected at motor points identified by electrical stimulation, or directly into or onto nerve. The potential complications of phenol or alcohol injection include pain, dysesthesias that can last from weeks to months, arrhythmias, variable duration of effect, and incomplete irreversibility. Subarachnoid block has been performed with intrathecal phenol or alcohol, although this therapy can result in severe weakness and reduced sphincter function.177
Botulinum Toxin Therapy (Botox, Dysport, Xeomin, Myobloc)
Botulinum toxin is a neurotoxin and the biologic product of the action of an anaerobic bacterium, Clostridium botulinum. The toxin is one of the world’s most deadly poisons. It is an extremely potent neuromuscular blocking agent when injected into the area of a motor point. It is approved by the FDA for the treatment of strabismus, blepharospasm, and facial, cervical, and laryngeal dystonia.18,83 Seven identified serotypes include botulinum toxin A through G. The neurotoxin itself is a 150-kDa single-chain protein that is secreted as a large complex with nontoxic neurotoxin-associated proteins (NAPs) (Figure 30-3). Interestingly, the tetanus neurotoxin secreted by Clostridium tetani is not known to have NAPs. The 150-kDa botulinum neurotoxin is configured and looped to form a di-chain consisting of a heavy chain and a light chain that are linked by a disulphide (S-S) bridge. Whereas the heavy chain is responsible for getting the whole molecule close to its target, it is the light chain that is responsible for the toxicity. The light chains bring about their toxicity by inhibiting the release of acetylcholine at the neuromuscular junction by affecting the SNARE (Soluable N-ethyl-maleimide sensitive factor Attachment REceptor) proteins. The SNARE proteins consist of vesicle-associated membrane protein (VAMP, also known as synaptobrevin-2), synaptosomal associated protein with a molecular mass of 25 kDa (SNAP-25), and syntaxin (a protein inserted into the plasma membrane primarily protruding into the cytosol). The serotypes (A through G) differ in their mechanism of action by damaging one or more of the SNARE proteins, ultimately bringing about neuromuscular blockade (Figure 30-4).
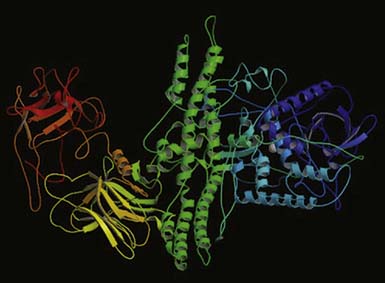
FIGURE 30-3 Crystallographic structure of botulinum neurotoxin (Botox).
(From Lacy DB, Tepp W, Cohen AC, et al: Crystal structure of botulinum neurotoxin type A and implications for toxicity, Nat Struct Biol 5:898-902, 1998.)
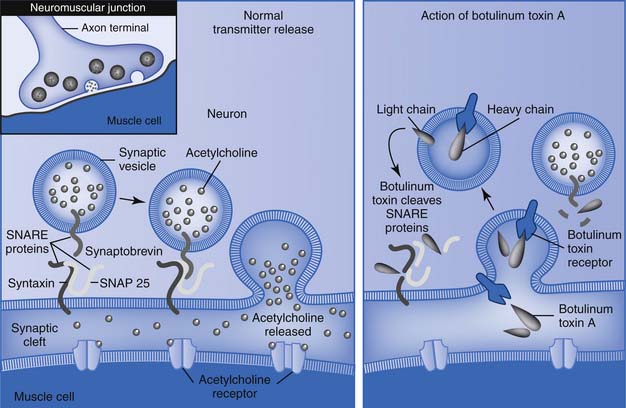
FIGURE 30-4 Mechanism of botulinum toxins.
(Redrawn from Arnon SS, Schechter R, Inglesby TV, et al: Botulinum toxin as a biological weapon: medical and public health management, JAMA 285:1059-1070, 2001.)
The botulinum toxin serotypes A and B are currently available in clinical practice. When injected intramuscularly the heavy chain of the toxin has an affinity for cholinergic nerve terminals. It is actively taken up by receptor-mediated endocytosis at the nerve terminal bouton of a motor nerve forming a toxin-containing vesicle. The light chain of the toxin is then actively extruded into the cytosol through the plasma membrane of the vesicle by a process of translocation. During this process there is cleavage of the disulphide (S-S) bridge to release the light chain from the heavy chain. The light chain of botulinum toxin A specifically cleaves the SNAP-25 protein, whereas botulinum toxin B cleaves the VAMP. In either case as the SNARE proteins are cleaved there is interference with the binding of the acetylcholine-filled vesicle with the terminal membrane, preventing its release via exocytosis. This neuromuscular junction blockade results in effective chemical denervation of muscle fibers and failure of contraction. Intrafusal muscle fibers can also be similarly paralyzed, resulting in decreased afferent spindle discharge and decreased reflex activity. Following blockage of the neuromuscular junction with botulinum toxin A, within days the motor neuron axon begins to sprout and forms synaptic contacts that peak over the next 5 to 10 weeks. Subsequently the parent axon reestablishes contact and begins to recover while the sprouts regress and become less active.115,191,218 The duration of clinical benefits with toxin A is variable but generally lasts 2 to 4 months.51,52
Around the world botulinum toxin serotype A is marketed as Botox, Dysport, Xeomin, and BTXA, whereas botulinum toxin B is marketed as Myobloc and NeuroBloc. At present only Botox and Myobloc are approved by the FDA, with a new drug application for Dysport recently submitted. Botulinum toxin serotype A (Botox) use has been approved by the FDA for the treatment of strabismus, blepharospasm, and primary axillary hyperhydrosis. Botulinum toxin serotype B (Myobloc) is approved for the treatment of cervical dystonia. Botulinum toxin A and B have been demonstrated to be safe and effective for other focal dystonias, such as spasmodic dysphonia (laryngeal dystonia). Botulinum toxin serotypes A and B also have been used in the treatment of spasticity.18,83 Caution with dosing is advised when switching between different products even within the same serotype because they do not have a uniform measure.
Generally a dose–effect relationship exists such that a greater toxin load per muscle results in a higher degree of weakness. A higher dose of botulinum toxin, for example, is typically required to weaken a larger spastic muscle, such as hamstring muscle, than a dystonic finger flexor. Therapy must be individualized for each patient, taking into account the body weight, muscle size, location of muscle, and degree of spasticity. It should also be noted that the serotype, concentration, and volume of the toxin can affect the amount of spread of the toxin and the intensity of the resulting muscle-weakening effect.
In 10 nonambulatory patients with spastic contraction of the thigh adductor muscles caused by multiple sclerosis, botulinum toxin improved sitting, positioning, and ease of attaining thigh abduction for urethral catheterization and perineal hygiene.224 Hyman et al.116 demonstrated efficacy of Botox in the treatment of hip adductor spasticity in multiple sclerosis, and Pittock et al.199 demonstrated effectiveness of Botox in the treatment of spastic equinovarus deformity after stroke. Electromyography-guided botulinum toxin (20 to 80 units) injections of the urethral rhabdosphincter are reported to reduce detrusor–sphincter dyssynergia in men with SCI.70 Transient hematuria was noted following the injection. In children with spastic plantar flexors caused by cerebral palsy, injection of a total dose of 4 units/kg resulted in significant improvement of ankle position and gait.38
A systematic review undertaken by van Kuijk et al.235 in 2002 reported that botulinum toxin treatment in poststroke spasticity was effective in reducing muscle tone (modified Ashworth Scale) and improving joint range, without improvement in functional abilities. An evidence-based review of stroke rehabilitation undertaken by Teasell et al.230 concluded that in the upper limb botulinum toxin alone or in combination with therapy significantly reduces spasticity. It is not clear whether the improvements are sustained, however, nor is there significant evidence that there is improved function and quality of life.217 In the lower limb, although there is strong evidence of reduction of spasticity,60 there is conflicting evidence of improved function. To enhance its effectiveness, an additional electrical stimulation protocol seems promising.16,113,119 In the literature one of the limitations in measuring functional outcome has been the lack of an appropriate functional measure tool. Use of goal-oriented outcome measures such as the GAS is promising. Further studies to evaluate goal-oriented functional outcomes are in progress.
Botulinum toxin A has been extensively used in clinical practice and is relatively safe. Side effects can occur as a result of the injection itself or the toxin. Rarely allergic reactions can occur, although systemic anaphylaxis is extremely rare. One death was reported in a woman following injection of Botox–lidocaine mixture for chronic back and neck pain, although a direct causal relationship to Botox was not established.143 Distant spread of the toxin can occur, and widespread single-fiber electromyography abnormalities have been shown away from the site of injection, although the magnitude of the increased jitter did not correlate with the dose of the toxin.136 A survey of American physicians reported 30 births after exposure to therapeutic botulinum toxin injections during pregnancy, with 27 normal deliveries, 2 miscarriages, and 1 premature birth.167 It is best to avoid treatment with botulinum toxins during pregnancy given the limited information. Rarely patients can develop antibodies to the toxins. The true prevalence is not known given the difficulties in variation in dose, concentration and frequency, and defining secondary nonresponse.
In recent years botulinum toxin A has become an important armament in the management of spasticity-related focal problems. In select situations it could be the first line of treatment. Keeping in mind it is only symptomatic management and not a cure, caution is advised in muscle selection, doses, and frequency of injections. The manufacturer of Botox recommends not to exceed 6 units/kg up to a maximum of 360 units in a 3-month period. In clinical practice many physicians use higher doses. A European consensus on the use of botulinum toxin A in adult spasticity recommends not exceeding a total dose of 600 units of Botox per treatment session.240
Chronic Infusion of Medications Into the Intrathecal Space
Intrathecal Baclofen (Lioresal)
Intrathecal baclofen therapy (ITB) therapy is the use of liquid baclofen delivered from a pump in the lower abdominal wall, through a catheter tunneled around to the spine, and then into the intrathecal space around the lumbar spinal cord (around T10 vertebral level). This allows a 100-fold increase in potency65 at the tissues most responsible for the spasticity, with little exposure of the brain to the medication, thus allowing for excellent lower body spasticity control without the significant sedating side effects of oral medications.
Several prospective, randomized studies have demonstrated the effectiveness of intrathecal baclofen for the treatment of spasticity.∗ Although this form of antispasticity therapy is highly effective, its use is limited by the invasive nature of the treatment, the cost of the implantable devices, and the risk for complications. However, for patients in whom severe spasticity is the cause for hospitalization, intrathecal baclofen can be cost-effective by reducing the need for inpatient hospitalization or 24-hour nursing care.17,179 The indication for considering a person for IBT is severe, intractable, problematic spasticity, resistant to tolerable doses of oral medication or other therapy. The spasticity must be problematic to the patient or givers, and IBT should never be instituted unless the spasticity is deemed to be problematic. The patient and family must be committed to the long-term follow-up care required, and be reliable to do so.
Because the catheter is usually threaded up to approximately the T10 vertebral body (where the lumbar spinal cord is situated), ITB is most beneficial in treating lower body spasticity such as for spinal cord injury, multiple sclerosis, and cerebral palsy diplegia. In those with both problematic upper and lower limb spasticity, the upper limb spasticity will not be as greatly affected. In these cases, one can use either concomitant treatment to the upper limbs with botulinum toxin, continued use of concomitant oral therapy, or threading the ITB treatment catheter up to the midthoracic area (T6–T7), affecting both upper and lower limb tone. This has been used in treating spasticity from quadriparetic cerebral palsy, tetraplegia from SCI, poststroke hemiplegia, and acquired brain injury.† Once it is decided that intrathecal baclofen is recommended, a test dose via lumbar puncture is given, starting at 50 mcg. The muscle tone of the patient should be evaluated hourly for 6 hours. A decrease in the Ashworth Scale score of a preselected muscle group by one or two grades is often cited as evidence of a threshold dose of intrathecal baclofen.160,180 If a therapeutic response is not obtained, then trials with 75 or 100 mcg could be conducted on following successive days until a therapeutic response is observed. Two methods are used to predict the initial 24-hour dose. One is to double the threshold dose and deliver over 24 hours. The other is to consider the duration of the antispasticity effect. If the effect of the test injection persisted for 8 hours, or one third of a 24-hour period, then the estimated 24-hour dose would be three times the test dose.
Intrathecal Morphine (Infumorph) and Midazolam (Versed)
The use of morphine by direct infusion into the epidural or intrathecal space has been reported to be a highly effective antispasticity and analgesic treatment.75 In a series of four patients with the combination of severe pain and spasticity, both conditions were controlled for up to 8 months with daily dosages of 2 to 4 mg. Tolerance, pruritus, nausea, hypotension, urinary retention, and respiratory depression can occur, however, with an intrathecal morphine bolus dose as low as 0.4 mg.47,123 Intrathecal midazolam (Versed) has also been reported to be effective but is limited by sedation.170
Surgical Interventions
Orthopedics and neurosurgery are the two most commonly involved surgical subspecialties involved in the treatment of spasticity. Common goals of surgery are usually to increase mobility, decrease the use of external aids, correct or prevent deformity, and ultimately maximize function.123 Musculoskeletal compensatory techniques used by the orthopedic surgeons include tendon lengthening, muscle–tendon transfer (e.g., the split anterior tibial transfer), contracture release, capsulotomy, osteotomy, resection arthroplasty, arthrodesis, epiphyseodesis, ankle fusion, and spine fusion.85,123,125,149 The neuroablative techniques used by the neurosurgeons involve the interruption of the spinal reflex arc by neurectomy, neurotomy, rhizotomy, selective rhizotomy, dorsal root entry zone lesion, and myelotomy (Bischoff or T-shaped). An anecdotal report details two stroke patients with painful spastic hemiparetic leg who experienced improved pain management following unilateral selective posterior rhizotomy.85 Other procedures include cordotomy, cordectomy, implantation of stimulators, or drug infusion devices. Ideally the procedure would be low in cost, extremely safe, require a small operation with complete relief of spasticity, and yet produce total preservation of voluntary movement, sensation, and sphincter and sexual function. A surgical therapy that could achieve this optimal outcome unfortunately remains a future goal.124
Conclusion
Spasticity, like the underlying neuropathology it represents, is in an evolving state of change. Initially the individual patient should be thoroughly assessed, the goal of treatment determined by agreement between the practitioner and the patient, and a comprehensive treatment plan initiated. This plan should include the avoidance of spasticity-aggravating factors, and frequent range of motion exercise.105 Patients receiving antispasticity medication should be reviewed periodically to assess response to treatment, as well as side effects and adverse effects; to monitor the optimal use of mobility aids and wheelchair seating (if applicable); and to monitor the accompanying neurologic impairment. Patients who are surgical candidates require a thorough initial investigation. This investigation should establish that skeletal growth and postoperative compliance have been considered, and that the best medical treatment, where appropriate, has failed to benefit the patient. In summary, aggressive diagnosis and management of spasticity, accompanied by the setting of appropriate goals established by dialogue between the physician, patient, and caregivers, can lead to substantial improvement in quality of life in affected individuals.
The use of any treatment modality should be tailored to the patient and the diagnosis. A drug that is recommended for a patient with SCI might not be the first-line treatment for a patient with a cerebral cause or spastic hypertonia caused by other aspects of the patient’s condition (e.g., relative intolerance for sedation as a side effect).70 Another consideration should be whether these agents, particularly those that have an effect on glutamate (necessary for learning), might slow neurorecovery in the first months following central nervous system injury or illness. For example, consider the observation that α-noradrenergic agonists, benzodiazepines, baclofen, and antiepileptic agents might impair neurorecovery in the first weeks and months after injury or illness.62,92 Considerable research in the different impairment populations, as well as the timing of interventions, remains an ongoing challenge in neurorehabilitation.
1. Abbruzzese G. The medical management of spasticity. Eur J Neurol. 2002;1(suppl 9):30-34.
2. American Academy of Neurology. Assessment: the clinical usefulness of botulinum toxin-A in treating neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1990;40:1332-1336.
3. Amtmann D., Weydt P., Johnson K.L., et al. Survey of cannabis use in patient with amyotrophic lateral sclerosis. Am J Hosp Palliat Care. 2004;21(2):95-104.
4. [Anonymous]. Environmental Status Scale, question 4, minimal record of disability for multiple sclerosis. New York: National Multiple Sclerosis Society; 1985.
5. [Anonymous]. Guide for the Uniform Data Set for Medical Rehabilitation, version 4.0 (adult FIM). Buffalo: UB Foundation Activities; 1993.
6. [Anonymous]. SF-36 Health Survey. Boston: Medical Outcome Trust; 1992.
7. Areosa S.A., Sherriff F., McShane R. Memantine for dementia. Cochrane Database Syst Rev. 2005:CD003154.
8. Arnon S.S., Schechter R., Inglesby T.V., et al. Botulinum toxin as a biological weapon: medical and public health management. JAMA. 2001;285(8):1059-1070.
9. Ashford S., Turner-Stokes L. Goal attainment for spasticity management using botulinum toxin. Physiother Res Int. 2006;11(1):24-34.
10. Ashworth B. Preliminary trial of carisoprodol in multiple sclerosis. Practitioner. 1964;192:540-542.
11. Ashworth N.L., Satkunam L.E., Deforge D. Treatment for spasticity in amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2004:CD004156.
12. Bajd T., Gregoric M., Vodovnik L., et al. Electrical stimulation in treating spasticity resulting from spinal cord injury. Arch Phys Med Rehabil. 1985;66:515-517.
13. Barbeau H., Richards C.L., Bedard P.J. Action of cyproheptadine in spastic paraparetic patients. J Neurol Neurosurg Psychiatry. 1982;45:923-926.
14. Barnes M.P. Spasticity: a rehabilitation challenge in the elderly. Gerontology. 2001;47(6):295-299.
15. Barolat G., Myklebust J.B., Wenninger W. Effects of spinal cord stimulation on spasticity and spasms secondary to myelopathy. Appl Neurophysiol. 1988;51:29-44.
16. Bayram S., Sivrioglu K., Karli N., Ozcan O. Low-dose botulinum toxin with short-term electrical stimulation in poststroke spastic drop foot: a preliminary study. Am J Phys Med Rehabil. 2006;85(1):75-81.
17. Becker W., Letts L. Intrathecal baclofen for adults with spinal spasticity. Can J Neurol Sci. 1995;22:122-129.
18. Bell K.R., Williams F. Use of botulinum toxin type A and type B for spasticity in upper and lower limbs. Phys Med Rehabil Clin North Am. 2003;14(4):821-835.
19. Ben-Menachem E. Vigabatrin. Epilepsia. 1995;36(suppl 2):S95-S104.
20. Bennegard G.M., Karlsson A.K. Higher glucose uptake in paralysed spastic leg. Spinal Cord. 2008;46(2):103-106.
21. Bensimon G., Lacomblez L., Meininger V., et al. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330(9):585-591.
22. Berger M., Bobbitt R.A., Carter W.B., et al. The Sickness Impact Profile: development and final revision of a health status measure. Med Care. 1981;19:787-805.
23. Bohannon R.W., Andrews A.W. Correlation of knee extensor muscle torque and spasticity with gait speed in patients with stroke. Arch Phys Med Rehabil. 1990;71:330-333.
24. Bohannon R.W., Smith M.B. Interrater reliability of a modified Ashworth Scale of muscle spasticity. Phys Ther. 1987;67(2):206-207.
25. Bonicalzi V, Canavero S. Lamotrigine effects on chronic pain: an open-label pilot study [abstract]. In Proc 8th World Congr of Pain 173, 1996.
26. Bradley L.J., Kirker G.B. Pregabalin in the treatment of spasticity: a retrospective case series. Disabil Rehabil. 2008;30(16):1230-1232.
27. Bradley P.B. Tranquilizers. I. Phenothiazine derivatives. In: Root W.S., Hofmann F.G., editors. Physiological pharmacology. New York: Academic Press, 1963.
28. Brar S.P., Smith M.B., Nelson L.M., et al. Evaluation of treatment protocols on minimal to moderate spasticity in multiple sclerosis. Arch Phys Med Rehabil. 1991;72:186-189.
29. Broderick C.P., Radnitz C.L., Bauman W.A. Diazepam usage in veterans with spinal cord injury. J Spinal Cord Med. 1997;20:406-409.
30. Buoni S., Zannolli R., Strambi M., et al. Combined treatment with vigabatrin and topiramate in West syndrome. J Child Neurol. 2004;19(5):385-386.
31. Burke D., Andrews C., Ashby P. Autogenic effects of static muscle stretch in spastic man. Arch Neurol. 1971;25:367-372.
32. Burry H.C. Objective measurement of spasticity. Dev Med Child Neurol. 1972;14:508-510.
33. Byrd B.F., Collins W., Primm R.K. Risk factors for severe bradycardia during oral clonidine therapy for hypertension. Arch Intern Med. 1988;148:729-733.
34. Cansore P., Musty R., Rein J., et al. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol. 1997;38(1):44-48.
35. Casale R., Glynn C., Buonocore M. Reduction of spastic hypertonia in patients with spinal cord injury: a double-blind comparison of intravenous orphenadrine citrate and placebo. Arch Phys Med Rehabil. 1995;76:660-665.
36. Cervera-Deval J., Morant-Guillen M.P., Fenollosa-Vasques P., et al. Social handicaps of multiple sclerosis and their relation to neurological alterations. Arch Phys Med Rehabil. 1994;75:1223-1227.
37. Childers M.K., Holland D. Psychomotor agitation following gabapentin use in brain injury. Brain Inj. 1997;11(7):537-540.
38. Chutorian A., Root L. Management of spasticity in children with botulinum-A toxin. Int Pediatr. 1994;9(suppl 1):35-43.
39. Clifford D.B. Tetrahydrocannabinol for tremor in multiple sclerosis. Ann Neurol. 1983;13:669-671.
40. Coffey R.J., Cahill D., Steers W., et al. for the Intrathecal Baclofen Multicenter Study Group: Intrathecal baclofen for intractable spasticity of spinal origin: results of a long-term multicenter study. J Neurosurg. 1993;78:226-232.
41. Cohen A.H., Rossignol S., Grillner S., editors. Neural control of rhythmic movements in vertebrates. New York: Wiley Interscience, 1988.
42. Cohen S.L., Raines A., Panagakow J., et al. Phenytoin and chlorpromazine in the treatment of spasticity. Arch Neurol. 1980;37:360-364.
43. Cole B., Finch E., Gowland C., et al, editors. Physical rehabilitation outcome measures. Baltimore: Williams & Wilkins, 1995.
44. Corcos D.M., Gottlieb G.L., Penn R.D., et al. Movement deficits caused by hyperexcitable stretch reflexes in spastic humans. Brain. 1986;109:1043-1058.
45. Costa E., Guidotti A. Molecular mechanisms in the receptor action of the benzodiazepines. Ann Rev Toxicol. 1979;19:531-545.
46. Coward D.M., Davies J., Herrling P., et al. Pharmacological properties of tizanidine (DS103-282). Stuttgart: FK Schattattauer Verlag; 1984.
47. Craig C.L., Zimbler S. Orthopedic procedures. In: Whyte G., editor. The practical management of spasticity in children and adults. Philadelphia: Lea & Febiger, 1990.
48. Cruikshank M., Eunson P. Intravenous diazepam infusion in the management of planned intrathecal baclofen withdrawal. Dev Med Child Neurol. 2007;49(8):626-628.
49. Cusick A., McIntyre S., Novak I., et al. A comparison of goal attainment scaling and the Canadian Occupational Performance Measure for paediatric rehabilitation research. Pediatr Rehabil. 2006;9(2):149-157.
50. Cutter N.C., Scott D.D., Johnson J.C., et al. Gabapentin effect of spasticity in multiple sclerosis: a placebo controlled, randomized trial. Arch Phys Med Rehabil. 2000;81:164-169.
51. Das T.K., Park D.M. Botulinum toxin in treating spasticity. Br J Clin Pract. 1989;43:401-403.
52. Das T.K., Park D.M. Effect of treatment with botulinum toxin on spasticity. Postgrad Med J. 1989;65:208-210.
53. Davidoff R.A. Antispasticity drugs: mechanisms of action. Ann Neurol. 1985;17:107-116.
54. Davies J. Selective depression of synaptic transmission of spinal neurones in the cat by a new centrally acting muscle relaxant, 5-choloro-4-(2-imidazolin-2-yl-amino)-2,1,3-benzothiadiazole (DS103 282). Br J Pharmacol. 1982;76:473-481.
55. Davies J., Johnston S.E. Selective antinociceptive effects of tizanidine (DS 103-282), a centrally acting muscle relaxant, on dorsal horn neurones in the feline spinal cord. Br J Pharmacol. 1984;82:409-421.
56. Davies J., Johnston S.E., Hill D.R., et al. Tizanidine (DS103-282), a centrally acting muscle relaxant, selectively depresses excitation of feline dorsal horn neurones to noxious peripheral stimuli by an action at alpha adrenoceptors. Neurosci Lett. 1984;48:197-202.
57. Delwaide P.J. Electrophysiological testing of spastic patients: its potential usefulness and limitations. In: Delwaide P.J., Young R.R., editors. Clinical neurophysiology in spasticity. Amsterdam: Elsevier, 1985.
58. Delwaide P.J. Étude expérimentale de l’hyperréflexie tendineuse en clinique neurologique. Brussels: Arscia; 1971.
59. Delwaide PJ, Gerard P. Reduction of non-reciprocal (Ib) inhibition: a key factor for interpreting spastic muscle stiffness. In International Congress on Stroke Rehabilitation, Berlin, November 1993.
60. Dengler R., Neyer U., Wohlfarth K., et al. Local botulinum toxin in the treatment of spastic drop foot. J Neurol. 1992;239:375-378.
61. Dewald J.P.A., Given J.D. Electrical stimulation and spasticity reduction: fact or fiction? In: Katz R., editor. Spasticity. State of the art reviews. Philadelphia: Hanley & Belfus, 1994.
62. Dikmen S.S., Temkin N.R., Miller B., et al. Neurobehavioral effects of phenytoin prophylaxis of posttraumatic seizures. JAMA. 1991;265:1271-1277.
63. Dimitrijevic M.M. Meshglove 2, modulation of residual upper limb motor control after stroke with whole-hand electrical stimulation. Scand J Rehabil Med. 1994;26:187-190.
64. Donovan W.H., Carter R.E., Rossi C.D., et al. Clonidine effect on spasticity: a clinical trial. Arch Phys Med Rehabil. 1988;69:193-194.
65. Dralle D., Muller H., Zierski J., et al. Intrathecal baclofen for spasticity. Lancet. 1985;2:1003.
66. Duncan G.W., Shahani B.T., Young R.R. An evaluation of baclofen treatment for certain symptoms in patients with spinal cord lesions. Neurology. 1976;26:441-446.
67. Dunevsky A., Perel A.B. Gabapentin for relief of spasticity associated with multiple sclerosis. Am J Phys Med Rehabil. 1998;77(5):451-454.
68. Dunn M., Davis R. The perceived effects of marijuana on spinal cord injured males. Paraplegia. 1974;12:175.
69. Duran M., Laporte J.R., Capella D. [News about the therapeutic use of cannabis and the endocannabinoid system.]. Med Clin (Barc). 2004;122(10):390-398.
70. Dykstra D.D., Sidi A.A. Treatment of detrusor-sphincter dyssynergia with botulinum A toxin: a double-blind study. Arch Phys Med Rehabil. 1990;71:24-26.
71. Ellis K.O., Carpenter J.F. Mechanisms of control of skeletal–muscle contraction by dantrolene sodium. Arch Phys Med Rehabil. 1974;55:362-369.
72. Elovic E. Principles of pharmaceutical management of spastic hypertonia. Phys Med Rehabil Clin North Am. 2001;12:793-816.
73. Elovic E.P., Simone L.K., Zafonte R. Outcome assessment for spasticity management in the patient with traumatic brain injury: the state of the art. J Head Trauma Rehabil. 2004;19(2):155-177.
74. Emre M. Review of clinical trials with Sirdalud in spasticity. In: Emre M., Benecke R., editors. Spasticity. London: Parthenon Publishing, 1989.
75. Erickson D.L., Blacklock J.B., Michaelson M., et al. Control of spasticity by implantable continuous flow morphine pump. Neurosurgery. 1985;16:215-217.
76. Esquenzai A., Hirai B.A. Assessment and management of gait dysfunction in patients with spastic stroke or brain injury. In: Katz R., editor. Spasticity: state of the art reviews. Philadelphia: Hanley & Belfus, 1994.
77. Feldman R.G., Kelly-Hayes M., Conomy J.P., et al. Baclofen for spasticity in multiple sclerosis: double-blind crossover and three-year study. Neurology. 1978;28:1094-1098.
78. Finch L., Barbeau H. Hemiplegic gait: new treatment strategies. Physiother Can. 1986;38:36-40.
79. Flewellen E.H., Nelson T.E., Jones W.P., et al. Dantrolene dose response in awake man: implications for management of malignant hyperthermia. Anesthesiology. 1983;59:275-280.
80. Formica A., Verger K., et al. Gabapentin for spasticity: a randomize, double-blind, placebo-controlled trial. Med Clin. 2005;124(3):81-85.
81. Francisco G.E., Boake C. Improvement in walking speed in poststroke spastic hemiplegia after intrathecal baclofen therapy: a preliminary study. Arch Phys Med Rehabil. 2003;84:1194-1199.
82. Francisco G.E., Kothari S., Huls C. GABA agonists and gabapentin for spastic hypertonia. Phys Med Rehabil Clin North Am. 2001;12(4):875-888. viii
83. Fried G.W., Fried K.M. Spinal cord injury and use of botulinum toxin in reducing spasticity. Phys Med Rehabil Clin North Am. 2003;14(4):901-910.
84. Frisbie J.H., Aguilera E.J. Diazepam and body weight in myelopathy patients. J Spinal Cord Med. 1995;18(3):200-202.
85. Fukuhara T., Kamata I. Selective posterior rhizotomy for painful spasticity in the lower limbs of hemiplegic patients after stroke: report of two cases. Neurosurgery. 2004;54(5):1268-1272.
86. Gatto E.M., Uribe Roca C., Raina G., et al. Vascular hemichorea/hemiballism and topiramate. Mov Disord. 2004;19(7):836-838.
87. Gelber D.A., Good D.C., Dromerick A., et al. Open-label dose-titration safety and efficacy study of tizanidine hydrochloride in the treatment of spasticity associated with chronic stroke. Stroke. 2001;32(8):1841-1846.
88. Geller A. Common addictions. Clin Symp. 1996;48:23-24.
89. Gerkin R., Curry S.C., Vance M.V., et al. First order elimination kinetics following baclofen overdose. Ann Emerg Med. 1986;15(7):115-118.
90. Giebler K.B. Physical modalities. In: Glenn M.B., Whyte J., editors. The practical management of spasticity in children and adults. Philadelphia: Lea & Febiger, 1990.
91. Goff B. Grading of spasticity, and its effect on voluntary movement. Physiotherapy. 1976;62:358-361.
92. Goldstein L.B. Pharmacologic modulation of recovery after stroke: clinical data. J Neurol Rehabil. 1991;5:129-140.
93. Gorgey A.S., Dudley G.A. Spasticity may defend skeletal muscle size and composition after incomplete spinal cord injury. Spinal Cord. 2008;46(2):96-102.
94. Grabb P.A., Guin-Renfroe S., Meythaler J.M. Midthoracic catheter tip placement for intrathecal baclofen administration in children with quadriparetic spasticity. Neurosurgery. 1999;45:833-837.
95. Gracely R.H., McGrath P., Dubner R. Rating scales of sensory and affective verbal pain descriptors. Pain. 1978;5:5-18.
96. Gracies J.M. Physical modalities other than stretch in spastic hypertonia. Phys Med Rehabil Clin North Am. 2001;12:747-768.
97. Grant S.M., Heel R.C. Vigabatrin: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in epilepsy and disorders of motor control. Drugs. 1991;41(6):889-926.
98. Greene W.B., Heckman J.D., editors. The clinical measurement of joint motion, Rosemont. American Academy of Orthopaedic Surgeons, 1994.
99. Gruenthal M., Mueller M., Olson W.L., et al. Gabapentin for the treatment of spasticity in patients with spinal cord injury. Spinal Cord. 1997;35(10):686-689.
100. Guberman A: Vigabatrin. J Neurol Sci. 1996;34:S13-S17.
101. Guillaume D., Van Havenbergh A., Vloeberghs M., et al. A clinical study of intrathecal baclofen using a programmable pump for intractable spasticity. Arch Phys Med Rehabil. 2005;86(11):2165-2171.
102. Guissard N., Duchateau J., Hainaut K. Muscle stretching and motoneuron excitability. Eur J Appl Physiol. 1988;58:47-52.
103. Gybels J., van Roost D. Spinal cord stimulation for spasticity. Adv Tech Stand Neurosurg. 1987;15:63-96.
104. Hagenbach U., Luz S., Ghafoor N., et al. The treatment of spasticity with Δ9-tetrahydrocannabinol in persons with spinal cord injury. Spinal Cord. 2007;45:551-562.
105. Halstead L.S., Seager S.W.J., Houston J.M., et al. Relief of spasticity in SCI men and women using rectal probe electrostimulation. Paraplegia. 1993;31:715-721.
106. Hankey G.J., Stewart-Wynne E.G., Perlman D. Intrathecal baclofen for severe spasticity. Med J Aust. 1986;145:465-466.
107. Hattan E., Angle M.R., Chalk C. Unexpected benefit of propofol in stiff-person syndrome. Neurology. 2008;70(18):1641-1642.
108. Hepp-Reymond M.-C., Marini G. Perspectives on motor behavior and its neural basis. Basel: Karger; 1997.
109. Herman R., Mayer N., Mecomber S.A. Clinical pharmaco-physiology of dantrolene sodium. Am J Phys Med. 1972;51:296-311.
110. Hesse S., Mauritz K.H. Management of spasticity. Curr Opin Neurol. 1997;10(6):498-501.
111. Hesse S., Reiter F., Konrad M., et al. Botulinum toxin type A and short-term electrical stimulation in the treatment of upper limb flexor spasticity after stroke: a randomized, double-blind, placebo-controlled trial. Clin Rehabil. 1998;12:381-388.
112. Hill D.R., Bowery N.G. 3H-Baclofen and 3H-GABA bind to bicuculline-insensitive GABAB sites in rat brain. Nature. 1981;290:149-152.
113. Hinderer S.R., Lehmann J.F., Price R., et al. Spasticity in spinal cord injured persons: quantitative effects of baclofen and placebo treatments. Am J Phys Med Rehabil. 1990;69(6):311-317.
114. Hinson V.K., Goetz C.G. Torsion dystonia in children. Curr Treat Options Neurol. 2003;5(4):291-297.
115. Holland R.I., Brown M.C. Nerve growth in botulinum toxin poisoned muscles. Neuroscience. 1981;6:1167-1179.
116. Hyman N., Barnes M., Bhakta B., et al. Botulinum toxin (Dysport) treatment of hip adductor spasticity in multiple sclerosis: a prospective, randomised, double blind, placebo controlled, dose ranging study. J Neurol Neurosurg Psychiatry. 2000;68:707-712.
117. Jensen M.P., Karoly P., Braver S. The measurement of pain intensity: a comparison of six methods. Pain. 1986;27:117-126.
118. Jerusalem F. A double-blind study on the antispastic action of beta-(4-chlophenyl)-gamma-amino butyric acid (CIBA) in multiple sclerosis. Nervenarzt. 1968;39:515-517.
119. Johnson C.A., Burridge J.H., Strike P.W., et al. The effect of combined use of botulinum toxin type A and functional electric stimulation in the treatment of spastic drop foot after stroke: a preliminary investigation. Arch Phys Med Rehabil. 2004;85:902-909.
120. Jones R.F., Burke D., Marosszeky J.E., et al. A new agent for the control of spasticity. J Neurol Neurosurg Psychol. 1970;33:464-468.
121. Kakebeeke T.H., Lechner H.E., Knapp P.A. The effect of passive cycling movements on spasticity after spinal cord injury: preliminary results. Spinal Cord. 2005;43(8):483-488.
122. Kameyama T., Nabeshima T., Sugimoto A., et al. Antinociceptive action of tizanidine in mice and rats. Naunyn Schmiedebergs Arch Pharmacol. 1985;330:93-96.
123. Karol L.A. Orthopedic management in myelomeningocele. Neurosurg Clin North Am. 1995;6(2):259-268.
124. Kasdon D.L., Abramovitz J.N. Neurosurgical approaches. In: Whyte G., editor. The practical management of spasticity in children and adults. Philadelphia: Lea & Febiger, 1990.
125. Katz R.T. Management of spasticity. Am J Phys Med Rehabil. 1988;67:108-116.
126. Khorasani A., Peruzzi W.T. Dantrolene treatment for abrupt intrathecal baclofen withdrawal. Anesth Analg. 1995;80:1054-1056.
127. Kirklan L.R. Baclofen dosage: a suggestion [letter]. Arch Phys Med Rehabil. 1984;65:214.
128. Knutsson E., Lindblom U., Martensson A. Differences in effects in gamma and alpha spasticity induced by the GABA derivative baclofen (Lioresal). Brain. 1973;96:29-46.
129. Knutsson E., Mårtensson A., Gransberg L. Antiparetic and antispastic effects induced by tizanidine in patients with spastic paresis. J Neurol Sci. 1982;53:187-204.
130. Kobinger W., Walland A. Investigations into the mechanisms of the hypotensive effect of 2,(2,6-dichlorophenylamino)2-imidazoline HCl. Eur J Pharmacol. 1967;2:155-162.
131. Kofler M., Kronenberg M.F., Rifici C., et al. Epileptic seizures associated with intrathecal baclofen application. Neurology. 1994;44:25-27.
132. Krach L.E. Pharmacotherapy of spasticity: oral medications and intrathecal baclofen. J Child Neurol. 2001;16(1):31-36.
133. Krawetz P., Nance P. Gait analysis of spinal cord injured (SCI) subjects: effects of injury level and spasticity. Arch Phys Med Rehabil. 1996;77:635-638.
134. Lacy D.B., Tepp W., Cohen A.C., et al. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5(10):898-902.
135. Lance J.W. Symposium synopsis. In: Feldman R.G., Young R.R., Koella W.P., editors. Spasticity: disordered motor control. Chicago: Yearbook Medical, 1980.
136. Lange D.J., Brin M.F., Warner C.L., et al. Distant effects of local injection of botulinum toxin. Muscle Nerve. 1987;10(6):552-555.
137. Laporte Y., Lloyd D.P.C. Nature and significance of the reflex connections established by large afferent fibers of muscular origin. Am J Physiol. 1952;169:609-621.
138. Lechner H.E., Kakebeeke T.H., Hegemann D., et al. The effect of hippotherapy on spasticity and on mental well-being of persons with spinal cord injury. Arch Phys Med Rehabil. 2007;88(10):1241-1248.
139. Lee K.C., Carson L., Kinnin E., et al. The Ashworth Scale: a reliable and reproducible method of measuring spasticity. J Neurol Rehabil. 1989;3:205-209.
140. Leone J.A., Kukulka C.G. Effects of tendon pressure on alpha motoneuron excitability in patients with stroke. Phys Ther. 1988;68(4):475-480.
141. Levine M.F., Hui-Chan C.W.Y. Relief of hemiparetic spasticity by TENS is associated with improvement in reflex and voluntary motor functions. EEG Clin Neurophysiol. 1992;85:131-142.
142. Levine M.G., Knott M., Kabat H. Relaxation of spasticity by electrical stimulation of antagonist muscles. Arch Phys Med Rehabil. 1952;33:668-673.
143. Li M., Goldberger B.A., Hopkins C. Fatal case of Botox-related anaphylaxis? J Forensic Sci. 2005;50(1):169-172.
144. Liddell E.G.T., Sherrington C. Reflexes in response to stretch (myotactic reflexes). Proc R Soc Lond B Biol Sci. 1924;96:212-242.
145. Lieber R.L., Steinman S., Barash I.A., et al. Structural and functional changes in spastic skeletal muscle. Muscle Nerve. 2004;29(5):615-627.
146. Macdonald R.L., Olsen R.W. GABA receptor channels. Annu Rev Neurosci. 1994;17:569-602.
147. Malec J., Harvey R.F., Cayner J.J. Cannabis effect on spasticity in spinal cord injury. Arch Phys Med Rehabil. 1982;63(3):116-118.
148. Mandac B.R., Hurvitz E.A., Nelson V.S. Hyperthermia associated with baclofen withdrawal and increased spasticity. Arch Phys Med Rehabil. 1993;74:96-97.
149. Massagli T.L. Spasticity and its management in children. Phys Med Rehabil Clin North Am. 1991;2(4):867-889.
150. Mathew A., Mathew M.C. Bedtime diazepam enhances well-being in children with spastic cerebral palsy. Pediatr Rehabil. 2005;8(1):63-66.
151. Matthew W.B. Ratio of maximum H reflex to maximum M response of spasticity. J Neurol Neurosurg Psychiatry. 1966;29:201-204.
152. Matthews P.B.C. Mammalian muscle receptors and their central actions. Baltimore: Williams & Wilkins; 1972.
153. Maupas E., Marque P., Roques C.F. Modulation of the transmission in group II heteronymous pathways by tizanidine in spastic hemiplegic patients. J Neurol Neurosurg Psychiatry. 2004;75(1):130-135.
154. Maurer M., Henn V., Dittrich A., et al. Delta 9-tetrahyrdo cannabinole shows antispastic and analgesics effects in a single case double-blind trial. Eur Arch Psychiatry Clin Neurosci. 1990;240(1):1-4.
155. Maynard F.M., Karunas R.S., Waring W.P. Epidemiology of spasticity following traumatic spinal cord injury. Arch Phys Med Rehabil. 1990;71:566-569.
156. Memin B., Pollack P., Hommel M., et al. Effects of botulinum toxin on spasticity. Rev Neurol (Paris). 1992;148(3):212-214.
157. Metz L. Multiple sclerosis: symptomatic therapies. Semin Neurol. 1998;18(3):389-395.
158. Meythaler J.M. Use of intrathecally delivered medications for spasticity and dystonia in acquired brain injury. In: Yaksh T.L., editor. Spinal drug delivery. New York: Elsevier, 1999.
159. Meythaler J.M., Clayton W., Davis L.K., et al. Orally delivered baclofen to control spastic hypertonia in acquired brain injury. J Head Trauma Rehabil. 2004;19:101-108.
160. Meythaler J.M., DeVivo M.J., Hadley M. Prospective study on the use of bolus intrathecal baclofen for spastic hypertonia due to acquired brain injury. Arch Phys Med Rehabil. 1996;77:461-466.
161. Meythaler J.M., Guin-Renfroe S., Brunner R.C., et al. Intrathecal baclofen for spastic hypertonia from stroke. Stroke. 2001;32:2099-2109.
162. Meythaler J.M., Guin-Renfroe S., Johnson A., et al. Prospective assessment of tizanidine for spasticity due to acquired brain injury. Arch Phys Med Rehabil. 2001;82:1155-1163.
163. Meythaler J.M., Guin-Renfroe S., Law C., et al. Continuously infused intrathecal baclofen over 12 months for spastic hypertonia in adolescents and adults with cerebral palsy. Arch Phys Med Rehabil. 2001;82:155-161.
164. Meythaler J.M., McCary A., Hadley M.N. Prospective assessment of continuous intrathecal infusion of baclofen caused by acquired brain injury: a preliminary report. J Neurosurg. 1997;87:415-419.
165. Meythaler J.M., Roper J.F., Davis L., et al. Cyproheptadine in intrathecal baclofen withdrawal: a case series. Arch Phys Med Rehabil. 2003;84:638-642.
166. Misra U.K., Pandey C.M. H reflex studies in neurolathyrism. Electroencephal Clin Neurophysiol. 1994;39:281-285.
167. Moser E., Ligon K.M., Singer C., et al. Botulinum toxin A (Botox) therapy during pregnancy. Neurology. 1997;(suppl 1):A399.
168. Mueller M.E., Gruenthal M., Olson W.L., et al. Gabapentin for relief of upper motor neuron symptoms in multiple sclerosis. Arch Phys Med Rehabil. 1997;78:521-524.
169. Muller H., Zierski J., Dralle D., et al. The effect of intrathecal baclofen in spasticity. In: Muller H., Zierski J., Penn R.D., editors. Local spinal therapy of spasticity. Berlin: Springer-Verlag, 1988.
170. Müller H., Zierski J. Clinical experience with spinal morphine, midazolam and tizanidine in spasticity. In: Müller H., Zierski J., Penn R., editors. Local-spinal therapy of spasticity. Berlin: Springer, 1988.
171. Nahas G.G., Sutin K.M., Harvey D.J., et al, editors. Marijuana and medicine. Totowa, BJ: Humana Press, 1999.
172. Nakamura-Craig M., Follenfant R.L. Effect of lamotrigine in the acute and chronic hyperalgesia induced by PGE2 and in the chronic hyperalgia in rats with streptozotocin-induced diabetes. Pain. 1995;63:33-37.
173. Nance P. A comparison of clonidine, cyproheptadine and baclofen in spastic spinal cord injured patients. J Am Paraplegia Soc. 1994;17:151-157.
174. Nance P., Meythaler J.M. Intrathecal drug therapy. Phys Med Rehabil Clin North Am. 1999;10(2):385-402.
175. Nance P.W. Alpha adrenergic and serotonergic agents in the treatment of spastic hypertonia. Phys Med Rehabil Clin North Am. 2001;12:889-906.
176. Nance PW. Effects of graded Jendrassik facilitation on type Ia and II mediated reflexes in able-bodied and spinal cord injured humans [abstract]. In Society for Neuroscience Abstracts; 1990.
177. Nathan P.W. Intrathecal phenol to relieve spasticity in paraplegia. Lancet. 1959;2:1099.
178. Nance PW. Reflex characteristics of spinal cord injured humans [abstract]. Soc for Neurosci Abst; 15:1241, 1989.
179. Nance P.W., Bugaresti J., Shellenberger K., et al. Efficacy and safety of tizanidine in the treatment of spasticity in patients with spinal cord injury. Neurology. 1994;44(suppl 9):S44-S52.
180. Nance P.W., Schryvers O.I., Schmidt B.J., et al. Intrathecal baclofen therapy for adults with spinal spasticity: therapeutic efficacy and effect on hospital admissions. Can J Neurol Sci. 1995;22:22-29.
181. Nance P.W., Shears A.H., Nance D.M. Clonidine in spinal cord injury. Can Med Assoc J. 1985;133:41-42.
182. Nance P.W., Shears A.H., Nance D.M. Reflex changes induced by clonidine in spinal cord injured patients. Paraplegia. 1989;27:296-301.
183. Nance P.W., Young R.R. Antispasticity medications. Phys Med Rehabil Clin North Am. 1999;10:337-355.
184. Nicholson B.D. Evaluation and treatment of central pain syndromes. Neurology. 2004;62(suppl 2):S30-S36.
185. Nielsen J., Klemar B., Hansen H.J. A new treatment of spasticity with repetitive magnetic stimulation in multiple sclerosis. J Neurol Neurosurg Psychol. 1995;58(2):254-255.
186. Norton B.J., Bomze H.A., Chaplin H. Approach to objective measurement of spasticity. Phys Ther. 1972;52:15-23.
187. Odeen I., Knutsson E. Evaluation of the effects of muscle stretch and weight load in patients with spastic paraplegia. Scand J Rehabil Med. 1981;13:117-121.
188. Olsen R.W. GABA–benzodiazepine–barbiturate receptor interactions. J Neurochem. 1981;37:1-13.
189. Orsnes G.B., Sorensen P.S., et alLaren T.K. Effect of baclofen on gait. Neurology. 2001;4(101):244-248.
190. O’Shaughnessy W.B. On the preparation of the Indian hemp or ganja. Trans Med Phys Soc Bombay. 1842;8:421-461.
191. Pamphlett R. Early terminal and nodal sprouting of motor axons after botulinum toxin. J Neurol Sci. 1989;92:181-192.
192. Park T.S., Owen J.H. Surgical management of spastic diplegia in cerebral palsy. N Engl J Med. 1992;326:745-749.
193. Pedersen E. Clinical assessment and pharmacologic therapy of spasticity. Arch Phys Med Rehabil. 1974;55:344-354.
194. Pedersen E., Arlien-Soborg P., Grynderup V., et al. GABA derivative in spasticity. Acta Neurol Scand. 1970;46:257-266.
195. Penn R.D., Savoy S.M., Corcos D., et al. Intrathecal baclofen for severe spinal spasticity. N Engl J Med. 1989;320:1517-1554.
196. Perry J. Pathological mechanisms. In: Perry J., editor. Gait analysis: normal and pathological function. Clifton Park: Delmar Learning, 1992.
197. Petajan J.H. Spasticity: effects of physical interventions. J Neurol Rehabil. 1990;4:219-225.
198. Pinder R.M., Brogden R.N., Speight T.M., et al. Dantrolene sodium. A review of its pharmacological properties and therapeutic effects. Drugs. 1977;13:2-23.
199. Pittock S.J., Moore A.P., Hardiman O., et al. A doubleblind randomised placebo-controlled evaluation of three doses of botulinum toxin type A (Dysport) in the treatment of spastic equinovarus deformity after stroke. Cerebrovasc Dis. 2003;15:289-300.
200. Pollock N., Baptiste S., Law M., et al. Occupational performance measures: a review based on the guidelines for client-centered practice of occupational therapy. Can J Occup Ther. 1990;57:77-81.
201. Priebe M.M., Sherwood A.M., Graves D.E., et al. Effectiveness of gabapentin in controlling spasticity: a quantitative study. Spinal Cord. 1997;35(3):171-175.
202. Remy-Neris O., Tiffreau V., Bouilland S., Bussel B. Intrathecal baclofen in subjects with spastic hemiplegia: assessment of the antispastic effect during gait. Arch Phys Med Rehabil. 2003;84:643-650.
203. Reynolds J.R. On the therapeutic uses and toxic effects of Cannabis indica. Lancet. 1890;1:637-638.
204. Robinson C.J., Kett N.A., Bolam J.M. Spasticity in spinal cord injured patients. 1. Short-term effects of surface electrical stimulation. Arch Phys Med Rehabil. 1988;69:598-604.
205. Robinson C.J., Kett N.A., Bolam J.M. Spasticity in spinal cord injured patients: 2. Initial measures and long-term effects of surface electrical stimulation. Arch Phys Med Rehabil. 1988;69:598-604.
206. Sabbahi M.A., DeLuca C.J., Powers W.R. Topical anesthesia: a possible treatment method for spasticity. Arch Phys Med Rehabil. 1981;62:310-314.
207. Salazar-Torres J., Pandyan A.D., Price C.I., et al. Does spasticity result from hyperactive stretch reflexes? Preliminary findings from a stretch reflex characterization study. Disabil Rehabil. 2004;26(12):756-760.
208. Sanger T.D. Severe resting clonus caused by thyrotoxicosis in a 16-year-old girl with hereditary spastic paraparesis. Mov Disord. 2004;19(6):712-713.
209. Satkunam L.E. Rehabilitation medicine. 3. Management of adult spasticity, CMAJ. 2003;169(11):1173-1179.
210. Sayers A.C., Burki H.R., Eichenberger E. The pharmacology of 5-chloro-4-(2-imidazolin-2-yl-amino)-2,1,3-benzothiadiazole (DS 103 282), a novel myotonolytic agent. Arzneimittel-Forsch. 1980;30:793-803.
211. Schomburg E.D., Steffens H. The effect of DOPA and clonidine on reflex pathways from group II afferents to alpha-motoneurons in the cat. Exp Brain Res. 1988;71:442-446.
212. Schwarz M., Turski L., Janiszewski W., et al. Is the muscle relaxant effect of diazepam in spastic mutant rats mediated through GABA-independent benzodiazepine receptors? Neurosci Lett. 1983;36:175-180.
213. Seib T., Price R., Reyes M.R., et al. The quantitative measurement of spasticity: effect of cutaneous electrical stimulation. Arch Phys Med Rehabil. 1994;75:746-750.
214. Serfaty M., Masterton G. Fatal poisonings attributed to benzodiazepines in Britain during the 1980s. Br J Psychiatry. 1993;163:386-393.
215. Shahani B.T., Young R.R. Management of flexor spasms with Lioresal. Arch Phys Med Rehabil. 1974;55:465-467.
216. Shefner J.M., Berman S.A., Young R.R. The effect of nicotine on recurrent inhibition in the spinal cord. Neurology. 1993;43:2647-2651.
217. Simpson D.M., Alexander D.N., O’Brien C.F., et al. Botulinum toxin type A in the treatment of upper extremity spasticity: a randomized, double-blind, placebo-controlled trial. Neurology. 1996;46:1306-1310.
218. Simpson L.L. Peripheral actions of the botulinum neurotoxins. Botulinum neurotoxin and tetanus toxin. San Diego, CA: Academic Press; 1989.
219. Sjolund B.H. Pain and rehabilitation after spinal cord injury: the case of sensory spasticity? Brain Res Brain Res Rev. 2002;40(1–3):250-256.
220. Skold C. Spasticity in spinal cord injury: self- and clinically rated intrinsic fluctuations and intervention-induced changes. Arch Phys Med Rehabil. 2000;81(2):144-149.
221. Smith C., Birnbaum G., Carter J.L., et al. Tizanidine treatment of spasticity caused by multiple sclerosis: results of a double-blind, placebo-controlled trial. Neurology. 1994;44(suppl 9):S34-S43.
222. Smith C.R., LaRocca N., Giesser B.S., et al. High-dose oral baclofen: experience in patients with multiple sclerosis. Neurology. 1991;41:1829-1831.
223. Smith P.F. Medicinal cannabis extracts for the treatment of multiple sclerosis. Curr Opin Invest Drugs. 2004;5(7):727-730.
224. Snow B.J., Tsui J.K., Bhatt M.H., et al. Treatment of spasticity with botulinum toxin: a double-blind study. Ann Neurol. 1990;28:512-515.
225. Stahl M.M., Saldeen P., Vinge E. Reversal of fetal benzodiazepine intoxication using flumazenil. Br J Obstet Gynaecol. 1993;100:185-188.
226. Starke K., Borowski E., Endo T. Preferential blockade of presynaptic alpha-adrenoceptors by yohimbine. Eur J Pharmacol. 1975;34:385-388.
227. Stein R., Nordal H.J., Oftedal S.I., et al. The treatment of spasticity in multiple sclerosis: a double-blind clinical trial of a new anti-spasticity drug tizanidine compared with baclofen. Acta Neurol Scand. 1987;75:190-194.
228. Study R.E., Barker J.L. Diazepam and (±) pentobarbital: fluctuation analysis reveals different mechanisms for potentiation of gamma-aminobutyric acid responses in cultured central neurons. Proc Natl Acad Sci U S A. 1981;78:7180-7184.
229. Taylor C.P., Angelotti T., Fauman E. Pharmacology and mechanism of action of pregabalin: the calcium channel alpha2-delta (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007;73(2):137-150.
230. Teasell R, Foley N, Salter K, et al. 10th edition of the stroke rehabilitation evidence-based review, Ottawa, Ontario, September 2007, Canadian Stroke Network.
231. Terrence D.V., Fromm G.H. Complications of baclofen withdrawal. Arch Neurol. 1981;38:588-589.
232. Topcu M., Yalnizoglu D., Saatci I., et al. Effect of topiramate on enlargement of head in Canavan disease: a new option for treatment of megalencephaly. Turk J Pediatr. 2004;46(1):67-71.
233. Unnerstall J.R., Kopajtic T.A., Kuhar M.J. Distribution of alpha-2 agonist binding sites in the rat and human central nervous system. Brain Res. 1984;7:69-101.
234. Van Hemet J.C.J. A double-blind comparison of baclofen and placebo in patients with spasticity of cerebral origin. In: Feldman R.G., Young R.R., Koella W.P., editors. Spasticity: disordered motor control. Chicago: Year Book Medical Publishers, 1980.
235. van Kuijk A.A., Geurts A.C., Bevaart B.J., et al. Treatment of upper extremity spasticity in stroke patients by focal neuronal or neuromuscular blockade: a systematic review of the literature. J Rehabil Med. 2002;34(2):51-61.
236. Wainberg M, Barbeau H. Modulatory action of cyproheptadine on the locomotor pattern of spastic paretic patients [abstract], Soc Neurosci Abstr; 139, 1986.
237. Wallace A., Montanez S., Lorio M., et al. Vigabatrin, unlike some anti-convulsants, does not hinder recovery of function. Epilepsia. 1996;37:27.
238. Ward A., Chaffman M.O., Sorkin E.M. Dantrolene: a review of its pharmacokinetic properties and therapeutic use in malignant hyperthermia, the neuroleptic malignant syndrome and an update of its use in muscle spasticity. Drugs. 1986;32:130-168.
239. Weingarden S.I., Belen J.G. Clonidine transdermal system for treatment of spasticity in spinal cord injury. Arch Phys Med Rehabil. 1992;73:876-877.
240. Wissel J., Ward A.B., Erztgaard P., et al. European consensus table on the use of botulinum toxin type A in adult spasticity. J Rehabil Med. 2009;41(1):13-25.
241. Wood K.M. The use of phenol as a neurolytic agent: a review. Pain. 1978;5:205-229.
242. Yablon S.A., Sipski M.L. Effect of transdermal clonidine on spinal spasticity: a case series. Am J Phys Med Rehabil. 1993;72:154-157.
243. Yarkony G.M., Roth E.J., Cybulski G.R., et al. Neuromuscular stimulation in spinal cord injury II: prevention of secondary complications. Arch Phys Med Rehabil. 1992;73:195-200.
244. Young R. Letters to the editor. J Neurol Neurosurg Psychol. 1982;45:1035-1039.
245. Young R.R. Spasticity: a review. Neurology. 1994;44(suppl 9):S12-S20.
246. Zackowski K.M., Dromerick A.W., Sahrmann S.A., et al. How do strength, sensation, spasticity and joint individuation relate to the reaching deficits of people with chronic hemiparesis. Brain. 2004;127(part 5):1035-1046.
247. Zajicek J., Fox P., Sanders H., et al. UK MS research group. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study). Lancet. 2003;362(9395):1517-1526.