Soluble Mediators of the Immune System
At the conclusion of this chapter, the reader should be able to:
• Name and compare the three complement activation pathways.
• Describe the mechanisms and consequences of complement activation.
• Explain the biological functions of the complement system.
• Name and describe alterations in complement levels.
• Briefly describe the assessment of complement levels.
• Compare other types of nonspecific mediators of the immune system, including cytokines, interleukins, tumor necrosis factor, hematopoietic growth factors, and chemokines.
• Discuss the clinical applications of C-reactive protein.
• Compare acute-phase reactant methods.
• Analyze an acute-phase protein case study.
• Correctly answer case study related multiple choice questions.
• Be prepared to participate in a discussion of critical thinking questions.
• Describe the principle, reporting results, sources of error, limitations, and clinical applications of the C-reactive protein procedure.
The Complement System
The complement system displays three overarching physiologic activities (Table 5-1). These are initiated in various ways through the following three pathways (Table 5-2):
Table 5-1
Three Main Physiologic Activities of the Complement System
| Activity | Responsible Complement Protein |
| Host Defense Against Infections | |
| Opsonization | Covalently bonded fragments of C3 and C4 |
| Chemotaxis and leukocyte activation | C5a, C3a, and C4a; anaphylatoxin leukocyte receptors |
| Lysis of bacterial and mammalian cells | C5-C9 membrane attack complex |
| Interface Between Innate and Adaptive Immunity | |
| Augmentation of antibody | C3b and C4b bound to immune complexes and to antigen |
| Responses | C3 receptors on B cells and antigen-presenting cells |
| Enhancement of immunologic memory | C3b and C4b bound to immune complexes and to antigen; C3 receptors on follicular dendritic cells |
| Disposal of Waste | |
| Clearance of immune complexes from tissues | C1q; covalently bonded fragments of C3 and C4 |
| Clearance of apoptotic cells | |

Adapted from Walport MJ: Complement, N Engl J Med 344:1058–1065, 2001.
Table 5-2
Initiators of Three Complement Activation Pathways
| Pathway | Initiators |
| Classic | Immune complexes Apoptotic cells Certain viruses and gram-negative bacteria C-reactive protein bound to ligand |
| Alternate | Various bacteria, fungi, viruses, or tumor cells |
| Mannose-binding lectin | Microbes with terminal mannose groups |
Adapted from Walport MJ: Complement, N Engl J Med 344:1058–1065, 2001.
The three pathways (Fig. 5-1) converge at the point of cleavage of C3 to C3b, the central event of the common final pathway, which in turn leads to the activation of the lytic complement sequence, C5 through C9, and cell destruction (Fig. 5-2).
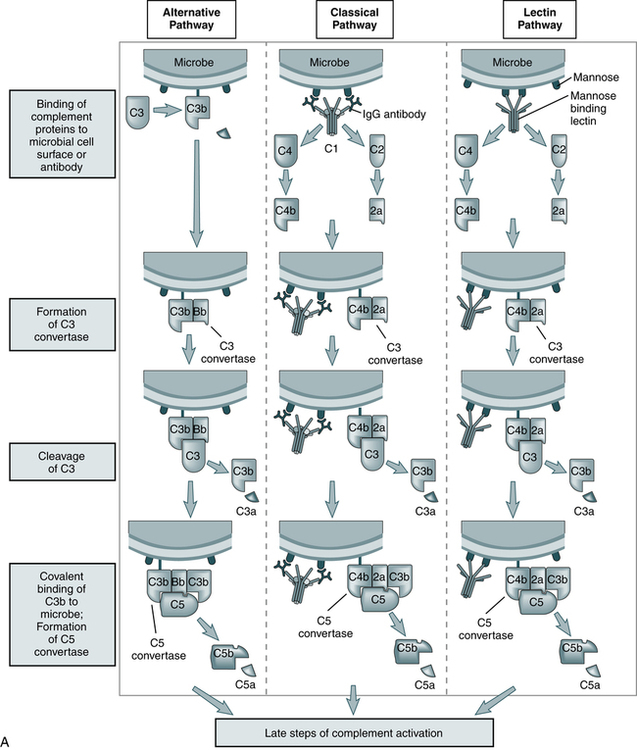
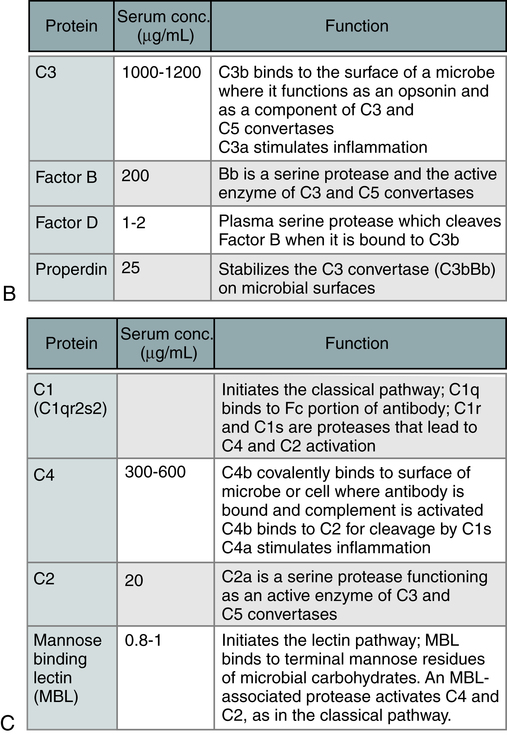
The steps in the activation of the alternative, classical, and lectin pathways are shown. Note the sequence of events is similar in all three pathways, although they differ in their requirement for antibody and in the proteins used. (From Abbas AK, Lichtman AH: Basic immunology: functions and disorders of the immune system, updated edition, ed 3, Philadelphia, 2011, Saunders.)
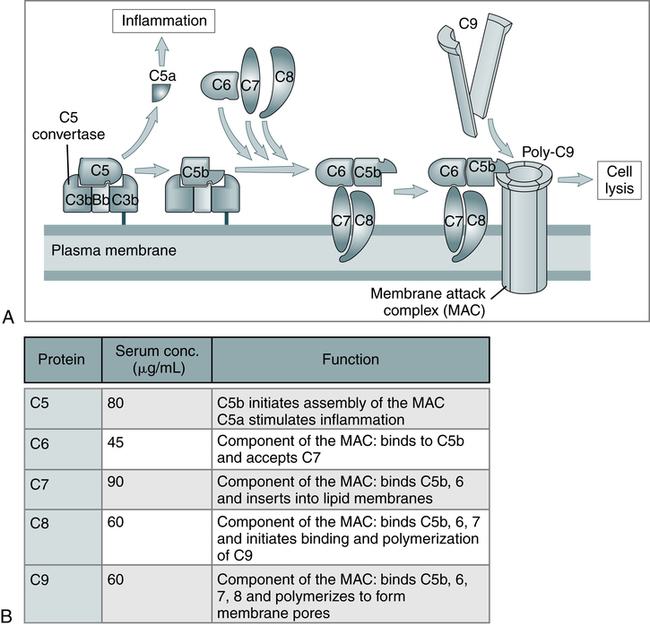
A, The late steps of complement activation start after the formation of the C5 convertase and are identical in the alternative and classical pathways. Products generated in the late steps induce inflammation (C5a) and cell lysis (the membrane attack complex [MAC]). B, The properties of the proteins of the late steps of complement activation are listed. (From Abbas AK, Lichtman AH: Basic immunology: functions and disorders of the immune system, updated edition, ed 3, Philadelphia, 2011, Saunders.)
Activation of Complement
• The classic pathway is initiated by the bonding of the C1 complex, consisting of C1q, C1r, and C1s, to antibodies bound to an antigen on the surface of a bacterial cell.
• The alternative pathway is initiated by contact with a foreign surface such as the polysaccharide coating of a microorganism and the covalent binding of a small amount of C3b to hydroxyl groups on cell surface carbohydrates and proteins. The pathway is activated by low-grade cleavage of C3 in plasma.
• The mannose-binding lectin pathway is initiated by binding of the complex of mannose-binding lectin and associated serine proteases (MASP1 and MASP2) to arrays of mannose groups on the surface of a bacterial cell.
Effects of Complement Activation
• Cell activation, such as production of inflammatory mediators.
• Cytolysis or hemolysis, if the cells are erythrocytes. The most important biologic role of complement in blood group serology is the production of cell membrane lysis of antibody-coated targets.
• Opsonization, which renders cells vulnerable to phagocytosis.
Classic Pathway
The classic pathway has three major stages:
Membrane Attack Complex
When fully assembled in the correct proportions, C7, C6, C5b, and C8 form the MAC (see Fig. 5-2, inset). The C5bC6 complex is hydrophilic but, with the addition of C7, it has additional detergent and phospholipid-binding properties as well. The presence of hydrophobic and hydrophilic groups within the same complex may account for its tendency to polymerize and form small protein micelles (a packet of chain molecules in parallel arrangement). It can attach to any lipid bilayer within its effective diffusion radius, which produces the phenomenon of reactive lysis on innocent so-called bystander cells. Once membrane bound, C5bC6C7 is relatively stable and can interact with C8 and C9.
BiologicAl Functions of Complement Proteins
The biological functions of the complement system fall into the following two general categories:
1. Cell lysis by the membrane attack complex (MAC)
2. Biological effects of proteolytic fragments of complement
The first category is the situation in which the MAC leads to osmotic lysis of a cell. The second category encompasses other effects of complement in immunity and inflammation that are mediated by the proteolytic fragments generated during complement activation. These fragments may remain bound to the same cell surfaces at which complement has been activated or may be released into the blood or extracellular fluid. In either situation, active fragments mediate their effects by binding to specific receptors expressed on various types of cells, including phagocytic leukocytes and the endothelium (Table 5-3).
Table 5-3
Selected Complement Components and Functions
| Complement Component(s) | Function |
| C5-C9 | Lysis of cells |
| C3B, IC3B | Opsonization in phagocytosis |
| C5A >C3A >>C4A | Anaphylatoxins/inflammation (vascular responses) |
| C5A | Polymorphonuclear leukocyte activation |
| Classic complement pathway, C3B, ?iC3b, C3dg | Immune complex removal B-lymphocyte activation |
Alterations in Complement Levels
Decreased Complement Levels
Low levels of complement suggest one of the following biological effects:
• Complement has been excessively activated recently.
• Complement is currently being consumed.
• A single complement component is absent because of a genetic defect.
Specific component deficiencies are associated with a variety of disorders (Table 5-4). Deficiencies of complement account for a small percentage of primary immunodeficiencies (<2%), but depression of complement levels frequently coexists with SLE and other disorders associated with an immunopathologic process (Box 5-1).
Table 5-4
Complement Deficiency in Human Beings
| Deficiency | Associated Disease |
| C1q | SLE-like syndrome; decreased secondary to agammaglobulinemia |
| C1r | SLE-like syndrome; dermatomyositis, vasculitis, recurrent infections and chronic glomerulo-nephritis, necrotizing skin lesions, arthritis |
| C1s | SLE, SLE-like syndrome |
| C1 INH | Hereditary angioedema, lupus nephritis |
| C2 | Recurrent pyogenic infections, SLE, SLE-like syndrome, discoid lupus, membranoproliferative glomerulonephritis, dermatomyositis, synovitis, purpura, Henoch-Schönlein purpura, hypertension, Hodgkin’s disease, chronic lymphocytic leukemia, dermatitis herpetiformis, polymyositis |
| C3 | Recurrent pyogenic infections, SLE-like syndrome, arthralgias, skin rash |
| C3 inactivator | Recurrent pyogenic infections, urticaria |
| C4 | SLE-like syndrome, SLE, dermatomyositis-like syndrome, vasculitis |
| C5 | Neisseria infections, SLE |
| C5 dysfunction | Leiner’s disease, gram-negative skin and bowel infection |
| C6 | Neisseria infections, SLE, Raynaud’s phenomenon, scleroderma-like syndrome, vasculitis |
| C7 | Neisseria infections, SLE, Raynaud’s phenomenon, scleroderma-like syndrome, vasculitis |
| C8 | Neisseria infections, xeroderma pigmentosa, SLE-like syndrome |
Modified from Colten HR, Rosen FS: Complement deficiencies annual review of immunology, 10:809-834, 1992 and Nusinow SR, Zuraw BL, Curd JG: The hereditary and acquired deficiencies of complement, Med Clin North Am 69:487-504, 1985.
Diagnostic Evaluation
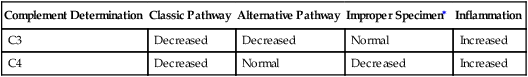
During immune complex reactions, certain complement proteins become physically bound to the tissue in which the immunologic reaction is occurring. These proteins can be demonstrated in tissue by appropriate immunopathologic stains. The most frequent evaluation of complement is by serum or plasma assay (Table 5-5). Complement components (e.g., C3 and C4) can be assessed by nephelometry. These assays are useful for the diagnosis and monitoring of patients.
Table 5-5
Interpretation of Complement Activation by Individual Components
| Complement Determination | Classic Pathway | Alternative Pathway | Improper Specimen∗ | Inflammation |
| C3 | Decreased | Decreased | Normal | Increased |
| C4 | Decreased | Normal | Decreased | Increased |
Assessment of Complement
The procedures discussed next can be used in diagnostic immunology.
C3PA (C3 Proactivator, Properdin Factor B)
The factor B component is consumed by activation of the alternative complement pathway. Assessment of C3PA indicates whether a decreased level of C3 results from the classic or alternative pathways of complement activation. Decreased levels of C3 and C4 demonstrate activation of the classic pathway. Decreased levels of C3 and C3PA with a normal level of C4 indicate complement activation via the alternative pathway (Table 5-5).
In SLE, both the classic and alternative pathways are activated.
Other Soluble Immune Response Mediators
Biological Response Modifiers
1. B lymphocytes that secrete specific antibodies
2. T lymphocytes that secrete soluble mediators, such as interleukin-2 (IL-2) and other ILs, granulocyte-monocyte colony-stimulating factor (GM-CSF), interferon-γ (IFN-γ) and tumor necrosis factor-β (TNF-β)
3. Natural killer (NK) lymphocytes that secrete IFN-α
4. Monocytes and macrophages that secrete IFN-α, IL-1, and other ILs, TNF-α, and GM-CSF and monocyte colony-stimulating factor (M-CSF)
• Active—use of microbial or chemical immunomodulators (adjuvants) in a specific or nonspecific form
• Adoptive—use of soluble mediators, such as ILs, to regulate components of the immune system
• Passive—transfer of preformed antibodies to tumorous recipients, such as monoclonal antibodies
• Restorative—application of soluble substances, such as interferons, for a wide range of diseases
Cytokines
Research is ongoing and the list of individual cytokines steadily increases. Cytokines are synthesized and secreted by the cells associated with innate and adaptive immunity in response to microbial and other antigen exposures (Tables 5-6 and 5-7).
Table 5-6
Examples of Cytokines of Innate and Adaptive Immunity
| Innate Immunity | Adaptive Immunity |
| Chemokines | IFN-γ |
| IFN type 1 (IFN-α, IFN-β) | IL-2 |
| IL-1 | IL-4 |
| IL-6 | IL-5 |
| IL-10 | IL-13 |
| IL-12 | Lymphotoxin (Lt) |
| IL-15 | TGF-β |
| IL-18 | |
| TNF |
IFN, Interferon; IL, interleukin; TGF, transforming growth factor; TNG, tumor necrosis factor.
Table 5-7
Comparative Features of Innate and Adaptive Immunity
| Type of Immunity | ||
| Innate | Adaptive | |
| Examples | TNF-α, IFN-β, IL-1, IL-12 | IFN-γ, IL-2, IL-4, IL-5 |
| Major cell source | Macrophages, NK cells | T lymphocytes |
| Major physiologic function | Mediators of innate immunity and inflammation (local and systemic) | Regulation of lymphocyte growth and differentiation Activation of effector cells (macrophages, eosinophils, mast cells) |
| Stimuli | LPS (endotoxin), bacterial peptidoglycans, viral RNA, T cell–derived cytokines (e.g., IFN-β) | Protein antigens |
| Quantity produced | Possibly high, detectable in serum | Usually low, usually undetectable in serum |
| Effects on body | Local and systemic | Usually local |
| Roles in disease | Systemic diseases | Local tissue injury |
| Inhibitors | Corticosteroids | Cyclosporine, FK-506 |
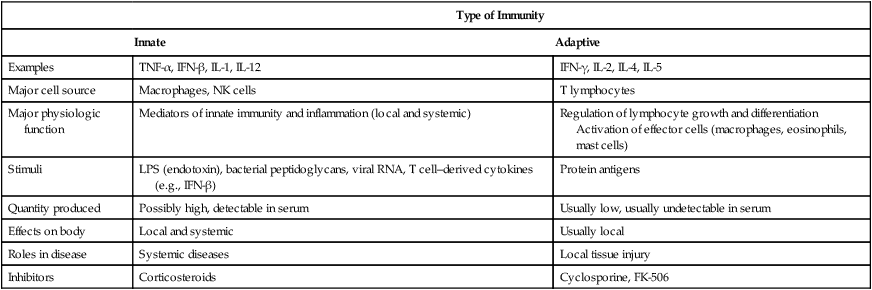
IFN, Interferon; IL, interleukin; LPS, lipopolysaccharides; TNF, tumor necrosis factor.
Adapted from Abbas AK, Lichtman AH, Pober JS: Cellular and molecular immunology, ed 4, Philadelphia, 2000, Saunders.
Cytokines are polypeptide products of activated cells that control a variety of cellular responses and thereby regulate the immune response. Many cytokines are released in response to specific antigens; however, cytokines are nonspecific in that their chemical structure is not determined by the stimulating antigen. Most cytokines have multiple activities and act on numerous cell types. Hematopoietic and lymphoid cell compartments are regulated by a complex network of interacting cytokines. The colony-stimulating factors (CSFs) and ILs have been shown to play important roles in normal proliferation, differentiation, and activation of several hematopoietic and lymphoid lineages (Tables 5-8 and 5-9).
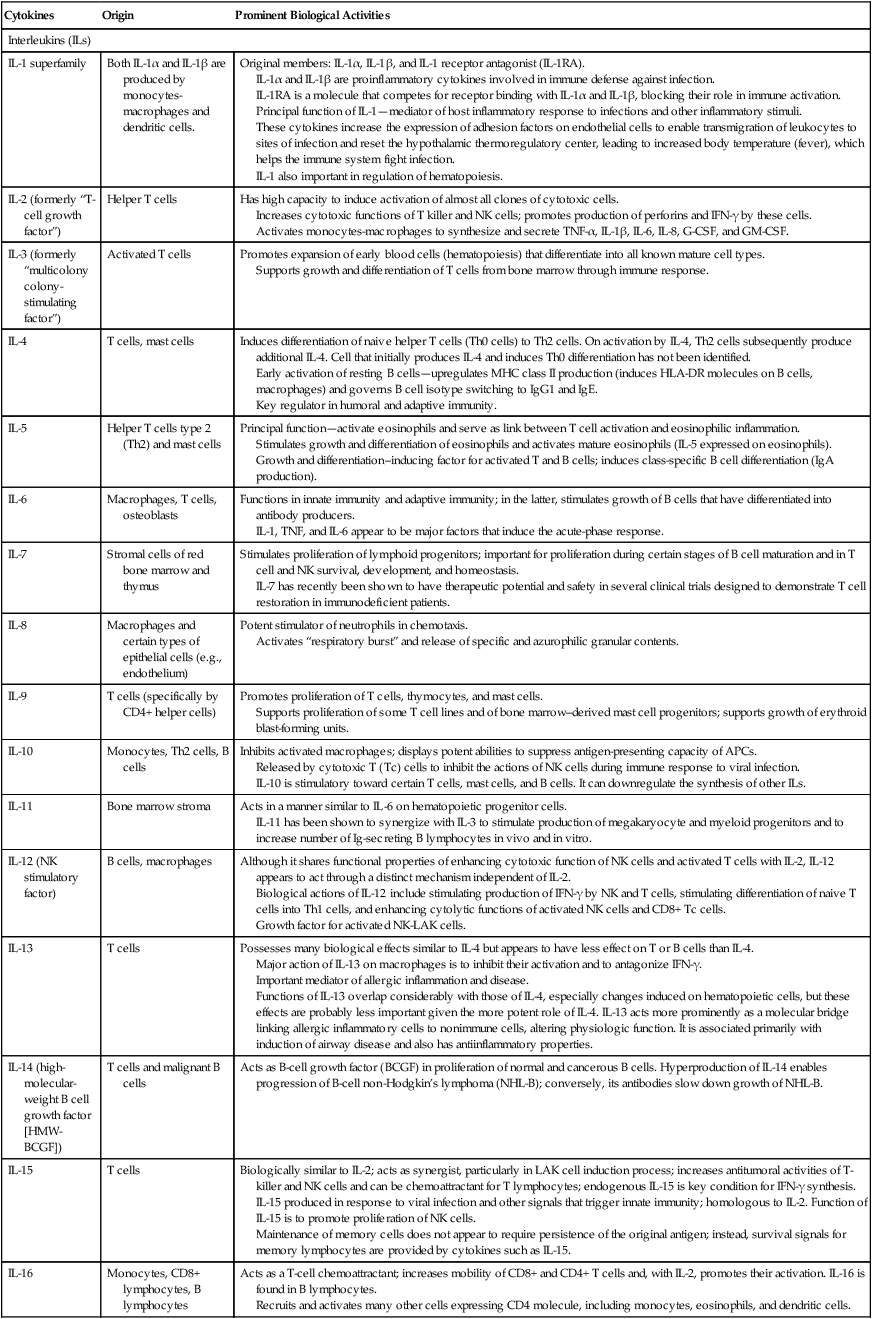
Table 5-8
Origin and Immunoregulatory Activity of Cytokines
| Cytokines | Origin | Prominent Biological Activities |
| Interleukins (ILs) | ||
| IL-1 superfamily | Both IL-1α and IL-1β are produced by monocytes-macrophages and dendritic cells. | Original members: IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1RA). IL-1α and IL-1β are proinflammatory cytokines involved in immune defense against infection. IL-1RA is a molecule that competes for receptor binding with IL-1α and IL-1β, blocking their role in immune activation. Principal function of IL-1—mediator of host inflammatory response to infections and other inflammatory stimuli. These cytokines increase the expression of adhesion factors on endothelial cells to enable transmigration of leukocytes to sites of infection and reset the hypothalamic thermoregulatory center, leading to increased body temperature (fever), which helps the immune system fight infection. IL-1 also important in regulation of hematopoiesis. |
| IL-2 (formerly “T-cell growth factor”) | Helper T cells | Has high capacity to induce activation of almost all clones of cytotoxic cells. Increases cytotoxic functions of T killer and NK cells; promotes production of perforins and IFN-γ by these cells. Activates monocytes-macrophages to synthesize and secrete TNF-α, IL-1β, IL-6, IL-8, G-CSF, and GM-CSF. |
| IL-3 (formerly “multicolony colony-stimulating factor”) | Activated T cells | Promotes expansion of early blood cells (hematopoiesis) that differentiate into all known mature cell types. Supports growth and differentiation of T cells from bone marrow through immune response. |
| IL-4 | T cells, mast cells | Induces differentiation of naive helper T cells (Th0 cells) to Th2 cells. On activation by IL-4, Th2 cells subsequently produce additional IL-4. Cell that initially produces IL-4 and induces Th0 differentiation has not been identified. Early activation of resting B cells—upregulates MHC class II production (induces HLA-DR molecules on B cells, macrophages) and governs B cell isotype switching to IgG1 and IgE. Key regulator in humoral and adaptive immunity. |
| IL-5 | Helper T cells type 2 (Th2) and mast cells | Principal function—activate eosinophils and serve as link between T cell activation and eosinophilic inflammation. Stimulates growth and differentiation of eosinophils and activates mature eosinophils (IL-5 expressed on eosinophils). Growth and differentiation–inducing factor for activated T and B cells; induces class-specific B cell differentiation (IgA production). |
| IL-6 | Macrophages, T cells, osteoblasts | Functions in innate immunity and adaptive immunity; in the latter, stimulates growth of B cells that have differentiated into antibody producers. IL-1, TNF, and IL-6 appear to be major factors that induce the acute-phase response. |
| IL-7 | Stromal cells of red bone marrow and thymus | Stimulates proliferation of lymphoid progenitors; important for proliferation during certain stages of B cell maturation and in T cell and NK survival, development, and homeostasis. IL-7 has recently been shown to have therapeutic potential and safety in several clinical trials designed to demonstrate T cell restoration in immunodeficient patients. |
| IL-8 | Macrophages and certain types of epithelial cells (e.g., endothelium) | Potent stimulator of neutrophils in chemotaxis. Activates “respiratory burst” and release of specific and azurophilic granular contents. |
| IL-9 | T cells (specifically by CD4+ helper cells) | Promotes proliferation of T cells, thymocytes, and mast cells. Supports proliferation of some T cell lines and of bone marrow–derived mast cell progenitors; supports growth of erythroid blast-forming units. |
| IL-10 | Monocytes, Th2 cells, B cells | Inhibits activated macrophages; displays potent abilities to suppress antigen-presenting capacity of APCs. Released by cytotoxic T (Tc) cells to inhibit the actions of NK cells during immune response to viral infection. IL-10 is stimulatory toward certain T cells, mast cells, and B cells. It can downregulate the synthesis of other ILs. |
| IL-11 | Bone marrow stroma | Acts in a manner similar to IL-6 on hematopoietic progenitor cells. IL-11 has been shown to synergize with IL-3 to stimulate production of megakaryocyte and myeloid progenitors and to increase number of Ig-secreting B lymphocytes in vivo and in vitro. |
| IL-12 (NK stimulatory factor) | B cells, macrophages | Although it shares functional properties of enhancing cytotoxic function of NK cells and activated T cells with IL-2, IL-12 appears to act through a distinct mechanism independent of IL-2. Biological actions of IL-12 include stimulating production of IFN-γ by NK and T cells, stimulating differentiation of naive T cells into Th1 cells, and enhancing cytolytic functions of activated NK cells and CD8+ Tc cells. Growth factor for activated NK-LAK cells. |
| IL-13 | T cells | Possesses many biological effects similar to IL-4 but appears to have less effect on T or B cells than IL-4. Major action of IL-13 on macrophages is to inhibit their activation and to antagonize IFN-γ. Important mediator of allergic inflammation and disease. Functions of IL-13 overlap considerably with those of IL-4, especially changes induced on hematopoietic cells, but these effects are probably less important given the more potent role of IL-4. IL-13 acts more prominently as a molecular bridge linking allergic inflammatory cells to nonimmune cells, altering physiologic function. It is associated primarily with induction of airway disease and also has antiinflammatory properties. |
| IL-14 (high-molecular-weight B cell growth factor [HMW-BCGF]) | T cells and malignant B cells | Acts as B-cell growth factor (BCGF) in proliferation of normal and cancerous B cells. Hyperproduction of IL-14 enables progression of B-cell non-Hodgkin’s lymphoma (NHL-B); conversely, its antibodies slow down growth of NHL-B. |
| IL-15 | T cells | Biologically similar to IL-2; acts as synergist, particularly in LAK cell induction process; increases antitumoral activities of T-killer and NK cells and can be chemoattractant for T lymphocytes; endogenous IL-15 is key condition for IFN-γ synthesis. IL-15 produced in response to viral infection and other signals that trigger innate immunity; homologous to IL-2. Function of IL-15 is to promote proliferation of NK cells. Maintenance of memory cells does not appear to require persistence of the original antigen; instead, survival signals for memory lymphocytes are provided by cytokines such as IL-15. |
| IL-16 | Monocytes, CD8+ lymphocytes, B lymphocytes | Acts as a T-cell chemoattractant; increases mobility of CD8+ and CD4+ T cells and, with IL-2, promotes their activation. IL-16 is found in B lymphocytes. Recruits and activates many other cells expressing CD4 molecule, including monocytes, eosinophils, and dendritic cells. |
| IL-17 | CD4+ lymphocytes | Induces granulopoiesis through G-CSF; can reinforce antibody-dependent tumor cell destruction; participates in regulation of many cytokines (IL-1, IL-4, IL-6, IL-10, IL-12, IFN-γ). Histamine and serotonin increase production of IL-17. IL-17 mimics many proinflammatory actions of TNF-α and TNF-β. |
| IL-18 | Macrophages | Acts as synergist with IL-12 in some effects, especially induction of IFN-γ production and inhibition of angiogenesis; high IFN-γ production under integrated effect of IL-18 and IL-12 suppresses tumor growth. IL-18 stimulates production of IFN-γ by NK cells and T cells, synergistic with IL-12. |
| IL-19 | Monocytes | Lipopolysaccharides (LPS) and GM-CSF stimulate synthesis of IL-19, which is then upregulated in monocytes. Biological function similar to that of IL-10; regulates functions of macrophages and suppresses activities of Th1 and Th2. |
| IL-20 | Activated keratinocytes, monocytes | Biological activities similar to those of IL-10 and can stimulate tumor growth. Regulates proliferation and differentiation of keratocytes during inflammation, particularly inflammation associated with the skin. Causes expansion of multipotential hematopoietic progenitor cells. |
| IL-21 | Various lymphocytes | Regulates hematopoiesis and immune response and influences development of lymphocytes; similar to IL-2 and IL-15 in antitumor defense system; promotes high production of T lymphocytes, fast growth and maturation of NK cells, and fast growth of B lymphocytes. Has potent regulatory effects on immune cells, interacting with cell surface IL-21 receptor, expressed in bone marrow cells and various lymphocytes. |
| IL-22 | Activated T cells | Similar to IL-10, but does not prohibit production of proinflammatory cytokines through monocytes in response to LPS; somewhat similar to IFN-α, -β, and -γ. |
| IL-23 | — | Newly discovered cytokine that shares some in vivo functions with IL-12. IL-23 is important part of inflammatory response against infection; as proinflammatory cytokine, it enhances T cell priming and stimulates production of proinflammatory molecules (IL-1, IL-6, TNF-α, NOS-2, chemokines), resulting in inflammation. |
| IL-24 | Activated monocytes-macrophages, Th2 cells | Appears to participate in cell survival and proliferation by inducing rapid activation of particular transcription factors called STAT-1 and STAT-3; predominantly released by and acts on nonhematopoietic (skin, lung, reproductive) tissues. Performs important roles in wound healing and cancer; cell death occurs in cancer cells and cell lines after exposure to IL-24. |
| IL-25 | Th2 cells, mast cells | Biologically characterized as a member of IL-17 cytokine family. Supports proliferation of cells in lymphoid lineage. Induces production of other cytokines (IL-4, IL-5, IL-13) in multiple tissues, which stimulate the expansion of eosinophils. Important molecule in controlling immunity of the gut; implicated in chronic inflammation associated with gastrointestinal tract; identified in chromosomal region associated with autoimmune diseases such as inflammatory bowel disease (IBD), although no direct evidence suggests that IL-25 plays a role in IBD. |
| IL-26 | Expressed in certain herpesvirus-transformed T cells, but not in primary stimulated T cells | Induces rapid phosphorylation of transcription factors STAT-1 and STAT-3, which enhance IL-10 and IL-8 secretion and expression of CD54 molecule on surface of epithelial cells. |
| IL-27 | — | Has important function in regulating activity of B and T lymphocytes; belongs to the IL-12 family. |
| IL-28 | — | Plays role in immune defense against viruses. |
| IL-29 | — | Plays important role in host defenses against microbes; its gene is highly upregulated in cells infected with virus. |
| IL-30 (also IL27p28) | New name of p28, a subunit of IL27 | Interleukin 30 (IL-30), a member of the long-chain four-helix bundle cytokine family, and EBI3 form the IL-27 heterdimer, which is expressed by APCs. IL-27 triggers expansion of antigen-specific naive CD4-positive T cells and promotes polarization toward a Th1 phenotype with expression of IFN-γ. IL-27 acts in synergy with IL-12 and binds to WSX1. |
| IL-31 | Produced preferentially by Th2 cells. Receptor subunits expressed in activated monocytes and unstimulated epithelial cells. |
Believed to play role in skin inflammation. |
| IL-32 | Monocytes-macrophages | Can induce cells of immune system (e.g., monocytes-macrophages) to secrete TNF-α in addition to chemokines such as IL-8. Induces expression of TNF-α and IL-8 in THP-1 monocytic cells. Expression of IL-32 is induced in human peripheral lymphocyte cells after mitogen stimulation, in human epithelial cells by IFN-γ, and in NK cells after exposure to IL-12–IL-18 combination. Involved in activation induced cell death. Expression of IL-32 is upregulated in T killer and NK-cells after cell activation, and IL-32β is predominant isoform in activated T cells. IL-32 is expressed specifically in T cells undergoing cell death; enforced expression of IL-32 induces apoptosis; downregulation rescues the cells from apoptosis. |
| IL-33 | Helper T cells | Induces type 2 cytokine production from Th cells. Mediates biological effects by interacting with orphan IL-1 receptor, activating intracellular molecules in certain signaling pathways that drive production of type 2 cytokines (e.g., IL-4, IL-5, IL-13) from polarized Th2 cells. Constitutive expression of IL-33 is found in smooth muscle cells and bronchial epithelial cells. Expression in primary lung or dermal fibroblasts and keratinocytes is inducible by treatment with TNF-α and IL-1β; these two cytokines only induce low-level expression in dendritic cells and macrophages. |
| Interferons (IFNs) | ||
| IFN-α | Leukocytes | Antiviral, increased MHC class I expression. |
| IFN-β | Fibroblasts, epithelial cells | Antiviral, increased MHC class I expression. |
| IFN-γ | T cells, NK cells | Major macrophage activator; induces MHC class II molecules on many cells and can synergize with TNF; augments NK cell activity; antagonist to IL-4. |
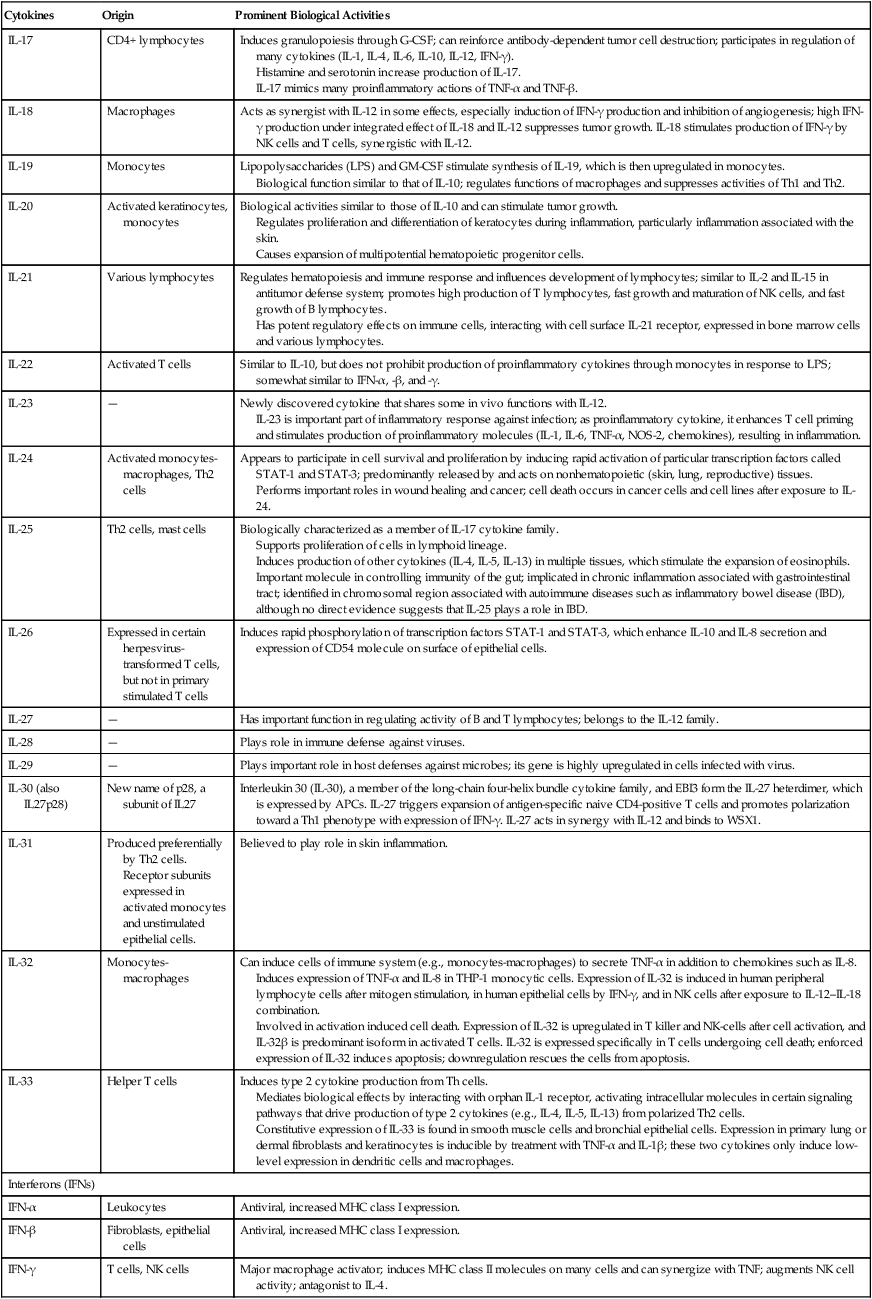
From Abbas AK, Lichtman AH: Basic Immunology ed 3 Update, Saunders, 2011, pp. 258-259 and http://www.gene.ucl.ac.uk/nomeclature/genefamily/il.php, 2007.
Table 5-9
Immunoregulatory Activity of Other Cytokines
| Factor | Target Cells | Prominent Biological Activities |
| Tumor Necrosis Factor (TNF) | ||
| TNF-α (cachectin) | Macrophages, NK cells | Local inflammation, endothelial activation |
| TNF-β (lymphotoxin) | T cells, B cells | Killing, endothelial activation |
| Tumor necrosis family | T cells, mast cells | |
| CD40 ligand | B-cell activation, class switching | |
| TNF Family | ||
| CD27 ligand | T cells | Stimulates T-cell proliferation |
| CD30 ligand | T cells | Stimulates T- and B-cell proliferation |
| Chemokines | ||
| Membrane cofactor protein (MCP-1) | Macrophages, others | Chemotactic for monocytes |
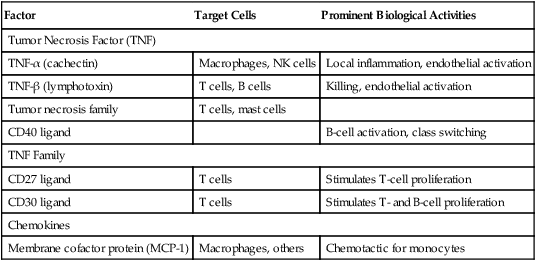
Adapted from Claman HN: The biology of the immune response, JAMA 268:2790–2796, 1992; Janeway C, Travers P: Immunobiology, ed 3, New York, 1997, Garland; and Abbas AK, Lichtman AH, Pober JS: Cellular and molecular immunology, ed 4, Philadelphia, 2000, Saunders.
• Secrete cytokines in rapid bursts, synthesized in response to cellular activation.
• Bind to specific membrane receptors on target cells.
• Regulate receptor expression in T and B cells, which drives positive amplification or negative feedback.
• Act on different cell types.
• Excite the same functional effects with multiple cytokines (redundancy).
• Act close to the site of synthesis on the same cell or on a nearby cell.
Hematopoietic Stimulators
Stem Cell Factor (c-kit Ligand)
Chemokines
Chemokines are a large family of structurally homologous cytokines that stimulate transendothelial leukocyte movement from the blood to the tissue site of infection and regulate the migration of polymorphonuclear leukocytes (PMNs) and mononuclear leukocytes within tissues (see Chapter 3). The largest family consists of CC chemokines that attract mononuclear cells to sites of chronic inflammation, such as monocyte chemoattractant protein 1 (MCP-1). A second family of chemokines consists of CXC chemokines, of which IL-8 (CXCL8) is the prototype. CXCL8 attracts PMNs to sites of acute inflammation, activates monocytes, and may direct the recruitment of these cells to vascular lesions. The third family, CX3, forms a cell adhesion receptor capable of arresting cells under physiologic flow conditions. TNF-α–converting enzyme can cleave CX3CL1 from the cell membrane.
Other functions of various chemokines include the following:
• Increasing the affinity of leukocyte integrins for their ligands on endothelium (e.g., intercellular adhesion molecule-1 [ICAM-1], ICAM-2, vascular cell adhesion molecule-1 [VCAM-1])
• Regulating the traffic of lymphocytes and other leukocytes through peripheral lymphoid tissues
• Maintaining normal migration of immune cells into lymphoid organs or other specialized cells to particular sites
Assessment of Cytokines
Defects in cytokine production can lead to autoimmunity (Table 5-10). Traditional methods for assessment of cytokines include the following:
Table 5-10
Defects in Cytokine Production that Can Lead to Autoimmunity
| Cytokine or Protein | Defect | Disorder |
| IL-1 receptor antagonist | Underexpression | Arthritis |
| IL-2 IL-7 IL-10 |
Overexpression | IBD |
| IL-2 receptor | Overexpression | IBD |
| IL-10 receptor | Overexpression | IBD |
| IL-3 | Overexpression | Demyelinating syndrome |
| TNF-α | OverexpressionUnderexpression | IBD, arthritis, vasculitis SLE |
| IFN-γ | Overexpression in skin | SLE |
| TGF-β | Underexpression | Systemic wasting syndrome, IBD |
| TGF-β receptor in T cells | Underexpression | SLE |
Adapted from Davidson A, Diamond B: Autoimmune diseases, N Engl J Med 345:340–350, 2001.
IL, interleukin; IBD, inflammatory bowel disease; TNF, tumor necrosis factor; SLE, systemic lupus erythematosus; IFN, interferon; TGF, transforming growth factor.
Newer methods of measurement include the following:
• Multiplexed assay using the FlowMetrix; quantifies multiple cytokines simultaneously
• Intracellular staining using flow cytometry
• Cord blood mononuclear cells stimulated by allergens (celELISA).
• Real-time PCR for lymph nodes or spleen
• Enzyme-linked immunosorbent spot (ELISPOT) assays
Acute-Phase Proteins
Overview
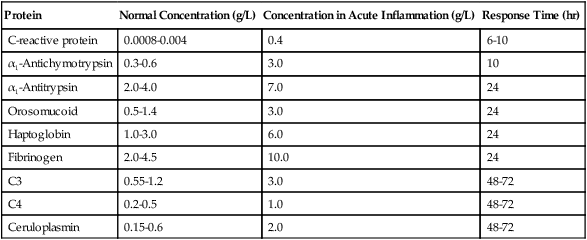
The main biological sign of inflammation is an increase in the ESR. In addition to the ESR, measurement of the plasma concentration of acute-phase reactants is usually a good indicator of local inflammatory activity and tissue damage. More than 20 acute-phase proteins have a definable role in inflammation (Box 5-2). These reactants constitute most of the serum glycoproteins (Table 5-11).
Table 5-11
Examples of Clinically Useful Acute-Phase Proteins
| Protein | Normal Concentration (g/L) | Concentration in Acute Inflammation (g/L) | Response Time (hr) |
| C-reactive protein | 0.0008-0.004 | 0.4 | 6-10 |
| α1-Antichymotrypsin | 0.3-0.6 | 3.0 | 10 |
| α1-Antitrypsin | 2.0-4.0 | 7.0 | 24 |
| Orosomucoid | 0.5-1.4 | 3.0 | 24 |
| Haptoglobin | 1.0-3.0 | 6.0 | 24 |
| Fibrinogen | 2.0-4.5 | 10.0 | 24 |
| C3 | 0.55-1.2 | 3.0 | 48-72 |
| C4 | 0.2-0.5 | 1.0 | 48-72 |
| Ceruloplasmin | 0.15-0.6 | 2.0 | 48-72 |
Assessment Methods
CASE STUDY
A 39-year-old woman was admitted for a cholecystectomy. She had a history of chronic cholecystitis; recent x-ray studies revealed stones in the gallbladder and a large stone in the biliary duct (Fig. 5-3). During surgery, a large stone was removed from the duct, and a cholangiogram showed no further obstructions of the hepatic or common bile ducts.
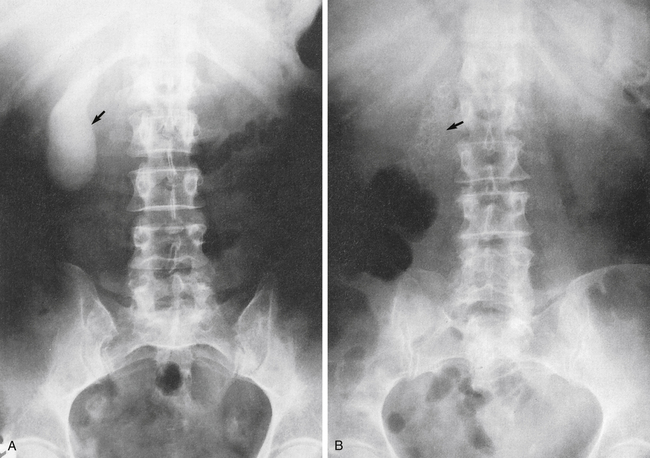
A, Normal gallbladder (arrow). B, Gallbladder filled with stones (arrow).
The patient became febrile 1 day after surgery. A 48-hour postoperative complete blood count (CBC) and CRP were ordered (Fig. 5-4). On the seventh postoperative day, she had abdominal pain and began vomiting. A CBC, ESR, CRP, and blood culture were ordered at that time. Immediately after drawing the blood work, the patient was started on a broad-spectrum antibiotic and discharged on hospital day 15.
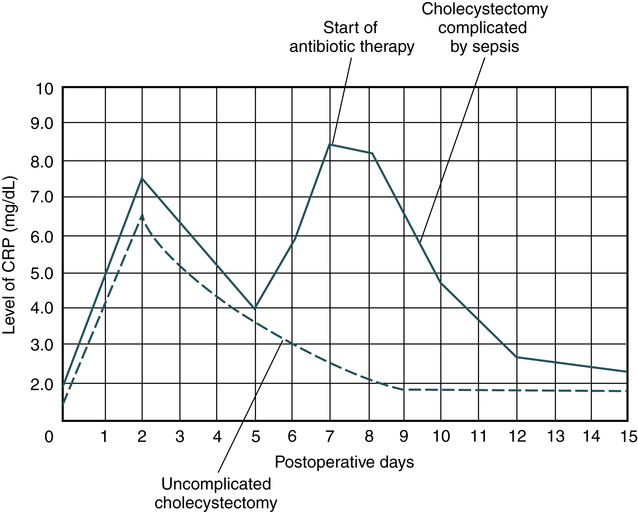
1. The patient’s C-Reactive Protein (C-RP) was elevated at 7 days post-operatively because:
See Appendix A for the answers to these questions.
 for discussion of the answers to these questions.
for discussion of the answers to these questions. C-Reactive Protein Rapid Latex Agglutination Test
C-Reactive Protein Rapid Latex Agglutination Test
Principle
See instructor  for the procedural protocol.
for the procedural protocol.
Chapter Highlights
• The complement system is a heat-labile series of 18 plasma proteins, many of which are enzymes or proteinases. Normally, complement components are present in the circulation in an inactive form.
• Complement is composed of three interrelated enzyme cascades—the classic, alternate, and mannose-binding lectin pathways.
• Complement levels may be abnormal in certain disease states. Increased complement levels are often associated with inflammatory conditions, trauma, and acute illness. Separate complement components (e.g., C3) are acute-phase proteins.
• The biological functions of the complement system fall into two general categories, cell lysis by the membrane attack complex or biological effects of proteolytic fragments of complement.
• Cytokines are a family of proteins that are synthesized and secreted by the cells associated with innate and adaptive immunity in response to microbial and other antigen exposure.
• Cytokines also participate in host defense. In innate immunity, cytokines mediate early inflammatory reactions to microbial organisms and stimulate adaptive immune responses. In contrast, in adaptive immunity, cytokines stimulate proliferation and differentiation of antigen-stimulated lymphocytes and activate specialized effector cells (e.g., macrophages).
• The interferons are a group of cytokines discovered in virally infected cultured cells. IFNs are one of the body’s natural defensive responses to foreign components (e.g., microbes, tumors, and antigens).
• Tumor necrosis factor is the principal mediator of the acute inflammatory response to gram-negative bacteria and other infectious microbes. TNF is responsible for many of the systemic complications of severe infections.
• Hematopoietic stimulators include stem cell factor, a cytokine that acts on immature stem cells.
• Chemokines are a large family of structurally homologous cytokines that stimulate transendothelial leukocyte movement from the blood to tissue site of infection and regulate the migration of polymorphonuclear leukocytes and mononuclear leukocytes within tissues. Chemokines appear to control the phased arrival of different cell populations at sites of inflammation.
• The acute-phase response is an innate body defense. This response is a nonspecific indicator of an inflammatory process.
• C-reactive protein is used clinically for monitoring infection, autoimmune disorders and, more recently, healing after a myocardial infarction. CRP levels parallel the course of the inflammatory response and return to lower undetectable levels as the inflammation subsides.