Shock
Perspective
In philosophic terms, shock can be viewed as a transition between life and death. Whether shock results from hemorrhage, sepsis, or cardiac failure, mortality rates exceed 20%.1,2 In scientific lexicon, shock results from the widespread failure of the circulatory system to oxygenate and nourish the body adequately. In the laboratory the scientist defines the metabolic effect of shock quantitatively, by examining the mechanisms by which shock alters mitochondrial energy transfer, evokes the production of toxic chemicals, and reduces their removal. At the bedside, however, the clinician identifies shock by linking the clinical impression, synthesized from the patient’s history of present illness, age, underlying health status, and general appearance, to quantitative data, including vital signs, blood chemistry, urine output, and direct measurements of oxygenation. When the clinical impression and the quantitative data suggest widespread organ hypoperfusion, emergent resuscitation is used to restore normal tissue oxygenation and substrate delivery to prevent deterioration into systemic inflammation, organ dysfunction, and death.
At the subcellular level, shock first affects the mitochondria. Mitochondria function at the lowest oxygen tension in the body, but paradoxically, they consume almost all the oxygen used by the body. More than 95% of aerobic chemical energy comes from mitochondrial combustion of fuel substrates (fats, carbohydrates, ketones) plus oxygen into carbon dioxide (CO2) and water. Mitochondria therefore have been referred to as the “canaries in the coal mine” because they are affected first in conditions of inadequate tissue perfusion.3,4 When mitochondria have inadequate oxygen, the cell catabolizes fuels to lactate, which inexorably accumulates and diffuses into the blood.
Classification
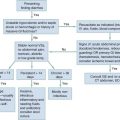
For years, shock has been classified into four broad categories based on Blalock’s 1934 description: hematologic, neurologic, vasogenic, and cardiogenic. This basic organization scheme remains useful today. Box 6-1 outlines five categories of shock that generally have specific mechanisms and treatments.
Epidemiology
The epidemiology of shock in the emergency department context remains speculative because shock is rarely listed as a primary coding diagnosis and depends on defining criteria. Arterial hypotension, defined as a systolic blood pressure (BP) below 100 mm Hg, is measured at least one time in 19% of ED patients5; however, diagnosed traumatic, cardiogenic, or septic shock is less common, constituting about 1 to 3% of all ED visits.
Specific Causes
The second phase of organ injury from hemorrhagic shock occurs during resuscitation. It has been said that the acute phase of hemorrhage “cocks the gun” by initiating the inflammatory cascade, and resuscitation “pulls the trigger” by accentuating the inflammation-induced organ injury from hemorrhagic shock. During resuscitation, neutrophils become most aggressive, binding to the lung endothelium and causing capillary leaks that characterize acute respiratory distress syndrome (ARDS). Inflammatory cytokines are liberated during resuscitation, and membrane injury occurs in many cells. In the liver, damage from inflammation and reactive oxygen species from neutrophils is compounded by persistent microischemia. During resuscitation from hemorrhagic shock, the normal balance of vasodilation by nitric oxide (NO) versus vasoconstriction by endothelins becomes distorted, producing patchy centrilobular ischemic damage in the liver, which may produce an immediate rise in blood transaminase levels. A growing body of evidence suggests that resuscitation from hemorrhage exerts greater injury on the heart than the actual hypotensive insult.6 Depending on the degree of hypotensive insult, the kidney may manifest acute spasm of the preglomerular arterioles, causing acute tubular necrosis. Systemic metabolic changes can impair fuel delivery to the heart and brain, secondary to depressed hepatic glucose output, impaired hepatic ketone production, and inhibited peripheral lipolysis.
Septic Shock
1. More patients are being treated at home for chronic immunocompromising diseases with indwelling catheters, which can serve as portals of entry into the vascular space for Staphylococcus aureus and coagulase-negative staphylococci.
2. The frequency of community-acquired infections caused by antibiotic-resistant gram-positive organisms has greatly increased in recent years, including infections caused by S. aureus, Streptococcus pneumoniae, and Streptococcus pyogenes.
Septic shock often causes three major effects that must be addressed during resuscitation: relative hypovolemia, cardiovascular depression, and induction of systemic inflammation. Septic shock produces relative hypovolemia from increased venous capacitance, which reduces right ventricular filling. Septic shock often causes absolute hypovolemia from gastrointestinal volume losses, tachypnea, sweating, and decreased ability to drink during development of the illness. Sepsis also induces capillary leak, which leads to relative loss of intravascular volume into third spaces. Recent evidence has shown that septic shock causes myocardial depression simultaneously with vasodepression and capillary leak. Direct measurements of cardiac contractility have shown that cardiac mechanical function becomes impaired early in the course of septic shock, even in the hyperdynamic stages. Multiple mechanisms may explain depressed heart function in sepsis, including actions of specific cytokines (most notably tumor necrosis factor alpha [TNF-α] and interleukin 1 beta [IL-1β]), overproduction of NO by nitric oxide synthase (iNOS),7 and possibly impairment in mitochondrial oxidative phosphorylation coincident with reduced mechanical efficiency.8,9 Evidence indicates that circulating mediators, myocardial cellular injury from inflammation, and deranged metabolism interact synergistically to injure the heart during septic shock. Systemic inflammation causes capillary leak in the lung, which may cause alveolar infiltration characteristic of ARDS early in the treatment of septic shock in up to 40% of patients. With the potential for early development of ARDS, more profound ventilation-perfusion (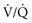 ) mismatching, and pneumonia or pulmonary aspiration, hypoxemia is more severe with septic shock than hemorrhagic shock.
) mismatching, and pneumonia or pulmonary aspiration, hypoxemia is more severe with septic shock than hemorrhagic shock.
Cardiogenic Shock
Cardiogenic shock results when more than 40% of the myocardium undergoes necrosis from ischemia, inflammation, toxins, or immune destruction. Otherwise, cardiogenic shock essentially produces the same circulatory and metabolic alterations as are observed with hemorrhagic shock. Undoubtedly, impaired baseline cardiac function can contribute to the development of circulatory shock secondary to infection, hemorrhage, or vasodilatory drug overdose. However, when shock results from a pure cardiac cause, severe left ventricular dysfunction will be evident on echocardiography early in the course. Patients with severe dysfunction are far more likely to have a cardiogenic cause of shock than patients with normal or moderate left ventricular dysfunction.10
Clinical Features
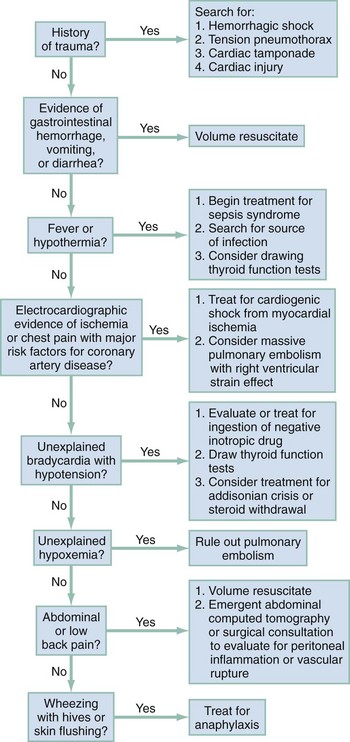
Patients in the ED frequently are in shock with no obvious cause. Rapid recognition of shock requires the integration of information from immediate history and physical examination, and shock can be strongly supported by the presence of a worsening base deficit or lactic acidosis. In general, patients with shock exhibit a stress response: they are ill appearing, asthenic, pale, often sweating, and usually tachypneic or grunting, and often have a weak and rapid pulse (Box 6-2). HR can be normal or low in shock, especially in cases complicated by prescribed drugs that depress HR or by profound hypoxemia. BP initially can be normal because of adrenergic reflexes. Although arterial BP as a sole measurement remains an unreliable marker of circulatory status, the finding of a single systolic BP less than 100 mm Hg in the ED is associated with a threefold increase in in-hospital mortality and a tenfold increase in sudden and unexpected death.5 The HR/systolic BP ratio may provide a better marker of shock than either measurement alone; a normal ratio is less than 0.8. Urine output provides an excellent indicator of organ perfusion and is readily available with insertion of a Foley catheter into the bladder. Measurement of urine output, however, requires 30 minutes to 1 hour for accurate determination of whether output is normal (>1.0 mL/kg/hr), reduced (0.5-1.0 mL/kg/hr), or severely reduced (<0.5 mL/kg/hr). Point measurements of the arterial or venous lactate concentration and the base deficit can be rapidly performed and provide accurate assessment of global perfusion status. A lactate concentration greater than 4.0 mM or a base deficit more negative than −4 mEq/L predicts the presence of circulatory insufficiency severe enough to cause subsequent multiple organ failure.11,12 Once the empirical criteria for circulatory shock have been discovered, the next step is to consider the cause of the shock. Figure 6-1 shows a potential sequence of decisions to help arrive at a diagnosis in a patient with undifferentiated shock.
The history, vital signs, and physical examination documented by prehospital providers afford valuable insight into a patient’s physiologic status before any medical intervention and can be useful in ED management. Studies suggest that both medical and trauma patients with prehospital hypotension have a threefold to fourfold higher in-hospital mortality rate than patients without hypotension.13,14
Laboratory, radiographic, and other ancillary data should be ordered to assess tissue and vital organ perfusion and to diagnose injury from trauma, find the source of infection with sepsis, or identify the cause of cardiac failure. A chest radiograph, electrocardiogram, finger-stick glucose measurement, complete blood count (CBC), urinalysis, serum electrolytes, and kidney and liver function tests are all indicated in the ED assessment. Arterial blood gases are ordered for a base deficit calculation and to correlate arterial gas tensions (oxygen [PaO2] and PaCO2) with those measured by pulse oximetry and capnography. Serum lactate measurement should be performed as early as possible in patients with suspected shock. Either venous or arterial lactate concentrations can be used.15,16 If peripheral venous lactate is used, the effect of time, storage temperature, and tourniquet use have no significant effect on in vitro lactate production by erythrocytes if the measurement is done within 15 minutes after the sample has been obtained.17 Cardiac and abdominal bedside ultrasound scanning can screen for inadequate central venous volume, occult hemoperitoneum, abdominal aortic aneurysm, left ventricular failure, and cardiac tamponade.18 A systematic ultrasound protocol can significantly improve the physician’s ability to accurately diagnose the cause of undifferentiated shock in ED patients, and the finding of hyperdynamic left ventricular function in patients with undifferentiated shock strongly suggests sepsis as the cause.19,20
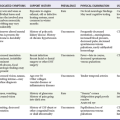
Consensus definitions of shock show the spectrum of hypoperfusion for the following three common causes of shock (Box 6-3):
1. Septic shock. The American College of Chest Physicians, European Society of Intensive Care Medicine, Society for Critical Care Medicine, American Thoracic Society, and Surgical Infection Society developed international consensus definitions to distinguish septic shock from its precursor conditions—systemic inflammatory response syndrome (SIRS), sepsis, and severe sepsis.21 Although this particular consensus definition requires persistent hypotension after fluid resuscitation to strictly define septic shock, initiation of treatment for empirically diagnosed severe sepsis or septic shock should not await the onset of hypotension. The incorporation of an indicator of tissue hypoperfusion into the clinical assessment may improve identification of hypoperfusion, particularly in subtle cases. Box 6-4 provides a list of variables that can assist with detecting tissue hypoperfusion.22
2. Hemorrhagic shock. The American College of Surgeons has divided hemorrhagic shock into four stages, depending on the severity of blood loss and the physiologic response to this loss, but such arbitrary divisions are of little value. A more useful approach defines hemorrhagic shock as being present when systemic hypoperfusion manifests as lactic acidosis with organ dysfunction.
3. Cardiogenic shock. Cardiogenic shock should be thought to be present whenever cardiac failure (ischemic, toxic, or obstructive) causes systemic hypoperfusion that manifests as lactic acidosis with organ dysfunction.
Management
Most patients with shock can be fully resuscitated with peripheral venous access established with two catheters of at least a size 18 gauge. Patients with cardiac failure or renal failure may benefit from closer measurement of dynamic variables of fluid responsiveness that can be measured from an arterial line (such as stroke volume variation or stroke volume index) or a central venous line (central venous pressure [CVP]).23 An 8.5-French catheter (Cordis sheath) allows for accurate measurement of the CVP and insertion of a pulmonary artery catheter or other monitoring device if needed. In children, a 3- or 5-French bilumen catheter can be placed in the femoral vein with few complications. To reduce the potential for limb damage from extravasation from a peripheral intravenous injection, vasoactive medications are optimally administered through a central venous catheter. If vasoactive medications are administered, additional peripheral intravenous catheters will be required for infusion of crystalloid and other treatments. Many patients with renal disease or cancer have indwelling catheters in place. In patients with empirical criteria for shock, this catheter should be used for intravenous access, unless satisfactory access has already been established at other anatomic sites. In EDs where the standard practice is not to use these ports at the request of other physicians, a specific hospital policy and training session should be developed to make an exception in the case of circulatory shock. In general, failure to administer fluids rapidly and in sufficient quantity outweighs considerations about preservation of the line for future therapy.
Quantitative Resuscitation
Quantitative resuscitation (also called goal-directed therapy, goal-oriented resuscitation, or hemodynamic optimization) was first described in 1988 and refers to the practice of resuscitating patients to predefined physiologic endpoints indicating that systemic perfusion and vital organ function have been restored. Since that time, many studies have evaluated the efficacy of such a therapeutic approach to shock, and a meta-analysis of these studies confirms its benefit for reducing mortality.24,25 For many years in the intensive care unit (ICU), physicians have relied on the use of the pulmonary artery catheter to help optimize left ventricular filling indices, but this practice is controversial. Several randomized controlled trials investigating the management of critically ill patients failed to demonstrate survival or length of stay benefit in patients managed with pulmonary artery catheters.26–30 Insufficient data have been published to support the use or avoidance of pulmonary artery catheters in ED populations, but their significant complication rate, coupled with uncertain or no benefit, argues strongly against their routine use.
The lactate clearance refers to serial measurements of venous or arterial lactate and is calculated according to the following formula31–33:

Lactate clearance has been shown to be equivalent to central venous oxygen saturation as an endpoint of early septic shock resuscitation.34 Given the increasing use of point-of-care testing platforms in the ED and the fact that lactate clearance measurements can be done from peripheral venous blood, in many patients this may be a preferred endpoint of resuscitation. If the lactate concentration has not decreased by 10 to 20% 2 hours after resuscitation has begun, additional steps are undertaken to improve systemic perfusion. Resuscitation should continue until the lactate concentration drops below 2 mM/L.
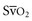 ) measurements reflect the balance between oxygen delivery and oxygen consumption. Previous studies have suggested that the
) measurements reflect the balance between oxygen delivery and oxygen consumption. Previous studies have suggested that the 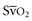 can be used as a surrogate for CI in targeting normalization of endpoints (
can be used as a surrogate for CI in targeting normalization of endpoints (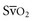 65%, or CI 2.5-3.5 L/min/m2) for therapeutic intervention in critically ill patients. Although
65%, or CI 2.5-3.5 L/min/m2) for therapeutic intervention in critically ill patients. Although 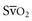 requires the use of a pulmonary artery catheter, the central venous oxygen saturation (Scv
requires the use of a pulmonary artery catheter, the central venous oxygen saturation (Scv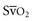 when changes or trends in the values are tracked over time.
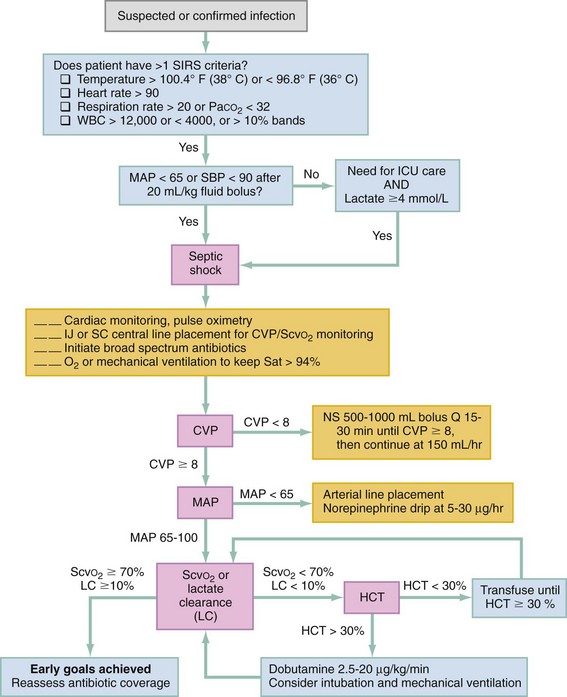
when changes or trends in the values are tracked over time.Quantitative resuscitation, which incorporates multiple indices of circulatory and oxygenation status, has been shown in meta-analyses to significantly reduce mortality and morbidity in ED patients with severe sepsis or septic shock when instituted as early in the disease course as is practical.25 In such an approach, patients are resuscitated within the first 6 hours of care to achieve normalization of markers of preload (CVP) and perfusion (mean BP) and adequate oxygen delivery (ScvO2 70% or lactate clearance 10%) (Fig. 6-2). The most well-known quantitative resuscitation strategy, termed early goal-directed therapy, has also been found effective in smaller prospective before-and-after studies of patients with sepsis.35–37 Three large multicenter validation studies of this resuscitation strategy in sepsis are underway.
Ventilation
Rapid sequence intubation is the preferred method of airway control in most patients with refractory shock (see Chapter 1). Intubation prevents aspiration, increases oxygenation, treats acute respiratory failure, provides initial treatment for metabolic or hypercarbic acidemia, and protects the patient who will be sent to an uncontrolled environment (e.g., for tests). Intubation also reduces the work of breathing, which, in the patient with hypoperfusion, further exacerbates lactic acidemia. Strenuous use of accessory respiratory muscles can increase oxygen consumption by 50 to 100% and decrease cerebral blood flow by 50%. More important, if the patient has increased airway resistance (e.g., bronchospasm with anaphylaxis) or a decrease in lung compliance (e.g., pulmonary edema, ARDS), a more negative intrathoracic pressure must be generated to fill the lungs with each inspiration. The greater suction effect is also exerted on the left ventricle, impeding its ability to eject and increasing functional afterload. Positive-pressure ventilation removes this impedance and can improve ventricular function and cardiac output up to 30%.
Volume Replacement
The goal in volume replacement is slightly elevated left ventricular end-diastolic volume, which is a difficult measurement to make in the ED. The CVP is most often used to estimate right ventricular filling pressure and is used in some quantitative resuscitation algorithms. Because both ventricles tend to stiffen during shock, a high CVP (10-15 cm H2O) is often needed to produce adequate filling volume. CVP measurement does not accurately reflect left ventricular end-diastolic volume, and a recent systematic review demonstrated the inability of CVP to predict the hemodynamic response to a fluid challenge.38 Thus, a presumed adequate CVP should be substantiated by increases in urine output and BP and decreasing lactate concentrations. Emerging literature suggests that the use of dynamic variables of fluid responsiveness that can be measured from an arterial line (such as stroke volume variation or stroke volume index) are superior to static variables (e.g., CVP), but their use in the ED has not been studied.23
Treating Specific Causes
Box 6-5 presents the general treatment approach for the three common causes of shock.
Hemorrhagic Shock
Standard treatment for hemorrhagic shock historically consisted of rapidly infusing several liters of isotonic crystalloid in adults or three successive 20-mL/kg boluses in children. Recent studies have endorsed the concept of either delayed resuscitation or hypotensive resuscitation for hemorrhagic shock. This is discussed in Chapters 36, 45, and 46. Controlling hemorrhage remains the cornerstone of treating hemorrhagic shock, and evidence continues to support immediate surgery when direct vascular control cannot otherwise be obtained (see Chapters 36 and 46).
Colloids, including albumin and hydroxyethyl hetastarch (Hespan), can be used as well, but at considerable increase in cost and without effect on morbidity or mortality.39 Colloids offer the theoretic advantage of a high osmotic pressure, which should help to maintain a normal intravascular volume after retransfusion from hemorrhage. A recent large multicenter trial conducted in the prehospital setting found that initial resuscitation fluid treatment with hypertonic saline or hypertonic saline and dextran, compared with normal saline, did not result in superior 28-day survival.40 If criteria for shock persist despite crystalloid infusion (see Box 6-2), packed red blood cells (PRBCs) should be infused (1-2 units in adults or 5-10 mL/kg in children). Type-specific blood should be used when the clinical scenario permits, but uncrossmatched blood should be immediately used for patients with arterial hypotension and uncontrolled hemorrhage. O-negative blood is used in women of childbearing age and O-positive blood in all others (see Chapter 7). Substantial evidence supports the use of leukodepleted blood, which has been filtered to remove donor neutrophils. Leukodepleted blood is used in countries outside the United States because it produces less retransfusion-related organ damage.
The infusion of hemoglobin-based blood substitutes (HBBSs) as alternatives to PRBCs for resuscitation of hemorrhagic shock has been extensively studied. A recent meta-analysis that included 16 trials involving five different HBBSs in various populations showed that the use of HBBS is associated with significant increased risk of death and myocardial infarction.41 Other artificial hemoglobin substitutes may be available in the future but at present show no benefit, and possibly harm, compared with PRBCs.
Septic Shock
Septic shock begins as an infectious nidus, which triggers a domino effect of cellular, microvascular, hematologic, and cardiovascular dysfunction. Treatment begins by establishing adequate ventilation to correct hypoxia and acidosis and to reduce systemic oxygen consumption and left ventricular work. This often requires endotracheal intubation and sedation for mechanical ventilation. The controversy regarding the use of etomidate in patients with septic shock is discussed in Chapter 1.
The third directive is to eradicate the infection with antimicrobial therapy and, where necessary, surgical drainage. The choice of antimicrobial agent can be directed by clinician experience and institutional minimal inhibitory concentration (MIC) data. Antimicrobials should be administered as soon as is practicable in a patient with septic shock. Evidence is scant, but it is intuitively appealing to administer antimicrobial medication at the earliest reasonable time.42–44 One recent large observational study found that about 60% of patients with septic shock received antibiotics within 3 hours of ED triage, which might be a reasonable target, depending on the patient’s presentation.45 When no focus can be found in septic shock, a semisynthetic penicillin with a β-lactamase inhibitor, in combination with an aminoglycoside plus vancomycin, is a rational empirical choice. When neutropenia is suspected in a patient with sepsis syndrome, the progression to refractory, fatal septic shock can be cataclysmic. Neutropenia should be suspected in patients who have recently undergone chemotherapy. Chemotherapy patients with sepsis represent a special challenge because the pathophysiology may be complicated by anemia, thrombocytopenia, dehydration from vomiting, and the effect of adjunctive steroid therapy. Chemotherapy patients often have indwelling catheters, which predispose them to more unusual causes of sepsis, including gram-positive bacteria and fungi (see Chapter 138).46
Septic shock refractory to volume restoration (urine output or BP remains low; lactate increases) requires vasopressor support. The primary goal of vasopressor support is to increase cardiac output and oxygen delivery to vital organs. Several recent randomized trials and a meta-analysis have suggested that norepinephrine (0.5-30 µg/min) is associated with improved efficacy and lower rates of adverse effects, making norepinephrine the vasopressor of choice for correction of hypotension in septic shock.47–49 Dobutamine may also be used with norepinephrine to increase cardiac output and maintain adequate oxygen delivery. A recent multicenter randomized controlled trial of 330 subjects reported that in cases in which simultaneous BP and inotropic support were necessary, there was not a difference in safety or efficacy between epinephrine (0.2 µg/kg/min starting dose) alone and norepinephrine plus dobutamine.50
The use of corticosteroids in the treatment of sepsis and septic shock has been investigated with mixed results. The results of two large randomized controlled trials confirm that there is no role for high-dose, short-course corticosteroid therapy in septic shock. Recently, two large multicenter randomized trials of low-dose hydrocortisone treatment failed to show survival benefit among all patients with septic shock.51,52 Use of low-dose hydrocortisone among patients who did not adequately respond to a corticotropin stimulation test was supported by one small study but refuted by a larger one.51,52 A recent meta-analysis suggested a modest 28-day mortality benefit of low-dose, short-course hydrocortisone treatment in septic shock.53 Most current guidelines recommend that low-dose hydrocortisone be administered only to patients receiving chronic steroid replacement and in patients with refractory shock despite adequate fluid and vasopressor support. Even this use is only marginally supported, if at all, by scientific evidence. Corticotropin stimulation testing is no longer considered of value.54
Cardiogenic Shock
The immediate treatment of cardiogenic shock focuses on improving myocardial contractility and pump function. Cardiogenic shock is traditionally defined as the combination of systemic signs of hypoperfusion with arterial systolic BP less than 90 mm Hg. If work of breathing is tiring the patient, if severe pulmonary edema is causing significant hypoxemia, or if respiratory failure is imminent, intubation and mechanical ventilation should be initiated, followed by emergent treatment of bradydysrhythmias or tachydysrhythmias and inotropic support. Etomidate and ketamine have the least risk for hemodynamic compromise and should be used (but in reduced doses) for intubation, accompanied by a full dose of succinylcholine. Before administration of vasoactive medications, if hypovolemia is present, it should be corrected by infusion of crystalloid or blood products. Vasopressor or inotropic agents improve myocardial contractility and arteriolar tone. The choice of which agent to use depends on signs and symptoms and the systolic blood pressure (SBP). If the SBP is below 100 mm Hg and signs and symptoms of shock are present, norepinephrine is the agent of choice. However, if the SBP is 70 to 100 mm Hg and there are no signs or symptoms of shock, dobutamine is the agent of choice.2,49 All of these agents should be started at the same doses as used for septic shock.
The dismal outcome of cardiogenic shock complicating acute myocardial infarction (MI) has been improved in recent years. Evidence suggests that emergent revascularization is not superior to medical management in reducing short-term mortality; however, significant improvements in mortality are seen at both 6 months and 1 year (see Chapter 81).55 At present the management of acute MI with cardiogenic shock proceeds as follows, and this constitutes optimal therapy: (1) ensure adequate ventilation and oxygenation, (2) treat emergent dysrhythmias, (3) initiate vasopressor or inotropic support, (4) administer aspirin if the patient is not allergic, and (5) initiate heparin anticoagulation and arrangement for emergent percutaneous coronary intervention (PCI).





References
1. Angus, D, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310.
2. Topalian, S, Ginsberg, F, Parrillo, JE. Cardiogenic shock. Crit Care Med. 2008;36:S66–S74.
3. Williams, RS. Canaries in the coal mine: Mitochondrial DNA and vascular injury from reactive oxygen species. Circ Res. 2000;86:915–916.
4. Watts, J, Kline, J. Bench to bedside: The role of mitochondrial medicine in the pathogenesis and treatment of cellular injury. Acad Emerg Med. 2003;10:985–997.
5. Jones, AE, Yiannibas, V, Johnson, CL, Kline, JA. Emergency department hypotension predicts sudden unexpected in-hospital mortality: A prospective cohort study. Chest. 2006;130:941–946.
6. McDonough, KH, Giaimo, ME, Quinn, M, Miller, HI. Effects of blood resuscitation versus dextran resuscitation after hemorrhage on intrinsic myocardial function. J Trauma. 2000;48:1122–1127.
7. Liaudet, L, Soriano, FG, Szabo, C. Biology of nitric oxide signaling. Crit Care Med. 2000;28:N37–N52.
8. Tatsumi, T, et al. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs contractile function in rat cardiac myocytes. J Am Coll Cardiol. 2000;35:1338–1346.
9. Watts, JA, Kline, JA, Thornton, LR, Grattam, RM, Brar, SS. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J Mol Cell Cardiol. 2004;36:141–150.
10. Moore, C, et al. Determination of left ventricular function by emergency physician echocardiography of hypotensive patients. Acad Emerg Med. 2002;9:186–193.
11. Porter, JM, Ivatury, RR. In search of the optimal end points of resuscitation in trauma patients: A review. J Trauma. 1998;44:908–914.
12. Shapiro, NI, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med. 2005;45:524–528.
13. Jones, A, et al. Nontraumatic out-of-hospital hypotension predicts in-hospital mortality. Ann Emerg Med. 2004;43:106–113.
14. Shapiro, N, et al. Isolated prehospital hypotension after traumatic injuries: A predictor of mortality? J Emerg Med. 2002;25:175–179.
15. Middleton, P, Kelly, AM, Brown, J, Robertson, M. Agreement between arterial and central venous values for pH, bicarbonate, base excess, and lactate. Emerg Med J. 2006;23:622–624.
16. Lavery, RF, et al. The utility of venous lactate to triage injured patients in the trauma center. J Am Coll Surg. 2000;190:656–664.
17. Jones, AE, Leonard, MM, Hernandez-Nino, J, Kline, JA. Determination of the effect of in vitro time, temperature, and tourniquet use on whole blood venous point-of-care lactate concentrations. Acad Emerg Med. 2007;14:587–591.
18. Bahner, D. Trinity: A hypotensive ultrasound protocol. J Diagn Med Sonogr. 2002;18:193–198.
19. Jones, A, Tayal, V, Sullivan, D, Kline, J. Randomized controlled trial of immediate versus delayed goal-directed ultrasound to identify the cause of nontraumatic hypotension in emergency department patients. Crit Care Med. 2004;32:1703–1708.
20. Jones, AE, Craddock, PA, Tayal, VS, Kline, JA. Diagnostic accuracy of left ventricular function for identifying sepsis among emergency department patients with nontraumatic symptomatic undifferentiated hypotension. Shock. 2005;24:513–517.
21. Levy, MM, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256.
22. Jones, AE, Puskarich, MA. Sepsis-induced tissue hypoperfusion. Crit Care Nurs Clin North Am. 2011;23:115–125.
23. Benington, S, Ferris, P, Nirmalan, M. Emerging trends in minimally invasive haemodynamic monitoring and optimization of fluid therapy. Eur J Anaesthesiol. 2009;26:893–905.
24. Kern, JW, Shoemaker, WC. Meta-analysis of hemodynamic optimization in high-risk patients. Crit Care Med. 2002;30:1686–1692.
25. Jones, AE, et al. The effect of a quantitative resuscitation strategy on mortality in patients with sepsis: A meta-analysis. Crit Care Med. 2008;36:2734–2739.
26. Rhodes, A, Cusack, RJ, Newman, PJ, Grounds, RM, Bennett, ED. A randomized, controlled trial of the pulmonary artery catheter in critically ill patients. Intensive Care Med. 2002;28:256–264.
27. Richard, C, et al. Early use of the pulmonary artery catheter and outcomes in patients with shock and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2003;290:2713–2720.
28. Harvey, S, et al. Assessment of the clinical effectiveness of pulmonary artery catheters in management of patients in intensive care (PAC-Man): A randomized controlled trial. Lancet. 2005;366:472–477.
29. Binanay, C, et al. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: The ESCAPE trial. JAMA. 2005;294:1625–1633.
30. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med. 2006;354:2213–2224.
31. Abramson, D, et al. Lactate clearance and survival following injury. J Trauma. 1993;35:584–588.
32. Nguyen, H, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med. 2004;32:1637–1642.
33. Arnold, RC, et al. Multi-center study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock. 2009;32:36–39.
34. Jones, AE, et al. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: A randomized clinical trial. JAMA. 2010;303:739–746.
35. Rivers, E, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. New Engl J Med. 2001;345:1368–1377.
36. Jones, AE, Focht, A, Horton, JM, Kline, JA. Prospective external validation of the clinical effectiveness of an emergency department-based early goal directed therapy protocol for severe sepsis and septic shock. Chest. 2007;132:425–432.
37. Micek, ST, et al. Before-after study of a standardized hospital order set for the management of septic shock. Crit Care Med. 2006;34:2707–2713.
38. Marik, PE, Baram, M, Vahid, B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares. Chest. 2008;134:172–178.
39. Finfer, S, et al. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247–2256.
40. Bulger, EM, et al. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: A randomized, placebo controlled trial. Ann Surg. 2011;253:431–441.
41. Natanson, C, Kern, SJ, Lurie, P, Banks, SM, Wolfe, SM. Cell-free hemoglobin-based blood substitutes and risk of myocardial infarction and death: A meta-analysis. JAMA. 2008;299:2304–2312.
42. Puskarich, MA, et al. Association between timing of antibiotic administration and mortality from septic shock in patients treated with a quantitative resuscitation protocol. Crit Care Med. 2011;39:2066–2071.
43. Kumar, A, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34:1589–1596.
44. Gaieski, DF, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045–1053.
45. Levy, MM, et al. The Surviving Sepsis Campaign: Results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010;38:367–374.
46. Sarkodee-Adoo, CB, Merz, WG, Karp, JE. Management of infections in patients with acute leukemia. Oncology (Williston Park). 2000;14:659–666.
47. Vasu, TS, et al. Norephinephrine or dopamine for septic shock: Systematic review of randomized clinical trials. J Intensive Care Med. 2012;27:172–178.
48. Patel, GP, et al. Efficacy and safety of dopamine versus norepinephrine in the management of septic shock. Shock. 2010;33:375–380.
49. De Backer, D, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med. 2010;362:779–789.
50. Annane, D, et al. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: A randomized trial. Lancet. 2007;370:676–684.
51. Sprung, CL, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124.
52. Annane, D, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871.
53. Annane, D, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: A systematic review. JAMA. 2009;301:2362–2375.
54. Dellinger, RP, et al. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327.
55. Hochman, JS, Sleeper, LA, White, HD, Dzavik, V. One-year survival following early revascularization for cardiogenic shock. JAMA. 2001;285:190–192.