Sickle cell syndromes
The sickle cell syndromes are a group of haemoglobinopathies which primarily affect the Afro-Caribbean population. The common feature of these diseases is inheritance of an abnormal haemoglobin β-chain gene – the gene is designated βS. Inheritance of two βS genes leads to a serious disorder termed sickle cell anaemia. A similar syndrome can result from inheritance of the βS gene with another abnormal β gene such as the haemoglobin C gene or β-thalassaemia gene. Inheritance of the βS gene with a normal β-chain gene (βA) causes the innocuous sickle cell trait (Fig 17.1).
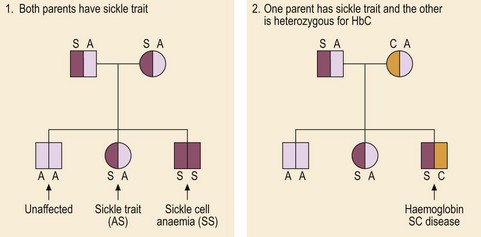
Fig 17.1 Inheritance of sickle cell syndromes.
Two pedigrees showing inheritance of sickle cell syndromes. In the first family one child is unaffected, one has sickle cell trait and one has sickle cell anaemia. In the second family one child has inherited the abnormal sickle gene and the HbC gene; this double heterozygosity leads to haemoglobin SC disease.
Pathophysiology
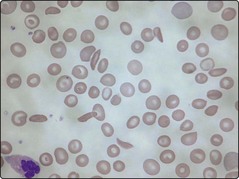
The abnormal βS gene has a high incidence in tropical and subtropical regions as the abnormal haemoglobin produced (HbS) gives some protection against falciparum malaria. HbS differs from normal haemoglobin (HbA) in that glutamic acid has been replaced by valine at the sixth amino acid from the N-terminus of the β-globin chain. The clinical features of sickle cell anaemia arise from the propensity of red cells containing haemoglobin S to undergo ‘sickling’. In the deoxygenated state HbS undergoes a conformational change leading to the creation of haemoglobin tetramers which aggregate to produce large polymers. The red cell loses its normal deformability and becomes characteristically sickle-shaped (Fig 17.2). Damage to the membrane leads to increased rigidity and the ultimate sequestration of the red cell in the reticuloendothelial system causing haemolytic anaemia. The inflexible sickle cells also become lodged in the microcirculation causing stasis and obstruction.
Clinical syndromes
This classic form of sickle cell syndrome is enormously variable in severity.
Vascular-occlusive crises
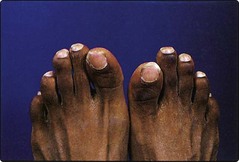
Acute, episodic, painful crises are a potentially disabling feature of sickle cell anaemia. They may be triggered by infection or cold. Patients complain of musculoskeletal pain which may be severe and require hospital admission. Hips, shoulders and vertebrae are most affected. Attacks are generally self-limiting but infarction of bone can occur and must be distinguished from salmonella osteomyelitis. Avascular necrosis of the femoral head is a crippling complication. Other organs are vulnerable to infarction; most serious is neurological damage which may manifest as seizures, transient ischaemic attacks (TIAs) and strokes. Vaso-occlusion in infancy is responsible for the ‘hand–foot syndrome’, a type of dactylitis damaging the small bones of hands and feet (Fig 17.3).




Diagnosis



Sickle cell trait (HbAS)
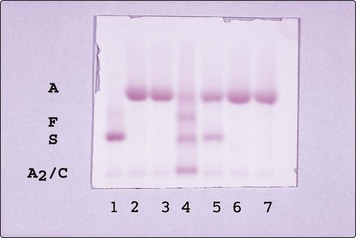
Sickle cell trait normally causes no clinical problems as there is enough HbA in red cells (approximately 60%) to prevent sickling. However, haematuria occasionally occurs as a result of renal papillary necrosis and additional care is required during pregnancy and anaesthesia. Diagnosis is by a sickling test and Hb electrophoresis (see Fig 17.4). Life expectancy is normal although there may be a slightly increased risk of sudden death during intense exercise in young adults.
Screening strategies
Sickle cell syndromes
 The sickle cell syndromes are a group of haemoglobinopathies which primarily affect people of African origin.
The sickle cell syndromes are a group of haemoglobinopathies which primarily affect people of African origin.
 Inheritance of two βS genes leads to the serious clinical disorder sickle cell anaemia (HbSS).
Inheritance of two βS genes leads to the serious clinical disorder sickle cell anaemia (HbSS).


 Sickle cell trait (HbAS) is an innocuous clinical disorder but genetic counselling is often needed.
Sickle cell trait (HbAS) is an innocuous clinical disorder but genetic counselling is often needed.
