CHAPTER 68 Sclerosing Cholangitis and Recurrent Pyogenic Cholangitis
Dr. Bruce Y. Tung contributed to this chapter in the previous edition of this book.
Sclerosing cholangitis encompasses a spectrum of cholestatic conditions that are characterized by patchy inflammation, fibrosis, and destruction of the intrahepatic and extrahepatic bile ducts. These conditions are typically chronic, progressive disorders in which persistent biliary damage may lead to biliary obstruction, biliary cirrhosis, and hepatic failure, with associated complications. The first description of sclerosing cholangitis is credited to Delbet in 1924.1 Although considered for many years to be an extremely rare disorder, the advent of endoscopic retrograde cholangiopancreatography (ERCP) in the 1970s has allowed an improved understanding of the true prevalence of this disorder and facilitated careful study of its natural history. Nevertheless, many aspects of sclerosing cholangitis remain poorly understood; most notably lacking are a detailed knowledge of its etiology and proven effective medical therapy.
A cholangiographic appearance of diffuse stricturing and segmental dilatation of the biliary system, designated sclerosing cholangitis, may be observed in many distinct conditions. The most frequent is primary sclerosing cholangitis (PSC), an idiopathic disorder that usually occurs in association with inflammatory bowel disease (IBD) but may develop independently. PSC may also be associated with a wide variety of fibrotic, autoimmune, and infiltrative disorders, although whether such associations imply a common pathogenesis or epiphenomena is unclear (Table 68-1). PSC is also associated with various immunodeficiency states; in such cases biliary abnormalities may be caused by infection with an opportunistic pathogen. The term secondary sclerosing cholangitis refers to a clinical and radiologic syndrome that is similar to PSC but develops as a consequence of a known pathogenesis or injury. Obstructive, toxic, ischemic, and neoplastic causes of secondary sclerosing cholangitis have been described (see Table 68-1). This chapter focuses on PSC and recurrent pyogenic cholangitis.
Table 68-1 Classification and Diseases Associated with Sclerosing Cholangitis
Secondary Sclerosing Cholangitis
PRIMARY SCLEROSING CHOLANGITIS
DIAGNOSIS
No standardized criteria for the diagnosis of PSC have been universally adopted. Early diagnostic criteria included diffuse intra- and extrahepatic bile duct strictures occurring in the absence of prior biliary surgery or cholelithiasis and after exclusion of cholangiocarcinoma.2 These criteria were later modified because of the recognition that the clinical spectrum of PSC is broader than initially appreciated, and strict adherence to the original criteria underestimates the prevalence of the disease. It is now apparent that a form of PSC, termed small-duct PSC, involves only the intrahepatic biliary tree, without obvious extrahepatic duct abnormalities.3 In addition, both cholelithiasis and choledocholithiasis may develop as a consequence of PSC, and their presence does not exclude a diagnosis of underlying PSC.4,5 Furthermore, cholangiocarcinoma is a relatively common complication of PSC, and both conditions frequently coexist.6
The diagnosis of PSC is based on typical cholangiographic findings in the setting of consistent clinical, biochemical, serologic, and histologic findings as well as exclusion of secondary causes of sclerosing cholangitis. The characteristic cholangiographic findings are multifocal stricturing and ectasia of the biliary tree. Areas of narrowing are interspersed with areas of normal or near-normal caliber and of post-stenotic dilatation. Although the majority of patients with PSC have coexisting abnormalities of the intra- and extrahepatic bile ducts, a small percentage have an isolated lesion. Patients with small-duct PSC may have a normal cholangiogram. Gallbladder abnormalities, including tumors, may exist in up to 41% of patients with PSC.7
ERCP is considered the standard for establishing a diagnosis of PSC but carries a risk for complications of up to 10% in patients with PSC.8,9 Magnetic resonance cholangiopancreatography (MRCP) has largely replaced ERCP for diagnostic cholangiography as a result of improvements in image quality and the noninvasive nature of MRCP (Fig. 68-1). In the few studies that have compared MRCP and ERCP in patients with PSC, MRCP has demonstrated comparable sensitivity for the detection of biliary structuring,10–13 although performance and interpretation of magnetic resonance cholangiograms vary with the technique and institution. ERCP has the advantage of combining high-resolution cholangiography with the potential for advanced diagnostic and therapeutic interventions, including brush cytology or intraductal biopsy for the diagnosis of cholangiocarcinoma, balloon or catheter dilation of strictures, biliary stent placement, sphincterotomy, and stone removal. Percutaneous transhepatic cholangiography (THC) may also yield diagnostic images and allow therapeutic intervention but requires percutaneous puncture and may be technically difficult if the intrahepatic bile ducts are not sufficiently dilated (see Chapter 70).
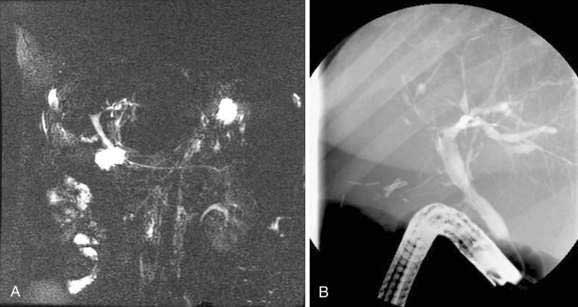
Patients with IBD and a cholestatic pattern of liver biochemical test elevations should undergo imaging of the hepatobiliary system because of the relatively high pretest probability of PSC. Ultrasonography or computed tomography (CT) may be useful for planning further diagnostic and therapeutic strategies in selected patients, but they are usually insufficient for a diagnosis of PSC because normal findings do not exclude the diagnosis. The decision as to which method of cholangiography to perform must be individualized. In most cases, ERCP is the initial test of choice for patients in whom a therapeutic intervention or the need for brush cytology is anticipated. In an asymptomatic patient with mild liver biochemical abnormalities who is unlikely to require therapeutic intervention, MRCP is the preferred initial test if the images are reliable.13 When MRCP is nondiagnostic and clinical suspicion for PSC remains, diagnostic ERCP is indicated.
Differential Diagnosis
In a patient with a cholangiographic appearance characteristic of sclerosing cholangitis, secondary causes of sclerosing cholangitis must be excluded (see Table 68-1). Patients with the acquired immunodeficiency syndrome (AIDS) and a CD4+ T-lymphocyte count below 100/mm3 can exhibit a cholangiographic appearance identical to that of PSC; this entity is termed AIDS cholangiopathy. Cryptosporidium, Microsporidium, cytomegalovirus, and other organisms have been isolated from the bile of affected patients.14,15 Exposure of the bile ducts to toxins such as intra-arterial floxuridine (FUDR)16 and formaldehyde administered to treat a hydatid cyst, when the cyst communicates with the biliary tract,17 can produce a similar cholangiographic appearance.
Primary biliary cirrhosis (PBC) is another chronic cholestatic condition that shares some clinical features with PSC (see Chapter 89); however, PBC predominantly affects middle-aged women, has no association with IBD, and is associated strongly with high titers of antimitochondrial antibodies. Whereas liver histologic findings in the two disorders overlap substantially,18 the distinction between the two is readily apparent on cholangiography. Patients with advanced PBC may demonstrate smooth tapering and narrowing of the intrahepatic bile ducts, but ductal irregularity or strictures are not seen and extrahepatic lesions do not occur. Antimitochondrial antibody-negative PBC (autoimmune cholangitis) may be difficult to distinguish from small-duct PSC because serologic profiles and cholangiographic findings may overlap, but the demographic and histologic features of the two disorders are distinct (see Chapter 89).
Autoimmune hepatitis may also be difficult to distinguish from PSC (see Chapter 88). In the pediatric population, PSC typically manifests with features of autoimmune hepatitis, and cholangiography is necessary to distinguish the two disorders (see Chapter 62).19 With use of a standardized scoring system for the diagnosis of autoimmune hepatitis, 7.5% of patients with PSC are characterized as “definite” or “probable” for the diagnosis of autoimmune hepatitis, thereby underscoring the need for cholangiography when PSC is suspected.20 Features suggestive of autoimmune hepatitis include female predominance, a hepatocellular rather than cholestatic pattern of liver biochemical test abnormalities, hypergammaglobulinemia, high titers of antinuclear and anti-smooth muscle antibodies, histologic evidence of periportal necroinflammation, and clinical response to glucocorticoid therapy. An overlap syndrome between PSC and autoimmune hepatitis has been described; it consists of a mixed cholestatic and hepatocellular pattern of liver biochemical test abnormalities, the presence in serum of autoantibodies including antineutrophil cytoplasmic antibodies (ANCA), cholangiography consistent with PSC, and histologic evidence of periductular fibrosis as well as periportal necroinflammation.21,22
A disorder of the pancreaticobiliary tree termed autoimmune pancreatitis, sclerosing pancreatocholangitis, or immunoglobulin (Ig) G4–associated cholangitis has been described.23 This disorder shares cholangiographic and clinical features with PSC but differs in its responsiveness to glucocorticoid therapy. Serum levels of IgG4 are often elevated in this disorder, and high numbers of IgG4 positive lymphocytes (>20 per high-powered field) are identified in pinch biopsies obtained from the major papilla or bile duct and may be diagnostic. Although specific diagnostic criteria for this disorder are still emerging, persons without IBD who present with symptoms and cholangiographic findings consistent with PSC should undergo measurement of serum IgG4 levels as well as endoscopy and biopsy of the major papilla to exclude IgG4-associated cholangitis (see Chapter 59).23–25
EPIDEMIOLOGY
Determination of the true incidence and prevalence of PSC is complicated by the variable presentation of the disease, inconsistent diagnostic criteria, and referral bias inherent in many published studies. Two population-based studies have provided the most accurate epidemiologic estimates of PSC in Western populations. On the basis of these studies performed in the United States and Norway, the incidence of PSC is estimated to be 0.9 to 1.3 per 100,000, and the point prevalence is estimated to be 8.5 to 13.6 per 100,000.26,27
Although PSC has been diagnosed in neonates and as late as the eighth decade of life, most patients present between the ages of 25 and 45 years, with a mean age of approximately 39 years.19,28–33 Approximately 70% of patients with PSC are men,27–31 but in the subset of patients without IBD, the male-to-female ratio is lower (0.72:1).34 Women with PSC are generally older at diagnosis.27,35 PSC is also associated with nonsmoking, an effect that cannot be explained entirely by the association between ulcerative colitis (UC) and nonsmoking.36,37
PRIMARY SCLEROSING CHOLANGITIS AND INFLAMMATORY BOWEL DISEASE
The relationship between PSC and IBD is striking and incompletely understood. Approximately 80% of all patients with PSC have concomitant IBD.27–29,31,38,39 Conversely, PSC is present in 2.4% to 4.0% of all patients with chronic UC and 1.4% to 3.4% of patients with Crohn’s disease.35,38,40,41 Of patients with both PSC and IBD, approximately 85% to 90% have UC and the remainder have Crohn’s colitis or ileocolitis. The association with IBD is stronger with more extensive colonic involvement; the prevalence of PSC is approximately 5.5% in those with pancolitis, in contrast to 0.5% in those with only distal colitis.35 PSC is not thought to occur in association with Crohn’s disease isolated to the small intestine. Racial differences in the association between PSC and IBD may exist; concomitant IBD is seen in only 21% of Japanese patients with PSC.42
Despite the strong association between PSC and UC, the two diseases often progress independently of each other.43 Although IBD is typically diagnosed before PSC, UC may be newly diagnosed years after liver transplantation for end-stage liver disease caused by PSC. Conversely, PSC may be diagnosed years after total proctocolectomy for UC.44,45
Whether PSC differs clinically in patients with and without concomitant IBD is unclear. Older reports demonstrated no histologic46 or cholangiographic47 differences between patients with or without IBD. One study,34 however, suggested that patients without IBD are more likely to be female, have disease isolated to the extrahepatic ducts, and be symptomatic at the time of diagnosis. Of the multiple multivariate analyses performed to identify risk factors for progression of PSC (see later), only one found that the presence of IBD has a significant independent effect on progression of PSC.29 Some patients without overt IBD may have subclinical histologic changes detected in the colon or may develop overt colitis at a later date.43 Therefore, a high index of suspicion for the emergence of IBD is warranted, and colonoscopy with random biopsies of the colonic mucosa is recommended for all patients with a new diagnosis of PSC.
ETIOLOGY AND PATHOGENESIS
Genetic Factors
The importance of genetic factors in the pathogenesis of PSC is demonstrated by familial occurrence of the disease and its associations with specific human leukocyte antigen (HLA) haplotypes. Although uncommon, familial clustering of cases of PSC have been reported.48,49 Furthermore, PSC is strongly associated with specific HLA haplotypes. Early studies described an overrepresentation of HLA B8 and DR3 in patients with PSC; these haplotypes are also associated with other autoimmune disorders such as myasthenia gravis and autoimmune hepatitis.50,51 These findings are not explained simply by the association between PSC and IBD because HLA B8 and DR3 are not overrepresented in patients with IBD but without PSC. The subsequent development of molecular genotyping demonstrated that the most common allele in patients with PSC is DRB3*0101, which encodes the DRw52a antigen. One study found this allele in 100% of 29 patients with PSC who underwent liver transplantation,52 but subsequent studies have demonstrated this allele in only 50% to 55% of patients with PSC.53–55 Currently, the extended HLA haplotypes that are most strongly associated with PSC are as follows:53,55,56
The strongest association maps to the HLA class I/III boundary on chromosome 6p21. Strong disease associations have been identified with the MICA*008 allele57 and the tumor necrosis factor α-2 allele.58,59 Despite the multiple HLA associations described, however, a single HLA-encoded gene that determines susceptibility to PSC appears unlikely. More likely are multiple HLA susceptibility loci, which may explain in part why PSC is a relatively rare disease even though the HLA haplotypes associated with PSC are relatively common in populations of Northern European descent.
Also controversial is whether specific haplotypes are associated with disease outcomes. One study suggested a poor prognosis in patients with PSC and HLA DR4,60 but this finding was not confirmed.55 A more recent multicenter study involving 256 patients with PSC showed that the heterozygous haplotype DR3,DQ2 was associated with a greater risk of liver transplantation or death and the DQ6 haplotype was associated with a decreased risk of disease progression.61
The relationship between several non-major histocompatibility complex (MHC) genes and susceptibility to PSC has also been investigated. An initial study reported an association with polymorphisms in the gene encoding matrix metalloproteinase 3 (MMP-3) and postulated a role for MMP-3 in progression of PSC because of its ability to regulate fibrosis and immune activation.62 A subsequent report, however, did not confirm an association between either MMP-1 or MMP-3 polymorphisms and PSC.63 Similarly, no associations between PSC and polymorphisms in the interleukin (IL)-1 or IL-10 genes have been noted.64
Immunologic Factors
Evidence suggests that the immune system plays a key role in the etiology and pathogenesis of PSC, including the multiple associations between PSC and other autoimmune disorders. The most frequently associated autoimmune disorders include type I diabetes mellitus and Graves’ disease, which are more common in patients with PSC and IBD than in patients with IBD alone.65 In addition, as described earlier, an overlap syndrome that includes features of both PSC and autoimmune hepatitis has been described.21,22,66 In rare cases, well-characterized autoimmune hepatitis may evolve into sclerosing cholangitis, suggesting that both diseases may be part of the same clinical spectrum.67 Unlike most other autoimmune disorders, however, PSC has an approximately 2:1 male predominance, is not associated with disease-specific autoantibodies, and does not exhibit a consistent clinical response to immunosuppressive therapy.
A wide range of serum autoantibodies are found in patients with PSC, although none is specific for the disease. Whether any of these associated antibodies plays a key role in the pathogenesis of the disease process or whether they represent simple epiphenomena is unclear. Antinuclear antibodies may be present in 24% to 53%, anti-smooth muscle antibodies in 13% to 20%, and an atypical perinuclear ANCA (pANCA) in 65% to 88% of patients with PSC.68–75 Antibodies directed against cardiolipin, bactericidal/permeability-increasing protein, cathepsin G, and lactoferrin have also been detected.74–75 Antibodies directed against an epitope shared by colonic and biliary epithelial cells have been demonstrated and may suggest a mechanism for the association between IBD and PSC.76 Autoantibodies that bind to human biliary epithelial cells (anti-BEC) have been shown to induce expression of IL-6 and the cell adhesion molecule CD44; this finding could represent a potential mechanism for the inflammatory bile duct destruction seen in patients with PSC.77
Abnormalities of both humoral and cellular immunity have been described in patients with PSC. They include an increase in circulating immune complexes, deficient clearance of immune complexes, and activation of the classical pathway of the complement system.78–80 Serum elevations of IL-8 and IL-10 also suggest exaggerated humoral immunity.81 Some of the abnormalities in cellular-mediated immunity that have been described include a decrease in circulating CD8+ cytotoxic T cells,82 increased numbers of γδ T cells in peripheral blood as well as portal areas of the liver,83 and overrepresentation of Vβ3 T-cell receptor gene segments in hepatic (but not peripheral) T-cell populations.84
Biliary Epithelial Cells
The role of biliary epithelial cells in the pathogenesis of PSC remains unclear. Biliary epithelial cells could serve as a trigger and a target for immune-mediated injury. Biliary epithelial cells have been shown to express MHC class II antigens85 and adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1)86 and could play a role as antigen-presenting cells to T lymphocytes. The expression of these molecules can be regulated on biliary epithelial cells by various cytokines, including IL-2 and interferon-γ.87 Biliary epithelial cells, however, may not express the co-stimulatory ligands necessary for activation of T lymphocytes.88 In addition, many of the same findings are seen in patients with PBC and extrahepatic bile duct obstruction as well, making it less likely that they play a primary pathogenic role in PSC.85
Infectious and Toxic Factors
The strong association between PSC and colitis has provoked the theory that penetration of infectious or toxic agents through an inflamed colon into the portal system may play an important role in the pathogenesis of PSC. Bile culture results have been positive in explanted livers in a majority of patients with PSC, although the number of bacterial strains has correlated inversely with the time since the last endoscopic intervention.89 In addition, bacterial endotoxin has been shown to accumulate in biliary epithelial cells in patients with PSC and PBC.90 In patients with AIDS cholangiopathy, a variety of organisms, including Cryptosporidium, Microsporidium, and cytomegalovirus, have been isolated from the bile.14,15 A study that evaluated serologic profiles in 41 patients with PSC found a higher percentage with Chlamydia lipopolysaccharide antibodies than in a large control population. No association was seen with any other microorganisms, including Mycoplasma and 22 viruses tested.91 Further study is necessary before a direct link between PSC and Chlamydia, or any other infectious agent, can be established.
A loss of normal colonic mucosal barrier because of inflammation could allow portal inflow of noninfectious toxins. Toxic damage leading to sclerosing cholangitis has been demonstrated in humans as well as animal models. Biliary exposure to caustic agents17 or hepatic artery infusion of chemotherapeutic agents such as FUDR16 can produce a cholangiographic appearance identical to that of PSC. In a rat model, administration of the biliary toxin α-naphthylisothiocyanate led to the development of a chronic cholangitis similar to sclerosing cholangitis in humans.92 The toxic injury hypothesis, however, does not explain why PSC is not associated with the severity of colonic inflammation in patients with IBD and why PSC may develop years after a patient has undergone total proctocolectomy.
Vascular Factors
Ischemia has been postulated to play a role in the pathogenesis of PSC because a similar cholangiographic appearance may be found after surgical trauma to the biliary vascular supply93 and after hepatic artery thrombosis or arterial fibrointimal hyperplasia after liver transplantation.94,95 In addition, PSC is associated with the presence of autoantibodies such as pANCA and anti-cardiolipin antibodies. These autoantibodies, in turn, are strongly associated with vasculitides such as Wegener’s granulomatosis, polyarteritis nodosa, and thrombotic syndromes. These associations suggest that immune-mediated vascular injury plays a role in the pathogenesis of PSC.
NATURAL HISTORY AND PROGNOSTIC MODELS
PSC is typically a progressive disease, although the natural history is incompletely understood.29–31,96–98 The disease may be considered to progress through the following four clinical phases, although some phases may not develop or be apparent in an individual patient:
Asymptomatic Primary Sclerosing Cholangitis
Asymptomatic patients with PSC make up 15% to 44% of cohorts examined in published studies29,31,32,98,99 Some reports have suggested that asymptomatic patients typically have a benign course of disease. Helzberg and colleagues98 reported on 11 asymptomatic patients with PSC who were followed for a mean of 37 months, and all 11 remained asymptomatic without evidence of progressive disease. By contrast, Porayko and colleagues99 followed 45 asymptomatic patients with PSC for a median of 6.25 years, and during the surveillance period, liver failure, resulting in liver transplantation or death, developed in 13 (31%). Overall, symptoms developed in 24 (53%), and progressive liver disease, demonstrated by new symptoms or signs, worsening cholangiographic findings, or progressive liver histologic abnormalities, developed in 34 (76%) patients. The Kaplan-Meier estimate of median survival free of liver failure in this study was 71% at seven years for the asymptomatic patients, significantly lower than the 96% expected on the basis of an age-, sex-, and race-matched U.S. control population. Differences in the rates of progression between these studies98,99 may be the result of differences in patient populations, the definition of “asymptomatic,” and the duration of clinical follow-up.
Symptomatic Primary Sclerosing Cholangitis
Patients with symptoms at the time of diagnosis generally have a worse prognosis than asymptomatic patients.29,30 The clinical stage is likely more advanced at the time of diagnosis in symptomatic patients, who have more severe biochemical derangements, more abnormalities on cholangiography, and a higher histologic stage on liver biopsy specimens than asymptomatic patients. Wiesner and colleagues29 compared the natural history of PSC in 37 asymptomatic patients with that in 137 patients who were symptomatic at the time of diagnosis. After a mean follow-up of six years, 55 (40%) of the symptomatic patients had died, compared with 4 (11%) in the asymptomatic group. The Kaplan-Meier estimate of median survival for the entire cohort was 11.9 years; for the symptomatic cohort, the estimated median survival was between 8 and 9 years. Farrant and colleagues30 described the natural history of PSC in 126 patients, of whom 84% were symptomatic. After a median follow-up of 5.8 years, the estimated median survival was 12 years. Similar findings were reported in a large study by Broome and colleagues.31 In 305 patients with PSC followed for a median of 5.25 years, of whom 44% were asymptomatic, the estimated median survival was 12 years. Patients who were symptomatic at the time of entry into the study had a significantly worse expected survival (9.3 years) than asymptomatic patients. A study of 174 patients with PSC by Ponsioen and colleagues97 suggested a better overall prognosis, with a median expected survival of 18 years. The reason for improved survival in this most recent study is not known, but patient data were predominantly from the 1990s, compared with data from the 1970s and 1980s in the other studies described. Although therapeutic advances were not dramatic in the interim, earlier diagnosis in the 1990s may have led to differences in patient selection that appeared to affect outcomes.
Small-Duct Primary Sclerosing Cholangitis
Patients who have histologic, biochemical, and clinical features of PSC but a normal cholangiogram are considered to have small-duct PSC, which accounts for 5% to 20% of all patients with PSC.3,100 Three studies have performed extended clinical follow-up in patients with small-duct PSC.100–102 In these studies, 12% to 17% of patients progressed to classic large-duct PSC over long-term follow-up, although the true rate may be higher because cholangiograms were not obtained routinely in all patients. Cholangiocarcinoma did not develop in any patient over a median follow-up of 63 to 126 months, and survival in the small-duct PSC group was better than that of matched control groups with classic PSC.100–102 Therefore, small-duct appears to represent an early stage of PSC, may progress to large-duct PSC in a small percentage of patients, and is associated with a better prognosis than classic PSC.
Prognostic Models
Multivariable prognostic models that have been developed to predict survival in patients with PSC are shown in Table 68-2. In an early multivariable analysis, hepatomegaly and a serum bilirubin level > 1.5 mg/dL were found to be independently associated with a poor prognosis in PSC. The patient’s age, histologic findings, presence of concomitant IBD, and pattern of cholangiographic involvement did not correlate independently with survival in this study.98 Wiesner and colleagues29 developed a prognostic model based on age, serum bilirubin level, hemoglobin value, presence or absence of IBD, and histologic stage. With this model, three risk groups (low, intermediate, high) were formed, and predicted survival curves were shown to be similar to observed survival curves. Farrant and colleagues30 developed a multivariable prognostic model in which hepatomegaly, splenomegaly, serum alkaline phosphatase level, histologic stage, and age at presentation were found to be important independent factors. Dickson and colleagues103 then presented a model developed from a multicenter collaboration in which data from 426 patients with PSC were pooled. In this analysis, the patient’s age, serum bilirubin level, histologic stage, and presence of splenomegaly were found to correlate independently with survival, and the model was validated against the observed survival data in a subgroup of the entire cohort. Broome and colleagues31 found the patient’s age, histologic stage, and serum bilirubin level to be independent predictors of survival in 305 patients with PSC, but this prognostic model was not validated independently. Most recently, Kim and colleagues,104 using easily obtainable clinical and biochemical factors, revised an earlier predictive model that did not require liver biopsy and did not rely on subjective physical findings such as splenomegaly or hepatomegaly. This revised natural history model (revised Mayo risk model) found the patient’s age, serum bilirubin level, serum aspartate aminotransferase (AST) level, and serum albumin level and a history of variceal bleeding to be independent predictors of survival. The model was generated from data on 529 patients from 5 centers and was validated using data from another center that had not been used in the development of the model.
Table 68-2 Independent Predictors of Survival and Prognostic Index Formulas Used in Natural History Models of Primary Sclerosing Cholangitis*

The Child (Child-Pugh) classification may also be used to predict survival in patients with PSC (see Chapter 90). Shetty and colleagues105 found that Kaplan-Meier seven-year survival rates for patients with Child class A, B, and C cirrhosis caused by PSC were 89.8%, 68%, and 24.9%, respectively. Subsequent evaluation, however, suggested that the Child classification is less accurate than the revised Mayo risk model, especially for patients with early-stage PSC.106
CLINICAL FEATURES
Symptoms
The initial clinical presentation of PSC can be quite varied and may run the gamut from asymptomatic elevations of serum alkaline phosphatase levels to decompensated cirrhosis with jaundice, ascites, hepatic encephalopathy, or variceal bleeding. The most common symptoms at the time of presentation include jaundice, fatigue, pruritus, and abdominal pain.19,28–33,107,108 Other associated symptoms may include fever, chills, night sweats, and weight loss (Table 68-3). The onset of these symptoms is typically insidious, although an acute hepatitis-like presentation has been described.40 Increasingly, PSC is diagnosed in an asymptomatic or minimally symptomatic stage. Large series have shown that 15% to 44% of patients with PSC are asymptomatic at the time of diagnosis,29,31,32,98,99 probably because of the routine liver biochemical screening in patients with IBD, as well as the widespread availability of MRCP and ERCP for evaluating elevated serum alkaline phosphatase levels.
Table 68-3 Most Common Symptoms and Signs at the Time of Diagnosis of Primary Sclerosing Cholangitis
| Symptoms | Rate (%) |
| Fatigue | 65-75 |
| Abdominal pain | 24-72 |
| Pruritus | 15-69 |
| Fever/night sweats | 13-45 |
| Asymptomatic | 15-44 |
| Weight loss | 10-34 |
| Signs | |
| Jaundice | 30-73 |
| Hepatomegaly | 34-62 |
| Splenomegaly | 32-34 |
| Hyperpigmentation | 14-25 |
| Ascites | 4-7 |
Data from references 19, 29–33, 98, 99, 107, 108.
Symptoms of PSC are often intermittent. Episodes of pruritus, jaundice, abdominal pain, and fever are typically interspersed with asymptomatic periods of varying duration.39,107,108 The intermittency of the symptoms is thought to reflect intermittent biliary obstruction caused by microlithiasis and sludge.5,109 This obstruction may predispose to cholestasis and induce an acute inflammatory reaction. Secondary bacterial infection may result in low-grade cholangitis and predispose to pigment stone formation.5
Physical Examination
Physical findings may be normal in patients with PSC, particularly those who are asymptomatic. When physical abnormalities are present, the most common include hepatomegaly, jaundice, and splenomegaly (see Table 68-3). Skin findings are common and include cutaneous hyperpigmentation, excoriations resulting from pruritus, and xanthomata. As liver disease progresses, spider angiomas, muscular atrophy, peripheral edema, ascites, and other signs of advanced liver disease may appear.28–30
Laboratory Findings
Chronic elevation of serum alkaline phosphatase levels, typically three to five times normal, is the biochemical hallmark of PSC. A normal alkaline phosphatase level, however, may be found in up to 6% of patients with cholangiographically proved PSC.110,111 In some cases, an advanced histologic stage has been demonstrated on a liver biopsy specimen despite normal serum alkaline phosphatase levels.110 Serum aminotransferase levels are typically elevated, although rarely above four to five times normal except in the pediatric population.112 The serum bilirubin level may be normal or elevated and often fluctuates. When the serum bilirubin level is elevated, the bilirubin is predominantly conjugated. Reductions in the serum albumin level and prolongation of the prothrombin time may reflect hepatic synthetic dysfunction with advanced liver disease. In addition, malnutrition and underlying IBD may lower serum albumin levels. Vitamin K malabsorption related to cholestasis may play a role in prolonging the prothrombin time. Other nonspecific consequences of cholestasis are elevations in serum copper, serum ceruloplasmin, and hepatic copper levels, increased urinary copper excretion, and elevated serum cholesterol levels.
Several immunologic markers and serum autoantibodies are found in the majority of patients with PSC, although none is specific for the disease. Hyperglobulinemia is frequent; serum IgM levels are elevated in up to 50% of patients, and IgG and IgA levels also may be elevated.28,29,28,107 Antinuclear antibodies, often in low titer, may be detected in 24% to 53% of patients. Anti-smooth muscle antibodies are found in 13% to 20% of patients, but antimitochondrial antibodies are found in less than 10%.26,28,70,73,75 Most commonly found in patients with PSC are pANCA,72 which are detected in 65% to 88% of patients and appear to react to a heterogeneous group of antigens.70,71,74,75 These antigens have been found to represent neutrophil nuclear envelope proteins predominantly, and the corresponding antibodies have been referred to as “antineutrophil nuclear antibodies” (ANNA).113 In contrast to Wegener’s granulomatosis, titers of pANCA do not appear to correlate with disease activity, severity, or response to medical therapy in patients with PSC.72 Furthermore, the presence of autoantibodies does not appear to differ in patients with and without IBD. Anti-cardiolipin antibodies are detected in 66% of patients with PSC, and the titer has been reported to correlate with disease severity.75 In general, despite the high frequency of autoantibodies in patients with PSC, a clear association between the presence of these antibodies, pathogenesis of the disease, and prognosis or response to treatment remains unproved. Measurement of autoantibodies is therefore of limited clinical value in patients with PSC.
Imaging Findings
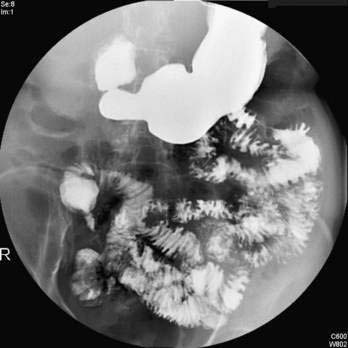
Cholangiography by ERCP, MRCP, or percutaneous THC establishes a diagnosis of PSC and provides information regarding the distribution and extent of disease. The characteristic cholangiographic findings include multifocal stricturing and ectasia of the biliary tree. Areas of narrowing are interspersed with areas of normal or near-normal caliber and areas of post-stenotic dilatation. The result is a classic “beaded” appearance to the biliary tree. The strictured segments are usually short, annular, or band-like in appearance (Fig. 68-2), although longer confluent strictures may be seen in more advanced disease. Localized segments of dilated ducts may have a saccular or diverticular appearance. Major areas of focal, tight narrowing known as dominant strictures may be seen and often involve the bifurcation of the hepatic duct.114 At times, diffuse involvement of the intrahepatic biliary tree may give a pruned appearance that is difficult to distinguish from the diffuse intrahepatic duct attenuation seen in patients with cirrhosis of any cause; irregularity of the duct wall or concomitant involvement of the extrahepatic bile duct supports a diagnosis of PSC.
Both the extrahepatic and intrahepatic bile ducts are abnormal in approximately 75% of cases. The intrahepatic ducts alone may be involved in 15% to 20% of cases.28,31,35,107 Abnormalities of the extrahepatic biliary tree in the absence of intrahepatic involvement are less common.98,99 The cystic duct and gallbladder may be involved in up to 15% of patients but may not be well visualized on routine cholangiography.115 Pancreatic duct irregularities similar to those seen in chronic pancreatitis may rarely be noted.116
PATHOLOGY
Gross and histologic specimens from the extrahepatic bile ducts demonstrate a diffusely thickened, fibrotic duct wall. The fibrosis is accompanied by a mixed inflammatory infiltrate that may involve the epithelium and biliary glands.117,118 Florid hyperplasia of the biliary glands with accompanying neural proliferation has been described.119 Examination of PSC explants removed at the time of liver transplantation has demonstrated areas of thin-walled saccular dilatation, termed “cholangiectasias,” that correspond to the beaded appearance on cholangiography.120
A wide range of liver biopsy findings may be seen in patients with PSC. For this reason, histologic findings are not typically diagnostic for PSC. The characteristic bile duct lesion is a fibro-obliterative process that may lead to an “onionskin” appearance of concentric fibrosis surrounding medium-sized bile ducts (Fig. 68-3); however, this finding is seen in less than one half of biopsy specimens.117,121,122 The smaller interlobular and septal bile duct branches may be entirely obliterated by this process, resulting in fibro-obliterative cholangitis. This finding is present in only 5% to 10% of biopsy specimens but is thought to be virtually pathognomonic of PSC.121 In this process, the biliary epithelium may degenerate and atrophy and be replaced entirely by fibrous cords. Other characteristic histopathologic findings may include bile duct proliferation, periductal inflammation, and ductopenia. The degree of inflammation can be quite variable but is typically a portal-based mixture of lymphocytes, plasma cells, and neutrophils with a periductal focus. Lymphoid follicles or aggregates may also be seen.121,122
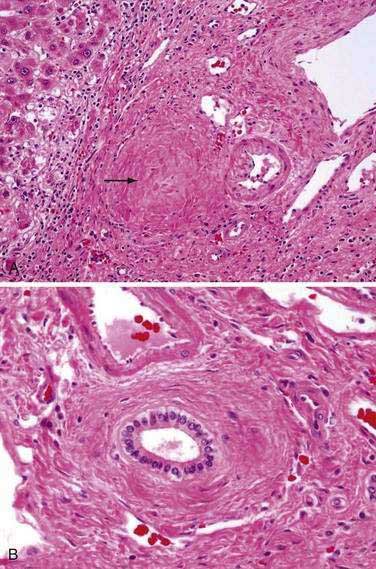
The histologic progression of PSC can be divided into four stages, analogous to a similar staging system in PBC46 (See Chapter 89). In stage I (portal stage) changes are confined to the portal tracts and consist of portal inflammation, connective tissue expansion, and cholangitis. Stage II (periportal stage) is characterized by expansion of inflammatory and fibrotic processes beyond the confines of the limiting plate, resulting in “piecemeal necrosis” (interface hepatitis) and periportal fibrosis. Depending on the degree of biliary obstruction, ductular proliferation and cholangitis may be of varying severity. Stage III (septal stage) is characterized by fibrous septa that bridge from one portal tract to the next. Bridging necrosis may occasionally be seen but is uncommon. Stage IV (cirrhotic stage) implies progression to biliary cirrhosis. The degree of inflammatory activity may subside as the stage of disease progresses, and focal bile ductular proliferation may be striking. A study that examined the time course of progression through the histologic stages revealed that for patients with stage II disease, 42%, 66%, and 93% progressed to a higher histologic stage at one, two, and five years, respectively.123 For patients initially with stage II disease, progression to biliary cirrhosis (stage IV) occurred in 14%, 25%, and 52%, respectively. Regression of stage was observed in 15% of patients and probably reflected sampling variability in the histologic assessment.
Many of the histologic findings of PSC are nonspecific and may be seen in other disorders. In particular, the histologic distinction between PSC and PBC may be difficult to discern. In one study, histologic examination could classify only 28% of patients who had one of the two diseases.18 When lymphocytic interface hepatitis is prominent, the distinction from autoimmune hepatitis may be challenging, especially because hypergammaglobulinemia and autoantibodies may be present in both conditions. In addition, an overlap syndrome with features of both PSC and autoimmune hepatitis has been described.21,22,66 When severe cholestasis develops, hepatic copper accumulation can be dramatic and may mimic that seen in Wilson disease.124
COMPLICATIONS
Cholestasis
The complications associated with all causes of chronic cholestasis may develop in patients with PSC (see also Chapters 20 and 89). Pruritus is one of the most common symptoms of PSC and may adversely affect a patient’s quality of life. Severe excoriations and debilitating symptoms may develop. The pathogenesis of pruritus in chronic cholestasis is poorly understood, and response to therapy is inconsistent (see Chapter 89). The accumulation of bile acids in the plasma and tissue of cholestatic patients has been cited as a potential cause of pruritus, and the pruritus of cholestasis is typically treated with oral administration of bile-acid binding resins such as cholestyramine. Not all patients with elevated serum bile acid levels itch, however. In addition, pruritus is frequently intermittent, despite the relative stability of serum bile acid levels. Several lines of evidence suggest that cholestasis is associated with an increased level of endogenous opioids. In animal models, cholestasis is associated with an increase in plasma levels of endogenous opioids.125 In humans, cholestatic patients may experience opiate withdrawal-like symptoms after the administration of an opioid antagonist. In addition, administration of naloxone and naltrexone, which have opioid antagonist properties, has been reported to relieve pruritus in cholestatic patients in small clinical trials.126,127
Nutritional deficiencies may complicate chronic cholestasis in patients with PSC. Intestinal absorption of the fat-soluble vitamins A, D, E, and K is particularly affected and is thought to be related to decreased intestinal concentrations of conjugated bile acids.128 Concomitant disease such as IBD, chronic pancreatitis, and celiac disease may also contribute to intestinal malabsorption. Clinical consequences include night blindness (vitamin A deficiency) and coagulopathy (vitamin K deficiency).
The importance of metabolic bone disease, also referred to as hepatic osteodystrophy, is often underrecognized in patients with PSC. Two forms of metabolic bone disease may develop: osteomalacia and osteoporosis. With improvements in nutritional management, osteomalacia (decreased bone mineralization) is now relatively rare, and most bone disease in cholestatic patients is osteoporosis. Bone mineral density is significantly lower in patients with PSC than in age- and sex-matched controls.129 The pathogenesis of bone density loss in PSC and other chronic cholestatic liver diseases is unknown. Intestinal malabsorption of vitamin D is probably not the primary abnormality because serum vitamin D levels are often normal and vitamin D repletion does not usually have a major impact on the severity of osteoporosis. In patients with concomitant IBD, the use of glucocorticoids may play a role in exacerbating bone loss in patients with PSC. Overall, severe osteoporosis is less common in patients with PSC than in those with PBC because a majority of patients with PSC are young men who have a higher baseline bone mineral density and a slower rate of bone loss than middle-aged women, who account for most cases of PBC.
Biliary Stones
Cholelithiasis and choledocholithiasis are more common in patients with PSC than in the general population. Gallstones are found in approximately 25% of patients with PSC and are often pigmented calcium bilirubinate stones.5 Biliary strictures may predispose to bile stasis and intraductal sludge and stone formation. Ultrasonography has only an intermediate sensitivity for detecting intraductal stones. Therefore, patients with PSC and worsening cholestasis or jaundice should undergo ERCP to distinguish biliary stone disease from the development of a dominant stricture or cholangiocarcinoma.
Cholangiocarcinoma
Cholangiocarcinoma is a feared complication of PSC and can arise from bile duct epithelium anywhere in the biliary tract (see Chapter 69). PSC should be considered a premalignant condition of the biliary tree, analogous to the relationship between UC and carcinoma of the colon. The reported frequency of cholangiocarcinoma in patients with PSC has ranged from 6% to 11% in natural history studies and from 7% to 36% in patients with PSC who undergo liver transplantation.130,131 Tumors are most commonly found in the common hepatic duct and perihilar region but may involve only the bile duct, intrahepatic ducts, cystic duct, or gallbladder. Cholangiocarcinoma remains the leading cause of death in patients with PSC.
The pathogenesis of cholangiocarcinoma in PSC is poorly understood. Although cholangiocarcinoma may complicate any stage of the disease, chronic inflammation is thought to predispose to epithelial dysplasia and an increased risk of malignant transformation. The role of chronic inflammation is supported by the observation that patients with chronic Clonorchis sinensis and other liver fluke infections are also at increased risk of cholangiocarcinoma (see Chapter 82).132 A role for proinflammatory cytokines in stimulating oxidative DNA damage and inactivation of DNA repair processes has been postulated.
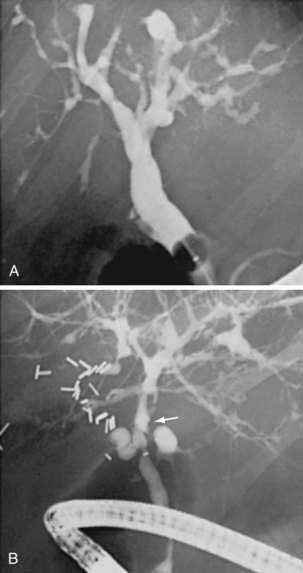
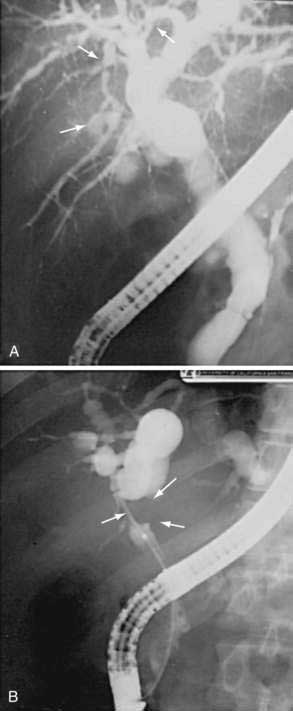
Biliary malignancy should be suspected when a patient with PSC exhibits rapid clinical deterioration with worsening jaundice, weight loss, and abdominal pain. Advanced PSC without cholangiocarcinoma may present with the identical clinical presentation. The diagnosis of cholangiocarcinoma presents a particular challenge in patients with PSC. A malignant biliary stricture may be indistinguishable cholangiographically from the underlying PSC (Fig. 68-4). Because of the tendency of cholangiocarcinoma to grow in sheets as opposed to a discrete mass, cross-sectional imaging with CT or magnetic resonance imaging (MRI) may be insensitive for detection of cholangiocarcinoma.
Several serum tumor markers of cholangiocarcinoma have been evaluated. Serum CA 19-9 has been the most commonly utilized tumor marker, with one small study reporting a sensitivity of 89% and specificity of 86% for a serum CA 19-9 level greater than 100 U/mL in diagnosing cholangiocarcinoma.133 A later study from the same group found a lower sensitivity for serum CA 19-9 but a correlation between the CA 19-9 level and tumor stage; no tumor was resectable in patients with a CA 19-9 level greater than 1000 U/mL.134 Another study described a biochemical index using CA 19-9 and carcinoembryonic antigen (CEA) levels (CA 19-9 + (CEA × 40) > 400), with a reported sensitivity of 86% for cholangiocarcinoma.135 More recent studies, however, suggested a poor sensitivity (33%) for this combined biochemical index despite a relatively high specificity (85%).136,137
Obtaining an adequate tissue sample presents a particular challenge in the diagnosis of cholangiocarcinoma. Dominant strictures resulting from PSC may be indistinguishable cholangiographically from those harboring cholangiocarcinoma. Brush cytology can be obtained at the time of ERCP, but the sensitivity of this approach is only 50% to 60% at best.138,139 False-positive results are also possible because chronically inflamed cells may take on a malignant cytologic appearance. The addition of a sampling technique to brush cytology, such as endobiliary biopsy or fine-needle aspiration (FNA), improves sensitivity,138–140 and when clinical or cholangiographic suspicion for cholangiocarcinoma is high, two tissue sampling techniques should be used. The sensitivity of cytologic examination is increased by use of specialized techniques such as fluorescent in situ hybridization and digital image analysis.
Endoscopic ultrasound (EUS) with FNA has an emerging role in the evaluation of suspected cholangiocarcinoma when brush cytology and other methods have failed to yield a diagnosis.141,142 The sensitivity of EUS-FNA for diagnosing cholangiocarcinoma in patients with PSC has been reported to be as high as 89%.141,142 High-frequency intraductal ultrasound (IDUS) is under study as a means of discriminating benign from malignant dominant bile duct strictures, and several studies143–145 have demonstrated that the addition of IDUS to ERCP improves the ability to distinguish benign from malignant dominant bile duct strictures in patients with PSC. Direct endoscopic visualization of bile duct strictures is also possible with the availability of choledoscopy (cholangioscopy) in clinical practice. A single small study has shown that the addition of choledoscopy to ERCP in patients with PSC and a dominant stricture enhanced detection of malignancy, with an overall sensitivity of 92%.146 Therefore, in patients with PSC, suspected cholangiocarcinoma, and negative brush cytology or endobiliary biopsy results, repeat ERCP plus EUS-FNA, IDUS, or choledoscopy improves the sensitivity of diagnosing cholangiocarcinoma.
The development of cholangiocarcinoma is an ominous sign, with a median survival of five months after diagnosis.6 In addition, recurrence of cholangiocarcinoma after liver transplantation is nearly universal, and cholangiocarcinoma is generally considered a contraindication to liver transplantation. Early identification of patients with PSC who are at high risk of cholangiocarcinoma is crucial so that liver transplantation may be undertaken before the bile duct cancer develops. One study reported a strong association between current or former smoking and the development of cholangiocarcinoma in patients with PSC.147 A second study, however, could not confirm an association with smoking.130 In addition, duration of PSC, distribution of biliary strictures, and past medical or surgical intervention do not appear to be associated with an increased risk of cholangiocarcinoma. Longer duration of IBD has been shown to be a risk factor for cholangiocarcinoma in some,148 but not all,130 studies. Overall, therefore, studies to date have not defined clear risk factors for cholangiocarcinoma that are clinically useful for identifying patients with PSC at particularly high risk. Nor has the optimal surveillance strategy been delineated.
Colonic Neoplasia
Most patients with PSC also have IBD; the majority have ulcerative pancolitis. UC is known to be associated with an increased risk of colonic dysplasia and carcinoma (see Chapter 112); the risk of colon cancer increases with the duration, extent, and severity of colitis. For patients with pancolitis, the cumulative risk of colon cancer is approximately 5% to 10% after 20 years and 12% to 20% after 30 years.149–151 A growing body of evidence suggests that patients with concomitant PSC and UC are at significantly higher risk for developing colonic neoplasia (dysplasia or carcinoma) than patients with UC alone.152–157 In one report, 132 patients with UC and PSC were compared with a randomly selected historical cohort of patients with UC but without PSC.155 Colonic carcinoma or dysplasia developed in significantly more patients with both UC and PSC than those with UC alone (25% versus 6%), and significantly more deaths from colorectal cancer were observed in the patients with PSC (4.5% versus 0%). A similarly designed study included 20 patients with both UC and PSC and 25 matched controls with UC alone.153 Colonic dysplasia was observed significantly more often in patients with both UC and PSC (45% versus 16%), although the time to the development of dysplasia was similar in the two groups. Patients with PSC and UC were also more likely than patients with UC alone to have synchronous sites of dysplasia in the colon. Another study examined 40 patients with PSC and UC matched with 80 control subjects with UC alone.152 The cumulative risk of colorectal dysplasia or carcinoma in patients with both UC and PSC was 9%, 31%, and 50% after 10, 20, and 25 years, respectively. These rates were significantly higher than those for the control group (2%, 5%, and 10%, respectively).
The mechanisms by which PSC confers an added risk of colonic neoplasia are not well understood. A high colonic concentration of secondary bile acids may play a role because patients with UC and colonic dysplasia or carcinoma have higher fecal bile acid concentrations than patients with UC who do not have dysplasia or carcinoma.158 This theory is supported by the higher incidence of right-sided colon cancer in patients with UC and PSC than in those with UC alone.155,156 In the study by Shetty and colleagues,155 76% of the colon cancers in patients with UC and PSC occurred proximal to the splenic flexure, compared with only 20% in patients with UC alone.155 Increased colonic secondary bile acid concentrations may also explain the apparent chemoprotective effect of ursodeoxycholic acid (UDCA) against the development of colonic neoplasia. Two studies have reported that UDCA use is associated with a lower risk of colonic dysplasia or cancer in patients with UC and PSC.159,160 UDCA may confer protection against colonic neoplasia by reducing colonic concentrations of secondary bile acids, as well as by affecting expression of protein kinase C isoforms, metabolism of arachidonic acid, and expression of cyclooxygenase-2.159–162 These data need to be confirmed in larger-scale, prospective trials before UDCA use can be recommended routinely for preventing colon cancer in patients with PSC. Whether UDCA has a chemoprotective effect in patients with UC but without PSC is unknown.
Patients with PSC who have UC should undergo annual colonoscopic surveillance for the detection of colonic dysplasia or cancer. As for colonoscopic surveillance in patients with UC alone, multiple mucosal biopsy specimens should be obtained (see Chapter 112).163 Most experts agree that colonoscopic surveillance should start immediately after the diagnosis of PSC. Surveillance should continue even after liver transplantation because these patients remain at increased risk for colonic neoplasia.164–166
Peristomal Varices
Varices at the stoma may develop in patients with PSC and portal hypertension who have previously undergone proctocolectomy with ileostomy for IBD.167,168 These varices may bleed spontaneously, and the bleeding may be dramatic. Treatment modalities that may initially be effective in achieving hemostasis include injection sclerotherapy,169 percutaneous transhepatic coil embolization,170 surgical stomal revision,171 and transjugular intrahepatic portosystemic shunt placement (see Chapter 90).172 Nevertheless, recurrent bleeding is common, and liver transplantation should be considered to relieve portal hypertension and treat the underlying liver disease.
TREATMENT
Medical Treatment of Underlying Disease
A wide variety of medications have been studied in patients with PSC (Table 68-4). Many of the published studies have been small and uncontrolled with limited follow-up. Because of the varied course of PSC, with spontaneous remissions and unpredictable flares, adequate clinical trials in patients with PSC require long-term follow-up. In addition, the defined study endpoints, whether clinical, biochemical, histologic, or a mathematical risk score, have varied greatly among published studies. To date, no medical treatment has been shown clearly to alter the course of PSC.
Table 68-4 Medical Therapy for Primary Sclerosing Cholangitis*
| NO PROVEN BENEFIT | POSSIBLE BENEFIT |
|---|---|
| Antibiotics Cholestyramine |
Ursodeoxycholic acid (20-30 mg/kg/day)173–177180 |
| Glucocorticoids183,184 | |
| Azathioprine | |
| Methotrexate187 | |
| Cyclosporine | |
| Tacrolimus188 | |
| Pentoxifylline189 | |
| Colchicine191 | |
| d-penicillamine192 | |
| Nicotine193,194 | |
| Perfenidone195 |
* Superscript numbers indicate references.
UDCA has been the most extensively studied drug in patients with PSC. At least five controlled clinical trials have been reported, with varying doses of UDCA.173–177 The mechanisms by which UDCA is thought to exert a beneficial effect in cholestatic conditions include protection of cholangiocytes against cytotoxic hydrophobic bile acids, stimulation of hepatobiliary secretion, protection of hepatocytes against bile-acid induced apoptosis, and induction of antioxidants (see also Chapter 89).178,179 In a randomized, controlled trial by Beuers and colleagues,173 patients with PSC treated with UDCA, 13 to 15 mg/kg/day for one year, had significant improvements in biochemical and histologic endpoints in comparison with those given placebo, but no effect on symptoms was noted. In the largest controlled trial of UDCA for PSC to date, Lindor and colleagues176 randomized 105 patients to UDCA, 13 to 15 mg/kg/day, or placebo for a median of 2.2 years. Significant biochemical improvements were seen with UDCA therapy, but no difference was seen in the primary endpoint, which was a composite of death, liver transplantation, histologic progression of at least two stages, hepatic decompensation, or quadrupling of the serum bilirubin level.
Because of the disappointing results with standard-dose UDCA, several groups have published results on the use of high-dose UDCA in the treatment of PSC. Mitchell and colleagues177 performed a two-year controlled trial of high-dose UDCA, 20 mg/kg/day, versus placebo in 26 patients with PSC. Compared with placebo, treatment with high-dose UDCA led to improvement in biochemical markers and a reduction in the progression of fibrosis and cholangiographic changes. In addition, high-dose UDCA was well tolerated, with no significant adverse events. Harnois and colleagues180 performed an open-label pilot study of UDCA, 25 to 30 mg/kg/day, for one year in 30 patients with PSC and compared changes in the revised Mayo risk score with those in patients from a previous randomized trial of standard-dose UDCA. After one year of treatment, improvement in the revised Mayo risk score in the high-dose UDCA group was significantly greater than that seen in placebo-treated patients from the prior study but no better than that in the patients treated with standard-dose UDCA.
With the encouraging results of these two small studies, a larger study was undertaken in which 219 patients with PSC were randomized to high-dose UDCA or placebo. After five years, no differences in symptoms, liver biochemical abnormalities, quality of life, or transplant-free survival were found between the two groups.181 A subsequent study in 31 patients randomized to low-dose, standard-dose, and high-dose UDCA found improvement in the revised Mayo risk score in all groups, with statistical significance only in the group receiving high-dose UDCA.182 Therefore, the precise benefit of UDCA in general, and high-dose UDCA in particular, remains uncertain.
Given the immunologic alterations in patients with PSC, immunosuppressive therapy would appear to be a reasonable consideration. Glucocorticoids, administered both orally and via nasobiliary lavage, have not shown a clear benefit in uncontrolled studies,183,184 although subgroups of patients may have a clinical response.185 Lack of long-term data demonstrating clear response and concerns about long-term adverse effects, including exacerbation of metabolic bone disease, have limited the use of glucocorticoids. Oral budesonide, a newer glucocorticoid with limited systemic toxicity, has been evaluated in an uncontrolled pilot study in 21 patients with PSC.186 After one year of therapy, treated subjects had minimal biochemical or histologic improvement and no change in revised Mayo risk score. In addition, significant loss of bone mass was seen with use of budesonide.
Other immunomodulators also have been evaluated. In a small prospective, controlled trial of methotrexate, no biochemical, histologic, or cholangiographic differences from therapy with placebo were seen after two years of treatment.187 In a small pilot study of tacrolimus therapy, significant biochemical improvement was observed after one year, but no change in cholangiographic or histologic severity was documented.188 Pentoxifylline, which inhibits tumor necrosis factor-α (TNF-α), led to no biochemical or symptomatic improvements in a one-year pilot study in 20 patients with PSC.189 Etanercept, a recombinant inhibitor of TNF-α, also showed no benefit in a small number of patients with PSC.190
Colchicine has been evaluated as a potential therapy for PSC because of its antifibrogenic potential. In a randomized, controlled clinical trial comparing colchicine therapy with placebo for three years, no differences were seen between the two groups in rates of mortality or liver transplantation or in symptoms or biochemical and histologic findings.191 D-penicillamine has also been studied in a randomized, controlled clinical trial192 because of the increased hepatic copper concentrations seen in patients with PSC and other chronic cholestatic conditions. In addition to its cupruretic effects, penicillamine may have antifibrogenic and immunosuppressive properties. Therapy with penicillamine therapy for three years, however, led to no difference in mortality or in biochemical or histologic progression as compared with therapy with placebo. In addition, penicillamine was associated with substantial toxicity. Other studies have failed to demonstrate a significant response to nicotine193,194 or the antifibrotic drug pirfenidone.195 Finally, combinations of various agents such as azathioprine, glucocorticoids, UDCA, and antibiotics have been studied in a limited fashion.196–198 The results of these studies have been mixed, with some showing no benefit and others demonstrating histologic improvement in small numbers of patients. A problem with combining agents is an increased risk of adverse drug reactions.
Medical Treatment of Complications
An important component in the medical care of patients with PSC is the management of complications of the disease, such as pruritus, nutritional deficiencies, and bacterial cholangitis. As discussed earlier, therapy with UDCA does not have a consistent effect on pruritus, although some patients may notice symptomatic improvement. Although treatment with antihistamines may improve pruritus, anion-exchange resins such as cholestyramine, colestipol hydrochloride, or colesevelam are typically more effective, although compliance is a problem with the use of bile acid resins. These drugs are relatively unpalatable, frequently produce constipation, and may interfere with the absorption of other medications. Rifampin appears to be an effective and safe alternative for patients who do not respond to the preceding measures.199,200 Opiate antagonists such as naloxone and naltrexone have also been shown to be effective for cholestatic pruritus, although self-limited episodes of opioid withdrawal-like symptoms may occur.126,127,201 Patients who are unresponsive to these measures and who do not obtain relief from endoscopic therapy of a dominant stricture (see later) may need to be considered for plasmapheresis (which has shown benefit in anecdotal reports) or even liver transplantation (see also Chapter 89).
Patients with PSC should be screened for nutritional deficiencies by measurement of fat-soluble vitamin levels and the prothrombin time. In most patients, vitamin supplements are given orally, but a parenteral route may be necessary in patients with severe intestinal fat malabsorption. Administration of vitamin A is usually effective for correcting subclinical vitamin A deficiency. Correction of vitamin D deficiency, with or without calcium supplements, is of unproven benefit in cholestatic liver disease but is generally recommended because of its safety.202 The use of bisphosphonates and other agents to promote bone formation requires further study in patients with PSC and osteoporosis. In patients with PSC, prolongation of the prothrombin time is more likely to be the result of advanced liver disease than of vitamin K deficiency, although a trial of oral vitamin K is warranted in patients with coagulopathy (see Chapters 89 and 92).
Endoscopic Management
Patients most likely to benefit from endoscopic intervention are those with one or more dominant stricture. These patients are more likely to present with specific symptoms such as worsening jaundice or pruritus, cholangitis, or abdominal pain. Multiple studies have reported significant improvements in clinical, biochemical, and cholangiographic endpoints in patients with a dominant stricture treated with endoscopic therapy,203–207 usually dilation with a balloon or graduated dilators, with or without temporary placement of a biliary stent. Sphincterotomy is often performed for improved access and to treat choledocholithiasis, if present. One retrospective study suggested that balloon dilation followed by stent placement offered no improvement and increased the risk of complications compared with balloon dilation alone.208 Because the study was not randomized, however, these findings may have been attributable to differences between the treatment groups.
Three studies have suggested that progression of the underlying disease process may be slowed by endoscopic therapy of a dominant stricture. Baluyut and colleagues209 performed graduated and balloon dilation, with or without stent placement, in 63 patients with PSC with a median follow-up of 34 months, and demonstrated an observed 5-year survival that was significantly better than survival predicted from the revised Mayo model score (see Table 68-3). Stiehl and colleagues210 performed endoscopic balloon dilation and occasional stent placement in 52 patients with PSC in whom a dominant stricture developed while the patients were on therapy with UDCA. Actuarial survival free of liver transplantation at three, five, and seven years was significantly better than that predicted from the multicenter model score (see Table 68-3). Finally, a retrospective chart review by Gluck and colleagues8 reported a single-center 20-year experience with endoscopic therapy for PSC. Endoscopic therapy for a dominant stricture was performed in 84 of 106 patients who underwent a total of 317 procedures during the 20-year observation period. The patients in whom endoscopic therapy was performed had a significantly higher survival rate than that predicted by the revised Mayo model score at years three and four. These studies were not randomized trials, and in some cases were retrospective, but they provide some supporting evidence to suggest that endoscopic management of a dominant stricture may alter the course of PSC.
Endoscopic therapy in PSC also has important limitations. Complications of ERCP, such as pancreatitis, cholangitis, worsening cholestasis, and perforation, occur at an overall rate of 7.3% to 10%.8,210,211 Patients with diffuse intrahepatic biliary stricturing and no dominant stricture are less likely to derive benefit from endoscopic intervention and may be at higher risk for post-ERCP cholangitis.208 If ERCP is performed in expert hands and only for specific indications such as worsening of jaundice, pruritus, or cholangitis—that is, in the subgroup of patients who are most likely to benefit from therapy—the risks in patients unlikely to benefit will be minimized (see Chapter 70).
Percutaneous Management
Percutaneous THC with balloon dilation, stenting, or both, can also be undertaken to treat biliary strictures in patients with PSC. This approach is typically recommended only when endoscopic intervention is contraindicated or unsuccessful because of the added risks of bleeding and bile peritonitis, as well as increased patient discomfort, associated with percutaneous intervention (see Chapter 70).
Surgical Management
Biliary Surgery
The role of biliary surgery in PSC has diminished considerably with improvements in endoscopic therapy and liver transplantation. Resections of a dominant stricture of the bile duct or near the hepatic bifurcation followed by hepaticojejunostomy or choledochojejunostomy have been the most commonly performed operations.212,213 Postoperative mortality is increased significantly in patients with PSC and cirrhosis.214 In addition, biliary surgery may complicate future liver transplantation. Currently, biliary surgery in patients with PSC is rarely indicated and should be reserved for the small subset of patients who have early-stage PSC and biliary strictures that are not amenable to endoscopic or percutaneous intervention.
Liver Transplantation
Liver transplantation (see also Chapter 95) is the only therapy that has been shown conclusively to improve the natural history of PSC. In addition, quality of life improves after liver transplantation.215,216 The procedure is recommended for patients with PSC in whom decompensated cirrhosis and complications of portal hypertension develop. Recurrent cholangitis or pruritus that is refractory to medical and endoscopic management rarely may also be indications for liver transplantation. Determination of the appropriate timing for liver transplantation in patients with PSC may be challenging, although use of available natural history models can be helpful. When a patient’s expected survival after liver transplantation exceeds survival predicted from the natural history models, liver transplantation should be undertaken, in the absence of contraindications. Intraoperatively, patients who undergo liver transplantation for PSC should have a Roux-en-Y choledochojejunostomy anastomosis, instead of a standard choledochocholedochostomy. This approach is recommended to allow removal of as much of the native biliary tree as possible, to reduce the risk of recurrent strictures and cholangiocarcinoma.217
Patient and graft survival after liver transplantation for PSC is excellent. A large single-center experience demonstrated 1-, 5-, and 10-year actuarial patient survival rates of 93.7%, 86.4%, and 69.8%, respectively. Corresponding graft survival rates were 83.4%, 79.0%, and 60.5%.218 Similar results have been reported in other series.219,220 Overall, survival rates after liver transplantation for PSC are significantly better than those for any other disease except PBC.221 Recipient factors that have been associated with a worse prognosis after liver transplantation for PSC are older age, decreased serum albumin level, renal failure, Child’s class C cirrhosis, and advanced United Network for Organ Sharing status.221,222
The presence of cholangiocarcinoma has a major impact on the outcome after liver transplantation for PSC. Early studies demonstrated that even in cases in which cholangiocarcinoma was discovered incidentally in the explant, recipient survival was poor, with a one-year survival rate of 30% in one series.223 On the basis of such studies, cholangiocarcinoma has generally been considered a contraindication to liver transplantation. Another report confirmed the poor post-liver transplantation outcome in patients with known cholangiocarcinoma but suggested a good survival rate for those who had a small cholangiocarcinoma found incidentally at the time of transplantation.220 Subsequent studies have demonstrated improved results of liver transplantation in patients with cholangiocarcinoma, with one- and five-year survival rates of 65% to 82% and 35% to 42%, respectively.224,225 Preoperative chemoradiation in highly selected patients with cholangiocarcinoma has shown promise in reducing the rate of tumor recurrence after liver transplantation (see Chapter 69).226,227
Biliary strictures commonly recur after liver transplantation for PSC and may represent recurrent PSC. In addition to recurrent PSC, potential causes of biliary strictures after liver transplantation include ABO blood group incompatibility, hepatic artery occlusion, chronic ductopenic graft rejection, Roux-en-Y–related cholangitis, and preservation-related ischemia. The diagnosis of recurrent PSC has been proposed to be confined to those patients who have a consistent cholangiographic pattern and compatible liver histology showing fibrous cholangitis, fibro-obliterative lesions, biliary fibrosis, or biliary cirrhosis, and who lack other risk factors for biliary strictures, such as hepatic artery occlusion, ABO incompatibility, or ductopenic graft rejection, or who develop non-anastomotic strictures within 90 days of transplantation.228 With these stringent criteria, the risk of recurrent PSC after liver transplantation ranges from 5.7% to 21.1%,219,220,229–230 and patient and graft survival do not appear to be adversely affected for the first five years of follow-up.220,230 No specific risk factors for the development of recurrent PSC have been clearly identified. In addition, no specific therapy has been shown to treat or prevent recurrent PSC effectively after liver transplantation.
The effect of liver transplantation on the course of underlying IBD and risk for subsequent colonic neoplasia remains controversial. The clinical course of IBD after liver transplantation has varied greatly among studies.45,219,232,233 Immunosuppression was thought possibly to ameliorate the clinical course of IBD but does not in most instances. A large single-center study showed that liver transplantation was associated with an increased risk for colectomy.234 The risk of colorectal neoplasia is generally agreed to be increased after liver transplantation in patients with UC,164–166219 although this finding has not been confirmed in all studies.234 Annual surveillance colonoscopy is recommended after liver transplantation for PSC in patients with UC.
RECURRENT PYOGENIC CHOLANGITIS
Recurrent pyogenic cholangitis (RPC) was originally defined by Cook and colleagues as a syndrome characterized by recurrent bacterial cholangitis, intrahepatic pigment stones, and biliary strictures, possibly leading to chronic liver disease and cholangiocarcinoma.235 RPC has also been called oriental cholangiohepatitis, “Hong Kong disease,” “biliary obstruction syndrome of the Chinese,” and hepatolithiasis.236
EPIDEMIOLOGY
Digby first described RPC in 1930 in Chinese patients in Hong Kong and was the first to recognize that RPC was clinically and pathologically distinct from biliary disease caused by gallstones in Western populations.237 Subsequently, most cases have occurred in patients from East Asia; men and women are affected equally. Patients in rural areas and those of lower socioeconomic status appear to be at increased risk, and the incidence is highest among persons between ages 30 and 40.238,239 In certain parts of Southeast Asia such as Taiwan, over one half of the cases of gallstone disease are estimated to be caused by RPC.240 In Singapore, Japan, and Hong Kong, 2% to 5% of biliary calculous disease is attributed to RPC.241
The incidence of RPC in East Asia appears to be decreasing. A study from the Queen Mary Hospital, a major referral center for RPC, reported that only 6.7% of a total of 490 patients who underwent surgery for hepatobiliary disease were classified as having RPC.241 By contrast, typical gallstone disease was the reason for surgery in 44%. These numbers likely reflect referral bias; a nationwide survey found that RPC or intrahepatic stones represented only about 20% of the total cases of biliary tract disease. The incidence of intrahepatic stones appears to be decreasing in Taiwan as well; one study reported that the incidence of hepatolithiasis decreased from 21% to 18% between 1981 and 1989.240 By contrast, the incidence and prevalence of RPC are increasing in Western countries, reflecting immigration patterns. One study from San Francisco found that the prevalence of RPC doubled between 1983 and 1995.242 Similarly, a review from a county hospital in Los Angeles reported that 57% of 18 patients with RPC seen over a seven-year period were of Asian descent, and 36% of the patients were of Hispanic descent.243
The reasons for the changing epidemiology of intrahepatic stones in East Asia are unclear. Several possible explanations have been proposed, including adoption of a Western-style diet with a higher protein content; improved hygiene, which may lead to reduced gastrointestinal infections and consequently decreased entry of bacteria into the portal circulation (an important cause of cholangitis); and, less likely, reduction in disease burden related to Clonorchis sinensis and Ascaris lumbricoides. A low-protein diet may lead to reduced biliary levels of glucaro-1:4-lactone, an inhibitor or bacterial β-glucuronidase, which helps promote the formation of calcium bilirubinate stones by deconjugating bilirubin into unconjugated bilirubin.244 Furthermore, a diet low in fat may be associated with reduced gallbladder contractility and thus promote stasis, which also is a factor in stone formation.245 The rising incidence of cholesterol gallstones in Asia suggests that environmental factors, such as adoption of a more Western-style diet, rather than genetic factors, are major factors in the pathogenesis of RPC.241
ETIOLOGY AND PATHOGENESIS
The etiology of RPC remains unknown. The most attractive hypothesis links biliary tract infection with the parasites Clonorchis sinensis, Opisthorchis species, and Ascaris lumbricoides (see Chapters 82 and 110). Infection with these parasites is endemic in the same geographic region where RPC is prevalent. C. sinensis, a trematode (liver fluke), is endemic in China, Japan, Taiwan, Korea, and Vietnam.246,247 O. felineus and O. viverrini are the two species most commonly implicated in opisthorchiasis. O. felineus has been described in Southeast Asia and in parts of the former Soviet Union, and A. lumbricoides, a roundworm, is a ubiquitous parasite and may infect over one billion people throughout the world.248,249 Both organisms colonize the biliary tree and lead to infection, biliary tract obstruction, and secondary bacterial cholangitis. The Clonorchis worm can survive for decades in the biliary tree and may lead to inflammatory changes in the bile ducts, as well as direct bile duct obstruction by the flukes, and shows a specific predilection for the left hepatic ducts.248 Analysis of biliary stones from patients with RPC has demonstrated evidence of ova.250
Patients with RPC, however, have not been shown to have an increased prevalence of infection with these parasites when compared with the general population, and approximately one half of patients with RPC demonstrate no evidence of infection.251–253 Furthermore, some parts of Asia with a high prevalence of RPC have low or undetectable rates of infection with C. sinensis.254 Nevertheless, it remains possible that parasitic infection may account for a significant proportion of cases of RPC. Moreover, currently available serologic tests lack precision to detect prior infection, and structural or functional changes in the bile ducts, gallbladder, or sphincter of Oddi caused by infection in the remote past may alter the biliary epithelia and thereby promote the later formation of intrahepatic stones.
Bacterial infections have also been proposed as a cause of RPC because of the high incidence of bacterial cholangitis in patients with RPC. Portal bacteremia, possibly related to gastrointestinal infection and bacterial translocation, has been associated with low socioeconomic status and malnutrition.239 The hypothesis that bacterial infections lead to RPC, however, does not explain the unique epidemiologic features of the disease.
Dietary factors and hygiene have also been implicated in the pathogenesis of RPC. Because cholecystokinin, a potent mediator of gallbladder contractility, is secreted in response to dietary fat, diets high in carbohydrate and low in saturated fat may be associated with reduced gallbladder contractility and promote stone formation. Furthermore, deconjugation of bilirubin by bacteria or endogenous enzymes, as noted earlier, may be related to dietary and environmental factors and may facilitate formation of pigment stones. Several investigators have suggested that patients with RPC may lack an inhibitor of bacterial β-glucuronidase.255,256
Another factor that may contribute to the pathogenesis of RPC is abnormal motor activity of the sphincter of Oddi. A study of biliary sphincter manometry in 15 patients with RPC and 15 normal control subjects257 demonstrated various abnormalities, including retrograde phasic waves, abnormal propagation of phasic contractions, elevated basal sphincter pressure, and increased frequency of phasic contractions in the sphincter among the patients with RPC (see Chapter 63). Almost one half of the patients had evidence of papillitis at endoscopy. The investigators suggested that papillitis may lead to altered function in the sphincter of Oddi, resulting in turn in delayed biliary drainage and recurrent cholangitis. Whether papillitis and sphincter of Oddi dysfunction are a cause or effect of RPC is difficult to ascertain from the study; recurrent bouts of cholangitis may, in fact, have led to changes in the biliary tract, and recurrent passage of small pigment stones and debris may have resulted in endoscopic and manometric changes.
CLINICAL FEATURES
Patients with RPC often present with symptoms of ascending cholangitis (45% of patients).258 Patients characteristically present with fever, right upper quadrant pain, and jaundice, also referred to as Charcot’s triad. A prior history of such attacks is elicited in the majority of patients, whereas up to 30% of patients present with an initial episode.242,250 Patients may also present with abdominal pain or pancreatitis. On physical examination, abdominal tenderness is a common finding. Hepatomegaly is present in approximately 20% of patients. A palpable gallbladder may be present in approximately 10% of patients and may point to emphysematous cholecystitis.250,251,253 Laboratory findings are compatible with biliary obstruction, with elevation of serum total and direct bilirubin, aminotransferase, and alkaline phosphatase levels; leukocytosis may be present.
Imaging findings in patients with RPC are characteristic (Figs. 68-5 and 68-6). The majority of patients (75% to 80%) have intrahepatic stones, with predominant involvement of the left hepatic duct. Up to 70% may also have associated stones in the gallbladder, and cholecystitis (calculous or acalculous) is a frequent associated finding.259–261 Dilatation of the bile ducts is found almost universally. The central bile ducts are dilated disproportionately, with abrupt tapering and attenuation of more peripheral bile ducts within the liver. The presence of bile duct calculi is usually associated with intrahepatic bile duct dilatation and downstream strictures. The left hepatic ducts are involved more often than are the right.250
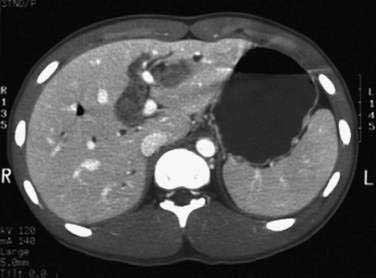
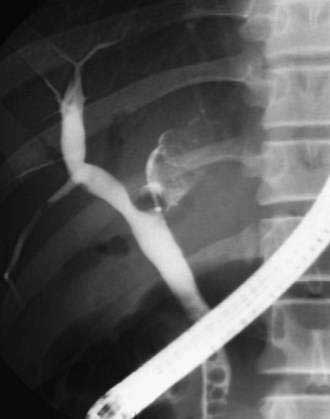
Cross-sectional imaging studies of the liver, such as ultrasonography and CT, as well as direct cholangiography, are useful for evaluating a patient with known or suspected RPC. Ultrasonography is a reasonable screening test early in the evaluation of the patient and may identify intra- and extrahepatic bile duct dilatation in the majority of cases. CT is helpful in delineating the complete segmental anatomy of the liver and is invaluable in planning surgical therapy (see Figure 68-5). CT may also demonstrate associated atrophic segments of the liver, abscesses, and cholangiocarcinoma.250 A study of more than 1300 patients with RPC from Korea described characteristic CT findings in 82 patients in whom cholangiocarcinoma was associated with RPC; cholangiocarcinoma tended to be located in atrophic segments associated with biliary calculi and was often accompanied by portal vein occlusion or narrowing.262
Direct cholangiography, whether performed by the percutaneous or endoscopic route, allows localization of intrahepatic stones and strictures and placement of drains or extraction of stones (see Figure 68-6). ERCP is the first choice for therapeutic intervention because it is less invasive than percutaneous THC.263 Removal of intrahepatic stones by this technique may be exceedingly difficult, however, because of the presence of a tight stricture and the sharp angulation of the intrahepatic bile ducts.
MRCP permits selective visualization of the biliary tree and has become the preferred test for diagnostic evaluation of the bile ducts. In a study from Korea,264 18 of 24 patients who had MRCP before undergoing surgical treatment for RPC also underwent direct cholangiography. Two examiners evaluated MRCP images and direct cholangiograms and compared them with surgical findings. All dilated bile ducts and 98% of focal duct strictures and intraductal stones were identified by MRCP. By contrast, direct cholangiography identified only 44% to 47% of segmental bile duct abnormalities. The authors concluded that MRCP was more sensitive than direct cholangiography for complete evaluation of the biliary tree. MRCP is an important complementary technique for examination of the biliary tree, especially in patients being considered for surgery because of its ability to visualize the bile ducts upstream from obstructed areas completely and to correlate the observed abnormalities with the segmental anatomy of the liver.265
Patients who present with an initial episode of cholangitis associated with intrahepatic stones and strictures should undergo evaluation for infection with Clonorchis and Opisthorchis species, particularly if the patient comes from or has traveled to an endemic area. The diagnosis of a parasitic infection is made by identification of eggs in fecal specimens; concentrated stool may be required. Eggs are present in stool after four weeks of infection.250 Duodenal or biliary fluid also may demonstrate eggs or intact worms. Peripheral eosinophilia may be present in cases of parasitic infection and may be associated with elevated serum IgE levels.246
PATHOLOGY
The characteristic findings in RPC include strictures and dilatation of the intra- and extrahepatic bile ducts. In classic cases, the left hepatic duct is more commonly and more severely affected than the right.250,251 With chronic disease, atrophy of the left lobe or the left lateral segment of the liver may occur and may be the site at which cholangiocarcinoma develops.250,251 The bile ducts are often obstructed with pigment stones, in addition to sludge and inspissated bile, and are frequently contaminated by bacteria and purulent material. The stones are composed predominantly of calcium bilirubinate, although cholesterol has been reported to be found in increasing proportions.239 Ova of parasites with a predilection for the biliary tree, such as C. senensis, have been described within the stones.250,252 An inflammatory infiltrate is frequently present in the walls of involved bile ducts and may be associated with periductal fibrosis and abscesses.250,251
TREATMENT
Antibiotic therapy should be initiated promptly, after cultures of blood and bile (if accessible) have been obtained. Abdominal ultrasonography is a reasonable initial imaging modality.259 In patients with evidence of cholangitis and dilated intra- and extrahepatic bile ducts, ERCP is the preferred interventional procedure. ERCP with sphincterotomy, with or without placement of a nasobiliary drain or an endobiliary drain, may adequately remove bile duct stones and traverse strictures. After adequate biliary drainage has been achieved, additional cross-sectional imaging with CT or MRI, with or without MRCP, may be considered.
Several studies have reviewed the success of various nonoperative interventions for initial management of patients with RPC presenting acutely. Sperling and colleagues258 compared outcomes in 41 patients with RPC based on whether they received immediate therapeutic ERCP, hepatobiliary surgery, or no intervention. Symptoms recurred in 62% of patients who underwent only diagnostic ERCP but one half as often in those treated with therapeutic ERCP or surgery. Therapeutic ERCP was particularly effective in patients with disease involving the extrahepatic bile ducts and was comparable in efficacy to surgery. Patients with disease involving both the right and left hepatic bile ducts tend to undergo more imaging studies, percutaneous cholangiograms, and endoscopic or surgical procedures.242
Hepaticojejunostomy has been a commonly used surgical procedure for the treatment of intrahepatic stones in patients with RPC. Kusano and colleagues,266 from Japan, reviewed the long-term outcomes of hepaticojejunostomy for intrahepatic hepatolithiasis in 159 patients over a 23-year period. Surgical approaches included hepatectomy (n = 94), biliary lithotripsy (n = 65), or hepaticojejunostomy (n = 72) (or combinations). Residual or recurrent stones were identified in approximately one third of patients after presumably complete removal. The rate of cholangitis was higher among patients who underwent a biliary-enteric anastomosis (22 of 72; 31%) than in those who did not (3 of 87; 3.4%). This study suggests that surgical therapy involving a biliary-enteric anastomosis may be associated with a higher rate of cholangitis than nonsurgical therapy.
Laparoscopic biliary bypass surgery has been proposed as a technically feasible and effective option for patients with RPC.267 A large retrospective case series from Canada of 42 patients who underwent surgical intervention for RPC demonstrated excellent postoperative outcomes for both liver resection and bile duct exploration with biliary bypass; the overall operative mortality rate was 0%, and the complication rate was 35% for resection and 30% for bile duct exploration.268
Treatment with an antihelminthic agent is indicated in patients with evidence of active parasitic infection. Praziquantel, 75 mg/kg in three divided doses for one day, is the treatment of choice (see also Chapter 82). The treatment is almost universally effective in both Clonorchis and Opisthorchis infections.269 Side effects of the medication include headache and gastrointestinal symptoms. Even patients without gastrointestinal symptoms but with Clonorchis or Opisthorchis in the stool should be treated to reduce the risk of cholangiocarcinoma.132
PROGNOSIS AND COMPLICATIONS
The natural history of RPC has been evaluated in a large series from Korea. The records of 193 patients with newly diagnosed RPC were reviewed to examine the rates and risk factors for recurrence.270 The mean follow-up period was 56 months (range, 1 to 242 months). Cumulative recurrence rates of cholangitis were 25% at three years and 37% at five years, with an overall rate of 45% during the follow-up period. Factors associated with cholangitis included recurrent stones (hazard ratio = 4.02; 95% CI: 1.3 to 12.4), residual stones (hazard ratio = 1.77; 95% CI: 1.1 to 3.0); prior hepatic resection was associated with a lower rate of recurrent cholangitis (hazard ratio = 0.28; 95% CI: 0.1 to 0.7). Strictures in the extrahepatic bile ducts were associated with a higher rate of cholangitis, whereas strictures in the intrahepatic ducts and disruption of the sphincter of Oddi (e.g., sphincterotomy) were not.
Cholangiocarcinoma is another long-term complication in patients with RPC (see Chapter 69). The frequency of cholangiocarcinoma has not been clearly delineated. One large study found that the frequency of cholangiocarcinoma among patients with RPC was approximately 3%.271 Other studies have reported the rate to be as high as 9%.272,273 Secondary biliary cirrhosis may develop and require liver transplantation.
Angulo P, Pearce DH, Johnson CD, et al. Magnetic resonance cholangiography in patients with biliary disease: Its role in primary sclerosing cholangitis. J Hepatol. 2000;33:520-7. (Ref 10.)
Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003;125:1364-9. (Ref 27.)
Bjornsson E. Immunoglobulin G4-associated cholangitis. Curr Opin Gastroenterol. 2008;24:389-94. (Ref 25.)
Bjornsson E, Chari ST, Smyrk TC, Lindor K. Immunoglobulin G4 associated cholangitis: Description of an emerging clinical entity based on review of the literature. Hepatology. 2007;45:1547-54. (Ref 23.)
Feldstein AE, Perrault J, El-Youssif M, et al. Primary sclerosing cholangitis in children: A long-term follow-up study. Hepatology. 2003;38:210-17. (Ref 33.)
Gluck M, Cantone N, Brandabur J, et al. A twenty-year experience with endoscopic therapy for symptomatic primary sclerosing cholangitis. J Clin Gastroenterol. 2008;42:1032-9. (Ref 8.)
Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: A 16-year prospective study. Hepatology. 2001;33:544-53. (Ref 66.)
Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol. 2008;14:3948-55. (Ref 24.)
Kaya M, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary sclerosing cholangitis: An evaluation of a modified scoring system. J Hepatol. 2000;33:537-42. (Ref 20.)
Mitchell SA, Grove J, Spurkland A, et al. Association of the tumour necrosis factor alpha -308 but not the interleukin 10 -627 promoter polymorphism with genetic susceptibility to primary sclerosing cholangitis. Gut. 2001;49:288-94. (Ref 59.)
Mitchell SA, Bansi DS, Hunt N, et al. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900-7. (Ref 177.)
Moff SL, Kamel IR, Eustace J, et al. Diagnosis of primary sclerosing cholangitis: A blinded comparative study using magnetic resonance cholangiography and endoscopic retrograde cholangiography. Gastrointest Endosc. 2006;64:219-23. (Ref 13.)
Olsson R, Boberg KM, de Muckadell OS, et al. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis: A 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464-72. (Ref 181.)
Said K, Glaumann H, Bergquist A. Gallbladder disease in patients with primary sclerosing cholangitis. J Hepatol. 2008;48:598-605. (Ref 7.)
Stiehl A, Rudolph G, Kloters-Plachky P, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: Outcome after endoscopic treatment. J Hepatol. 2002;36:151-6. (Ref 9.)
Textor HJ, Flacke S, Pauleit D, et al. Three-dimensional magnetic resonance cholangiopancreatography with respiratory triggering in the diagnosis of primary sclerosing cholangitis: Comparison with endoscopic retrograde cholangiography. Endoscopy. 2002;34:984-90. (Ref 11.)
Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89-95. (Ref 159.)
1. Delbet M. Retrecissement du choladogue cholecysto-duodenostomie. Bull Mem Soc Nation Chirugie. 1924;50:1144-6.
2. Warren KW, Athanassiades S, Monge JI. Primary sclerosing cholangitis. A study of forty-two cases. Am J Surg. 1966;111:23-38.
3. Boberg KM, Schrumpf E, Fausa O, et al. Hepatobiliary disease in ulcerative colitis. An analysis of 18 patients with hepatobiliary lesions classified as small-duct primary sclerosing cholangitis. Scand J Gastroenterol. 1994;29:744-52.
4. Pokorny CS, McCaughan GW, Gallagher ND, et al. Sclerosing cholangitis and biliary tract calculi—primary or secondary? Gut. 1992;33:1376-80.
5. Kaw M, Silverman WB, Rabinovitz M, et al. Biliary tract calculi in primary sclerosing cholangitis. Am J Gastroenterol. 1995;90:72-5.
6. Rosen CB, Nagorney DM, Wiesner RH, et al. Cholangiocarcinoma complicating primary sclerosing cholangitis. Ann Surg. 1991;213:21-5.
7. Said K, Glaumann H, Bergquist A. Gallbladder disease in patients with primary sclerosing cholangitis. J Hepatol. 2008;48:598-605.
8. Gluck M, Cantone N, Brandabur J, et al. A twenty-year experience with endoscopic therapy for symptomatic primary sclerosing cholangitis. J Clin Gastroenterol. 2008;42:1032-9.
9. Stiehl A, Rudolph G, Kloters-Plachky P, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: Outcome after endoscopic treatment. J Hepatol. 2002;36:151-6.
10. Angulo P, Pearce DH, Johnson CD, et al. Magnetic resonance cholangiography in patients with biliary disease: Its role in primary sclerosing cholangitis. J Hepatol. 2000;33:520-7.
11. Textor HJ, Flacke S, Pauleit D, et al. Three-dimensional magnetic resonance cholangiopancreatography with respiratory triggering in the diagnosis of primary sclerosing cholangitis: Comparison with endoscopic retrograde cholangiography. Endoscopy. 2002;34:984-90.
12. Vitellas KM, El-Dieb A, Vaswani KK, et al. MR cholangiopancreatography in patients with primary sclerosing cholangitis: Interobserver variability and comparison with endoscopic retrograde cholangiopancreatography. Am J Roentgenol. 2002;179:399-407.
13. Moff SL, Kamel IR, Eustace J, et al. Diagnosis of primary sclerosing cholangitis: A blinded comparative study using magnetic resonance cholangiography and endoscopic retrograde cholangiography. Gastrointest Endosc. 2006;64:219-23.
14. Benhamou Y, Caumes E, Gerosa Y, et al. AIDS-related cholangiopathy. Critical analysis of a prospective series of 26 patients. Dig Dis Sci. 1993;38:1113-18.
15. Pol S, Romana CA, Richard S, et al. Microsporidia infection in patients with the human immunodeficiency virus and unexplained cholangitis. N Engl J Med. 1993;328:95-9.
16. Barnett KT, Malafa MP. Complications of hepatic artery infusion: A review of 4580 reported cases. Int J Gastrointest Cancer. 2001;30:147-60.
17. Castellano G, Moreno-Sanchez D, Gutierrez J, et al. Caustic sclerosing cholangitis. Report of four cases and a cumulative review of the literature. Hepatogastroenterology. 1994;41:458-70.
18. Wiesner RH, LaRusso NF, Ludwig J, et al. Comparison of the clinicopathologic features of primary sclerosing cholangitis and primary biliary cirrhosis. Gastroenterology. 1985;88:108-14.
19. Wilschanski M, Chait P, Wade JA, et al. Primary sclerosing cholangitis in 32 children: Clinical, laboratory, and radiographic features, with survival analysis. Hepatology. 1995;22:1415-22.
20. Kaya M, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary sclerosing cholangitis: An evaluation of a modified scoring system. J Hepatol. 2000;33:537-42.
21. Gohlke F, Lohse AW, Dienes HP, et al. Evidence for an overlap syndrome of autoimmune hepatitis and primary sclerosing cholangitis. J Hepatol. 1996;24:699-705.
22. McNair AN, Moloney M, Portmann BC, et al. Autoimmune hepatitis overlapping with primary sclerosing cholangitis in five cases. Am J Gastroenterol. 1998;93:777-84.
23. Bjornsson E, Chari ST, Smyrk TC, Lindor K. Immunoglobulin G4 associated cholangitis: Description of an emerging clinical entity based on review of the literature. Hepatology. 2007;45:1547-54.
24. Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol. 2008;14:3948-55.
25. Bjornsson E. Immunoglobulin G4-associated cholangitis. Curr Opin Gastroenterol. 2008;24:389-94.
26. Boberg KM, Aadland E, Jahnsen J, et al. Incidence and prevalence of primary biliary cirrhosis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scand J Gastroenterol. 1998;33:99-103.
27. Bambha K, Kim WR, Talwalkar J, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology. 2003;125:1364-9.
28. Chapman RW, Arborgh BA, Rhodes JM, et al. Primary sclerosing cholangitis: A review of its clinical features, cholangiography, and hepatic histology. Gut. 1980;21:870-7.
29. Wiesner RH, Grambsch PM, Dickson ER, et al. Primary sclerosing cholangitis: Natural history, prognostic factors and survival analysis. Hepatology. 1989;10:430-6.
30. Farrant JM, Hayllar KM, Wilkinson ML, et al. Natural history and prognostic variables in primary sclerosing cholangitis. Gastroenterology. 1991;100:1710-17.
31. Broome U, Olsson R, Loof L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996;38:610-15.
32. Okolicsanyi L, Fabris L, Viaggi S, et al. Primary sclerosing cholangitis: Clinical presentation, natural history and prognostic variables: An Italian multicentre study. Eur J Gastroenterol Hepatol. 1996;8:685-91.
33. Feldstein AE, Perrault J, El-Youssif M, et al. Primary sclerosing cholangitis in children: A long-term follow-up study. Hepatology. 2003;38:210-17.
34. Rabinovitz M, Gavaler JS, Schade RR, et al. Does primary sclerosing cholangitis occurring in association with inflammatory bowel disease differ from that occurring in the absence of inflammatory bowel disease? A study of sixty-six subjects. Hepatology. 1990;11:7-11.
35. Olsson R, Danielsson A, Jarnerot G, et al. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology. 1991;100:1319-23.
36. Loftus EVJr, Sandborn WJ, Tremaine WJ, et al. Primary sclerosing cholangitis is associated with nonsmoking: A case-control study. Gastroenterology. 1996;110:1496-502.
37. Van Erpecum KJ, Smits SJ, van de Meeberg PC, et al. Risk of primary sclerosing cholangitis is associated with nonsmoking behavior. Gastroenterology. 1996;110:1503-6.
38. Schrumpf E, Elgjo K, Fausa O, et al. Sclerosing cholangitis in ulcerative colitis. Scand J Gastroenterol. 1980;15:689-97.
39. Tobias R, Wright J, Kottle R, et al. Primary sclerosing cholangitis associated with inflammatory bowel disease in Cape Town, 1975-1981. S Afr Med J. 1983;63:229-35.
40. Shepherd HA, Selby WS, Chapman RW, et al. Ulcerative colitis and persistent liver dysfunction. Q J Med. 1983;52:503-13.
41. Rasmussen HH, Fallingborg JF, Mortensen PB, et al. Hepatobiliary dysfunction and primary sclerosing cholangitis in patients with Crohn’s disease. Scand J Gastroenterol. 1997;32:604-10.
42. Takikawa H, Manabe T. Primary sclerosing cholangitis in Japan—analysis of 192 cases. J Gastroenterol. 1997;32:134-7.
43. Broome U, Lofberg R, Lundqvist K, et al. Subclinical time span of inflammatory bowel disease in patients with primary sclerosing cholangitis. Dis Colon Rectum. 1995;38:1301-5.
44. Lundqvist K, Broome U. Differences in colonic disease activity in patients with ulcerative colitis with and without primary sclerosing cholangitis: A case control study. Dis Colon Rectum. 1997;40:451-6.
45. Papatheodoridis GV, Hamilton M, Mistry PK, et al. Ulcerative colitis has an aggressive course after orthotopic liver transplantation for primary sclerosing cholangitis. Gut. 1998;43:639-44.
46. Ludwig J, Barham SS, LaRusso NF, et al. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology. 1981;1:632-40.
47. MacCarty RL, LaRusso NF, Wiesner RH, et al. Primary sclerosing cholangitis: Findings on cholangiography and pancreatography. Radiology. 1983;149:39-44.
48. Quigley EM, LaRusso NF, Ludwig J, et al. Familial occurrence of primary sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1983;85:1160-65.
49. Jorge AD, Esley C, Ahumada J. Family incidence of primary sclerosing cholangitis associated with immunologic diseases. Endoscopy. 1987;19:114-17.
50. Schrumpf E, Fausa O, Forre O, et al. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol. 1982;17:187-91.
51. Chapman RW, Varghese Z, Gaul R, et al. Association of primary sclerosing cholangitis with HLA-B8. Gut. 1983;24:38-41.
52. Prochazka EJ, Terasaki PI, Park MS, et al. Association of primary sclerosing cholangitis with HLA-DRw52a. N Engl J Med. 1990;322:1842-4.
53. Farrant JM, Doherty DG, Donaldson PT, et al. Amino acid substitutions at position 38 of the DR beta polypeptide confer susceptibility to and protection from primary sclerosing cholangitis. Hepatology. 1992;16:390-5.
54. Zetterquist H, Broome U, Einarsson K, et al. HLA class II genes in primary sclerosing cholangitis and chronic inflammatory bowel disease: No HLA-DRw52a association in Swedish patients with sclerosing cholangitis. Gut. 1992;33:942-6.
55. Olerup O, Olsson R, Hultcrantz R, et al. HLA-DR and HLA-DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1995;108:870-8.
56. Donaldson PT, Norris S. Immunogenetics in PSC. Best Pract Res Clin Gastroenterol. 2001;15:611-27.
57. Norris S, Kondeatis E, Collins R, et al. Mapping MHC-encoded susceptibility and resistance in primary sclerosing cholangitis: The role of MICA polymorphism. Gastroenterology. 2001;120:1475-82.
58. Bernal W, Moloney M, Underhill J, et al. Association of tumor necrosis factor polymorphism with primary sclerosing cholangitis. J Hepatol. 1999;30:237-41.
59. Mitchell SA, Grove J, Spurkland A, et al. Association of the tumour necrosis factor alpha -308 but not the interleukin 10 -627 promoter polymorphism with genetic susceptibility to primary sclerosing cholangitis. Gut. 2001;49:288-94.
60. Mehal WZ, Lo YM, Wordsworth BP, et al. HLA DR4 is a marker for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1994;106:160-7.
61. Boberg KM, Spurkland A, Rocca G, et al. The HLA-DR3,DQ2 heterozygous genotype is associated with an accelerated progression of primary sclerosing cholangitis. Scand J Gastroenterol. 2001;36:886-90.
62. Satsangi J, Chapman RW, Haldar N, et al. A functional polymorphism of the stromelysin gene (MMP-3) influences susceptibility to primary sclerosing cholangitis. Gastroenterology. 2001;121:124-30.
63. Wiencke K, Louka AS, Spurkland A, et al. Association of matrix metalloproteinase-1 and -3 promoter polymorphisms with clinical subsets of Norwegian primary sclerosing cholangitis patients. J Hepatol. 2004;41:209-14.
64. Donaldson PT, Norris S, Constantini PK, et al. The interleukin-1 and interleukin-10 gene polymorphisms in primary sclerosing cholangitis: No associations with disease susceptibility/resistance. J Hepatol. 2000;32:882-6.
65. Saarinen S, Olerup O, Broome U. Increased frequency of autoimmune diseases in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:3195-9.
66. Gregorio GV, Portmann B, Karani J, et al. Autoimmune hepatitis/sclerosing cholangitis overlap syndrome in childhood: A 16-year prospective study. Hepatology. 2001;33:544-53.
67. Abdo AA, Bain VG, Kichian K, et al. Evolution of autoimmune hepatitis to primary sclerosing cholangitis: A sequential syndrome. Hepatology. 2002;36:1393-9.
68. Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology. 1980;79:200-6.
69. Chapman RW, Cottone M, Selby WS, et al. Serum autoantibodies, ulcerative colitis and primary sclerosing cholangitis. Gut. 1986;27:86-91.
70. Klein R, Eisenburg J, Weber P, et al. Significance and specificity of antibodies to neutrophils detected by western blotting for the serological diagnosis of primary sclerosing cholangitis. Hepatology. 1991;14:1147-52.
71. Seibold F, Weber P, Klein R, et al. Clinical significance of antibodies against neutrophils in patients with inflammatory bowel disease and primary sclerosing cholangitis. Gut. 1992;33:657-62.
72. Lo SK, Fleming KA, Chapman RW. A 2-year follow-up study of anti-neutrophil antibody in primary sclerosing cholangitis: Relationship to clinical activity, liver biochemistry and ursodeoxycholic acid treatment. J Hepatol. 1994;21:974-8.
73. Gur H, Shen G, Sutjita M, et al. Autoantibody profile of primary sclerosing cholangitis. Pathobiology. 1995;63:76-82.
74. Roozendaal C, Van Milligen de Wit AW, Haagsma EB, et al. Antineutrophil cytoplasmic antibodies in primary sclerosing cholangitis: Defined specificities may be associated with distinct clinical features. Am J Med. 1998;105:393-9.
75. Angulo P, Peter JB, Gershwin ME, et al. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol. 2000;32:182-7.
76. Mandal A, Dasgupta A, Jeffers L, et al. Autoantibodies in sclerosing cholangitis against a shared peptide in biliary and colon epithelium. Gastroenterology. 1994;106:185-92.
77. Xu B, Broome U, Ericzon BG, et al. High frequency of autoantibodies in patients with primary sclerosing cholangitis that bind biliary epithelial cells and induce expression of CD44 and production of interleukin 6. Gut. 2002;51:120-7.
78. Bodenheimer HCJr, LaRusso NF, Thayer WRJr, et al. Elevated circulating immune complexes in primary sclerosing cholangitis. Hepatology. 1983;3:150-4.
79. Minuk GY, Angus M, Brickman CM, et al. Abnormal clearance of immune complexes from the circulation of patients with primary sclerosing cholangitis. Gastroenterology. 1985;88:166-70.
80. Senaldi G, Donaldson PT, Magrin S, et al. Activation of the complement system in primary sclerosing cholangitis. Gastroenterology. 1989;97:1430-4.
81. Bansal AS, Thomson A, Steadman C, et al. Serum levels of interleukins 8 and 10, interferon gamma, granulocyte-macrophage colony stimulating factor and soluble CD23 in patients with primary sclerosing cholangitis. Autoimmunity. 1997;26:223-9.
82. Lindor KD, Wiesner RH, Katzmann JA, et al. Lymphocyte subsets in primary sclerosing cholangitis. Dig Dis Sci. 1987;32:720-5.
83. Martins EB, Graham AK, Chapman RW, et al. Elevation of gamma delta T lymphocytes in peripheral blood and livers of patients with primary sclerosing cholangitis and other autoimmune liver diseases. Hepatology. 1996;23:988-93.
84. Broome U, Grunewald J, Scheynius A, et al. Preferential V beta3 usage by hepatic T lymphocytes in patients with primary sclerosing cholangitis. J Hepatol. 1997;26:527-34.
85. Chapman RW, Kelly PM, Heryet A, et al. Expression of HLA-DR antigens on bile duct epithelium in primary sclerosing cholangitis. Gut. 1988;29:422-7.
86. Adams DH, Hubscher SG, Shaw J, et al. Increased expression of intercellular adhesion molecule 1 on bile ducts in primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology. 1991;14:426-31.
87. Himeno H, Saibara T, Onishi S, et al. Administration of interleukin-2 induces major histocompatibility complex class II expression on the biliary epithelial cells, possibly through endogenous interferon-gamma production. Hepatology. 1992;16:409-17.
88. Leon MP, Bassendine MF, Wilson JL, et al. Immunogenicity of biliary epithelium: Investigation of antigen presentation to CD4+ T cells. Hepatology. 1996;24:561-7.
89. Olsson R, Bjornsson E, Backman L, et al. Bile duct bacterial isolates in primary sclerosing cholangitis: A study of explanted livers. J Hepatol. 1998;28:426-32.
90. Sasatomi K, Noguchi K, Sakisaka S, et al. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J Hepatol. 1998;29:409-16.
91. Ponsioen CY, Defoer J, Ten Kate FJ, et al. A survey of infectious agents as risk factors for primary sclerosing cholangitis: Are Chlamydia species involved? Eur J Gastroenterol Hepatol. 2002;14:641-8.
92. Tjandra K, Sharkey KA, Swain MG. Progressive development of a Th1-type hepatic cytokine profile in rats with experimental cholangitis. Hepatology. 2000;31:280-90.
93. Terblanche J, Allison HF, Northover JM. An ischemic basis for biliary strictures. Surgery. 1983;94:52-7.
94. Guichelaar MM, Benson JT, Malinchoc M, et al. Risk factors for and clinical course of non-anastomotic biliary strictures after liver transplantation. Am J Transplant. 2003;3:885-90.
95. Sebagh M, Farges O, Kalil A, et al. Sclerosing cholangitis following human orthotopic liver transplantation. Am J Surg Pathol. 1995;19:81-90.
96. Lebovics E, Palmer M, Woo J, et al. Outcome of primary sclerosing cholangitis. Analysis of long-term observation of 38 patients. Arch Intern Med. 1987;147:729-31.
97. Ponsioen CY, Vrouenraets SM, Prawirodirdjo W, et al. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a Dutch population. Gut. 2002;51:562-6.
98. Helzberg JH, Petersen JM, Boyer JL. Improved survival with primary sclerosing cholangitis. A review of clinicopathologic features and comparison of symptomatic and asymptomatic patients. Gastroenterology. 1987;92:1869-75.
99. Porayko MK, Wiesner RH, LaRusso NF, et al. Patients with asymptomatic primary sclerosing cholangitis frequently have progressive disease. Gastroenterology. 1990;98:1594-602.
100. Angulo P, Maor-Kendler Y, Lindor KD. Small-duct primary sclerosing cholangitis: A long-term follow-up study. Hepatology. 2002;35:1494-500.
101. Bjornsson E, Boberg KM, Cullen S, et al. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002;51:731-5.
102. Broome U, Glaumann H, Lindstom E, et al. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC). J Hepatol. 2002;36:586-9.
103. Dickson ER, Murtaugh PA, Wiesner RH, et al. Primary sclerosing cholangitis: Refinement and validation of survival models. Gastroenterology. 1992;103:1893-901.
104. Kim WR, Therneau TM, Wiesner RH, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc. 2000;75:688-94.
105. Shetty K, Rybicki L, Carey WD. The Child-Pugh classification as a prognostic indicator for survival in primary sclerosing cholangitis. Hepatology. 1997;25:1049-53.
106. Kim WR, Poterucha JJ, Wiesner RH, et al. The relative role of the Child-Pugh classification and the Mayo natural history model in the assessment of survival in patients with primary sclerosing cholangitis. Hepatology. 1999;29:1643-8.
107. Stockbrugger RW, Olsson R, Jaup B, et al. Forty-six patients with primary sclerosing cholangitis: Radiological bile duct changes in relationship to clinical course and concomitant inflammatory bowel disease. Hepatogastroenterology. 1988;35:289-94.
108. Aadland E, Schrumpf E, Fausa O, et al. Primary sclerosing cholangitis: A long-term follow-up study. Scand J Gastroenterol. 1987;22:655-64.
109. Grijm R, Huibregtse K, Bartelsman J, et al. Therapeutic investigations in primary sclerosing cholangitis. Dig Dis Sci. 1986;31:792-8.
110. Balasubramaniam K, Wiesner RH, LaRusso NF. Primary sclerosing cholangitis with normal serum alkaline phosphatase activity. Gastroenterology. 1988;95:1395-8.
111. Cooper JF, Brand EJ. Symptomatic sclerosing cholangitis in patients with a normal alkaline phosphatase: Two case reports and a review of the literature. Am J Gastroenterol. 1988;83:308-11.
112. El-Shabrawi M, Wilkinson ML, Portmann B, et al. Primary sclerosing cholangitis in childhood. Gastroenterology. 1987;92:1226-35.
113. Terjung B, Spengler U, Sauerbruch T, et al. “Atypical p-ANCA” in IBD and hepatobiliary disorders react with a 50-kilodalton nuclear envelope protein of neutrophils and myeloid cell lines. Gastroenterology. 2000;119:310-22.
114. Cameron JL, Gayler BW, Sanfey H, et al. Sclerosing cholangitis. Anatomical distribution of obstructive lesions. Ann Surg. 1984;200:54-60.
115. Brandt DJ, MacCarty RL, Charboneau JW, et al. Gallbladder disease in patients with primary sclerosing cholangitis. AJR Am J Roentgenol. 1988;150:571-4.
116. Schimanski U, Stiehl A, Stremmel W, et al. Low prevalence of alterations in the pancreatic duct system in patients with primary sclerosing cholangitis. Endoscopy. 1996;28:346-9.
117. Lefkowitch J. Primary sclerosing cholangitis. Arch Intern Med. 1982;142:1157-60.
118. Ludwig J. Surgical pathology of the syndrome of primary sclerosing cholangitis. Am J Surg Pathol. 1989;13:43-9.
119. Katabi N, Albores-Saavedra J. The extrahepatic bile duct lesions in end-stage primary sclerosing cholangitis. Am J Surg Pathol. 2003;27:349-55.
120. Harrison RF, Hubscher SG. The spectrum of bile duct lesions in end-stage primary sclerosing cholangitis. Histopathology. 1991;19:321-7.
121. Ludwig J, Czaja AJ, Dickson ER, et al. Manifestations of nonsuppurative cholangitis in chronic hepatobiliary diseases: Morphologic spectrum, clinical correlations and terminology. Liver. 1984;4:105-16.
122. Barbatis C, Grases P, Shepherd HA, et al. Histological features of sclerosing cholangitis in patients with chronic ulcerative colitis. J Clin Pathol. 1985;38:778-83.
123. Angulo P, Larson DR, Therneau TM, et al. Time course of histological progression in primary sclerosing cholangitis. Am J Gastroenterol. 1999;94:3310-13.
124. Gross JBJr, Ludwig J, Wiesner RH, et al. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology. 1985;89:272-8.
125. Swain MG, Rothman RB, Xu H, et al. Endogenous opioids accumulate in plasma in a rat model of acute cholestasis. Gastroenterology. 1992;103:630-5.
126. Bergasa NV, Alling DW, Talbot TL, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double-blind, randomized, controlled trial. Ann Intern Med. 1995;123:161-7.
127. Wolfhagen FH, Sternieri E, Hop WC, et al. Oral naltrexone treatment for cholestatic pruritus: A double-blind, placebo-controlled study. Gastroenterology. 1997;113:1264-9.
128. Jorgensen RA, Lindor KD, Sartin JS, et al. Serum lipid and fat-soluble vitamin levels in primary sclerosing cholangitis. J Clin Gastroenterol. 1995;20:215-19.
129. Angulo P, Therneau TM, Jorgensen A, et al. Bone disease in patients with primary sclerosing cholangitis: Prevalence, severity and prediction of progression. J Hepatol. 1998;29:729-35.
130. Chalasani N, Baluyut A, Ismail A, et al. Cholangiocarcinoma in patients with primary sclerosing cholangitis: A multicenter case-control study. Hepatology. 2000;31:7-11.
131. Burak K, Angulo P, Pasha TM, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:523-6.
132. Schwartz DA. Cholangiocarcinoma associated with liver fluke infection: A preventable source of morbidity in Southeast Asian immigrants. Am J Gastroenterol. 1986;81:76-9.
133. Nichols JC, Gores GJ, LaRusso NF, et al. Diagnostic role of serum CA 19-9 for cholangiocarcinoma in patients with primary sclerosing cholangitis. Mayo Clin Proc. 1993;68:874-9.
134. Patel AH, Harnois DM, Klee GG, et al. The utility of CA 19-9 in the diagnoses of cholangiocarcinoma in patients without primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:204-7.
135. Ramage JK, Donaghy A, Farrant JM, et al. Serum tumor markers for the diagnosis of cholangiocarcinoma in primary sclerosing cholangitis. Gastroenterology. 1995;108:865-9.
136. Bjornsson E, Kilander A, Olsson R. CA 19-9 and CEA are unreliable markers for cholangiocarcinoma in patients with primary sclerosing cholangitis. Liver. 1999;19:501-8.
137. Hultcrantz R, Olsson R, Danielsson A, et al. A 3-year prospective study on serum tumor markers used for detecting cholangiocarcinoma in patients with primary sclerosing cholangitis. J Hepatol. 1999;30:669-73.
138. Ponchon T, Gagnon P, Berger F, et al. Value of endobiliary brush cytology and biopsies for the diagnosis of malignant bile duct stenosis: Results of a prospective study. Gastrointest Endosc. 1995;42:565-72.
139. Sugiyama M, Atomi Y, Wada N, et al. Endoscopic transpapillary bile duct biopsy without sphincterotomy for diagnosing biliary strictures: A prospective comparative study with bile and brush cytology. Am J Gastroenterol. 1996;91:465-7.
140. Jaiwala J, Fogel E, Sherman S, et al. Triple-tissue sampling at ERCP in malignant biliary obstruction. Gastrointest Endosc. 2000;51:583-90.
141. Eloubeidi MA, Chen VK, Jhala NC, et al. Endoscopic ultrasound-guided fine needle aspiration biopsy of suspected cholangiocarcinoma. Clin Gastroenterol Hepatol. 2004;2:209-13.
142. Fritscher-Ravens A, Broering DC, Knoefel WT, et al. EUS-guided fine-needle aspiration of suspected hilar cholangiocarcinoma in potentially operable patients with negative brush cytology. Am J Gastroenterol. 2004;99:45-51.
143. Tischendorf JJ, Meier PN, Schneider A, et al. Transpapillary intraductal ultrasound in the evaluation of dominant bile duct stenoses in patients with primary sclerosing cholangitis. Scand J Gastroenterol. 2007;42:1011-17.
144. Stavropoulos S, Larghi A, Verna E, et al. Intraductal ultrasound for the evaluation of patients with biliary strictures and no abdominal mass on computed tomography. Endoscopy. 2005;37:715-21.
145. Chak A, Canto M, Stevens P, et al. Clinical applications of a new through-the-scope ultrasound probe: Prospective comparison with an ultrasound endoscope. Gastrointest Endosc. 1997;45:291-5.
146. Tischendorf JJ, Kruger M, Trautwein C, et al. Cholangioscopic characterization of dominant bile duct stenoses in patients with primary sclerosing cholangitis. Endoscopy. 2006;38:665-9.
147. Bergquist A, Glaumann H, Persson B, et al. Risk factors and clinical presentation of hepatobiliary carcinoma in patients with primary sclerosing cholangitis: A case-control study. Hepatology. 1998;27:311-16.
148. Boberg KM, Bergquist A, Mitchell S, et al. Cholangiocarcinoma in primary sclerosing cholangitis: Risk factors and clinical presentation. Scand J Gastroenterol. 2002;37:1205-11.
149. Gyde SN, Prior P, Allan RN, et al. Colorectal cancer in ulcerative colitis: A cohort study of primary referrals from three centres. Gut. 1988;29:206-17.
150. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228-33.
151. Lennard-Jones JE, Melville DM, Morson BC, et al. Precancer and cancer in extensive ulcerative colitis: Findings among 401 patients over 22 years. Gut. 1990;31:800-6.
152. Broome U, Lofberg R, Veress B, et al. Primary sclerosing cholangitis and ulcerative colitis: Evidence for increased neoplastic potential. Hepatology. 1995;22:1404-8.
153. Brentnall TA, Haggitt RC, Rabinovitch PS, et al. Risk and natural history of colonic neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis. Gastroenterology. 1996;110:331-8.
154. Marchesa P, Lashner BA, Lavery IC, et al. The risk of cancer and dysplasia among ulcerative colitis patients with primary sclerosing cholangitis. Am J Gastroenterol. 1997;92:1285-8.
155. Shetty K, Rybicki L, Brzezinski A, et al. The risk for cancer or dysplasia in ulcerative colitis patients with primary sclerosing cholangitis. Am J Gastroenterol. 1999;94:1643-9.
156. Lindberg BU, Broome U, Persson B. Proximal colorectal dysplasia or cancer in ulcerative colitis. The impact of primary sclerosing cholangitis and sulfasalazine: Results from a 20-year surveillance study. Dis Colon Rectum. 2001;44:77-85.
157. Soetikno RM, Lin OS, Heidenreich PA, et al. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: A meta-analysis. Gastrointest Endosc. 2002;56:48-54.
158. Hill MJ, Melville DM, Lennard-Jones JE, et al. Faecal bile acids, dysplasia, and carcinoma in ulcerative colitis. Lancet. 1987;2:185-6.
159. Tung BY, Emond MJ, Haggitt RC, et al. Ursodiol use is associated with lower prevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001;134:89-95.
160. Pardi DS, Loftus EVJr, Kremers WK, et al. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology. 2003;124:889-93.
161. Wali RK, Frawley BPJr, Hartmann S, et al. Mechanism of action of chemoprotective ursodeoxycholate in the azoxymethane model of rat colonic carcinogenesis: Potential roles of protein kinase C-alpha, -beta II, and -zeta. Cancer Res. 1995;55:5257-64.
162. Ikegami T, Matsuzaki Y, Shoda J, et al. The chemopreventive role of ursodeoxycholic acid in azoxymethane-treated rats: Suppressive effects on enhanced group II phospholipase A2 expression in colonic tissue. Cancer Lett. 1998;134:129-39.
163. Rubin CE, Haggitt RC, Burmer GC, et al. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology. 1992;103:1611-20.
164. Bleday R, Lee E, Jessurun J, et al. Increased risk of early colorectal neoplasms after hepatic transplant in patients with inflammatory bowel disease. Dis Colon Rectum. 1993;36:908-12.
165. Loftus EVJr, Aguilar HI, Sandborn WJ, et al. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology. 1998;27:685-90.
166. Vera A, Gunson BK, Ussatoff V, et al. Colorectal cancer in patients with inflammatory bowel disease after liver transplantation for primary sclerosing cholangitis. Transplantation. 2003;75:1983-8.
167. Peck JJ, Boyden AM. Exigent ileostomy hemorrhage. A complication of proctocolectomy in patients with chronic ulcerative colitis and primary sclerosing cholangitis. Am J Surg. 1985;150:153-8.
168. Wiesner RH, LaRusso NF, Dozois RR, et al. Peristomal varices after proctocolectomy in patients with primary sclerosing cholangitis. Gastroenterology. 1986;90:316-22.
169. Morgan TR, Feldshon SD, Tripp MR. Recurrent stomal variceal bleeding. Successful treatment using injection sclerotherapy. Dis Colon Rectum. 1986;29:269-70.
170. Samaraweera RN, Feldman L, Widrich WC, et al. Stomal varices: Percutaneous transhepatic embolization. Radiology. 1989;170:779-82.
171. Beck DE, Fazio VW, Grundfest-Broniatowski S. Surgical management of bleeding stomal varices. Dis Colon Rectum. 1988;31:343-6.
172. Shibata D, Brophy DP, Gordon FD, et al. Transjugular intrahepatic portosystemic shunt for treatment of bleeding ectopic varices with portal hypertension. Dis Colon Rectum. 1999;42:1581-5.
173. Beuers U, Spengler U, Kruis W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: A placebo-controlled trial. Hepatology. 1992;16:707-14.
174. Stiehl A, Walker S, Stiehl L, et al. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3-year pilot study with a placebo-controlled study period. J Hepatol. 1994;20:57-64.
175. De Maria N, Colantoni A, Rosenbloom E, et al. Ursodeoxycholic acid does not improve the clinical course of primary sclerosing cholangitis over a 2-year period. Hepatogastroenterology. 1996;43:1472-9.
176. Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med. 1997;336:691-5.
177. Mitchell SA, Bansi DS, Hunt N, et al. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900-7.
178. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525-31.
179. Mitsuyoshi H, Nakashima T, Sumida Y, et al. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem Biophys Res Commun. 1999;263:537-42.
180. Harnois DM, Angulo P, Jorgensen RA, et al. High-dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol. 2001;96:1558-62.
181. Olsson R, Boberg KM, de Muckadell OS, et al. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis: A 5-year multicenter, randomized, controlled study. Gastroenterology. 2005;129:1464-72.
182. Cullen SN, Rust C, Fleming K, et al. High dose ursodeoxycholic aid for the treatment of primary sclerosing cholangitis is safe and effective. J Hepatol. 2008;48:792.
183. Allison MC, Burroughs AK, Noone P, et al. Biliary lavage with corticosteroids in primary sclerosing cholangitis. A clinical, cholangiographic and bacteriological study. J Hepatol. 1986;3:118-22.
184. Lindor KD, Wiesner RH, Colwell LJ, et al. The combination of prednisone and colchicine in patients with primary sclerosing cholangitis. Am J Gastroenterol. 1991;86:57-61.
185. Boberg KM, Egeland T, Schrumpf E. Long-term effect of corticosteroid treatment in primary sclerosing cholangitis patients. Scand J Gastroenterol. 2003;38:991-5.
186. Angulo P, Batts KP, Jorgensen RA, et al. Oral budesonide in the treatment of primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:2333-7.
187. Knox TA, Kaplan MM. A double-blind controlled trial of oral-pulse methotrexate therapy in the treatment of primary sclerosing cholangitis. Gastroenterology. 1994;106:494-9.
188. Van Thiel DH, Carroll P, Abu-Elmagd K, et al. Tacrolimus (FK 506), a treatment for primary sclerosing cholangitis: Results of an open-label preliminary trial. Am J Gastroenterol. 1995;90:455-9.
189. Bharucha AE, Jorgensen R, Lichtman SN, et al. A pilot study of pentoxifylline for the treatment of primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:2338-42.
190. Epstein MP, Kaplan MM. A pilot study of etanercept in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2004;49:1-4.
191. Olsson R, Broome U, Danielsson A, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology. 1995;108:1199-203.
192. LaRusso NF, Wiesner RH, Ludwig J, et al. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology. 1988;95:1036-42.
193. Angulo P, Bharucha AE, Jorgensen RA, et al. Oral nicotine in treatment of primary sclerosing cholangitis: A pilot study. Dig Dis Sci. 1999;44:602-7.
194. Vleggaar FP, van Buuren HR, van Berge Henegouwen GP, et al. No beneficial effects of transdermal nicotine in patients with primary sclerosing cholangitis: Results of a randomized double-blind placebo-controlled cross-over study. Eur J Gastroenterol Hepatol. 2001;13:171-5.
195. Angulo P, MacCarty RL, Sylvestre PB, et al. Pirfenidone in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2002;47:157-61.
196. Schramm C, Schirmacher P, Helmich-Becker I, et al. Combined therapy with azathioprine, prednisolone and ursodiol in patients with primary sclerosing cholangitis. Ann Int Med. 1999;131:943-6.
197. van Hiigstraten HJ, Vleggar FP, Boland GJ, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: A randomized double-blind pilot study. Am J Gastroenterol. 2000;95:2015-22.
198. Farkkila MA, Karkkaninen P, Karvonen AL, et al. Combination of ursodeoxycholic acid and metronidazole in PSC: Reduction of inflammation in liver correlates with decrease of the new Mayo risk score. [abstract]. Gastroenterol. 2003;S124:A707.
199. Ghent CN, Carruthers SG. Treatment of pruritus in primary biliary cirrhosis with rifampin. Results of a double-blind, crossover, randomized trial. Gastroenterology. 1988;94:488-93.
200. Podesta A, Lopez P, Terg R, et al. Treatment of pruritus of primary biliary cirrhosis with rifampin. Dig Dis Sci. 1991;36:216-20.
201. Terg R, Coronel E, Sorda J, et al. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37:717-22.
202. Herlong HF, Recker RR, Maddrey WC. Bone disease in primary biliary cirrhosis: Histologic features and response to 25-hydroxyvitamin D. Gastroenterology. 1982;83:103-8.
203. Lee JG, Schutz SM, England RE, et al. Endoscopic therapy of sclerosing cholangitis. Hepatology. 1995;21:661-7.
204. Smith MT, Sherman S, Lehman GA. Endoscopic management of benign strictures of the biliary tree. Endoscopy. 1995;27:253-66.
205. Van Milligen de Wit AW, van Bracht J, Rauws EA, et al. Endoscopic stent therapy for dominant extrahepatic bile duct strictures in primary sclerosing cholangitis. Gastrointest Endosc. 1996;44:293-9.
206. Wagner S, Gebel M, Meier P, et al. Endoscopic management of biliary tract strictures in primary sclerosing cholangitis. Endoscopy. 1996;28:546-51.
207. Ponsioen CY, Lam K, van Milligen de Wit AW, et al. Four years experience with short term stenting in primary sclerosing cholangitis. Am J Gastroenterol. 1999;94:2403-7.
208. Kaya M, Petersen BT, Angulo P, et al. Balloon dilation compared to stenting of dominant strictures in primary sclerosing cholangitis. Am J Gastroenterol. 2001;96:1059-66.
209. Baluyut AR, Sherman S, Lehman GA, et al. Impact of endoscopic therapy on the survival of patients with primary sclerosing cholangitis. Gastrointest Endosc. 2001;53:308-12.
210. Stiehl A, Rudolph G, Kloters-Plachky P, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: Outcome after endoscopic treatment. J Hepatol. 2002;36:151-6.
211. Van den Hazel SJ, Wolfhagen EH, van Buuren HR, et al. Prospective risk assessment of endoscopic retrograde cholangiography in patients with primary sclerosing cholangitis. Dutch PSC Study Group. Endoscopy. 2000;32:779-82.
212. Martin FM, Rossi RL, Nugent FW, et al. Surgical aspects of sclerosing cholangitis. Results in 178 patients. Ann Surg. 1990;212:551-6.
213. Myburgh JA. Surgical biliary drainage in primary sclerosing cholangitis. The role of the Hepp-Couinaud approach. Arch Surg. 1994;129:1057-62.
214. Cameron JL, Pitt HA, Zinner MJ, et al. Resection of hepatic duct bifurcation and transhepatic stenting for sclerosing cholangitis. Ann Surg. 1988;207:614-22.
215. Gross CR, Malinchoc M, Kim WR, et al. Quality of life before and after liver transplantation for cholestatic liver disease. Hepatology. 1999;29:356-64.
216. Saldeen K, Friman S, Olausson M, et al. Follow-up after liver transplantation for primary sclerosing cholangitis: Effects on survival, quality of life, and colitis. Scand J Gastroenterol. 1999;34:535-40.
217. Welsh FK, Wigmore SJ. Roux-en-Y choledochojejunostomy is the method of choice for biliary reconstruction in liver transplantation for primary sclerosing cholangitis. Transplantation. 2004;77:602-4.
218. Graziadei IW, Wiesner RH, Marotta PJ, et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121-7.
219. Narumi S, Roberts JP, Emond JC, et al. Liver transplantation for sclerosing cholangitis. Hepatology. 1995;22:451-7.
220. Goss JA, Shackleton CR, Farmer DG, et al. Orthotopic liver transplantation for primary sclerosing cholangitis. A 12-year single center experience. Ann Surg. 1997;225:472-81.
221. Roberts MS, Angus DC, Bryce CL, et al. Survival after liver transplantation in the United States: A disease-specific analysis of the UNOS database. Liver Transpl. 2004;10:886-97.
222. Ricci P, Therneau TM, Malinchoc M, et al. A prognostic model for the outcome of liver transplantation in patients with cholestatic liver disease. Hepatology. 1997;25:672-7.
223. Nashan B, Schlitt HJ, Tusch G, et al. Biliary malignancies in primary sclerosing cholangitis: Timing for liver transplantation. Hepatology. 1996;23:1105-11.
224. Robles R, Figueras J, Turrion VS, et al. Spanish experience in liver transplantation for hilar and peripheral cholangiocarcinoma. Ann Surg. 2004;239:265-71.
225. Brandsaeter B, Isoniemi H, Broome U, et al. Liver transplantation for primary sclerosing cholangitis: Predictors and consequences of hepatobiliary malignancy. J Hepatol. 2004;40:815-22.
226. De Vreede I, Steers JL, Burch PA, et al. Prolonged disease-free survival after orthotopic liver transplantation plus adjuvant chemo-irradiation for cholangiocarcinoma. Liver Transpl. 2000;6:309-16.
227. Sudan D, DeRoover A, Chinnakotla S, et al. Radiochemotherapy and transplantation allow long-term survival for nonresectable hilar cholangiocarcinoma. Am J Transplant. 2002;2:774-9.
228. Graziadei IW. Recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2002;8:575-81.
229. Kubota T, Thomson A, Clouston AD, et al. Clinicopathologic findings of recurrent primary sclerosing cholangitis after orthotopic liver transplantation. J Hepatobiliary Pancreat Surg. 1999;6:377-81.
230. Graziadei IW, Wiesner RH, Batts KP, et al. Recurrence of primary sclerosing cholangitis following liver transplantation. Hepatology. 1999;29:1050-6.
231. Kugelmas M, Spiegelman P, Osgood MJ, et al. Different immunosuppressive regimens and recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2003;9:727-32.
232. Gavaler JS, Delemos B, Belle SH, et al. Ulcerative colitis disease activity as subjectively assessed by patient-completed questionnaires following orthotopic liver transplantation for sclerosing cholangitis. Dig Dis Sci. 1991;36:321-8.
233. MacLean AR, Lilly L, Cohen Z, et al. Outcome of patients undergoing liver transplantation for primary sclerosing cholangitis. Dis Colon Rectum. 2003;46:1124-8.
234. Dvorchik I, Subotin M, Demetris AJ, et al. Effect of liver transplantation on inflammatory bowel disease in patients with primary sclerosing cholangitis. Hepatology. 2002;35:380-4.
235. Cook J, Hou PC, Ho HC, et al. Recurrent pyogenic cholangeitis. Br J Surg. 1954;42:188-203.
236. Mage S, Morel AS. Surgical experience with cholangiohepatitis (Hong Kong disease) in Canton Chinese. Ann Surg. 1965;162:187-90.
237. Digby K. Common duct stones of liver origin. Br J Surg. 1930;17:578.
238. Nagase M, Hikase Y, Soloway RD, et al. Gallstones in Western Japan. Factors affecting the prevalence of intrahepatic stones. Gastroenterology. 1980;78:684-90.
239. Kim MH, Sekijima J, Lee SP. Primary intrahepatic stones. Am J Gastroenterol. 1995;90:540-8.
240. Nakayama F, Soloway RD, Nakama T, et al. Hepatolithiasis in East Asia. Retrospective study. Dig Dis Sci. 1986;31:21-6.
241. Lo CM, Fan ST, Wong J. The changing epidemiology of recurrent pyogenic cholangitis. Hong Kong Med J. 1997;3:302-4.
242. Harris HW, Kumwenda ZL, Sheen-Chen SM, et al. Recurrent pyogenic cholangitis. Am J Surg. 1998;176:34-7.
243. Cosenza CA, Durazo F, Stain SC, et al. Current management of recurrent pyogenic cholangitis. Am Surgeon. 1999;65:939-43.
244. Matsushiro T, Suzuki N, Sato T, et al. Effects of diet on glucaric acid concentration in bile and the formation of calcium bilirubinate gallstones. Gastroenterology. 1977;72:630-3.
245. Ashkin JR, Lyon DT, Shull SD, et al. Factors affecting delivery of bile to the duodenum in man. Gastroenterology. 1978;74:560-5.
246. Liu LX, Harinasuta KT. Liver and intestinal flukes. Gastroenterol Clin North Am. 1996;25:627-36.
247. Rim HJ. Clonorchiasis: An update. J Helminthol. 2005;79:269-81.
248. Khuroo MS, Zargar SA. Biliary ascariasis. A common cause of biliary and pancreatic disease in an endemic area. Gastroenterology. 1985;88:418-23.
249. Rana SS, Bhasin DK, Nanda M, Singh K. Parasitic infestations of the biliary tract. Curr Gastroenterol Rep. 2007;9:156-64.
250. Lim JH. Oriental cholangiohepatitis: Pathologic, clinical and radiologic features. Am J Roentgenol. 1991;157:1-8.
251. Chou ST, Chan CW. Recurrent pyogenic cholangitis: A necropsy study. Pathology. 1980;12:415-28.
252. Yellin AE, Donovan AJ. Biliary lithiasis and helminthiasis. Am J Surg. 1981;142:128-36.
253. Carmona RH, Crass RA, Lim RCJr, et al. Oriental cholangitis. Am J Surg. 1984;148:117-24.
254. Nakayama F, Koga A. Hepatolithiasis: Present status. World J Surg. 1984;8:9-14.
255. Ostrow JD. The etiology of pigment gallstones. Hepatology. 1984;4:215S-22S.
256. Spivak W, Divenuto D, Yuey W. Non-enzymatic hydrolysis of bilirubin mono- and diglucuronide to unconjugated bilirubin in model and native bile systems. Potential role in the formation of gallstones. Biochem J. 1987;242:323-9.
257. Khuroo MS, Zargar SA, Yattoo GN, et al. Oddi’s sphincter motor activity in patients with recurrent pyogenic cholangitis. Hepatology. 1993;17:53-8.
258. Sperling RM, Koch J, Sandhu JS, et al. Recurrent pyogenic cholangitis in Asian immigrants to the United States: Natural history and role of therapeutic ERCP. Dig Dis Sci. 1997;42:865-71.
259. Lim JH, Ko YT, Lee DH, et al. Oriental cholangiohepatitis: Sonographic findings in 48 cases. Am J Roentgenol. 1990;155:511-14.
260. Reynolds WR, Brinkman JD, Haney BD, et al. Oriental cholangiohepatitis. Mil Med. 1994;159:158-60.
261. Fan ST, Choi TK, Wong J. Recurrent pyogenic cholangitis: Current management. World J Surg. 1991;15:248-53.
262. Kim JH, Kim TK, Eun HW, et al. CT findings of cholangiocarcinoma associated with recurrent pyogenic cholangitis. Am J Roentgenol. 2006;187:1571-7.
263. Choi TK, Wong J. Endoscopic retrograde cholangiopancreatography and endoscopic papillotomy in recurrent pyogenic cholangitis. Clin Gastroenterol. 1986;15:393-415.
264. Park MS, Yu JS, Kim KW, et al. Recurrent pyogenic cholangitis: Comparison between MR cholangiography and direct cholangiography. Radiology. 2001;220:677-82.
265. Jain M, Agarwal A. MRCP findings in recurrent pyogenic cholangitis. Eur J Radiol. 2008;66:79-83.
266. Kusano T, Isa T, Muto Y, et al. Long-term results of hepaticojejunostomy for hepatolithiasis. Am Surg. 2001;67:442-6.
267. Tang CN, Siu WT, Ha JP, et al. Laparoscopic biliary bypass—a single centre experience. Hepatogastroenterology. 2007;54:503-7.
268. Al-Sukhni W, Gallinger S, Pratzer A, et al. Recurrent pyogenic cholangitis with hepatolithiasis—the role of surgical therapy in North America. J Gastrointest Surg. 2008;12:496-503.
269. Pungpak S, Radomyos P, Radomyos BE, et al. Treatment of Opisthorchis viverrini and intestinal fluke infections with Praziquantel. Southeast Asian J Trop Med Public Health. 1998;29:246-9.
270. Hwang JH, Yoon YB, Kim YT, et al. Risk factors for recurrent cholangitis after initial hepatolithiasis treatment. J Clin Gastroenterol. 2004;38:364-7.
271. Jan YY, Chen MF, Wang CS, et al. Surgical treatment of hepatolithiasis: Long-term results. Surgery. 1996;120:509-14.
272. Kubo S, Kinoshita H, Hirohashi K, et al. Hepatolithiasis associated with cholangiocarcinoma. World J Surg. 1995;19:637-41.
273. Cheng YF, Lee TY, Sheen-Chen SM, et al. Treatment of complicated hepatolithiasis with intrahepatic biliary stricture by ductal dilatation and stenting: Long-term results. World J Surgery. 2000;24:712-16.