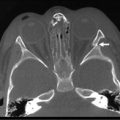
Solid Organ Transplantation in Children
Liver Transplantation
Indications for Transplantation
Table 45-1 reviews the leading diagnoses that lead to LT. These disease entities define the bimodal age distribution of pediatric transplant recipients. Infants and children with biliary atresia and, occasionally, rapidly progressive hepatic failure secondary to metabolic abnormalities, such as neonatal tyrosinemia, hemochromatosis, or neonatal hepatic vascular tumors, are the patients who require transplantation early in life. Patients with hepatic tumors, metabolic disturbances, fulminant hepatic failure, and cirrhosis present as older children and adolescents requiring LT.
TABLE 45-1
Indications for Liver Transplantation at Cincinnati Children’s Hospital Medical Center, 1986–2007
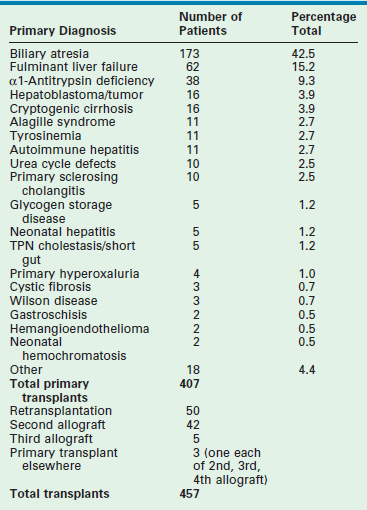
Biliary Atresia
Children with extrahepatic biliary atresia constitute at least 50% of the pediatric LT population. Successful biliary drainage achieving an anicteric state following the Kasai portoenterostomy is the most important factor affecting preservation of liver function and long-term survival.1 Primary transplantation without portoenterostomy is not recommended in patients with biliary atresia unless the initial presentation is greater than 120 days of age and the liver biopsy shows advanced cirrhosis.2,3 We believe that the Kasai portoenterostomy should be the primary surgical intervention for all other infants with extrahepatic biliary atresia. Patients with progressive disease following a Kasai procedure should be offered early orthotopic liver transplantation (OLT). The sequential use of these two procedures optimizes overall survival and organ use.3
Patients with extrahepatic biliary atresia who are seen for transplantation form several cohorts. Infants with a failed Kasai have recurrent cholangitis, ascites, rapidly progressive portal hypertension, malnutrition, and progressive hepatic synthetic failure, and often require OLT within the first two years of life. Children with the successful establishment of biliary drainage have an improved prognosis, but this alone does not preclude the development of cirrhosis and portal hypertension leading to hypersplenism, variceal hemorrhage, ascites, and occasionally hepatopulmonary syndrome. These patients may require LT later in childhood. Patients with mild hepatocellular enzyme and bilirubin elevation, and mild portal hypertension can be observed with ongoing medical therapy. Approximately 20% of all patients with biliary atresia will not require OLT at some point in their life.4,5
Alagille Syndrome
Alagille syndrome (angiohepatic dysplasia) is an autosomal dominant genetic disorder that manifests as bile duct paucity which leads to progressive cholestasis and pruritus, xanthomas, malnutrition, and growth failure. Liver failure occurs late, if at all. Specific criteria for LT are difficult to quantify. Preoperative evaluation must include assessment for congenital cardiac disease and renal insufficiency, both of which are associated with this syndrome. Hepatocellular carcinoma (HCC) has also been seen in occasional patients.6,7
Experience using external biliary diversion or internal ileal bypass accompanied by ursodeoxycholic acid therapy has demonstrated a significant decrease in both pruritus and complications of hypercholesterolemia.8 Both of these procedures may ameliorate or decrease the rate of ongoing parenchymal destruction and cirrhosis, obviating the need for LT. Quality of life issues such as intractable pruritus, hypercholesterolemia, severe growth retardation, and intractable bone disease are criteria for consideration for LT.9–11
Metabolic Disease
An important indication for hepatic transplantation in older children is hepatic-based metabolic disease. In these patients, LT is not only lifesaving but also accomplishes phenotypic and functional cure. A review of these diseases and their mode of presentation are given in Box 45-1 and Table 45-2.
TABLE 45-2
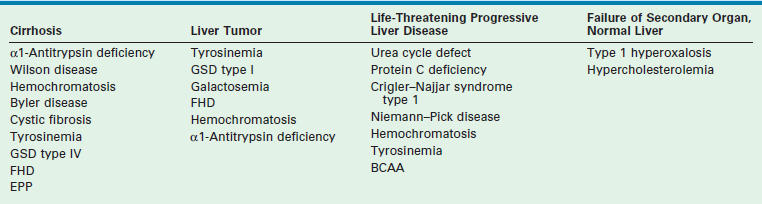
aClassification of inherited metabolic disorders according to clinical modes of presentation.
Reprinted from Balistreri WF, Ohi R, Todani T, et al. Hepatobiliary, Pancreatic and Splenic Disease in Children: Medical and Surgical Management. Amsterdam: Elsevier Science; 1997. p. 395–9.
Hepatic replacement to correct the metabolic defect should be considered before other organ systems are affected, and before complications develop that would preclude transplantation, such as in patients with tyrosinemia, in whom there is a high risk of HCC.12 Although results of transplantation are excellent in the metabolic disease subgroup, replacement of the entire liver in order to correct single enzyme deficiencies is an inefficient, but presently necessary procedure. Current research centers around hepatocyte transplantation and gene therapy.13–17 These efforts may better serve this patient population in the future. Patients with primarily extrahepatic manifestations of their disease, such as cystic fibrosis, are occasionally helped by LT, although their prognosis is most often determined by their primary illness.18
Fulminant Hepatic Failure
Patients with fulminant hepatic failure without recognized antecedent liver disease present diagnostic and prognostic difficulties. Rapid clinical deterioration frequently makes establishment of a definitive diagnosis impossible before there is an urgent need for transplantation. Acute viral hepatitis of undefined etiology makes up the largest group, followed by drug toxicity and toxin exposure. Previously unrecognized metabolic disease must also be considered. Recently, an immune-based defect has been recognized as a cause of fulminant liver failure.19 This population needs to be identified as these children may require a combination of bone marrow and LT to achieve long-term survival. When acceptable clinical and metabolic stability make liver biopsy safe, diagnostic information allowing directed treatment of the primary liver disease is helpful. The presence of ongoing coagulopathy often dictates the need for an open approach to biopsy.
The prognosis of patients with fulminant liver failure is difficult to predict, and neurologic outcome is potentially suboptimal.16,20,21 Use of intracranial pressure (ICP) monitoring in patients with progressive encephalopathy has allowed early recognition and treatment for increased ICP. Monitoring should be instituted for patients with advancing grade III encephalopathy, and in all patients with grade IV encephalopathy. Intracranial monitoring is continued intraoperatively and for 24 to 48 hours after OLT, because significant increases in ICP can develop postoperatively. Failure to maintain a cerebral perfusion pressure greater than 50 mmHg and an ICP less than 20 mmHg has been associated with very poor neurologic outcomes.21 Also, survival following LT is significantly decreased in patients who reach grade IV encephalopathy. Efforts to identify and perform LT in children before this deterioration occurs are of utmost importance. When candidates are identified before they develop irreversible neurologic abnormalities, the results of transplantation are dramatically improved.
Liver Tumors
Transplantation for hepatoblastoma is recommended for individuals who, after the administration of several cycles of chemotherapy, have a neoplasm confined to the liver that is unresectable.22,23 Children who had prior isolated metastasis that disappeared while undergoing preoperative chemotherapy can be considered in select instances.24 A favorable response to pretransplant chemotherapy suggests a more favorable long-term outcome.25 In the current Children’s Oncology Group (COG) trial AHEP 0731, early referral for transplant evaluation is being evaluated for children who present with large lesions that appear unresectable.
Transplantation for HCC is complicated by less successful chemotherapy options and frequent extrahepatic involvement. The reported two-year survival rates are only 20–30%.26 Most deaths are due to recurrent HCC within the allograft or to extrahepatic tumor involvement. When primary HCC is discovered incidentally within the cirrhotic native liver at the time of hepatectomy, the overall prognosis is unaffected by the tumor.27
Donor Considerations
Donor Options
The single factor limiting the availability of LT is the supply of donor organs. The number of patients awaiting LT has increased by eleven fold since 1991.28,29 Available donor resources have not kept pace. As a consequence, the waiting time to transplant for all pediatric age groups has increased significantly, with young children and infants most affected (Figs 45-1 and 45-2). This severely limited supply of available donor organs has driven the advancement of many innovative liver transplant surgical procedures. The development of reduced-size LT allowed significant expansion of the donor pool for infants and small children. This not only improved the availability of donor organs but also allowed access to donors with improved stability and organ function. Evolution of these operative techniques has resulted in the development of both split-LT and live donor (LD) transplantation.
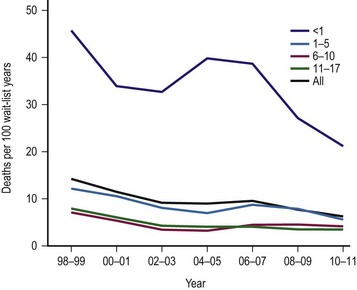
FIGURE 45-1 Graph depicting the mortality rate for adults and children on the liver transplant waiting list from 1999–2010. Note the largest number of deaths occur in infants less than 1 year of age. The numbers represent the age of the patient (in years). (Source: Scientific Registry Transplant Recipients—2011 Annual Report).
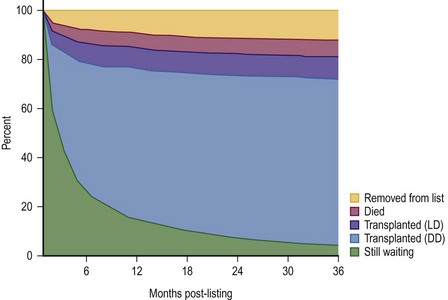
FIGURE 45-2 This diagram describes what has happened to children who have been listed for liver transplantation. Fortunately, most of the patients have undergone either live donor (LD) or deceased donor (DD) transplantation, although about 5% of patients are still on the transplant list after 36 months. (Source: Scientific Registry Transplant Recipients—2011 Annual Report).
In the hands of experienced transplant teams, these procedures all have similar success to whole organ transplantation. Furthermore, access to these donor options has reduced the waiting list mortality rate to less than 5%.
Organ Allocation
In 1998, the ‘final rule’ established by the Health Resources and Service Administration (HRSA) mandated the formation of a system for candidate stratification based on a continuous severity score reflecting 90 day waiting list mortality, i.e., outcome.30 The system for pediatric patients, the Pediatric End-Stage Liver Disease (PELD) score, was created using an analysis of the prospective registry of children listed for transplantation by the consortium Studies of Pediatric Liver Transplantation (SPLIT).31 The parameters selected included total bilirubin, international normalized ratio (INR), albumin, age <1 year, and evidence of failure to thrive (Box 45-2). The primary function of PELD is the stratification of candidates for LT by risk of 90 day waiting list mortality, allowing optimal use of donor organs. When death rates for all children listed were analyzed, PELD was an accurate predictor of mortality risk and demonstrated progressive risk until high scores were reached (>35) (Fig. 45-3).32
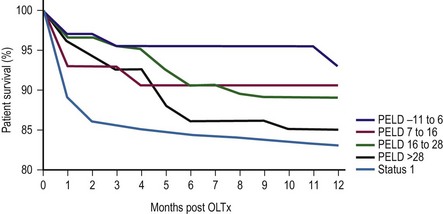
FIGURE 45-3 Pediatric End-Stage Liver Disease score predictive of survival after transplantation. (Redrawn from Barshes NR, Lee TC, Udell IW, et al. The PELD model as a predictor of survival benefit and of post transplant survival in pediatric liver transplant recipients. Liver Transpl 2006;12:475–80.)
Donor Selection
Assessment of donor organ suitability is undertaken by evaluating clinical information, static biochemical tests, and dynamic tests of hepatocellular function. Static biochemical tests identify preexisting functional abnormalities or organ trauma, but do not serve as good benchmarks to differentiate among acceptable and poor donor allografts. Donor liver biopsy is helpful in questionable cases to identify preexisting liver disease or donor liver steatosis. The shortage of donor organs has led to expanded efforts to use individuals of advanced age and marginal stability, termed ‘extended criteria donors’ (ECD).33 The evolving donor risk index (DRI) is used as a guide that quantifies relative risk of graft failure.34 In the future, organ allocation may be based on maximal life years gained, an approach being utilized in kidney allocation.35
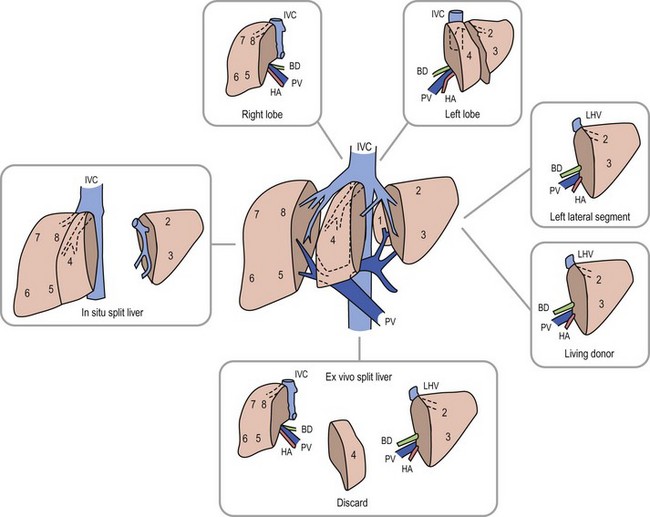
Anatomic replacement of the native liver in the orthotopic position requires selection or surgical preparation of the donor liver to fill, but not exceed available space in the recipient. When using full-sized allografts, a donor weight range from 50–125% of the recipient weight is usually appropriate, taking into consideration body habitus and factors that would increase the abdominal size in the recipient such as ascites and hepatosplenomegaly. The right lobe graft, using segments 5 to 8, and the right trisegmentectomy graft using segments 4 to 8 can be accommodated when the weight difference is no greater than 2 : 1 between the donor-to-recipient (D:R). The thickness of the right lobe makes this allograft of limited usefulness in small recipients. Right lobe grafts from LDs have become widely used in adults. The left lobe, using segments 1 to 4, is applicable with a D:R disparity from 2.5 : 1 to 5 : 1, and a left lateral segment (segments 2 and 3) can be used with up to a 10 : 1 D:R weight difference.
The successful experience with in situ division of the living donor left lateral segment is a basis for the in situ split procedure. Two variations of the procedure are utilized depending on the needs of the recipients, a right–left lobe split or a right trisegmentectomy–left lateral segment split. For the right trisegmentectomy–left lateral segment split, the left lateral segment is prepared similar to a living related donor graft. The viability of segment 4 can be examined at the time of the division and is usually incorporated with the right lobe graft to increase the cellular mass of the allograft. For a left-right lobe graft, the parenchymal resection follows the anatomic lobar plane through the gallbladder fossa to the vena cava.36,37 A crush and tie technique is preferred to achieve good closure of the vascular and biliary structures. The bile duct, portal vein, and hepatic artery are divided at the right or left confluence. The vena cava is left incorporated with the allograft in both right and left lobe preparation. Vena caval reduction by posterior caval wall resection and closure is occasionally necessary. Resection of the inferior protruding portion of the caudate lobe is necessary during left lobe preparation to reduce the likelihood of arterial angulation, which can result in arterial thrombosis. This also facilitates shortening of the inferior vena cava to fit in a small recipient.38 When using left lateral segment (LLS) allografts, the parenchymal dissection follows the right margin of the falciform ligament with preservation of the left hilar structures. Direct implantation of the left hepatic vein into the combined orifice of the right and middle/left hepatic veins in the recipient vena cava is preferred; the donor vena cava is not retained with this segmental allograft. Further reduction of the LLS graft to a monosegmental graft may be necessary in very small recipients. Resection of the distal LLS is technically easier than an anatomic segment II/III division. Because this procedure adds considerably to the donor procurement time, and the necessary skill of the donor team, it is more demanding and occasionally difficult to successfully orchestrate. This technique is, however, despite these considerations, the preferred method for split-liver donor preparation.39,40
The benefits of split-liver transplantation are best achieved when ideal donors are selected. Strict restrictions on age, vasopressor administration, pre-donation hepatic function, and limited donor hospitalization have been used to select optimal donor candidates. When these donors are selected, the results from both in situ and ex-situ techniques are similar, with both techniques now having patient survival for both allografts of 90–93% and graft survival rates of 86–89%.41
The use of LDs has increased as the safety and success of this approach has been demonstrated (Fig. 45-4).42–44 One of the critical elements of LD transplantation is the proper selection of a donor, usually a parent or relative. This procedure is performed on the assumption that donor safety can be assured and that the donor’s liver function is normal. Careful attention to proper living donor consent is important. Parental concerns to help their ill child make true informed consent are a challenge. A dedicated ‘donor advocate’ not directly associated with the transplant team should assist with this process. Independent medical assessment of the donor is essential. United Network Organ Sharing (UNOS) has recently established clear criteria for this process.45 After a satisfactory medical and psychological examination by a physician not directly involved with the transplant program, computed tomography (CT) scanning is used to measure the volume of the potential donor segment to assure that it will meet the metabolic needs, but not exceed the space available in the recipient. If acceptable, CT angiography or arteriography is undertaken to assess the hepatic arterial anatomy, thereby excluding potential donors with multiple arteries to segments 2 and 3, and facilitating minimal hilar vascular dissection at the time of LT. Experience has shown that, when donors were deemed unacceptable, 90% of patients were excluded on the basis of history, examination, laboratory screening, and ABO type. Donor safety has been excellent in all pediatric LD series.46–48
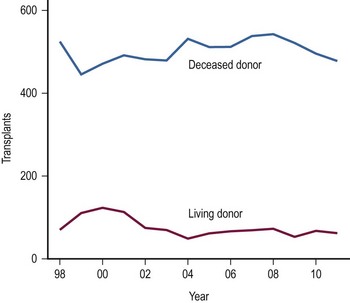
FIGURE 45-4 The number of living donor and deceased donor liver transplants in infants and children performed between 1998–2011. (Source: Scientific Registry Transplant Recipients—2011 Annual Report).
In most pediatric cases, the LLS donated from an adult is used as the graft. In situ dissection of the LLS, preserving the donor vascular integrity until the parenchymal division is completed, is undertaken. At the time of harvest, the left hepatic vein is divided from the vena cava, and the left branch of the portal vein and hepatic artery are removed with the allograft.46 Vascular continuity of the hepatic arterial branches to segment 4 is maintained, if possible. Increased experience has been gained using the right lobe as an LD allograft for larger recipients such as adolescents and adults.42,49,50 This more extensive operation has proven to be a challenge to the donor and recipient alike, with complication and mortality rates significantly exceeding that of left lateral segmentectomy. The number of right lobe LD recipients now greatly exceeds the number of children receiving LD grafts;51 however, several publicized donor deaths and increased interest in ‘split-liver’ cadaveric procurement have slowed the enthusiasm and growth of right lobe donation.
The selection of a donor segment with an appropriate parenchymal mass for adequate function is critical to success. However, the minimal mass necessary for recovery is not yet established. Any calculation must take into account loss of function following preservation damage, acute rejection, and technical problems. When the D:R weight range falls within the normal 8 : 1 to 10 : 1 ratio, risk is minimal. Estimates of donor graft to recipient body weight ratio (GRWR) may prove to be a more accurate predictor of adequate graft volume. When the GRWR is less than 0.7%, overall allograft and patient survival suffered. In extreme cases in which small-for-size grafts are used, excessive portal flow can lead to hemorrhagic necrosis of the graft. Large-for-size allografts (GRWR >5.0%) have a less deleterious effect.52 A review of these donor anatomic options is shown in Figure 45-5.
The Transplant Procedure
Control of hemorrhage is essential during the recipient hepatectomy, requiring meticulous technique. Coagulation factor assays (V, VII, VIII, fibrinogen, platelets, prothrombin time, partial thromboplastin time) allow specific blood product supplementation to improve clotting function. Use of venovenous bypass is reserved for recipients >40 kg who demonstrate hemodynamic instability at the time of venous interruption.
Before re-establishing circulation to the allograft, anesthetic adjustments must be made to address the large volume of blood needed to refill the liver as well as hypothermic solutions released upon reperfusion. Inotropic support using dopamine (5–10 µg/kg/min) is also started. Calcium and sodium bicarbonate are administered to combat the effects of hyperkalemia from any remaining preservation solution or from systemic acidosis due to aortic and vena caval occlusion. Sufficient blood volume expansion, administered as packed red blood cells to raise the central venous pressure (CVP) to 15–20 cmH2O and the hematocrit to 40%, minimizes the development of hypotension on unclamping and prevents dilutional anemia. Cooperative communication between the surgical and anesthesia teams facilitates a smooth sequential reestablishment of vena caval, portal venous, and then arterial recirculation to the allograft.
Biliary tract reconstruction in patients with biliary atresia or in those weighing less than 25 kg is achieved through an end-to-side choledochojejunostomy using interrupted dissolving monofilament sutures. A multifenestrated Silastic internal biliary stent is placed before completing the anastomosis (Fig. 45-6). In most cases, the prior Roux-en-Y limb can be used, with a 30–35 cm length being preferred. Primary bile duct reconstruction without stenting is used in older patients with whole organ allografts.
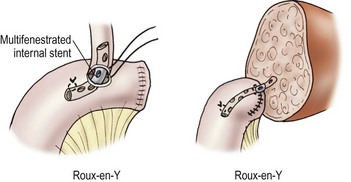
FIGURE 45-6 Bile duct reconstruction is shown using the common hepatic duct in whole organ transplants (left) and segmental hepatic ducts into a Roux-en-Y intestinal limb for reduced-sized liver transplants (right). An internal multifenestrated stent is used in both situations. (From Ryckman F. Liver transplantation. In: Ziegler MM, Azizkhan RG, Weber T, editors. Operative Pediatric Surgery. New York: McGraw-Hill; 2003. p. 1275.)
Immunosuppressive Management
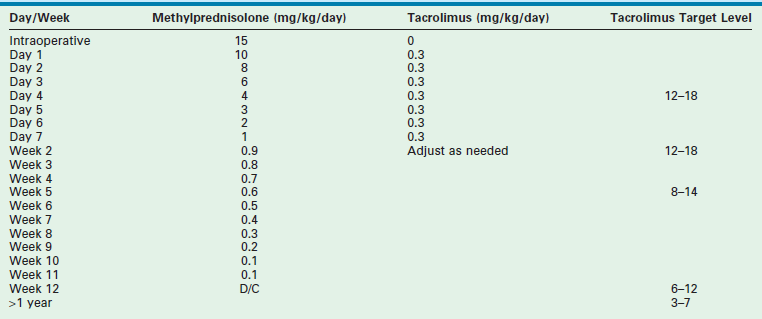
Most centers use an immunosuppressive protocol based on the administration of multiple complementary medications. All use corticosteroids and cyclosporine or tacrolimus. Additional antimetabolites (azathioprine, mycophenolate) are used when more intensive treatment is needed. Prior protocols using polyclonal or monoclonal induction therapy have been abandoned in most cases due to the extent of the immunosuppressive potency. A sample protocol is given in Table 45-3.
Postoperative Complications
Most postoperative complications present with cholestasis, increasing hepatocellular enzyme levels, and on occasion fever, lethargy, and anorexia. Therapy directed at the specific causes of the allograft dysfunction is essential. Empiric therapy for presumed complications is fraught with misdiagnoses, morbidity, and mortality. A flow diagram outlining this evaluation is shown in Figure 45-7.
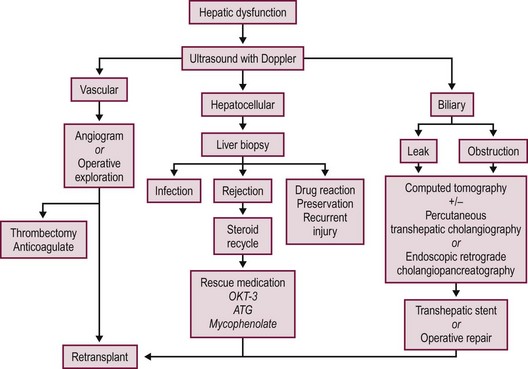
FIGURE 45-7 Schematic flow diagram for management of postoperative liver allograft dysfunction. ATG, antithymocyte globulin; OKT-3, monoclonal antibody.
Primary Nonfunction
Primary nonfunction (PNF) of the hepatic allograft implies the absence of metabolic and synthetic activity following LT. Complete nonfunction requires immediate retransplantation before irreversible coagulopathy and cerebral edema occur. Lesser degrees of allograft dysfunction occur more frequently, and are associated with several donor, recipient, and operative factors (Box 45-3). The status of the donor liver contributes significantly to the potential for PNF. Ischemic injury secondary to anemia, hypotension, hypoxia, or trauma is often difficult to ascertain in the history of multiple trauma victims. Donor liver steatosis has also been recognized as a factor contributing to severe dysfunction or nonfunction in the donor liver.53,54 Macrovesicular steatosis on donor liver biopsy is somewhat more common in adult than pediatric donors and, when severe, is recognized grossly by the enlarged yellow, greasy consistency of the donor liver. The risk of PNF increases as the degree of fatty infiltration increases.54 Histologic findings are classified as mild if less than 30% of the hepatocytes have fatty infiltration, moderate if 30 to 60% are involved, and severe if more than 60% of the hepatocytes have fatty infiltration. Livers with severe fatty infiltration should be discarded, and donors with moderate involvement are used with some concern, with the degree of steatosis and the condition of the recipient determining use of the allograft.
The use of ABO-incompatible donors has been controversial. Allograft and patient survival rates in adult recipients have not been comparable to those achieved using ABO-identical or compatible donors.55,56 However, pediatric recipients of ABO-incompatible allografts have achieved survival rates equivalent to those using ABO-compatible and ABO-identical donors with either cadaveric donors (CDs) or LDs.57–59
Vascular Thrombosis
Hepatic artery thrombosis (HAT) occurs in children three to four times more frequently than in adult transplant series, occurring most often within the first 30 days following transplantation.60,61 Factors influencing the development of HAT are listed in Box 45-4. HAT presents with a variable clinical picture that may include: (1) fulminant allograft failure; (2) biliary disruption or obstruction; or (3) systemic sepsis. Doppler ultrasound (US) imaging has been accurate in identifying arterial thrombosis, and is used as the primary screening modality to assess vascular flow following transplantation or whenever complications arise. Acute HAT with allograft failure most often requires immediate retransplantation. Successful thrombectomy and allograft salvage is possible if reconstruction is undertaken before allograft necrosis.62 Biliary complications are particularly common following HAT. Ischemic injury to the biliary tree or anastomosis can result in intraparenchymal biloma formation or cholestasis. The development of septicemia or multifocal abscesses in sites of ischemic necrosis secondary to gram-negative enteric bacteria, Enterococcus, anaerobic bacteria, or fungi can also occur. Antibiotic therapy directed toward these organisms, along with operative or percutaneous drainage, is indicated when specific abscesses are identified. Drainage and biliary stenting may control bile leakage and infection until retransplantation is undertaken.
Prevention of HAT requires meticulous arterial reconstruction at the time of transplantation. Anatomic reconstruction is preferred in whole organ allografts; direct implantation of the celiac axis into the infrarenal aorta is recommended for all reduced-size liver allografts. All complex vascular reconstructions of the donor hepatic artery should be undertaken ex vivo whenever possible using microsurgical techniques before transplantation. When vascular grafts are required, they should also be implanted onto the infrarenal aorta.63 No systemic anticoagulation is routinely used by our group, but aspirin (20–40 mg/day) is administered to all children for 100 days.
Portal vein thrombosis (PVT) is uncommon in whole organ allografts unless prior portosystemic shunting has altered the flow within the splanchnic vascular bed or unless severe portal vein stenosis in the recipient has impaired flow to the allograft. Preexisting PVT in the recipient can be overcome by thrombectomy, portal vein replacement, or extra anatomic venous bypass. In biliary atresia recipients, portal vein hypoplasia is best corrected by anastomosis of the portal vein to the confluence of the splenic and superior mesenteric veins in the recipient. When there is inadequate portal vein length on the donor organ, iliac vein interposition grafts are used. Early thrombosis following LT requires immediate anastomotic revision and thrombectomy. Discrepancies in venous size imposed by reduced-size allografts can be modified to allow anastomotic construction.64,65 Deficiencies of anticoagulant proteins, such as protein C and S, and antithrombin III deficiency in the recipient must also be excluded as a contributing cause for vascular thrombosis.66 Failure to recognize PVT can lead to either allograft demise or, on a more chronic basis, to significant portal hypertension with hemorrhagic sequelae or intractable ascites.
Biliary Complications
Complications related to biliary tract reconstruction occur in approximately 10% of pediatric liver transplant recipients. Their spectrum and treatment is determined by the status of the hepatic artery and the type of allograft used. Although whole and reduced-size allografts have an equivalent risk of biliary tract complications, the spectrum of complications differs.67,68
Primary bile duct reconstruction is the preferred biliary tract reconstruction in adults, but it is less commonly used in children. It has the advantage of preserving the sphincter of Oddi, decreasing the incidence of enteric reflux and subsequent cholangitis, and not requiring an intestinal anastomosis. Experience using primary choledocho-choledochostomy without a T-tube has been favorable.69 Late complications following any type of primary ductal reconstruction include anastomotic stricture, biliary sludge formation, and recurrent cholangitis. Endoscopic dilation and internal stenting of anastomotic strictures has been successful in early postoperative cases. Roux-en-Y choledochojejunostomy is the preferred treatment for recurrent stenosis or postoperative leak.
Reconstruction of the bile ducts in patients with reduced-size allografts is more challenging. Division of the bile duct in close proximity to the cut-surface margin of the allograft, with careful preservation of the biliary duct collateral circulation, decreases but does not eliminate a ductal stricture secondary to ischemia. In our early experience, in 14% of patients with left lobe reduced-size allografts, a short segmental stricture developed requiring anastomotic revision (Fig. 45-8). Operative revision of the biliary anastomosis and reimplantation of the bile ducts into the Roux-en-Y is necessary. Percutaneous transhepatic cholangiography is essential to define the intrahepatic ductal anatomy before operative revision, and temporary catheter decompression of the obstructed bile ducts promotes treatment of cholangitis and allows elective reconstruction. Operative reconstruction is accompanied by transhepatic passage of exteriorized multifenestrated biliary ductal stents, which remain in place until reconstructive success is documented. Late stenosis is unlikely. Dissection away from the vasobiliary sheath in the donor has significantly decreased the incidence of this complication.
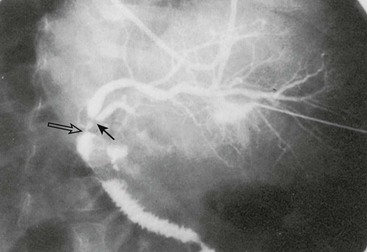
FIGURE 45-8 Segmental bile duct stricture at the junction of the left lateral and left medial segmental bile ducts in a left lobe reduced-size allograft. Solid arrow, bile duct stricture; open arrow, Roux-en-Y loop and bile duct anastomosis. (From Ryckman FC. Liver transplantation in children. In: Suchy FJ, editor. Liver Disease in Children. St. Louis: CV Mosby; 1994. p. 941.)
Acute Cellular Rejection
Allograft rejection is characterized by the histologic triad of endothelialitis, portal triad lymphocyte infiltration with bile duct injury, and hepatic parenchymal cell damage.70 Allograft biopsy is essential to establish the diagnosis before treatment. The rapidity of the rejection process and its response to therapy dictates the intensity and duration of antirejection treatment.
Acute rejection occurs in approximately two-thirds of patients following LT.71 The primary treatment is a short course of high-dose steroids. Bolus doses administered over several days with a rapid taper to baseline therapy is successful in 80% of cases.72 When refractory or recurrent rejection occurs, antilymphocyte therapy using the monoclonal antibody OKT-3® or thymoglobulin® is successful in 90% of cases.38
Chronic Rejection
Uniform diagnosis and management of chronic rejection is complicated by the lack of a consistent definition or clinical course. Chronic rejection occurs in 5–10% of transplanted patients. Its incidence appears to be decreasing in all transplant groups, perhaps related to better overall immunosuppressive strategies. There is some suggestion that the use of tacrolimus based immunosuppression is a key element in this apparent decrease.73,74 Risk factors for its development are many, and no factor predicts the outcome of treatment. The chronic rejection rate is significantly lower in recipients of LD grafts compared with cadaveric grafts.75 In that study, African-American recipients had a significantly higher rate of chronic rejection than did Caucasian recipients. In addition, the number of acute rejection episodes, transplantation for autoimmune disease, occurrence of post-transplant lymphoproliferative disease (PTLD), and cytomegalovirus (CMV) infection were also significant risk factors for chronic rejection. The primary clinical manifestation is a progressive increase in biliary ductal enzymes (alkaline phosphatase, GGT) and progressive cholestasis. Chronic rejection can be initially asymptomatic or may follow unsuccessful treatment for acute rejection. It can occur within weeks of transplantation or later in the postoperative course.
Chronic rejection usually follows one of two clinical forms.76 In the first, the injury is primarily to the biliary epithelium and the clinical course is slowly progressive with preservation of synthetic function. Histologically, either interlobular bile duct destruction in the absence of ischemic injury or hepatocellular necrosis is seen. In full expression, this form is characterized as acute vanishing bile duct syndrome when severe ductopenia is seen in at least 20 portal tracts.77,78 The eventual spontaneous resolution in up to one-half of affected patients with tacrolimus therapy has led to the development of enhanced immunosuppression protocols for this patient subgroup.76 Retransplantation is occasionally necessary, but it is rarely needed emergently.
The second subtype is characterized by the early development of progressive ischemic injury to both bile ducts and hepatocytes, leading to ductopenia and ischemic necrosis with fibrosis. The clinical picture of cholestasis is accompanied by significant synthetic dysfunction with superimposed vascular thrombosis or biliary stricture formation. The vascular endothelial injury responsible for the progressive ischemic changes is characterized by the development of subintimal foam cells or fibrointimal hypertrophy. The clinical course is relentlessly progressive, and nearly always requires retransplantation. Unfortunately, recurrence of chronic rejection in the retransplanted allograft is common.77
Recent studies have focused on donor specific antibody mediated abnormalities that may be the pathophysiologic basis for chronic rejection.79 The immunologic nature of this process is emphasized by the primary target role of the biliary and vascular endothelium, the only tissues in the liver that express class II antigen. Other interdependent cofactors such as CMV infection, human leukocyte antigen (HLA) mismatching, positive B-cell crossmatching, and differing racial demographics of the donor to recipient have all failed to show consistent correlation with the development of chronic rejection.76,77
Renal Insufficiency
The long-term success of LT has been related to the effective immunosuppression with calcineurin inhibitors (CNI), such as cyclosporine and tacrolimus. However, nephrotoxicity associated with their long-term use has become a major problem which can affect up to 70% of all nonrenal transplant recipients. Renal insufficiency can present in many ways following CNI administration and LT. When this occurs during the initial post-transplant weeks, it is most often related to transient excessive blood levels, and is reversible with appropriate dose correction. Impaired glomerular filtration rate (GFR) seen in pediatric recipients with stable graft function represents a more serious problem. Up to 20% may have a drop in their GFR to below 50 mL/min/1.73m2, and 5% may progress to end-stage renal disease (ESRD). Adult studies have shown a progressive increase in chronic renal failure from 0.9% at year 1 to 8.6% at year 13 post OLTx.80 Similarly, ESRD rose from 1.6% at year 1 to 9.5% at year 13 after LT, yielding a total incidence of renal dysfunction of 18%. The presence of an elevated serum creatinine pre-LT, at one-year post-transplant, and the presence of hepato-renal syndrome prior to transplant were all identified risk factors.80,81 Cyclosporine and tacrolimus both appear to be similar in risk.
In a review from our program, in children who were more than three years post-LT, we found that 32% had a GFR <70 mL/minute/1.73 m2.82 The factors primarily related to lower GFR were the presence of an elevated creatinine at one year after LT and the length of time following transplantation. Our data supported the concept of a continued decline in renal function following LT.83,84 Considering the expected long survival for children undergoing LT, the possibility of progressive asymptomatic renal insufficiency leading to severe kidney disease poses a significant challenge.
Efforts to reverse ongoing renal insufficiency using protocols that include instituting non-nephrotoxic agents, such as mycophenolate mofetil (MMF) while decreasing the CNI dose, have shown limited success in improving GFR while protecting against the risks of acute rejection at the time of immunosuppressive drug conversion.85 Efforts have also been undertaken to use the new class of monoclonal anti-CD25 antibodies for induction therapy coupled with MMF and steroids in an effort to avoid administration of CNI during the first post-transplant week. These agents appear to afford sufficient protection against rejection to successfully allow the late administration of CNI. Whether these efforts will prevent the later development of renal insufficiency is yet unproven.86 Efforts to completely eliminate CNI use have been complicated by acute or ductopenic rejection. Current efforts suggest that earlier staged reduction of CNI prior to the development of severe GFR reduction will decrease, but not eliminate this complication. Once established, chronic renal failure does not appear to resolve with CNI dose adjustment. Although CNI toxicity is now well appreciated, the association of both hepatic and renal disease in many metabolic diseases of childhood may also contribute to the GFR abnormalities seen after LT.
Infection
Fungal sepsis represents a significant potential problem in the early post-transplant period. Aggressive protocols for pretransplant prophylaxis are based on the concept that fungal infections originate from organisms colonizing the receipient’s gastrointestinal (GI) tract. In two studies, selective bowel decontamination was successful in eliminating pathogenic Gram-negative bacteria from the GI tract in 87% of adult patients.87,88 Also, in all cases, Candida was eliminated. However, these protocols have not been practical in pediatric recipients because there is a long waiting time for pediatric organs and the taste of the antibiotics is not well accepted. However, these regimens are commonly used in the preoperative preparation for combined liver/small intestinal transplantation. Fungal infection most often occurs in patients requiring multiple operative procedures and those who have had multiple antibiotic courses. Development of fungemia or urosepsis requires retinal, cardiac, and renal investigation, and antifungal therapy should be promptly initiated. Severe fungal infection has a mortality rate greater than 80%, making early treatment essential. All patients undergoing LT should receive antifungal prophylaxis with fluconazole.
The majority of early and severe viral infections are caused by viruses of the Herpesviridae family, including Epstein–Barr virus (EBV), CMV, and herpes simplex virus (HSV). CMV transmission dynamics are well studied and serve as a prototype for herpesvirus transmission in the transplant population. The likelihood that CMV infection will develop is influenced by the preoperative CMV status of the donor and recipient.89,90 Seronegative recipients receiving seropositive donor organs are at greatest risk, with seropositive donor-to-recipient combinations at the next greatest risk. Use of various immune-based prophylactic protocols including IV IgG or hyper immune anti-CMV IgG, coupled with acyclovir or ganciclovir/valganciclovir, have all achieved success in decreasing the incidence of symptomatic CMV infection, although seroconversion in seronegative recipients from seropositive donor organs inevitably occurs.
EBV infection occurring in the perioperative period represents a significant risk to the pediatric recipient.91 It has a varying presentation including a mononucleosis-like syndrome, hepatitis-simulating rejection, extranodal lymphoproliferative infiltration with bowel perforation, peritonsillar or lymph node enlargement, and encephalopathy. In small children, its primary portal of entry is often the tonsils, making asymptomatic tonsillar hypertrophy a common initial presentation.92 EBV infection can occur as a primary infection or following reactivation of a past infection. When serologic evidence of active infection exists, an acute reduction in immunosuppression is indicated. It has become clear that continuous surveillance is necessary as the presentation is often nonspecific and the prognosis is related to early diagnosis. Screening using quantitative PCR testing to determine the EBV blood viral load appears to be the best current predictor of risk. However, viral loads have been identified in asymptomatic patients and patients recovering from PTLD, limiting the specificity of this approach. The balance between viral load measured by quantitative PCR and specific cellular immune response, perhaps mediated by CD8 T-cells specific to EBV, may explain this lack of specificity to viral load alone.93–95
Many pediatric transplant centers now use serially measured quantitative EBV DNA PCR as an indication for primary immunosuppression modulation. We recommend monthly EBV DNA PCR counts to monitor increased genomic expression. Increasing viral load levels warrant more frequent monitoring on a weekly or every two week schedule. In the EBV seronegative pretransplant patients, >40 genomes/105 peripheral blood leukocytes (PBL), and >200 genomes/105 PBL identify patients needing reduction in primary immunosuppression by 25 to 100%. Institution of antiviral therapy with ganciclovir and CMV IgG is also used in most cases, although only nonrandomized observational studies support their use. Both agents are active in vitro against linear replicating forms of the EBV, but have no activity against the circular episome in immortalized B-cells. Treatment should be continued until symptoms of lymphadenopathy have resolved and viral EBV DNA PCR has returned to baseline.91,96 It should however be cautioned that PTLD can develop and progress without increases in EBV-PCR viral load.97
PTLD, a potentially fatal abnormal proliferation of B-lymphocytes, can occur in any situation in which immunosuppression is used. The importance of PTLD in pediatric LT is a result of the intensity of the immunosuppression required, its lifetime duration, and the absence of prior exposure to EBV infection in 60–80% of pediatric recipients. PTLD is the most common tumor in children following transplantation representing 50% of all tumors compared to 15% in adults. About 80% of cases occur within the first two years following transplantation.94 Multiple studies analyzing immunosuppressive therapy and the development of PTLD have shown a progressive increase in its incidence with (1) the increase in total immunosuppressive load; (2) EBV naïve recipients; and (3) the intensity of active viral load.98 No single immunosuppressive agent has been directly related to PTLD, although high-dose cyclosporine, tacrolimus, polyclonal antilymphocyte sera (MALG, ALG), and monoclonal antibodies (OKT-3) have all been implicated. Prolonged treatment with anti–T-cell agents and the increased duration, intensity, and total immunosuppressive load are the origin of the immunity that creates the background for neoplasia.
The second pathogenic feature influencing PTLD appears to be EBV infection. Primary or reactivation infections usually precede the recognition of PTLD. Active EBV infection, whether primary or reactivation, involves B-cell proliferation. A simultaneous increase in cytotoxic T-cell activity is the normal host’s mechanism for preventing EBV dissemination. Loss of this natural protection as a result of the administration of T-cell inhibitory immunotherapy allows polyclonal B-cell proliferation to progress following EBV viral replication and release. These EBV proliferating cells express specific viral antigens which represent possible targets for the immune system, thereby explaining the well described regression of PTLD after immunosuppressive tapering. With time, transformation of a small population of cells results in a malignant monoclonal aggressive B-cell lymphoma.93,99–101
Treatment of PTLD is stratified according to the immunologic cell typing and clinical presentation.102 Documented PTLD requires an immediate decrease or discontinuation of immunosuppression and institution of anti-EBV therapy. We prefer to use IV ganciclovir for initial antiviral therapy owing to the high incidence of concurrent CMV infection. Acyclovir is used for long-term treatment. The development of newer antiviral alternatives such as valganciclovir may offer better long-term options in the future.103 Patients with polyclonal B-cell proliferation frequently show regression with this treatment.91,96 If tumor cells express B-cell marker CD 20 on histology, the anti-CD 20 monoclonal antibody Rituximab can be administered weekly. Although associated in many cases with significant reduction in tumor mass, patients have frequently experienced reversible neutropenia requiring granulocyte colony stimulating factor (GCSF) and hypogammaglobulinemia requiring supplementation.104 Acute liver rejection has frequently been seen during Rituximab treatment. Patients with aggressive monoclonal malignancies have poor survival even with immunosuppressive reduction, acyclovir, and conventional chemotherapy or radiation therapy. These additional treatment modalities often precipitate the development of fatal systemic infection. Efforts to reconstitute the EBV specific cellular immunity using partially HLA matched EBV specific cytotoxic T-cells may offer improved treatment outcome for advanced cases. The future development of anti-EBV vaccine may decrease the present significant risks of this unique complication of pediatric transplantation.105 When treatment is successful, careful follow-up to identify recurrent disease or delayed central nervous system involvement is essential.
Retransplantation
The vast majority of retransplantation procedures in infants and children are done as a result of acute allograft demise caused by HAT or PNF. Acute rejection, chronic rejection, and biliary complications are more uncommon causes. Many of these complications are associated with concurrent sepsis, which further complicates reoperation and compromises success. Survival following transplantation is directly related to prompt identification of appropriate patients and acquisition of a suitable organ. When retransplantation is promptly undertaken for early graft failure, patient survival rate, in our experience, is good (73%). However, when retransplantation is undertaken for chronic allograft failure, often complicated by multiple organ system insufficiency, the survival rate is lower (45%).106
Similar findings were reported by UNOS Region I in their combined experience. Patients undergoing retransplantation for acute organ failure experienced twice the overall survival rate as those undergoing retransplantation for chronic disease.107 In addition, acute retransplantation survival was significantly influenced by the time to acquire a retransplant organ, with a greater than three-day wait decreasing the survival rate from 52% to 20%. The overall incidence of retransplantation is 15% in our series and ranges in others’ experience from 8–29%. This incidence is similar for primary whole organ allografts and reduced-size allografts. Reduced-size allografts are frequently used when retransplantation is required in view of their greater availability and their decreased incidence of allograft-threatening complications.38,108,109 These findings emphasize the need for early identification of children requiring retransplantation and expeditious reoperation before the development of multisystem organ failure or sepsis.
Outcome Following Transplantation
Although complications following LT are frequent and severe, the overall results are rewarding. Improvements in organ preservation, operative management, immunosuppression, and treatment of postoperative complications have all contributed to the excellent survival rate that is currently seen. Factors influencing the survival of children undergoing transplantation are detailed in Box 45-5. Most successful transplant programs have reached overall one-year survival rates of 90%, with greatly decreased risk thereafter.110,111 Similar if not better results have resulted from LD transplantation, especially for small recipients.112–114 Improved survival in these small recipients likely results from a decrease in life-threatening and graft-threatening complications, such as HAT and PNF, in the reduced-size donor organ.
Patients with fulminant hepatic failure have an overall survival rate that is significantly lower than other diagnostic groups, with metabolic disease having the highest survival rate. Prior operations and multiple episodes of subacute bacterial peritonitis prior to LT influence the development of complications, especially bowel perforation, but do not adversely affect overall survival in most cases. However, the most important factor determining survival is the severity of the patient’s illness at the time of initial transplantation.38,115 When stratified for illness by PELD scores, Pediatric Risk of Mortality (PRISM) score, and the UNOS score, the PRISM score was the most accurate in predicting both survival and morbidity during the perioperative period.116 Current efforts to use technical variant grafts have experienced similar survival rates as those for whole organ recipients (Fig. 45-9).108,117
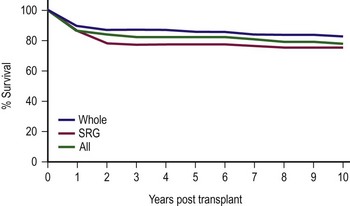
FIGURE 45-9 Ten-year patient and allograft survival subdivided by whole and surgically reduced grafts (SRG). (Data from Cincinnati Children’s Hospital Medical Center, Liver Care Center.)
The significant success now achieved following LT cannot overshadow the need for improved management of post-transplant consequences of immunosuppression and pre-LT chronic disease. The most significant factors contributing to long-term failure of the allograft or patient death in our and others’ program are consequences of immunosuppressive medications: late infection, PTLD, and chronic rejection.111,118–120 Our ability to overcome these challenges will determine the life-long success of transplantation for our youngest recipients. The overriding objective of LT in children is complete rehabilitation with improved quality of life. Factors contributing to successful achievement of this goal include an improved nutritional status with appropriate growth and development, as well as enhanced motor and cognitive skills that allow social reintegration.
Optimal postoperative nutrition significantly facilitates recovery and rehabilitation. This initially requires 100–130 calories/kg/day in recipients weighing less than 10 kg. Hepatic synthetic function, gut absorption, and appetite all improve following successful pediatric LT. Despite these improvements, growth disturbances do not immediately resolve.121–123 In the first year after transplantation, very little catch-up growth occurs. During the second and third year, patients usually show significant catch-up growth, with the potential for catch-up growth being directly correlated with the degree of preoperative growth retardation. Decreased corticosteroid administration enhances this growth. Also, growth is further improved by the use of alternate-day steroids or complete steroid withdrawal in patients with stable allograft function two years following LT.124 The ‘steroid-sparing’ effects of new immunosuppressive agents, such as tacrolimus, could diminish this unwanted consequence of immune modulation.
Although most pediatric LT recipients are returning to normal age-appropriate activities, recent studies indicate that they may experience subtle functional difficulties.125–127 Neuropsychological function studies of children following transplantation demonstrate multiple deficits involving learning and memory, abstraction, concept formation, visual–spatial function, and motor function. The well-documented neurologic and cerebral abnormalities associated with chronic cirrhosis precede transplantation, but certainly influence these results.126,128–130
The long-term impact that transplantation has on the psychosocial and financial health of the entire family unit is also the subject of much investigation. Long-term pediatric LT survivors need in-depth, multicenter longitudinal studies to clarify these issues. Health Related Quality of Life (HRQOL) surveys of families after LT demonstrate improved health and physical perception compared to the pretransplant status.131 However, as can be imagined, the overall impact on the family is significant. The health and quality of life in pediatric LT recipients was lower in these surveys than that reported for healthy children, but similar to that for children with other chronic illness. Age at transplantation, the time elapsed since transplantation, hospitalizations within the previous year, maternal education, and race were significant predictors of physical health. Age at transplantation and maternal education predicted psychosocial function. Both younger children and adolescents continue to have self-confidence and body image concerns, and their participation in family activities and sports is often limited. Caregivers and mothers are heavily impacted by the need for care of their chronically ill children prior to transplant and during their recovery. Many have experienced job losses, career changes, and significant family stress. The importance in providing these caregivers with critical support and psychological help is essential for the entire family to survive and flourish following the difficult times that inevitably follow LT.131
Intestinal Transplantation
Intestinal failure can be a significant problem in the pediatric population. Total parenteral nutrition (TPN) remains the first-line therapy in children who have a loss of enteral autonomy. However, complications of long-term TPN, such as cholestatic liver disease, venous thromboses, and recurrent catheter sepsis, may preclude its continued use. When complications of long-term TPN become life threatening and the bowel length is too short for enteral alimentation, the alternative becomes intestinal transplantation. Although the exact incidence of intestinal failure in children is unclear, over two-thirds of the patients who are currently on the national intestinal transplantation waiting list are children, with the majority of active patients being 5 years of age or younger.132
Advances in immunosuppressive regimens, the technical aspects of the transplant operation, and the surveillance and treatment of transplant-related complications have continued to improve the outcome of patients who have required intestinal transplantation.133,134 With the improved outcomes, the role of intestinal transplantation has evolved from a heroic last effort to salvage patients with no remaining treatment options to a standard part of the armamentarium in the management of patients with intestinal failure. Since the initial approval of federal reimbursement for intestinal transplantation by the Centers for Medicare and Medicaid Services in October 2000,135 continued success with intestinal transplantation in children has shifted focus from short-term patient survival to optimizing long-term allograft function and patient survival.
Indications for Transplantation
The causes of intestinal failure can be divided into three broad categories: short bowel syndrome (SBS), intestinal dysmotility syndromes, and absorptive disorders. SBS, usually caused by the loss of intestinal length due to an intra-abdominal catastrophe or in the setting of a congenital gastrointestinal disorder, is the most common cause in children. Disease processes necessitating an operation that result in SBS range from necrotizing enterocolitis, intestinal atresia, and gastroschisis in the newborn to Crohn disease and traumatic injury to the main intestinal blood supply in the older population. Midgut volvulus, another frequent cause of SBS, can occur at any age, although the majority of cases occur in infants.
TPN is the standard treatment for patients who experience acute intestinal failure. Bowel rehabilitation should be aggressively pursued because intestinal adaptation can result in eventual enteral autonomy. Intestinal rehabilitation programs that utilize a combination of TPN, gradual re-introduction of enteral feeds, intestinal antimotility agents, and treatment of small bowel bacterial overgrowth, in association with hepatoprotective lipid minimization strategies and ethanol lock usage to reduce the incidence of catheter-related bloodstream infections, are successful in achieving enteral autonomy in many patients.136,137 Autologous intestinal reconstruction procedures, such as the serial transverse enteroplasty (STEP) and longitudinal intestinal lengthening and tailoring (LILT), may be beneficial in selected patients. In general, survival and complete return of gastrointestinal function may be predicted when the post-resection length of the intestine exceeds 5% of normal for gestational age if the ileocecal valve is present, or is greater than 10% of normal if the ileocecal valve is absent.138
The current Medicare-approved indications for intestinal transplantation are shown in Box 45-6.135 Progressive liver dysfunction and TPN-associated cholestatic liver disease result in significant mortality. Unless the TPN can be stopped, liver failure is a strong indication for combined liver/intestinal or multivisceral transplantation. Studies have shown that the presence of hyperbilirubinemia greater than 3 mg/dL or bridging fibrosis and cirrhosis found on liver biopsy in an infant dependent on TPN is associated with a 1-year survival of less than 30%.139 The remaining criteria of limited central venous access, multiple catheter-related bloodstream infections, and frequent episodes of severe dehydration reflect complications from chronic TPN use, and in the absence of significant concurrent liver dysfunction, are indications for isolated intestinal transplantation.
Currently, with improved outcomes after intestinal transplantation, some centers advocate early intestinal transplantation before the onset of TPN-induced complications.140 These groups propose that patient and graft survival after intestinal transplantation will be optimized if recipients undergo transplantation prior to the onset of secondary organ damage, especially liver disease. Furthermore, the quality of life in patients who have undergone successful intestinal transplantation may be better than that of patients who require long-term TPN administration. This issue remains controversial and requires further study because TPN is a well-established and effective means of treatment for patients with intestinal failure. In addition, with recent advances in hepatoprotective approaches to TPN administration and improved care related to central venous catheters, children with intestinal failure on TPN are now able to be maintained complication-free for longer periods of time, thereby allowing further delay in the need for intestinal transplantation. Despite the lack of consensus with regard to optimal timing of intestinal transplantation, it has become clear that early and timely patient referral for pretransplant evaluation is critical to ensuring the best opportunity for long-term survival.141
Operative Considerations
The three types of intestinal allografts include multivisceral, liver-small bowel composite, and isolated small bowel. The type of intestinal transplant utilized is dictated by the needs of each patient. The biggest limitation to intestinal transplantation in the pediatric population is the need for size-matched grafts. Patients with intestinal failure generally have limited abdominal domain (due in part to a lack of intestine volume), which necessitates near-identical-size donors. Advances in the use of reduced-size liver allografts have been applied to the liver-small bowel composite allograft as a means of increasing the flexibility of donor-to-recipient size matching.142 However, nearly 50% of patients on the intestinal transplant waiting list die before undergoing transplantation owing to the lack of appropriate donors, and the highest risk subgroup are children less than 1 year old.143 In the past, it was believed that recipients who were CMV-negative should not receive intestinal grafts from CMV-positive donors because of a high incidence of severe, potentially life-threatening CMV infection after transplantation.144 However, with current antiviral therapy regimens, it appears this barrier has been overcome.145 In early series, a higher risk of infectious complications was reported in patients receiving allografts that included donor colon. More recently, however, morbidity and mortality rates in recipients of colon-inclusive allografts have not been found to be higher.146 Children with total intestinal aganglionosis and chronic idiopathic intestinal pseudo-obstruction most commonly receive colon-inclusive allografts.
The Transplant Procedure
Liver–Small Bowel Composite Allograft
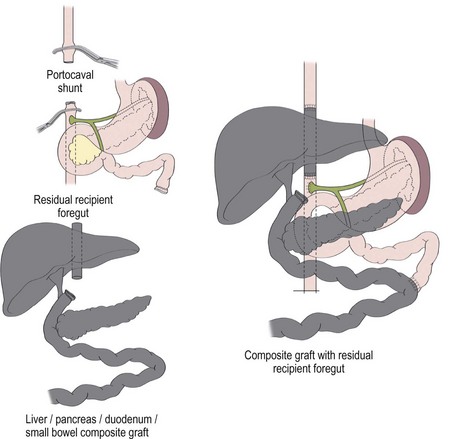
A liver–small bowel composite allograft is a modification of the multivisceral allograft in which the stomach is removed during procurement. This form of transplantation is indicated in patients with intestinal failure and impending or overt TPN-induced liver failure, and is the most common type of intestinal transplant currently utilized in children (Fig. 45-10). The recipient’s liver and residual small intestine are removed, while the native stomach, duodenum, pancreas, and spleen are left intact. A portocaval shunt from the native portal vein to the inferior vena cava is necessary to provide venous outflow from the recipient’s foregut organs (Fig. 45-11). As in the multivisceral allograft, the donor celiac artery and SMA are the source of arterial inflow to the transplanted organs. The donor portal vein and biliary tree remain intact, having not undergone dissection during procurement. As a result, no portal vein or bile duct reconstruction is needed. The pancreas is also left intact to protect the peri-biliary ductal vessels and to prevent the possibility of pancreatic leak from a divided surface. Venous outflow from the transplanted organs is once again provided by the donor liver placed in a standard orthotopic position. If the liver is too large, an ex vivo hepatic lobectomy can be performed, usually removing the right lobe of the liver (Fig. 45-12). Gastrointestinal continuity from the patient’s native stomach and duodenum to the newly transplanted small bowel is achieved by anastomosis of the native duodenum to the donor jejunum. If the recipient has any colon remaining, a donor ileum to recipient colon anastomosis is created distal to the ileostomy.
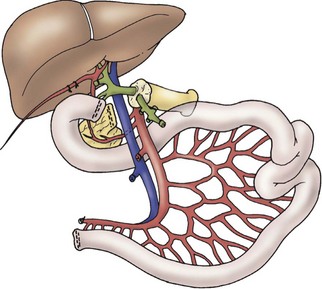
FIGURE 45-10 Schematic diagram of liver/intestine composite allograft. (From Abu-Elmagd K, Reyes J, Todo S, et al. Clinical intestinal transplantation: New perspectives and immunologic considerations. J Am Coll Surg 1998;186:512–27.)
Isolated Small Bowel Allograft
Transplantation of the small intestine alone entails procurement of only the jejunum and ileum. During procurement, the SMA and vein are divided just below the third portion of the duodenum at the root of the mesentery, generating an allograft of jejunum and ileum (Fig. 45-13). This type of transplant is indicated in patients with intestinal failure, but without liver dysfunction. Arterial inflow is provided by anastomosis of the SMA to the recipient’s aorta. Venous drainage of the transplanted intestines is either into the inferior vena cava or to the native superior mesenteric vein. Initially it was believed that venous drainage into the native portal circulation was beneficial to the liver, but recent studies suggest minimal benefit.147 Therefore, currently, the most common approach to venous reconstruction is an end-to-side anastomosis of the donor superior mesenteric vein to the native inferior vena cava. Gastrointestinal continuity is restored by anastomosis of the recipient’s native proximal bowel to the transplanted jejunum. Once again, if residual colon is present, a donor ileum to native colon anastomosis is created downstream from the ileostomy.
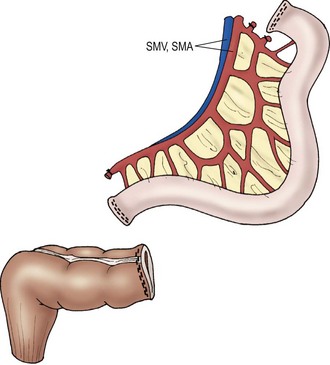
FIGURE 45-13 Schematic diagram of isolated small intestinal transplant. SMV, superior mesenteric vein; SMA, superior mesenteric artery. (Adapted from Abu-Elmagd K, Fung J, Bueno J, et al. Logistics and technique for procurement of intestinal, pancreatic, and hepatic grafts from the same donor. Ann Surg 2000;232:680–7.)
Postoperative Complications
Immunosuppression and Allograft Rejection
The most significant recent advances in the management of intestinal transplant recipients have occurred in the development of immunosuppression regimens. Rejection remains the most common complication after intestinal transplantation, and acute rejection occurs in approximately 60% of pediatric recipients within the first three months after transplantation.134 Overwhelming rejection was one of the most frequent causes of allograft loss during the early transplant experience due, in part, to the limited number of immunosuppressive agents available. Prior to the development of tacrolimus as a maintenance immunosuppression approach, high-dose immunosuppression was necessary to prevent rejection. Infections, PTLD, and adverse effects related to high-dose immunosuppression were frequent causes of a poor outcome. The goal of current immunosuppressive regimens is to use just enough immunosuppression to prevent rejection, but not so much that infections and PTLD occur. In an effort to achieve tolerance, many centers utilize a lymphocyte-depleting strategy, such as thymoglobulin or alemtuzumab, to induce early elimination of graft-specific T-lymphocytes. The management of immunosuppression in patients who undergo intestinal transplantation remains the most challenging aspect of the postoperative care.
Surveillance endoscopy and biopsy is initiated within five days of transplantation to evaluate for acute rejection, and is performed twice per week for the first month. Significant progress in the definition of the histologic characteristics of acute small bowel rejection has been achieved.148 If acute rejection is diagnosed, a short course of high-dose corticosteroids is administered. If rejection is severe or persists despite high-dose corticosteroid administration, antilymphocyte antibody agents (thymoglobulin or alemtuzumab) are required. Chronic rejection remains the most common cause of late allograft dysfunction and failure.
Graft-versus-Host Disease (GVHD)
GVHD occurs in approximately 5–10% of intestinal transplant recipients, most commonly in the youngest pediatric recipients. Its high incidence is related to the large burden of donor lymphocytes that are cotransplanted with the allograft. Acute GVHD characteristically affects the skin, native liver, and native gastrointestinal tract, and carries a high mortality. Monitoring of donor-derived T-cell chimerism is being utilized to identify patients at risk for GVHD. Management strategies for GVHD after intestinal transplantation include high-dose corticosteroids and lymphocyte depletion with alemtuzumab.149
Outcome Following Transplantation
Early outcomes of patients who underwent intestinal transplantation was poor with few long-term survivors. With advances in operative techniques, immunosuppression strategies, allograft surveillance, and monitoring and treatment of infections and other transplant-related complications, significant improvements in patient survival have occurred in the past 15 years. Currently, according to the ultrasound Scientific Registry of Transplant Recipients, approximately 80% of pediatric recipients survive 1 year and 65% survive 3 years after transplantation. Short-term allograft survival has likewise increased significantly. Despite improvements in short-term outcomes, the long-term outcomes have remained largely unchanged over the past decade. Current impediments to long-term allograft survival are primarily related to chronic rejection, particularly in isolated small bowel transplantation. Chronic rejection is responsible for the greatest proportion of allograft loss 2 years after transplantation.134 Further advances in surveillance and immunosuppressive strategies are needed to optimize long-term allograft function and survival.
Renal Transplantation
Chronic renal failure is uncommon in infants. The estimate incidence of ESRD in infants is 0.2 per million total population of infants younger than 1 year of age.150,151 Renal aplasia/dysplasia, obstructive and complex urological malformations, and focal segmental glomerulosclerosis (FSGS) are the most common causes of ESRD in children younger than 5 years of age (Table 45-4). In the past antenatal renal failure caused by obstructive malformations resulted in fetal demise or pulmonary insufficiency incompatible with post natal survival. With the evolution of fetal diagnosis and in-utero therapy, the pulmonary insufficiency can be attenuated, resulting in a population of neonates with adequate pulmonary reserve, but perinatal renal insufficiency.
TABLE 45-4
Primary Diagnoses in Children Requiring Renal Transplantation
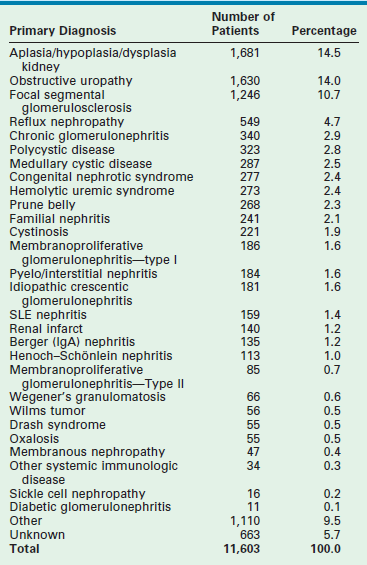
Data from the North American Pediatric Renal Transplant Cooperative Study 2010 Annual Report.
Glomerulonephritis and lupus nephritis, as well as recurrent pyelonephritis, are the common causes of ESRD in older children.152 As the number of patients who have undergone renal transplantation in childhood has increased, chronic rejection following renal transplantation has recently become a cause of ESRD.
Knowledge of the etiology of the ESRD is important to allow assessment of the potential for recurrence within a transplant allograft and consideration of LD transplantation. Patients with a ‘structural/congenital’ etiology, without an immunologic component, also enjoy better graft survival rates than those patients with glomerulonephritis.153
Pretransplant Management
Dialysis
Hemodialysis can be used when there is an unsuitable peritoneal cavity due to prior surgery or multiple peritoneal infections. However, the construction and maintenance of adequate long-term vascular access sites in small infants and children is difficult. Use of central venous catheters rather than arteriovenous fistulas is our preferred mode for temporary hemodialysis access in infants and small children, although infection and vascular thromboses complicate this therapy. Access via the internal jugular veins is preferred over subclavian routes to avoid obstruction of the venous outflow from the upper extremity, which compromises future arteriovenous fistula sites. Although dialysis and its complications, such as infection, have a great influence on the complexity of care, they do not affect the ultimate results of renal transplantation.154
Nutritional Support
The need for vigorous nutritional support of the infant with uremia is well demonstrated by the growth retardation seen in infants and children with ESRD. The etiology of this growth disturbance is multifactorial, including anorexia that leads to protein and calorie insufficiency, renal osteodystrophy, aluminum toxicity, uremic acidosis, impaired somatomedin activity, and growth hormone and insulin resistance.155 Because the most intense period of a child’s growth occurs during the first 2 years of life, careful nutritional support during that time is essential.
The mean weight at the time of renal transplantation has improved from 2.2 SD to 1.6 SD below the appropriate age-adjusted and gender-adjusted mean for normal children in a recent North American Pediatric Renal Transplant Cooperative Study (NAPRTCS). This growth deficit was greater (2.8 SD) in children younger than 5 years of age. Transplantation afforded a 0.8 SD increase in growth over the first post-transplant year. However, this growth then reached a stable plateau. After 2 to 3 years, the mean weight values were comparable to those of normal children.152 Children 6 years of age and older showed no improvement in their height deficit 5 years after transplantation.156,157 These limitations to ‘catch-up’ growth emphasize the need for early transplantation in young ESRD patients. If epiphyseal closure has occurred (bone age >12 years), additional bone growth is often not achieved.151,158,159 Normalization of growth rarely occurs with hemodialysis or peritoneal dialysis.
The importance of efforts to normalize nutritional parameters is emphasized by the adverse impact of uremia on the infant’s developing nervous system. The significance of this problem was emphasized in a study in which progressive encephalopathy, developmental delay, microcephaly, hypotonia, seizures, and dyskinesia developed in 20 of 23 children with ESRD before 1 year of age.160 All of these patients had significant growth impairment. Monitoring of the head circumference has been suggested to identify the infant at risk, with the intent to initiate dialysis, nutritional support, or transplantation if this parameter deviates from the normal curve.151
Preoperative Preparation
In preparation for renal transplantation, an extensive evaluation of the urinary tract and immunologic status of the patient is important. The increased frequency of urinary tract abnormalities as the primary cause of ESRD in infants and children necessitates the investigation of the urinary tract for sites of obstruction, the presence of ureteral reflux, and the functional state and capacity of the urinary bladder.161 This investigation is best accomplished by obtaining an ultrasound or intravenous pyelogram evaluating the upper urinary tract and a VCUG to assess the bladder and reflux. Any concerns about bladder function or structure requires urodynamics and cystoscopy.
In patients with long-standing oliguric ESRD, the bladder may be very small. In the absence of obstructive or neuromuscular pathology, enlargement of the bladder with normal urinary production is expected. Any operative correction of urethral obstruction or augmentation of bladder size should be performed well in advance of transplantation. Preoperative sterilization of the urinary tract and the development of unobstructed urinary outflow should be the ultimate goals of evaluation and reconstruction. Although complex anomalies of the urogenital tract often require many operative procedures to augment, reconstruct, or create an acceptable lower urinary tract, most children can undergo successful reconstruction with continent urinary reservoirs without the need for intestinal conduits.162
Selection of the appropriate donor source for transplantation is a decision for the transplant team and family to consider together. A related immediate family member has the advantage of a low incidence of postoperative ATN, improved histologic matching, and extended organ function. In addition, any operative procedures required for preparation of the recipient, as well as the transplant procedure, can be scheduled around the needs of the patient, simplifying preoperative care and potentially avoiding the complications of dialysis. Parents form the majority of donors. The 2010 NAPRTCS report indicates that 40% of children receive an LD kidney from a parent.153 Thorough evaluation of the potential donor to exclude intrinsic renal anomalies, vascular anomalies, and systemic illness is important.
Deceased donor (DD) kidneys are currently used for 49% of renal transplants.153 The unpredictability of donor organ availability and the need to establish a negative antibody cross-match for DD transplantation make surgical planning impossible. The size of a potential allograft, DD or related LD, is also important. Kidneys from small adult donors can be transplanted into infants as small as 5 kg with good success.163 DD organs from pediatric donors 5 years of age or older also result in an excellent survival rate. However, a progressive decrease in one-year graft survival has been noted when kidneys from donors younger than 3 to 4 years of age are used.164,165 This decrease in graft and patient survival is related to the donor organ source. Children 2 to 5 years of age have a similar survival rate as the overall pediatric population when LDs are used. Recognition of this potential risk has led to reluctance by most centers to use donors younger than 2 years of age. An effect of donor age on graft survival has been attributed to an increased rate of both graft thrombosis and acute rejection.152
In the past, the decision to use a DD as opposed to an LD was influenced by the possibility of disease recurrence within the transplanted kidney. The incidence of disease recurrence following transplantation and the risk of graft loss are listed in Table 45-5.166 With new treatment strategies for recurrent disease, the risk of graft loss has been attenuated, and as a result, LD kidneys are now preferred. This decision to favor LD kidneys is influenced by the recent improvements in outcomes which show similar one-year graft survival for DD and LD for all age groups.
TABLE 45-5
Recurrence Rates and Graft Loss from Recurrent Disease in Children
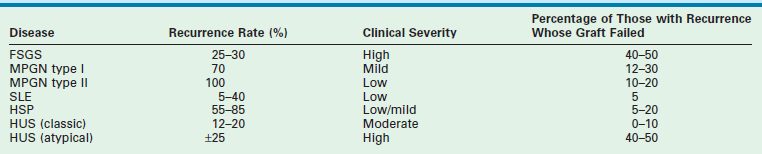
From Fine RN, Ettenger R. In: Morris PJ, editor. Kidney Transplantation: Principles and Practice. 4th ed. Philadelphia: WB Saunders; 1994. p. 418.
Pre-emptive Transplantation
The desire to perform preemptive renal transplantation before undertaking dialysis is often fueled by the patient’s or parents’ desire to avoid the dialysis procedures, potential infections, or cardiovascular complications, and the psychological impairment inherent with dialysis. A NAPRTCS review found 26% of primary transplantations were performed without prior dialysis.167 Most cases used LDs rather than DDs. There was no difference in patient or graft survival in this group when compared with patients who underwent dialysis before transplantation. Pre-emptive transplantation is not possible when uncontrolled hypertension, massive proteinuria, or recurrent infection require prior native kidney removal, or when oliguric renal failure requires immediate dialysis.
Transplant Procedure
Renal transplantation in infants and small children can be undertaken through a generous retroperitoneal approach or a transabdominal approach with placement of the allograft posterior to the right or left colon. An extraperitoneal approach to the retroperitoneum allows the possibility for peritoneal dialysis and should be used if possible. The arterial anastomosis is constructed end-to-side into the distal aorta or common iliac artery, and venous outflow of the allograft is via the inferior vena cava or common iliac vein. Ureteral implantation using the Lich extravesical ureteroneocystostomy avoids the need for a cystotomy and minimizes postoperative blood clots within the bladder, which can obstruct the urinary catheter. When larger donor kidneys are used in small recipients, the vessels must be shortened to avoid redundancy when the kidney is positioned in the retroperitoneum. The internal iliac artery is not used in children so that pelvic blood flow is preserved (Fig. 45-14). Ureteral ‘double-J’ stents are used when small ureter size may lead to obstruction.
Postoperative Management
Post-transplant management requires careful observation for technical complications, rejection, recurrence of the primary renal disease, and prevention of immunosuppression-related complications. Frequent fluid and electrolyte monitoring is necessary immediately following transplantation because larger kidneys can excrete the equivalent of the infant’s blood volume within a single hour. Careful attention to serum concentrations of calcium, phosphorus, magnesium, and electrolytes is also needed. Urine output is initially replaced isovolumetrically, then tapered as the high-output state decreases. Glucose-free replacement fluids minimize hyperglycemia and osmotic diuresis in the recipient. Selection of appropriate electrolyte concentrations is guided by urinary electrolyte excretion. Central venous filling pressures should be maintained at 7–10 cmH2O to ensure adequate intravascular volume. In patients with high-output renal failure, urine losses from both the native and transplant kidneys need to be replaced to avoid hypoperfusion and thrombosis. Maintenance of catheter patency is essential, and any episode of decreased urinary output should be rapidly investigated to exclude urinary catheter occlusion and bladder distention. An algorithm for the evaluation of early postoperative oliguria is shown in Figure 45-15.
Postoperative Complications
Vascular thrombosis still accounts for graft loss in up to 13% of initial transplants and 19% of repeat transplants in children.166 Graft thrombosis is significantly more frequent in children younger than 2 years of age and is directly related to the age of both the donor and the recipient. In addition, prolonged cold ischemic preservation time (>24 hours) and the related presence of ATN with delayed graft function also increase this risk. Prior transplantation and more than five pretransplant blood transfusions have also been shown to be independent risk factors.166 Immediate post-transplant Doppler ultrasound vascular imaging is helpful in confirming suitable allograft blood flow following abdominal closure, especially when large allografts are implanted into small recipients. Adequate hydration is important to maintain perfusion. Anticoagulation has not been used in most series.
Hypertension
Hypertension following renal transplantation is common. One month after transplantation, 72% of all patients require treatment, although this percentage decreases to 53% at 30 months.168 Careful attention to the pretransplant control of hypertension and dietary management improves the post-transplant management. Hypertension presents a significant risk to renal function when using small allografts. Hypertension in the early postoperative period is most often due to fluid overload or acute rejection, but it can also originate in the native kidneys.
Immunosuppression
Many immunosuppressive regimens are available and all share similar strategy. Most regimens include corticosteroids, cyclosporine or tacrolimus, and azathioprine or MMF. Polyclonal or monoclonal antilymphocyte antibodies are used when ATN is anticipated or for retransplantation in highly pre-sensitized patients. Significant efforts to decrease or discontinue steroids have been attempted to enhance growth and development. At four years after transplantation, 31% of LD and 23% of DD recipients were receiving alternate-day prednisone.152
Overall, there has been a decrease in the frequency of acute rejection with 12 month probabilities in LD of 32% and DD of 36%. The risk of rejection is similar for LD and DD recipients in the first few post-transplant weeks. Factors that increase the likelihood of rejection or long-term graft loss include receiving a graft from a DD rather than a related LD, receiving a graft from a donor younger than 5 years of age, having the graft in cold storage for more than 24 hours, being an African-American recipient, and delayed graft function from ATN.152 The ability to treat rejection has also improved with complete reversal of acute rejection in 65% of episodes. The rate of success in treating rejection declines with each successive rejection episode, increased recipient age, and late rejection episodes.152 Most rejection episodes can be treated with steroid administration alone (78%); monoclonal anti–T-cell agents such as orthoclone OKT-3 are needed in 32%. In patients who remain rejection free for the 1st post-transplant year, the risk of rejection in the following year is 20%.154
Outcome Following Transplantation
The overall results of renal transplantation in children are steadily improving. Overall one-year transplant graft survival rates of 88–100% have been reported for LD allografts, with results for DD allografts being 50 to 72%.151,163–166 In the a recent report, 1-, 2-, and 5-year graft survival rates were as follows: DD, 78%, 72%, and 59%; LD, 90%, 86%, and 85%, respectively (Fig. 45-16).154
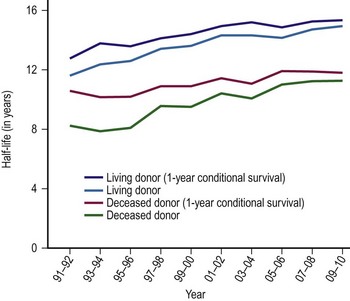
FIGURE 45-16 This graph demonstrates the allograft survival characteristics over 5 years in children as measured by half lives in years versus year transplanted comparing living donor and deceased donor renal transplants. One-year conditional survival is the half-life after the recipient survives the first year. This parameter eliminates patients with early graft failure from any cause. (Source: Scientific Registry Transplant Recipients—2011 Annual Report.)
Chronic rejection has become the most common cause of graft failure, accounting for 27% of all graft losses.157 With improved immunosuppressive treatments, acute rejection accounts for only 15% of failures. Recurrence of the original disease caused graft failure in 6%, and vascular thrombosis accounted for 12% of graft failures. Long-term graft survival following pediatric renal transplantation continues to deteriorate after 10 years despite low patient mortality rates. Death of the recipient with a functioning graft is an uncommon problem. In one study, when this did occur, death resulted primarily from infection (40%) or cardiovascular causes (21%).169 Young recipients (0 to 1 year of age) and patients with early graft failure were at the highest risk. Progressive loss of renal function may be secondary to complications of hypertension, hyperfiltration, hypercholesterolemias, chronic indolent immunologic damage leading to chronic rejection, and progressive primary renal disease, which all contribute to long-term graft loss.153,166
Pancreas Transplanation
Children have rarely been candidates for pancreas transplantation. In the past, the results following pancreas transplantation have not justified the risks associated with immunosuppression and operation. However, recent improvements in the operative technique and follow-up have improved. Overall, 1-year patient survival rate exceeds 90%, and graft survival with complete insulin independence exceeds 70% in patients in whom combined kidney and pancreas transplantation is undertaken. The survival rate is approximately 50% in isolated pancreas transplantation.170,171
The addition of pancreas transplantation with kidney replacement for diabetic nephropathy does not subject the patient to additional immunosuppressive risks, and it is better accepted. The use of isolated pancreas transplantation is reserved for patients who have extremely labile glucose control or experience hypoglycemic unawareness syndrome.170 As the results of this operation improve in the future, its role in children will need to be reviewed.
Pancreatic islet transplantation is also possible in children. However, its role in the treatment of juvenile diabetes in childhood is still limited. This procedure has been undertaken in children following pancreatic resection when the cellular autotransplant does not require immunosuppressive treatment, with excellent results.172 Further expansion of this approach awaits evidence that hypoglycemic correction retards the systemic complications of diabetes.
References
1. DeRusso, PA, Ye, W, Shepherd, R, et al. Growth failure and outcomes in infants with biliary atresia: A report from the Biliary Atresia Research Consortium. Hepatology. 2007; 46:1632–1638.
2. Kasai, M, Mochizuki, I, Ohkohchi, N, et al. Surgical limitation for biliary atresia: Indication for liver transplantation. J Pediatr Surg. 1989; 24:851–854.
3. Ryckman, F, Fisher, R, Pedersen, S, et al. Improved survival in biliary atresia patients in the present era of liver transplantation. J Pediatr Surg. 1993; 28:382–385.
4. Zitelli, BJ, Malatack, JJ, Gartner, JC, Jr., et al. Evaluation of the pediatric patient for liver transplantation. Pediatrics. 1986; 78:559–565.
5. Nio, M, Ohi, R, Hayashi, Y, et al. Current status of 21 patients who have survived more than 20 years since undergoing surgery for biliary atresia. J Pediatr Surg. 1996; 31:381–384.
6. Ryckman, FC. New issues concerning the etiology of an old problem. J Am Coll Surg. 1996; 183:637–639.
7. Reily, D. Familial Intrahepatic Cholestasis Syndromes. In: Suchy FJ, ed. Liver Disease in Children. 1st ed. St. Louis: CV Mosby; 1994:443–459.
8. Ng, VL, Ryckman, FC, Porta, G, et al. Long-term outcome after partial external biliary diversion for intractable pruritus in patients with intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2000; 30:152–156.
9. Cardona, J, Houssin, D, Gauthier, F, et al. Liver transplantation in children with Alagille syndrome—a study of twelve cases. Transplantation. 1995; 60:339–342.
10. Hoffenberg, EJ, Narkewicz, MR, Sondheimer, JM, et al. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr. 1995; 127:220–224.
11. Tzakis, AG, Reyes, J, Tepetes, K, et al. Liver transplantation for Alagille’s syndrome. Arch Surg. 1993; 128:337–339.
12. Ryckman FC, Alonso MH, eds. Transplantation for Hepatic Malignancy in Children, 1st ed, Philadelphia: WB Sanders, 1996.
13. Jan, D, Poggi, F, Laurent, J, et al. Liver transplantation: New indications in metabolic disorders? Transplant Proc. 1994; 26:189–190.
14. Mito, M, Kusano, M, Kawaura, Y. Hepatocyte transplantation in man. Transplant Proc. 1992; 24:3052–3053.
15. Jan, D, Laurent, J, Lacaille, F, et al. Liver transplantation in children with inherited metabolic disorders. Transplant Proc. 1995; 27:1706–1707.
16. Strom, S, Fisher, R. Hepatocyte transplantation: New possibilities for therapy. Gastroenterology. 2003; 124:568–571.
17. Strom, SC, Fisher, RA, Rubinstein, WS, et al. Transplantation of human hepatocytes. Transplant Proc. 1997; 29:2103–2106.
18. Fridell, JA, Bond, GJ, Mazariegos, GV, et al. Liver transplantation in children with cystic fibrosis: A long-term longitudinal review of a single center’s experience. J Pediatr Surg. 2003; 38:1152–1156.
19. Stapp, J, Wilkerson, S, Stewart, D, et al. Fulminant neonatal liver failure in siblings: Probable congenital hemophagocytic lymphohistiocytosis. Pediatr Dev Pathol. 2006; 9:239–244.
20. Strom, SC, Fisher, RA, Thompson, MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation. 1997; 63:559–569.
21. Lidofsky, SD, Bass, NM, Prager, MC, et al. Intracranial pressure monitoring and liver transplantation for fulminant hepatic failure. Hepatology. 1992; 16:1–7.
22. Reyes, JD, Carr, B, Dvorchik, I, et al. Liver transplantation and chemotherapy for hepatoblastoma and hepatocellular cancer in childhood and adolescence. J Pediatr. 2000; 136:795–804.
23. Tiao, GM, Bobey, N, Allen, S, et al. The current management of hepatoblastoma: A combination of chemotherapy, conventional resection, and liver transplantation. J Pediatr. 2005; 146:204–211.
24. Perilongo, G, Brown, J, Shafford, E, et al. Hepatoblastoma presenting with lung metastases: treatment results of the first cooperative, prospective study of the International Society of Paediatric Oncology on childhood liver tumors. Cancer. 2000; 89:1845–1853.
25. Pimpalwar, AP, Sharif, K, Ramani, P, et al. Strategy for hepatoblastoma management: Transplant versus nontransplant surgery. J Pediatr Surg. 2002; 37:240–245.
26. Austin, MT, Leys, CM, Feurer, ID, et al. Liver transplantation for childhood hepatic malignancy: A review of the United Network for Organ Sharing (UNOS) database. J Pediatr Surg. 2006; 41:182–186.
27. Iwatsuki, S, Gordon, RD, Shaw, BW, Jr., et al. Role of liver transplantation in cancer therapy. Ann Surg. 1985; 202:401–407.
28. Davies, DB, Harper, A. The OPTN waiting list, 1988–2003. Clin Transpl. 2004; 27–40.
29. Austin, MT, Feurer, ID, Pinson, CW. Access to pediatric liver transplantation: Does regional variation play a role? J Gastrointest Surg. 2006; 10:387–394.
30. Organ Procurement and Transplantation Network—HRSA. Final rule with comment period. Fed Regist. 1998; 63:16296–16338.
31. McDiarmid, SV, Anand, R, Lindblad, AS. Development of a pediatric end-stage liver disease score to predict poor outcome in children awaiting liver transplantation. Transplantation. 2002; 74:173–181.
32. Barshes, NR, Lee, TC, Udell, IW, et al. The pediatric end-stage liver disease (PELD) model as a predictor of survival benefit and posttransplant survival in pediatric liver transplant recipients. Liver Transpl. 2006; 12:475–480.
33. Tector, AJ, Mangus, RS, Chestovich, P, et al. Use of extended criteria livers decreases wait time for liver transplantation without adversely impacting posttransplant survival. Ann Surg. 2006; 244:439–450.
34. Schaubel, DE, Sima, CS, Goodrich, NP, et al. The survival benefit of deceased donor liver transplantation as a function of candidate disease severity and donor quality. Am J Transplant. 2008; 8:419–425.
35. Wolfe, RA, McCullough, KP, Leichtman, AB. Predictability of survival models for waiting list and transplant patients: Calculating LYFT. Am J Transplant. 2009; 9:1523–1527.
36. Broelsch, CE, Emond, JC, Whitington, PF, et al. Application of reduced-size liver transplants as split grafts, auxiliary orthotopic grafts, and living related segmental transplants. Ann Surg. 1990; 212:368–375.
37. Emond, JC, Whitington, PF, Thistlethwaite, JR, et al. Reduced-size orthotopic liver transplantation: Use in the management of children with chronic liver disease. Hepatology. 1989; 10:867–872.
38. Ryckman, FC, Flake, AW, Fisher, RA, et al. Segmental orthotopic hepatic transplantation as a means to improve patient survival and diminish waiting-list mortality. J Pediatr Surg. 1991; 26:422–427.
39. Emond, JC, Whitington, PF, Thistlethwaite, JR, et al. Transplantation of two patients with one liver. Analysis of a preliminary experience with ‘split-liver’ grafting. Ann Surg. 1990; 212:14–22.
40. Reyes, J. Adaptation of split liver grafts in pediatric patients. Pediatr Transplant. 2001; 5:148–152.
41. Deshpande, RR, Bowles, MJ, Vilca-Melendez, H, et al. Results of split liver transplantation in children. Ann Surg. 2002; 236:248–253.
42. Broelsch, CE, Frilling, A, Testa, G, et al. Living donor liver transplantation in adults. Eur J Gastroenterol Hepatol. 2003; 15:3–6.
43. Broelsch, CE, Burdelski, M, Rogiers, X, et al. Living donor for liver transplantation. Hepatology. 1994; 20:49S–55S.
44. Rogiers, X, Burdelski, M, Broelsch, CE. Liver transplantation from living donors. Br J Surg. 1994; 81:1251–1253.
45. Klein, AS, Messersmith, EE, Ratner, LE, et al. Organ donation and utilization in the United States, 1999–2008. Am J Transplant. 2010; 10:973–986.
46. Broelsch, CE, Whitington, PF, Emond, JC, et al. Liver transplantation in children from living related donors. Surgical techniques and results. Ann Surg. 1991; 214:428–437.
47. Otte, JB. Auxiliary partial orthotopic liver transplantation for acute liver failure in children. Pediatr Transplant. 1999; 3:252–256.
48. Otte, JB. Donor complications and outcomes in live-liver transplantation. Transplantation. 2003; 75:1625–1626.
49. Tissieres, P, Prontera, W, Chevret, L, et al. The pediatric risk of mortality score in infants and children with fulminant liver failure. Pediatr Transplant. 2003; 7:64–68.
50. Fan, ST, Lo, CM, Liu, CL. Technical refinement in adult-to-adult living donor liver transplantation using right lobe graft. Ann Surg. 2000; 231:126–131.
51. Humar, A, Beissel, J, Crotteau, S, et al. Whole liver versus split liver versus living donor in the adult recipient: An analysis of outcomes by graft type. Transplantation. 2008; 85:1420–1424.
52. Lo, CM, Fan, ST, Liu, CL, et al. Minimum graft size for successful living donor liver transplantation. Transplantation. 1999; 68:1112–1116.
53. Zamboni, F, Franchello, A, David, E, et al. Effect of macrovescicular steatosis and other donor and recipient characteristics on the outcome of liver transplantation. Clin Transplant. 2001; 15:53–57.
54. Imber, CJ, St Peter, SD, Handa, A, et al. Hepatic steatosis and its relationship to transplantation. Liver Transpl. 2002; 8:415–423.
55. Mor, E, Skerrett, D, Manzarbeitia, C, et al. Successful use of an enhanced immunosuppressive protocol with plasmapheresis for ABO-incompatible mismatched grafts in liver transplant recipients. Transplantation. 1995; 59:986–990.
56. Farges, O, Kalil, AN, Samuel, D, et al. The use of ABO-incompatible grafts in liver transplantation: a life-saving procedure in highly selected patients. Transplantation. 1995; 59:1124–1133.
57. Tanaka, A, Tanaka, K, Kitai, T, et al. Living related liver transplantation across ABO blood groups. Transplantation. 1994; 58:548–553.
58. Cacciarelli, TV, So, SK, Lim, J, et al. A reassessment of ABO incompatibility in pediatric liver transplantation. Transplantation. 1995; 60:757–760.
59. Yandza, T, Lambert, T, Alvarez, F, et al. Outcome of ABO-incompatible liver transplantation in children with no specific alloantibodies at the time of transplantation. Transplantation. 1994; 58:46–50.
60. Warnaar, N, Polak, WG, de Jong, KP, et al. Long-term results of urgent revascularization for hepatic artery thrombosis after pediatric liver transplantation. Liver Transpl. 2010; 16:847–855.
61. Kim, HB. Urgent revascularization for hepatic artery thrombosis: Maybe good for the few, definitely good for the many. Liver Transpl. 2010; 16:812–814.
62. Langnas, AN, Marujo, W, Stratta, RJ, et al. Hepatic allograft rescue following arterial thrombosis. Role of urgent revascularization. Transplantation. 1991; 51:86–90.
63. Stevens, LH, Emond, JC, Piper, JB, et al. Hepatic artery thrombosis in infants. A comparison of whole livers, reduced-size grafts, and grafts from living-related donors. Transplantation. 1992; 53:396–399.
64. Kirsch, JP, Howard, TK, Klintmalm, GB, et al. Problematic vascular reconstruction in liver transplantation. Part II. Portovenous conduits. Surgery. 1990; 107:544–548.
65. Stieber, AC, Zetti, G, Todo, S, et al. The spectrum of portal vein thrombosis in liver transplantation. Ann Surg. 1991; 213:199–206.
66. Harper, AM, Edwards, EB, Ellison, MD. The OPTN waiting list, 1988-2000. Clin Transpl. 2001; 73–85.
67. Peclet, MH, Ryckman, FC, Pedersen, SH, et al. The spectrum of bile duct complications in pediatric liver transplantation. J Pediatr Surg. 1994; 29:214–219.
68. Heffron, TG, Emond, JC, Whitington, PF, et al. Biliary complications in pediatric liver transplantation. A comparison of reduced-size and whole grafts. Transplantation. 1992; 53:391–395.
69. Rouch, DA, Emond, JC, Thistlethwaite, JR, Jr., et al. Choledochocholedochostomy without a T tube or internal stent in transplantation of the liver. Surg Gynecol Obstet. 1990; 170:239–244.
70. Snover, DC, Sibley, RK, Freese, DK, et al. Orthotopic liver transplantation: A pathological study of 63 serial liver biopsies from 17 patients with special reference to the diagnostic features and natural history of rejection. Hepatology. 1984; 4:1212–1222.
71. Mor, E, Solomon, H, Gibbs, JF, et al. Acute cellular rejection following liver transplantation: Clinical pathologic features and effect on outcome. Semin Liver Dis. 1992; 12:28–40.
72. Adams, DH, Neuberger, JM. Treatment of acute rejection. Semin Liver Dis. 1992; 12:80–88.
73. Jain, A, Mazariegos, G, Pokharna, R, et al. The absence of chronic rejection in pediatric primary liver transplant patients who are maintained on tacrolimus-based immunosuppression: A long-term analysis. Transplantation. 2003; 75:1020–1025.
74. Jain, A, Mazariegos, G, Kashyap, R, et al. Pediatric liver transplantation. A single center experience spanning 20 years. Transplantation. 2002; 73:941–947.
75. Gupta, P, Hart, J, Cronin, D, et al. Risk factors for chronic rejection after pediatric liver transplantation. Transplantation. 2001; 72:1098–1102.
76. Freese, DK, Snover, DC, Sharp, HL, et al. Chronic rejection after liver transplantation: A study of clinical, histopathological and immunological features. Hepatology. 1991; 13:882–891.
77. Ludwig, J, Wiesner, RH, Batts, KP, et al. The acute vanishing bile duct syndrome (acute irreversible rejection) after orthotopic liver transplantation. Hepatology. 1987; 7:476–483.
78. Demetris, A, Adams, D, Bellamy, C, et al. Update of the International Banff Schema for Liver Allograft Rejection: Working recommendations for the histopathologic staging and reporting of chronic rejection. An International Panel. Hepatology. 2000; 31:792–799.
79. O’Leary, JG, Kaneku, H, Susskind, BM, et al. High mean fluorescence intensity donor-specific anti-HLA antibodies associated with chronic rejection Postliver transplant. Am J Transplant. 2011; 11:1868–1876.
80. Gonwa, TA, Mai, ML, Melton, LB, et al. End-stage renal disease (ESRD) after orthotopic liver transplantation (OLTX) using calcineurin-based immunotherapy: Risk of development and treatment. Transplantation. 2001; 72:1934–1939.
81. Fisher, NC, Nightingale, PG, Gunson, BK, et al. Chronic renal failure following liver transplantation: A retrospective analysis. Transplantation. 1998; 66:59–66.
82. Campbell, K, Yazigi, N, Ryckman, F, et al. Renal Function in Long-Term Pediatric Liver Transplant Survivors. American Transplantation Congress. 2003.
83. Campbell, K, Ng, V, Martin, S, et al. Glomerular filtration rate following pediatric liver transplantation—the SPLIT experience. Am J Transplant. 2010; 10:2673–2682.
84. Campbell, KM, Yazigi, N, Ryckman, FC, et al. High prevalence of renal dysfunction in long-term survivors after pediatric liver transplantation. J Pediatr. 2006; 148:475–480.
85. Aw, MM, Samaroo, B, Baker, AJ, et al. Calcineurin-inhibitor related nephrotoxicity- reversibility in paediatric liver transplant recipients. Transplantation. 2001; 72:746–749.
86. Heffron, TG, Pillen, T, Smallwood, GA, et al. Pediatric liver transplantation with daclizumab induction. Transplantation. 2003; 75:2040–2043.
87. Wiesner, RH, Hermans, PE, Rakela, J, et al. Selective bowel decontamination to decrease gram-negative aerobic bacterial and Candida colonization and prevent infection after orthotopic liver transplantation. Transplantation. 1988; 45:570–574.
88. Andrews, W, Siegel, J, Renaro, T. Prevention and treatment of selected fungal and viral infections in pediatric liver transplant recipients. Clin Transplant. 1991; 5:204–207.
89. Patel, R, Snydman, DR, Rubin, RH, et al. Cytomegalovirus prophylaxis in solid organ transplant recipients. Transplantation. 1996; 61:1279–1289.
90. Fox, AS, Tolpin, MD, Baker, AL, et al. Seropositivity in liver transplant recipients as a predictor of cytomegalovirus disease. J Infect Dis. 1988; 157:383–385.
91. Holmes, RD, Sokol, RJ. Epstein-Barr virus and post-transplant lymphoproliferative disease. Pediatr Transplant. 2002; 6:456–464.
92. Broughton, S, McClay, JE, Murray, A, et al. The effectiveness of tonsillectomy in diagnosing lymphoproliferative disease in pediatric patients after liver transplantation. Arch Otolaryngol Head Neck Surg. 2000; 126:1444–1447.
93. Smets, F, Latinne, D, Bazin, H, et al. Ratio between Epstein-Barr viral load and anti-Epstein-Barr virus specific T-cell response as a predictive marker of posttransplant lymphoproliferative disease. Transplantation. 2002; 73:1603–1610.
94. Smets, F, Sokal, EM. Epstein-Barr virus-related lymphoproliferation in children after liver transplant: Role of immunity, diagnosis, and management. Pediatr Transplant. 2002; 6:280–287.
95. Sokal, EM, Antunes, H, Beguin, C, et al. Early signs and risk factors for the increased incidence of Epstein-Barr virus-related posttransplant lymphoproliferative diseases in pediatric liver transplant recipients treated with tacrolimus. Transplantation. 1997; 64:1438–1442.
96. Holmes, RD, Orban-Eller, K, Karrer, FR, et al. Response of elevated Epstein-Barr virus DNA levels to therapeutic changes in pediatric liver transplant patients: 56-month follow up and outcome. Transplantation. 2002; 74:367–372.
97. Axelrod, DA, Holmes, R, Thomas, SE, et al. Limitations of EBV-PCR monitoring to detect EBV associated post-transplant lymphoproliferative disorder. Pediatr Transplant. 2003; 7:223–227.
98. Penn, I. Post-transplant malignancy: The role of immunosuppression. Drug Saf. 2000; 23:101–113.
99. Sokal, EM, Caragiozoglou, T, Lamy, M, et al. Epstein-Barr virus serology and Epstein-Barr virus-associated lymphoproliferative disorders in pediatric liver transplant recipients. Transplantation. 1993; 56:1394–1398.
100. Jabs, WJ, Hennig, H, Kittel, M, et al. Normalized quantification by real-time PCR of Epstein-Barr virus load in patients at risk for posttransplant lymphoproliferative disorders. J Clin Microbiol. 2001; 39:564–569.
101. Guthery, SL, Heubi, JE, Bucuvalas, JC, et al. Determination of risk factors for Epstein-Barr virus-associated posttransplant lymphoproliferative disorder in pediatric liver transplant recipients using objective case ascertainment. Transplantation. 2003; 75:987–993.
102. Hanto, DW, Frizzera, G, Gajl-Peczalska, KJ, et al. Epstein-Barr virus, immunodeficiency, and B cell lymphoproliferation. Transplantation. 1985; 39:461–472.
103. Bueno, J, Ramil, C, Green, M. Current management strategies for the prevention and treatment of cytomegalovirus infection in pediatric transplant recipients. Paediatr Drugs. 2002; 4:279–290.
104. Serinet, MO, Jacquemin, E, Habes, D, et al. Anti-CD20 monoclonal antibody (Rituximab) treatment for Epstein-Barr virus-associated, B-cell lymphoproliferative disease in pediatric liver transplant recipients. J Pediatr Gastroenterol Nutr. 2002; 34:389–393.
105. Haque, T, Wilkie, GM, Taylor, C, et al. Treatment of Epstein-Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet. 2002; 360:436–442.
106. Tiao, GM, Alonso, M, Bezerra, J, et al. Liver transplantation in children younger than 1 year—the Cincinnati experience. J Pediatr Surg. 2005; 40:268–273.
107. Washburn, WK, Bradley, J, Cosimi, AB, et al. A regional experience with emergency liver transplantation. Transplantation. 1996; 61:235–239.
108. Langnas, AN, Marujo, WC, Inagaki, M, et al. The results of reduced-size liver transplantation, including split livers, in patients with end-stage liver disease. Transplantation. 1992; 53:387–391.
109. Esquivel, CO, Nakazato, P, Cox, K, et al. The impact of liver reductions in pediatric liver transplantation. Arch Surg. 1991; 126:1278–1285.
110. Studies of Pediatric Liver Transplantation (SPLIT). Year 2000 outcomes. Transplantation. 2001; 72:463–476.
111. Fridell, JA, Jain, A, Reyes, J, et al. Causes of mortality beyond one year after primary pediatric liver transplant under tacrolimus. Transplantation. 2002; 74:1721–1724.
112. Mack, CL, Ferrario, M, Abecassis, M, et al. Living donor liver transplantation for children with liver failure and concurrent multiple organ system failure. Liver Transpl. 2001; 7:890–895.
113. Emre, S. Living-donor liver transplantation in children. Pediatr Transplant. 2002; 6:43–46.
114. Bucuvalas, JC, Ryckman, FC. The long- and short-term outcome of living-donor liver transplantation. J Pediatr. 1999; 134:259–261.
115. Bilik, R, Greig, P, Langer, B, et al. Survival after reduced-size liver transplantation is dependent on pretransplant status. J Pediatr Surg. 1993; 28:1307–1311.
116. PRCarroll, CL, Goodman, DM, Superina, RA, et al. Timed Pediatric Risk of Mortality Scores predict outcomes in pediatric liver transplant recipients. Pediatr Transplant. 2003; 7:289–295.
117. Otte, JB, de Ville de Goyet, J, Sokal, E, et al. Size reduction of the donor liver is a safe way to alleviate the shortage of size-matched organs in pediatric liver transplantation. Ann Surg. 1990; 211:146–157.
118. Ryckman, FC, Alonso, MH, Bucuvalas, JC, et al. Long-term survival after liver transplantation. J Pediatr Surg. 1999; 34:845–849.
119. Wallot, MA, Mathot, M, Janssen, M, et al. Long-term survival and late graft loss in pediatric liver transplant recipients—a 15-year single-center experience. Liver Transpl. 2002; 8:615–622.
120. Sudan, DL, Shaw, BW, Jr., Langnas, AN. Causes of late mortality in pediatric liver transplant recipients. Ann Surg. 1998; 227:289–295.
121. Sarna, S, Sipila, I, Jalanko, H, et al. Factors affecting growth after pediatric liver transplantation. Transplant Proc. 1994; 26:161–164.
122. Sarna, S, Sipila, I, Vihervuori, E, et al. Growth delay after liver transplantation in childhood: Studies of underlying mechanisms. Pediatr Res. 1995; 38:366–372.
123. Balistreri, WF, Bucuvalas, JC, Ryckman, FC. The effect of immunosuppression on growth and development. Liver Transpl Surg. 1995; 1:64–73.
124. Chin, SE, Shepherd, RW, Cleghorn, GJ, et al. Survival, growth and quality of life in children after orthotopic liver transplantation: A 5 year experience. J Paediatr Child Health. 1991; 27:380–385.
125. Zitelli, BJ, Miller, JW, Gartner, JC, Jr., et al. Changes in life-style after liver transplantation. Pediatrics. 1988; 82:173–180.
126. Stewart, SM, Hiltebeitel, C, Nici, J, et al. Neuropsychological outcome of pediatric liver transplantation. Pediatrics. 1991; 87:367–376.
127. Stewart, SM, Uauy, R, Waller, DA, et al. Mental and motor development, social competence, and growth one year after successful pediatric liver transplantation. J Pediatr. 1989; 114:574–581.
128. Tarter, RE, Hays, AL, Sandford, SS, et al. Cerebral morphological abnormalities associated with non-alcoholic cirrhosis. Lancet. 1986; 2:893–895.
129. Bernthal, P, Hays, A, Tarter, RE, et al. Cerebral CT scan abnormalities in cholestatic and hepatocellular disease and their relationship to neuropsychologic test performance. Hepatology. 1987; 7:107–114.
130. Tarter, RE, Sandford, SL, Hays, AL, et al. Hepatic injury correlates with neuropsychologic impairment. Int J Neurosci. 1989; 44:75–82.
131. Bucuvalas, JC, Britto, M, Krug, S, et al. Health-related quality of life in pediatric liver transplant recipients: A single-center study. Liver Transpl. 2003; 9:62–71.
132. Mazariegos, GV, Steffick, DE, Horslen, S, et al. Intestine transplantation in the United States, 1999–2008. Am J Transplant. 2010; 10:1020–1034.
133. Mazariegos, GV, Squires, RH, Sindhi, RK. Current perspectives on pediatric intestinal transplantation. Curr Gastroenterol Rep. 2009; 11:226–233.
134. Nayyar, N, Mazariegos, G, Ranganathan, S, et al. Pediatric small bowel transplantation. Semin Pediatr Surg. 2010; 19:68–77.
135. (HCFA) HCFA. ‘Combined Liver and Intestinal and Multi-visceral Transplantation (#CAG-00036) Decision Memorandum’. 2000.
136. Youssef, NN, Mezoff, AG, Carter, BA, et al. Medical update and potential advances in the treatment of pediatric intestinal failure. Curr Gastroenterol Rep. 2012; 14:243–252.
137. Ching, YA, Gura, K, Modi, B, et al. Pediatric intestinal failure: nutrition, pharmacologic, and surgical approaches. Nutr Clin Pract. 2007; 22:653–663.
138. Touloukian, RJ, Smith, GJ. Normal intestinal length in preterm infants. J Pediatr Surg. 1983; 18:720–723.
139. Bueno, J, Ohwada, S, Kocoshis, S, et al. Factors impacting the survival of children with intestinal failure referred for intestinal transplantation. J Pediatr Surg. 1999; 34:27–32.
140. Fishbein, TM, Matsumoto, CS. Intestinal replacement therapy: Timing and indications for referral of patients to an intestinal rehabilitation and transplant program. Gastroenterol. 2006; 130:S147–S151.
141. Avitzur, Y, Grant, D. Intestine transplantation in children: Update 2010. Pediatr Cl N Am. 2010; 57:415–431.
142. de Ville de Goyet, J, Mitchell, A, Mayer, AD, et al. En block combined reduced-liver and small bowel transplants: From large donors to small children. Transplantation. 2000; 69:555–559.
143. Fryer, J, Pellar, S, Ormond, D, et al. Mortality in candidates waiting for combined liver-intestine transplants exceeds that for other candidates waiting for liver transplants. Liver Transpl. 2003; 9:748–753.
144. Furukawa, H, Manez, R, Kusne, S, et al. Cytomegalovirus disease in intestinal transplantation. Transpl P. 1995; 27:1357–1358.
145. Eid, AJ, Razonable, RR. New developments in the management of cytomegalovirus infection after solid organ transplantation. Drugs. 2010; 70:965–981.
146. Kato, T, Selvaggi, G, Gaynor, JJ, et al. Inclusion of donor colon and ileocecal valve in intestinal transplantation. Transplantation. 2008; 86:293–297.
147. Berney, T, Kato, T, Nishida, S, et al. Portal versus systemic drainage of small bowel allografts: Comparative assessment of survival, function, rejection, and bacterial translocation. J Am Coll Surg. 2002; 195:804–813.
148. Ruiz, P, Takahashi, H, Delacruz, V, et al. International grading scheme for acute cellular rejection in small-bowel transplantation: single-center experience. Transpl P. 2010; 42:47–53.
149. Shin, CR, Nathan, J, Alonso, M, et al. Incidence of acute and chronic graft-versus-host disease and donor T-cell chimerism after small bowel or combined organ transplantation. J Pediatr Surg. 2011; 46:1732–1738.
150. Potter, DE, Holliday, MA, Piel, CF, et al. Treatment of end-stage renal disease in children: A 15-year experience. Kidney Int. 1980; 18:103–109.
151. Fine, R. Renal Transplantation in Children. In: Morris P, ed. Kidney Transplantation: Principals and Practice. Orlando: Grune & Stratton; 1984:509–546.
152. McEnery, PT, Stablein, DM, Arbus, G, Tejani, A. Renal transplantation in children. A report of the North American Pediatric Renal Transplant Cooperative Study. N Engl J Med. 1992; 326:1727–1732.
153. North American Pediatric Renal Transplant Cooperative Study. 2010 Annual Report.
154. Warady, BA, Hebert, D, Sullivan, EK, et al. Renal transplantation, chronic dialysis, and chronic renal insufficiency in children and adolescents. The 1995 Annual Report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol. 1997; 11:49–64.
155. Hanna, JD, Krieg, RJ, Jr., Scheinman, JI, Chan, JC. Effects of uremia on growth in children. Semin Nephrol. 1996; 16:230–241.
156. Benfield, MR. Current status of kidney transplant: Update 2003. Pediatr Clin N Am. 2003; 50:1301–1334.
157. Benfield, MR, McDonald, RA, Bartosh, S, et al. Changing trends in pediatric transplantation: 2001 Annual Report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Transplant. 2003; 7:321–335.
158. Englund, M, Berg, U, Tyden, G. A longitudinal study of children who received renal transplants 10–20 years ago. Transplantation. 2003; 76:311–318.
159. Fine, RN. Growth following solid-organ transplantation. Pediatr Transplant. 2002; 6:47–52.
160. Grushkin, CM, Fine, RN. Growth in children following renal transplantation. Am J Dis Child. 1973; 125:514–556.
161. Rotundo, A, Nevins, TE, Lipton, M, et al. Progressive encephalopathy in children with chronic renal insufficiency in infancy. Kidney Int. 1982; 21:486–491.
162. Najarian, J, Ascher, NL, Mauer, SM. Kidney Transplantation. In: Welch K, Randolph J, Ravitch M, eds. Pediatric Surgery. Chicago: Year Book Medical; 1986:360–373.
163. Turcotte, J, Campbell, DA, Dafoe, D. Pediatric Renal Transplantation. In: Cerilli G, ed. Organ Transplantation and Replacement. Philadelphia: JB Lippincott; 1988:349–360.
164. Ildstad, ST, Tollerud, DJ, Noseworthy, J, et al. The influence of donor age on graft survival in renal transplantation. J Pediatr Surg. 1990; 25:134–139.
165. Ildstad, ST, Tollerud, DJ, Noseworthy, J, et al. Renal transplantation in pediatric recipients. Transpl P. 1989; 21:1936–1937.
166. Singh, A, Stablein, D, Tejani, A. Risk factors for vascular thrombosis in pediatric renal transplantation: A special report of the North American Pediatric Renal Transplant Cooperative Study. Transplantation. 1997; 63:1263–1267.
167. Bereket, G, Fine, RN. Pediatric renal transplantation. Pediatr Clin N Am. 1995; 42:1603–1628.
168. Mahmoud, A, Said, MH, Dawahra, M, et al. Outcome of preemptive renal transplantation and pretransplantation dialysis in children. Pediatr Nephrol. 1997; 11:537–541.
169. Tejani, A, Sullivan, EK, Alexander, S, et al. Posttransplant deaths and factors that influence the mortality rate in North American children. Transplantation. 1994; 57:547–553.
170. Sutherland, DE. The case for pancreas transplantation. Diabetes Metab. 1996; 22:132–138.
171. Stratta, RJ, Larsen, JL, Cushing, K. Pancreas transplantation for diabetes mellitus. Annu Rev Med. 1995; 46:281–298.
172. Bellin, MD, Sutherland, DE. Pediatric islet autotransplantation: Indication, technique, and outcome. Curr Diabetes Rep. 2010; 10:326–331.