Sedative and Anxiolytic Drugs
Sedation is best considered as a continuum between normal consciousness and general anaesthesia. The most frequently cited description of the varying levels of sedation utilized in clinical practice is that from the American Society of Anesthesiologists (Table 7.1).
TABLE 7.1
Continuum of Depth of Sedation: Definition of General Anaesthesia and Levels of Sedation/ Analgesia. American Society of Anesthesiologists, 2009
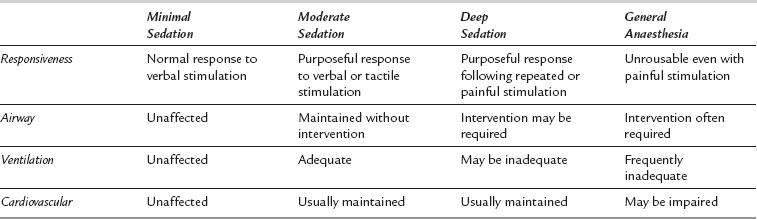
The difference between sedative and anaesthetic drugs is largely one of usage. Many anaesthetic drugs may be used at reduced dosage to produce sedation and, similarly, agents used primarily as sedatives will produce a form of anaesthesia if given in sufficiently high doses. The usual target is to produce conscious sedation, i.e. to titrate drug therapy so the patient is free of anxiety, free of pain and responding purposefully to command. In adults, this corresponds to the levels of sedation from anxiolysis to moderate sedation as described in Table 7.1.
INDICATIONS FOR THE USE OF SEDATIVE DRUGS
Sedative Drugs
Most sedative drugs may be categorized into one of three main groups: benzodiazepines, antipsychotics and α2-adrenoceptor agonists. Drugs classified more usually as intravenous anaesthetic agents, particularly propofol and ketamine, are also used as sedatives in subanaesthetic doses; the pharmacology of these drugs is discussed in Chapter 3. Similarly, remifentanil, which is used increasingly as part of a sedative regimen, is described fully in Chapter 5. Inhaled anaesthetics (see Ch 2) are also used occasionally as sedatives (e.g. sevoflurane to an end-tidal concentration of 0.3–0.5 kPa, or nitrous oxide).
BENZODIAZEPINES
Systemic Effects
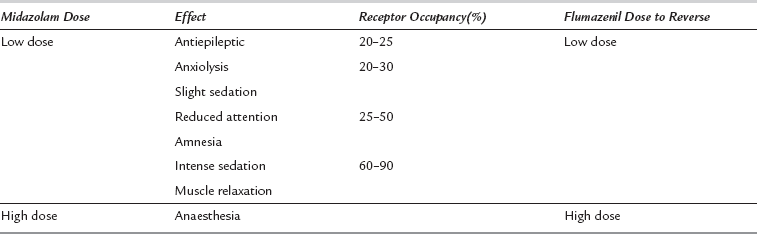
Sedation occurs as a dose-dependent depression of cerebral activity with mild sedation at low receptor occupancy progressing to a state similar to general anaesthesia when most receptor sites are occupied (Table 7.2). Benzodiazepines have a high therapeutic index (ratio of effective to lethal dose) because, in overdose, the differences in receptor density result in greater sensitivity to cortical than to medullary depression. However, upper airway obstruction and loss of protective reflexes occur before profound sedation ensues, and are a major hazard following inadvertent oversedation or self-poisoning.
Diazepam








Midazolam














α2-ADRENOCEPTOR AGONISTS
Clonidine
Systemic Effects
CNS Effects: Clonidine produces sedation and anxiolysis. It also reduces requirements for both intravenous and volatile anaesthetic agents. There is a ceiling to the reduction of MAC effect, because of the potential for activity at α1-receptors at high dose. More selective α2-adrenoceptor agonists reduce MAC to a much greater extent.
CVS Effects: The cardiovascular effects of clonidine probably involve both α2-adrenoceptors and imidazoline receptors. Administration leads to decreases in heart rate and arterial pressure. Clonidine is known to lower the ‘set point’ around which arterial pressure is regulated. The α2-agonist effects are a reduction in sympathetic tone and an increase in parasympathetic tone. The resulting decreases in heart rate, myocardial contractility and systemic vascular resistance lead to a reduction in myocardial oxygen requirements. This may be advantageous in attenuating stress-induced haemodynamic responses. However, undesirable cardiovascular depression has been the major limiting factor in developing the use of clonidine as a sedative.