CHAPTER 8 Sciatica and Nerve Root Pain in Disc Herniation and Spinal Stenosis
A Basic Science Review and Clinical Perspective
The clinical symptoms seen in association with lumbar disc herniation and spinal stenosis1,2 are based on pathophysiologic involvement of spinal nerve roots. There has been an increasing interest in this topic during the past decade, and more recent research has been aimed at defining basic pathophysiologic events at the tissue, cellular, or subcellular level that are involved in the generation of sciatica and nerve root pain. This chapter reviews the current knowledge about these mechanisms and discusses these mechanisms in relation to the clinical features of lumbar disc herniation and spinal stenosis.
Pathophysiologic Mechanisms in Relation to Clinical Symptoms
The symptoms of nerve root pathophysiology may be divided into two main categories: pain and nerve dysfunction.2 Nerve root pain is typically radiating in nature and is usually related to a specific nerve root or roots. Nerve dysfunction may be present in motor and sensory modalities, producing motor weakness and sensory disturbances. One may assume that pain and nerve dysfunction are due to different pathophysiologic events, but they are tightly linked through mechanisms that are discussed in this chapter.
Two specific mechanisms at the “tissue level” may be defined: (1) mechanical deformation of the nerve roots and (2) biologic or biochemical activity of the disc tissue with effects on the roots. The mechanical deformation theory is the oldest concept of nerve root injury induced by herniated disc tissue and dates back to the turn of the 20th century with clinical observations on injuries in the lumbosacral junction with subsequent leg pain and includes the more recent seminal observations of Mixter and Barr.1–5 The theory that biologic activity of the disc tissue may injure the nerve roots was demonstrated experimentally in 1993.6 The experimental knowledge regarding these two mechanisms is discussed separately.
Mechanical Effects on Nerve Roots
Enclosed by the vertebral bones, the spinal nerve roots are relatively well protected from external trauma. The nerve roots do not possess the same amounts and organization of protective connective tissue sheaths as do the peripheral nerves, however. The spinal nerve roots may be particularly sensitive to mechanical deformation secondary to intraspinal disorders, such as disc herniations and protrusions, spinal stenosis, degenerative disorders, and tumors.7–9 There has been moderate research interest in the past regarding nerve root compression. Gelfan and Tarlov10 in 1956 and Sharpless11 in 1975 performed some initial experiments on the effects of compression on nerve impulse conduction. Although no calibration was performed on the compression devices used, the results of both studies indicated that nerve roots were more susceptible to compression than peripheral nerves. Interest in nerve root pathophysiology has increased considerably more recently, and numerous studies are reviewed here.
Experimental Compression of Nerve Roots
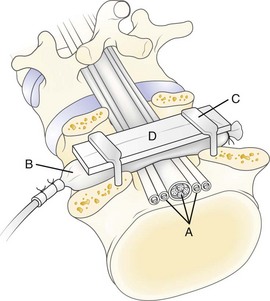
In 1991, a model was presented that for the first time allowed for experimental, graded compression of cauda equina nerve roots at known pressure levels.7,8 In this model, the cauda equina of pigs was compressed by an inflatable balloon that was fixed to the spine (Fig. 8–1). The cauda equina could also be observed through the translucent balloon. This model made it possible to study the flow in the intrinsic nerve root blood vessels at various pressure levels12 because the blood flow and vessel diameters of the intrinsic vessels could be observed simultaneously through the balloon with the use of a vital microscope. The average occlusion pressure for the arterioles was found to be slightly below and directly related to the systolic blood pressure. The blood flow in the capillary networks was intimately dependent on the blood flow of the adjacent venules. This finding corroborates the assumption that venular stasis may induce capillary stasis and changes in the microcirculation of the nerve tissue, which has been suggested as one mechanism in carpal tunnel syndrome.13 The mean occlusion pressures for the venules showed large variations. A pressure of 5 to 10 mm Hg was found to be sufficient for inducing venular occlusion. Because of retrograde stasis, it is assumed that the capillary blood flow also would be affected in such situations.
In the same experimental setup, the effects of gradual decompression, after initial acute compression maintained for only a short while, were studied.14 The average pressure for starting the blood flow was seen to be slightly lower at decompression than at compression for arterioles, capillaries, and venules. With this protocol, there was not a full restoration of the blood flow, however, until the compression was reduced from 5 to 0 mm Hg. This observation stresses further the previous impression that vascular impairment is present even at low pressure levels.
Because the nutrition of the nerve root is affected, a compression-induced impairment of the vasculature may be one mechanism for nerve root dysfunction. The nerve roots also have a considerable nutritional supply, however, via diffusion from the cerebrospinal fluid.15 To assess the compression-induced effects on the total contribution to the nerve roots, an experiment was designed in which 3H-labeled methylglucose was allowed to be transported to the nerve tissue in the compressed segment via the blood vessels and via the cerebrospinal fluid diffusion after systemic injection.16 The results showed that no compensatory mechanism from cerebrospinal fluid diffusion could be expected at the low pressure levels. On the contrary, 10 mm Hg compression was sufficient to induce a 20% to 30% reduction of the transport of methylglucose to the nerve roots compared with control.
It is known from experimental studies on peripheral nerves that compression also may induce an increase in the vascular permeability, leading to intraneural edema formation.17 Such edema may increase the endoneurial fluid pressure, which may impair the endoneurial capillary blood flow and jeopardize the nutrition of the nerve roots.18–20 Because the edema usually persists for some time after the removal of a compressive agent, edema may negatively affect the nerve root for a longer period than the compression itself. The presence of intraneural edema is also related to subsequent formation of intraneural fibrosis21 and may contribute to the slow recovery seen in some patients with nerve compression disorders. To assess if intraneural edema also may form in nerve roots as the result of compression, the distribution of Evans blue–labeled albumin in the nerve tissue was analyzed after compression at various pressures and at various durations.22 The study showed that edema was formed even at low-pressure levels. The predominant location was at the edges of the compression zone.
The function of the nerve roots has been studied by direct electrical stimulation and recordings either on the nerve itself or in the corresponding muscular segments.23–26 During a 2-hour compression period, a critical pressure level for inducing a reduction of minimal alveolar pressure or amplitude was between 50 mm Hg and 75 mm Hg. Higher pressure levels (100 to 200 mm Hg) induced a total conduction block with varying degrees of recovery after compression release. To study the effects of compression on sensory nerve fibers, the electrodes in the sacrum were instead used to record a compound nerve action potential after stimulating the sensory nerves in the tail (i.e., distal to the compression zone). The results showed that the sensory fibers were slightly more susceptible to compression than the motor fibers.25,26 Also, the nerve roots were more susceptible to compression injury if the blood pressure was reduced pharmacologically.24 This finding further implies the importance of the blood supply to maintain the functional properties of the nerve roots.
Onset Rate of Compression
A rapid onset rate of less than 1 second has been found to induce more pronounced edema formation,22 methylglucose transport,16 and impulse propagation23 than a slow onset rate of approximately 20 seconds. Regarding methylglucose transport, the results show that the levels within the compression zone are more pronounced at a rapid onset rate than at a slow onset rate at corresponding pressure levels. There was also a striking difference between the two onset rates when considering the segments outside the compression zones. In the slow onset series, the levels approached baseline values closer to the compression zone than in the rapid onset series; this may indicate the presence of a more pronounced edge-zone edema in the rapid onset series, with a subsequent reduction of the nutritional transport also in the nerve tissue adjacent to the compression zone.
For the rapid onset compression, which is likely to be more closely related to spine trauma or disc herniation than to spinal stenosis, it has been seen that a pressure of 600 mm Hg maintained only for 1 second is sufficient to induce a gradual impairment of nerve conduction during the 2 hours studied after the compression was ended.27 Overall, the mechanisms for these pronounced differences between the different onset rates are unclear, but they may be related to differences in displacement rates of the compressed nerve tissue toward the uncompressed parts, owing to the viscoelastic properties of the nerve tissue.9 Such phenomena may lead not only to structural damage to the nerve fibers but also to structural changes in the blood vessels with subsequent edema formation. The gradual formation of intraneural edema may also be closely related to the described observations of a gradually increasing difference in nerve conduction impairment between the two onset rates.22,23 In the case of spinal stenosis, the rate may be a great deal slower, and pain or nerve dysfunction may not be seen until after considerable ischemic injury.
Double or Multiple Levels of Nerve Root Compression
Patients with double or multiple levels of spinal stenosis seem to have more pronounced symptoms than patients with stenosis only at one level.28 The presented model was modified to address this clinical issue. Using two balloons at two adjacent disc levels, which resulted in a 10-mm uncompressed nerve segment between the balloons, induced a much more pronounced impairment of nerve impulse conduction than had been previously found at corresponding pressure levels.29 A pressure of 10 mm Hg in two balloons induced a 60% reduction of nerve impulse amplitude during 2 hours of compression, whereas 50 mm Hg in one balloon showed no reduction.
The mechanism for the difference between single and double compression may not simply be based on the fact that the nerve impulses have to pass more than one compression zone at double level compression. There may also be a mechanism based on the local vascular anatomy of the nerve roots. In contrast to peripheral nerves, there are no regional nutritive arteries from surrounding structures to the intraneural vascular system in spinal nerve roots.7,30–33 Compression at two levels might induce a nutritionally impaired region between the two compression sites. In this way the segment affected by the compression would be widened from one balloon diameter (10 mm) to two balloon diameters, including the nerve segment (30 mm) in between. This hypothesis was partly confirmed in an experiment on continuous analyses of the total blood flow in the uncompressed nerve segment located between two compression balloons. The results showed that a 64% reduction of total blood flow in the uncompressed segment was induced when both balloons were inflated to 10 mm Hg.34 At a pressure close to the systemic blood pressure, there was complete ischemia in the nerve segment. Data from a study on the nutritional transport to the nerve tissue in double-level compression showed that there is a reduction of this transport to the uncompressed nerve segment located between the two compression balloons that was similar to the reduction within the two compression sites.35 There is experimental evidence that the nutrition to the nerve segment located between two compression sites in nerve roots is severely impaired, although this nerve segment itself is uncompressed.
Regarding nerve conduction, it was also evident that the effects were enhanced if the distance between the compression balloons was increased from one vertebral segment to two vertebral segments.29 This was not the case, however, in the nutritional transport study where the methylglucose levels in the compression zones and in the uncompressed intermediate segment were similar between double compression over one and two vertebral segments.35 This similarity indicates that the nutrition to the uncompressed nerve segment located between two compression sites is affected almost to the same extent as at the compression sites, regardless of the distance between the compression sites but that functional impairment may be directly related to the distance between the two compression sites. The impairment of the nutrition to the nerve segment between the two compression balloons seems to be a more important mechanism than the fact that the nerve impulses have to overcome two compression sites in double-level compression.
By using electrical nerve root stimulation to increase metabolic rate and simulate a walking situation in the double-level compression model, an initial short-term increase in cauda equina blood flow was seen that rapidly decreased.36 Such observations further support the pathophysiologic significance of double-level cauda equina compression in spinal stenosis.
Chronic Experimental Nerve Root Compression
Delamarter and colleagues37 presented a model on the dog cauda equina in which they applied a constricting plastic band. The band was tightened around the thecal sac to induce a 25%, 50%, or 75% reduction of the cross-sectional area. The band was left in its place for various times. Analyses were performed and showed structural and functional changes that were proportional to the degree of constriction.
To induce a slower onset and more controlled compression, Cornefjord and colleagues38 used a constrictor to compress the nerve roots in the pig. The constrictor was initially intended for inducing vascular occlusion in experimental ischemic conditions in dogs. The constrictor consists of an outer metal shell that on the inside is covered with a material called ameroid that expands when in contact with fluids. Because of the metal shell, the ameroid expands inward with a maximum of expansion after 2 weeks, resulting in a compression of a nerve root placed in the central opening of the constrictor. Compression of the first sacral nerve root in the pig resulted in a significant reduction of nerve conduction velocity and axonal injuries.38 It has also been found that there is an increase in substance P in the nerve root and the dorsal root ganglion after such compression.39 Substance P is a neurotransmitter that is related to pain transmission. The study may provide experimental evidence that compression of nerve roots produces pain. The constrictor model has also been used to study blood flow changes in the nerve root vasculature.40 It could be observed that the blood flow is not reduced just outside the compression zone but significantly reduced in parts of the nerve roots located inside the constrictor.
One important aspect in clinical nerve root compression conditions is that the compression level is probably unstable and varies as the result of changes in posture and movements.41,42 In 1995, Konno and colleagues43 introduced a model in which the pressure could be changed after some time of initial chronic compression. An inflatable balloon was introduced under the lamina of the seventh lumbar vertebrae in the dog. The normal anatomy and the effects of acute compression using compressed air were first evaluated in previous studies.44 By inflating the balloon at a known pressure slowly over 1 hour with a viscous substance that would harden in the balloon, a compression of the cauda equina could be induced with a known initial pressure level. The compression was verified by myelography. Because the balloon under the lamina comprised a twin set of balloons, the second balloon component could be connected to a compressed air device and could be used to add compression to the already chronically compressed cauda equina.
Spinal Stenosis: Experimental-Clinical Correlation
If nerve compression is of an extremely low onset rate as in spinal stenosis, there may be an adaptation of the nerve tissue to the applied pressure. In cadaveric experiments, Schönström and colleagues45 found that when a hose clamp was tightened around a human cadaveric cauda equina specimen there was a critical cross-sectional area of the dural sac when the first signs of pressure increase among nerve roots were recorded by a catheter placed in the compression zone. This cross-sectional area was approximately 75 mm2, which was also found to correlate with a corresponding measurement on computed tomography (CT) in patients with spinal stenosis.46 When the hose clamp was tightened further, the pressure increased. Owing to creep phenomena in the nerve tissue, the pressure decreased with time, however. When the pressure did not normalize within 10 minutes, the “sustained size” was registered and was found to be in the range of 45 to 50 mm2.45 This study indicates that even in acute compression there is an adaptation of the nerve tissue to the applied pressure. From a longer perspective, this probably means that the nerve may also be reorganized in its microstructural elements, which would result in a nerve with a smaller diameter. Under such circumstances, with gradually decreasing nerve diameter, the nerve pressure acting on the nerve would be reduced to some degree.
There is a correlation between the animal experimental observations regarding critical pressures for functional and nutritional changes in nerve roots under compression on one side and the measurements of pressure levels among nerve roots in human cadaveric lumbar spines after experimental constriction of the dural sac. An acute pressure increase among cauda equina nerve roots to 50 mm Hg was induced when the cross-sectional area of the dural sac was reduced to 63 mm2, and a pressure of 100 mm Hg was induced at a cross-sectional area of 57 mm2.45 Such pressure levels correlate with in vivo observations regarding physiologic changes in cauda equina nerve roots after experimental compression.7,12,22
Epidural pressure measurements have been performed, evaluating the relationship between epidural pressure and posture.42 It was found that the local epidural pressure at the stenotic level was low in lying and sitting postures and high in standing postures. Pressure was increased with extension but decreased with flexion of the spine. The highest epidural pressure, 117 mm Hg, was found in standing with extension. Measurements have also been reported regarding changes in epidural pressure during walking in patients with lumbar spinal stenosis.47 The pressure changed during walking with a wave pattern of increase and decrease. Such observations correlate with the previously mentioned experimental observations regarding intermittent cauda equina compression.41
Mechanical Nerve Root Deformation and Pain
Some experimental observations indicate that mechanical nerve root deformation per se may induce impulses that cause pain. Howe and colleagues48 found that mechanical stimulation of nerve roots or peripheral nerves resulted in nerve impulses of short duration and that these impulses were prolonged if the nerve tissue had been exposed to mechanical irritation by a chromic gut ligature for 2 to 4 weeks. Corresponding results were obtained in an in vitro system using rabbit nerve roots.49 In this setup, it was also evident that the dorsal root ganglion was more susceptible to mechanical stimulation than the nerve roots. The dorsal root ganglion has elicited special interest in this regard, and an increase in the level of neurotransmitters related to pain transmission has been found in the dorsal root ganglion in response to whole-body vibration of rabbits.50 A similar increase has been seen in the dorsal root ganglion and nerve root after local constriction of the same nerve root.39 In vivo models of pain behavior have shown that mechanical nerve deformation superimposed on inflammation is painful, whereas either factor alone might not cause severe pain.51–56 The magnitude of nerve root compression pressure (measured intraoperatively) correlates with neurologic deficit but not with degree of straight-leg raising test.57
Neuropathologic Changes and Pain
There is considerable research evidence regarding the relationship of pain to neuropathologic changes.58 Much of what is known has been studied in relationship to mechanical and inflammatory injury of the sciatic nerve in the rat. Entrapment of a peripheral nerve produces pathologic change in proportion to the degree of compression and its duration,59 as is known to be the case for nerve root compression. In an electron microscopic study,59 minor degrees of nerve compression were associated with ischemic injury to Schwann cells, resulting in their necrosis and in demyelination. Severe nerve compression was associated with injury to the axon, resulting in wallerian degeneration.
Subsequent experiments established the relationship of pain to these forms of neuropathologic change.60 These studies established that mild levels of ischemia producing demyelination were generally not painful, whereas severe ischemia-producing wallerian degeneration resulted in hyperalgesia. The pathology of the chronic constriction injury model of neuropathic pain is based on this relationship and the added insult of inflammation caused by the chromic gut ligatures used to compress the nerve.61 It is now recognized that the cytokine-driven processes of wallerian degeneration are the dominant neuropathologic factors linking nerve injury and pain60,62,63 and that the degree and extent of wallerian degeneration relate directly to the magnitude and duration of hyperalgesia.64
Biologic and Biochemical Effects on Nerve Roots
The clinical picture of sciatica with a characteristic distribution of pain and nerve dysfunction but in the absence of herniated disc material at radiologic examination and at surgery has indicated that mechanical nerve root compression may not be the only factor that is responsible for sciatic pain. It has been suggested that the disc tissue per se may have some injurious properties.9 Not until 1993 was it confirmed experimentally, however, that local, epidural application of autologous nucleus pulposus with no mechanical deformation induces significant changes in structure and function of the adjacent nerve roots.6
Biologic Effects of Nucleus Pulposus
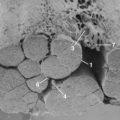
In 1993, Olmarker and colleagues6 published a study that showed that autologous nucleus pulposus can induce a reduction in nerve conduction velocity and light microscopic structural changes in a pig cauda equina model of nerve root injury. These axonal changes had a focal distribution, however, and the quantity of injured axons was too low to be responsible for the significant neurophysiologic dysfunction observed. A follow-up study of areas of the nerve roots exposed to nucleus pulposus that appeared to be normal by light microscopy revealed that there were significant injuries of Schwann cells with vacuolization and disintegration of Schmidt-Lanterman incisures (Fig. 8–2).65 Schmidt-Lanterman incisures are essential for the normal exchange of ions between the axon and the surrounding tissues. An injury to this structure would be likely to interfere with the normal impulse conduction properties of the axons, although these models’ changes may not fully explain the neurophysiologic dysfunction observed.
The pathophysiologic potential of the nucleus pulposus was emphasized further in an experiment using a dog model in which it was seen that a surgical incision of the anulus fibrosus, with minimal leakage of nucleus pulposus, was enough to induce significant changes in structure and function of the adjacent nerve root.66 It has also been seen that epidural application of the autologous nucleus pulposus within 2 hours induces an intraneural edema67,68 that leads to a reduction of the intraneural blood flow.68 Histologic changes of the nerve roots are present after 3 hours,69 and a subsequent reduction of the nerve conduction velocity starts 3 to 24 hours after application.6,69 The nucleus pulposus may also interfere with the nutrition to the intraspinal nerve tissue. After application to the dorsal root ganglion, it was found that the intraneural blood flow was dramatically decreased and that there was a simultaneous increase of the tissue fluid pressure.68
Methylprednisolone reduces the pathophysiologic events of the nucleus pulposus–induced nerve root injury if given within 24 hours. To establish if the presence of autologous nucleus pulposus could initiate a leukotactic response from the surrounding tissues, a study was initiated that assessed the potential inflammatogenic properties of the nucleus pulposus.70 Autologous nucleus pulposus and autologous retroperitoneal fat were placed in separate perforated titanium chambers and placed subcutaneously, together with a sham chamber, in the pig. The number of leukocytes was assessed 7 days later for the chambers. The nucleus pulposus–containing chambers had a number of leukocytes that exceeded the two others by 150%. In another experiment, autologous nucleus pulposus and muscle were placed in Gore-Tex tubes subcutaneously in rabbits.71 After 2 weeks, there was an accumulation of macrophages and T-helper and T-suppresser cells in the tube with nucleus pulposus that persisted the full observation time of 4 weeks.
Kawakami and colleagues72 showed that neuropathic pain in an experimental setting seems to be mediated by infiltrating leukocytes, a finding consistent with the previous observations of neuroimmunologic inflammatory changes and pain.73 In rats made leukopenic by using nitrogen mustard, the pain response was absent after application of nucleus pulposus, whereas normal rats with nucleus pulposus application displayed a pathologic response to stimulation. The same group also showed that inhibition of cyclooxygenase-2 might reduce nucleus pulposus–induced pain behavior.74 Taken together, these data further support the impression that autologous nucleus pulposus may elicit inflammatory reactions when outside the intervertebral disc space and that such reactions may not be restricted to resorption of the herniated tissue but also may be intimately involved in the pathophysiology of sciatica.
Nucleus Pulposus and Sciatic Pain
Pain is much more difficult to assess than nerve conduction in controlled experimental studies. The available literature indicates that pain may be induced by mechanical factors and nucleus pulposus–mediated factors. The role of the nucleus pulposus in this context is interesting in view of patients with obvious symptoms of disc herniation but with no visible herniation at radiologic examination or surgery.75,76 The potential of nucleus pulposus material to induce pain has also been indicated in clinical studies that showed that noncontained herniations (the nucleus pulposus was in contact with the epidural space) were much more painful and had a more pronounced straight-leg raising test result than contained herniations.77–79
Studies on rats using pain behavior assessment indicated that the nucleus pulposus is involved in pain production. Pain behavior in this context refers to response thresholds to thermal and mechanical stimulation. Kawakami and colleagues52,53 showed that a three-level laminectomy and application of homologous nucleus pulposus or anulus fibrosus taken from three intervertebral discs in another rat, applied at three nerve roots, produces pain behavior. Other studies54 suggest a dose-response relationship between pain behavior and the amount of nucleus pulposus material in the epidural space. The combination of nucleus pulposus herniation and mechanical injury produces pain.54 This observation is consistent with the neuropathologic understanding of pain and the consequences of combined mechanical and inflammatory injury to nerve fibers that are superimposed to increase the number of fibers injured and the corresponding increase in proinflammatory cytokines.63,64 The same pathophysiologic response was observed in a study assessing walking patterns, in which it was seen that only the combination of displacement and disc incision produced detectable changes.56 Also, a pain behavior study assessing changes in spontaneous behavior showed that only the combined action of displacement and disc incision produced changes, whereas displacement or disc incision per se did not produce changes.80
These experimental studies on pain behavior suggest that the presence of nucleus pulposus has sensitized the nerve tissue. Minor compression of peripheral nerves is not painful, and touching of a normal nerve root during local anesthesia is not painful.81 Touching of a nerve root exposed to a disc herniation often reproduces the sciatic pain, however.81 Although the combination of a mechanical component and the presence of nucleus pulposus seems to be a prerequisite to produce changes in the in vivo situation, more recent neurophysiologic studies have shown that the mere application of nucleus pulposus may induce increased neuronal pain transmission.82 This finding reflects that pain behavior assessment is a gross instrument to detect pain and that nucleus pulposus may induce pain in the absence of a mechanical component as well.
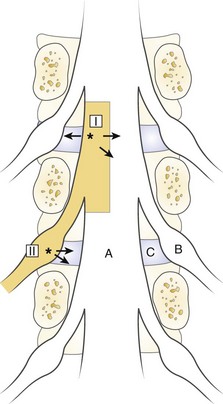
The spinal dura mater is known to contain nerve endings, and stimulation of the dura has been suggested as a mechanism for sciatic pain.9,81,83,84 Irritation or stimulation of the dura as one important factor for sciatica is an interesting theory that could explain many clinical features. One may assume that the dura is segmentally innervated, the sensory nerves travel in a caudal-lateral direction, and the dura is drained to the corresponding nerve root by the nerve of Luschka.85–88 Stimulation of the dura at a point where the dorsolateral herniations appear (I in Fig. 8–3) should be recorded by the corresponding nerve root.89 At this location, the irritation may spread medially to the contralateral segment, however, producing bilateral symptoms, or laterally, producing symptoms from levels above. Similarly, a lateral disc herniation (II in Fig. 8–3) could produce symptoms in the lower level.
Other Consequences of Herniated Nucleus Pulposus
Histologic observations have indicated that nerve root changes caused by nucleus pulposus are focal and mainly found in the center of the nerve roots, resembling a mononeuritis simplex that is induced by nerve infarction secondary to embolism of the intraneural vessels.6,65,90 Particularly in view of the work of Jayson and colleagues91–94 indicating an impairment of the venous outflow from the nerve roots owing to periradicular vascular changes, one must consider vascular impairment as one factor. Large molecules deposited in the epidural space can be found in the intraneural vessels of the adjacent nerve roots within seconds after application.95 Epidurally placed substances can penetrate the relatively impermeable dura, cross over the cerebrospinal fluid, and diffuse through the root sheath and into the axons.
The inflammatory components of nucleus pulposus may be involved in vascular and rheologic phenomena, such as coagulation, and may be involved in nerve root vascular embolism. It has been observed that the presence of nucleus pulposus may induce thrombus formation in microvessels.70 Inflammatory mediators may also exert a direct effect on the myelin sheaths, as indicated by an electron microscopic study of nerve roots exposed to autologous nucleus pulposus in the pig.65 There were significant injuries of Schwann cells with vacuolization and disintegration of Schmidt-Lanterman incisures, which closely resembles the injury pattern of inflammatory nerve disease.96,97 As previously described, results from studies have also indicated that epidural application of nucleus pulposus induces an increase of the vascular permeability and a subsequent reduction of the blood flow in the adjacent nerve roots, which suggests vascular impairment as being of pathophysiologic importance.
It has also been suggested that because the nucleus pulposus is avascular and “hidden” from the systemic circulation, a presentation of the nucleus pulposus could result in an autoimmune reaction directed to antigens present in the nucleus pulposus and that bioactive substances from this reaction may injure the nerve tissue.98–105 One may also hypothesize that there could be autoimmune reactions not only to the disc but also to components from the nerve tissue that are released as the result of injury, such as basic myelin proteins. A study also assessed the possible presence of immune complexes in herniated disc tissue obtained at surgery as an indicator of immunoactivation.106 IgG was found in close relation to the disc cells in herniated disc material. No IgG was found, however, in the residual disc that was evacuated at the time of surgery. No immune complexes were found in control disc material obtained at spine surgery for other causes than pain. Although inconclusive, this study may indicate that immunologic activation may be present in some cases of sciatica.
Chemical Components of Nucleus Pulposus
The nucleus pulposus is composed mainly of proteoglycans, collagen, and cells.107,108 The proteoglycan component has gained the most attention and has been suggested to have a direct irritating effect on nerve tissue.104,109,110 Neither the collagen nor the cells have previously been suggested to be of pathophysiologic importance. More recent studies of the cells of the nucleus pulposus have shown, however, that these cells are capable of producing metalloproteinases such as collagenase or gelatinase and interleukin (IL)-6 and prostaglandin E2 and do so spontaneously in culture. Using the same pig model previously described, the possible role of the nucleus pulposus cells for the nucleus pulposus–induced nerve injury has been assessed.111 In a blinded fashion, autologous nucleus pulposus was subjected to 24 hours of freezing at −20°C, digestion by hyaluronidase, or just heating the box at 37°C for 24 hours. The treated nucleus pulposus was reapplied after 24 hours, and analyses were performed 7 days later. In animals in which the nucleus pulposus had been frozen and the cells killed, there were no changes in nerve conduction velocity, whereas in the other two series the results were similar to application of unaltered nucleus pulposus.
It seems reasonable to believe that the cells are responsible in some way for inducing the nerve injury. This assumption was supported further by observations indicating that application of cultured pig disc cells to the cauda equina reproduced the reduction in nerve conduction velocity.111 Application of disc cell membranes also reproduced this reduction, however, indicating that the responsible substances probably are membrane bound.
Substances such as IgG, hydrogen ions, nitric oxide, and phospholipase A2 have previously been suggested to be responsible for the pathophysiologic reactions.104,112–116 Another substance produced by the disc cells that has similar pathophysiologic effects as nucleus pulposus is tumor necrosis factor (TNF)-α.117
Cytokines as Mediators of Nerve Dysfunction and Pain
TNF is known to be a regulatory proinflammatory cytokine that has specific biologic effects and the ability to upregulate and act synergistically with other cytokines such as IL-1β and IL-6.118–127 Immediately after nerve injury, TNF is released and upregulated by Schwann cells at the site of nerve injury124; this is followed by release and upregulation of TNF in many other endoneurial cells, including endothelial cells, fibroblasts, and mast cells. TNF is also produced by chondrocytes and disc cells.117,125–128 This local production of TNF is the stimulus that results in macrophage attraction to the injury site,62 which contributes massively to the concentration of proinflammatory cytokines in the injured tissue. Several studies have shown that blocking TNF production or delaying the invasion of macrophages to the site of nerve injury results in reduced or delayed neuropathologic change and reduced hyperalgesia.73,129
When performing a meta-analysis on the biologic and pathophysiologic effects induced by TNF and by nucleus pulposus, one may find that there is almost a perfect match. TNF is known to induce axonal and myelin injury similar to that observed after nucleus pulposus application,130–136 intravascular coagulation,137–139 and increased vascular permeability.139 TNF is also known to be neurotoxic133,135,140,141 and to induce painful behavioral changes130,142 and ectopic nerve activity when applied locally.131,143 TNF is sequestered in a membrane-bound form and is activated after shedding by certain enzymes. Matrix metalloproteinases (MMPs) are particularly important in this regard. MMP-9 and MMP-2 are upregulated immediately after a nerve injury.144 MMPs process the inactive, membrane-bound form of TNF and its receptors to the biologically active form and are directly associated with breakdown of the blood-brain and blood-nerve barriers. MMP-9 and TNF receptors are also retrogradely transported from the site of nerve injury to the corresponding dorsal root ganglion and spinal cord,145 where they may have a direct role in gene regulation. This may relate to the observation that cell membranes of disc cells are sufficient to mediate the nucleus pulposus–induced effects.111
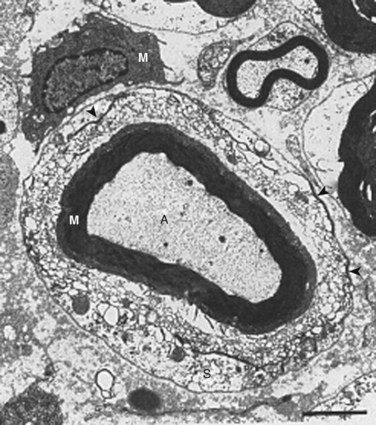
TNF induces activation of endothelial adhesion molecules such as intercellular and vascular cell adhesion molecules, adhering circulating immune cells to the vessel walls (Fig. 8–4).121,146,147 As a consequence of the TNF-induced increased vascular permeability, these cells migrate into the endoneurial space where the axons are located.148–150 The cells release their content of TNF and other cytokines, which may induce accumulation of ion channels locally in the axonal membranes.151–153 The channels may allow for an increased passage of sodium and potassium, which may result in spontaneous discharges and in discharges of ectopic impulses after mechanical stimulation. TNF by itself can cause spontaneous electrical activity in A-delta and C nociceptors.143 Such discharges, whether they come from a pain fiber or a nerve fiber transmitting other sensory information, are interpreted as pain by the brain.154–157 Such a mechanism may relate to the sensitization of the nerve roots seen in the experimental and clinical studies just discussed and to motion-evoked sciatic pain, such as the straight-leg raising test.
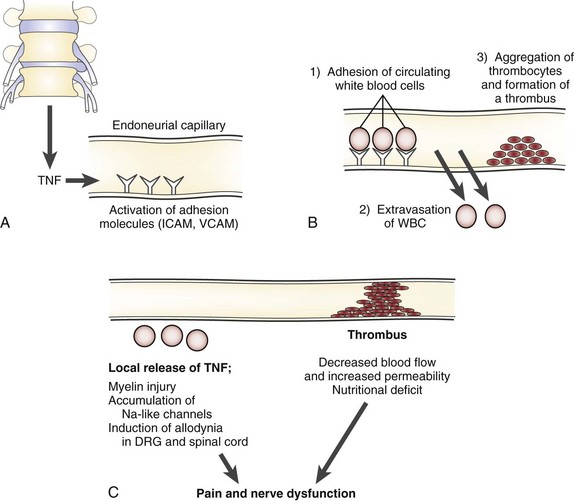
Previous studies have also indicated that local application of nucleus pulposus may disintegrate the myelin sheath65,66; this is also a known effect of TNF.130,148,158–160 In particular, this injury seems to affect Schmidt-Lanterman incisures, which are responsible for the ion exchange between the axon and the surrounding tissues.161–164 This injury could also contribute to the formation of ectopic impulses and to the sensitization to mechanical stimulus. Experimental and clinical studies have shown that nerve root compression and disc herniation can induce increased concentrations of neurofilament in the cerebrospinal fluid.165,166 Increased levels of serum antibodies against one or more nervous system–associated glycosphingolipids have been shown in patients with sciatica and disc herniation, indicating a possible autoimmune response.167
More recent work regarding molecular events in the pathophysiology of neuropathic pain has suggested a potential role of TNF for inducing allodynia.131,168–173 TNF may mediate the formation of allodynia in the dorsal root ganglion and at the spinal cord level because of its local upregulation, which occurs via a positive feedback loop caused by TNF itself. This cycle seems to be broken by a direct effect of TNF on the upregulation of anti-inflammatory cytokines such as IL-10, which eventually leads to a reduction of TNF and the physiologic balance of proinflammatory and anti-inflammatory cytokines. Such regulation seems to be induced by mechanical injury to peripheral parts of the axons and by a direct effect of TNF exposure and further enhances the impression that TNF may be an important mediator of neuropathic pain. TNF is a potent activator of cells; because it is retrogradely transported from the site of nerve injury to the dorsal root ganglion and spinal cord, it may be this proinflammatory stimulus that activates central glia and neurons.145
Apart from directly affecting the endoneurially located axons, TNF may also indirectly interfere with the axons by compromising the nutritional transport. TNF may induce intravascular coagulation, similar to nucleus pulposus, after local application137,174–176; this reduces the local blood flow in the intraneural capillaries.75 A nutritional reduction induces ischemia in the nerve root, which may induce neuroischemic pain.
TNF was found in disc cells; when TNF was inhibited with a nonspecific cytokine inhibitor, the nucleus pulposus–induced reduction in nerve conduction velocity after experimental application of nucleus pulposus in a pig model was completely blocked.117 When using more specific TNF inhibitors, such as a monoclonal antibody to TNF (infliximab) and a soluble TNF receptor (etanercept), the inhibition was equally effective.174 Investigations have shown that infliximab may attenuate immunoreactivity of brain-derived neurotrophic factor and may prevent neurologic and histologic changes in dorsal root ganglion in rats after experimental disc herniation.177–179 Application of selected cytokines in the pig model showed that TNF reduced the nerve conduction velocity per se.180 IL-1β and interferon-γ induced only a slight reduction of nerve conduction velocity.
Application of certain cytokines to intraspinal nerves may also increase the somatosensory neural response.181 Discharges from wide-dynamic-range neurons after stimulation of a receptor field of a dorsal root ganglion exposed to nucleus pulposus increased significantly after application.182 This increase may be related to the sensitization of the sensory system caused by proinflammatory cytokines and the production of low-grade spontaneous electrophysiologic activity in nociceptors by TNF,143 which by itself is an important factor that contributes to sensitization. Administering an antibody specific for TNF efficiently inhibited this effect. An in vivo study assessing changes in spontaneous behavior clearly showed that changes induced by the combined action of mechanical deformation and disc incision were markedly inhibited by intraperitoneal injection of a monoclonal antibody specific for TNF.55
TNF seems to be an important mediator for the observed effects on nerve function and for pain induced by local application of nucleus pulposus. Additional support for this hypothesis comes from previous work that showed that blockade of TNF upregulation in macrophages by thalidomide129 and downregulation of TNF by IL-10 administration183 reduced the magnitude and duration of hyperalgesia after nerve injury. Because cytokine interactions are complex, other cytokines such as IL-1β and IL-6 may be involved as well.180,181,184,185 Because these cytokines are induced by TNF, as well as inducing TNF, their exact role has not been completely evaluated.
The possible role of brain-derived neurotrophic factor in nerve root pathophysiology and experimental disc herniation has been analyzed.186 The appearance and distribution of macrophages and TNF in the dorsal root ganglion of rats after experimental disc herniation187,188 and the relationship between nerve growth factor and pain behavioral changes have been described.189 It has also been shown that disc-related cytokines can inhibit axonal outgrowth from dorsal root ganglion cells in vitro.190
Clinical Use of Cytokine Inhibitors for Treatment of Sciatica
On the basis of the experimental findings that TNF may mimic nucleus pulposus–induced nerve dysfunction and pain, pilot clinical trials regarding the possible use of TNF inhibition for the treatment of sciatica have been initiated. Karppinen and colleagues191 administered a monoclonal antibody specific for TNF (infliximab [Remicade]) to 10 volunteers waiting for surgery for radiologically verified disc herniations with severe sciatica. In this open-label study, infliximab reduced pain assessed by visual analog scale by 50% at 1 hour after infusion. After 2 weeks, 60% of the patients were pain-free. At 3 months after the single infusion, 90% were pain-free. No adverse drug reactions were noted, and no patients required surgery. A 1-year follow-up192 of the 10 patients treated with infliximab showed that the beneficial effect of a single infusion of 3 mg/kg of infliximab for disc herniation–induced sciatica was sustained in most patients. The study authors also noted that infliximab did not seem to interfere with spontaneous resorption of disc herniations.
Genevay and colleagues193 administered a TNF inhibitor in the form of a soluble TNF receptor (etanercept [Enbrel]) by three subcutaneous injections to 10 patients with severe sciatica. The patients had a 70% reduction of leg pain assessed by visual analog scale 10 days after starting the treatment. At 6 weeks, the reduction was 83%. The results were statistically significantly better than for 10 patients treated with three intravenous injections of methylprednisolone.
There is one randomized study published by Cohen and colleagues194 regarding treatment of sciatica by local epidural injections of the TNF inhibitor etanercept. The investigators randomly assigned 24 patients with subacute radiculopathy into three groups each consisting of 8 patients. The patients in each group received either 2 mg, 4 mg, or 6 mg on two occasions, and two of the eight patients were saline controls. All etanercept-treated patients had significant improvement 1 month after treatment compared with saline-treated patients regarding leg and back pain. The effects persisted 6 months after treatment in all but one patient. The authors concluded that “etanercept holds promise as a treatment for lumbosacral radiculopathy.”194 Genevay and colleagues195 published the results of a multicenter, double-blind, placebo-controlled trial on the use of the TNF inhibitor adalimumab (Humira) subcutaneously injected in 31 patients with severe, acute sciatica caused by disc herniation. Two injections were given 7 days apart; 30 control patients received placebo injections in the same manner. The results showed that there was a significantly more favorable evolution of leg pain in the adalimumab group than in the placebo group, but the effect size was relatively small. There were twice as many patients in the adalimumab group who fulfilled the criteria for “responders,” and there were significantly fewer surgical discectomies in this group compared with the placebo-treated controls.
Taken together, these observations indicate a potential clinical effect of TNF inhibition for the treatment of sciatica. It may be surprising that TNF inhibition seems to be so much superior to anti-inflammatory treatment by nonsteroidal anti-inflammatory drugs or methylprednisolone or even morphine. One may conclude that it is more efficient to act at the responsible mediators directly than aiming at general anti-inflammatory effects. This clinical comparison strongly supports the TNF hypothesis of neuropathic pain.63,117,174 Sciatica has a neuropathic pain component, and nonspecific anti-inflammatory medication and morphine are less efficient in such conditions. Nevertheless, further studies must be undertaken before any definite conclusions regarding its efficacy for the treatment of sciatica may be drawn.196,197
Acknowledgments
1 Mixter WJ, Barr JS. Rupture of the intervertebral disc with involvement of the spinal canal. N Engl J Med. 1934;211:210-215.
This article is about the discovery of the herniated disc.
2 Olmarker K, Holm S, Rosenqvist AL, Rydevik B. Experimental nerve root compression. A model of acute, graded compression of the porcine cauda equina and an analysis of neural and vascular anatomy. Spine (Phila Pa 1976). 1991;16:61-69.
This articles discusses the first time a model for graded compression of nerve roots was introduced.
3 Olmarker K, Rydevik B, Nordborg C. Autologous nucleus pulposus induces neurophysiologic and histologic changes in porcine cauda equina nerve roots. Spine. 1993;1(18):1425-1432.
This study demonstrated for the first time the injurious effects of autologus nucleus puposus.
4 Kawakami M, Weinstein JN, Chatani K, et al. Experimental lumbar radiculopathy. Behavioral and histologic changes in a model of radicular pain after spinal nerve root irritation with chromic gut ligatures in the rat. Spine. 1994;15(19):1795-1802.
This study was the first to examine nerve root pain in an experimental model.
5 Olmarker K, Myers RR. Pathogenesis of sciatic pain: role of herniated nucleus pulposus and deformation of spinal nerve root and dorsal root ganglion. Pain. 1998;78:99-105.
6 Olmarker K, Larsson K. Tumor necrosis factor alpha and nucleus-pulposus-induced nerve root injury. Spine. 1998;23:2538-2544.
This was the first study to link a specific molecule to the pathophysiology of sciatica.
7 Cohen SP, Bogduk N, Dragovich A, et al. Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety study of transforaminal epidural etanercept for the treatment of sciatica. Anesthesiology. 2009;110:1116-1126.
1 Mixter WJ, Barr JS. Rupture of the intervertebral disc with involvement of the spinal canal. N Engl J Med. 1934;211:210-215.
2 Olmarker K, Hasue M. Classification and pathophysiology of spinal pain syndromes. In: Weinstein JN, Rydevik B, Sonntag VKH, editors. Essentials of the Spine. New York: Raven Press; 1995:11-25.
3 Bailey P, Casamajor L. Osteo-arthritis of the spine as a cause of compression of the spinal cord and its roots. J Nerv Ment Dis. 1911;38:588-609.
4 Goldthwait JE. The lumbo-sacral articulation: An explanation of many cases of “lumbago” and “sciatica” and paraplegia. Boston Med Surg J. 1911;164:365-372.
5 Sachs B, Fraenkel J. Progressive ankylotic rigidity of the spine. J Nerv Ment Dis. 1900;27:1-15.
6 Olmarker K, Rydevik B, Nordborg C. Autologous nucleus pulposus induces neurophysiologic and histologic changes in porcine cauda equina nerve roots. Spine (Phila Pa 1976). 1993;18:1425-1432.
7 Olmarker K. Spinal nerve root compression: Nutrition and function of the porcine cauda equina compressed in vivo. Acta Orthop Scand Suppl. 1991;242:1-27.
8 Olmarker K, Holm S, Rosenqvist AL, et al. Experimental nerve root compression: A model of acute, graded compression of the porcine cauda equina and an analysis of neural and vascular anatomy. Spine (Phila Pa 1976). 1991;16:61-69.
9 Rydevik B, Brown MD, Lundborg G. Pathoanatomy and pathophysiology of nerve root compression. Spine (Phila Pa 1976). 1984;9:7-15.
10 Gelfan S, Tarlov IM. Physiology of spinal cord, nerve root and peripheral nerve compression. Am J Physiol. 1956;185:217-229.
11 Sharpless SK. Susceptibility of spinal nerve roots to compression block: The research status of spinal manipulative therapy. Goldstein M, editor. NIH-Workshop: NINCDS Monograph. 1975:155-161.
12 Olmarker K, Rydevik B, Holm S, et al. Effects of experimental graded compression on blood flow in spinal nerve roots: A vital microscopic study on the porcine cauda equina. J Orthop Res. 1989;7:817-823.
13 Sunderland S. The nerve lesion in the carpal tunnel. J Neurol Neurosurg Psychiatry. 1976;39:615-626.
14 Olmarker K, Holm S, Rydevik B, et al. Restoration of blood flow during gradual decompression of a compressed segment of the porcine cauda equina: A vital microscopic study. Neuroorthopaedics. 1991;10:83-87.
15 Rydevik B, Holm S, Brown MD, et al. Diffusion from the cerebrospinal fluid as a nutritional pathway for spinal nerve roots. Acta Physiol Scand. 1990;138:247-248.
16 Olmarker K, Rydevik B, Hansson T, et al. Compression-induced changes of the nutritional supply to the porcine cauda equina. J Spinal Disord. 1990;3:25-29.
17 Rydevik B, Lundborg G. Permeability of intraneural microvessels and perineurium following acute, graded experimental nerve compression. Scand J Plast Reconstr Surg. 1977;11:179-187.
18 Myers RR, Murakami H, Powell HC. Reduced nerve blood flow in edematous neuropathies: A biomechanical mechanism. Microvasc Res. 1986;32:145-151.
19 Myers RR, Mizisin AP, Powell HC, et al. Reduced nerve blood flow in hexachlorophene neuropathy: Relationship to elevated endoneurial fluid pressure. J Neuropathol Exp Neurol. 1982;41:391-399.
20 Myers R, Powell H. Galactose neuropathy: Impact of chronic endoneurial edema on nerve blood flow. Ann Neurol. 1984;16:587-594.
21 Rydevik B, Lundborg G, Nordborg C. Intraneural tissue reactions induced by internal neurolysis: An experimental study on the blood-nerve barrier, connective tissues and nerve fibres of rabbit tibial nerve. Scand J Plast Reconstr Surg. 1976;10:3-8.
22 Olmarker K, Rydevik B, Holm S. Edema formation in spinal nerve roots induced by experimental, graded compression: An experimental study on the pig cauda equina with special reference to differences in effects between rapid and slow onset of compression. Spine (Phila Pa 1976). 1989;14:569-573.
23 Olmarker K, Holm S, Rydevik B. Importance of compression onset rate for the degree of impairment of impulse propagation in experimental compression injury of the porcine cauda equina. Spine (Phila Pa 1976). 1990;15:416-419.
24 Garfin SR, Cohen MS, Massie JB, et al. Nerve-roots of the cauda equina: The effect of hypotension and acute graded compression on function. J Bone Joint Surg Am. 1990;72:1185-1192.
25 Pedowitz RA, Garfin SR, Massie JB, et al. Effects of magnitude and duration of compression on spinal nerve root conduction. Spine (Phila Pa 1976). 1992;17:194-199.
26 Rydevik BL, Pedowitz RA, Hargens AR, et al. Effects of acute, graded compression on spinal nerve root function and structure: An experimental study of the pig cauda equina. Spine (Phila Pa 1976). 1991;16:487-493.
27 Olmarker K, Lind B, Holm S, et al. Continued compression increases impairment of impulse propagation in experimental compression of the porcine cauda equina. Neuroorthopaedics. 1991;11:75-81.
28 Porter RW, Ward D. Cauda equina dysfunction: The significance of two-level pathology. Spine (Phila Pa 1976). 1992;17:9-15.
29 Olmarker K, Rydevik B. Single- versus double-level nerve root compression: An experimental study on the porcine cauda equina with analyses of nerve impulse conduction properties. Clin Orthop Relat Res. 1992;279:35-39.
30 Lundborg G. Structure and function of the intraneural microvessels as related to trauma, edema formation, and nerve function. J Bone Joint Surg Am. 1975;57:938-948.
31 Parke WW, Watanabe R. The intrinsic vasculature of the lumbosacral spinal nerve roots. Spine (Phila Pa 1976). 1985;10:508-515.
32 Parke WW, Gammell K, Rothman RH. Arterial vascularization of the cauda equina. J Bone Joint Surg Am. 1981;63:53-62.
33 Petterson CA, Olsson Y. Blood supply of spinal nerve roots: An experimental study in the rat. Acta Neuropathol (Berl). 1989;78:455-461.
34 Takahashi K, Olmarker K, Holm S, et al. Double-level cauda equina compression: An experimental study with continuous monitoring of intraneural blood flow in the porcine cauda equina. J Orthop Res. 1993;11:104-109.
35 Cornefjord M, Takahashi K, Matsui Y, et al. Impairment of nutritional transport at double-level cauda equina compression: An experimental study. Neuroorthopaedics. 1992;13:107-112.
36 Baker AR, Collins TA, Porter RW, et al. Laser Doppler study of porcine cauda equina blood flow: The effect of electrical stimulation of the rootlets during single and double site, low pressure compression of the cauda equina. Spine (Phila Pa 1976). 1995;20:660-664.
37 Delamarter RB, Bohlman HH, Dodge LD, et al. Experimental lumbar spinal stenosis: Analysis of the cortical evoked potentials, microvasculature, and histopathology. J Bone Joint Surg Am. 1990;72:110-120.
38 Cornefjord M, Sato K, Olmarker K, et al. A model for chronic nerve root compression studies: Presentation of a porcine model for controlled, slow-onset compression with analyses of anatomic aspects, compression onset rate, and morphologic and neurophysiologic effects. Spine (Phila Pa 1976). 1997;22:946-957.
39 Cornefjord M, Olmarker K, Farley DB, et al. Neuropeptide changes in compressed spinal nerve roots. Spine (Phila Pa 1976). 1995;20:670-673.
40 Sato K, Olmarker K, Cornefjord M, et al. Changes of intraradicular blood flow in chronic nerve root compression: An experimental study on pigs. Neuroorthopaedics. 1994;16:1-7.
41 Konno S, Olmarker K, Byrod G, et al. Intermittent cauda equina compression: An experimental study of the porcine cauda equina with analyses of nerve impulse conduction properties. Spine (Phila Pa 1976). 1995;20:1223-1226.
42 Takahashi K, Miyazaki T, Takino T, et al. Epidural pressure measurements: Relationship between epidural pressure and posture in patients with lumbar spinal stenosis. Spine (Phila Pa 1976). 1995;20:650-653.
43 Konno S, Yabuki S, Sato K, et al. A model for acute, chronic, and delayed graded compression of the dog cauda equina: Presentation of the gross, microscopic, and vascular anatomy of the dog cauda equina and accuracy in pressure transmission of the compression model. Spine (Phila Pa 1976). 1995;20:2758-2764.
44 Sato K, Konno S, Yabuki S, et al. A model for acute, chronic, and delayed graded compression of the dog cauda equina: Neurophysiologic and histologic changes induced by acute, graded compression. Spine (Phila Pa 1976). 1995;20:2386-2391.
45 Schönström N, Bolender NF, Spengler DM, et al. Pressure changes within the cauda equina following constriction of the dural sac: An in vitro experimental study. Spine (Phila Pa 1976). 1984;9:604-607.
46 Schönström NS, Bolender NF, Spengler DM. The pathomorphology of spinal stenosis as seen on CT scans of the lumbar spine. Spine (Phila Pa 1976). 1985;10:806-811.
47 Takahashi K, Kagechika K, Takino T, et al. Changes in epidural pressure during walking in patients with lumbar spinal stenosis. Spine (Phila Pa 1976). 1995;20:2746-2749.
48 Howe JF, Loeser JD, Calvin WH. Mechanosensitivity of dorsal root ganglia and chronically injured axons: A physiological basis for the radicular pain of nerve root compression. Pain. 1977;3:25-41.
49 Cavanaugh JM, Ozaktay AC, Yamashita T, et al. Mechanisms of low back pain: A neurophysiologic and neuroanatomic study. Clin Orthop Relat Res. 1997;335:166-180.
50 Weinstein J, Pope M, Schmidt R, et al. Neuropharmacologic effects of vibration on the dorsal root ganglion: An animal model. Spine (Phila Pa 1976). 1988;13:521-525.
51 Chatani K, Kawakami M, Weinstein JN, et al. Characterization of thermal hyperalgesia, c-fos expression, and alterations in neuropeptides after mechanical irritation of the dorsal root ganglion. Spine (Phila Pa 1976). 1995;20:277-289. discussion 290
52 Kawakami M, Weinstein JN, Spratt KF, et al. Experimental lumbar radiculopathy: Immunohistochemical and quantitative demonstrations of pain induced by lumbar nerve root irritation of the rat. Spine (Phila Pa 1976). 1994;19:1780-1794.
53 Kawakami M, Weinstein JN, Chatani K, et al. Experimental lumbar radiculopathy: Behavioral and histologic changes in a model of radicular pain after spinal nerve root irritation with chromic gut ligatures in the rat. Spine (Phila Pa 1976). 1994;19:1795-1802.
54 Olmarker K, Myers RR. Pathogenesis of sciatic pain: Role of herniated nucleus pulposus and deformation of spinal nerve root and dorsal root ganglion. Pain. 1998;78:99-105.
55 Olmarker K, Nutu M, Storkson R. Changes in spontaneous behavior in rats exposed to experimental disc herniation are blocked by selective TNF-alpha inhibition. Spine (Phila Pa 1976). 2003;28:1635-1641.
56 Olmarker K, Iwabuchi M, Larsson K, et al. Walking analysis of rats subjected to experimental disc herniation. Eur Spine J. 1998;7:394-399.
57 Takahashi K, Shima I, Porter RW. Nerve root pressure in lumbar disc herniation. Spine (Phila Pa 1976). 1999;24:2003-2006.
58 Myers R, Shubayev VI, Campana WM. Neuropathology of painful neuropathies. In: Sommer C, editor. Pain in Peripheral Nerve Disease. Basel: Karger; 2001:8-30.
59 Powell HC, Myers RR. Pathology of experimental nerve compression. Lab Invest. 1986;55:91-100.
60 Myers RR, Yamamoto T, Yaksh TL, et al. The role of focal nerve ischemia and Wallerian degeneration in peripheral nerve injury producing hyperesthesia. Anesthesiology. 1993;78:308-316.
61 Sommer C, Galbraith JA, Heckman HM, et al. Pathology of experimental compression neuropathy producing hyperesthesia. J Neuropathol Exp Neurol. 1993;52:223-233.
62 Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: From Augustus Waller’s observations to neuroinflammation. J Peripher Nerv Syst. 2002;7:13-27.
63 Myers R, Wagner R, Sorkin LS. Hyperalgesic action of cytokines on peripheral nerves. In: Watkins LR, Maier SF, editors. Cytokines and Pain. Basel: Birkhäuser Verlag; 1999:133-157.
64 Myers RR, Heckman HM, Powell HC. Axonal viability and the persistence of thermal hyperalgesia after partial freeze lesions of nerve. J Neurol Sci. 1996;139:28-38.
65 Olmarker K, Nordborg C, Larsson K, et al. Ultrastructural changes in spinal nerve roots induced by autologous nucleus pulposus. Spine (Phila Pa 1976). 1996;21:411-414.
66 Kayama S, Konno S, Olmarker K, et al. Incision of the anulus fibrosus induces nerve root morphologic, vascular, and functional changes: An experimental study. Spine (Phila Pa 1976). 1996;21:2539-2543.
67 Byrod G, Otani K, Brisby H, et al. Methylprednisolone reduces the early vascular permeability increase in spinal nerve roots induced by epidural nucleus pulposus application. J Orthop Res. 2000;18:983-987.
68 Yabuki S, Kikuchi S, Olmarker K, et al. Acute effects of nucleus pulposus on blood flow and endoneurial fluid pressure in rat dorsal root ganglia. Spine (Phila Pa 1976). 1998;23:2517-2523.
69 Byrod G, Rydevik B, Nordborg C, et al. Early effects of nucleus pulposus application on spinal nerve root morphology and function. Eur Spine J. 1998;7:445-449.
70 Olmarker K, Blomquist J, Stromberg J, et al. Inflammatogenic properties of nucleus pulposus. Spine (Phila Pa 1976). 1995;20:665-669.
71 Takino T, Takahashi K, Miyazaki T, et al: Immunoreactivity of nucleus pulposus. Presented at International Society for the Study of the Lumbar Spine, Helsinki, Finland, 1995.
72 Kawakami M, Tamaki T, Matsumoto T, et al. Role of leukocytes in radicular pain secondary to herniated nucleus pulposus. Clin Orthop Relat Res. 2000;376:268-277.
73 Myers RR, Heckman HM, Rodriguez M. Reduced hyperalgesia in nerve-injured WLD mice: Relationship to nerve fiber phagocytosis, axonal degeneration, and regeneration in normal mice. Exp Neurol. 1996;141:94-101.
74 Kawakami M, Matsumoto T, Hashizume H, et al. Epidural injection of cyclooxygenase-2 inhibitor attenuates pain-related behavior following application of nucleus pulposus to the nerve root in the rat. J Orthop Res. 2002;20:376-381.
75 Macnab I. Negative disc exploration: An analysis of the causes of nerve-root involvement in sixty-eight patients. J Bone Joint Surg Am. 1971;53:891-903.
76 Crock HV. Observations on the management of failed spinal operations. J Bone Joint Surg Br. 1976;58:193-199.
77 Jonsson B, Stromqvist B. Clinical appearance of contained and noncontained lumbar disc herniation. J Spinal Disord. 1996;9:32-38.
78 Ito T, Takano Y, Yuasa N. Types of lumbar herniated disc and clinical course. Spine (Phila Pa 1976). 2001;26:648-651.
79 Nygaard OP, Mellgren SI, Osterud B. The inflammatory properties of contained and noncontained lumbar disc herniation. Spine (Phila Pa 1976). 1997;22:2484-2488.
80 Olmarker K, Storkson R, Berge OG. Pathogenesis of sciatic pain: A study of spontaneous behavior in rats exposed to experimental disc herniation. Spine (Phila Pa 1976). 2002;27:1312-1317.
81 Kuslich SD, Ulstrom CL, Michael CJ. The tissue origin of low back pain and sciatica: A report of pain response to tissue stimulation during operations on the lumbar spine using local anesthesia. Orthop Clin North Am. 1991;22:181-187.
82 Anzai H, Hamba M, Onda A, et al. Epidural application of nucleus pulposus enhances nociresponses of rat dorsal horn neurons. Spine (Phila Pa 1976). 2002;27:E50-E55.
83 Olmarker K, Rydevik B. Pathophysiology of sciatica. Orthop Clin North Am. 1991;22:223-234.
84 El-Mahdi MA, Abdel Latif FY, Janko M. The spinal nerve root “innervation,” and a new concept of the clinicopathological interrelations in back pain and sciatica. Neurochirurgia (Stuttg). 1981;24:137-141.
85 Edgar MA, Nundy S. Innervation of the spinal dura. J Neurol Neurosurg Psychiatry. 1966;29:530-534.
86 Kaplan EB. Recurrent meningeal branch of the spinal nerve. Bull Hosp Joint Dis. 1947.
87 von Luschka H: Die Nerven des Menschen, 1850.
88 Rudinger N. Die Gelenkennerven des menschlichen Körpers. Erlangen: Ferdinand Enke; 1857.
89 Olmarker K. Experimental basis of sciatica. J Orthop Sci. 1996;1:230-242.
90 Dyck PJ, Karnes J, Lais A, et al. Pathologic alterations of the peripheral nervous system of humans. In: Dyck PJ, Thomas PK, Lambert EH, et al, editors. Peripheral Neuropathy. Philadelphia: WB Saunders; 1984:828-930.
91 Hoyland JA, Freemont AJ, Jayson MI. Intervertebral foramen venous obstruction: A cause of periradicular fibrosis? Spine (Phila Pa 1976). 1989;14:558-568.
92 Cooper RG, Freemont AJ, Hoyland JA, et al. Herniated intervertebral disc-associated periradicular fibrosis and vascular abnormalities occur without inflammatory cell infiltration. Spine (Phila Pa 1976). 1995;20:591-598.
93 Jayson MI, Keegan A, Million R, et al. A fibrinolytic defect in chronic back pain syndromes. Lancet. 1984;2:1186-1187.
94 Klimiuk PS, Pountain GD, Keegan AL, et al. Serial measurements of fibrinolytic activity in acute low back pain and sciatica. Spine (Phila Pa 1976). 1987;12:925-928.
95 Byrod G, Olmarker K, Konno S, et al. A rapid transport route between the epidural space and the intraneural capillaries of the nerve roots. Spine (Phila Pa 1976). 1995;20:138-143.
96 Dalcanto MC, Wisniewski HM, Johnson AB, et al. Vesicular disruption of myelin in autoimmune demyelination. J Neurol Sci. 1975;24:313-319.
97 Hahn AF, Gilbert JJ, Feasby TE. Passive transfer of demyelination by experimental allergic neuritis serum. Acta Neuropathol (Berl). 1980;49:169-176.
98 Bisla RS, Marchisello PJ, Lockshin MD, et al. Auto-immunological basis of disk degeneration. Clin Orthop Relat Res. 1976;121:205-211.
99 Bobechko WP, Hirsch C. Auto-immune response to nucleus pulposus in the rabbit. J Bone Joint Surg Br. 1965;47:574-580.
100 Gertzbein SD, Tile M, Gross A, Falk R. Autoimmunity in degenerative disc disease of the lumbar spine. Orthop Clin North Am. 1975;6:67-73.
101 Gertzbein SD. Degenerative disk disease of the lumbar spine: Immunological implications. Clin Orthop Relat Res. 1977;129:68-71.
102 Gertzbein SD, Tait JH, Devlin SR. The stimulation of lymphocytes by nucleus pulposus in patients with degenerative disk disease of the lumbar spine. Clin Orthop Relat Res. 1977;123:149-154.
103 LaRocca H. New horizons in research on disc disease. Orthop Clin North Am. 1971;2:521-531.
104 Naylor A. The biophysical and biochemical aspects of intervertebral disc herniation and degeneration. Ann R Coll Surg Engl. 1962;31:91-114.
105 Geiss A, Larsson K, Rydevik B, et al. Autoimmune properties of nucleus pulposus: An experimental study in pigs. Spine (Phila Pa 1976). 2007;32:168-173.
106 Satoh K, Konno S, Nishiyama K, et al. Presence and distribution of antigen-antibody complexes in the herniated nucleus pulposus. Spine (Phila Pa 1976). 1999;24:1980-1984.
107 Bayliss MT, Johnstone B. Biochemistry of the intervertebral disc. In: Jayson MIV, editor. The Lumbar Spine and Back Pain. Edinburgh: Churchill-Livingstone; 1992:111-131.
108 Eyre D, Benya P, Buckwalter J. Intervertebral disc. In: Frymoyer JW, Gordon SL, editors. New Perspectives on Low Back Pain. Rosemont: American Academy of Orthopaedic Surgeons; 1988:149-207.
109 Marshall LL, Trethewie ER. Chemical irritation of nerve-root in disc prolapse. Lancet. 1973;2:320.
110 Marshall LL, Trethewie ER, Curtain CC. Chemical radiculitis: A clinical, physiological and immunological study. Clin Orthop Relat Res. 1977;129:61-67.
111 Kayama S, Olmarker K, Larsson K, et al. Cultured, autologous nucleus pulposus cells induce structural and functional changes in spinal nerve roots. Spine (Phila Pa 1976). 1998;23:2155-2158.
112 Brisby H, Byrod G, Olmarker K, et al. Nitric oxide as a mediator of nucleus pulposus-induced effects on spinal nerve roots. J Orthop Res. 2000;18:815-820.
113 Diamant B, Karlsson J, Nachemson A. Correlation between lactate levels and pH in discs of patients with lumbar rhizopathies. Experientia. 1968;24:1195-1196.
114 Nachemson A. Intradiscal measurements of pH in patients with lumbar rhizopathies. Acta Orthop Scand. 1969;40:23-42.
115 Pennington JB, McCarron RF, Laros GS. Identification of IgG in the canine intervertebral disc. Spine (Phila Pa 1976). 1988;13:909-912.
116 Saal JS, Franson RC, Dobrow R, et al. High levels of inflammatory phospholipase A2 activity in lumbar disc herniations. Spine (Phila Pa 1976). 1990;15:674-678.
117 Olmarker K, Larsson K. Tumor necrosis factor alpha and nucleus-pulposus-induced nerve root injury. Spine (Phila Pa 1976). 1998;23:2538-2544.
118 Chao CC, Hu S, Ehrlich L, et al. Interleukin-1 and tumor necrosis factor-alpha synergistically mediate neurotoxicity: Involvement of nitric oxide and of N-methyl-D-aspartate receptors. Brain Behav Immun. 1995;9:355-365.
119 Gadient RA, Cron KC, Otten U. Interleukin-1 beta and tumor necrosis factor-alpha synergistically stimulate nerve growth factor (NGF) release from cultured rat astrocytes. Neurosci Lett. 1990;117:335-340.
120 Bluthe RM, Dantzer R, Kelley KW. Interleukin-1 mediates behavioural but not metabolic effects of tumor necrosis factor alpha in mice. Eur J Pharmacol. 1991;209:281-283.
121 McHale JF, Harari OA, Marshall D, et al. TNF-alpha and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J Immunol. 1999;163:3993-4000.
122 Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259-1265.
123 McGee DW, Bamberg T, Vitkus SJ, et al. A synergistic relationship between TNF-alpha, IL-1 beta, and TGF-beta 1 on IL-6 secretion by the IEC-6 intestinal epithelial cell line. Immunology. 1995;86:6-11.
124 Wagner R, Myers RR. Schwann cells produce tumor necrosis factor alpha: Expression in injured and non-injured nerves. Neuroscience. 1996;73:625-629.
125 Satomi N, Haranaka K, Kunii O. Research on the production site of tumor necrosis factor (TNF). Jpn J Exp Med. 1981;51:317-322.
126 Bachwich PR, Lynch JP3rd, Larrick J, et al. Tumor necrosis factor production by human sarcoid alveolar macrophages. Am J Pathol. 1986;125:421-425.
127 Robbins DS, Shirazi Y, Drysdale BE, et al. Production of cytotoxic factor for oligodendrocytes by stimulated astrocytes. J Immunol. 1987;139:2593-2597.
128 Sayers TJ, Macher I, Chung J, et al. The production of tumor necrosis factor by mouse bone marrow-derived macrophages in response to bacterial lipopolysaccharide and a chemically synthesized monosaccharide precursor. J Immunol. 1987;138:2935-2940.
129 Sommer C, Marziniak M, Myers RR. The effect of thalidomide treatment on vascular pathology and hyperalgesia caused by chronic constriction injury of rat nerve. Pain. 1998;74:83-91.
130 Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7:2897-2901.
131 Igarashi T, Kikuchi S, Shubayev V, et al. 2000 Volvo Award winner in basic science studies: Exogenous tumor necrosis factor-alpha mimics nucleus pulposus-induced neuropathology: Molecular, histologic, and behavioral comparisons in rats. Spine (Phila Pa 1976). 2000;25:2975-2980.
132 Liberski PP, Yanagihara R, Nerurkar V, et al. Further ultrastructural studies of lesions produced in the optic nerve by tumor necrosis factor alpha (TNF-alpha): A comparison with experimental Creutzfeldt-Jakob disease. Acta Neurobiol Exp (Warsz). 1994;54:209-218.
133 Madigan MC, Sadun AA, Rao NS, et al. Tumor necrosis factor-alpha (TNF-alpha)-induced optic neuropathy in rabbits. Neurol Res. 1996;18:176-184.
134 Redford EJ, Hall SM, Smith KJ. Vascular changes and demyelination induced by the intraneural injection of tumour necrosis factor. Brain. 1995;118(Pt 4):869-878.
135 Selmaj K, Raine CS. Tumor necrosis factor mediates myelin damage in organotypic cultures of nervous tissue. Ann N Y Acad Sci. 1988;540:568-570.
136 Stoll G, Jung S, Jander S, et al. Tumor necrosis factor-alpha in immune-mediated demyelination and Wallerian degeneration of the rat peripheral nervous system. J Neuroimmunol. 1993;45:175-182.
137 Nawroth P, Handley D, Matsueda G, et al. Tumor necrosis factor/cachectin-induced intravascular fibrin formation in meth A fibrosarcomas. J Exp Med. 1988;168:637-647.
138 van der Poll T, Jansen PM, Van Zee KJ, et al. Tumor necrosis factor-alpha induces activation of coagulation and fibrinolysis in baboons through an exclusive effect on the p55 receptor. Blood. 1996;88:922-927.
139 Watts ME, Arnold S, Chaplin DJ. Changes in coagulation and permeability properties of human endothelial cells in vitro induced by TNF-alpha or 5,6 MeXAA. Br J Cancer. 1996;74(Suppl 27):S164-S167.
140 Viviani B, Corsini E, Galli CL, et al. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271-276.
141 Wuthrich RP, Jevnikar AM, Takei F, et al. Intercellular adhesion molecule-1 (ICAM-1) expression is upregulated in autoimmune murine lupus nephritis. Am J Pathol. 1990;136:441-450.
142 Sommer C, Schmidt C, George A, et al. A metalloprotease-inhibitor reduces pain associated behavior in mice with experimental neuropathy. Neurosci Lett. 1997;237:45-48.
143 Sorkin LS, Xiao WH, Wagner R, et al. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255-262.
144 Shubayev VI, Myers RR. Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res. 2000;855:83-89.
145 Shubayev VI, Myers RR. Axonal transport of TNF-alpha in painful neuropathy: Distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001;114:48-56.
146 Mattila P, Majuri ML, Mattila PS, et al. TNF alpha-induced expression of endothelial adhesion molecules, ICAM-1 and VCAM-1, is linked to protein kinase C activation. Scand J Immunol. 1992;36:159-165.
147 Pober JS. Effects of tumour necrosis factor and related cytokines on vascular endothelial cells. Ciba Found Symp. 1987;131:170-184.
148 Creange A, Barlovatz-Meimon G, Gherardi RK. Cytokines and peripheral nerve disorders. Eur Cytokine Netw. 1997;8:145-151.
149 Munro JM, Pober JS, Cotran RS. Tumor necrosis factor and interferon-gamma induce distinct patterns of endothelial activation and associated leukocyte accumulation in skin of Papio anubis. Am J Pathol. 1989;135:121-133.
150 Oku N, Araki R, Araki H, et al. Tumor necrosis factor-induced permeability increase of negatively charged phospholipid vesicles. J Biochem (Tokyo). 1987;102:1303-1310.
151 Kagan BL, Baldwin RL, Munoz D, et al. Formation of ion-permeable channels by tumor necrosis factor-alpha. Science. 1992;255:1427-1430.
152 Baldwin RL, Stolowitz ML, Hood L, et al. Structural changes of tumor necrosis factor alpha associated with membrane insertion and channel formation. Proc Natl Acad Sci U S A. 1996;93:1021-1026.
153 Wei Y, Babilonia E, Pedraza PL, et al. Acute application of TNF stimulates apical 70-pS K+ channels in the thick ascending limb of rat kidney. Am J Physiol Renal Physiol. 2003;285:F491-F497.
154 Woolf CJ. The pathophysiology of peripheral neuropathic pain-abnormal peripheral input and abnormal central processing. Acta Neurochir Suppl (Wien). 1993;58:125-130.
155 Attal N, Bouhassira D. Mechanisms of pain in peripheral neuropathy. Acta Neurol Scand Suppl. 1999;173:12-24. discussion 48-52
156 Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23-37.
157 Wall PD. Neuropathic pain and injured nerve: central mechanisms. Br Med Bull. 1991;47:631-643.
158 Selmaj K, Raine CS, Cross AH. Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694-700.
159 Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339-346.
160 Villarroya H, Violleau K, Ben Younes-Chennoufi A, et al. Myelin-induced experimental allergic encephalomyelitis in Lewis rats: Tumor necrosis factor alpha levels in serum and cerebrospinal fluid immunohistochemical expression in glial cells and macrophages of optic nerve and spinal cord. J Neuroimmunol. 1996;64:55-61.
161 Ghabriel MN, Allt G. Schmidt-Lanterman incisures: I. A quantitative teased fibre study of remyelinating peripheral nerve fibres. Acta Neuropathol (Berl). 1980;52:85-95.
162 Shanklin WM, Azzam NA. Histological and histochemical studies on the incisures of Schmidt-Lanterman. J Comp Neurol. 1964;123:5-10.
163 Robertson JD. The ultrastructure of Schmidt-Lanterman clefts and related shearing defects of the myelin sheath. J Biophys Biochem Cytol. 1958;4:39-46.
164 Todd BA, Inman C, Sedgwick EM, et al. Ionic permeability of the frog sciatic nerve perineurium: Parallel studies of potassium and lanthanum penetration using electrophysiological and electron microscopic techniques. J Neurocytol. 2000;29:551-567.
165 Brisby H, Olmarker K, Rosengren L, et al. Markers of nerve tissue injury in the cerebrospinal fluid in patients with lumbar disc herniation and sciatica. Spine (Phila Pa 1976). 1999;24:742-746.
166 Cornefjord M, Nyberg F, Rosengren L, et al. Cerebrospinal fluid biomarkers in experimental spinal nerve root injury. Spine (Phila Pa 1976). 2004;29:1862-1868.
167 Brisby H, Balague F, Schafer D, et al. Glycosphingolipid antibodies in serum in patients with sciatica. Spine (Phila Pa 1976). 2002;27:380-386.
168 Schafers M, Sorkin LS, Geis C, et al. Spinal nerve ligation induces transient upregulation of tumor necrosis factor receptors 1 and 2 in injured and adjacent uninjured dorsal root ganglia in the rat. Neurosci Lett. 2003;347:179-182.
169 Schafers M, Lee DH, Brors D, et al. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. 2003;23:3028-3038.
170 Schafers M, Svensson CI, Sommer C, et al. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517-2521.
171 Winkelstein BA, Rutkowski MD, Sweitzer SM, et al. Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. J Comp Neurol. 2001;439:127-139.
172 Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980-9989.
173 DeLeo JA, Rutkowski MD, Stalder AK, et al. Transgenic expression of TNF by astrocytes increases mechanical allodynia in a mouse neuropathy model. Neuroreport. 2000;11:599-602.
174 Olmarker K, Rydevik B. Selective inhibition of tumor necrosis factor-alpha prevents nucleus pulposus-induced thrombus formation, intraneural edema, and reduction of nerve conduction velocity: Possible implications for future pharmacologic treatment strategies of sciatica. Spine (Phila Pa 1976). 2001;26:863-869.
175 Spillert CR, Sun S, Ponnudurai R, et al. Tumor necrosis factor-induced necrosis: A monocyte-mediated hypercoagulable effect. J Natl Med Assoc. 1995;87:508-509.
176 Aderka D. Role of tumor necrosis factor in the pathogenesis of intravascular coagulopathy of sepsis: Potential new therapeutic implications. Isr J Med Sci. 1991;27:52-60.
177 Onda A, Murata Y, Rydevik B, et al. Infliximab attenuates immunoreactivity of brain-derived neurotrophic factor in a rat model of herniated nucleus pulposus. Spine (Phila Pa 1976). 2004;29:1857-1861.
178 Murata Y, Onda A, Rydevik B, et al. Selective inhibition of tumor necrosis factor-alpha prevents nucleus pulposus-induced histologic changes in the dorsal root ganglion. Spine (Phila Pa 1976). 2004;29:2477-2484.
179 Murata Y, Olmarker K, Takahashi I, et al. Effects of selective tumor necrosis factor-alpha inhibition to pain-behavioral changes caused by nucleus pulposus-induced damage to the spinal nerve in rats. Neurosci Lett. 2005;382:148-152.
180 Aoki Y, Rydevik B, Kikuchi S, et al. Local application of disc-related cytokines on spinal nerve roots. Spine (Phila Pa 1976). 2002;27:1614-1617.
181 Ozaktay AC, Cavanaugh JM, Asik I, et al. Dorsal root sensitivity to interleukin-1 beta, interleukin-6 and tumor necrosis factor in rats. Eur Spine J. 2002;11:467-475.
182 Onda A, Yabuki S, Kikuchi S. Effects of neutralizing antibodies to tumor necrosis factor-alpha on nucleus pulposus-induced abnormal nociresponses in rat dorsal horn neurons. Spine (Phila Pa 1976). 2003;28:967-972.
183 Wagner R, Janjigian M, Myers RR. Anti-inflammatory interleukin-10 therapy in CCI neuropathy decreases thermal hyperalgesia, macrophage recruitment, and endoneurial TNF-alpha expression. Pain. 1998;74:35-42.
184 Wehling P, Cleveland SJ, Heininger K, et al. Neurophysiologic changes in lumbar nerve root inflammation in the rat after treatment with cytokine inhibitors: Evidence for a role of interleukin-1. Spine (Phila Pa 1976). 1996;21:931-935.
185 Brisby H, Olmarker K, Larsson K, et al. Proinflammatory cytokines in cerebrospinal fluid and serum in patients with disc herniation and sciatica. Eur Spine J. 2002;11:62-66.
186 Onda A, Murata Y, Rydevik B, et al. Immunoreactivity of brain-derived neurotrophic factor in rat dorsal root ganglion and spinal cord dorsal horn following exposure to herniated nucleus pulposus. Neurosci Lett. 2003;352:49-52.
187 Murata Y, Onda A, Rydevik B, et al. Distribution and appearance of tumor necrosis factor-alpha in the dorsal root ganglion exposed to experimental disc herniation in rats. Spine (Phila Pa 1976). 2004;29:2235-2241.
188 Murata Y, Rydevik B, Takahashi K, et al. Macrophage appearance in the epineurium and endoneurium of dorsal root ganglion exposed to nucleus pulposus. J Peripher Nerv Syst. 2004;9:158-164.
189 Onda A, Murata Y, Rydevik B, et al. Nerve growth factor content in dorsal root ganglion as related to changes in pain behavior in a rat model of experimental lumbar disc herniation. Spine (Phila Pa 1976). 2005;30:188-193.
190 Larsson K, Rydevik B, Olmarker K. Disc related cytokines inhibit axonal outgrowth from dorsal root ganglion cells in vitro. Spine (Phila Pa 1976). 2005;30:621-624.
191 Karppinen J, Korhonen T, Malmivaara A, et al. Tumor necrosis factor-alpha monoclonal antibody, infliximab, used to manage severe sciatica. Spine (Phila Pa 1976). 2003;28:750-753.
192 Korhonen T, Karppinen J, Malmivaara A, et al. Efficacy of infliximab for disc herniation-induced sciatica: One-year follow-up. Spine (Phila Pa 1976). 2004;29:2115-2119.
193 Genevay S, Stingelin S, Gabay C. Efficacy of etanercept in the treatment of acute severe sciatica. Ann Rheum Dis. 2004;63:1120-1123.
194 Cohen SP, Bogduk N, Dragovich A, et al. Randomized, double-blind, placebo-controlled, dose-response, and preclinical safety of transforaminal epidural etanercept for the treatment of sciatica. Anesthesiology. 2009;110:1116-1126.
195 Genevay S, Viatte S, Finck A, et al. Adalimumab in severe and acute sciatica: A multicentre, randomised, double-blind, placebo controlled trial. Arthritis Rheum. April 6, 2010. [E-pub ahead of print]
196 Cooper RG, Freemont AJ. TNF-alpha blockade for herniated intervertebral disc-induced sciatica: A way forward at last? [editorial]. Rheumatology. 2004;43:119-121.
197 Genevay S, Gabay C. Is disk-related sciatica a TNFα-dependent inflammatory disease? Joint Bone Spine. 2005;72:4-6.