Selected agents of pulmonary value
After reading this chapter, the reader will be able to:
1. Define key terms and definitions pertaining to selected agents of pulmonary value
2. Discuss the indication for α1-proteinase inhibitor therapy
3. Recognize α1-proteinase inhibitor deficiency in a patient
4. List α1-proteinase inhibitors that are available
5. List three types of formulations for nicotine replacement
6. Recognize the advantages and disadvantages of nicotine replacement
7. Discuss the indication for nitric oxide
8. Describe the effect of inhaled nitric oxide on a patient
9. List the two toxic products of nitric oxide
10. List the two inhaled prostacyclin analogues available in the United States
Chapter 16 presents three groups of drugs that are used for the direct treatment or prevention of respiratory disease: α1-proteinase inhibitors (APIs), used in the treatment of congenital α1-antitrypsin deficiency; nicotine replacement and other agents used in smoking cessation; and pulmonary vasodilators, used for pulmonary hypertension states in newborns and for acute respiratory distress syndrome (ARDS) in adults. Two inhaled synthetic analogues of prostacyclin (PGI2) for the treatment of pulmonary hypertension are described as well.
α1-proteinase inhibitor (human)
α1-antitrypsin deficiency
α1-AT deficiency is a genetic defect that can lead to the development of severe panacinar emphysema. This autosomal recessive disorder is characterized by serum API levels less than 35% of normal and manifests as panacinar emphysema at age 30 to 50 years. API deficiency is estimated to account for approximately 2% of all cases of emphysema in the United States. It is estimated that there are 60,000 to 100,000 Americans with severe α1-AT deficiency.1,2 Studies done in the United States vary in their estimates of the prevalence among newborns of α1-AT deficiency, ranging from 1 in 2857 to 1 in 5097.3 Among whites, the genetic disorder α1-AT deficiency is as common as cystic fibrosis.4 In about 50% of cases of emphysema that result from API deficiency, there is accompanying chronic bronchitis with mucus hypersecretion, perhaps as a result of secretory cell metaplasia caused by unchecked proteases in the epithelial lining fluid.5 Emphysema caused by API deficiency is worse in the lower lung zones and can be markedly accelerated by cigarette smoking.1
The basic pathology of emphysema resulting from API deficiency is an imbalance between proteases, especially neutrophil elastase (NE) and antiproteases, especially API. The main substrate for API is neutrophil elastase. The pathogenesis of emphysema is described as a process of alveolar wall destruction caused by insufficient protection from the protease neutrophil elastase, an enzyme that can cleave all forms of connective tissue and degrade elastic fiber in the lungs by solubilizing elastin. With inadequate API levels in the lung to balance the protease activity, emphysema occurs at a significantly earlier age than is normally seen. A presentation of severe emphysema at an unexpectedly young age, such as the third or fourth decade, leads to a high suspicion of a genetic defect causing inadequate API levels in the blood and subsequently in the lungs. The main role of another protease inhibitor, secretory leukocyte protease inhibitor (SLPI), which is secreted by bronchial glands and goblet cells, is to protect the airway epithelium against proteolytic injury. However, Wewers and associates6 provided evidence that API (α1-AT) is the predominant antiprotease protecting against neutrophil elastase.
Genetics
API is a 54-kDa glycoprotein, encoded by a single gene on chromosome 14. The alleles of the API gene can be categorized as follows:5
• API normal: Normal serum levels of normal-functioning API
• API deficient: Lower than normal serum concentrations of API with altered electrophoretic properties
• API null: Undetectable API levels in the serum
• API dysfunctional: Normal amounts of abnormally functioning API
Persons with normal alleles for API (designated by the letter M for the alleles) are termed PI*MM, for protease inhibitor with a pair of the normal alleles. They are homozygous for the normal allele. Normal values for serum API are 150 to 350 mg/dL based on comparison with a commercial standard preparation and 20 to 48 μM based on comparison with a purified laboratory standard. The commercially available preparations are about 40% higher compared with the purified laboratory standards. Results referenced to the commercial standard are expressed as milligrams per deciliter, whereas comparisons with the highly purified (true) standard are given in micromolar units. Commercial standard values can be converted to true standard values by multiplying the commercial value by 0.71.5,6
About 95% of persons in the severely deficient category are homozygous for the Z allele and are designated as PI*ZZ. Serum levels of API in these individuals range from 2.5 to 7 μM, or a mean of about 16% of normal.5 The Z allele is rare in Asians and African Americans. Alleles that do not express API at all are quite rare, and such individuals are designated as PI type null-null. PI type null-null individuals have an absence of measurable API in the serum. Wewers and colleagues6 described the treatment of a patient with the null-null phenotype and no measurable API serum levels. They were able to show that intravenously administered augmentation therapy with α1-AT (API) led to normal API levels in the blood and in the lung epithelial lining fluid.
The major risk factor for developing emphysema among PI*ZZ individuals seems to be cigarette smoking, in which emphysema appears much earlier than in nonsusceptible individuals, as previously noted. Other features seen with airflow obstruction in PI*ZZ individuals include a history of pneumonia, episodes of increased cough and sputum production, and a parental history of emphysema.2
Indication for drug therapy
API therapy is indicated for long-term replacement therapy in individuals with congenital deficiency of API, with clinically demonstrable panacinar emphysema. At present four agents are available: Augmentation therapy and maintenance are indicated only for patients who have established API deficiency.7 Results from controlled, long-term trials to show that long-term therapy halts the progression of emphysema are unavailable because of inherent difficulties in such trials, including the need for large numbers of patients.1 API therapy has been provided only to adult subjects. Given the nature of the disease and the action of the drugs, the drugs cannot reverse damage or improve lung function. These drugs are extremely expensive, costing $25,000 to $40,000 per year for therapy. A cost-effectiveness analysis of Prolastin-C concluded that α1-AT replacement therapy is cost-effective in individuals who have severe α1-AT deficiency and severe chronic obstructive pulmonary disease (COPD).8
The American Thoracic Society (ATS) stated that API augmentation therapy should be used for patients with a serum concentration of API less than 11 μM, or 80 mg/dL.2,9 API therapy is not indicated for patients with emphysema related to cigarette smoking who have normal or heterozygous phenotypes.5 It is not indicated for individuals with liver disease associated with API deficiency, unless they also have lung disease. ATS guidelines suggest using augmentation therapy if lung function studies become abnormal and if serial studies show deterioration.
Dosage and administration
The recommended dosage of API is 60 mg/kg of body weight, given once weekly. The dose is given intravenously at a rate of 0.08 mL/kg/min or greater, depending on patient comfort, and usually takes about 15 to 70 minutes for total infusion. Table 16-1 provides a summary of Aralast NP, Prolastin-C, Zemaira, and Glassia.
TABLE 16-1
α1-Proteinase Inhibitors Currently Available
| BRAND NAME | STRENGTH (mg) |
| Aralast NP* | 400 and 800 (active) |
| Prolastin-C* | 500 and 1000 |
| Zemaira* | 1000‡ |
| Glassia† | 1000 |
*Powder form; reconstitution must take place before administration.
 RESPIRATORY CARE ASSESSMENT OF THERAPY OF α1-PROTEINASE INHIBITOR (HUMAN)
RESPIRATORY CARE ASSESSMENT OF THERAPY OF α1-PROTEINASE INHIBITOR (HUMAN)Smoking cessation drug therapy
Nicotine and lobeline are naturally occurring alkaloids that are capable of stimulating acetylcholine receptors at the autonomic ganglia of the sympathetic and the parasympathetic systems and cholinergic nicotinic receptors at skeletal muscle sites (see Chapter 5) and in the brain. The structures of these two agents are shown in Figure 16-1. The affinity of nicotine for ganglionic and neuromuscular receptor sites led to the use of the term nicotinic to distinguish these receptors from muscarinic receptors because all of these receptors use acetylcholine as a neurotransmitter.
In addition to stimulating nicotinic receptors at the autonomic ganglia, neuromuscular junctions, and adrenal medulla, nicotine binds to receptors in the central nervous system (CNS). This action causes respiratory stimulation, tremors, convulsions, nausea, and emesis. The last two effects are often seen when nicotine is first inhaled as tobacco smoke, although tolerance rapidly occurs. Nicotine is the chief alkaloid in tobacco products, and addiction to nicotine is the basis for tobacco dependence. In a seasoned smoker, within seconds of inhaling from a cigarette, the internal carotid arteries carry a large bolus of nicotine to the brain, where it binds to nicotine receptors.10 This binding causes secretion of dopamine, which causes a feeling of pleasure and cognitive arousal. Nicotine also increases levels of norepinephrine, β-endorphin, acetylcholine, serotonin, and other substances in the CNS, all of which increase the sensation of euphoria and well-being; enhance concentration, alertness, and memory; and decrease tension and anxiety. Sensitivity and responsiveness to nicotine in the CNS are genetically determined and constitute the basis for forming the physiologic addiction to nicotine. Without the proper genetic substrate, a smoker cannot become nicotine dependent. About 10% of smokers lack this substrate and are not physiologically dependent; 90% have the substrate and are nicotine addicted to various degrees.10
Cigarette smoking is a preventable cause of cardiovascular and lung disease and accelerates the rate of decline of lung function that occurs with aging, as shown in the Lung Health Study.11 The Lung Health Study concluded that aggressive smoking intervention and cessation reduce the age-related decline in forced expiratory volume in 1 second (FEV1) among middle-aged smokers. Withdrawal from the nicotine in tobacco products is difficult because the stimulatory and reward effects are lost, and physical symptoms occur. The latter include craving for nicotine, nervousness, irritability, anxiety, drowsiness, sleep disturbance, impaired concentration, and increased appetite with attendant weight gain. Nicotine replacement therapy, in various dosing formulations, is intended to aid with smoking cessation by allowing initial replacement and then gradual withdrawal of the nicotine found in tobacco. Because nicotine is well absorbed from the skin and mucosa, a transdermal patch, a chewable gum formulation, a nasal spray, and an inhaler have been developed.
Indication for use
Nicotine replacement agents are indicated as an aid to smoking cessation to relieve nicotine withdrawal symptoms. Replacement therapy should be used as part of a comprehensive smoking cessation program to increase compliance and reduce relapse. Smokers with signs of strong physical dependence on nicotine may benefit the most from nicotine replacement therapy. Signs of strong physical dependence are listed in Box 16-1.10
Drug formulations
Smoking cessation drug therapy includes various formulations of agents. The U.S. Department of Health and Human Services Public Health Service 2008 update on guidelines to treat tobacco dependence categorizes pharmacotherapy into first-line and second-line agents.11 First-line medications include nicotine replacement, bupropion (Zyban), and varenicline (Chantix). Second-line agents include clonidine (Catapres) and nortriptyline (Pamelor). Table 16-2 lists pharmaceutical details on the various agents in use at the time of this edition. Goodfellow and Waugh13 reviewed tobacco prevention and smoking cessation agents. Details on nicotine substitute agents can be found in manufacturers’ literature.14
TABLE 16-2
Smoking Cessation Drug Formulations
| CATEGORY | BRAND NAME | DOSAGE |
| Nicotine transdermal system | NicoDerm CQ | 21 mg/day for first 6 weeks, 14 mg/day for next 2 weeks, 7 mg/day for last 2 weeks |
| Nicotrol | 15 mg/day for first 12 weeks, 10 mg/day for next 2 weeks, 5 mg/day for last 2 weeks | |
| Nicotine polacrilex | Nicorette (gum) | 2 mg if <25 cigarettes/day: 9 pieces/day, maximum 24 pieces/day; 4 mg if ≥25 cigarettes/day: 9 pieces/day, maximum of 24 pieces/day |
| Commit (lozenge) | 2 mg and 4 mg, no more than 5 lozenges in 6 hours, maximum 20 lozenges/day | |
| Nicotrol NS | 0.5 mg/spray, one each nostril (1.0 mg); 1 or 2 doses/hour (2 sprays with nasal spray, 1 each nostril, is 1 dose), up to 5 doses/hour, or 40 doses/day | |
| Nicotrol Inhaler | 4 mg/use; recommended dosage 24-64 mg (6-16 cartridges) per day, up to 12 weeks, with gradual reduction over 12 weeks | |
| Bupropion | Zyban SR, XL | 150-mg sustained-release tablets; begin at 150 mg/day for 3 days; increase to 150 mg/day bid, with maximum of 300 mg/day, interval of 8 hours between doses; continue treatment for 7-12 weeks |
| Varenicline | Chantix | 1 week titration of 0.5 mg once daily for first 3 days, twice daily for remainder of week; begin 1 mg twice daily for 11 weeks |
| Clonidine | Catapres | 0.10-mg tablet daily, increasing by 0.10 mg as needed; 0.10-mg transdermal patch daily, increasing to 0.20-mg patch as needed |
| Nortriptyline | Aventyl, Pamelor | 25-mg tablet daily, increasing to 100 mg daily |
Nicotine polacrilex (nicotine resin complex)
The nicotine inhaler offers smokers a “simulated cigarette”; the kit contains a 10-mg/cartridge unit dose, which delivers 4 mg/use; a mouthpiece; blister trays of nicotine cartridges; and a plastic case. The use of a mouthpiece resembling a cigarette holder allows delivery of the nicotine in a manner similar to smoking a cigarette, with oral gratification. This system delivers less nicotine than the other systems. All of the nicotine is absorbed across the oropharyngeal membranes.10 The inhaler may be most useful in a smoker with low dependency, as an adjunct to the patch to treat sudden cravings, or in combination with bupropion.
Bupropion (zyban)
Bupropion is an antidepressant found in Wellbutrin; it is also a nonnicotine aid to smoking cessation. The drug is a relatively weak inhibitor of neuronal uptake of norepinephrine, serotonin, and dopamine, which is the basis for its antidepressant effect. The exact mechanism by which bupropion aids in smoking cessation is unknown. Bupropion (Zyban) may relieve nicotine withdrawal by slowing the normal reuptake of dopamine or preventing its breakdown in the CNS. It has been shown that mood and emotional state are related to the need for smoking and nicotine, although bupropion is effective in smoking cessation even if the smoker is not depressed.10 Symptoms of nicotine dependence among smokers are correlated with the magnitude of symptoms of depression. Subjects who are negative or depressed are less likely to be able to quit smoking. This finding would indicate that the antidepressant effect of bupropion assists in smoking cessation. Jorenby and associates15 found little difference between a placebo group and a nicotine patch group in smoking cessation at 12 months (15.6% versus 16.4%). A group receiving bupropion alone achieved a 30.3% cessation rate, and the combination of bupropion and nicotine patch gave the highest cessation rate of 35.5%. This study suggests that bupropion added to nicotine substitutes in a program of smoking cessation is helpful. Only about 6% of smokers succeed in quitting with no replacement therapy.15
Varenicline (chantix)
Varenicline is a selective α4β2 nicotinic acetylcholine receptor partial agonist developed for explicit use in smoking cessation. Varenicline (Chantix) works by attaching to α4β2 receptors, inhibiting the activation of this receptor by nicotine. The sensation produced by smoking is blocked, breaking the cycle of nicotine addition.16
In clinical trials, varenicline produced a 39% quit rate compared with 20% for bupropion and 11% for placebo after 12 weeks.17 In another study, varenicline produced quit rates of 44% and 49%, respectively, at lower and higher doses of the drug compared with placebo (12%).18
Precautions
Clonidine (catapres)
Clonidine (Catapres) is an antihypertensive agent that has been prescribed to reduce symptoms of opioid and alcohol withdrawl.11 At present, clonidine is not approved by the U.S. Food and Drug Administration (FDA) for use as a smoking cessation aid. It can be taken orally or is available as a transdermal patch. Common side effects include drowsiness, fatigue, depression, nausea, and weight gain. More importantly, this agent should not be stopped abruptly; this can cause nervousness, tremors, headache, and increased blood pressure.
Nortriptyline (aventyl, pamelor)
Nortriptyline is a tricyclic antidepressant approved by the FDA to treat depression. It is not FDA approved for use as a smoking cessation aid. However, similar to bupropion, it is thought to have an effect on tobacco dependence because of its antidepressant mode of action.12 As with other antidepressants, common side effects include dizziness, insomnia, and blurred vision.
Nitric oxide
Nitric oxide (NO) is a product of endothelial cells that acts as a nitrovasodilator. It was investigated for its ability to reduce pulmonary vascular resistance in various disease states, such as persistent pulmonary hypertension of the newborn (PPHN) and ARDS. Furchgott and Zawadzki19 showed that endothelial cells in blood vessels elaborate a short-lived vasodilator, which was termed endothelium-derived relaxing factor (EDRF). The neurotransmitter acetylcholine, which can normally dilate blood vessels, has no effect or vasoconstricts if applied to blood vessels without endothelium. Subsequently, the substance EDRF was identified by Palmer and colleagues20 and Ignarro and colleagues21 as NO. This endogenously produced vasodilator can be inhaled as a gas to cause pulmonary vasodilation.
Indication for use
NO is approved for use in neonates with hypoxic respiratory failure to reduce pulmonary artery pressure and to increase oxygenation in newborns with pulmonary hypertension and hypoxia. Off-label use of NO in adults with ARDS has been reported; however, data on its effectiveness are conflicting.14
Dosage and administration
NO, supplied in two sizes of gas cylinder, is available at 100 ppm and 800 ppm. The recommended dose is 20 ppm. The treatment should be maintained up to 14 days or until the underlying oxygenation problem has resolved and the neonate can be successfully weaned from NO. In the Neonatal Inhaled Nitric Oxide Study (NINOS) trial, most patients who failed to improve on 20 ppm and whose dose was increased to 80 ppm had no response at the higher concentration.22 The risk of methemoglobinemia and elevated nitrogen dioxide (NO2) levels increases significantly at doses greater than 20 ppm, as discussed subsequently.
In clinical trials of NO, the delivery system used was the INOvent system (Ikaria, Clinton, N.J.), which gives a constant concentration of NO during the respiratory cycle with minimal NO2 generation. The following summarizes guidelines for the safe administration of NO, based on several sources listed in the references, including a statement by the American Academy of Pediatrics:3,23,24
• Blending and delivery systems should be designed and tested for accurate NO delivery, minimum NO2 production, and capability of administering NO in constant concentration ranges in parts per million or less throughout the respiratory cycle.
• The delivery system should be calibrated using a precisely defined mixture of NO and NO2.
• Sample gas for analysis should be drawn before the Y-piece, proximal to the patient.
• Inhaled NO and NO2 should be monitored continuously, using chemiluminescence or electrochemical analyzers.
• Oxygen levels in the inspired gas should be measured.
• Blood methemoglobin levels should be measured frequently.
• The minimal effective concentration of NO should be used.
• Weaning from NO should be gradual to prevent arterial desaturation and pulmonary hypertension.
• Because inhaled NO is used in respiratory failure, institutions that offer NO therapy generally should have extracorporeal membrane oxygenation (ECMO) capability in the event NO therapy fails. Alternatively, a plan for timely transfer of infants to a collaborating ECMO center should be established prospectively, and transfer should be accomplished without interruption of NO therapy.
Pharmacology of nitric oxide
The formation, mode of action, and fate of endogenous NO are diagrammed in Figure 16-2. NO is formed endogenously in vascular endothelial cells of the respiratory tract from the precursor amino acid l-arginine by several isoforms of the enzyme nitric oxide synthase (NOS). NOS requires the cosubstrates nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen (O2). In the reaction, nitrogen is contributed by the arginine; oxygen, by the oxygen molecule; and a free electron, by NADPH. NOS is categorized as constitutive NOS (cNOS), including that found in endothelial cells (ecNOS) and in neurons (nNOS), and as inducible NOS (iNOS).23 The vascular relaxation caused by acetylcholine is due to stimulation of cNOS, which results in an increase in NO. Histamine, leukotrienes, and bradykinin are other mediators that increase cNOS-mediated NO and promote vasodilation and lowering of blood pressure.
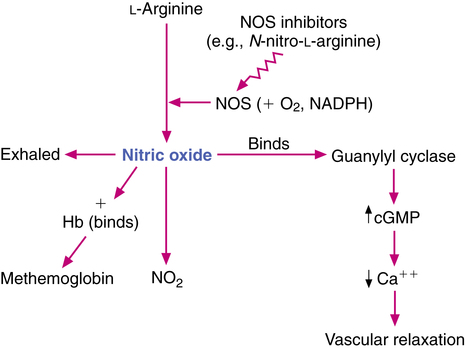
Proinflammatory cytokines, such as interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), and interleukin-1 (IL-1), can induce iNOS to increase endogenous levels of NO. Glucocorticoids block the induction of iNOS and inhibit the formation of NO.22 NO is the active form of nitrovasodilators such as nitroglycerin and sodium nitroprusside.17 NO has also been identified as at least one of the neurotransmitters in the nonadrenergic, noncholinergic (NANC) inhibitory nervous system (see Chapter 5).23 The endogenous production of NO can be inhibited by l-arginine analogues, which inhibit NOS—for example, N-nitro-l-arginine. The NO molecule is small and lipophilic, and it has a very short duration of action of 0.1 to 5 seconds in physiologic systems.21,25
NO is generated in vascular endothelial cells and diffuses rapidly into myocytes in the endothelium, binding to guanylyl cyclase. Guanylyl cyclase (also termed guanylate cyclase) stimulates the production of cyclic guanosine-3′, 5′-monophosphate (cGMP), which causes a decrease in intracellular calcium and consequent vascular or nonvascular smooth muscle relaxation. The NO-induced increase in cGMP within the cells also inhibits platelet adherence and aggregation and polymorphonuclear leukocyte chemotaxis.24 NO readily diffuses into the blood vessel itself and into endothelial cells and enters the red blood cells to bind rapidly with hemoglobin, forming methemoglobin and becoming inactivated in the process. NO is also converted in the red blood cells to nitrate, and some endogenous NO is exhaled from the lung.26 Because NO diffuses so readily into the bloodstream and is inactivated by being bound to hemoglobin, its action is limited to the pulmonary vascular endothelium, whether generated endogenously within the lung or inhaled as an exogenous gas. It is a selective pulmonary vasodilator. The end products of NO that enter the systemic circulation are predominantly methemoglobin and nitrate. Nitrite is the predominant NO metabolite excreted in the urine, accounting for more than 70% of the inhaled dose. Aranda and Pearl27 published a more detailed review of the biology of NO.
Effect on pulmonary circulation
With normal pulmonary hemodynamics (normal vascular resistance), inhalation of NO produces no effect on pulmonary arterial pressure or gas exchange.19 However, Frostell and associates28 reported that hypoxic pulmonary vasoconstriction caused by breathing 12% oxygen in healthy adults increased mean pulmonary arterial pressure from 14.7 ± 0.8 mm Hg to 19.8 ± 0.9 mm Hg.26 This increase in pulmonary arterial pressure was reversed by adding 40 ppm of NO to the gas mixture. No change occurred in systemic vascular resistance because NO was inactivated locally by hemoglobin. Taylor and associates29 found that 5 ppm of NO successfully improved oxygenation in the short-term for patients with acute lung injury.
In persistent PPHN, pulmonary vascular resistance is elevated, which causes right-to-left shunting through the patent ductus arteriosus and foramen ovale. Inhaled NO dilates pulmonary blood vessels in regions of the lung where ventilation is delivered; this redistributes pulmonary blood flow from areas of low ventilation to areas with better ventilation. The improved ventilation-perfusion matching leads to an improved partial pressure of oxygen in arterial blood (Pao2). Because NO is rapidly and locally inactivated by hemoglobin, no systemic vasodilation or hypotension occurs.30
Toxicity
Toxicity with exposure to NO can be caused by the NO itself, by the formation of the nitrite, NO2, and the formation of methemoglobin. NO can be a mediator of lung injury, for example, with paraquat poisoning, in which inhibition of NOS reduces the amount of lung injury.31 NO2 is a strong oxidizer that causes lipid peroxidation in cells. The amount of NO2 produced depends on the amount of NO and the amount of surrounding O2. The higher the Fio2, the greater the amount of oxidation of NO to NO2. Similarly, the higher the concentration of NO, the shorter the time to achieve oxidation to NO2. The lethal effect of NO2 is due to pulmonary edema, and short-term exposure to more than 150 ppm of NO2 is usually fatal.26 In the usual doses of NO, such as 0.5% to 4%, methemoglobinemia is not usually a problem, although this should be monitored.
Synthetic analogues of prostacyclin (PGI2)
Iloprost (ventavis)
Indication for use
Ventavis is indicated for the treatment of pulmonary arterial hypertension in patients with New York Heart Association (NYHA) class III or IV symptoms. NYHA is a functional and therapeutic classification of physical activity in patients with cardiac dysfunction. The classification, I through IV, describes the limitations of physical activity; NYHA III and IV are higher and pose considerable restriction on patient activity. Ventavis is not intended for pediatric use; it is intended for adults 18 years old and older. In clinical studies, Ventavis has been shown to improve NYHA functional class, improve exercise capacity, and increase walking distance.32
Treprostinil (tyvaso)
Treprostinil was first approved by the FDA as an injectable form in 2002. Since that time, research has been ongoing to create and test an inhaled version.33 Inhaled treprostinil (Tyvaso) was approved for use in the United States in 2009. Tyvaso is a prostacyclin analogue that causes vasodilation of the pulmonary and systemic arterial vascular beds and inhibits platelet aggregation. It is administered using the Tyvaso Inhalation System (United Therapeutics Corp., Silver Spring, Md.), which is an ultrasonic, pulsed-delivery device.
Precautions
 RESPIRATORY CARE ASSESSMENT OF SMOKING CESSATION DRUG THERAPY
RESPIRATORY CARE ASSESSMENT OF SMOKING CESSATION DRUG THERAPY
During treatment and short term
• Monitor abstinence rates at intervals of 3, 6, or 12 months.
• Monitor for symptoms of nicotine overdosage (possible if subjects continue smoking while using nicotine substitutes), such as nausea, salivation, abdominal pain, vomiting, diarrhea, cold sweat, headache, dizziness, disturbed vision and hearing, mental confusion, or marked weakness.
• Assess bupropion use for improvement in emotional attitude, including reduction in irritability, anxiety, difficulty in concentrating, or depression.
• Assess varenicline use for improvement in nicotine withdrawal symptoms.
• Assess clonidine and nortriptyline for possible use as smoking cessation aids.
• Monitor clonidine for adverse actions related to cardiovascular status.
 RESPIRATORY CARE ASSESSMENT OF NITRIC OXIDE
RESPIRATORY CARE ASSESSMENT OF NITRIC OXIDE
During treatment and short term
• Evaluate therapy for a reduction in the oxygenation index (OI = mean airway pressure in cm H2O × Fio2/Pao2).
• Evaluate the effect of nitric oxide and monitor the Pao2 and the overall level of ventilatory support (Fio2, inspiratory pressure and time, end-expiratory pressure, rate).
• Monitor preductal and postductal pulse oximetry (Spo2) to evaluate shunting.
• If available, review the echocardiogram to evaluate right-to-left shunting.
• Monitor inspired nitric oxide and nitrogen dioxide, along with methemoglobin.
• Monitor cardiovascular status and stability, including the level of intravenous fluids and vasoactive medications needed.
 RESPIRATORY CARE ASSESSMEMT OF SYNTHETIC ANALOGUES OF PROSTACYCLIN (PGI2)
RESPIRATORY CARE ASSESSMEMT OF SYNTHETIC ANALOGUES OF PROSTACYCLIN (PGI2) SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
1. For which disease state is an α1-proteinase inhibitor (API) indicated?
2. What is the route of administration for an α1-proteinase inhibitor?
3. What is the mode of action of α1-proteinase inhibitors in treating emphysema associated with inadequate API levels?
4. Is treatment with an α1-proteinase inhibitor indicated for age-related emphysema or in general for individuals who smoke and have emphysema later in life?
5. Identify three pharmaceutical formulations of nicotine that are used as smoking cessation aids.
6. What is the usual effect of nicotine, whether in a smoking cessation aid or in cigarettes, on blood pressure?
7. Name two nonnicotine agents used in the treatment of smoking cessation.
8. What is the effect of inhaled nitric oxide?
9. Identify two potentially toxic by-products of inhaled nitric oxide.
10. What is the usual dose of inhaled nitric oxide?
11. Identify two disease states in which nitric oxide has been used to reverse pulmonary hypertension.
12. What is the greatest hazard in terms of pulmonary health with the delivery of Ventavis?
 CLINICAL SCENARIO
CLINICAL SCENARIO