Chapter 63 Soft Tissue Sarcoma
Soft tissue sarcomas (STS) are rare malignancies derived from mesenchymal tissue and comprise approximately 1% of adult cancers in the United States, resulting in almost 10,500 new cases annually; approximately 5700 occur in males and 4800 affect females.1,2 No obvious association with ethnicity seems evident. The disease is characterized by heterogeneity in anatomic site, histologic tumor subtype, and risk of metastatic progression. All age groups are affected, with pediatric sarcomas frequently occurring during infancy whereas the incidence of adult-type sarcomas increases in the final decades of life3; the median age at onset of STS is generally from 50 to 55 years. Just over half originate in the limbs (extremities), whereas approximately 40% arise in the retroperitoneum/visceral tissues and torso (truncal), with the remainder affecting the head and neck (10%).4 Because of different conventions in attribution of certain sarcomas to the abdomen and thorax there has been a recent increase in incidence in these sites.
Etiology and Epidemiology
Environmental Etiologic Factors
A perplexing issue about STS is that in most cases no clearly defined etiologic factor is evident. Notwithstanding this, several factors have been identified that either predispose or are associated with the development of STS. Potential risk factors need to be further assessed in STS such as herbicide exposure and constitutional and hormonal factors during childhood, puberty, and adulthood. STS were identified in a group of non–AIDS-defining neoplasms linked to various concurrent viral infections in the human immunodeficiency virus (HIV)-infected host, including children.5,6 No clear demographic features have been identified in normal populations, although one Italian study questioned whether body weight contributed to increased risk.7
The factor with perhaps the greatest notoriety is radiation resulting from accidental, therapeutic, or diagnostic exposure. Radiation-associated STS have demonstrated inferior survival compared with sporadic STS, even if one adjusts for known prognostic factors such as histologic type, size, age, margin status, and site.8 The secondary risk of cancer has been enshrined in the collective oncology consciousness from as long ago as 1922.9 However, certain conditions in themselves are associated with intrinsic host predisposition to radiation-induced neoplasia, most notably retinoblastoma. Although rare in the cancer incidence statistics, children are an obvious concern because of the available latent period that can span many decades.
Although the risk associations with RT are well appreciated, the molecular mechanisms and real antecedent cause of these lesions are poorly understood. Germline mutations in tumor suppressor genes and/or accumulated damage to DNA repair genes may lead to neoplasia, but it remains unknown if most individuals have a genetic defect that underpinned the development of the first cancer, for which RT was administered, and, in turn, led to the induction of the RT-induced cancer.10 If this is the case, there may be subgroups of individuals at considerably greater risk, whereas others have relatively low risk.10 One hypothesis is that incomplete damage in normal tissues results in mutagenic responses and disorganized reparative proliferation that can eventually trigger tumor induction.11 This may provide a basis for the observation that RT-induced sarcomas commonly originate on the perimeter of the previous RT target volume and not necessarily in the high-dose region.
RT use in the primary management of breast cancer remains a problem from the standpoint of sarcoma induction. Karlsson and colleagues12 studied 122,991 women with breast cancer in Sweden from 1958 to 1992 and found 116 STS (40 angiosarcomas and 76 other lesions). Mery and associates13 reported the risk factor for development of STS for 563,155 women with breast cancer who presented from 1973 to 2003. A total of 948 patients developed STS; patients who received RT had a higher incidence of all STS (1.5-fold), in particular angiosarcoma (9-fold) and malignant fibrous histiocytoma. The incidence peaked at 10 years and remained elevated up to 20 years after RT. These authors also found that patients who underwent partial mastectomies had a 7-fold higher risk of developing secondary angiosarcoma when compared with those who underwent total mastectomies, after adjusting for RT and lymph node dissection. Having breast cancer also may incur additional risk because lymphedema after the treatment of breast cancer is independently associated with the development of lymphangiosarcoma.14 In this setting, the lymphangiosarcoma generally develops beyond the immediate irradiated volume in addition to the area that was targeted.15
Trauma is a controversial factor. Often, a minor episode of injury may draw attention to a preexisting mass or awkward malignant masses may be more prone to trauma and consequent injury. Pukkala and collegues16 studied cancer incidences in world class athletes in Finland. Smoking-related cancers were less frequent, whereas the incidence of other cancers was not reduced. The authors speculated whether injuries during active sport may predispose to sarcoma, and, in turn, there has been concern that operative trauma, including arthroplasty, may increase the risk of STS. However, Scandinavian studies17,18 on more that 100,000 patients who had undergone total hip or knee arthroplasty showed no increased risk of sarcoma and there was no sarcoma case presenting at the site of operation. Desmoid tumors are commonly seen in the anterior abdominal wall after pregnancy, although the biologic and traumatic basis for this is unclear.19
Several chemical carcinogens, including thorotrast, vinyl chloride, and arsenic, are established in the causation of hepatic angiosarcomas. Workers exposed to phenoxyherbicides, chlorophenols, and dioxins had higher mortality rates from cancer compared with controls, and the increase was highest for STS.20–22 There have been conflicting reports about occupational exposure to phenoxyacetic acids detectable in chlorophenols (frequent in wood preservatives) and certain herbicides.23–26
Molecular Pathogenetic Mechanisms
Contemporary appreciation of the molecular pathogenesis and behavior of STS suggests that these lesions are divisible into two major genetic groups: (1) those possessing specific genetic alterations and usually simple karyotypes, such as reciprocal chromosomal translocations (e.g., SS18-SSX1 or SS18-SSX2 in synovial sarcoma); and (2) sarcomas with nonspecific genetic alterations and complex unbalanced karyotypes.27 However, these concepts are considerably more complex and are evolving rapidly,28 and only a brief discussion is presented here.
Simple Karyotypic Sarcomas with Specific Genetic Alterations
The first group, those characterized by specific recurrent chromosomal translocations, comprises approximately one third of all sarcomas, and the resulting product of the specific gene fusions usually encode aberrant chimeric transcription factors. These translocations are often the only cytogenetic abnormalities and appear to be pathogenetically important. However, although successful cloning of most genes involved in recurrent translocations has taken place, there remains a paucity of information about the downstream targets mediating oncogenic transformation. In general, the promiscuous pairing of a proximal gene that contributes a promoter and functional domain, with a distal gene possessing a DNA binding domain that confers target specificity, determines the phenotype of various STS.27 Most translocations result in the production of a tumor-specific RNA that encodes a novel transcription factor that plays a role in oncogenesis in mesenchymal cells.
One uncommon mechanism results in a chimeric autocrine growth factor and is evident in dermatofibrosarcoma protuberans (DFSP) in which the t(17;22)(q22;q13) translocation involving the COL1A1-PDGFB genes provides a therapeutic target for response to the tyrosine kinase inhibitor imatinib. Mechanisms such as this represent opportunities for future management. At this time, however, they are typically used in a diagnostic manner to identify a particular subtype of tumor. This is the case for synovial sarcomas, nearly all of which exhibit the t(X;18)(p11;q11) translocation and can be distinguished from other tumors with similar morphology, such as a malignant peripheral nerve sheath tumor. Another example is the fusion product PAX3-FOXO1 arising from the t(2;13)(q35;q14) translocation as well as the t(1;13)(p36;q14) translocation expressed in alveolar rhabdomyosarcoma27 (Table 63-1).
TABLE 63-1 Cytogenetic Abnormalities and Fusion Genes in Soft Tissue Sarcoma
| Histologic Subtype | Usual Translocations | Genes Involved |
|---|---|---|
| Alveolar sarcoma of soft parts | t(X;17)(p11;q25) | TFE3-ASPL |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11) | FUS-ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWS-ATF1 |
| Congenital fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1-PDGFB |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWS-WT1 |
| Endometrial stromal sarcoma | t(7;17)(p15;q21) | JAZF1-JJAZ1 |
| Ewing’s/peripheral primitive neuroectodermal tumor | t(11;22)(q24;q12) | EWS-FLI1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) | EWS-NR4A3 |
| Fibromyxoid sarcoma, low grade | t(7;16)(q33;p11) | FUS-CREB3L2 |
| Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) | TPM3-ALK |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-DDIT3 |
| Rhabdomyosarcoma (alveolar) | t(2;13)(q35;q14)t(1;13)(p36,q14) | PAX3(or7)-FOXO1 |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1 or SSX2 |
Note: Gene nomenclature evolves at least as rapidly as pathologic terminology.
Modified from Antonescu CR: The role of genetic testing in soft tissue sarcoma, Histopathology 48:13-21, 2006.
Complex Karyotypic Sarcomas without Specific Genetic Alterations
The remaining two thirds of STS lack a recurrent genetic signature and are characterized by numerous aberrations, including chromosomal losses and gains. Most adult spindle cell tumors, leiomyosarcoma, and pleomorphic sarcomas belong to this group. At the molecular level, this sarcoma subset features a high prevalence of TP53 checkpoint alterations, including TP53 inactivating mutations, homozygous deletion of CDKN2A, MDM2 amplifications, and so on. Unfortunately, most cases with complex karyotypes and multiple aberrations do not have a consistent correlation with pathologic or clinical parameters. There also appears to be no indication in most of these lesions that detectable genetic alterations might be useful either for diagnosis or prognosis.29
Genetic Conditions Associated with Sarcomas
Given the genetic information just presented, it is not surprising that STS are associated with several genetic conditions. Patients with neurofibromatosis have a 5% to 10% lifetime risk of developing a malignant neurofibrosarcoma arising in a neurofibroma,30–33 most commonly in the central nervous system. These patients frequently present the additional diagnostic dilemma of distinguishing between potential metastases or other benign neurofibromas found on staging investigations.
A significant risk for STS is found in individuals with somatic mutations in the TP53 tumor suppressor gene, including Li-Fraumeni syndrome, which is characterized by a familial cluster of sarcoma (12%), breast cancer (25%), leukemia, adrenal carcinoma, brain tumors (12%), lung cancer, skin cancer, and pancreatic cancer.34,35
Long-term survivors of retinoblastoma with the associated RB gene abnormality often develop tumors later in life, with a risk of 10% to 15%.36,37 For hereditary retinoblastoma, the cumulative incidence of a second cancer at 50 years after diagnosis is 50% compared with only 5% for nonhereditary retinoblastoma.38 Wong and associates38 showed that the relative risk of STS increases with dose beginning at 5 Gy, rising to 10.7-fold at dose levels exceeding 60 Gy.
Gardner’s syndrome, a subset of familial adenomatous polyposis, is associated with the development of intra-abdominal desmoids.39
Biologic Characteristics and Molecular Biology
As already mentioned, sarcomas have extensive cytogenic abnormalities that may aid in their differential diagnosis and may ultimately provide therapeutic targets. Unfortunately, molecular prognostication and prediction of treatment response are proving more elusive than was originally anticipated. For example, as previously mentioned, the presence of the translocation t(X;18)(p11;q11) has been used to confirm the diagnosis of synovial cell sarcoma for patients with poorly differentiated lesions that cannot be otherwise characterized. Irrespective of their histologic appearance, almost all synovial sarcomas contain the t(X;18)(SS18-SSX) translocation involving the more prevalent SSX1 (associated with monophasic histology)40 or SSX2 (associated with biphasic histology) gene, two closely related genes from chromosome Xp11, and the SS18 gene from chromosome 18q11. The result is the formation of a chimeric gene thought to function by encoding a transcription-activating protein. It was suggested that the transcripts of these fusion genes had specific prognostic significance,41–43 but results have been contradictory.44
Tissue hypoxia appears associated with the development of distant metastases independently from depth, size, and grade.45 Expression of osteopontin (OPN), a phosphorylated glycoprotein, appears to be induced by tumor hypoxia.46,47 Elevated serum or plasma levels of OPN have been correlated with tumor progression and metastasis in a variety of cancers.48,49 One study based on tumor and serum samples from 93 adult patients with STS found that an elevated OPN protein level in serum and tumor is a significant negative prognostic factor. Elevated OPN levels were associated with higher stage, higher grade, subtype, larger tumor size, and an increased rate of relapse.50 In addition, elevated OPN levels have been associated with decreased overall survival (OS), suggesting its potential use as a prognostic marker.51 Presumably one mechanism could be represented by hypoxia-induced molecular aberrations linked to expression of OPN.
Pathology and Pathways of Spread
Pathology
Classic Pathology Classification
While originating from the mesoderm and in some lesions from ectoderm, a beguiling feature of the legacy of the nomenclature of sarcoma pathology is the traditional view of histologic subtypes originating from similar-appearing normal tissue counterparts. The most common classification scheme for STS is still based on histogenesis, as described by the World Health Organization (WHO) and others52,53,54 (Table 63-2). However, as the degree of histologic differentiation becomes less apparent, the determination of a putative cellular origin becomes increasingly difficult.
TABLE 63-2 Histologic Classification of Soft Tissue Sarcoma*
| Fibrous Tumors |
* Benign lesions other than fibromatosis and atypical lipoma are not included in the present tabulation.
Adapted from references 52, 53, and 54.
This view of pathogenesis and histogenesis is now being contested. Although some benign lesions (especially lipomas) may resemble mature differentiated normal tissue, many STS arise in tissues that do not typically possess the putative normal tissues mentioned. For example, synovial sarcomas are only rarely found in synovial tissues. Because the necessary genetic information is contained in all diploid cells, a more plausible explanation may be that a given set of genes may program differentiation in any mesenchymal cell, thereby giving origin to almost any mesenchymal neoplasm.52,53,54 It seems unlikely that there is a single precursor primitive mesenchymal stem cell for each sarcoma.
Classification by Functional Genomics
With advances in molecular characterization of STS, and the emerging dissatisfaction with traditional methods of classification, it is not unexpected that recent proposals have explored gene expression profiling for these tumors (Fig. 63-1) to attempt to identify a genome-based classification scheme.55,56,57,58,59,60 For example, Segal and colleagues59 examined RNA sarcoma samples and found that synovial sarcomas, round-cell/myxoid liposarcomas, clear cell sarcomas, and gastrointestinal stromal tumors (GIST) displayed distinct and homogeneous gene expression profiles. In a separate study, Nielsen and colleagues58 observed that KIT appeared to be one of the prominent discriminator genes for malignant fibrous histiocytoma. More recently, Nakayama and co-workers57 have shown that many neoplasms initially (mis)classified as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma based on their morphology and immunoprofile could be reclassified as myxofibrosarcoma based on gene expression, suggesting that gene profiling will be a useful tool to aid histologic diagnosis of malignant fibrous histiocytoma. Two genes, GPR64 and TNXB, were particularly expressed by myxofibrosarcomas but not by undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma, thus allowing distinction between these two histotypes.
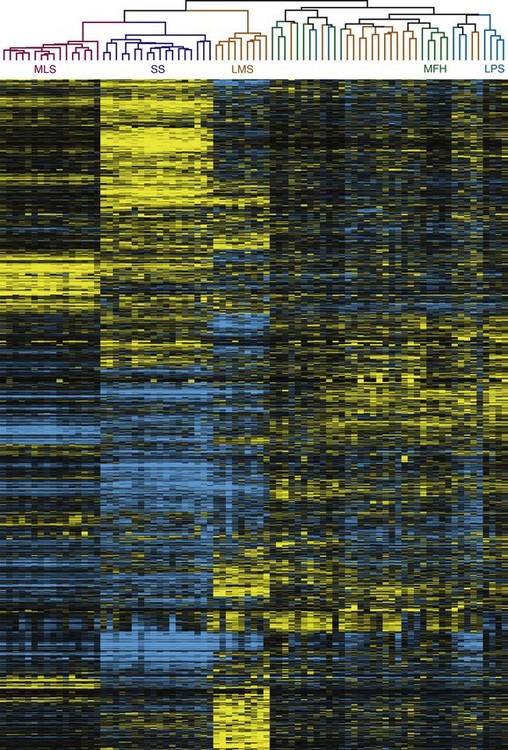
New molecular technologies such as comparative genomic hybridization array–based techniques and gene expression analyses show promise for the identification of genes, gene products, and signaling pathways involved in the pathogenesis, progression, and metastatic dissemination of sarcomas that may provide new strategies for the enhancement of local/systemic control and the reduction of the toxic effects of sarcoma management.56 In particular, nanotechnology offers countless opportunities for novel diagnostic and therapeutic approaches to selectively target tumors at the cellular level.60
Grading of Sarcoma
Several grading schemes have been proposed and validated as efficacious and are discussed in detail elsewhere.61 The three-tier system proposed by the French Federation of Cancer Centres is precisely defined, easy to use, and the most widely employed. The French system relies on a relatively balanced evaluation of parameters (differentiation score, mitoses, necrosis), but its greatest limitation lies in the assignment of a differentiation score. Roughly defined as the extent to which a lesion resembles normal tissue, differentiation score has little applicability for tumors that ostensibly have no normal tissue counterpart. It is also recognized that no system performs perfectly in all sarcomas. In one review, Deyrup and Weiss61 highlighted problems, including the fact that some STS do not lend themselves well to grading. These include (1) those in which grade provides no additional information (e.g., well-differentiated liposarcoma/atypical lipomatous neoplasm, Ewing’s sarcoma); (2) “ungradable” histologic subtypes (e.g., epithelioid sarcoma, clear cell sarcoma, angiosarcoma); and (3) sarcomas that have traditionally been graded but where this characteristic does not appear to differentiate these lesions prognostically (e.g., malignant peripheral nerve sheath tumor).
In addition to the French system, there is a U.S. National Cancer Institute (NCI) system.62 Both use degrees of necrosis and mitotic rate, but the American system considers histologic type or subtype, location, cellularity, and nuclear pleomorphism. The situation is further complicated by the potential use of different tiers of grade within existing classification systems (e.g., two-, three- and four-grade classification systems). The TNM stage classification of the American Joint Committee on Cancer (AJCC) now employs a three-tiered grading system based largely on the wish to incorporate the French system.63
Pathologic Features and Natural History
Selected Pathologic Subtypes
Malignant Fibrous Histiocytoma
Since the late 1970s malignant fibrous histiocytoma has borne the mantle of most common STS of middle and late adulthood and therefore warrants specific mention in the chapter. Originally described in the 1960s, the term was used to describe a group of sarcomas deemed to be derived from a mixed histiocytic and fibroblastic lineage. The existence of ‘‘malignant fibrous histiocytoma’’ as a distinct entity is now considered controversial and at best is regarded as a heterogeneous group of tumors without a specific known line of differentiation. Most of these tumors were considered to be either a variable storiform and/or undifferentiated pleomorphic phenotype or one of four rarer additional types: myxoid (the next most common subtype), giant cell, inflammatory, and angiomatoid (a rare variant and the only one to survive contemporary reclassification). Reclassification of many tumors in this group has afforded better prognostication, and the old term “malignant fibrous histiocytoma” now is generally considered obsolete.54
Current nomenclature recognizes the entities of undifferentiated high-grade pleomorphic sarcoma (previously storiform-pleomorphic “malignant fibrous histiocytoma”) and myxofibrosarcoma (formerly myxoid “malignant fibrous histiocytoma”), which is now the most common sarcoma of older adults, peaking in incidence in the seventh and eighth decades and presenting most frequently in the lower extremity. Most of these tumors are deep seated, developing in fascia and skeletal muscle, and have a high potential for local recurrence.54,56 Low-grade myxofibrosarcoma is known to have a particularly infiltrative growth pattern, and imaging evaluation with particular attention to tail-like extensions should be considered for management.64 This type must be differentiated from other sarcomas containing myxoid areas, such as pleomorphic liposarcoma. The feature that sets malignant fibrous histiocytoma lesions apart is the absence of a defined line of differentiation from which epithelial, melanotic, and lymphoid differentiations have been excluded.52
The natural history is extraordinarily variable, reflecting the heterogeneity of these lesions. The most common site for metastases from pleomorphic sarcomas is the lung (90%), bone (8%), and liver (1%), with rare regional lymph node metastases.54
Angiosarcoma
Angiosarcoma is currently included in the seventh edition of the TNM stage classification.63 Particularly unique is the propensity of superficial angiosarcoma to occur in the dermal tissues of the head and neck manifesting typically on the scalp (about 50%) or facial skin. These neoplasms commonly present as purple, bruiselike lesions in elderly white men and are rarely seen in patients of African origin.65 The macules usually become nodular, may coalesce, and may ulcerate. Frank bleeding is an ongoing and major problem and often results in chronic oozing through dressings. A particularly difficult aspect of management is the apparent multifocal nature that makes judgment about the area of risk almost impossible from the standpoint of accurate definition of margins for both surgery or RT. The clinical examination must be meticulous because it is the only real means of identifying the deceptive areas of multifocal involvement that may exist.66 Frequently, individual patches coalesce into flat masses of substantial size. Involvement of the eyelid and periorbital tissues is particularly troublesome. Metastasis is common if patients survive sufficiently long and occurs most typically in regional lymph nodes and the lung.
The majority of angiosarcomas are considered to originate from the endothelium of the blood vasculature.67 Koch and colleagues68 have outlined the pathologic features, which can range from well to poorly differentiated lesions. They are typically composed of an accumulation of an irregular or sinusoidal pattern of vessel with vascular spaces lined by a single row of atypical endothelium that may be several layers thick. Highly cellular lesions can manifest as sheets of cells with vascular space obliteration that contribute solid components as vascular channels effaced by the crowding of cells. High-grade, poorly differentiated lesions can be composed of undifferentiated cells and disordered architecture, making them difficult to discern from other histologies, although the hallmark clinical presentation and behavior generally leave little doubt about the diagnosis. On immunohistochemistry, these tumors are usually positive for factor VIII–related antigen, vimentin, CD34, and CD31.
It is suggested that angiogenesis and vascular permeability play a central role in the development of angiosarcomas. This is likely influenced by vascular endothelial growth factor A (VEGF-A), which plays a significant role in angiogenesis and vascular permeability. VEGF-A induces angiogenesis by acting through a tyrosine kinase receptor predominantly found on vascular endothelial cells, including VEGF receptor-1. VEGF-A acts to enhance the growth of tumors by promoting a more extensive blood vessel supply and increases the probability of hematogenous metastases. Similarly, it has been proposed that angiogenesis and vascular permeability play a central role in development of angiosarcomas, suggesting a role for VEGF-A in the pathogenesis of angiosarcomas,67 thereby opening the potential for molecular targeted agents in the future. Bevacizumab, a humanized monoclonal antibody to VEGF, is an attractive agent to consider in angiosarcoma, given its ability to inhibit tumor growth. Koontz and coworkers69 have reported promising results in two patients using neoadjuvant bevacizumab combined with RT.
Rhabdomyosarcoma
Rhabdomyosarcoma is especially important because it is the fifth most common cancer in childhood,70 but it also occurs in the adult, where the outcome is significantly less favorable.71 One feature concerns the “spindle cell rhabdomyosarcoma” that is only rarely described in the adult compared with children and carries a relatively favorable prognosis. In the adult it appears to have a predilection for the head and neck and may be unusually aggressive.72,73 The rhabdomyosarcoma classification recognizes embryonal, botryoid, alveolar, and pleomorphic subtypes for both childhood and adult cases. Rhabdomyosarcoma can be recognized on light microscopy by the presence of cross striations within cytoplasmic fibrils of spindle-shaped cells that typically demonstrate immunostaining for myogenic markers.74 Seventy percent of rhabdomyosarcomas can be classified as embryonal and 20% alveolar, and the remainder are variants, including the uncommon pleomorphic subtype. The pleomorphic subtype is almost never seen in the pediatric population and is not considered in those classifications; there is debate about whether it truly represents part of the disease process and it may fall into the “malignant fibrous histiocytoma” type categories described earlier.
A diagnosis of alveolar rhabdomyosarcoma is made if the tumor has alveolar-like spaces that are filled with round malignant, eosinophilic cells. Presence of any amount of alveolar morphology in the lesion qualifies it to be alveolar rhabdomyosarcoma, and alveolar morphology has been identified as a poor prognosticator. There is also a solid variant of this tumor that lacks alveolar spaces but is still included in this subtype. The presence of sheets of anaplastic cells warrants a diagnosis of pleomorphic rhabdomyosarcoma. Today the diagnosis of the less favorable alveolar subtype is generally based on the identification of the characteristic chromosomal translocations t(2;13)(q35;q14) and t(1;13)(p36,q14) and their fusion products (see Table 63-1).27
Both translocations may be associated with different clinical phenotypes.75 The translocations and the gene fusion products are characteristic, and the PAX3 gene family involved in the disease fingerprint may have a role in muscle development.76,77
Rhabdomyosarcoma is also one of the sarcomas associated with a risk of lymph node metastasis. Irrespective of age, the alveolar and embryonal subtypes manifest high rates of response to chemotherapy and RT. The initial evaluation of rhabdomyosarcoma is similar to regular STS but should ordinarily include bone marrow examination. Sampling of cerebrospinal fluid should be considered in patients with parameningeal lesions. Rarely, but distinctly, there is a tendency for these lesions to develop synchronous or metachronous metastases in one or both breasts (Fig. 63-2).
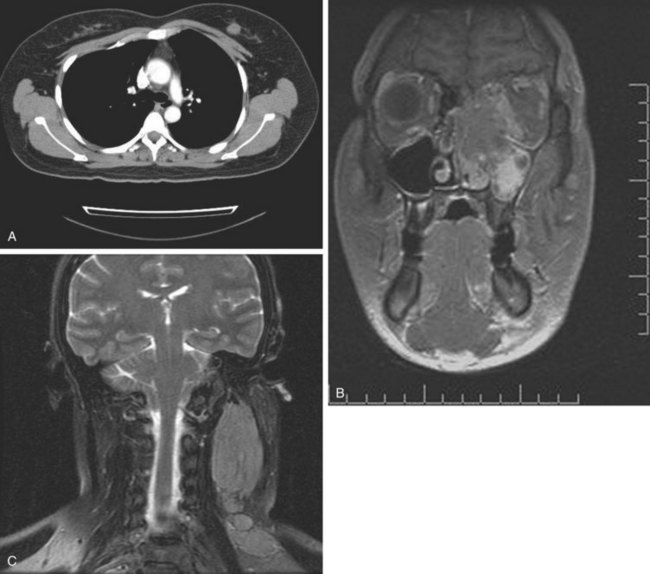
Liposarcoma
Liposarcoma is the second most commonly encountered subtype of STS, and myxoid liposarcoma (MLS) is the most common variant.78 Classic MLS has a t(12:16) translocation, resulting in the TLS-CHOP fusion shared with the more aggressive round cell variant and not seen in predominantly myxoid, well-differentiated liposarcoma.79 It also has an unusual pattern of recurrence that includes a predilection for soft tissue metastases.80–83,84
Apparent multifocal presentation or recurrence at two or more anatomically separate soft tissue sites (with predilection for the retroperitoneum and mediastinum) and bone metastases are a striking yet unpredictable feature of myxoid liposarcoma and contrasts to other STS (Fig. 63-3). Surgery and RT may result in relatively long disease-free intervals (e.g., many years). Uncertainty had existed about whether such multifocal lesions represent an unusual pattern of metastasis or multiple separate primary tumors until Antonescu and associates85 showed that all cases of multifocal myxoid liposarcoma bore the identical molecular lineage of the parent primary tumor.
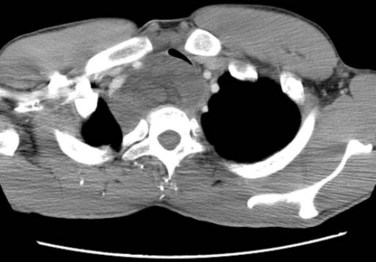
Figure 63-3 CT showing isolated mediastinal relapse with tracheal obstruction occurring in a patient who had presented 7 years previously with a myxoid liposarcoma in the popliteal fossa similar to the lesion shown in Figure 63-9. The initial limb lesion was treated with preoperative RT and maintained local control. This patient underwent airway management, preoperative RT, and surgical resection and was disease free with 15 months of follow-up. Isolated soft tissue relapse in this fashion is a hallmark of the behavior of myxoid liposarcoma and may result in long-term remission or salvage of such lesions.
More favorable survival in a large cohort of liposarcoma patients compared with other histologic subtypes has been reported in a series exceeding 1000 cases. The observation was independent of other prognostic factors, including grade, depth, and tumor size.86 Myxoid liposarcoma also appears to manifest an unusually sensitive response to RT87,88 (Fig. 63-4). Two theories, which are not mutually exclusive, of the underlying mechanism have been postulated. RT might cause adipocyte-like maturation/differentiation of tumor cells,88 although the time scale of the relatively immediate clinical response suggests it may be the less plausible explanation. Alternatively, radiation-induced damage to the tumor vasculature may lead to tumor cell death.89 This radiosensitivity may explain the propensity for unexpectedly favorable local control (>95%) after adjuvant RT.84,90,91
Dermatofibrosarcoma Protuberans
The recent observations of response to imatinib (STI 571, Gleevec) in dermatofibrosarcoma protuberans predicted by the presence of the t(17;22) translocation provides possibilities for molecular target therapies in the future.92 However, for this tumor one should recall the excellent control rates seen with surgery alone or combined with RT; and these targeted approaches are generally applicable to situations in which surgery and RT are not feasible approaches. For inoperable cases, imatinib has demonstrated antitumor effects in advanced disease harboring t(17;22) and is also active in fibrosarcomatous disease. Two distinct phase II trials of imatinib (400 to 800 mg/day) were conducted by the European Organization for Research and Treatment of Cancer (EORTC) and by the Southwest Oncology Group (SWOG). These trials were combined for analysis to provide more statistical power to assess the clinical response rates of locally advanced and/or metastatic dermatofibrosarcoma protuberans to imatinib and to assess the safety of this therapy.93 Sixteen and eight patients were enrolled onto the EORTC and SWOG trials, respectively, with the objective response rate approaching 50%. These promising response rates in these small studies suggest that median time to progression did not differ between patients taking 400 mg/day and those taking that same dose twice a day.
Synovial Sarcoma
Typically occurring in the para-articular areas of the tendon sheaths and joints of young adults, at least 50% of synovial sarcomas are in the lower limbs (especially the knee), and most of the remainder originate in the upper limbs.94 Calcification may be apparent. There is little resemblance between synovial membranes and synovial sarcoma, and because the tumor rarely arises from synovial tissue it has been suggested that the name of this subtype should be modified.95,96 Local treatment follows the general principles of treatment of STS with conservation surgery and RT. This histologic subtype exhibits more favorable responses to chemotherapy than most other histologies.97 Nevertheless, the role of chemotherapy still remains unproven and there is a prevailing view that it should not be delivered outside a clinical trials setting.98,99 A synovial sarcoma–specific nomogram has been developed by Canter and colleagues40 based on preoperative variables that may facilitate informed decision making with regard to selecting patients most likely to benefit from adjuvant/neoadjuvant chemotherapy and, in particular, for the typical size cut points clinicians use for consideration of neoadjuvant chemotherapy. The disease also has a reputation for higher risk of lymph node metastasis (<2% to 14%),100,101 although elective nodal management is not indicated. An increased potential for distant metastases has been observed with larger tumors (>5 cm), local relapse, and age older than 20 years.102
Gastrointestinal Stromal Tumors
GIST are rare STS that represent the first solid tumor exhibiting a consistent favorable response to molecular targeted therapy. These tumors may arise anywhere in the gastrointestinal tract but occur predominantly in the stomach or small intestine. They can also develop from omentum and retroperitoneum. Metastases occur predominantly within the peritoneal cavity and the liver. GIST harbor so-called gain-of-function mutations in the KIT or platelet-derived growth factor–alpha receptor (PDGFR-α) genes in 85% and 5% of cases, respectively.103,104 Through these mutations the receptors are constitutively activated. The advent of the tyrosine kinase inhibitor imatinib was a major breakthrough in the management of advanced GIST because the response rates were dramatic and because effective treatment options previously had not been available.105,106 Patients whose tumors contain exon 11 KIT mutations are more likely to respond to imatinib than those whose tumors have an exon 9 KIT mutations or those with no detectable kinase mutation.107 The unique behavior of this entity has resulted in the introduction of a new stage classification for clinical use that differs from that employed in regular STS (see later in the section on staging).
Clear Cell Sarcoma (Malignant Melanoma of Soft Parts)
For many years this unusual tumor has also been referred to as malignant melanoma of soft parts or clear cell sarcoma of tendons and aponeuroses. Like true melanoma it has a predilection to develop lymph node metastases but in contrast presents as a deep soft tissue mass rather than a cutaneous lesion. Clear cell sarcoma has a distinct chromosomal translocation, t(12;22)(q13;q12), involving EWS and ATF1 genes. Because of the presence of intracellular melanin, and immunoreactivity for S-100 and HMB45 it has been suggested that this entity is better considered a subtype of melanoma and not a soft tissue sarcoma. Recent analysis by genomic profiling and cluster analysis appears to confirm this view.108
Pathways of Spread
Local Disease
STS generally respect barriers to tumor spread in the axial plane of the extremity such as bone, interosseous membrane, and major fascial planes, and this feature should be exploited in planning tissue-preserving approaches to management. Thus the margins of RT should be wide in the cephalocaudal direction but in the cross section there may be much greater security in defining nontarget structures. Extremity lesions arising in extracompartmental spaces such as the axilla, femoral triangle, sartorial canal, and popliteal space have the ability to extend proximally and distally at considerable distances with less anatomic restraint and early involvement of the neurovascular bundle.109 The RT margins should reflect this and include undisturbed tissue planes and barriers to tumor incursion. For nonextremity lesions, the direction of sarcoma growth is also along the involved musculature but care must be taken to ensure that the fascial planes are appropriately recognized and encompassed in surgical or RT target volumes.110
A complete description of all the body’s anatomic planes and fascial compartments is beyond the scope of this chapter. Nonetheless, they guide the principles used to manage these lesions. The importance of the osteofascial compartments of the extremities is that, from an anatomic standpoint, they operate as functional units and are protected from each others’ risks of tumor contamination in the absence of inappropriate violation of the subdividing fascial septa. The latter may occur with misplaced surgical intrusion, including inappropriate placement of drain or biopsy tracts.111
Regional Lymph Node Involvement
In STS there is generally no need for any specific regional lymph node treatment. Important exceptions to this generalization include epithelioid sarcoma, clear cell sarcoma, angiosarcoma, and rhabdomyosarcoma100,101,112 (Table 63-3). More recently, regional lymph node metastases were detected in 28 of 1597 cases treated within a single institution. Results are similar to that reported in Table 63-3, with epithelioid sarcoma and clear cell sarcoma subtypes having the highest lymph node involvement.112 Traditionally, lymph node involvement has been associated with an adverse prognosis rivaling that of distant metastasis. However, data suggest that isolated lymph node metastasis may not be as deleterious a factor.113,114 This is reflected in the most recent version of the TNM staging system (see later section on staging).63
Clinical Manifestations, Patient Evaluation, and Staging
Patient Evaluation
Multidisciplinary Management
Consistency in approach to these rare diseases allows comparison of treatment approaches from center to center as well as ensuring quality of care. An essential component to accomplish this is the implementation of multidisciplinary teams consisting of surgeons, radiation oncologists, medical oncologists, pathologists, radiologists, and personnel from support disciplines that allow for the optimal care of these complex cases.115 With a multidisciplinary team, all members ideally would see the patient and participate in formulating a treatment plan before implementation of any component of therapy.
Part of the multidisciplinary approach must also include an appreciation of the principles for safe acquisition of diagnostic tissues as well as review of pathology by an experienced pathologist. Lehnhardt and associates116 showed that expert pathology opinion was essential for optimal treatment of sarcoma and remains a source of concern even when expertise is present. Discordance in diagnosis (tumor entity and grading) was apparent in excess of 70% of cases among pathologists in private clinics, slightly less in general hospital pathology departments, was evident in 66% of cases in an academic environment, and was only improved to 30% in a sarcoma reference center even when punch or needle biopsies were not being considered. Inexperience of the pathologist was considered a strong factor leading to discrepant diagnosis, thereby warranting a recommendation for routine expert second opinion.116
Another practical but important factor is that the radiation oncologist should ideally be aware of and preferably have an opportunity to see the patient before any surgery being performed. Otherwise, imaging and pathology details may be lacking or inadequate and precisely appreciating the original tumor characteristic for accurate postoperative adjuvant treatment may not be possible. The consequence of a fragmented RT service with significantly compromised outcome was evident in a study that addressed an extremity subsite of STS in one center where the local failure was 28% compared with that in two other centers (local failure 5% to 10%) where treatment and consultation were restricted to sarcoma specialist radiation oncologists working in the same multidisciplinary setting as the surgeons.117
A recent report118 evaluated outcome in 4205 operative cases of STS and showed that high-volume centers (those in the upper one third of case of volume in a geographic region) reported better survival outcomes. Smaller centers may have more difficulty formulating a multidisciplinary team with all necessary components of care, including access to an effective multidisciplinary cancer conference that meets frequently and regularly.
Initial Assessment
The overall initial evaluation of a patient with a soft tissue sarcoma is summarized in the diagnostic algorithm (Fig. 63-5). All soft tissue masses deep to the investing fascia should be considered to be sarcoma until proven otherwise. The type of biopsy, or a prior inappropriate excision, may jeopardize the form and outcome of local treatment by contaminating tissues that were uninvolved initially. Before embarking on treatment, histologic confirmation of the diagnosis is necessary. To minimize the risk of undesirable outcome, prebiopsy cross-sectional imaging is necessary to avoid compromising the planning of management.
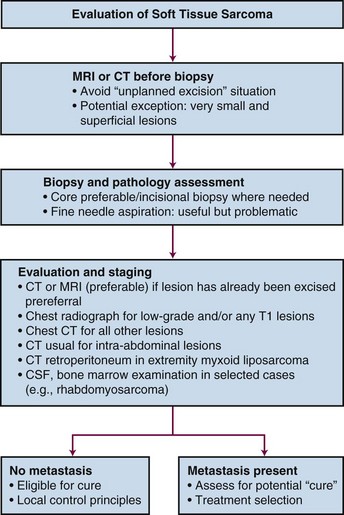
Imaging
Accurate, highly sophisticated imaging to delineate tumor extent for diagnostic, surgical, and RT planning is essential to the management of sarcoma. Plain radiography, bone scintigraphy, angiography, computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography (PET) are generally not helpful in determining the diagnosis, but all may contribute in different ways to delineation of the extent of local involvement. The authors have recently discussed the imaging characteristics of STS in detail.119
A “blinded” study by the Radiation Diagnostic Oncology Group120 that compared MRI and CT in patients with malignant bone and soft tissue tumors showed no specific advantage of MRI over CT. Although the diagnostic evaluation may be equally served by both modalities, the treatment planning requirements (for both surgery and RT) frequently require additional information, including advantages through MRI/CT image fusion techniques.
PET has an emerging role in the evaluation of STS.119,121,122 It uses radiotracers specific for biologic processes to produce images of regional tissue metabolism with 18F-fluorodeoxyglucose (FDG), the most commonly employed radiotracer for this purpose. New tracers are being evaluated that may refine the differing roles for PET that include evaluation of (1) the grade and extent of local disease, (2) the detection of malignant transformation in neurofibromatosis, (3) the presence of regional or distant metastases, and (4) the response to initial (i.e., neoadjuvant) treatment as a surrogate to prognosticate on future outcome. It does seem that high- versus low-grade lesions may be distinguished and may even provide an imaging technique for biopsy guidance; in addition, response prediction to induction treatments seems to be possible.119,121–123,124
Despite the preceding discussion, the contribution of PET to routine management is currently uncertain. It is clear, however, that radiotracers specific for glucose metabolic imaging or imaging of tumor cell proliferation cannot yet replace biopsy and histopathologic evaluation for sarcoma grading.123
Establishing the Diagnosis
The biopsy should be carefully designed to facilitate subsequent surgical removal and adjuvant therapy. The biopsy track should be excised afterward or alternatively may be included in the RT target volume of preoperative treatment. The open approach provides the highest yield because sufficient tissue for traditional histologic analysis as well as immunohistochemistry, electron microscopy, molecular assessment, and cytogenetics is made available but has cost implications in addition to other potential limitations. For example, tumor cells are capable of spread and implantation along tissue planes facilitated by extravasated blood from the surgical site and, consequently, the biopsy should be performed through the smallest incision possible with absolute hemostasis. Disadvantages of this approach may include complications and potential delay in initiation of neoadjuvant therapies if there are wound problems or the possibility for poorly designed incisional biopsies to breach natural barriers to tumor growth, such as muscle fascia and periosteum that provide local containment of tumor.125
Alternatives to open biopsy include fine-needle aspiration biopsy (FNAB) or core needle biopsy. FNAB can be useful, but these tumors are rare, complex, and difficult to diagnose even in experienced hands and FNAB only should be used when a cytopathologist with extensive experience in sarcoma is available. Despite this, there is a growing experience in diagnosis of sarcomas with FNAB and it has been advocated by some groups if the expertise is available.125,126 More generally, it is certainly useful for establishing the presence of recurrence (both local and metastatic).
Core needle biopsy under local anesthesia provides a more consistent tissue specimen and cell preparation compared with FNAB. Because diagnosis and grading are increasingly based on tissue obtained by core needle biopsy, new challenges have been presented to pathologists, especially if neoadjuvant therapy is used, because this eliminates the possibility of accurate grading later when the lesion is resected. A close dialogue with clinicians to resolve ambiguities may enhance accuracy.61
In the event that an open biopsy is required, an incisional biopsy of deep tumors is preferred over excisional biopsy to minimize the difficulty of the “unplanned” excision.127–129 This unplanned approach may increase the rate of local recurrence in sarcomas of the extremities.127
Staging Classifications
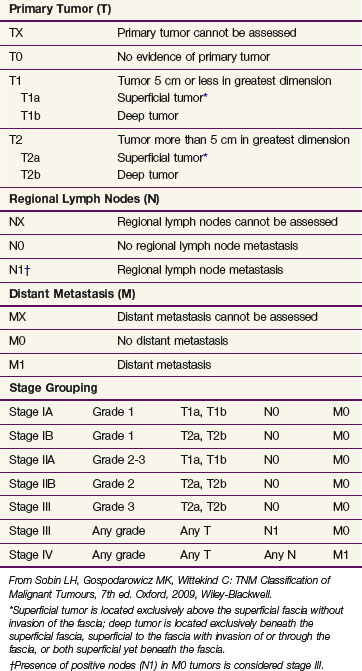
The Union for International Cancer Control (UICC)/American Joint Committee on Cancer (AJCC) TNM (7th edition) staging system63 is the most widely employed staging system for STS and is unusual within the overall TNM classification system in that it incorporates histologic grade with anatomic disease characteristics. In general, the system is based on an ascending hierarchy of risk depending on whether a lesion is deemed to have no adverse features for relapse or one, two, or three factors present represented by size partitioned at the 5-cm break point, grade (three-tiered system), and lesion depth as “a” (superficial tumor arising outside the investing fascia) or “b” (a deep tumor that arises beneath the fascia or invades the fascia) (Table 63-4). All STS subtypes are included except hollow visceral sarcomas, Kaposi sarcoma, fibromatosis (desmoid tumor), and sarcoma arising from the dura mater, brain, and parenchymatous organs.
Proposed Staging for Retroperitoneal Sarcomas
Retroperitoneal sarcomas are currently classified by the TNM system. However, another classification system has been developed based on grade, completeness of resection, and distant metastases.130 For class 1 (low grade/grossly complete resection/no metastases) the stage-specific 5-year survival rate was 89%. For class 2 (high grade/grossly complete resection/no metastases) the 5-year survival was 40%. For class 3 (any grade/incomplete resection/no metastases) it was 26%; and for class 4 (distant metastases) it was 17%. These results were obtained from a cohort of patients treated in a single tertiary referral center (n = 107). Five-year OS was 55%. This classification is also problematic from a methodologic standpoint because it requires one parameter (the completeness of resection) to become available after the main treatment has been undertaken and thus cannot be applied at baseline when the patient first presents with sarcoma. Validation is required before it can be considered for general use.
Gastrointestinal Stromal Tumors
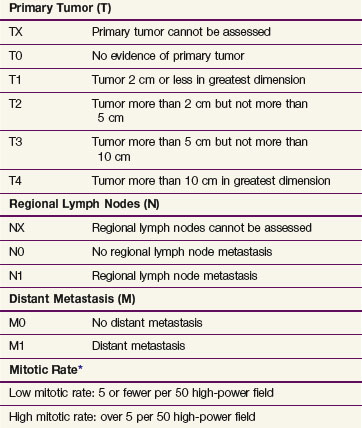
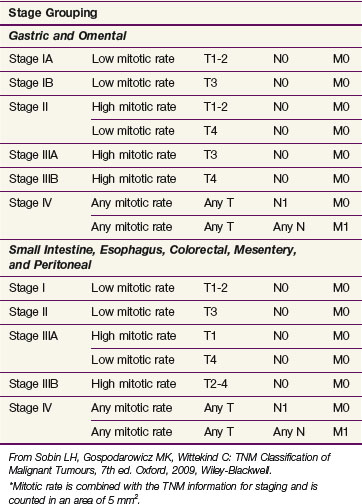
For the first time GIST is included in the TNM staging system with separate stage grouping schemes provided for gastric, omental, and small intestinal tumors. This new staging system uses tumor size, dissemination status, and mitotic rate as the staging parameters. Tumor size is partitioned at the 2-, 5-, and 10-cm breakpoints. Mitotic rates are categorized as low and high. Of interest is the use of two stage grouping schemes depending on whether the tumor has arisen in the stomach or omentum as opposed to other sites within the abdomen or esophagus (Table 63-5).
Primary Therapy
Results of Treatment
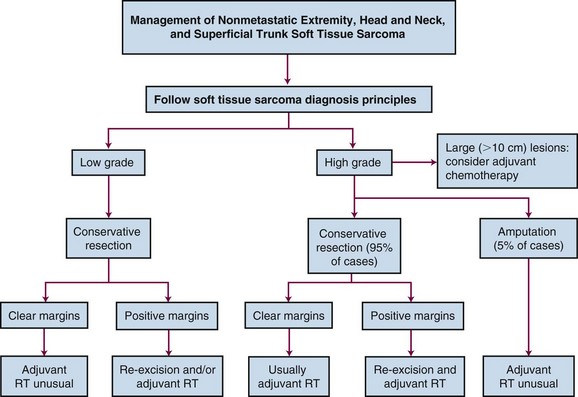
The 5-year survival of patients with STS overall is approximately 60%.131 The benchmark for local control in extremity sarcomas is 90% (Table 63-6). There has been little progress in reducing the risk of metastases. An overall approach to the local management of nonmetastatic extremity, head and neck, and superficial trunk (including body wall) STS is summarized in Figure 63-6. In general, the approach is confined to surgery with or without RT and chemotherapy is recommended in selected cases, including chemoresponsive lesions and in some high-risk cases, although the evidence for the latter is less substantial. One study spanning 40 years analyzed the treatment-related mortality of 629 long-term survivors of STS (absence of disease recurrence) who were treated with RT and function-preserving surgery. Overall, STS survivors did not appear to experience significant excess mortality as a result of aggressive cancer treatment compared with controls from the U.S. general population. Notably, females younger than the age of 50 years at the time of diagnosis with nonextremity primary tumors have higher all-cause mortality potentially due to cancer.132
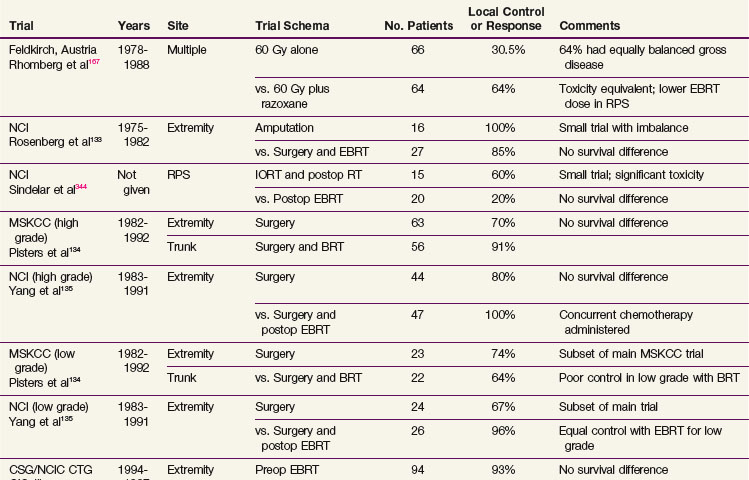
Evidence About Local Management (Randomized Trials)
The origins of contemporary extremity STS management can be traced to the landmark trial at the NCI where patients with high-grade sarcoma of the extremities were randomized to receive amputation versus a limb-sparing operation followed by adjuvant RT.133 In addition to this NCI trial there have been a number of others addressing several important controversies, which are summarized for convenience in Table 63-6. Notably, two trials134,135 have compared conservative surgery alone with similar surgery with adjuvant RT (one addressing brachytherapy and the second postoperative external beam RT [EBRT]). Another trial addressed the two most frequently used local adjuvant approaches, preoperative versus postoperative EBRT, in extremity STS.74
Philosophy of Local Management
Surgery is the mainstay of the management of STS and was formerly used alone, albeit often in the form of an amputation. Better understanding of the fascial containment and routes of spread, as well as advancements in surgical techniques such as microvascular free flap reconstruction,136 have led to tissue preservation in most patients. Inappropriate violation of fascia should not take place because tumors can ordinarily be considered to involve either the superficial or the deep compartment but rarely both. If the deep compartment is involved or has been contaminated, reexcision, RT, or frequently both may be required. RT needs to generously include the deep compartment together with the original subcutaneous tissue site if it was involved or contaminated. Relatively few STS penetrate cortical bone. In the absence of bone invasion, the periosteum is an adequate surgical margin for STS treated with wide excision and RT.137
Surgical Management Issues
Extent of Surgery and Causes of Different “Resection Margins”
Of more importance than the description of the type of surgery is the actual extent of viable residual disease remaining after surgery. This is not a precise variable but is best represented by expert pathologic assessment of the surgical “resection margin.” Characterization of the margin guides the need for adjuvant RT for those tumors with insufficient normal tissue surrounding tumor to reliably prevent local recurrence. Having positive margins nearly doubles the risk of local recurrence and increases the risk of distant recurrence and disease-related death.138 Stoeckle and associates139 reported a fourfold difference in local recurrence between resections with negative versus positive margins in their single-center prospective evaluation of 205 patients who underwent surgery for STS of the extremity or trunk wall. Moreover, Gronchi and colleagues140 found that the quality of surgical margins independently predicted local control and survival but not distant metastases after evaluation of 997 consecutive patients affected by primary extremity STS. These authors reported that survival was directly influenced by local recurrence in pelvic and shoulder girdle sites and maintained that higher surgical aggressiveness is necessary in these situations.
We have shown that positive resection margins may have different causes with potential for different outcome. However, two categories of positive surgical margins are associated with a higher risk of local recurrence despite the use of adjuvant RT: (1) patients who present after pre-referral unplanned excision and who have a positive margin on subsequent reexcision (local recurrence rate: 31.6%) and (2) those with unanticipated positive margins occurring during primary sarcoma resection (local recurrence rate: 37.5%).141 Therefore the policy in STS surgery of attempting to achieve negative surgical margins is sound and this is best accomplished by performing surgery at an experienced referral center. Nonetheless, in this context some caveats exist. For example, patients with microscopically positive margin low-grade liposarcomas have a low risk of local failure (recurrence rate: 4.2%), as do patients in whom a positive margin is anticipated before surgery to preserve critical structures, and RT is able to sterilize the minimal residual disease (recurrence rate: 3.6%). In the latter setting it appears that a small, isolated positive resection margin, planned from the outset by the multidisciplinary team with the intention of sparing a critical anatomic structure (e.g., at a major nerve, vessel, or bone), has a favorable outcome, provided adjuvant RT is appropriately applied, compared with the more extensive margin-positive situations noted earlier in which significant local contamination may be present.141
Although recent data indicate that the unplanned surgery (without reexcision) situation can often be managed successfully with immediate RT,142 we remain concerned that substantial contamination means that subsequent RT will be less successful. In addition, the burden of residual disease may often be underestimated.141 Chandrasekar and colleagues143 found that of 316 reexcisions after an unplanned excision of a soft tissue sarcoma, 188 patients had residual tumor (59%). Furthermore, patients had a higher likelihood of developing a local recurrence if the reexcision specimen contained residual tumor. Cahlon and associates144 reported the long-term outcomes in extremity STS after pathologically negative re-resection without adjuvant RT. An overall local recurrence rate of 9% was reported for 200 patients. However, patients of older age and/or stage III disease had an increased rate of local recurrence, prompting the authors to comment that treatment decisions especially with regard to the adjuvant use of RT should not be based solely on the finding of a negative re-resection.
Surgery Alone
Conservation Approaches
There is undoubtedly a subgroup of patients who do not need RT.145–148 A more recent prospective single-center trial has provided data demonstrating selected patients with primary T1 STS of the extremity and trunk can be treated by R0 surgery alone with acceptable local control and excellent long-term survival.149 The problem with these data is that most are retrospective and emanate from large centers with experience in sarcoma management and case selection is present. The challenge remains in how to select those cases that can be managed by a single-modality approach versus those who also require RT.
Amputation
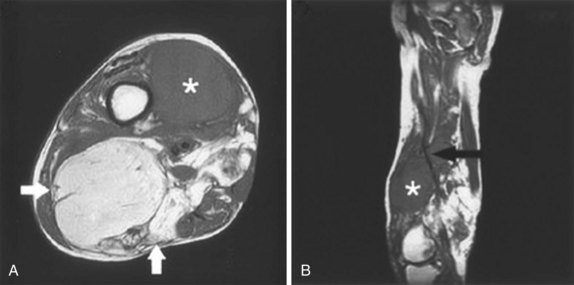
Even in a milieu in which limb preservation with adjuvant RT is a preferred policy, there will always remain a group of patients who are better treated with amputation.150 At Memorial Sloan-Kettering Cancer Center (MSKCC) from 1968 to 1990 the amputation rate fell from 50% to below 5%151 and at the Princess Margaret Hospital amputation was never popular and only used in 7 of 226 patients with extremity STS treated between 1980 and 1988,152 both experiences providing strong evidence that the indications for this approach should be rare.153 Amputation only should be performed (1) when it is impossible to achieve adequate surgical clearance because the lesion involves major neurovascular structures or multiple compartments (Fig. 63-7); (2) when potential major RT complications would result from dose and volume considerations; and (3) in some lower extremity distal lesions when a below-knee amputation prosthesis may be more serviceable than a limb damaged by extensive surgery and RT.154
Radiation Therapy Management Issues
Rationale for Adjuvant Radiation Therapy
Adjuvant RT is generally used to permit more conservative tissue resections and therefore less-extensive operations than would have ordinarily been the case. External beam photons delivered preoperatively or postoperatively or by brachytherapy are the usual modes of administration. Strander and colleagues,155 in a systematic overview involving 4579 patients, employed data from 5 randomized trials, 6 prospective studies, 25 retrospective studies, and 3 additional articles. Adjuvant RT improved the local control rate in combination with conservative surgery in the treatment of STS of the extremities and trunk in patients with negative, marginal, or minimal microscopic positive surgical margins. A local control rate of 90% is expected. More recently, Kim and colleagues156 reported no significant difference between patients who received RT with R0 margins compared with those treated with close or positive margins combined with RT in terms of local failure free and OS and concluded that margin status did not predict for local failure when adjuvant RT is used. Similarly, the Scandinavian group found that RT improves local control regardless of surgical margin and grade in extremity and trunk wall STS, with the effect being most pronounced in deep-seated, high-grade tumors even if removed with a wide surgical margin.157 Improvement is obtained with RT after intralesional surgery, but the local control rate is less satisfactory.
For STS in other anatomic sites, such as retroperitoneum, head and neck, breast, and uterus, the benefit in local control with adjuvant RT is less compelling.155 In the situation in which positive margins remain after surgery, postoperative RT doses greater than 64 Gy, superficial tumor location, and extremity site have been associated with improved local control.158 For circumstances in which preoperative RT is given and positive margins are found on surgical resection, the effectiveness of a postoperative RT boost is debatable. We retrospectively analyzed 216 patients with positive surgical margins treated with preoperative RT for extremity STS. Fifty-two of these patients were treated with preoperative RT alone (50 Gy) and were compared with 41 who received preoperative RT plus a postoperative boost (80% received 16 Gy postoperatively for a total dose of 66 Gy). The patients who received a postoperative boost had lower 5-year estimated local recurrence-free survival rates (73.8% vs. 90.4% for preoperative RT only). The postoperative boost after preoperative RT did not provide an advantage in preventing local recurrence.159 We therefore avoid scheduling a postoperative boost owing to the increased risk of difficult-to-treat RT morbidities with the higher radiation doses involved and because of previous clinical experience that patients who could not receive a postoperative boost because of wound healing complications or other contraindications did not seem to be at a higher risk of local recurrence.
Details of the Randomized Trials of Adjuvant Radiation Therapy
As mentioned earlier, adjuvant RT with conservative surgical resection has been evaluated in two randomized clinical trials.134,135 Yang and associates135 at the NCI randomized high-grade extremity lesions after limb-sparing surgery to receive adjuvant RT or no further treatment after the surgery. Concurrent chemotherapy was also used depending on grade. The local control for those receiving RT was 99% compared with 70% in the control group (p = .0001). The results were similar for high- and low-grade tumors. Adjuvant RT, using brachytherapy, was also evaluated in a randomized trial at MSKCC with a similar effect in high-grade lesions.134 No improvement in local control was evident with brachytherapy in the low-grade tumors (see Table 63-6), which was attributed to more slowly cycling cells that are thereby excluded from the radiosensitive phases of the cycle during brachytherapy.134
Scheduling: Preoperative versus Postoperative Radiation Therapy
The two most common methods of RT delivery are preoperative or postoperative external beam. A Canadian randomized trial compared preoperative and postoperative RT in extremity STS.74,160 Wound complications were twice as common with preoperative RT as with postoperative RT (35% vs. 17%), although the increased risk was almost entirely confined to patients with sarcomas of the lower extremity.74 RT toxicity rates in the Canadian trial after 2 years differed between the two arms of the study with a tendency to manifest greater fibrosis in the postoperative arm of the study.161 This analysis suggested strongly that larger treatment volumes were predictive of greater joint stiffness and fibrosis and may also contribute to limb edema. Patients with late treatment responses such as fibrosis, joint stiffness, or limb edema had significantly lower limb function scores at later time points.161
With 3.3 years median follow-up, the initial report of the Canadian trial showed identical local control between the two arms of the trial and identified an early improvement in OS (p = .0481) in the preoperative RT arm74 that was not substantiated with 5-year results.160 The 5-year results for preoperative versus postoperative RT, respectively, were as follows: local control, 93% versus 92%; metastatic relapse free, 67% versus 69%; recurrence-free survival, 58% versus 59%; OS, 73% versus 67% (p = .48); and cause-specific survival, 78% versus 73% (p = .64). Only resection margins were significant for local control. Tumor size and grade were the only significant factors for metastatic relapse, OS, and cause-specific survival. Grade was the only consistent predictor of recurrence-free survival.160
Recently, a literature-based meta-analysis of oncologic outcomes of preoperative versus postoperative RT in localized resectable STS analyzed five eligible studies that included a total of 1098 patients. This analysis suggested that the risk of local recurrence may be lower after preoperative RT and that the risk of metastatic spread is not increased with the delay in surgical resection necessary to complete preoperative RT.162 Furthermore, Sampath and associates163 assessed the impact of RT sequencing on OS and cause-specific survival. A retrospective analysis was conducted using the National Oncology database and found 1087 patients who received RT with surgery. OS was significantly improved with preoperative RT versus postoperative RT, with a median survival of 124 months and 90 months, respectively (p = .02). Cause-specific survival was also significantly higher with preoperative RT.
Radiation Therapy Alone
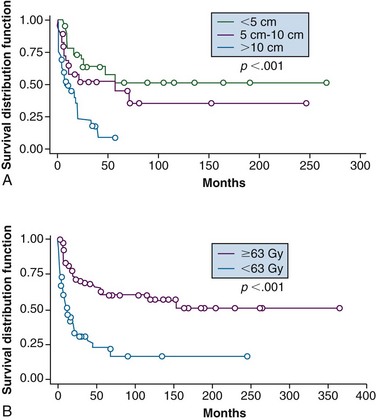
Although every effort should be made to attempt resection, there remain patients in whom definitive RT may be considered owing to a combination of unresectability and/or medical comorbidity or to achieve palliation when surgery is not considered appropriate. Tepper and colleagues164 treated 51 patients with definitive photon beam irradiation to a total dose of 64 to 66 Gy. The 5-year local control and survival rates were 33% and 25%, respectively. Local control was better for tumors less than 5 cm (87.5%) than in tumors 5 to 10 cm (53%) or greater than 10 cm (30%). More recently, Kepka and associates165 updated this series that now contains 112 patients who underwent RT for gross disease. Local control at 5 years was 51%, 45%, and 9% for tumors less than 5 cm, 5 to 10 cm, and greater than 10 cm, respectively (Fig. 63-8A). Patients who received doses of less than 63 Gy had inferior 5-year outcomes compared with those receiving higher doses, including local control of 22% versus 60% (Fig. 63-8B); disease-free survival (DFS) of 10% versus 36%, and OS of 14% versus 52%. These researchers also noted a rise in complications at doses of 68 Gy or more, which appears to provide a therapeutic window.165
Potentially, higher doses of RT with concurrent dose-intensified chemotherapy regimens, possibly delivered with conformal RT volumes to further protect normal tissues, provide an opportunity to enhance local control in this setting. Preliminary data using a concurrent ifosfamide-based protocol exists and may provide an opportunity to address unresectable disease.166
Rhomberg and associates167 prospectively evaluated the radiation sensitizer razoxane in a randomized controlled trial. Among 82 patients with gross disease, RT combined with razoxane demonstrated an increased response rate compared with photon irradiation alone (74% vs. 49%) with improved local control (64% vs. 30%, p <.05). Acute skin reactions were enhanced in the “sensitizer” arm, but late toxicity was not increased. Jakob and colleagues168 similarly used temozolomide preoperatively in combination with intensity-modulated irradiation (IMRT) for large STS at problematic sites where clear resection margins could not be achieved. An R0 resection was achieved in 7 of 15 cases. The antitumoral effect of temozolomide seems to depend on methylguanine methyltransferase promoter methylation status; and although present in subgroups of STS there are no data available from phase II studies testing the activity of temozolomide in STS. These data will need confirmation with a larger patient population.
Radiation Therapy Modality
External Beam Irradiation, Including Intensity-Modulated Irradiation
EBRT may include photons or particle beam (electrons, protons, pions, carbon ions, or neutrons) but in practice is confined to photon treatments.74,135
An alternative approach is the use of intraoperative radiation therapy (IORT) delivered with electron beam. The major interest for IORT has been in retroperitoneal sarcoma where the proximity to bowel limits RT delivery and surgical margins are often compromised because of anatomic constraints. IORT combined with RT for retroperitoneal management has yielded promising results in terms of improved local control, but follow-up has been modest with small patient numbers.169 Some reports also describe IORT for extremity sarcomas,170,171 but formal clinical trials have not compared the relative merits of this approach. Its use may be governed as much by its availability at a given center as by any special advantage that it may confer.
Recently, external beam approaches have evolved significantly to explore the use of IMRT because of the increased capacity to selectively target regions of risk while sparing normal tissues in the non–target-vulnerable regions. In the setting in which adjuvant doses to intricate volumes are relatively high and normal tissue protection is desirable, steep dose gradients are achievable between target and normal tissues with IMRT technology. A retrospective chart review comparing a three-dimensional conformal technique to IMRT for 10 thigh tumors172 provided evidence that IMRT is capable of reducing dose to the femur using a 5-mm planning tumor volume (PTV) margin at the interface of the bone and soft tissues, without compromising target coverage. One of our previous studies, also using a 5-mm PTV margin, found that IMRT substantially lowered the dose to the future tissues required for tumor resection and wound closure, with improved target conformality for lower extremity sarcomas.173 This provided evidence for a phase II clinical trial that we recently concluded that examined the effects of preoperative IMRT on wound complication rates by using precisely shaped radiation target volumes for patients with lower limb STS.174
Until recently, particle beam approaches were the only method of treating complex tumors, such as those that present at the skull base, with high-dose RT. Stereotactic fractionated RT or IMRT may provide similar outcomes. IMRT has provided a 3-year actuarial local control of 95% and regional node control rate of 90% in 28 younger patients aged 1 to 29 years (median age 8 years) with rhabdomyosarcoma despite the use of relatively close RT margins (1.5 cm).175 Combs and colleagues176 have also reported excellent outcomes using IMRT and fractionated stereotactic RT in children with head and neck rhabdomyosarcoma with a low incidence of treatment-related side effects. More recent publications reported the results of IMRT as an adjuvant treatment for primary STS of the extremity in a population of high-risk patients for local recurrence.177,178 Despite the majority of patients presenting with adverse prognostic factors such as tumor size greater than 10 cm, location deep to the fascia, and high-grade disease with positive or close surgical resection margins, the 3- and 5-year actuarial local recurrence rate was 94%.177 Moreover, common RT treatment–related toxicities, such as edema, joint stiffness, fibrosis, nerve damage, and bone fractures, were of lower grade and less debilitating than seen with conventional treatment. Additionally, the high rate of local control was maintained irrespective of poor prognostic factors traditionally associated with conventional treatment techniques/brachytherapy, such as upper extremity site, and in situations in which tumors arise in geometrically complex anatomic areas. Although the sample size was small (n = 41) with short follow-up, the clinical outcomes reported with the use of IMRT were encouraging.
Another study from MSKCC compared the local control of IMRT versus brachytherapy for primary high-grade extremity STS. Low-dose-rate (LDR) brachytherapy was given to 71patients and IMRT was used in 63 patients. Despite higher rates of adverse prognostic features in the IMRT group, IMRT was the only predictor of improved local control (p = .04) (92% local control for IMRT compared with 81% for brachytherapy). These results encourage the use of IMRT; however, it remains to be seen if these results continue to be observed with longer follow-up (mean follow-up was 46 months for the IMRT group and 47 months for the brachytherapy group).179 Furthermore, because of the lack of trials comparing EBRT to brachytherapy, it is also fair to question whether these results are truly contingent on the use of IMRT or if similar findings could be achieved using conventional EBRT approaches.
In the near future it is expected that trials investigating the merits of IMRT will become more popular. For example, a phase II study initiated by the City of Hope Comprehensive Cancer Center is currently investigating the use of IMRT for the treatment of patients undergoing surgery for stage IB, II, or III STS. This trial is studying the side effects and effectiveness of preoperative IMRT.180 Moreover, image-guided RT will also increase in frequency as online assessment tools are streamlined for clinical use. The recently completed Radiation Therapy Oncology Group (RTOG)-0630 trial is a phase II trial investigating image-guided preoperative RT for primary STS of the extremity to determine the effect of reduced radiation volume on lymphedema, subcutaneous fibrosis, and joint stiffness.181
Brachytherapy
Brachytherapy has several advantages over EBRT, including its prompt initiation after surgery when clonogen numbers are at a minimum, its easier administration with chemotherapy when this is part of the treatment approach because the skin is relatively protected, and the shorter overall treatment time (typically 4 to 6 days vs. 5 to 6.5 weeks for EBRT), which is attractive and acceptable to patients. However, brachytherapy results, when the therapy is not administered in centers with high referral populations of patients treated with brachytherapy, may fall short of those expected with EBRT. There is also the lack of efficacy for low-grade tumors noted earlier, a feature not apparent with EBRT.135 Brachytherapy may also not provide optimal results when the anatomy of the lesion is unsuited for ideal implant geometry, such as in the proximal limb girdle regions.182
The American Brachytherapy Society has recommended that brachytherapy should not be used as a sole treatment modality in several situations: (1) if the CTV cannot be adequately encompassed in the implant geometry; (2) where the proximity of critical anatomy is anticipated to prevent administration of meaningful dose; (3) where the resection margins are positive; and (4) if there is skin involvement.183 In such situations use of EBRT alone or with brachytherapy may be used. Thus an alternative approach is a combination of EBRT with a brachytherapy boost. There is greater security from the comprehensive coverage typical of EBRT and additional radiobiologic and dosimetric advantages of perioperative brachytherapy. This has resulted in high control rates comparable to full-dose adjuvant EBRT. Typical approaches could employ 45 Gy with EBRT followed by 20 Gy delivered using brachytherapy, realizing local control rates of approximately 90%.184–188 This approach seems useful if satisfactory geometry is achievable and the tumor is located in sites where external irradiation as a sole modality has a relatively high probability of producing major toxicity.
Evidence supporting HDR brachytherapy use is limited to small case series, and comparisons are difficult owing to nonstandardization of target volumes, dose prescription points, and delivered dose. In particular, there are no robust large series evaluating HDR brachytherapy and concern remains over reduced local control, particularly in proximally located tumors. There may even be evidence that modern external beam approaches using IMRT may offer advantages over brachytherapy179 (see previous section, External Beam Irradiation, Including Intensity-Modulated Irradiation.)
Radiation Therapy Dose
External Beam Dose Fractionation for Resectable Disease
EBRT prescriptions for postoperative RT depend on the tumor grade and involvement of the surgical margin but typically employ 60 and 66 Gy for low- and high-grade tumors, respectively, with a “shrinking field” technique after 50 Gy at 1.8 to 2.0 Gy per fraction (Table 63-7). Not infrequently, doses may need to be limited to 45 to 50 Gy in certain anatomic areas containing critical structures (e.g., head and neck, intra-abdominal sites) to minimize morbidity. In these situations the preoperative approach probably has an advantage. For preoperative RT our policy has been to encompass gross tumor, including peritumoral edema where possible, to a dose of 50 Gy in 25 fractions over 5 weeks to be followed by surgery 4 to 6 weeks later.
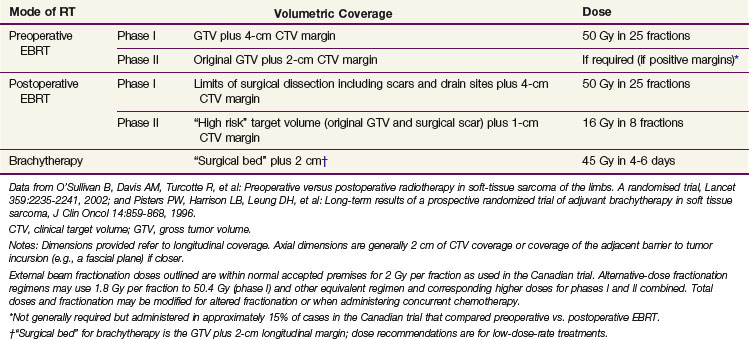
Dose for Gross Unresectable Disease
Alternative EBRT strategies have also used modified dose fractionation approaches in combination with chemotherapy. Thus far it is not clear what additional advantages these approaches may provide and they have not been widely adopted despite being originally reported two decades previously.189,190
Our interpretation of Kepka and colleagues’ results (as discussed earlier) is that unresected disease treated with the goal of long-term control should receive at least 70 Gy in 35 fractions or equivalent when this is anatomically feasible, bearing in mind adjacent normal tissues that may demand dose modification; doses may also need to be higher when employing smaller dose per fraction strategies to ameliorate late-responding tissue effects.165 In this situation the therapeutic ratio may be further enhanced by the addition of systemic treatments to augment the RT response.167 Other strategies are also discussed in a later section.
Brachytherapy Dose
The optimal adjuvant doses for postoperative brachytherapy remain undefined.183 Doses will vary depending on the dose rate and whether it is being used as a sole modality or in conjunction with EBRT (Table 63-8). The prescription point for the implant may also vary but is generally recommended at an isodose line 5 to 10 mm from the plane of the implant. There is general agreement about the dose for LDR monotherapy (see Table 63-8) and doses of 45 to 50 Gy at about 0.45 Gy/hr seem acceptable with the skin dose not exceeding 20 to 25 Gy when large areas of skin are treated.183,191 When LDR brachytherapy is used with EBRT (typically 45 to 50 Gy) doses of 15 to 25 Gy are generally administered over 2 to 3 days.183 Fractionated HDR brachytherapy is more problematic, in part because it is a new technique with little clinical experience in a rare disease; in addition, there are radiobiologic issues governing serious late toxicity. For example, caution should be exercised when placing catheters in contact with neurovascular structures and the dose per fraction should probably be curtailed. In general, doses of approximately 36 Gy in 10 fractions using a 6-hour interfraction interval have been suggested, although some authors have used higher doses.191 Guided by the more abundant breast cancer experience, a dose of 32 to 34 Gy in twice-daily fractions of 3.4 to 4.0 Gy over 4 to 5 days192–194 seems useful and safe, although even here there have reports of fat necrosis.194
TABLE 63-8 Guidelines Based on Published Literature for Adjuvant Dose Selections for Postoperative LDR and HDR Brachytherapy for Soft Tissue Sarcoma for Different Approaches
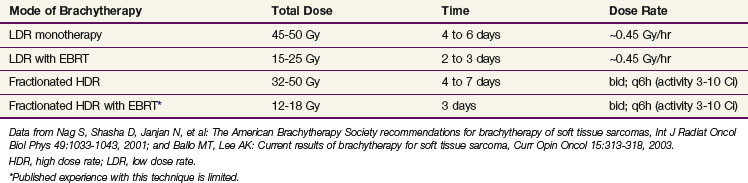
An additional comment concerns the use of HDR IORT, a novel and rarely used approach but one that has been reported for the treatment of both primary and recurrent sarcoma. Doses of 10 to 15 Gy as single treatment prescribed at 0.5 cm were used to supplement EBRT doses of 45 to 50 Gy depending on surgical margins for retroperitoneal sarcoma.195 The large single doses required make this a problematic approach in the vicinity of the vulnerable anatomy of the retroperitoneum. Niewald and colleagues evaluated their experience using an iridium-192 source for HDR IORT combined with EBRT (23 to 56 Gy dependent on resection status) for 29 primary STS tumors and nine recurrences. The majority were located in the extremities (27 of 38), with the remainder in the retroperitoneum or chest area. Ten of 36 patients in whom sufficient data were available experienced a local recurrence with a 5-year actuarial local control rate of 63%. Five-year OS was 57%. These authors noted that the complication rates were considerable, including late skin toxicity and wound healing problems, and, although not severe, should be taken into consideration when therapy decisions are made.196 Other authors have observed similar wound healing complications with HDR brachytherapy.197,198 To decrease these wound complications, various techniques have been employed, including staged flap reconstruction, which avoids flap irradiation, combined with negative pressure wound therapy, which shows promise.199
Factors Influencing Choice of Radiation Therapy Dose
Ongoing problems of fibrosis that appear to be volume related were discussed earlier.161 In addition we have noted a fracture rate that appears dose related (doses of 60 Gy or greater to bone) in a series of 363 patients with lower extremity sarcomas treated with EBRT and surgery, but without chemotherapy, and followed for a median of 50 months.200 A total of 27 postirradiation fractures were identified (6.3%). Seventeen of these patients had postoperative RT (with 15 patients receiving 66 Gy and 2 receiving 60 Gy), and 3 had preoperative RT with a postoperative boost (total dose, 66 Gy). Twenty patients had received 60 to 66 Gy (fracture rate 20/192, or 10%), and 3 had received 50 Gy (fracture rate 3/172, or 2%). The results also demonstrate that female gender confers a higher risk of fracture, and others have shown that chemotherapy aggravates the risk.201,202,203
More recently, volumetric RT dosimetry details for 21 patients with bone fractures were retrospectively compared with a random sample of 53 patients without fractures to determine fracture risk factors.204 We found that the risk of radiation-induced fracture appeared to be reduced if the volume of bone irradiated to 40 Gy or greater was kept below 64%. Fracture incidence was lower when the mean dose to bone was less than 37 Gy or the maximum dose anywhere along the length of bone was less than 59 Gy. There was also a trend toward smaller mean RT volumes for patients without fractures.
Because of these known dose-related issues and more recently acquired volumetric dosimetry information facilitating the choice of dose objectives for RT planning, it may be more useful to tailor the EBRT (or brachytherapy) strategy to the needs of the anatomic setting and the volume of tissue that needs to be irradiated, especially if there is need to encompass vulnerable normal tissue structures. Such modifications may include combining moderate dose levels of both brachytherapy and EBRT, choosing preoperative versus postoperative external beam approaches to reduce risks of fracture, peripheral nerve injury, and other late RT toxicities, or using techniques such as IMRT.205
Radiation Target Volume
The local growth characteristics of STS have suggested that tumor cells may be located in the surrounding soft tissues at some distance from the main tumor mass.206,207 The original observation led to the development of the concept of a “reactive zone” surrounding the lesion, situated between the tumor margin/pseudocapsule and more remote normal tissues. The reactive zone was described as consisting of a granulation tissue–like proliferation whose characteristics included edema and neovascularity and, in addition, might contain satellite tumor cells.207 MRI has documented high T2-weighted signal changes surrounding STS. These radiographic abnormalities are thought to be caused by increased water content and have, therefore, been labeled “peritumoral edema.” However, it is not clear whether these MRI findings are actually the result of tissue edema and correspond to the reactive zone noted earlier or also contain tumor cells.208
With this in mind, we reported a small series (n = 15) of patients in which we identified if satellite tumor cells can be identified histologically in the tissues surrounding STS and whether their presence correlates with increased T2-weighted signal intensity on MRI (Fig. 63-9). Sarcoma cells were identified histologically in the tissues beyond the tumor in 10 of 15 patients who had not received previous RT. In six cases, tumor cells were located within 1 cm of the tumor margin, and in four cases malignant cells were found at a distance greater than 1 cm and up to a maximum of 4 cm. The location of tumor cells beyond the margin did not correlate with tumor size, nor did it correlate with the location or extent of peritumoral changes.209
In the discussion that follows, dose-volume guidelines for both EBRT and brachytherapy are summarized (see Table 63-7) based on parameters used in two randomized controlled trials and bearing in mind anatomic and imaging characteristics of the lesions and normal tissues.
External Beam Volumes
Within the framework of uncertainty noted earlier, our policy has been to use a 4-cm CTV margin (approximating a 5-cm field margin after the PTV and penumbra are considered) beyond any imaging abnormalities, including “peritumoral edema,” or areas of surgical disruption irrespective of grade or size of the tumor. However, practice is variable and there have been no randomized trials in this area. Our policy reflects the practice undertaken in a randomized trial that provided very acceptable 5-year local control rates74,160 (see Table 63-6). IMRT combined with daily three-dimensional image-guided RT and a clinically proven immobilization method has afforded a reduction of our PTV margin for extremity sarcoma treatment in an evidence-based fashion to 5 mm.210,211
Other work in this area includes the description by Kim and colleagues212 of a preoperative three-dimensional RT volume for extremity STS. The CTV included the T1-weighted postgadolinium-defined gross tumor volume with 1.0 to 1.5 cm radial and 3.5 cm longitudinal margins. The PTV expansion was 5 to 7 mm where 95% of the prescribed dose was delivered with acceptable local control, DFS, and OS. In addition, the recently completed RTOG-0630 trial defined the preoperative RT volume with slightly smaller margins. For intermediate- to high-grade tumors greater than or equal to 8 cm, the CTV included the gross tumor and clinical microscopic margins with suspicious edema as defined by MRI T2-weighted imaging, plus 3-cm margins in the longitudinal dimension. The volume could be shortened to include the end of a compartment. The radial margin was defined as 1.5 cm, including any portion of the tumor not confined by an intact barrier to spread. For lower-grade tumors the CTV still included the GTV and suspicious edema, plus 2-cm margins in the longitudinal directions. The radial margin from the lesion was 1 cm, including any portion of the tumor not confined by an intact barrier to tumor spread.181
A randomized phase III trial of volume of postoperative RT given to adult patients with extremity STS is underway by the United Kingdom National Cancer Research Network (NCRN)214 that is comparing a 5-cm longitudinal margin from GTV or 1 cm from the surgical scar, whichever is longer in the craniocaudal direction, and a 2-cm axial margin (standard arm) versus a 2-cm longitudinal margin to GTV and a 2-cm axial margin (experimental arm) to assess if a reduced volume of postoperative RT will increase limb function without compromising local control.213 Patients in both arms of the study will receive 66 Gy over 6.5 weeks with a volume reduction after 25 fractions (50 Gy) in the standard arm to the same volume as used from the outset in the experimental arm. Limb function is to be assessed using the Toronto Extremity Salvage Score (TESS).
Brachytherapy Volumes
The American Brachytherapy Society has recommended at least a 2- to 5-cm longitudinal margin beyond the CTV and at least 1 cm beyond the lateral edge of the CTV183 (Fig. 63-10). These are based on general agreement by experts, although there is no consensus as to the exact size of the margin beyond the tumor bed. The CTV may be difficult to define but in general is represented by the volume of tissue considered at risk for microscopic extension of tumor and includes the tumor bed visualized on radiographic studies and under direct inspection intraoperatively.183 The brachytherapy protocol at MSKCC uses margins of 2 cm around the surgical bed.215,216
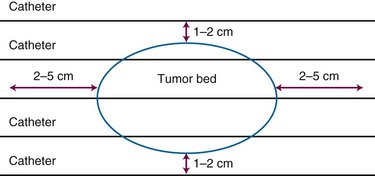
Other Local Management Approaches
Combined Preoperative Chemotherapy and Radiation Therapy
Enhancing the local effect of RT with concurrent chemotherapy while also promptly addressing the distant metastasis risk before the surgery is an attractive concept. Strategies include the use of perfusion techniques (see later) as well as a sequential intravenous chemotherapy and RT strategy in patients with localized, high-grade, large (>8 cm) extremity STS described by DeLaney and colleagues.217 The protocol involved three courses of doxorubicin (Adriamycin), ifosfamide, mesna, and dacarbazine (MAID) interdigitated with two 22-Gy courses of RT (11 fractions each) for a total preoperative radiation dose of 44 Gy. This was followed by surgical resection. An additional 16 Gy (8 fractions) boost dose was delivered if the surgical margins were microscopically positive. The outcomes of 48 patients treated with this regimen appeared superior to those of a matched cohort of historical controls. The 5-year OS were 87% versus 58% (p = .0003) for the MAID versus control patients cohorts with the difference linked to an improvement in the distant metastases rate (5-year freedom from distant metastasis rate of 75% versus 44% (p = .0016), and the local control was similar in both groups. Febrile neutropenia occurred in 25% of patients receiving the chemotherapy/RT sequential therapy; 14 (29%) of the patients treated with MAID also experienced wound complications. Late fatal myelodysplasia was seen in 1 patient who received chemotherapy.
Because of the promising results seen in this single-institution pilot study, the RTOG initiated a phase II trial to evaluate this neoadjuvant regimen in a multi-institutional intergroup setting. Kraybill and colleagues218 reported the short-term results of this phase II study in the management of high-risk, high-grade STS of the extremities and body wall. Patients received neoadjuvant modified MAID, interdigitated preoperative RT (44 Gy administered in split courses), and three cycles of postoperative modified MAID. Estimated 3-year rates for DFS, distant DFS, and OS were 56.6%, 64.5%, and 75.1%, respectively. There were three deaths associated with this protocol (5%). Two were secondary to acute myelogenous leukemia, and the third patient died secondary to leukopenic sepsis and respiratory failure. Grade IV toxicity was experienced by 84% of patients. Five patients required amputations, two of which were related to treatment toxicity. The authors noted the substantial toxicity of this trial, precluding its unmodified use outside of a clinical trial setting. Most recently, the long-term results have been published with a median follow-up for surviving patients of 7.7 years and all but four of the surviving patients having had more than 5 years’ follow-up. The 5-year estimated rates of disease-free and distant DFS, 56.1% and 64.1% respectively, are favorable for disease of this size and grade.219 In addition, the incidence of significant toxicity was markedly reduced after 1 year and almost completely resolved after 2 years.
Additional protocols have been developed with adjuvant and neoadjuvant chemotherapy because of the biology of STS and poor outcome. However, concern remains regarding efficacy and toxicity issues concerning these protocols.28,98,220,221
Neoadjuvant Chemotherapy with Regional Hyperthermia
The rationale behind using regional hyperthermia combined with chemotherapy is the theory that heat kills cells by direct thermal toxicity and will concentrate the action of chemotherapy within the heated volume, increase drug efficacy, and induce tumoricidal immune responses. Recently, the results of a randomized phase III multicenter clinical trial were reported that compared the effects of neoadjuvant chemotherapy with and without regional hyperthermia in addition to local therapy (surgery and RT when indicated) for localized intermediate/high-risk STS.222 Despite nine centers being involved, only three contributed the majority of patients to the trial (310 of 341). Local recurrence rates were high for both extremity and nonextremity tumors; the addition of hyperthermia seemed to benefit patients whose tumors were inadequately managed by surgery alone and who were treated at centers with experience administering this technique.223 The precise indications for the use of RT were not defined in the report of this study, although it has been suggested that RT may have been restricted to R1 and R2 resections, which may explain the high rate of local recurrence in this series.223
Isolated Limb Perfusion with Extracorporeal Bypass
Isolated limb perfusion (ILP) has been popularized in Europe and was developed predominantly as a means of addressing the more prevalent local control problem of limb threat from satellitosis and in-transit metastases complicating malignant melanoma. The essential requirement is limb arterial and venous access to isolate the limb from the remainder of the body through an extracorporeal circulation system. A heart-lung machine is used to reoxygenate the blood recirculating from the limb. The blood in the extracorporeal circuit is also often warmed to 39°C to 40°C to enhance the effect of chemotherapy.224 After isolation of the extremity a chemotherapeutic agent is perfused into the limb. Several have been used with the most effective or popular being melphalan with tumor necrosis factor–alpha (TNF-α) at a highly lethal dose under normal circumstances.225 TNF-α was initially approved in Europe after a multicenter trial225 in patients with locally advanced STS deemed unresectable by an independent review committee; the response rate to isolated limb perfusion with TNF-α plus melphalan was 76%, and the limb was saved in 71% of patients. Both components of the regimen (melphalan and TNF-α) appear important, and omission of TNF-α has resulted in a decrease in the tissue dose of melphalan, probably from its effects on the tumor vasculature with subsequent treatment failure.226 Recent efforts, including a randomized trial (see Table 63-6) have focused on attempts to reduce the dose of TNF to try to reduce toxicity.227,228 Hyperthermic ILP with lower dose TNF-α (1 mg) has been shown to be as effective in terms of response rates and patient outcomes for a group of patients with locally advanced or recurrent STS,229 including progressive desmoid-type fibromatosis. It has the benefit of lower systemic toxicity rates compared with protocols using higher TNF-α doses. Other authors230 have reported results using doxorubicin instead of melphalan and higher temperature (42° C).
The major component of the selection for the ILP approach hinges on the decision that patients would otherwise require amputation with marked loss of function if treated in conventional ways. Following this, patients who develop resectable disease usually undergo surgery. The response rates for multiple primary STS and melanoma appear very similar: overall response of 95% and 96% for both diseases and complete response of 69% (in melanoma) and 61% (in multiple primary STS).224 Grünhagen and associates231 reported the outcome and prognostic factor analysis of 217 consecutive ILPs with TNF-α and melphalan for limb-threatening STS. The overall response rate was 75%, and limb salvage was achieved in 87% of the perfused limbs. The same authors have also reported efficacy of this treatment for locally advanced extremity desmoid tumors in a small study of 11 patients.232 Amputation was avoided in all cases, and the authors suggest it should be considered when resection without important functional sacrifice may be deemed improbable.
Recently, others have reported long-term damage of ILP to the healthy surrounding normal tissue. Hoven-Gondrie and colleagues233 evaluated the long-term morbidity of the combined treatment of ILP and delayed surgical resection followed by EBRT for 32 patients. Serious late toxic effects were seen in two thirds of the patients. Similarly, additional reports have described late morbidity, including critical leg ischemia necessitating limb amputation as well as pathologic fractures 5 to 10 years after treatment.234,235
Limb Perfusion without Extracorporeal Bypass
Techniques of percutaneous instillation of chemotherapy into the vessels of the affected limb have been employed for decades. Eilber and colleagues190 first described the use of preoperative intra-arterial chemotherapy with doxorubicin combined with RT. This infusional approach is conceptually attractive because it provides high doses of chemotherapy directly into the affected limb but, as noted later, was later found equivalent to intravenous chemotherapy. A feature of the protocol described by Eilber and colleagues is the use of higher dose per fraction (3.5 Gy per fraction) compared with more conventional preoperative approaches. The total dose used in the protocol also has varied over the years with doses of 17.5 Gy, 28 Gy, and the original protocol of 35 Gy preceded by a 3-day infusion of intra-arterial doxorubicin. In the first of two randomized trials, the 17.5-Gy regimen was evaluated in combination with additional postoperative chemotherapy but it had no additional benefit compared with no further chemotherapy.236 In the second trial (n = 99), the intra-arterial approach with 28 Gy was compared with an intravenous approach and they were found not to be different.237
More recently, Hegazy and associates238 reported the results of 40 patients with locally advanced extremity STS (28 lower limbs, 12 upper limbs) who received preoperative isolated limb infusion with doxorubicin and external irradiation for limb-threatening STS. Isolated limb infusion was started 3 to 7 days before EBRT to a total dose of 35 Gy in 10 fractions. After a 3- to 7-week interval before surgery, tumor response was seen in 85% of patients, histologic response was seen in 80%, and limb salvage was achieved in 82.5% at a median follow-up of 15 months. One advantage advocated by Hegazy and associates238 is its use in limb-threatening presentations as opposed to limiting it to conventional adjuvant use. These promising results for this indication require validation in larger patient cohorts.
Particle Radiation Therapy
There is substantial interest in the use of heavier charged particles, in particular, carbon ions, because of their excellent physical dose distribution and higher relative biologic effectiveness in some situations. Serizawa and colleagues239 have reported the results of carbon ion RT for 24 patients with unresectable high-grade retroperitoneal sarcomas: 16 had primary disease and 8 had recurrent disease. The dose ranged from 52.8 to 73.6 GyE (Gy-equivalent) in 16 fixed fractions over 4 weeks with OS at 2 and 5 years of 75% and 50%, respectively. Local control rates were 77% and 69% at 2 and 5 years. Of interest were the low toxicities seen, with no gastrointestinal tract complications encountered and no observed toxicity greater than grade II. Others240,241 have reported similar survival for low-grade chondrosarcomas, locally advanced sarcomas not suitable for surgery, and recurrent tumors with low toxicity rates.
Spot scanning proton beam therapy has been used in the curative treatment of STS located in the vicinity of critical structures such as the spinal cord, optic apparatus, bowel, and kidney with comparable local control to EBRT approaches as well as acceptable toxicity levels. Experience is limited.242,243
Promising results were achieved in a phase II study of high-dose photon/proton RT with modern surgical techniques in the management of nonmetastatic spine and paraspinal sarcomas, particularly for primary spine tumors. For a total of 50 patients with a median follow-up of 48 months, the 5-year actuarial local control, recurrence-free survival, and OS were 78%, 63%, and 87%, respectively. Three sacral neuropathies appeared after 77.12 to 77.4 Gy relative biologic effectiveness.244
Adjuvant Chemotherapy
Meta-analysis of Randomized Trials
The results from 14 randomized trials performed over three decades were comprehensively combined from 1568 patients in a landmark meta-analysis124 that used individual patient data. This study addressed the role of doxorubicin-based adjuvant chemotherapy in STS. In the meta-analysis, the figures for distant relapse-free interval and overall recurrence-free survival were significantly in favor of the use of adjuvant chemotherapy. However, and most importantly, the difference in OS, the ultimate aim of adjuvant chemotherapy, was not significantly different between the chemotherapy and the control groups (p = .12).
Only three trials245–247 in the meta-analysis had a relatively large sample size. The inclusion of cases with minimal or only small risk of metastatic failure may have diluted the power of the studies to detect an effect that could only realistically apply to those patients who have a genuine risk of metastasis.
Studies Since the Individual Patient Data Meta-analysis
Since the publication of the meta-analysis, an important study was reported from Italy. The inclusion criteria for this trial were appropriately restricted to very adverse tumors (large high-grade lesions) and a very intensive cytotoxic regimen (five cycles of 4′-epidoxorubicin 60 mg/m2 days 1 and 2 and ifosfamide 1.8 g/m2 days 1 through 5, with hydration, mesna, and granulocyte colony-stimulating factor) compared with no systemic treatment.248 DFS and OS were statistically improved at preliminary analysis. However, by the time of publication, the metastatic rate in both study arms was virtually identical (44% and 45%), and it can be expected that the ultimate survival rate will also be the same.248 Unfortunately, this raises the question whether the potential value of aggressive chemotherapy may largely be in delaying the manifestation of distant metastasis but not its prevention.
Recently, a literature-based meta-analysis was performed including the 14 trials mentioned earlier with an additional 4 randomized trials included, for a total of 1953 patients.249 A combination of doxorubicin and ifosfamide was used in 5 trials. The mean follow-up was 4.9 years, and the absolute improvement for OS was 6% in favor of chemotherapy (40% vs. 46%, p = .003). Chemotherapy also reduced local recurrence by 4% (19% vs. 15%) and distant recurrence by 9% (37% vs. 28%). These results need to be verified in a further individual patient data meta-analysis.
Recent Analyses of Multicenter Data
Recently, several attempts have been made to pool data from large established cancer centers in the United States. One such study included all patients treated or not treated with adjuvant or neoadjuvant chemotherapy at M.D. Anderson Cancer Center and MSKCC but showed no statistical difference in OS for patients treated with chemotherapy versus no adjuvant chemotherapy.250 In contrast, combined nonrandomized data from the University of California, Los Angeles, and MSKCC showed that adjuvant chemotherapy may be useful for some STS subsets, including myxoid/round cell liposarcoma.220 Moreover, data from the MSKCC and the Dana-Farber Cancer Center institutional databases have suggested a significant improvement in disease-specific survival in patients with high-grade extremity STS greater than 10 cm.251 In interpreting such data it is important to recognize biases that may exist, and it seems clear that randomized trials should continue.
Influence of Chemotherapy on Local Control
The largest trial in the meta-analysis (EORTC 62771) compared the outcome of adjuvant CYVADIC chemotherapy (utilizing cyclophosphamide, vincristine, doxorubicin [Adriamycin], and dacarbazine [DTIC]) with controls in 468 patients.246 The relapse-free survival was significantly better after the CYVADIC regimen (56% vs. 43% for controls; p = .007), and local recurrence was significantly reduced (17% vs. 31%; p = .01). This favorable result in this subset analysis was confined to head, neck, and trunk tumors. In contrast, distant metastases rates were similar in both study arms as were OS (63% vs. 56%; p = .64). Thus adjuvant chemotherapy may improve local control in high-risk groups of STS, where local control has traditionally not been as satisfactory.
More recently, the EORTC Soft Tissue and Bone Sarcoma Group (STBSG) reported the preliminary results of a large adjuvant trial of chemotherapy with doxorubicin and ifosfamide (EORTC-STBSG 62931)252 that failed to demonstrate any significant difference in the relapse free survival or OS. More patients received RT in this recent trial compared with the EORTC-STBSG 62771 trial (73% vs. 42%), and, notably, outcome was improved in the EORTC-STBSG 62931 trial participants. Whether the composition of the trial will permit the final report to again address the interplay between anatomic complexity (especially in head and neck sites where surgery is frequently compromised because of functional and cosmetic considerations) and RT with or without chemotherapy and local control remains uncertain.
Summary of Evidence About Adjuvant Chemotherapy
Overall, even contemporary trials seem to provide conflicting results, leading to the conclusion that adjuvant chemotherapy has not proven to improve the outcome for patients with STS.28 For the high-risk patients, those presenting with large, high-grade and deep lesions (stage III disease), a benefit to adjuvant chemotherapy appears to be small if present at all. For this reason we tend to individualize treatment for a given clinical setting. Younger patients with relatively chemotherapy-sensitive subtypes such as myxoid/round cell liposarcoma and synovial sarcoma may experience benefit, and it has also been our policy to advise chemotherapy in patients presenting with lymph node involvement and in adverse presentations of head and neck or torso sarcoma when surgery and RT delivery are compromised.