[level-membership-for-critical-care-medicine-category]
6 Shock, Cardiac Arrest, and Resuscitation
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Shock
Shock pearls
• Shock is a common cause of morbidity and mortality in pediatric patients with acute illness and injury.
• Early recognition of shock is essential for survival. Signs of shock in children may be more subtle than those in the adult and hypotension may not develop until late in the clinical course, when cardiovascular collapse is imminent.
• Treatment focuses on restoration of adequate oxygen delivery. Fluid therapy, inotropes, vasodilators, vasopressors, hydrocortisone, thyroid replacement, and extracardiac support devices may be required to accomplish this goal.
• Further shock treatment depends on the cause of the cardiovascular dysfunction and complications caused by the period of inadequate oxygen delivery.
Introduction
Shock is commonly defined as a clinical state characterized by an inadequate delivery of oxygen and metabolic substrates to meet the metabolic demands of the cells and tissues of the body.53,245,246 Inadequate delivery of oxygen results in cellular hypoxia, anaerobic metabolism, lactic acidosis, activation of the host inflammatory response, and eventual vital organ dysfunction.
Cardiovascular physiology and shock pathophysiology
Cellular Basis of Shock
Adenosine triphosphate (ATP) is the energy currency of the cell. Shock is a state of acute energy failure in which there is insufficient ATP production to support systemic cellular function.53 During stress and periods of increased energy demand, glucose is produced from glycogenolysis and gluconeogenesis. Fat metabolism is the secondary source of energy in this state. Long-chain fatty acids are oxidized, and carnitine is used to shuttle acetyl coenzyme A (CoA) into mitochondria. Protein catabolism can also contribute acetyl CoA to the Krebs cycle for energy production. However, this method of energy production is inefficient; aerobic metabolism provides 20-fold the energy produced by anaerobic metabolism. Glucose is oxidized to pyruvate via glycolysis (also called the Embden-Meyerhof pathway), generating only two molecules of ATP in the process.
In practical terms, using this operational definition, a state of shock may result from inadequate substrate delivery (glycopenia) or mitochondrial dysfunction (cellular dysoxia).53 Oxygen delivery to the cells and tissues depends primarily on three factors: hemoglobin (Hb) concentration, cardiac output (CO), and the relative proportion of oxygenated hemoglobin (i.e., percent saturation [SaO2]). Oxygen is transported in the blood combined with hemoglobin, although a relatively small amount is freely dissolved in the plasma fraction of the blood. When fully saturated at normal body temperature, each gram of hemoglobin can carry about 1.34 to 1.36 mL of oxygen. The normal arterial oxygen content is calculated as follows:
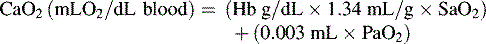
Oxygen delivery (DO2) is a product of arterial oxygen content and CO:
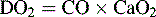
Adequate glucose delivery depends on the presence of adequate blood glucose concentration, normal blood flow (or CO), and an adequate concentration of insulin for cells with insulin-responsive glucose transporters (e.g., cardiomyocytes). Glycopenic shock can be caused by hypoglycemia and by extreme insulin resistance.53 Finally, even when oxygen delivery and glucose delivery are adequate, shock may occur as a result of mitochondrial dysfunction. For example, cyanide poisons the oxidative phosphorylation chain, preventing production of ATP. Cellular dysoxia (also known as cytopathic hypoxia) may theoretically occur from one or a combination of several mechanisms, including diminished delivery of a key substrate (e.g., pyruvate) to the Krebs cycle of the electron transport chain or uncoupling of oxidative phosphorylation.
Determinants of Oxygen Delivery
Normal CO
As defined previously, oxygen delivery is the product of CO and arterial oxygen content (CaO2). CO is the product of heart rate (HR) and stroke volume (SV; Fig. 6-1). SV, in turn, is dependent on preload, afterload, and contractility. Furthermore, blood pressure (BP) is determined by the product of CO and systemic vascular resistance (SVR).
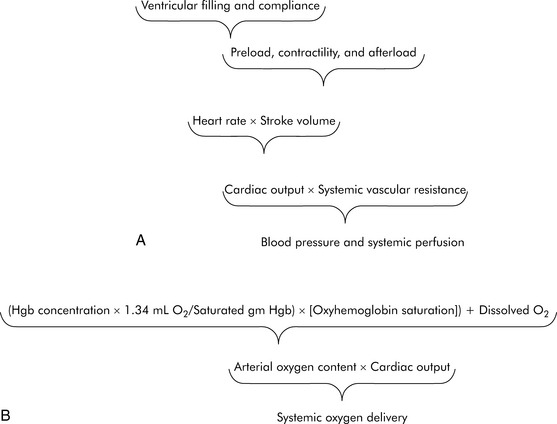
Myocardial performance can be affected by changes in oxygenation, perfusion, serum ionized calcium concentration, acid-base, and electrolyte balance, sympathetic or vagal stimulation, and drugs.244 These factors can affect CO by altering either HR or SV.
Factors Influencing SV
Ventricular Preload
Preload is the amount of myocardial fiber stretch that is present before contraction. The significance of ventricular preload was first appreciated by Howell (in 1894), Frank (in 1894), and Starling (in 1914)242 in a series of experiments performed on isolated normal myocardial muscle preparations. Howell, Frank, and Starling observed that normal myocardium generates greater tension during contraction if it is stretched before contraction. This increase in the force of contraction occurs as a result of optimization of overlap between actin and myosin filaments in the sarcomere. These observations became known as the Frank-Starling law of the heart, which states that an increase in ventricular work, systolic tension, and SV results from an increase in presystolic stretch (preload). The graphic representation of the relationship between ventricular end-diastolic myocardial fiber length (usually approximated by ventricular end-diastolic pressure [VEDP]) and SV is the Frank-Starling curve, which is a ventricular (myocardial) function curve (Fig. 6-2).
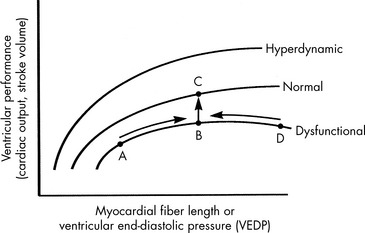
To a point, as VEDP is increased, the force of contraction and myocardial fiber shortening should increase, and SV should rise.98 If, however, the ventricle is filled beyond a critical point, overlap of actin and myosin filaments is no longer optimal; ventricular dilation can result, and stroke volume decreases.47 Extremely high VEDPs (higher than approximately 25 cm H2O pressure when capillary permeability is normal, and lower pressures when capillary permeability is increased) result in pulmonary and systemic edema, and high pressures will compromise coronary and subendocardial blood flow.
A family of ventricular function curves can be constructed to illustrate the response of normal, depressed, or enhanced myocardial response to increased VEDP (see Fig. 6-2). If the patient demonstrates poor myocardial function, the ventricular function curve will be relatively flat, and a high VEDP will be required to produce even a modest improvement in myocardial function. If myocardial function is normal, a small increase in VEDP can produce a significant rise in SV or CO. If ventricular function is hyperdynamic, even nominal increases in VEDP will produce significant increases in SV or CO.
A goal of the treatment of any patient with cardiovascular dysfunction is to maximize SV and CO while minimizing adverse effects of fluid administration, such as pulmonary edema. An increase in SV and CO can be achieved by moving the patient to the highest point of an individual ventricular function curve (see Fig. 6-2) through judicious fluid administration. Improvement in SV and CO also can be achieved by altering the ventricular compliance, using vasodilator therapy. Further increase in SV and CO also can be achieved through improvement in cardiac contractility; this raises the ventricular function curve (see Fig. 6-2). Such an improvement can be attained by eliminating factors that normally depress myocardial function or by administering inotropic agents or vasodilators (discussed under Afterload).
RVEDP and CVP often can be estimated with careful clinical assessment of the level of hydration, liver size, palpation of the infant’s fontanelle, determination of presence (or absence) of systemic edema, and evaluation of the cardiac size on chest radiograph.275 Dry mucous membranes, a sunken fontanelle, and the absence of hepatomegaly are findings consistent with a normal or low central venous pressure. Hepatomegaly and periorbital edema usually are present once the CVP is elevated significantly. Systemic edema also may be noted despite a normal or low CVP if capillary leak or hypoalbuminemia is present. A high RVEDP and heart failure is often associated with cardiac enlargement on chest radiograph.
Left VEDP (LVEDP) is equal to left atrial pressure unless mitral valve disease is present. Reliable estimation of LVEDP is not possible through clinical assessment alone.275 Although the presence of pulmonary edema frequently is assumed to indicate the presence of a high LVEDP (exceeding 20 to 25 mm Hg), pulmonary edema may be observed at any (even a low) LVEDP if capillary leak is present.
Ventricular Compliance
Ventricular compliance refers to the distensibility of the ventricle. It is defined as the change in ventricular volume (in milliliters) for a given change in pressure (in millimeters of mercury), or ΔV/ΔP, and can be depicted graphically by a ventricular compliance curve (Fig. 6-3). The opposite of compliance is stiffness (ΔP/ΔV).
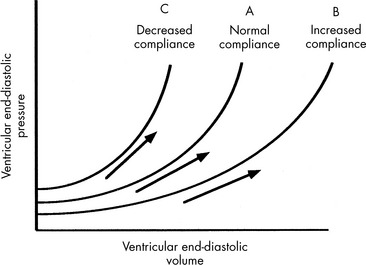
If the ventricle is extremely compliant, a large volume of fluid may be administered without producing a significant increase in VEDP (see Fig. 6-3, curve B). If the ventricle is dysfunctional (as occurs with restrictive cardiomyopathy) or hypertrophied, ventricular compliance usually is reduced (see Fig. 6-3, curve C). In this case, even a small volume of administered intravenous fluids will produce a significant rise in VEDP.268 The more dysfunctional and noncompliant the ventricle, the higher the resting VEDP and the VEDP needed to optimize SV and ventricular performance (see Fig. 6-2).
Ventricular compliance is not constant over all ranges of VEDP. Any ventricle is maximally compliant at low filling pressures. As the ventricle is filled, compliance is reduced because ventricular stretch may be maximal.55,169 Rapid volume infusion tends to raise VEDP more rapidly than gradual volume infusion. Compliant ventricles usually demonstrate a substantial improvement in SV when an intravenous fluid bolus is administered.
Vasodilator therapy will improve ventricular compliance. When these drugs are administered the compliance curve is altered, so a greater end-diastolic volume may be present without a substantial increase in VEDP (see Fig. 6-3). SV may then be increased without a rise in VEDP.
Compliance also is affected by ventricular size, pericardial space, and HR.55 Infants have small and relatively noncompliant ventricles, so the infant’s VEDP may rise sharply with minimal fluid volume administration. If the same volume (on a per-kilogram basis) is administered to an older child, a smaller change in VEDP will result because the ventricles are larger and more compliant in older children.
Ventricular Contractility
Although contractility can be measured in the laboratory, it is not easily isolated and measured in the clinical setting. The most common method of evaluating contractility at the bedside is echocardiographic evaluation of fiber-shortening times and measurement of the shortening fraction of left ventricular diameter. Shortening fraction is calculated by determining the difference between the end-diastolic and end-systolic dimensions. Normal shortening fraction is approximately 28% to 44%.206
If a thermodilution CO pulmonary artery catheter is in place, or if reliable Doppler CO estimations can be obtained, the nurse can create a ventricular function curve (see Fig. 6-2). If CO improves with no change in VEDP or HR, ventricular contractility or compliance has probably improved.
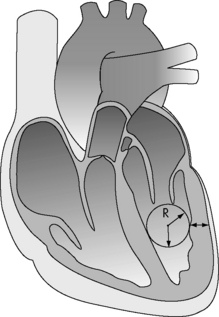
Afterload
The major determinants of ventricular afterload or wall stress are: ventricular lumen radius, ventricular wall thickness (note that hypertrophy decreases afterload), and the ventricular intracavitary ejection pressure (Fig. 6-4). In the absence of left ventricular outflow tract obstruction (e.g., aortic stenosis), left ventricular ejection pressure will equal aortic and systemic arterial pressure. In the absence of right ventricular outflow tract obstruction (e.g., pulmonary stenosis), right ventricular ejection pressure will equal pulmonary arterial pressure. Systemic and pulmonary artery pressures in turn are determined by blood flow and resistance. Therefore in the absence of ventricular outflow tract obstruction or significant alterations in ventricular size or wall thickness, ventricular afterload is related primarily to the impedance provided by the pulmonary and systemic arterial circulations.
Oxygen Delivery
The ultimate function of the heart and lungs is to deliver oxygenated blood to the tissues. Systemic DO2 is the volume of oxygen (in milliliters) delivered to the tissues per minute. DO2(I) is the volume of oxygen delivered to the tissues per minute, indexed to body surface area (BSA), so that the units are milliliters per minute per square meter. DO2 is the product of arterial oxygen content (the amount of oxygen in arterial blood in milliliters per deciliter) the CO (in liters per minute), and a factor of 10. DO2(I) is the product of arterial oxygen content (in milliliters per deciliter), the cardiac index (in liters per minute per square meter), and a factor of 10 (see Fig. 6-1).
Under resting conditions with normal distribution of CO, oxygen delivery is more than adequate to meet the total oxygen requirements the tissues need to maintain aerobic metabolism, which is referred to as oxygen consumption (VO2). Excess oxygen delivery or oxygen reserve serves as a buffer, so a modest reduction in oxygen delivery will be more than adequately compensated by increased extraction of the delivered oxygen, without any significant reduction in oxygen consumption. During stress or vigorous exercise, oxygen consumption markedly increases, as does oxygen delivery. Therefore, under most conditions, the metabolic demands of the cells and tissues of the body dictate the level of oxygen delivery. However, little oxygen is stored in the cells and tissues of the body; therefore as oxygen delivery falls with critical illness, oxygen extraction must necessarily increase to meet metabolic demands, and oxygen consumption remains relatively constant (i.e., it is delivery independent; Fig. 6-5). However, there is a critical level of oxygen delivery at which the body’s compensatory mechanisms are no longer able to meet metabolic needs (i.e., the point at which oxygen extraction is maximal). Once oxygen delivery falls below this level, oxygen consumption must also fall and is said to become delivery dependent (see Fig. 6-5, Normal critical delivery threshold).
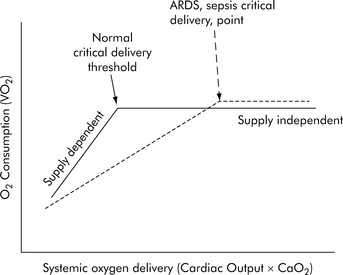
If either arterial oxygen content or CO falls without a commensurate and compensatory increase in the other component, oxygen delivery will fall (Fig. 6-6, A–H). For example, if arterial oxygen content falls (e.g., caused by a fall in hemoglobin concentration or its saturation), oxygen delivery can be maintained by a commensurate rise in CO. If CO falls, however, oxygen delivery will fall in direct correlation, because there is no way for arterial oxygen content to increase when CO falls.

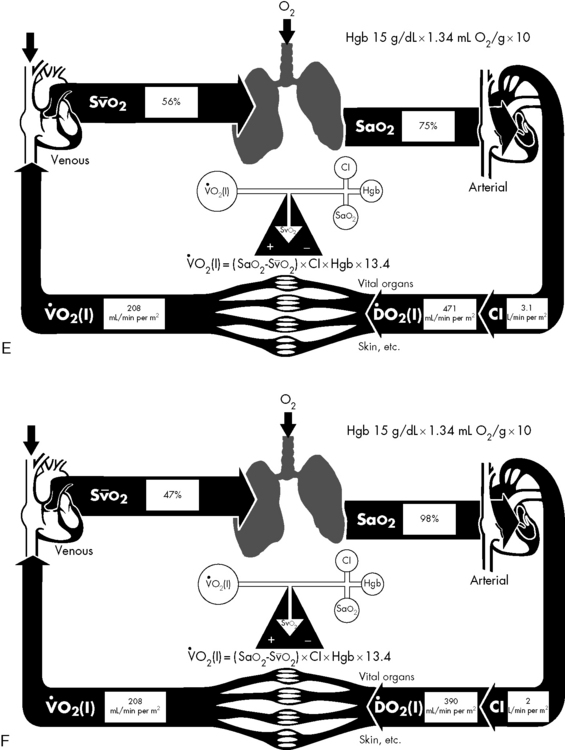
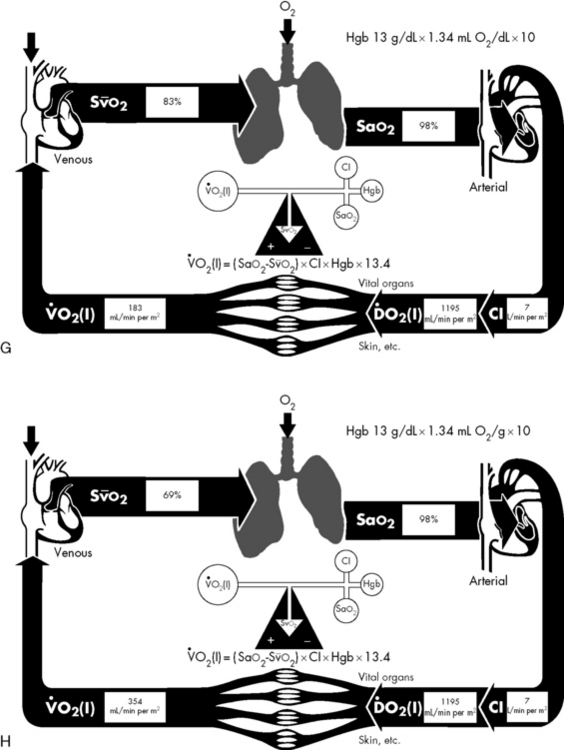
Oxygen Content
The optimal hemoglobin threshold for erythrocyte (red blood cell) transfusion in critically ill children is unknown.160 Whereas transfusion is indicated in conditions of hemodynamic instability and anemia (hemoglobin <10 g/dL), once stability is achieved the threshold hemoglobin level for transfusion may need to be lowered. A recent multicenter trial demonstrated that in stable, critically ill children a hemoglobin threshold of 7 g/dL for red blood cell transfusion decreased transfusion requirements without increasing adverse outcome.160
Cardiac Output
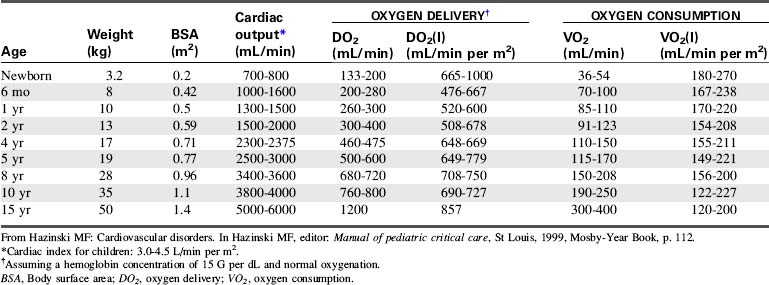
CO is the volume of blood ejected by the heart in 1 minute. CO is the product of HR and SV. CO may be recorded in liters per minute or milliliters per minute, although in children it is often normalized to BSA and recorded as the cardiac index (in milliliters per minute per square meter BSA). Normal cardiac index averages 3 to 4.5 mL/min per m2 BSA (Table 6-1).
Clinical Assessment of CO
Compensatory vasoconstriction can initially maintain BP (BP = CO × SVR). In fact, infants and children have high SVR and vasoactive capacity such that hypotension is a late sign of shock.53,245 This survival mechanism is designed to counterbalance the limited cardiac reserve in children.
Calculation and Estimation of CO
CO can be calculated using a thermodilution catheter placed in the pulmonary artery. Injection of an iced quantity of fluid into the right atrium will produce a temperature change in the pulmonary artery that is inversely related to the CO. Such thermodilution CO calculations can be performed using a pulmonary artery catheter with thermistor or a separate thermistor placed into the pulmonary artery at the time of cardiac surgery. Additional information about thermodilution CO calculation can be found in Chapter 21.
The CO can be continuously evaluated if the pulmonary artery catheter contains an oximeter for continuous evaluation of pulmonary artery (mixed venous) oxyhemoglobin saturation. Pulmonary artery catheterization is not commonly performed in pediatric critical care, although it may be indicated in certain situations that complicate shock resuscitation.276
Pulse contour intermittent and continuous CO (PiCCO®) monitoring is possible using a femoral or axillary arterial catheter and a central venous catheter. Algorithms use pulse contour analysis combined with intermittent thermodilution calculations. The arterial waveform contours are analyzed to assess SV and thereby cardiac index (CI). In a pediatric validation study, the PiCCO system provided continuous calculation of CI without the need to perform thermodilution injections with every calculation.90
Esophageal Doppler studies of blood flow velocity in the descending aorta have been used in adults for many years, and pediatric probes are now available. In the past, the higher HRs in children compromised reliability of this technique of CO evaluation. Fairly reliable CO calculations have recently been reported in children,270 although this method of CO evaluation is very operator dependent.3
CO and blood volume can be calculated from a form of ultrasound dilution. This technology uses a computer and arterial and venous sensors attached to a specialized extracorporeal tubing loop that is placed between an indwelling central venous and arterial catheter. A small bolus (0.5 to 1.0 mL/kg) of isotonic saline is injected into the central venous catheter to create a dilution curve. The saline dilutes the total blood protein concentration and alters blood velocity detected by the arterial sensor in the extracorporeal loop. The change in blood velocity over time can be graphed, and results in a dilution curve that is similar to that created during thermodilution CO calculations. In addition to calculating the CO and index, the ultrasound dilution computer calculates SV index, active circulating volume, SVR, and ejection fraction. This technology has been validated in neonates and children.157
Noninvasive measurement of CO in children is novel and evolving.128 Doppler ultrasonography is a simple, noninvasive method of assessing blood flow and is an accepted noninvasive method of deriving CO.118 Data obtained with Doppler ultrasonography correlate well with accepted standard hemodynamic methods in animal studies, adults, and neonates; formal validation is underway in children.37,65,128
Because adequate organ perfusion is the goal of supportive and therapeutic critical care, monitors that directly assess organ oxygenation offer the best possibility of improved recognition and treatment of circulatory abnormalities to reduce multiorgan dysfunction and related morbidity and mortality.260 Some of these monitors are presented in the next several sections
Relationship of Mixed Venous Oxygen Saturation to CO
A true mixed venous oxygen saturation is evaluated from blood in the pulmonary artery, because it includes a true mixed sample of systemic venous blood from the superior vena cava, inferior vena cava, and coronary circulations. The mixed venous oxygen saturation can be continuously monitored through the use of a pulmonary artery catheter that contains a pulmonary artery oximeter. If hemoglobin concentration and saturation and oxygen consumption or extraction are stable (these assumptions often cannot be made about the critically ill patient), the mixed venous oxygen saturation will vary directly with the CO (see Fig. 6-6).
When oxygen delivery falls, tissues compensate by increasing the amount of oxygen extraction from circulating blood.196 Mixed venous oxygen saturation is thus used as a measure of the balance between oxygen delivery and oxygen demand, and it will fall as the result of either decreased oxygen delivery (i.e., decreased arterial oxygen content; decreased CO, index, or both) or increased oxygen extraction (from increased demand, decreased delivery, or both). The normal mixed venous oxygen saturation is greater than 70%.196 Mixed venous oxygen saturation levels between 50% and 75% reflect compensatory increased extraction in cases of increased oxygen demand or decreased oxygen delivery, whereas levels between 30% and 50% reflect the beginning of lactic acidosis indicating exhaustion of compensatory extraction. A further decline to levels between 25% and 30% is indicative of severe lactic acidosis, and levels less than 25% probably reflect cellular death.196
The mixed venous oxygen saturation has been shown to be an effective indicator of the balance between oxygen supply and demand, and it is a useful guide for management in adults. However, its use is limited in pediatrics by the need for a pulmonary artery or central venous catheter for intermittent or continuous measurement; this is often not feasible, especially early in resuscitation.183
Use of Central Venous Oxygen Saturation Monitoring
The desire for easier venous oxygen saturation measurement has led to an assessment of central venous oxygen saturation (ScvO2). Unlike the pulmonary artery catheter samples that are composed of blood from all parts of the systemic venous circulation, including blood from the coronary sinus, ScvO2 is obtained from the superior vena cava, so it assesses only the oxygen delivery-oxygen consumption balance in the upper body.196 Many studies have examined how the two are related. Oxygen saturation of superior vena cava blood is approximately 70%, which is slightly lower than the saturation of blood in the inferior vena cava (75%). Coronary sinus blood, with an oxygen saturation of approximately 30% to 40%, is then added to the combined superior vena cava and inferior vena cava blood, so that a true mixed venous sample in the pulmonary artery (after mixing is complete) averages approximately 70% to 75% and corresponds to a mixed venous oxygen tension of 38 to 40 mm Hg. Thus the ScvO2 is normally approximately 2% to 3% higher than mixed venous oxygen saturation in healthy individuals.183 In shock states, however, the difference can range from 5% to 18%.37,183 Some studies have shown that although the two are not interchangeable, there is a good correlation in the trending of one to the other, and the ScvO2 correlates well with changes in systemic perfusion.81,132,183
Limitations of continuous ScvO2 monitoring are significant. Because sympathetic tone raises vascular resistance in splanchnic-mesenteric beds as CO falls, in shock states the effects of desaturated blood from those regions on ScvO2 may be blunted. As an extreme example, the renal blood flow may fall to 20% of its normal value because of intense renal vasoconstriction, with renal vein saturation falling from 85% to 25% while the ScvO2 remains above 60%.132
Near-Infrared Spectroscopy (NIRS)
NIRS is relatively new technology that is used to access regional tissue oxygenation as an indicator of tissue perfusion. Near-infrared light (700 to 1000 nm bandwidth) is transmitted through muscle, bone, tissue, and skin, and reflected light is then read by a sensor.183 The technology is similar to that used for pulse oximetry and continuous monitoring of ScvO2. This technology uses the difference in light absorption between deoxygenated hemoglobin versus oxygenated hemoglobin to calculate a percent saturation level. Because most blood is not in arteries but in capillaries and veins, the greatest quantitative contribution to the calculated light absorption of hemoglobin is from venous and capillary blood. Pulse oximeters attempt to subtract the nonpulsatile component of the light signal (i.e., contributed from veins and capillaries) to examine the absorption spectrum of arterial blood, whereas NIRS technology focuses on the total light signal. An NIRS device approved by the U.S. Food and Drug Administration (INVOS; Somanetics, Troy, MI) uses a dual-detector system to subtract a shallow light path from a deep light path, theoretically allowing the derivation of the average oxyhemoglobin saturation in a volume of tissue approximately 2.5 to 3.0 cm deep under the skin and the sensor. The device displays an approximation of venous-weighted hemoglobin saturation in tissue deep below the sensor. The parameter is displayed as a relative number from 0% to 100% using an algorithm calibrated from in vivo and in vitro cerebral models, and is termed regional oxygen saturation (rSO2).132
Because venous-weighted capillary blood represents the limiting oxygen tension for diffusion, NIRS provides a window to evaluate the oxygen economy in the monitored tissue bed or regional tissue oxyhemoglobin saturation.91,132 The use of NIRS technology to monitor oxygenation in the brain, muscle, liver, and kidney has been extensively described.91,132,133,228 Changes in tissue oxygen tension monitored by NIRS are sensitive indicators of perfusion-metabolism coupling, and regional NIRS monitoring can guide resuscitation from shock.66,132
The indication approved by the U.S. Food and Drug Administration for the INVOS device is for trend monitoring of regional saturation in the cerebral circulation, which has been widely modeled as a compartment with 75% venous blood, thus allowing validation of the device in an accepted clinical model. In animal models, brain rSO2 less than 40% is associated with intracellular anaerobic metabolism and depletion of high-energy phosphates.158 Clinical data in children and adults support the hypothesis that cerebral rSO2 less than 40% to 50%, or a change in baseline of more than 20%, is associated with hypoxic-ischemic neural injury.132,133,228
Blood Pressure
Arterial BP may be the most widely used and most widely misinterpreted parameter related to CO. Arterial BP is generated by the kinetic energy imparted by the heart and the interaction of blood flow, viscosity, systemic vascular tone, and downstream (venous) pressure. The major determinants of mean arterial BP are the SVR and CO. Peripheral and central arterial pressure differences result from differential impedances in the vascular system and are increased with nonuniform vasoconstriction. Noninvasive BP measurement by auscultation using a cuff and sphygmomanometer (mercury manometer) and noninvasive oscillometric BP measurements correlate well with indwelling intraarterial measurements under conditions of normal vascular resistance and blood flow, but “cuff” and invasive pressures frequently diverge under conditions of low and high SVR.132 Automated determination by cuff oscillometry shows good reproducibility and allows for determination of the mean pressure.
Although BP is rapidly and routinely measured, its relationship to CO or systemic perfusion is confounded by simultaneous changes in SVR. SVR tends to change in the opposite direction from CO; the sympathetic nervous system is activated by a falling BP, and sympathetic outflow is reduced and the parasympathetic system is activated with a rising BP. These responses are designed to minimize the variability in BP, making this parameter a late indicator of failing circulation. Similarly, resuscitation to a BP end point is often incomplete if the goal is to restore adequate CO and perfusion.276
Blood flow through a regional vascular bed is directly proportional to the organ perfusion pressure (ΔP), which is calculated as the difference between the arterial inflow pressure (Pa) and the venous outflow pressure (Pv): ΔP = Pa − Pv.53 With reasonable approximation and ignoring local extravascular effects, the inflow arterial pressure can be estimated to be the mean arterial pressure (MAP), and the outflow venous pressure can be estimated to be CVP:
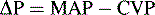
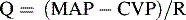
Under ideal laminar flow conditions, vascular resistance is independent of flow and pressure; therefore an increase in vascular resistance will decrease blood flow, and a decrease in vascular resistance will increase blood flow for any given ΔP. Control mechanisms in the body generally maintain arterial and venous BPs within a narrow range; therefore changes in organ and tissue blood flow are primarily regulated by changes in vascular resistance. Resistance to blood flow within a vascular network is determined by the size of the vessels, the organization of the vascular network, physical characteristics of the blood, and extravascular forces acting on the vasculature.53
Shock states
Shock is a progressive condition of circulatory failure that results in inadequate CO and oxygen and substrate delivery to the tissues. Shock may be present in the patient with a low, normal, or high CO or index.245
Hypovolemic Shock
Etiology
Hypovolemia is the most common cause of shock in infants and children.245 Hypovolemic shock is best defined as a sudden decrease in the intravascular (blood) volume, relative to vascular capacity (i.e., inadequate intravascular volume relative to the vascular space), to such an extent that effective tissue perfusion cannot be maintained. Causes include hemorrhage from gastrointestinal disease or trauma, fluid and electrolyte loss, endocrine disease, and plasma loss.
Clinical Signs and Symptoms
Reliable indicators of early, compensated hypovolemic shock in children are persistent tachycardia, cutaneous vasoconstriction, and diminution of the pulse pressure (difference between systolic and diastolic BPs). Clinical evidence of decreased tissue perfusion includes skin mottling, prolonged capillary refill, and cool extremities. Systemic arterial BP is frequently normal, which is the result of increased SVR, making BP monitoring of limited value in managing the patient with compensated hypovolemic shock.132,245 Neurologic status is initially normal or only minimally impaired.
Although capillary refill has long been advocated as an important means of quickly and reliably assessing the level of hydration and circulatory status of acutely ill or injured children, there are several important limitations to the use of capillary refill in the assessment of circulatory status in children.23,112,297 It is necessary to account for the ambient temperature, in addition to the site of measurement, when interpreting the results.
On rare occasions, BP may be paradoxically elevated in children with hypovolemia.33 Recognition of this phenomenon is extremely important because some healthcare providers may be reluctant to provide appropriate fluid resuscitation for these patients because of fear of exacerbating the hypertension. Treatment of the hypertension, especially with β-blockers or calcium channel antagonists, may precipitate hypotension. Dehydrated, volume-depleted children with paradoxic hypertension should be given a trial of volume expansion; such therapy will cause little harm and could ameliorate the hypertension.
With continued loss of intravascular volume or with delayed or inadequate intravascular volume replacement, the intravascular fluid losses surpass the body’s compensatory abilities, causing circulatory failure and organ dysfunction. Pronounced systemic vasoconstriction and hypovolemia produce ischemia and hypoxia in the visceral and cutaneous circulations. Cellular metabolism and function are altered in these areas, resulting in damage to the blood vessels, kidneys, liver, pancreas, and bowel. SV and CO are decreased. The patient ultimately becomes hypotensive, acidotic, lethargic, or comatose, and oliguric or anuric. It is important to emphasize that arterial BP falls only after compensatory mechanisms are exhausted; this may be long after the precipitating event and only after a severe reduction in CO.243 Terminal phases of hypovolemic shock are characterized by myocardial dysfunction and widespread cell death.
Ischemia can develop in nonvital organs as a result of reduced circulating blood volume and preferential vasoconstriction. In skeletal muscle during shock, the normal intermittent perfusion pattern in capillary networks has been observed to transform to a marked maldistribution of flow, with cessation of blood flow in most capillaries.198,199 In addition to a low and irregular flow state in capillaries, recent findings have demonstrated a progressive narrowing of the capillary lumen during shock. The nearly 25% decrease in lumen diameter after 1 hour of shock principally results from swelling of endothelial cells; this significantly increases the capillary resistance to flow and contributes to further flow retardation.198,199 In addition, in a variety of organs impaired microcirculatory blood flow has been observed with reperfusion after a period of ischemia (no reflow or slow reflow). The causes of no reflow include red blood cell aggregation, leukocyte trapping, and edema of tissue and capillary endothelial cells.199
In any form of shock, including hypovolemic shock, the end result of hypoperfusion and tissue ischemia is cellular oxygen and nutrient deficiency that can affect the integrity of cell function and structure. Anaerobic metabolism results from the decrease in oxygen delivery, leading to glycogen depletion and lactate production. An increase in cytosolic calcium is also evident, which leads to an increase in membrane phospholipid hydrolysis and lysosomal membrane damage.295 This process eventually progresses to irreversible cellular injury and a host of inflammatory responses, which stimulate further tissue inflammation and injury. Ischemia or reperfusion can also induce expression of inflammatory genes in endothelial cells. In some hypovolemic animal models, fluid resuscitation leads to a decrease in the inflammatory cascade that was triggered by organ ischemia secondary to hypoperfusion.295 Because this inflammatory response is not likely to protect or repair organs from hypovolemic insult, downregulation of this inflammation seen with fluid resuscitation is likely to be beneficial.
Serum tonicity is important in both the presentation and the management of dehydration. In approximately 80% of cases of dehydration, patients have normal serum osmolality; 15% are hyperosmolar, and 5% are hypoosmolar.243 The serum sodium concentration can be used as a rough estimate of serum osmolality and dehydration, which is classified as hyponatremic, isonatremic, or hypernatremic (Box 6-1).
Box 6-1 Classification of Hypovolemic Shock Based on Serum Sodium
Hyponatremic: serum sodium <130 mEq/dL
The approach for infants or children with diarrheal dehydration can be organized using the five-point assessment141,243 (Table 6-2). Initial management is guided by a simple history and physical examination, followed by assignment of the patient to one of three groups based on severity of illness.
| Point of Assessment | Method |
| Volume deficit | History and physical |
| Osmolar disturbance | Serum sodium |
| Acid-base disturbance | Serum pH, PCO2, bicarbonate |
| Potassium disturbance | Serum potassium |
| Renal function | Serum blood urea nitrogen, creatinine |
| Urine analysis, specific gravity, sodium |
Adapted from Kallen RJ: The management of diarrheal dehydration in infants using parenteral fluids. Pediatr Clin North Am 37:265, 1990.
Table 6-3 lists the signs and symptoms that can be used to group patients by severity of dehydration. Of the 10 findings listed, none is sufficiently accurate to be used in isolation.113 The presence of fewer than three signs corresponds with a fluid deficit of less than 5%, whereas children with a deficit of 5% to 9% generally have three or more clinical findings. At least six or seven findings should be present to diagnose a deficit of 10% or more.
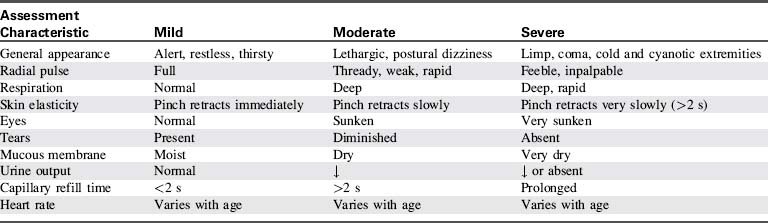
A recent study has suggested that it may be possible to rely on a relatively restricted subset of clinical indicators: general appearance, capillary refill, mucous membranes, and tears. Of these four findings, the presence of any two indicates a deficit of 5% or more, and three or more findings indicates a deficit of at least 10%.113
Children usually have healthy compensatory mechanisms that initially maintain peripheral perfusion and BP despite volume loss. HR increases to maintain CO despite deceased SV (see table of Normal Blood Pressures in Children on pages inside front cover). For this reason, tachycardia is an early sign of acute blood loss with impending shock. Total blood volume in children is generally estimated as 7% to 8% of body weight or 70 to 80 mL/kg. With blood loss of 10% to 15% (class I hemorrhage as defined by Advanced Trauma Life Support), the injured child easily compensates with a 10% to 20% increase in HR (Table 6-4). BP and peripheral capillary refill are usually unchanged. Even with a 20% to 25% loss of blood volume (class II hemorrhage), there is often little change in the systolic BP as increasing tachycardia and peripheral vasoconstriction mediated through the sympathetic nervous system and other mechanisms serve to maintain perfusion pressure. It is for this reason that one must avoid the tendency to equate hypotension with shock in the pediatric trauma patient. Hypotension is a late finding with ongoing hemorrhage and often signals that the child is near the point of complete decompensation.243 Furthermore, studies in adults and children with trauma showed that systolic BP is a predictor of mortality and that prehospital hypotension is associated with an increased risk of mortality.120,185,274 There is a need for studies to validate the appropriate BP target and to better define hypotension in critically ill children.120
Table 6-4 Classification of Hemorrhage by Blood Volume Lost
| Degree of Hemorrhage | Blood Volume Lost (%) | Signs |
| Class I | <15 | Minimal tachycardia; normal respiration, BP, capillary refill |
| Class II | 15-30 | Tachycardia; tachypnea; diminished pulse pressure; systolic BP unchanged; prolonged capillary refill; minimal decrease in urine output; anxiety |
| Class III | 30-40 | Tachycardia; tachypnea; decreased BP; decreased urine output; mental status changes |
| Class IV | >40 | Hypotension; anuria; loss of consciousness |
BP, Blood pressure.
Adapted from Morgan WM 3rd, O’Neill JA Jr: Hemorrhagic and obstructive shock in pediatric patients. New Horiz 6:150−154, 1998.
Management
Vascular access is achieved using the largest, most easily accessible vein. Peripheral venotomy can be performed in the veins of the hand, foot, arm, leg, or scalp, but these vessels tend to be small and difficult to cannulate in the hypovolemic child, so attempts to achieve vascular access should be limited before attempting intraosseous access.243 If peripheral access is difficult and vascular access is critical, early intraosseous access is encouraged.57a,259
If time allows and clinical experts are available, vascular access can be obtained by central venous cannulation or saphenous cutdown; however, the latter route is less frequently used because intraosseous access is so readily available. The femoral vein is often the most easily accessible in an emergency situation.243
The amount of volume required to resuscitate a child in hypovolemic shock is variable. However, the most common error made in resuscitation of a child in hypovolemic shock is inadequate or delayed volume administration.119,271 The variance in disease processes that lead to hypovolemic shock may require different treatment and will be discussed separately.
Fluid Therapy: Diarrhea and Dehydration
Diarrhea can severely disrupt the balance of fluids and electrolytes, and it is a leading cause of infant mortality worldwide.290 Dehydration and shock as a consequence of diarrhea are more likely to occur in infants or small children than in adults.
Administering large volumes of isotonic saline (0.9% sodium chloride) frequently can cause the development of hyperchloremic acidosis; this occurs because isotonic saline contains 154 mEq/L of chloride, and its administration results in a dilutional decrease of serum bicarbonate and an increase in serum chloride concentration. Both of these changes will produce metabolic acidosis. Resuscitation with lactated Ringer’s solution provides a more balanced electrolyte replacement and is less likely to produce metabolic acidosis. The infusion of lactated Ringer’s solution in general does not influence the circulating serum lactate concentration.74
The use of colloid is not indicated in the initial resuscitation of the patient with shock secondary to dehydration. Albumin and other colloids have been used effectively for volume replacement in patients who have large volumes of nonfunctional extracellular or third space fluid loss or low albumin states.123
The end point of fluid resuscitation should be normalization of HR and respiratory rate, an increase in arterial BP, an increase in pulse pressure and peripheral perfusion, establishing adequate urine output, and a decrease in the metabolic acidosis. However, if shock persists after the first fluid infusion is completed, a second infusion of 20 mL/kg should be started. If the patient does not improve after two or three infusions of isotonic crystalloid solution (40 to 60 mL/kg), more aggressive monitoring and therapy are clearly required and the child must be evaluated for complicating factors. Causes of unresponsive shock in a patient with adequate oxygenation include unrecognized pneumothorax or pericardial effusion, intestinal ischemia (e.g., volvulus, intussusception, necrotizing enterocolitis), sepsis, myocardial dysfunction, adrenocortical insufficiency, pulmonary hypertension, and potential occult bleeding caused by inflicted trauma.243
Children with dehydration secondary to burns or septic shock frequently require large amounts of fluid administration, because they have ongoing capillary leak; they often require concurrent inotropic support, vasopressor support, or both.50 To guide therapy, it is important to determine whether these children have failure of the vasculature, cardiac function, or a combination, leading to requirements for support in addition to volume administration. The cardiac failure seen with thermal injuries, trauma, or sepsis must be evaluated further to distinguish cardiac dysfunction from decreased CO resulting from external compression or obstruction (e.g., pericarditis, tamponade).92
Fluid Therapy in Traumatic or Hemorrhagic Shock
Shock in the injured child is almost always secondary to hypovolemia, so acute blood loss must always be considered first. With hemorrhagic shock, the source of blood loss may be external and readily apparent, such as the laceration of a major vessel in an extremity or a large scalp laceration. The scalp is often a source of significant blood loss in infants and children, because of its inherent vascularity and because the head is relatively large (with a relatively large percentage of the child’s BSA) in young children. More commonly, however, the source of blood loss is internal and therefore less apparent. Extremity (i.e., femur fracture) and intraabdominal injuries are common after blunt trauma in children, especially with motor vehicle crashes,243 and can result in significant occult blood loss.
Intraabdominal solid organ injury is a common cause of occult blood loss, and such injuries are probably the most common cause of traumatic shock in children.99,152 Single or complex fractures of the liver and spleen often result in significant hemorrhage. Associated injury to the inferior vena cava is rare, but if present it usually leads to life-threatening hemorrhage. Mesenteric injury is often seen as part of the “lap belt syndrome,” but does not usually result in sufficient blood loss to cause shock. Blunt injury of the kidney can also cause significant blood loss into a retroperitoneal hematoma. Fracture of the bony pelvis is relatively common in older children and may lead to massive hemorrhage, at times requiring urgent stabilization.
With any signs of hypovolemic shock (e.g., tachycardia), a fluid bolus of 20 mL/kg of isotonic crystalloid solution should be given. Transfusion of packed red blood cells or whole blood is indicated for the patient with hemorrhagic shock who shows signs of persistent intravascular volume depletion despite the rapid administration of 60 mL/kg (three boluses) of crystalloid. Packed red blood cells should be given in boluses of 10 mL/kg. Type-specific blood is used if available, but typing should not delay resuscitation. Un–cross-matched O-negative universal donor blood should be immediately available. Aggressive fluid resuscitation is paramount, even when there is associated closed-head injury, because ongoing hypoperfusion leads to secondary hypoxic end-organ injury, especially in the brain.5,14,54,60,302
Although hypovolemia is the most frequent cause of shock, occasionally other causes of hemodynamic compromise may be present. Obstructive shock owing to tension pneumothorax or pericardial tamponade, myocardial confusion, and spinal shock can complicate the management of pediatric trauma.243 Blunt cardiac injury can cause myocardial contusion, myocardial concussion, aneurysm, septal defects, chamber rupture, valvular rupture, and damage to the pericardium.40,78,249 Each of these entities has separate presentations, although the lesions are often concurrent. The majority of blunt injuries to the heart are myocardial contusions; devastating events such as ventricular rupture are rare.78,249
There are important clinical considerations in the management of children with blunt cardiac injury:
• If a child with a suspected blunt cardiac injury is hemodynamically stable and has a normal sinus rhythm, then the development of serious arrhythmia or cardiac failure is not likely. In other words, high risk for a patient is usually evident immediately.78
• Trauma patients with a suspected cardiac injury should receive prompt evaluation of cardiac function with echocardiography.40,78 Any patient with unexplained hypotension or diminished peripheral perfusion should be studied using echocardiography.
• Elevated cardiac isoenzyme values and electrocardiography are nonspecific for clinically significant myocardial injury.78
Crystalloid Versus Colloid
The advantages of using crystalloids for resuscitation are their ready availability and their cost effectiveness. The cost of 5% albumin is manyfold that of the common crystalloid solutions. The main disadvantage is that crystalloids reduce colloid oncotic pressure and can predispose to pulmonary and peripheral edema.61
There is now a reasonable evidence base to support the use of isotonic crystalloids (e.g., normal saline) as the best initial choice of fluid for intravenous resuscitation of shock. Two randomized controlled studies of fluid resuscitation in pediatric septic shock failed to demonstrate any difference in outcome, regardless of whether crystalloid or colloid (i.e., albumin) fluid therapy was used in initial resuscitation.303,319 A well-powered study investigating fluid resuscitation of adults in shock has also failed to show benefit of the use of colloids over crystalloids. Subgroups of patients in the adult study (e.g., patients with traumatic brain injuries) had a better outcome with the use of crystalloids instead of colloids.96 A physiology-based argument has existed for many years that whereas crystalloids may be clinically effective for fluid resuscitation, their reduced osmolar drag relative to colloid therapy leads to increased capillary leak and movement of water extravascularly. As a result, the “3:1 rule” suggests that a significantly greater volume of crystalloid than colloid would be necessary to replace the depleted intravascular compartment. A consistent finding from all three of the previously cited studies is that there is no difference between the volume of crystalloid and colloid necessary to resuscitate the patient in shock. This is not to say that there is no role for colloids in fluid resuscitation. Children with advanced septic shock might still benefit from colloid therapy, and those with advanced hemorrhagic shock will ultimately require the use of blood products to replace both blood cells and coagulation factors.
Distributive Shock: Definitions
Neurogenic shock results from autonomic dysfunction occurring secondary to injury to the spinal cord. Loss of peripheral vascular tone and subsequent increased venous capacitance lead to a relative hypovolemia caused by expansion of the vascular space. Children with neurogenic shock often fail to improve with fluid resuscitation, but instead respond to treatment with selective α-adrenergic vasoactive infusions (Table 6-5; see discussion under Cardiogenic Shock, Treatment, later in this chapter).
Table 6-5 Pediatric Vasoactive Drugs for the Treatment of Shock*†
| Dose | Effects | Cautions |
| Sympathomimetics | ||
| Dobutamine, 2-20 mcg/kg per min | Selective β-adrenergic effects; increases cardiac contractility and also increases heart rate (this latter effect is variable); β2 effects produce peripheral vasodilatation; no dopaminergic or α-adrenergic effects | Extreme tachyarrhythmias have been reported, particularly in infants; hypotension may develop; may produce pulmonary venoconstriction |
| Dopamine, 1-5 mcg/kg per min | Dopaminergic effects predominate, including increase in glomerular filtration rate and urine volume | Can produce extreme tachyarrhythmias; can result in increase in pulmonary artery pressure; inhibits thyroid-stimulating hormone and aldosterone secretion |
| Dopamine, 2-10 mcg/kg per min | Dopaminergic effects persist and β1 effects are seen, especially an increase in heart rate | As above |
| Dopamine, 8-20 mcg/kg per min | α-Adrenergic effects dominate | As above |
| Epinephrine, 0.05-0.15 mcg/kg per min | Endogenous catecholamine, which produces α, β1, and β2 adrenergic effects; at low doses, β1 effects dominate | Will increase myocardial work and oxygen consumption at any dose; splanchnic constriction will occur at even low doses |
| Epinephrine, 0.2-0.3 mcg/kg per min | α-Adrenergic (vasoconstrictive) effects dominate | As above |
| Isoproterenol, 0.05-0.1 mcg/kg per min | β-Adrenergic effects; β1 effects may result in rapid increase in heart rate; β2 effects may produce peripheral vasodilatation and also may effectively treat bronchoconstriction | Monitor for tachyarrhythmias, hypotension; will increase myocardial oxygen consumption |
| Norepinephrine, 0.05-1.0 mcg/kg per min | Endogenous catecholamine with α- and β-adrenergic effects; produces potent peripheral and renal vasoconstriction; can increase blood pressure | May produce tachyarrhythmias, increased myocardial work, and increased oxygen consumption; may result in hepatic and mesenteric ischemia |
| Vasopressin | ||
| Vasopressin, 0.2 to 2 milliunits/kg per min (0.0002 to 0.002 unit/kg per min) | Antidiuretic hormone analogue that acts on vasopressin receptors; produces peripheral and splanchnic vasoconstriction; also used to treat GI hemorrhage for this reason | May cause hypertension, bradycardia |
| Phosphodiesterase inhibitors and inodilators | ||
| Inamrinone, Loading dose: 0.075 to 0.1 mg/kg (75 to 100 mcg/kg) slowly, Infusion: 5-10 mcg/kg per min | Nonadrenergic inotropic agent that produces phosphodiesterase inhibition and increase in intracellular cyclic-adenosine monophosphate cAMP intracellular calcium uptake also is delayed; these effects result in improved cardiac contractility and vasodilatation | Monitor for arrhythmias (especially accelerated junctional rhythm, junctional tachycardia, and ventricular ectopy); may produce hypotension (especially if patient is hypovolemic), liver and gastrointestinal dysfunction, thrombocytopenia, and abdominal pain; experience in children is limited and recent |
| Milrinone, Loading Dose: 0.05 mg/kg (50 mcg/kg) over 10-60 min, Infusion: 0.25-0.75 mcg/kg per min | As above | Reduce dose when renal dysfunction present |
| Vasodilators | ||
| Nitroglycerin, 0.25-0.5 mcg/kg per min; increase as tolerated to maximum 10 mcg/kg per min (adolescents, 5 mcg/min; note, not per kg per min) | Arterial and venodilator | Is adsorbed by polyvinylchloride tubing; use special infusion set |
| Nitroprusside, 0.3-0.5 mcg/kg per min; titrate up to 8 mcg/kg per min | Arterial and venodilator | Light-sensitive; use specialized infusion set or cover tubing when infusion slow; may produce thiocyanate and cyanide toxicity, particularly for higher doses or prolonged infusion |
GI, Gastrointestinal; IV, intravenous.
* Infusion rate = mL/hr = [Weight (kg) × Dose (mcg/kg per min) × 60 min/h] ÷ Concentration (mcg/mL).
† For additional information, see Cardiogenic Shock, Treatment in this chapter.
Septic Shock
Etiology and Definitions
Septic shock is the most complex and controversial type of shock. Septic shock comprises a cascade of metabolic, hemodynamic, and clinical changes resulting from invasive infection and the release of microbial toxins into the bloodstream. Historically, a distinction was made between the clinical findings and the type of invading microorganism (e.g., gram-positive versus gram-negative shock). However, on closer analysis, it became apparent that the systemic response was independent of the type of invading organism (e.g., bacteria, virus, fungus, rickettsia), rather it was a host-dependent response.51,243 The morbidity and mortality are primarily the result of endogenous proteins and phospholipids synthesized by the patient.51 Sepsis is the culmination of complex interactions between the infecting microorganism and the host immune, inflammatory, and coagulation responses.11,46,265
One of the factors hampering progress in the understanding of sepsis and septic shock has been the variability of definitions used and patient populations studied. As an approach to standardizing the definitions of sepsis and organ failure, the American College of Chest Physicians and the Society of Critical Care Medicine held a consensus conference to define sepsis and organ failure more precisely.9 This group proposed the term systemic inflammatory response syndrome (SIRS), recognizing that the inflammatory response can be precipitated by processes other than infection. The group included in the definition of SIRS abnormalities of temperature, HR, respiratory rate, and white blood cell count. Whereas the definitions used were applied to adults, age-appropriate values must be used to apply the same descriptions to children.
Recently, consensus definitions for the pediatric sepsis continuum were developed and published.109 The pediatric sepsis continuum includes SIRS, infection, sepsis, severe sepsis, septic shock, and multiple organ dysfunction syndrome.
Initial definitions for these points on the sepsis continuum have been proposed and are listed in Box 6-2. A few comments need to be made concerning the use of these definitions. First, it is important to realize that the definitions are sensitive but not specific screening tools to be used primarily for research purposes, and any direct application to the clinical setting must take into account the dynamic and continuous nature of the sepsis disease process and the static and categorical nature of the definitions.39
Systemic Inflammatory Response Syndrome
• ALTERATION IN TEMPERATURE: Core body temperature of >38.5°C or <36°C.
• ALTERATION IN HEART RATE (characterized by any of the three open bullets, below):
 Tachycardia in the absence of external stimulus, chronic drugs, or painful stimuli, defined as follows:
Tachycardia in the absence of external stimulus, chronic drugs, or painful stimuli, defined as follows:

• ALTERATION IN RESPIRATORY RATE (characterized by either of the open bullets, below):


• ALTERATION IN WHITE BLOOD CELL COUNT: Leukocyte count elevated or depressed for age (not secondary to chemotherapy-induced leukopenia) or >10% immature neutrophils.
Infection (one of the following)
Modified from Goldstein B, et al: International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med 6:2−8, 2005.
Third, the clinical parameters used to define SIRS and organ dysfunction are greatly affected by the normal physiologic changes that occur as children develop,39,109 and the baseline organ function of children with chronic diseases must be considered. The consensus definitions propose six age groups for age-specific vital signs and laboratory parameters: newborn (0 to 7 days), neonate (1 week to 1 month), infant (1 month to 1 year), toddler and preschooler (2 to 5 years), school-aged child (6 to 12 years), and adolescent and young adult (13 to 18 years). The age groups were determined by a combination of age-specific risks for invasive infections, age-specific antibiotic treatment recommendations, and developmental cardiorespiratory physiologic changes.39,109 Evidence-based data combined both physiologic and laboratory normative information to support these age groupings. Furthermore, most of the normative data were reported to identify the 5th and 95th percentile ranges. The relationship between values and ranges obtained in normal children and appropriate values and ranges in critically ill children with sepsis is unclear.
Finally, the panel chose not to advocate for the use of a single multiple-organ dysfunction score, but developed criteria for organ dysfunction based on those used in the Pediatric Logistic Organ Dysfunction score or the pediatric multiple organ dysfunction syndrome39,159,239 (Box 6-3).
Box 6-3 Criteria for Organ Dysfunction in Children with Severe Sepsis or Septic Shock
Cardiovascular dysfunction
Respiratory* (one of the following four bulleted conditions)
• PaCO2/FiO2 < 300 in absence of cyanotic heart disease or preexisting lung disease
• PaCO2 > 65 torr or 20 mm Hg over baseline PaCO2
• Proven need† or >50% inspired oxygen to maintain saturation >92%
• Need for nonelective invasive or noninvasive mechanical ventilation‡
Modified from Goldstein B, et al: International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med 6:2−8, 2005.
* Acute respiratory distress syndrome must include a PaO2/FiO2 ratio ≤200 mm Hg, bilateral infiltrates, acute onset, and no evidence of left heart failure. Acute lung injury is defined identically except the PaO2/FiO2 ratio must be ≤300 mm Hg.
† Proven need assumes oxygen requirement was tested by decreasing flow with subsequent increase in flow if required.
‡ In postoperative patients, this requirement can be met if the patient has developed an acute inflammatory or infectious process in the lungs that prevents them from being extubated.
These definitions for pediatric sepsis are a significant step forward in facilitating clinical trials in pediatric sepsis, but the definitions go beyond use for research; they can facilitate earlier clinical recognition and treatment of sepsis in children.239 Further refinements may allow differentiation between morbidity arising from infection and morbidity caused by the host response to infection. The PIRO system stratifies patients on the basis of their predisposing conditions (P), the nature and extent of the infection (I), the nature and magnitude of the host response (R), and the degree of concomitant organ dysfunction (O)164,231 (Table 6-6).
Table 6-6 Basic Elements of PIRO Stratification of Patients with Sepsis
| P | Predisposing factors | Innate: Genetic polymorphisms and deficiencies of immune response genes affecting innate immune response, coagulation system complement receptors, Toll-like receptors, and intracellular signaling. Acquired: Burns, trauma, acquired immune deficiencies |
| I | Infection | Site, quantity, intrinsic virulence, and local versus systemic infection caused by specific microbial pathogens |
| R | Response | Differential responses based on hyperresponsiveness versus hyporesponsiveness-immunosuppression; response modifiers such as age, sex, nutritional status, diabetes, other preexisting diseases, and physiologic status of host |
| O | Organ dysfunction | Number, severity, and pattern of organ dysfunction in response to systemic infection; primary versus secondary organ injury; and organ injury owing to sepsis versus preexisting organ dysfunction |
Modified from Opal SM: Concept of PIRO as a new conceptual framework to understand sepsis. Pediatr Crit Care Med 6(3):S55–S60, 2005.
Pathophysiology
Organ dysfunction is the final tissue sequelae in response to severe sepsis and is the ultimate determinant of survival. Mortality is much higher in patients with sepsis who develop progressive multiple organ failure than in those who develop a single or no organ dysfunction in response to sepsis.164,231 What remains unclear is why some patients develop one type of organ dysfunction and others develop another organ dysfunction, despite seemingly similar septic stimuli. Some patients develop profound coagulation abnormalities in the early phases of sepsis, whereas others develop severe acute respiratory dysfunction syndrome, and others experience acute renal failure. The sequence and pattern of organ dysfunction after severe sepsis can also vary between patients and provide some insights into the likelihood of survival and response to therapy.
Some patients have a marked systemic response, often referred to as hyperinflammation, induced by certain microbial pathogens.231 The clearest examples of this occur in previously healthy children who develop meningococcal sepsis or in patients with streptococcal toxic shock syndrome after a relatively mild soft-tissue injury.232,241 The other extreme exists in febrile, neutropenic patients with cancer or in bone marrow transplant patients who have a markedly impaired systemic host response to microbial infection and often die of overwhelming infection with an inadequate host response.11,135,231 There are numerous gradations in the spectrum between these two extremes, yet it is often difficult to clinically distinguish between patients with hyperinflammation and those with inadequate inflammatory responses.
A key element in the pathogenesis of sepsis is activation of the cytokine network. Cytokines are host produced, pleomorphic immunoregulatory peptides.305,316 The most widely investigated cytokines are tumor necrosis factor, interleukin-1, and interleukin-8, which are generally proinflammatory, and interleukin-6 and interleukin-10, which tend to be antiinflammatory. A trigger, such as a microbial toxin, stimulates the production of tumor necrosis factor and interleukin-1, which in turn promote endothelial cell-leukocyte adhesion, release of proteases and arachidonic acid metabolites, and activation of clotting.51,316 Interleukin-1 and tumor necrosis factor are synergistic, share many biologic functions, and interact to promote positive feedback cascades that result in fever, vasodilation, cardiovascular failure, and lactic acidosis (see Septic Mediators in the Chapter 6 Supplement on the Evolve Website).51
The cytokines stimulate the production of many important effector molecules, including proinflammatory cytokines, antiinflammatory cytokines, and nitric oxide. Increased pro-inflammatory cytokines, antiinflammatory cytokines, and nitric oxide correlate with outcome and the development of multiple organ failure in pediatric sepsis and septic shock.51,289,320
It is now clear that inflammation is just one of the many contributors to septic physiology. Other factors include enhanced coagulation and impaired fibrinolysis (Table 6-7).8,135,194 Because of the multiple factors involved, the clinical pattern and presentation of septic shock can vary a great deal and depend on the dynamic interplay of the invading organism, elapsing time, and host response.
| Agent | Site of Action |
| Antiendotoxin antibodies | Neutralize endotoxin |
| E5 (murine) | |
| HA-1A (human) | |
| Bactericidal permeability-increasing protein | Neutralize endotoxin |
| TNF antibodies | Block TNF |
| IL-1 receptor antagonists | Inhibit action of IL-1 on cellular receptors |
| IL-1 antibodies | Block IL-1 receptor interaction |
| IL-6 and IL-8 antibodies | Block IL-6, IL-8 receptor interaction |
| Bradykinin-receptor antagonists | Prevent vasoactive effects of bradykinin |
| Platelet activating factor antagonist | Block platelet activation and platelet aggregation |
| Cyclooxygenase inhibitors (ibuprofen) | Block pyrogen, thromboxane, and prostaglandin production |
| NO synthase inhibitors | Block production of NO, restore vascular tone |
| Inhibitors of leukocyte-adhesion molecules | Prevent endothelium-leukocyte interaction |
| Corticosteroids | Promote and antiinflammatory response |
| Antiinflammatory cytokines (IL-6, IL-10) | Inhibit inflammatory response; reduce production of proinflammatory cytokines |
IL, Interleukin; NO, nitric oxide; TNF, tumor-necrosis factor.
Clinical Signs and Symptoms
Abnormal hemodynamic responses are the hallmarks of septic shock. The clinical cascade may unfold over several days or a few hours. In many patients, the early stages consist of a hyperdynamic state with an elevated CO, decreased SVR, and a widened pulse pressure, with episodic hypotension and warm extremities with brisk capillary refill.35,37,51,52 In this hyperdynamic stage, the patient may also demonstrate high fever, mental confusion, and hyperventilation. Although these patients are typically tachycardic and tachypneic, the vital signs and clinical examination may not reflect the severity of disease. Mental status changes, a sensitive indicator of hypoperfusion, may present in a clinical spectrum that begins with fussiness, irritability, or hallucinations and, if untreated, evolves to lack of response to parents or noxious stimuli, and eventual coma.243
If sepsis is unchecked and uncorrected, cardiovascular function steadily deteriorates, which is characterized by a decline in CO, hypotension, and worsening metabolic acidosis. Paradoxically, hypotension may develop in the presence of normal or elevated CO. One fundamental abnormality in patients with septic shock is an altered relationship of SVR and CO.292 The patient’s status deteriorates when cardiac compensation for falling SVR is lost. Some patients die of refractory hypotension associated with extremely low SVR.191,292
A progression from high to low CO can occur over any time period.35 As CO falls, tissue perfusion worsens, leading to anaerobic metabolism and accumulation of lactic acid.195 Progressive lactic acidemia can be a sign of impending death.21,22,292 Infants or children with heart disease and any other patients with limited cardiac reserve can progress rapidly to this hypodynamic picture.
Survival following septic shock has been related to the host’s ability to establish and maintain a hyperdynamic cardiovascular state.44,253,254 In this hyperdynamic shock, CO is increased and the SVR is low. Clinical studies of adults with septic shock have documented both right and left ventricular dilation with decreased contractility and reduced ejection fraction.44,238 In these patients, CO is maintained only through ventricular dilation (an increase in end-diastolic volume maintains SV at near-normal levels despite the fall in ejection fraction) and tachycardia (see Fig. 6-6). These septic shock studies in adults have not been replicated in children. Infants and children may not be fully capable of using or maintaining the same compensatory physiologic mechanisms. For example, infants and children have higher resting HRs, which can limit the efficiency of a chronotropic protective response. Because the pediatric ventricle is less compliant than the adult ventricle, the pediatric ventricle may be less able to dilate.
In the early stages of septic shock, hypovolemia or myocardial dysfunction (resulting from preexisting or intercurrent ventricular disease) can blunt or eliminate the hyperdynamic response to sepsis.191,292 Several factors can contribute to the relative hypovolemia that commonly develops during septic shock. Increased capillary permeability, dilation of arteries and veins with subsequent peripheral pooling of intravascular blood volume, inappropriate polyuria, and poor oral intake all combine to reduce the effective blood volume (i.e., inadequate intravascular volume relative to the vascular space).243 Fluid loss secondary to fever, diarrhea, vomiting, or sequestered third space fluid also contributes to the relative hypovolemia.
In some patients without complicating preexisting heart disease, a relative depression in left ventricular contractility exists even in early stages.44,153,238,264,291 Myocardial contractility is most likely depressed because of the inhibitory effects of one or more circulating substances (so-called myocardial depressant factor or factors). In patients who survive, this myocardial depression is transient.
Contrary to adult experience, low CO—not low SVR—is associated with mortality in pediatric septic shock.51,254 Ceneviva et al.56 reported outcome data associated with aggressive volume resuscitation and goal-directed therapies in children with septic shock. In this study, 50 children with fluid refractory septic shock were hemodynamically characterized using measurements and calculations obtained through use of pulmonary artery catheters. The majority of children (58%) in septic shock showed a low CO and high SVR state (group I); 20% showed the classic adult high CO and low SVR state (group II); and 22% had decreased CO and SVR (group III).
Patients in group I responded to inotropic therapy with or without a vasodilator. Patients in group II responded to vasopressor therapy, but some needed the addition of an inotrope for evolving myocardial dysfunction. Patients in group III responded to combined vasopressor and inotropic therapy. The overall 28-day survival rate was 80% (group I, 72%; group II, 90%; group III, 91%). This study demonstrates that therapies directed at cardiac dysfunction and systemic vasoconstriction (i.e., vasodilators) are likely to be needed more in pediatric than in adult septic shock.56
Cardiovascular abnormalities described in sepsis include alterations of both systolic and diastolic ventricular function.212,291 Although diastolic performance in sepsis has been less characterized, echocardiographic studies have demonstrated a pattern of abnormal left ventricular relaxation, which is more severe in nonsurvivors.
Septic shock is also characterized by abnormal utilization of oxygen.122,124,254 Although sepsis causes a hypermetabolic stress associated with increased oxygen demand, deterioration characteristically occurs when oxygen consumption falls during a period of increased cardiovascular function and oxygen delivery. This fall in oxygen consumption is the result of decreased oxygen extraction and reflects a severe impairment of oxidative metabolism at a time of major metabolic and physiologic stress. The pathophysiology of inadequate oxygen consumption in septic shock remains unclear. The most widely accepted theories to account for this oxygen debt are the redistribution of blood flow, with a consequent decrease in nutrient capillary flow, and the development of a cellular metabolic blockade at the mitochondrial level such that delivered oxygen cannot be used. Progressive deterioration in oxygen consumption and oxygen extraction portends a poor prognosis for patients in septic shock.124
Management
Antibiotic Treatment
The removal or control of microorganisms by surgical debridement or drainage and antibiotic therapy is a crucial component of the treatment of septic shock. Antibiotic treatment is appropriate in patients with circulatory shock whenever an infectious etiology is suspected. Antimicrobial drugs are necessary but not sufficient for the treatment of sepsis, and paradoxically, they may precipitate septic changes by liberating microbial products (e.g., release of endotoxin following lysis of gram-negative bacteria).316
Airway and Ventilation
Detection of septic shock is facilitated by the nearly universal presence of tachypnea.316 Sepsis places extreme demands on the cardiopulmonary system, requiring a high-minute ventilation precisely when the compliance of the respiratory system is diminished, airway resistance is increased, and muscle efficiency is impaired. Timely intubation and mechanical ventilation reduce respiratory muscle oxygen demand and the risk of aspiration and cerebral anoxia from catastrophic respiratory arrest.
Volume Resuscitation
Restoration of preload by volume resuscitation is the first therapeutic measure for a decreased CO.38,50–52,72,119 Early and effective expansion of the circulating blood volume may enhance oxygen delivery and prevent progression of the septic shock state. The American College of Critical Care Medicine-Pediatric Advanced Life Support guidelines recommend rapid, stepwise interventions with the following therapeutic end points in the first hour: reduction in extreme tachycardia, capillary refill of less than 2 s, normal pulses with no differential between peripheral and central pulses, warm extremities, urine output greater than 1 mL/kg per hour, and normal mental status.38,72 Further hemodynamic optimization using metabolic endpoints to treat global tissue hypoxia include an ScvO2 obtained from the superior vena cava greater than or equal to 70%, a cardiac index greater than 3.3, and less than 6.0 L/min per m2 BSA with normal perfusion pressure for age.38,52,72
Studies in children have shown that early aggressive volume repletion, even without addressing myocardial dysfunction, significantly improves outcome.72,119,227 More importantly, when resuscitation was inadequate and shock persisted, the odds of mortality doubled with each hour of persistent shock.137 Goal-directed hemodynamic optimization represents an organized approach to volume repletion, attainment of a target BP (vascular tone) and a more definitive resolution of a pathologic oxygen supply dependency or global tissue hypoxia through the normalization of ScvO2 as a surrogate end point.72
A study in adult patients with severe sepsis and septic shock confirmed that global tissue hypoxia, defined by an ScvO2 less than 70%, can still be present even if physical examination, vital signs, central venous pressure, and urinary output are considered adequate.262 In addressing this global tissue hypoxia using an early goal-directed approach, further reductions in the inflammatory response, morbidity, and mortality were achieved compared with conventional therapy.262 Whereas the early goal-directed therapy patients received more fluids, red blood cell transfusions, and inotropes over the first 6 hours for a higher rate of attaining an ScvO2 greater than or equal to 70%, there was essentially no difference in these therapies after the first 72 hours. Because of this early hemodynamic optimization, significant reductions in vasopressor, mechanical ventilation, and pulmonary artery catheter use were also realized.262 Similarly, a pediatric study using a goal-directed approach demonstrated more fluid, more red blood cell transfusion, and more inotropic support in the first 6 hours of treatment in the group using ScvO2 ≥70% as an end point.72 After 72 hours the amount of crystalloid administered and the percentage of red blood cells in transfusion were not different between the two groups. These observations were similar to those found in adults. These studies support the concept that early resuscitation rather than later resuscitation is beneficial.52,56,227 In addition, there was a statistically significant reduction in the number of children with ScvO2 less than 70% at 72 hours in the intervention group (i.e., early and aggressive fluid resuscitation produced better oxygen delivery to utilization ratio). This finding supports the concept that goal-directed therapy can reduce global oxygen debt over the long term and the short term in patients with septic shock. In addition to the favorable reduction in mortality, a decrease in pulmonary (increased number of ventilator-free days), neurologic, and renal dysfunction was also seen.
Isotonic crystalloid solutions can be used initially to restore intravascular volume in the presence of septic shock if they increase the blood volume and CO. Debate over the relative advantages and disadvantages of crystalloid and colloid fluids for the treatment of sepsis and septic shock continues.52 A metaanalysis has shown no difference in mortality between patients resuscitated with normal saline or albumin, and a multidisciplinary consensus statement concluded that crystalloid is the preferred solution in patients with sepsis.89 Three randomized controlled trials compared the use of colloid to crystalloid resuscitation in children with dengue shock.82,220,319 No difference in mortality between the colloid or crystalloid resuscitation groups was shown.
Theoretically, transfusion of packed red blood cells to maintain a normal hemoglobin concentration is the most effective means of increasing arterial oxygen content and systemic oxygen availability. Initial guidelines in patients with severe sepsis target a hemoglobin concentration greater than or equal to 10 g/dL.52
The optimal hemoglobin for a critically ill child with severe sepsis is not known.71 A recent multicenter trial reported similar outcomes in stable, critically ill children managed with a transfusion threshold of 7.0 g/dL and those with a threshold of 9.5 g/dL; however, the study excluded children with congenital heart disease or hemodynamic instability.160 Whether a lower transfusion trigger is safe or appropriate in the initial resuscitation has not been determined.71
Excessive tachycardia, severe mixed venous desaturation, cardiac systolic dysfunction, and failure to resolve lactic acidosis or failure to improve gastric intramucosal pH may indicate the need to increase hemoglobin concentration to levels of 10 g/dL.72,180,243,292
Although fluid administration and antimicrobial therapy remain the cornerstones of the therapeutic approach to sepsis and septic shock, patients often require therapy with inotropic agents, vasoactive drugs, or both.38,51 Children can have predominant cardiac failure, predominant vascular failure, or a combination of cardiac and vascular failure.35,37,52,56,72 Therapeutic approaches differ with each condition.
Myocardial depression complicates septic shock and can prevent the development of optimal CO despite adequate intravascular volume. The catecholamine and noncatecholamine inotropes may both be effective in reversing myocardial depression and improving contractility (see Table 6-5; Figs. 6-7 and 6-8).38
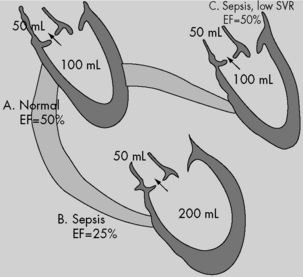
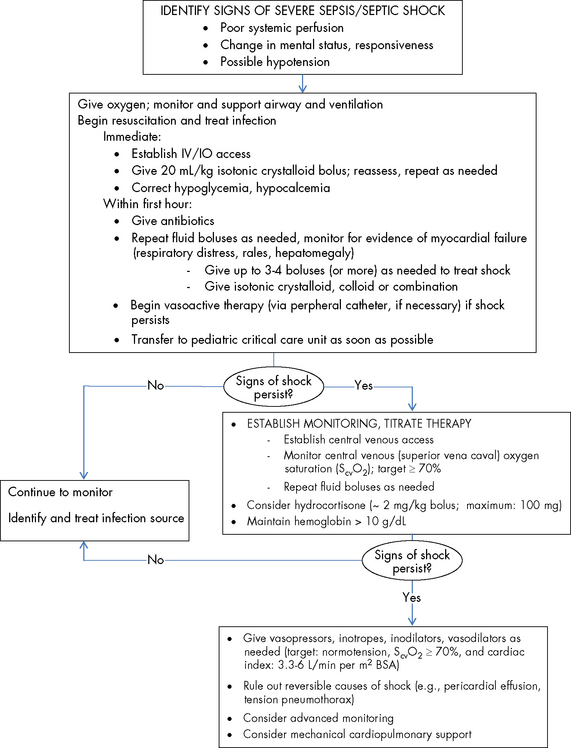
Fig. 6-8 Suggestions for hemodynamic support in pediatric septic shock.
Based on recommendations of Brierley J, Carcillo JA, Choong K, Cornell T, et al: Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med 37:666–688, 2009. BP, Blood pressure; CI, cardiac index; SVRI, systemic vascular resistance index.
Inotropic Support
Children in a normotensive, hypodynamic state after volume resuscitation require an inotrope to increase CO and to reverse shock. Dobutamine or moderate-dose dopamine can be used as the first line of inotropic support. Patients with dobutamine-resistant septic shock often respond to the addition of epinephrine with or without vasodilators. When patients remain in a normotensive low CO state, despite adequate volume resuscitation and epinephrine and vasodilator therapy, then the use of milrinone should be considered.25,174
Catecholamines exert their β-adrenergic receptor effects by elevating intracellular calcium transit through increasing production of intracellular cAMP. However, benefits from the application of catecholamines in septic shock may not be sufficient. Myocardial hyporesponsiveness to catecholamine administration during septic shock has been documented by several investigators.52,247,291 This hyporesponsivity of the myocardium to catecholamines may be due to cytokine-induced uncoupling of the β-adrenergic receptor system or due to deranged intracellular calcium metabolism.25
The bipyridines (milrinone, inamrinone) have no interaction with the adrenergic receptors, but exert their effects through the inhibition of phosphodiesterases, which degrade cAMP. Milrinone has been shown to improve cardiovascular function in pediatric patients with hypodynamic septic shock who are adequately volume resuscitated and are being treated with catecholamines.25,174
Vasopressors
Dopamine is an α-adrenergic agonist at higher concentrations. It mediates vasoconstriction indirectly by causing the release of norepinephrine from sympathetic vesicles.272
Norephinephrine often reverses dopamine-refractory septic shock.202 Norephinephrine can also improve urine output and renal function by increasing perfusion pressure in hypotensive patients.
Both high-dose dopamine and norepinephrine have been recommended as the first-line vasopressor agents in the treatment of refractory shock. There is controversy about whether one agent is superior to the other.70 Dopamine and norepinephrine have different effects on the kidney, the splanchnic region, and the pituitary axis, but the clinical implications of these differences are uncertain. Consensus guidelines and expert recommendations suggest that either agent may be used as a first-choice vasopressor in patients with shock.71
Recently, a multicenter, randomized trial assigned adult patients with shock to receive either dopamine or norepinephrine as first-line vasopressor therapy to restore and maintain BP.70 The type of shock present most frequently was septic shock (62.2%), followed by cardiogenic shock (16.7%) and hypovolemic shock (15.7%). The primary outcome was mortality at 28 days after randomization; secondary end points included adverse events and number of days without the need for organ support. Although the mortality did not differ significantly between the group of patients treated with dopamine and the group treated with norepinephrine, this study raises serious concerns about the safety of dopamine therapy, because dopamine, as compared with norepinephrine, was associated with more arrhythmias and with an increased rate of death in the subgroup of patients with cardiogenic shock.70
Although epinephrine is used for resuscitation and to treat anaphylaxis, its β2-adrenergic effects can cause hyperglycemia, acidosis, and other adverse effects; therefore it may not be ideal for treating shock.170 However, a recent study reported no difference in clinical in-patient outcomes or safety in a prospective comparison of norepinephrine with or without dobutamine to epinephrine for septic shock.12
In children with a warm vasodilated state after volume resuscitation, norepinephrine can be used to increase diastolic BP while maintaining good perfusion.51,202 However, these cases of vasodilatory shock are characterized by hypotension caused by peripheral vasodilation and by a poor response to therapy with vasopressor drugs.162 Vasodilatory shock is caused by the inappropriate activation of vasodilator mechanisms and the failure of vasoconstrictor mechanisms. Unregulated nitric oxide synthesis through activation of soluble guanylate cyclase and generation of cyclic guanosine monophosphate (cGMP) causes dephosphorylation of myosin and hence vasorelaxation. In addition, nitric oxide synthesis and metabolic acidosis activate the potassium channels in the plasma membrane of vascular smooth muscle. The resulting hyperpolarization of the membrane prevents the calcium that mediates norepinephrine- and angiotensin II–induced vasoconstriction from entering the cell. As a result, hypotension and vasodilation persist, despite high plasma concentrations of these hormones. In marked contrast, the plasma concentrations of vasopressin are low, despite the presence of hypotension. This finding is unexpected, because plasma vasopressin concentrations are markedly elevated early in septic shock and hemorrhagic shock. However, the initial massive release of hormone may result in subsequent depletion, such that plasma concentrations of vasopressin are eventually too low to maintain arterial pressure. Although the pressor response to exogenous vasopressin in vasodilatory shock may be due to several different mechanisms, the ability of this hormone to block potassium channels in vascular smooth muscle and interfere with nitric oxide signaling are probably important contributors.
Elucidation of key components of the pathogenesis of vasodilatory shock—potassium channel activation, an increase in nitric oxide synthesis, and vasopressin deficiency—has suggested new possibilities for therapy. Unfortunately, a recent phase-three trial of a nonselective inhibitor of nitric oxide synthase in patients with septic shock was halted because of side effects.63
In recent years, vasopressin has been increasingly used to treat the hypotension associated with shock.24,162,170 Vasopressin can be particularly effective in reversing mediator-induced vasodilatory shock in patients with sepsis or anaphylaxis.170,173,323
The use of vasopressin for hypotension in the setting of vasodilatory shock (warm shock) seems appropriate, and the limited use of vasopressin in this setting appears to be safe. However, for both adults and children, the appropriate patient population and dose regimen have not yet been determined.173
In a recent report, admission and serial plasma vasopressin levels were appropriately elevated in children with septic shock.176 These findings are in contrast with those reported in adults, suggesting that the role of vasopressin administration in children with septic shock is limited until prospective studies are performed. Although the addition of vasopressin to norepinephrine therapy in adult patients with septic shock appears to produce mortality rates similar to those of mortality with norepinephrine alone and it is safe, there is no compelling advantage to using vasopressin rather than norephinephrine.240,266
Vasopressin could have variable effects depending on the cardiovascular profile of shock in any given patient. For example, vasopressin could produce favorable effects in a high CO and low SVR shock state, but could be deleterious if used alone in a patient with severe left ventricular dysfunction. Other reported adverse effects of vasopressin and norepinephrine include decreased CO,181,266 mesenteric ischemia,151,266,306 hyponatremia, skin necrosis, and digital ischemia.83,125,140
The use of vasopressors should be titrated to end points of normal perfusion pressure or SVR. Strict end-point measures should be used to avoid unnecessary vasoconstriction to key organs. A frequent adverse affect of α-agonist treatment is localized severe vasoconstriction after extravasation of the infusion; therefore these infusions are best administered into central venous or intraosseous catheters.272
Corticosteroid Therapy
Corticosteroid therapy has been used in varied doses for sepsis and related syndromes for more than 50 years, with no clear benefits on mortality.13 Since 1998, studies in adults have consistently used prolonged low-dose corticosteroid therapy, and analysis of this subgroup suggests a beneficial drug effect on short-term mortality.13 The Corticosteroid Therapy of Septic Shock (CORTICUS) study group found that hydrocortisone did not improve survival or reversal of shock in patients with septic shock, either overall or in patients who did not have a response to corticotrophin, although hydrocortisone hastened reversal of shock but not survival in those patients in whom shock was ultimately reversed.283
Over the past decade the concept of relative adrenal insufficiency has been proposed in the critical care unit (PCCU) setting and generally refers to patients with vasopressor-resistant hypotension.277 In critically ill adults who had a normal baseline cortisol level (≥20 mcg/dL) and incremental rise of cortisol of less than or equal to 9 mcg/dL after administration of 250 mcg of intravenous adrenocorticotropic hormone (ACTH), treatment with stress doses of hydrocortisone was associated with improved survival.10
Pizarro et al.252 studied the baseline and peak incremental response (increase above baseline) to 250 mcg of ACTH in critically ill children with septic shock to learn whether this test would predict shock resistant to vasopressor in the pediatric setting. All children (18%) with a basal serum cortisol of less than 20 mcg/dL had catecholamine-resistant shock. Eighty percent of those with a low incremental cortisol response to ACTH (≤9 mcg/dL at 30 or 60 min) compared with 20% who had “normal” incremental cortisol response had catecholamine-resistant shock. The study concludes that absolute and relative adrenal insufficiency is common in children with septic shock and may contribute to the development of catecholamine-resistant shock—that is, it is associated with an increased vasopressor requirement.252 However, doubts still persist regarding the efficacy of replacement therapy with low-dose steroids in children with catecholamine-resistant septic shock, and further study is needed to determine whether treatment of such patients changes morbidity, mortality, or both.252,327
The most recent consensus guidelines for treatment of pediatric septic shock suggest that hydrocortisone therapy should be reserved for use in children with catecholamine resistance and suspected or proven adrenal insufficiency.38 Patients at risk for adrenal insufficiency include children with severe septic shock and purpura, children who have previously received steroid therapies for chronic illness, and children with pituitary or adrenal abnormalities.71,252,261,327 Children who have clear risk factors for adrenal insufficiency should be treated with stress-dose steroids (hydrocortisone, 50 mg/m2 BSA per 24 hours).71
Children who have received etomidate or ketoconazole may also be at risk for adrenal insufficiency.327 The use of etomidate as an anesthetic induction agent in critically ill patients is controversial, because this drug inhibits the 11β-hydroxylase enzyme that converts 11β-deoxycortisol into cortisol in the adrenal gland.190 A single dose of etomidate has been demonstrated to inhibit cortisol production for up to 48 hours, prompting the suggestion of steroid supplementation during this period.204,307
Adrenal insufficiency in severe pediatric sepsis is associated with a poor prognosis.68,71 No strict definitions exist, but absolute adrenal insufficiency in the case of catecholamine-resistant septic shock is assumed at a random total cortisol concentration of less than 18 mcg/dL.71 Relative adrenal insufficiency has been defined as a peak serum cortisol level increase of 9 mcg/dL or less above baseline, evaluated 30- or 60-minutes after administration of 250 mcg of corticotrophin (a 30- to 60-minutes post-ACTH stimulation test).71 The treatment of relative adrenal insufficiency in children with septic shock is controversial.71,327 A retrospective study from a large administrative database recently reported that the use of any corticosteroid in children with severe sepsis was associated with increased mortality.192 Given the lack of data in children and the potential risk, steroids should not be used in children who do not have risk factors or who do not meet minimal criteria for adrenal insufficiency. A randomized, controlled trial in children is eagerly awaited.
There are potential complications to glucocorticoid administration in critically ill children, including antianabolic effects, attenuated immunity, depressed wound healing, calcium mobilization, impaired insulin action, and hyperglycemia.327 Although adjunctive corticosteroids for severe sepsis may hasten resolution of unstable hemodynamics, this may occur at the metabolic risk of hyperglycemia.327 Risk of hyperglycemia and associated increased mortality secondary to the gluconeogenic effects of cortisol deserve close scrutiny in children.93,284,327
Immunotherapy
Because septic shock can result from endogenous proteins and phospholipids synthesized by the patient, it has been hypothesized that diminution or elimination of the host response will lessen morbidity and mortality.243 Although this hypothesis continues to be explored, the results of immunotherapy have, in general, been discouraging.193,243,258 Some experimental therapies and their targets are listed in Table 6-7.
Administration of polyvalent intravenous immunoglobulin was recently shown to have no effect on outcome in an international, multicenter study of proven or suspected neonatal sepsis.39a In a recent randomized controlled study, administration of polyclonal immunoglobulin in pediatric patients with sepsis syndrome (n = 100) significantly reduced mortality and length of stay, also resulting in fewer complications, especially disseminated intravascular coagulation.87
Treatment of Coagulation Dysfunction
In patients diagnosed with sepsis, severe sepsis, or septic shock, cytokine-mediated endothelial injury and tissue factor activation initiate a cascade of events that culminate in the development of coagulation dysfunction characterized as procoagulant and antifibrinolytic.226 This abnormal state predisposes the patient to develop microvascular thrombosis, tissue ischemia, and organ hypoperfusion. Multiple organ dysfunction syndrome may be a product of this perturbation in coagulation regulation. Treatments aimed at correcting this coagulation dysfunction have met with limited success.226
Coagulation proteases and their cofactors modify the outcome of severe inflammation by engaging signaling-competent cell surface receptors. The central effector protease of the protein C pathway, activated protein C, interacts with the endothelial cell protein C receptor, protease-activated receptors, and other receptors to affect hemostasis and immune cell function.311 The immune regulatory capacity of the protein C pathway and its individual components might be at least as important as its well-documented effects on coagulation.311
The use of recombinant human activated protein C, although indicated in some adults with sepsis-induced organ dysfunction, is not recommended for use in children with septic shock.71 A randomized clinical trial of recombinant human activated protein C in pediatric severe sepsis patients was stopped by recommendation of the Data Monitoring Committee for futility, with evidence of increased bleeding complications.71,213
The keystone of the treatment of hemostatic abnormalities in patients with sepsis is to treat the underlying infection using appropriate antibiotics and source control.165,166 However, additional supportive treatment, specifically aimed at the coagulation abnormalities, may be required.
Low levels of platelets and coagulation factors may increase the risk of bleeding. However, plasma or platelet substitution therapy should not be instituted on the basis of laboratory results alone and is indicated only in patients with active bleeding and in those requiring an invasive procedure or otherwise at risk for bleeding complications.165 The use of agents that are capable of restoring the dysfunctional anticoagulation pathways in patients with disseminated intravascular coagulation has been studied.165 The use of antithrombin concentrate, activated protein C, and recombinant factor VIIa are not currently recommended for use in pediatric septic shock.110,200
Systemic inflammation will invariably lead to activation of the coagulation system, but components of the coagulation system may markedly modulate the inflammatory response. Increasing evidence points to extensive cross-talk between the coagulation and inflammation systems at various points, with tissue factor, thrombin, components of the protein C pathway and fibrinolytic activators, and inhibitors playing pivotal roles. Increased insight into the molecular mechanisms that play a role in the close relationship between inflammation and coagulation could lead to the identification of new targets for therapies that can modify excessive activation or dysregulation of these systems.166
Extracorporeal Membrane Oxygenation (ECMO)
ECMO is used in septic shock in children but its effect is not clear. Survival from refractory shock or respiratory failure associated with sepsis is 80% in neonates and 50% in children.71 In one study of 12 ECMO patients with meningococcal sepsis, 8 of 12 patients survived, with six functionally normal at a median of 1 year of follow-up.108 One multivariate analysis of long-term survivors of ECMO found no significant difference between the performance of children with sepsis and those treated for other causes (i.e., without sepsis).205
Cardiogenic Shock
Etiology and Definition
Cardiac shock is the pathophysiologic state in which an abnormality of cardiac function is responsible for failure to maintain adequate tissue oxygenation.58,245 The final common pathway is depressed CO, which in most instances results from decreased myocardial contractility. Cardiogenic shock or severe congestive heart failure (CHF) during infancy and childhood represents a diagnostic and therapeutic challenge because of its myriad etiologies (Box 6-4). In the following section, the abbreviation CHF is used for severe CHF or cardiogenic shock.
Pathophysiology
In contrast to hypovolemic shock, compensatory responses can have deleterious effects in patients with cardiogenic shock.45,58,269,273 Compensatory responses are nonspecific and imprecise, and in patients with cardiogenic shock these responses may contribute to the progression of shock by further depressing cardiac function.
The Sympathetic Nervous System
The baroreceptor-mediated increase in sympathetic tone that occurs with ventricular dysfunction has several consequences, including increased myocardial contractility, tachycardia, and arterial vasoconstriction, and it can produce increased cardiac afterload as well as venoconstriction and increased cardiac preload.269
Increased local and circulating concentrations of norepinephrine can contribute to myocyte hypertrophy; this can occur directly through stimulation of α1- and β-adrenergic receptors or secondarily by activating the renin-angiotensin-aldosterone system. Norepinephrine is directly toxic to myocardial cells, an effect mediated through calcium overload, the induction of apoptosis, or both.216,269 Norepinephrine-induced death of myocytes can be prevented by concomitant, nonselective β-adrenergic blockade or combined β- and α-adrenergic blockade. In the past, β-adrenergic blockade was thought to be contraindicated in patients with heart failure. However, if patients can tolerate short-term β-adrenergic blockade, ventricular function subsequently improves.58,229,273,315 There are few data on the use of β-blockers in children with heart failure.273
Renin-Angiotensin-Aldosterone System
The activity of the renin-angiotensin-aldosterone system is increased in most patients with heart failure.269,273 As with plasma norepinephrine, the degree of increase in plasma renin activity provides a prognostic index in these patients.269 Patients with mild heart failure may have little or no increase in either plasma renin activity or the plasma aldosterone concentration. However, normal plasma renin and aldosterone values would be inappropriate in these patients because of their increased intracellular fluid and total blood volumes.251,310 Among patients with severe heart failure, the concentrations of plasma renin and aldosterone are high.
Aldosterone has an important role in the pathophysiology of heart failure. Aldosterone promotes the retention of sodium, loss of magnesium and potassium, sympathetic activation, parasympathetic inhibition, myocardial and vascular fibrosis, baroreceptor dysfunction, vascular damage, and impaired arterial compliance.251,310 Many physicians have assumed that inhibition of the renin-angiotensin-aldosterone system by an angiotensin converting enzyme (ACE) inhibitor will adequately suppress the formation of aldosterone. In addition, treatment with an aldosterone-receptor blocker in conjunction with an ACE inhibitor has been considered relatively contraindicated because of the potential for serious hyperkalemia. However, there is increasing evidence to suggest that ACE inhibitors only transiently suppress the production of aldosterone.251,310 Furthermore, treatment with the aldosterone-receptor blocker spironolactone, in conjunction with an ACE inhibitor, is effective and well tolerated and it does not lead to serious hyperkalemia.7,251
Other adverse consequences of long-term activation of the renin-angiotensin-aldosterone system in patients with CHF include a progressive remodeling of the heart and vasculature, which is mediated in part by the induction of various cytokines and growth factors.310 Most of these actions support or increase arterial BP and maintain glomerular filtration. Vasoconstriction and the release of aldosterone in response to angiotensin occur in seconds or minutes, in keeping with their roles of supporting the circulation after hemorrhage, dehydration, or postural change. Other actions, such as vascular growth and ventricular hypertrophy, require days or weeks.111
An ACE inhibitor is now considered first-line therapy for patients with heart failure.315 These drugs have improved survival in patients with chronic heart failure and all degrees of severity. Moreover, ACE inhibition reverses left ventricular hypertrophy, a common harbinger of heart failure.43,250 The role of angiotensin-converting enzyme inhibitors in the treatment of heart failure in children is much less clear.273
The amino acid sequence of angiotensin II has long been known, and compounds that competitively inhibit its action have been studied almost as long. The recently released angiotensin-receptor antagonist losartan appears to be beneficial in some patients with heart disease.111
Nonosmotic Release of Arginine Vasopressin
Water retention in excess of sodium retention can occur in patients with heart failure and lead to hyponatremia. In fact, hyponatremia is an ominous prognostic indicator in patients with heart failure.269 Hyponatremia may be partly due to the increased water intake caused by the increased thirst associated with heart failure. However, increased water intake alone rarely causes hyponatremia. In patients with heart failure and hyponatremia, hypoosmolality, which inhibits the release of arginine vasopressin in normal subjects, is associated with persistently high plasma concentrations of arginine vasopressin. This observation suggests a pivotal role for arginine vasopressin in hyponatremia. In preliminary studies of patients with heart failure, a nonpeptide, orally active antagonist of arginine vasopressin proved effective in reversing impaired urinary diluting capacity, increasing solute-free water excretion, and correcting hyponatremia.269
Natriuretic Hormone System
The natriuretic hormone system participates in the regulation of fluid balance and vascular resistance in healthy humans and in those afflicted with a variety of disease states.64 The natriuretic hormone system is composed of several structurally and functionally related peptides, primarily produced by the myocardium, vascular endothelium, and kidneys; they influence volume homeostasis and vascular tone by binding to dedicated receptors throughout the cardiovascular and renal system.64 These peptides induce natriuresis, diuresis, and vasodilation and specifically act to counter the effects of the renin-angiotensin-aldosterone system.217
The physiologic properties of the natriuretic peptides made them attractive candidates for heart failure therapy. Indeed, BNP (nesiritide) was found to be beneficial, and it was approved for treatment of acute decompensated CHF in the United States in 2001. However, responsiveness to natriuretic peptides decreases as heart failure worsens, even as the plasma concentrations of the peptides rise.168
Soon after the discovery of the physiologic role of these peptides, it became clear that the natriuretic peptides serve as biochemical markers for heart disease or shock.30,75,97,168,186,201,217 In comparative studies, BNP and its terminal prohormone fragment, N-terminal pro-BNP, were found to be superior to ANP as markers in heart failure and myocardial infarction.217
Endothelial Hormones
A crucial vascular structure is the endothelium, because it is strategically located between the circulating blood and the vascular smooth muscle and it is a source of a variety of mediators regulating vascular tone and growth as well as platelet function and coagulation.182 The endothelium is both a target for and a mediator of cardiovascular disease.
Prostacyclin and prostaglandin E are vasodilating hormones produced from arachidonic acid in many cells (see Septic Shock, Mediators of the Septic Cascade, and Evolve Fig. 6-1 in the Chapter 6 Supplement on the Evolve Website). Angiotensin II, norepinephrine, and renal nerve stimulation increase the synthesis of these vasodilating prostaglandins, which then attenuate the vasoconstrictor effects of these three stimuli.58 These vasodilatory prostaglandins may thus counterbalance the neurohormone-induced renal vasoconstriction that occurs in heart failure.
Nitric oxide is an even more potent vasodilator than prostacyclin and prostaglandin E. Endothelial cells contain a constitutive nitric oxide synthase, the activity of which may be blunted in heart failure.182,210 Thus, the constrictor action of the endogenous vasoconstrictors whose concentrations are elevated in heart failure—including angiotensin II, norepinephrine, and arginine vasopressin—may be enhanced by a concomitant decrease in nitric oxide synthesis in endothelial cells.
The endothelins are peptides of 21 amino acids that are produced in a wide variety of cells. Endothelin-1 is the only member produced in endothelial cells, and it is also produced in vascular smooth muscle cells.167 Hypoxia, shear stress, and hormones associated with the development of CHF stimulate the production of endothelin-1.167
Endothelin-1 can stimulate aldosterone secretion and decrease kidney perfusion and function, both of which contribute to the retention of sodium and water and increased intravascular volume. Endothelin-1 also stimulates hypertrophy of the ventricles and increases sympathetic activity and arterial vasoconstriction. Endothelin-1 is one of the most potent vasoconstrictors, and plasma endothelin concentrations are increased in some patients with heart failure.167,235 High plasma endothelin-1 concentrations are associated with a poor prognosis in patients with heart failure.235
Locally produced endothelin-1 has been implicated in closure of the ductus arteriosus at birth.167 Inhibition of the production and action of endothelin-1 by prostacyclin or prostaglandin E may prevent closure of the ductus.
Cytokines
It is becoming increasingly apparent that proinflammatory cytokines play an important role in modulating the structure and function of the diseased heart.59 The plasma concentrations of some cytokines, such as tumor necrosis factor alpha (TNF-α), are increased in patients with heart failure.59 TNF-α, which is produced under a variety of stresses, exerts negative inotropic effects, and can produce left ventricular remodeling, pulmonary edema, cardiomyopathy, and is a major mediator of apoptosis (cell death).59,318 Various cytokines and TNF-α, in particular, represent new targets for therapeutic intervention in patients with heart failure.
Ventricular Dilation and Hypertrophy
The ventricle responds to abnormal loading conditions by chamber dilation and hypertrophy. Ventricular dilation causes an increase in end-diastolic ventricular volume, which results in an increased SV and an improvement in CO. Another compensatory mechanism is hypertrophy of the myocardium.100 However, both these mechanisms increase oxygen requirements and make the heart more susceptible to ischemia.
Myocardial hypertrophy is an early milestone during the clinical course of heart failure and is an important risk factor for subsequent cardiac morbidity and mortality.136 In response to a variety of mechanical, hemodynamic, hormonal, and pathologic stimuli, the heart adapts to increased demands of cardiac work by increasing muscle mass through the initiation of a hypertrophic response. However, initiation of this hypertrophic response can result in heart muscle failure and the loss of myocytes as a result of programmed cell death (apoptosis).58,318
Diastolic Heart Failure
Another form of CHF and cardiogenic shock (see Fig. 6-3) is caused by diastolic dysfunction.16,115,312 Impaired myocardial relaxation changes the pressure-to-volume relationship during diastole and increases ventricular pressure at any volume. This lack of myocardial relaxation is hemodynamically unfavorable because increased left VEDP will be transmitted to the lungs and result in pulmonary edema and dyspnea. Such patients exhibit heart failure, but have normal left ventricular systolic function.100 Heart failure in the presence of a normal cardiac silhouette on the chest radiograph should suggest the possibility of diastolic dysfunction.
Diastolic heart failure is an insidious disease. Insults to the myocardium are followed by a series of compensatory changes that are beneficial in the short run, but have long-term deleterious effects. Structured remodeling and other factors, including myocardial ischemia, left ventricular hypertrophy, increased HR, and abnormal calcium flux can impair diastolic function and cause an increase in left ventricular filling pressure.16,115,312
Both ischemia and hypertrophy impair relaxation in early diastole. Ischemia reduces the supply of high-energy phosphates required for rapid removal of calcium from the cytoplasm and hypertrophy slows the rate of actin-myosin dissociation.312 Hypertrophy also decreases left ventricular compliance in all phases of diastole.312
When approaching a patient with cardiogenic shock, it is important to characterize both systolic and diastolic function. Therapy designed to improve systolic function may impair myocardial diastolic function.312
Right Ventricular Failure
Right ventricular (RV) failure can be defined as the inability of the right ventricle to provide adequate blood flow through the pulmonary circulation at a normal central venous pressure.114 RV failure may complicate the management of many pediatric critical care unit (PCCU) patients. Respiratory failure causing hypoxic pulmonary vasoconstriction, pulmonary hypertension, and consequent RV dysfunction is often seen in critically ill patients. Sepsis can cause right-sided heart failure directly by inducing RV dysfunction.161 Pulmonary embolism (PE) can result in RV failure. RV failure may be an independent risk factor for morbidity and mortality in patients with left ventricular failure.114 Finally, pulmonary arterial hypertension in children contributes significantly to morbidity and mortality in diverse pediatric cardiac, lung, hematologic, and other diseases.2 Without therapy, high pulmonary vascular resistance contributes to progressive RV failure, low CO, and death.2,41 Fundamental management strategies for pulmonary hypertension are listed in Box 6-5 (see also Chapter 8).225,293
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
6 Shock, Cardiac Arrest, and Resuscitation
Shock
Shock pearls
• Shock is a common cause of morbidity and mortality in pediatric patients with acute illness and injury.
• Early recognition of shock is essential for survival. Signs of shock in children may be more subtle than those in the adult and hypotension may not develop until late in the clinical course, when cardiovascular collapse is imminent.
• Treatment focuses on restoration of adequate oxygen delivery. Fluid therapy, inotropes, vasodilators, vasopressors, hydrocortisone, thyroid replacement, and extracardiac support devices may be required to accomplish this goal.
• Further shock treatment depends on the cause of the cardiovascular dysfunction and complications caused by the period of inadequate oxygen delivery.
Introduction
Shock is commonly defined as a clinical state characterized by an inadequate delivery of oxygen and metabolic substrates to meet the metabolic demands of the cells and tissues of the body.53,245,246 Inadequate delivery of oxygen results in cellular hypoxia, anaerobic metabolism, lactic acidosis, activation of the host inflammatory response, and eventual vital organ dysfunction.
Cardiovascular physiology and shock pathophysiology
Cellular Basis of Shock
Adenosine triphosphate (ATP) is the energy currency of the cell. Shock is a state of acute energy failure in which there is insufficient ATP production to support systemic cellular function.53 During stress and periods of increased energy demand, glucose is produced from glycogenolysis and gluconeogenesis. Fat metabolism is the secondary source of energy in this state. Long-chain fatty acids are oxidized, and carnitine is used to shuttle acetyl coenzyme A (CoA) into mitochondria. Protein catabolism can also contribute acetyl CoA to the Krebs cycle for energy production. However, this method of energy production is inefficient; aerobic metabolism provides 20-fold the energy produced by anaerobic metabolism. Glucose is oxidized to pyruvate via glycolysis (also called the Embden-Meyerhof pathway), generating only two molecules of ATP in the process.
In practical terms, using this operational definition, a state of shock may result from inadequate substrate delivery (glycopenia) or mitochondrial dysfunction (cellular dysoxia).53 Oxygen delivery to the cells and tissues depends primarily on three factors: hemoglobin (Hb) concentration, cardiac output (CO), and the relative proportion of oxygenated hemoglobin (i.e., percent saturation [SaO2]). Oxygen is transported in the blood combined with hemoglobin, although a relatively small amount is freely dissolved in the plasma fraction of the blood. When fully saturated at normal body temperature, each gram of hemoglobin can carry about 1.34 to 1.36 mL of oxygen. The normal arterial oxygen content is calculated as follows:
Oxygen delivery (DO2) is a product of arterial oxygen content and CO:
Adequate glucose delivery depends on the presence of adequate blood glucose concentration, normal blood flow (or CO), and an adequate concentration of insulin for cells with insulin-responsive glucose transporters (e.g., cardiomyocytes). Glycopenic shock can be caused by hypoglycemia and by extreme insulin resistance.53 Finally, even when oxygen delivery and glucose delivery are adequate, shock may occur as a result of mitochondrial dysfunction. For example, cyanide poisons the oxidative phosphorylation chain, preventing production of ATP. Cellular dysoxia (also known as cytopathic hypoxia) may theoretically occur from one or a combination of several mechanisms, including diminished delivery of a key substrate (e.g., pyruvate) to the Krebs cycle of the electron transport chain or uncoupling of oxidative phosphorylation.
Determinants of Oxygen Delivery
Normal CO
As defined previously, oxygen delivery is the product of CO and arterial oxygen content (CaO2). CO is the product of heart rate (HR) and stroke volume (SV; Fig. 6-1). SV, in turn, is dependent on preload, afterload, and contractility. Furthermore, blood pressure (BP) is determined by the product of CO and systemic vascular resistance (SVR).
Myocardial performance can be affected by changes in oxygenation, perfusion, serum ionized calcium concentration, acid-base, and electrolyte balance, sympathetic or vagal stimulation, and drugs.244 These factors can affect CO by altering either HR or SV.
Factors Influencing SV
Ventricular Preload
Preload is the amount of myocardial fiber stretch that is present before contraction. The significance of ventricular preload was first appreciated by Howell (in 1894), Frank (in 1894), and Starling (in 1914)242 in a series of experiments performed on isolated normal myocardial muscle preparations. Howell, Frank, and Starling observed that normal myocardium generates greater tension during contraction if it is stretched before contraction. This increase in the force of contraction occurs as a result of optimization of overlap between actin and myosin filaments in the sarcomere. These observations became known as the Frank-Starling law of the heart, which states that an increase in ventricular work, systolic tension, and SV results from an increase in presystolic stretch (preload). The graphic representation of the relationship between ventricular end-diastolic myocardial fiber length (usually approximated by ventricular end-diastolic pressure [VEDP]) and SV is the Frank-Starling curve, which is a ventricular (myocardial) function curve (Fig. 6-2).
To a point, as VEDP is increased, the force of contraction and myocardial fiber shortening should increase, and SV should rise.98 If, however, the ventricle is filled beyond a critical point, overlap of actin and myosin filaments is no longer optimal; ventricular dilation can result, and stroke volume decreases.47 Extremely high VEDPs (higher than approximately 25 cm H2O pressure when capillary permeability is normal, and lower pressures when capillary permeability is increased) result in pulmonary and systemic edema, and high pressures will compromise coronary and subendocardial blood flow.
A family of ventricular function curves can be constructed to illustrate the response of normal, depressed, or enhanced myocardial response to increased VEDP (see Fig. 6-2). If the patient demonstrates poor myocardial function, the ventricular function curve will be relatively flat, and a high VEDP will be required to produce even a modest improvement in myocardial function. If myocardial function is normal, a small increase in VEDP can produce a significant rise in SV or CO. If ventricular function is hyperdynamic, even nominal increases in VEDP will produce significant increases in SV or CO.
A goal of the treatment of any patient with cardiovascular dysfunction is to maximize SV and CO while minimizing adverse effects of fluid administration, such as pulmonary edema. An increase in SV and CO can be achieved by moving the patient to the highest point of an individual ventricular function curve (see Fig. 6-2) through judicious fluid administration. Improvement in SV and CO also can be achieved by altering the ventricular compliance, using vasodilator therapy. Further increase in SV and CO also can be achieved through improvement in cardiac contractility; this raises the ventricular function curve (see Fig. 6-2). Such an improvement can be attained by eliminating factors that normally depress myocardial function or by administering inotropic agents or vasodilators (discussed under Afterload).
RVEDP and CVP often can be estimated with careful clinical assessment of the level of hydration, liver size, palpation of the infant’s fontanelle, determination of presence (or absence) of systemic edema, and evaluation of the cardiac size on chest radiograph.275 Dry mucous membranes, a sunken fontanelle, and the absence of hepatomegaly are findings consistent with a normal or low central venous pressure. Hepatomegaly and periorbital edema usually are present once the CVP is elevated significantly. Systemic edema also may be noted despite a normal or low CVP if capillary leak or hypoalbuminemia is present. A high RVEDP and heart failure is often associated with cardiac enlargement on chest radiograph.
Left VEDP (LVEDP) is equal to left atrial pressure unless mitral valve disease is present. Reliable estimation of LVEDP is not possible through clinical assessment alone.275 Although the presence of pulmonary edema frequently is assumed to indicate the presence of a high LVEDP (exceeding 20 to 25 mm Hg), pulmonary edema may be observed at any (even a low) LVEDP if capillary leak is present.
Ventricular Compliance
Ventricular compliance refers to the distensibility of the ventricle. It is defined as the change in ventricular volume (in milliliters) for a given change in pressure (in millimeters of mercury), or ΔV/ΔP, and can be depicted graphically by a ventricular compliance curve (Fig. 6-3). The opposite of compliance is stiffness (ΔP/ΔV).
If the ventricle is extremely compliant, a large volume of fluid may be administered without producing a significant increase in VEDP (see Fig. 6-3, curve B). If the ventricle is dysfunctional (as occurs with restrictive cardiomyopathy) or hypertrophied, ventricular compliance usually is reduced (see Fig. 6-3, curve C). In this case, even a small volume of administered intravenous fluids will produce a significant rise in VEDP.268 The more dysfunctional and noncompliant the ventricle, the higher the resting VEDP and the VEDP needed to optimize SV and ventricular performance (see Fig. 6-2).
Ventricular compliance is not constant over all ranges of VEDP. Any ventricle is maximally compliant at low filling pressures. As the ventricle is filled, compliance is reduced because ventricular stretch may be maximal.55,169 Rapid volume infusion tends to raise VEDP more rapidly than gradual volume infusion. Compliant ventricles usually demonstrate a substantial improvement in SV when an intravenous fluid bolus is administered.
Vasodilator therapy will improve ventricular compliance. When these drugs are administered the compliance curve is altered, so a greater end-diastolic volume may be present without a substantial increase in VEDP (see Fig. 6-3). SV may then be increased without a rise in VEDP.
Compliance also is affected by ventricular size, pericardial space, and HR.55 Infants have small and relatively noncompliant ventricles, so the infant’s VEDP may rise sharply with minimal fluid volume administration. If the same volume (on a per-kilogram basis) is administered to an older child, a smaller change in VEDP will result because the ventricles are larger and more compliant in older children.
Ventricular Contractility
Although contractility can be measured in the laboratory, it is not easily isolated and measured in the clinical setting. The most common method of evaluating contractility at the bedside is echocardiographic evaluation of fiber-shortening times and measurement of the shortening fraction of left ventricular diameter. Shortening fraction is calculated by determining the difference between the end-diastolic and end-systolic dimensions. Normal shortening fraction is approximately 28% to 44%.206
If a thermodilution CO pulmonary artery catheter is in place, or if reliable Doppler CO estimations can be obtained, the nurse can create a ventricular function curve (see Fig. 6-2). If CO improves with no change in VEDP or HR, ventricular contractility or compliance has probably improved.
Afterload
The major determinants of ventricular afterload or wall stress are: ventricular lumen radius, ventricular wall thickness (note that hypertrophy decreases afterload), and the ventricular intracavitary ejection pressure (Fig. 6-4). In the absence of left ventricular outflow tract obstruction (e.g., aortic stenosis), left ventricular ejection pressure will equal aortic and systemic arterial pressure. In the absence of right ventricular outflow tract obstruction (e.g., pulmonary stenosis), right ventricular ejection pressure will equal pulmonary arterial pressure. Systemic and pulmonary artery pressures in turn are determined by blood flow and resistance. Therefore in the absence of ventricular outflow tract obstruction or significant alterations in ventricular size or wall thickness, ventricular afterload is related primarily to the impedance provided by the pulmonary and systemic arterial circulations.
Oxygen Delivery
The ultimate function of the heart and lungs is to deliver oxygenated blood to the tissues. Systemic DO2 is the volume of oxygen (in milliliters) delivered to the tissues per minute. DO2(I) is the volume of oxygen delivered to the tissues per minute, indexed to body surface area (BSA), so that the units are milliliters per minute per square meter. DO2 is the product of arterial oxygen content (the amount of oxygen in arterial blood in milliliters per deciliter) the CO (in liters per minute), and a factor of 10. DO2(I) is the product of arterial oxygen content (in milliliters per deciliter), the cardiac index (in liters per minute per square meter), and a factor of 10 (see Fig. 6-1).
Under resting conditions with normal distribution of CO, oxygen delivery is more than adequate to meet the total oxygen requirements the tissues need to maintain aerobic metabolism, which is referred to as oxygen consumption (VO2). Excess oxygen delivery or oxygen reserve serves as a buffer, so a modest reduction in oxygen delivery will be more than adequately compensated by increased extraction of the delivered oxygen, without any significant reduction in oxygen consumption. During stress or vigorous exercise, oxygen consumption markedly increases, as does oxygen delivery. Therefore, under most conditions, the metabolic demands of the cells and tissues of the body dictate the level of oxygen delivery. However, little oxygen is stored in the cells and tissues of the body; therefore as oxygen delivery falls with critical illness, oxygen extraction must necessarily increase to meet metabolic demands, and oxygen consumption remains relatively constant (i.e., it is delivery independent; Fig. 6-5). However, there is a critical level of oxygen delivery at which the body’s compensatory mechanisms are no longer able to meet metabolic needs (i.e., the point at which oxygen extraction is maximal). Once oxygen delivery falls below this level, oxygen consumption must also fall and is said to become delivery dependent (see Fig. 6-5, Normal critical delivery threshold).
If either arterial oxygen content or CO falls without a commensurate and compensatory increase in the other component, oxygen delivery will fall (Fig. 6-6, A–H). For example, if arterial oxygen content falls (e.g., caused by a fall in hemoglobin concentration or its saturation), oxygen delivery can be maintained by a commensurate rise in CO. If CO falls, however, oxygen delivery will fall in direct correlation, because there is no way for arterial oxygen content to increase when CO falls.
Oxygen Content
The optimal hemoglobin threshold for erythrocyte (red blood cell) transfusion in critically ill children is unknown.160 Whereas transfusion is indicated in conditions of hemodynamic instability and anemia (hemoglobin <10 g/dL), once stability is achieved the threshold hemoglobin level for transfusion may need to be lowered. A recent multicenter trial demonstrated that in stable, critically ill children a hemoglobin threshold of 7 g/dL for red blood cell transfusion decreased transfusion requirements without increasing adverse outcome.160
Cardiac Output
CO is the volume of blood ejected by the heart in 1 minute. CO is the product of HR and SV. CO may be recorded in liters per minute or milliliters per minute, although in children it is often normalized to BSA and recorded as the cardiac index (in milliliters per minute per square meter BSA). Normal cardiac index averages 3 to 4.5 mL/min per m2 BSA (Table 6-1).
Clinical Assessment of CO
Compensatory vasoconstriction can initially maintain BP (BP = CO × SVR). In fact, infants and children have high SVR and vasoactive capacity such that hypotension is a late sign of shock.53,245 This survival mechanism is designed to counterbalance the limited cardiac reserve in children.
Calculation and Estimation of CO
CO can be calculated using a thermodilution catheter placed in the pulmonary artery. Injection of an iced quantity of fluid into the right atrium will produce a temperature change in the pulmonary artery that is inversely related to the CO. Such thermodilution CO calculations can be performed using a pulmonary artery catheter with thermistor or a separate thermistor placed into the pulmonary artery at the time of cardiac surgery. Additional information about thermodilution CO calculation can be found in Chapter 21.
The CO can be continuously evaluated if the pulmonary artery catheter contains an oximeter for continuous evaluation of pulmonary artery (mixed venous) oxyhemoglobin saturation. Pulmonary artery catheterization is not commonly performed in pediatric critical care, although it may be indicated in certain situations that complicate shock resuscitation.276
Pulse contour intermittent and continuous CO (PiCCO®) monitoring is possible using a femoral or axillary arterial catheter and a central venous catheter. Algorithms use pulse contour analysis combined with intermittent thermodilution calculations. The arterial waveform contours are analyzed to assess SV and thereby cardiac index (CI). In a pediatric validation study, the PiCCO system provided continuous calculation of CI without the need to perform thermodilution injections with every calculation.90
Esophageal Doppler studies of blood flow velocity in the descending aorta have been used in adults for many years, and pediatric probes are now available. In the past, the higher HRs in children compromised reliability of this technique of CO evaluation. Fairly reliable CO calculations have recently been reported in children,270 although this method of CO evaluation is very operator dependent.3
CO and blood volume can be calculated from a form of ultrasound dilution. This technology uses a computer and arterial and venous sensors attached to a specialized extracorporeal tubing loop that is placed between an indwelling central venous and arterial catheter. A small bolus (0.5 to 1.0 mL/kg) of isotonic saline is injected into the central venous catheter to create a dilution curve. The saline dilutes the total blood protein concentration and alters blood velocity detected by the arterial sensor in the extracorporeal loop. The change in blood velocity over time can be graphed, and results in a dilution curve that is similar to that created during thermodilution CO calculations. In addition to calculating the CO and index, the ultrasound dilution computer calculates SV index, active circulating volume, SVR, and ejection fraction. This technology has been validated in neonates and children.157
Noninvasive measurement of CO in children is novel and evolving.128 Doppler ultrasonography is a simple, noninvasive method of assessing blood flow and is an accepted noninvasive method of deriving CO.118 Data obtained with Doppler ultrasonography correlate well with accepted standard hemodynamic methods in animal studies, adults, and neonates; formal validation is underway in children.37,65,128
Because adequate organ perfusion is the goal of supportive and therapeutic critical care, monitors that directly assess organ oxygenation offer the best possibility of improved recognition and treatment of circulatory abnormalities to reduce multiorgan dysfunction and related morbidity and mortality.260 Some of these monitors are presented in the next several sections
Relationship of Mixed Venous Oxygen Saturation to CO
A true mixed venous oxygen saturation is evaluated from blood in the pulmonary artery, because it includes a true mixed sample of systemic venous blood from the superior vena cava, inferior vena cava, and coronary circulations. The mixed venous oxygen saturation can be continuously monitored through the use of a pulmonary artery catheter that contains a pulmonary artery oximeter. If hemoglobin concentration and saturation and oxygen consumption or extraction are stable (these assumptions often cannot be made about the critically ill patient), the mixed venous oxygen saturation will vary directly with the CO (see Fig. 6-6).
When oxygen delivery falls, tissues compensate by increasing the amount of oxygen extraction from circulating blood.196 Mixed venous oxygen saturation is thus used as a measure of the balance between oxygen delivery and oxygen demand, and it will fall as the result of either decreased oxygen delivery (i.e., decreased arterial oxygen content; decreased CO, index, or both) or increased oxygen extraction (from increased demand, decreased delivery, or both). The normal mixed venous oxygen saturation is greater than 70%.196 Mixed venous oxygen saturation levels between 50% and 75% reflect compensatory increased extraction in cases of increased oxygen demand or decreased oxygen delivery, whereas levels between 30% and 50% reflect the beginning of lactic acidosis indicating exhaustion of compensatory extraction. A further decline to levels between 25% and 30% is indicative of severe lactic acidosis, and levels less than 25% probably reflect cellular death.196
The mixed venous oxygen saturation has been shown to be an effective indicator of the balance between oxygen supply and demand, and it is a useful guide for management in adults. However, its use is limited in pediatrics by the need for a pulmonary artery or central venous catheter for intermittent or continuous measurement; this is often not feasible, especially early in resuscitation.183
Use of Central Venous Oxygen Saturation Monitoring
The desire for easier venous oxygen saturation measurement has led to an assessment of central venous oxygen saturation (ScvO2). Unlike the pulmonary artery catheter samples that are composed of blood from all parts of the systemic venous circulation, including blood from the coronary sinus, ScvO2 is obtained from the superior vena cava, so it assesses only the oxygen delivery-oxygen consumption balance in the upper body.196 Many studies have examined how the two are related. Oxygen saturation of superior vena cava blood is approximately 70%, which is slightly lower than the saturation of blood in the inferior vena cava (75%). Coronary sinus blood, with an oxygen saturation of approximately 30% to 40%, is then added to the combined superior vena cava and inferior vena cava blood, so that a true mixed venous sample in the pulmonary artery (after mixing is complete) averages approximately 70% to 75% and corresponds to a mixed venous oxygen tension of 38 to 40 mm Hg. Thus the ScvO2 is normally approximately 2% to 3% higher than mixed venous oxygen saturation in healthy individuals.183 In shock states, however, the difference can range from 5% to 18%.37,183 Some studies have shown that although the two are not interchangeable, there is a good correlation in the trending of one to the other, and the ScvO2 correlates well with changes in systemic perfusion.81,132,183
Limitations of continuous ScvO2 monitoring are significant. Because sympathetic tone raises vascular resistance in splanchnic-mesenteric beds as CO falls, in shock states the effects of desaturated blood from those regions on ScvO2 may be blunted. As an extreme example, the renal blood flow may fall to 20% of its normal value because of intense renal vasoconstriction, with renal vein saturation falling from 85% to 25% while the ScvO2 remains above 60%.132
Near-Infrared Spectroscopy (NIRS)
NIRS is relatively new technology that is used to access regional tissue oxygenation as an indicator of tissue perfusion. Near-infrared light (700 to 1000 nm bandwidth) is transmitted through muscle, bone, tissue, and skin, and reflected light is then read by a sensor.183 The technology is similar to that used for pulse oximetry and continuous monitoring of ScvO2. This technology uses the difference in light absorption between deoxygenated hemoglobin versus oxygenated hemoglobin to calculate a percent saturation level. Because most blood is not in arteries but in capillaries and veins, the greatest quantitative contribution to the calculated light absorption of hemoglobin is from venous and capillary blood. Pulse oximeters attempt to subtract the nonpulsatile component of the light signal (i.e., contributed from veins and capillaries) to examine the absorption spectrum of arterial blood, whereas NIRS technology focuses on the total light signal. An NIRS device approved by the U.S. Food and Drug Administration (INVOS; Somanetics, Troy, MI) uses a dual-detector system to subtract a shallow light path from a deep light path, theoretically allowing the derivation of the average oxyhemoglobin saturation in a volume of tissue approximately 2.5 to 3.0 cm deep under the skin and the sensor. The device displays an approximation of venous-weighted hemoglobin saturation in tissue deep below the sensor. The parameter is displayed as a relative number from 0% to 100% using an algorithm calibrated from in vivo and in vitro cerebral models, and is termed regional oxygen saturation (rSO2).132
Because venous-weighted capillary blood represents the limiting oxygen tension for diffusion, NIRS provides a window to evaluate the oxygen economy in the monitored tissue bed or regional tissue oxyhemoglobin saturation.91,132 The use of NIRS technology to monitor oxygenation in the brain, muscle, liver, and kidney has been extensively described.91,132,133,228 Changes in tissue oxygen tension monitored by NIRS are sensitive indicators of perfusion-metabolism coupling, and regional NIRS monitoring can guide resuscitation from shock.66,132
The indication approved by the U.S. Food and Drug Administration for the INVOS device is for trend monitoring of regional saturation in the cerebral circulation, which has been widely modeled as a compartment with 75% venous blood, thus allowing validation of the device in an accepted clinical model. In animal models, brain rSO2 less than 40% is associated with intracellular anaerobic metabolism and depletion of high-energy phosphates.158 Clinical data in children and adults support the hypothesis that cerebral rSO2 less than 40% to 50%, or a change in baseline of more than 20%, is associated with hypoxic-ischemic neural injury.132,133,228
Blood Pressure
Arterial BP may be the most widely used and most widely misinterpreted parameter related to CO. Arterial BP is generated by the kinetic energy imparted by the heart and the interaction of blood flow, viscosity, systemic vascular tone, and downstream (venous) pressure. The major determinants of mean arterial BP are the SVR and CO. Peripheral and central arterial pressure differences result from differential impedances in the vascular system and are increased with nonuniform vasoconstriction. Noninvasive BP measurement by auscultation using a cuff and sphygmomanometer (mercury manometer) and noninvasive oscillometric BP measurements correlate well with indwelling intraarterial measurements under conditions of normal vascular resistance and blood flow, but “cuff” and invasive pressures frequently diverge under conditions of low and high SVR.132 Automated determination by cuff oscillometry shows good reproducibility and allows for determination of the mean pressure.
Although BP is rapidly and routinely measured, its relationship to CO or systemic perfusion is confounded by simultaneous changes in SVR. SVR tends to change in the opposite direction from CO; the sympathetic nervous system is activated by a falling BP, and sympathetic outflow is reduced and the parasympathetic system is activated with a rising BP. These responses are designed to minimize the variability in BP, making this parameter a late indicator of failing circulation. Similarly, resuscitation to a BP end point is often incomplete if the goal is to restore adequate CO and perfusion.276
Blood flow through a regional vascular bed is directly proportional to the organ perfusion pressure (ΔP), which is calculated as the difference between the arterial inflow pressure (Pa) and the venous outflow pressure (Pv): ΔP = Pa − Pv.53 With reasonable approximation and ignoring local extravascular effects, the inflow arterial pressure can be estimated to be the mean arterial pressure (MAP), and the outflow venous pressure can be estimated to be CVP:
Under ideal laminar flow conditions, vascular resistance is independent of flow and pressure; therefore an increase in vascular resistance will decrease blood flow, and a decrease in vascular resistance will increase blood flow for any given ΔP. Control mechanisms in the body generally maintain arterial and venous BPs within a narrow range; therefore changes in organ and tissue blood flow are primarily regulated by changes in vascular resistance. Resistance to blood flow within a vascular network is determined by the size of the vessels, the organization of the vascular network, physical characteristics of the blood, and extravascular forces acting on the vasculature.53
Shock states
Shock is a progressive condition of circulatory failure that results in inadequate CO and oxygen and substrate delivery to the tissues. Shock may be present in the patient with a low, normal, or high CO or index.245
Hypovolemic Shock
Etiology
Hypovolemia is the most common cause of shock in infants and children.245 Hypovolemic shock is best defined as a sudden decrease in the intravascular (blood) volume, relative to vascular capacity (i.e., inadequate intravascular volume relative to the vascular space), to such an extent that effective tissue perfusion cannot be maintained. Causes include hemorrhage from gastrointestinal disease or trauma, fluid and electrolyte loss, endocrine disease, and plasma loss.
Clinical Signs and Symptoms
Reliable indicators of early, compensated hypovolemic shock in children are persistent tachycardia, cutaneous vasoconstriction, and diminution of the pulse pressure (difference between systolic and diastolic BPs). Clinical evidence of decreased tissue perfusion includes skin mottling, prolonged capillary refill, and cool extremities. Systemic arterial BP is frequently normal, which is the result of increased SVR, making BP monitoring of limited value in managing the patient with compensated hypovolemic shock.132,245 Neurologic status is initially normal or only minimally impaired.
[/not-level-membership-for-critical-care-medicine-category]
