315 |
Interstitial Lung Diseases |
Patients with interstitial lung diseases (ILDs) come to medical attention mainly because of the onset of progressive exertional dyspnea or a persistent nonproductive cough. Hemoptysis, wheezing, and chest pain may be present. Often, the identification of interstitial opacities on chest x-ray focuses the diagnostic approach on one of the ILDs.
ILDs represent a large number of conditions that involve the parenchyma of the lung—the alveoli, the alveolar epithelium, the capillary endothelium, and the spaces between those structures—as well as the perivascular and lymphatic tissues. The disorders in this heterogeneous group are classified together because of similar clinical, roentgenographic, physiologic, or pathologic manifestations. These disorders often are associated with considerable rates of morbidity and mortality, and there is little consensus regarding the best management of most of them.
ILDs have been difficult to classify because >200 known individual diseases are characterized by diffuse parenchymal lung involvement, either as the primary condition or as a significant part of a multiorgan process, as may occur in the connective tissue diseases (CTDs). One useful approach to classification is to separate the ILDs into two groups based on the major underlying histopathology: (1) those associated with predominant inflammation and fibrosis and (2) those with a predominantly granulomatous reaction in interstitial or vascular areas (Table 315-1). Each of these groups can be subdivided further according to whether the cause is known or unknown. For each ILD there may be an acute phase, and there is usually a chronic one as well. Rarely, some are recurrent, with intervals of subclinical disease.
|
MAJOR CATEGORIES OF ALVEOLAR AND INTERSTITIAL INFLAMMATORY LUNG DISEASE |

Sarcoidosis (Chap. 390), idiopathic pulmonary fibrosis (IPF), and pulmonary fibrosis associated with CTDs (Chaps. 378, 382, 388, and 427) are the most common ILDs of unknown etiology. Among the ILDs of known cause, the largest group includes occupational and environmental exposures, especially the inhalation of inorganic dusts, organic dusts, and various fumes or gases (Chap. 311). A multidisciplinary approach—requiring close communication between clinician, radiologist, and when appropriate, pathologist—is often required to make the diagnosis. High-resolution computed tomography (HRCT) scanning improves the diagnostic accuracy and may eliminate the need for tissue examination in many cases, especially in IPF. For other forms, tissue examination, usually obtained by thoracoscopic lung biopsy, is critical to confirmation of the diagnosis.
PATHOGENESIS
The ILDs are nonmalignant disorders and are not caused by identified infectious agents. The precise pathway(s) leading from injury to fibrosis is not known. Although there are multiple initiating agent(s) of injury, the immunopathogenic responses of lung tissue are limited, and the mechanisms of repair have common features (Fig. 315-1).
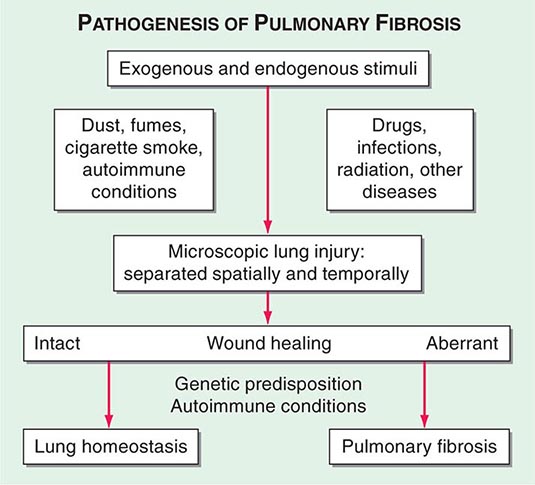
FIGURE 315-1 Proposed mechanism for the pathogenesis of pulmonary fibrosis. The lung is naturally exposed to repetitive injury from a variety of exogenous and endogenous stimuli. Several local and systemic factors (e.g., fibroblasts, circulating fibrocytes, chemokines, growth factors, and clotting factors) contribute to tissue healing and functional recovery. Dysregulation of this intricate network through genetic predisposition, autoimmune conditions, or super-imposed diseases can lead to aberrant wound healing, with the result of pulmonary fibrosis. Alternatively, excessive injury to the lung may overwhelm even intact reparative mechanisms and lead to pulmonary fibrosis. (From S Garantziotis et al: J Clin Invest 114:319, 2004.)
As mentioned above, the two major histopathologic patterns are a granulomatous pattern and a pattern in which inflammation and fibrosis predominate.
Granulomatous Lung Disease This process is characterized by an accumulation of T lymphocytes, macrophages, and epithelioid cells organized into discrete structures (granulomas) in the lung parenchyma. The granulomatous lesions can progress to fibrosis. Many patients with granulomatous lung disease remain free of severe impairment of lung function or, when symptomatic, improve after treatment. The main differential diagnosis is between sarcoidosis (Chap. 390) and hypersensitivity pneumonitis (Chap. 310).
Inflammation and Fibrosis The initial insult is an injury to the epithelial surface that causes inflammation in the air spaces and alveolar walls. If the disease becomes chronic, inflammation spreads to adjacent portions of the interstitium and vasculature and eventually causes interstitial fibrosis. Important histopathologic patterns found in the ILDs include usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia, respiratory bronchiolitis/desquamative interstitial pneumonia, organizing pneumonia, diffuse alveolar damage (acute or organizing), and lymphocytic interstitial pneumonia. The development of irreversible scarring (fibrosis) of alveolar walls, airways, or vasculature is the most feared outcome in all of these conditions because it is often progressive and leads to significant derangement of ventilatory function and gas exchange.
HISTORY
Duration of Illness Acute presentation (days to weeks), although unusual, occurs with allergy (drugs, fungi, helminths), acute interstitial pneumonia (AIP), eosinophilic pneumonia, and hypersensitivity pneumonitis. These conditions may be confused with atypical pneumonias because of diffuse alveolar opacities on chest x-ray. Subacute presentation (weeks to months) may occur in all ILDs but is seen especially in sarcoidosis, drug-induced ILDs, the alveolar hemorrhage syndromes, cryptogenic organizing pneumonia (COP), and the acute immunologic pneumonia that complicates systemic lupus erythematosus (SLE) or polymyositis. In most ILDs, the symptoms and signs form a chronic presentation (months to years). Examples include IPF, sarcoidosis, pulmonary Langerhans cell histiocytosis (PLCH), pneumoconioses, and CTDs. Episodic presentations are unusual and include eosinophilic pneumonia, hypersensitivity pneumonitis, COP, vasculitides, pulmonary hemorrhage, and Churg-Strauss syndrome.
Age Most patients with sarcoidosis, ILD associated with CTD, lymphangioleiomyomatosis (LAM), PLCH, and inherited forms of ILD (familial IPF, Gaucher disease, Hermansky-Pudlak syndrome) present between the ages of 20 and 40 years. Most patients with IPF are older than 60 years.
Gender LAM and pulmonary involvement in tuberous sclerosis occur exclusively in premenopausal women. In addition, ILD in Hermansky-Pudlak syndrome and in the CTDs is more common in women; an exception is ILD in rheumatoid arthritis (RA), which is more common in men. IPF is more common in men. Because of occupational exposures, pneumoconioses also occur more frequently in men.
Family History Familial lung fibrosis has been associated with mutations in the surfactant protein C gene, the surfactant protein A2 gene, telomerase reverse transcriptase (TERT), telomerase RNA component (TERC), and the promoter of a mucin gene (MUC5B). Familial lung fibrosis is characterized by several patterns of interstitial pneumonia, including nonspecific interstitial pneumonia, desquamative interstitial pneumonia, and UIP. Older age, male sex, and a history of cigarette smoking have been identified as risk factors for familial lung fibrosis. Family associations (with an autosomal dominant pattern) have been identified in tuberous sclerosis and neurofibromatosis. Familial clustering has been identified increasingly in sarcoidosis. The genes responsible for several rare ILDs have been identified, i.e., alveolar microlithiasis, Gaucher disease, Hermansky-Pudlak syndrome, and Niemann-Pick disease, along with the genes for surfactant homeostasis in pulmonary alveolar proteinosis and for control of cell growth and differentiation in LAM.
Smoking History Two-thirds to 75% of patients with IPF and familial lung fibrosis have a history of smoking. Patients with PLCH, respiratory bronchiolitis/desquamative interstitial pneumonia (DIP), Goodpasture syndrome, respiratory bronchiolitis, and pulmonary alveolar proteinosis are usually current or former smokers.
Occupational and Environmental History A strict chronologic listing of the patient’s lifelong employment must be sought, including specific duties and known exposures. In hypersensitivity pneumonitis (see Fig. 310-1), respiratory symptoms, fever, chills, and an abnormal chest roentgenogram are often temporally related to a hobby (pigeon breeder’s disease) or to the workplace (farmer’s lung) (Chap. 310). Symptoms may diminish or disappear after the patient leaves the site of exposure for several days; similarly, symptoms may reappear when the patient returns to the exposure site.
Other Important Past History Parasitic infections may cause pulmonary eosinophilia, and therefore a travel history should be taken in patients with known or suspected ILD. History of risk factors for HIV infection should be elicited because several processes may occur at the time of initial presentation or during the clinical course, e.g., HIV infection, organizing pneumonia, AIP, lymphocytic interstitial pneumonitis, and diffuse alveolar hemorrhage.
Respiratory Symptoms and Signs Dyspnea is a common and prominent complaint in patients with ILD, especially the idiopathic interstitial pneumonias, hypersensitivity pneumonitis, COP, sarcoidosis, eosinophilic pneumonias, and PLCH. Some patients, especially those with sarcoidosis, silicosis, PLCH, hypersensitivity pneumonitis, lipoid pneumonia, or lymphangitis carcinomatosis, may have extensive parenchymal lung disease on chest imaging studies without significant dyspnea, especially early in the course of the illness. Wheezing is an uncommon manifestation of ILD but has been described in patients with chronic eosinophilic pneumonia, Churg-Strauss syndrome, respiratory bronchiolitis, and sarcoidosis. Clinically significant chest pain is uncommon in most ILDs. However, substernal discomfort is common in sarcoidosis. Sudden worsening of dyspnea, especially if associated with acute chest pain, may indicate a spontaneous pneumothorax, which occurs in PLCH, tuberous sclerosis, LAM, and neurofibromatosis. Frank hemoptysis and blood-streaked sputum are rarely presenting manifestations of ILD but can be seen in the diffuse alveolar hemorrhage (DAH) syndromes, LAM, tuberous sclerosis, and the granulomatous vasculitides. Fatigue and weight loss are common in all ILDs.
PHYSICAL EXAMINATION
The findings are usually not specific. Most commonly, physical examination reveals tachypnea and bibasilar end-inspiratory dry crackles, which are common in most forms of ILD associated with inflammation but are less likely to be heard in the granulomatous lung diseases. Crackles may be present in the absence of radiographic abnormalities on the chest radiograph. Scattered late inspiratory high-pitched rhonchi—so-called inspiratory squeaks—are heard in patients with bronchiolitis. The cardiac examination is usually normal except in the middle or late stages of the disease, when findings of pulmonary hypertension and cor pulmonale may become evident (Chap. 304). Cyanosis and clubbing of the digits occur in some patients with advanced disease.
LABORATORY
Antinuclear antibodies and anti-immunoglobulin antibodies (rheumatoid factors) are identified in some patients, even in the absence of a defined CTD. A raised lactate dehydrogenase (LDH) level is a nonspecific finding common to ILDs. Elevation of the serum level of angiotensin-converting enzyme is common in ILDs, especially sarcoidosis. Serum precipitins confirm exposure when hypersensitivity pneumonitis is suspected, although they are not diagnostic of the process. Antineutrophil cytoplasmic or anti-basement membrane antibodies are useful if vasculitis is suspected. The electrocardiogram is usually normal unless pulmonary hypertension is present; then it demonstrates right-axis deviation, right ventricular hypertrophy, or right atrial enlargement or hypertrophy. Echocardiography also reveals right ventricular dilation and/or hypertrophy in the presence of pulmonary hypertension.
CHEST IMAGING STUDIES
Chest X-Ray ILD may be first suspected on the basis of an abnormal chest radiograph, which most commonly reveals a bibasilar reticular pattern. A nodular or mixed pattern of alveolar filling and increased reticular markings also may be present. Subgroups of ILDs exhibit nodular opacities with a predilection for the upper lung zones (sarcoidosis, PLCH, chronic hypersensitivity pneumonitis, silicosis, berylliosis, RA [necrobiotic nodular form], ankylosing spondylitis). The chest x-ray correlates poorly with the clinical or histopathologic stage of the disease. The radiographic finding of honeycombing correlates with pathologic findings of small cystic spaces and progressive fibrosis; when present, it portends a poor prognosis. In most cases, the chest radiograph is nonspecific and usually does not allow a specific diagnosis.
Computed Tomography HRCT is superior to the plain chest x-ray for early detection and confirmation of suspected ILD (Fig. 315-2). In addition, HRCT allows better assessment of the extent and distribution of disease, and it is especially useful in the investigation of patients with a normal chest radiograph. Coexisting disease is often best recognized on HRCT scanning, e.g., mediastinal adenopathy, carcinoma, or emphysema. In the appropriate clinical setting, HRCT may be sufficiently characteristic to preclude the need for lung biopsy in IPF, sarcoidosis, hypersensitivity pneumonitis, asbestosis, lymphangitic carcinoma, and PLCH. When a lung biopsy is required, HRCT scanning is useful for determining the most appropriate area from which biopsy samples should be taken.
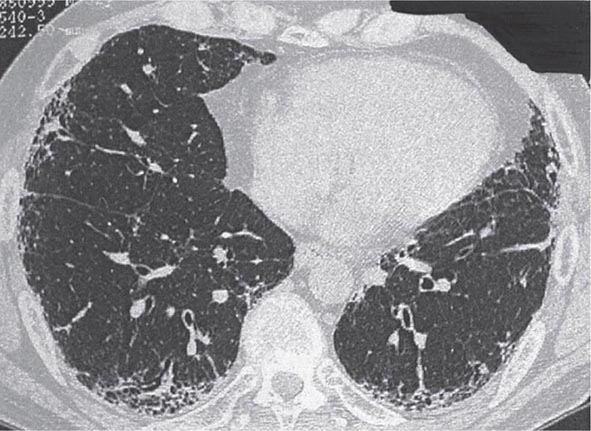
FIGURE 315-2 Idiopathic pulmonary fibrosis. High-resolution computed tomography image shows bibasal, peripheral predominant reticular abnormality with traction bronchiectasis and honeycombing. The lung biopsy showed the typical features of usual interstitial pneumonia.
PULMONARY FUNCTION TESTING
Spirometry and Lung Volumes Measurement of lung function is important in assessing the extent of pulmonary involvement in patients with ILD. Most forms of ILD produce a restrictive defect with reduced total lung capacity (TLC), functional residual capacity, and residual volume (Chap. 306e). Forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) are reduced, but these changes are related to the decreased TLC. The FEV1/FVC ratio is usually normal or increased. Lung volumes decrease as lung stiffness worsens with disease progression. A few disorders produce interstitial opacities on chest x-ray and obstructive airflow limitation on lung function testing (uncommon in sarcoidosis and hypersensitivity pneumonitis but common in tuberous sclerosis and LAM). Pulmonary function studies have been proved to have prognostic value in patients with idiopathic interstitial pneumonias, particularly IPF and nonspecific interstitial pneumonia (NSIP).
Diffusing Capacity A reduction in the diffusing capacity of the lung for carbon monoxide (DLCO) is a common but nonspecific finding in most ILDs. This decrease is due in part to effacement of the alveolar capillary units but, more important, to mismatching of ventilation and perfusion (![]() /
/![]() ). Lung regions with reduced compliance due to either fibrosis or cellular infiltration may be poorly ventilated but may still maintain adequate blood flow, and the ventilation-perfusion mismatch in these regions acts like true venous admixture. The severity of the reduction in DLCO does not correlate with disease stage.
). Lung regions with reduced compliance due to either fibrosis or cellular infiltration may be poorly ventilated but may still maintain adequate blood flow, and the ventilation-perfusion mismatch in these regions acts like true venous admixture. The severity of the reduction in DLCO does not correlate with disease stage.
Arterial Blood Gas The resting arterial blood gas may be normal or reveal hypoxemia (secondary to a mismatching of ventilation to perfusion) and respiratory alkalosis. A normal arterial O2 tension (or saturation by oximetry) at rest does not rule out significant hypoxemia during exercise or sleep. Carbon dioxide (CO2) retention is rare and is usually a manifestation of end-stage disease.
CARDIOPULMONARY EXERCISE TESTING
Because hypoxemia at rest is not always present and because severe exercise-induced hypoxemia may go undetected, it is useful to perform exercise testing with measurement of arterial blood gases to detect abnormalities of gas exchange. Arterial oxygen desaturation, a failure to decrease dead space appropriately with exercise (i.e., a high VD/VT [dead space/tidal volume] ratio [Chap. 306e]), and an excessive increase in respiratory rate with a lower than expected recruitment of tidal volume provide useful information about physiologic abnormalities and extent of disease. Serial assessment of resting and exercise gas exchange is an excellent method for following disease activity and responsiveness to treatment, especially in patients with IPF. Increasingly, the 6-min walk test is used to obtain a global evaluation of submaximal exercise capacity in patients with ILD. The walk distance and level of oxygen desaturation tend to correlate with the patient’s baseline lung function and mirror the patient’s clinical course.
FIBEROPTIC BRONCHOSCOPY AND BRONCHOALVEOLAR LAVAGE (BAL)
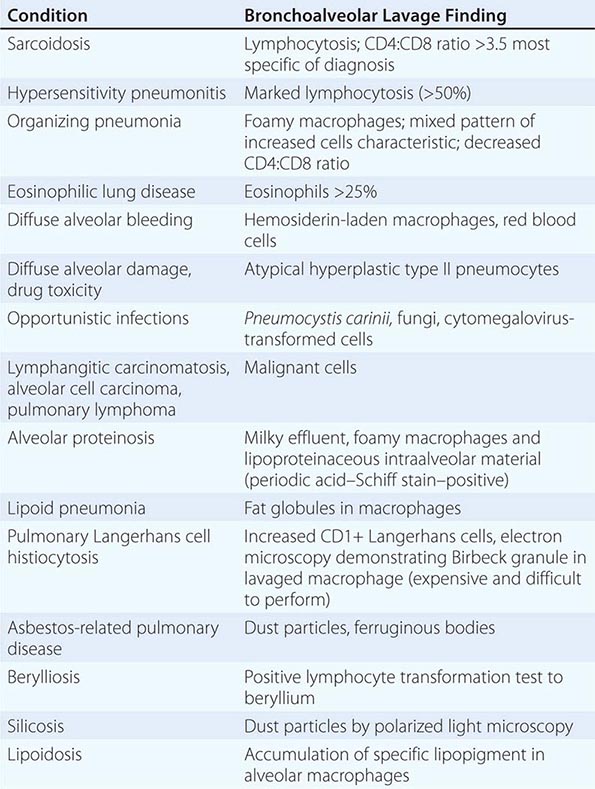
In selected diseases (e.g., sarcoidosis, hypersensitivity pneumonitis, DAH syndrome, cancer, pulmonary alveolar proteinosis), cellular analysis of BAL fluid may be useful in narrowing the differential diagnostic possibilities among various types of ILD (Table 315-2). The role of BAL in defining the stage of disease and assessment of disease progression or response to therapy remains poorly understood, and the usefulness of BAL in the clinical assessment and management remains to be established.
|
DIAGNOSTIC VALUE OF BRONCHOALVEOLAR LAVAGE IN INTERSTITIAL LUNG DISEASE |
TISSUE AND CELLULAR EXAMINATION
Lung biopsy is the most effective method for confirming the diagnosis and assessing disease activity. The findings may identify a more treatable process than originally suspected, particularly chronic hypersensitivity pneumonitis, COP, respiratory bronchiolitis–associated ILD, or sarcoidosis. Biopsy should be obtained before the initiation of treatment. A definitive diagnosis avoids confusion and anxiety later in the clinical course if the patient does not respond to therapy or experiences serious side effects from it.
Fiberoptic bronchoscopy with multiple transbronchial lung biopsies (four to eight biopsy samples) is often the initial procedure of choice, especially when sarcoidosis, lymphangitic carcinomatosis, eosinophilic pneumonia, Goodpasture syndrome, or infection is suspected. If a specific diagnosis is not made by transbronchial biopsy, surgical lung biopsy by video-assisted thoracic surgery or open thoracotomy is indicated. Adequate-sized biopsies from multiple sites, usually from two lobes, should be obtained. Relative contraindications to lung biopsy include serious cardiovascular disease, honeycombing and other roentgenographic evidence of diffuse end-stage disease, severe pulmonary dysfunction, and other major operative risks, especially in the elderly.
INDIVIDUAL FORMS OF INTERSTITIAL LUNG DISEASE
IDIOPATHIC PULMONARY FIBROSIS
IPF is the most common form of idiopathic interstitial pneumonia. Separating IPF from other forms of lung fibrosis is an important step in the evaluation of all patients presenting with ILD. IPF has a distinctly poor response to therapy and a bad prognosis.
Clinical Manifestations Exertional dyspnea, a nonproductive cough, and inspiratory crackles with or without digital clubbing may be present on physical examination. HRCT lung scans typically show patchy, predominantly basilar, subpleural reticular opacities, often associated with traction bronchiectasis and honeycombing (Fig. 315-2). A definite UIP pattern on HRCT is highly accurate for the presence of a UIP pattern on surgical lung biopsy. Atypical findings that should suggest an alternative diagnosis include extensive ground-glass abnormality, nodular opacities, upper or midzone predominance, and prominent hilar or mediastinal lymphadenopathy. Pulmonary function tests often reveal a restrictive pattern, a reduced DLCO, and arterial hypoxemia that is exaggerated or elicited by exercise.
Histologic Findings Confirmation of the presence of the UIP pattern on histologic examination is essential to confirm this diagnosis. Transbronchial biopsies are not helpful in making the diagnosis of UIP, and surgical biopsy usually is required. The histologic hallmark and chief diagnostic criterion of UIP is a heterogeneous appearance at low magnification with alternating areas of normal lung, interstitial inflammation, foci of proliferating fibroblasts, dense collagen fibrosis, and honeycomb changes. These histologic changes affect the peripheral, subpleural parenchyma most severely. The interstitial inflammation is usually patchy and consists of a lymphoplasmacytic infiltrate in the alveolar septa, associated with hyperplasia of type 2 pneumocytes. The fibrotic zones are composed mainly of dense collagen, although scattered foci of proliferating fibroblasts are a consistent finding. The extent of fibroblastic proliferation is predictive of disease progression. Areas of honeycomb change are composed of cystic fibrotic air spaces that frequently are lined by bronchiolar epithelium and filled with mucin. Smooth-muscle hyperplasia is commonly seen in areas of fibrosis and honeycomb change. A fibrotic pattern with some features similar to UIP may be found in the chronic stage of several specific disorders, such as pneumoconioses (e.g., asbestosis), radiation injury, certain drug-induced lung diseases (e.g., nitrofurantoin), chronic aspiration, sarcoidosis, chronic hypersensitivity pneumonitis, organized chronic eosinophilic pneumonia, and PLCH. Commonly, other histopathologic features are present in these situations, thus allowing separation of these lesions from the UIP-like pattern. Consequently, the term usual interstitial pneumonia is used for patients in whom the lesion is idiopathic and not associated with another condition.
NONSPECIFIC INTERSTITIAL PNEUMONIA
This condition defines a subgroup of the idiopathic interstitial pneumonias that can be distinguished clinically and pathologically from UIP, DIP, AIP, and COP. Importantly, many cases with this histopathologic pattern occur in the context of an underlying disorder, such as a CTD, drug-induced ILD, or chronic hypersensitivity pneumonitis.
Clinical Manifestations Patients with idiopathic NSIP have clinical, serologic, radiographic, and pathologic characteristics highly suggestive of autoimmune disease and meet the criteria for undifferentiated CTD. Idiopathic NSIP is a subacute restrictive process with a presentation similar to that of IPF but usually at a younger age, most commonly in women who have never smoked. It is often associated with a febrile illness. HRCT shows bilateral, subpleural ground-glass opacities, often associated with lower lobe volume loss (Fig. 315-3). Patchy areas of airspace consolidation and reticular abnormalities may be present, but honeycombing is unusual.
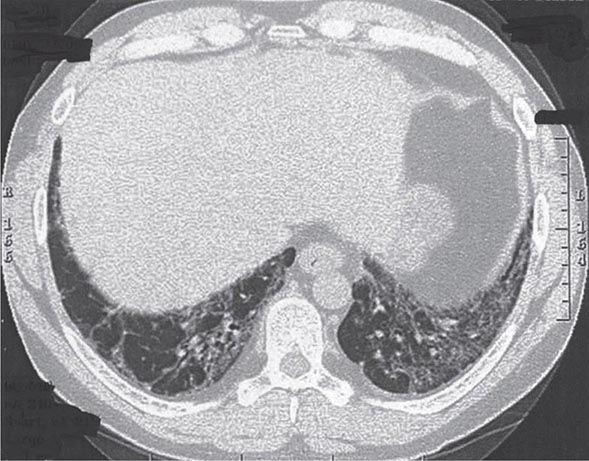
FIGURE 315-3 Nonspecific interstitial pneumonia. High-resolution computed tomography through the lower lung shows volume loss with extensive ground-glass abnormality, reticular abnormality, and traction bronchiectasis. There is sparing on the lung immediately adjacent to the pleura. Histology showed a combination of inflammation and mild fibrosis.
Histologic Findings The key histopathologic feature of NSIP is the uniformity of interstitial involvement across the biopsy section, and this may be predominantly cellular or fibrosing. There is less temporal and spatial heterogeneity than in UIP, and little or no honeycombing is found. The cellular variant is rare.
Treatment The majority of patients with NSIP have a good prognosis (5-year mortality rate estimated at <15%), with most showing improvement after treatment with glucocorticoids, often used in combination with azathioprine or mycophenolate mofetil.
ACUTE INTERSTITIAL PNEUMONIA (HAMMAN-RICH SYNDROME)
Clinical Manifestations AIP is a rare, fulminant form of lung injury characterized histologically by diffuse alveolar damage on lung biopsy. Most patients are older than 40 years. AIP is similar in presentation to the acute respiratory distress syndrome (ARDS) (Chap. 322) and probably corresponds to the subset of cases of idiopathic ARDS. The onset is usually abrupt in a previously healthy individual. A prodromal illness, usually lasting 7–14 days before presentation, is common. Fever, cough, and dyspnea are common manifestations at presentation. Diffuse, bilateral, air-space opacification is present on the chest radiograph. HRCT scans show bilateral, patchy, symmetric areas of ground-glass attenuation. Bilateral areas of air-space consolidation also may be present. A predominantly subpleural distribution may be seen.
Histologic Findings The diagnosis of AIP requires the presence of a clinical syndrome of idiopathic ARDS and pathologic confirmation of organizing diffuse alveolar damage. Therefore, lung biopsy is required to confirm the diagnosis.
Treatment Most patients have moderate to severe hypoxemia and develop respiratory failure. Mechanical ventilation is often required. The mortality rate is high (>60%), with most patients dying within 6 months of presentation. Recurrences have been reported. However, those who recover often have substantial improvement in lung function. The main treatment is supportive. It is not clear that glucocorticoid therapy is effective.
CRYPTOGENIC ORGANIZING PNEUMONIA
Clinical Manifestations COP is a clinicopathologic syndrome of unknown etiology. The onset is usually in the fifth and sixth decades. The presentation may be of a flulike illness with cough, fever, malaise, fatigue, and weight loss. Inspiratory crackles are frequently present on examination. Pulmonary function is usually impaired, with a restrictive defect and arterial hypoxemia being most common. The roentgenographic manifestations are distinctive, revealing bilateral, patchy, or diffuse alveolar opacities in the presence of normal lung volume. Recurrent and migratory pulmonary opacities are common. HRCT shows areas of air-space consolidation, ground-glass opacities, small nodular opacities, and bronchial wall thickening and dilation. These changes occur more frequently in the periphery of the lung and in the lower lung zone.
Histologic Findings Lung biopsy shows granulation tissue within small airways, alveolar ducts, and airspaces, with chronic inflammation in the surrounding alveoli. Foci of organizing pneumonia are a nonspecific reaction to lung injury found adjacent to other pathologic processes or as a component of other primary pulmonary disorders (e.g., cryptococcosis, granulomatosis with polyangiitis [Wegener], lymphoma, hypersensitivity pneumonitis, and eosinophilic pneumonia). Consequently, the clinician must carefully reevaluate any patient found to have this histopathologic lesion to rule out these possibilities.
Treatment Glucocorticoid therapy induces clinical recovery in two-thirds of patients. A few patients have rapidly progressive courses with fatal outcomes despite glucocorticoids.
ILD ASSOCIATED WITH CIGARETTE SMOKING
Desquamative Interstitial Pneumonia • CLINICAL MANIFESTATIONS DIP is a rare but distinct clinical and pathologic entity found almost exclusively in cigarette smokers. The histologic hallmark is the extensive accumulation of macrophages in intraalveolar spaces with minimal interstitial fibrosis. The peak incidence is in the fourth and fifth decades. Most patients present with dyspnea and cough. Lung function testing shows a restrictive pattern with reduced DLCO and arterial hypoxemia. The chest x-ray and HRCT scans usually show diffuse hazy opacities.
HISTOLOGIC FINDINGS A diffuse and uniform accumulation of macrophages in the alveolar spaces is the hallmark of DIP. The macrophages contain golden, brown, or black pigment of tobacco smoke. There may be mild thickening of the alveolar walls by fibrosis and scanty inflammatory cell infiltration.
TREATMENT Clinical recognition of DIP is important because the process is associated with a better prognosis (10-year survival rate is ~70%) in response to smoking cessation. There are no clear data showing that systemic glucocorticoids are effective in DIP.
Respiratory Bronchiolitis–Associated ILD • CLINICAL MANIFESTATIONS Respiratory bronchiolitis–associated ILD (RB-ILD) is considered to be a subset of DIP and is characterized by the accumulation of macrophages in peribronchial alveoli. The clinical presentation is similar to that of DIP. Crackles are often heard on chest examination and occur throughout inspiration; sometimes they continue into expiration. The process is best seen on HRCT lung scanning, which shows bronchial wall thickening, centrilobular nodules, ground-glass opacity, and emphysema with air trapping. There is a spectrum of CT features in asymptomatic smokers (and elderly asymptomatic individuals) that may not necessarily represent clinically relevant disease.
HISTOLOGIC FINDINGS The histologic findings in RB-ILD include alveolar macrophage accumulation in respiratory bronchioles, with a variable chronic inflammatory cell infiltrate in bronchiolar and surrounding alveolar walls and occasional peribronchial alveolar septal fibrosis. The pulmonary parenchyma may show presence of smoking-related emphysema.
TREATMENT RB-ILD appears to resolve in most patients after smoking cessation alone.
Pulmonary Langerhans Cell Histiocytosis • CLINICAL MANIFESTATIONS This is a rare, smoking-related, diffuse lung disease that primarily affects men between the ages of 20 and 40 years. The clinical presentation varies from an asymptomatic state to a rapidly progressive condition. The most common clinical manifestations at presentation are cough, dyspnea, chest pain, weight loss, and fever. Pneumothorax occurs in ~25% of patients. Hemoptysis and diabetes insipidus are rare manifestations. The radiographic features vary with the stage of the disease. The combination of ill-defined or stellate nodules (2–10 mm in diameter), reticular or nodular opacities, bizarre-shaped upper zone cysts, preservation of lung volume, and sparing of the costophrenic angles are characteristics of PLCH. HRCT that reveals a combination of nodules and thin-walled cysts is virtually diagnostic of PLCH. The most common pulmonary function abnormality is a markedly reduced DLCO, although varying degrees of restrictive disease, airflow limitation, and diminished exercise capacity may occur.
HISTOLOGIC FINDINGS The characteristic histopathologic finding in PLCH is the presence of nodular sclerosing lesions that contain Langerhans cells accompanied by mixed cellular infiltrates. The nodular lesions are poorly defined and are distributed in a bronchiolocentric fashion with intervening normal lung parenchyma. As the disease advances, fibrosis progresses to involve adjacent lung tissue, leading to pericicatricial air space enlargement, which accounts for the concomitant cystic changes.
TREATMENT Discontinuance of smoking is the key treatment, resulting in clinical improvement in one-third of patients. Most patients with PLCH experience persistent or progressive disease. Death due to respiratory failure occurs in ~10% of patients.
ILD ASSOCIATED WITH CONNECTIVE TISSUE DISORDERS
Clinical findings suggestive of a CTD (musculoskeletal pain, weakness, fatigue, fever, joint pain or swelling, photosensitivity, Raynaud’s phenomenon, pleuritis, dry eyes, dry mouth) should be sought in any patient with ILD. The CTDs may be difficult to rule out since the pulmonary manifestations occasionally precede the more typical systemic manifestations by months or years. The most common form of pulmonary involvement is the nonspecific interstitial pneumonia histopathologic pattern. However, determining the precise nature of lung involvement in most of the CTDs is difficult due to the high incidence of lung involvement caused by disease-associated complications of esophageal dysfunction (predisposing to aspiration and secondary infections), respiratory muscle weakness (atelectasis and secondary infections), complications of therapy (opportunistic infections), and associated malignancies. For the majority of CTDs, with the exception of progressive system sclerosis, recommended initial treatment for ILD includes oral glucocorticoids often in association with an immunosuppressive agent (usually oral or intravenous cyclophosphamide or oral azathioprine) or mycophenolate mofetil.
Progressive Systemic Sclerosis (PSS) • CLINICAL MANIFESTATIONS (See also Chap. 382) Clinical evidence of ILD is present in about one-half of patients with PSS, and pathologic evidence in three-quarters. Pulmonary function tests show a restrictive pattern and impaired diffusing capacity, often before any clinical or radiographic evidence of lung disease appears. The HRCT features of lung disease in PSS range from predominant ground-glass attenuation to a predominant reticular pattern and are mostly similar to idiopathic NSIP.
HISTOLOGIC FINDINGS NSIP is the histopathologic pattern in most patients (~75%); the UIP pattern is rare (<10%).
TREATMENT Therapy is similar to that in idiopathic NSIP. UIP in PSS has a better outcome than IPF. The most widely used initial treatment regimen is low-dose glucocorticoid therapy and an immunosuppressive agent, usually oral or pulse cyclophosphamide. There are no convincing data showing this regime to be efficacious, and there is concern that the risk of renal crisis rises substantially with corticosteroids. Pulmonary vascular disease alone or in association with pulmonary fibrosis, pleuritis, or recurrent aspiration pneumonitis is strikingly resistant to current modes of therapy.
Rheumatoid Arthritis • CLINICAL MANIFESTATIONS (See also Chap. 380) ILD associated with RA is more common in men. Pulmonary manifestations of RA include pleurisy with or without effusion, ILD in up to 20% of cases, necrobiotic nodules (nonpneumoconiotic intrapulmonary rheumatoid nodules) with or without cavities, Caplan syndrome (rheumatoid pneumoconiosis), pulmonary hypertension secondary to rheumatoid pulmonary vasculitis, organized pneumonia, and upper airway obstruction due to cricoarytenoid arthritis.
HISTOLOGIC FINDINGS There are two primary histopathologic patterns of ILD that are observed in patients with ILD associated with RA: NSIP pattern and UIP pattern.
TREATMENT Little data exist to support the management of ILD in RA. Initial treatment of rheumatoid ILD, if required, is typically with oral glucocorticoids, which should be tried for 1–3 months. The potential benefit of anti-tumor necrosis factor α (TNF-α) therapy has been clouded by concerns about the development of a rapid and occasionally fatal lung disease in patients with RA-associated ILD treated with anti-TNF-α therapy.
Systemic Lupus Erythematosus • CLINICAL MANIFESTATIONS (See also Chap. 378) Lung disease is a common complication in SLE. Pleuritis with or without effusion is the most common pulmonary manifestation. Other lung manifestations include the following: atelectasis, diaphragmatic dysfunction with loss of lung volumes, pulmonary vascular disease, pulmonary hemorrhage, uremic pulmonary edema, infectious pneumonia, and organized pneumonia. Acute lupus pneumonitis characterized by pulmonary capillaritis leading to alveolar hemorrhage is uncommon. Chronic, progressive ILD is uncommon (<10%). It is important to exclude pulmonary infection. Although pleuropulmonary involvement may not be evident clinically, pulmonary function testing, particularly DLCO, reveals abnormalities in many patients with SLE.
HISTOLOGIC FINDINGS The most common pathologic patterns seen include NSIP, UIP, LIP, and, on occasion, organizing pneumonia and amyloidosis.
TREATMENT There have been no controlled trials of treatment for ILD in SLE. Treatment involves the use of a glucocorticoid, either alone or, more often, in combination with an additional immunomodulating agent.
Polymyositis and Dermatomyositis (PM/DM) • CLINICAL MANIFESTATIONS (See also Chap. 388) ILD occurs in ~10% of patients with PM/DM. Diffuse reticular or nodular opacities with or without an alveolar component occur radiographically, with a predilection for the lung bases (NSIP pattern). ILD occurs more commonly in the subgroup of patients with an anti-Jo-1 antibody that is directed to histidyl tRNA synthetase. Weakness of respiratory muscles contributing to aspiration pneumonia may be present. A rapidly progressive illness characterized by diffuse alveolar damage may cause respiratory failure.
HISTOLOGIC FINDINGS NSIP predominates over UIP, organizing pneumonia, or other patterns of interstitial pneumonia.
TREATMENT The optimal treatment is unknown. The most widely used initial treatment is oral glucocorticoids. Fulminant disease may require high-dose intravenous methylprednisolone (1.0 g/d) for 3–5 days.
Sjögren Syndrome • CLINICAL MANIFESTATIONS (See also Chap. 383) General dryness and lack of airway secretion cause the major problems of hoarseness, cough, and bronchitis.
HISTOLOGIC FINDINGS Lung biopsy is frequently required to establish a precise pulmonary diagnosis. Fibrotic NSIP is most common. Lymphocytic interstitial pneumonitis, lymphoma, pseudolymphoma, bronchiolitis, and bronchiolitis obliterans are associated with this condition.
TREATMENT Glucocorticoids have been used in the management of ILD associated with Sjögren syndrome with some degree of clinical success.
DRUG-INDUCED ILD
Clinical Manifestations Many classes of drugs have the potential to induce diffuse ILD, which is manifest most commonly as exertional dyspnea and nonproductive cough. A detailed history of the medications taken by the patient is needed to identify drug-induced disease, including over-the-counter medications, oily nose drops, and petroleum products (mineral oil). In most cases, the pathogenesis is unknown, although a combination of direct toxic effects of the drug (or its metabolite) and indirect inflammatory and immunologic events are likely. The onset of the illness may be abrupt and fulminant, or it may be insidious, extending over weeks to months. The drug may have been taken for several years before a reaction develops (e.g., amiodarone), or the lung disease may occur weeks to years after the drug has been discontinued (e.g., carmustine). The extent and severity of disease are usually dose-related.
Histologic Findings The patterns of lung injury vary widely and depend on the agent.
Treatment Treatment consists of discontinuation of any possible offending drug and supportive care.
EOSINOPHILIC PNEUMONIA
(see Chap. 310)
PULMONARY ALVEOLAR PROTEINOSIS (PAP)
Clinical Manifestations Although not strictly an ILD, PAP resembles and is therefore considered with these conditions. It has been proposed that a defect in macrophage function, more specifically an impaired ability to process surfactant, may play a role in the pathogenesis of PAP. PAP is an autoimmune disease with a neutralizing antibody of immunoglobulin G isotype against granulocyte-macrophage colony-stimulating factor (GM-CSF). These findings suggest that neutralization of GM-CSF bioactivity by the antibody causes dysfunction of alveolar macrophages, which results in reduced surfactant clearance. There are three distinct classes of PAP: acquired (>90% of all cases), congenital, and secondary. Congenital PAP is transmitted in an autosomal recessive manner and is caused by homozygosity for a frameshift mutation (121ins2) in the SP-B gene, which leads to an unstable SP-B mRNA, reduced protein levels, and secondary disturbances of SP-C processing. Secondary PAP is rare among adults and is caused by lysinuric protein intolerance, acute silicosis and other inhalational syndromes, immunodeficiency disorders, and malignancies (almost exclusively of hematopoietic origin) and hematopoietic disorders.
The typical age of presentation is 30–50 years, and males predominate. The clinical presentation is usually insidious and is manifested by progressive exertional dyspnea, fatigue, weight loss, and low-grade fever. A nonproductive cough is common, but occasionally expectoration of “chunky” gelatinous material may occur. Polycythemia, hypergammaglobulinemia, and increased LDH levels are common. Markedly elevated serum levels of lung surfactant proteins A and D have been found in PAP. In the absence of any known secondary cause of PAP, an elevated serum anti-GM-CSF titer is highly sensitive and specific for the diagnosis of acquired PAP. BAL fluid levels of anti-GM-CSF antibodies correlate better with the severity of PAP than do serum titers. Radiographically, bilateral symmetric alveolar opacities located centrally in middle and lower lung zones result in a “bat-wing” distribution. HRCT shows a ground-glass opacification and thickened intralobular structures and interlobular septa.
Histologic Findings This diffuse disease is characterized by the accumulation of an amorphous, periodic acid–Schiff–positive lipoproteinaceous material in the distal air spaces. There is little or no lung inflammation, and the underlying lung architecture is preserved.
Treatment Whole-lung lavage(s) through a double-lumen endotracheal tube provides relief to many patients with dyspnea or progressive hypoxemia and also may provide long-term benefit.
PULMONARY LYMPHANGIOLEIOMYOMATOSIS
Clinical Manifestations Pulmonary LAM is a rare condition that afflicts premenopausal women and should be suspected in young women with “emphysema,” recurrent pneumothorax, or chylous pleural effusion. It is often misdiagnosed as asthma or chronic obstructive pulmonary disease. Whites are affected much more commonly than are members of other racial groups. The disease accelerates during pregnancy and abates after oophorectomy. Common complaints at presentation are dyspnea, cough, and chest pain. Hemoptysis may be life threatening. Spontaneous pneumothorax occurs in 50% of patients; it may be bilateral and necessitate pleurodesis. Meningioma and renal angiomyolipomas (hamartomas), characteristic findings in the genetic disorder tuberous sclerosis, are also common in patients with LAM. Chylothorax, chyloperitoneum (chylous ascites), chyluria, and chylopericardium are other complications. Pulmonary function testing usually reveals an obstructive or mixed obstructive-restrictive pattern, and gas exchange is often abnormal. HRCT shows thin-walled cysts surrounded by normal lung without zonal predominance.
Histologic Findings Pathologically, LAM is characterized by the proliferation of atypical pulmonary interstitial smooth muscle and cyst formation. The immature-appearing smooth-muscle cells react with monoclonal antibody HMB45, which recognizes a 100-kDa glycoprotein (gp100) originally found in human melanoma cells.
Treatment Progression is common, with a median survival of 8–10 years from diagnosis. No therapy is of proven benefit in LAM. Sirolimus, an inhibitor of the mammalian target of rapamycin (mTOR), appears to be an active agent for LAM. After 12 months, it stabilized lung function (FVC, FEV1, and functional residual capacity) and was associated with a reduction in symptoms and improvement in quality of life. Adverse effects (e.g., mucositis, diarrhea, nausea, hypercholesterolemia, acneiform rash, peripheral edema) were more common in the sirolimus group, but serious adverse effects were not increased. Subjects were followed off sirolimus for an additional 12 months, during which time pulmonary function declined at the same rate as in the placebo group. Progesterone and luteinizing hormone–releasing hormone analogues have been used. Oophorectomy is no longer recommended, and estrogen-containing drugs should be discontinued. Lung transplantation offers the only hope for cure despite reports of recurrent disease in the transplanted lung.
SYNDROMES OF ILD WITH DIFFUSE ALVEOLAR HEMORRHAGE
Clinical Manifestations The clinical onset is often abrupt, with cough, fever, and dyspnea. Severe respiratory distress requiring ventilatory support may be evident at initial presentation. Although hemoptysis is expected, it can be absent at the time of presentation in one-third of the cases. For patients without hemoptysis, new alveolar opacities, a falling hemoglobin level, and hemorrhagic BAL fluid point to the diagnosis. The chest radiograph is nonspecific and most commonly shows new patchy or diffuse alveolar opacities. Recurrent episodes of DAH may lead to pulmonary fibrosis, resulting in interstitial opacities on the chest radiograph. An elevated white blood cell count and falling hematocrit are common. Evidence for impaired renal function caused by focal segmental necrotizing glomerulonephritis, usually with crescent formation, also may be present. Varying degrees of hypoxemia may occur and are often severe enough to require ventilatory support. DLCO may be increased, resulting from the increased hemoglobin within the alveoli compartment.
Histologic Findings Injury to arterioles, venules, and the alveolar septal (alveolar wall or interstitial) capillaries can result in hemoptysis secondary to disruption of the alveolar-capillary basement membrane. This results in bleeding into the alveolar spaces, which characterizes DAH. Pulmonary capillaritis, characterized by a neutrophilic infiltration of the alveolar septae, may lead to necrosis of these structures, loss of capillary structural integrity, and the pouring of red blood cells into the alveolar space. Fibrinoid necrosis of the interstitium and red blood cells within the interstitial space are sometimes seen. Bland pulmonary hemorrhage (i.e., DAH without inflammation of the alveolar structures) also may occur.
Evaluation of either lung or renal tissue by immunofluorescent techniques indicates an absence of immune complexes (pauci-immune) in granulomatosis with polyangiitis (Wegener), microscopic polyangiitis, pauci-immune glomerulonephritis, and isolated pulmonary capillaritis. A granular pattern is found in the CTDs, particularly SLE, and a characteristic linear deposition is found in Goodpasture syndrome. Granular deposition of IgA-containing immune complexes is present in Henoch-Schönlein purpura.
Treatment The mainstay of therapy for the DAH associated with systemic vasculitis, CTD, Goodpasture syndrome, and isolated pulmonary capillaritis is IV methylprednisolone, 0.5–2 g daily in divided doses for up to 5 days, followed by a gradual tapering, and then maintenance on an oral preparation. Prompt initiation of therapy is important, particularly in the face of renal insufficiency, because early initiation of therapy has the best chance of preserving renal function. The decision to start other immunosuppressive therapy (cyclophosphamide or azathioprine) acutely depends on the severity of illness.
Goodpasture Syndrome • CLINICAL MANIFESTATIONS Pulmonary hemorrhage and glomerulonephritis are features in most patients with this disease. Autoantibodies to renal glomerular and lung alveolar basement membranes are present. This syndrome can present and recur as DAH without an associated glomerulonephritis. In such cases, circulating anti-basement membrane antibody is often absent, and the only way to establish the diagnosis is by demonstrating linear immunofluorescence in lung tissue.
HISTOLOGIC FINDINGS The underlying histology may be bland hemorrhage or DAH associated with capillaritis.
TREATMENT Plasmapheresis has been recommended as adjunctive treatment.
INHERITED DISORDERS ASSOCIATED WITH ILD
Pulmonary opacities and respiratory symptoms typical of ILD can develop in related family members and in several inherited diseases. These diseases include the phakomatoses, tuberous sclerosis and neurofibromatosis (Chap. 118), and the lysosomal storage diseases, Niemann-Pick disease and Gaucher disease (Chap. 432e). The Hermansky-Pudlak syndrome is an autosomal recessive disorder in which granulomatous colitis and ILD may occur. It is characterized by oculocutaneous albinism, bleeding diathesis secondary to platelet dysfunction, and the accumulation of a chromolipid, lipofuscin material in cells of the reticuloendothelial system. A fibrotic pattern is found on lung biopsy, but the alveolar macrophages may contain cytoplasmic ceroid-like inclusions.
ILD WITH A GRANULOMATOUS RESPONSE IN LUNG TISSUE OR VASCULAR STRUCTURES
Inhalation of organic dusts, which cause hypersensitivity pneumonitis, or of inorganic dust, such as silica, which elicits a granulomatous inflammatory reaction leading to ILD, produces diseases of known etiology (Table 315-1) that are discussed in Chaps. 310 and 311. Sarcoidosis (Chap. 390) is prominent among granulomatous diseases of unknown cause in which ILD is an important feature.
Granulomatous Vasculitides (See also Chap. 385) The granulomatous vasculitides are characterized by pulmonary angiitis (i.e., inflammation and necrosis of blood vessels) with associated granuloma formation (i.e., infiltrates of lymphocytes, plasma cells, epithelioid cells, or histiocytes, with or without the presence of multinucleated giant cells, sometimes with tissue necrosis). The lungs are almost always involved, although any organ system may be affected. Granulomatosis with polyangiitis (Wegener) and Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) primarily affect the lung but are associated with a systemic vasculitis as well. The granulomatous vasculitides generally limited to the lung include necrotizing sarcoid granulomatosis and benign lymphocytic angiitis and granulomatosis. Granulomatous infection and pulmonary angiitis due to irritating embolic material (e.g., talc) are important known causes of pulmonary vasculitis.
LYMPHOCYTIC INFILTRATIVE DISORDERS
This group of disorders features lymphocyte and plasma cell infiltration of the lung parenchyma. The disorders either are benign or can behave as low-grade lymphomas. Included is angioimmunoblastic lymphadenopathy with dysproteinemia, a rare lymphoproliferative disorder characterized by diffuse lymphadenopathy, fever, hepatosplenomegaly, and hemolytic anemia, with ILD in some cases.
Lymphocytic Interstitial Pneumonitis This rare form of ILD occurs in adults, some of whom have an autoimmune disease or dysproteinemia. It has been reported in patients with Sjögren syndrome and HIV infection.
Lymphomatoid Granulomatosis • CLINICAL MANIFESTATIONS Pulmonary lymphomatoid granulomatosis generally presents predominantly in men between the ages of 30 and 50, although patients can be affected at any age. The effects of race and geography on disease incidence are not known, although a higher diagnosis rate is reported in Western countries. Although it may affect virtually any organ, it is most frequently characterized by pulmonary (>90%), skin, and central nervous system involvement. The most common presenting symptoms and signs include cough, fever, rash/nodules, malaise, weight loss, neurologic abnormalities, dyspnea, and chest pain.
HISTOLOGIC FINDINGS This multisystem disorder of unknown etiology is an angiocentric malignant (T cell) lymphoma characterized by a polymorphic lymphoid infiltrate, an angiitis, and granulomatosis.
TREATMENT The clinical course of lymphomatoid granulomatosis ranges from remission without treatment to death from malignant lymphoma within 2 years. The choice of a treatment strategy should be based upon the presence of symptoms, history of using an inciting medication, extent of extrapulmonary involvement, and careful assessment of the histopathologic grade of the lesion. Referral to a hematology oncology specialist for consultation is recommended.
BRONCHOCENTRIC GRANULOMATOSIS
Clinical Manifestations Rather than a specific clinical entity, bronchocentric granulomatosis (BG) is a descriptive histologic term that is applied to an uncommon and nonspecific pathologic response to a variety of airway injuries. There is evidence that BG is caused by a hypersensitivity reaction to Aspergillus or other fungi in patients with asthma. About one-half of the patients described have had chronic asthma with severe wheezing and peripheral blood eosinophilia. In patients with asthma, BG probably represents one pathologic manifestation of allergic bronchopulmonary aspergillosis or another allergic mycosis. In patients without asthma, BG has been associated with RA and a variety of infections, including tuberculosis, echinococcosis, histoplasmosis, coccidioidomycosis, and nocardiosis. The chest roentgenogram reveals irregularly shaped nodular or mass lesions with ill-defined margins, which are usually unilateral and solitary, with upper lobe predominance.
Histologic Findings Bronchocentric granulomatosis is characterized by peribronchial and peribronchiolar necrotizing granulomatous inflammation. Destruction of airway walls and adjacent parenchyma leads to granulomatous replacement of mucosa and submucosa by palisading, epithelioid, and multinucleated histiocytes. Bronchocentric granulomatosis does not typically involve the pulmonary arteries.
Treatment Glucocorticoids are the treatment of choice, often with an excellent outcome, although recurrences may occur as therapy is tapered or stopped.
GLOBAL CONSIDERATIONS
![]() Limited epidemiologic data exist describing the prevalence or incidence of ILD in the general population. With a few exceptions, e.g., sarcoidosis and certain occupational and environmental exposures, there appear to be no significant differences in the prevalence or incidence of ILD among various populations. For sarcoidosis, there are important environmental, racial, and genetic differences (Chap. 390).
Limited epidemiologic data exist describing the prevalence or incidence of ILD in the general population. With a few exceptions, e.g., sarcoidosis and certain occupational and environmental exposures, there appear to be no significant differences in the prevalence or incidence of ILD among various populations. For sarcoidosis, there are important environmental, racial, and genetic differences (Chap. 390).
316 |
Disorders of the Pleura |
PLEURAL EFFUSION
The pleural space lies between the lung and the chest wall and normally contains a very thin layer of fluid, which serves as a coupling system. A pleural effusion is present when there is an excess quantity of fluid in the pleural space.
Etiology Pleural fluid accumulates when pleural fluid formation exceeds pleural fluid absorption. Normally, fluid enters the pleural space from the capillaries in the parietal pleura and is removed via the lymphatics in the parietal pleura. Fluid also can enter the pleural space from the interstitial spaces of the lung via the visceral pleura or from the peritoneal cavity via small holes in the diaphragm. The lymphatics have the capacity to absorb 20 times more fluid than is formed normally. Accordingly, a pleural effusion may develop when there is excess pleural fluid formation (from the interstitial spaces of the lung, the parietal pleura, or the peritoneal cavity) or when there is decreased fluid removal by the lymphatics.
Diagnostic Approach Patients suspected of having a pleural effusion should undergo chest imaging to diagnose its extent. Chest ultrasound has replaced the lateral decubitus x-ray in the evaluation of suspected pleural effusions and as a guide to thoracentesis. When a patient is found to have a pleural effusion, an effort should be made to determine the cause (Fig. 316-1). The first step is to determine whether the effusion is a transudate or an exudate. A transudative pleural effusion occurs when systemic factors that influence the formation and absorption of pleural fluid are altered. The leading causes of transudative pleural effusions in the United States are left ventricular failure and cirrhosis. An exudative pleural effusion occurs when local factors that influence the formation and absorption of pleural fluid are altered. The leading causes of exudative pleural effusions are bacterial pneumonia, malignancy, viral infection, and pulmonary embolism. The primary reason for making this differentiation is that additional diagnostic procedures are indicated with exudative effusions to define the cause of the local disease.
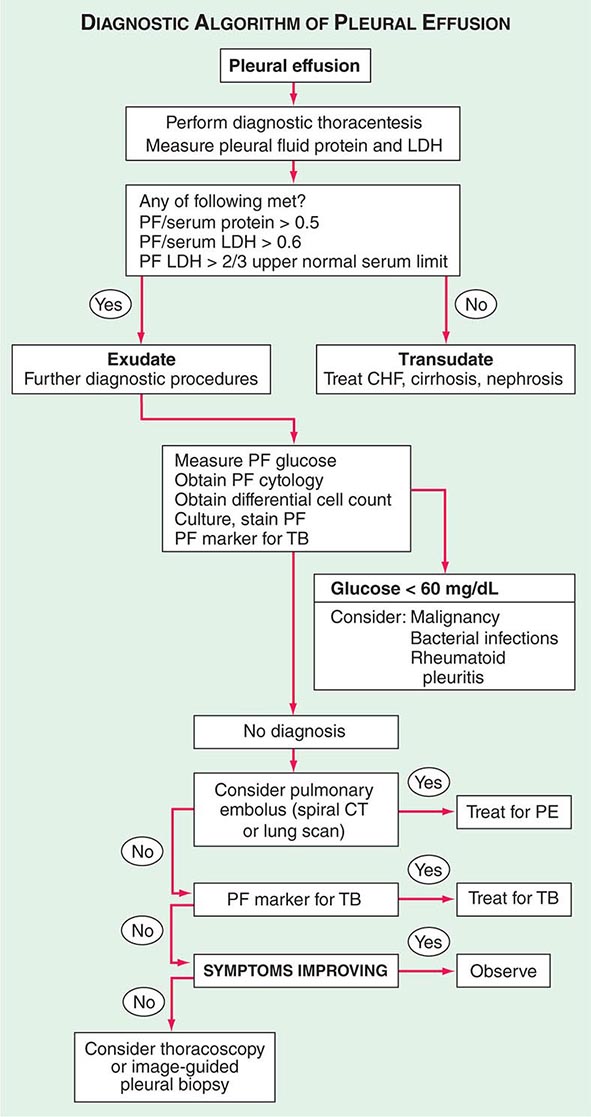
FIGURE 316-1 Approach to the diagnosis of pleural effusions. CHF, congestive heart failure; CT, computed tomography; LDH, lactate dehydrogenase; PE, pulmonary embolism; PF, pleural fluid; TB, tuberculosis.
Transudative and exudative pleural effusions are distinguished by measuring the lactate dehydrogenase (LDH) and protein levels in the pleural fluid. Exudative pleural effusions meet at least one of the following criteria, whereas transudative pleural effusions meet none:
1. Pleural fluid protein/serum protein >0.5
2. Pleural fluid LDH/serum LDH >0.6
3. Pleural fluid LDH more than two-thirds the normal upper limit for serum
These criteria misidentify ~25% of transudates as exudates. If one or more of the exudative criteria are met and the patient is clinically thought to have a condition producing a transudative effusion, the difference between the protein levels in the serum and the pleural fluid should be measured. If this gradient is >31 g/L (3.1 g/dL), the exudative categorization by these criteria can be ignored because almost all such patients have a transudative pleural effusion.
If a patient has an exudative pleural effusion, the following tests on the pleural fluid should be obtained: description of the appearance of the fluid, glucose level, differential cell count, microbiologic studies, and cytology.
Effusion Due to Heart Failure The most common cause of pleural effusion is left ventricular failure. The effusion occurs because the increased amounts of fluid in the lung interstitial spaces exit in part across the visceral pleura; this overwhelms the capacity of the lymphatics in the parietal pleura to remove fluid. In patients with heart failure, a diagnostic thoracentesis should be performed if the effusions are not bilateral and comparable in size, if the patient is febrile, or if the patient has pleuritic chest pain to verify that the patient has a transudative effusion. Otherwise the patient’s heart failure is treated. If the effusion persists despite therapy, a diagnostic thoracentesis should be performed. A pleural fluid N-terminal pro-brain natriuretic peptide (NT-proBNP) >1500 pg/mL is virtually diagnostic that the effusion is secondary to congestive heart failure.
Hepatic Hydrothorax Pleural effusions occur in ~5% of patients with cirrhosis and ascites. The predominant mechanism is the direct movement of peritoneal fluid through small openings in the diaphragm into the pleural space. The effusion is usually right-sided and frequently is large enough to produce severe dyspnea.
Parapneumonic Effusion Parapneumonic effusions are associated with bacterial pneumonia, lung abscess, or bronchiectasis and are probably the most common cause of exudative pleural effusion in the United States. Empyema refers to a grossly purulent effusion.
Patients with aerobic bacterial pneumonia and pleural effusion present with an acute febrile illness consisting of chest pain, sputum production, and leukocytosis. Patients with anaerobic infections present with a subacute illness with weight loss, a brisk leukocytosis, mild anemia, and a history of some factor that predisposes them to aspiration.
The possibility of a parapneumonic effusion should be considered whenever a patient with bacterial pneumonia is initially evaluated. The presence of free pleural fluid can be demonstrated with a lateral decubitus radiograph, computed tomography (CT) of the chest, or ultrasound. If the free fluid separates the lung from the chest wall by >10 mm, a therapeutic thoracentesis should be performed. Factors indicating the likely need for a procedure more invasive than a thoracentesis (in increasing order of importance) include the following:
1. Loculated pleural fluid
2. Pleural fluid pH <7.20
3. Pleural fluid glucose <3.3 mmol/L (<60 mg/dL)
4. Positive Gram stain or culture of the pleural fluid
5. Presence of gross pus in the pleural space
If the fluid recurs after the initial therapeutic thoracentesis and if any of these characteristics are present, a repeat thoracentesis should be performed. If the fluid cannot be completely removed with the therapeutic thoracentesis, consideration should be given to inserting a chest tube and instilling the combination of a fibrinolytic agent (e.g., tissue plasminogen activator, 10 mg) and deoxyribonuclease (5 mg) or performing a thoracoscopy with the breakdown of adhesions. Decortication should be considered when these measures are ineffective.
Effusion Secondary to Malignancy Malignant pleural effusions secondary to metastatic disease are the second most common type of exudative pleural effusion. The three tumors that cause ~75% of all malignant pleural effusions are lung carcinoma, breast carcinoma, and lymphoma. Most patients complain of dyspnea, which is frequently out of proportion to the size of the effusion. The pleural fluid is an exudate, and its glucose level may be reduced if the tumor burden in the pleural space is high.
The diagnosis usually is made via cytology of the pleural fluid. If the initial cytologic examination is negative, thoracoscopy is the best next procedure if malignancy is strongly suspected. At the time of thoracoscopy, a procedure such as pleural abrasion should be performed to effect a pleurodesis. An alternative to thoracoscopy is CT- or ultrasound-guided needle biopsy of pleural thickening or nodules. Patients with a malignant pleural effusion are treated symptomatically for the most part, since the presence of the effusion indicates disseminated disease and most malignancies associated with pleural effusion are not curable with chemotherapy. The only symptom that can be attributed to the effusion itself is dyspnea. If the patient’s lifestyle is compromised by dyspnea and if the dyspnea is relieved with a therapeutic thoracentesis, one of the following procedures should be considered: (1) insertion of a small indwelling catheter or (2) tube thoracostomy with the instillation of a sclerosing agent such as doxycycline (500 mg).
Mesothelioma Malignant mesotheliomas are primary tumors that arise from the mesothelial cells that line the pleural cavities; most are related to asbestos exposure. Patients with mesothelioma present with chest pain and shortness of breath. The chest radiograph reveals a pleural effusion, generalized pleural thickening, and a shrunken hemithorax. The diagnosis is usually established with image-guided needle biopsy or thoracoscopy.
Effusion Secondary to Pulmonary Embolization The diagnosis most commonly overlooked in the differential diagnosis of a patient with an undiagnosed pleural effusion is pulmonary embolism. Dyspnea is the most common symptom. The pleural fluid is almost always an exudate. The diagnosis is established by spiral CT scan or pulmonary arteriography (Chap. 300). Treatment of a patient with a pleural effusion secondary to pulmonary embolism is the same as it is for any patient with pulmonary emboli. If the pleural effusion increases in size after anticoagulation, the patient probably has recurrent emboli or another complication, such as a hemothorax or a pleural infection.
Tuberculous Pleuritis (See also Chap. 202) In many parts of the world, the most common cause of an exudative pleural effusion is tuberculosis (TB), but tuberculous effusions are relatively uncommon in the United States. Tuberculous pleural effusions usually are associated with primary TB and are thought to be due primarily to a hypersensitivity reaction to tuberculous protein in the pleural space. Patients with tuberculous pleuritis present with fever, weight loss, dyspnea, and/or pleuritic chest pain. The pleural fluid is an exudate with predominantly small lymphocytes. The diagnosis is established by demonstrating high levels of TB markers in the pleural fluid (adenosine deaminase >40 IU/L or interferon γ >140 pg/mL). Alternatively, the diagnosis can be established by culture of the pleural fluid, needle biopsy of the pleura, or thoracoscopy. The recommended treatments of pleural and pulmonary TB are identical (Chap. 202).
Effusion Secondary to Viral Infection Viral infections are probably responsible for a sizable percentage of undiagnosed exudative pleural effusions. In many series, no diagnosis is established for ~20% of exudative effusions, and these effusions resolve spontaneously with no long-term residua. The importance of these effusions is that one should not be too aggressive in trying to establish a diagnosis for the undiagnosed effusion, particularly if the patient is improving clinically.
Chylothorax A chylothorax occurs when the thoracic duct is disrupted and chyle accumulates in the pleural space. The most common cause of chylothorax is trauma (most frequently thoracic surgery), but it also may result from tumors in the mediastinum. Patients with chylothorax present with dyspnea, and a large pleural effusion is present on the chest radiograph. Thoracentesis reveals milky fluid, and biochemical analysis reveals a triglyceride level that exceeds 1.2 mmol/L (110 mg/dL). Patients with chylothorax and no obvious trauma should have a lymphangiogram and a mediastinal CT scan to assess the mediastinum for lymph nodes. The treatment of choice for most chylothoraxes is insertion of a chest tube plus the administration of octreotide. If these modalities fail, a pleuroperitoneal shunt should be placed unless the patient has chylous ascites. Alternative treatments are ligation of the thoracic duct and percutaneous transabdominal thoracic duct blockage. Patients with chylothoraxes should not undergo prolonged tube thoracostomy with chest tube drainage because this will lead to malnutrition and immunologic incompetence.
Hemothorax When a diagnostic thoracentesis reveals bloody pleural fluid, a hematocrit should be obtained on the pleural fluid. If the hematocrit is more than one-half of that in the peripheral blood, the patient is considered to have a hemothorax. Most hemothoraxes are the result of trauma; other causes include rupture of a blood vessel or tumor. Most patients with hemothorax should be treated with tube thoracostomy, which allows continuous quantification of bleeding. If the bleeding emanates from a laceration of the pleura, apposition of the two pleural surfaces is likely to stop the bleeding. If the pleural hemorrhage exceeds 200 mL/h, consideration should be given to thoracoscopy or thoracotomy.
Miscellaneous Causes of Pleural Effusion There are many other causes of pleural effusion (Table 316-1). Key features of some of these conditions are as follows: If the pleural fluid amylase level is elevated, the diagnosis of esophageal rupture or pancreatic disease is likely. If the patient is febrile, has predominantly polymorphonuclear cells in the pleural fluid, and has no pulmonary parenchymal abnormalities, an intraabdominal abscess should be considered.
|
DIFFERENTIAL DIAGNOSES OF PLEURAL EFFUSIONS |
The diagnosis of an asbestos pleural effusion is one of exclusion. Benign ovarian tumors can produce ascites and a pleural effusion (Meigs’ syndrome), as can the ovarian hyperstimulation syndrome. Several drugs can cause pleural effusion; the associated fluid is usually eosinophilic. Pleural effusions commonly occur after coronary artery bypass surgery. Effusions occurring within the first weeks are typically left-sided and bloody, with large numbers of eosinophils, and respond to one or two therapeutic thoracenteses. Effusions occurring after the first few weeks are typically left-sided and clear yellow, with predominantly small lymphocytes, and tend to recur. Other medical manipulations that induce pleural effusions include abdominal surgery; radiation therapy; liver, lung, or heart transplantation; and the intravascular insertion of central lines.
PNEUMOTHORAX
Pneumothorax is the presence of gas in the pleural space. A spontaneous pneumothorax is one that occurs without antecedent trauma to the thorax. A primary spontaneous pneumothorax occurs in the absence of underlying lung disease, whereas a secondary pneumothorax occurs in its presence. A traumatic pneumothorax results from penetrating or nonpenetrating chest injuries. A tension pneumothorax is a pneumothorax in which the pressure in the pleural space is positive throughout the respiratory cycle.
Primary Spontaneous Pneumothorax Primary spontaneous pneumothoraxes are usually due to rupture of apical pleural blebs, small cystic spaces that lie within or immediately under the visceral pleura. Primary spontaneous pneumothoraxes occur almost exclusively in smokers; this suggests that these patients have subclinical lung disease. Approximately one-half of patients with an initial primary spontaneous pneumothorax will have a recurrence. The initial recommended treatment for primary spontaneous pneumothorax is simple aspiration. If the lung does not expand with aspiration or if the patient has a recurrent pneumothorax, thoracoscopy with stapling of blebs and pleural abrasion is indicated. Thoracoscopy or thoracotomy with pleural abrasion is almost 100% successful in preventing recurrences.
Secondary Pneumothorax Most secondary pneumothoraxes are due to chronic obstructive pulmonary disease, but pneumothoraxes have been reported with virtually every lung disease. Pneumothorax in patients with lung disease is more life-threatening than it is in normal individuals because of the lack of pulmonary reserve in these patients. Nearly all patients with secondary pneumothorax should be treated with tube thoracostomy. Most should also be treated with thoracoscopy or thoracotomy with the stapling of blebs and pleural abrasion. If the patient is not a good operative candidate or refuses surgery, pleurodesis should be attempted by the intrapleural injection of a sclerosing agent such as doxycycline.
Traumatic Pneumothorax Traumatic pneumothoraxes can result from both penetrating and nonpenetrating chest trauma. Traumatic pneumothoraxes should be treated with tube thoracostomy unless they are very small. If a hemopneumothorax is present, one chest tube should be placed in the superior part of the hemithorax to evacuate the air and another should be placed in the inferior part of the hemithorax to remove the blood. Iatrogenic pneumothorax is a type of traumatic pneumothorax that is becoming more common. The leading causes are transthoracic needle aspiration, thoracentesis, and the insertion of central intravenous catheters. Most can be managed with supplemental oxygen or aspiration, but if these measures are unsuccessful, a tube thoracostomy should be performed.
Tension Pneumothorax This condition usually occurs during mechanical ventilation or resuscitative efforts. The positive pleural pressure is life-threatening both because ventilation is severely compromised and because the positive pressure is transmitted to the mediastinum, resulting in decreased venous return to the heart and reduced cardiac output.
Difficulty in ventilation during resuscitation or high peak inspiratory pressures during mechanical ventilation strongly suggest the diagnosis. The diagnosis is made by physical examination showing an enlarged hemithorax with no breath sounds, hyperresonance to percussion, and shift of the mediastinum to the contralateral side. Tension pneumothorax must be treated as a medical emergency. If the tension in the pleural space is not relieved, the patient is likely to die from inadequate cardiac output or marked hypoxemia. A large-bore needle should be inserted into the pleural space through the second anterior intercostal space. If large amounts of gas escape from the needle after insertion, the diagnosis is confirmed. The needle should be left in place until a thoracostomy tube can be inserted.
317 |
Disorders of the Mediastinum |
The mediastinum is the region between the pleural sacs. It is separated into three compartments (Table 317-1). The anterior mediastinum extends from the sternum anteriorly to the pericardium and brachiocephalic vessels posteriorly. It contains the thymus gland, the anterior mediastinal lymph nodes, and the internal mammary arteries and veins. The middle mediastinum lies between the anterior and posterior mediastina and contains the heart; the ascending and transverse arches of the aorta; the venae cavae; the brachiocephalic arteries and veins; the phrenic nerves; the trachea, the main bronchi, and their contiguous lymph nodes; and the pulmonary arteries and veins. The posterior mediastinum is bounded by the pericardium and trachea anteriorly and the vertebral column posteriorly. It contains the descending thoracic aorta, the esophagus, the thoracic duct, the azygos and hemiazygos veins, and the posterior group of mediastinal lymph nodes.
|
THE THREE COMPARTMENTS OF THE MEDIASTINUM |

MEDIASTINAL MASSES
The first step in evaluating a mediastinal mass is to place it in one of the three mediastinal compartments, since each has different characteristic lesions (Table 317-1). The most common lesions in the anterior mediastinum are thymomas, lymphomas, teratomatous neoplasms, and thyroid masses. The most common masses in the middle mediastinum are vascular masses, lymph node enlargement from metastases or granulomatous disease, and pleuropericardial and bronchogenic cysts. In the posterior mediastinum, neurogenic tumors, meningoceles, meningomyeloceles, gastroenteric cysts, and esophageal diverticula are commonly found.
Computed tomography (CT) scanning is the most valuable imaging technique for evaluating mediastinal masses and is the only imaging technique that should be done in most instances. Barium studies of the gastrointestinal tract are indicated in many patients with posterior mediastinal lesions, because hernias, diverticula, and achalasia are readily diagnosed in this manner. An iodine-131 scan can efficiently establish the diagnosis of intrathoracic goiter.
A definite diagnosis can be obtained with mediastinoscopy or anterior mediastinotomy in many patients with masses in the anterior or middle mediastinal compartments. A diagnosis can be established without thoracotomy via percutaneous fine-needle aspiration biopsy or endoscopic transesophageal or endobronchial ultrasound-guided biopsy of mediastinal masses in most cases. An alternative way to establish the diagnosis is video-assisted thoracoscopy. In many cases, the diagnosis can be established and the mediastinal mass removed with video-assisted thoracoscopy.
ACUTE MEDIASTINITIS
Most cases of acute mediastinitis either are due to esophageal perforation or occur after median sternotomy for cardiac surgery. Patients with esophageal rupture are acutely ill with chest pain and dyspnea due to the mediastinal infection. The esophageal rupture can occur spontaneously or as a complication of esophagoscopy or the insertion of a Blakemore tube. Appropriate treatment consists of exploration of the mediastinum with primary repair of the esophageal tear and drainage of the pleural space and the mediastinum.
The incidence of mediastinitis after median sternotomy is 0.4–5.0%. Patients most commonly present with wound drainage. Other presentations include sepsis and a widened mediastinum. The diagnosis usually is established with mediastinal needle aspiration. Treatment includes immediate drainage, debridement, and parenteral antibiotic therapy, but the mortality rate still exceeds 20%.
CHRONIC MEDIASTINITIS
The spectrum of chronic mediastinitis ranges from granulomatous inflammation of the lymph nodes in the mediastinum to fibrosing mediastinitis. Most cases are due to histoplasmosis or tuberculosis, but sarcoidosis, silicosis, and other fungal diseases are at times causative. Patients with granulomatous mediastinitis are usually asymptomatic. Those with fibrosing mediastinitis usually have signs of compression of a mediastinal structure such as the superior vena cava or large airways, phrenic or recurrent laryngeal nerve paralysis, or obstruction of the pulmonary artery or proximal pulmonary veins. Other than antituberculous therapy for tuberculous mediastinitis, no medical or surgical therapy has been demonstrated to be effective for mediastinal fibrosis.
PNEUMOMEDIASTINUM
In this condition, there is gas in the interstices of the mediastinum. The three main causes are (1) alveolar rupture with dissection of air into the mediastinum; (2) perforation or rupture of the esophagus, trachea, or main bronchi; and (3) dissection of air from the neck or the abdomen into the mediastinum. Typically, there is severe substernal chest pain with or without radiation into the neck and arms. The physical examination usually reveals subcutaneous emphysema in the suprasternal notch and Hamman’s sign, which is a crunching or clicking noise synchronous with the heartbeat and is best heard in the left lateral decubitus position. The diagnosis is confirmed with the chest radiograph. Usually no treatment is required, but the mediastinal air will be absorbed faster if the patient inspires high concentrations of oxygen. If mediastinal structures are compressed, the compression can be relieved with needle aspiration.
318 |
Disorders of Ventilation |
DEFINITION AND PHYSIOLOGY
In health the arterial level of carbon dioxide (PaCO2) IS maintained between 37 and 43 mmHg at sea level. All disorders of ventilation result in abnormal measurements of PaCO2. This chapter reviews chronic ventilatory disorders.
The continuous production of CO2 by cellular metabolism necessitates its efficient elimination by the respiratory system. The relationship between CO2 production and PaCO2 is described by the equation: PaCO2 = (k) (![]() CO2)/
CO2)/![]() A, where
A, where ![]() CO2 represents the carbon dioxide production, k is a constant, and
CO2 represents the carbon dioxide production, k is a constant, and ![]() A is fresh gas alveolar ventilation (Chap. 306e).
A is fresh gas alveolar ventilation (Chap. 306e). ![]() A can be calculated as minute ventilation × (1 – Vd/Vt), where the dead space fraction Vd/Vt represents the portion of a tidal breath that remains within the conducting airways at the conclusion of inspiration and so does not contribute to alveolar ventilation. As such, all disturbances of PaCO2 must reflect altered CO2 production, minute ventilation, or dead space fraction.
A can be calculated as minute ventilation × (1 – Vd/Vt), where the dead space fraction Vd/Vt represents the portion of a tidal breath that remains within the conducting airways at the conclusion of inspiration and so does not contribute to alveolar ventilation. As such, all disturbances of PaCO2 must reflect altered CO2 production, minute ventilation, or dead space fraction.
Diseases that alter ![]() CO2 are often acute (e.g., sepsis, burns, or pyrexia), and their contribution to ventilatory abnormalities and/or respiratory failure is reviewed elsewhere. Chronic ventilatory disorders typically involve inappropriate levels of minute ventilation or increased dead space fraction. Characterization of these disorders requires a review of the normal respiratory cycle.
CO2 are often acute (e.g., sepsis, burns, or pyrexia), and their contribution to ventilatory abnormalities and/or respiratory failure is reviewed elsewhere. Chronic ventilatory disorders typically involve inappropriate levels of minute ventilation or increased dead space fraction. Characterization of these disorders requires a review of the normal respiratory cycle.
The spontaneous cycle of inspiration and expiration is automatically generated in the brainstem. Two groups of neurons located within the medulla are particularly important: the dorsal respiratory group (DRG) and the ventral respiratory column (VRC). These neurons have widespread projections including the descending projections into the contralateral spinal cord where they perform many functions. They initiate activity in the phrenic nerve/diaphragm, project to the upper airway muscle groups and spinal respiratory neurons, and innervate the intercostal and abdominal muscles that participate in normal respiration. The DRG acts as the initial integration site for many of the afferent nerves relaying information about PaO2, PaCO2, pH, and blood pressure from the carotid and aortic chemoreceptors and baroreceptors to the central nervous system (CNS). In addition, the vagus nerve relays information from stretch receptors and juxtapulmonary-capillary receptors in the lung parenchyma and chest wall to the DRG. The respiratory rhythm is generated within the VRC as well as the more rostrally located parafacial respiratory group (pFRG), which is particularly important for the generation of active expiration. One particularly important area within the VRC is the so-called pre-Bötzinger complex. This area is responsible for the generation of various forms of inspiratory activity, and lesioning of the pre-Bötzinger complex leads to the complete cessation of breathing. The neural output of these medullary respiratory networks can be voluntarily suppressed or augmented by input from higher brain centers and the autonomic nervous system. During normal sleep, there is an attenuated response to hypercapnia and hypoxemia, resulting in mild nocturnal hypoventilation that corrects upon awakening.
Once neural input has been delivered to the respiratory pump muscles, normal gas exchange requires an adequate amount of respiratory muscle strength to overcome the elastic and resistive loads of the respiratory system (Fig. 318-1A) (Chap. 306e). In health, the strength of the respiratory muscles readily accomplishes this, and normal respiration continues indefinitely. Reduction in respiratory drive or neuromuscular competence or substantial increase in respiratory load can diminish minute ventilation, resulting in hypercapnia (Fig. 318-1B). Alternatively, if normal respiratory muscle strength is coupled with excessive respiratory drive, then alveolar hyperventilation ensues and leads to hypocapnia (Fig. 318-1C).
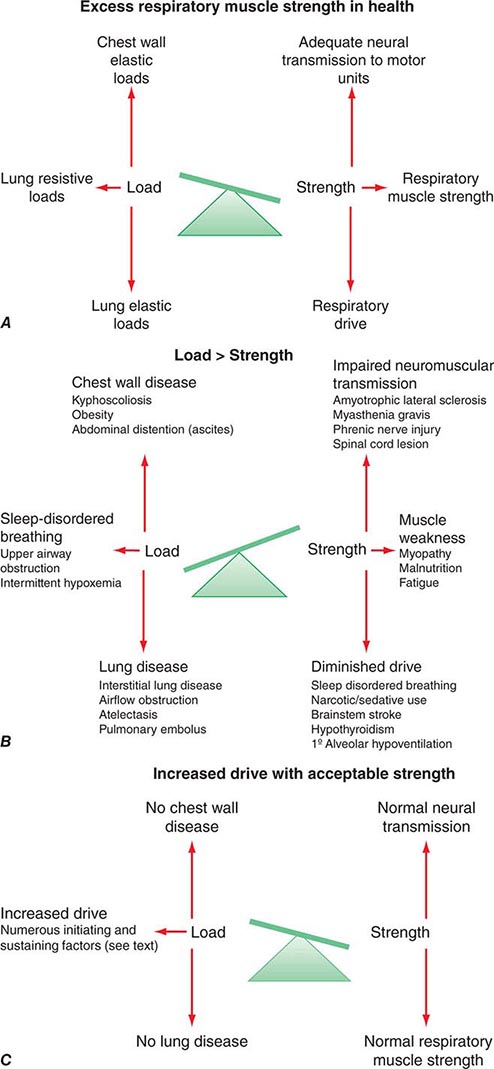
FIGURE 318-1 Examples of balance between respiratory system strength and load. A. Excess respiratory muscle strength in health. B. Load greater than strength. C. Increased drive with acceptable strength.
HYPOVENTILATION
CLINICAL FEATURES
Diseases that reduce minute ventilation or increase dead space fall into four major categories: parenchymal lung and chest wall disease, sleep-disordered breathing, neuromuscular disease, and respiratory drive disorders (Fig. 318-1B). The clinical manifestations of hypoventilation syndromes are nonspecific (Table 318-1) and vary depending on the severity of hypoventilation, the rate at which hypercapnia develops, the degree of compensation for respiratory acidosis, and the underlying disorder. Patients with parenchymal lung or chest wall disease typically present with shortness of breath and diminished exercise tolerance. Episodes of increased dyspnea and sputum production are hallmarks of obstructive lung diseases such as chronic obstructive pulmonary disease, whereas progressive dyspnea and cough are common in interstitial lung diseases. Excessive daytime somnolence, poor-quality sleep, and snoring are common among patients with sleep-disordered breathing. Sleep disturbance and orthopnea are also described in neuromuscular disorders. As neuromuscular weakness progresses, the respiratory muscles, including the diaphragm, are placed at a mechanical disadvantage in the supine position due to the upward movement of the abdominal contents. New-onset orthopnea is frequently a sign of reduced respiratory muscle force generation. More commonly, however, extremity weakness or bulbar symptoms develop prior to sleep disturbance in neuromuscular diseases such as amyotrophic lateral sclerosis (ALS) or muscular dystrophy. Patients with respiratory drive disorders do not have symptoms distinguishable from other causes of chronic hypoventilation.
|
SIGNS AND SYMPTOMS OF HYPOVENTILATION |
The clinical course of patients with chronic hypoventilation from neuromuscular or chest wall disease follows a characteristic sequence: an asymptomatic stage where daytime PaO2 and PaCO2 are normal followed by nocturnal hypoventilation, initially during rapid eye movement (REM) sleep and later in non-REM sleep. Finally, if vital capacity drops further, daytime hypercapnia develops. Symptoms can develop at any point along this time course and often depend on the pace of respiratory muscle functional decline. Regardless of cause, the hallmark of all alveolar hypoventilation syndromes is an increase in alveolar PCO2 (PaCO2) and therefore in PaCO2. The resulting respiratory acidosis eventually leads to a compensatory increase in plasma bicarbonate concentration. The increase in PaCO2 results in an obligatory decrease in PaO2, often resulting in hypoxemia. If severe, the hypoxemia manifests clinically as cyanosis and can stimulate erythropoiesis and thus induce secondary erythrocytosis. The combination of chronic hypoxemia and hypercapnia may also induce pulmonary vasoconstriction, leading eventually to pulmonary hypertension, right ventricular hypertrophy, and right heart failure.
DIAGNOSIS
Elevated plasma bicarbonate in the absence of volume depletion is suggestive of hypoventilation. An arterial blood gas demonstrating elevated PaCO2 with a normal pH confirms chronic alveolar hypoventilation. The subsequent evaluation to identify an etiology should initially focus on whether the patient has lung disease or chest wall abnormalities. Physical examination, imaging studies (chest x-ray and/or computed tomography [CT] scan), and pulmonary function tests are sufficient to identify most lung/chest wall disorders leading to hypercapnia. If these evaluations are unrevealing, then the clinician should screen for obesity hypoventilation syndrome (OHS), the most frequent sleep disorder leading to chronic hypoventilation, which is typically accompanied by obstructive sleep apnea (OSA). Several screening tools have been developed to identify patients at risk for OSA. The Berlin Questionnaire has been validated in a primary care setting and identifies patients likely to have OSA. The Epworth Sleepiness Scale (ESS) and the STOP-Bang questionnaires have not been validated in outpatient primary care settings but are quick and easy to use. The ESS measures daytime sleepiness, with a score of ≥10 indentifying individuals who warrant additional investigation. The STOP-Bang survey has been used in preoperative clinics to identify patients at risk of having OSA. In this population, it has 93% sensitivity and 90% negative predictive value.
If the ventilatory apparatus (lungs, airways, chest wall) is not responsible for chronic hypercapnia, then the focus should shift to respiratory drive and neuromuscular disorders. There is an attenuated increase in minute ventilation in response to elevated CO2 and/or low O2 in respiratory drive disorders. These diseases are difficult to diagnose and should be suspected when patients with hypercapnia are found to have normal respiratory muscle strength, normal pulmonary function, and normal alveolar-arterial PO2 difference. Hypoventilation is more marked during sleep in patients with respiratory drive defects, and polysomnography often reveals central apneas, hypopneas, or hypoventilation. Brain imaging (CT scan or magnetic resonance imaging [MRI]) can sometimes identify structural abnormalities in the pons or medulla that result in hypoventilation. Chronic narcotic use or significant hypothyroidism can depress the central respiratory drive and lead to chronic hypercapnia as well.
Respiratory muscle weakness has to be profound before lung volumes are compromised and hypercapnia develops. Typically physical examination reveals decreased strength in major muscle groups prior to the development of hypercapnia. Measurement of maximum inspiratory and expiratory pressures or forced vital capacity (FVC) can be used to monitor for respiratory muscle involvement in diseases with progressive muscle weakness. These patients also have increased risk for sleep-disordered breathing, including hypopneas, central and obstructive apneas, and hypoxemia. Nighttime oximetry and capnometry during polysomnography are helpful in better characterizing sleep disturbances in this patient population.
HYPOVENTILATION SYNDROMES
OBESITY HYPOVENTILATION SYNDROME
The diagnosis of OHS requires body mass index (BMI) ≥30 kg/m2 and chronic daytime alveolar hypoventilation, defined as PaCO2 ≥45 mmHg at sea level in the absence of other known causes of hypercapnia. In almost 90% of cases, the sleep-disordered breathing is in the form of OSA. Several international studies in different populations confirm that the overall prevalence of OSA syndrome, defined by an apnea-hypopnea index (AHI) ≥5 and daytime sleepiness, is approximately 3–4% in middle-aged men and 2% in middle-aged women. Thus, the population at risk for the development of OHS continues to rise as the worldwide obesity epidemic persists. Although no population-based prevalence studies of OHS have been performed, some estimates suggest there may be as many as 500,000 individuals with OHS in the United States.
Some, but not all, studies suggest that severe obesity (BMI >40 kg/m2) and severe OSA (AHI >30 events per hour) are risk factors for the development of OHS. The pathogenesis of hypoventilation in these patients is the result of multiple physiologic variables and conditions including OSA, increased work of breathing, respiratory muscle impairment, ventilation-perfusion mismatching, and depressed central ventilatory responsiveness to hypoxemia and hypercapnia. These defects in central respiratory drive often improve with treatment, which suggests that decreased ventilatory responsiveness is a consequence rather than a primary cause of OHS. The treatment of OHS is similar to that for OSA: weight reduction and nocturnal NIPPV. There is evidence that weight loss alone lowers PaCO2 in patients with OHS. However, treatment with NIPPV should never be delayed while the patient attempts to lose weight. Continuous positive airway pressure (CPAP) improves daytime hypercapnia and hypoxemia in more than half of patients with OHS and concomitant OSA. Bilevel positive airway pressure should be reserved for patients not able to tolerate high levels of CPAP support or patients who remain hypoxemic despite resolution of obstructive respiratory events. NIPPV with bilevel positive airway pressure should be strongly considered if hypercapnia persists after several weeks of CPAP therapy with objectively proven adherence. Patients with OHS and no evidence of OSA are typically started on bilevel positive airway pressure, as are patients presenting with acute decompensated OHS. Finally, comorbid conditions that impair ventilation, such as chronic obstructive pulmonary disease, should be aggressively treated in conjunction with coexisting OHS.
CENTRAL HYPOVENTILATION SYNDROME
This syndrome can present later in life or in the neonatal period where it is often called Ondine’s curse or congenital central hypoventilation syndrome. Abnormalities in the gene encoding PHOX2b, a transcription factor with a role in neuronal development, have been implicated in the pathogenesis of congenital central hypoventilation syndrome. Regardless of the age of onset, these patients have absent respiratory response to hypoxia or hypercapnia, mildly elevated PaCO2 while awake, and markedly elevated PaCO2 during non-REM sleep. Interestingly these patients are able to augment their ventilation and “normalize” PaCO2 during exercise and during REM sleep. These patients typically require NIPPV or mechanical ventilation as therapy and should be considered for phrenic nerve or diaphragmatic pacing at centers with experience performing these procedures.
HYPERVENTILATION
CLINICAL FEATURES
Hyperventilation is defined as ventilation in excess of metabolic requirements (CO2 production) leading to a reduction in PaCO2. The physiology of patients with chronic hyperventilation is poorly understood, and there is no typical clinical presentation. Symptoms can include dyspnea, paresthesias, tetany, headache, dizziness, visual disturbances, and atypical chest pain. Because symptoms can be so diverse, patients with chronic hyperventilation present to a variety of health care providers, including internists, neurologists, psychologists, psychiatrists, and pulmonologists.
It is helpful to think of hyperventilation as having initiating and sustaining factors. Some investigators believe that an initial event leads to increased alveolar ventilation and a drop in PaCO2 to ~20 mmHg. The ensuing onset of chest pain, breathlessness, paresthesia, or altered consciousness can be alarming. The resultant increase in minute volume to relieve these acute symptoms only serves to exacerbate symptoms that are often misattributed by the patient and health care workers to cardiopulmonary disorders. An unrevealing evaluation for causes of these symptoms often results in patients being anxious and fearful of additional attacks. It is important to note that anxiety disorders and panic attacks are not synonymous with hyperventilation. Anxiety disorders can be both an initiating and sustaining factor in the pathogenesis of chronic hyperventilation, but these are not necessary for the development of chronic hypocapnia.
DIAGNOSIS
Respiratory symptoms associated with acute hyperventilation can be the initial manifestation of systemic illnesses such as diabetic ketoacidosis. Causes of acute hyperventilation need to be excluded before a diagnosis of chronic hyperventilation is considered. Arterial blood gas sampling that demonstrates a compensated respiratory alkalosis with a near normal pH, low PaCO2, and low calculated bicarbonate is necessary to confirm chronic hyperventilation. Other causes of respiratory alkalosis, such as mild asthma, need to be diagnosed and treated before chronic hyperventilation can be considered. A high index of suspicion is required because increased minute ventilation can be difficult to detect on physical examination. Once chronic hyperventilation is established, a sustained 10% increase in alveolar ventilation is enough to perpetuate hypocapnia. This increase can be accomplished with subtle changes in the respiratory pattern, such as occasional sigh breaths or yawning two to three times per minute.
ACKNOWLEDGMENT
We would like to acknowledge Eliot A. Phillipson for earlier versions of this chapter and Jan-Marino Ramirez for his careful critique and helpful suggestions.
319 |
Sleep Apnea |
Obstructive sleep apnea/hypopnea syndrome (OSAHS) and central sleep apnea (CSA) are both classified as sleep-related breathing disorders. OSAHS and CSA share some risk factors and physiological bases but also have unique features. Each disorder is associated with impaired ventilation during sleep and disruption of sleep, and each diagnosis requires careful elicitation of the patient’s history, physical examination, and physiological testing. OSAHS, the more common disorder, causes daytime sleepiness, impairs daily function, and is a major contributor to cardiovascular disease in adults and to behavioral problems in children. CSA is less common and may occur in combination with obstructive sleep apnea, as a primary condition, or secondary to a medical condition or medication. CSA impairs overnight gas exchange and may result in symptoms of either insomnia or excessive sleepiness.
OBSTRUCTIVE SLEEP APNEA/HYPOPNEA SYNDROME (OSAHS)
Definition OSAHS is defined on the basis of nocturnal and daytime symptoms as well as sleep study findings. Diagnosis requires the patient to have (1) either symptoms of nocturnal breathing disturbances (snoring, snorting, gasping, or breathing pauses during sleep) or daytime sleepiness or fatigue that occurs despite sufficient opportunities to sleep and is unexplained by other medical problems; and (2) five or more episodes of obstructive apnea or hypopnea per hour of sleep (the apnea-hypopnea index [AHI], calculated as the number of episodes divided by the number of hours of sleep) documented during a sleep study. OSAHS also may be diagnosed in the absence of symptoms if the AHI is above 15. Each episode of apnea or hypopnea represents a reduction in breathing for at least 10 sec. OSAHS is often identified when associated with a ≥3% drop in oxygen saturation and/or a brain cortical arousal. OSAHS severity is based on the frequency of breathing disturbances (AHI), the amount of oxygen desaturation with respiratory events, the duration of apneas and hypopneas, the degree of sleep fragmentation, and the level of daytime sleepiness.
Pathophysiology During inspiration, intraluminal pharyngeal pressure becomes increasingly negative, creating a “suctioning” force. Because the pharyngeal airway has no bone or cartilage, airway patency is dependent on the stabilizing influence of the pharyngeal dilator muscles. Although these muscles are continuously activated during wakefulness, neuromuscular output declines with sleep onset. In patients with a collapsible airway, the reduction in neuromuscular output results in transient episodes of pharyngeal collapse (manifesting as an “apnea”) or near collapse (manifesting as a “hypopnea”). The episodes of collapse are terminated when ventilatory reflexes are activated and cause arousal, thus stimulating an increase in neuromuscular activity and opening of the airway. The airway may collapse at various levels: the soft palate (most common), tongue base, lateral pharyngeal walls, and/or epiglottis (Fig. 319-1). OSAHS may be most severe during REM (rapid eye movement) sleep, when neuromuscular output to the skeletal muscles is particularly low, and in the supine position due to gravitational forces.
FIGURE 319-1 Common sites of airway collapse. For example, the palate, tongue, and/or epiglottis (Ep) can be posteriorly displaced, and the lateral pharyngeal walls (LW) can collapse.
Individuals with a small pharyngeal lumen require relatively high levels of neuromuscular innervation to maintain patency during wakefulness and thus are predisposed to excessive airway collapsibility during sleep. The airway lumen may be narrowed with enlargement of soft tissue structures (tongue, palate, and uvula) due to fat deposition, increased lymphoid tissue, or genetic variation. Craniofacial factors such as mandibular retroposition or micrognathia, reflecting genetic variation or developmental influences, also can reduce lumen dimensions. In addition, lung volumes influence the caudal traction on the pharynx and consequently the stiffness of the pharyngeal wall. Accordingly, low lung volume in the recumbent position, which is particularly pronounced in the obese, contributes to collapse. A high degree of nasal resistance (e.g., due to nasal septal deviation or polyps) can contribute to airway collapse by increasing the negative intraluminal suction pressure. High-level nasal resistance also may trigger mouth opening during sleep, which breaks the seal between the tongue and the teeth and allows the tongue to fall posteriorly and occlude the airway.
Pharyngeal muscle activation is integrally linked to ventilatory drive. Thus, factors related to ventilatory control, particularly ventilatory sensitivity, arousal threshold, and neuromuscular responses to CO2, contribute to the pathogenesis of OSAHS. A buildup in CO2 during sleep activates both the diaphragm and the pharyngeal muscles, which stiffen the upper airway and can counteract inspiratory suction pressures and maintain airway patency to an extent that depends on the anatomic predisposition to collapse. However, pharyngeal collapse can occur when the ventilatory control system is overly sensitive to CO2, with resultant wide fluctuations in ventilation and ventilatory drive and in upper airway instability. Moreover, increasing levels of CO2 during sleep result in central nervous system arousal, causing the individual to move from a deeper to a lighter level of sleep or to awaken. A low arousal threshold (i.e., awaken to a low level of CO2 or ventilatory drive) can preempt the CO2-mediated process of pharyngeal muscle compensation and prevent airway stabilization. A high arousal threshold, conversely, may prevent appropriate termination of apneas, prolonging apnea duration and oxyhemoglobin desaturation severity. Finally, any impairment in the ability of the muscles to compensate during sleep can contribute to collapse of the pharynx. The relative contributions of risk factors vary among individuals. Approaches to the measurement of these factors in clinical settings, with consequent enhancement of “personalized” therapeutic interventions, are being actively investigated.
Risk Factors and Prevalence The major risk factors for OSAHS are obesity and male sex. Additional risk factors include mandibular retrognathia and micrognathia, a positive family history of OSAHS, genetic syndromes that reduce upper airway patency (e.g., Down syndrome, Treacher-Collins syndrome), adenotonsillar hypertrophy (especially in children), menopause (in women), and various endocrine syndromes (e.g., acromegaly, hypothyroidism).
Approximately 40–60% of cases of OSAHS are attributable to excess weight. Obesity predisposes to OSAHS through the narrowing effects of upper airway fat on the pharyngeal lumen. Obesity also reduces chest wall compliance and decreases lung volumes, resulting in a loss of caudal traction on upper airway structures. Obese individuals are at a fourfold or greater risk for OSAHS than their normal-weight counterparts. A 10% weight gain is associated with a >30% increase in AHI. Even modest weight loss or weight gain can influence the risk and severity of OSAHS. However, the absence of obesity does not exclude this diagnosis.
The prevalence of OSAHS is two- to fourfold higher among men than among women. Factors that predispose men to OSAHS include android patterns of obesity (resulting in upper-airway fat deposition) and relatively great pharyngeal length, which exacerbates collapsibility. Premenopausal women are relatively protected from OSAHS by the influence of sex hormones on ventilatory drive. The decline in sex differences in older age is associated with an increased OSAHS prevalence in women after menopause.
Variations in craniofacial morphology that reduce the size of the posterior airway space increase OSAHS risk. The contribution of hard-tissue structural features to OSAHS is most evident in nonobese patients. Identification of features such as retrognathia can influence therapeutic decision-making.
OSAHS has a strong genetic basis, as evidenced by its significant familial aggregation and heritability. For a first-degree relative of a patient with OSAHS, the odds ratio of having OSAHS is approximately twofold higher than that for someone without an affected relative.
OSAHS prevalence varies with age, from 2–15% among middle-aged adults to >20% among elderly individuals. There is a peak due to lymphoid hypertrophy among children between the ages of 3 and 8 years; with airway growth and lymphoid tissue regression during later childhood, prevalence declines. Then, as obesity prevalence increases in middle life and women enter menopause, OSAHS again increases.
The prevalence of OSAHS may be especially high among patients with diabetes or hypertension. Individuals of Asian ancestry appear to be at increased risk of OSAHS at relatively low levels of body mass index, possibly because of the influence of craniofacial risk factors that narrow the nasopharynx. In the United States, African Americans, especially children and young adults, are at higher risk for OSAHS than their Caucasian counterparts. In a majority of adults with OSAHS, the disorder is undiagnosed.
Course of the Disorder The precise onset of OSAHS is usually hard to identify. A person may snore for many years, often beginning in childhood, before OSAHS is identified. Weight gain may precipitate an increase in symptoms, which in turn may lead the patient to pursue an evaluation. OSAHS may become less severe with weight loss, particularly after bariatric surgery. Marked increases and decreases in the AHI are uncommon unless accompanied by weight change.
APPROACH TO THE PATIENT:
Obstructive Sleep Apnea/Hypopnea Syndrome (OSAHS)
An evaluation for OSAHS should be considered in patients with symptoms of OSAHS and one or more risk factors. Screening also should be considered in patients who report symptoms consistent with OSAHS and who are at high risk for OSAHS-related morbidities, such as hypertension, diabetes mellitus, and cardiac and cerebrovascular diseases.
SYMPTOMS AND HISTORY
When possible, a sleep history should be obtained in the presence of a bed partner. Snoring is the most common complaint; however, its absence does not exclude the diagnosis, as pharyngeal collapse may occur without tissue vibration. Gasping or snorting during sleep may also be reported, reflecting termination of individual apneas with abrupt airway opening. Dyspnea is unusual, and its absence generally distinguishes OSAHS from paroxysmal nocturnal dyspnea, nocturnal asthma, and acid reflux with laryngospasm. Patients also may describe frequent awakening or sleep disruption, which is more common among women and older adults. The most common daytime symptom is sleepiness. This symptom can be difficult to elicit and may be hard to distinguish from exercise-related fatigue, deconditioning, and malaise. In contrast to true sleepiness, the latter symptoms generally improve with rest. Other symptoms include a dry mouth, nocturnal heartburn, diaphoresis of the chest and neck, nocturia, morning headaches, trouble concentrating, irritability, and mood disturbances. Several questionnaires that evaluate snoring frequency, self-reported apneas, and daytime sleepiness can facilitate OSAHS screening. The predictive ability of a questionnaire can be enhanced by a consideration of whether the patient is male or has risk factors such as obesity or hypertension.
PHYSICAL FINDINGS
Physical findings often reflect the etiologic factors for the disorder as well as comorbid conditions, particularly vascular disease. On examination, patients may exhibit hypertension and regional (central) obesity, as indicated by a large waist and neck circumference. The oropharynx may reveal a small orifice with crowding due to an enlarged tongue, a low-lying soft palate with a bulky uvula, large tonsils, a high arched palate, and/or micro/retrognathia. Since high-level nasal resistance can increase pharyngeal collapsibility, the nasal cavity should be inspected for polyps, septal deviation, and other signs of obstruction. Because patients with heart failure are at increased risk for both OSAHS and CSA, a careful cardiac examination should be conducted to detect possible left- or right-sided cardiac dysfunction. Evidence of cor pulmonale suggests severe OSAHS or a comorbid cardiopulmonary condition. A neurologic evaluation is needed to evaluate for conditions such as neuromuscular and cerebrovascular diseases, which increase OSAHS risk.
LABORATORY FINDINGS
Diagnostic Findings Since symptoms and signs do not accurately predict the severity of sleep-related breathing disturbances, specific diagnosis and categorization of OSAHS severity require objective measurement of breathing during the period of sleep. The gold standard for diagnosis of OSAHS is an overnight polysomnogram (PSG). A negative in-laboratory PSG rules out OSAHS except in unusual circumstances—e.g., with insufficient REM sleep or supine sleep. Home sleep tests that record only a few respiratory and cardiac channels commonly are used as a cost-effective means for diagnosing patients without significant comorbidity who have a high pretest probability of OSAHS. However, a home study may yield a false-negative result if sleep time is not accurately estimated, and further evaluation may therefore be required.
The key physiological information collected during a sleep study for OSAHS assessment includes measurement of breathing (changes in airflow, respiratory excursion), oxygenation (hemoglobin oxygen saturation), body position, and cardiac rhythm. In addition, PSGs and some home sleep studies measure sleep continuity and sleep stages (by electroencephalography, chin electromyography, and electro-oculography), limb movements (by leg sensors), and snoring intensity. This information is used to quantify the frequency and subtypes of abnormal respiratory events during sleep as well as associated changes in oxygen saturation, arousals, and sleep stage distributions. Tables 319-1 and 319-2 define the respiratory events scored and the severity guidelines employed during a sleep study. Figure 319-2 shows examples of sleep-related respiratory events. A typical sleep study report provides quantitative data such as the AHI and the profile of oxygen saturation over the night (mean, nadir, time at low levels). Reports may also include the respiratory disturbance index, which includes the number of respiratory effort–related arousals in addition to the number of apneas plus hypopneas. In-laboratory PSG also quantifies sleep latency (time from “lights off” to first sleep onset), sleep efficiency (percentage of time asleep relative to time in bed), arousal index (number of cortical arousals per hour of sleep), time in each sleep stage, and periodic limb movement index. OSAHS severity can be further characterized according to the degree of sleep fragmentation associated with respiratory disturbances. Relevant metrics include the frequency of cortical micro-arousals or awakenings per sleep hour, reduction in sleep continuity (low sleep efficiency), reduction of time in deeper stages of sleep (stage N3 and REM sleep) and increases in light sleep (stage N1). The detection of autonomic arousals, such as surges in blood pressure, changes in heart rate, and abnormalities in cardiac rhythm, also provides relevant information on OSAHS severity.
|
RESPIRATORY EVENT DEFINITIONS |
FIGURE 319-3 Example of flow limitation. The inspiratory flow pattern in a patent airway is rounded and peaks in the middle. In contrast, a partially obstructed airway exhibits an early peak followed by mid-inspiratory flattening, yielding a scooped-out appearance.
|
OBSTRUCTIVE SLEEP APNEA/HYPOPNEA SYNDROME (OSAHS): QUANTIFICATION AND SEVERITY SCALE |
aEach level of AHI can be further quantified by level of sleepiness and associated hypoxemia.
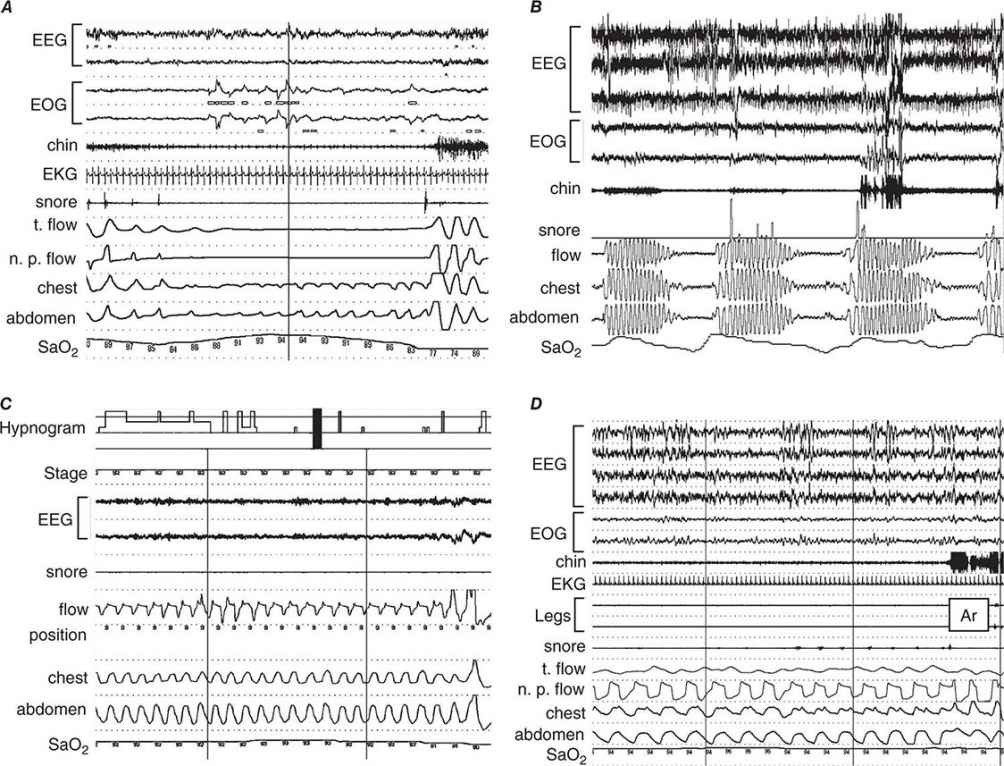
FIGURE 319-2 A. Obstructive apnea. There are 30 sec of no airflow, as shown in the nasal pressure (n. p. flow) and thermistor-measured flow (t. flow). Note the presence of chest-abdomen motion, indicating respiratory effort against an occluded airway. B. Central apnea in a patient with Cheyne-Stokes respiration due to congestive heart failure. The flat chest-abdomen tracings indicate the absence of inspiratory effort during the central apneas. C. Hypopnea. Partial obstruction of the pharyngeal airway can limit ventilation, leading to desaturation (a mild decrease in this patient, from 93% to 90%) and arousal. D. Respiratory effort–related arousal (RERA). Minimal flow reduction terminated by an arousal (Ar) without desaturation constitutes a RERA. EEG, electroencephalogram; EOG, electro-oculogram; EKG, electrocardiogram.
Other Laboratory Findings Various imaging studies, including cephalometric radiography, MRI, CT, and fiberoptic endoscopy, can be used to identify anatomic risk factors for OSAHS. Cardiac testing may yield evidence of impaired systolic or diastolic ventricular function or abnormal cardiac structure. Overnight blood pressure monitoring often displays a “non-dipping” pattern (absence of the typical 10-mmHg fall during sleep from blood pressure while awake). Arterial blood gas measurements made during wakefulness are usually normal. Waking hypoxemia or hypercarbia suggests coexisting lung disease or hypoventilation syndrome. Patients with severe nocturnal hypoxemia may have elevated hemoglobin values. A multiple sleep latency test or a maintenance of wakefulness test can be useful in quantifying sleepiness and helping to distinguish OSAHS from narcolepsy.
Health Consequences and Comorbidities OSAHS is a major contributor to cardiac, cerebrovascular, and metabolic disorders as well as to premature death. It is the most common medical cause of daytime sleepiness and negatively influences quality of life. This broad range of health effects is attributable to the impact of sleep fragmentation, cortical arousal, and intermittent hypoxemia on vascular, cardiac, metabolic, and neurologic functions. OSAHS-related respiratory events stimulate sympathetic overactivity, leading to acute blood pressure surges during sleep, endothelial damage, and nocturnal as well as daytime hypertension. OSAHS-related hypoxemia also stimulates release of acute-phase proteins and reactive oxygen species that exacerbate insulin resistance and lipolysis and cause an augmented prothrombotic and proinflammatory state. Inspiratory effort against an occluded airway causes large intrathoracic negative pressure swings, altering cardiac preload and afterload and resulting in cardiac remodeling and reduced cardiac function. Hypoxemia and sympathetic-parasympathetic imbalance also may cause electrical remodeling of the heart and myocyte injury.
HYPERTENSION OSAHS can raise blood pressure to prehypertensive and hypertensive ranges, increase the prevalence of a non-dipping overnight blood pressure pattern, and increase the risk of resistant hypertension. Elevations in blood pressure are due to augmented sympathetic nervous system activation as well as alterations in the rennin–angiotensin–aldosterone system and fluid balance. Treatment of OSAHS with nocturnal continuous positive airway pressure (CPAP) has been shown to reduce 24-h ambulatory blood pressure. Although the overall impact of CPAP on blood pressure levels is relatively modest (averaging 2–4 mmHg), larger improvements are observed among patients with high AHIs and sleepiness.
CARDIOVASCULAR, CEREBROVASCULAR, AND METABOLIC DISEASES Among the most serious health consequences of OSAHS is its impact on cardiac and metabolic functions. Strong epidemiologic evidence indicates that OSAHS significantly increases the risk of coronary artery disease, heart failure with and without reduced ejection fraction, atrial and ventricular arrhythmias, atherosclerosis and coronary artery disease, stroke, and diabetes. Treatment of OSAHS has been shown to reduce several markers of cardiovascular risk, improve insulin resistance, decrease the recurrence rate of atrial fibrillation, and improve various outcomes in patients with active cardiovascular disease. Large-scale trials are under way to evaluate the role of OSAHS treatment in reducing cardiac event rates and in prolonging the survival of patients with cardiac disease.
SLEEPINESS More than 50% of patients with moderate to severe OSAHS report daytime sleepiness. Patients with OSAHS symptoms have a twofold increased risk of occupational accidents. Individuals with elevated AHIs are involved in motor vehicle crashes as much as seven times more often than persons with normal AHIs. Randomized controlled trials have shown that treatment of OSAHS with nasal CPAP therapy alleviates sleepiness as measured by either questionnaire or objective testing. However, the degree of improvement varies widely. Residual sleepiness may be due to several factors, including suboptimal treatment adherence, insufficient sleep time, other sleep disorders, or prior hypoxic-mediated damage in brain areas involved in alertness. Visceral adipose tissue, whose amounts are increased in patients with OSAHS, releases somnogenic cytokines that may contribute to sleepiness. Thus, even after treatment, it is important to assess and monitor patients for residual sleepiness and to evaluate the necessity of optimizing treatment adherence, improving sleep patterns, and identifying other disorders contributing to sleepiness.
QUALITY OF LIFE AND MOOD Reductions in health-related quality of life are common in patients with OSAHS, with the largest decrements on the physical and vitality subscales. Treatment with CPAP often results in improvement in these patient-reported outcomes. Depression, in particular symptoms of somatic depression (irritability, fatigue, lack of energy) is commonly reported in OSAHS.
|
OBSTRUCTIVE SLEEP APNEA/HYPOPNEA SYNDROME-(OSAHS)– |
A comprehensive approach to the management of OSAHS is needed to reduce risk factors and comorbidities. The clinician should seek to identify and address lifestyle and behavioral factors as well as comorbidities that may be exacerbating OSAHS. As appropriate, treatment should aim to reduce weight; optimize sleep duration (7–9 hours); regulate sleep schedules (with similar bedtimes and wake times across the week); encourage the patient to avoid sleeping in the supine position; treat nasal allergies; increase physical activity; eliminate alcohol ingestion within 3 h of bedtime; and minimize use of sedating medications. Patients should be counseled to avoid drowsy driving.
CPAP is the standard medical therapy with the highest level of evidence for efficacy. Delivered through a nasal or nasal-oral mask, CPAP works as a mechanical splint to hold the airway open, thus maintaining airway patency during sleep. An overnight CPAP titration study, performed either in a laboratory or with a home “autotitrating” device, is required to determine the optimal pressure setting that reduces the number of apneas/hypopneas during sleep, improves gas exchange, and reduces arousals. Rates of adherence to CPAP treatment are highly variable (average, 50–80%) and may be improved with support by a skilled health care team who can address side effects (Table 319-3). Despite the limitations of CPAP, controlled studies have demonstrated its beneficial effect on blood pressure, alertness, mood, and insulin sensitivity. Uncontrolled studies also indicate a favorable effect on cardiovascular outcomes, cardiac ejection fraction, atrial fibrillation recurrence, and mortality risk.
|
SIDE EFFECTS OF CONTINUOUS POSITIVE AIRWAY PRESSURE–(CPAP)–AND THEIR TREATMENTS |
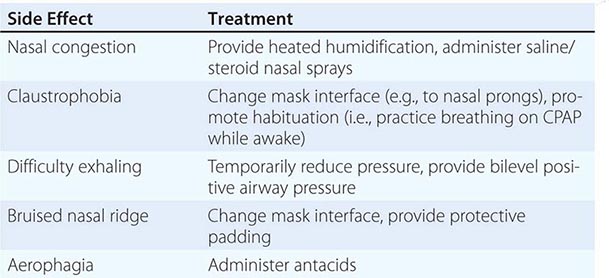
Oral appliances for OSAHS work by advancing the mandible, thus opening the airway by repositioning the lower jaw and pulling the tongue forward. These devices generally work better when customized for patient use; maximal adaptation can take several weeks. Efficacy studies show that these devices can reduce the AHI by ≥50% in two-thirds of individuals, although these data are based largely on patients with mild OSAHS. Side effects of oral appliances include temporomandibular joint pain and tooth movement. Oral appliances are most often used for treating patients with mild OSAHS or patients who do not tolerate CPAP. However, since adherence to the use of oral appliances sometimes exceeds CPAP adherence, these devices are under investigation for treatment of more severe disease.
Upper airway surgery for OSAHS is less effective than CPAP and is mostly reserved for the treatment of patients who snore, have mild OSAHS, and cannot tolerate CPAP. Uvulopalatopharyngoplasty (removal of the uvula and the margin of the soft palate) is the most common surgery and, although results vary greatly, has a success rate similar to or slightly lower than treatment with oral appliances. Upper airway surgery is less effective in severe OSAHS and in obese patients. Success rates may be higher for multilevel surgery (involving more than one site/structure) performed by an experienced surgeon, but the selection of patients is an important factor and relies on careful targeting of culprit areas for surgical resection. Bariatric surgery is an option for obese patients with OSAHS and can improve not only OSAHS but also other obesity-associated health conditions. Other procedures that can decrease snoring but have minimal effects on OSAHS include injection of the soft palate (resulting in stiffening), radiofrequency ablation, laser-assisted uvulopalatoplasty, and palatal implants.
Supplemental oxygen can improve oxygen saturation, but there is little evidence that it improves OSAHS symptoms or the AHI.
CENTRAL SLEEP APNEA
CSA, which is less common than OSAHS, may occur in isolation or, more often, in combination with obstructive events in the form of “mixed” apneas. CSA is often caused by an increased sensitivity to PCO2, which leads to an unstable breathing pattern that manifests as hyperventilation alternating with apnea. A prolonged circulation delay between the pulmonary capillaries and carotid chemoreceptors is also a contributing cause; thus individuals with congestive heart failure are at risk for CSA. With prolonged circulation delay, there is a crescendo-decrescendo breathing pattern known as Cheyne-Stokes respiration (Fig. 319–2B). Other risk factors for CSA include opioid medications (which appear to have a dose-dependent effect on CSA) and hypoxia (e.g., breathing at high altitude). In some individuals, CPAP—particularly at high pressures—seems to induce central apnea; this condition is referred to as complex sleep apnea. Rarely, CSA may be caused by blunted chemosensitivity due to congenital disorders (congenital central hypoventilation syndrome) or acquired factors. Treatment of CSA is difficult and depends on the underlying cause. Limited data suggest that supplemental oxygen can reduce the frequency of central apneas, particularly in patients with hypoxemia. Cheyne-Stokes respiration is treated by optimizing therapy for heart failure and, in some cases, using CPAP with or without supplemental oxygen. Adaptive servoventilation, a form of ventilatory support that dynamically changes inspiratory support levels across periods of apnea and hypopnea, can minimize large fluctuations in PCO2 that produce central apnea and can be effective for the treatment of CSA.
320e |
Lung Transplantation |
Lung transplantation is a therapeutic consideration for many patients with nonmalignant end-stage lung disease, and it prolongs survival and improves quality of life in appropriately selected recipients. Since 1985 almost 40,000 procedures have been recorded worldwide, and since 2009 more than 3000 transplants have been reported annually.
INDICATIONS
The indications span the gamut of lung diseases, but in some respects the distribution of indications differs among countries. According to aggregate international data, the most common indications in the last few years have been chronic obstructive pulmonary disease (COPD), ~29%; idiopathic pulmonary fibrosis (IPF), ~28%; cystic fibrosis (CF), ~16%; α1-antitrypsin deficiency emphysema, ~3.5%; and idiopathic pulmonary arterial hypertension (IPAH), ~3%. Other diseases have made up the balance of primary indications, and retransplantation has accounted for ~3% of procedures.
RECIPIENT SELECTION
Transplantation should be considered when other therapeutic options have been exhausted and when the patient’s prognosis is expected to improve as a result of the procedure. Survival rates after transplantation can be compared with predictive indices for the patient’s disease, but each patient’s clinical course must be integrated into the assessment as well. Moreover, quality of life is a primary motive for transplantation for many patients, and the prospect of improved quality-adjusted survival is often attractive even if the survival advantage itself may be marginal.
Disease-specific consensus guidelines for referring patients for evaluation and for proceeding with transplantation are summarized in Table 320e-1 and are linked to clinical, physiologic, radiographic, and pathologic features that influence the prognosis of the respective diseases. Candidates for lung transplantation are also thoroughly screened for comorbidities that might affect the outcome adversely. Conditions such as systemic hypertension, diabetes mellitus, gastroesophageal reflux, and osteoporosis are not unusual; however, if uncomplicated and adequately managed, they do not disqualify patients from transplantation. The upper age limit is ~70 years at most centers, but the median age of recipients has been increasing steadily over the last decade. In the United States in 2009, 22% of recipients were ≥65 years old.
|
DISEASE-SPECIFIC GUIDELINES FOR REFERRAL AND TRANSPLANTATION |
Abbreviations: BODE, body mass index (B), airflow obstruction (O), dyspnea (D), exercise capacity (E); DLCO, diffusing capacity for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; HRCT, high-resolution computed tomography; ICU, intensive care unit; NYHA, New York Heart Association; PaCO2, partial pressure of carbon dioxide in arterial blood; SpO2, arterial oxygen saturation by pulse oximetry; UIP, usual interstitial pneumonitis.
Source: Summarized from JB Orens et al: J Heart Lung Transplant 25:745, 2006. For BODE index, BR Celli et al: N Engl J Med 350:1005, 2004.
Standard exclusions include HIV infection, chronic active hepatitis B or C infection, uncontrolled or untreatable pulmonary or extrapulmonary infection, uncured malignancy, active cigarette smoking, drug or alcohol dependency, irreversible physical deconditioning, chronic nonadherence with medical care, significant disease of another vital organ (e.g., heart, liver, or kidney), and psychiatric or psychosocial situations that could substantially interfere with post-transplantation management. Other problems that may compromise outcome constitute relative contraindications. Some typical issues are ventilator-dependent respiratory failure, previous thoracic surgical procedures, obesity, and coronary artery disease. Chronic infection with antibiotic-resistant Pseudomonas species, Burkholderia species, Aspergillus species, or nontuberculous mycobacteria is a unique concern in some patients with CF. The potential impact of these and other factors must be judged in the clinical context to determine an individual candidate’s suitability for transplantation.
WAITING LIST AND ORGAN ALLOCATION
Organ allocation policies are influenced by medical, ethical, geographic, and political factors, with systems varying from country to country. Regardless of the system, potential recipients are placed on a waiting list and must be matched for blood group compatibility and, with some latitude, for lung size with an acceptable donor. Most lungs are procured from deceased donors after total brain failure (“brain death”), but only ~15–20% of brain-death organ donors yield either one or two lungs suitable for transplantation. Lungs from donors after cardiac death have been utilized to a limited extent (~2% of lung donors in the United States in 2009). Recently, ex vivo lung perfusion has been used by some centers to assess donor lungs that are marginal or high-risk for implantation by standard criteria; if the results of ex vivo testing are satisfactory, these lungs have been transplanted successfully.
In the United States, a lung allocation scoring system is used to prioritize patients on the waiting list. The lung allocation score (LAS) for a patient is based on the patient’s risk of death during 1 year on the waiting list and the patient’s likelihood of survival for 1 year after transplantation. The LAS can range from 0 to 100, and precedence for transplantation is ranked from highest to lowest scores. Both the lung disease and its severity affect a patient’s LAS; parameters in the LAS must be updated biannually but can be submitted for recalculation whenever the patient’s condition changes. The median LAS for all candidates on the waiting list is usually ~35, but the LAS tends to be higher among patients with IPF and CF than among patients with COPD and IPAH.
Under this system in the United States, the median waiting time for transplantation has been ~135 days. The overall death rate on the waiting list has been ~6.5%, but death rates vary substantially with the diagnosis (e.g., COPD, ~3%; IPF, ~7%) and with the LAS (e.g., 40–49, ~7%; 50–59, ~15%; ≥60, ~25%). The indications for transplantation depend not only on the prevalence and natural history of the various lung diseases but also on the LAS typically associated with these diseases. While patients with IPF constitute ~20% of the waiting list, they make up ~34% of recipients because their allocation scores are typically higher than those of patients with other diseases.
TRANSPLANT PROCEDURE
Bilateral transplantation is mandatory for CF and other forms of bronchiectasis because the risk of spillover infection from a remaining native lung precludes single-lung transplantation. Heart-lung transplantation is obligatory for Eisenmenger syndrome with complex anomalies that cannot readily be repaired in conjunction with lung transplantation and for concomitant end-stage lung and heart disease. However, cardiac replacement is not necessary for cor pulmonale because right ventricular function will recover when pulmonary vascular afterload is normalized by lung transplantation.
Either bilateral or single-lung transplantation is an option for other diseases unless there is a special consideration, but bilateral transplantation has been utilized increasingly for most indications. Recently, ~65% of procedures in the United States have been bilateral, and ~70% of transplants for COPD, ~55% of those for IPF, and ~95% of those for IPAH in the international registry have been bilateral.
Living-donor lobar transplantation has had a limited role in adult lung transplantation but is now rarely performed. It has been used predominantly for teenagers or young adults with CF and has usually been reserved for patients who were unlikely to survive the wait for a deceased-donor organ.
POSTTRANSPLANTATION MANAGEMENT
Induction therapy with an antilymphocyte globulin or an interleukin 2 receptor antagonist is utilized by ~55% of centers, and a three-drug maintenance immunosuppressive regimen that includes a calcineurin inhibitor (cyclosporine or tacrolimus), a purine synthesis antagonist (azathioprine or a mycophenolic acid precursor), and prednisone is traditional. Subsequently, other drugs (e.g., sirolimus) may be substituted into the regimen for various reasons. Prophylaxis against Pneumocystis jirovecii pneumonia is standard, and prophylaxis against cytomegalovirus (CMV) infection and fungal infection is part of many protocols. The dose of cyclosporine, tacrolimus, or sirolimus is adjusted by blood-level monitoring. All of these agents are metabolized by the hepatic cytochrome P450 system, and interactions with medications that affect this pathway can significantly alter their clearance and blood level.
Routine management focuses on monitoring of the allograft, regulation of immunosuppressive therapy, and expeditious detection of problems or complications. Regular contact with a nurse coordinator, physician follow-up, chest radiography, blood tests, and spirometry are customary, and periodic surveillance bronchoscopies are employed in some programs. If recovery is uncomplicated, lung function rapidly improves and then stabilizes by 3–6 months after transplantation. Subsequently, the variation in spirometric measurements is small, and a sustained decline of ≥10–15% signals a potentially significant problem.
OUTCOMES
Survival Major registries publish survival rates (Table 320e-2) and other outcomes annually (www.ishlt.org; www.srtr.org). In the international registry, the survival half-life for recipients with IPF is 4.4 years; IPAH, 5 years; COPD, 5.3 years; and CF, 7.5 years. However, age and transplantation procedure have a significant impact on outcome. For recipients 18–59 years of age, the survival half-life is 5–6 years, but this figure decreases to 4.4 years among patients 60–65 years old and to 3.6 years for those >65 years old. Survival rates at >15 years have been significantly higher after bilateral transplantation than after unilateral transplantation for COPD, α1-antitrypsin deficiency emphysema, IPF, and IPAH.
|
RECIPIENT SURVIVAL, BY PRETRANSPLANTATION DIAGNOSIS (1990–2010) |
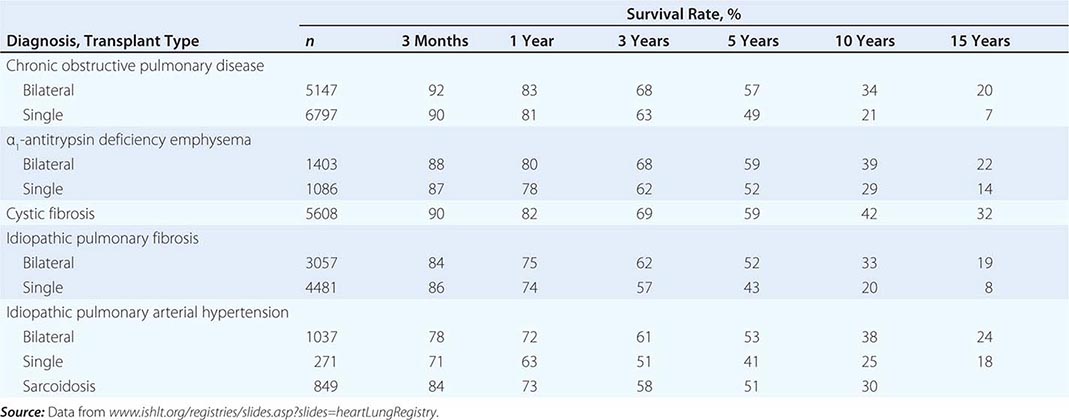
The main sources of perioperative mortality include technical complications of the operation, primary graft dysfunction, and infections. Acute rejection and CMV infection are common problems in the first year, but neither is usually fatal. Beyond the first year, chronic rejection and non-CMV infections cause the majority of deaths.
Risk factors for mortality have been analyzed in the international and U. S. registries. In these analyses, factors associated with an increased risk of death, especially in the first year after transplantation, have included the following: recipients hospitalized at the time of transplantation; recipients supported by mechanical ventilation, extracorporeal membrane oxygenation, inotropic drugs, or dialysis at the time of transplantation; and recipients undergoing retransplantation. However, other factors have contributed as well. The mortality risk has been higher at centers with <20–30 transplantations per year.
Function Regardless of the disease, successful transplantation impressively restores cardiopulmonary function. After bilateral transplantation, pulmonary function tests are typically normal; after unilateral transplantation, a mild abnormality characteristic of the remaining diseased lung is still apparent. Formal exercise testing usually demonstrates some impairment in maximal work rate and maximal oxygen uptake, but few recipients report any limitation to activities of daily living.
Quality of Life Both overall and health-related quality-of-life scores are enhanced. With multidimensional profiles, improvements extend across most domains and are sustained longitudinally unless chronic rejection or some other complication develops. Other problems that detract from quality of life include renal dysfunction and drug side effects.
Cost The cost of transplantation depends on the health care system, other health care policies, and economic factors that vary from country to country. In the United States in 2011, the average billed charge for the period from 30 days before bilateral lung transplantation until 180 days after discharge from the transplantation admission was $797,300. The total cost included the following charges: all care during 30 days before transplantation, $21,400; organ procurement, $90,300; hospital transplantation admission, $458,500; physician fees during transplantation admission, $56,300; all inpatient and outpatient care for 180 days after discharge, $142,600; and all outpatient drugs (including immunosuppressants) for 180 days after discharge, $28,200.
COMPLICATIONS
Lung transplantation can be complicated by a variety of problems (Table 320e-3). Aside from predicaments that are unique to transplantation, side effects and toxicities of immunosuppressive medications can cause new medical problems or aggravate preexisting conditions.
|
MAJOR POTENTIAL COMPLICATIONS OF LUNG TRANSPLANTATION AND IMMUNOSUPPRESSION |
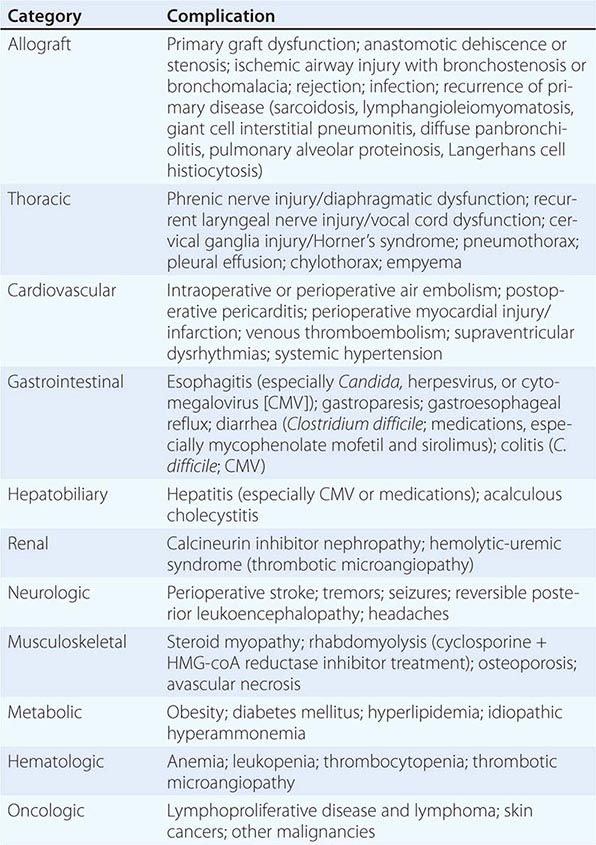
Graft Dysfunction Primary graft dysfunction (PGD), an acute lung injury, is a manifestation of multiple potential insults to the donor organ that are inherent in the transplantation process. The principal clinical features are diffuse pulmonary infiltrates and hypoxemia within 72 h of transplantation; however, the presentation can be mimicked by pulmonary venous obstruction, hyperacute rejection, pulmonary edema, and pneumonia.
The severity is variable, and a standardized grading system has been established. Up to 50% of lung transplant recipients may have some degree of PGD, and ~10–20% have severe PGD. The treatment follows the conventional supportive paradigm for acute lung injury. Inhalation of nitric oxide and extracorporeal membrane oxygenation have been used in severe cases; retransplantation has also been performed, but when undertaken in the first 30 days this procedure is associated with a poor survival rate (~30% at 1 year). Most recipients with mild PGD recover, but the mortality rate for severe PGD has been ~40–60%. PGD is also associated with longer postoperative ventilator support, longer intensive care unit and hospital stays, higher costs, and excess morbidity, and severe PGD is a risk factor for the later development of chronic rejection (see below).
Airway Complications The bronchial blood supply to the donor lung is disrupted during procurement. Bronchial revascularization during transplantation is technically feasible in some cases, but it is not widely practiced. Consequently, after implantation, the donor bronchus is dependent on retrograde bronchial blood flow from the pulmonary circulation and is vulnerable to ischemia.
The spectrum of airway problems includes anastomotic necrosis and dehiscence, occlusive granulation tissue, anastomotic or bronchial stenosis, and bronchomalacia. The incidence has been in the range of 7–18%, but the associated mortality rate has been low. These problems usually can be managed bronchoscopically with techniques such as simple endoscopic debridement, laser photoresection, balloon dilation, and bronchial stenting.
Rejection Rejection is the main deterrent to higher medium- and long-term survival rates. In this immunologic response to alloantigen recognition, both cell-mediated and antibody-mediated (humoral) cascades can play a role. Cellular rejection is effected by T lymphocyte interactions with donor alloantigens, mainly in the major histocompatibility complex (MHC), whereas humoral rejection is driven by antibodies to donor MHC alloantigens or possibly to non-MHC antigens on epithelial or endothelial cells.
Rejection is often categorized as acute or chronic without reference to the underlying mechanism. Acute rejection is cell-mediated, and its incidence is highest in the first 6–12 months after transplantation. In contrast, chronic rejection generally emerges later, and both alloimmune and non-alloimmune fibroproliferative reactions may contribute to its pathogenesis.
Acute Cellular Rejection With current immunosuppressive regimens, ~30–40% of recipients experience acute rejection in the first year. Acute cellular rejection (ACR) can be clinically silent or can be manifested by nonspecific symptoms or signs that may include cough, low-grade fever, dyspnea, hypoxemia, inspiratory crackles, interstitial infiltrates, and declining lung function; however, clinical impressions are not reliable. The diagnosis is confirmed by transbronchial biopsies showing the characteristic lymphocytic infiltrates around arterioles or bronchioles, and a standardized pathologic scheme is used to grade the biopsies.
Minimal ACR on a surveillance biopsy in a clinically stable recipient is often left untreated, but higher grades generally are treated regardless of the clinical situation. Treatment usually includes a short course of high-dose glucocorticoids and adjustment of the maintenance immunosuppressive regimen. Most episodes respond to this approach; however, more intensive therapy is sometimes necessary for persistent or recurrent episodes.
Chronic Rejection This complication is the main impediment to long-term survival and is the source of substantial morbidity because of its impact on lung function and quality of life. Clinically, chronic rejection is characterized physiologically by airflow limitation and pathologically by bronchiolitis obliterans; the process is designated bronchiolitis obliterans syndrome (BOS). Transbronchial biopsies are relatively insensitive for detecting bronchiolitis obliterans, and pathologic confirmation is not required for diagnosis. Thus, after other causes of graft dysfunction have been excluded, the diagnosis of BOS is based primarily on a sustained decrement (≥20%) in forced expiratory volume in 1 s (FEV1), although smaller declines in FEV1 (≥10%) or in midexpiratory flow rate (FEF25–75%) may presage BOS. Spirometric criteria for diagnosis and staging of BOS have been standardized.
The prevalence of BOS approaches 50% by 5 years after transplantation. Antecedent ACR is the main risk factor, but PGD, CMV pneumonitis, other community-acquired respiratory viral infections, and gastroesophageal reflux have been implicated as well. BOS can present acutely and imitate infectious bronchitis, or it can manifest as an insidious decline in lung function. The chest radiograph is typically unchanged; CT may reveal mosaic perfusion, air trapping, ground-glass opacities, or bronchiolectasis. Bronchoscopy is indicated to rule out other processes, but transbronchial biopsies identify bronchiolitis obliterans in a minority of cases.
BOS usually is treated with augmented immunosuppression, but there is no consensus about therapy. Strategies include changes in the maintenance drug regimen, including the addition of azithromycin, antilymphocyte globulin, photopheresis, and total lymphoid irradiation. Although therapy may stabilize lung function, the overall results of treatment have been disappointing; the median survival period after onset has been ~3–4 years. Retransplantation is a consideration if clinical circumstances and other comorbidities are not prohibitive, but survival rates have been inferior to those with primary transplantation.
Humoral Rejection Consensus on the role of antibody-mediated rejection is still evolving. Hyperacute rejection is caused by preformed HLA antibodies in the recipient, but it is minimized by pretransplantation antibody screening coupled with virtual or direct cross-matching with any potential donor. Donor-specific HLA antibodies develop after transplantation in up to 50% of recipients, and their presence has been associated with an increased risk of both ACR and BOS and with poorer overall survival. However, the mechanisms by which these antibodies could contribute to ACR or BOS or could otherwise be detrimental have not been unraveled. Formal criteria for antibody-mediated rejection have been defined for renal transplantation, but few cases in lung transplantation fulfill these criteria. Nonetheless, episodes of acute lung allograft dysfunction occasionally have been attributed directly to antibody-mediated injury. If treatment is indicated, potential therapies include plasmapheresis and administration of IV immune globulin, rituximab, bortezomib, or eculizumab.
Infection The lung allograft is especially susceptible to infection, which has been one of the leading causes of death in recipients. In addition to a blunted immune response from immunosuppressive drugs, other normal defenses are compromised: the cough reflex is diminished, and mucociliary clearance is impaired in the transplanted lung. The spectrum of infections includes both opportunistic and non-opportunistic pathogens.
Bacterial bronchitis or pneumonia can occur at any time but is very common in the perioperative period. Later, bronchitis occurs frequently in recipients with BOS, and Pseudomonas aeruginosa or methicillin-resistant Staphylococcus aureus is often the culprit.
CMV is the most common cause of viral infection. Although gastroenteritis, colitis, and hepatitis can occur, CMV viremia and CMV pneumonia are the main illnesses. Most episodes occur in the first 6 months, and treatment with ganciclovir is effective unless resistance develops. Other community-acquired viruses, such as influenza, parainfluenza, and respiratory syncytial viruses, also contribute to respiratory complications. The most problematic fungal infections are caused by Aspergillus species. The spectrum encompasses simple pulmonary colonization, tracheobronchitis, invasive pulmonary aspergillosis, and disseminated aspergillosis, and the clinical scenario dictates treatment.
Other Complications Other potential complications are listed in Table 320e-3. Many of them are related to side effects or toxicities of immunosuppressive drugs. Management of these general medical problems is guided by standard practices, but the complex milieu of transplantation requires close collaboration and good communication among health care providers.




