[level-membership-for-surgery-category]31
Selected Complex Malformations that Frequently Require Maxillofacial Reconstruction
Evaluation & Treatment
Klippel–Feil Anomaly
Background
The Klippel–Feil anomaly (KFA) is characterized by the congenital fusion of two or more cervical vertebrae. In its most severe form, there is massive cervical vertebral fusion with a short neck, limited head movement, and a low posterior hairline.2,12,15,21,23,29,37,39,55 Gorlin and Cohen have pointed out that the term vertebral fusion is not accurate because this condition results from the failure of the normal segmentation process.24 Vertebral fusion can be traced to the third embryonic week when the segmentation of the mesodermal somites normally takes place.
KFA was described as early as the 16th century, and similar anatomic findings in the second and third cervical vertebrae have been found in an Egyptian mummy that has been dated to be from about 500 B.C. In 1912, Maurice Klippel and Andre Feil described the postmortem findings of a 46-year-old French tailor who had a short, immobile neck with massive fusion of cervical and upper thoracic vertebrae.34 In 1919, Feil added reports of 13 additional patients, 12 of whose cases had been reported previously.
In 1967, Gunderson and colleagues categorized KFA in accordance with three morphologic types of cervical vertebral fusion.27 Type I consists of the massive fusion of many cervical and upper thoracic vertebrae into bony locks. Type II involves fusion at only one or two interspaces. Type III comprises both cervical fusion and lower thoracic or lumbar fusion.
It is important to remember that KFA is morphologically and etiologically heterogeneous. This was further documented by Gunderson, who used Feil’s classification system to review inheritance patterns.28 Type I cases have almost all been sporadic, with a female predilection. Type II is transmitted as an autosomal dominant disorder. For Type III, an autosomal recessive mode of inheritance has been suggested.
Various authors differ with regard to their interpretation of what constitutes KFA.9,61–63 A malformation with the triad of a short neck, a low posterior hairline, and a painless restriction of cervical motion characterizes just over 50% of patients. The functional limitation of the neck range of motion is the most consistent finding. Despite frequent sparing of the atlanto–axial joint, rotation is typically impaired more than flexion or extension is, with the latter exhibiting a range of more than 90 degrees, even with only one open disc space.
Diverse ocular anomalies are frequently found, with the impairment of extraocular movement being the most frequent.57Convergent strabismus and, less commonly, horizontal nystagmuses are seen. From 25% to as many as 50% of affected children exhibit hearing loss that may be sensorineural, mixed, or conductive.11,16,39,51,60,64 Clefting of the secondary palate is present in 15% to 20% of patients.10,58,59 The combination of hearing loss and cleft palate explains the hypernasality that has been documented in approximately 15% to 20% of patients with KFA. Congenital heart defects occur in 4.2% of patients, which is in stark contrast with the prevalence of 0.6% among all live births in the general population.50 Ventricular septal defect is the most common heart anomaly documented,43 and vascular anomalies may also occur.5,6 Congenital urinary anomalies are a frequent finding, with unilateral renal agenesis occurring in 28% of patients.14 Genital anomalies (e.g., vaginal agenesis) also occur at a higher frequency in these patients than they do in the general population.40,42 Neurologic disturbances that consist of involuntary dyskinesis, spasticity or hyperreflexia, syringomyelia, syringobulbia, disc protrusion, osteophytes, and narrowing at the level of the craniovertebral junction have been reported..3,20,22,36,44,48 Nagib and colleagues found patients with KFA with two unsegmented blocks of bone, cervical stenosis, or cranial involvement to be at highest risk for neurologic disturbances.45–47 Those with only one unsegmented bone were found to be at low risk for cervical injuries. The flared trapezius muscles associated with KFA give the individual an appearance that may be similar to that seen in patients with Turner or Noonan syndrome.8,13,18,38 The cervical vertebral anomalies and other defects of the spine that occur within the oculo–auriculo–vertebral spectrum may also be confused with those of KFA.4,7,8,25,32,33,35,49,52,54,56
Author’s Approach to Facial Reconstruction in Patients with Klippel–Feil Anomaly
The surgical management of KFA requires the accurate diagnosis of each specific problem (see Fig. 1-2).17,26,31 Middle-ear infections should be treated promptly, and any hearing deficit should be aided whenever possible. Clefting of the secondary palate should be repaired in the usual way. Hypernasality as a result of inadequate soft palate motion after initial palate repair should be recognized and treated with a pharyngoplasty. Extraocular muscle abnormalities are managed with eye patching, spectacles, and eye-muscle surgery, as appropriate. The recognition of cardiovascular and genitourinary malformations should lead to medical treatment and surgery. Cervical spine instability requires the decompression of any nerve impingement and the fusion of unstable segments. Unfortunately, the typical fixed neck curvature observed in patients with KFA cannot safely be straightened.
The aim of the reconstructive procedures used to correct a webbed neck is to create as normal a neck contour as possible with a symmetrical posterior hairline while avoiding obvious scarring.1 It is important to recognize that not all patients with webbed necks present with the same deformity. The clinician must assess the quality and quantity of available neck skin as well as the height and straightness of the cervical spine. Type I KFA consists of an extremely short skeletal neck that results in the increased width of the neck soft tissues. A spectrum of neck soft-tissue rearrangement procedures has been suggested to improve the webbed-neck deformity. Menick and colleagues modified the “butterfly” excision of redundant skin to avoid a midline scar.41 They did this by extending the more traditional inferior flaps into the scalp. Unfortunately, with this design, the scars are displaced laterally, and they become visible at the hairline, close to the original site of the web. Some surgeons feel that a midline posterior scar is often preferred aesthetically to one that can be seen along the lateral neck region. The placement of tissue expanders followed by flap rearrangements has also been carried out with some success. Thus, to review, procedures designed to correct the webbed neck suffer from an inability to elevate the hairline, noticeable scarring, and the intrinsically inadequate height of the skeletal structures of the neck.
The craniofacial skeletal deformities of patients with KFA typically present in one of two patterns.19,30,53 The first is in association with a congenital asymmetrical cervical spine fusion. In these patients, a variable degree of non-synostotic anterior plagiocephaly is present. The ipsilateral fronto–orbito–zygomatic flattening and retrusion may be significant enough that anterior cranial vault, orbital, and zygomatic reconstruction carried out through an intracranial approach will be indicated to adequately improve morphology (Fig. 31-1). These individuals will also have an asymmetrical dentofacial deformity that requires Le Fort I (maxillary) osteotomy, bilateral sagittal split ramus osteotomies of the mandible, and an osseous genioplasty procedure for three-dimensional repositioning (Fig. 31-2). The most unsettling aspect of the craniofacial reconstruction relates to the fact that the neck cannot be straightened. It will remain crooked, and so decisions about the best aesthetic positioning of the osteotomized craniofacial skeletal units must also be adjusted.
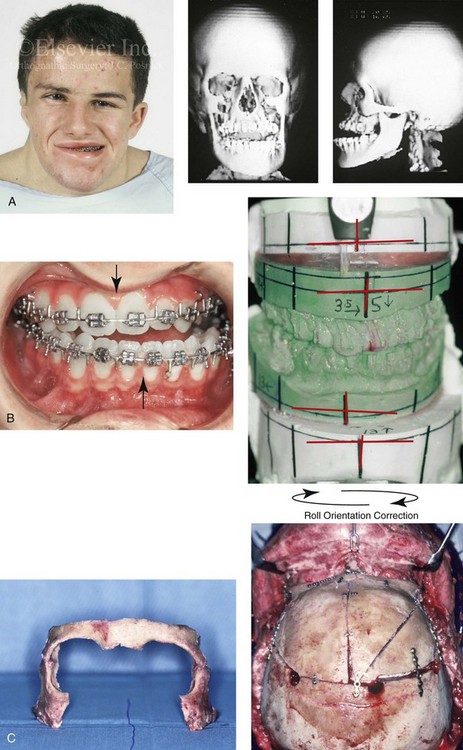
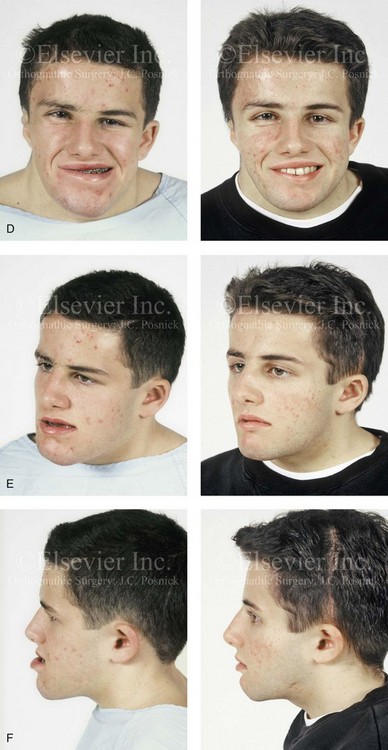
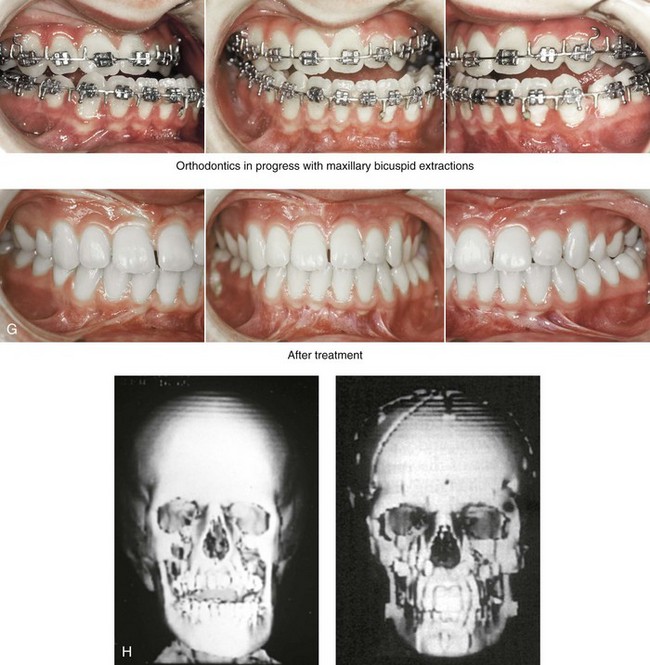
Figure 31-1 A 17-year-old boy with Klippel–Feil anomaly has a fixed head tilt as a result of a painless cervical spine fusion and a partial facial nerve palsy on the right side. There is marked asymmetry and deformity of the craniofacial region that involves the upper facial skeleton (i.e., the cranial vault, the orbits, and the zygomas) and the lower facial skeleton (i.e., the maxilla, the mandible, and the chin). The reconstruction was compromised by the fixed head tilt and the CN VII deficit. The patient underwent cranio–orbito–zygomatic (intracranial) reconstruction through a coronal scalp incision followed by orthognathic surgery that included a Le Fort I osteotomy, bilateral sagittal split ramus osteotomies, and an osseous genioplasty. A, Frontal facial and computed tomography scan views before surgery. B, Occlusal view before surgery and articulated dental cast that indicates analytic model planning. C, Intraoperative view of removed orbital and zygomatic units and intraoperative bird’s-eye view of the anterior cranial vault after cranio–orbito–zygomatic reconstruction. D, Frontal views with smile before and after reconstruction. Note that the fixed head tilt limited the the correction of facial symmetry. E, Oblique views before and after reconstruction. F, Profile views before and after reconstruction. G, Occlusal views with orthodontics in progress (including upper bicuspid extractions) and then after reconstruction. H, Three-dimensional computed tomography scan views before and after reconstruction.

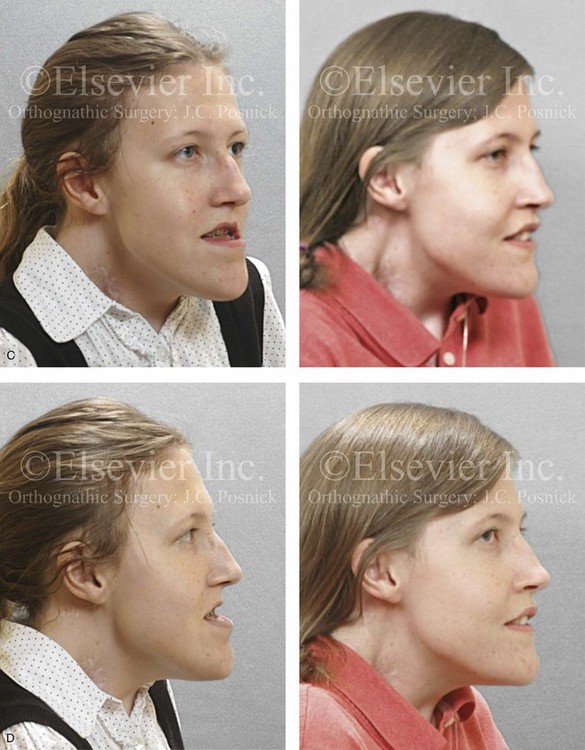
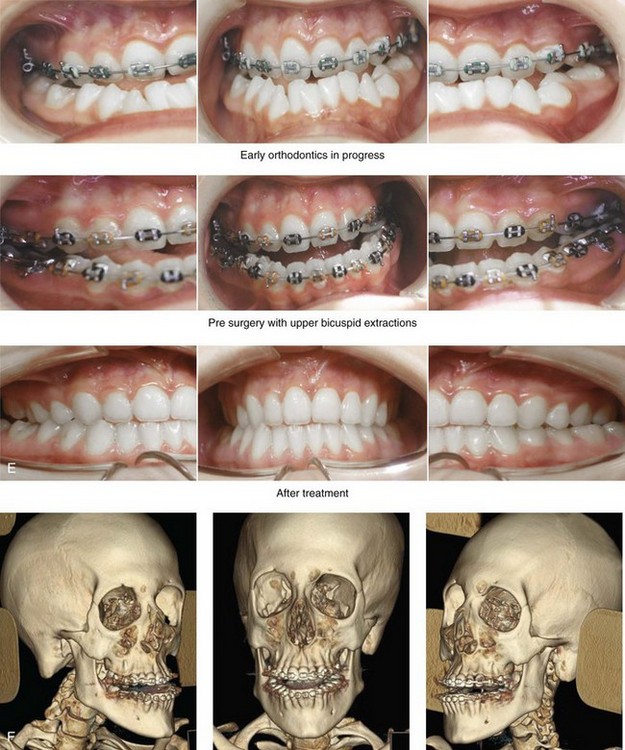

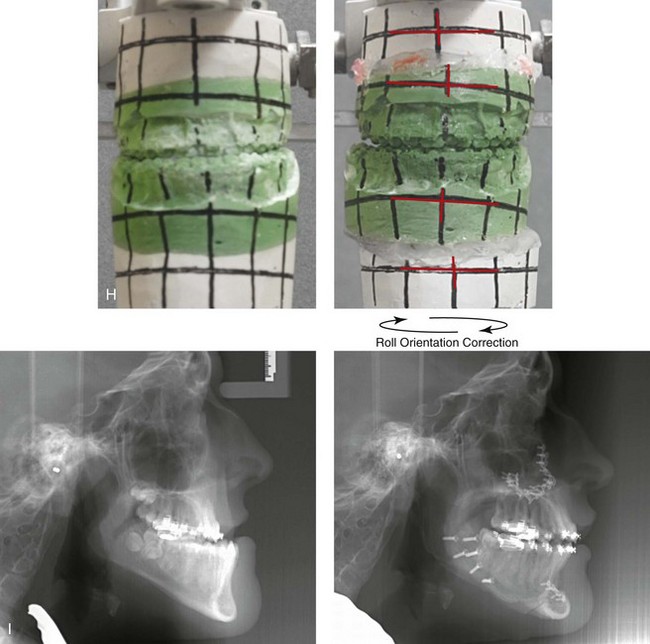
Figure 31-2 A teenage girl with the Klippel–Feil and Poland congenital anomalies has a fixed head tilt as a result of a painless cervical spine fusion. At birth, she was noted to have a significant diaphragmatic herniation of the stomach and spleen into the thoracic cavity. She underwent procedures for the correction of the diaphragmatic hernia and then required esophageal dilatations. She also required decompression and fusion for the cervical anomalies to limit injury to the upper spinal cord. She was referred to this surgeon as a teenager and underwent evaluations that included restorative dentistry, orthodontics, speech pathology, otolaryngology, pediatric surgery, pediatric orthopedic spine surgery, and pediatrics professionals. The assessment of pulmonary capacity, swallowing and dysphagia, and safe range of motion of the neck were of particular importance. There was marked asymmetry and deformity of the maxillomandibular region. The plans for reconstruction were affected by the fixed head tilt. The patient underwent a combined orthodontic and surgical approach, and her procedures included: maxillary Le Fort I osteotomy (horizontal advancement and cant correction); sagittal split ramus osteotomies (horizontal advancement and asymmetry improvement); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty and inferior turbinate reduction. Intraoperative cervical monitoring was carried out to limit the chance of spinal cord injury. A, Frontal views in repose before and after reconstruction. B, Frontal views with smile before and after reconstruction. C, Oblique facial views before and after reconstruction. D, Profile views before and after reconstruction. E, Occlusal views before treatment, with orthodontics in progress and with upper bicuspid extractions, and then after reconstruction. F, Computed tomography scan views before reconstruction. G and H, Articulated dental models that indicate analytic model planning. I, Cephalometric radiographs before and after reconstruction.
The second pattern of presentation is in association with a congenital symmetric cervical spine fusion. The cranio–orbito–zygomatic region is symmetrical and has relatively good proportions.53 No reconstruction procedures are generally required in the upper facial skeleton. With reference to the maxillomandibular region, the constant open mouth posture of the mandible generally results in vertical maxillary excess and a constricted maxillary arch width. The mandible is usually retrognathic, with a vertically long retrusive chin and an open-bite malocclusion. A degree of maxillomandibular asymmetry is expected. The neck–chin angle is markedly obtuse. These individuals will benefit from orthognathic surgery in combination with orthodontic treatment. The orthognathic surgery will generally require a Le Fort I osteotomy in segments to intrude the maxilla vertically, advance it horizontally, and widen it transversely. The preferred approach also involves bilateral sagittal split ramus osteotomies of the mandible with horizontal advancement and counterclockwise rotation to match the new maxillary position in combination with a vertical reduction and advancement genioplasty (see Chapter 15).
Down Syndrome (Trisomy 21 Syndrome)
Background
Speculation about the historic prevalence of Down syndrome has included references to depictions of the syndrome in 15th- and 16th-century paintings.69,81 Martínez-Frías described what seems to be the earliest observation of the syndrome in a sculpted terracotta head from 580 A.D.89 The terracotta piece comes from the Toltec culture of Mexico, and its facial features, which include macroglossia, help to define its subject as a person with Down syndrome. The features of this Down syndrome were initially described by Langdon Down in 1866, and the condition represents a combination of phenotypic features that includes a degree of mental deficiency and characteristic facial features.72
The birth prevalence of trisomy 21 syndrome is generally stated to be 1 in 650 live births, but it is known to vary in different populations from 1 in 600 to 1 in 2000 live births. During the 1970s, 15% of patients who had been institutionalized for mental deficiency were found to have trisomy 21 syndrome.75
About 95% of all cases of Down syndrome arise from non-disjunction. Approximately 80% are of maternal origin, with 20% being of paternal origin. The risk increases with increasing maternal age as well as with paternal age and particularly when the parents are more than 35 years old. It has been estimated that approximately 4.8% of Down syndrome cases arise from an unbalanced translocation, which may occur de novo or by parental transmission. Inherited D/G translocations occur in 93% of maternal cases and 7% of paternal cases; however, for G/G translocations, 50% are of maternal origin, and 50% are of paternal origin. It has been estimated that 65% to 80% of trisomy 21 conceptuses result in spontaneous abortions.75
Detectable mosaicism (found in about 3% of trisomy 21 cases) and the Down syndrome phenotype may only be partially expressed.75 The use of the DNA samples from individuals who have partial trisomy 21 with or without features of the phenotype has been helpful for understanding the nature of the disorder. The area between loci D21S58 and D21S42 has been identified as being associated with mental retardation and with most of the facial features of Down syndrome. The facial features that have been identified in this region include oblique palpebral fissures; epicanthal folds; a flat nasal bridge; a protruding tongue; short, broad hands with clinodactyly of the fifth finger; a gap between first and second toes; hypotonia; short stature; Brushfield spots; and characteristic dermatoglyphic findings, including a single palmar crease and an increased number of ulnar loops.88
Individuals with Down syndrome have a specific set of major congenital malformations, including heart anomalies (30% to 40%) and gastrointestinal tract anomalies such as duodenal stenosis or atresia, imperforate anus, and Hirschsprung disease.75 The development of Alzheimer disease occurs at a much earlier age in individuals with trisomy 21 than it does in the general population. The frequency of symptomatic gallbladder disease is 25% higher among adults with Down syndrome as compared with the general population.
Down syndrome is characterized by extensive phenotypic variability.65,67,71,98 Although cognitive impairment, muscle hypotonia at birth, and dysmorphic features occur to some extent in all individuals, most associated traits occur in only a fraction of affected individuals. In the patient with Down syndrome, a number of characteristic facial features that vary from patient to patient are frequently recognized:
2. Oblique (anti-mongoloid) slanting of the eyelids
3. Strabismus as a result of extraocular muscle dysfunction
4. Flatness of the dorsum of the nose
5. Hypoplasia of the upper jaw that results in an Angle Class III malocclusion
In 1967, Otermin Aguirre presented a series of patients with Down syndrome who underwent reconstructive surgery in an attempt to normalize their facial morphology.93 During the same year, Hohler attempted to normalize the face of a young girl with Down syndrome by augmenting the hypoplastic dorsum of the nose and the receding chin with alloplastic implants.77 In 1977, Lemperle and Spitalny systematically tried to normalize the faces of children with Down syndrome.82–85 Similar results were published by Olbrisch,91,92 Patterson and colleagues,94 Wexler and Peled,103,104 and Rozner,99 in addition to several others.76,90,94,97
Despite the initial enthusiasm for facial reconstruction in children with Down syndrome during the late 1960s through the mid 1980s, a critical review of the procedures used to normalize the face was not carried out until the mid to late 1980s. In 1989, Katz and Kravetz presented a balanced, thoughtful review and a discussion of the literature regarding the “effectiveness of facial surgery for persons with Down syndrome.”79 The research of Katz and Kravetz may be summarized as follows: If improved functioning, appearance, and social acceptability are measured by parent and treating physician satisfaction, then the outcome of facial plastic surgery for people with Down syndrome seems positive. If function, appearance, and social acceptability are evaluated by people who were not involved in making the decision to conduct the facial plastic surgery—and particularly when control subjects are included in the evaluation—then the outcome of facial plastic surgery for people with Down syndrome is not so positive. For most patients with Down syndrome, the surgeons’ and parents’ well-intentioned attempts to normalize the child’s facial morphology could not be realized.96
Macroglossia with protrusion of the large tongue is a frequent feature of Down syndrome. Dystonia or hypotonia of the tongue is also a factor.66,74 During the 1970s and the early 1980s, several investigators suggested that partial glossectomy in the patient with Down syndrome could improve the clarity of speech.68,70 However, studies that used objective criteria to document speech results after partial glossectomy in patients with Down syndrome showed no significant differences in the number of articulation errors between audiotape recordings made preoperatively and 6 months postoperatively in a series of patients or between the recordings of the same patients and recordings of an additional series of Down syndrome control patients who did not undergo surgery. A further study by Margar-Bacal and colleagues looked at changes in the aesthetic appearance and intelligibility of speech after partial glossectomy in patients with Down syndrome.87 The aesthetic appearance of speech (i.e., the visual acceptability of the patient while he or she is speaking) was judged from visual information only. Judgments of speech intelligibility were made separately from the auditory portion of the videotapes. The acceptability and intelligibility of the children were also judged together during audiovisual presentation. Statistical analysis showed that speech was significantly more acceptable aesthetically after surgery. No significant difference was found in speech intelligibility postoperatively as compared with preoperatively.
An Angle Class III malocclusion as a result of the midface hypoplasia with overclosure of the mandible is a frequent finding in patients with Down syndrome.95 This was objectively studied by Farkas with the use of anthropometric measurements in which he documented that 60% of children with Down syndrome showed a disproportionately small depth to the middle third of the face (i.e., the anthropometric measurement from the subnasal point to the tragus).73 The congenital absence of permanent teeth, poor root formation, and hypoplasia of the enamel are frequent complicating factors.
Author’s Approach to Facial Reconstruction in Individual’s with Down Syndrome
When a child with Down syndrome approaches skeletal maturity (i.e., 14 to 16 years of age for girls, 16 to 18 years of age for boys) and a significant jaw deformity with malocclusion is present, then a comprehensive approach of orthodontic treatment and orthognathic surgery can effectively alter the facial proportions (aesthetics) and improve head and neck functions (i.e., lip posture, chewing, speech articulation, and breathing) (Fig. 31-3). A formal sleep study is indicated before surgery to clarify the extent of upper airway obstruction and the degree of maxillomandibular advancement required to manage it.1,78,80,86,100–102 The orthodontic treatment may be compromised as a result of poor dental root formation, congenitally missing teeth, or limited compliance. The coordination of care with an appropriate dental team (e.g., a restorative dentist, a periodontist, an orthodontist) is useful before the initiation of any treatment.
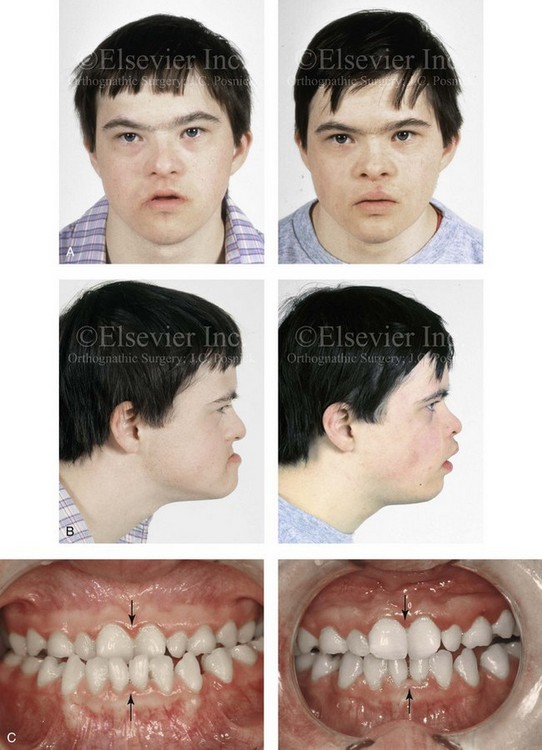
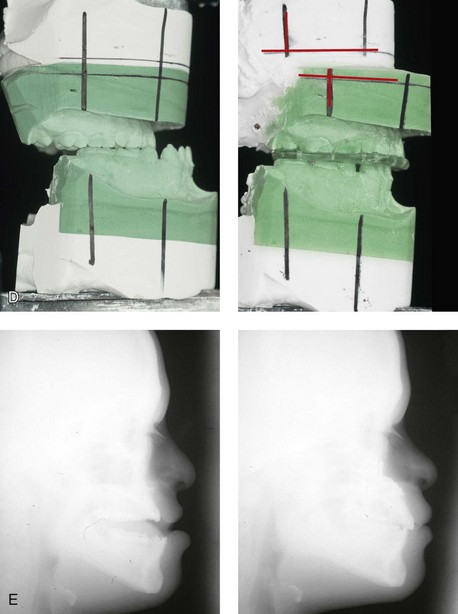
Figure 31-3 A 16-year-old boy with Down syndrome presented with a jaw deformity and malocclusion. He underwent a combined orthodontic and surgical approach. The surgery was limited to a Le Fort I osteotomy (horizontal advancement and vertical lengthening). A, Frontal views before and after reconstruction. B, Profile views before and after reconstruction. C, Occlusal views before and after reconstruction. Note that the orthodontic and restorative dental objectives were limited by congenital malformations of the dental crowns and dental roots. D, Articulated dental casts that indicate analytic model planning. E, Lateral cephalometric radiographs before and after reconstruction.
The correction of the dentofacial deformity and the management of the airway frequently requires maxillary Le Fort I osteotomy (horizontal advancement and vertical lengthening), often with sagittal split ramus osteotomies for alignment to the new maxillary position (i.e., advancement but not setback) and an osseous genioplasty (horizontal advancement and vertical lengthening). Septoplasty and inferior turbinate reduction should be simultaneously performed if required to improve nasal airflow (see Chapter 10).
Neurofibromatosis
Background
In 1849, Robert Smith, a professor of surgery at Dublin Medical School, reported clinical and necropsy findings in two cases of individuals who were presumed to have neurofibromatosis; he cited 75 references to similar patients from the earlier medical literature.130 Unfortunately, he did not recognize that the tumors (neurofibromas) contained neural elements. In 1882, von Recklinghausen published his findings, which convinced the medical world that neurofibromatosis was a distinct entity of neural origin.165 Today, neurofibromatosis is known to be etiologically heterogeneous. Cohen and Hayden were the first to differentiate Proteus syndrome from neurofibromatosis in 1979.119,162 Types I and II are the forms that are frequently encountered by the craniomaxillofacial surgeon.
Neurofibromatosis Type II (Acoustic Type)
The hallmark of Type II neurofibromatosis is the presence of bilateral acoustic neuromas.147,150 Symptoms are usually caused by pressure on the vestibulocochlear and facial nerve complex. The first symptom is usually hearing loss that often begins during the teenage years or the early 20s. Occasionally, hearing loss may occur as early as the first decade or as late as the seventh decade of life. Cafe-au-lait spots and cutaneous neurofibromas are also present, but they are seen less frequently than they are among patients with neurofibromatosis Type I.121 Tumors of the central nervous system are especially common, with Schwann cell tumors occurring most frequently. Multiple tumors of meningeal or glial origin may also occur.147 Neurofibromatosis Type II has autosomal dominant inheritance with penetrance of more than 95%. The responsible gene has been mapped to chromosome 22.
Neurofibromatosis Type I (von Recklinghausen Type)
Joseph Merrick, the Elephant Man, has often been thought to have had von Recklinghausen disease. However, after considering several diagnostic possibilities, Cohen concluded that Merrick’s skeletal findings are most consistent with Proteus syndrome.119,130,162
This classic form of neurofibromatosis accounts for approximately 90% of all affected cases. Major features include six or more cafe-au-lait spots, cutaneous neurofibromas, and Lisch nodules.130 Axillary freckling develops in about 66% of all patients. Neurofibromatosis type I, which is the classic form of neurofibromatosis, is the one described by von Recklinghausen; it is also the most frequent form, occurring in 1 of every 2500 to 3000 births.146,160,165,166 Inheritance is autosomal dominant, with almost 100% penetrance. Approximately 50% of cases represent new mutations, and an increase in paternal age at the time of conception has been found to be an associated feature. Although most evidence points to neurofibromatosis as a disorder of neural crest derivation, some controversy remains about whether the neural and mesenchymal components are interrelated or if they may arise independent of each other.110,116,153
Type I is caused by a mutation of the neurofibromin 1 (NF1) gene. Approximately 5% to 20% of all patients with Type I neurofibromatosis patients carry a heterozygous deletion of approximately 1.5 Mb that involves the NF1 gene and the contiguous change line in its flanking region.157 Miller and Hall found that patients born to affected mothers have more severe disease than those born to affected fathers.152 The mutation rate in the NF1 gene is one of the highest known in humans, with approximately 50% of all patients with NF1 presenting with novel mutations.124
The natural history of neurofibromatosis Type I has been reviewed by several authors.138 More than 40% of patients have some clinical manifestations at birth, and more than 60% have manifestations by the second year of life. Cafe-au-lait spots usually develop first, with multiple lesions present within the first year of life. In approximately 50% of patients, axillary freckling appears later. Cutaneous neurofibromas appear around the onset of puberty and increase in number throughout life.138 Lisch nodules begin to appear during early childhood and have been observed in almost all affected adults. Plexiform neurofibromas occur in 30% of these patients.
Approximately 33% of all patients develop one or more complications in association with the disease.105–108,115 It has been estimated that some form of malignancy develops in 6% of patients with neurofibromatosis type I who are more than 18 years old.109,117,118 Other important complications that may occur are neurologic and include epilepsy, aqueduct stenosis, and spinal neurofibromas. Learning disabilities of various kinds affect 25% of patients.137,154,163
Any part of the eye may be involved.111,126,128,132 Lisch nodules of various sizes may be found anywhere in the iris. These lesions are melanocytic hamartomas, and they are found only in patients with neurofibromatosis. Phakoma, congenital glaucoma, corneal opacity, detached retina, optic atrophy, and congenital ptosis of the eyelids have all been reported. Intraorbital lesions may also produce proptosis and eye-muscle palsies.
The literature concerning the presenting head and neck dysmorphology and the suggested treatment of orbitotemporal neurofibromatosis is extensive.* The plexiform neurofibroma that involves the orbit, the eyelid, and the temporal region may also have an intracranial component. It generally appears during childhood as a swelling of the upper eyelid, and it slowly and progressively worsens (Fig. 31-4). Further involvement of the subcutaneous tissues of the eyelids with varying degrees of ptosis and proptosis of the globe also occurs. For patients in whom progression continues, there is further plexiform neurofibromatosis of the temporal area, pulsating proptosis, continued enlargement of the eyelids with extensive mechanical ptosis, and the inability to open the eye, which results in severely diminished visual acuity or blindness. The globe itself is involved with neurofibroma, and buphthalmos may be present. The eye may be painful as a result of infection and epiphora.
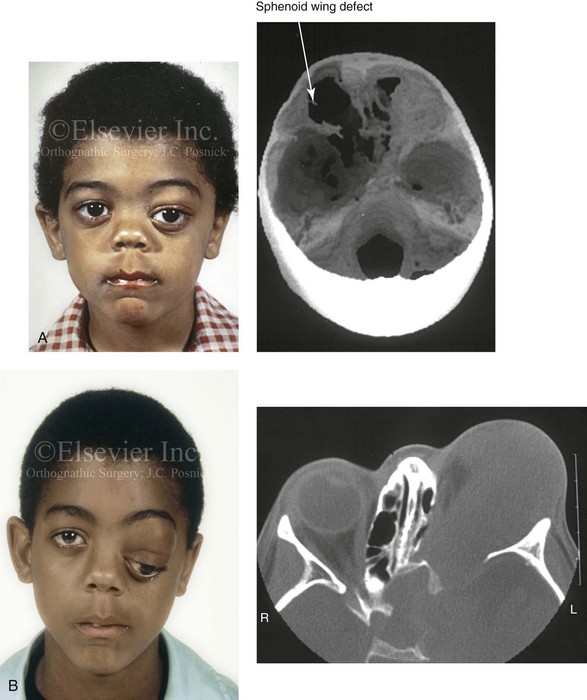
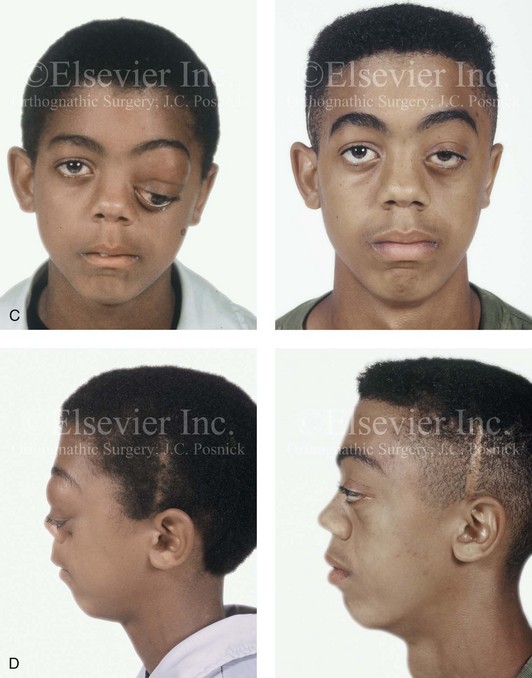
Figure 31-4 A child with neurofibromatosis involving the left cranio–orbital region. Aplasia of the sphenoid wing and the gradual growth of plexiform neurofibroma occurred, which extended from the cavernous sinus through the widened superior orbital fissure, through the cone of the eye, and into the upper eyelid. Despite an intracranial debulking procedure carried out by another surgeon during the patient’s middle childhood years, the boy presented to this surgeon with residual and recurrent plexiform neurofibroma that resulted in the distortion and proptosis of the left eye. Vision remained in the affected eye, and the decision was made to preserve it. The patient underwent intracranial reconstruction of the cranio–orbito–zygomatic skeleton, including the debulking of the soft-tissue tumor and the vertical and horizontal debulking of the upper eyelid. Intracranial debulking with eye presentation was accomplished. A, Frontal view during early childhood and a computed tomography scan of the anterior skull base indicating the defect in the sphenoid wing. B, Frontal view at 10 years of age. Despite a debulking procedure carried out by another surgeon, residual growth has reoccurred. An axial computed tomography scan view through the midorbits indicates the extent of plexiform neurofibromatosis from the cavernous sinus through the upper eyelid. C, Frontal views before and after reconstruction. Note that the surgical correction of the upper eyelid ptosis was simultaneously carried out but it was limited to maintain adequate corneal protection. D, Profile views before and after reconstruction.
Author’s Approach to Reconstruction in Patients with Orbitotemporal Neurofibromatosis
It is recognized that the facial reconstruction of patients with this condition is often difficult as a result of the multilevel involvement of the skeleton, the soft tissues, and the viscera (i.e., brain, eyes, tongue, soft palate, sinus, and teeth). A basic premise of treatment is to preserve the seeing eye whenever feasible.114,127,135,136,155,158,159,161 The prevention of brain injury and the improvement of the tongue, lips, soft palate, and jaw function are high priorities. Jackson and colleagues have suggested a classification system for orbitotemporal neurofibromatosis that they have found useful for treatment planning139:
1. Orbital soft-tissue involvement with a seeing eye
2. Orbital soft-tissue and significant bony involvement with a seeing eye
3. Orbital soft-tissue and significant bony involvement with a blind or absent eye
It has been pointed out by Jackson and others that conservative surgical debulking procedures that leave considerable amounts of the plexiform neurofibroma leave patients prone to continued growth and the need for further surgery.141 Whenever feasible, more complete resection of the tumor with immediate reconstruction is preferred.
When the temporal–cheek region is extensively involved, the facial nerve presents an extra challenge. If possible, the facial nerve is dissected free, the tumor is debulked, and the soft-tissue flaps are redraped (see Fig. 31-4). When the facial nerve is extensively infiltrated by a tumor that grotesquely deforms the face, it may be necessary or preferable to resect the nerve as part of the debulking procedure. Occasionally we have used an approach that involves the radical debulking of the tumor, the planned sacrifice of the involved facial nerve, and the simultaneous reconstruction of the cheek–facial nerve defect with a reinnervated free muscle transfer.
Despite the less than ideal restoration of function and enhancement of facial aesthetics that is often achieved with the presenting orbitotemporal, temporal–cheek, and maxillomandibular neurofibromatosis deformities, patients and families are generally pleased with the efforts (see Fig. 31-4). Longitudinal computed tomography scanning and magnetic resonance imaging reassessment are useful to follow the disease process, because residual neurofibromatosis generally remains, and malignant transformation may occur.
Hemihyperplasia of the Face
Background
Hemihyperplasia was first described by Meckel in 1822.192 Many comprehensive reviews have been published since that time.173,174,181,182 In patients with hemihyperplasia, the enlarged area may run the gamut from a single digit to a single limb to unilateral facial enlargement to involvement of half of the body.168–170,179,183,187,189,198,199,203,206–208 In some patients, the defect is limited to a single tissue type (e.g., muscular, vascular, skeletal, nervous system).204
The etiology and pathogenesis of hemihyperplasia are poorly understood, with a variable range of clinical abnormalities having been described.171,175,178 This fact—in combination with the large number of reported sporadic cases—is suggestive of etiologic heterogeneity. When the face is involved, asymmetry is usually evident at birth, and it may become accentuated or at least more of a concern with age, especially at puberty.180,188,190,192,196,200–202 The involved bones have been found to be unilaterally enlarged, often with accelerated bone age on the affected side. A variety of non-neoplastic abnormalities of the limbs, teeth, skin, central nervous system, cardiovascular system, liver, kidneys, and genitalia have also been observed.
Various neoplasms have been reported in association with hemihyperplasia, including Wilms tumor, adrenal cortical carcinoma, and hepatoblastoma to name but a few.172,176,177,184–186,191 In the study by Hoyme and colleagues of 104 cases of isolated hemihyperplasia, tumors developed in 3.8% of patients.186 The neoplasms associated with hemihyperplasia have been found to have an embryonal origin. Patients with hemihyperplasia should be screened with ultrasound for tumors every 6 months for the first 4 years of life and probably less frequently thereafter until the age of 7 years.176,184
Any and all ipsilateral facial structures may be involved, including the bones of the cranium, the orbit, the zygoma, the maxilla, and the mandible.193–195 Premature development and eruption of the teeth occur, as does macrodontia of varying degrees. Unilateral enlargement of the tongue and its papillae is also a frequently observed feature. The overall facial soft tissues are increased in thickness, size, and volume. The eyeball and even the brain may be enlarged on the affected side.
Hemihyperplasia of the face should be distinguished from hemimandibular hyperplasia (HMH), which is a three-dimensional enlargement of the whole hemimandible but not of the other facial structures.193–195 Furthermore, in patients with HMH, no other aspects of the body are enlarged, and no predisposition to tumors has been identified. HMH was first reported by Adams in 1836.182 Epidemiologic data suggest that there is equal incidence of HMH in both sexes and in all ethnic groups. Obwegeser drew the distinction between HMH and hemimandibular elongation.193–195 He also clearly differentiated these conditions from hemihyperplasia of the face (see Chapter 22).
Author’s Approach to Reconstruction in Patients with Hemihyperplasia
Procedures to improve facial symmetry and Euclidian proportions for the individual with hemihyperplasia of the face are offered when feasible (Fig. 31-5).197 Overzealous attempts to debulk the soft-tissue enlargement (e.g., resection of the parotid gland, resection or liposuction of the deep layer soft tissues of the cheek) may result in damage to the facial nerve or introduce an “operated” look and are thus generally discouraged.205 Surgery should be directed at regions and tissue layers that are most amenable to safe improvement. Orthodontic treatment in combination with orthognathic surgery is often a fruitful approach. A maxillary Le Fort I osteotomy to bring the maxillary dental midline back to the facial midline; to correct canting; and to adjust the vertical dimension, horizontal projection, and arch width is frequently useful. This is combined with bilateral sagittal split ramus osteotomies of the mandible to reposition the jaw and also to recontour the inferior border. An oblique osteotomy of the chin for repositioning and also to recontour the inferior border is carried out (see Fig. 31-5).
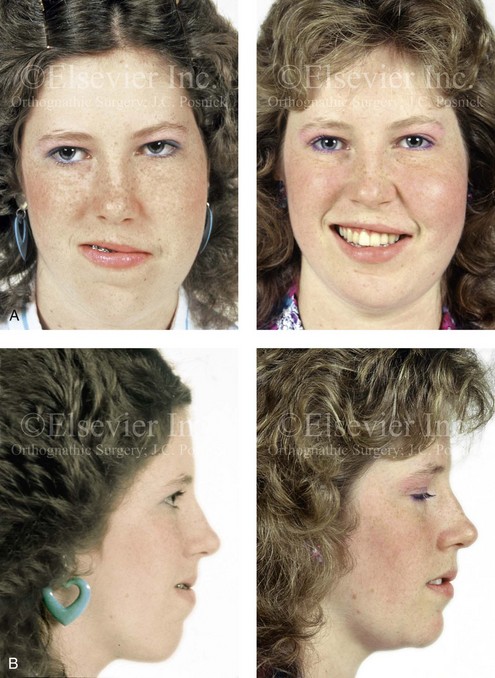
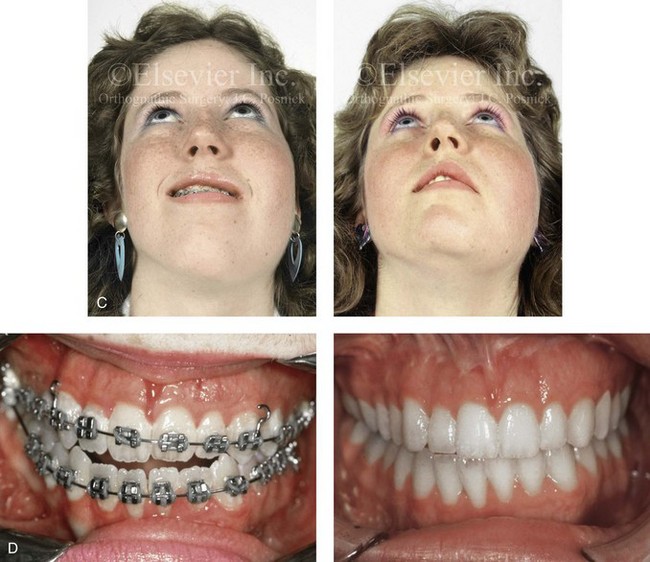
Figure 31-5 A 16-year-old girl with left hemihyperplasia of the face that affects both the soft tissues and the skeletal structures. There were distortions of the maxilla and the mandible that were treated through a combined orthodontic and surgical approach. The patient underwent a Le Fort I osteotomy (vertical intrusion and improvement of canting and asymmetry), bilateral sagittal split ramus osteotomies (improvement of asymmetry), and an osseous genioplasty (vertical reduction and horizontal advancement). Note that improvements in the facial skeleton were possible but that the soft-tissue fullness remains. A, Frontal views before and after reconstruction. B, Profile views before and after reconstruction. C, Worm’s-eye views before and after reconstruction confirm residual soft-tissue fullness. D, Occlusal views before and after reconstruction. Note: Maxillo-mandibular reconstruction does not address the soft tissue enlargement.
Vascular Malformations
Background
Vascular anomalies fall into two groups: hemangiomas and vascular malformations. Mulliken and Glowacki were the first to clarify a biologic classification of vascular anomalies on the basis of endothelial properties.226,231,232 Those authors described how hemangiomas have a proliferative endothelium whereas vascular malformations have a stable endothelium.218 Hemangiomas have a natural history that is characterized by initial proliferation, stability, and then at least partial involution (Figs. 31-6 and 31-7). Vascular malformations are present at birth, although they may not be clinically evident until sometime later during childhood. In general, vascular malformations grow commensurately with the child, but they may expand suddenly as a result of trauma, infection, or hormonal influences (Figs. 31-8 through 31-11). Vascular malformations generally occur sporadically, although inherited autosomal dominant entities do exist. The International Society for the Study of Vascular Anomalies classification system is based on Mulliken and Glowacki’s work.226 Vascular tumors include hemangiomas, Kaposiform hemangioendotheliomas, tufted angiomas, pyogenic granulomas, and hemangiopericytomas. Vascular malformations include capillary, venous, lymphatic, and arteriovenous conditions; malformations can also be mixed and include combinations of these. From a clinical perspective, it is useful to think in terms of the flow characteristics of the malformations (i.e., either slow flow or fast flow) and the type of vessel drainage.213–215,217,223–228
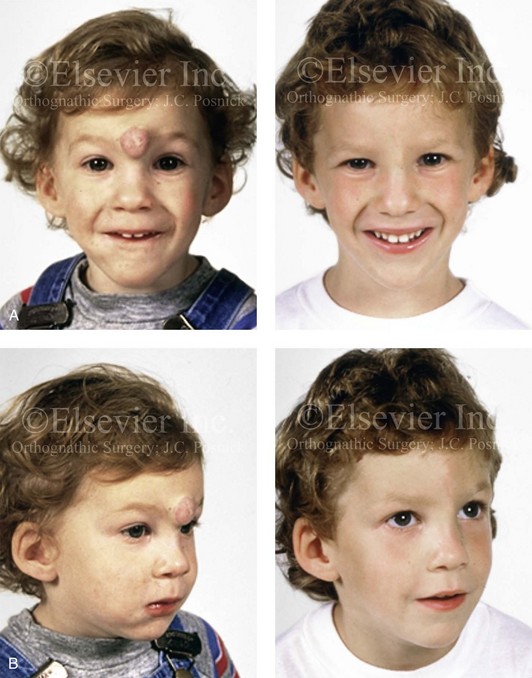
Figure 31-6 A 5-year-old boy with a hemangioma that involves the soft tissues of the forehead and the upper nose (glabella region) is shown before and 2 years after undergoing a direct vertical midline excision. Note that the soft-tissue growth did not significantly resorb (compress) the underlying skeleton. A, Frontal views before and 4 years after reconstruction. B, Oblique views before and 4 years after reconstruction.
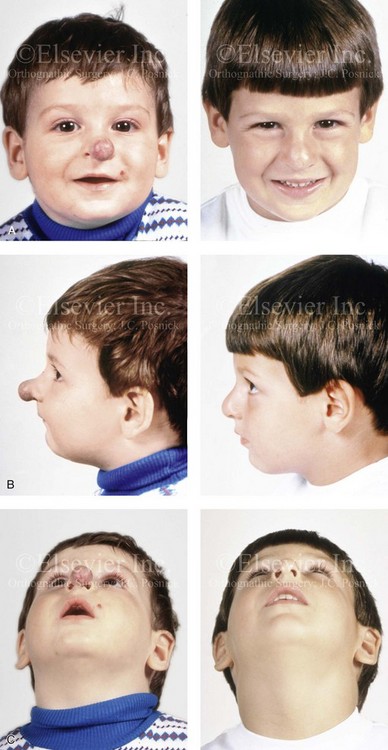
Figure 31-7 A 5-year-old boy with a hemangioma that involves the soft tissues of the tip of the nose. He is shown before and after undergoing surgical debulking through a columella-splitting open rhinoplasty technique. The soft-tissue growth displaced but did not destroy the lower lateral cartilages (LLC) or septum; the LLC were reconstructed with the use of a suture pexy technique after tumor debulking. A, Frontal views before and after reconstruction. B, Profile views before and after reconstruction. C, Worm’s-eye views before and after reconstruction.
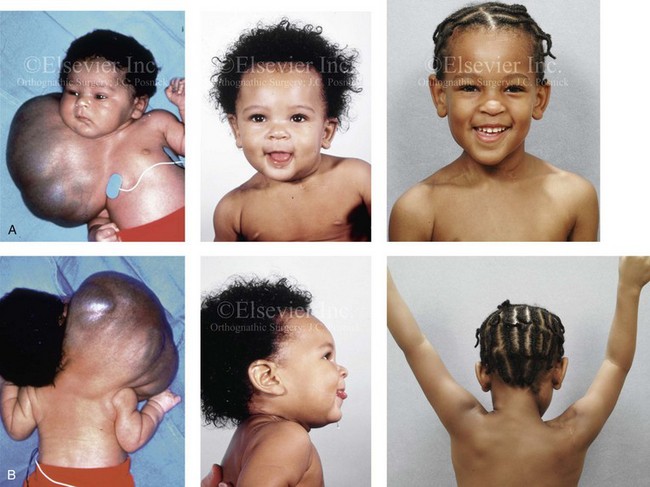
Figure 31-8 A newborn child with a massive low-flow mixed (lymphatic–venous) vascular malformation that involved the right side of the face, neck, chest wall, upper arm, and upper back. The malformation was recognized through ultrasonic evaluation during fetal development, and the child was delivered by Cesarean section with a good airway. At 6 weeks of age, he underwent a planned single staged surgical debulking procedure. He retains good cranial nerve and brachial plexus function. Portions of the sternocleidomastoid, trapezius, and platysma muscles were sacrificed in the process. No significant deformation of the craniofacial skeleton resulted. A, Frontal facial views before surgery, at 7 months of age, and then at 5 years after resection. B, Posterior facial views before surgery, at 7 months of age, and then 5 years after resection. The patient is shown with both arms extended, which indicates satisfactory neuromotor control.
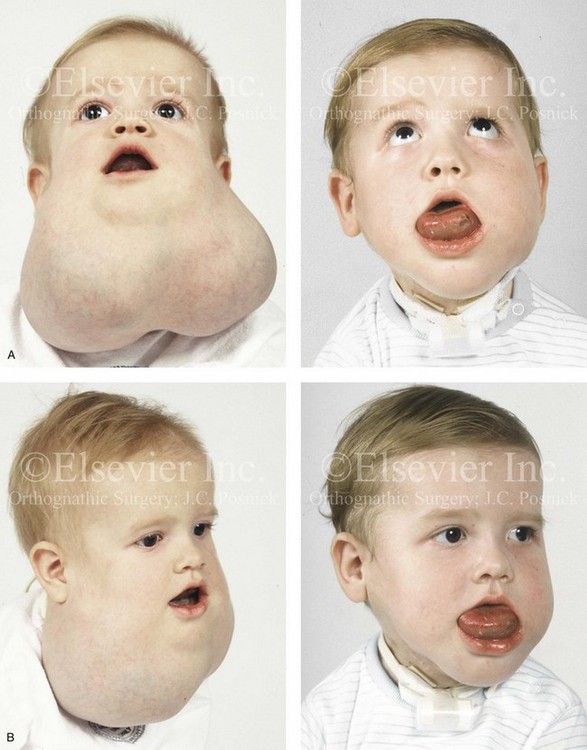
Figure 31-9 A child was born with a massive mixed low-flow (venous–lymphatic) vascular malformation of both sides of the neck and the floor of the mouth. He is shown before and after single staged surgical debulking through a bilateral neck (apron) incision. With debulking of the mass, a tracheostomy could be placed to improve oxygenation. Note that the postoperative residual venous engorgement of the tongue remains problematic. Severe deformities of the maxillomandibular skeleton are expected. A, Frontal views before and after debulking and tracheostomy. B, Oblique views before and after surgery.
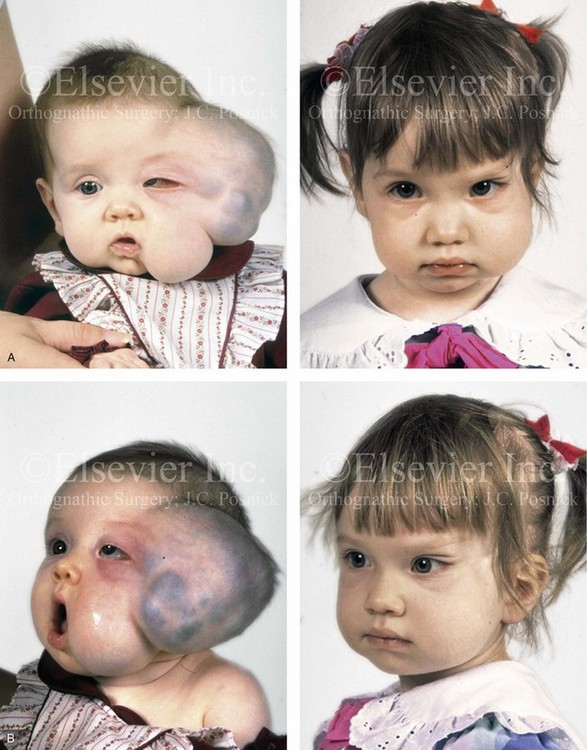
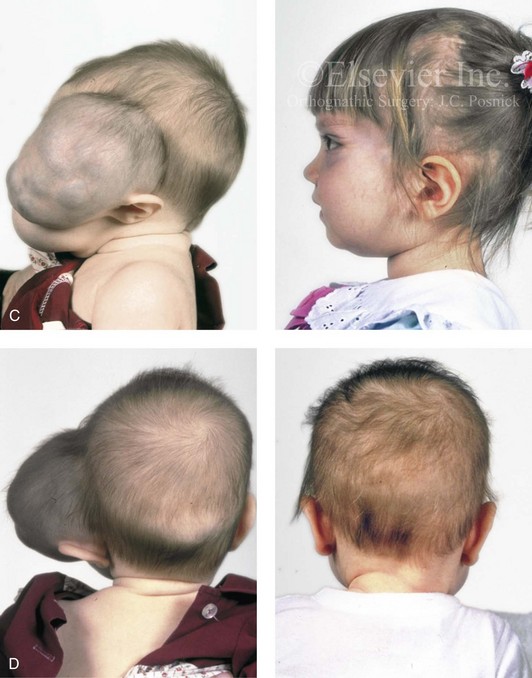
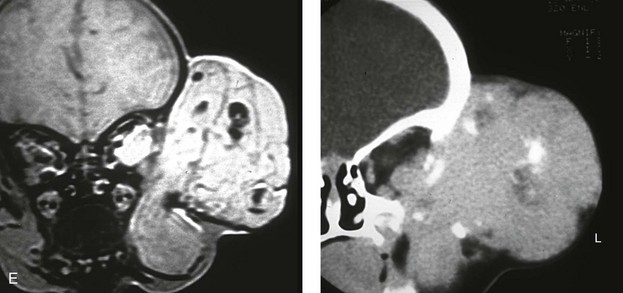
Figure 31-10 A child who was born with a massive low-flow (venous) vascular malformation that involved the left side of the face underwent single staged resection/reconstruction when she was 6 months old. She is shown before and 1 year after surgical debulking, with facial nerve preservation carried out through a preauricular and coronal scalp incision. A degree of temporal bone compression resulted from the tumor mass. A, Frontal views before and after debulking and reconstruction. B, Oblique views before and after reconstruction. C, Profile views before and after reconstruction. D, Posterior views before and after reconstruction. Note that the external ear was repositioned after the tumor was resected. E, Preoperative radiographic studies are shown. A magnetic resonance image and a computed tomography scan demonstrate the vascular malformations of the left face and orbit.
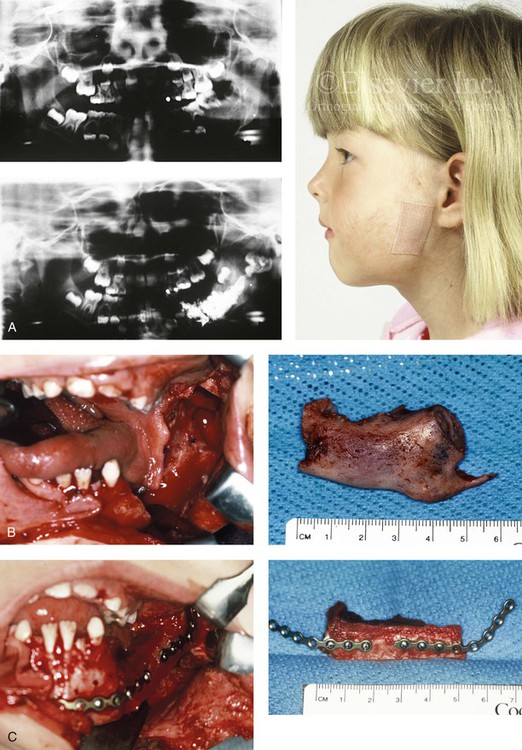
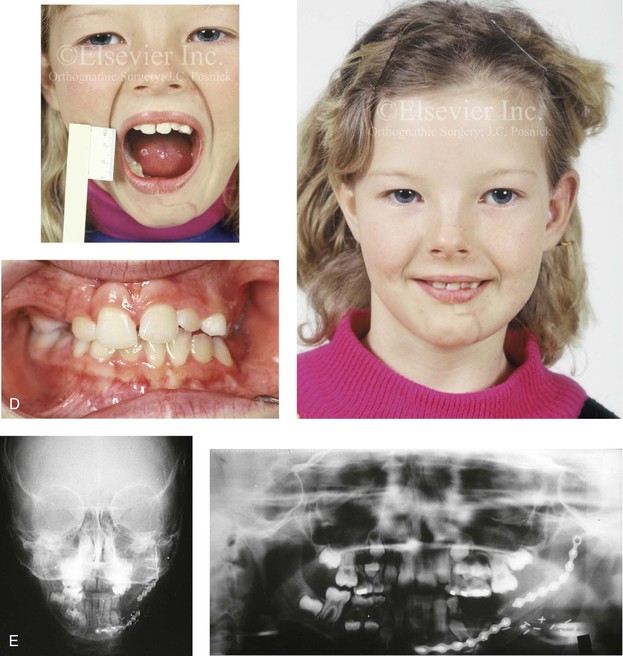
Figure 31-11 A 6-year-old girl had a high-flow (arteriovenous) vascular malformation that involved the left mandible. She had suffered multiple bleeding episodes before and after embolization and the direct injection of “glue” into the medullary cavity of the left mandible. She is shown before and 2 years after the surgical excision of the left mandible and associated soft tissues with immediate reconstruction involving a vascularized fibula bone flap. A, Profile view before surgery with bandage covering the chronic fistula tract. Preoperative Panorex radiographs before and after direct (glue) injection into the medullary cavity of the mandible. B, Involved left body–ascending ramus of the mandible excised as a specimen. Intraoral view of the mandible after resection and specimen removal is shown. C, View of autogenous fibular bone flap just before and after placement into the mandibular defect. Extended miniplates are attached to the graft and then secured to the native mandible. The vessels were then anastomosed for revascularization. D, Frontal views 2 years after excision and immediate reconstruction demonstrating satisfactory mouth opening, good facial symmetry, and facial nerve function. Occlusal view after reconstruction. E, Anteroposterior and Panorex facial radiographs after reconstruction. A (photo), B (right), C (right), D (top), From Posnick JC, Wells MD, Zuker RM: Use of the free fibula flap in the immediate reconstruction of pediatric mandibular tumors: report of cases, J Oral Maxillofac Surg 51:189–196, 1993.
Complications from vascular malformations include venostasis, ischemia, coagulopathy, disseminated intravascular coagulopathy, heart failure, and even death.209 Skeletal anomalies can occur as a result of intraosseous involvement, skeletal expansion, or skeletal compression (erosion). Any head and neck function can be affected by the presence of a vascular anomaly’s tumor-like mass.
The diagnostic imaging of vascular malformations primarily relies on radiographic modalities.212 Phleboliths are generally pathognomonic for venous malformations. Color Doppler ultrasound is an invaluable tool for the assessment of blood flow velocities and patterns of vascular lesions. Computed tomography plays an important role in the understanding of skeletal involvement. Magnetic resonance imaging is a useful non-invasive and non-ionizing test for the accurate depiction of soft-tissue relationships and allows slow-flow malformations to be accurately differentiated from high-flow malformations. Angiography and venography can play important diagnostic and therapeutic roles in the management of specific vascular malformations. Advantages of venography include the clarification of lesion size and extent and of venous drainage patterns. Angiography can clarify arteriovenous shunting, the presence of macrofistulae and microfistulae, and the location of the lesion’s nidus. Therapeutic embolization via angiography with the use of particular materials or coils can be carried out.
Lymphatic Malformations
Lymphatic malformations are slow-flow anomalies.223,225,226,228 Macrocystic lesions are easier to deal with than microcysts. Microcystic lesions do not resolve spontaneously, and they are often harder to manage surgically as a result of their close integration within the normal head and neck soft tissues that must be preserved. Only half of lymphatic lesions are recognized at birth, but the majority are evident by approximately 2 years of age. Skeletal and soft-tissue overgrowth are typical in the craniomaxillofacial region. Cervicofacial lymphatic malformations tend to present with skeletal distortions (e.g., mandibular overgrowth, malocclusion). There may be intraosseous involvement, but skeletal expansion and erosion (remodeling) as a result of mass effect are typical. Symptoms of localized swelling, pain, and erythema associated with infection are common. Significant complications include intralesional bleeding and infection that are often associated with rapid expansion and then moderate regression. Rapid swelling can lead to airway obstruction. Macrocysts may undergo involution if they are ruptured by trauma, or they may be treated via the injection of sclerosing agents or aspiration. However, surgical removal should be considered, when feasible. Complete resection is seldom possible because lymphatic malformations often surround and incorporate normal anatomy that is not deemed appropriate for resection (see Figs. 31-8 and 31-9).220–222,229,230 Orthognathic procedures would frequently be beneficial but are not often carried out.
Venous Malformations
Venous malformations are the most common form seen in the head and neck region.223,225,226,228 They are present at the time of birth, but they may not have been recognized, or they may have been misdiagnosed.210,219 Histologically, they are composed of variably sized, thickened, dilated, spongy channels that are devoid of smooth muscle and lined by endothelial cells. Clinically, they are compressible subcutaneous masses that are often deep blue in color. They generally grow commensurate with the child but, as with lymphatic malformations, trauma, infection, and hormonal changes can cause rapid expansion. Venous malformations can involve any tissue layer (i.e., skin, mucosa, muscle, brain, bone, or viscera). Thrombosis within the malformation commonly leads to phlebolith formation. When present, phleboliths are pathognomonic and assist with the accurate diagnosis. Venous malformations are usually unilateral and result in significant facial asymmetry and dysfunction. They may affect speech, swallowing, chewing, breathing, vision, hearing, cognition, and jaw or neck range of motion. They may be intraosseous or have secondary effects on the bone (i.e., expansion, remodeling, or resorption). Treatment to reduce the size of venous malformations includes the injection of sclerosing agents in an attempt to damage the endothelial cells via intravascular thrombosis and fibrosis as well as partial surgical excision (see Figs. 31-8, 31-9, and 31-10).220,221,222,229
Arteriovenous Malformations
Arteriovenous malformations are high-flow malformations with a direct connection between an artery and a vein, with no intervening capillary bed.223,225,226,228 These malformations are also present at birth, but they may have gone unrecognized or have been misdiagnosed as a more straightforward hemangioma. The confirmation of clinical diagnosis is helpful with color Doppler examination, which can determine flow rate, volume, and reversal of flow. Magnetic resonance imaging can determine the extent of the lesion and its relationship with the surrounding anatomy. Angiography can further clarify the lesion and be used for the therapeutic embolization of an arteriovenous malformations. If an arteriovenous malformation is well localized and can be reliably excised with satisfactory reconstruction, this is always a first choice (see Fig. 31-11). Unfortunately, larger, more extensive lesions are not surgically resectable. Superselective embolization performed by an experienced interventional radiologist with the placement of an ablative agent (e.g., absolute alcohol, sodium tetradecyl sulfate) to induce the destruction of the endothelium may be useful. Embolization that involves coils or particles is another technique for temporarily occluding flow. The ligation of feeding vessels or proximal embolization is generally avoided, because rapid collateralization tends to occur.220,221,222,229
Capillary Malformations
Capillary malformations, which were previously referred to as port-wine stains, are intradermal vascular anomalies.223,225,226,228 They occur in less than 1% of newborns, and they have an equal distribution in male and female individuals.216 They typically present as either pink or red cutaneous discolorations. They have a tendency to darken, thicken, and become nodular with age. Several conditions are associated with capillary malformations, including Sturge–Weber syndrome, which presents with capillary malformations in the trigeminal nerve distribution in combination with vascular anomalies of the leptomeninges, often with a seizure disorder, developmental delay, and eye involvement (i.e., glaucoma and retinal detachment).211 Proteus syndrome may also be associated with cutaneous vascular malformations.215 When feasible, the treatment of capillary malformations with a pulsed dye laser should be considered.
Velocardiofacial Syndrome
Background
Velocardiofacial syndrome (VCFS) is a genetic condition that is characterized by abnormal pharyngeal arch development that results in defective development of the thymus, the parathyroid glands, and the conotruncal region of the heart. Shprintzen and colleagues first described this syndrome in1978.249 Since then, almost 200 different individual clinical features have been associated with VCFS.233,240,247 No single individual has been found to have all of the described features. Affected individuals will present with unique facial features, and they may also have cardiac defects, palatal abnormalities, hypernasal speech, hypotonia, defective thymic development, and a variety of learning disabilities.248
Approximately 75% of individuals with VCFS have cardiac anomalies. The cardiac defects are usually of the conotruncal type.234,242 The most common variations include an interrupted aortic arch (50%), truncus arteriosus (35%), and tetralogy of Fallot (16%). Palatal abnormalities predispose these individuals to speech difficulties. Defective thymic development is associated with impaired immune function. Learning disabilities include overt developmental delay, psychiatric disorders, and musculoskeletal defects.238 Interestingly, ophthalmologic abnormalities occur in approximately 70% of individuals with VCFS; these may include bilateral cataracts, tortuous retinal vessels, and small optic discs.
Approximately 10% of individuals with VCFS will have DiGeorge syndrome, which consists of at least two of the three following features239:
Approximately 15% to 20% of patients with VCFS will be born with Pierre Robin sequence (see Chapter 4). Many individuals are mistakenly categorized as having CHARGE syndrome.
Pathophysiology
VCFS is now known to be a congenital disorder that is caused by a microdeletion at the Q11.2 band, which is located on the long arm (Q) of chromosome 22.235,243 The microdeletion is known to cause an abnormality of morphogenesis that affects the migration of the neural crest cells and therefore the early development of branchial arches. In approximately 90% of affected individuals, the disorder occurs as a result of a new mutation. In the other 10% of cases, the disorder is inherited from a parent in an autosomal dominant fashion. Affected individuals have a 50% chance of passing VCSF to each offspring.244
Epidemiology
The prevalence of VCFS is the United States is approximately 1 in 2000 live births. Internationally, the syndrome occurs in 1 in 4000 live births. The frequency of VCFS in newborns with isolated cleft palate is approximately 8%. There seems to be no gender predilection. The syndrome is present at birth, but it is often unrecognized until childhood or later.237 Although the heart defect or the cleft palate is generally detected soon after birth, the learning disorders and psychiatric illnesses may not become apparent until the school-aged years.236 Even then the correct diagnosis is not always made.
DiGeorge syndrome, which involves a total absence of the thymus and a severe T-cell immunodeficiency, is found in less than 0.5% of patients with VCFS.236,241,245 Most affected individuals have only partial defects, with impaired thymic development and variable defects in their T-cell numbers.
Author’s Approach to Reconstruction in Patients with Velocardiofacial Syndrome
Evaluation by clinicians who have experience with clefting usually centers around the issue of hypernasal speech and whether or not a palatal procedure is indicated. Concern about the location of the carotid arteries (when considering the use of a pharyngeal flap) is always part of the discussion.246
A percentage of teenagers with VCFS will present with a jaw deformity and malocclusion. These patients will benefit from a comprehensive orthodontic and surgical approach. They generally have a long face growth pattern (see Chapter 21) and require osteotomies of the maxilla, the mandible, and the chin region (Fig. 31-12).
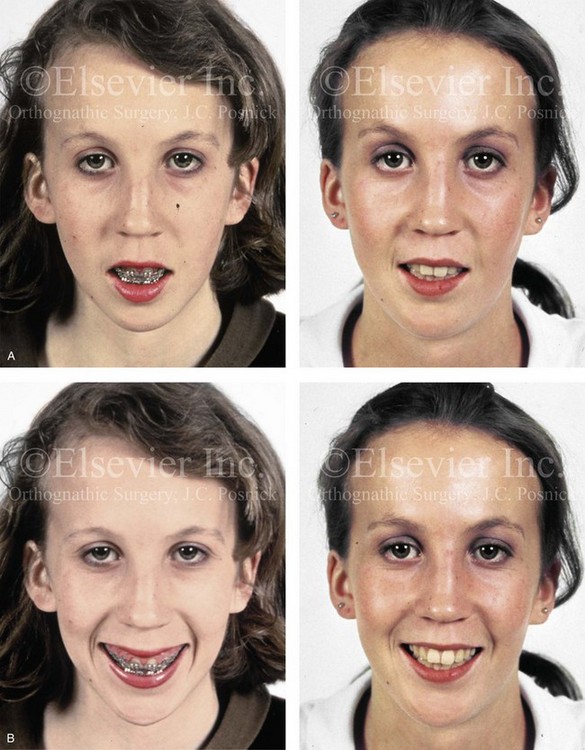
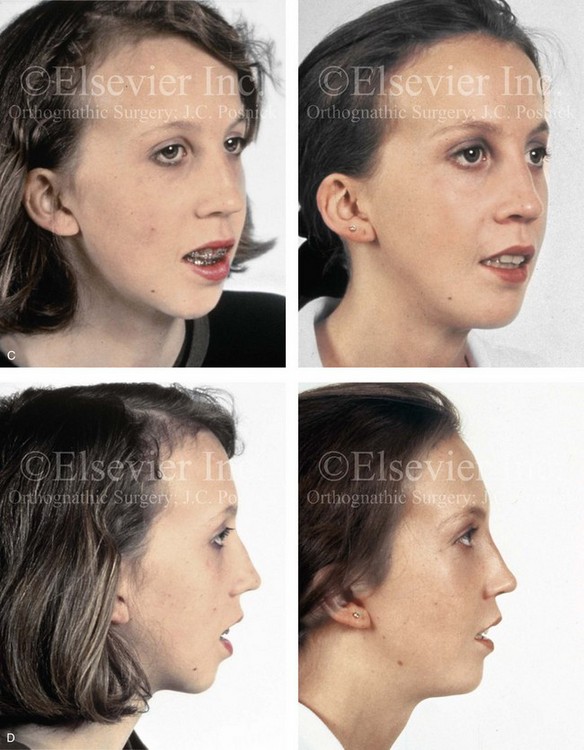
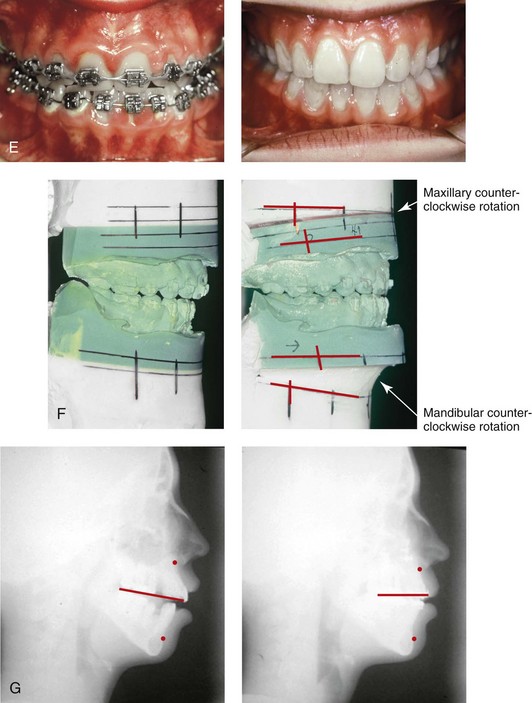
Figure 31-12 A 14-year-old girl who was born with velocardiofacial syndrome. She had mild learning disabilities, an adequately functioning velopharyngeal valve, and mild sibilant distortions with articulation as a result of malocclusion. She had a long face growth pattern that included a skeletal Class II anterior open-bite malocclusion. She underwent orthodontic treatment that included four bicuspid extractions and orthognathic surgery. The patient’s procedures included Le Fort I osteotomy (anterior vertical intrusion with counterclockwise rotation) and bilateral sagittal split ramus osteotomies (horizontal advancement with counterclockwise rotation). A, Frontal views in repose before and after reconstruction. B, Frontal views with smile before and after reconstruction. C, Oblique views before and after reconstruction. D, Profile views before and after reconstruction. E, Occlusal views before and after reconstruction. F, Articulated dental casts that indicate analytic model planning. G, Lateral cephalometric radiographs before and after reconstruction.
References
1. Agris, J, Dingman, RO, Varon, J. Correction of webbed neck defects. Ann Plast Surg. 1983; 2:299.
2. Baba, H, Maezawa, Y, Furusawa, N, et al. The cervical spine in the Klippel–Feil syndrome: A report of 57 cases. Int Orthop. 1995; 19:204.
3. Baird, PA, Robinson, GC, Buckler, WS. Klippel–Feil syndrome: A study of mirror movements detected by electromyography. Am J Dis Child. 1967; 113:546.
4. Bauman, GI. Absence of the cervical spine: Klippel–Feil syndrome. JAMA. 1932; 98:129.
5. Bavinck, JN, Weaver, DD. Subclavian artery supply disruption sequence: Hypothesis of a vascular etiology for Poland, Klippel–Feil, and Moebius anomalies. Am J Med Genet. 1986; 23:903.
6. Brill, CB, Peyster, RG, Keller, MS, et al. Isolation of the right subclavian artery with subclavian steal in a child with Klippel–Feil anomaly: An example of the subclavian artery supply disruption sequence. Am J Med Genet. 1987; 26:933–940.
7. Brown, MW, Templeton, AW, Hodges, FJ, 3rd. The incidence of acquired and congenital fusions in the cervical spine. Am J Roentgenol. 1964; 92:1255.
8. Chandler, FA. Webbed neck (pterygium colli). Am J Dis Child. 1937; 53:798.
9. Chemke, J, Nisani, R, Fischel, RE. Absent ulna in the Klippel–Feil syndrome: An unusual associated malformation. Clin Genet. 1980; 17:167.
10. Cohney, BC. The association of cleft palate with the Klippel–Feil syndrome. Plast Reconstr Surg. 1963; 31:179.
11. Daniilidis, J, Maganaris, T, Dimitriadis, A, et al. Stapes gusher and Klippel–Feil syndrome. Laryngoscopy. 1978; 88:1178.
12. Da Silva, EO. Autosomal recessive Klippel–Feil syndrome. J Med Genet. 1982; 19:130.
13. De Bruin, M. Pterygium colli congenitum (congenital webbing). Am J Dis Child. 1928; 36:333.
14. Duncan, PA. Embryologic pathogenesis of renal agenesis associated with cervical vertebral anomalies (Klippel–Feil phenotype). Birth Defects Orig Artic Ser. 1977; 13:91.
15. Erskine, CA. An analysis of the Klippel–Feil syndrome. Arch Pathol. 1946; 41:269.
16. Everberg, G, et al. Wildervanck syndrome: Klippel–Feil syndrome associated with deafness and retraction of the eyeball. Br J Radiol. 1963; 36:562.
17. Fietti, VG, Jr., Fielding, W. The Klippel–Feil syndrome: Early roentgenographic appearance and progression of the deformity: A report of two cases. J Bone Joint Surg Am. 1976; 58:891.
18. Foucar, HO. Pterygium colli and allied conditions. Can Med Assoc J. 1948; 59:251.
19. Fragoso, R, Cid-Garcia, A, Hernandez, A, et al. Frontonasal dysplasia in the Klippel–Feil syndrome: A new associated malformation. Clin Genet. 1982; 22:270.
20. Gardner, WJ. Klippel–Feil syndrome, iniencephalus, anencephalus, hindbrain hernia, and mirror movements: Overdistention of the neural tube. Childs Brain. 1979; 5:361.
21. Gienapp, Rvon. Zur erbbiologie der Klippel–Feilschen krankheit. Nervenarzt. 1950; 21:74.
22. Gilmour, JR. The essential identity of the Klippel–Feil syndrome and iniencephaly. J Pathol Bacteriol. 1941; 53:117.
23. Ginrup, PA, Ginrup, L. Klippel–Feil syndrome. Danish Med Bull. 1964; 11:50.
24. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
25. Gray, SW, Romaine, CB, Skandalakis, JE. Congenital fusion of the cervical vertebrae. Surg Gynecol Obstet. 1964; 118:373.
26. Guille, JT, Miller, A, Bowen, JR, et al. The natural history of Klippel–Feil syndrome: Clinical, roentgenographic, and magnetic resonance imaging findings at adulthood. J Pediatr Orthop. 1995; 15:17.
27. Gunderson, CH, Greenspan, RH, Glaser, GH, et al. The Klippel–Feil syndrome: Genetic and clinical evaluation of cervical fusion. Medicine. 1967; 46:491.
28. Gunderson, CH, Solitaire, GB. Mirror movements in patients with the Klippel–Feil syndrome. Arch Neurol. 1968; 18:675.
29. Heiner, H. Uber die kombination von medianer gaumenspalte und Klippel–Feil syndrom. Dtsch Stomatol. 1960; 10:92.
30. Helmi, C, Pruzansky, S. Craniofacial and extracranial malformations in the Klippel–Feil syndrome. Cleft Palate J. 1980; 17:65.
31. Hensinger, RN, Lang, JE, MacEwen, GD. Klippel–Feil syndrome: A constellation of associated anomalies. J Bone Joint Surg Am. 1974; 56:1246.
32. Jarcho, S, Levin, PM. Hereditary malformation of the vertebral bodies. Bull Johns Hopkins Hosp. 1938; 62:216.
33. Juberg, RC, Gershank, JJ. Cervical vertebral fusion (Klippel–Feil) syndrome with consanguineous parents. J Med Genet. 1976; 13:246.
34. Klippel, M, Feil, A. Un cas d’absence des vertebres cervicales. Nouv Iconogr Salpet. 1912; 25:223.
35. Lauerman, WC. Maurice Klippel. Spine. 1995; 20:157.
36. Lowry, RB. The Klippel–Feil anomalad as part of the fetal alcohol syndrome. Teratology. 1977; 16:53.
37. Luftman, II, Weintraub, S. Klippel–Feil syndrome in a full-term newborn infant. NY State J Med. 1951; 51:2035.
38. Mahajan, PV, Bharucha, BA. Evaluation of short neck: New neck length percentiles and linear correlations with height and sitting height. Indian Pediatr. 1994; 31:1193.
39. McLay, K, Maran, AGD. Deafness and the Klippel–Feil syndrome. J Otolaryngol. 1969; 83:175.
40. Mecklenburg, RS, Krueger, PM. Extensive genitourinary anomalies associated with Klippel–Feil syndrome. Am J Dis Child. 1974; 128:92.
41. Menick, FJ, Furnas, DW, Achauer, BM. Lateral cervical advancement flaps for the correction of webbed-neck deformity. Plast Reconstr Surg. 1984; 73:223.
42. Moore, WB, Matthews, TJ, Rabinowitz, R. Genitourinary anomalies associated with Klippel–Feil syndrome. J Bone Joint Surg Am. 1975; 57:355.
43. Morrison, SG, Perry, LW, Scott, LP, 3rd. Congenital torticollis (Klippel–Feil syndrome) and cardiovascular anomalies. Am J Dis Child. 1968; 115:614.
44. Mosberg, WH. The Klippel–Feil syndrome: Etiology and treatment of neurologic signs. J Nerv Ment Dis. 1953; 117:479.
45. Nagib, MG, Larson, DA, Maxwell, RE, et al. Neuroschisis of the cervical spinal cord in a patient with Klippel–Feil syndrome. Neurosurgery. 1987; 20:629.
46. Nagib, MG, Maxwell, RE, Chou, SN. Identification and management of high-risk patients with Klippel–Feil syndrome. J Neurosurg. 1984; 61:523.
47. Nagib, MG, Maxwell, RE, Chou, SN. Klippel–Feil syndrome in children: Clinical features and management. Childs Nerv Syst. 1985; 1:255.
48. Neidengard, L, Carter, TE, Smith, DW. Klippel–Feil malformation complex in fetal alcohol syndrome. Am J Dis Child. 1978; 132:929.
49. Nobel, TP, Frawley, JM. Klippel–Feil syndrome: Numerical reduction of cervical vertebrae. Ann Surg. 1925; 82:728.
50. Nora, JJ, et al. Klippel–Feil syndrome with congenital heart disease. Am J Dis Child. 1961; 102:858.
51. Palant, DI, Carter, BL. Klippel–Feil syndrome and deafness: A study with polytomography. Am J Dis Child. 1972; 123:218.
52. Pizzutillo, PD, Woods, M, Nicholson, L, et al. Risk factors in Klippel–Feil syndrome. Spine. 1994; 19:2110.
53. Posnick, JC. Other craniofacial syndromes: Klippel–Feil anomaly, neurofibromatosis, Down syndrome, micro-ophthalmia, hemifacial hyperplasia (hemi-hypertrophy). In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:503–527.
54. Raas-Rothschild, A, Goodman, RM, Grunbaum, M, et al. Klippel–Feil anomaly with sacral agenesis: Additional subtype, type IV. J Craniofac Genet Dev Biol. 1988; 8:297.
55. Schwarze, K. Zur frage des Klippel–Feilschen fehlers der wirbelsule. Arch Orthop Unfallchir. 1941; 41:47.
56. Shoul, MJ, Ritvo, M. Clinical and roentgenological manifestations of Klippel–Feil syndrome (congenital fusion of the cervical vertebrae brevicollis): Report of 8 additional cases and review of the literature. Am J Roentgenol. 1952; 68:369.
57. Slate, RK, Posnick, JC, Armstrong, DC, Buncic, JR. Cervical spine subluxation associated with congenital muscular torticollis and craniofacial asymmetry. Plast Reconstr Surg. 1993; 91(7):1187–1195.
58. Sommerfeld, RM, Schweiger, JW. Cleft palate with Klippel–Feil. syndrome. Oral Surg. 1969; 27:737.
59. Stadnicki, G, Rassumowski, D. The association of cleft palate with the Klippel–Feil. syndrome. Oral Surg. 1972; 33:335.
60. Stewart, EJ, O’Reilly, BF. Klippel–Feil syndrome and conductive deafness. J Laryngol Otol. 1989; 103:947.
61. Tveter, KJ, Kluge, T. Cor triloculare biatrium associated with Klippel–Feil syndrome. Acta Paediatr Scand. 1965; 54:489.
62. Weber, V, Schneider, HJ, Haberlandt, W. Pathogenetische und genetische fragen beim Klippel–Feil syndrom–vergleichende betrachtungen bei zwei vettern. Fortschr Geb/Roentgenstr/Nuklearmed. 1973; 119:209.
63. Willemsen, WN. Combination of the Mayer–Rokitansky–Kuster and Klippel–Feil syndrome—A case report and literature review. Eur J Obstet Gynecol Reprod Biol. 1982; 13:229.
64. Windle-Taylor, PC, Emery, PJ, Phelps, PD. Ear deformities associated with the Klippel–Feil syndrome. Ann Otol Rhinol Laryngol. 1981; 90:210.
65. Adams, MM, Erickson, JD, Layde, PM, et al. Down syndrome: Recent trends in the United States. JAMA. 1981; 246:758.
66. Ardran, GM, Harker, P, Kemp, FH. Tongue size in Down’s syndrome. J Ment Defic Res. 1972; 16:160.
67. Balkany, TJ, Downs, MP, Jafek, BW, et al. Hearing loss in Down syndrome. Clin Pediatr. 1979; 18:116.
68. Bjuggren, G, Jensen, R, Strombeck, JO. Macroglossia and its surgical treatment: Indications and postoperative experiences from the orthodontic, phoniatric, and surgical points of view. Scand J Plast Reconstr Surg. 1968; 2:116.
69. Brousseau, K. Mongolism. Baltimore, Md: Williams & Wilkins; 1928.
70. Dios, PD, Feijoo, JF, Ferreiro, MC, et al. Functional consequences of partial glossectomy. J Oral Maxillofac Surg. 1994; 52:12.
71. Donaldson, JD, Redmond, WM. Surgical management of obstructive sleep apnea in children with Down syndrome. J Otolaryngol. 1988; 17:398.
72. Down, JL. Observation on an ethnic classification of idiots. Ment Retard. 1866; 33(1):54–56.
73. Farkas, LG, Munro, IR, Kolar, JC. Abnormal measurements and disproportions in the face of Down syndrome patients: Preliminary report of an anthropometric study. Plast Reconstr Surg. 1985; 75:159.
74. Gisel, EG, Lange, LJ, Niman, CW. Tongue movements in 4 and 5 year old Down syndrome children during eating: A comparison with normal children. Am J Occup Ther. 1984; 38:660.
75. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
76. Hinderer, U. Malar implants for improvement of the facial appearance. Plast Reconstr Surg. 1975; 56:157.
77. Hohler, H. Changes in facial expression as a result of plastic surgery in mongoloid children. Aesthetic Plast Surg. 1977; 1:245.
78. Hultcrantz, E, Svanholm, H. Down syndrome and sleep apnea—a therapeutic challenge. Int J Pediatr Otorhinolaryngol. 1991; 21:263.
79. Katz, S, Kravetz, S. Facial plastic surgery for persons with Down syndrome: research finding and their professional such implications. Am J Ment Retard. 1989; 94(2):101–110.
80. Lefavre, J-F, Cohen, SR, Burstein, FD, et al. Down syndrome: Identification and surgical management of obstructive sleep apnea. Plast Reconstr Surg. 1997; 99:629.
81. Lejeune, J, Turpin, R, Gautier, M. Le mongolisme, maladie chromosomique (trisomie). Bull Acad Natl Med. 1959; 143:256.
82. Lemperle, G. Plastic surgery in children with Down syndrome. In: Lane D, Stratford B, eds. Current approaches to Down syndrome. Eastbourne, UK: Holt, Rinehart & Winston; 1985:131.
83. Lemperle, G, Nievergelt, J. Plastisch–chirurgische korrekturen im geicht von kindern mit Down–syndrom. In Plastische Chirurgie bei Menschen mit Down Syndrom. Lebenshilfe Marburg. 1983; 9:19.
84. Lemperle, G, Radu, D. Facial plastic surgery in children with Down syndrome. Plast Reconstr Surg. 1980; 66:337.
85. Lemperle, G, Spitalny, HH. Long-term experience with silicone implants in the face. In: Caronni EP, ed. Craniofacial surgery. Boston: Little, Brown; 1985:490.
86. Marcus, CL, Keens, TG, Bautista, DB, et al. Obstructive sleep apnea in children with Down syndrome. Pediatrics. 1991; 88:132.
87. Margar-Bacal, F, Witzel, MA, Munro, IR. Speech intelligibility after partial glossectomy in children with Down syndrome. Plast Reconstr Surg. 1987; 79:44.
88. Michejda, M, Menolascino, FJ. Skull base abnormalities in Down syndrome. Ment Retard. 1975; 13:24.
89. Martinez-Frias, ML. The real earliest historical evidence of Down syndrome. Am J Med Genet A. 2005; 132A(2):231.
90. Mearig, JS. Facial surgery and an active modification approach for children with Down syndrome: Some psychological and ethical issues. Rehabil Lit. 1985; 46:72.
91. Olbrisch, RR. Plastic surgical management of children with Down syndrome: Indications and results. Br J Plast Surg. 1982; 35:195.
92. Olbrisch, RR. Plastic surgery in 250 children with Down syndrome: Indications and results. Transactions of the Eighth International Congress on Plastic and Reconstructive Surgery. Montreal: International Plastic Surgery Society; 1983.
93. Otermin, A. Mongolism (Down syndrome) and plastic surgery. Prensa Meica Argentina. 1967; 60:27.
94. Patterson, RS, Munro, IR, Farkas, LG. Transconjunctival lateral canthopexy in Down syndrome patients: A nonstigmatizing approach. Plast Reconstr Surg. 1987; 79:714.
95. Posnick, JC. Other craniofacial syndromes: Klippel–Feil anomaly, neurofibromatosis, Down syndrome, micro-ophthalmia, hemifacial hyperplasia (hemi-hypertrophy). In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:503–527.
96. Pueschel S, Rynders J, eds. Down syndrome: Advances in biomedicine and the behavioral sciences. Cambridge, Mass: Ware Press, 1982.
97. Rand, Y, Mintsker, Y, Feuerstein, R. Reconstructive plastic surgery in Down syndrome children and adults: Parents’ evaluations—follow-up data. Jerusalem, Hadassah-Wizo-Canada Research Institute. 1984.
98. Rolfe, CR, Montague, JC, Jr., Tirman, RM, et al. Pilot perceptual and physiological investigation of hypernasality in Down syndrome adults. Folia Phoniatr. 1979; 31:177.
99. Rozner, L. Facial plastic surgery for Down syndrome. Lancet. 1983; 1:132.
100. Silverman, M. Airway obstruction and sleep disruption in Down syndrome. Br Med J. 1988; 296:1618.
101. Strome, M. Down syndrome: A modern otorhinolaryngological perspective. Laryngoscope. 1981; 91:1581.
102. Strome, M. Obstructive sleep apnea in Down syndrome children: A surgical approach. Laryngoscope. 1986; 96:1340.
103. Wexler, MR, Peled, IJ. Plastic surgery in Down syndrome. Down Syndrome. 1983; 6:7.
104. Wexler, MR, Peled, IJ, Rand, Y, et al. Rehabilitation of the face in patients with Down syndrome. Plast Reconstr Surg. 1986; 77:383.
105. Abuelo, DN, Meryash, DL. Neurofibromatosis with fully expressed Noonan syndrome. Am J Med Genet. 1988; 29:937.
106. Afifi, AK, Dolan, KD, Van Gilder, JC, et al. Ventriculomegaly in neurofibromatosis. Neurofibromatosis. 1988; 1:299–305.
107. Baden, E, Pierce, HE, Jackson, WF. Multiple neurofibromatosis with oral lesions: Review of the literature and report of a case. Oral Surg Oral Med Oral Pathol. 1955; 8:268.
108. Baden, E, Jones, JR, Khedekar, R, et al. Neurofibromatosis of the tongue: A light and electronmicroscopic study with review of the literature from 1849 to 1981. J Oral Med. 1984; 39:157.
109. Bader, JL. Neurofibromatosis and cancer. Ann NY Acad Sci. 1986; 486:57.
110. Barker, D, Wright, E, Nguyen, K, et al. Gene for von Recklinghausen neurofibromatosis is in the pericentric region of chromosome 17. Science. 1987; 236:1100.
111. Benedict, PH, Szabo, G, Fitzpatrick, TB, et al. Melanotic macules in Albright syndrome and in neurofibromatosis. JAMA. 1968; 205:618.
112. Binet, FF, Kieffer, SA, Martin, SH, et al. Orbital dysplasia in neurofibromatosis. Radiology. 1969; 93:829.
113. Bloem, JJ, van der Meulen, JC. Neurofibromatosis in plastic surgery. Br J Plast Surg. 1978; 31:50.
114. Brooks, B, Lehman, EP. The bone changes in Recklinghausen’s neurofibromatosis. Surg Gynecol Obstet. 1924; 38:587.
115. Buntin, PT, Fitzgerald, JF. Gastrointestinal neurofibromatosis. Am J Dis Child. 1970; 119:521.
116. Carey, JC, Laub, JM, Hall, BD. Penetrance and variability in neurofibromatosis: A genetic study of 60 families. Birth Defects Orig Artic Ser. 1979; 15:271.
117. Chao, DH-C. Congenital neurocutaneous syndromes in childhood: I. Neurofibromatosis. J Pediatr. 1959; 55:189.
118. Clark, RD, Hutter, JJ, Jr. Familial neurofibromatosis and juvenile chronic myelogenous leukemia. Hum Genet. 1982; 60:230.
119. Cohen, MM, Jr., Hayden, PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979; 15:291.
120. Crawford, AH. Neurofibromatosis in children. Acta Orthop Scand Suppl. 1986; 218:1.
121. Crowe, FW, Schull, WJ. Diagnostic importance of cafe-au-lait spot in neurofibromatosis. Arch Intern Med. 1953; 91:758.
122. D’Ambrosio, JA, Langlais, RP, Young, RS. Jaw and skull changes in neurofibromatosis. Oral Surg. 1988; 66:391.
123. Davis, WB, Edgerton, MT, Hoffmeister, SF. Neurofibromatosis of the head and neck. Plast Reconstr Surg. 1954; 14:186.
124. Diehl, SR, Boehnke, M, Erickson, RP, et al. A refined genetic map of the region of chromosome 17 surrounding the von Recklinghausen neurofibromatosis (NFI) gene. Am J Hum Genet. 1989; 44:33.
125. Dreyfuss, U, Ben-Arieh, JY, Hirshowitz, B. Liposarcoma: A rare complication in neurofibromatosis: Case report. Plast Reconstr Surg. 1978; 61:287.
126. Dunn, DW. Neurofibromatosis in childhood. Curr Probl Pediatr. 1987; 17:445.
127. Farag, MZ. Vascular malformation of the parotid gland in von Recklinghausen’s disease. J Laryngol Otol. 1983; 97:571.
128. Fienman, NL, Yakovac, W. Neurofibromatosis in childhood. J Pediatr. 1970; 76:339.
129. Freeman, AG. Proptosis and neurofibromatosis. Lancet. 1987; 1:1032.
130. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
131. Grabb, WC, Reed, O, Dingman, RMO, et al. Facial hamartomas in children: Neurofibroma, lymphangioma, and hemangioma. Plast Reconstr Surg. 1980; 66:509.
132. Grant, WM, Walton, DS. Distinctive gonioscopic findings in glaucoma due to neurofibromatosis. Arch Ophthalmol. 1968; 79:127.
133. Griffith, BH, Lewis, YL, Jr., McKinney, P. Neurofibromas of the head and neck. Surg Gynecol Obstet. 1985; 160:534.
134. Hall, BD. Congenital lid ptosis associated with neurofibromatosis [letter to the editor]. Am J Med Genet. 1986; 25:595.
135. Holt, JF. Neurofibromatosis in children. Am J Roentgenol. 1978; 130:615.
136. Holt, JF, Wright, EM. The radiologic features of neurofibromatosis. Radiology. 1948; 51:647.
137. Horwich, A, Riccardi, VM, Francke, U. Brief clinical report: Aqueductal stenosis leading to hydrocephalus—an unusual manifestation of neurofibromatosis. J Med Genet. 1983; 14:577.
138. Hunt, JC, Pugh, DG. Skeletal lesions in neurofibromatosis. Radiology. 1961; 76:1.
139. Jackson, IT, Carbonnel, A, Potparic, Z, et al. Orbitotemporal neurofibromatosis: Classification and treatment. Plast Reconstr Surg. 1993; 92:1.
140. Jackson, IT, Laws, ER, Martin, RD. The surgical management of orbital neurofibromatosis. Plast Reconstr Surg. 1983; 71:751.
141. Jacobs, MH. Oral manifestations in von Recklinghausen’s disease (neurofibromatosis). Am J Orthod Oral Surg. 1946; 32:28.
142. Janecka, IP. Correction of ocular dystopia. Plast Reconstr Surg. 1996; 97:892.
143. Kaplan, P, Rosenblatt, B. A distinctive facial appearance in neurofibromatosis von Recklinghausen. Am J Med Genet. 1985; 21:463.
144. Koblin, I, Reil, B. Changes in the facial skeleton in cases of neurofibromatosis. J Maxillofac Surg. 1975; 3:23.
145. Kragh, LV, Soule, EH, Masson, JK. Neurofibromatosis (von Recklinghausen’s disease) of the head and neck: cosmetic and reconstructive aspects. Plast Reconstr Surg Transplant Bull. 1960; 25:565.
146. Laue, L, Comite, F, Hench, K, et al. Precocious puberty associated with neurofibromatosis and optic gliomas. Am J Dis Child. 1985; 139:1097.
147. Lewis, RA, Gerson, LP, Axelson, KA, et al. Von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology. 1984; 91:929.
148. Maceri, DS, Saxon, KG. Neurofibromatosis of the head and neck. Head Neck Surg. 1984; 6:842.
149. Marchac, D. Intracranial enlargement of the orbital cavity and palpebral remodelling for orbitopalpebral neurofibromatosis. Plast Reconstr Surg. 1984; 73:534.
150. Martuza, RL, Eldridge, R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med. 1988; 318:684.
151. Miller, M, Hall, JG. Possible maternal effect in severity of neurofibromatosis. Lancet. 1978; 2(8099):1071–1073.
152. Munro, IR, Martin, RD. The management of gigantic benign craniofacial tumors: The reverse facial osteotomy. Plast Reconstr Surg. 1980; 65:777.
153. Opitz, JM, Weaver, DD. The neurofibromatosis-Noonan syndrome. Am J Med Genet. 1985; 21:477.
154. Parker, DA, Skalko, RG. Congenital asymmetry: Report of 10 cases with associated developmental abnormalities. Pediatrics. 1969; 44:584.
155. Pollack, MA, Shprintzen, RJ. Velopharyngeal insufficiency in neurofibromatosis. Int J Pediatr Otorhinolaryngol. 1981; 3:257.
156. Posnick, JC. Other craniofacial syndromes: Klippel–Feil anomaly, neurofibromatosis, Down syndrome, micro-ophthalmia, hemifacial hyperplasia (hemi-hypertrophy). In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:503–527.
157. Riccardi, VM, Kleiner, B, Lubs, ML. Neurofibromatosis: Variable expression is not intrinsic to the mutant gene. Birth Defects Orig Artic Ser. 1979; 15:283.
158. Rittersma, J, ten Kate, LP, Westerink, P. Neurofibromatosis with mandibular deformities. Oral Surg Oral Med Oral Pathol. 1972; 33:718.
159. Shapiro, SD, Abramovitch, K, Van Dis, ML, et al. Neurofibromatosis: Oral and radiographic manifestations. Oral Surg. 1984; 58:493.
160. Sorensen, SA, Mulvihill, JJ, Nielsen, A. On the natural history of von Recklinghausen neurofibromatosis. Ann N Y Acad Sci. 1986; 486:30.
161. Spencer, WG, Shattock, SG. A case of macroglossia neurofibromatosa. Proc R Soc Med. 1908; 1:8.
162. Tibbles, JA, Cohen, MM, Jr. The Proteus syndrome: The Elephant Man diagnosed. Br Med J (Clin Res Ed). 1986; 293:683.
163. Van Damme, PA, Freihofer, HPM, De Wilde, CM. Neurofibroma in the articular disc of the temporomandibular joint: A case report. J Craniomaxillofac Surg. 1996; 24:310.
164. Van der Meulen, JC. Orbital neurofibromatosis. Clin Plast Surg. 1987; 14:123.
165. Von Recklinghausen, FD. Uber die multiplen fibrome der haut und ihre Bezichung zu den multiplen neuromen: festschrift fur Rudolph Virchow. Berlin: Hirschwald; 1882.
166. Weber, FP. Neurofibromatosis of the tongue in a child, together with a note on the classification of incomplete and anomalous cases of Recklinghausen disease. Br J Child Dis. 1910; 7:13.
167. Westerhof, W, Delleman, JW, Wolters, E, et al. Neurofibromatosis and hypertelorism. Arch Dermatol. 1984; 120:1579.
168. Andermann E, et al: A syndrome of hemihypertrophy, hemihypesthesia, hemiareflexia and scoliosis in three unrelated girls. Fifth International Conference on Birth Defects, Montreal, Quebec, Canada, August 21–27, 1977.
169. Arnold, EB. Case of hemiacromegaly. Int J Orthodont. 1933; 22:1228.
170. Bell, RA, McTigue, DJ. Complex congenital hemihypertrophy: A case report and literature review. J Pedod. 1984; 8:300.
171. Bergman, JA. Primary hemifacial hypertrophy: Review and report of a case. Arch Otolaryngol. 1973; 97:490.
172. Boxer, LA, Smith, DL. Wilm’s tumor prior to onset of hemihypertrophy. Am J Dis Child. 1970; 120:564.
173. Cohen, MM, Jr. Overgrowth syndromes. In: El-Shafie M, Klippel CH, eds. Associated congenital malformations. Baltimore, Md: Williams & Wilkins; 1981:71–104.
174. Cohen, MM, Jr. A comprehensive and critical assessment of overgrowth and overgrowth syndromes. Adv Hum Genet. 1989; 18:181.
175. Eisenberg, RL, Pfister, RC. Medullary sponge kidney associated with congenital hemihypertrophy (asymmetry). Am J Roentgenol. 1972; 116:773.
176. Fraumeni, JF, Jr., Geiser, CF, Manning, MD. Wilms’ tumor and congenital hemihypertrophy: Report of five new cases and review of literature. Pediatrics. 1967; 40:886.
177. Fraumeni, JF, Miller, RW. Adrenocortical neoplasms with hemihypertrophy, brain tumors, and other disorders. J Pediatr. 1967; 70:129.
178. Furnas, DW, Soper, RT, Nickman, NJ, et al. Congenital hemihypertrophy of the face: Impersonator of childhood facial tumors. J Pediatr Surg. 1970; 5:344.
179. Gesell, A. Hemihypertrophy and twinning: Further study of the nature of hemihypertrophy with report of a new case. Am J Med Sci. 1927; 173:542.
180. Gonzalez-Crussi, F, Lee, SC, McKinney, M. The pathology of congenital localized gigantism. Plast Reconstr Surg. 1977; 59:411.
181. Gorlin, RJ, Cohen, MM, Jr., Levin, LS. Syndromes of the head and neck, ed 3. New York: Oxford University Press; 1990.
182. Gorlin, RJ, Meskin, LH. Congenital hemihypertrophy. J Pediatr. 1962; 61:870.
183. Haicken, BN. Congenital hemihypertrophy. Am J Dis Child. 1970; 120:373.
184. Harkin, JC, Reed, RJ. Tumors of the peripheral nervous system. In: Atlas of Tumor Pathology, series 2, fascicle 3. Washington, DC: Armed Forces Institute of Pathology; 1969.
185. Hennessy, WT, Cromie, WJ, Duckett, JW. Congenital hemihypertrophy and associated abdominal lesions. Urology. 1981; 18:576.
186. Hoyme HE, et al: The incidence of neoplasia in children with isolated congenital hemihypertrophy. David Smith Meeting on Malformations and Morphogenesis, Burlington, Vt, August 11–14, 1986.
187. Khanna, JN, Andrade, NN. Hemifacial hypertrophy: Report of two cases. Int J Oral Maxillofac Surg. 1989; 18:294.
188. Kogon, SL, Jarvis, AM, Daley, TD, et al. Hemifacial hypertrophy affecting the maxillary dentition. Oral Surg Oral Med Oral Pathol. 1984; 58:549.
189. Lenstrup, E. Eight cases of hemihypertrophy. Acta Pediatr Scand. 1926; 6:205.
190. Loh, HS. Congenital hemifacial hypertrophy. Br Dent J. 1982; 153:111.
191. Meadows, AT. Wilms’ tumor in three children of a woman with hemihypertrophy. N Engl J Med. 1974; 291:23.
192. Meckel, JF. Uber die seitliche asymmetrie im tierichen korper, anatomische physiologische beobachtungen und untersuchungen. Renger: Haile; 1822.
193. Obwegeser, HL. Descriptive terminology for jaw anomalies. Oral Surg Oral Med Oral Pathol. 1993; 75:138–140.
194. Obwegeser, HL. Mandibular growth anomalies: Terminology-aetiology-diagnosis-treatment. Berlin, Heidelberg, NY: Springer-Verlag; 2001.
195. Obwegeser, HL, Mekek, M. Hemimandibular hyperplasia—hemimandibular elongation. J Maxillofac Surg. 1986; 14:183–208.
196. Pollock, RA, Newman, MH, Burdi, AR, et al. Congenital hemifacial hyperplasia: An embryologic hypothesis and case report. Cleft Palate J. 1985; 22:173.
197. Posnick, JC. Other craniofacial syndromes: Klippel–Feil anomaly, neurofibromatosis, Down syndrome, micro-ophthalmia, hemifacial hyperplasia (hemi-hypertrophy). In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:503–527.
198. Reed, EA. Congenital total hemihypertrophy. Arch Neurol Psychiatry. 1925; 14:824.
199. Ringrose, RE, Jabbour, JT, Keele, DK. Hemihypertrophy. Pediatrics. 1965; 36:434.
200. Rudolph, DC, Norvold, RW. Congenital partial hemihypertrophy involving marked malocclusions. J Dent Res. 1944; 23:133.
201. Sculerati, N, Jacobs, JB. Congenital facial hemihypertrophy: Report of a case with airway compromise. Head Neck Surg. 1985; 8:124.
202. Spielman, WR, Marano, PD, Kolodny, SC, et al. True hemifacial hypertrophy: Report of case. J Oral Surg. 1971; 29:592.
203. Tommasi, AF, Jitomirski, F. Crossed congenital hemifacial hemihyperplasia. Oral Surg Oral Med Oral Pathol. 1989; 67:190.
204. Viljoen, D, Pearn, J, Beighton, P. Manifestations and natural history of idiopathic hemihypertrophy: A review of eleven cases. Clin Genet. 1984; 26:81.
205. Viteporn, S. Surgical-orthodontic treatment in hemifacial hypertrophy: A case report. Int J Adult Orthodon Orthognath Surg. 1993; 8:55.
206. Wagner, R, see Kottmeier, HL. Uber hemihypertrophia und heniatrophia corporis totalis nebst spontane extremitatenganrane bei saugliegen im anschluss zu einem ungewohnlichen fall. Acta Paediatr Scand. 1938; 20:543.
207. Wakefield, EG, Hines, EA, Jr. Congenital hemihypertrophy: A report of eight cases. Am J Med Sci. 1933; 195:493.
208. Ward, J, Lerner, HH. A review of the subject of congenital hemihypertrophy and a complete case report. J Pediatr. 1947; 31:403.
209. Arneja, JS, Gosain, AK. Vascular malformations. Plast Reconstr Surg. 2008; 121:195e–206e.
210. Berenguer, B, Burrows, PE, Zurakowski, D, et al. Sclerotherapy of craniofacial venous malformations: Complications and results. Plast Reconstr Surg. 1999; 104:1.
211. Burns, AJ, Kaplan, LC, Mulliken, JB. Is there association between hemangioma and syndromes with dysmorphic features? Pediatrics. 1991; 88:1257–1267.
212. Burrows, PE, Mulliken, JB, Fellows, KE, et al. Childhood hemangiomas and vascular malformations: Angiographic differentiation. AJR Am J Roentgenol. 1983; 141:483.
213. Cohen, MM, Jr. Vasculogenesis, angiogenesis, hemangiomas, and vascular malformations. Am J Med Genet. 2002; 108:265–274.
214. Cohen, MM, Jr. Vascular update: Morphogenesis, tumors, malformations, and molecular dimensions. Am J Med Genet. 2006; 140A:2013–2038.
215. Cohen, MM, Jr., Neri, G, Weksberg, R. Overgrowth syndromes. New York: Oxford University Press; 2002.
216. Enjolras, O, Mulliken, JB. The current management of vascular birthmarks. Pediatr Dermatol. 1993; 10:311.
217. Enjolras, O, Mulliken, JB. Vascular tumors and vascular malformations. Adv Dermatol. 1998; 13:375–423.
218. Folkman, J, D’Amore, PA. Blood vessel formation: What is its molecular basis? Cell. 1996; 87:1153.
219. Hein, KD, Mulliken, JB, Kozakewich, HPW, et al. Venous malformations of skeletal muscle. Plast Reconstr Surg. 2002; 110:1625.
220. Jackson, IT, Carreno, R, Potparic, Z, et al. Hemangiomas, vascular malformations, and lymphovenous malformations: Classification and methods of treatment. Plast Reconstr Surg. 1993; 91:1216.
221. Jackson, IT, Keskin, M, Yavuzer, R, et al. Compartmentalization of massive vascular malformations. Plast Reconstr Surg. 2005; 115:10.
222. Marler, JJ, Mulliken, JB. Current management of hemangiomas and vascular malformations. Clin Plast Surg. 2005; 32:99.
223. Mulliken, JB. Cutaneous vascular anomalies. Semin Vasc Surg. 1993; 6:204–218.
224. Mulliken, JB. Vascular anomalies. In: Aston SJ, Beasley RW, Thorne CHM, eds. Grabb and Smith’s plastic surgery. ed 5. Philadelphia: Lippincott-Raven; 1997:191.
225. Mulliken, JB, Fishman, SJ, Burrows, PE. Vascular anomalies. Curr Probl Surg. 2000; 37:517.
226. Mulliken, JB, Glowacki, J. Hemangiomas and vascular malformations in infants and children: A classification based on endothelial characteristics. Plast Reconstr Surg. 1982; 69:412–422.
227. Mulliken, JB, Murray, JE, Castaneda, AR, et al. Management of a vascular malformation of the face using total circulatory arrest. Surg Gynecol Obstet. 1978; 146:168.
228. Mulliken, JB, Young, AE. Vascular birthmarks: Hemangiomas and malformations. Philadelphia: Saunders; 1998.
229. Posnick, JC. Hemangiomas and vascular malformations of the head and neck: Evaluation and management. In: Posnick JC, ed. Craniofacial and maxillofacial surgery in children and young adults. Philadelphia: WB Saunders Co; 2000:564–596.
230. Pribaz, JJ, Morris, DJ, Mulliken, JB. Three-dimensional folded free-flap reconstruction of complex facial defects using intraoperative modelling. Plast Reconstr Surg. 1994; 93:285.
231. Takahashi, K, Mulliken, JB, Kozakewich, HPW, et al. Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest. 1994; 93:2357.
232. Vikkula, M, Boon, LM, Mulliken, JB. Molecular genetics of vascular malformations. Matrix Biol. 2001; 20:327.
233. Antshel, KM, Fremont, W, Kates, WR. The neurocognitive phenotype in velo-cardio-facial syndrome: a developmental perspective. Dev Disabil Res Rev. 2008; 14:43–51.
234. Carotti, A, Digilio, MC, Piacentini, G, et al. Cardiac defects and results of cardiac surgery in 22q11. 2 deletion syndrome. Dev Disabil Res Rev. 2008; 14:35–42.
235. Cuneo, BF. 22q11. 2 deletion syndrome: DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes. Curr Opin Pediatr. 2001; 13:465–472.
236. De Smedt, B, Swillen, A, Ghesquiere, P, et al. Pre-academic and early academic achievement in children with velocardiofacial syndrome (del22q11. 2) of borderline or normal intelligence. Genet Couns. 2003; 14:15–29.
237. Digilio, MC, Angioni, A, De Santis, M, et al. Spectrum of clinical variability in familial deletion 22q11. 2: from full manifestation to extremely mild clinical anomalies. Clin Genet. 2003; 63:308–313.
238. Dyce, O, McDonald-McGinn, D, Kirschner, RE, et al. Otolaryngologic manifestations of the 22q11. 2 deletion syndrome. Arch Otolaryngol Head Neck Surg. 2002; 128:1408–1412.
239. Eberle, P, Berger, C, Junge, S, et al. Persistent low thymic activity and non-cardiac mortality in children with chromosome 22q11. 2 microdeletion and partial DiGeorge syndrome. Clin Exp Immunol. 2009; 155:189–198.
240. Goldberg, R, Motzkin, B, Marion, R, et al. Velo-cardio-facial syndrome: a review of 120 patients. Am J Med Genet. 1993; 45:313–319.
241. Goldmuntz, E. DiGeorge syndrome: new insights. Clin Perinatol. 2005; 32:963–978.
242. Goldmuntz, E, Clark, BJ, Mitchell, LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998; 32:492–498.
243. Kelly, D, Goldberg, R, Wilson, D, et al. Confirmation that the velo-cardio-facial syndrome is associated with haplo-insufficiency of genes at chromosome 22q11. Am J Med Genet. 1993; 45:308–312.
244. McDonald-McGinn, DM, Zackai, EH. Genetic counseling for the 22q11. 2 deletion. Dev Disabil Res Rev. 2008; 14:69–74.
245. McLean-Tooke, A, Spickett, GP, Gennery, AR. Immunodeficiency and autoimmunity in 22q11. 2 deletion syndrome. Scand J Immunol. 2007; 66:1–7.
246. Mehendale, FV, Sommerlad, BC. Surgical significance of abnormal internal carotid arteries in velocardiofacial syndrome in 43 consecutive hynes pharyngoplasties. Cleft Palate Craniofac J. 2004; 41:368–674.
247. Ryan, AK, Goodship, JA, Wilson, DL, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997; 34:798–804.
248. Shprintzen, RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev. 2008; 14:3–10.
249. Shprintzen, RJ, Goldberg, RB, Lewin, ML, et al. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978; 15:56–62.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]31
Selected Complex Malformations that Frequently Require Maxillofacial Reconstruction
Evaluation & Treatment
Klippel–Feil Anomaly
Background
The Klippel–Feil anomaly (KFA) is characterized by the congenital fusion of two or more cervical vertebrae. In its most severe form, there is massive cervical vertebral fusion with a short neck, limited head movement, and a low posterior hairline.2,12,15,21,23,29,37,39,55 Gorlin and Cohen have pointed out that the term vertebral fusion is not accurate because this condition results from the failure of the normal segmentation process.24 Vertebral fusion can be traced to the third embryonic week when the segmentation of the mesodermal somites normally takes place.
KFA was described as early as the 16th century, and similar anatomic findings in the second and third cervical vertebrae have been found in an Egyptian mummy that has been dated to be from about 500 B.C. In 1912, Maurice Klippel and Andre Feil described the postmortem findings of a 46-year-old French tailor who had a short, immobile neck with massive fusion of cervical and upper thoracic vertebrae.34 In 1919, Feil added reports of 13 additional patients, 12 of whose cases had been reported previously.
In 1967, Gunderson and colleagues categorized KFA in accordance with three morphologic types of cervical vertebral fusion.27 Type I consists of the massive fusion of many cervical and upper thoracic vertebrae into bony locks. Type II involves fusion at only one or two interspaces. Type III comprises both cervical fusion and lower thoracic or lumbar fusion.
It is important to remember that KFA is morphologically and etiologically heterogeneous. This was further documented by Gunderson, who used Feil’s classification system to review inheritance patterns.28 Type I cases have almost all been sporadic, with a female predilection. Type II is transmitted as an autosomal dominant disorder. For Type III, an autosomal recessive mode of inheritance has been suggested.
Various authors differ with regard to their interpretation of what constitutes KFA.9,61–63 A malformation with the triad of a short neck, a low posterior hairline, and a painless restriction of cervical motion characterizes just over 50% of patients. The functional limitation of the neck range of motion is the most consistent finding. Despite frequent sparing of the atlanto–axial joint, rotation is typically impaired more than flexion or extension is, with the latter exhibiting a range of more than 90 degrees, even with only one open disc space.
Diverse ocular anomalies are frequently found, with the impairment of extraocular movement being the most frequent.57Convergent strabismus and, less commonly, horizontal nystagmuses are seen. From 25% to as many as 50% of affected children exhibit hearing loss that may be sensorineural, mixed, or conductive.11,16,39,51,60,64 Clefting of the secondary palate is present in 15% to 20% of patients.10,58,59 The combination of hearing loss and cleft palate explains the hypernasality that has been documented in approximately 15% to 20% of patients with KFA. Congenital heart defects occur in 4.2% of patients, which is in stark contrast with the prevalence of 0.6% among all live births in the general population.50 Ventricular septal defect is the most common heart anomaly documented,43 and vascular anomalies may also occur.5,6 Congenital urinary anomalies are a frequent finding, with unilateral renal agenesis occurring in 28% of patients.14 Genital anomalies (e.g., vaginal agenesis) also occur at a higher frequency in these patients than they do in the general population.40,42 Neurologic disturbances that consist of involuntary dyskinesis, spasticity or hyperreflexia, syringomyelia, syringobulbia, disc protrusion, osteophytes, and narrowing at the level of the craniovertebral junction have been reported..3,20,22,36,44,48 Nagib and colleagues found patients with KFA with two unsegmented blocks of bone, cervical stenosis, or cranial involvement to be at highest risk for neurologic disturbances.45–47 Those with only one unsegmented bone were found to be at low risk for cervical injuries. The flared trapezius muscles associated with KFA give the individual an appearance that may be similar to that seen in patients with Turner or Noonan syndrome.8,13,18,38 The cervical vertebral anomalies and other defects of the spine that occur within the oculo–auriculo–vertebral spectrum may also be confused with those of KFA.4,7,8,25,32,33,35,49,52,54,56
Author’s Approach to Facial Reconstruction in Patients with Klippel–Feil Anomaly
The surgical management of KFA requires the accurate diagnosis of each specific problem (see Fig. 1-2).17,26,31 Middle-ear infections should be treated promptly, and any hearing deficit should be aided whenever possible. Clefting of the secondary palate should be repaired in the usual way. Hypernasality as a result of inadequate soft palate motion after initial palate repair should be recognized and treated with a pharyngoplasty. Extraocular muscle abnormalities are managed with eye patching, spectacles, and eye-muscle surgery, as appropriate. The recognition of cardiovascular and genitourinary malformations should lead to medical treatment and surgery. Cervical spine instability requires the decompression of any nerve impingement and the fusion of unstable segments. Unfortunately, the typical fixed neck curvature observed in patients with KFA cannot safely be straightened.
The aim of the reconstructive procedures used to correct a webbed neck is to create as normal a neck contour as possible with a symmetrical posterior hairline while avoiding obvious scarring.1 It is important to recognize that not all patients with webbed necks present with the same deformity. The clinician must assess the quality and quantity of available neck skin as well as the height and straightness of the cervical spine. Type I KFA consists of an extremely short skeletal neck that results in the increased width of the neck soft tissues. A spectrum of neck soft-tissue rearrangement procedures has been suggested to improve the webbed-neck deformity. Menick and colleagues modified the “butterfly” excision of redundant skin to avoid a midline scar.41 They did this by extending the more traditional inferior flaps into the scalp. Unfortunately, with this design, the scars are displaced laterally, and they become visible at the hairline, close to the original site of the web. Some surgeons feel that a midline posterior scar is often preferred aesthetically to one that can be seen along the lateral neck region. The placement of tissue expanders followed by flap rearrangements has also been carried out with some success. Thus, to review, procedures designed to correct the webbed neck suffer from an inability to elevate the hairline, noticeable scarring, and the intrinsically inadequate height of the skeletal structures of the neck.
The craniofacial skeletal deformities of patients with KFA typically present in one of two patterns.19,30,53 The first is in association with a congenital asymmetrical cervical spine fusion. In these patients, a variable degree of non-synostotic anterior plagiocephaly is present. The ipsilateral fronto–orbito–zygomatic flattening and retrusion may be significant enough that anterior cranial vault, orbital, and zygomatic reconstruction carried out through an intracranial approach will be indicated to adequately improve morphology (Fig. 31-1). These individuals will also have an asymmetrical dentofacial deformity that requires Le Fort I (maxillary) osteotomy, bilateral sagittal split ramus osteotomies of the mandible, and an osseous genioplasty procedure for three-dimensional repositioning (Fig. 31-2). The most unsettling aspect of the craniofacial reconstruction relates to the fact that the neck cannot be straightened. It will remain crooked, and so decisions about the best aesthetic positioning of the osteotomized craniofacial skeletal units must also be adjusted.
Figure 31-1 A 17-year-old boy with Klippel–Feil anomaly has a fixed head tilt as a result of a painless cervical spine fusion and a partial facial nerve palsy on the right side. There is marked asymmetry and deformity of the craniofacial region that involves the upper facial skeleton (i.e., the cranial vault, the orbits, and the zygomas) and the lower facial skeleton (i.e., the maxilla, the mandible, and the chin). The reconstruction was compromised by the fixed head tilt and the CN VII deficit. The patient underwent cranio–orbito–zygomatic (intracranial) reconstruction through a coronal scalp incision followed by orthognathic surgery that included a Le Fort I osteotomy, bilateral sagittal split ramus osteotomies, and an osseous genioplasty. A, Frontal facial and computed tomography scan views before surgery. B, Occlusal view before surgery and articulated dental cast that indicates analytic model planning. C, Intraoperative view of removed orbital and zygomatic units and intraoperative bird’s-eye view of the anterior cranial vault after cranio–orbito–zygomatic reconstruction. D, Frontal views with smile before and after reconstruction. Note that the fixed head tilt limited the the correction of facial symmetry. E, Oblique views before and after reconstruction. F, Profile views before and after reconstruction. G, Occlusal views with orthodontics in progress (including upper bicuspid extractions) and then after reconstruction. H, Three-dimensional computed tomography scan views before and after reconstruction.
Figure 31-2 A teenage girl with the Klippel–Feil and Poland congenital anomalies has a fixed head tilt as a result of a painless cervical spine fusion. At birth, she was noted to have a significant diaphragmatic herniation of the stomach and spleen into the thoracic cavity. She underwent procedures for the correction of the diaphragmatic hernia and then required esophageal dilatations. She also required decompression and fusion for the cervical anomalies to limit injury to the upper spinal cord. She was referred to this surgeon as a teenager and underwent evaluations that included restorative dentistry, orthodontics, speech pathology, otolaryngology, pediatric surgery, pediatric orthopedic spine surgery, and pediatrics professionals. The assessment of pulmonary capacity, swallowing and dysphagia, and safe range of motion of the neck were of particular importance. There was marked asymmetry and deformity of the maxillomandibular region. The plans for reconstruction were affected by the fixed head tilt. The patient underwent a combined orthodontic and surgical approach, and her procedures included: maxillary Le Fort I osteotomy (horizontal advancement and cant correction); sagittal split ramus osteotomies (horizontal advancement and asymmetry improvement); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty and inferior turbinate reduction. Intraoperative cervical monitoring was carried out to limit the chance of spinal cord injury. A, Frontal views in repose before and after reconstruction. B, Frontal views with smile before and after reconstruction. C, Oblique facial views before and after reconstruction. D, Profile views before and after reconstruction. E, Occlusal views before treatment, with orthodontics in progress and with upper bicuspid extractions, and then after reconstruction. F, Computed tomography scan views before reconstruction. G and H, Articulated dental models that indicate analytic model planning. I, Cephalometric radiographs before and after reconstruction.
The second pattern of presentation is in association with a congenital symmetric cervical spine fusion. The cranio–orbito–zygomatic region is symmetrical and has relatively good proportions.53 No reconstruction procedures are generally required in the upper facial skeleton. With reference to the maxillomandibular region, the constant open mouth posture of the mandible generally results in vertical maxillary excess and a constricted maxillary arch width. The mandible is usually retrognathic, with a vertically long retrusive chin and an open-bite malocclusion. A degree of maxillomandibular asymmetry is expected. The neck–chin angle is markedly obtuse. These individuals will benefit from orthognathic surgery in combination with orthodontic treatment. The orthognathic surgery will generally require a Le Fort I osteotomy in segments to intrude the maxilla vertically, advance it horizontally, and widen it transversely. The preferred approach also involves bilateral sagittal split ramus osteotomies of the mandible with horizontal advancement and counterclockwise rotation to match the new maxillary position in combination with a vertical reduction and advancement genioplasty (see Chapter 15).
Down Syndrome (Trisomy 21 Syndrome)
Background
Speculation about the historic prevalence of Down syndrome has included references to depictions of the syndrome in 15th- and 16th-century paintings.69,81 Martínez-Frías described what seems to be the earliest observation of the syndrome in a sculpted terracotta head from 580 A.D.89 The terracotta piece comes from the Toltec culture of Mexico, and its facial features, which include macroglossia, help to define its subject as a person with Down syndrome. The features of this Down syndrome were initially described by Langdon Down in 1866, and the condition represents a combination of phenotypic features that includes a degree of mental deficiency and characteristic facial features.72
The birth prevalence of trisomy 21 syndrome is generally stated to be 1 in 650 live births, but it is known to vary in different populations from 1 in 600 to 1 in 2000 live births. During the 1970s, 15% of patients who had been institutionalized for mental deficiency were found to have trisomy 21 syndrome.75
About 95% of all cases of Down syndrome arise from non-disjunction. Approximately 80% are of maternal origin, with 20% being of paternal origin. The risk increases with increasing maternal age as well as with paternal age and particularly when the parents are more than 35 years old. It has been estimated that approximately 4.8% of Down syndrome cases arise from an unbalanced translocation, which may occur de novo or by parental transmission. Inherited D/G translocations occur in 93% of maternal cases and 7% of paternal cases; however, for G/G translocations, 50% are of maternal origin, and 50% are of paternal origin. It has been estimated that 65% to 80% of trisomy 21 conceptuses result in spontaneous abortions.75
Detectable mosaicism (found in about 3% of trisomy 21 cases) and the Down syndrome phenotype may only be partially expressed.75 The use of the DNA samples from individuals who have partial trisomy 21 with or without features of the phenotype has been helpful for understanding the nature of the disorder. The area between loci D21S58 and D21S42 has been identified as being associated with mental retardation and with most of the facial features of Down syndrome. The facial features that have been identified in this region include oblique palpebral fissures; epicanthal folds; a flat nasal bridge; a protruding tongue; short, broad hands with clinodactyly of the fifth finger; a gap between first and second toes; hypotonia; short stature; Brushfield spots; and characteristic dermatoglyphic findings, including a single palmar crease and an increased number of ulnar loops.88
Individuals with Down syndrome have a specific set of major congenital malformations, including heart anomalies (30% to 40%) and gastrointestinal tract anomalies such as duodenal stenosis or atresia, imperforate anus, and Hirschsprung disease.75 The development of Alzheimer disease occurs at a much earlier age in individuals with trisomy 21 than it does in the general population. The frequency of symptomatic gallbladder disease is 25% higher among adults with Down syndrome as compared with the general population.
Down syndrome is characterized by extensive phenotypic variability.65,67,71,98 Although cognitive impairment, muscle hypotonia at birth, and dysmorphic features occur to some extent in all individuals, most associated traits occur in only a fraction of affected individuals. In the patient with Down syndrome, a number of characteristic facial features that vary from patient to patient are frequently recognized:
2. Oblique (anti-mongoloid) slanting of the eyelids
3. Strabismus as a result of extraocular muscle dysfunction
4. Flatness of the dorsum of the nose
5. Hypoplasia of the upper jaw that results in an Angle Class III malocclusion
In 1967, Otermin Aguirre presented a series of patients with Down syndrome who underwent reconstructive surgery in an attempt to normalize their facial morphology.93 During the same year, Hohler attempted to normalize the face of a young girl with Down syndrome by augmenting the hypoplastic dorsum of the nose and the receding chin with alloplastic implants.77 In 1977, Lemperle and Spitalny systematically tried to normalize the faces of children with Down syndrome.82–85 Similar results were published by Olbrisch,91,92 Patterson and colleagues,94 Wexler and Peled,103,104 and Rozner,99 in addition to several others.76,90,94,97
Despite the initial enthusiasm for facial reconstruction in children with Down syndrome during the late 1960s through the mid 1980s, a critical review of the procedures used to normalize the face was not carried out until the mid to late 1980s. In 1989, Katz and Kravetz presented a balanced, thoughtful review and a discussion of the literature regarding the “effectiveness of facial surgery for persons with Down syndrome.”79 The research of Katz and Kravetz may be summarized as follows: If improved functioning, appearance, and social acceptability are measured by parent and treating physician satisfaction, then the outcome of facial plastic surgery for people with Down syndrome seems positive. If function, appearance, and social acceptability are evaluated by people who were not involved in making the decision to conduct the facial plastic surgery—and particularly when control subjects are included in the evaluation—then the outcome of facial plastic surgery for people with Down syndrome is not so positive. For most patients with Down syndrome, the surgeons’ and parents’ well-intentioned attempts to normalize the child’s facial morphology could not be realized.96
Macroglossia with protrusion of the large tongue is a frequent feature of Down syndrome. Dystonia or hypotonia of the tongue is also a factor.66,74 During the 1970s and the early 1980s, several investigators suggested that partial glossectomy in the patient with Down syndrome could improve the clarity of speech.68,70 However, studies that used objective criteria to document speech results after partial glossectomy in patients with Down syndrome showed no significant differences in the number of articulation errors between audiotape recordings made preoperatively and 6 months postoperatively in a series of patients or between the recordings of the same patients and recordings of an additional series of Down syndrome control patients who did not undergo surgery. A further study by Margar-Bacal and colleagues looked at changes in the aesthetic appearance and intelligibility of speech after partial glossectomy in patients with Down syndrome.87 The aesthetic appearance of speech (i.e., the visual acceptability of the patient while he or she is speaking) was judged from visual information only. Judgments of speech intelligibility were made separately from the auditory portion of the videotapes. The acceptability and intelligibility of the children were also judged together during audiovisual presentation. Statistical analysis showed that speech was significantly more acceptable aesthetically after surgery. No significant difference was found in speech intelligibility postoperatively as compared with preoperatively.
An Angle Class III malocclusion as a result of the midface hypoplasia with overclosure of the mandible is a frequent finding in patients with Down syndrome.95 This was objectively studied by Farkas with the use of anthropometric measurements in which he documented that 60% of children with Down syndrome showed a disproportionately small depth to the middle third of the face (i.e., the anthropometric measurement from the subnasal point to the tragus).73 The congenital absence of permanent teeth, poor root formation, and hypoplasia of the enamel are frequent complicating factors.
Author’s Approach to Facial Reconstruction in Individual’s with Down Syndrome
When a child with Down syndrome approaches skeletal maturity (i.e., 14 to 16 years of age for girls, 16 to 18 years of age for boys) and a significant jaw deformity with malocclusion is present, then a comprehensive approach of orthodontic treatment and orthognathic surgery can effectively alter the facial proportions (aesthetics) and improve head and neck functions (i.e., lip posture, chewing, speech articulation, and breathing) (Fig. 31-3). A formal sleep study is indicated before surgery to clarify the extent of upper airway obstruction and the degree of maxillomandibular advancement required to manage it.1,78,80,86,100–102 The orthodontic treatment may be compromised as a result of poor dental root formation, congenitally missing teeth, or limited compliance. The coordination of care with an appropriate dental team (e.g., a restorative dentist, a periodontist, an orthodontist) is useful before the initiation of any treatment.
Figure 31-3 A 16-year-old boy with Down syndrome presented with a jaw deformity and malocclusion. He underwent a combined orthodontic and surgical approach. The surgery was limited to a Le Fort I osteotomy (horizontal advancement and vertical lengthening). A, Frontal views before and after reconstruction. B, Profile views before and after reconstruction. C, Occlusal views before and after reconstruction. Note that the orthodontic and restorative dental objectives were limited by congenital malformations of the dental crowns and dental roots. D, Articulated dental casts that indicate analytic model planning. E, Lateral cephalometric radiographs before and after reconstruction.
The correction of the dentofacial deformity and the management of the airway frequently requires maxillary Le Fort I osteotomy (horizontal advancement and vertical lengthening), often with sagittal split ramus osteotomies for alignment to the new maxillary position (i.e., advancement but not setback) and an osseous genioplasty (horizontal advancement and vertical lengthening). Septoplasty and inferior turbinate reduction should be simultaneously performed if required to improve nasal airflow (see Chapter 10).
Neurofibromatosis
Background
In 1849, Robert Smith, a professor of surgery at Dublin Medical School, reported clinical and necropsy findings in two cases of individuals who were presumed to have neurofibromatosis; he cited 75 references to similar patients from the earlier medical literature.130 Unfortunately, he did not recognize that the tumors (neurofibromas) contained neural elements. In 1882, von Recklinghausen published his findings, which convinced the medical world that neurofibromatosis was a distinct entity of neural origin.165 Today, neurofibromatosis is known to be etiologically heterogeneous. Cohen and Hayden were the first to differentiate Proteus syndrome from neurofibromatosis in 1979.119,162 Types I and II are the forms that are frequently encountered by the craniomaxillofacial surgeon.
Neurofibromatosis Type II (Acoustic Type)
The hallmark of Type II neurofibromatosis is the presence of bilateral acoustic neuromas.147,150 Symptoms are usually caused by pressure on the vestibulocochlear and facial nerve complex. The first symptom is usually hearing loss that often begins during the teenage years or the early 20s. Occasionally, hearing loss may occur as early as the first decade or as late as the seventh decade of life. Cafe-au-lait spots and cutaneous neurofibromas are also present, but they are seen less frequently than they are among patients with neurofibromatosis Type I.121 Tumors of the central nervous system are especially common, with Schwann cell tumors occurring most frequently. Multiple tumors of meningeal or glial origin may also occur.147 Neurofibromatosis Type II has autosomal dominant inheritance with penetrance of more than 95%. The responsible gene has been mapped to chromosome 22.
Neurofibromatosis Type I (von Recklinghausen Type)
Joseph Merrick, the Elephant Man, has often been thought to have had von Recklinghausen disease. However, after considering several diagnostic possibilities, Cohen concluded that Merrick’s skeletal findings are most consistent with Proteus syndrome.119,130,162
This classic form of neurofibromatosis accounts for approximately 90% of all affected cases. Major features include six or more cafe-au-lait spots, cutaneous neurofibromas, and Lisch nodules.130 Axillary freckling develops in about 66% of all patients. Neurofibromatosis type I, which is the classic form of neurofibromatosis, is the one described by von Recklinghausen; it is also the most frequent form, occurring in 1 of every 2500 to 3000 births.146,160,165,166 Inheritance is autosomal dominant, with almost 100% penetrance. Approximately 50% of cases represent new mutations, and an increase in paternal age at the time of conception has been found to be an associated feature. Although most evidence points to neurofibromatosis as a disorder of neural crest derivation, some controversy remains about whether the neural and mesenchymal components are interrelated or if they may arise independent of each other.110,116,153
Type I is caused by a mutation of the neurofibromin 1 (NF1) gene. Approximately 5% to 20% of all patients with Type I neurofibromatosis patients carry a heterozygous deletion of approximately 1.5 Mb that involves the NF1 gene and the contiguous change line in its flanking region.157 Miller and Hall found that patients born to affected mothers have more severe disease than those born to affected fathers.152 The mutation rate in the NF1 gene is one of the highest known in humans, with approximately 50% of all patients with NF1 presenting with novel mutations.124
The natural history of neurofibromatosis Type I has been reviewed by several authors.138 More than 40% of patients have some clinical manifestations at birth, and more than 60% have manifestations by the second year of life. Cafe-au-lait spots usually develop first, with multiple lesions present within the first year of life. In approximately 50% of patients, axillary freckling appears later. Cutaneous neurofibromas appear around the onset of puberty and increase in number throughout life.138 Lisch nodules begin to appear during early childhood and have been observed in almost all affected adults. Plexiform neurofibromas occur in 30% of these patients.
Approximately 33% of all patients develop one or more complications in association with the disease.105–108,115 It has been estimated that some form of malignancy develops in 6% of patients with neurofibromatosis type I who are more than 18 years old.109,117,118 Other important complications that may occur are neurologic and include epilepsy, aqueduct stenosis, and spinal neurofibromas. Learning disabilities of various kinds affect 25% of patients.137,154,163
Any part of the eye may be involved.111,126,128,132 Lisch nodules of various sizes may be found anywhere in the iris. These lesions are melanocytic hamartomas, and they are found only in patients with neurofibromatosis. Phakoma, congenital glaucoma, corneal opacity, detached retina, optic atrophy, and congenital ptosis of the eyelids have all been reported. Intraorbital lesions may also produce proptosis and eye-muscle palsies.
The literature concerning the presenting head and neck dysmorphology and the suggested treatment of orbitotemporal neurofibromatosis is extensive.* The plexiform neurofibroma that involves the orbit, the eyelid, and the temporal region may also have an intracranial component. It generally appears during childhood as a swelling of the upper eyelid, and it slowly and progressively worsens (Fig. 31-4). Further involvement of the subcutaneous tissues of the eyelids with varying degrees of ptosis and proptosis of the globe also occurs. For patients in whom progression continues, there is further plexiform neurofibromatosis of the temporal area, pulsating proptosis, continued enlargement of the eyelids with extensive mechanical ptosis, and the inability to open the eye, which results in severely diminished visual acuity or blindness. The globe itself is involved with neurofibroma, and buphthalmos may be present. The eye may be painful as a result of infection and epiphora.
Figure 31-4 A child with neurofibromatosis involving the left cranio–orbital region. Aplasia of the sphenoid wing and the gradual growth of plexiform neurofibroma occurred, which extended from the cavernous sinus through the widened superior orbital fissure, through the cone of the eye, and into the upper eyelid. Despite an intracranial debulking procedure carried out by another surgeon during the patient’s middle childhood years, the boy presented to this surgeon with residual and recurrent plexiform neurofibroma that resulted in the distortion and proptosis of the left eye. Vision remained in the affected eye, and the decision was made to preserve it. The patient underwent intracranial reconstruction of the cranio–orbito–zygomatic skeleton, including the debulking of the soft-tissue tumor and the vertical and horizontal debulking of the upper eyelid. Intracranial debulking with eye presentation was accomplished. A, Frontal view during early childhood and a computed tomography scan of the anterior skull base indicating the defect in the sphenoid wing. B, Frontal view at 10 years of age. Despite a debulking procedure carried out by another surgeon, residual growth has reoccurred. An axial computed tomography scan view through the midorbits indicates the extent of plexiform neurofibromatosis from the cavernous sinus through the upper eyelid. C, Frontal views before and after reconstruction. Note that the surgical correction of the upper eyelid ptosis was simultaneously carried out but it was limited to maintain adequate corneal protection. D, Profile views before and after reconstruction.
[/not-level-membership-for-surgery-category]
