HIGH-ANION GAP ACIDOSES
Lactic Acidosis An increase in plasma l-lactate may be secondary to poor tissue perfusion (type A)—circulatory insufficiency (shock, cardiac failure), severe anemia, mitochondrial enzyme defects, and inhibitors (carbon monoxide, cyanide)—or to aerobic disorders (type B)—malignancies, nucleoside analogue reverse transcriptase inhibitors in HIV, diabetes mellitus, renal or hepatic failure, thiamine deficiency, severe infections (cholera, malaria), seizures, or drugs/toxins (biguanides, ethanol, methanol, propylene glycol, isoniazid, and fructose). Unrecognized bowel ischemia or infarction in a patient with severe atherosclerosis or cardiac decompensation receiving vasopressors is a common cause of lactic acidosis. Pyroglutamic acidemia has been reported in critically ill patients receiving acetaminophen, which is associated with depletion of glutathione. D-Lactic acid acidosis, which may be associated with jejunoileal bypass, short bowel syndrome, or intestinal obstruction, is due to formation of D-lactate by gut bacteria.
Ketoacidosis • DIABETIC KETOACIDOSIS (DKA) This condition is caused by increased fatty acid metabolism and the accumulation of ketoacids (acetoacetate and β-hydroxybutyrate). DKA usually occurs in insulin-dependent diabetes mellitus in association with cessation of insulin or an intercurrent illness such as an infection, gastroenteritis, pancreatitis, or myocardial infarction, which increases insulin requirements temporarily and acutely. The accumulation of ketoacids accounts for the increment in the AG and is accompanied most often by hyperglycemia (glucose >17 mmol/L [300 mg/dL]). The relationship between the ΔAG and ΔHCO3– is typically ~1:1 in DKA. It should be noted that, because insulin prevents production of ketones, bicarbonate therapy is rarely needed except with extreme acidemia (pH < 7.1), and then in only limited amounts. Patients with DKA are typically volume depleted and require fluid resuscitation with isotonic saline. Volume overexpansion with IV fluid administration is not uncommon, however, and contributes to the development of a hyperchloremic acidosis during treatment of DKA. The mainstay for treatment of this condition is IV regular insulin and is described in Chap. 417 in more detail.
ALCOHOLIC KETOACIDOSIS (AKA) Chronic alcoholics can develop ketoacidosis when alcohol consumption is abruptly curtailed and nutrition is poor. AKA is usually associated with binge drinking, vomiting, abdominal pain, starvation, and volume depletion. The glucose concentration is variable, and acidosis may be severe because of elevated ketones, predominantly β-hydroxybutyrate. Hypoperfusion may enhance lactic acid production, chronic respiratory alkalosis may accompany liver disease, and metabolic alkalosis can result from vomiting (refer to the relationship between ΔAG and ΔHCO3–). Thus, mixed acid-base disorders are common in AKA. As the circulation is restored by administration of isotonic saline, the preferential accumulation of β-hydroxybutyrate is then shifted to acetoacetate. This explains the common clinical observation of an increasingly positive nitroprusside reaction as the patient improves. The nitroprusside ketone reaction (Acetest) can detect acetoacetic acid but not β-hydroxybutyrate, so that the degree of ketosis and ketonuria can not only change with therapy, but can be underestimated initially. Patients with AKA usually present with relatively normal renal function, as opposed to DKA, where renal function is often compromised because of volume depletion (osmotic diuresis) or diabetic nephropathy. The AKA patient with normal renal function may excrete relatively large quantities of ketoacids in the urine and, therefore, may have a relatively normal AG and a discrepancy in the ΔAG/ΔHCO3– relationship.
Drug- and Toxin-Induced Acidosis • SALICYLATES (See also Chap. 472e) Salicylate intoxication in adults usually causes respiratory alkalosis or a mixture of high-AG metabolic acidosis and respiratory alkalosis. Only a portion of the AG is due to salicylates. Lactic acid production is also often increased.
ALCOHOLS Under most physiologic conditions, sodium, urea, and glucose generate the osmotic pressure of blood. Plasma osmolality is calculated according to the following expression: Posm = 2Na+ + Glu + BUN (all in mmol/L), or, using conventional laboratory values in which glucose and BUN are expressed in milligrams per deciliter: Posm = 2Na+ + Glu/18 + BUN/2.8. The calculated and determined osmolality should agree within 10–15 mmol/kg H2O. When the measured osmolality exceeds the calculated osmolality by >10–15 mmol/kg H2O, one of two circumstances prevails. Either the serum sodium is spuriously low, as with hyperlipidemia or hyperproteinemia (pseudohyponatremia), or osmolytes other than sodium salts, glucose, or urea have accumulated in plasma. Examples of such osmolytes include mannitol, radiocontrast media, ethanol, isopropyl alcohol, ethylene glycol, propylene glycol, methanol, and acetone. In this situation, the difference between the calculated osmolality and the measured osmolality (osmolar gap) is proportional to the concentration of the unmeasured solute. With an appropriate clinical history and index of suspicion, identification of an osmolar gap is helpful in identifying the presence of poison-associated AG acidosis. Three alcohols may cause fatal intoxications: ethylene glycol, methanol, and isopropyl alcohol. All cause an elevated osmolal gap, but only the first two cause a high-AG acidosis.
ETHYLENE GLYCOL (See also Chap. 472e) Ingestion of ethylene glycol (commonly used in antifreeze) leads to a metabolic acidosis and severe damage to the CNS, heart, lungs, and kidneys. The increased AG and osmolar gap are attributable to ethylene glycol and its metabolites, oxalic acid, glycolic acid, and other organic acids. Lactic acid production increases secondary to inhibition of the tricarboxylic acid cycle and altered intracellular redox state. Diagnosis is facilitated by recognizing oxalate crystals in the urine, the presence of an osmolar gap in serum, and a high-AG acidosis. Although use of a Wood’s lamp to visualize the fluorescent additive to commercial antifreeze in the urine of patients with ethylene glycol ingestion, this is rarely reproducible. The combination of a high AG and high osmolar gap in a patient suspected of ethylene glycol ingestion should be taken as evidence of ethylene glycol toxicity. Treatment should not be delayed while awaiting measurement of ethylene glycol levels in this setting.
METHANOL (See also Chap. 472e) The ingestion of methanol (wood alcohol) causes metabolic acidosis, and its metabolites formaldehyde and formic acid cause severe optic nerve and CNS damage. Lactic acid, ketoacids, and other unidentified organic acids may contribute to the acidosis. Due to its low molecular mass (32 Da), an osmolar gap is usually present.
PROPYLENE GLYCOL Propylene glycol is the vehicle used in IV administration of diazepam, lorazepam, phenobarbital, nitroglycerine, etomidate, enoximone, and phenytoin. Propylene glycol is generally safe for limited use in these IV preparations, but toxicity has been reported, most often in the setting of the intensive care unit in patients receiving frequent or continuous therapy. This form of high-gap acidosis should be considered in patients with unexplained high-gap acidosis, hyperosmolality, and clinical deterioration. Propylene glycol, like ethylene glycol and methanol, is metabolized by alcohol dehydrogenase. With intoxication by propylene glycol, the first response is to stop the offending infusion. Additionally, fomepizole should also be administered in acidotic patients.
ISOPROPYL ALCOHOL Ingested isopropanol is absorbed rapidly and may be fatal when as little as 150 mL of rubbing alcohol, solvent, or deicer is consumed. A plasma level >400 mg/dL is life-threatening. Isopropyl alcohol is metabolized by alcohol dehydrogenase to acetone. The characteristic features differ from ethylene glycol and methanol in that the parent compound, not the metabolites, causes toxicity, and an AG acidosis is not present because acetone is rapidly excreted. Both isopropyl alcohol and acetone increase the osmolal gap, and hypoglycemia is common. Alternative diagnoses should be considered if the patient does not improve significantly within a few hours. Patients with hemodynamic instability with plasma levels above 400 mg/dL should be considered for hemodialysis.
PYROGLUTAMIC ACID Acetaminophen-induced high-AG metabolic acidosis is uncommon but is being recognized more often in either patients with acetaminophen overdose or malnourished or critically ill patients receiving acetaminophen in typical dosage. 5-Oxoproline accumulation after acetaminophen should be suspected in the setting of an unexplained high-AG acidosis without elevation of the osmolar gap in patients receiving acetaminophen. The first step in treatment is to immediately discontinue the drug. Additionally, sodium bicarbonate IV should be given. Although N-acetylcysteine has been suggested, it is not known if it hastens the metabolism of 5-oxoproline by increasing intracellular glutathione concentrations in this setting.
Renal Failure (See also Chap. 335) The hyperchloremic acidosis of moderate renal insufficiency is eventually converted to the high-AG acidosis of advanced renal failure. Poor filtration and reabsorption of organic anions contribute to the pathogenesis. As renal disease progresses, the number of functioning nephrons eventually becomes insufficient to keep pace with net acid production. Uremic acidosis is characterized, therefore, by a reduced rate of NH4+ production and excretion. The acid retained in chronic renal disease is buffered by alkaline salts from bone. Despite significant retention of acid (up to 20 mmol/d), the serum [HCO3–] does not decrease further, indicating participation of buffers outside the extracellular compartment. Chronic metabolic acidosis results in significant loss of bone mass due to reduction in bone calcium carbonate. Chronic acidosis also increases urinary calcium excretion, proportional to cumulative acid retention.
NON–ANION GAP METABOLIC ACIDOSES
Alkali can be lost from the gastrointestinal tract from diarrhea or from the kidneys (renal tubular acidosis, RTA). In these disorders (Table 66-5), reciprocal changes in [Cl–] and [HCO3–] result in a normal AG. In pure non–AG acidosis, therefore, the increase in [Cl–] above the normal value approximates the decrease in [HCO3–]. The absence of such a relationship suggests a mixed disturbance.
|
CAUSES OF NON–ANION GAP ACIDOSIS |
Abbreviations: ACE-I, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; PHA, pseudohypoaldosteronism; RTA, renal tubular acidosis.
Proximal RTA (type 2 RTA) (Chap. 339) is most often due to generalized proximal tubular dysfunction manifested by glycosuria, generalized aminoaciduria, and phosphaturia (Fanconi syndrome). With a low plasma [HCO3–], the urine pH is acid (pH <5.5). The fractional excretion of [HCO3–] may exceed 10–15% when the serum HCO3– >20 mmol/L. Because HCO3– is not reabsorbed normally in the proximal tubule, therapy with NaHCO3 will enhance renal potassium wasting and hypokalemia.
The typical findings in acquired or inherited forms of classic distal RTA (type 1 RTA) include hypokalemia, non-AG metabolic acidosis, low urinary NH4+ excretion (positive UAG, low urine [NH4+]), and inappropriately high urine pH (pH > 5.5). Most patients have hypocitraturia and hypercalciuria, so nephrolithiasis, nephrocalcinosis, and bone disease are common. In generalized distal RTA (type 4 RTA), hyperkalemia is disproportionate to the reduction in glomerular filtration rate (GFR) because of coexisting dysfunction of potassium and acid secretion. Urinary ammonium excretion is invariably depressed, and renal function may be compromised, for example, due to diabetic nephropathy, obstructive uropathy, or chronic tubulointerstitial disease.
Hyporeninemic hypoaldosteronism typically causes non-AG metabolic acidosis, most commonly in older adults with diabetes mellitus or tubulointerstitial disease and renal insufficiency. Patients usually have mild to moderate CKD (GFR, 20–50 mL/min) and acidosis, with elevation in serum [K+] (5.2–6.0 mmol/L), concurrent hypertension, and congestive heart failure. Both the metabolic acidosis and the hyperkalemia are out of proportion to impairment in GFR. Nonsteroidal anti-inflammatory drugs, trimethoprim, pentamidine, and angiotensin-converting enzyme (ACE) inhibitors can also cause non-AG metabolic acidosis in patients with renal insufficiency (Table 66-5).
METABOLIC ALKALOSIS
Metabolic alkalosis is manifested by an elevated arterial pH, an increase in the serum [HCO3–], and an increase in PaCO2 as a result of compensatory alveolar hypoventilation (Table 66-1). It is often accompanied by hypochloremia and hypokalemia. The arterial pH establishes the diagnosis, because it is increased in metabolic alkalosis and decreased or normal in respiratory acidosis. Metabolic alkalosis frequently occurs in association with other disorders such as respiratory acidosis or alkalosis or metabolic acidosis.
PATHOGENESIS
Metabolic alkalosis occurs as a result of net gain of [HCO3–] or loss of nonvolatile acid (usually HCl by vomiting) from the extracellular fluid. For HCO3– to be added to the extracellular fluid, it must be administered exogenously or synthesized endogenously, in part or entirely by the kidneys. Because it is unusual for alkali to be added to the body, the disorder involves a generative stage, in which the loss of acid usually causes alkalosis, and a maintenance stage, in which the kidneys fail to compensate by excreting HCO3–.
Maintenance of metabolic alkalosis represents a failure of the kidneys to eliminate HCO3– in the usual manner. The kidneys will retain, rather than excrete, the excess alkali and maintain the alkalosis if (1) volume deficiency, chloride deficiency, and K+ deficiency exist in combination with a reduced GFR; or (2) hypokalemia exists because of autonomous hyperaldosteronism. In the first example, alkalosis is corrected by administration of NaCl and KCl, whereas, in the latter, it may be necessary to repair the alkalosis by pharmacologic or surgical intervention, not with saline administration.
DIFFERENTIAL DIAGNOSIS
To establish the cause of metabolic alkalosis (Table 66-6), it is necessary to assess the status of the extracellular fluid volume (ECFV), the recumbent and upright blood pressure, the serum [K+], and the renin-aldosterone system. For example, the presence of chronic hypertension and chronic hypokalemia in an alkalotic patient suggests either mineralocorticoid excess or that the hypertensive patient is receiving diuretics. Low plasma renin activity and normal urine [Na+] and [Cl–] in a patient who is not taking diuretics indicate a primary mineralocorticoid excess syndrome. The combination of hypokalemia and alkalosis in a normotensive, nonedematous patient can be due to Bartter’s or Gitelman’s syndrome, magnesium deficiency, vomiting, exogenous alkali, or diuretic ingestion. Determination of urine electrolytes (especially the urine [Cl–]) and screening of the urine for diuretics may be helpful. If the urine is alkaline, with an elevated [Na+] and [K+] but low [Cl–], the diagnosis is usually either vomiting (overt or surreptitious) or alkali ingestion. If the urine is relatively acid and has low concentrations of Na+, K+, and Cl–, the most likely possibilities are prior vomiting, the posthypercapnic state, or prior diuretic ingestion. If, on the other hand, neither the urine sodium, potassium, nor chloride concentrations are depressed, magnesium deficiency, Bartter’s or Gitelman’s syndrome, or current diuretic ingestion should be considered. Bartter’s syndrome is distinguished from Gitelman’s syndrome because of hypocalciuria and hypomagnesemia in the latter disorder.
|
CAUSES OF METABOLIC ALKALOSIS |
Abbreviations: DCT, distal convoluted tubule; ECFV, extracellular fluid volume; TALH, thick ascending limb of Henle’s loop.
Alkali Administration Chronic administration of alkali to individuals with normal renal function rarely causes alkalosis. However, in patients with coexistent hemodynamic disturbances, alkalosis can develop because the normal capacity to excrete HCO3– may be exceeded or there may be enhanced reabsorption of HCO3–. Such patients include those who receive HCO3– (PO or IV), acetate loads (parenteral hyperalimentation solutions), citrate loads (transfusions), or antacids plus cation-exchange resins (aluminum hydroxide and sodium polystyrene sulfonate). Nursing home patients receiving tube feedings have a higher incidence of metabolic alkalosis than nursing home patients receiving oral feedings.
METABOLIC ALKALOSIS ASSOCIATED WITH ECFV CONTRACTION, K+ DEPLETION, AND SECONDARY HYPERRENINEMIC HYPERALDOSTERONISM
Gastrointestinal Origin Gastrointestinal loss of H+ from vomiting or gastric aspiration results in retention of HCO3–. During active vomiting, the filtered load of bicarbonate is acutely increased to exceed the reabsorptive capacity of the proximal tubule for HCO3– so that the urine becomes alkaline and high in potassium. When vomiting ceases, the persistence of volume, potassium, and chloride depletion causes maintenance of the alkalosis because of an enhanced capacity of the nephron to reabsorb HCO3–. Correction of the contracted ECFV with NaCl and repair of K+ deficits corrects the acid-base disorder by restoring the ability of the kidney to excrete the excess bicarbonate.
Renal Origin • DIURETICS (See also Chap. 279) Drugs that induce chloruresis, such as thiazides and loop diuretics (furosemide, bumetanide, torsemide, and ethacrynic acid), acutely diminish the ECFV without altering the total body bicarbonate content. The serum [HCO3–] increases because the reduced ECFV “contracts” the [HCO3–] in the plasma (contraction alkalosis). The chronic administration of diuretics tends to generate an alkalosis by increasing distal salt delivery, so that K+ and H+ secretion are stimulated. The alkalosis is maintained by persistence of the contraction of the ECFV, secondary hyperaldosteronism, K+ deficiency, and the direct effect of the diuretic (as long as diuretic administration continues). Repair of the alkalosis is achieved by providing isotonic saline to correct the ECFV deficit.
SOLUTE LOSING DISORDERS: BARTTER’S SYNDROME AND GITELMAN’S SYNDROME See Chap. 339.
NONREABSORBABLE ANIONS AND MAGNESIUM DEFICIENCY Administration of large quantities of nonreabsorbable anions, such as penicillin or carbenicillin, can enhance distal acidification and K+ secretion by increasing the transepithelial potential difference. Mg2+ deficiency results in hypokalemic alkalosis by enhancing distal acidification through stimulation of renin and hence aldosterone secretion.
POTASSIUM DEPLETION Chronic K+ depletion may cause metabolic alkalosis by increasing urinary acid excretion. Both NH4+ production and absorption are enhanced and HCO3– reabsorption is stimulated. Chronic K+ deficiency upregulates the renal H+, K+-ATPase to increase K+ absorption at the expense of enhanced H+ secretion. Alkalosis associated with severe K+ depletion is resistant to salt administration, but repair of the K+ deficiency corrects the alkalosis.
AFTER TREATMENT OF LACTIC ACIDOSIS OR KETOACIDOSIS When an underlying stimulus for the generation of lactic acid or ketoacid is removed rapidly, as with repair of circulatory insufficiency or with insulin therapy, the lactate or ketones are metabolized to yield an equivalent amount of HCO3–. Other sources of new HCO3– are additive with the original amount generated by organic anion metabolism to create a surfeit of HCO3–. Such sources include (1) new HCO3– added to the blood by the kidneys as a result of enhanced acid excretion during the preexisting period of acidosis, and (2) alkali therapy during the treatment phase of the acidosis. Acidosis-induced contraction of the ECFV and K+ deficiency act to sustain the alkalosis.
POSTHYPERCAPNIA Prolonged CO2 retention with chronic respiratory acidosis enhances renal HCO3– absorption and the generation of new HCO3– (increased net acid excretion). Metabolic alkalosis results from the effect of the persistently elevated [HCO3–] when the elevated PaCO2 is abruptly returned toward normal.
METABOLIC ALKALOSIS ASSOCIATED WITH ECFV EXPANSION, HYPERTENSION, AND HYPERALDOSTERONISM
Increased aldosterone levels may be the result of autonomous primary adrenal overproduction or of secondary aldosterone release due to renal overproduction of renin. Mineralocorticoid excess increases net acid excretion and may result in metabolic alkalosis, which may be worsened by associated K+ deficiency. ECFV expansion from salt retention causes hypertension. The kaliuresis persists because of mineralocorticoid excess and distal Na+ absorption causing enhanced K+ excretion, continued K+ depletion with polydipsia, inability to concentrate the urine, and polyuria.
Liddle’s syndrome (Chap. 339) results from increased activity of the collecting duct Na+ channel (ENaC) and is a rare monogenic form of hypertension due to volume expansion manifested as hypokalemic alkalosis and normal aldosterone levels.
Symptoms With metabolic alkalosis, changes in CNS and peripheral nervous system function are similar to those of hypocalcemia (Chap. 423); symptoms include mental confusion; obtundation; and a predisposition to seizures, paresthesia, muscular cramping, tetany, aggravation of arrhythmias, and hypoxemia in chronic obstructive pulmonary disease. Related electrolyte abnormalities include hypokalemia and hypophosphatemia.
RESPIRATORY ACIDOSIS
Respiratory acidosis can be due to severe pulmonary disease, respiratory muscle fatigue, or abnormalities in ventilatory control and is recognized by an increase in PaCO2 and decrease in pH (Table 66-7). In acute respiratory acidosis, there is an immediate compensatory elevation (due to cellular buffering mechanisms) in HCO3–, which increases 1 mmol/L for every 10-mmHg increase in PaCO2. In chronic respiratory acidosis (>24 h), renal adaptation increases the [HCO3–] by 4 mmol/L for every 10-mmHg increase in PaCO2. The serum HCO3– usually does not increase above 38 mmol/L.
|
RESPIRATORY ACID-BASE DISORDERS |
The clinical features vary according to the severity and duration of the respiratory acidosis, the underlying disease, and whether there is accompanying hypoxemia. A rapid increase in PaCO2 may cause anxiety, dyspnea, confusion, psychosis, and hallucinations and may progress to coma. Lesser degrees of dysfunction in chronic hypercapnia include sleep disturbances; loss of memory; daytime somnolence; personality changes; impairment of coordination; and motor disturbances such as tremor, myoclonic jerks, and asterixis. Headaches and other signs that mimic raised intracranial pressure, such as papilledema, abnormal reflexes, and focal muscle weakness, are due to vasoconstriction secondary to loss of the vasodilator effects of CO2.
Depression of the respiratory center by a variety of drugs, injury, or disease can produce respiratory acidosis. This may occur acutely with general anesthetics, sedatives, and head trauma or chronically with sedatives, alcohol, intracranial tumors, and the syndromes of sleep-disordered breathing including the primary alveolar and obesity-hypoventilation syndromes (Chaps. 318 and 319). Abnormalities or disease in the motor neurons, neuromuscular junction, and skeletal muscle can cause hypoventilation via respiratory muscle fatigue. Mechanical ventilation, when not properly adjusted and supervised, may result in respiratory acidosis, particularly if CO2 production suddenly rises (because of fever, agitation, sepsis, or overfeeding) or alveolar ventilation falls because of worsening pulmonary function. High levels of positive end-expiratory pressure in the presence of reduced cardiac output may cause hypercapnia as a result of large increases in alveolar dead space (Chap. 306e). Permissive hypercapnia is being used with increasing frequency because of studies suggesting lower mortality rates than with conventional mechanical ventilation, especially with severe CNS or heart disease. The respiratory acidosis associated with permissive hypercapnia may require administration of NaHCO3 to increase the arterial pH to 7.25, but overcorrection of the acidemia may be deleterious.
Acute hypercapnia follows sudden occlusion of the upper airway or generalized bronchospasm as in severe asthma, anaphylaxis, inhalational burn, or toxin injury. Chronic hypercapnia and respiratory acidosis occur in end-stage obstructive lung disease. Restrictive disorders involving both the chest wall and the lungs can cause respiratory acidosis because the high metabolic cost of respiration causes ventilatory muscle fatigue. Advanced stages of intrapulmonary and extrapulmonary restrictive defects present as chronic respiratory acidosis.
The diagnosis of respiratory acidosis requires the measurement of PaCO2 and arterial pH. A detailed history and physical examination often indicate the cause. Pulmonary function studies (Chap. 306e), including spirometry, diffusion capacity for carbon monoxide, lung volumes, and arterial PaCO2 and O2 saturation, usually make it possible to determine if respiratory acidosis is secondary to lung disease. The workup for nonpulmonary causes should include a detailed drug history, measurement of hematocrit, and assessment of upper airway, chest wall, pleura, and neuromuscular function.
RESPIRATORY ALKALOSIS
Alveolar hyperventilation decreases PaCO2 and increases the HCO3–/PaCO2 ratio, thus increasing pH (Table 66-7). Nonbicarbonate cellular buffers respond by consuming HCO3–. Hypocapnia develops when a sufficiently strong ventilatory stimulus causes CO2 output in the lungs to exceed its metabolic production by tissues. Plasma pH and [HCO3–] appear to vary proportionately with PaCO2 over a range from 40–15 mmHg. The relationship between arterial [H+] concentration and PaCO2 is ~0.7 mmol/L per mmHg (or 0.01 pH unit/mmHg), and that for plasma [HCO3–] is 0.2 mmol/L per mmHg. Hypocapnia sustained for >2–6 h is further compensated by a decrease in renal ammonium and titratable acid excretion and a reduction in filtered HCO3– reabsorption. Full renal adaptation to respiratory alkalosis may take several days and requires normal volume status and renal function. The kidneys appear to respond directly to the lowered PaCO2 rather than to alkalosis per se. In chronic respiratory alkalosis a 1-mmHg decrease in PaCO2 causes a 0.4-to 0.5-mmol/L drop in [HCO3–] and a 0.3-mmol/L decrease (or 0.003 increase in pH) in [H+].
The effects of respiratory alkalosis vary according to duration and severity but are primarily those of the underlying disease. Reduced cerebral blood flow as a consequence of a rapid decline in PaCO2 may cause dizziness, mental confusion, and seizures, even in the absence of hypoxemia. The cardiovascular effects of acute hypocapnia in the conscious human are generally minimal, but in the anesthetized or mechanically ventilated patient, cardiac output and blood pressure may fall because of the depressant effects of anesthesia and positive-pressure ventilation on heart rate, systemic resistance, and venous return. Cardiac arrhythmias may occur in patients with heart disease as a result of changes in oxygen unloading by blood from a left shift in the hemoglobin-oxygen dissociation curve (Bohr effect). Acute respiratory alkalosis causes intracellular shifts of Na+, K+, and PO42– and reduces free [Ca2+] by increasing the protein-bound fraction. Hypocapnia-induced hypokalemia is usually minor.
Chronic respiratory alkalosis is the most common acid-base disturbance in critically ill patients and, when severe, portends a poor prognosis. Many cardiopulmonary disorders manifest respiratory alkalosis in their early to intermediate stages, and the finding of normocapnia and hypoxemia in a patient with hyperventilation may herald the onset of rapid respiratory failure and should prompt an assessment to determine if the patient is becoming fatigued. Respiratory alkalosis is common during mechanical ventilation.
The hyperventilation syndrome may be disabling. Paresthesia; circumoral numbness; chest wall tightness or pain; dizziness; inability to take an adequate breath; and, rarely, tetany may be sufficiently stressful to perpetuate the disorder. Arterial blood-gas analysis demonstrates an acute or chronic respiratory alkalosis, often with hypocapnia in the range of 15–30 mmHg and no hypoxemia. CNS diseases or injury can produce several patterns of hyperventilation and sustained PaCO2 levels of 20–30 mmHg. Hyperthyroidism, high caloric loads, and exercise raise the basal metabolic rate, but ventilation usually rises in proportion so that arterial blood gases are unchanged and respiratory alkalosis does not develop. Salicylates are the most common cause of drug-induced respiratory alkalosis as a result of direct stimulation of the medullary chemoreceptor (Chap. 472e). The methylxanthines, theophylline, and aminophylline stimulate ventilation and increase the ventilatory response to CO2. Progesterone increases ventilation and lowers arterial PaCO2 by as much as 5–10 mmHg. Therefore, chronic respiratory alkalosis is a common feature of pregnancy. Respiratory alkalosis is also prominent in liver failure, and the severity correlates with the degree of hepatic insufficiency. Respiratory alkalosis is often an early finding in gram-negative septicemia, before fever, hypoxemia, or hypotension develops.
The diagnosis of respiratory alkalosis depends on measurement of arterial pH and PaCO2. The plasma [K+] is often reduced and the [Cl–] increased. In the acute phase, respiratory alkalosis is not associated with increased renal HCO3– excretion, but within hours net acid excretion is reduced. In general, the HCO3– concentration falls by 2.0 mmol/L for each 10-mmHg decrease in PaCO2. Chronic hypocapnia reduces the serum [HCO3–] by 4.0 mmol/L for each 10-mmHg decrease in PaCO2. It is unusual to observe a plasma HCO3– <12 mmol/L as a result of a pure respiratory alkalosis.
When a diagnosis of respiratory alkalosis is made, its cause should be investigated. The diagnosis of hyperventilation syndrome is made by exclusion. In difficult cases, it may be important to rule out other conditions such as pulmonary embolism, coronary artery disease, and hyperthyroidism.
SECTION 8 |
ALTERATIONS IN SEXUAL FUNCTION AND REPRODUCTION |
67 |
Sexual Dysfunction |
Male sexual dysfunction affects 10–25% of middle-aged and elderly men, and female sexual dysfunction occurs with similar frequency. Demographic changes, the popularity of newer treatments, and greater awareness of sexual dysfunction by patients and society have led to increased diagnosis and associated health care expenditures for the management of this common disorder. Because many patients are reluctant to initiate discussion of their sex lives, physicians should address this topic directly to elicit a history of sexual dysfunction.
MALE SEXUAL DYSFUNCTION
PHYSIOLOGY OF MALE SEXUAL RESPONSE
Normal male sexual function requires (1) an intact libido, (2) the ability to achieve and maintain penile erection, (3) ejaculation, and (4) detumescence. Libido refers to sexual desire and is influenced by a variety of visual, olfactory, tactile, auditory, imaginative, and hormonal stimuli. Sex steroids, particularly testosterone, act to increase libido. Libido can be diminished by hormonal or psychiatric disorders and by medications.
Penile tumescence leading to erection depends on an increased flow of blood into the lacunar network accompanied by complete relaxation of the arteries and corporal smooth muscle. The microarchitecture of the corpora is composed of a mass of smooth muscle (trabecula) that contains a network of endothelial-lined vessels (lacunar spaces). Subsequent compression of the trabecular smooth muscle against the fibroelastic tunica albuginea causes a passive closure of the emissary veins and accumulation of blood in the corpora. In the presence of a full erection and a competent valve mechanism, the corpora become noncompressible cylinders from which blood does not escape.
The central nervous system (CNS) exerts an important influence by either stimulating or antagonizing spinal pathways that mediate erectile function and ejaculation. The erectile response is mediated by a combination of central (psychogenic) innervation and peripheral (reflexogenic) innervation. Sensory nerves that originate from receptors in the penile skin and glans converge to form the dorsal nerve of the penis, which travels to the S2-S4 dorsal root ganglia via the pudendal nerve. Parasympathetic nerve fibers to the penis arise from neurons in the intermediolateral columns of the S2-S4 sacral spinal segments. Sympathetic innervation originates from the T11 to the L2 spinal segments and descends through the hypogastric plexus.
Neural input to smooth-muscle tone is crucial to the initiation and maintenance of an erection. There is also an intricate interaction between the corporal smooth-muscle cell and its overlying endothelial cell lining (Fig. 67-1). Nitric oxide, which induces vascular relaxation, promotes erection and is opposed by endothelin 1 (ET-1) and Rho kinase, which mediate vascular contraction. Nitric oxide is synthesized from l-arginine by nitric oxide synthase and is released from the nonadrenergic, noncholinergic (NANC) autonomic nerve supply to act postjunctionally on smooth-muscle cells. Nitric oxide increases the production of cyclic 3′,5′-guanosine monophosphate (cyclic GMP), which induces relaxation of smooth muscle (Fig. 67-2). Cyclic GMP is gradually broken down by phosphodiesterase type 5 (PDE-5). Inhibitors of PDE-5, such as the oral medications sildenafil, vardenafil, and tadalafil, maintain erections by reducing the breakdown of cyclic GMP. However, if nitric oxide is not produced at some level, PDE-5 inhibitors are ineffective, as these drugs facilitate, but do not initiate, the initial enzyme cascade. In addition to nitric oxide, vasoactive prostaglandins (PGE1, PGF2α) are synthesized within the cavernosal tissue and increase cyclic AMP levels, also leading to relaxation of cavernosal smooth-muscle cells.
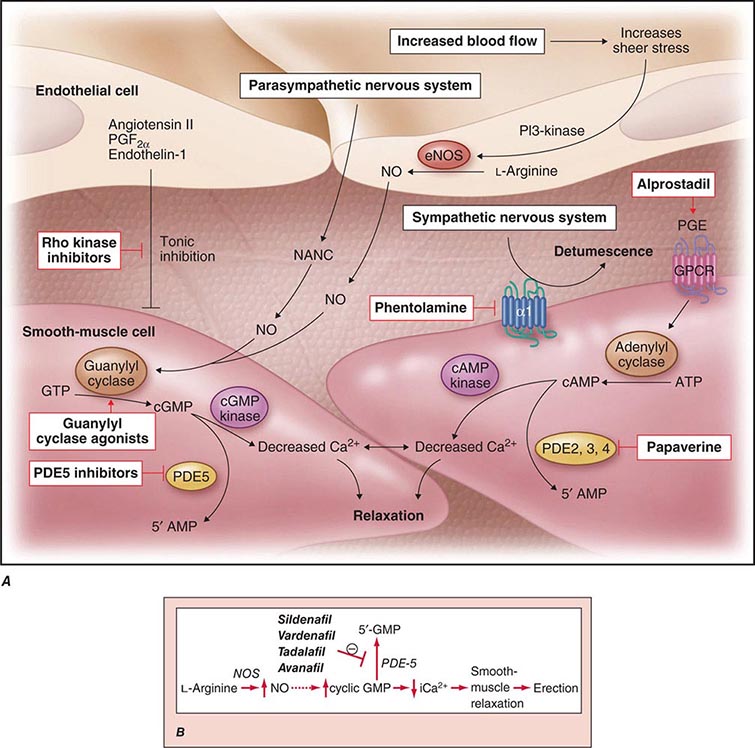
FIGURE 67-1 Pathways that regulate penile smooth-muscle relaxation and erection. A. Outflow from the parasympathetic nervous system leads to relaxation of the cavernous sinusoids in two ways, both of which increase the concentration of nitric oxide (NO) in smooth-muscle cells. First, NO is the neurotransmitter in nonadrenergic, noncholinergic (NANC) fibers; second, stimulation of endothelial nitric oxide synthase (eNOS) through cholinergic output causes increased production of NO. The NO produced in the endothelium then diffuses into the smooth-muscle cells and decreases its intracellular calcium concentration through a pathway mediated by cyclic guanosine monophosphate (cGMP), leading to relaxation. A separate mechanism that decreases the intracellular calcium level is mediated by cyclic adenosine monophosphate (cAMP). With increased cavernosal blood flow, as well as increased levels of vascular endothelial growth factor (VEGF), the endothelial release of NO is further sustained through the phosphatidylinositol 3 (PI3) kinase pathway. Active treatments (red boxes) include drugs that affect the cGMP pathway (phosphodiesterase [PDE] type 5 inhibitors and guanylyl cyclase agonists), the cAMP pathway (alprostadil), or both pathways (papaverine), along with neural-tone mediators (phentolamine and Rho kinase inhibitors). Agents that are being developed include guanylyl cyclase agonists (to bypass the need for endogenous NO) and Rho kinase inhibitors (to inhibit tonic contraction of smooth-muscle cells mediated through endothelin). α1, α-adrenergic receptor; GPCR, G-protein–coupled receptor, GTP, guanosine triphosphate; PGE, prostaglandin E; PGF, prostaglandin F. B. Biochemical pathways of NO synthesis and action. Sildenafil, vardenafil, and tadalafil enhance erectile function by inhibiting phosphodiesterase type 5 (PDE-5), thereby maintaining high levels of cyclic 3′,5′-guanosine monophosphate (cyclic GMP). iCa2+, intracellular calcium; NOS, nitric oxide synthase. (Part A from K McVary: N Engl J Med 357:2472, 2007; with permission.)
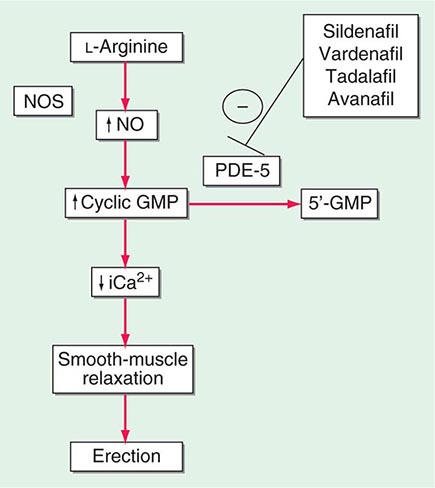
FIGURE 67-2 Biochemical pathways modified by phosphodiesterase type 5 (PDE-5) inhibitors. Sildenafil, vardenafil, tadalafil and avanafil enhance erectile function by inhibiting PDE-5, thereby maintaining high levels of cyclic 3′,5′-guanosine monophosphate (cyclic GMP). iCa2+, intracellular calcium; NO, nitric oxide; NOS, nitric oxide synthase.
Ejaculation is stimulated by the sympathetic nervous system; this results in contraction of the epididymis, vas deferens, seminal vesicles, and prostate, causing seminal fluid to enter the urethra. Seminal fluid emission is followed by rhythmic contractions of the bulbocavernosus and ischiocavernosus muscles, leading to ejaculation. Premature ejaculation usually is related to anxiety or a learned behavior and is amenable to behavioral therapy or treatment with medications such as selective serotonin reuptake inhibitors (SSRIs). Retrograde ejaculation results when the internal urethral sphincter does not close; it may occur in men with diabetes or after surgery involving the bladder neck.
Detumescence is mediated by norepinephrine from the sympathetic nerves, endothelin from the vascular surface, and smooth-muscle contraction induced by postsynaptic α-adrenergic receptors and activation of Rho kinase. These events increase venous outflow and restore the flaccid state. Venous leak can cause premature detumescence and is caused by insufficient relaxation of the corporal smooth muscle rather than a specific anatomic defect. Priapism refers to a persistent and painful erection and may be associated with sickle cell anemia, hypercoagulable states, spinal cord injury, or injection of vasodilator agents into the penis.
ERECTILE DYSFUNCTION
Epidemiology Erectile dysfunction (ED) is not considered a normal part of the aging process. Nonetheless, it is associated with certain physiologic and psychological changes related to age. In the Massachusetts Male Aging Study (MMAS), a community-based survey of men age 40–70, 52% of responders reported some degree of ED. Complete ED occurred in 10% of respondents, moderate ED in 25%, and minimal ED in 17%. The incidence of moderate or severe ED more than doubled between the ages of 40 and 70. In the National Health and Social Life Survey (NHSLS), which included a sample of men and women age 18–59, 10% of men reported being unable to maintain an erection (corresponding to the proportion of men in the MMAS reporting severe ED). Incidence was highest among men in the age group 50–59 (21%) and men who were poor (14%), divorced (14%), and less educated (13%).
The incidence of ED is also higher among men with certain medical disorders, such as diabetes mellitus, obesity, lower urinary tract symptoms secondary to benign prostatic hyperplasia (BPH), heart disease, hypertension, decreased high-density lipoprotein (HDL) levels, and diseases associated with general systemic inflammation (e.g., rheumatoid arthritis). Cardiovascular disease and ED share etiologies as well as pathophysiology (e.g., endothelial dysfunction), and the degree of ED appears to correlate with the severity of cardiovascular disease. Consequently, ED represents a “sentinel symptom” in patients with occult cardiovascular and peripheral vascular disease.
Smoking is also a significant risk factor in the development of ED. Medications used in treating diabetes or cardiovascular disease are additional risk factors (see below). There is a higher incidence of ED among men who have undergone radiation or surgery for prostate cancer and in those with a lower spinal cord injury. Psychological causes of ED include depression, anger, stress from unemployment, and other stress-related causes.
Pathophysiology ED may result from three basic mechanisms: (1) failure to initiate (psychogenic, endocrinologic, or neurogenic), (2) failure to fill (arteriogenic), and (3) failure to store adequate blood volume within the lacunar network (venoocclusive dysfunction). These categories are not mutually exclusive, and multiple factors contribute to ED in many patients. For example, diminished filling pressure can lead secondarily to venous leak. Psychogenic factors frequently coexist with other etiologic factors and should be considered in all cases. Diabetic, atherosclerotic, and drug-related causes account for >80% of cases of ED in older men.
VASCULOGENIC The most common organic cause of ED is a disturbance of blood flow to and from the penis. Atherosclerotic or traumatic arterial disease can decrease flow to the lacunar spaces, resulting in decreased rigidity and an increased time to full erection. Excessive outflow through the veins despite adequate inflow also may contribute to ED. Structural alterations to the fibroelastic components of the corpora may cause a loss of compliance and inability to compress the tunical veins. This condition may result from aging, increased cross-linking of collagen fibers induced by nonenzymatic glycosylation, hypoxemia, or altered synthesis of collagen associated with hypercholesterolemia.
NEUROGENIC Disorders that affect the sacral spinal cord or the autonomic fibers to the penis preclude nervous system relaxation of penile smooth muscle, thus leading to ED. In patients with spinal cord injury, the degree of ED depends on the completeness and level of the lesion. Patients with incomplete lesions or injuries to the upper part of the spinal cord are more likely to retain erectile capabilities than are those with complete lesions or injuries to the lower part. Although 75% of patients with spinal cord injuries have some erectile capability, only 25% have erections sufficient for penetration. Other neurologic disorders commonly associated with ED include multiple sclerosis and peripheral neuropathy. The latter is often due to either diabetes or alcoholism. Pelvic surgery may cause ED through disruption of the autonomic nerve supply.
ENDOCRINOLOGIC Androgens increase libido, but their exact role in erectile function is unclear. Individuals with castrate levels of testosterone can achieve erections from visual or sexual stimuli. Nonetheless, normal levels of testosterone appear to be important for erectile function, particularly in older males. Androgen replacement therapy can improve depressed erectile function when it is secondary to hypogonadism; however, it is not useful for ED when endogenous testosterone levels are normal. Increased prolactin may decrease libido by suppressing gonadotropin-releasing hormone (GnRH), and it also leads to decreased testosterone levels. Treatment of hyperprolactinemia with dopamine agonists can restore libido and testosterone.
DIABETIC ED occurs in 35–75% of men with diabetes mellitus. Pathologic mechanisms are related primarily to diabetes-associated vascular and neurologic complications. Diabetic macrovascular complications are related mainly to age, whereas microvascular complications correlate with the duration of diabetes and the degree of glycemic control (Chap. 417). Individuals with diabetes also have reduced amounts of nitric oxide synthase in both endothelial and neural tissues.
PSYCHOGENIC Two mechanisms contribute to the inhibition of erections in psychogenic ED. First, psychogenic stimuli to the sacral cord may inhibit reflexogenic responses, thereby blocking activation of vasodilator outflow to the penis. Second, excess sympathetic stimulation in an anxious man may increase penile smooth-muscle tone. The most common causes of psychogenic ED are performance anxiety, depression, relationship conflict, loss of attraction, sexual inhibition, conflicts over sexual preference, sexual abuse in childhood, and fear of pregnancy or sexually transmitted disease. Almost all patients with ED, even when it has a clear-cut organic basis, develop a psychogenic component as a reaction to ED.
MEDICATION-RELATED Medication-induced ED (Table 67-1) is estimated to occur in 25% of men seen in general medical outpatient clinics. The adverse effects related to drug therapy are additive, especially in older men. In addition to the drug itself, the disease being treated is likely to contribute to sexual dysfunction. Among the antihypertensive agents, the thiazide diuretics and beta blockers have been implicated most frequently. Calcium channel blockers and angiotensin converting-enzyme inhibitors are cited less frequently. These drugs may act directly at the corporal level (e.g., calcium channel blockers) or indirectly by reducing pelvic blood pressure, which is important in the development of penile rigidity. α-Adrenergic blockers are less likely to cause ED. Estrogens, GnRH agonists, H2 antagonists, and spironolactone cause ED by suppressing gonadotropin production or by blocking androgen action. Antidepressant and antipsychotic agents—particularly neuroleptics, tricyclics, and SSRIs—are associated with erectile, ejaculatory, orgasmic, and sexual desire difficulties.
|
DRUGS ASSOCIATED WITH ERECTILE DYSFUNCTION |
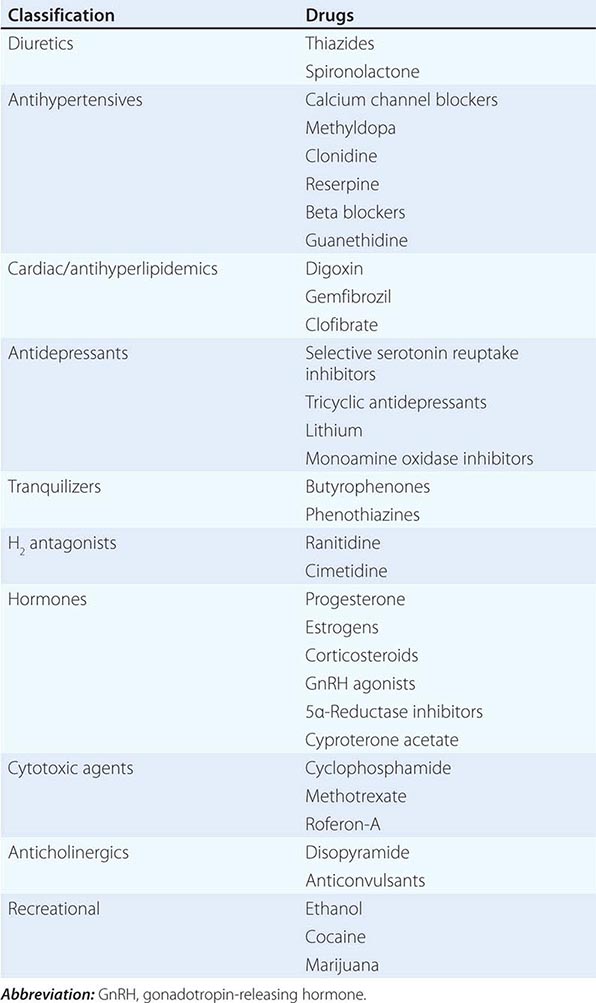
If there is a strong association between the institution of a drug and the onset of ED, alternative medications should be considered. Otherwise, it is often practical to treat the ED without attempting multiple changes in medications, as it may be difficult to establish a causal role for a drug.
APPROACH TO THE PATIENT:
Erectile Dysfunction
A good physician-patient relationship helps unravel the possible causes of ED, many of which require discussion of personal and sometimes embarrassing topics. For this reason, a primary care provider is often ideally suited to initiate the evaluation. However, a significant percentage of men experience ED and remain undiagnosed unless specifically questioned about this issue. By far the two most common reasons for underreporting of ED are patient embarrassment and perceptions of physicians’ inattention to the disease. Once the topic is initiated by the physician, patients are more willing to discuss their potency issues. A complete medical and sexual history should be taken in an effort to assess whether the cause of ED is organic, psychogenic, or multifactorial (Fig. 67-3).
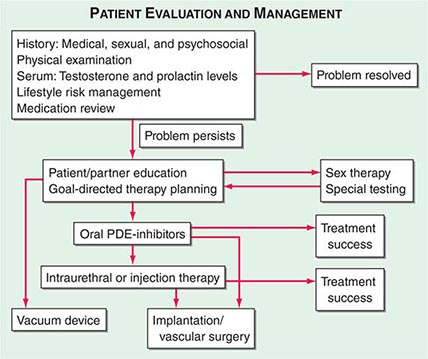
FIGURE 67-3 Algorithm for the evaluation and management of patients with erectile dysfunction. PDE, phosphodiesterase.
Both the patient and his sexual partner should be interviewed regarding sexual history. ED should be distinguished from other sexual problems, such as premature ejaculation. Lifestyle factors such as sexual orientation, the patient’s distress from ED, performance anxiety, and details of sexual techniques should be addressed. Standardized questionnaires are available to assess ED, including the International Index of Erectile Function (IIEF) and the more easily administered Sexual Health Inventory for Men (SHIM), a validated abridged version of the IIEF.
The initial evaluation of ED begins with a review of the patient’s medical, surgical, sexual, and psychosocial histories. The history should note whether the patient has experienced pelvic trauma, surgery, or radiation. In light of the increasing recognition of the relationship between lower urinary tract symptoms and ED, it is advisable to evaluate for the presence of symptoms of bladder outlet obstruction. Questions should focus on the onset of symptoms, the presence and duration of partial erections, and the progression of ED. A history of nocturnal or early morning erections is useful for distinguishing physiologic ED from psychogenic ED. Nocturnal erections occur during rapid eye movement (REM) sleep and require intact neurologic and circulatory systems. Organic causes of ED generally are characterized by a gradual and persistent change in rigidity or the inability to sustain nocturnal, coital, or self-stimulated erections. The patient should be questioned about the presence of penile curvature or pain with coitus. It is also important to address libido, as decreased sexual drive and ED are sometimes the earliest signs of endocrine abnormalities (e.g., increased prolactin, decreased testosterone levels). It is useful to ask whether the problem is confined to coitus with one partner or also involves other partners; ED not uncommonly arises in association with new or extramarital sexual relationships. Situational ED, as opposed to consistent ED, suggests psychogenic causes. Ejaculation is much less commonly affected than erection, but questions should be asked about whether ejaculation is normal, premature, delayed, or absent. Relevant risk factors should be identified, such as diabetes mellitus, coronary artery disease (CAD), and neurologic disorders. The patient’s surgical history should be explored with an emphasis on bowel, bladder, prostate, and vascular procedures. A complete drug history is also important. Social changes that may precipitate ED are also crucial to the evaluation, including health worries, spousal death, divorce, relationship difficulties, and financial concerns.
Because ED commonly involves a host of endothelial cell risk factors, men with ED report higher rates of overt and silent myocardial infarction. Therefore, ED in an otherwise asymptomatic male warrants consideration of other vascular disorders, including CAD.
The physical examination is an essential element in the assessment of ED. Signs of hypertension as well as evidence of thyroid, hepatic, hematologic, cardiovascular, or renal diseases should be sought. An assessment should be made of the endocrine and vascular systems, the external genitalia, and the prostate gland. The penis should be palpated carefully along the corpora to detect fibrotic plaques. Reduced testicular size and loss of secondary sexual characteristics are suggestive of hypogonadism. Neurologic examination should include assessment of anal sphincter tone, investigation of the bulbocavernosus reflex, and testing for peripheral neuropathy.
Although hyperprolactinemia is uncommon, a serum prolactin level should be measured, as decreased libido and/or ED may be the presenting symptoms of a prolactinoma or another mass lesion of the sella (Chap. 403). The serum testosterone level should be measured, and if it is low, gonadotropins should be measured to determine whether hypogonadism is primary (testicular) or secondary (hypothalamic-pituitary) in origin (Chap. 411). If not performed recently, serum chemistries, complete blood count (CBC), and lipid profiles may be of value, as they can yield evidence of anemia, diabetes, hyperlipidemia, or other systemic diseases associated with ED. Determination of serum prostate-specific antigen (PSA) should be conducted according to recommended clinical guidelines (Chap. 115).
Additional diagnostic testing is rarely necessary in the evaluation of ED. However, in selected patients, specialized testing may provide insight into pathologic mechanisms of ED and aid in the selection of treatment options. Optional specialized testing includes (1) studies of nocturnal penile tumescence and rigidity, (2) vascular testing (in-office injection of vasoactive substances, penile Doppler ultrasound, penile angiography, dynamic infusion cavernosography/cavernosometry), (3) neurologic testing (biothesiometry-graded vibratory perception, somatosensory evoked potentials), and (4) psychological diagnostic tests. The information potentially gained from these procedures must be balanced against their invasiveness and cost.
|
TREATMENT |
MALE SEXUAL DYSFUNCTION |
PATIENT EDUCATION
Patient and partner education is essential in the treatment of ED. In goal-directed therapy, education facilitates understanding of the disease, the results of the tests, and the selection of treatment. Discussion of treatment options helps clarify how treatment is best offered and stratify first- and second-line therapies. Patients with high-risk lifestyle issues such as obesity, smoking, alcohol abuse, and recreational drug use should be counseled on the role those factors play in the development of ED.
Therapies currently employed for the treatment of ED include oral PDE-5 inhibitor therapy (most commonly used), injection therapies, testosterone therapy, penile devices, and psychological therapy. In addition, limited data suggest that treatments for underlying risk factors and comorbidities—for example, weight loss, exercise, stress reduction, and smoking cessation—may improve erectile function. Decisions regarding therapy should take into account the preferences and expectations of patients and their partners.
ORAL AGENTS
Sildenafil, tadalafil, vardenafil, and avanafil are the only approved and effective oral agents for the treatment of ED. These four medications have markedly improved the management of ED because they are effective for the treatment of a broad range of causes, including psychogenic, diabetic, vasculogenic, post-radical prostatectomy (nerve-sparing procedures), and spinal cord injury. They belong to a class of medications that are selective and potent inhibitors of PDE-5, the predominant phosphodiesterase isoform found in the penis. They are administered in graduated doses and enhance erections after sexual stimulation. The onset of action is approximately 30–120 min, depending on the medication used and other factors, such as recent food intake. Reduced initial doses should be considered for patients who are elderly, are taking concomitant alpha blockers, have renal insufficiency, or are taking medications that inhibit the CYP3A4 metabolic pathway in the liver (e.g., erythromycin, cimetidine, ketoconazole, and possibly itraconazole and mibefradil), as they may increase the serum concentration of the PDE-5 inhibitors (PDE-5i) or promote hypotension.
Initially, there were concerns about the cardiovascular safety of PDE-5i drugs. These agents can act as a mild vasodilator, and warnings exist about orthostatic hypotension with concomitant use of alpha blockers. The use of PDE-5i is not contraindicated in men who are also receiving alpha blockers, but they must be stabilized on this blood pressure medication prior to initiating therapy. Concerns also existed that use of PDE-5i would increase cardiovascular events. However, the safety of these drugs has been confirmed in several controlled trials with no increase in myocardial ischemic events or overall mortality compared to the general population.
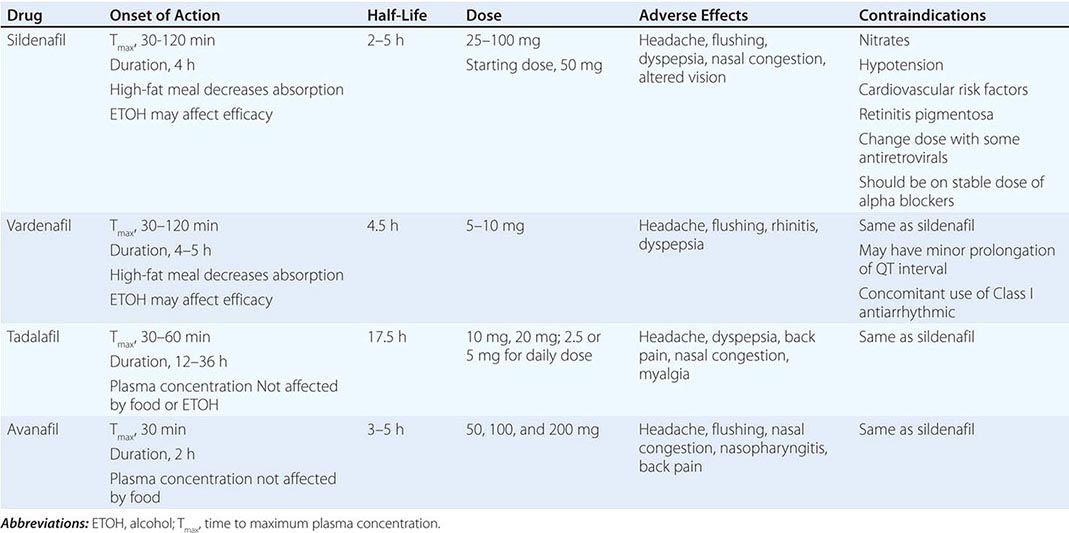
Several randomized trials have demonstrated the efficacy of this class of medications. There are no compelling data to support the superiority of one PDE-5i over another. Subtle differences between agents have variable clinical relevance (Table 67-2).
|
CHARACTERISTICS OF PDE-5i MEDICATIONS |
Patients may fail to respond to a PDE-5i for several reasons (Table 67-3). Some patients may not tolerate PDE-5i secondary to adverse events from vasodilation in nonpenile tissues expressing PDE-5 or from the inhibition of homologous nonpenile isozymes (i.e., PDE-6 found in the retina). Abnormal vision attributed to the effects of PDE-5i on retinal PDE-6 is of short duration, reported only with sildenafil and not thought to be clinically significant. A more serious concern is the possibility that PDE-5i may cause nonarteritic anterior ischemic optic neuropathy; although data to support that association are limited, it is prudent to avoid the use of these agents in men with a prior history of nonarteritic anterior ischemic optic neuropathy.
|
ISSUES TO CONSIDER IF PATIENTS REPORT FAILURE OF PDE-5i TO IMPROVE ERECTILE DYSFUNCTION |
Abbreviations: NO, nitric oxide; PDE-5i, phosphodiesterase type 5 inhibitor.
Testosterone supplementation combined with a PDE-5i may be beneficial in improving erectile function in hypogonadal men with ED who are unresponsive to PDE-5i alone. These drugs do not affect ejaculation, orgasm, or sexual drive. Side effects associated with PDE-5i include headaches (19%), facial flushing (9%), dyspepsia (6%), and nasal congestion (4%). Approximately 7% of men using sildenafil may experience transient altered color vision (blue halo effect), and 6% of men taking tadalafil may experience loin pain. PDE-5i is contraindicated in men receiving nitrate therapy for cardiovascular disease, including agents delivered by the oral, sublingual, transnasal, and topical routes. These agents can potentiate its hypotensive effect and may result in profound shock. Likewise, amyl/butyl nitrate “poppers” may have a fatal synergistic effect on blood pressure. PDE-5i also should be avoided in patients with congestive heart failure and cardiomyopathy because of the risk of vascular collapse. Because sexual activity leads to an increase in physiologic expenditure (5–6 metabolic equivalents [METS]), physicians have been advised to exercise caution in prescribing any drug for sexual activity to those with active coronary disease, heart failure, borderline hypotension, or hypovolemia and to those on complex antihypertensive regimens.
Although the various forms of PDE-5i have a common mechanism of action, there are a few differences among the four agents (Table 67-2). Tadalafil is unique in its longer half-life, whereas avanafil appears to have the most rapid onset of action. All four drugs are effective for patients with ED of all ages, severities, and etiologies. Although there are pharmacokinetic and pharmacodynamic differences among these agents, clinically relevant differences are not clear.
ANDROGEN THERAPY
Testosterone replacement is used to treat both primary and secondary causes of hypogonadism (Chap. 411). Androgen supplementation in the setting of normal testosterone is rarely efficacious in the treatment of ED and is discouraged. Methods of androgen replacement include transdermal patches and gels, parenteral administration of long-acting testosterone esters (enanthate and cypionate), and oral preparations (17 α-alkylated derivatives) (Chap. 411). Oral androgen preparations have the potential for hepatotoxicity and should be avoided.
Men who receive testosterone should be reevaluated after 1–3 months and at least annually thereafter for testosterone levels, erectile function, and adverse effects, which may include gynecomastia, sleep apnea, development or exacerbation of lower urinary tract symptoms or BPH, prostate cancer, lowering of HDL, erythrocytosis, elevations of liver function tests, and reduced fertility. Periodic reevaluation should include measurement of CBC and PSA and digital rectal exam. Therapy should be discontinued in patients who do not respond within 3 months.
VACUUM CONSTRICTION DEVICES
Vacuum constriction devices (VCDs) are a well-established noninvasive therapy. They are a reasonable treatment alternative for select patients who cannot take sildenafil or do not desire other interventions. VCDs draw venous blood into the penis and use a constriction ring to restrict venous return and maintain tumescence. Adverse events with VCD include pain, numbness, bruising, and altered ejaculation. Additionally, many patients complain that the devices are cumbersome and that the induced erections have a nonphysiologic appearance and feel.
INTRAURETHRAL ALPROSTADIL
If a patient fails to respond to oral agents, a reasonable next choice is intraurethral or self-injection of vasoactive substances. Intraurethral prostaglandin E1 (alprostadil), in the form of a semisolid pellet (doses of 125–1000 μg), is delivered with an applicator. Approximately 65% of men receiving intraurethral alprostadil respond with an erection when tested in the office, but only 50% achieve successful coitus at home. Intraurethral insertion is associated with a markedly reduced incidence of priapism in comparison to intracavernosal injection.
INTRACAVERNOSAL SELF-INJECTION
Injection of synthetic formulations of alprostadil is effective in 70–80% of patients with ED, but discontinuation rates are high because of the invasive nature of administration. Doses range between 1 and 40 μg. Injection therapy is contraindicated in men with a history of hypersensitivity to the drug and men at risk for priapism (hypercoagulable states, sickle cell disease). Side effects include local adverse events, prolonged erections, pain, and fibrosis with chronic use. Various combinations of alprostadil, phentolamine, and/or papaverine sometimes are used.
SURGERY
A less frequently used form of therapy for ED involves the surgical implantation of a semirigid or inflatable penile prosthesis. The choice of prosthesis is dependent on patient preference and should take into account body habitus and manual dexterity, which may affect the ability of the patient to manipulate the device. Because of the permanence of prosthetic devices, patients should be advised to first consider less invasive options for treatment. These surgical treatments are invasive, are associated with potential complications, and generally are reserved for treatment of refractory ED. Despite their high cost and invasiveness, penile prostheses are associated with high rates of patient and partner satisfaction.
SEX THERAPY
A course of sex therapy may be useful for addressing specific interpersonal factors that may affect sexual functioning. Sex therapy generally consists of in-session discussion and at-home exercises specific to the person and the relationship. Psychosexual therapy involves techniques such as sensate focus (nongenital massage), sensory awareness exercises, correction of misconceptions about sexuality, and interpersonal difficulties therapy (e.g., open communication about sexual issues, physical intimacy scheduling, and behavioral interventions). These approaches may be useful in patients who have psychogenic or social components to their ED, although data from randomized trials are scanty and inconsistent. It is preferable if therapy includes both partners if the patient is involved in an ongoing relationship.
FEMALE SEXUAL DYSFUNCTION
Female sexual dysfunction (FSD) has traditionally included disorders of desire, arousal, pain, and muted orgasm. The associated risk factors for FSD are similar to those in males: cardiovascular disease, endocrine disorders, hypertension, neurologic disorders, and smoking (Table 67-4).
|
RISK FACTORS FOR FEMALE SEXUAL DYSFUNCTION |
Abbreviation: GnRH, gonadotropin-releasing hormone.
EPIDEMIOLOGY
Epidemiologic data are limited, but the available estimates suggest that as many as 43% of women complain of at least one sexual problem. Despite the recent interest in organic causes of FSD, desire and arousal phase disorders (including lubrication complaints) remain the most common presenting problems when surveyed in a community-based population.
PHYSIOLOGY OF THE FEMALE SEXUAL RESPONSE
The female sexual response requires the presence of estrogens. A role for androgens is also likely but less well established. In the CNS, estrogens and androgens work synergistically to enhance sexual arousal and response. A number of studies report enhanced libido in women during preovulatory phases of the menstrual cycle, suggesting that hormones involved in the ovulatory surge (e.g., estrogens) increase desire.
Sexual motivation is heavily influenced by context, including the environment and partner factors. Once sufficient sexual desire is reached, sexual arousal is mediated by the central and autonomic nervous systems. Cerebral sympathetic outflow is thought to increase desire, and peripheral parasympathetic activity results in clitoral vasocongestion and vaginal secretion (lubrication).
The neurotransmitters for clitoral corporal engorgement are similar to those in the male, with a prominent role for neural, smooth-muscle, and endothelial released nitric oxide (NO). A fine network of vaginal nerves and arterioles promotes a vaginal transudate. The major transmitters of this complex vaginal response are not certain, but roles for NO and vasointestinal polypeptide (VIP) are suspected. Investigators studying the normal female sexual response have challenged the long-held construct of a linear and unmitigated relationship between initial desire, arousal, vasocongestion, lubrication, and eventual orgasm. Caregivers should consider a paradigm of a positive emotional and physical outcome with one, many, or no orgasmic peak and release.
Although there are anatomic differences as well as variation in the density of vascular and neural beds in males and females, the primary effectors of sexual response are strikingly similar. Intact sensation is important for arousal. Thus, reduced levels of sexual functioning are more common in women with peripheral neuropathies (e.g., diabetes). Vaginal lubrication is a transudate of serum that results from the increased pelvic blood flow associated with arousal. Vascular insufficiency from a variety of causes may compromise adequate lubrication and result in dyspareunia. Cavernosal and arteriole smooth-muscle relaxation occurs via increased nitric oxide synthase (NOS) activity and produces engorgement in the clitoris and the surrounding vestibule. Orgasm requires an intact sympathetic outflow tract; hence, orgasmic disorders are common in female patients with spinal cord injuries.
68 |
Hirsutism |
Hirsutism, which is defined as androgen-dependent excessive male-pattern hair growth, affects approximately 10% of women. Hirsutism is most often idiopathic or the consequence of androgen excess associated with the polycystic ovarian syndrome (PCOS). Less frequently, it may result from adrenal androgen overproduction as occurs in nonclassic congenital adrenal hyperplasia (CAH) (Table 68-1). Rarely, it is a sign of a serious underlying condition. Cutaneous manifestations commonly associated with hirsutism include acne and male-pattern balding (androgenic alopecia). Virilization refers to a condition in which androgen levels are sufficiently high to cause additional signs and symptoms, such as deepening of the voice, breast atrophy, increased muscle bulk, clitoromegaly, and increased libido; virilization is an ominous sign that suggests the possibility of an ovarian or adrenal neoplasm.
|
CAUSES OF HIRSUTISM |
HAIR FOLLICLE GROWTH AND DIFFERENTIATION
Hair can be categorized as either vellus (fine, soft, and not pigmented) or terminal (long, coarse, and pigmented). The number of hair follicles does not change over an individual’s lifetime, but the follicle size and type of hair can change in response to numerous factors, particularly androgens. Androgens are necessary for terminal hair and sebaceous gland development and mediate differentiation of pilosebaceous units (PSUs) into either a terminal hair follicle or a sebaceous gland. In the former case, androgens transform the vellus hair into a terminal hair; in the latter case, the sebaceous component proliferates and the hair remains vellus.
There are three phases in the cycle of hair growth: (1) anagen (growth phase), (2) catagen (involution phase), and (3) telogen (rest phase). Depending on the body site, hormonal regulation may play an important role in the hair growth cycle. For example, the eyebrows, eyelashes, and vellus hairs are androgen-insensitive, whereas the axillary and pubic areas are sensitive to low levels of androgens. Hair growth on the face, chest, upper abdomen, and back requires higher levels of androgens and is therefore more characteristic of the pattern typically seen in men. Androgen excess in women leads to increased hair growth in most androgen-sensitive sites except in the scalp region, where hair loss occurs because androgens cause scalp hairs to spend less time in the anagen phase.
Although androgen excess underlies most cases of hirsutism, there is only a modest correlation between androgen levels and the quantity of hair growth. This is due to the fact that hair growth from the follicle also depends on local growth factors, and there is variability in end organ (PSU) sensitivity. Genetic factors and ethnic background also influence hair growth. In general, dark-haired individuals tend to be more hirsute than blond or fair individuals. Asians and Native Americans have relatively sparse hair in regions sensitive to high androgen levels, whereas people of Mediterranean descent are more hirsute.
CLINICAL ASSESSMENT
Historic elements relevant to the assessment of hirsutism include the age at onset and rate of progression of hair growth and associated symptoms or signs (e.g., acne). Depending on the cause, excess hair growth typically is first noted during the second and third decades of life. The growth is usually slow but progressive. Sudden development and rapid progression of hirsutism suggest the possibility of an androgen-secreting neoplasm, in which case virilization also may be present.
The age at onset of menstrual cycles (menarche) and the pattern of the menstrual cycle should be ascertained; irregular cycles from the time of menarche onward are more likely to result from ovarian rather than adrenal androgen excess. Associated symptoms such as galactorrhea should prompt evaluation for hyperprolactinemia (Chap. 403) and possibly hypothyroidism (Chap. 405). Hypertension, striae, easy bruising, centripetal weight gain, and weakness suggest hypercortisolism (Cushing’s syndrome; Chap. 406). Rarely, patients with growth hormone excess (i.e., acromegaly) present with hirsutism. Use of medications such as phenytoin, minoxidil, and cyclosporine may be associated with androgen-independent excess hair growth (i.e., hypertrichosis). A family history of infertility and/or hirsutism may indicate disorders such as nonclassic CAH (Chap. 406). Lipodystrophy is often associated with increased ovarian androgen production that occurs as a consequence of insulin resistance. Patients with lipodystrophy have a preponderance of central fat distribution together with scant subcutaneous adipose tissue in the upper and lower extremities.
Physical examination should include measurement of height and weight and calculation of body mass index (BMI). A BMI >25 kg/m2 is indicative of excess weight for height, and values >30 kg/m2 are often seen in association with hirsutism, probably the result of increased conversion of androgen precursors to testosterone. Notation should be made of blood pressure, as adrenal causes may be associated with hypertension. Cutaneous signs sometimes associated with androgen excess and insulin resistance include acanthosis nigricans and skin tags. Body fat distribution should also be noted.
An objective clinical assessment of hair distribution and quantity is central to the evaluation in any woman presenting with hirsutism. This assessment permits the distinction between hirsutism and hypertrichosis and provides a baseline reference point to gauge the response to treatment. A simple and commonly used method to grade hair growth is the modified scale of Ferriman and Gallwey (Fig. 68-1), in which each of nine androgen-sensitive sites is graded from 0 to 4. Approximately 95% of white women have a score below 8 on this scale; thus, it is normal for most women to have some hair growth in androgen-sensitive sites. Scores above 8 suggest excess androgen-mediated hair growth, a finding that should be assessed further by means of hormonal evaluation (see below). In racial/ethnic groups that are less likely to manifest hirsutism (e.g., Asian women), additional cutaneous evidence of androgen excess should be sought, including pustular acne and thinning scalp hair.
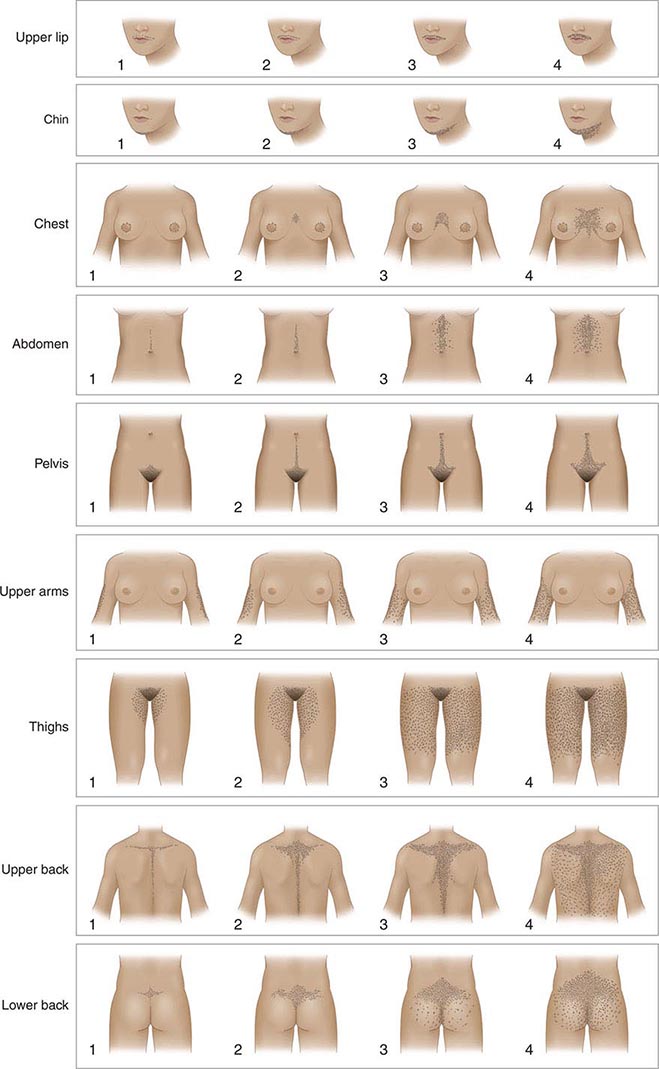
FIGURE 68-1 Hirsutism scoring scale of Ferriman and Gallwey. The nine body areas that have androgen-sensitive areas are graded from 0 (no terminal hair) to 4 (frankly virile) to obtain a total score. A normal hirsutism score is <8. (Modified from DA Ehrmann et al: Hyperandrogenism, hirsutism, and polycystic ovary syndrome, in LJ DeGroot and JL Jameson [eds], Endocrinology, 5th ed. Philadelphia, Saunders, 2006; with permission.)
HORMONAL EVALUATION
Androgens are secreted by the ovaries and adrenal glands in response to their respective tropic hormones: luteinizing hormone (LH) and adrenocorticotropic hormone (ACTH). The principal circulating steroids involved in the etiology of hirsutism are testosterone, androstenedione, and dehydroepiandrosterone (DHEA) and its sulfated form (DHEAS). The ovaries and adrenal glands normally contribute about equally to testosterone production. Approximately half of the total testosterone originates from direct glandular secretion, and the remainder is derived from the peripheral conversion of androstenedione and DHEA (Chap. 411).
Although it is the most important circulating androgen, testosterone is in effect the penultimate androgen in mediating hirsutism; it is converted to the more potent dihydrotestosterone (DHT) by the enzyme 5α-reductase, which is located in the PSU. DHT has a higher affinity for, and slower dissociation from, the androgen receptor. The local production of DHT allows it to serve as the primary mediator of androgen action at the level of the pilosebaceous unit. There are two isoenzymes of 5α-reductase: Type 2 is found in the prostate gland and in hair follicles, and type 1 is found primarily in sebaceous glands.
One approach to the evaluation of hirsutism is depicted in Fig. 68-2. In addition to measuring blood levels of testosterone and DHEAS, it is important to measure the level of free (or unbound) testosterone. The fraction of testosterone that is not bound to its carrier protein, sex hormone–binding globulin (SHBG), is biologically available for conversion to DHT and binding to androgen receptors. Hyperinsulinemia and/or androgen excess decrease hepatic production of SHBG, resulting in levels of total testosterone within the high-normal range, whereas the unbound hormone is elevated more substantially. Although there is a decline in ovarian testosterone production after menopause, ovarian estrogen production decreases to an even greater extent, and the concentration of SHBG is reduced. Consequently, there is an increase in the relative proportion of unbound testosterone, and it may exacerbate hirsutism after menopause.
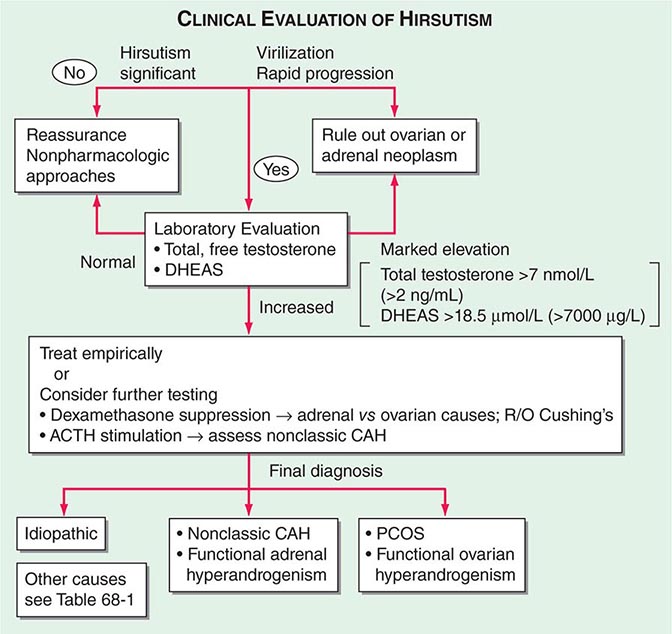
FIGURE 68-2 Algorithm for the evaluation and differential diagnosis of hirsutism. ACTH, adrenocorticotropic hormone; CAH, congenital adrenal hyperplasia; DHEAS, sulfated form of dehydroepiandrosterone; PCOS, polycystic ovarian syndrome.
A baseline plasma total testosterone level >12 nmol/L (>3.5 ng/mL) usually indicates a virilizing tumor, whereas a level >7 nmol/L (>2 ng/mL) is suggestive. A basal DHEAS level >18.5 μmol/L (>7000 μg/L) suggests an adrenal tumor. Although DHEAS has been proposed as a “marker” of predominant adrenal androgen excess, it is not unusual to find modest elevations in DHEAS among women with PCOS. Computed tomography (CT) or magnetic resonance imaging (MRI) should be used to localize an adrenal mass, and transvaginal ultrasound usually suffices to identify an ovarian mass if clinical evaluation and hormonal levels suggest these possibilities.
PCOS is the most common cause of ovarian androgen excess (Chap. 412). An increased ratio of LH to follicle-stimulating hormone (FSH) is characteristic in carefully studied patients with PCOS. However, because of the pulsatile nature of gonadotropin secretion, this finding may be absent in up to half of women with PCOS. Therefore, measurement of plasma LH and FSH is not needed to make a diagnosis of PCOS. Transvaginal ultrasound classically shows enlarged ovaries and increased stroma in women with PCOS. However, cystic ovaries also may be found in women without clinical or laboratory features of PCOS.
It has been suggested that the measurement of circulating levels of antimüllerian hormone (AMH) may help in making the diagnosis of PCOS; however, this remains controversial. AMH levels reflect ovarian reserve and correlate with follicular number. Measurement of AMH can be useful when considering premature ovarian insufficiency in a patient who presents with oligomenorrhea, in which case a subnormal level of AMH will be present.
Because adrenal androgens are readily suppressed by low doses of glucocorticoids, the dexamethasone androgen-suppression test may broadly distinguish ovarian from adrenal androgen overproduction. A blood sample is obtained before and after the administration of dexamethasone (0.5 mg orally every 6 h for 4 days). An adrenal source is suggested by suppression of unbound testosterone into the normal range; incomplete suppression suggests ovarian androgen excess. An overnight 1-mg dexamethasone suppression test, with measurement of 8:00 a.m. serum cortisol, is useful when there is clinical suspicion of Cushing’s syndrome (Chap. 406).
Nonclassic CAH is most commonly due to 21-hydroxylase deficiency but also can be caused by autosomal recessive defects in other steroidogenic enzymes necessary for adrenal corticosteroid synthesis (Chap. 406). Because of the enzyme defect, the adrenal gland cannot secrete glucocorticoids (especially cortisol) efficiently. This results in diminished negative feedback inhibition of ACTH, leading to compensatory adrenal hyperplasia and the accumulation of steroid precursors that subsequently are converted to androgen. Deficiency of 21-hydroxylase can be reliably excluded by determining a morning 17-hydroxyprogesterone level <6 nmol/L (<2 μg/L) (drawn in the follicular phase). Alternatively, 21-hydroxylase deficiency can be diagnosed by measurement of 17-hydroxyprogesterone 1 h after the administration of 250 μg of synthetic ACTH (cosyntropin) intravenously.
69 |
Menstrual Disorders and Pelvic Pain |
Menstrual dysfunction can signal an underlying abnormality that may have long-term health consequences. Although frequent or prolonged bleeding usually prompts a woman to seek medical attention, infrequent or absent bleeding may seem less troubling and the patient may not bring it to the attention of the physician. Thus, a focused menstrual history is a critical part of every encounter with a female patient. Pelvic pain is a common complaint that may relate to an abnormality of the reproductive organs but also may be of gastrointestinal, urinary tract, or musculoskeletal origin. Depending on its cause, pelvic pain may require urgent surgical attention.
MENSTRUAL DISORDERS
DEFINITION AND PREVALENCE
Amenorrhea refers to the absence of menstrual periods. Amenorrhea is classified as primary if menstrual bleeding has never occurred in the absence of hormonal treatment or secondary if menstrual periods cease for 3–6 months. Primary amenorrhea is a rare disorder that occurs in <1% of the female population. However, between 3 and 5% of women experience at least 3 months of secondary amenorrhea in any specific year. There is no evidence that race or ethnicity influences the prevalence of amenorrhea. However, because of the importance of adequate nutrition for normal reproductive function, both the age at menarche and the prevalence of secondary amenorrhea vary significantly in different parts of the world.
Oligomenorrhea is defined as a cycle length >35 days or <10 menses per year. Both the frequency and the amount of vaginal bleeding are irregular in oligomenorrhea, and moliminal symptoms (premenstrual breast tenderness, food cravings, mood lability), suggestive of ovulation, are variably present. Anovulation can also present with intermenstrual intervals <24 days or vaginal bleeding for >7 days. Frequent or heavy irregular bleeding is termed dysfunctional uterine bleeding if anatomic uterine and outflow tract lesions or a bleeding diathesis has been excluded.
Primary Amenorrhea The absence of menses by age 16 has been used traditionally to define primary amenorrhea. However, other factors, such as growth, secondary sexual characteristics, the presence of cyclic pelvic pain, and the secular trend toward an earlier age of menarche, particularly in African-American girls, also influence the age at which primary amenorrhea should be investigated. Thus, an evaluation for amenorrhea should be initiated by age 15 or 16 in the presence of normal growth and secondary sexual characteristics; age 13 in the absence of secondary sexual characteristics or if height is less than the third percentile; age 12 or 13 in the presence of breast development and cyclic pelvic pain; or within 2 years of breast development if menarche, defined by the first menstrual period, has not occurred.
Secondary Amenorrhea or Oligomenorrhea Anovulation and irregular cycles are relatively common for up to 2 years after menarche and for 1–2 years before the final menstrual period. In the intervening years, menstrual cycle length is ~28 days, with an intermenstrual interval normally ranging between 25 and 35 days. Cycle-to-cycle variability in an individual woman who is ovulating consistently is generally +/– 2 days. Pregnancy is the most common cause of amenorrhea and should be excluded early in any evaluation of menstrual irregularity. However, many women occasionally miss a single period. Three or more months of secondary amenorrhea should prompt an evaluation, as should a history of intermenstrual intervals >35 or <21 days or bleeding that persists for >7 days.
DIAGNOSIS
Evaluation of menstrual dysfunction depends on understanding the interrelationships between the four critical components of the reproductive tract: (1) the hypothalamus, (2) the pituitary, (3) the ovaries, and (4) the uterus and outflow tract (Fig. 69-1; Chap. 412). This system is maintained by complex negative and positive feedback loops involving the ovarian steroids (estradiol and progesterone) and peptides (inhibin B and inhibin A) and the hypothalamic (gonadotropin-releasing hormone [GnRH]) and pituitary (follicle-stimulating hormone [FSH] and luteinizing hormone [LH]) components of this system (Fig. 69-1).
FIGURE 69-1 Role of the hypothalamic-pituitary-gonadal axis in the etiology of amenorrhea. Gonadotropin-releasing hormone (GnRH) secretion from the hypothalamus stimulates follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion from the pituitary to induce ovarian folliculogenesis and steroidogenesis. Ovarian secretion of estradiol and progesterone controls the shedding of the endometrium, resulting in menses, and, in combination with the inhibins, provides feedback regulation of the hypothalamus and pituitary to control secretion of FSH and LH. The prevalence of amenorrhea resulting from abnormalities at each level of the reproductive system (hypothalamus, pituitary, ovary, uterus, and outflow tract) varies depending on whether amenorrhea is primary or secondary. PCOS, polycystic ovarian syndrome.
Disorders of menstrual function can be thought of in two main categories: disorders of the uterus and outflow tract and disorders of ovulation. Many of the conditions that cause primary amenorrhea are congenital but go unrecognized until the time of normal puberty (e.g., genetic, chromosomal, and anatomic abnormalities). All causes of secondary amenorrhea also can cause primary amenorrhea.
Disorders of the Uterus or Outflow Tract Abnormalities of the uterus and outflow tract typically present as primary amenorrhea. In patients with normal pubertal development and a blind vagina, the differential diagnosis includes obstruction by a transverse vaginal septum or imperforate hymen; müllerian agenesis (Mayer-Rokitansky-Kuster-Hauser syndrome), which has been associated with mutations in the WNT4 gene; and androgen insensitivity syndrome (AIS), which is an X-linked recessive disorder that accounts for ~10% of all cases of primary amenorrhea (Chap. 411). Patients with AIS have a 46,XY karyotype, but because of the lack of androgen receptor responsiveness, those with complete AIS have severe underandrogenization and female external genitalia. The absence of pubic and axillary hair distinguishes them clinically from patients with müllerian agenesis, as does an elevated testosterone level. Asherman’s syndrome presents as secondary amenorrhea or hypomenorrhea and results from partial or complete obliteration of the uterine cavity by adhesions that prevent normal growth and shedding of the endometrium. Curettage performed for pregnancy complications accounts for >90% of cases; genital tuberculosis is an important cause in regions where it is endemic.
Disorders of Ovulation Once uterus and outflow tract abnormalities have been excluded, other causes of amenorrhea involve disorders of ovulation. The differential diagnosis is based on the results of initial tests, including a pregnancy test, an FSH level (to determine whether the cause is likely to be ovarian or central), and assessment of hyperandrogenism (Fig. 69-2).
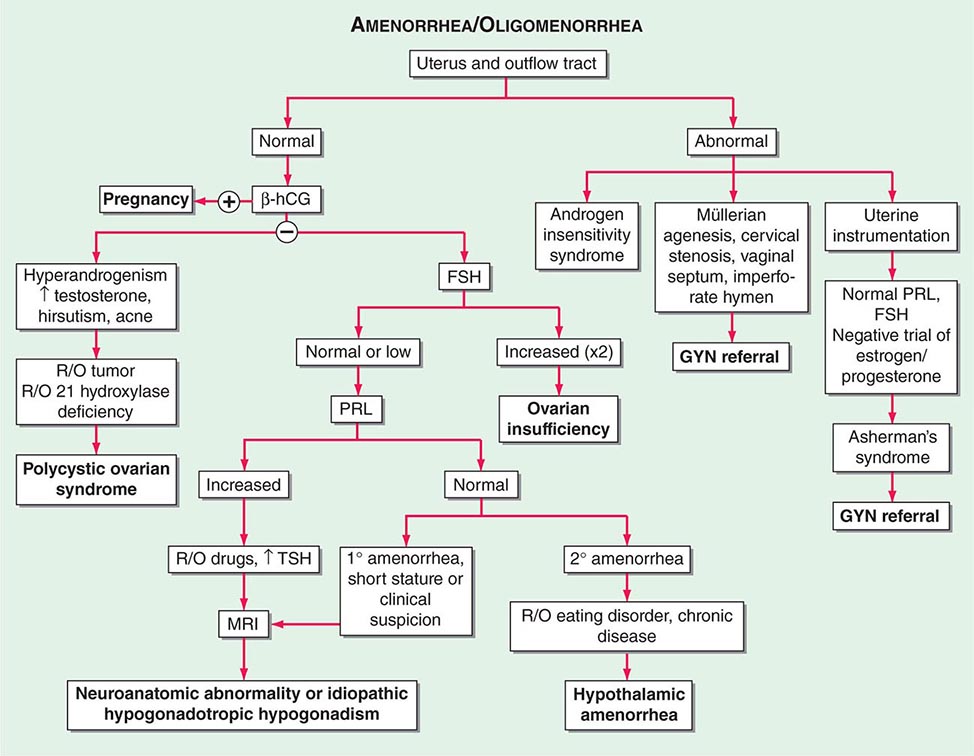
FIGURE 69-2 Algorithm for evaluation of amenorrhea. β-hCG, human chorionic gonadotropin; FSH, follicle-stimulating hormone; GYN, gynecologist; MRI, magnetic resonance imaging; PRL, prolactin; R/O, rule out; TSH, thyroid-stimulating hormone.
HYPOGONADOTROPIC HYPOGONADISM Low estrogen levels in combination with normal or low levels of LH and FSH are seen with anatomic, genetic, or functional abnormalities that interfere with hypothalamic GnRH secretion or normal pituitary responsiveness to GnRH. Although relatively uncommon, tumors and infiltrative diseases should be considered in the differential diagnosis of hypogonadotropic hypogonadism (Chap. 403). These disorders may present with primary or secondary amenorrhea. They may occur in association with other features suggestive of hypothalamic or pituitary dysfunction, such as short stature, diabetes insipidus, galactorrhea, and headache. Hypogonadotropic hypogonadism also may be seen after cranial irradiation. In the postpartum period, it may be caused by pituitary necrosis (Sheehan’s syndrome) or lymphocytic hypophysitis. Because reproductive dysfunction is commonly associated with hyperprolactinemia from neuroanatomic lesions or medications, prolactin should be measured in all patients with hypogonadotropic hypogonadism (Chap. 403).
Isolated hypogonadotropic hypogonadism (IHH) occurs in women, although it is three times more common in men. IHH generally presents with primary amenorrhea, although 50% have some degree of breast development, and one to two menses have been described in ~10%. IHH is associated with anosmia in about 50% of women (termed Kallmann’s syndrome). Genetic causes of IHH have been identified in ~60% of patients (Chaps. 411 and 412).
Functional hypothalamic amenorrhea (HA) is caused by a mismatch between energy expenditure and energy intake. Recent studies suggest that variants in genes associated with IHH may increase susceptibility to these environmental inputs, accounting in part for the clinical variability in this disorder. Leptin secretion may play a key role in transducing the signals from the periphery to the hypothalamus in HA. The hypothalamic-pituitary-adrenal axis also may play a role. The diagnosis of HA generally can be made on the basis of a careful history, a physical examination, and the demonstration of low levels of gonadotropins and normal prolactin levels. Eating disorders and chronic disease must be specifically excluded. An atypical history, headache, signs of other hypothalamic dysfunction, or hyperprolactinemia, even if mild, necessitates cranial imaging with computed tomography (CT) or magnetic resonance imaging (MRI) to exclude a neuroanatomic cause.
HYPERGONADOTROPIC HYPOGONADISM Ovarian failure is considered premature when it occurs in women <40 years old and accounts for ~10% of secondary amenorrhea. Primary ovarian insufficiency (POI) has generally replaced the terms premature menopause and premature ovarian failure in recognition that this disorder represents a continuum of impaired ovarian function. Ovarian insufficiency is associated with the loss of negative-feedback restraint on the hypothalamus and pituitary, resulting in increased FSH and LH levels. FSH is a better marker of ovarian failure as its levels are less variable than those of LH. Antimüllerian hormone (AMH) levels will also be low in patients with POI, but are more frequently used in management of infertility. As with natural menopause, POI may wax and wane, and serial measurements may be necessary to establish the diagnosis.
Once the diagnosis of POI has been established, further evaluation is indicated because of other health problems that may be associated with POI. For example, POI occurs in association with a variety of chromosomal abnormalities, including Turner’s syndrome, autoimmune polyglandular failure syndromes, radio- and chemotherapy, and galactosemia. The recognition that early ovarian failure occurs in premutation carriers of the fragile × syndrome is important because of the increased risk of severe mental retardation in male children with FMR1 mutations. In the majority of cases, a cause for POI is not determined. Although there are increasing reports of genetic mutations in individuals and families with POI, testing for other than chromosomal abnormalities and FMR1 mutations is not recommended.
Hypergonadotropic hypogonadism occurs rarely in other disorders, such as mutations in the FSH or LH receptors. Aromatase deficiency and 17α-hydroxylase deficiency are associated with decreased estrogen and elevated gonadotropins and with hyperandrogenism and hypertension, respectively. Gonadotropin-secreting tumors in women of reproductive age generally present with high, rather than low, estrogen levels and cause ovarian hyperstimulation or dysfunctional bleeding.
POLYCYSTIC OVARIAN SYNDROME (PCOS) PCOS is diagnosed based on a combination of clinical or biochemical evidence of hyperandrogenism, amenorrhea or oligomenorrhea, and the ultrasound appearance of polycystic ovaries. Approximately half of patients with PCOS are obese, and abnormalities in insulin dynamics are common, as is metabolic syndrome. Symptoms generally begin shortly after menarche and are slowly progressive. Lean oligo-ovulatory patients with PCOS generally have high LH levels in the presence of normal to low levels of FSH and estradiol. The LH/FSH ratio is less pronounced in obese patients in whom insulin resistance is a more prominent feature.
PELVIC PAIN
The mechanisms that cause pelvic pain are similar to those that cause abdominal pain (Chap. 20) and include inflammation of the parietal peritoneum, obstruction of hollow viscera, vascular disturbances, and pain originating in the abdominal wall. Pelvic pain may reflect pelvic disease per se but also may reflect extrapelvic disorders that refer pain to the pelvis. In up to 60% of cases, pelvic pain can be attributed to gastrointestinal problems, including appendicitis, cholecystitis, infections, intestinal obstruction, diverticulitis, and inflammatory bowel disease. Urinary tract and musculoskeletal disorders are also common causes of pelvic pain.
APPROACH TO THE PATIENT:
Pelvic Pain
As with all types of abdominal pain, the first priority is to identify life-threatening conditions (shock, peritoneal signs) that may require emergent surgical management. The possibility of pregnancy should be identified as soon as possible by menstrual history and/or testing. A thorough history that includes the type, location, radiation, and status with respect to increasing or decreasing severity can help identify the cause of acute pelvic pain. Specific associations with vaginal bleeding, sexual activity, defecation, urination, movement, or eating should be specifically sought. Determination of whether the pain is acute versus chronic and cyclic versus noncyclic will direct further investigation (Table 69-1). However, disorders that cause cyclic pain occasionally may cause noncyclic pain, and the converse is also true.
|
CAUSES OF PELVIC PAIN |
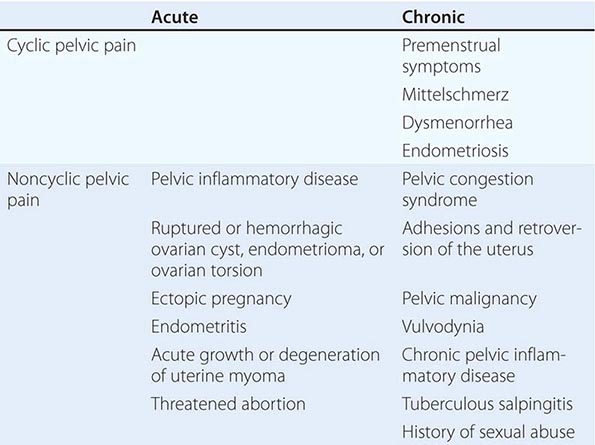
ACUTE PELVIC PAIN
Pelvic inflammatory disease most commonly presents with bilateral lower abdominal pain. It is generally of recent onset and is exacerbated by intercourse or jarring movements. Fever is present in about half of these patients; abnormal uterine bleeding occurs in about one-third. New vaginal discharge, urethritis, and chills may be present but are less specific signs. Adnexal pathology can present acutely and may be due to rupture, bleeding or torsion of cysts, or, much less commonly, neoplasms of the ovary, fallopian tubes, or paraovarian areas. Fever may be present with ovarian torsion. Ectopic pregnancy is associated with right- or left-sided lower abdominal pain, with clinical signs generally appearing 6–8 weeks after the last normal menstrual period. Amenorrhea is present in ~75% of cases and vaginal bleeding in ~50% of cases. Orthostatic signs and fever may be present. Risk factors include the presence of known tubal disease, previous ectopic pregnancies, a history of infertility, diethylstilbestrol (DES) exposure of the mother in utero, or a history of pelvic infections. Threatened abortion may also present with amenorrhea, abdominal pain, and vaginal bleeding. Although more common than ectopic pregnancy, it is rarely associated with systemic signs. Uterine pathology includes endometritis and, less frequently, degenerating leiomyomas (fibroids). Endometritis often is associated with vaginal bleeding and systemic signs of infection. It occurs in the setting of sexually transmitted infections, uterine instrumentation, or postpartum infection.
A sensitive pregnancy test, complete blood count with differential, urinalysis, tests for chlamydial and gonococcal infections, and abdominal ultrasound aid in making the diagnosis and directing further management.
CHRONIC PELVIC PAIN
Some women experience discomfort at the time of ovulation (mittelschmerz). The pain can be quite intense but is generally of short duration. The mechanism is thought to involve rapid expansion of the dominant follicle, although it also may be caused by peritoneal irritation by follicular fluid released at the time of ovulation. Many women experience premenstrual symptoms such as breast discomfort, food cravings, and abdominal bloating or discomfort. These moliminal symptoms are a good marker of prior ovulation, although their absence is less helpful.
Dysmenorrhea Dysmenorrhea refers to the crampy lower abdominal midline discomfort that begins with the onset of menstrual bleeding and gradually decreases over the next 12–72 h. It may be associated with nausea, diarrhea, fatigue, and headache and occurs in 60–93% of adolescents, beginning with the establishment of regular ovulatory cycles. Its prevalence decreases after pregnancy and with the use of oral contraceptives.
Primary dysmenorrhea results from increased stores of prostaglandin precursors, which are generated by sequential stimulation of the uterus by estrogen and progesterone. During menstruation, these precursors are converted to prostaglandins, which cause intense uterine contractions, decreased blood flow, and increased peripheral nerve hypersensitivity, resulting in pain.
Secondary dysmenorrhea is caused by underlying pelvic pathology. Endometriosis results from the presence of endometrial glands and stroma outside the uterus. These deposits of ectopic endometrium respond to hormonal stimulation and cause dysmenorrhea, which begins several days before menses. Endometriosis also may be associated with painful intercourse, painful bowel movements, and tender nodules in the uterosacral ligament. Fibrosis and adhesions can produce lateral displacement of the cervix. Transvaginal pelvic ultrasound is part of the initial workup and may detect an endometrioma within the ovary, rectovaginal or bladder nodules, or ureteral involvement. The CA125 level may be increased, but it has low negative predictive value. Definitive diagnosis requires laparoscopy. Symptomatology does not always predict the extent of endometriosis. The prevalence is lower in black and Hispanic women than in Caucasians and Asians. Other secondary causes of dysmenorrhea include adenomyosis, a condition caused by the presence of ectopic endometrial glands and stroma within the myometrium. Cervical stenosis may result from trauma, infection, or surgery.
SECTION 9 |
ALTERATIONS IN THE SKIN |
70 |
Approach to the Patient with a Skin Disorder |
The challenge of examining the skin lies in distinguishing normal from abnormal findings, distinguishing significant findings from trivial ones, and integrating pertinent signs and symptoms into an appropriate differential diagnosis. The fact that the largest organ in the body is visible is both an advantage and a disadvantage to those who examine it. It is advantageous because no special instrumentation is necessary and because the skin can be biopsied with little morbidity. However, the casual observer can be misled by a variety of stimuli and overlook important, subtle signs of skin or systemic disease. For instance, the sometimes minor differences in color and shape that distinguish a melanoma (Fig. 70-1) from a benign nevomelanocytic nevus (Fig. 70-2) can be difficult to recognize. A variety of descriptive terms have been developed that characterize cutaneous lesions (Tables 70-1, 70-2, and Tables 70-3; Fig. 70-3), thereby aiding in their interpretation and in the formulation of a differential diagnosis (Table 70-4). For example, the finding of scaling papules, which are present in psoriasis or atopic dermatitis, places the patient in a different diagnostic category than would hemorrhagic papules, which may indicate vasculitis or sepsis (Figs. 70-4 and 70-5, respectively). It is also important to differentiate primary from secondary skin lesions. If the examiner focuses on linear erosions overlying an area of erythema and scaling, he or she may incorrectly assume that the erosion is the primary lesion and that the redness and scale are secondary, whereas the correct interpretation would be that the patient has a pruritic eczematous dermatitis with erosions caused by scratching.
FIGURE 70-1 Superficial spreading melanoma. This is the most common type of melanoma. Such lesions usually demonstrate asymmetry, border irregularity, color variegation (black, blue, brown, pink, and white), a diameter >6 mm, and a history of change (e.g., an increase in size or development of associated symptoms such as pruritus or pain).
FIGURE 70-2 Nevomelanocytic nevus. Nevi are benign proliferations of nevomelanocytes characterized by regularly shaped hyperpigmented macules or papules of a uniform color.
|
DESCRIPTION OF PRIMARY SKIN LESIONS |
|
DESCRIPTION OF SECONDARY SKIN LESIONS |
|
COMMON DERMATOLOGIC TERMS |
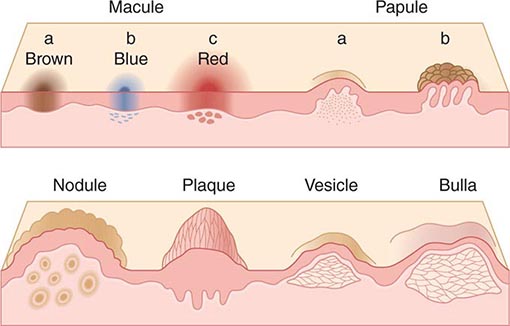
FIGURE 70-3 A schematic representation of several common primary skin lesions (see Table 70-1).
|
SELECTED COMMON DERMATOLOGIC CONDITIONS |
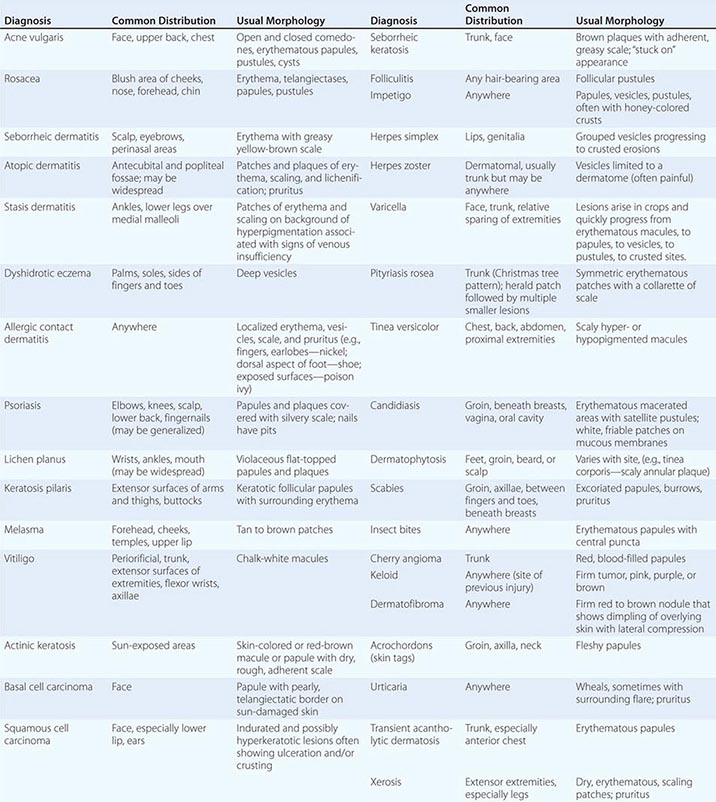
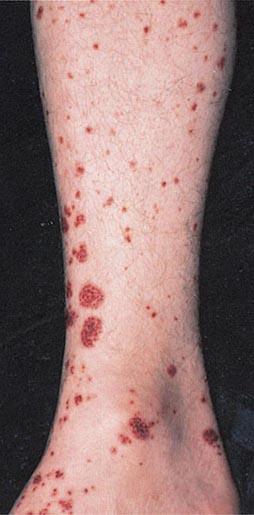
FIGURE 70-4 Necrotizing vasculitis. Palpable purpuric papules on the lower legs are seen in this patient with cutaneous small-vessel vasculitis. (Courtesy of Robert Swerlick, MD; with permission.)

FIGURE 70-5 Meningococcemia. An example of fulminant meningococcemia with extensive angular purpuric patches. (Courtesy of Stephen E. Gellis, MD; with permission.)
Skin Disorder
In examining the skin it is usually advisable to assess the patient before taking an extensive history. This approach ensures that the entire cutaneous surface will be evaluated, and objective findings can be integrated with relevant historical data. Four basic features of a skin lesion must be noted and considered during a physical examination: the distribution of the eruption, the types of primary and secondary lesions, the shape of individual lesions, and the arrangement of the lesions. An ideal skin examination includes evaluation of the skin, hair, and nails as well as the mucous membranes of the mouth, eyes, nose, nasopharynx, and anogenital region. In the initial examination, it is important that the patient be disrobed as completely as possible to minimize chances of missing important individual skin lesions and permit accurate assessment of the distribution of the eruption. The patient should first be viewed from a distance of about 1.5–2 m (4–6 ft) so that the general character of the skin and the distribution of lesions can be evaluated. Indeed, the distribution of lesions often correlates highly with diagnosis (Fig. 70-6). For example, a hospitalized patient with a generalized erythematous exanthem is more likely to have a drug eruption than is a patient with a similar rash limited to the sun-exposed portions of the face. Once the distribution of the lesions has been established, the nature of the primary lesion must be determined. Thus, when lesions are distributed on elbows, knees, and scalp, the most likely possibility based solely on distribution is psoriasis or dermatitis herpetiformis (Figs. 70-7 and 70-8, respectively). The primary lesion in psoriasis is a scaly papule that soon forms erythematous plaques covered with a white scale, whereas that of dermatitis herpetiformis is an urticarial papule that quickly becomes a small vesicle. In this manner, identification of the primary lesion directs the examiner toward the proper diagnosis. Secondary changes in skin can also be quite helpful. For example, scale represents excessive epidermis, while crust is the result of a discontinuous epithelial cell layer. Palpation of skin lesions can yield insight into the character of an eruption. Thus, red papules on the lower extremities that blanch with pressure can be a manifestation of many different diseases, but hemorrhagic red papules that do not blanch with pressure indicate palpable purpura characteristic of necrotizing vasculitis (Fig. 70-4).
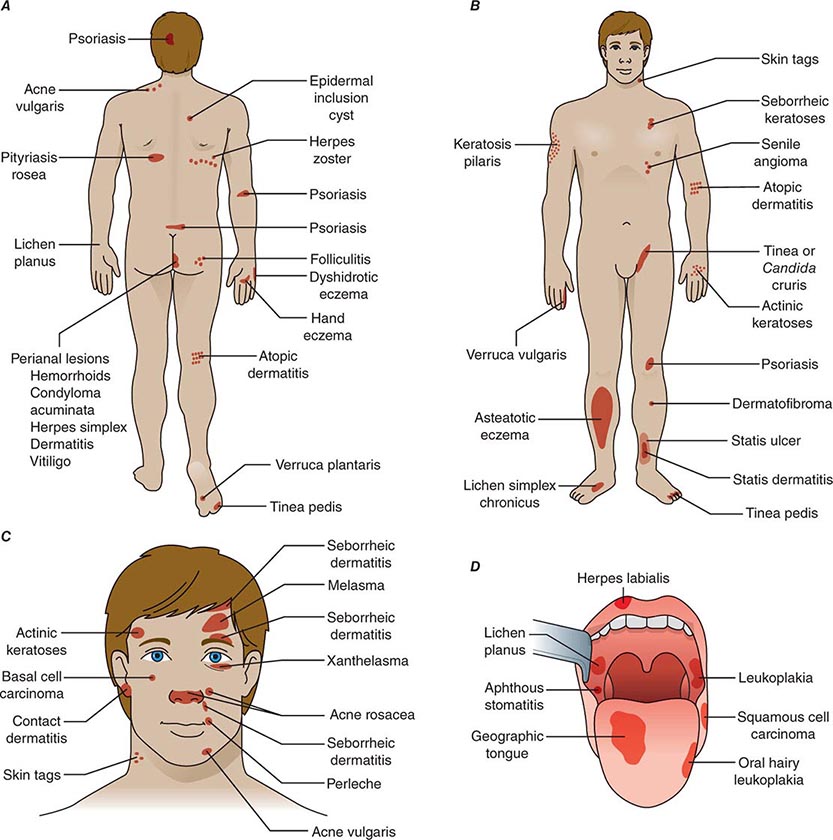
FIGURE 70-6 Distribution of some common dermatologic diseases and lesions.
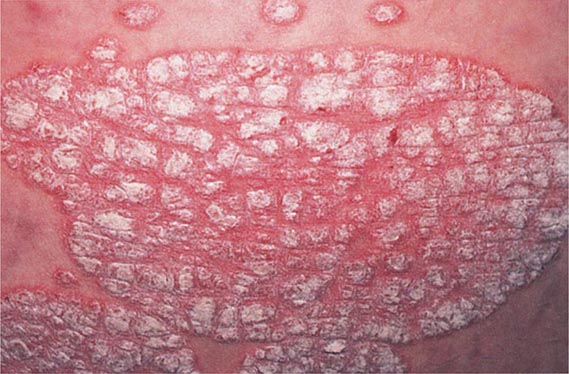
FIGURE 70-7 Psoriasis. This papulosquamous skin disease is characterized by small and large erythematous papules and plaques with overlying adherent silvery scale.
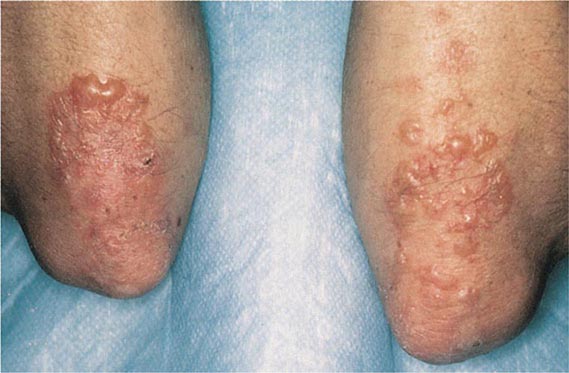
FIGURE 70-8 Dermatitis herpetiformis. This disorder typically displays pruritic, grouped papulovesicles on elbows, knees, buttocks, and posterior scalp. Vesicles are often excoriated due to associated pruritus.
The shape of lesions is also an important feature. Flat, round, erythematous papules and plaques are common in many cutaneous diseases. However, target-shaped lesions that consist in part of erythematous plaques are specific for erythema multiforme (Fig. 70-9). Likewise, the arrangement of individual lesions is important. Erythematous papules and vesicles can occur in many conditions, but their arrangement in a specific linear array suggests an external etiology such as allergic contact dermatitis (Fig. 70-10) or primary irritant dermatitis. In contrast, lesions with a generalized arrangement are common and suggest a systemic etiology.
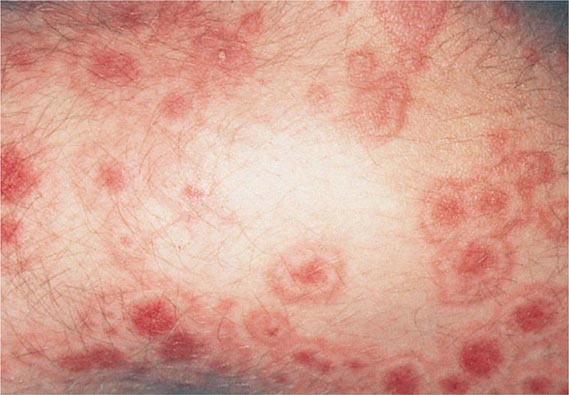
FIGURE 70-9 Erythema multiforme. This eruption is characterized by multiple erythematous plaques with a target or iris morphology. It usually represents a hypersensitivity reaction to drugs (e.g., sulfonamides) or infections (e.g., HSV). (Courtesy of the Yale Resident’s Slide Collection; with permission.)
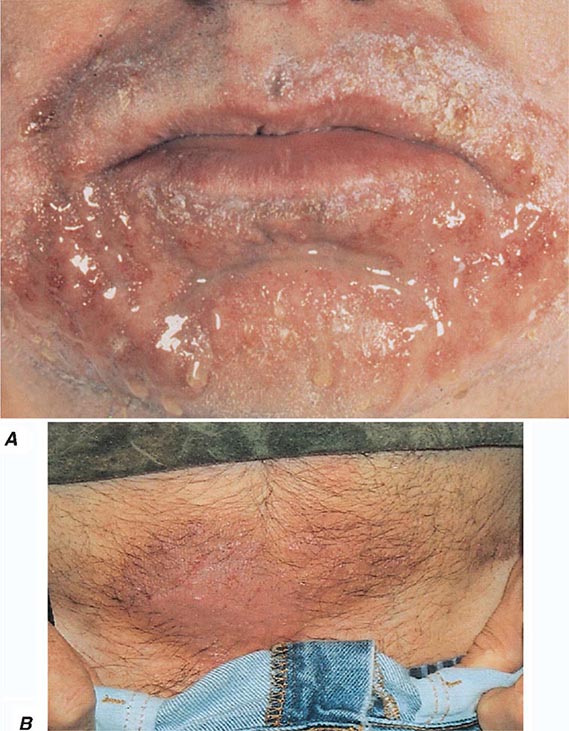
FIGURE 70-10 Allergic contact dermatitis (ACD). A. An example of ACD in its acute phase, with sharply demarcated, weeping, eczematous plaques in a perioral distribution. B. ACD in its chronic phase, with an erythematous, lichenified, weeping plaque on skin chronically exposed to nickel in a metal snap. (B, Courtesy of Robert Swerlick, MD; with permission.)
As in other branches of medicine, a complete history should be obtained to emphasize the following features:
1. Evolution of lesions
a. Site of onset
b. Manner in which the eruption progressed or spread
c. Duration
d. Periods of resolution or improvement in chronic eruptions
2. Symptoms associated with the eruption
a. Itching, burning, pain, numbness
b. What, if anything, has relieved symptoms
c. Time of day when symptoms are most severe
3. Current or recent medications (prescribed as well as over-the-counter)
4. Associated systemic symptoms (e.g., malaise, fever, arthralgias)
5. Ongoing or previous illnesses
6. History of allergies
7. Presence of photosensitivity
8. Review of systems
9. Family history (particularly relevant for patients with melanoma, atopy, psoriasis, or acne)
10. Social, sexual, or travel history
DIAGNOSTIC TECHNIQUES
Many skin diseases can be diagnosed on the basis of gross clinical appearance, but sometimes relatively simple diagnostic procedures can yield valuable information. In most instances, they can be performed at the bedside with a minimum of equipment.
Skin Biopsy A skin biopsy is a straightforward minor surgical procedure; however, it is important to biopsy a lesion that is most likely to yield diagnostic findings. This decision may require expertise in skin diseases and knowledge of superficial anatomic structures in selected areas of the body. In this procedure, a small area of skin is anesthetized with 1% lidocaine with or without epinephrine. The skin lesion in question can be excised or saucerized with a scalpel or removed by punch biopsy. In the latter technique, a punch is pressed against the surface of the skin and rotated with downward pressure until it penetrates to the subcutaneous tissue. The circular biopsy is then lifted with forceps, and the bottom is cut with iris scissors. Biopsy sites may or may not need suture closure, depending on size and location.
KOH Preparation A potassium hydroxide (KOH) preparation is performed on scaling skin lesions where a fungal infection is suspected. The edge of such a lesion is scraped gently with a no. 15 scalpel blade. The removed scale is collected on a glass microscope slide and then treated with 1 or 2 drops of a solution of 10–20% KOH. KOH dissolves keratin and allows easier visualization of fungal elements. Brief heating of the slide accelerates dissolution of keratin. When the preparation is viewed under the microscope, the refractile hyphae are seen more easily when the light intensity is reduced and the condenser is lowered. This technique can be used to identify hyphae in dermatophyte infections, pseudohyphae and budding yeasts in Candida infections, and “spaghetti and meatballs” yeast forms in tinea versicolor. The same sampling technique can be used to obtain scale for culture of selected pathogenic organisms.
Tzanck Smear A Tzanck smear is a cytologic technique most often used in the diagnosis of herpesvirus infections (herpes simplex virus [HSV] or varicella zoster virus [VZV]) (see Figs. 217-1 and 217-3). An early vesicle, not a pustule or crusted lesion, is unroofed, and the base of the lesion is scraped gently with a scalpel blade. The material is placed on a glass slide, air-dried, and stained with Giemsa or Wright’s stain. Multinucleated epithelial giant cells suggest the presence of HSV or VZV; culture, immunofluorescence microscopy, or genetic testing must be performed to identify the specific virus.
Diascopy Diascopy is designed to assess whether a skin lesion will blanch with pressure as, for example, in determining whether a red lesion is hemorrhagic or simply blood-filled. Urticaria (Fig. 70-11) will blanch with pressure, whereas a purpuric lesion caused by necrotizing vasculitis (Fig. 70-4) will not. Diascopy is performed by pressing a microscope slide or magnifying lens against a lesion and noting the amount of blanching that occurs. Granulomas often have an opaque to transparent, brown-pink “apple jelly” appearance on diascopy.
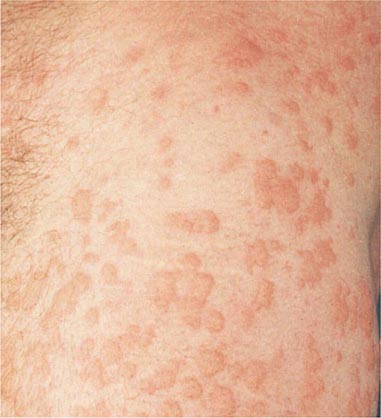
FIGURE 70-11 Urticaria. Discrete and confluent, edematous, erythematous papules and plaques are characteristic of this whealing eruption.
Wood’s Light A Wood’s lamp generates 360-nm ultraviolet (“black”) light that can be used to aid the evaluation of certain skin disorders. for example, a Wood’s lamp will cause erythrasma (a superficial, intertriginous infection caused by Corynebacterium minutissimum) to show a characteristic coral pink color, and wounds colonized by Pseudomonas will appear pale blue. Tinea capitis caused by certain dermatophytes (e.g., Microsporum canis or M. audouinii) exhibits a yellow fluorescence. Pigmented lesions of the epidermis such as freckles are accentuated, while dermal pigment such as postinflammatory hyperpigmentation fades under a Wood’s light. Vitiligo (Fig. 70-12) appears totally white under a Wood’s lamp, and previously unsuspected areas of involvement often become apparent. A Wood’s lamp may also aid in the demonstration of tinea versicolor and in recognition of ash leaf spots in patients with tuberous sclerosis.
FIGURE 70-12 Vitiligo. Characteristic lesions display an acral distribution and striking depigmentation as a result of loss of melanocytes.
Patch Tests Patch testing is designed to document sensitivity to a specific antigen. In this procedure, a battery of suspected allergens is applied to the patient’s back under occlusive dressings and allowed to remain in contact with the skin for 48 h. The dressings are removed, and the area is examined for evidence of delayed hypersensitivity reactions (e.g., erythema, edema, or papulovesicles). This test is best performed by physicians with special expertise in patch testing and is often helpful in the evaluation of patients with chronic dermatitis.
71 |
Eczema, Psoriasis, Cutaneous Infections, Acne, and Other Common Skin Disorders |
ECZEMA AND DERMATITIS
Eczema is a type of dermatitis, and these terms are often used synonymously (e.g., atopic eczema or atopic dermatitis [AD]). Eczema is a reaction pattern that presents with variable clinical findings and the common histologic finding of spongiosis (intercellular edema of the epidermis). Eczema is the final common expression for a number of disorders, including those discussed in the following sections. Primary lesions may include erythematous macules, papules, and vesicles, which can coalesce to form patches and plaques. In severe eczema, secondary lesions from infection or excoriation, marked by weeping and crusting, may predominate. In chronic eczematous conditions, lichenification (cutaneous hypertrophy and accentuation of normal skin markings) may alter the characteristic appearance of eczema.
ATOPIC DERMATITIS
AD is the cutaneous expression of the atopic state, characterized by a family history of asthma, allergic rhinitis, or eczema. The prevalence of AD is increasing worldwide. Some of its features are shown in Table 71-1.
|
CLINICAL FEATURES OF ATOPIC DERMATITIS |
![]() The etiology of AD is only partially defined, but there is a clear genetic predisposition. When both parents are affected by AD, >80% of their children manifest the disease. When only one parent is affected, the prevalence drops to slightly over 50%. A characteristic defect in AD that contributes to the pathophysiology is an impaired epidermal barrier. In many patients, a mutation in the gene encoding filaggrin, a structural protein in the stratum corneum, is responsible. Patients with AD may display a variety of immunoregulatory abnormalities, including increased IgE synthesis; increased serum IgE levels; and impaired, delayed-type hypersensitivity reactions.
The etiology of AD is only partially defined, but there is a clear genetic predisposition. When both parents are affected by AD, >80% of their children manifest the disease. When only one parent is affected, the prevalence drops to slightly over 50%. A characteristic defect in AD that contributes to the pathophysiology is an impaired epidermal barrier. In many patients, a mutation in the gene encoding filaggrin, a structural protein in the stratum corneum, is responsible. Patients with AD may display a variety of immunoregulatory abnormalities, including increased IgE synthesis; increased serum IgE levels; and impaired, delayed-type hypersensitivity reactions.
The clinical presentation often varies with age. Half of patients with AD present within the first year of life, and 80% present by 5 years of age. About 80% ultimately coexpress allergic rhinitis or asthma. The infantile pattern is characterized by weeping inflammatory patches and crusted plaques on the face, neck, and extensor surfaces. The childhood and adolescent pattern is typified by dermatitis of flexural skin, particularly in the antecubital and popliteal fossae (Fig. 71-1). AD may resolve spontaneously, but approximately 40% of all individuals affected as children will have dermatitis in adult life. The distribution of lesions in adults may be similar to those seen in childhood; however, adults frequently have localized disease manifesting as lichen simplex chronicus or hand eczema (see below). In patients with localized disease, AD may be suspected because of a typical personal or family history or the presence of cutaneous stigmata of AD such as perioral pallor, an extra fold of skin beneath the lower eyelid (Dennie-Morgan folds), increased palmar skin markings, and an increased incidence of cutaneous infections, particularly with Staphylococcus aureus. Regardless of other manifestations, pruritus is a prominent characteristic of AD in all age groups and is exacerbated by dry skin. Many of the cutaneous findings in affected patients, such as lichenification, are secondary to rubbing and scratching.
FIGURE 71-1 Atopic dermatitis. Hyperpigmentation, lichenification, and scaling in the antecubital fossae are seen in this patient with atopic dermatitis. (Courtesy of Robert Swerlick, MD; with permission.)
LICHEN SIMPLEX CHRONICUS
Lichen simplex chronicus may represent the end stage of a variety of pruritic and eczematous disorders, including AD. It consists of a circumscribed plaque or plaques of lichenified skin due to chronic scratching or rubbing. Common areas involved include the posterior nuchal region, dorsum of the feet, and ankles. Treatment of lichen simplex chronicus centers on breaking the cycle of chronic itching and scratching. High-potency topical glucocorticoids are helpful in most cases, but, in recalcitrant cases, application of topical glucocorticoids under occlusion, or intralesional injection of glucocorticoids may be required.
CONTACT DERMATITIS
Contact dermatitis is an inflammatory skin process caused by an exogenous agent or agents that directly or indirectly injure the skin. In irritant contact dermatitis (ICD), this injury is caused by an inherent characteristic of a compound—for example, a concentrated acid or base. Agents that cause ACD induce an antigen-specific immune response (e.g., poison ivy dermatitis). The clinical lesions of contact dermatitis may be acute (wet and edematous) or chronic (dry, thickened, and scaly), depending on the persistence of the insult (see Fig. 70-10).
Irritant Contact Dermatitis ICD is generally well demarcated and often localized to areas of thin skin (eyelids, intertriginous areas) or to areas where the irritant was occluded. Lesions may range from minimal skin erythema to areas of marked edema, vesicles, and ulcers. Prior exposure to the offending agent is not necessary, and the reaction develops in minutes to a few hours. Chronic low-grade irritant dermatitis is the most common type of ICD, and the most common area of involvement is the hands (see below). The most common irritants encountered are chronic wet work, soaps, and detergents. Treatment should be directed toward the avoidance of irritants and the use of protective gloves or clothing.
Allergic Contact Dermatitis ACD is a manifestation of delayed-type hypersensitivity mediated by memory T lymphocytes in the skin. Prior exposure to the offending agent is necessary to develop the hypersensitivity reaction, which may take as little as 12 h or as much as 72 h to develop. The most common cause of ACD is exposure to plants, especially to members of the family Anacardiaceae, including the genus Toxicodendron. Poison ivy, poison oak, and poison sumac are members of this genus and cause an allergic reaction marked by erythema, vesiculation, and severe pruritus. The eruption is often linear or angular, corresponding to areas where plants have touched the skin. The sensitizing antigen common to these plants is urushiol, an oleoresin containing the active ingredient pentadecylcatechol. The oleoresin may adhere to skin, clothing, tools, and pets, and contaminated articles may cause dermatitis even after prolonged storage. Blister fluid does not contain urushiol and is not capable of inducing skin eruption in exposed subjects.
HAND ECZEMA
Hand eczema is a very common, chronic skin disorder in which both exogenous and endogenous factors play important roles. It may be associated with other cutaneous disorders such as AD, and contact with various agents may be involved. Hand eczema represents a large proportion of cases of occupation-associated skin disease. Chronic, excessive exposure to water and detergents, harsh chemicals, or allergens may initiate or aggravate this disorder. It may present with dryness and cracking of the skin of the hands as well as with variable amounts of erythema and edema. Often, the dermatitis will begin under rings, where water and irritants are trapped. Dyshidrotic eczema, a variant of hand eczema, presents with multiple, intensely pruritic, small papules and vesicles on the thenar and hypothenar eminences and the sides of the fingers (Fig. 71-2). Lesions tend to occur in crops that slowly form crusts and then heal.
FIGURE 71-2 Dyshidrotic eczema. This example is characterized by deep-seated vesicles and scaling on palms and lateral fingers, and the disease is often associated with an atopic diathesis.
The evaluation of a patient with hand eczema should include an assessment of potential occupation-associated exposures. The history should be directed to identifying possible irritant or allergen exposures.
NUMMULAR ECZEMA
Nummular eczema is characterized by circular or oval “coinlike” lesions, beginning as small edematous papules that become crusted and scaly. The etiology of nummular eczema is unknown, but dry skin is a contributing factor. Common locations are the trunk or the extensor surfaces of the extremities, particularly on the pretibial areas or dorsum of the hands. Nummular eczema occurs more frequently in men and is most common in middle age. The treatment of nummular eczema is similar to that for AD.
ASTEATOTIC ECZEMA
Asteatotic eczema, also known as xerotic eczema or “winter itch,” is a mildly inflammatory dermatitis that develops in areas of extremely dry skin, especially during the dry winter months. Clinically, there may be considerable overlap with nummular eczema. This form of eczema accounts for a large number of physician visits because of the associated pruritus. Fine cracks and scale, with or without erythema, characteristically develop in areas of dry skin, especially on the anterior surfaces of the lower extremities in elderly patients. Asteatotic eczema responds well to topical moisturizers and the avoidance of cutaneous irritants. Overbathing and the use of harsh soaps exacerbate asteatotic eczema.
STASIS DERMATITIS AND STASIS ULCERATION
Stasis dermatitis develops on the lower extremities secondary to venous incompetence and chronic edema. Patients may give a history of deep venous thrombosis and may have evidence of vein removal or varicose veins. Early findings in stasis dermatitis consist of mild erythema and scaling associated with pruritus. The typical initial site of involvement is the medial aspect of the ankle, often over a distended vein (Fig. 71-3).
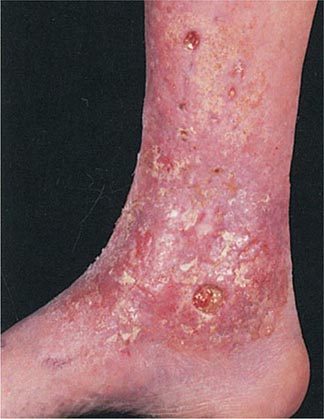
FIGURE 71-3 Stasis dermatitis. An example of stasis dermatitis showing erythematous, scaly, and oozing patches over the lower leg. Several stasis ulcers are also seen in this patient.
Stasis dermatitis may become acutely inflamed, with crusting and exudate. In this state, it is easily confused with cellulitis. Chronic stasis dermatitis is often associated with dermal fibrosis that is recognized clinically as brawny edema of the skin. As the disorder progresses, the dermatitis becomes progressively pigmented due to chronic erythrocyte extravasation leading to cutaneous hemosiderin deposition. Stasis dermatitis may be complicated by secondary infection and contact dermatitis. Severe stasis dermatitis may precede the development of stasis ulcers.
SEBORRHEIC DERMATITIS
Seborrheic dermatitis is a common, chronic disorder characterized by greasy scales overlying erythematous patches or plaques. Induration and scale are generally less prominent than in psoriasis, but clinical overlap exists between these diseases (“sebopsoriasis”). The most common location is in the scalp, where it may be recognized as severe dandruff. On the face, seborrheic dermatitis affects the eyebrows, eyelids, glabella, and nasolabial folds (Fig. 71-4). Scaling of the external auditory canal is common in seborrheic dermatitis. In addition, the postauricular areas often become macerated and tender. Seborrheic dermatitis may also develop in the central chest, axilla, groin, submammary folds, and gluteal cleft. Rarely, it may cause widespread generalized dermatitis. Pruritus is variable.

FIGURE 71-4 Seborrheic dermatitis. Central facial erythema with overlying greasy, yellowish scale is seen in this patient. (Courtesy of Jean Bolognia, MD; with permission.)
Seborrheic dermatitis may be evident within the first few weeks of life, and within this context it typically occurs in the scalp (“cradle cap”), face, or groin. It is rarely seen in children beyond infancy but becomes evident again during adult life. Although it is frequently seen in patients with Parkinson’s disease, in those who have had cerebrovascular accidents, and in those with HIV infection, the overwhelming majority of individuals with seborrheic dermatitis have no underlying disorder.
PAPULOSQUAMOUS DISORDERS (TABLE 71-2)
|
PAPULOSQUAMOUS DISORDERS |

PSORIASIS
Psoriasis is one of the most common dermatologic diseases, affecting up to 2% of the world’s population. It is an immune-mediated disease clinically characterized by erythematous, sharply demarcated papules and rounded plaques covered by silvery micaceous scale. The skin lesions of psoriasis are variably pruritic. Traumatized areas often develop lesions of psoriasis (the Koebner or isomorphic phenomenon). In addition, other external factors may exacerbate psoriasis, including infections, stress, and medications (lithium, beta blockers, and antimalarial drugs).
The most common variety of psoriasis is called plaque-type. Patients with plaque-type psoriasis have stable, slowly enlarging plaques, which remain basically unchanged for long periods of time. The most commonly involved areas are the elbows, knees, gluteal cleft, and scalp. Involvement tends to be symmetric. Plaque psoriasis generally develops slowly and runs an indolent course. It rarely remits spontaneously. Inverse psoriasis affects the intertriginous regions, including the axilla, groin, submammary region, and navel; it also tends to affect the scalp, palms, and soles. The individual lesions are sharply demarcated plaques (see Fig. 70-7), but they may be moist and without scale due to their locations.
Guttate psoriasis (eruptive psoriasis) is most common in children and young adults. It develops acutely in individuals without psoriasis or in those with chronic plaque psoriasis. Patients present with many small erythematous, scaling papules, frequently after upper respiratory tract infection with β-hemolytic streptococci. The differential diagnosis should include pityriasis rosea and secondary syphilis.
In pustular psoriasis, patients may have disease localized to the palms and soles, or the disease may be generalized. Regardless of the extent of disease, the skin is erythematous, with pustules and variable scale. Localized to the palms and soles, it is easily confused with eczema. When it is generalized, episodes are characterized by fever (39°–40°C [102.2°–104.0°F]) lasting several days, an accompanying generalized eruption of sterile pustules, and a background of intense erythema; patients may become erythrodermic. Episodes of fever and pustules are recurrent. Local irritants, pregnancy, medications, infections, and systemic glucocorticoid withdrawal can precipitate this form of psoriasis. Oral retinoids are the treatment of choice in nonpregnant patients.
Fingernail involvement, appearing as punctate pitting, onycholysis, nail thickening, or subungual hyperkeratosis, may be a clue to the diagnosis of psoriasis when the clinical presentation is not classic.
According to the National Psoriasis Foundation, up to 30% of patients with psoriasis have psoriatic arthritis (PsA). There are five subtypes of PsA: symmetric, asymmetric, distal interphalangeal predominant (DIP), spondylitis, and arthritis mutilans. Symmetric arthritis resembles rheumatoid arthritis, but is usually milder. Asymmetric arthritis can involve any joint and may present as “sausage digits.” DIP is the classic form, but occurs in only about 5% of patients with PsA. It may involve fingers and toes. Spondylitis also occurs in about 5% of patients with PsA. Arthritis mutilans is severe and deforming. It affects primarily the small joints of the hands and feet. It accounts for fewer than 5% of PsA cases.
An increased risk of metabolic syndrome, including increased morbidity and mortality from cardiovascular events, has been demonstrated in psoriasis patients. Appropriate screening tests should be performed. The etiology of psoriasis is still poorly understood, but there is clearly a genetic component to the disease. In various studies, 30–50% of patients with psoriasis report a positive family history. Psoriatic lesions contain infiltrates of activated T cells that are thought to elaborate cytokines responsible for keratinocyte hyperproliferation, which results in the characteristic clinical findings. Agents inhibiting T cell activation, clonal expansion, or release of proinflammatory cytokines are often effective for the treatment of severe psoriasis (see below).
|
TREATMENT |
PSORIASIS |
Treatment of psoriasis depends on the type, location, and extent of disease. All patients should be instructed to avoid excess drying or irritation of their skin and to maintain adequate cutaneous hydration. Most cases of localized, plaque-type psoriasis can be managed with mid-potency topical glucocorticoids, although their long-term use is often accompanied by loss of effectiveness (tachyphylaxis) and atrophy of the skin. A topical vitamin D analogue (calcipotriene) and a retinoid (tazarotene) are also efficacious in the treatment of limited psoriasis and have largely replaced other topical agents such as coal tar, salicylic acid, and anthralin.
Ultraviolet (UV) light, natural or artificial, is an effective therapy for many patients with widespread psoriasis. Ultraviolet B (UVB), narrowband UVB, and ultraviolet A (UVA) light with either oral or topical psoralens (PUVA) is used clinically. UV light’s immunosuppressive properties are thought to be responsible for its therapeutic activity in psoriasis. It is also mutagenic, potentially leading to an increased incidence of nonmelanoma and melanoma skin cancer. UV-light therapy is contraindicated in patients receiving cyclosporine and should be used with great care in all immunocompromised patients due to the increased risk of skin cancer.
Various systemic agents can be used for severe, widespread psoriatic disease (Table 71-3). Oral glucocorticoids should not be used for the treatment of psoriasis due to the potential for development of life-threatening pustular psoriasis when therapy is discontinued. Methotrexate is an effective agent, especially in patients with psoriatic arthritis. The synthetic retinoid acitretin is useful, especially when immunosuppression must be avoided; however, teratogenicity limits its use.
|
FDA-APPROVED SYSTEMIC THERAPY FOR PSORIASIS |
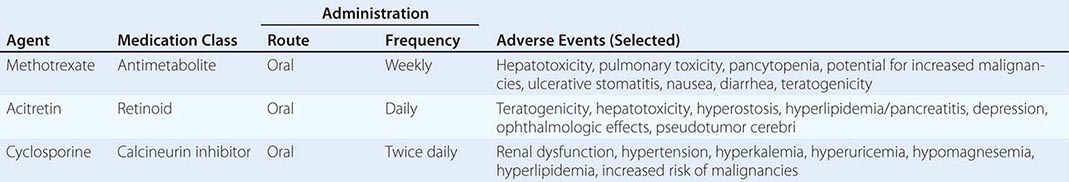
The evidence implicating psoriasis as a T cell–mediated disorder has directed therapeutic efforts to immunoregulation. Cyclosporine and other immunosuppressive agents can be very effective in the treatment of psoriasis, and much attention is currently directed toward the development of biologic agents with more selective immunosuppressive properties and better safety profiles (Table 71-4). Experience with these biologic agents is limited, and information regarding combination therapy and adverse events continues to emerge. Use of tumor necrosis factor (TNF-α) inhibitors may worsen congestive heart failure (CHF), and they should be used with caution in patients at risk for or known to have CHF. Further, none of the immunosuppressive agents used in the treatment of psoriasis should be initiated if the patient has a severe infection; patients on such therapy should be routinely screened for tuberculosis. There have been reports of progressive multifocal leukoencephalopathy in association with treatment with the TNF-α inhibitors. Malignancies, including a risk or history of certain malignancies, may limit the use of these systemic agents.
|
BIOLOGICS APPROVED FOR PSORIASIS OR PSORIATIC ARTHRITIS |
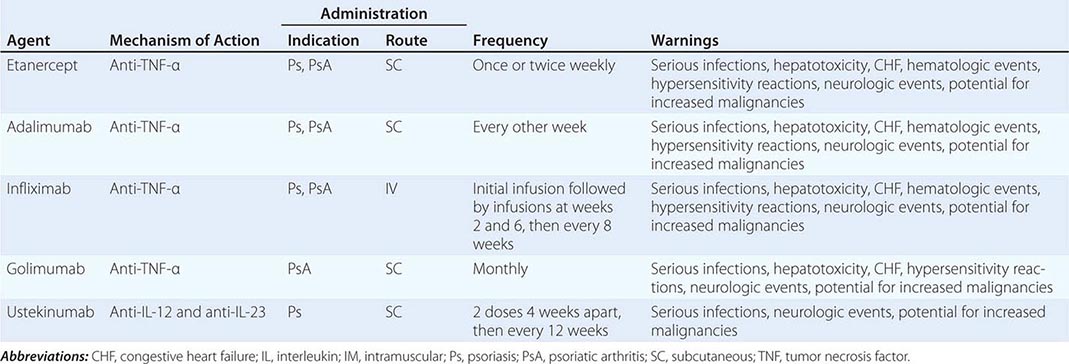
LICHEN PLANUS
Lichen planus (LP) is a papulosquamous disorder that may affect the skin, scalp, nails, and mucous membranes. The primary cutaneous lesions are pruritic, polygonal, flat-topped, violaceous papules. Close examination of the surface of these papules often reveals a network of gray lines (Wickham’s striae). The skin lesions may occur anywhere but have a predilection for the wrists, shins, lower back, and genitalia (Fig. 71-5). Involvement of the scalp (lichen planopilaris) may lead to scarring alopecia, and nail involvement may lead to permanent deformity or loss of fingernails and toenails. LP commonly involves mucous membranes, particularly the buccal mucosa, where it can present on a spectrum ranging from a mild, white, reticulate eruption of the mucosa to a severe, erosive stomatitis. Erosive stomatitis may persist for years and may be linked to an increased risk of oral squamous cell carcinoma. Cutaneous eruptions clinically resembling LP have been observed after administration of numerous drugs, including thiazide diuretics, gold, antimalarial agents, penicillamine, and phenothiazines, and in patients with skin lesions of chronic graft-versus-host disease. In addition, LP may be associated with hepatitis C infection. The course of LP is variable, but most patients have spontaneous remissions 6 months to 2 years after the onset of disease. Topical glucocorticoids are the mainstay of therapy.
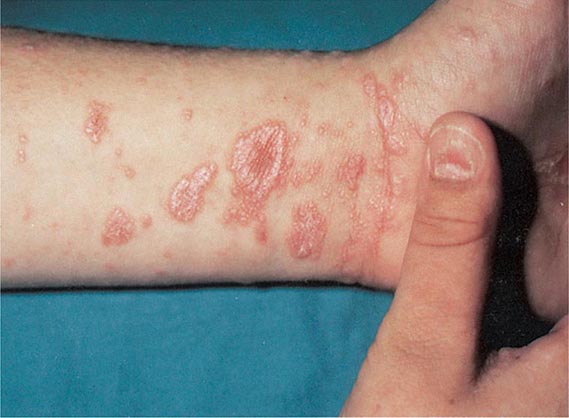
FIGURE 71-5 Lichen planus. An example of lichen planus showing multiple flat-topped, violaceous papules and plaques. Nail dystrophy, as seen in this patient’s thumbnail, may also be a feature. (Courtesy of Robert Swerlick, MD; with permission.)
PITYRIASIS ROSEA
Pityriasis rosea (PR) is a papulosquamous eruption of unknown etiology occurring more commonly in the spring and fall. Its first manifestation is the development of a 2- to 6-cm annular lesion (the herald patch). This is followed in a few days to a few weeks by the appearance of many smaller annular or papular lesions with a predilection to occur on the trunk (Fig. 71-6). The lesions are generally oval, with their long axis parallel to the skinfold lines. Individual lesions may range in color from red to brown and have a trailing scale. PR shares many clinical features with the eruption of secondary syphilis, but palm and sole lesions are extremely rare in PR and common in secondary syphilis. The eruption tends to be moderately pruritic and lasts 3–8 weeks. Treatment is directed at alleviating pruritus and consists of oral antihistamines; mid-potency topical glucocorticoids; and, in some cases, UVB phototherapy.
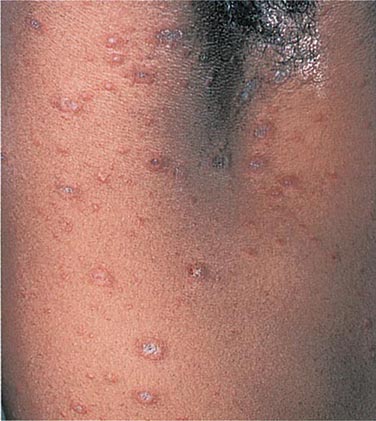
FIGURE 71-6 Pityriasis rosea. In this patient with pityriasis rosea, multiple round to oval erythematous patches with fine central scale are distributed along the skin tension lines on the trunk.
CUTANEOUS INFECTIONS (TABLE 71-5)
|
COMMON SKIN INFECTIONS |
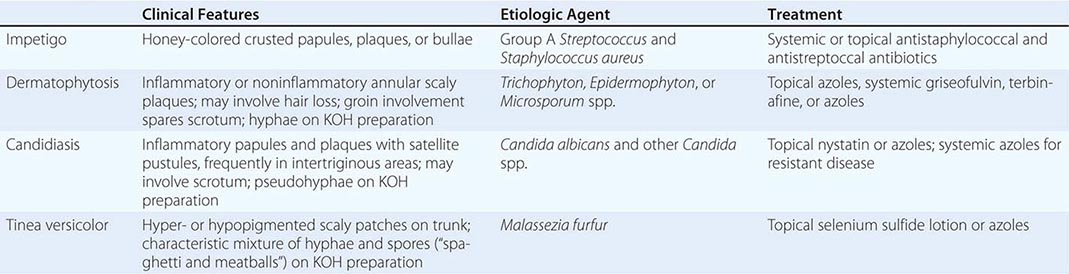
IMPETIGO, ECTHYMA, AND FURUNCULOSIS
Impetigo is a common superficial bacterial infection of skin caused most often by S. aureus (Chap. 172) and in some cases by group A β-hemolytic streptococci (Chap. 173). The primary lesion is a superficial pustule that ruptures and forms a characteristic yellow-brown honey-colored crust (see Fig. 173-3). Lesions may occur on normal skin (primary infection) or in areas already affected by another skin disease (secondary infection). Lesions caused by staphylococci may be tense, clear bullae, and this less common form of the disease is called bullous impetigo. Blisters are caused by the production of exfoliative toxin by S. aureus phage type II. This is the same toxin responsible for staphylococcal scalded-skin syndrome, often resulting in dramatic loss of the superficial epidermis due to blistering. The latter syndrome is much more common in children than in adults; however, it should be considered along with toxic epidermal necrolysis and severe drug eruptions in patients with widespread blistering of the skin. Ecthyma is a deep non-bullous variant of impetigo that causes punched-out ulcerative lesions. It is more often caused by a primary or secondary infection with Streptococcus pyogenes. Ecthyma is a deeper infection than typical impetigo and resolves with scars. Treatment of both ecthyma and impetigo involves gentle debridement of adherent crusts, which is facilitated by the use of soaks and topical antibiotics in conjunction with appropriate oral antibiotics.
Furunculosis is also caused by S. aureus, and this disorder has gained prominence in the last decade because of CA-MRSA. A furuncle, or boil, is a painful, erythematous nodule that can occur on any cutaneous surface. The lesions may be solitary but are most often multiple. Patients frequently believe they have been bitten by spiders or insects. Family members or close contacts may also be affected. Furuncles can rupture and drain spontaneously or may need incision and drainage, which may be adequate therapy for small solitary furuncles without cellulitis or systemic symptoms. Whenever possible, lesional material should be sent for culture. Current recommendations for methicillin-sensitive infections are β-lactam antibiotics. Therapy for CA-MRSA was discussed previously (see “Atopic Dermatitis”). Warm compresses and nasal mupirocin are helpful therapeutic additions. Severe infections may require IV antibiotics.
ERYSIPELAS AND CELLULITIS
See Chap. 156.
DERMATOPHYTOSIS
Dermatophytes are fungi that infect skin, hair, and nails and include members of the genera Trichophyton, Microsporum, and Epidermophyton (Chap. 243). Tinea corporis, or infection of the relatively hairless skin of the body (glabrous skin), may have a variable appearance depending on the extent of the associated inflammatory reaction. Typical infections consist of erythematous, scaly plaques, with an annular appearance that accounts for the common name “ringworm.” Deep inflammatory nodules or granulomas occur in some infections, most often those inappropriately treated with mid- to high-potency topical glucocorticoids. Involvement of the groin (tinea cruris) is more common in males than in females. It presents as a scaling, erythematous eruption sparing the scrotum. Infection of the foot (tinea pedis) is the most common dermatophyte infection and is often chronic; it is characterized by variable erythema, edema, scaling, pruritus, and occasionally vesiculation. The infection may be widespread or localized but generally involves the web space between the fourth and fifth toes. Infection of the nails (tinea unguium or onychomycosis) occurs in many patients with tinea pedis and is characterized by opacified, thickened nails and subungual debris. The distal-lateral variant is most common. Proximal subungual onychomycosis may be a marker for HIV infection or other immunocompromised states. Dermatophyte infection of the scalp (tinea capitis) continues to be common, particularly affecting inner-city children but also affecting adults. The predominant organism is Trichophyton tonsurans, which can produce a relatively noninflammatory infection with mild scale and hair loss that is diffuse or localized. T. tonsurans can also cause a markedly inflammatory dermatosis with edema and nodules. This latter presentation is a kerion.
The diagnosis of tinea can be made from skin scrapings, nail scrapings, or hair by culture or direct microscopic examination with potassium hydroxide (KOH). Nail clippings may be sent for histologic examination with periodic acid–Schiff (PAS) stain.
TINEA VERSICOLOR
Tinea versicolor is caused by a nondermatophytic, dimorphic fungus, Malassezia furfur, a normal inhabitant of the skin. The expression of infection is promoted by heat and humidity. The typical lesions consist of oval scaly macules, papules, and patches concentrated on the chest, shoulders, and back but only rarely on the face or distal extremities. On dark skin the lesions often appear as hypopigmented areas, while on light skin they are slightly erythematous or hyperpigmented. A KOH preparation from scaling lesions will demonstrate a confluence of short hyphae and round spores (“spaghetti and meatballs”). Lotions or shampoos containing sulfur, salicylic acid, or selenium sulfide will clear the infection if used daily for 1–2 weeks and then weekly thereafter. These preparations are irritating if left on the skin for >10 min; thus, they should be washed off completely. Treatment with some oral antifungal agents is also effective, but they do not provide lasting results and are not FDA approved for this indication. A very short course of ketoconazole has been used, as have itraconazole and fluconazole. The patient must sweat after taking the medication if it is to be effective. Griseofulvin is not effective and terbinafine is not reliably effective for tinea versicolor.
CANDIDIASIS
Candidiasis is a fungal infection caused by a related group of yeasts whose manifestations may be localized to the skin and mucous membranes or, rarely, may be systemic and life-threatening (Chap. 240). The causative organism is usually Candida albicans. These organisms are normal saprophytic inhabitants of the gastrointestinal tract but may overgrow due to broad-spectrum antibiotic therapy, diabetes mellitus, or immunosuppression and cause disease. Candidiasis is a very common infection in HIV-infected individuals (Chap. 226). The oral cavity is commonly involved. Lesions may occur on the tongue or buccal mucosa (thrush) and appear as white plaques. Fissured, macerated lesions at the corners of the mouth (perléche) are often seen in individuals with poorly fitting dentures and may also be associated with candidal infection. In addition, candidal infections have an affinity for sites that are chronically wet and macerated, including the skin around nails (onycholysis and paronychia), and in intertriginous areas. Intertriginous lesions are characteristically edematous, erythematous, and scaly, with scattered “satellite pustules.” In males, there is often involvement of the penis and scrotum as well as the inner aspect of the thighs. In contrast to dermatophyte infections, candidal infections are frequently painful and accompanied by a marked inflammatory response. Diagnosis of candidal infection is based upon the clinical pattern and demonstration of yeast on KOH preparation or culture.
WARTS
Warts are cutaneous neoplasms caused by papillomaviruses. More than 100 different human papillomaviruses (HPVs) have been described. A typical wart, verruca vulgaris, is sessile, dome-shaped, and usually about a centimeter in diameter. Its surface is hyperkeratotic, consisting of many small filamentous projections. The HPV types that cause typical verruca vulgaris also cause typical plantar warts, flat warts (verruca plana), and filiform warts. Plantar warts are endophytic and are covered by thick keratin. Paring of the wart will generally reveal a central core of keratinized debris and punctate bleeding points. Filiform warts are most commonly seen on the face, neck, and skinfolds and present as papillomatous lesions on a narrow base. Flat warts are only slightly elevated and have a velvety, nonverrucous surface. They have a propensity for the face, arms, and legs and are often spread by shaving.
Genital warts begin as small papillomas that may grow to form large, fungating lesions. In women, they may involve the labia, perineum, or perianal skin. In addition, the mucosa of the vagina, urethra, and anus can be involved as well as the cervical epithelium. In men, the lesions often occur initially in the coronal sulcus but may be seen on the shaft of the penis, the scrotum, or the perianal skin or in the urethra.
Appreciable evidence has accumulated indicating that HPV plays a role in the development of neoplasia of the uterine cervix and anogenital skin (Chap. 117). HPV types 16 and 18 have been most intensely studied and are the major risk factors for intraepithelial neoplasia and squamous cell carcinoma of the cervix, anus, vulva, and penis. The risk is higher among patients immunosuppressed after solid organ transplantation and among those infected with HIV. Recent evidence also implicates other HPV types. Histologic examination of biopsied samples from affected sites may reveal changes associated with typical warts and/or features typical of intraepidermal carcinoma (Bowen’s disease). Squamous cell carcinomas associated with HPV infections have also been observed in extragenital skin (Chap. 105), most commonly in patients immunosuppressed after organ transplantation. Patients on long-term immunosuppression should be monitored for the development of squamous cell carcinoma and other cutaneous malignancies.
HERPES SIMPLEX
See Chap. 216.
HERPES ZOSTER
See Chap. 217.
ACNE
ACNE VULGARIS
Acne vulgaris is a self-limited disorder primarily of teenagers and young adults, although perhaps 10–20% of adults may continue to experience some form of the disorder. The permissive factor for the expression of the disease in adolescence is the increase in sebum production by sebaceous glands after puberty. Small cysts, called comedones, form in hair follicles due to blockage of the follicular orifice by retention of keratinous material and sebum. The activity of bacteria (Propionibacterium acnes) within the comedones releases free fatty acids from sebum, causes inflammation within the cyst, and results in rupture of the cyst wall. An inflammatory foreign-body reaction develops as result of extrusion of oily and keratinous debris from the cyst.
The clinical hallmark of acne vulgaris is the comedone, which may be closed (whitehead) or open (blackhead). Closed comedones appear as 1- to 2-mm pebbly white papules, which are accentuated when the skin is stretched. They are the precursors of inflammatory lesions of acne vulgaris. The contents of closed comedones are not easily expressed. Open comedones, which rarely result in inflammatory acne lesions, have a large dilated follicular orifice and are filled with easily expressible oxidized, darkened, oily debris. Comedones are usually accompanied by inflammatory lesions: papules, pustules, or nodules.
The earliest lesions seen in adolescence are generally mildly inflamed or noninflammatory comedones on the forehead. Subsequently, more typical inflammatory lesions develop on the cheeks, nose, and chin (Fig. 71-7). The most common location for acne is the face, but involvement of the chest and back is common. Most disease remains mild and does not lead to scarring. A small number of patients develop large inflammatory cysts and nodules, which may drain and result in significant scarring. Regardless of the severity, acne may affect a patient’s quality of life. With adequate treatment, this effect may be transient. In the case of severe, scarring acne, the effects can be permanent and profound. Early therapeutic intervention in severe acne is essential.
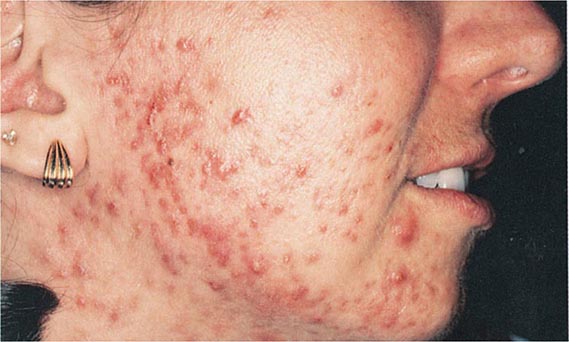
FIGURE 71-7 Acne vulgaris. An example of acne vulgaris with inflammatory papules, pustules, and comedones. (Courtesy of Kalman Watsky, MD; with permission.)
Exogenous and endogenous factors can alter the expression of acne vulgaris. Friction and trauma (from headbands or chin straps of athletic helmets), application of comedogenic topical agents (cosmetics or hair preparations), or chronic topical exposure to certain industrial compounds may elicit or aggravate acne. Glucocorticoids, topical or systemic, may also elicit acne. Other systemic medications such as oral contraceptive pills, lithium, isoniazid, androgenic steroids, halogens, phenytoin, and phenobarbital may produce acneiform eruptions or aggravate preexisting acne. Genetic factors and polycystic ovary disease may also play a role.
ACNE ROSACEA
Acne rosacea, commonly referred to simply as rosacea, is an inflammatory disorder predominantly affecting the central face. Persons most often affected are Caucasians of northern European background, but rosacea also occurs in patients with dark skin. Rosacea is seen almost exclusively in adults, only rarely affecting patients <30 years old. Rosacea is more common in women, but those most severely affected are men. It is characterized by the presence of erythema, telangiectases, and superficial pustules (Fig. 71-8) but is not associated with the presence of comedones. Rosacea rarely involves the chest or back.
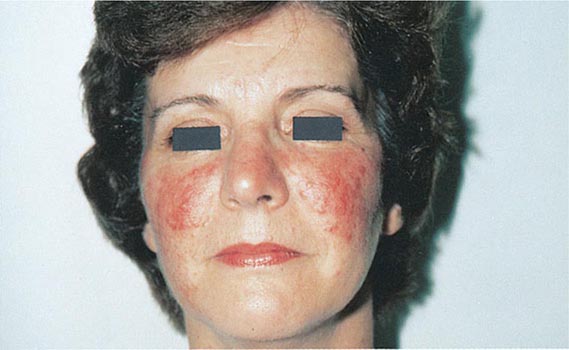
FIGURE 71-8 Acne rosacea. Prominent facial erythema, telangiectasia, scattered papules, and small pustules are seen in this patient with acne rosacea. (Courtesy of Robert Swerlick, MD; with permission.)
There is a relationship between the tendency for facial flushing and the subsequent development of acne rosacea. Often, individuals with rosacea initially demonstrate a pronounced flushing reaction. This may be in response to heat, emotional stimuli, alcohol, hot drinks, or spicy foods. As the disease progresses, the flush persists longer and longer and may eventually become permanent. Papules, pustules, and telangiectases can become superimposed on the persistent flush. Rosacea of very long standing may lead to connective tissue overgrowth, particularly of the nose (rhinophyma). Rosacea may also be complicated by various inflammatory disorders of the eye, including keratitis, blepharitis, iritis, and recurrent chalazion. These ocular problems are potentially sight-threatening and warrant ophthalmologic evaluation.
SKIN DISEASES AND SMALLPOX VACCINATION
Although smallpox vaccinations were discontinued several decades ago for the general population, they are still required for certain military personnel and first responders. In the absence of a bioterrorism attack and a real or potential exposure to smallpox, such vaccination is contraindicated in persons with a history of skin diseases such as AD, eczema, and psoriasis, who have a higher incidence of adverse events associated with smallpox vaccination. In the case of such exposure, the risk of smallpox infection outweighs the risk of adverse events from the vaccine (Chap. 261e).
72 |
Skin Manifestations of Internal Disease |
It is a generally accepted concept in medicine that the skin can develop signs of internal disease. Therefore, in textbooks of medicine, one finds a chapter describing in detail the major systemic disorders that can be identified by cutaneous signs. The underlying assumption of such a chapter is that the clinician has been able to identify the specific disorder in the patient and needs only to read about it in the textbook. In reality, concise differential diagnoses and the identification of these disorders are actually difficult for the nondermatologist because he or she is not well-versed in the recognition of cutaneous lesions or their spectrum of presentations. Therefore, this chapter covers this particular topic of cutaneous medicine not by simply focusing on individual diseases, but by describing the various presenting clinical signs and symptoms that point to specific disorders. Concise differential diagnoses will be generated in which the significant diseases will be distinguished from the more common cutaneous disorders that have minimal or no significance with regard to associated internal disease. The latter disorders are reviewed in table form and always need to be excluded when considering the former. For a detailed description of individual diseases, the reader should consult a dermatologic text.
PAPULOSQUAMOUS SKIN LESIONS
(Table 72-1) When an eruption is characterized by elevated lesions, either papules (<1 cm) or plaques (>1 cm), in association with scale, it is referred to as papulosquamous. The most common papulosquamous diseases—tinea, psoriasis, pityriasis rosea, and lichen planus—are primary cutaneous disorders (Chap. 71). When psoriatic lesions are accompanied by arthritis, the possibility of psoriatic arthritis or reactive arthritis (formerly known as Reiter’s syndrome) should be considered. A history of oral ulcers, conjunctivitis, uveitis, and/or urethritis points to the latter diagnosis. Lithium, beta blockers, HIV or streptococcal infections, and a rapid taper of systemic glucocorticoids are known to exacerbate psoriasis. Comorbidities in patients with psoriasis include cardiovascular disease and metabolic syndrome.
|
SELECTED CAUSES OF PAPULOSQUAMOUS SKIN LESIONS |
aDiscussed in detail in Chap. 71; cardiovascular disease and the metabolic syndrome are comorbidities in psoriasis; primarily in Europe, hepatitis C virus is associated with oral lichen planus. bAssociated with chronic sun exposure more often than exposure to arsenic; usually one or a few lesions. cSee also Red Lesions in “Papulonodular Skin Lesions.” dAlso cutaneous lesions of HTLV-1-associated adult T cell leukemia/lymphoma. eSee also Red-Brown Lesions in “Papulonodular Skin Lesions.”
Whenever the diagnosis of pityriasis rosea or lichen planus is made, it is important to review the patient’s medications because the eruption may resolve by simply discontinuing the offending agent. Pityriasis rosea–like drug eruptions are seen most commonly with beta blockers, angiotensin-converting enzyme (ACE) inhibitors, and metronidazole, whereas the drugs that can produce a lichenoid eruption include thiazides, antimalarials, quinidine, beta blockers, and ACE inhibitors. In some populations, there is a higher prevalence of hepatitis C viral infection in patients with lichen planus. Lichen planus–like lesions are also observed in chronic graft-versus-host disease.
In its early stages, the mycosis fungoides (MF) form of cutaneous T cell lymphoma (CTCL) may be confused with eczema or psoriasis, but it often fails to respond to the appropriate therapy for those inflammatory diseases. MF can develop within lesions of large-plaque parapsoriasis and is suggested by an increase in the thickness of the lesions. The diagnosis of MF is established by skin biopsy in which collections of atypical T lymphocytes are found in the epidermis and dermis. As the disease progresses, cutaneous tumors and lymph node involvement may appear.
In secondary syphilis, there are scattered red-brown papules with thin scale. The eruption often involves the palms and soles and can resemble pityriasis rosea. Associated findings are helpful in making the diagnosis and include annular plaques on the face, nonscarring alopecia, condyloma lata (broad-based and moist), and mucous patches as well as lymphadenopathy, malaise, fever, headache, and myalgias. The interval between the primary chancre and the secondary stage is usually 4–8 weeks, and spontaneous resolution without appropriate therapy occurs.
ERYTHRODERMA
(Table 72-2) Erythroderma is the term used when the majority of the skin surface is erythematous (red in color). There may be associated scale, erosions, or pustules as well as shedding of the hair and nails. Potential systemic manifestations include fever, chills, hypothermia, reactive lymphadenopathy, peripheral edema, hypoalbuminemia, and high-output cardiac failure. The major etiologies of erythroderma are (1) cutaneous diseases such as psoriasis and dermatitis (Table 72-3); (2) drugs; (3) systemic diseases, most commonly CTCL; and (4) idiopathic. In the first three groups, the location and description of the initial lesions, prior to the development of the erythroderma, aid in the diagnosis. For example, a history of red scaly plaques on the elbows and knees would point to psoriasis. It is also important to examine the skin carefully for a migration of the erythema and associated secondary changes such as pustules or erosions. Migratory waves of erythema studded with superficial pustules are seen in pustular psoriasis.
|
CAUSES OF ERYTHRODERMA |
a Discussed in detail in Chap. 71.
|
ERYTHRODERMA (PRIMARY CUTANEOUS DISORDERS) |
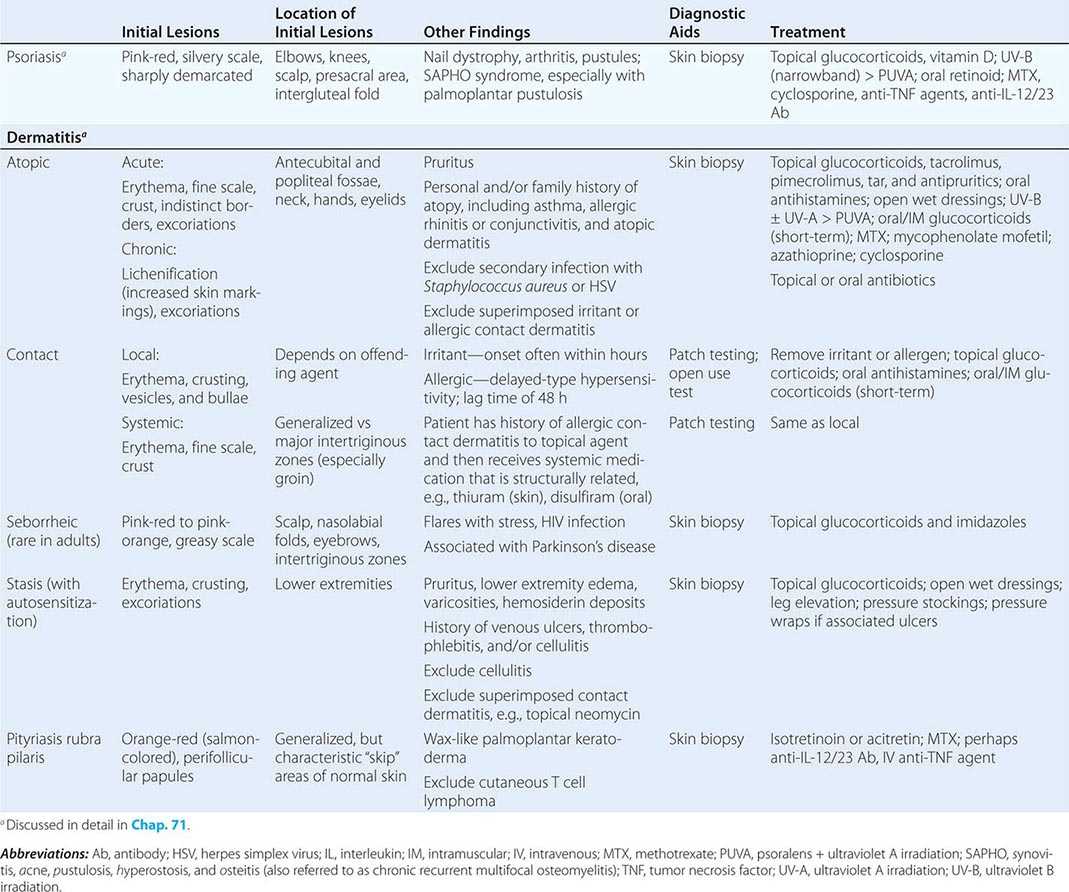
Drug-induced erythroderma (exfoliative dermatitis) may begin as an exanthematous (morbilliform) eruption (Chap. 74) or may arise as diffuse erythema. A number of drugs can produce an erythroderma, including penicillins, sulfonamides, carbamazepine, phenytoin, and allopurinol. Fever and peripheral eosinophilia often accompany the eruption, and there may also be facial swelling, hepatitis, myocarditis, thyroiditis, and allergic interstitial nephritis; this constellation is frequently referred to as drug reaction with eosinophilia and systemic symptoms (DRESS) or drug-induced hypersensitivity reaction (DIHS). In addition, these reactions, especially to aromatic anticonvulsants, can lead to a pseudolymphoma syndrome (with adenopathy and circulating atypical lymphocytes), while reactions to allopurinol may be accompanied by gastrointestinal bleeding.
The most common malignancy that is associated with erythroderma is CTCL; in some series, up to 25% of the cases of erythroderma were due to CTCL. The patient may progress from isolated plaques and tumors, but more commonly, the erythroderma is present throughout the course of the disease (Sézary syndrome). In the Sézary syndrome, there are circulating clonal atypical T lymphocytes, pruritus, and lymphadenopathy. In cases of erythroderma where there is no apparent cause (idiopathic), longitudinal evaluation is mandatory to monitor for the possible development of CTCL. There have been isolated case reports of erythroderma secondary to some solid tumors—lung, liver, prostate, thyroid, and colon—but it is primarily during a late stage of the disease.
ALOPECIA
(Table 72-4) The two major forms of alopecia are scarring and nonscarring. Scarring alopecia is associated with fibrosis, inflammation, and loss of hair follicles. A smooth scalp with a decreased number of follicular openings is usually observed clinically, but in some patients, the changes are seen only in biopsy specimens from affected areas. In nonscarring alopecia, the hair shafts are absent or miniaturized, but the hair follicles are preserved, explaining the reversible nature of nonscarring alopecia.
|
CAUSES OF ALOPECIA |
aMost patients with trichotillomania, pressure-induced alopecia, or early stages of traction alopecia. bWhile the majority of patients with discoid lesions have only cutaneous disease, these lesions do represent one of the 11 American College of Rheumatology criteria (1982) for systemic lupus erythematosus. cCan involve underlying muscles and osseous structures.
The most common causes of nonscarring alopecia include androgenetic alopecia, telogen effluvium, alopecia areata, tinea capitis, and the early phase of traumatic alopecia (Table 72-5). In women with androgenetic alopecia, an elevation in circulating levels of androgens may be seen as a result of ovarian or adrenal gland dysfunction or neoplasm. When there are signs of virilization, such as a deepened voice and enlarged clitoris, the possibility of an ovarian or adrenal gland tumor should be considered.
|
NONSCARRING ALOPECIA (PRIMARY CUTANEOUS DISORDERS) |
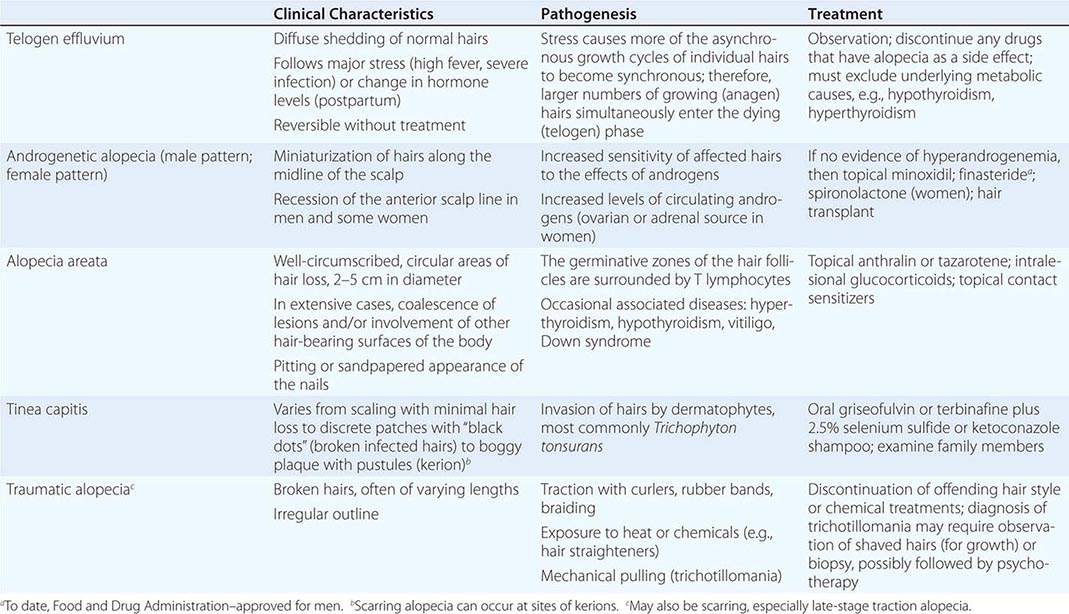
Exposure to various drugs can also cause diffuse hair loss, usually by inducing a telogen effluvium. An exception is the anagen effluvium observed with antimitotic agents such as daunorubicin. Alopecia is a side effect of the following drugs: warfarin, heparin, propylthiouracil, carbimazole, isotretinoin, acitretin, lithium, beta blockers, interferons, colchicine, and amphetamines. Fortunately, spontaneous regrowth usually follows discontinuation of the offending agent.
Less commonly, nonscarring alopecia is associated with lupus erythematosus and secondary syphilis. In systemic lupus there are two forms of alopecia—one is scarring secondary to discoid lesions (see below), and the other is nonscarring. The latter form coincides with flares of systemic disease and may involve the entire scalp or just the frontal scalp, with the appearance of multiple short hairs (“lupus hairs”) as a sign of initial regrowth. Scattered, poorly circumscribed patches of alopecia with a “moth-eaten” appearance are a manifestation of the secondary stage of syphilis. Diffuse thinning of the hair is also associated with hypothyroidism and hyperthyroidism (Table 72-4).
Scarring alopecia is more frequently the result of a primary cutaneous disorder such as lichen planus, folliculitis decalvans, chronic cutaneous (discoid) lupus, or linear scleroderma (morphea) than it is a sign of systemic disease. Although the scarring lesions of discoid lupus can be seen in patients with systemic lupus, in the majority of patients, the disease process is limited to the skin. Less common causes of scarring alopecia include sarcoidosis (see “Papulonodular Skin Lesions,” below) and cutaneous metastases.
In the early phases of discoid lupus, lichen planus, and folliculitis decalvans, there are circumscribed areas of alopecia. Fibrosis and subsequent loss of hair follicles are observed primarily in the center of these alopecic patches, whereas the inflammatory process is most prominent at the periphery. The areas of active inflammation in discoid lupus are erythematous with scale, whereas the areas of previous inflammation are often hypopigmented with a rim of hyperpigmentation. In lichen planus, perifollicular macules at the periphery are usually violet-colored. A complete examination of the skin and oral mucosa combined with a biopsy and direct immunofluorescence microscopy of inflamed skin will aid in distinguishing these two entities. The peripheral active lesions in folliculitis decalvans are follicular pustules; these patients can develop a reactive arthritis.
FIGURATE SKIN LESIONS
(Table 72-6) In figurate eruptions, the lesions form rings and arcs that are usually erythematous but can be skin-colored to brown. Most commonly, they are due to primary cutaneous diseases such as tinea, urticaria, granuloma annulare, and erythema annulare centrifugum (Chaps. 71 and 73). An underlying systemic illness is found in a second, less common group of migratory annular erythemas. It includes erythema migrans, erythema gyratum repens, erythema marginatum, and necrolytic migratory erythema.
|
CAUSES OF FIGURATE SKIN LESIONS |
aMigratory erythema with erosions; favors lower extremities and girdle area.
Abbreviation: CDC, Centers for Disease Control and Prevention.
In erythema gyratum repens, one sees numerous mobile concentric arcs and wavefronts that resemble the grain in wood. A search for an underlying malignancy is mandatory in a patient with this eruption. Erythema migrans is the cutaneous manifestation of Lyme disease, which is caused by the spirochete Borrelia burgdorferi. In the initial stage (3–30 days after tick bite), a single annular lesion is usually seen, which can expand to ≥10 cm in diameter. Within several days, up to half of the patients develop multiple smaller erythematous lesions at sites distant from the bite. Associated symptoms include fever, headache, photophobia, myalgias, arthralgias, and malar rash. Erythema marginatum is seen in patients with rheumatic fever, primarily on the trunk. Lesions are pink-red in color, flat to minimally elevated, and transient.
There are additional cutaneous diseases that present as annular eruptions but lack an obvious migratory component. Examples include CTCL, subacute cutaneous lupus, secondary syphilis, and sarcoidosis (see “Papulonodular Skin Lesions,” below).
ACNE
(Table 72-7) In addition to acne vulgaris and acne rosacea, the two major forms of acne (Chap. 71), there are drugs and systemic diseases that can lead to acneiform eruptions.
|
CAUSES OF ACNEIFORM ERUPTIONS |
aEGFR, epidermal growth factor receptor.
Patients with the carcinoid syndrome have episodes of flushing of the head, neck, and sometimes the trunk. Resultant skin changes of the face, in particular telangiectasias, may mimic the clinical appearance of acne rosacea.
PUSTULAR LESIONS
Acneiform eruptions (see “Acne,” above) and folliculitis represent the most common pustular dermatoses. An important consideration in the evaluation of follicular pustules is a determination of the associated pathogen, e.g., normal flora, Staphylococcus aureus, Pseudomonas aeruginosa (“hot tub” folliculitis), Malassezia, dermatophytes (Majocchi’s granuloma), and Demodex spp. Noninfectious forms of folliculitis include HIV- or immunosuppression-associated eosinophilic folliculitis and folliculitis secondary to drugs such as glucocorticoids, lithium, and epidermal growth factor receptor (EGFR) inhibitors. Administration of high-dose systemic glucocorticoids can result in a widespread eruption of follicular pustules on the trunk, characterized by lesions in the same stage of development. With regard to underlying systemic diseases, nonfollicular-based pustules are a characteristic component of pustular psoriasis (sterile) and can be seen in septic emboli of bacterial or fungal origin (see “Purpura,” below). In patients with acute generalized exanthematous pustulosis (AGEP) due primarily to medications (e.g., cephalosporins), there are large areas of erythema studded with multiple sterile pustules in addition to neutrophilia.
TELANGIECTASIAS
(Table 72-8) To distinguish the various types of telangiectasias, it is important to examine the shape and configuration of the dilated blood vessels. Linear telangiectasias are seen on the face of patients with actinically damaged skin and acne rosacea, and they are found on the legs of patients with venous hypertension and generalized essential telangiectasia. Patients with an unusual form of mastocytosis (telangiectasia macularis eruptiva perstans) and the carcinoid syndrome (see “Acne,” above) also have linear telangiectasias. Lastly, linear telangiectasias are found in areas of cutaneous inflammation. For example, lesions of discoid lupus frequently have telangiectasias within them.
|
CAUSES OF TELANGIECTASIAS |
aBecoming less common.
Poikiloderma is a term used to describe a patch of skin with: (1) reticulated hypo- and hyperpigmentation, (2) wrinkling secondary to epidermal atrophy, and (3) telangiectasias. Poikiloderma does not imply a single disease entity—although it is becoming less common, it is seen in skin damaged by ionizing radiation as well as in patients with autoimmune connective tissue diseases, primarily dermatomyositis (DM), and rare genodermatoses (e.g., Kindler syndrome).
In systemic sclerosis (scleroderma) the dilated blood vessels have a unique configuration and are known as mat telangiectasias. The lesions are broad macules that usually measure 2–7 mm in diameter but occasionally are larger. Mats have a polygonal or oval shape, and their erythematous color may appear uniform, but, upon closer inspection, the erythema is the result of delicate telangiectasias. The most common locations for mat telangiectasias are the face, oral mucosa, and hands—peripheral sites that are prone to intermittent ischemia. The limited form of systemic sclerosis, often referred to as the CREST (calcinosis cutis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) variant (Chap. 382), is associated with a chronic course and anticentromere antibodies. Mat telangiectasias are an important clue to the diagnosis of this variant as well as the diffuse form of systemic sclerosis because they may be the only cutaneous finding.
Periungual telangiectasias are pathognomonic signs of the three major autoimmune connective tissue diseases: lupus erythematosus, systemic sclerosis, and DM. They are easily visualized by the naked eye and occur in at least two-thirds of these patients. In both DM and lupus, there is associated nailfold erythema, and in DM, the erythema is often accompanied by “ragged” cuticles and fingertip tenderness. Under 10× magnification, the blood vessels in the nailfolds of lupus patients are tortuous and resemble “glomeruli,” whereas in systemic sclerosis and DM, there is a loss of capillary loops and those that remain are markedly dilated.
In hereditary hemorrhagic telangiectasia (Osler-Rendu-Weber disease), the lesions usually appear during adolescence (mucosal) and adulthood (cutaneous) and are most commonly seen on the mucous membranes (nasal, orolabial), face, and distal extremities, including under the nails. They represent arteriovenous (AV) malformations of the dermal microvasculature, are dark red in color, and are usually slightly elevated. When the skin is stretched over an individual lesion, an eccentric punctum with radiating legs is seen. Although the degree of systemic involvement varies in this autosomal dominant disease (due primarily to mutations in either the endoglin or activin receptor–like kinase gene), the major symptoms are recurrent epistaxis and gastrointestinal bleeding. The fact that these mucosal telangiectasias are actually AV communications helps to explain their tendency to bleed.




