Sleep and sleep pharmacology
After reading this chapter, the reader will be able to:
1. Define terms that pertain to sleep and sleep pharmacology
2. Describe sleep, its individual stages, and their electrophysiologic correlates
3. Comprehend the basic neurophysiologic mechanisms that promote brain arousal and wakefulness
4. Comprehend the basic neurophysiologic mechanisms that promote sleep onset and maintenance
5. Describe basic circadian processes and their interaction with the sleep-wake cycle
6. Recognize several sleep disorders that are amenable to pharmacotherapy
7. Describe the rationale for using certain classes of drugs to treat specific sleep-related disorders
Compounds whose parent structure is uric acid. These compounds depress central nervous system activity. Long-acting barbiturates such as pentobarbital have been used to treat epilepsy. Barbital was used during the early 20th century to facilitate sleep in individuals with insomnia.
Compounds whose parent structure is a fusion of a diazepine ring with a benzene ring. Benzodiazepines enhance activity of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). Benzodiazepines, which reduce anxiety and promote muscle relaxation, also promote sleep. The earliest benzodiazepines were chlordiazepoxide (Librium) and diazepam (Valium). Benzodiazepines for insomnia are now being replaced by nonbenzodiazepines such as zolpidem (Ambien), eszopiclone (Lunesta), and others.
Circadian rhythms “Circa” is Latin for “about,” and “diem
” is Latin for “day.” Circadian rhythms refer to the approximately 24-hour cycle of biochemical, physiologic, and behavioral processes.
Measurement and recording of the gross electrical activity of the brain. During EEG recordings, electrodes are typically placed across multiple scalp regions. The electrodes are connected to amplifiers and filters that detect, magnify, and record the electrical activity of the brain.
Presence of excessive sleepiness. Daytime sleepiness is so great that it leads to inappropriate daytime napping or sleep. Excessive sleepiness is not alleviated by prolonged sleep times or by napping.
Class of drugs used to induce sleep.
Group of sleep disorders manifested by undesirable motor, sensory, or behavioral phenomena that occur during sleep. The International Classification of Sleep Disorders, Revised (ICSD-R) lists 24 parasomnias. More commonly encountered parasomnias include confusional arousals, sleep terrors, and sleepwalking.
Measurement and recording of EEG activity, typically coupled with measurement and recording of cardiorespiratory activity and eye movements, during sleep.
History of treatment of sleep disorders
Humans spend about a third of their life sleeping, and sleep disorders, which affect a large proportion of the general population and occur in all age groups, represent a major public health and global economic burden.1 It is estimated that 50 million to 70 million adults in the United States have a chronic sleep disorder that interferes with their daily functioning and adversely affects their health and quality of life. Until the mid-1960s, the field of sleep medicine focused primarily on describing and treating insomnia, parasomnias (e.g., sleep walking, night terrors) and hypersomnias, such as narcolepsy. Patients experiencing these symptoms typically sought consultations from neurologists, psychiatrists, psychologists, or their family physician. Until more recently, treatments were generally based on empiric pharmaceutical intervention and behavioral modification protocols or psychotherapy or both.
It is beyond the scope of this chapter to address the entire spectrum of sleep disorders classified in the International Classification of Sleep Disorders, Revised (ICSD-R) or to provide an exhaustive summary of all the pharmacologic, neutraceutical, or cognitive behavioral therapies employed in this field. This chapter describes the classes of drugs that are likely to be encountered when treating patients with sleep disorders. First, a broad but brief overview of the history and evolution of sleep research and sleep pharmacology is presented followed by a brief overview of the brain mechanisms underlying the processes of wakefulness and sleep, including circadian aspects. Some key sleep-related disorders are described, and typical compounds that may be used to treat those disorders are reviewed. Adult sleep apnea syndrome, which is primarily treated with mechanical devices such as oral appliances or application of continuous positive airway pressure (CPAP), are not discussed. Apnea and bradycardia of prematurity, which are treated primarily with respiratory stimulants, are discussed in Chapter 8.
In addition to “sleeping pills,” many nondepressant medications are used in sleep medicine. In disorders such as restless legs syndrome (RLS) and PLMD, sleep onset is often delayed and fragmented. First-line therapy for these disorders includes dopamine agonists and, in some cases, opiates. Respiratory stimulants are another class of medications sometimes employed in the treatment of sleep disorders. Medroxyprogesterone and acetazolamide have been used in an effort to enhance ventilation in patients with high altitude–induced central sleep apnea or obesity-hypoventilation syndrome. More recently, the antidepressant mirtazapine has been shown to reduce apnea severity in animal models emulating sleep apnea as well as in some patients.2 An effective pharmaceutical treatment for obstructive sleep apnea does not yet exist.
Progression of sleep
Sleep was originally thought to be a passive rather than an active process. Canton, a physiologist at the Royal Infirmary in Liverpool, is credited with the discovery of electroencephalography (EEG), an important milestone in the understanding of sleep as an active process. His experimental observations on cortical currents in rabbits and monkeys were published in the British Journal of Medicine.3 Using a mirror galvanometer, Canton observed an increase in the amplitude of waves measured from the cortex during states of sleep as opposed to a decrement in cortical amplitudes during wakefulness. Canton was the first to perform sleep EEG in mammals.4 Subsequently, several observations including those of von Economo and the experiments by Morruzi and colleagues clearly showed that sleep is not a passive phenomenon but involves several brain regions, especially the diencephalon and the brainstem, which actively control sleep and states of arousal.5–8 In 1957, Dement and Kleitman reported the cyclical alternating pattern of non–rapid eye movement (NREM) and REM sleep.9
When determining the most appropriate pharmaceutical intervention for a sleep disorder, it is important first to consider how sleep is defined and measured and the normative values of time spent in sleep and in each sleep stage (i.e., sleep architecture). Mammalian sleep can be defined as a cyclical, reversible behavioral state of perceptual disengagement from, and unresponsiveness to, the external environment. Within normal human monophasic sleep, sleep is polygraphically characterized into two distinct stages based on a constellation of behavioral and electrophysiologic parameters. These two stages are NREM and REM sleep. NREM sleep is categorized further into stages N1-N3 (formerly known as stages 1 to 4); N1 is the lightest and stage N3 is the deepest sleep stage. Figures 23-1 to 23-4 show examples of EEG-defined wakefulness followed by examples of stages N1 to N3.
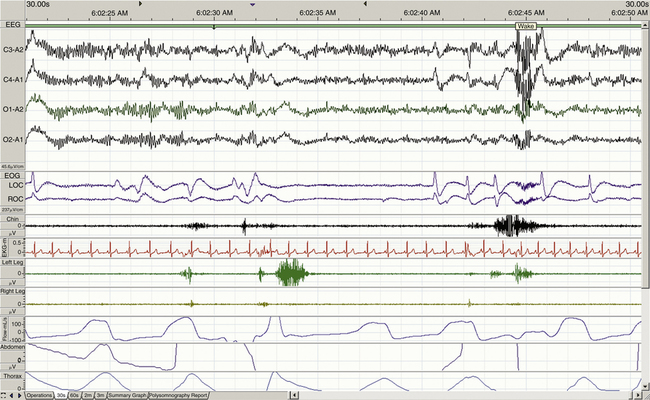
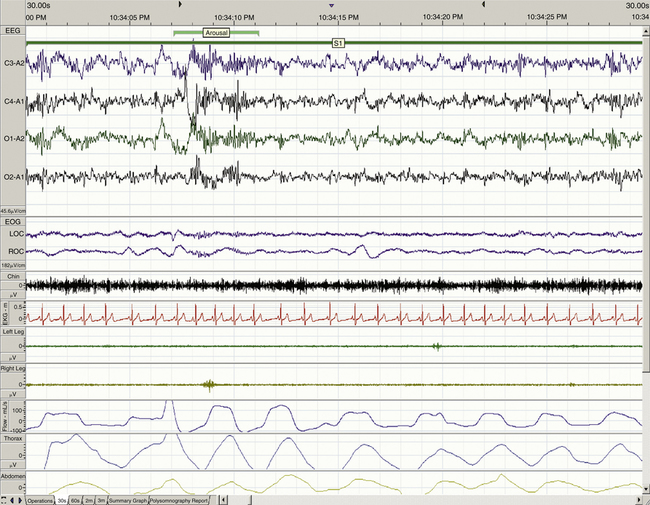
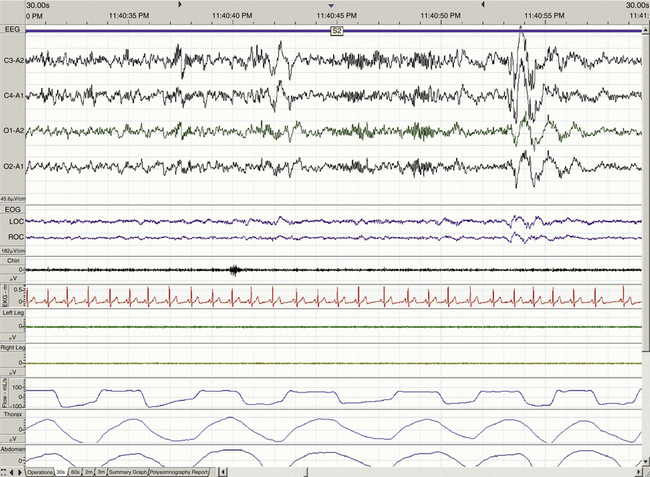
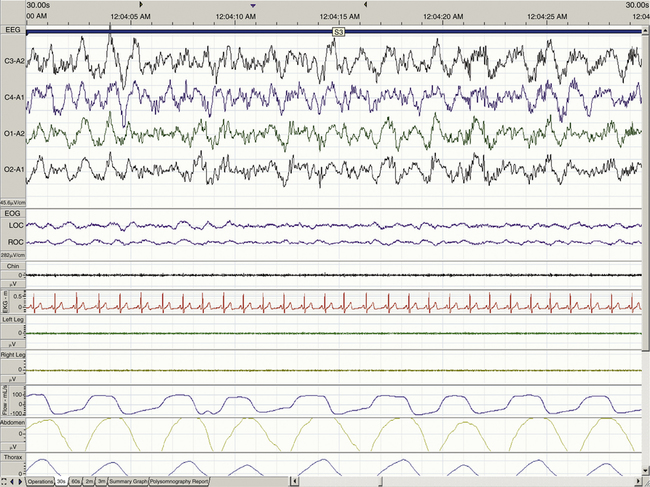
The term for REM sleep is derived from the periodic bursts of REMs during sleep. REM sleep has both tonic (persistent) and phasic (episodic) components. During tonic REM sleep, the EEG tracing shows a similar pattern to that of N1, but it may also exhibit increased activity in the theta frequency range (3 to 7 Hz) and sawtooth-type waves. REM sleep is also accompanied by a generalized muscle atonia, with the exception of the extraocular muscles and the diaphragm. Figure 23-5 shows an example of EEG-defined REM sleep.
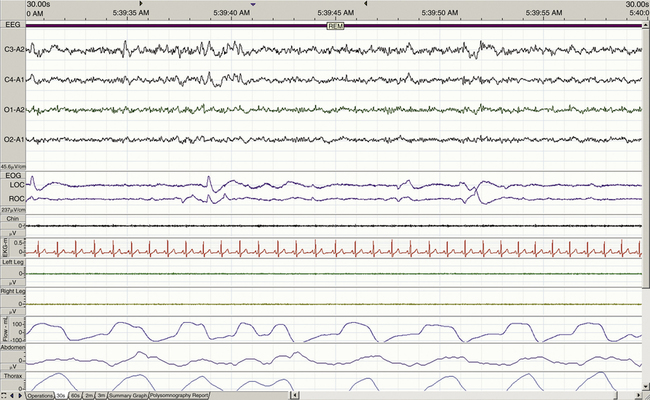
Aserinsky and Kleitman were the first to observe the electrophysiologic characteristics of REM sleep and, in particular, the rapid, jerky, and binocularly symmetric eye movements in this sleep stage.10 EEG patterns similar to wakefulness were noted, showing the characteristic fast desynchronized rhythms in the cortical EEG, and the term paradoxical sleep was introduced by Jouvet and Michel in 1960; the term active sleep was used by other researchers. These terms are used interchangeably in the literature, although subtle differences exist. Additionally, autonomic activation occurs during this state as respiratory and heart rates are increased. Dream recall is also common when subjects are awakened during this stage, whereas dream recall during NREM sleep is relatively rare (Table 23-1).
TABLE 23-1
Electroencephalographic Correlates of Sleep Stages
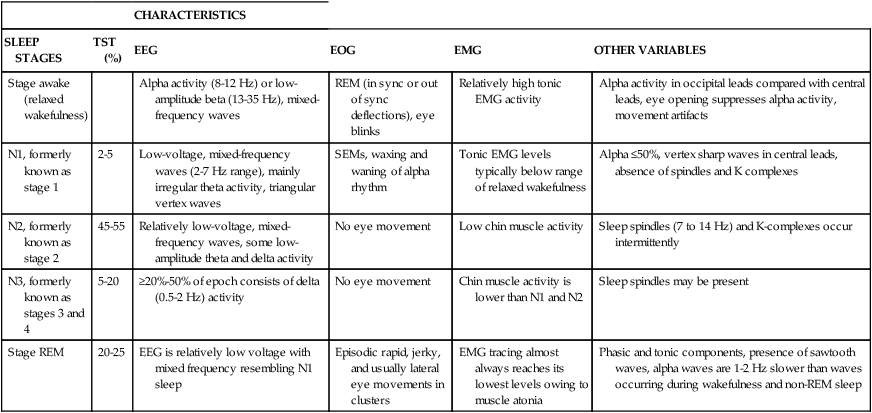
| CHARACTERISTICS | |||||
| SLEEP STAGES | TST (%) | EEG | EOG | EMG | OTHER VARIABLES |
| Stage awake (relaxed wakefulness) | Alpha activity (8-12 Hz) or low-amplitude beta (13-35 Hz), mixed-frequency waves | REM (in sync or out of sync deflections), eye blinks | Relatively high tonic EMG activity | Alpha activity in occipital leads compared with central leads, eye opening suppresses alpha activity, movement artifacts | |
| N1, formerly known as stage 1 | 2-5 | Low-voltage, mixed-frequency waves (2-7 Hz range), mainly irregular theta activity, triangular vertex waves | SEMs, waxing and waning of alpha rhythm | Tonic EMG levels typically below range of relaxed wakefulness | Alpha ≤50%, vertex sharp waves in central leads, absence of spindles and K complexes |
| N2, formerly known as stage 2 | 45-55 | Relatively low-voltage, mixed-frequency waves, some low-amplitude theta and delta activity | No eye movement | Low chin muscle activity | Sleep spindles (7 to 14 Hz) and K-complexes occur intermittently |
| N3, formerly known as stages 3 and 4 | 5-20 | ≥20%-50% of epoch consists of delta (0.5-2 Hz) activity | No eye movement | Chin muscle activity is lower than N1 and N2 | Sleep spindles may be present |
| Stage REM | 20-25 | EEG is relatively low voltage with mixed frequency resembling N1 sleep | Episodic rapid, jerky, and usually lateral eye movements in clusters | EMG tracing almost always reaches its lowest levels owing to muscle atonia | Phasic and tonic components, presence of sawtooth waves, alpha waves are 1-2 Hz slower than waves occurring during wakefulness and non-REM sleep |
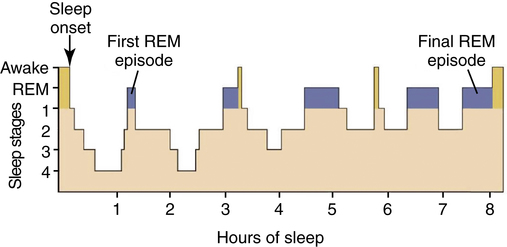
Sleep stages occur in cycles that repeat approximately every 90 to 120 minutes. A normal sleep cycle begins with N1 and proceeds through N3. Sleep rapidly passes through the same stages in reverse order before REM sleep is initiated, usually first occurring about 90 minutes after sleep onset. Although significant interindividual variation in sleep need is noted, adult humans typically sleep about 7 to 9 hours per night and spend almost one-third of their life sleeping. Figure 23-6 provides a graphic representation of the cyclical distribution of sleep states across a single night in a normal healthy adult.
Neurophysiologic mechanisms
Arousal and wakefulness
In a series of postmortem examinations of the brains of patients who had died as a result of the outbreak of encephalitis lethargica after World War I, von Economo8 observed that lesions in the rostral midbrain and posterior hypothalamus had a profound effect on sleep and wakefulness. He derived two significant correlates from his observations. The first was that lesions in the preoptic and basal forebrain (BF) areas caused severe insomnia. The second was that lesions in the posterior and lateral hypothalamus (LH) caused severe hypersomnia. On the basis of these data, von Economo hypothesized the existence of a group of sleep-promoting neurons around the hypothalamic optic chiasm and, conversely, a group of wake-promoting neurons in the area of the posterior hypothalamus. Both observations have been proved to be essentially correct, but it was only toward the end of the 20th century that the hypothalamic influences on sleep and wakefulness were integrated into the mechanisms of vigilance state control. Before that, the emphasis had been on brainstem mechanisms and the ascending reticular activating system (ARAS).
Ascending reticular activating system
Physiologic analysis of the mechanisms of EEG arousal and wakefulness began with the classic studies of Bremer, who, in 1935, showed that if the brainstem of a cat was completely transected at the level of the midbrain (i.e., to produce the cerveau isolé), the result was that the cat maintained a persistent state of sleep. Different interpretations of this result were possible until Moruzzi and Magoun6 demonstrated the existence of an active arousal center below the level of the transaction, in the pons (i.e., the brainstem). Subsequent lesion and electrical stimulation studies identified the brainstem core, or pontine reticular system, as a critical component of arousal and wakefulness. This system was termed the ascending reticular activating system and was morphologically defined by cell bodies that projected from the brainstem to innervate the midbrain and cortex.
A more recent advance has been the identification of the importance of a cholinergic activating system in EEG arousal. This is one of the major components of the ARAS, and its identification depended on methods that were developed for labeling neurons that contain specific neurotransmitters. Steriade and colleagues11 identified cells located near the pons-midbrain junction that increased their discharge rate about 60 seconds before the first change to an aroused state was noted on the EEG. These neurons were found to project to the thalamus, and the change in their discharge rate was the first indication of arousal. Subsequent work identified these neurons as containing the neurotransmitter acetylcholine and being localized to the laterodorsal pontine tegmentum/pedunculopontine tegmentum (LDT/PPT) region.
Cholinergic systems are not the exclusive substrate of EEG arousal, however, and evidence that multiple systems are involved in arousal and wakefulness came from the inability of lesions of any single one of these systems to disrupt EEG arousal on a permanent basis.12 Other brainstem reticular neuronal projections to the thalamus using glutamate neurotransmission and noradrenergic and serotoninergic projections from the locus caeruleus and raphe nuclei also play important roles in maintaining wakefulness. In addition to brainstem nuclei, a cholinergic input to the cortex that ascends from the BF nuclei, especially the nucleus basalis of Meynert, plays an important role. Histaminergic neurons localized in the tuberomammillary nucleus (TMN) of the posterior hypothalamus also promote wakefulness. Discovery of the orexin/hypocretin system in 1999 (see section on Narcolepsy later) led to another CNS arousal system being identified. It is probably the latter hypothalamic systems that were affected in the brains examined by von Economo.8
Thalamic mechanisms of arousal
Most AAS projections that mediate EEG arousal and wakefulness synapse in the thalamus, which is an essential center for the organization of EEG arousal and for maintaining activation at a cortical level. Thalamic mechanisms at a cellular level influence the differences between wakefulness and NREM sleep. A detailed consideration of these thalamic mechanisms is beyond the scope of this chapter; they depend primarily on the cells in the thalamus that project to the cortex (i.e., thalamocortical neurons).13 Thalamocortical neurons differ in their rate and pattern of discharge depending on the vigilance state. When the arousal-related glutamatergic, cholinergic, noradrenergic, and serotoninergic projections are active, they drive the thalamocortical neurons to discharge in single spike mode. This discharge keeps the EEG in an active state, which contributes to arousal and wakefulness.
Sleep onset and processes that maintain sleep
An important consideration is the mechanism that begins this process of reducing the drive from the activating (wakefulness) systems.14 In other words, how does sleep begin? Electrophysiologic recordings of cells in the BF and anterior hypothalamic regions showed that some of these neurons discharge only during sleep, and this was hypothesized to be an active sleep-promoting mechanism. Confirmation came from studies by Sherin and colleagues,15 who used anatomic techniques to detect neurons in the ventrolateral preoptic (VLPO) area that were selectively active during NREM sleep. Subsequent immunohistochemical studies identified the neurotransmitters contained in these cells as inhibitory γ-aminobutyric acid (GABA) and glycine. Anatomic work showed neurons containing GABA and glycine projected not only to wakefulness-promoting histaminergic neurons in the TMN and other hypothalamic centers including the BF, but also to all the brainstem nuclei important in EEG arousal.16 This group of cells coordinates the inhibition of activity in all components of the AAS to facilitate sleep onset. Current studies continue to investigate the interaction of these neurons with other systems that are important in sleep and in particular how other cells within a region around the VLPO area, named the extended VLPO area, are involved with initiating the onset of REM sleep.
Two-process model of sleep regulation
Homeostatic sleep drive.
Sleep propensity is determined by homeostatic and circadian sleep drives. The homeostatic component of sleep need is the sleepiness that follows prolonged wakefulness, and there is now considerable evidence to support the role of adenosine as a mediator of this component. This role of adenosine seems to depend primarily on its inhibitory action on the BF wakefulness-promoting neurons. Commonsense evidence for a sleep-enhancing effect of adenosine comes from the ubiquitous use of coffee and tea to increase alertness because these beverages contain caffeine, an adenosine receptor antagonist.17 McCarley and colleagues18,19 hypothesized that during prolonged wakefulness adenosine accumulates selectively in the BF and promotes the transition from wakefulness to sleep by inhibiting the wakefulness-promoting BF neurons through its action at the adenosine A1 receptor. Regulation of the levels of extracellular adenosine depends primarily on metabolic rate: Increased metabolism leads to reduced high-energy phosphate stores and increased adenosine, which, via an equilibrative transporter, leads to increased extracellular adenosine. The BF wakefulness-promoting neurons inhibit the VLPO area, and the adenosine-mediated inhibition of BF cells is one mechanism by which the VLPO cells begin to discharge as sleepiness increases and the sleep episode begins.
Circadian process.
In addition to the homeostatic need for sleep, sleepiness depends on a second major influence, circadian phase. Humans, similar to many other species, continue to show regular, circadian sleep-wake cycles and other physiologic and hormonal rhythms in the absence of a 24-hour light/dark (LD) cycle. These rhythms must depend on an internal “clock” or pacemaker that is self-sustaining in the absence of external time cues and can be reset by changes in the environment. The mechanisms of this internal clock have been subject to research over many years, and significant progress has been made using genetic data obtained from a wide variety of species, including the bread mold Neurospora, the fruit fly Drosophila, and the mouse.20 The diversity of the species from which these results have been obtained emphasizes a remarkable conservation of function in time-keeping in biologic systems during evolution.
Combining the characteristics of the endogenous sleep-wake rhythm with those of a circadian oscillator has led to testable mathematical models of sleep propensity. One of the most significant of these models was developed by Borbely,21 who based his model on a two-process single oscillator model. In this model, sleep is seen as the net result of two processes. One, process S, or sleep propensity, builds during wakefulness and declines exponentially during sleep and is indexed by the delta power of the EEG. As noted earlier, adenosine is a likely candidate for the endogenous mediator of process S. The second process, process C, is an endogenous circadian oscillator that closely parallels core body temperature. The output from the endogenous clock is probably the mediator of process C.
Circadian sleep-wake and physiologic rhythms are treated as sinusoidal variables in this model, assumptions that are not supported by actual data. The sleep-wake state is essentially a binary process, and the actual shape of the variation in physiologic variables across the nycthemeron is asymmetric and is modulated, under normal conditions, by changes related to activity and sleep onset. The recording of core body temperature under several different conditions, including normal expression of the sleep-wake cycle, sleep deprivation with constant activity over 24 hours, and continuous bed rest with minimal activity but normal sleep-wake behavior, might provide more accurate modeling data after appropriate subtractive manipulation.22 This point is addressed in more detail subsequently.
Circadian processes and chronobiology
Circadian timing system
Biologic rhythms vary systematically across the 24 hours of the nycthemeron. In particular, circadian rhythms are driven by endogenous pacemakers that have periods (τ, tau) approximating 24 hours.23 As noted previously, these are self-sustained, internally generated biologic signals that, in the natural environment, are normally synchronized or entrained to the 24-hour LD cycle. Both sleep and temporal organization are evolutionarily conserved behaviors, although they can change during the life span of an organism.24 Such evolutionarily conserved, intrinsic temporal order is crucial for human health and well-being, and disturbances in these rhythms result in behavioral, physiologic, psychological, biochemical, and endocrinologic abnormalities.
Studies over many years have attempted to derive an accurate estimation of the period of the endogenous pacemaker. To do so required the subjects not only to be placed under “free-running” conditions in which the environment was completely devoid of any time cues, but also under conditions in which the period of the endogenous pacemaker could not be entrained to the rest-activity cycle. These considerations led to the adoption of the forced desynchrony protocol, originally developed by Kleitman in 1938.9 In this type of study, subjects were kept on a rest-activity cycle that was sufficiently long (e.g., 28 hours) to prevent the entrainment of the endogenous pacemaker to this rhythm. Results were variable, however, until Czeisler and colleagues25 in 1999 changed the protocol so that their subjects were exposed to very dim light (about 10 to 15 lux) throughout. In this way, Czeisler and colleagues were able to determine the period of the human circadian pacemaker at 24.18 hours; they also reported that healthy older subjects had the same periodicity, with the same stability and precision, as younger subjects.
Under normal circumstances, circadian rhythms become synchronized or entrained to the environmental LD cycle, which acts as a pervasive and prominent synchronizer, or zeitgeber.26 Light signals are perceived in the retina and are transmitted via a monosynaptic pathway, the retinohypothalamic tract (RHT), to the suprachiasmatic nucleus (SCN). Non–image forming effects of retinal light exposure range from effects on various physiologic measures, such as shifts in the circadian rhythms of melatonin and body temperature, to effects on psychological measures—for example, high environmental light intensity increases arousal and alertness.
Although the mechanism by which light exerts these alerting effects is unknown, a more recently discovered network of blue light–sensitive retinal ganglion cells (RGCs)27,28 is likely part of the input system for the physiologic effects. In animals29,30 and in humans,31 the suppression of melatonin and shifts in circadian rhythms are particularly sensitive to a short-wavelength, blue component of light. The blue light–sensitive RGCs express the photopigment melanopsin and a neuropeptide, pituitary adenylate cyclase activating polypeptide (PACAP).27,28,32 Significantly, the RGCs project to brain regions implicated in sleep mechanisms, including the SCN and the VLPO area.28,33 This finding suggests that they might mediate the effect of light on sleepiness.34
Suprachiasmatic nucleus: the central oscillator
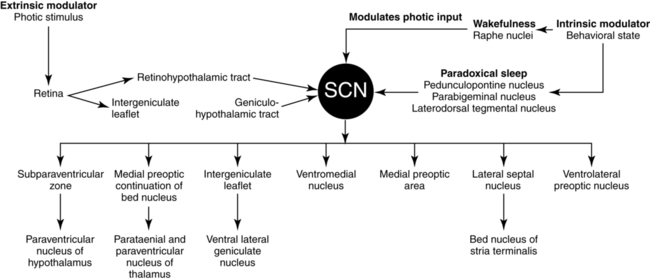
The SCN, which is localized to the anterior hypothalamus, acts as the “biologic clock” to coordinate the circadian rhythm.35–40 The SCN receives photic information via the glutamatergic RHT and the geniculohypothalamic tract (GHT), which contains NPY and nonphotic information via serotoninergic neurons originating in the dorsal raphe nucleus (DRN). SCN neurons, which project to the dorsomedial and posterior hypothalamic areas and the VLPO area, actively promote and maintain wakefulness during the day and sleep at night (Figure 23-7). The SCN is involved in the regulation of the timing of sleep-wake states and in the expression of the sleep-wake cycle and may play a role in the coordination of specific sleep stages.37,40–42 The role of the SCN in the control of sleep has been studied extensively in several species. In squirrel monkeys, a diurnal species similar to humans, the circadian signal produced by the SCN promotes wakefulness during the subjective day and consolidation of sleep at night. Lesions of the SCN have been found to disrupt the consolidation of both sleep and wakefulness as a result of a disrupted circadian rhythm.43 Neurons in the SCN express two melatonin receptors (MT1 and MT2) that have different functional roles.
Chronopharmacology
Chronobiology is concerned with the mechanisms of periodic biologic influences on health and disease44; pharmacology refers to the medical discipline concerned with the biochemical and physiologic aspects of drug effects, including absorption, distribution, metabolism, elimination, toxicity, and specific mechanisms of drug action. The effectiveness of drugs, also a critical aspect of pharmacology, depends on pharmacodynamics (i.e., what the drug does to the body) and pharmacokinetics (i.e., what the body does to the drug). These considerations involve the quantitative aspects of drug absorption, distribution, and excretion that are crucial for the design of rational dosage regimens.
Chronopharmacology, or the study of time-dependent variations in pharmacology,45 was developed from the inclusion of chronobiologic principles in the study of pharmacology. Traditionally, drug delivery has assumed that a chemical is absorbed predictably from the site of administration. A second-generation drug delivery goal has been the achievement of a continuous constant rate (i.e., zero-order) delivery of drugs. However, living organisms are not “zero-order” in their response to drugs. As mentioned earlier, living organisms are predictable resonating dynamic systems governed by intrinsic oscillators, so they require different amounts of drug at different times within the circadian cycle to maximize the desired and undesired (i.e., chronotoxicity) effects of the drug. Two concepts are important when considering changes in drug efficacy over the 24-hour period. The first is circadian changes in drug bioavailability (i.e., chronokinetics), and the second is circadian changes in the susceptibility to the drug (chronesthesy). In brief, clinical chronopharmacology, or chronotherapeutics, is the purposeful alteration of drug levels to match biologic rhythms and to optimize therapeutic outcomes and minimize side effects.
Melatonin as a chronobiotic and chronohypnotic agent
Drugs that directly influence circadian mechanisms are often referred to as chronobiotics.46 The prototype for this type of drug is melatonin (N-acetyl-5-methoxytryptamine), a pineal hormone that has been identified as an important endogenous regulatory factor, with levels that vary with circadian time. In humans, melatonin is an important signal for maintaining endogenous rhythms in synchrony with the environmental LD cycle. Melatonin is exclusively secreted during the subjective night, and its plasma level increases during the evening and declines in the early morning.47 The finding that melatonin is secreted primarily during the night and the close relationship between the nocturnal increase in endogenous melatonin and the timing of human sleep have suggested that melatonin might be important in sleep regulation. The onset of melatonin secretion occurs approximately 2 hours before bedtime and has been shown to correlate with the onset of evening sleepiness. In other words, the transition phase from wakefulness and arousal to high sleep propensity coincides with the nocturnal increase in endogenous melatonin. As noted earlier, SCN neurons express high concentrations of both melatonin receptors, MT1 and MT2, although both receptors are also found widely expressed throughout the CNS. Signaling via MT1 leads primarily to inhibition of activity in SCN neurons.48,49 In contrast, the principal MT2-mediated actions in the SCN are related to circadian phase shifts and constitute activation of protein kinase C and increased cell activity.50,51
It is possible that melatonin contributes to sleep initiation by inhibiting the circadian wakefulness-generating mechanisms, an effect that could be mediated by MT1 receptors in the SCN. Melatonin release is pulsatile during light sleep, and the hormone could function to induce deeper sleep and prevent awakening by continuing to inhibit arousal at the level of the SCN. Overall, the hypnotic and chronobiotic effects of melatonin might be mediated in the SCN and possibly through the MT1 receptor.52–54 Induction and maintenance of sleep at the appropriate circadian phase, which could be MT1-mediated, is different from shifting the phase of sleep, which could be MT2-mediated, as a result of exogenous change in the zeitgeber.
It is relevant that the hypnotic effect of melatonin depends on the circadian phase of administration. Stone and coworkers,55 using a double-blind placebo-controlled study, found that melatonin administered at night (23:30 hours) had no significant effect on sleep in healthy individuals, whereas melatonin administered in the evening (18:00 hours) exerted a hypnotic activity. Despite such evidence for the hypnotic action of melatonin, its efficacy in promoting sleep is still controversial, especially because most results show only borderline significance or are otherwise difficult to evaluate because of methodologic inconsistencies.56 However, the relatively poor outcomes in these studies in terms of sleep efficiency or total sleep time may be due to the short half-life of melatonin in plasma (less than 30 minutes). It is possible that a melatonin receptor agonist with a longer half-life and occupying receptors in the SCN for a longer duration could be more effective than melatonin in promoting sleep in insomniac patients.57
Ramelteon is a melatonin receptor agonist that has been shown to be selective for MT1 and MT2 receptors but without affinity for the melatonin-binding site, quinone reductase 2, previously denoted MT3.58,59 Ramelteon has no affinity for other major CNS receptors, including binding sites for neurotransmitters, neuropeptides, regulatory enzymes, or ion channels.58,59 However, various additional non–membrane binding sites of melatonin remain to be tested.60
Sleep disorders: causes and treatments
Insomnia
Insomnia is characterized by difficulty in falling asleep (i.e., a sleep latency of greater than 30 minutes), insufficient sleep (i.e., total sleep time of less than 5.5 to 6 hours), multiple nocturnal awakenings, early morning awakening with inability to resume sleep, or nonrestorative sleep. Common daytime complaints include somnolence, fatigue, irritability, and difficulty concentrating and performing everyday tasks. In addition, subjects with a diagnosis of insomnia are at higher risk for illness and for injury caused by drowsiness while driving. Because insomnia is associated with difficulty in concentration, it is a major risk factor for accidents.61 Insomnia is a common disorder that affects 30% to 35% of the U.S. adult population and is chronic in about 10%.62 The risk of insomnia is greatest in elderly adults. The adverse physiologic and psychological sequelae of insomnia have a major negative impact on the quality of life in affected individuals.63 Almost 10% of people with chronic insomnia have daytime consequences of fatigue, irritability, and impaired concentration that affect health, mood, and normal functioning. With reduced productivity and an increased risk of accidents, the overall economic burden of insomnia is estimated to be 1% of gross domestic product.64,65 Insomnia is also experienced as a stressor by patients who have major depressive disorders, and disturbed sleep has been identified as a hallmark of depression; this is not always recognized in clinical practice.
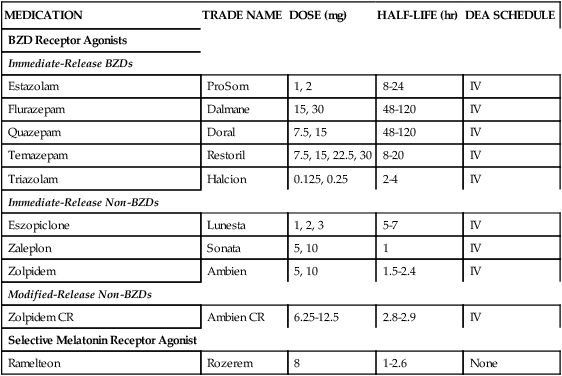
Pharmacologic treatment of insomnia in the last few decades has been based on several classes of medication (Table 23-2). Benzodiazepines were introduced in the 1970s and rapidly increased in popularity because of their efficacy and relative safety compared with barbiturates, carbamates, chloral derivatives, and methaqualone. In recent years, however, prescriptions for benzodiazepines have declined because of their associated side-effect profile, including the tendency of benzodiazepines to promote dependence, the occurrence of rebound insomnia after withdrawal of short-acting and intermediate-acting derivatives, and the loss of efficacy after a few weeks of treatment. The reduction in benzodiazepine use has also coincided with the introduction of a structurally dissimilar group of nonbenzodiazepine derivatives, including the cyclopyrrolone agents zopiclone and eszopiclone, the imidazopyridine derivative zolpidem, and the pyrazolopyrimidine compound zaleplon.
TABLE 23-2
Medications Approved by U.S. Food and Drug Administration for Treatment of Insomnia
| MEDICATION | TRADE NAME | DOSE (mg) | HALF-LIFE (hr) | DEA SCHEDULE |
| BZD Receptor Agonists | ||||
| Immediate-Release BZDs | ||||
| Estazolam | ProSom | 1, 2 | 8-24 | IV |
| Flurazepam | Dalmane | 15, 30 | 48-120 | IV |
| Quazepam | Doral | 7.5, 15 | 48-120 | IV |
| Temazepam | Restoril | 7.5, 15, 22.5, 30 | 8-20 | IV |
| Triazolam | Halcion | 0.125, 0.25 | 2-4 | IV |
| Immediate-Release Non-BZDs | ||||
| Eszopiclone | Lunesta | 1, 2, 3 | 5-7 | IV |
| Zaleplon | Sonata | 5, 10 | 1 | IV |
| Zolpidem | Ambien | 5, 10 | 1.5-2.4 | IV |
| Modified-Release Non-BZDs | ||||
| Zolpidem CR | Ambien CR | 6.25-12.5 | 2.8-2.9 | IV |
| Selective Melatonin Receptor Agonist | ||||
| Ramelteon | Rozerem | 8 | 1-2.6 | None |
Restless legs syndrome and periodic limb movement disorder
More recent epidemiologic and genetic linkage studies distinguish two forms of RLS: early onset, or primary, RLS, and late onset, or secondary, RLS. The early onset form typically begins in childhood or young adulthood with gradual progression in symptom severity. Early onset RLS is inherited in an autosomal dominant fashion, shows genetic anticipation, is twice as prevalent in females, and is associated with at least two separate genetic loci.66 In addition, PLMD is more typically associated with this form of RLS.
Late onset RLS is characterized by a later age of symptom onset, usually older than 45 years; an equal female to male ratio; a rapidly progressive course; and a relationship with anemia and other associated identifiable causes such as diabetes, kidney disease, neuropathy and even nervous system trauma.66 In addition to these intrinsic pathophysiologic mechanisms, certain extrinsic factors are associated with increased frequency and severity of symptoms. H2 histamine antagonists such as ranitidine or cimetidine have been reported to worsen symptoms of RLS.67 Caffeine, alcohol, and some antidepressants including fluoxetine also are reported to worsen the frequency and severity of RLS or PLMD symptoms.68,69 Epidemiologic studies suggest that the onset of RLS increases with age, with a prevalence rate of 2% in children, 3% in 30-year-olds, and up to 20% in 80-year-olds. Genetic studies and linkage analyses also show that early onset RLS is a heritable trait, but the pathophysiologic mechanisms of RLS remain unclear.66
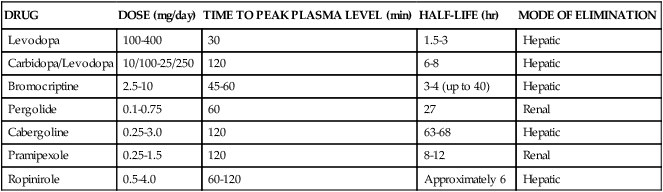
The first link made between dopamine and RLS was based on the observation that many patients derived relief from dopamine augmenting drugs. This was acknowledged in the “Practice Parameters for the treatment of RLS and PLM,”70 which stated that dopaminergic agents are the best-studied and most successful agents for treatment of RLS and PLMD. Following multiple clinical trials with dopamine-enhancing compounds, levodopa with decarboxylase inhibitors and dopamine agonists such as pergolide were found to be the most effective for treatment of RLS and PLMD.70 Despite the promise that dopamine-enhancing compounds can reduce the symptoms of RLS, it should be noted that their use for RLS is currently approved only for adults; data are lacking with regard to their use for RLS in pediatric populations and during pregnancy. When prescribing any type of dopaminergic medication for RLS symptom relief, the potential for side effects, such as nausea, gastrointestinal distress, reduced blood pressure, and sleepiness, should be taken into account and discussed with a sleep medicine physician. In addition, because dopamine modulates mood, cognition, wakefulness, and sleep, any dopamine precursor, agonist, or antagonist can feasibly result in acute thought and behavioral changes. Given that most RLS patients need very low doses of dopaminergic medications for symptomatic relief, the likelihood of a serious or adverse outcome is remote (Table 23-3).
TABLE 23-3
Pharmacologic Management of Restless Legs Syndrome
| DRUG | DOSE (mg/day) | TIME TO PEAK PLASMA LEVEL (min) | HALF-LIFE (hr) | MODE OF ELIMINATION |
| Levodopa | 100-400 | 30 | 1.5-3 | Hepatic |
| Carbidopa/Levodopa | 10/100-25/250 | 120 | 6-8 | Hepatic |
| Bromocriptine | 2.5-10 | 45-60 | 3-4 (up to 40) | Hepatic |
| Pergolide | 0.1-0.75 | 60 | 27 | Renal |
| Cabergoline | 0.25-3.0 | 120 | 63-68 | Hepatic |
| Pramipexole | 0.25-1.5 | 120 | 8-12 | Renal |
| Ropinirole | 0.5-4.0 | 60-120 | Approximately 6 | Hepatic |
Narcolepsy
The earliest clinical descriptions of narcolepsy were documented by the German physician Westphal in 1877 and by Fisher in 1878. However, the French physician Gelineau, writing in 1880, is generally acknowledged as the first clinician to recognize narcolepsy as a distinct clinical entity. Initial descriptions included characterization of excessive daytime sleepiness and “sleep attacks.” Although all three of these early descriptions documented the appearance of sleep attacks, they did not discern the symptoms of sudden muscle weakness triggered by an emotional event (i.e., cataplexy) as separate from the sleep attack event. This is most evident from the first description of narcolepsy as a clinical entity: “I propose to give the name of narcolepsy (‘narco = somnolence’ and ‘lepsy = seized by’) to a rare neurosis or at least little known until now, characterized by a mandatory need to sleep, sudden and of short duration, that recurs at more or less close intervals.”71 In 1902, Loewenfeld recognized that the emotion-induced muscle weakness was a separate feature of the disorder and first used the term cataplexy to describe it.
Today, narcolepsy is characterized by a tetrad of clinical symptoms. Two features of the tetrad—persistent excessive daytime sleepiness and cataplexy—were documented in the original descriptions of the disorder and remain the only two clinical features essential for diagnosis. The recognition of two additional features—hypnagogic hallucinations (the onset of dreams while still awake) and sleep paralysis (a temporary loss of muscle tone or an inability to perform voluntary movements either at sleep onset or on awakening)—were added later.72 Another common symptom of narcolepsy is fragmented sleep with multiple arousals and awakenings at night.
Most cases of narcolepsy are sporadic and occur without evidence of genetic inheritance. Until more recently, the etiology of narcolepsy was unknown, but it was associated with the specific human leukocyte antigen (HLA) allele, DQB10602, and often in combination with HLA-DR2 (DRB115). In 1998, two research groups first described, clustered around the lateral and perifornical hypothalamus, a group of cells that contained a previously unknown neurotransmitter.73,74 These cells contained orexin/hypocretin, and based on the neuroanatomy they were hypothesized to be involved in the regulation of sleep and wakefulness. However, the physiology suggested a major role in food intake and energy balance, which was the focus of research during the first 12 months. Within 1 year of the discovery, however, two independent groups discovered in 1999 that the clinical symptoms of narcolepsy were associated with a loss or dysfunction of orexin/hypocretin-containing cells.75,76
Parasomnias
Parasomnias are undesirable motor, sensory, or behavioral phenomena that occur primarily during sleep.77 These phenomena range from normal to abnormal and from benign to potentially lethal and can be associated with normal developmental processes or neurodegeneration. The ICSD-R lists 24 parasomnias encompassing NREM sleep or arousal disorders, REM sleep-related disorders, sleep-wake transition disorders, and other parasomnias. The focus here is on more common NREM and REM sleep parasomnias. NREM sleep parasomnias, also termed arousal disorders, include confusional arousals, sleep terrors, and sleepwalking. Arousal disorders can be either primary or secondary if they are associated with an identifiable pathology such as a seizure disorder, obstructive sleep apnea, nocturnal cardiac ischemia, or nocturnal paroxysmal dystonia. The pathophysiologic mechanisms underlying arousal disorders are still unknown, but current hypotheses suggest that they may result from the brain being simultaneously in a state of partial wakefulness and NREM sleep. This state leads to an ability to perform complex motor or verbal actions without conscious awareness of the actions.
 SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
1. What is the International Classification of Sleep Disorders, Revised, and what information does it contain?
2. What are the electroencephalographic correlates of wakefulness and sleep stages N1, N2, N3, and REM?
3. What type of drug had been used for centuries to promote sleep onset and maintenance?
4. What class or classes of drugs have replaced opium?
5. How many people in the United States experience chronic sleep disorders?
6. Who was von Economo, and what theories guided his neuroanatomic exploration of brain regions involved in the processes of initiating and maintaining wakefulness and sleep?
7. What is the reticular activating system?
8. How do neurons within the ventrolateral preoptic (VLPO) area affect sleep?
9. What are the suprachiasmatic nuclei (SCN), and what are intrinsic discharge properties exhibited by many of its neurons?
10. Define chronobiology and chronopharmacology.
11. Describe pharmacologic treatments for insomnia.
12. What is the difference between restless legs syndrome (RLS) and periodic limb movement disorder (PLMD)?
13. What is the tetrad of clinical symptoms that defines narcolepsy, and what types of drugs are used to treat this disorder?
14. Give two examples of a parasomnia, and name the class of drugs routinely used in the treatment of these sleep disorders.
15. Insomnia is characterized by what types of principal complaint, and what drug classes are used to treat this disorder?
 CLINICAL SCENARIO
CLINICAL SCENARIO