[level-membership-for-pulmolory-and-respiratory-category]
14 Sarcomas and Sarcomatoid Neoplasms of the Lungs and Pleural Surfaces
This chapter summarizes the clinicopathologic information pertaining to such lesions. It devotes exclusive attention to malignant lesions; benign mesenchymal neoplasms of the lung and pleura are discussed in Chapter 19. Pseudotumors are likewise considered in another portion of this book. Nevertheless, those pathologic categories and other lesions will indeed be mentioned here in the context of differential diagnosis. The entities that are discussed are arranged in order of frequency, to give the reader a sense of their relative incidences.
Sarcomatoid Carcinoma of the Lung
Recent changes in the classification of lung tumors have included sarcoma-like tumors. Five subtypes of those lesions are now codified; pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and pulmonary blastoma are predicated on the particulars of their microscopic appearances.1,2 We consider all of these neoplasms to be part of the same tumor family, that of sarcomatoid carcinomas (SCs), as discussed in more detail subsequently. Although SCs are rare in an absolute sense, they represent the most common mesenchymal-like malignancies of the airways.3,4 True sarcomas are very infrequently seen in the tracheobronchial tree.4–13 Therefore, one usually considers a cytologically atypical spindle cell tumor of the lung to be an SC unless thorough immunohistological and ultrastructural studies indicate otherwise.5 This review will discuss neoplasms in this general category, using information taken from the pertinent literature as well as the personal experience of the authors.
Historical and Terminologic Considerations
Over time, morphologically similar tumors have been given a variety of designations in the upper and lower respiratory tracts. These diagnostic terms have included blastoma, sarcomatoid carcinoma, spindle cell carcinoma, squamous cell carcinoma with pseudosarcomatous stroma, pseudosarcoma, and carcinosarcoma, based largely on the specific microscopic attributes of the lesions in question and the conceptual leanings of the authors describing them.14–43
More than 70 years ago, Saphir and Vass44 assessed the literature then extant on carcinosarcomas and concluded that they represented primary epithelial malignancies that had undergone divergent differentiation (tumor metaplasia). Their paper cited several lesions of the lung. Thereafter, opposing publications on histogenesis espoused the opinion that biphasic neoplasms of the airways were “collision” tumors, or that they reflected the proliferation of non-neoplastic mesenchymal tissue components that were induced by the carcinomatous elements.45–47 At the turn of the last century, Krompecher48 and others49 had held to the same theories as those of Saphir and Vass. In the past two decades, results of studies using electron microscopy, immunohistology, and “molecular” assays of clonality have tended to support the latter foresighted views of those pioneers convincingly. Hence, it is believed currently that blastomas, carcinosarcomas, carcinomas with pseudosarcomatous stroma, and SCs comprise a single morphological spectrum of basically epithelial tumors, regardless of their anatomic locations.34–36 Biphasic sarcomatoid carcinoma and monophasic sarcomatoid carcinoma have been proposed as replacements for the former designations of carcinosarcoma and spindle cell carcinoma, respectively.33,34,37 In the penultimate iteration of the World Health Organization nosologic scheme, such lesions were included in the category designated “carcinoma with pleomorphic, sarcomatoid, or sarcomatous elements.”36
Clinicopathologic Features of Pulmonary Sarcomatoid Carcinomas
Clinical Attributes
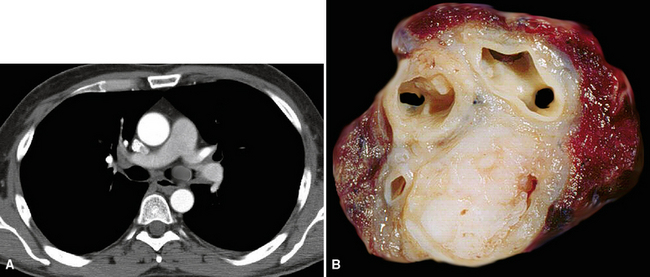
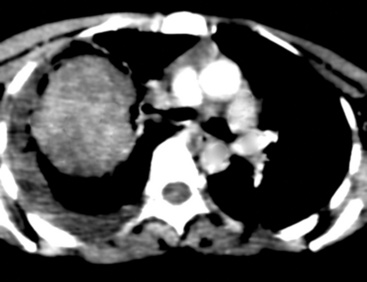
SCs arise in the large bronchi and peripheral lung fields much more often than in the trachea, although the authors have indeed seen some lesions that originated above the carina. The majority of individuals with pulmonary SC are men, and most have a history of heavy smoking.14,23,24 The average patient is 60 years of age.33 Clinical signs and symptoms produced by these tumors are directly associated with the tumor’s specific location. Endoluminal lesions in large tubular airways characteristically cause refractory or recurrent pneumonia in the corresponding distal parenchyma, or they are associated with progressive dyspnea, cough, hemoptysis, and audible expiratory rhonchi over the affected lung field.14–17,23,45–47,50,51 In contrast, SC in the peripheral lung often manifests no symptoms or, alternatively, leads to chest pain caused by invasion of the pleura and extrapulmonary soft tissue.23 As might be expected, central endobronchial tumors are smaller than peripheral SCs; their average sizes are 6 cm and greater than 10 cm, respectively46,52 (Fig. 14-1).
Despite their anaplastic nature, SCs of the lung are surgically resectable in roughly 90% of cases,23,33 and approximately one half of patients with such neoplasms present with stage I disease. Paradoxically, however, the prognosis of pulmonary SC is still dismal. Overall 5-year survival is 20%, with a slightly better figure being associated with small, central endobronchial lesions.23,33,52 Metastatic SC of the lung involves the same organ sites that are affected by more usual forms of lung cancer: namely, the opposite lung, liver, bones, adrenal glands, and brain.46,51 The metastases may exhibit either carcinomatous or sarcoma-like histologic configurations, or both.46 Adjuvant radiation treatment and chemotherapy have been used in many cases of pulmonary SC, but these measures have provided little benefit in general.14,33,38
Macroscopic Features
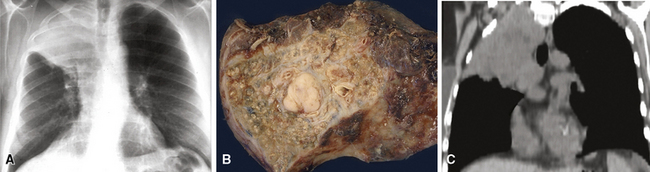
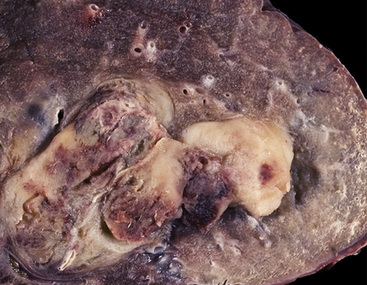
Grossly, the lesions of pulmonary SC that are greater than 5 cm in size tend to exhibit central necrosis and hemorrhage, and they also demonstrate irregular permeation of the surrounding lung parenchyma33,34 (Fig. 14-2). Tumors that are smaller and located within bronchial confines often exhibit a polypoid appearance and are attached to the subjacent mucosa by a relatively narrow stalk of tissue.52 SCs in the peripheral lung parenchyma may have the gross appearance of conventional adenocarcinomas.34
Histologic Characteristics
Homologous Biphasic Sarcomatoid Carcinoma
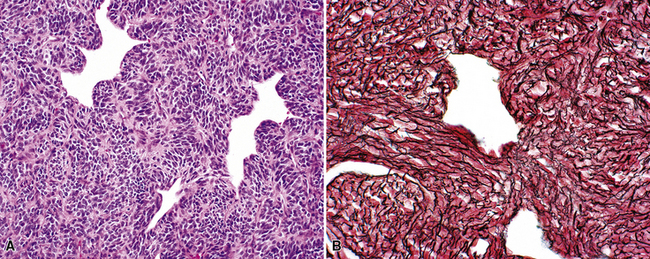
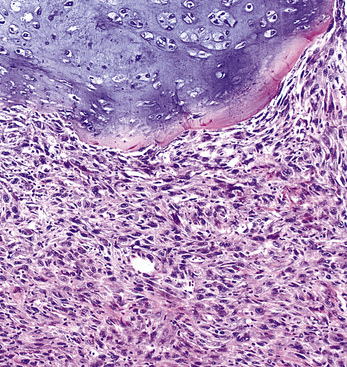
Variants of SC that are also called spindle cell carcinomas are constituted microscopically by a predominance of nondescript spindled and pleomorphic cells, admixed with a minor, obviously carcinomatous, component. The latter portion of such lesions is generally inconspicuous and variably distributed; in roughly 40% of cases, such foci are very rare and require extensive sampling to document their presence. The general appearance of the carcinomatous elements is that of a well- to moderately differentiated squamous malignancy in most instances, whereas adenosquamous, adenocarcinomatous, large cell undifferentiated, or neuroendocrine carcinoma is seen in a minority of cases14,53–56 (Fig. 14-3). Rare examples of this tumor type show mixtures of several carcinoma morphotypes.54 Zones of transition between epithelial and sarcoma-like components are usually evident, at least focally.
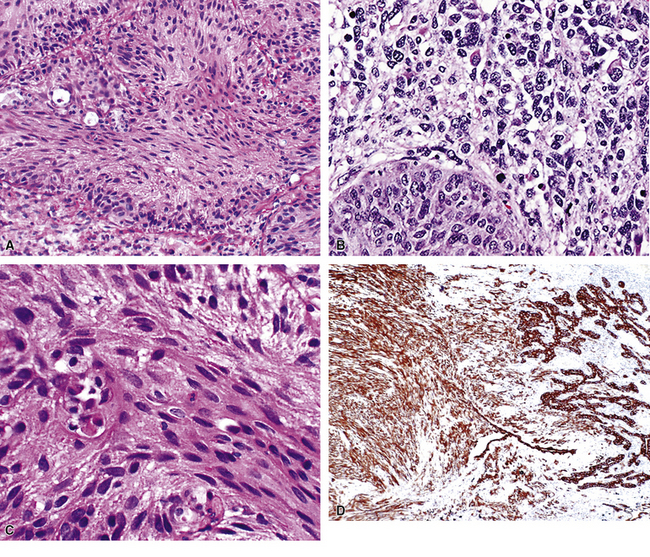
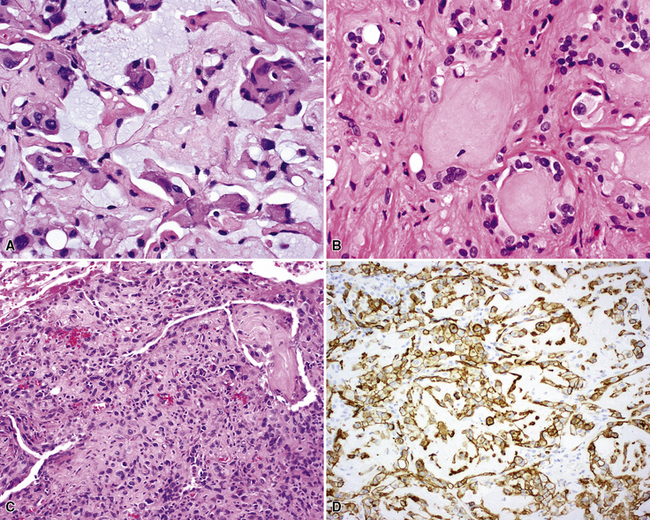
The sarcomatoid elements of this subtype of SC lack specialized differentiation into identifiable myogenic, chondro-osseous, or vasoformative tissues by standard light microscopy, and, as such, are homologous (organ-appropriate) to the lung. They are composed of markedly heterogeneous cells with nuclear atypia and variable growth patterns. The corresponding microscopic images range from those of fibromatosis-like or low-grade fibrosarcoma-like areas, with relatively bland nuclear features, sparse mitoses, moderate-to-rich matrical collagen deposition, and a “herringbone” pattern, to others in which pleomorphic giant cells are mixed with fusiform elements showing dense cellularity, coarse chromatin, prominent nucleoli, and numerous mitoses14–16,23,33,45 (Fig. 14-4). The last of these descriptions is closely similar to that attending spindle cell–pleomorphic malignant fibrous histiocytoma (MFH) of the soft tissues.54 Neoplastic spindle cells often infiltrate the submucosa of small- and medium-sized bronchi, which nonetheless tend to retain their mural cartilage plates and mucosal integrity.33,34
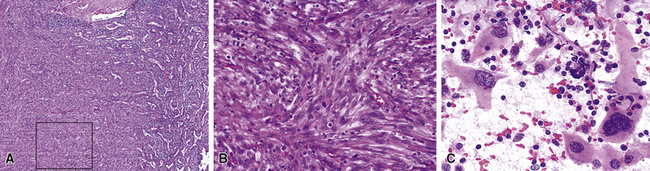
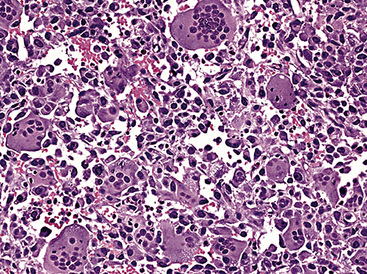
Sarcoma-like elements in some biphasic spindle cell carcinomas may include cytologically bland, osteoclast-like giant cells52,57,58 (Fig. 14-5). The latter are admixed with atypical fusiform cells or more uniform polygonal tumor cells. Another variant histologic pattern is that in which bluntly fusiform tumor cells surround discrete zones of necrosis, producing a necrotizing granuloma-like image. Rare examples of SC may demonstrate extravasated erythrocytes between relatively bland spindled tumor cells, simulating the characteristics of Kaposi sarcoma (KS) or even nodular fasciitis.33
Heterologous Biphasic Sarcomatoid Carcinoma
Other biphasic sarcomatoid neoplasms differ from the descriptions just given, in regard to their content of focal myogenous, vasogenic, chondro-osseous, or lipogenic differentiation.59 Thus, they are analogous to the heterologous form of malignant mixed müllerian tumors of the uterus, ovaries, and other female genital sites.60,61 Those neoplasms may exhibit microscopic foci that simulate embryonal or adult-type pleomorphic rhabdomyosarcoma, which contain proliferations of closely apposed compact round cells with a slightly myxoid background, or large “strap” cells with cytoplasmic eosinophilia and cross-striations, respectively14–16,33,34,46,52 (Fig. 14-6). Other heterologous SCs contain components that closely imitate the histologic features of osteosarcoma or chondrosarcoma.23,34 In light of this information, it is easy to understand why lesions with such microscopic features were believed to be carcinosarcomas in the past and are still so designated by some observers today. The obviously carcinomatous elements in these lesions usually take the form of squamous carcinoma, but lesions with glandular or neuroendocrine differentiation have also been reported.14,54,55 Transitional zones between obvious epithelial foci, nondescript sarcomatoid areas, and myosarcoma-like components are often evident.
With regard to the relationship between biologic behavior and histologic appearance, there is no difference in the clinical evolution of homologous and heterologous biphasic pulmonary SCs. A distinction is made between those lesions only to reflect their synonymity with sarcomatoid epithelial tumors in other body sites.60,61
Monophasic Sarcomatoid Carcinomas
Some SCs display no conventional light microscopic evidence of epithelial differentiation whatsoever. A carcinomatous nature for these neoplasms is discerned only after immunohistochemical or ultrastructural evaluations have been done, but it is usually suspected beforehand because of the clinical and gross characteristics of the lesions.33
Most tumors in this category are constituted exclusively of cell populations like those in the sarcoma-like components of biphasic SCs. These potentially include foci that have an unremarkable spindle cell or pleomorphic image (Fig. 14-7) as well as areas imitating rhabdomyosarcoma, osteosarcoma, or other morphologic appearances that do not correspond to native tissues in the non-neoplastic lung. Because of the monomorphic nature of the tumor variants under discussion here, which lack any attributes of conventional lung carcinomas histologically, the corresponding diagnosis suggested by the World Health Organization criteria for pulmonary neoplasms62 (based only on hematoxylin and eosin stains and conventional histologic examination) would be that of a primary pulmonary sarcoma. The latter point has made the existence of monophasic SC of the lung somewhat contentious. Nevertheless, we have no doubt of its validity as a reproducible pathologic entity, and other authors appear to concur.35
Indeed, one might go so far as to state that virtually all malignant pulmonary tumors that are exclusively composed of heterologous (organ-inappropriate) elements—such as osteoblastic tissues63,64—are, in reality, monophasic SCs. That statement pertains even if no immunohistochemical evidence of epithelial differentiation can be found, because the ultimate biological evolution of such lesions is identical to that of conventional lung cancers.
Special Variants of Sarcomatoid Carcinoma of the Lung
Pulmonary Blastoma
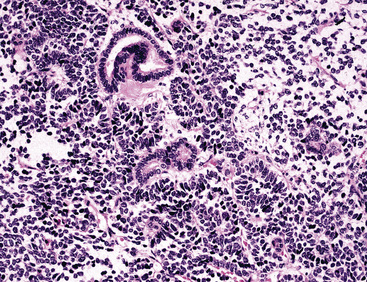
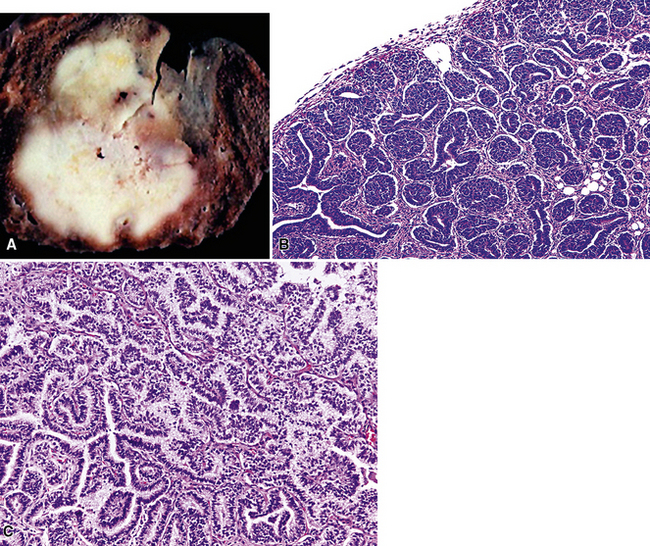
Since its initial description by Barnett and Barnard65 and a later discussion by Spencer,66 pulmonary blastoma (PB) has been regarded by some observers as the pulmonary counterpart of primitive childhood tumors of other organs.67–73 This view has been fostered in part by confusion of PB with pleuropulmonary blastoma (PPB; discussed subsequently), the latter of which is primarily seen in adolescent patients.74–80 PB is a biphasic neoplasm, containing a mixture of tubular epithelial cell profiles and compact groupings of nondescript bluntly fusiform cells with a blastema-like configuration39,74,77 (Fig. 14-8). These resemble the elements of renal Wilms tumors.81 On the other hand, PPB altogether lacks epithelial differentiation and may instead show divergent mesenchymal differentiation into myogenous or chondro-osseous tissues.77 Moreover, PB shows no particular disease associations, whereas PPB is linked in a familial fashion to a number of other malignant neoplasms and non-neoplastic disorders.79
If one carefully excludes examples of PPB from consideration, it becomes clear that PB is seen overwhelmingly in adults, and its clinical characteristics are superimposable on those of ordinary lung cancers and other pulmonary SCs. This realization allows one to more easily embrace an alternative view of the nature of PB that was advanced in the past by Souza and colleagues,82 Stackhouse and associates,14 and Millard,83 among others. Those authors held the opinion that PB is merely a special, usually peripheral form of pulmonary SC (carcinosarcoma), rather than a blastemal neoplasm that contains truly embryonal tissues. We also espouse the latter premise.
Returning to the particular microscopic attributes of PB, it should be noted that this tumor may demonstrate the same range of epithelial and mesenchymoid differentiation that is seen in other biphasic pulmonary SCs.39,69,84 The epithelial elements in PB resemble fetal pulmonary pseudoglands (a misnomer), composed of stratified columnar cells with glycogen-rich clear cytoplasm and high nuclear-to-cytoplasmic ratios.69,84–86 Luminal mucin may be present in those cellular arrays, and squamous morules are sometimes also evident.69 Interestingly, “occult” neuroendocrine differentiation is a rather common finding in the epithelial components of PB, with potential histochemical argyrophilia and immunoreactivity for neuroendocrine markers.87,88 There is a significant sharing of microscopic features between classic PB and the tumor described as “pulmonary endodermal tumor resembling fetal lung” or alternatively as “well-differentiated adenocarcinoma simulating fetal lung” (Fig. 14-9). It differs in its relative lack of a malignant stromal component and more frequent synthesis of a particular oncofetal polypeptide—α-fetoprotein.39,85,87–90
The elements of PB showing mesenchymoid differentiation are, as stated earlier, usually nondescript morphologically and blastema-like or fibroblast-like. However, examples of this tumor have been documented in which heterologous rhabdomyoblastoid, leiomyosarcomatoid, or apparent chondro-osseous tissues were present.69,84 This observation serves to further solidify the linkage of PB to other sarcomatoid pulmonary carcinomas, as do reports of some tumors in which “typical” PB was admixed with homologous or heterologous biphasic SC, as described earlier.82,91–93
The clinical behavior of PB is difficult to determine with certainty, because of the aforementioned contamination of some series with cases of PPB. However, mortality figures of 30% to 70% have been reported, with death usually being due to distant metastases.67,69,84 Secondary deposits of PB may have a purely epithelial, purely mesenchymal-like, or biphasic appearance, as is true of other SCs.
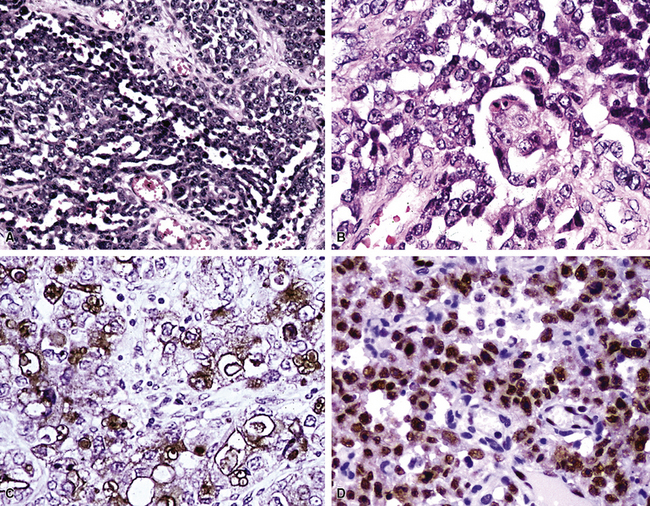
Pseudoangiosarcomatous (Pseudovascular) Carcinoma
The authors have studied several lung tumors in which obvious squamous cell carcinoma was admixed with areas demonstrating interanastomosing channels mantled by anaplastic, plump, epithelioid cells, focally grouped into pseudopapillae. Because the open spaces in these areas contained erythrocytes and focally formed blood lakes, the histologic appearance was that of biphasic SC in which an angiosarcomatoid component was admixed with overt squamous carcinoma.94 (Fig. 14-10). These neoplasms are believed to represent the pulmonary counterparts of pseudovascular adenoid squamous cell carcinoma, as seen in the skin, breast, thyroid gland, and other organs.34,94–99 This is a tumor type that is known to simulate true angiosarcoma but lacks actual endothelial differentiation. Thus, pseudoangiosarcomatous carcinoma (PASC) would be an apt synonym.96
Primary pulmonary angiosarcoma is, comparatively, a very rare lesion, comprising only 10% of true sarcomas of the lung in one report from the Mayo Clinic.2 Mainly anecdotal reports of this tumor exist in the remaining literature, and not all of them satisfy rigorous diagnostic criteria.100–108 Metastases to the lung from angiosarcomas arising in extrapulmonary sites are much more common, including examples that have originated in the heart, great vessels, or extrathoracic viscera.100
Similar to reports on previously cited pseudovascular carcinomas in other body sites, two publications have specifically considered squamous cell carcinomas of the lung that imitated angiosarcomas. The first, by Banerjee and coworkers,96 showed that such tumors produced clinical symptoms and signs resembling those of ordinary types of lung cancer. They presented in the fifth to seventh decades of life; were associated with cigarette smoking, complaints of cough, weight loss, and dyspnea; and were visible on chest radiographs as well-defined central or peripheral parenchymal masses. In another series by Nappi and colleagues,94 the tumors were essentially identical microscopically to pseudovascular squamous carcinomas of the breast and skin. Important differences between PASCs and true pleuropulmonary angiosarcomas include an absence of atypical endothelial cells in stromal blood vessels surrounding the tumor mass in PASCs; less infiltrative growth through the interstitium of the lung; and, perhaps most importantly, the presence of small foci of morphologically obvious squamous cell carcinoma in most PASCs.94
Inflammatory Sarcomatoid Carcinoma
Much attention has been given to a group of space-occupying lesions in the lung that carry the popular but inaccurate designation of inflammatory pseudotumors (IPs).101–114 These proliferations may occur in children or adults and have been divided into fibrohistiocytic, plasma cell granulomatous, and focal organizing pneumonia types, based on their individual clinicopathological features. The nomenclature used for this group of lesions has been well summarized by Koss114 and Matsubara and associates.106 There is still some controversy over whether all such lesions are neoplastic, or whether some might be reactive in nature.115 However, the bulk of available information indicates that IPs are relatively innocuous clinical imitators of overtly malignant neoplasms.114 Occasional cases have been linked causally to specific infectious organisms,116,117 but the etiologic factors associated with most pulmonary IPs are uncertain.
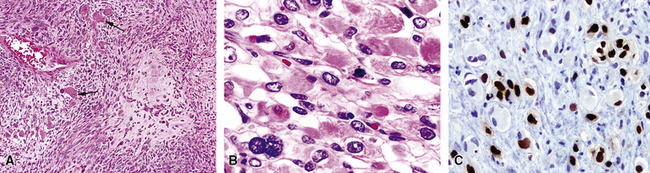
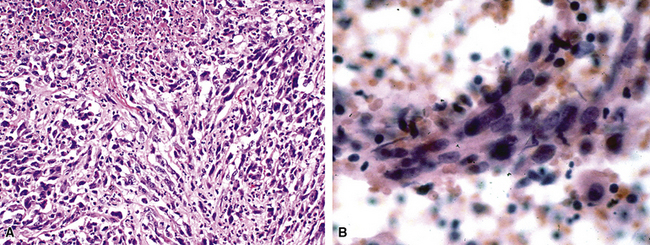
In contrast, primary SC is generally regarded as a neoplasm that may simulate pleuropulmonary mesenchymal malignancies, and it is typically not mentioned in discussions on the pathologic differential diagnosis of IPs. This is so because SC usually demonstrates obvious cytologic anaplasia and lacks a significant component of inflammatory cells. Indeed, the morphologic distinction between IPs and all bronchogenic carcinomas (including SC) has been portrayed by some authors as an uncomplicated process.105,108 However, we have observed several examples of pulmonary SC that exhibited surprisingly bland morphologic appearances, and which, as a result, were separable from IP only by thorough study and adjunctive pathologic techniques. These have been designated as examples of inflammatory sarcomatoid carcinoma (ISC).118,119
Examples of ISC are composed of variably densely apposed spindle cells with only modest pleomorphism, arranged haphazardly or in fascicular and storiform patterns. The stroma is at least partially myxoid in some cases and may be prominently so. The tumors demonstrate an irregular, spiculated interface with the surrounding lung. The adjacent parenchyma exhibits interstitial fibrosis, and small nodular infiltrates of mature lymphocytes are admixed with dense collagenous tissue at the periphery of ISCs (Fig. 14-11). Focally hyalinized, keloidal-type collagen is admixed with the tumor cells in the central portions of some of these tumors, with or without small foci of central necrosis. Vascular invasion and luminal obliteration by neoplastic cells may be apparent. Similarly, bronchial submucosal infiltration is another potential observation. ISCs do not contain appreciable stromal neutrophils, eosinophils, or xanthoma cells, but a moderate number of lymphocytes and plasma cells can be seen.
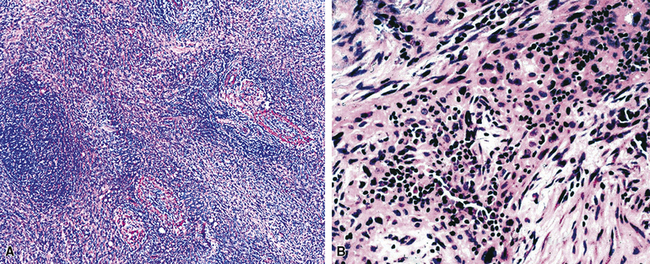
Cytologically, the nuclei of the tumor cells in ISCs are relatively uniform in size and spindle-shaped, with coarse chromatin and occasional small nucleoli (Fig. 14-12). Cytoplasm is moderate in amount and amphophilic. Generally, there are no more than 2 mitoses per 10 high-power fields (×400) on average, and pathologic division figures are absent. Thorough sampling of the tumor tissue in cases of ISC typically demonstrates minute foci of cohesive epithelioid cells, suggesting the diagnosis of squamous carcinoma on conventional histologic grounds. However, some ISCs lack such foci and are recognizable as carcinomas only with special pathologic evaluations (see Fig. 14-12B).118
Pleurotropic (Pseudomesotheliomatous) Sarcomatoid Carcinoma
Occasional examples of SC are distinctive not because of their histologic attributes but because of their macroscopic appearances. In particular, a small subset of these neoplasms arises in the very periphery of the pulmonary parenchyma and grows preferentially into the pleura that encases the lungs.120,121 This produces clinical symptoms and signs that are indistinguishable from those of malignant mesothelioma; hence, the names pleurotropic or pseudomesotheliomatous carcinoma.121,122 Moreover, the microscopic features of pleurotropic SC are basically superimposable with those of biphasic or sarcomatoid mesotheliomas.
Results of Adjunctive Pathologic Studies
Accounts of the electron microscopic and immunohistologic characteristics of pulmonary SC have not been altogether uniform. Some authors have preferred the view that such data in fact confirm the existence of true carcinosarcomas,123–125 whereas others have thought that this information instead supports the concept of a pathologic continuum that is predicated on carcinoma in pure form.15,17,38,126–130 We strongly prefer the second of these opinions.
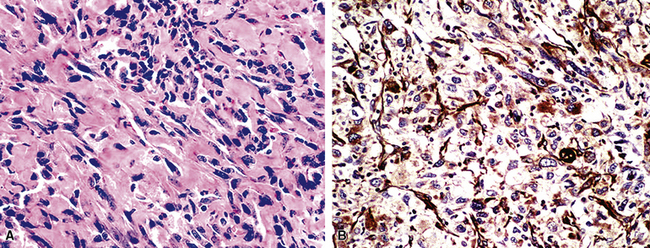
It is true that the sarcoma-like elements in SC of the airways do not uniformly exhibit the ultrastructural presence of intercellular junctions and tonofibrils, or immunoreactivity for keratin or epithelial membrane antigen (EMA) in fusiform and pleomorphic tumor cells. In fact, these generic markers of epithelial differentiation may be seen only extremely focally in such neoplastic components, and we have even seen isolated examples in which cell membrane–based EMA reactivity was obvious, but keratin positivity was altogether absent. Humphrey and coworkers observed cytoplasmic tonofibrils or keratin positivity in the sarcomatoid elements of only three of eight pulmonary SCs.15 However, it should be noted that the latter study was performed with a single heteroantiserum to high-molecular-weight keratin, representing a relatively insensitive means of immunodetection. In our previously published experience with SCs of the respiratory tract,33 81% were ultrastructurally or immunohistochemically proven (with a mixture of monoclonal antikeratin antibodies [AE1/AE3/CAM5.2/MAK-6]) to be wholly epithelial in nature, and other authors have recorded similar findings at immunohistochemical and genetic levels of investigation.126–132
The fact that features of epithelial differentiation are present at all in the sarcomatoid elements of these neoplasms strongly supports the premise that respiratory tract SC is a basically carcinomatous lesion “in transition.”133 This concept has been well-accepted in reference to dedifferentiated sarcomas of the soft tissue, in which clonal evolution is thought to account for a change in the morphology as well as the immunophenotype of the progenitor lesion.134 Lessons learned in the latter sphere—as well as molecular biologic assessments of clonality in SCs135—have direct corollaries in the context under discussion here. We have observed the coexpression of vimentin (a “primordial” intermediate filament) in all examples of keratin-positive SC of the airways, and a minority of these lesions are additionally labeled for desmin (the intermediate filament of myogenous cells) and muscle-specific actin in the same cells that contain the other two filament proteins (Fig. 14-13).33 Collagen type IV is also seen surrounding individual tumor cells in most instances. Markers of neuroendocrine differentiation, such as chromogranin-A, CD57, and synaptophysin, likewise may be seen in selected lesions in their overtly epithelial components,53,55 and S100 protein is apparent in foci resembling chondroid tissue by conventional microscopy.4 In contrast, Friend leukemia-virus integration protein-1 (FLI-1) and CD31 are typically absent in PASC, whereas one or both of those endothelial determinants would be expected in true pleuropulmonary angiosarcomas.95,136
These accrued observations coincide with ultrastructural findings reported by Battifora in two cases of SC, which demonstrated the coincidence of desmosomes, tonofibrils, and collagen production in the same neoplastic cells, implying the presence of multilinear differentiation.137 Thus, SC can be viewed basically as an epithelial neoplasm with divergent mesenchymal differentiation, in which carcinoma cells acquire the potential to express a mesenchymal phenotype at light microscopic, ultrastructural, and immunohistologic levels. The pathogenetic bases for this peculiarity are currently unknown, but the practical deduction to be gleaned from this construct is that all SCs of the respiratory tract should be treated clinically as poorly differentiated carcinomas.
Wakely has reviewed the characteristics of pulmonary spindle cell tumors as seen in fine needle aspiration biopsy specimens138 (see Fig. 14-7). He concluded that adjunctive pathologic studies, such as those discussed above, were virtually mandatory before definitive diagnoses could be reached in that context.
Differential Diagnosis of Sarcomatoid Carcinoma
The differential diagnosis of SCs of the airways principally centers on the exclusion of true sarcomas, which are discussed later in this chapter. As a particular word of caution, it should be noted that synovial sarcoma (SS) and sarcomatoid mesothelioma may be very closely similar to SC as seen with the electron microscope or in immunophenotypic evaluations. The marked propensity for SS to affect children, adolescents, and young adults, its typical t(X;18) (p11.2;q11.2) cytogenetic aberration,139,140 and nuclear labeling for transducin-like enhancer [of Split]-1 (TLE1) protein141–144 (not seen in carcinomas) are crucial points in its distinction from SC of the upper airways. Of course, nuances of histologic appearances and radiographic characteristics are also valuable in this specific differential diagnostic setting. Similarly, roentgenologic findings are more helpful than morphologic observations in making the distinction between SC and spindle cell or biphasic mesothelioma. The ultrastructural profiles of the latter two lesions are again very similar,1,4,145 and, aside from selective reactivity for calretinin and podoplanin146 in mesotheliomas, the same comment applies to their immunophenotypes.
True Primary Sarcomas of the Lung
Kaposi Sarcoma
The natural history of KS is a sad testimony to the global impact of acquired immunodeficiency syndrome (AIDS). Before the 1980s, KS was a relatively rare neoplasm outside of Africa and the Mediterranean basin. Moreover, with relatively uncommon exceptions, this lesion was a cutaneous proliferation that uncommonly involved the viscera.147 However, today, with particular regard to the intrathoracic organs, KS is—in most large metropolitan areas of the world—the most common of all pulmonary sarcomas.148 Whereas initial presentation of this tumor in the bronchopulmonary tract was an almost-unknown phenomenon prior to the advent of AIDS, it is currently a well-recognized variation of the latter disease.149
Clinical Summary
In the context just mentioned, many patients with KS of the lung are homosexual men,150–156 but they also include other high-risk groups for AIDS such as intravenous drug abusers. Most individuals with KS generally have other symptoms and signs of AIDS, such as weight loss, fever, night sweats, fatigue, lymphadenopathy, and opportunistic infections. However, fever may be directly caused by KS in the lungs. There has been a case report of an AIDS patient with persistent pyrexia for which no source of infection was found but that finally resolved after radiation therapy.157 Cutaneous KS is usually detected early in its clinical evolution, but identical tumors of the bronchial mucosa and lung parenchyma typically have grown to a volume sufficient to produce symptoms and therefore are relatively advanced at the time of diagnosis.158 Presenting complaints specific to the neoplasm include dyspnea, stridor (when endobronchial lesions are present), cough, and hemoptysis, which may be massive.149
On bronchoscopic examination, nodular or flat bluish-red discolorations in the mucosa are seen, some of which may be actively bleeding. This bronchoscopic appearance is usually considered diagnostic, and endobronchial lesions are not generally biopsied. The diagnostic yield of transbronchial biopsies is usually low, and unless they are deep enough, KS of the lung will be missed because the mucosa itself is uninvolved.159 Open lung biopsies are more productive, but they are not absolutely sensitive.
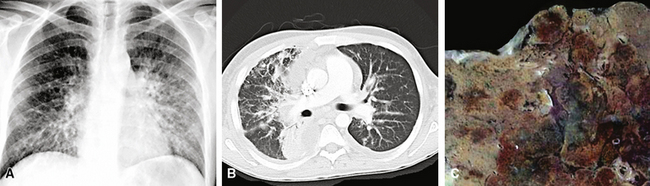
Radiographic findings on chest x-rays may be nonspecific, showing only ill-defined interstitial infiltrates (Fig. 14-14). An alveolar filling pattern is usually evident only if the patient has suffered hemoptysis and aspirated blood, but pleural effusions or pneumothorax may be seen in cases where the lesion involves the serosal surfaces as well as the lung parenchyma.160,161 Mediastinal adenopathy is not common, but, if it is present, this can be very helpful in separating KS from Pneumocystis jiroveci infection, because the latter does not cause adenopathy. Computed tomography (CT) scans and magnetic resonance imaging (MRI) generally provide no more information than chest radiography. In summary, the presence of bilateral pleural effusions and bilateral interstitial infiltrates with ill-defined nodularity is suggestive of pulmonary KS, especially in a patient with known tumor elsewhere.150,151
Pathologic Findings
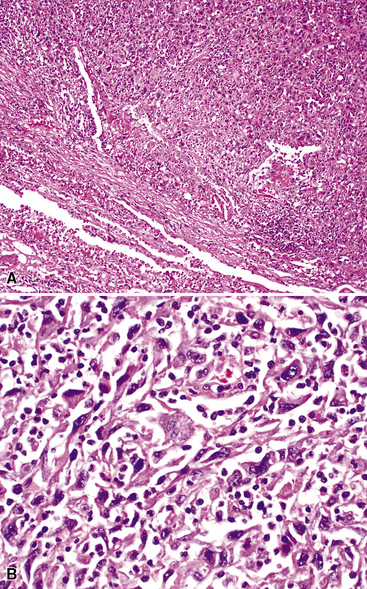
As alluded to above, it is distinctly uncommon for the pathologist to be able to make a definitive diagnosis of KS of the lung on a transbronchial biopsy specimen. Usually, a wedge biopsy is necessary, as obtained via video-guided thoracoscopy or a limited thoracotomy.159 On gross examination, this type of specimen exhibits numerous hemangiomatoid or ecchymosis-like zones of bluish-red discoloration in the parenchyma, with ill-defined borders.
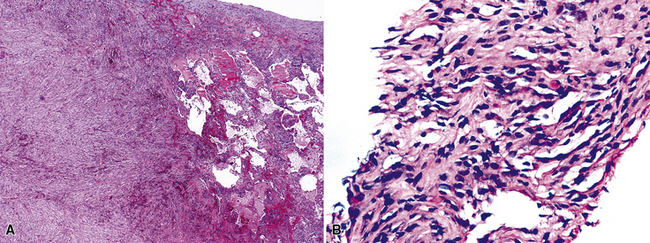
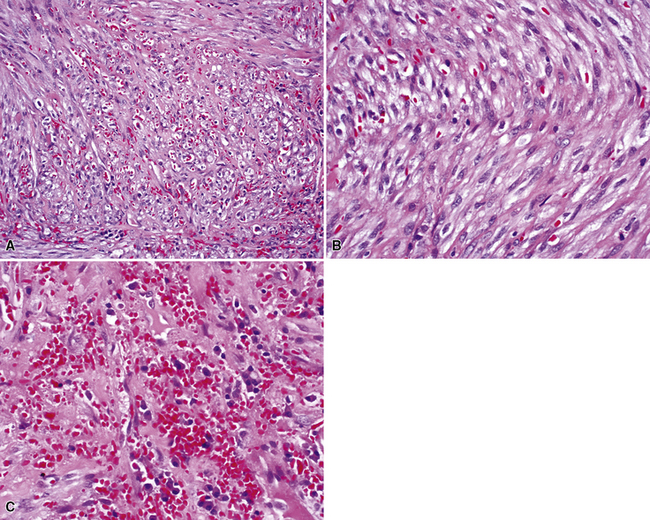
In the lung, KS shows a tendency to grow along pre-existing fibrous intrapulmonary septa, and it also concentrates around small tubular airways and blood vessels (Fig. 14-15). The tumor comprises a mixture of ectatic, thin-walled blood vessels that “dissect” or push through the pulmonary interstitial collagen, together with haphazardly arranged fascicles of spindle cells that show only modest nuclear atypia and may contain cytoplasmic vacuoles.149,159 Extravasated erythrocytes and hemosiderin pigment are common in and around the tumor masses (Fig. 14-16). Pleural KS “layers” itself over the submesothelial mantle of connective tissue, effacing the mesothelium itself in doing so.
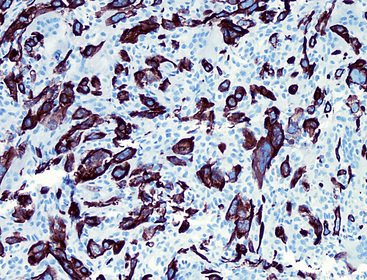
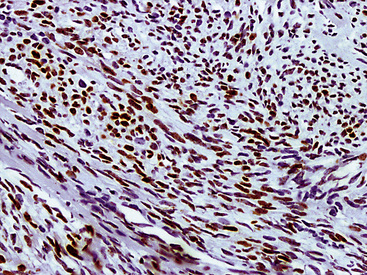
The differential diagnosis of KS from vascular granulation tissue and other spindle cell proliferations in the lung is greatly enhanced by immunohistochemical analyses. KS is reactive for latent nuclear antigen-1 of human herpesvirus-8 in approximately 85% of cases162 (Fig. 14-17). It also labels for FLI-1163 and podoplanin.164
Therapy and Prognosis
Regardless of its occurrence in AIDS or in non–human immunodeficiency virus (HIV)-related cases, the presence of KS in the lung is prognostically ominous. Virtually all patients with visceral disease die within 2 years, from infection if not from KS itself.148,154,156 Because of the multiplicity of KS, surgical resection is not a realistic option in the management of patients with this neoplasm. Chemotherapy is considered the treatment of choice, with a relatively good response rate and relatively rapid improvement within 2 to 4 weeks.25 Chemotherapy regimens in the few published therapeutic trials designed specifically for pulmonary KS have primarily included adriamycin, bleomycin, and vincristine.149,165–167 Gill and colleagues found an 85% response with combination chemotherapy in a group of 13 patients.166 Patients who benefited from this treatment included those who achieved at least partial responses. Complete response is defined by the following three criteria:
A partial response is characterized by the same three points, except that the degree of resolution is not total.165,166
Despite fairly good results with combination chemotherapy, patients with pulmonary KS do not show long survival. In the trial reported by Gill and colleagues, the median survival for responders was slightly but significantly longer than that of nonresponders (10 versus 6 months, respectively). However, considerable overlap between the two groups was present.166
Other more experimental (and inconclusive) approaches have included the administration of zidovudine, interferon, and other antiviral compounds.147,153,154 Radiotherapy may provide palliation of symptoms but is noncurative. The most important piece of data in prognosticating cases of KS of the lung is the serologic HIV status of the patient, inasmuch as AIDS is currently a uniformly lethal, albeit chronic, illness.
Fibrosarcoma
Primary fibrosarcoma of the lung (FSL), like its soft tissue counterpart, is defined as a fibroblastic spindle cell neoplasm without any evidence of specialized cellular differentiation. Although it has been cited in the past—along with leiomyosarcoma—as the most common primary pulmonary sarcoma,168 FSL was, and probably still is, overdiagnosed.169 Two separate studies from the Mayo Clinic cited two different time-dependent incidence figures for FSL. From 1950 to 1978, it constituted 50% of all primary pulmonary sarcomas170 but only 20% in the decade 1980 to 1990.171 As considered previously, it is our belief that the great majority of pulmonary “fibrosarcomas” are actually SCs. Only those tumors that have been subjected to rigorous and specialized pathologic examination should be accepted as bona fide examples of this rare sarcoma variant.
Clinical Summary
Guccion and Rosen studied 13 cases of FSL, which were divided into endobronchial and intrapulmonary types.168 This classification scheme was said to have clinical and prognostic importance. In conjunction with a review of 48 reported cases in the literature, the authors just cited found that the majority of endobronchial FSLs occurred in children and young adults, whereas parenchymal tumors predominated in middle-aged and elderly patients. In contrast, Pettinato and associates reported three parenchymal tumors in two newborns and a 6-month old infant.172 There was roughly an equal distribution by gender among endobronchial lesions; however, most intraparenchymal neoplasms occurred in men. All cases of endobronchial FSL in a series reported from the Armed Forces Institute of Pathology (AFIP) had symptoms of cough, hemoptysis, or chest pain; some of the parenchymal cases did as well.168 Thoracic imaging studies of FSL usually show discrete, homogenous masses. However, one reported pulmonary fibrosarcoma simulated a bronchogenic cyst clinically and radiographically.173 Gladish and coworkers13 have observed that fibrosarcoma is much more likely to arise in the soft tissue of the chest wall and secondarily involve the lung than it is to show the converse of that relationship.
Pathologic Findings
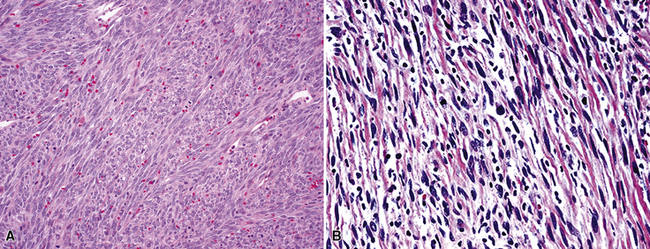
FSL is histologically identical to its soft tissue counterparts and characteristically shows sheets and intertwining fascicles of spindle-shaped cells with a typical interdigitating growth pattern and discernible stromal collagenogenesis (Fig. 14-18). The tumor cells contain oval to elongated, hyperchromatic nuclei, and scant amphophilic cytoplasm with ill-defined cellular borders. In addition, some areas may have a slightly epithelioid appearance, in which the tumor cells are more ovoid than spindled, and others may show significant pleomorphism that merges with the image of MFH (Fig. 14-19). Mitotic activity is variable.
Electron microscopic and immunohistochemical studies are required to confirm the fibroblastic nature of these neoplasms. The tumor cells in FSL are characterized by abundant rough endoplasmic reticulum and free ribosomes, as well as the production of extracellular collagen fibers that may be aligned at right angles to the tumor cell membranes. There should be no detectable myofilaments, pericellular basal lamina, or intercellular junctions in lesions thought to represent FSL. Because there are no specific immunologic markers for fibroblasts, the diagnosis of fibrosarcoma is one of ultimate immunohistologic exclusion. Tumor cells in FSL generally stain only for vimentin, a primitive intermediate filament protein, and they lack all epithelial, myogenous, neural, and endothelial markers.172,174,175
Therapy and Prognosis
Although resection is the treatment of choice, many surgically treated fibrosarcomas of the lungs do recur, and survival after this event is short, with patient fatality usually occurring within 2 years.168 Three cases seen at the Mayo Clinic between 1980 and 1990 occurred in young women whose lesions all recurred within 15 months following surgical excision.171 In contrast, primary bronchopulmonary fibrosarcomas in children appear to have a relatively favorable prognosis and behave only as low-grade malignancies.172,173 The five patients with pediatric FSL reported by Pettinato and colleagues all had complete surgical removal of their tumors, and four were disease-free after 4 to 9 years. The fifth case in that series had insignificant follow-up.172 The efficacy of adjunctive chemotherapy and irradiation has not yet been proven.
Primary Pulmonary Hyalinizing Spindle Cell Tumor with Giant Rosettes
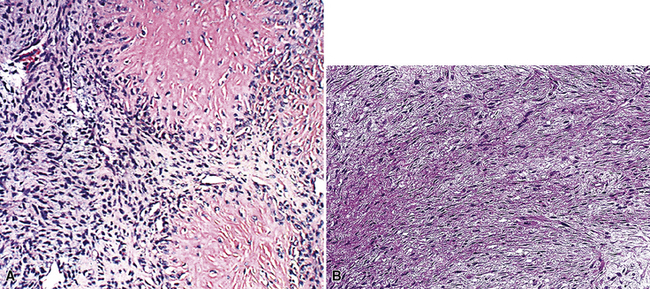
A rare lesion that is probably biologically related to FSL is one called hyalinizing spindle cell tumor with giant rosettes (HSCT). This entity was originally documented as a low-grade sarcoma in the deep soft tissues of adults,176 but at least two cases have been reported as primary pulmonary examples.177,178 HSCT has a distinctive morphologic appearance, featuring the multifocal presence of large rosette-like structures amid a bland spindle cell proliferation (Fig. 14-20). The lesion has infiltrative borders, with no necrosis and only limited mitotic activity.
HSCT has a partial kinship with Evans tumor (low-grade fibromyxoid sarcoma)179 of soft tissue, on behavioral, morphologic, and cytogenetic grounds. Both of those tumors manifest a t(7;16) (q33;p11) chromosomal translocation, producing fusion of the FUS and CREB3L2 genes.178 Unlike FSL, HSCT demonstrates some immunophenotypic variability, often labeling for alpha-isoform actin in its spindle cell population. The cells in the giant rosettes may manifest immunoreactivity for S100 protein and CD45RO,176 the latter of which is more typically a hematopoietic determinant. The precise biologic potential of HSCT in the lung is uncertain, because of the anecdotal nature of reported cases.
Primary Pulmonary Leiomyosarcoma
The most common anatomic locations for leiomyosarcomas in general are the uterus, gastrointestinal tract, and soft tissue, in order of relative frequency. The extremely uncommon primary pulmonary leiomyosarcoma (PPLMS) presumably originates from bronchial or pulmonary vascular smooth muscle. Only three cases of leiomyosarcoma were found among roughly 10,000 primary malignancies of the lung at one large American medical center between 1980 and 1990.171 Because secondary pulmonary involvement by malignant smooth muscle tumors is a relatively frequent event, the diagnosis of PPLMS absolutely requires exclusion of an occult extrathoracic neoplasm presenting with a single “herald” metastasis to the lung.
Clinical Summary
A series of 19 PPLMSs seen at the AFIP was divided into those neoplasms that were predominantly endobronchial and others that were intraparenchymal, in analogy to pulmonary fibrosarcomas.168 The majority of these tumors in children are endobronchial in nature,180,181 whereas those in adults are not. In contrast to leiomyosarcomas of the soft tissue, which occur most commonly in women, patients in the aforementioned report from the AFIP were almost exclusively men. However, another survey that reviewed 92 cases of PPLMS in the literature found a male-to-female ratio of 2.5, suggesting that the paramilitary study just cited was biased demographically by its affiliation with the armed services.182 In contrast to carcinoma of the lung, leiomyosarcoma is not associated with cigarette smoking or other potential inhalant carcinogens. Most patients with PPLMS (particularly its endobronchial form) are symptomatic, often complaining of cough, hemoptysis, or chest pain. However, intraparenchymal lesions may be discovered incidentally on chest radiographs. Roentgenographically, PPLMS usually takes the form of a discrete mass (Fig. 14-21), sometimes with cavitation or cyst formation that is best seen by CT of the thorax.168,183,184
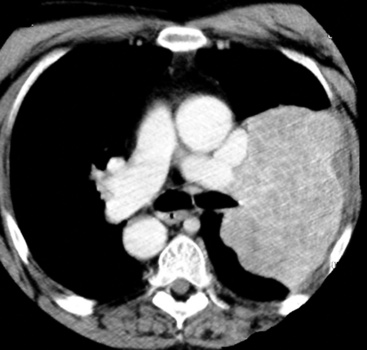
Pathologic Findings
Parenchymal tumors range from 3 to 15 cm in maximum dimension. They are well circumscribed, white to yellowish tan, and variably firm. Cut surfaces of these neoplasms commonly show hemorrhagic and necrotic areas. Endobronchial tumors are often smaller than the intraparenchymal lesions, presumably because of confinement by the bronchial walls.185
Microscopy discloses histologic features that mirror those of leiomyosarcomas elsewhere in the body. On low-power magnification, there are interlacing fascicles of spindled cells arranged haphazardly, yielding a “whorled” appearance (Fig. 14-22). The neoplastic cells have cigar-shaped nuclei with blunt ends, a moderate amount of cytoplasm, and indistinct cell borders (Fig. 14-23). Fascicles cut in cross section demonstrate characteristic intracellular perinuclear lucencies.185–188 Prominent myxoid stromal change may be observed.189
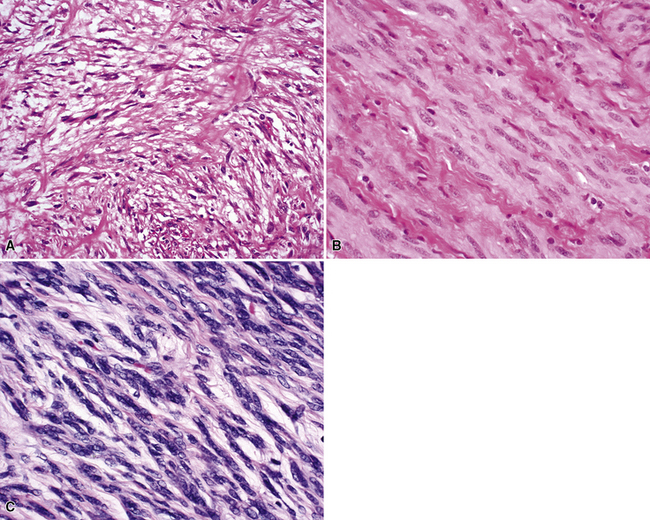
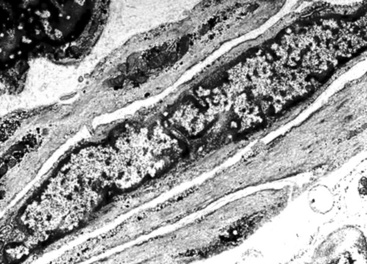
The differential diagnosis of PPLMS includes fibrosarcoma and malignant peripheral nerve sheath tumor, as well as SC. Electron microscopy and immunohistochemistry are again helpful in confirming the smooth muscle nature of a spindle cell neoplasm.187 Ultrastructural features of leiomyogenous differentiation include cytoplasmic dense bodies punctuating skeins of thin filaments, subplasmalemmal dense plaques, plasmalemmal pinocytotic vesicles, and pericellular basal lamina (Fig. 14-24). Immunoreactivity for desmin, muscle-specific actin, calponin, caldesmon, or smooth muscle actin is also characteristic of the tumor cells in PPLMS.190
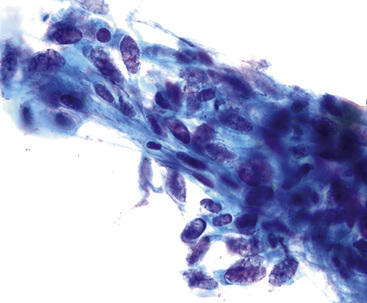
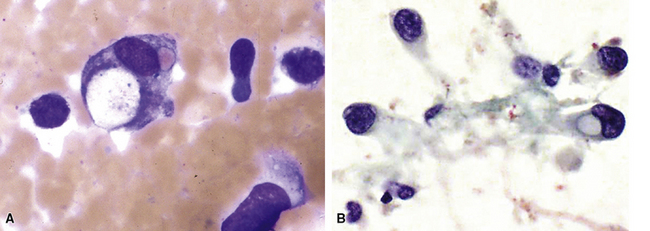
Transthoracic fine needle aspiration of possible PPLMS can be attempted if the lesion is large and peripherally located. The cytologic preparations from that procedure typically show a dyshesive population of relatively monotonous spindle cells, with blunt-ended fusiform nuclei. Mitotic figures and nuclear pleomorphism are variably seen; some lesions also may exhibit a more epithelioid cytologic image191–193 (Fig. 14-25).
Therapy and Prognosis
The natural history of PPLMS and its responses to various therapeutic regimens are difficult to predict because of the rarity of this neoplasm. However, there is generally a consensus that surgical resection is the treatment of choice182,185–187 and that it produces a survival rate of 45% to 50% at 5 years. Survival for as long as 15 to 30 years has been documented in PPLMS cases.168,170 However, pulmonary leiomyosarcomas appear to be relatively resistant to irradiation and chemotherapy.182,188 Various prognostic variables have been discussed in connection with these lesions.168,170 Endobronchial tumors are thought to be less aggressive than parenchymal neoplasms, largely because the former tend to be smaller and are diagnosed earlier. It follows, therefore, that tumor size is an important indicator of biologic behavior for all pulmonary leiomyosarcomas. The scope of mitotic activity may affect prognosis as well. In an AFIP series on PPLMS, a mitotic rate of 8 or less per 10 high-power fields was associated with infrequent metastasis and a generally favorable clinical outcome.168
Epithelioid Hemangioendothelioma
In 1975, Dail and Liebow reported the first cases of an unusual pulmonary neoplasm that they termed intravascular bronchioloalveolar tumor (IVBAT). This name reflected their original hypothesis that the lesion in question was an epithelial tumor—specifically, a bronchioloalveolar carcinoma variant showing prominent vascular invasion.194,195 Four years thereafter, Corrin and associates alternatively proposed an endothelial origin for this tumor based on the results of ultrastructural studies.196 Subsequent evaluations by other authors have confirmed the vascular histogenesis of the IVBAT. Indeed, in 1982, Weiss and Enzinger described a series of soft tissue tumors that were histologically identical to IVBAT, and these authors were the first to use the term epithelioid hemangioendothelioma (EH) to emphasize their distinctively epithelioid (or histiocytoid) cytologic features.197 In addition to the lungs and soft tissues, EH also primarily occurs in the bone and liver.198
Clinical Summary
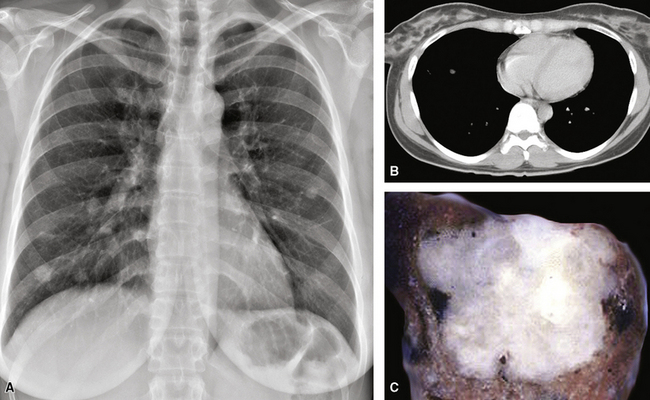
EH of the lung is a neoplasm that arises predominantly in female patients; women account for roughly 80% of all cases.195,199,200 It primarily occurs in young adults, with approximately 50% of affected individuals being younger than 40 years of age; only 10% are older than 50 years of age at diagnosis.198,201 Many affected persons are asymptomatic, and their tumors are detected incidentally on chest radiographs. Patients who have tumor-related complaints usually present with pleuritic pain, dyspnea, and cough. Case reports have also documented alveolar hemorrhage as a presenting sign of pulmonary EH,202,203 and it may simulate thromboembolic disease symptomatically as well.204 Chest radiographs commonly show numerous, small nodular lesions throughout both lung fields (Fig. 14-26). Therefore, EH enters the roentgenographic differential diagnosis of multiple pulmonary nodules in asymptomatic young women, together with metastatic germ cell tumors, chondroid pulmonary hamartomas, multiple arteriovenous malformations of the lung, deposits of “benign metastasizing leiomyoma,” and malignant lymphoma.205
Pathologic Findings
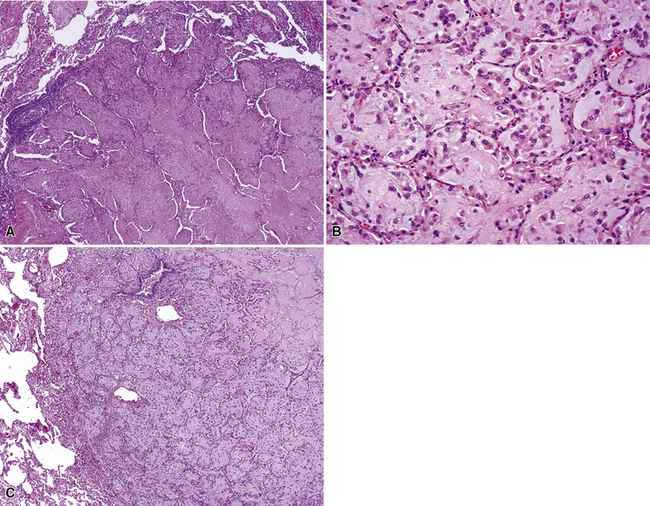
The pathologic diagnosis of EH is almost always made by open lung biopsy, inasmuch as transbronchial biopsy is usually ineffectual because of sampling constraints. Most nodules of EH are discrete and usually measure less than 2 cm. They are grayish-white to tan and have a chondroid macroscopic consistency. More nodules are typically seen on histologic examination than are apparent grossly. Microscopically, EH is typified by multiple oval or round nodules with hypocellular, sclerotic, or necrotic centers195,199 (Fig. 14-27). These are surrounded by rims of viable, more cellular tissue that is associated with a myxohyaline fibrous stroma; exceptionally, metaplastic bone formation may be apparent.206 The neoplastic cell population is composed of plump, epithelioid cells, which are the histologic hallmark of EH (Fig. 14-28). They have centrally located, round-to-oval nuclei, with ample amounts of eosinophilic cytoplasm. Often, intracytoplasmic vacuoles are evident, which should raise the possibility of endothelial differentiation on light microscopy. Saqi and colleagues have described “rhabdoid” cellular differentiation in EH as well.207
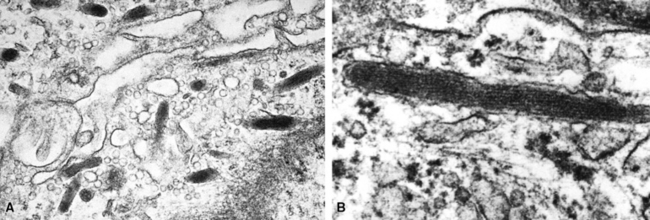
Ultrastructural and immunohistochemical evaluation can be of great assistance in confirming the endothelial origin of EH.196,198,208–211 Briefly, ultrastructural features of this tumor include cytoplasmic vacuoles, Weibel-Palade bodies (Fig. 14-29), cell membranous pinocytotic vesicles, and pericellular basal lamina.210 Histochemical and immunologic markers of endothelial differentiation—such as Ulex europaeus I lectin, anti-CD31, anti-FLI-1, and anti-CD34—are helpful in labeling the neoplastic cells in virtually all examples of EH.206,212 Weinreb and coworkers have also described labeling for CD10 in a majority of EH cases.213
Fine needle aspiration biopsy of EH yields a loosely cohesive population of epithelioid cells that may be binucleated or multinucleated. Variably sized cytoplasmic vacuoles are demonstrable in a proportion of the tumor cells (Fig. 14-30).214
Therapy and Prognosis
Surgery is usually not feasible as effective treatment for EH because of its tendency to show intrapulmonary multicentricity. Unfortunately, irradiation and chemotherapy likewise have been of little benefit.195,199 Nevertheless, EH is generally an indolent neoplasm that is classified as a borderline malignancy. It is associated with a protracted clinical course and potential survival of several years after diagnosis.208 Most patients do eventually succumb to the tumor and die of respiratory failure secondary to progressive parenchymal replacement; in one series reported by Einsfelder and Kuhnen, 36% of patients were dead or likely to die of their tumors after 52 months’ follow-up.206 Adverse prognostic factors that predict a more rapid decline in pulmonary function include prominent symptoms at the time of presentation; radiographic demonstration of extensive intravascular, endobronchial, or pleural spread of the tumor;195 and the presence of fusiform tumor cells.200
A particularly vexing clinical problem is represented by those patients who have EH synchronously in several organs, including the lungs. In such cases, one is never certain whether tumor multifocality or metastasis is operative. Pragmatically, each involved organ is usually treated as if it harbors an independent primary tumor in this scenario.215
The general predilection of EH for women, a reported association of primary hepatic EH with oral contraceptives, and the lack of effective therapy for this tumor have prompted some investigators to explore the possibility of treatment involving hormonal modulation. These tumors have been examined for possible expression of estrogen and progesterone receptor proteins as well as other estradiol-binding moieties. Ohori and colleagues analyzed five cases of pulmonary EH for steroid hormone receptors by immunohistochemical methods, using paraffin-embedded material.216 Only one case showed apparent binding of estradiol. Our own unpublished experience with the immunohistologic characteristics of pulmonary EH has disclosed no reactivity with monoclonal antibodies against estrogen and progesterone receptor proteins. Thus, we believe that hormonal therapies are unlikely to produce significant results in this setting.
Hemangiopericytoma and Intrapulmonary Solitary Fibrous Tumor
As first described in 1942 by Stout and Murray,217 hemangiopericytoma (HPC) is an uncommon, potentially malignant neoplasm that shows apparent differentiation toward the phenotype of pericytes. These are cells with long cytoplasmic processes that surround capillaries and serve a vasoregulatory function. HPC occurs most commonly in the deep muscles of the thigh, the pelvic fossa, and the retroperitoneum. However, 5% to 10% of all hemangiopericytomas are said to present as primary pulmonary tumors.185,218 It must be remembered that the lungs and the bones are the anatomic sites that most frequently harbor metastases of HPC,219 and therefore a primary extrapulmonary tumor must be excluded before a diagnosis of a primary HPC can be rendered safely.
It should also be understood that the currently recommended classification scheme for mesenchymal neoplasms has merged HPC with solitary fibrous tumor (SFT).220,221 Hence, for all intents and purposes, the two entities are considered to be closely related if not identical, and the abbreviation of HPC-SFT will be used in reference to that tumor group.
Clinical Summary
Pulmonary HPC-SFT affects men and women equally, and most commonly arises in middle adulthood. The peak incidence of this lesion is in the fifth decade of life, although individuals as young as 4 years of age and as old as 73 years of age have been reported.222,223 Some tumors are detected incidentally on radiographic studies without causing pulmonary symptoms; 6 of 18 cases in one series fit this scenario.224 Alternatively, presenting symptoms may include hemoptysis, chest pain, cough, and dyspnea, and, more rarely, pulmonary osteoarthropathy.224
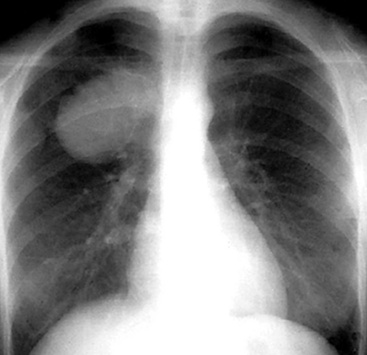
Various radiographic imaging studies have been used in studying this neoplasm. Although angiography has usually not been performed, vascular contrast studies of HPC-SFT generally show a characteristic intralesional “blush.”225 No other pathognomonic features are evident in chest x-rays, CT scans, and MRIs of the lung.223,226 Plain film x-rays typically show a discrete, homogeneously dense mass with lobulated contours. On CT images, however, HPC-SFT is heterogeneous. Central low-density areas are evident that correspond to necrotic foci, and an apparent capsule may be seen at the interface with surrounding lung parenchyma. MRIs also show intratumoral heterogeneity with respect to tissue density and are apparently more sensitive in depicting intralesional hemorrhage. These images were found to be the most useful in delineating the potential plane of surgical separation between an HPC-SFT and surrounding soft tissue in one report.227 In summary, a radiologic diagnosis of pulmonary HPC-SFT may be suspected in a middle-aged person lacking pulmonary symptoms, but whose radiographic imaging studies reveal a large, lobulated, sharply marginated, variably dense mass (Fig. 14-31) that does not cause compression atelectasis.226
Pathologic Findings
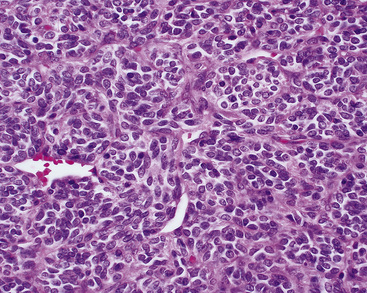
HPC-SFTs of the lung can attain large sizes, and lesions measuring up to 18 cm have been documented. The typical gross appearance of this tumor is that of a well-circumscribed, yellow to tan-brown mass with a pseudocapsule, as well as areas of internal necrosis and hemorrhage. Histologic sections typically show a relatively monomorphous cellular proliferation surrounding thin-walled, anastomosing vascular channels lined by a single endothelial layer. These blood vessels often (but not always) assume gaping, “staghorn,” or “antler-like” configurations (Figs. 14-32 and 14-33). The population of neoplastic cells is uniform, with oval compact nuclei and ill-defined cytoplasm.185 Mitotic activity and areas of necrosis and hemorrhage are noted frequently. Vascular invasion of large pulmonary vessels, however, is uncommon. With regard to the latter features, some pathologists have, in the past, rendered a diagnosis of benign hemangiopericytoma if necrosis, hemorrhage, and mitoses were absent. In our view, this approach is highly inadvisable. We have seen cases of pulmonary HPC-SFT with exceedingly bland histologic profiles in which metastasis nonetheless supervened. Accordingly, it is advised that each report on this tumor should carry the statement that HPC-SFT is at least potentially malignant behaviorally.219,224
Pulmonary HPC-SFT has sometimes been overdiagnosed because other neoplasms may show foci that resemble the former lesion. In this regard, it is notable that in their seminal report, Stout and Murray admonished others to make the diagnosis of hemangiopericytoma by ultimate exclusion.217 The histologic differential diagnosis includes SC, SS, fibrous histiocytomas, leiomyosarcoma, and mesenchymal chondrosarcoma.219 Distinctions among these neoplasms are best made by ancillary studies.
Electron microscopy can confirm the pericytic nature of HPC through the demonstration of polygonal cells with cytoplasmic processes, pinocytotic vesicles, basal lamina, and a paucity of other organelles.174,219 HPC-SFT is a neoplasm that demonstrates a relatively restricted group of immunoreactants; these include vimentin, collagen type IV, CD34, CD99, CD57, and bcl-2 protein54,228 (Fig. 14-34). Endothelial stains such as Ulex europaeus I, CD31, and FLI-1 highlight the lining of intralesional vascular spaces but do not label the surrounding tumor cells. Silver impregnation techniques highlight a complex reticulin matrix with individual cell investment.
Therapy and Prognosis
As in the management of soft tissue HPC-SFT, complete surgical excision is the mainstay of therapy for primary pulmonary tumors of this type. However, it is known that intraoperative rupture of pulmonary HPC-SFT may occur (especially those tumors that are adherent to the chest wall). As expected, this complication results in early local recurrence, as reported by Van Damme and associates, and should therefore be avoided at all costs.223 Chemotherapy and irradiation have not been shown to be consistently effective adjuvant modalities, but they may have some role to play in management.229,230 A study by Jha and coworkers on the general role of radiation therapy in treating HPC-SFT showed that postoperative irradiation was useful for local tumor control, salvage therapy after local recurrence, and palliation.231 Tumors less than 5 cm in maximum dimension exhibit a better response than those that are larger than 10 cm.232
As mentioned above, hemangiopericytomas in general are well known for their unpredictable biologic behavior. Postoperative survival has ranged from 10 weeks to 18 years.223,232 Even with apparently complete surgical resection, HPC-SFT recurs locally within 2 years in approximately 50% of cases,224,227,233 and later recurrences are seen as well; distant metastasis, however, is uncommon.228
Clinical and histologic features that have been cited as prognostically useful219,222,227 include the presence of symptoms at presentation, mitotic activity of four or more mitoses per 10 high-power microscopic fields, spontaneous tumor necrosis, vascular invasion, and tumor size greater than 5 cm. In one series, metastases were seen in one third of tumors that measured greater than 5 cm, and in two thirds of those greater than 10 cm.222 However, Yousem and Hochholzer did not find any single histologic or clinical feature that was statistically significant in reliably predicting the clinical course of primary pulmonary HPC-SFT.224
Malignant Fibrous Histiocytoma
MFH (now commonly called pleomorphic sarcoma, not further specified) is a common, extensively studied soft tissue sarcoma of older adults that develops most frequently in the extremities and the retroperitoneum. In a series of 200 cases by Weiss and Enzinger,234 the lungs were the most common site of metastases. Thus, exclusion of an occult soft tissue tumor is once again necessary before a diagnosis of primary pulmonary malignant fibrous histiocytoma (PPMFH) can be made. A review of Mayo Clinic cases found only four examples among 10,134 tumors arising in the lung.171 Currently, there are fewer than 75 reported cases of PPMFH in the English literature.234–245
Clinical Summary
In general, MFH is a neoplasm of patients who are in late middle age, with a median of 54 years. However, its occasional occurrence in children and young adults has also been reported.235,244 No consistent predilection for either gender is seen. Previous irradiation is a pathogenetic risk factor for tumors arising in soft tissue, and the literature similarly contains sporadic reports of PPMFH presenting in patients who have received radiation therapy previously. Clinical and radiographic features of this tumor are nonspecific, and a distinction from the much more common epithelial tumors of the lungs absolutely requires tissue examination. The majority of patients present with symptoms of cough, chest pain, hemoptysis, or dyspnea. Chest x-rays generally show a solitary mass with a nondescript appearance and a relatively homogeneous density on CT or MRI studies.243
Pathologic Findings
Most examples of PPMFH are intraparenchymal, but occasional endobronchial lesions have also been observed.235 There is apparently no predilection for any particular lobe of either lung. These tumors are usually large, ranging up to 25 cm in maximum dimension,246 with an average size of 6 to 7 cm. They are well circumscribed, lobular, and white-tan, and not uncommonly they contain central necrosis or cavitation on macroscopic examination.
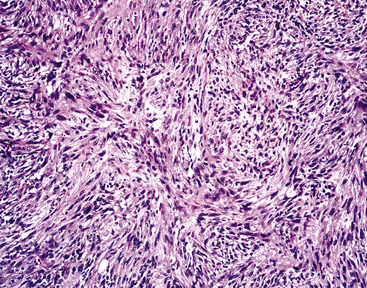
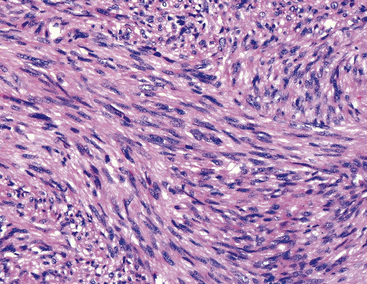
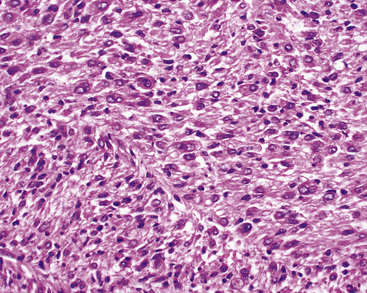
Histologically, PPMFH is characterized by fusiform and pleomorphic elements that are arranged in storiform, fascicular, or medullary patterns (Fig. 14-35). As the name “MFH” implies, this tumor was originally thought to be composed of malignant fibroblast-like and the histiocytoid cells; however, it now appears that there is little if any relationship between the neoplastic elements and true histiocytes. Fusiform tumor cells contain elongated nuclei and relatively scant cytoplasm, and the histiocytoid cells have round-to-oval nuclei with a moderate quantity of amphophilic cytoplasm (Fig. 14-36). A hallmark of most lesions in this category is the presence of large, bizarre, often multinucleated cells with irregular contours. Mitoses, including atypical forms, are easily found and number from 5 to 30 per 10 high-power microscopic fields.174
The differential diagnosis of PPMFH by light microscopy includes primary or secondary pleomorphic sarcomas (e.g., dedifferentiated leiomyosarcoma and pleomorphic rhabdomyosarcoma), metastatic malignant melanoma, and SC.13 Immunohistochemical and electron microscopic studies can be used to separate these pathologic entities.236–240,246 Ultrastructurally, PPMFH shows fibroblastic and histiocyte-like differentiation, with abundant rough endoplasmic reticulum, numerous lysosomes, and a variable number of small cytoplasmic lipid droplets. Desmosomes, tonofibrils, elongated cell processes, myogenous filament skeins, and cytoplasmic dense bodies are absent. PPMFH expresses vimentin but is devoid of other specialized markers of myogenous, neural, or epithelial differentiation on immunohistologic analyses.174
Therapy and Prognosis
The rarity of PPMFH again serves as an impediment to assessments of optimal treatment for this tumor. Surgical resection is currently the recommended treatment of choice, even if the lesion in question shows limited extrapulmonary spread to the intrathoracic great vessels or soft tissue.247 Adjunctive chemotherapy and irradiation have not proven to be effective in the few published cases of PPMFH in which these treatments have been used.235,245,246 In one series of 22 examples,235 7 of 15 patients who underwent radical surgical resection suffered relapses and died from metastatic disease. There was recurrence in the lungs and pleura, as well as metastasis to the liver and brain; almost all of these events occurred within 12 months of diagnosis. However, survival for as long as 5 to 10 years has been documented in a few patients with PPMFH.235,237
Rhabdomyosarcoma
For practical purposes, primary rhabdomyosarcoma of the lung (RMSL) is a tumor that is confined to the pediatric population. In adults, tumors resembling rhabdomyosarcoma are almost invariably examples of SC,1,4 and this fact should be borne in mind in interpreting all but the most recent literature on this topic. Moreover, this is another sarcoma type that much more commonly arises outside of the lungs, and the probability that one is dealing with metastasis to the pulmonary parenchyma is therefore important to remember.
Clinical Summary
To date, there are less than 30 well-documented examples of bona fide, “pure” intrapulmonary rhabdomyosarcoma in the pertinent literature. They all occurred in patients who were in the first two decades of life, and most were in children younger than 10 years of age.248–251 At least one lesion, documented by Choi and colleagues, was seen in a child with neurofibromatosis.252 Symptoms and signs of RMSL may be nonspecific, including cough, wheezing, and dyspnea, or the patient may present with spontaneous pneumothorax.253 The latter relates to a peculiar tendency of RMSL to associate itself with cystic lesions of the lungs, particularly as one component of a PPB (see subsequent discussion).249,250,254 When these cysts rupture, pneumothorax results. The underlying lesions in such cases of RMSL have included not only PPB, but also congenital cystic adenomatoid malformations and peripheral bronchogenic cysts.254
Pathologic Findings
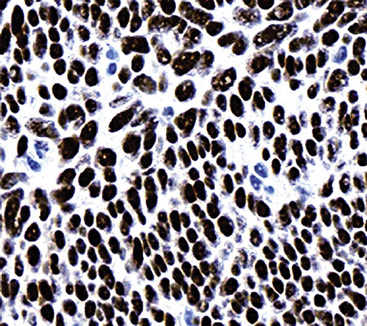
Pulmonary rhabdomyosarcomas have most often assumed an embryonal or an alveolar growth pattern249,250 (Figs. 14-37 and 14-38), although pleomorphic tumors, which are usually encountered in the soft tissues of adults, have been reported in the lung as well.255 These neoplasms are typically composed of small round cells that are configured in one of three ways. These include “solid” sheetlike clusters with no further distinguishing morphologic attributes, dyscohesive groups with internal pseudolumina or alveoli, and “botryoid” proliferations in which a polypoid lesion (usually within a bronchial lumen) shows a zonation into hypocellular and hypercellular cellular strata (so-called “cambium” layers).256 In contradistinction to other small round cell tumors of children, RMSL demonstrates a moderate degree of cellular pleomorphism and anisonucleosis. Nuclear chromatin is usually coarse and clumped, cytoplasm is scanty and amphophilic or eosinophilic, and mitoses and apoptotic cells are easily found. Small foci of spontaneous necrosis are also present.
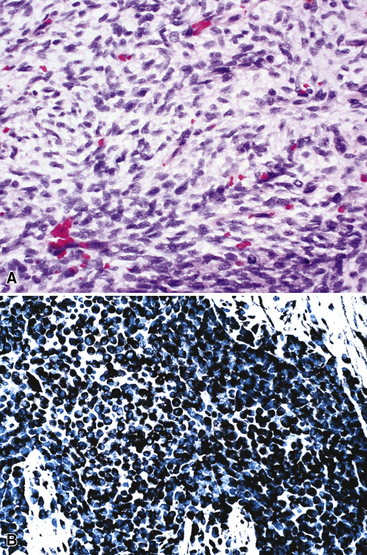
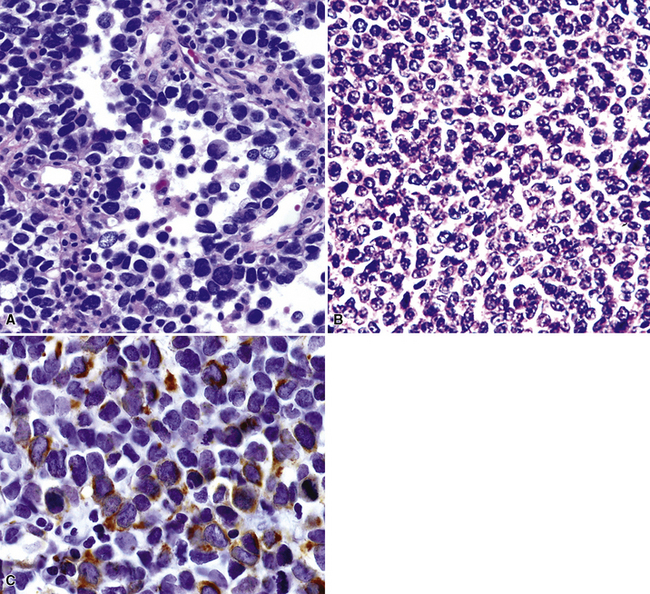
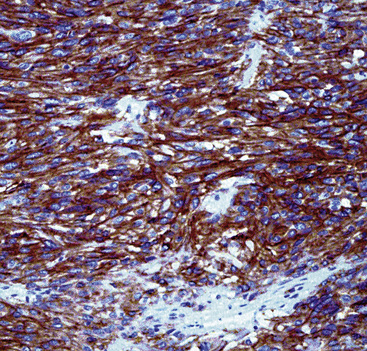
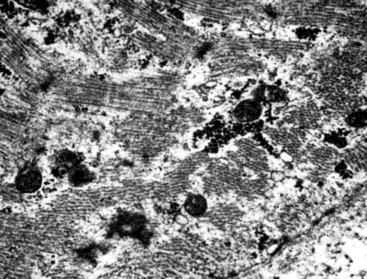
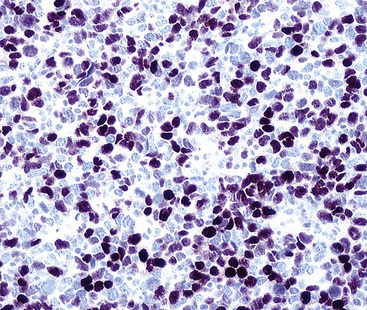
Particularly in those lesions that have an embryonal “solid” appearance histologically, special pathologic studies are nearly always necessary to procure a definitive diagnosis. Moreover, these analyses should also be done in all putative cases in adults, for reasons mentioned earlier. Histochemical stains show that striated muscle tumors contain abundant glycogen, as determined with the periodic acid/Schiff (PAS) method with and without diastase digestion. Electron microscopically, rhabdomyosarcoma is characterized by the focal presence of intermediate filament whorls in the cytoplasm, sometimes with the addition of thick filaments in aggregates that resemble primitive muscular Z-bands (Fig. 14-39). Furthermore, cytoplasmic glycogen is present, and pericellular basal lamina can be visualized. This constellation of fine structural attributes excludes other small cell tumors from diagnostic consideration.257 By immunohistology, RMSL is found to express one or more myogenous determinants, such as desmin, tropomyosin, titin, muscle-specific actin, “fast” myosin, myo-D1, myogenin (Fig. 14-40), or Z-band protein.258–260 Vimentin is also uniformly found, but markers of epithelial differentiation, such as keratin and EMA, must be absent in order to make the diagnosis of RMSL.
Therapy and Prognosis
The great majority of reported cases of RMSL have been treated surgically, with the usual addition of postoperative irradiation and standard chemotherapy such as that used by the InterGroup Rhabdomyosarcoma Study.248–255 However, there are no controlled studies to determine whether this protocol is the optimal approach to management. Once again, the extreme rarity of the lesion in question interferes with the design of the most efficacious therapeutic regimen.
Prognostically, the fact that RMSL is a visceral manifestation of rhabdomyosarcoma is an adverse clinical variable, along with the probability that such tumors may attain a size of several centimeters before coming to clinical attention. Pathologic features that have been associated with unfavorable tumoral behavior include the focal or global presence of an alveolar growth pattern and the existence of areas that resemble adult-type pleomorphic rhabdomyosarcoma.261
Chondrosarcoma of the Respiratory Tract
Chondrosarcomas are uncommon but well-documented in the supporting tissues of both the upper and lower airways. Indeed, although cartilaginous malignancies are usually observed in the proximal long bones of adults, visceral lesions of this type have been reported in a variety of locations. As one would expect, there are few if any examples of primary pulmonary chondrosarcoma (PPCS) that involve the most distal portion of the respiratory tract, because cartilaginous support for the bronchi ends at the level of subsegmental bronchi.262–264 Accordingly, most of these lesions affect the trachea and major bronchial divisions,265–267 and chondrosarcomas that are seen in the peripheral lung fields should be carefully examined radiologically to make certain that they are not extensions of contiguous bony lesions in the sternum, vertebral bodies, or ribs.
Clinical Summary
Patients with PPCS are adults, with no predilection of the tumor for either gender. They may present with slowly evolving stridor, wheezing, cough, vague chest pain, or episodes of hemoptysis.264,265 Systemic complaints are not encountered. Tracheobronchoscopy usually demonstrates a smooth, nodular, glistening mass that stretches and attenuates the overlying mucosa but does not ulcerate it. Attempted biopsy of the mass through the bronchoscope is usually unsuccessful because of the firm consistency of the tumor and difficulty of sampling submucosal lesions in general.262
Radiographically, there may be no visible abnormalities on plain films if the neoplasm is predominantly or exclusively intraluminal in a large airway. Other examples of PPCS are manifested simply as sharply circumscribed, lobulated masses (Fig. 14-41) that may contain flecks of central calcification or cystic change.267 The latter findings are more graphically displayed in CT or MRI studies.265
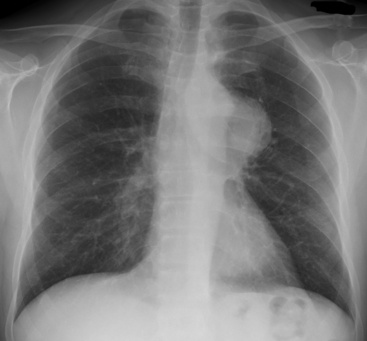
Pathologic Findings
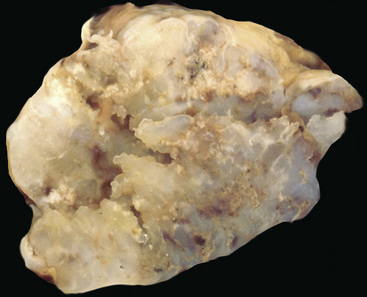
Chondrosarcomas of the lung differ significantly from pulmonary chondroid hamartomas on macroscopic and microscopic grounds. The latter of those two lesions are sharply marginated grossly; they typically entrap small tubular airways and are composed of extremely well-differentiated chondrocytes. In contrast, a PPCS has an irregular peripheral macroscopic interface with the lung (Fig. 14-42); it exhibits at least modest nuclear pleomorphism, nuclear crowding, and cellular binucleation; and it does not contain respiratory epithelial profiles262,264 (Fig. 14-43). Despite the statements just made, most chondrosarcomas of the lung are well-differentiated tumors. Hence, a striking degree of nondescript spindle cell growth or cellular anaplasia in a cartilage-forming tumor should instead invoke concerns over a probable diagnosis of SC with divergent chondroid areas.1,4 Mesenchymal chondrosarcomas,263 represented by small cell neoplasms of childhood with a resemblance to Ewing sarcoma (see later discussion) (Fig. 14-44) are rare and special variants that differ from the descriptions just given. They comprise sheets of small lymphocyte-like cells, often punctuated by hemangiopericytoid blood vessels, with interposed islands of embryonal-type cartilage. Another singular subtype of chondrosarcoma is the dedifferentiated form, in which low-grade chondroid tissue is juxtaposed to a highly anaplastic pleomorphic tumor component (Fig. 14-45). It has been reported only rarely as a primary pulmonary lesion.268,269
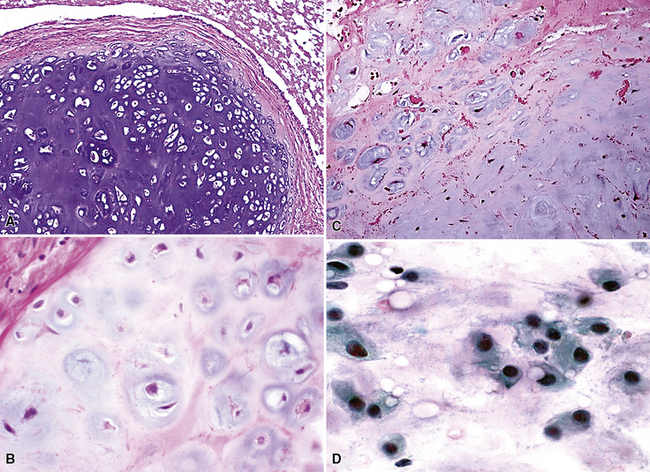
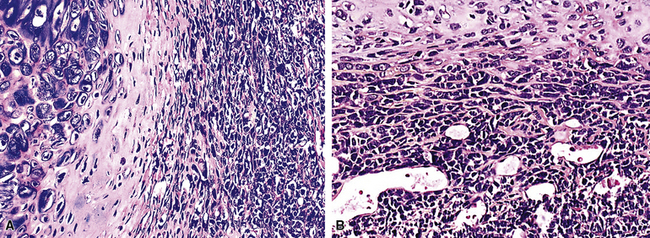
Therapy and Prognosis
Primary chondrosarcomas of the airway are best treated by surgical ablation. Because these are, in most cases, slowly growing and generally low-grade malignancies, such intervention carries with it a good chance of long-term survival if the tumor can be completely extirpated.265 Chemotherapy and irradiation are ineffectual in treating PPCS and probably incur more morbidity than is acceptable in the treatment of an indolent sarcoma. On the other hand, those interventions would be appropriate to consider for examples of dedifferentiated PPCS.
Primary Pulmonary Synovial Sarcoma
Although SS is primarily a peripheral soft tissue tumor, it is also potentially seen as a primary pleuropulmonary lesion. Published reports of approximately 100 cases have now attested to that fact.190,270–278 Because of the range of histologic and radiographic appearances associated with SS, it enters into differential diagnosis with several other neoplasms in the lung and pleura.
Clinical Findings
The basic symptoms and signs that are associated with primary pulmonary SS are no different than those attending bronchogenic carcinomas, except they are seen in a younger age group (mean, 38.5 years). Chest pain, cough, shortness of breath, and hemoptysis may be encountered, but, in one series, one fourth of all patients were asymptomatic.270 Men and women are relatively equally likely to develop SS of the lung. Roentgenographically, there is usually little to point the radiologist toward a specific diagnosis of SS in the lung (Fig. 14-46), which may arise in any segment of any pulmonary lobe. However, some cases show flocculent or particulate calcification on plain film radiographs or CT scans. Cystic change may also be noted, and a minority of lesions is clearly associated with a major bronchus.270–277
Pathologic Findings
The gross and microscopic spectrum of SS of the lung parallels that seen in similar tumors of peripheral soft tissue.279 Macroscopically, one sees a fleshy, ill-circumscribed mass in virtually any intrapulmonary location (Fig. 14-47). Classically, biphasic SS is composed of fascicles of compact spindle cells arranged in interweaving or “herringbone” fascicles, punctuated by clefts or overtly glandlike spaces that are lined by cuboidal to low columnar tumor cells (Fig. 14-48). The latter inclusions may contain mucoid matrical material and may also demonstrate squamous or goblet cell metaplasia. Nuclei of the neoplastic cells in both components are generally monomorphic, with dispersed chromatin and inconspicuous nucleoli. Cytoplasm is sparse in the fusiform cellular elements. In contrast, a moderate quantity of amphophilic, eosinophilic, or vacuolated cytoplasm is apparent in glandlike foci. Mitotic activity is greatly variable, may be surprisingly scant, and only rarely features the presence of “atypical” division figures. Intercellular calcifications (sometimes with a psammomatous configuration) may be scattered randomly throughout the tumor (Fig. 14-49), be clustered in discrete foci, or be lacking altogether. The finding of necrosis is likewise highly variable. Tumoral stroma can be overtly collagenous, delicate and fibrovascular, or myxoid. Besides the prototypic form of biphasic SS, another with an admixture of solid polygonal cell clusters and spindle cell zones (Fig. 14-50) is recognized.279
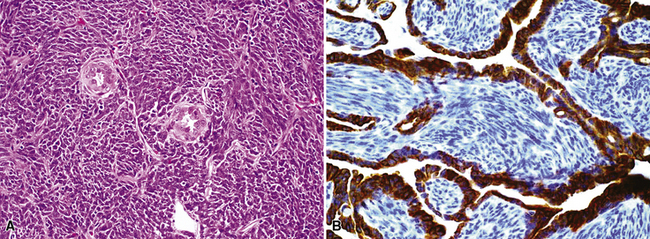
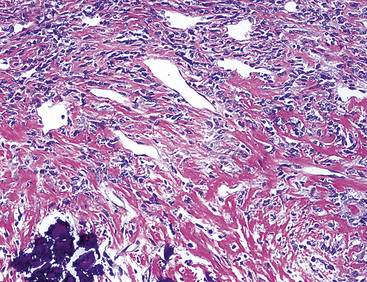
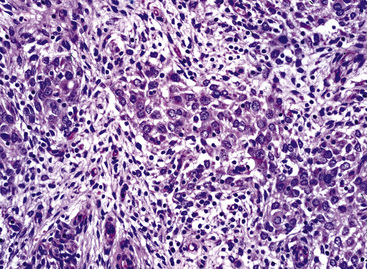
The existence of monophasic SS is now undeniable. This variant may be composed entirely of fusiform elements, epithelioid polygonal cells that may or may not demonstrate obvious glandlike differentiation, or sheets of small round cells.270 Monophasic spindle cell SS is more common by far (Fig. 14-51), and recognition of it as a distinct entity has resulted in reclassification of many fibrosarcomas of the soft tissues and other sites. It is now realized that the great majority of sarcomas with a “herringbone” spindle cell constituency are actually synovial rather than fibroblastic, as traditionally taught.258 Another key feature to the recognition of monophasic spindle cell SS is the presence of a “staghorn” pattern of intratumoral vascularity.279 Divergent differentiation, simulating osteogenic, neural, or squamous lesions, is another pathologic facet of monophasic SS that may cause diagnostic consternation.270
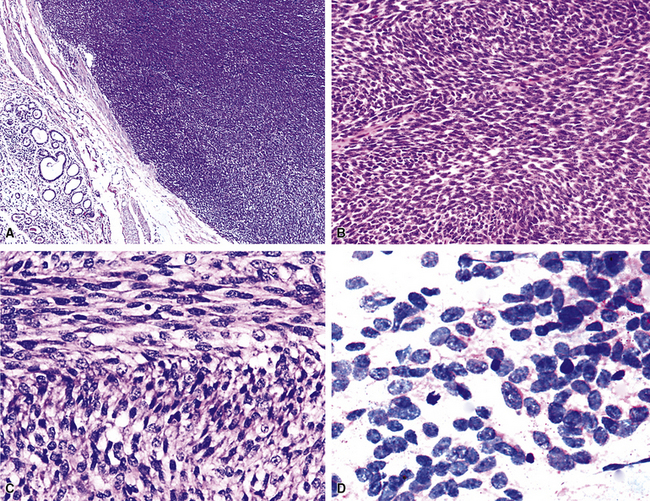
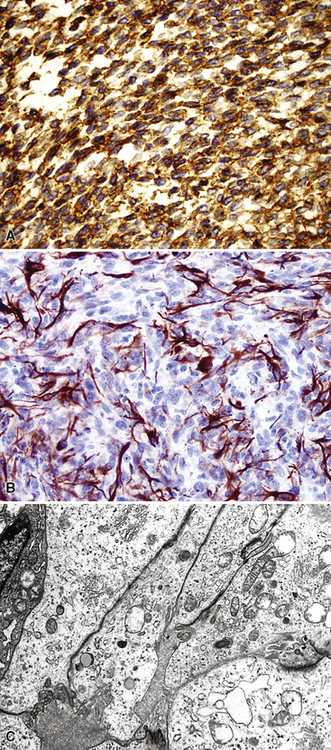
Electron microscopy and immunohistology have shown that SS is actually an epithelial proliferation, with ultrastructurally well-formed junctional complexes between the tumor cells.258,279 Reticulin stains are often useful in outlining epithelioid cell clusters when they are present but indistinct. Immunostains for keratin and EMA (Fig. 14-52) can be used to similar advantage; these determinants are also commonly seen together with vimentin in the spindle cells of biphasic or monophasic SS. CD99, calretinin, and bcl-2 protein are often observed in SS as well.279 Another extremely useful marker is TLE-1; it is a nuclear protein that is observed in virtually all cases of SS141–144 (Fig. 14-53); there is some sharing of reactivity for TLE1 with neural neoplasms,143 but, when used in an immunohistochemical panel setting, that should not pose a diagnostic problem. It is clear that SS shows a characteristic t(X;18) (p11.2;q11.2) chromosomal translocation (Fig. 14-54), which can be assessed using traditional cytogenetic techniques or fluorescence or chromogenic in situ hybridization.272,273 This karyotypic aberration produces a selective fusion transcript known as SYT-SSX, and the polymerase chain reaction can be used to detect it diagnostically, using suitable primer pairs of nucleotides.144,277

The major differential diagnostic alternatives to primary SS in the lung are metastatic SS from soft tissue sites, HPC-SFT, fibrosarcoma, mesothelioma, and SC. Among these possibilities, only SS shows the aforementioned t(X;18) chromosomal abnormality, making cytogenetic evaluation, with or without other adjunctive studies, highly desirable in this context. A decision tree that can be used in the differential diagnosis of SS is shown in Figure 14-55.
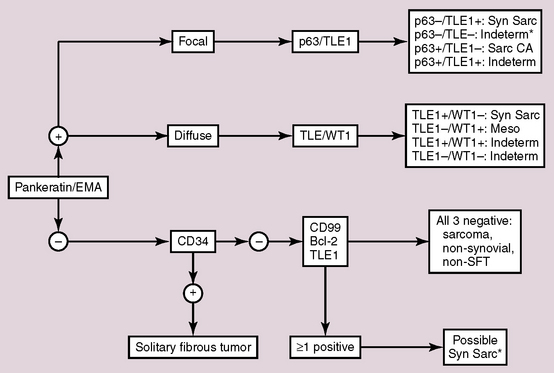
Therapy and Prognosis
The long-term outlook for patients with SS of the lung is guarded at best. In a series reported by Zeren and associates, 14 of 18 patients had died of their tumors or were likely to do so at a mean follow-up period of 12.5 years.270 In general, this neoplasm has the ability to recur locally or demonstrate distant metastasis many years after its initial diagnosis; indeed, follow-up shows that tumor-related mortality continues to accrue up to 20 years after presentation.279
Essary and coworkers280 also reported that primary pulmonary SS is a more aggressive lesion than its soft tissue counterpart. They posited that this was due to its usual large size at diagnosis and because of a common difficulty in resecting the tumor completely.
Other Primary Pulmonary Sarcomas
In addition to those tumors that have been previously considered, other sarcoma morphotypes may be encountered in the lungs. These include liposarcoma281–283 (Fig. 14-56), angiosarcoma,245,284 malignant peripheral nerve sheath tumor,185,190,285,286 osteosarcoma,245,287 angiomatoid fibrous histiocytoma288 (Fig. 14-57), and alveolar soft parts sarcoma289–292 (Fig. 14-58). Fewer than 20 cases each of these neoplasms have been documented in the literature, making it impossible to present their clinicopathologic attributes as if they were thoroughly studied and well-characterized.
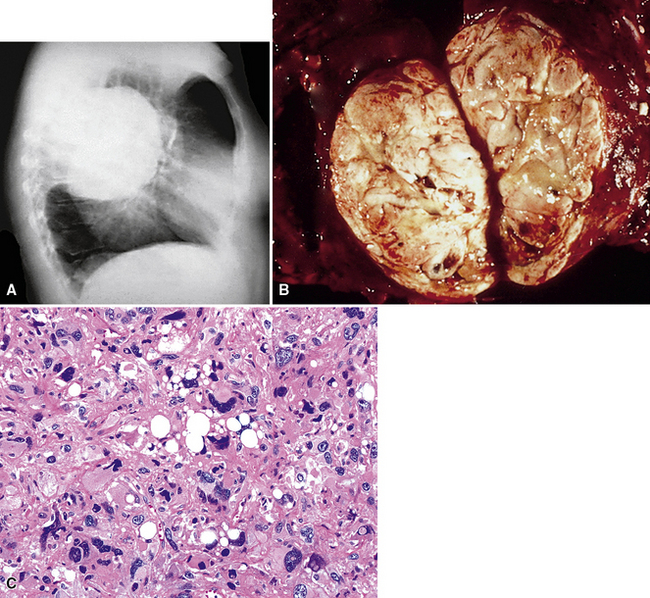
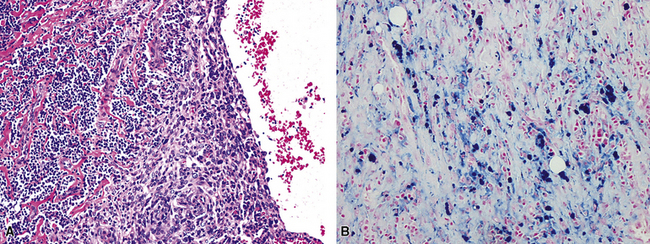
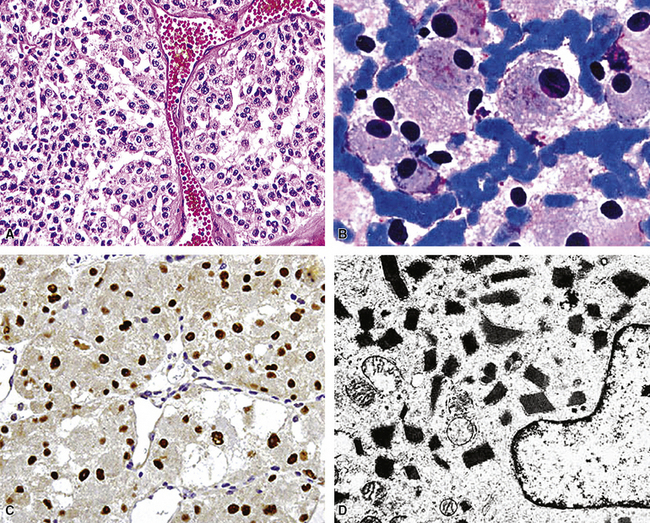
However, a few generalizations do appear to be appropriate. First, liposarcomas and malignant schwannomas of the lung have usually been documented as tumors that have an endobronchial component, potentially producing airway obstruction. In contrast, this feature is not part of the profile of either angiosarcoma or osteosarcoma. Secondly, both angiosarcoma-like and osteosarcoma-like epithelial neoplasms are vastly more common in the respiratory tract than true sarcomas with those respective microscopic patterns.4 Thus, the pathologist must be certain to address the likelihood of SC under such circumstances. Finally, therapy for those rare lesions in this category that have proven to be primary in the lungs is completely anecdotal and has been based on extrapolation from treatment used for histologically similar neoplasms in osseous and soft tissue sites.
Primary Malignant Melanomas of the Lung
Melanomas arising primarily in the lung are extraordinarily rare; the literature contains sparse reports on this topic.293–303 The largest series is from the AFIP, consisting of eight cases seen over a period of many years.293 Although rigorous criteria have been proposed for primary malignant melanoma of the lung (PMML), this interpretation cannot be established with absolute certainty because of the well-known capacity for spontaneous regression of primary melanomas in mucosal or cutaneous sites. To consider a diagnosis of PMML seriously, there obviously must be no prior history of a potentially malignant pigmented tumor of the skin or ocular uveal tract. Moreover, clinical examination for possibly occult melanomas in the integument, nail beds, eyes, nasal cavity, paranasal sinuses, oral cavity, esophagus, anus, rectum, vulva, and leptomeninges should not show any extrapulmonary lesions. In fact, some authors have suggested that a case of PMML can be regarded as bona fide only retrospectively, after a postmortem examination has excluded another source of a primary melanoma.294–296 Hence, it is self-evident that this diagnosis can never be considered irrefutable during life.
As to the fundamental question of the origin of primary melanoma in the respiratory tract, some have stressed that the tracheobronchial tree is, in fact, of endodermal derivation—similar to the oral cavity and the esophagus—where well-documented primary melanomas have originated.296 Nonetheless, these tumors are generally thought to be neuroectodermal, and their presence in endodermal or mesodermal sites is therefore problematic with reference to classic histogenetic theory. We instead subscribe to the “stem cell” theory of neoplasia, wherein embryologic constructs are essentially irrelevant, to explain the phenomenon under discussion here. In that vein, it is interesting that some benign proliferations of the lung also demonstrate partial or global melanocytic differentiation—namely, clear-cell “sugar” tumor, angiomyolipoma, and lymphangioleiomyomatosis.304–308
Clinical Summary
The ages of reported patients with PMML have ranged from 29 to 80 years. Because of the rarity of this lesion, no meaningful statements can be made regarding gender-related incidence figures. Some patients with bronchopulmonary melanoma have been asymptomatic, whereas others have presented with hemoptysis, dyspnea, or cough.293–301 A location in the lumen of the trachea may be associated with asthma-like symptoms. Plain film radiographic examinations generally have shown abnormalities only if the tumors were in the pulmonary parenchyma; in other words, endobronchial tumors are visible only on CT or MRI.
Pathologic Findings
In several reported cases of PMML, the lesions have been centered in large airways, including the trachea and major bronchi.293,299,301,309–311 Grossly, these tumors are generally polypoid, endoluminal masses that are partially or completely obstructing, or they present as nodules within the lung parenchyma, which range from 1 to 4.5 cm in greatest dimension. In addition, they are characteristically colored, in shades of brown or black, but may occasionally be amelanotic (tan-gray).
Histologically, PMMLs are composed of heterogeneously pigmented and variably pleomorphic cells that range from epithelioid to fusiform in configuration with occasional gigantiform figures; overtly sarcoma-like lesions are certainly part of the repertoire of these neoplasms (Fig. 14-59). Indeed, divergent differentiation into chondro-osseous–like tissue has been reported in melanomas.312
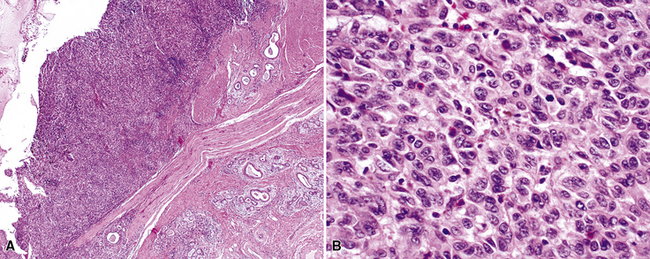
Several times over the years, we have made mistakes in the microscopic interpretation of melanoma (usually metastatic) in the lungs. If pigment is absent or sparse in the tumor cells, no meaningful historical data are supplied by the surgeon, and the lesion being studied is solitary; the stage is set for a possible error. Maeda and colleagues313 and Yamada and associates314 have discussed the particular resemblance of amelanotic melanoma to large cell undifferentiated lung carcinoma, and we concur with their conclusions. It is certainly not a necessary step to subject all undifferentiated pulmonary neoplasms to immunohistologic evaluations. Nonetheless, if such studies are obtained, and there is no reactivity in the tumor cells for pankeratin (often used as an “internal control” in carcinoma cases), the alternative possibility of melanoma should be considered.
An important microscopic feature that supports a primary origin in the respiratory tract is the presence of a cytologically atypical in situ melanocytic proliferation in adjacent bronchial mucosa, which may be metaplastic293,296 (Fig. 14-60). To confirm the presence of melanocytic differentiation293 and exclude the differential diagnoses of anaplastic carcinoma or sarcoma, electron microscopic studies showing the presence of cytoplasmic premelanosomes (Fig. 14-61) or immunohistochemical negativity for keratin and labeling for S100 protein, HMB-45 antigen, Melan-A/MART-1, PNL2, or tyrosinase are useful.
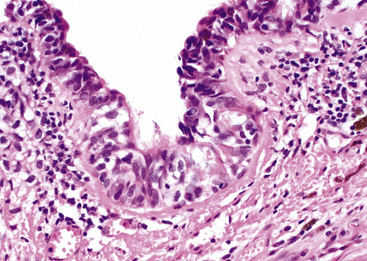
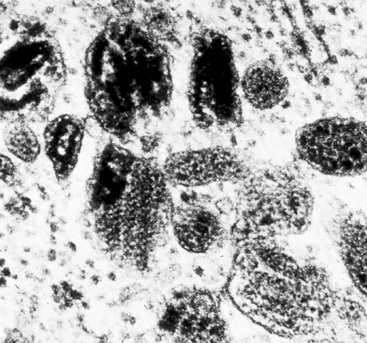
Therapy and Prognosis
Before assigning a diagnosis of probable PMML, one must undertake a thorough dermatologic examination, as well as extensive radiologic and endoscopic assessments.315–317 Those measures are designed to detect an occult primary mucocutaneous melanoma in another location. De Wilt and coworkers also have considered the scenario in which pathologists confront a possible diagnosis of intrapulmonary melanoma in intraoperative consultation.318 If the tumor being analyzed is amelanotic, it will likely resemble a high-grade carcinoma in frozen sections and touch preparations. Even if the patient has a known history of melanoma, the surgeon should be counseled to perform a conservative—but adequate—excisional procedure that would be appropriate for primary lung cancer under those circumstances.
In most documented cases of PMML, patients have fared extremely poorly, with the majority dying within 1 year of diagnosis.293,296–298,300 Reid and Mehta reported one individual who survived 11 years after surgery299; however, there was no mention in the latter study of clinical evaluations that were designed to exclude other primary sites of origin, and no in situ melanocytic proliferation was found in the pulmonary resection specimen. Thus, the latter case is doubtful as a verifiable PMML. Most patients have undergone surgical resections of their tumors, although a few have received irradiation or chemotherapy.294 In extension of therapeutic results obtained in cases of other primary melanomas of the viscera, it must be concluded that long-term survival of patients with PMML is an idiosyncratic and unlikely event.
Sarcomas of the Pulmonary Arterial Trunk
Although it is technically not part of the respiratory tract, the pulmonary arterial trunk is an appropriate topic for this discussion that centers on intrathoracic mesenchymal malignancies. For more than 75 years, it has been known that this vascular segment may serve as the point of origin for sarcomas with diverse histologic features and clinical manifestations that are just as variable.319 To date, approximately 200 cases of pulmonary trunk sarcoma (PTS) have been documented.13,319–322
Clinical Summary
Patients with PTS are adults in middle life or beyond, with no predilection for either gender. They present with a panoply of potential symptoms and signs, the most common of which simulate the findings of right-sided cardiac failure or pulmonary thromboembolic disease.323 Patients often complain of intractable cough, progressive dyspnea, and dull chest pain that may increase with exertion; cardiac tamponade has rarely been observed.321 Neck veins may be distended, a loud precordial systolic heart murmur may be audible at the upper left sternal border, and the patient may manifest the complete clinical scenario of anasarca.324,325 However, cardiac imaging studies demonstrate no evidence of ventricular hypokinesis or perfusion abnormalities.309
In the era before modern angiography, echocardiography, CT, and specialized radionuclide scans, the diagnosis of PTS was usually made for the first time at autopsy.319 However, current imaging modalities are now capable of revealing the tumor in question rather easily309 (Fig. 14-62). It takes the form of a partially obstructing, endoluminal mass at the level of the right ventricular outlet or above it, and it may extend over a span of several centimeters. Attachment of the lesion to the arterial wall is variable in character and may be sessile or pedunculated. The neoplasm is typically somewhat heterogeneous in density and greatly heterogeneous in size, from case to case.
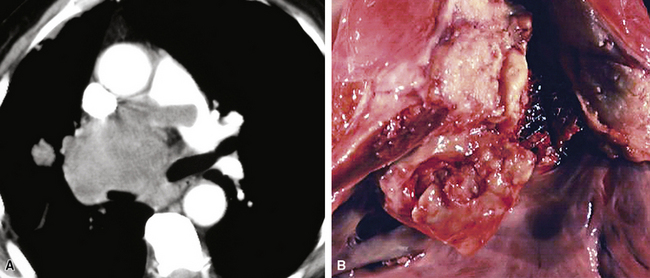
Pathologic Findings
The preoperative diagnosis of PTS is typically one that may not involve the pathologist, inasmuch as biopsy of an intravascular mass in the right ventricular outlet is a challenging procedure. Thus, his or her first encounter with such lesions may be in the frozen section laboratory during a definitive surgical procedure. In this context, it is important to realize that a firm diagnosis of a particular sarcoma type (or even of a malignancy) may not be an easy proposition. Some PTSs take the form of rather paucicellular myxoid proliferations with surprisingly bland cytologic characteristics (Fig. 14-63), whereas others are anaplastic tumors that defy easy classification under the microscope319 (Fig. 14-64). The proffered pathologic interpretations in the literature on PTS include such diagnoses as undifferentiated sarcoma, angiosarcoma, leiomyosarcoma, rhabdomyosarcoma, fibromyxosarcoma, fibrosarcoma, chondrosarcoma, osteosarcoma, hemangioendothelioma, malignant fibrous histiocytoma, and malignant mesenchymoma. 325–335 What one is able to glean from this apparently confusing list is that the histologic spectrum and pathologic grades of PTS are broadly distributed, such that no two individual lesions look quite the same under the microscope. Beyond that, pathologists take a great deal of interest in speculating on the mechanistic reasons for this diversity, but this issue has admittedly little clinical import at the present time.
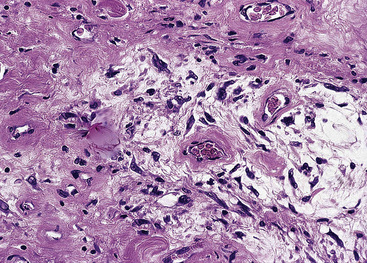
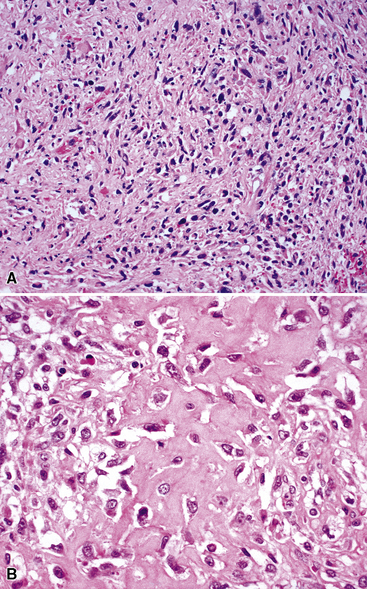
With respect to differential diagnosis, the majority of PTSs have the characteristics of high-grade spindle cell or pleomorphic sarcomas, which, in current parlance, would usually be grouped together under the rubric “MFH.” However, some low-grade fibromyxoid variants can closely simulate an intracardiac myxoma or organizing mural thrombus.319 Close attention to morphologic detail is the only certain method for distinguishing between such possibilities.
With particular regard to cytologic preparations of thoracic sarcomas, including PTS, the relative lack of architectural detail seen in fine needle aspiration biopsies has an “equalizing” effect. With the exception of biphasic synovial sarcoma, spindle cell sarcomas of various lineages (Fig. 14-65), pleomorphic sarcomas (Fig. 14-66), and small round cell tumors (Fig. 14-67) have superimposable microscopic images in such preparations. Thus, ancillary diagnostic methods are necessary to make specific diagnoses in those neoplastic categories.
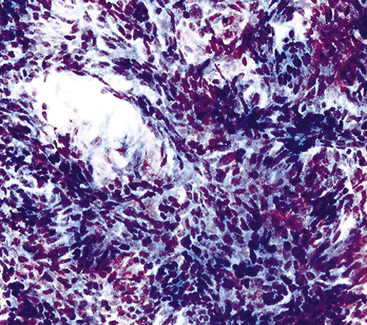
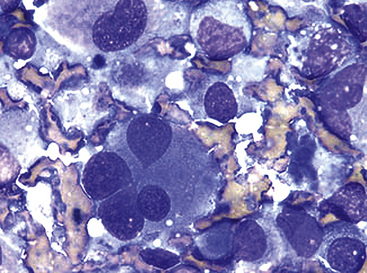
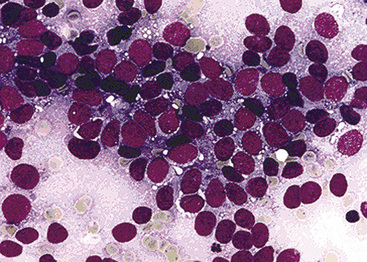
Therapy and Prognosis
Because of the dominance of autopsy reports in the earliest literature on PTS, the recommended therapy for this tumor must still be considered evolutionary. At the present time, providing that a firm radiologic diagnosis can be made or a frozen section interpretation of sarcoma can be rendered, the surgeon may perform an en bloc resection of the pulmonary trunk and its luminal tumor contents, followed by interposition of a synthetic graft.310 This is probably the most definitive approach to operative therapy, inasmuch as it is difficult to determine the boundaries of intramural tumor growth by visual inspection. The latter point makes more limited vascular resections and reconstructions a tenuous enterprise.
Extension of the tumor through the wall of the pulmonary artery is the single most important piece of pathologic information in cases of PTS, inasmuch as histologic grading generally does not appear to correlate with tumor behavior in a consistent fashion.319,325 An exception to the latter statement is embodied in a report by Tavora and colleagues, who suggested that low-grade myofibroblastic PTS had a distinctly better prognosis than other histotypes.336 In addition, the surgeon’s or radiologist’s estimation of whether the lesion is pedunculated or sessile has considerable importance. Tumors with a narrow stalk tend to “flutter” in the stream of ejected blood in the ventricular outflow tract, and pieces of the neoplasm may be embolized into the lungs.337 This phenomenon is not as common with lesions that assume a broadly based sessile macroscopic growth pattern.
Tumors of the Pleura
Sarcomatoid Malignant Mesothelioma (see also Chapter 20)
Clinical Summary
Regardless of histologic subtype—sarcomatoid or otherwise—the clinical features of intrathoracic mesothelioma are the same.338–340 Malignant mesothelioma (MM) of the pleura typically affects adult men, although women and children are certainly represented in the patient population with this neoplasm.340,341 The most common presenting symptoms and signs are pleuritic-type chest pain and progressive shortness of breath, with a pleural effusion on chest radiographs. An influenza-like syndrome is occasionally reported in association with pleural mesothelioma. The lesion most commonly takes the form of multiple nodules and plaques in the serosal surfaces but may occasionally be represented by a solitary localized mass (Fig. 14-68). Later in the clinical course, encasement of the lung by confluent neoplastic tissue is seen (Fig. 14-69).
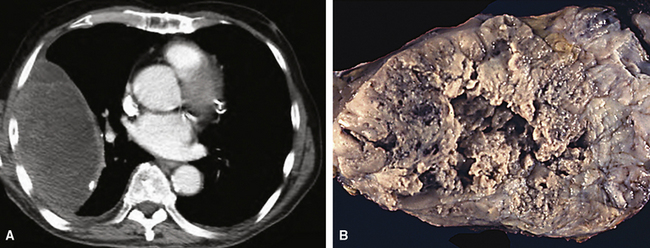
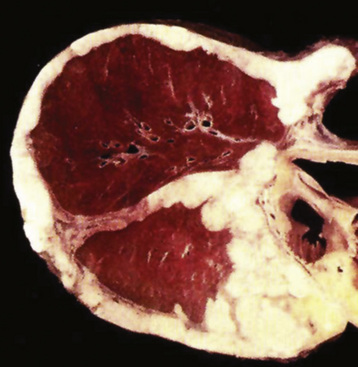
Like examples of peritoneal mesothelioma that have been linked causally to chronic recurrent peritonitis in the context of familial Mediterranean fever,342 the authors have observed several pleural tumors that arose in the background of chronic pleuritis in patients with a connective tissue disease (e.g. lupus erythematosus). Roughly 50% to 70% of pleural mesotheliomas can be objectively related to prior occupational-level asbestos exposure.343 In those instances, 85% to 90% of patients show the presence of (1) pleural plaques or pleural calcifications or (2) quantitative pulmonary asbestos fiber burdens clearly in excess of the background, serving as tangible markers of such exposure.344–350 Those findings are vastly more reliable than patients’ historical accounts of occupational conditions and should be sought in all instances before concluding objectively that a given mesothelioma is indeed asbestos-related etiologically.
Other accepted pathogenetic factors in mesothelioma cases include inhalation exposure to erionite, chronic infection of the pleural spaces (e.g., in tuberculous pleuritis), and prior therapeutic irradiation of the thorax.351,352 Substantial recent interest has centered on the potential role of simian virus-40 in this setting,353 but conclusions about whether this agent is indeed causative in any way are premature. At least 30% of pleural mesotheliomas are idiopathic.343
Pathologic Findings
In considering the pathologic appearances of overtly malignant mesothelial tumors, a surprising variety of patterns has emerged over time, and these have expanded the traditional categorical outline, which previously included “epithelioid,” “biphasic,” and “sarcomatoid” mesotheliomas.340,354,355 The epithelioid subgroup has now been enlarged to include mesothelial malignancies that have a wholly clear cell, oncocytoid (“deciduoid”) or granular cell, tubulopapillary, large polygonal cell, polyhedral stromal mucin-producing, “medullary” epithelioid, or even small cell appearance (see Chapter 20).354 The differential diagnostic potentialities raised by such images are numerous, including metastatic non–small cell carcinomas of various primary origins, metastatic melanoma, pleural sarcomas with an epithelioid or small round cell appearance, and even metastatic small cell neuroendocrine carcinoma. In reference to biphasic MM—with epithelioid and sarcoma-like elements—primary SS of the pleura is an important diagnostic alternative.356 Indeed, in the absence of data showing the characteristic t(X;18) chromosomal translocation of SS, which is associated with production of SYT-SSX fusion transcripts,357 or immunoreactivity for TLE1 (see earlier discussion), its separation from MM can be extremely challenging. This is so because the immunophenotypes of the two tumors are so similar.358
Monophasic sarcomatoid mesothelioma potentially simulates a range of spindle cell sarcoma morphotypes that may affect the pleura (Figs. 14-70 and 14-71), again including monophasic SS, but also fibrosarcoma, MFH, rhabdomyosarcoma, chondrosarcoma, osteosarcoma, leiomyosarcoma, and malignant peripheral nerve sheath tumor.1,359–363 Pseudomesotheliomatous secondary pleural carcinomas with a sarcomatoid phenotype also enter the differential diagnosis in a meaningful fashion, as referenced earlier in this discussion. The histomorphologic findings in that particular differential diagnostic group of tumors have been summarized previously.
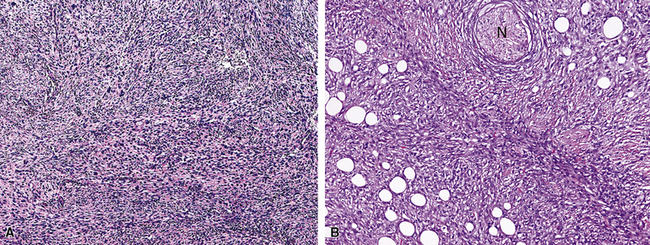
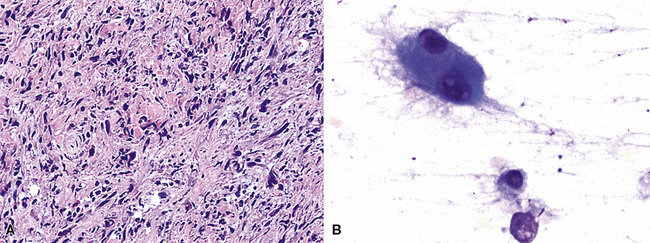
In specific reference to sarcomatoid mesotheliomas, the basic histologic image of such lesions is virtually identical to that associated with SC of the lung.364–369 Indeed, cases in which a tumor mass involves both the peripheral lung and the pleura require immunohistologic or molecular evaluation in order for a distinction to be made with definition between those tumor types.370 As such, sarcomatoid mesotheliomas are composed exclusively by overtly malignant spindle cells and pleomorphic elements, with or without such heterotopic tissue as osteoid, cartilage, muscle, or osteoclast-like giant cells.
Lymphohistiocytic mesothelioma371,372 was formerly classified as a sarcomatoid subtype, but that lesion is now generally considered to be a form of epithelioid mesothelioma with a lymphoepithelioma-like image. Another special variant of sarcomatoid mesothelioma merits further consideration: namely, desmoplastic mesothelioma.373–379 That tumor is characterized by relatively bland neoplastic spindle cells that are set in a densely collagenized fibrohyaline matrix with a so-called “patternless pattern” of growth (Fig. 14-72). Differential diagnosis of that tumor type is with fibrous (or fibrohyaline) pleuritis (“fibrous pleurisy”); the presence of focally dense and atypical cellular growth, necrosis, obvious invasion of lung or soft tissue, or metastasis points to a diagnosis of mesothelioma.378 On the other hand, lymphohistiocytoid mesotheliomas may be confused with malignant lymphoma, inflammatory myofibroblastic (pseudo)tumor of the pleura, or metastatic lymphoepithelioma-like carcinoma.371,372
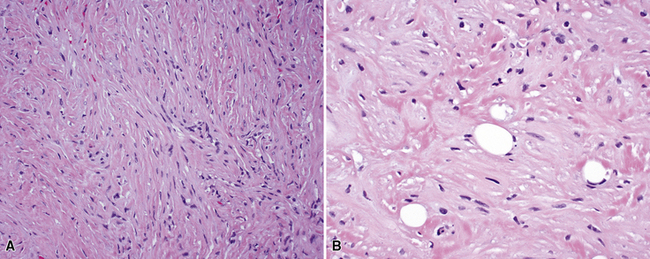
This information brings one to a consideration of adjunctive pathologic studies in the objectification of a diagnosis of MM. In regard to this important topic, it should be remembered that there are definite roles for a number of laboratory analyses, including but not limited to histochemistry, immunohistology, electron microscopy, fluorescent or chromogenic in situ hybridization, the polymerase chain reaction using appropriately chosen primers, and traditional cytogenetic evaluations.338–350,380,381
Although most attention has been paid in recent years to the immunohistochemical separation of epithelioid MM from metastatic adenocarcinoma,382,383 the panel of markers used for that purpose is generally not helpful in the differential diagnosis of nonepithelioid (i.e., sarcomatoid) MM variants, with selected exceptions. Standard approaches to separating MM from adenocarcinoma include immunostains for keratin (either “pankeratin,” or keratin 5/6, or both), EMA, thrombomodulin, HBME-1, calretinin, Wilms tumor gene product 1 (WT1), podoplanin,146 tumor-associated glycoprotein-72 (recognized by B72.3), carcinoembryonic antigen, CD15, Ber-EP4, BG8, and MOC-31, with expected reactivity in MM primarily including any of the first seven of those determinants.384 Electron microscopy is still extremely useful in this particular context, inasmuch as the long, branching, bushy microvilli that one associates with mesothelial cells are best represented in epithelioid MM.385
Neither immunohistology nor electron microscopy is nearly as helpful in the realm of biphasic or sarcomatoid tumors, and an entirely different set of morphologic and immunophenotypic variables must be assessed in those lesions. For example, keratin, WT1, podoplanin, and calretinin assume much greater value in the differential diagnosis of sarcomatoid MM (Fig. 14-73).146,384,386 The principal interpretative alternatives are those of true sarcoma, sarcomatoid pseudomesotheliomatous carcinoma, or malignant solitary fibrous tumor; sarcoma-like mesothelioma differs from the latter two entities in that it may divergently express specialized mesenchymal markers such as desmin and muscle actin isoforms.387,388
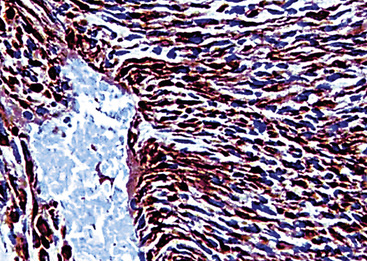
Figure 14-73 Diffuse immunoreactivity is seen in this sarcomatoid mesothelioma for podoplanin, with antibody D2-40.
Fluorescent in situ hybridization or polymerase chain reaction for SYT-SSX transcripts may be necessary to separate some examples of SS (which are also reactive for keratin and potentially for calretinin) from MM with certainty.357,358 Electron microscopic analyses are likewise not very helpful in the differential diagnosis of sarcomatoid MM, because that variant of mesothelioma tends to lose specialized ultrastructural features of epithelial cells (Fig. 14-74).369,389 Rarely, localized sarcomatoid mesotheliomas also may resemble solitary fibrous tumors of the pleura390; in those cases, immunoreactivity for CD34, or CD99, or both, tends to exclude a diagnosis of MM.391
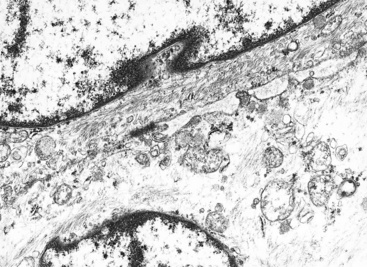
Therapy and Prognosis
The natural history of malignant pleural mesothelioma is an adverse one. The usual survival of patients with that tumor is less than 15 months, with death occurring because of cardiorespiratory embarrassment or pulmonary superinfection.338,339,392 A peculiarity of this neoplasm is its tendency to grow through surgical defects in the chest wall, either represented by thoracotomy incisions or thoracostomy sites. Metastases outside the thorax are relatively rare, although they have been reported in a small minority of cases in such sites as liver, bones, and skin.393 Treatment is generally supportive, inasmuch as irradiation and chemotherapy produce little survival benefit.394 Extrapleural pneumonectomy is still a controversial surgical approach to mesothelioma; its proponents claim a definite decrease in mortality in the operative group compared with stage-matched and age-matched controls managed by other means.395 However, those observations have not been supported by other studies.396
Primary Pleural Sarcoma
Sarcomas are as rare in the pleura as they are in the lungs. Most neoplasms that take the generic appearance of malignant mesenchymal tumors in the serosae of the thorax are, in actuality, epithelial lesions. They may either represent metastatic SCs or variants of MM, as considered earlier. In addition, there are only a limited number of definable clinicopathologic entities to consider in this specific anatomic location. These include fibrosarcoma, malignant solitary (localized) fibrous tumor of the pleura, leiomyosarcoma, SS, Askin malignant thoracopulmonary small round cell tumor, PPB, KS, EH, and angiosarcoma. Extraordinarily rare examples of granulocytic sarcoma (extramedullary tumefactive acute myeloid leukemia),397 malignant peripheral nerve sheath tumor (Fig. 14-75),362 mesenchymal chondrosarcoma,398 extraskeletal myxoid chondrosarcoma (Fig. 14-76),399 liposarcoma,400,401 and extraosseous osteosarcoma402,403 have been documented as apparently primary pleural tumors, but information on such lesions is anecdotal.
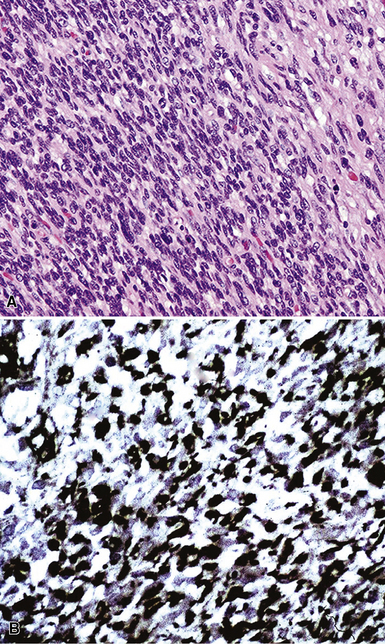
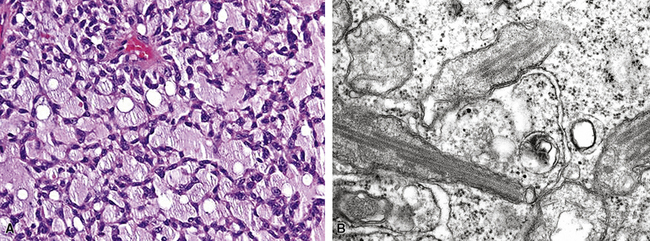
Pleural Fibrosarcoma and Malignant Solitary (Localized) Fibrous Tumor
A review of the literature on serosal neoplasms reveals few examples of well-documented primary pleural fibrosarcoma (PPFS).404,405 The latter is only arbitrarily distinguished from malignant solitary (localized) fibrous tumor (MSFT) of the pleura406,407 by its clinical growth pattern, which is diffuse rather than localized. However, in other respects, these two tumor entities are virtually identical to one another; in fact, some examples of PPFS have apparently evolved from solitary fibrous tumors of the pleura.408–413 Although some authors prefer to separate malignant fibrous tumors of the pleura into “true” fibrosarcoma and MFH-like tumors,414 all of these lesions will herein be considered as a single group because of their closely similar clinicopathologic attributes.
Clinical Summary
Pleural fibrosarcoma and MSFT arise in adult patients over a wide range of ages (15–75 years), with a male-to-female ratio of 3:1. They may be associated with dull or pleuritic chest pain, dyspnea, cough, systemic flu-like symptoms, and digital clubbing.414,415 In addition, a small proportion of patients may manifest paraneoplastic hypoglycemia (Doege-Potter syndrome) because of the production of an insulin-like peptide by the tumor cells.415,416 It appears that pleural sarcomas have no association with prior asbestos exposure (in contradistinction to a proportion of MMs).367 Other potential etiologies of these lesions are unsettled at the present time, but some authors have reported a putative pathogenetic linkage to chronic tuberculous pleuritis and prior pyothorax.417,418
Radiographic studies in cases of PPFS commonly demonstrate the presence of a unilateral pleural effusion, which may be massive.415,419 In addition, a dominant mass and diffuse but irregular thickening of the pleura are usually evident, and are especially visible with CT or MRI studies.419 Based on clinical data, it is not possible to distinguish PPFS from diffuse MM, and tissue procurement is mandatory for this purpose. On the other hand, MSFT are typically well-circumscribed, pleural-based masses on chest x-rays; they usually show rounded contours, but may occasionally be lobulated415 (Fig. 14-77). Most measure between 1 and 10 cm in maximal dimension. In contrast to benign solitary fibrous pleural tumors, MSFTs are less often pedunculated and usually attain a larger size. Moreover, the latter lesion has a higher likelihood of involving the parietal pleura or mediastinum, or of demonstrating “inverting” growth into the subjacent lung parenchyma.408,415
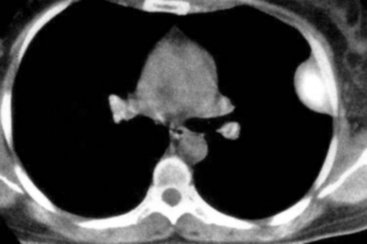
Pathologic Findings
The macroscopic appearance of PPFS is virtually identical to that of diffuse MM (i.e., as a “rind” of solid tissue that encases the lung and restricts its movement). These tumors commonly extend into interlobar fissures and intrapulmonary interstitial septa as well.404–416 On the other hand, MSFTs are sessile or pedunculated localized masses that are most often seen in the upper portions of either hemithorax. They have bosselated, fleshy, tan-gray cut surfaces, usually with foci of spontaneous necrosis and hemorrhage408,415 (Fig. 14-78).
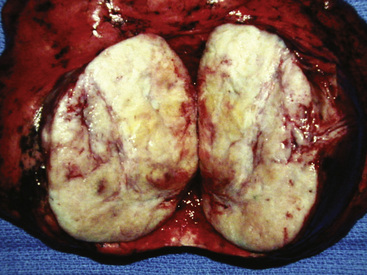
Figure 14-78 Gross photograph of excised malignant solitary fibrous tumor of the pleura demonstrating internal foci of necrosis.
Microscopically, one sees a dense proliferation of spindle cells with high nuclear-to-cytoplasmic ratios, coarse chromatin, nuclear irregularity, and prominent nucleoli. Mitotic activity is typically brisk, and foci of spontaneous hemorrhage and necrosis may be evident as well (Fig. 14-79). The neoplastic cells may be arranged in a storiform fashion and show moderate-to-marked pleomorphic cytologic features, calling to mind the histologic attributes of MFH (see earlier discussion).404,415 In other cases, they are aligned in a fascicular “herringbone” configuration, as in pulmonary fibrosarcomas (Fig. 14-80). The subjacent lung is involved by tumor only if it extends downward from the pleura via the intrasegmental fibrous septa, and there is no association with pleural fibrohyaline plaques, the presence of intraparenchymal asbestos fibers, or asbestosis. MSFT may contain areas that resemble benign solitary fibrous pleural tumors, in which more bland spindle cell aggregates are enmeshed in hyalinized, keloidal-type collagen.415 A “staghorn” stromal vascular pattern is common in such areas as well.
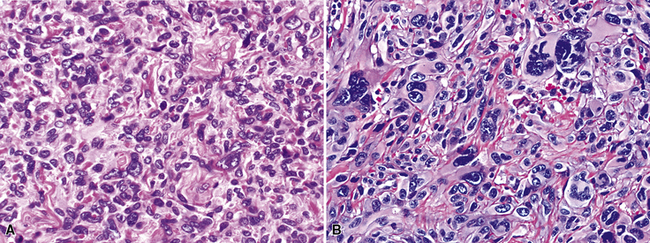
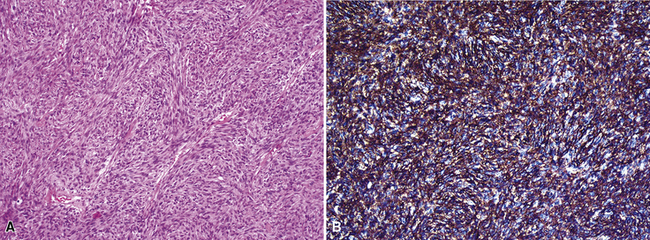
Electron microscopy shows only primitive, fibroblast-like characteristics of the neoplastic cells. They are loosely apposed and surrounded in part by collagen fibers. Cytoplasmic contents are rudimentary and include the basic metabolic organelles as well as abundant free polyribosomes and rough endoplasmic reticulum (Fig. 14-81). There is no ultrastructural evidence of epithelial or myogenous differentiation. Similarly, immunohistologic assessment of PPFS and MSFT demonstrates reactivity for vimentin alone, to the exclusion of actin, desmin, keratin, and EMA.404–409 In contrast, true mesotheliomas (including sarcomatoid variants) uniformly express epithelial markers.386 In contrast to benign SFTs, some MSFTs may lack immunoreactivity for CD34, bcl-2 protein, and CD99.413
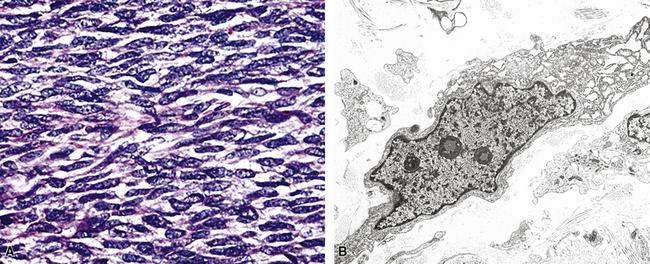
Therapy and Prognosis
PPFS is not often treatable by surgical means because of its diffuse nature. The only operative procedure that can be attempted in such circumstances is extrapleural pneumonectomy, which generally is associated with a very high level of morbidity and mortality. Radiotherapy and chemotherapy (including intrapleural instillation of pharmaceuticals) may play a role in palliation of symptoms, but unfortunately they are not curative treatments. Death is due to progressive respiratory compromise, and PPFS also may involve the pericardium and produce cardiac embarrassment.420 Actuarial 1-year survival was only 39% in one series where multimodality therapy was used.414
MSFT, on the other hand, is amenable to complete surgical resection in a high proportion of cases; in a series from the AFIP, 45% of such lesions were cured by excision alone.415 Most of these were pedunculated, well-localized masses that involved only a small area of the pleural surface, and the authors of the latter report therefore suggested that resectability was the single most favorable prognostic feature in MSFT cases. Those lesions that do go on to recur may seed the ipsilateral pleural surfaces or involve the contralateral pleura, the lung parenchyma, and other viscera. Interestingly, relapses often still take the form of localized masses, and even patients with persistent tumor may go on to survive for extended periods of time.367 Irradiation and chemotherapy do not appear to offer any benefits in this setting, and they may even shorten the survival of patients with MSFT.415
Primary Pleural Leiomyosarcoma
Leiomyosarcomas are extremely rare as primary pleural neoplasms, with fewer than 25 well-documented cases in the literature.421,422 Only one series of such tumors has been reported, by Moran and associates.421
Clinical Summary
Patients with primary pleural leiomyosarcoma present in a similar fashion to those with mesothelioma, except that a greater proportion have had asymptomatic lesions. Radiographically, pleural effusions have not been observed consistently in association with such tumors, the majority of which appeared as solitary, solid, unilateral masses measuring up to 18 cm in greatest dimension (Fig. 14-82).423 Some examples have encased the lung completely, simulating malignant mesothelial tumors.421
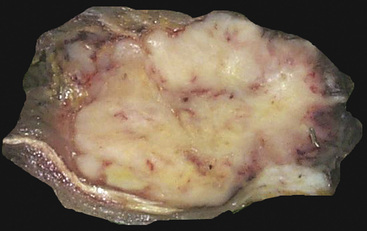
Pathologic Findings
As in other anatomic sites, pleural leiomyosarcomas are characterized by fascicles and whorls of atypical spindle cells, featuring fusiform nuclei, fibrillary eosinophilic cytoplasm, nuclear pleomorphism, and mitotic activity (Fig. 14-83). Necrosis is also frequently encountered as well. The tumors invade the lung parenchyma, or the soft tissues of the chest wall, or both.
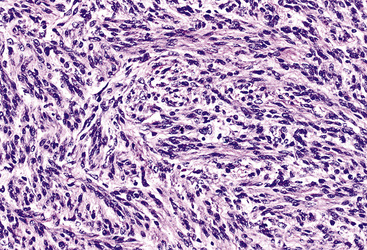
Ultrastructural analyses have shown typical findings of smooth muscle differentiation in such tumors, including plasmalemmal dense plaques, pinocytotic vesicles, cytoplasmic thin filaments punctuated by dense bodies, and pericellular basal lamina (Fig. 14-84). Immunohistologically, pleural leiomyosarcomas are reactive for vimentin, desmin, muscle-specific actin, caldesmon, calponin, and alpha-isoform actin (Fig. 14-85),421 yielding potential immunophenotypic overlap with SC or mesothelioma. However, in our experience, keratin, EMA, and calretinin are uniformly absent, providing points of difference from the latter two epithelial tumors.
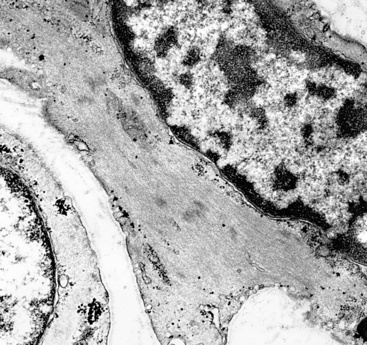
Therapy and Prognosis
Because of the rarity of these lesions, only anecdotal information is available on the biology of primary pleural leiomyosarcomas. Moran and associates advocated surgical ablation but found that two of five such lesions they studied could not be resected completely.421 The merits of adjuvant therapeutic modalities are as yet unstudied.
Askin Tumor (Primitive Neuroectodermal Tumor) and Desmoplastic Small Round Cell Tumor
In 1979, Askin and colleagues described a peculiar thoracic neoplasm that was seemingly limited to children, adolescents, and young adults.424 This lesion arises from the pleura or the extrapleural intercostal soft tissue and was originally named the malignant small cell tumor of the thoracopulmonary region. Since then, it has become known more simply as Askin tumor, or, alternatively—because the neoplasm has been shown to exhibit neuroepithelial differentiation—thoracopulmonary primitive neuroectodermal tumor (TPNET).425–436 Prior to its seminal description, it is likely that this lesion was included among cases of Ewing sarcoma of the thorax or peripheral neuroblastoma.437 Primary primitive neuroectodermal tumor of the lung is considered elsewhere in this monograph, but Askin tumor is technically considered to be separate from that entity and will therefore be discussed at this point. A related neoplasm is known as desmoplastic small round cell tumor (DSRCT) of the serosal surfaces. It was originally described in the peritoneum and is more common there by far, but several examples have been described in the pleura as well.438–440
Clinical Summary
Askin tumor and DSRCT demonstrate a peak incidence during the second decade of life (mean age, 15 years), and they show a slight male predilection. Isolated cases of TPNETs in infants and in older adults have also been documented.438–443 These neoplasms may present as asymptomatic masses in the chest wall or produce symptoms of cough, unilateral chest pain, and dyspnea or tachypnea. Pleural effusion is a common complication and may be detected on physical examination or by radiography of the thorax.425 Although Askin tumors and DSRCTs have been confused with classic neuroblastoma in some reports, they are not associated with elevations of catecholamine metabolite levels in the urine or blood, and they do not produce the opsoclonus-myoclonus syndrome.433,441
Chest x-rays and other imaging studies typically show a large mass that may be pleural-based or centered in the thoracic soft tissue, with secondary extension into the pleural space (Fig. 14-86). TPNETs and DSRCTs often reach greater than 10 cm at the time of initial diagnosis, and they demonstrate ill-defined interfaces with the subjacent lung or surrounding tissues.444
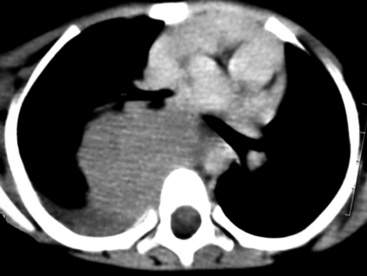
Pathologic Findings
Askin tumor is one of the prototypical small round cell neoplasms of children and may be confused with several other tumor entities by the pathologist.433 At a macroscopic level, TPNET is lobulated with fleshy, relatively soft, tan-gray cut surfaces (Fig. 14-87) that may show foci of hemorrhage and necrosis.424 Microscopically, it exhibits cellular monomorphism, with round-to-oval nuclei, even distribution of chromatin, indistinct nucleoli, and variable mitotic activity (Fig. 14-88). Stromal blood vessels are numerous and form a discernible network within the tumor mass; matrical hemorrhage also may be manifest.424–441 One of the most characteristic findings of TPNET on conventional microscopy is the presence of neural-type cellular “rosettes,” wherein tumor cells are disposed radially around small virtual tissue spaces424,425 (Fig. 14-89). Histochemically, Askin tumor may or may not contain abundant glycogen with the PAS method (Fig. 14-90), although in the original series on this lesion, only PAS-negative neoplasms were accepted to facilitate separation from classic Ewing sarcoma.
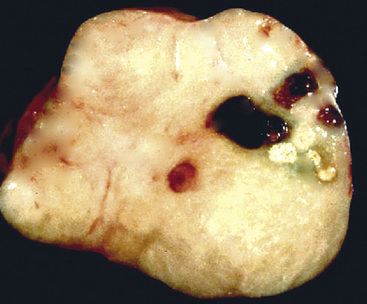
Figure 14-87 Partial excision of the lesion yielding a fleshy mass demonstrating internal foci of necrosis.
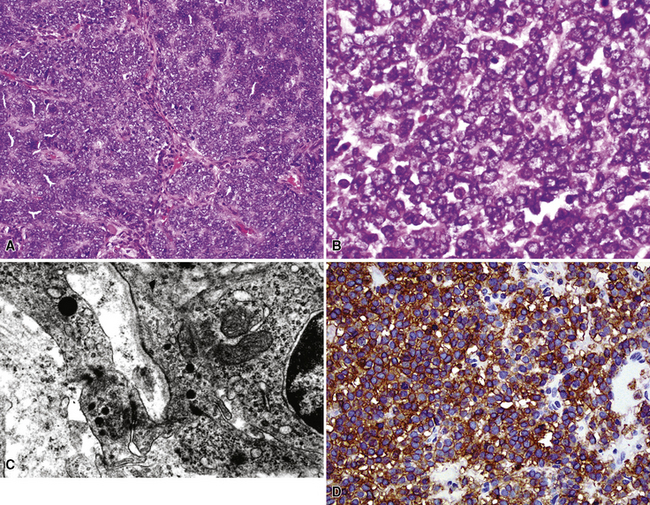
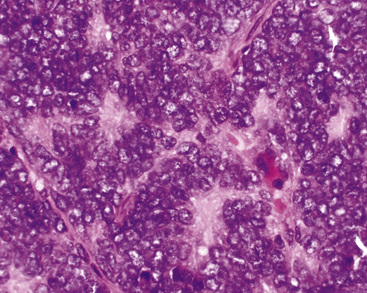
Figure 14-89 Primitive intercellular rosettes are present in this Askin tumor (primitive neuroectodermal tumor).
DSRCT differs from the description just given in that it features aggregates of small monomorphic tumor cells that are set in a much more fibrogenic stroma than that seen in Askin tumor (Fig. 14-91). The growth pattern is also more organoid than in conventional TPNET.438–440 Although no obvious evidence of myogenous differentiation is apparent at a conventional morphologic level, immunostains typically show coreactivity for vimentin, desmin, and keratin in DSRCT (Fig. 14-92),439 and ultrastructural studies also support the presence of bifid epithelial-myogenic differentiation.
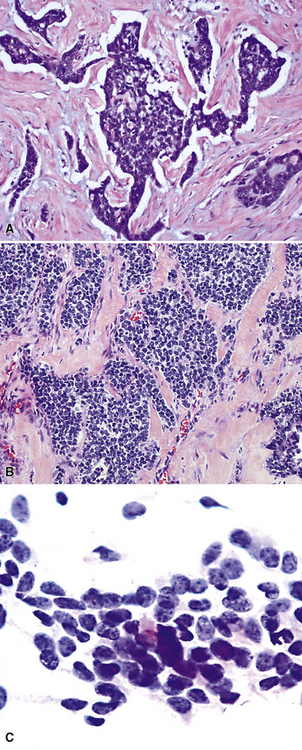
Figure 14-91 A and B, Desmoplastic small round cell tumor of the pleura, in which angular cell groups composed of cells like those of “ordinary” primitive neuroectodermal tumor (see Figs. 14-88 and 14-89) are set in a markedly fibrous stroma. They are dyshesive and monomorphic in a fine needle aspiration biopsy specimen (C).
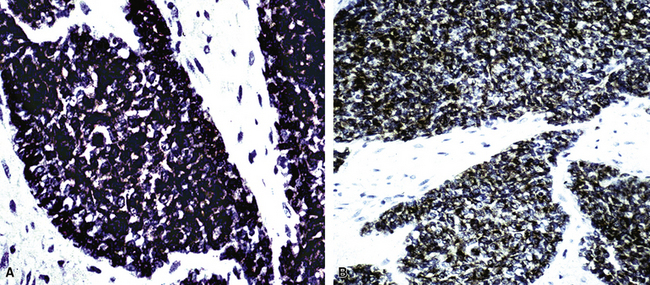
Special studies of biopsy or resection specimens are mandatory to recognize TPNET and DSRCT properly and exclude other diagnostic possibilities. Those include mesenchymal chondrosarcoma, small cell synovial sarcoma, hemangiopericytoma, and metastatic small cell neuroendocrine carcinoma; the last of these possibilities is unlikely in the usual patient group with TPNET. Along with other peripheral neuroepithelial neoplasms, TPNET and DSRCT demonstrate characteristic t(11:22) chromosomal translocations425,445 (Fig. 14-93). By electron microscopy, they demonstrate blunt cytoplasmic processes that contain dense-core granules or microtubules; these characteristics are seen in classic neuroblastoma as well, but not in other small round cell tumors.433
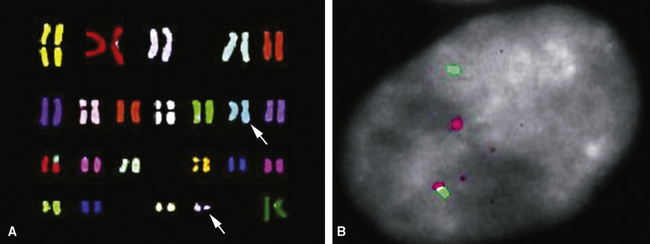
Immunohistochemically, TPNET is related to classic Ewing tumor–PNET in that it shows consistent reactivity for synaptophysin as well as for CD99.446 DSRCT is more variable with regard to its positivity for both of those markers.447,448 Askin tumor may be distinguished from classic neuroblastoma immunophenotypically; the former lesion is reactive for both beta2-microglobulin and CD99,439 whereas the latter tumor is not. Among primitive neuroectodermal tumor, DSRCT, and neuroblastoma, only DSRCT labels for WT1 (Fig. 14-94).449,450
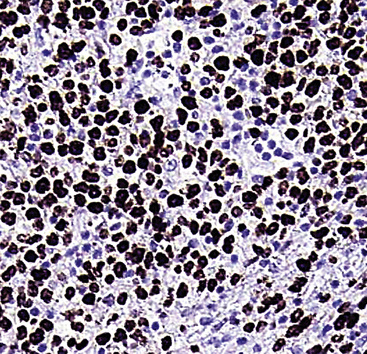
Therapy and Prognosis
The most important prognostic procedure in cases of TPNET or DSRCT is that of accurate staging. Using a scheme devised by the National Cancer Institute, stage I tumors are defined as those measuring less than 5 cm in maximum diameter that can be completely excised, stage II lesions are less than 5 cm and are grossly resectable but show positive microscopic margins, stage III neoplasms are greater than 5 cm and are nonresectable, and stage IV primitive neuroectodermal tumors have metastasized to extrapleural sites.425 Low stage has shown a direct correlation with long-term survival after surgical removal and intensive cyclical postoperative treatment with irradiation and chemotherapy, using protocols similar to those used for Ewing sarcoma.426,451 Stage III and IV TPNETs and DSRCTs are probably best managed nonsurgically, because there are no data to support a role for debulking surgery in such circumstances.425
A sobering aspect of the therapy for TPNET and DSRCT is that it undeniably subjects patients who become survivors to the risk of a second malignancy. Intensive radiation to the chest wall may be followed years later by a postradiation sarcoma (or mesothelioma) in approximately 1% of cases, and Farhi and coworkers have described several examples of postchemotherapy myelodysplastic syndrome and acute leukemia in this context.452
Pleuropulmonary Blastoma
Until 1988, a group of anaplastic mesenchymal tumors of the peripheral lung and pleura in children had been grouped together under the rubric “pediatric pulmonary blastoma.” Nonetheless, Manivel and colleagues74 showed that such lesions differed from typical PBs in adults, which comprise a subset of SCs. The childhood tumors were found to be more often primary in the pleura; they also showed a histologic resemblance to soft tissue sarcomas. Because of these important points of difference from adult PBs, the pediatric lesions were reclassified as PPBs.
Clinical Summary
PPBs arise most often in the first decade of life, without a distinct preference for males or females. However, isolated examples have been reported in adult patients as well.74–80,453–463 Cough, chest pain, weight loss, dyspnea or tachypnea, and spontaneous pneumothorax are the most common presenting complaints.74,459 Evidence of a pleural effusion may also be found on physical examination, and a small subset of patients present with acute, rapidly progressive respiratory embarrassment.464 It is clear that this neoplasm may be part of certain “cancer families,” in which other soft tissue sarcomas, variants of Wilms tumor, and cystic nephromas may be seen in other members of the kindred.78,79,459,460 Other familial and patient-specific associations have been noted between PPB and sex-cord stromal tumors of the gonads, seminomatous germ cell tumors, intestinal polyps, and thyroid hyperplasia.465 The operative gene defect that ultimately yields PPB has now been identified. It is represented by a constitutive mutation in the DICER1 gene on chromosome 12, which encodes an endoribonuclease that is critical to the generation of small regulatory ribonucleic acid molecules.466
Radiologic studies typically demonstrate the presence of a large, irregularly outlined mass in the thorax, which may have its epicenter in the pleura, the mediastinal soft tissue, or the peripheral lung parenchyma. These lesions can be massive—sometimes effacing an entire hemithorax—and they demonstrate internal variation in density on CT or MRI (Fig. 14-95). Some examples may show focal internal calcification, and types I and II PPB (see subsequent discussion) demonstrate obvious internal cyst formation (Fig. 14-96).74,459,461,462
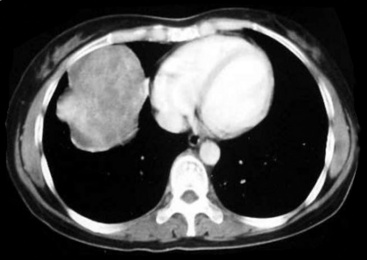
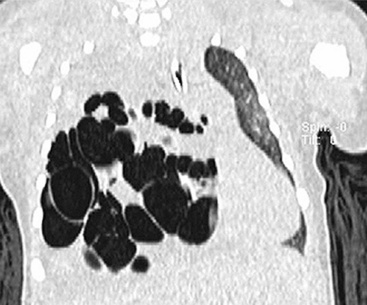
Pathologic Findings
Dehner and associates77,459 have subclassified PPBs into three groups, based on the extent of cystic change that they demonstrate. Type I tumors are predominantly cystic, type II lesions are mixed solid and cystic, and type III PPBs are predominantly solid (Figs. 14-97 to 14-99). Cystic foci are lined by modified respiratory epithelium that is typically bland cytologically, and the surrounding stroma is variably myxoid and relatively hypocellular. It is now believed that PPB is associated with “type 4” cystic congenital adenomatoid malformations (CCAMs) of the lung, whereas bronchioloalveolar carcinoma of the lung in children is linked to CCAM type 1467,468 (see Chapter 4).
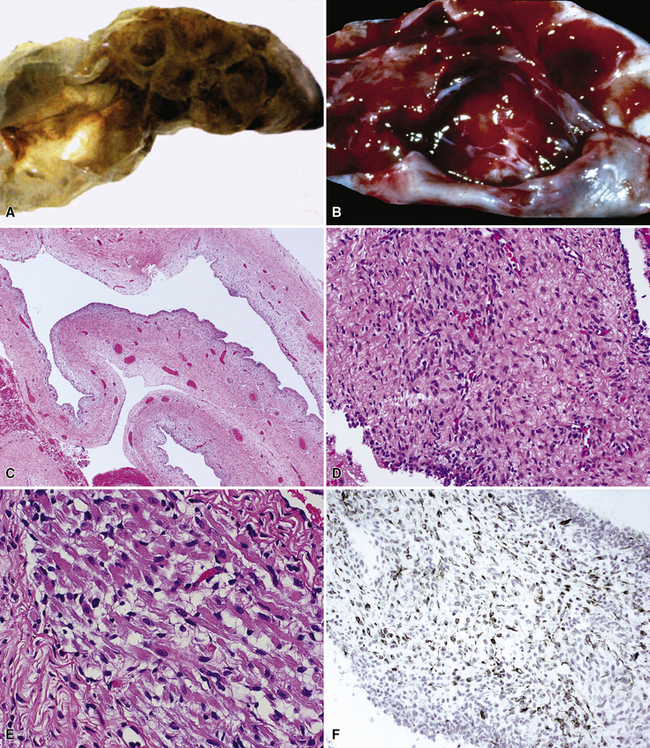
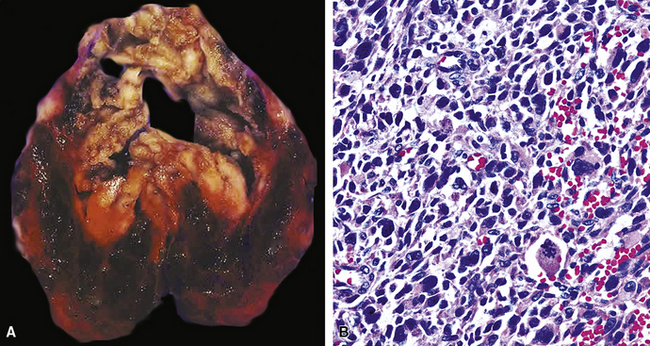
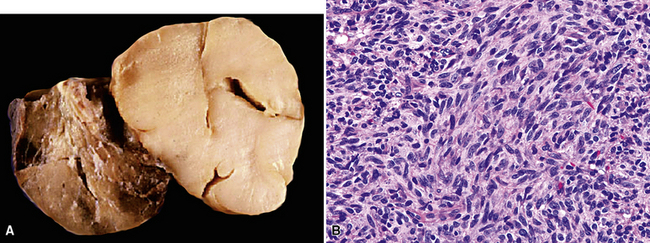
Microscopically, one sees a heterogeneous mixture of growth patterns that are admixed with one another in various solid regions of these tumors. Some foci resemble MFH (Fig. 14-100); others take on a rhabdomyosarcomatous appearance; and still other areas have the features of fibrosarcoma, liposarcoma, chondrosarcoma, or osteosarcoma.74,459 In the past, some observers applied the term “malignant mesenchymoma” to PPB, but such a designation has generally fallen from favor in current nosology. Importantly, epithelial foci are absent in PPB, in contrast to their dominance in so-called “adult pulmonary blastoma.”74,77,459 Immunohistochemical and ultrastructural studies demonstrate findings that are in accord with the aforementioned microscopic features, and they again fail to reveal epithelial characteristics in these lesions.74,469
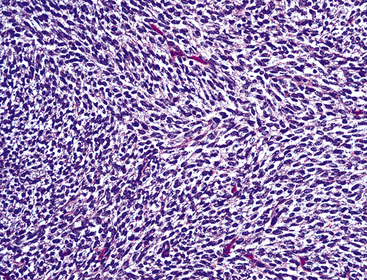
Fine needle aspiration biopsies typically show dyshesive, pleomorphic cells from PPBs (Fig. 14-101). Immunohistologic studies are required to assess the presence of lineage-related markers in such elements.
Hill and coworkers have suggested that cystic type I PPB evolves into types II or III over time in a sizable proportion of cases. Type I tumors show the presence of primitive mesenchymal tissues mantling intralesional cysts, beneath a cytologically bland lining of respiratory epithelium. Rhabdomyosarcoma-like and chondrosarcoma-like elements are present in 49% and 40% of cases, respectively (Figs. 14-102 and 14-103). The latter elements become even more common in types II and III PPB.470
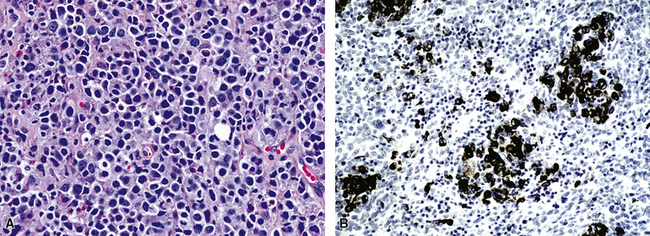
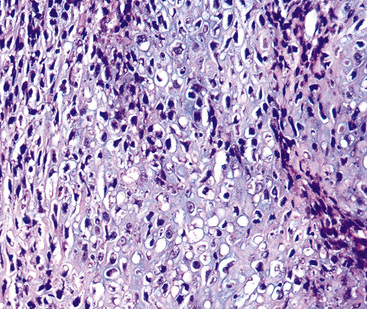
Therapy and Prognosis
PPB is a rare tumor; therefore, organized protocol studies of therapy are still in evolution. In general, however, it is obvious that this neoplasm is a highly aggressive lesion that requires every effort at surgical extirpation, followed by intensive radiation and chemotherapy.459,465 Because of the histologic characteristics of the tumor, which are like those of de novo soft tissue sarcomas, it would seem appropriate to use drug combinations that are directed toward the various histologic components of PPB (e.g., rhabdomyosarcoma, MFH, osteosarcoma). Surgical debulking of the tumor mass should also be considered in individual cases where complete resection is not thought to be feasible. Dehner and colleagues have related morphologic findings in PPBs to prognosis; predominantly cystic lesions have the best outlook, whereas type II and type III neoplasms are aggressive and often prove fatal within 2 years of diagnosis.459 Interestingly, Wright has also reported a case wherein successive recurrences of a PPB showed progressive transformation from type I to type III morphology.463 Priest and associates have reported a singular tendency for PPB to demonstrate metastases to the brain (Fig. 14-104). That complication is observed in 11% of type II cases and 54% of type III cases.471
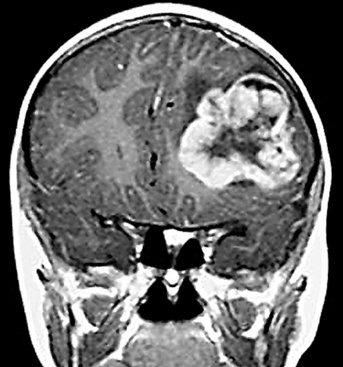
Vascular Sarcomas of xthe Pleura
As mentioned previously, angiosarcoma, KS, and EH may take origin in the pleura as well as in the pulmonary parenchyma. The general clinicopathologic attributes of these lesions have been described above. It is notable that the most common initial sign of angiosarcoma and KS of the pleura is the presence of a bloody pleural effusion (Fig. 14-105).284 Gross examination of the tumor at thoracotomy or thoracoscopy shows multiple soft, hemorrhagic, red-violet, nodular pleural implants in examples of angiosarcoma and KS. On the other hand, EH is virtually identical to MM at a macroscopic level (Fig. 14-106), and histologic study is necessary to distinguish between them.472,473 As is true of their intrapulmonary counterparts, KS and angiosarcoma of the pleura are associated with a dismal prognosis,474,475 whereas patients with EH may survive for prolonged periods.472
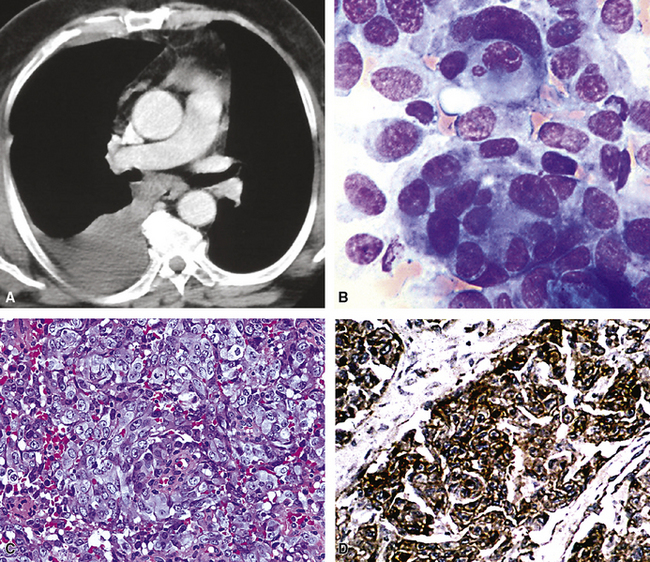
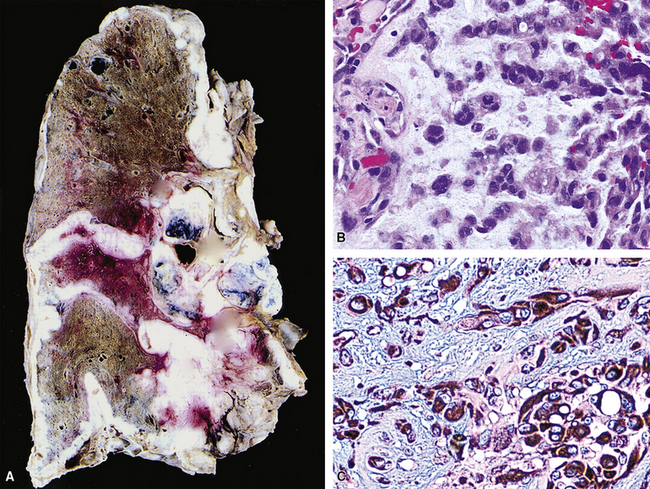
1 Huang J.C., Ritter J.H., Wick M.R. Malignant nonepithelial neoplasms of the lungs and pleural surfaces. In: Aisner J., et al, editors. Comprehensive Textbook of Thoracic Oncology. Baltimore: Williams & Wilkins; 1996:815-849.
2 Franks T.J., Galvin J.R. Sarcomatoid carcinoma of the lung: histologic criteria and common lesions in the differential diagnosis. Arch Pathol Lab Med. 2010;134:49-54.
3 Pelosi G., Sonzogni A., De Pas T., et al. Pulmonary sarcomatoid carcinomas: a practical overview. Int J Surg Pathol. 2010;18:103-120.
4 Wick M.R., Ritter J.H., Humphrey P.A. Sarcomatoid carcinomas of the lung: a clinicopathologic review. Am J Clin Pathol. 1997;108:40-53.
5 Litzky L.A. Pulmonary sarcomatous tumors. Arch Pathol Lab Med. 2008;132:1104-1117.
6 Steele R.H. Lung tumors: a personal review. Diagn Histopathol. 1983;6:119-123.
7 Nascimento A.G., Unni K.K. Sarcomas of the lung. Mayo Clin Proc. 1982;57:355-359.
8 Guccion J.G., Rosen S.H. Bronchopulmonary leiomyosarcomas and fibrosarcomas: a study of 32 cases and review of the literature. Cancer. 1972;30:835-847.
9 Przygodzki R.M., Moran C.A., Suster S., Koss M.N. Primary pulmonary rhabdomyosarcomas: a clinicopathologic and immunohistochemical study of three cases. Mod Pathol. 1995;8:658-661.
10 Suster S. Primary sarcomas of the lung. Semin Diagn Pathol. 1995;12:140-157.
11 Zeren H., Moran C.A., Suster S., et al. Primary pulmonary sarcomas with features of monophasic synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of 25 cases. Hum Pathol. 1995;26:474-480.
12 Gaertner E., Zeren E.H., Fleming M.V., et al. Biphasic synovial sarcomas arising in the pleural cavity: a clinicopathologic study of five cases. Am J Surg Pathol. 1996;20:36-45.
13 Gladish G.W., Sabloff B.M., Munden R.F., et al. Primary thoracic sarcomas. Radiographics. 2002;22:621-637.
14 Stackhouse E.M., Harrison E.G.Jr, Ellis F.H.Jr. Primary mixed malignancies of lung: carcinosarcoma and blastoma. J Thorac Cardiovasc Surg. 1969;57:385-399.
15 Humphrey P.A., Scroggs M.W., Roggli V.L., et al. Pulmonary carcinoma with a sarcomatoid element. Hum Pathol. 1988;19:155-165.
16 Ishida T., Tatsishi M., Kaneko S., et al. Carcinosarcoma and spindle-cell carcinoma of the lung. J Thorac Cardiovasc Surg. 1990;100:844-852.
17 Matsui K., Kitagawa M. Spindle cell carcinoma of the lung: a clinicopathologic study of three cases. Cancer. 1991;67:2361-2367.
18 Leventon G.S., Evans H.L. Sarcomatoid squamous cell carcinoma of the mucous membranes of the head and neck. Cancer. 1981;48:994-1003.
19 Piscioli F., Aldovini D., Bondi A., et al. Squamous cell carcinoma with sarcoma-like stroma of the nose and paranasal sinuses: report of two cases. Histopathology. 1984;8:633-639.
20 Lane N. Pseudosarcoma (polypoid sarcoma-like masses) associated with squamous cell carcinoma of the mouth, fauces, and larynx: report of ten cases. Cancer. 1957;10:19-41.
21 Lasser K.H., Naeim F., Higgins J., et al. “Pseudosarcoma” of the larynx. Am J Surg Pathol. 1979;3:397-404.
22 Lambert P.R., Ward P.H., Berci G. Pseudosarcoma of the larynx: a comprehensive analysis. Arch Otolaryngol. 1980;106:700-708.
23 Davis M.P., Eagan R.T., Weiland L.H., et al. Carcinosarcoma of the lung: Mayo Clinic experience and response to chemotherapy. Mayo Clin Proc. 1984;59:598-603.
24 Chang Y.L., Lee Y.C., Shih J.Y., et al. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small-cell carcinoma. Lung Cancer. 2001;34:91-97.
25 Fung C.H., Lo J.W., Yonan T.N., et al. Pulmonary blastoma: an ultrastructural study with brief review of literature and discussion of pathogenesis. Cancer. 1977;39:153-163.
26 Heffner D.K., Hyams V.J. Teratocarcinosarcoma (malignant teratoma?) of the nasal cavity and paranasal sinuses: a clinicopathologic study of 20 cases. Cancer. 1984;53:2140-2154.
27 Patterson S.D., Ballard R.W. Nasal blastoma: a light and electron microscopic study. Ultrastruct Pathol. 1980;1:487-494.
28 Minckler D.S., Meligro C.H., Norris H.T. Carcinosarcoma of the larynx. Cancer. 1970;26:195-200.
29 Goellner J.R., Devine K.D., Weiland L.H. Pseudosarcoma of the larynx. Am J Clin Pathol. 1973;59:312-326.
30 Farrell D.J., Cooper P.N., Malcolm A.J. Carcinosarcoma of the lung associated with asbestosis. Histopathology. 1995;27:484-486.
31 Berho M., Moran C.A., Suster S. Malignant mixed epithelial/mesenchymal neoplasms of the lung. Semin Diagn Pathol. 1995;12:123-139.
32 Reynolds S., Jenkins G., Akosa A., et al. Carcinosarcoma of the lung: an unusual cause of empyema. Respir Med. 1995;89:73-75.
33 Nappi O., Glasner S.D., Swanson P.E., et al. Biphasic and monophasic sarcomatoid carcinomas of the lung: a reappraisal of “carcinosarcomas” and “spindle cell carcinomas”. Am J Clin Pathol. 1994;102:331-340.
34 Nappi O., Wick M.R. Sarcomatoid neoplasms of the respiratory tract. Semin Diagn Pathol. 1993;10:137-147.
35 Nakajima M., Kasai T., Hashimoto H., et al. Sarcomatoid carcinoma of the lung: a clinicopathologic study of 37 cases. Cancer. 1999;86:608-616.
36 Brambilla E., Travis W.D., Colby T.V., et al. The new World Health Organization classification of lung tumors. Eur Respir J. 2001;18:1059-1068.
37 Terzi A., Gorla A., Piubello Q., et al. Biphasic sarcomatoid carcinoma of the lung: report of 5 cases and review of the literature. Eur J Surg Oncol. 1997;23:457.
38 Ro J.Y., Chen J.L., Lee J.S., et al. Sarcomatoid carcinoma of the lung: immunohistochemical and ultrastructural studies of 14 cases. Cancer. 1992;69:376-386.
39 Koss M.N. Pulmonary blastomas. Cancer Treat Res. 1995;72:349-362.
40 Miller R.R., Champagne K., Murray R.C. Primary pulmonary germ cell tumor with blastomatous differentiation. Chest. 1994;106:1595-1596.
41 Huwer H., Kalweit G., Straub U., et al. Pulmonary carcinosarcoma: diagnostic problems and determinants of prognosis. Eur J Cardiothorac Surg. 1996;10:403-407.
42 Melissari M., Giordano G., Gabrielli M. Immunohistochemical and ultrastructural study of a case of carcinosarcoma (biphasic sarcomatoid carcinoma) of the lung with rhabdomyoblastic differentiation. Pathologica. 1997;89:412-419.
43 Pankowski J., Grodzki T., Janowski H., et al. Carcinosarcoma of the lung: report of three cases. J Cardiovasc Surg. 1998;39:121-125.
44 Saphir O., Vass A. Carcinosarcoma. Am J Cancer. 1938;33:331-359.
45 Sarma D.P., Deshotels S.J.Jr. Carcinosarcoma of the lung. J Surg Oncol. 1982;19:216-218.
46 Bergmann M., Ackerman L.V., Kemler R.L. Carcinosarcoma of the lung: review of the literature and report of two cases treated by pneumonectomy. Cancer. 1951;4:919-929.
47 Kakos G.S., Williams T.E.Jr, Assor D., et al. Pulmonary carcinosarcoma: etiologic, therapeutic, and prognostic considerations. J Thorac Cardiovasc Surg. 1971;61:777-783.
48 Krompecher E. Der drusernartige Oberflachen-Epitheliakrebscarcinom epitheliale Adenoides. Beitr Pathol. 1900;28:1-41.
49 Herxheimer G., Reinke F. Carcinoma sarcomatodes: pathologie des Krebses. Ergeb Allg Pathol Pathol Anat. 1912;16:280-282.
50 Moore T.C. Carcinosarcoma of the lung. Surgery. 1961;50:886-893.
51 Cabarcos A., Gomez-Dorronsoro M., Lobo-Beristain J.L. Pulmonary carcinosarcoma: a case study and review of the literature. Br J Dis Chest. 1985;79:83-90.
52 Ludwigsen E. Endobronchial carcinosarcoma. Virchows Arch A Pathol Anat. 1977;373:293-302.
53 Rainosek D.E., Ro J.Y., Ordonez N.G., et al. Sarcomatoid carcinoma of the lung: a case with atypical carcinoid and rhabdomyosarcomatous components. Am J Clin Pathol. 1994;102:360-364.
54 Fishback N.F., Travis W.D., Moran C.A., et al. Pleomorphic (spindle/giant cell) carcinoma of the lung: a clinicopathologic correlation of 78 cases. Cancer. 1994;73:2936-2945.
55 Tsubota Y.T., Kawaguchi T., Hoso T., et al. A combined small cell and spindle cell carcinoma of the lung: report of a unique case with immunohistochemical and ultrastructural studies. Am J Surg Pathol. 1992;16:1108-1115.
56 Khalifa M., Hruby G., Ehrlich L., et al. Combined large cell neuroendocrine carcinoma and spindle cell carcinoma of the lung. Ann Diagn Pathol. 2001;5:240-245.
57 Oyasu R., Battifora H.A., Buckingham W.B., et al. Metaplastic squamous cell carcinoma of bronchus simulating giant cell tumor of bone. Cancer. 1977;39:1119-1128.
58 Love G.L., Droca P.J. Bronchogenic sarcomatoid squamous cell carcinoma with osteoclast-like giant cells. Hum Pathol. 1983;14:1004-1006.
59 Kitazawa R., Kitazawa S., Nishimura Y., et al. Lung carcinosarcoma with liposarcoma element: autopsy case. Pathol Int. 2006;56:449-452.
60 Colombi R.P. Sarcomatoid carcinomas of the female genital tract (malignant mixed müllerian tumors). Semin Diagn Pathol. 1993;10:169-175.
61 George E., Manivel J.C., Dehner L.P., et al. Malignant mixed müllerian tumors: an immunohistochemical study of 47 cases with histogenetic considerations and clinical correlation. Hum Pathol. 1991;22:215-223.
62 Travis W.D., Colby T.V., Corrin B., et al. Histological Typing of Lung and Pleural Tumours (International Histological Classification of Tumours). Geneva, Switzerland: World Health Organization, 1999;1-55.
63 Wagner M.S., Reyes C.V. Primary chondroblastic osteosarcoma of the lung. Sarcoma. 1999;3:193-195.
64 Niimi R., Matsumine A., Kusuzaki K., et al. Primary osteosarcoma of the lung: a case report and review of the literature. Med Oncol. 2008;25:251-255.
65 Barnett N.R., Barnard W.G. Some unusual thoracic tumors. Br J Surg. 1945;32:447-457.
66 Spencer H. Pulmonary blastoma. J Pathol Bacteriol. 1961;82:161-165.
67 Francis D., Jacobsen M. Pulmonary blastoma. Curr Top Pathol. 1983;73:265-294.
68 Ohtomo K., Araki T., Yashiro N., et al. Pulmonary blastoma in children. Radiology. 1983;147:101-104.
69 Gal A.A., Marchevsky A.M., Koss M.N. Unusual tumors of the lung. In: Marchevsky A.M., editor. Surgical Pathology of Lung Neoplasms. New York: Marcel Dekker; 1990:325-388.
70 Jetley N.K., Bhatnagar V., Krishna A., et al. Pulmonary blastoma in a neonate. J Pediatr Surg. 1988;23:1009-1010.
71 Jimenez J.F. Pulmonary blastoma in childhood. J Surg Oncol. 1987;334:87-93.
72 Senac M.O.Jr, Wood B.P., Isaacs H., et al. Pulmonary blastoma: a rare childhood malignancy. Radiology. 1991;179:743-746.
73 Cohen M., Emms M., Kaschula R.OC. Childhood pulmonary blastoma: a pleuropulmonary variant of the adult pulmonary blastoma. Pediatr Pathol. 1991;11:737-739.
74 Manivel J.C., Priest J.R., Watterson J., et al. Pleuropulmonary blastoma: the so-called pulmonary blastoma of childhood. Cancer. 1988;62:1516-1526.
75 Hachitanda Y., Aoyama C., Sato J.K., et al. Pleuropulmonary blastoma in childhood: a tumor of divergent differentiation. Am J Surg Pathol. 1993;17:382-391.
76 Seballos R.M., Klein R.L. Pulmonary blastoma in children: report of two cases and review of the literature. J Pediatr Surg. 1994;29:1553-1556.
77 Dehner L.P. Pleuropulmonary blastoma is the pulmonary blastoma of childhood. Semin Diagn Pathol. 1994;11:144-151.
78 Delahunt B., Thomson K.J., Ferguson A.F., et al. Familial cystic nephroma and pleuropulmonary blastoma. Cancer. 1993;71:1338-1342.
79 Sciot R., Dal-Cin P., Brock P., et al. Pleuropulmonary blastoma (pulmonary blastoma of childhood): genetic link with other embryonal malignancies? Histopathology. 1994;24:559-563.
80 Schmaltz C., Sauter S., Opitz O., et al. Pleuropulmonary blastoma: a case report and review of the literature. Med Pediatr Oncol. 1995;25:479-484.
81 Re G.G., Hazen-Martin D.J., Sens D.A., et al. Nephroblastoma (Wilms’ tumor): a model system of aberrant renal development. Semin Diagn Pathol. 1994;11:125-135.
82 Souza R.C., Peasley E.D., Takaro T. Pulmonary blastomas: a distinctive group of carcinosarcomas of the lung. Ann Thorac Surg. 1965;1:259-268.
83 Millard M. Lung, pleura, and mediastinum, 6th ed. Anderson W.AD., editor. Pathology, vol. 2,. CV Mosby, St. Louis, 1971;875-997.
84 Koss M.N., Hochholzer L., O’Leary T. Pulmonary blastomas. Cancer. 1991;67:2368-2381.
85 Takahasi H., Tanaka K., Uchida Y., et al. A case of pulmonary blastoma composed of histological features of both pulmonary blastoma and pulmonary endodermal tumor resembling fetal lung. J Jpn Assn Thorac Surg. 1995;43:1217-1222.
86 Babycos P.B., Daroca P.J.Jr. Polypoid pulmonary endodermal tumor resembling fetal lung: report of a case. Mod Pathol. 1995;8:303-306.
87 Kodama T., Shimosato Y., Watanabe S., et al. Six cases of well-differentiated adenocarcinoma simulating fetal lung tissues in pseudoglandular stage: comparison with pulmonary blastoma. Am J Surg Pathol. 1984;8:735-744.
88 Kradin R.L., Young R.H., Dickersin R.G., et al. Pulmonary blastoma with argyrophil cells lacking sarcomatous features (pulmonary endodermal tumor resembling fetal lung). Am J Surg Pathol. 1982;6:165-172.
89 Manning J.T., Ordonez N.G., Rosenberg H.S., et al. Pulmonary endodermal tumor resembling fetal lung. Arch Pathol Lab Med. 1985;109:48-50.
90 Muller-Hermelink H.K., Kaiserling E. Pulmonary adenocarcinoma of fetal type: alternating differentiation argues in favor of a common endodermal stem cell. Virchows Arch A. 1986;409:195-210.
91 Olenick S.J., Fan C.C., Ryoo J.W. Mixed pulmonary blastoma and carcinosarcoma. Histopathology. 1994;25:171-174.
92 Roth J.A., Elquezabel A. Pulmonary blastoma evolving into carcinosarcoma: a case study. Am J Surg Pathol. 1978;2:407-413.
93 Jacobsen M., Francis D. Pulmonary blastoma: a clinicopathologic study of eleven cases. Acta Pathol Microbiol Immunol Scand [A]. 1980;88:151-160.
94 Nappi O., Swanson P.E., Wick M.R. Pseudovascular adenoid squamous cell carcinoma of the lung: clinicopathologic of three cases and comparison with true pleuropulmonary angiosarcoma. Hum Pathol. 1994;25:373-378.
95 Ritter J.H., Mills S.E., Nappi O., et al. Angiosarcoma-like neoplasms of epithelial organs: true endothelial tumors or variants of carcinoma? Semin Diagn Pathol. 1995;12:270-282.
96 Banerjee S.S., Eyden B.P., Wells S., et al. Pseudoangiosarcomatous carcinoma: a clinicopathological study of seven cases. Histopathology. 1992;21:13-23.
97 Nappi O., Wick M.R., Pettinato G., et al. Pseudovascular adenoid squamous cell carcinoma of the skin: a neoplasm that may be mistaken for angiosarcoma. Am J Surg Pathol. 1992;16:429-438.
98 Eusebi V., Lamovec J., Cattani M.G., et al. Acantholytic variant of squamous cell carcinoma of the breast. Am J Surg Pathol. 1986;10:855-861.
99 Mills S.E., Gaffey M.J., Watts J.C., et al. Angiomatoid carcinoma and “angiosarcoma” of the thyroid gland: a spectrum of endothelial differentiation. Am J Clin Pathol. 1994;102:322-330.
100 Umiker W., Iverson L. Postinflammatory “tumors” of the lung. J Thorac Surg. 1954;28:55-63.
101 Titus J., Harrison E.G.Jr, Clagett O., et al. Xanthomatous and inflammatory pseudotumors of the lung. Cancer. 1962;15:522-538.
102 Mandelbaum I., Brashear R.E., Hull M.T. Surgical treatment and course of pulmonary pseudotumor (plasma cell granuloma). J Thorac Cardiovasc Surg. 1981;82:77-82.
103 Berardi R.S., Lee S.S., Chen H.P., et al. Inflammatory pseudotumors of the lung. Surg Gynecol Obstet. 1983;156:89-96.
104 Chen H.P., Lee S.S., Berardi R.S. Inflammatory pseudotumor of the lung: ultrastructural and light microscopic study of a myxomatous variant. Cancer. 1984;54:861-865.
105 Maples M.D., Adkins R.B.Jr, Graham B.S., et al. Pseudotumor of the lung. Am Surg. 1985;51:84-88.
106 Matsubara O., Tan-Liu N.S., Kenney R.M., et al. Inflammatory pseudotumors of the lung: progression from organizing pneumonia to fibrous histiocytoma or to plasma cell granuloma in 32 cases. Hum Pathol. 1988;19:807-814.
107 Machicao C.N., Sorensen K., Abdul K.F., et al. Transthoracic needle aspiration biopsy in inflammatory pseudotumors of the lung. Diagn Cytopathol. 1989;5:400-403.
108 Ishida T., Oka T., Nishino T., et al. Inflammatory pseudotumor of the lung in adults: radiographic and clinicopathological analysis. Ann Thorac Surg. 1989;48:90-95.
109 Barbareschi M., Ferrero S., Aldovini D., et al. Inflammatory pseudotumor of the lung: immunohistochemical analysis on four new cases. Histol Histopathol. 1990;5:205-211.
110 Kobzik L. Benign pulmonary lesions that may be misdiagnosed as malignant. Semin Diagn Pathol. 1990;7:129-138.
111 Vujanic G.M., Dojcinov D. Inflammatory pseudotumor of the lung in children. Pediatr Hematol Oncol. 1991;8:121-129.
112 Daudi F.A., Lees G.M., Higa T.E. Inflammatory pseudotumors of the lung: two cases and a review. Can J Surg. 1991;34:461-464.
113 Nonomura A., Mizukami Y., Matsubara F., et al. Seven patients with plasma cell granuloma (inflammatory pseudotumor) of the lung, including two with intrabronchial growth: an immunohistochemical and electron microscopic study. Intern Med. 1992;31:756-765.
114 Koss M.N. Tumor-like conditions of lung. Adv Pathol Lab Med. 1994;7:123-150.
115 Pettinato G., Manivel J.C., DeRosa N., et al. Inflammatory myofibroblastic tumor (plasma cell granuloma): clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol. 1990;94:538-546.
116 Lipton J.H., Fong T.C., Gill M.J., et al. Q fever inflammatory pseudotumor of the lung. Chest. 1987;92:756-757.
117 Bishopric G.A., D’Agay M.F., Schlemmer B., et al. Pulmonary pseudotumor due to Corynebacterium equi in a patient with the acquired immunodeficiency syndrome. Thorax. 1988;43:486-487.
118 Wick M.R., Ritter J.H., Nappi O. Inflammatory sarcomatoid carcinoma of the lung: report of three cases and clinicopathologic comparison with inflammatory pseudotumors in adult patients. Hum Pathol. 1995;26:1014-1021.
119 Antic T., Kapur U., Vigneswaran W.T., et al. Inflammatory sarcomatoid carcinoma: a case report and discussion of a malignant tumor with benign appearance. Arch Pathol Lab Med. 2005;129:1334-1337.
120 Hartmann C.A., Schutze H. Mesothelioma-like tumors of the pleura: a review of 72 autopsy cases. Cancer Res Clin Oncol. 1994;120:331-347.
121 Shah I.A., Salvatore J.R., Kummet T., et al. Pseudomesotheliomatous carcinoma involving pleura and peritoneum: a clinicopathologic and immunohistochemical study of three cases. Ann Diagn Pathol. 1999;3:148-159.
122 Inomata M., Kawagishi Y., Yamada T., et al. Two cases of pulmonary sarcomatoid carcinoma mimicking malignant mesothelioma. J Jpn Resp Soc. 2010;48:33-38.
123 Huszar M., Herczeg E., Lieberman Y., et al. Distinctive immunofluorescent labeling of epithelial and mesenchymal elements of carcinosarcoma with antibodies specific for different intermediate filaments. Hum Pathol. 1984;15:532-538.
124 Zimmerman K.G., Sobonya R.E., Payne C.M. Histochemical and ultrastructural features of an unusual pulmonary carcinosarcoma. Hum Pathol. 1981;12:1046-1051.
125 Koss M.N., Hochholzer L., Frommelt R.A. Carcinosarcomas of the lung: a clinicopathologic study of 66 patients. Am J Surg Pathol. 1999;23:1514-1526.
126 Yousem S.A., Wick M.R., Randhawa P., et al. Pulmonary blastoma: an immunohistochemical comparison to fetal lung in its pseudoglandular stage. Am J Clin Pathol. 1990;93:167-175.
127 Matsui K., Kitagawa M., Miwa A. Lung carcinoma with spindle cell components: sixteen cases examined by immunohistochemistry. Hum Pathol. 1992;23:1289-1297.
128 Addis B.J., Corrin B. Pulmonary blastoma, carcinosarcoma, and spindle-cell carcinoma: an immunohistochemical study of keratin intermediate filaments. J Pathol. 1985;147:291-301.
129 Suster S., Huszar M., Herczeg E. Spindle-cell carcinoma of the lung: immunocytochemical and ultrastructural study of a case. Histopathology. 1987;11:871-878.
130 Berean K., Truong L.D., Dudley A.W.Jr, et al. Immunohistochemical characterization of pulmonary blastoma. Am J Clin Pathol. 1988;89:773-777.
131 Pelosi G., Scarpa A., Manzotti M., et al. K-ras gene mutational analysis supports a monoclonal origin of biphasic pleomorphic carcinoma of the lung. Mod Pathol. 2004;17:538-546.
132 Hountis P., Moraitis S., Dedeilias P., et al. Sarcomatoid lung carcinomas: a case series. Cases J. 2009;2:7900-7910.
133 Wick M.R., Swanson P.E. Carcinosarcomas—current perspectives and a historical review of nosological concepts. Semin Diagn Pathol. 1993;10:118-127.
134 Brooks J.J. The significance of double phenotypic patterns and markers in human sarcomas. Am J Pathol. 1986;125:113-123.
135 Thompson L., Chang B., Barsky S.H. Monoclonal origins of malignant mixed tumors (carcinosarcomas). Am J Surg Pathol. 1996;20:277-287.
136 Rossi S., Orvieto E., Furlanetto A., et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
137 Battifora H. Spindle-cell carcinoma: ultrastructural evidence of squamous origin and collagen production by the tumor cells. Cancer. 1976;37:2275-2282.
138 Wakely P.E.Jr. Pulmonary spindle cell lesions: correlation of aspiration cytopathology and histopathology. Ann Diagn Pathol. 2001;5:216-228.
139 Roberts C.A., Seemayer T.A., Neff J.R., et al. Translocation (X;18) in primary synovial sarcoma of the lung. Cancer Genet Cytogenet. 1996;88:49-52.
140 Kaplan M.A., Goodman M.D., Satish J., et al. Primary pulmonary sarcoma with morphologic features of monophasic synovial sarcoma and chromosome translocation t(X;18). Am J Clin Pathol. 1996;105:195-199.
141 Terry J., Saito T., Subramanian S., et al. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31:240-246.
142 Jagdis A., Rubin B.P., Tubbs R.R., et al. Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma. Am J Surg Pathol. 2009;33:1743-1751.
143 Kosemehmetoglu K., Vrana J.A., Folpe A.L. TLE1 expression is not specific for synovial sarcoma: a whole section study of 163 soft tissue and bone neoplasms. Mod Pathol. 2009;22:872-878.
144 Knösel T., Heretsch S., Altendorf-Hofmann A., et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t(X;18): analysis of 319 cases. Eur J Cancer. 2010;46:1170-1176.
145 Kung I.T., Thallas V., Spencer E.J., et al. Expression of muscle actins in diffuse mesotheliomas. Hum Pathol. 1995;26:565-570.
146 Padgett D.M., Cathro H.P., Wick M.R., et al. Podoplanin is a better immunohistochemical marker for sarcomatoid mesothelioma than calretinin. Am J Surg Pathol. 2008;32:123-127.
147 Wick M.R. Kaposi’s sarcoma unrelated to the acquired immunodeficiency syndrome: a review. Curr Opin Oncol. 1991;3:377-383.
148 Ognibene F.P., Shelhamer J.H. Kaposi’s sarcoma. Clin Chest Med. 1988;9:459-465.
149 Garay S.M., Belenko M., Fazzini E., et al. Pulmonary manifestations of Kaposi’s sarcoma. Chest. 1987;91:39-43.
150 White D.A., Matthay R.A. Noninfectious pulmonary complications of infection with the human immunodeficiency virus. Am Rev Respir Dis. 1989;140:1763-1787.
151 Meduri G.U., Stover D.E., Lee M., et al. Pulmonary Kaposi’s sarcoma in the acquired immune deficiency syndrome: clinical, radiographic, and pathologic manifestations. Am J Med. 1986;81:11-18.
152 Purdy L.J., Colby T.V., Yousem S.A., et al. Pulmonary Kaposi’s sarcoma: premortem histologic diagnosis. Am J Surg Pathol. 1986;10:301-311.
153 Cadranel J., Naccache J., Wislez M., et al. Pulmonary malignancies in the immunocompromised patient. Respiration. 1999;66:289-309.
154 Katariya K., Thurer R.J. Malignancies associated with the immunocompromised state. Chest Surg Clin N Am. 1999;9:63-77.
155 Smith C., Lilly S., Mann K.P., et al. AIDS-related malignancies. Ann Med. 1998;30:323-344.
156 Hannon F.B., Easterbrook P.J., Padley S., et al. Bronchopulmonary Kaposi’s sarcoma in 106 HIV-1-infected patients. Int J STD AIDS. 1998;9:518-525.
157 Bach M.C., Bagwell S.P., Fanning J.P. Primary pulmonary Kaposi’s sarcoma in the acquired immunodeficiency syndrome: a cause of persistent pyrexia. Am J Med. 1988;85:274-275.
158 Hanno R., Owen L.G., Callen J.P. Kaposi’s sarcoma with extensive silent internal involvement. Int J Dermatol. 1979;18:718-721.
159 Gal A.A., Koss M.N., Hartmann B., et al. A review of pulmonary pathology in the acquired immune deficiency syndrome. Surg Pathol. 1988;1:325-346.
160 O’Brien R.F., Cohn D.L. Serosanguineous pleural effusions in AIDS-associated Kaposi’s sarcoma. Chest. 1989;96:460-466.
161 Floris C., Sulis M.L., Bernascani M., et al. Pneumothorax in pleuropulmonary Kaposi’s sarcoma related to acquired immune deficiency syndrome. Am J Med. 1989;87:123-124.
162 Cheuk W., Wong K.O., Wong C.S., et al. Immunostaining for human herpesvirus 8 latent nuclear antigen-1 helps distinguish Kaposi sarcoma from its mimickers. Am J Clin Pathol. 2004;121:335-342.
163 Folpe A.L., Chand E.M., Goldblum J.R., et al. Expression of Fli-1, a nuclear transcription factor, distinguishes vascular neoplasms from potential mimics. Am J Surg Pathol. 2001;25:1061-1066.
164 Weninger W., Partanen T.A., Breiteneder-Geleff S., et al. Expression of vascular endothelial growth factor receptor-3 and podoplanin suggests a lymphatic endothelial cell origin of Kaposi’s sarcoma tumor cells. Lab Invest. 1999;79:243-251.
165 Ireland-Gill A., Espina B.M., Akil B., et al. Treatment of acquired immunodeficiency syndrome-related Kaposi’s sarcoma using bleomycin-containing combination chemotherapy regimens. Semin Oncol. 1992;19(2 suppl 5):32-37.
166 Gill P.S., Akil B., Colletti P., et al. Pulmonary Kaposi’s sarcoma: clinical findings and results of therapy. Am J Med. 1989;87:57-61.
167 Ognibene F.P., Steis R.G., Macher A.M., et al. Kaposi’s sarcoma causing pulmonary infiltrates and respiratory failure in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;102:471-475.
168 Guccion J.G., Rosen S.H. Bronchopulmonary leiomyosarcoma and fibrosarcoma: a study of 32 cases and review of the literature. Cancer. 1972;30:836-847.
169 Enzinger F.M., Weiss S.W. Fibrosarcoma. In Soft Tissue Tumors, 2nd ed., St. Louis: CV Mosby; 1988:201-222.
170 Nascimento A.G., Unni K.K., Bernatz P.E. Sarcomas of the lung. Mayo Clin Proc. 1982;57:355-359.
171 Miller D.L., Allen M.S. Rare pulmonary neoplasms. Mayo Clin Proc. 1993;68:492-498.
172 Pettinato G., Manivel J.C., Saldana M.J., et al. Primary bronchopulmonary fibrosarcoma of childhood and adolescence: reassessment of a low-grade malignancy. Hum Pathol. 1989;20:463-471.
173 Goldthorn J.F., Duncan M.H., Kosloske A.M., et al. Cavitating primary pulmonary fibrosarcoma in a child. J Thorac Cardiovasc Surg. 1986;91:932-934.
174 Wick M.R., Manivel J.C. Primary sarcomas of the lung. In: Williams C.J., Krikorian J.G., Green M.R., Raghavan D., editors. Textbook of Uncommon Cancer. New York: John Wiley & Sons; 1988:335-381.
175 Logrono R., Filipowicz E.A., Eyzaguirre E.J., et al. Diagnosis of primary fibrosarcoma of the lung by fine needle aspiration and core biopsy. Arch Pathol Lab Med. 1999;123:731-735.
176 Lane K.L., Shannon R.J., Weiss S.W. Hyalinizing spindle cell tumor with giant rosettes: a distinctive tumor closely resembling low-grade fibromyxoid sarcoma. Am J Surg Pathol. 1997;21:1481-1488.
177 Magro G., Fraggetta F., Manusia M., et al. Hyalinizing spindle cell tumor with giant rosettes: a previously undescribed lesion of the lung. Am J Surg Pathol. 1998;22:1431-1433.
178 Kim L., Yoon Y.H., Choi S.J., et al. Hyalinizing spindle cell tumor with giant rosettes arising in the lung: report of a case with FUS-CREB3L2 fusion transcripts. Pathol Int. 2007;57:153-157.
179 Vernon S.E., Bejarano P.A. Low-grade fibromyxoid sarcoma: a brief review. Arch Pathol Lab Med. 2006;130:1358-1360.
180 Beluffi G., Bertolotti P., Mietta A., et al. Primary leiomyosarcoma of the lung in a girl. Pediatr Radiol. 1986;16:240-244.
181 Jimenez J.F., Uthman E.O., Townsend J.W., et al. Primary bronchopulmonary leiomyosarcoma in childhood. Arch Pathol Lab Med. 1986;110:348-351.
182 Yellin A., Rosenman Y., Lieberman Y. Review of smooth muscle tumours of the lower respiratory tract. Br J Dis Chest. 1984;78:337-351.
183 Lillo-Gil R., Albrechtsson U., Jakobsson B. Pulmonary leiomyosarcoma appearing as a cyst: report of one case and review of the literature. Thorac Cardiovasc Surg. 1985;33:250-252.
184 Yu H., Ren H., Miao Q., et al. Pulmonary leiomyosarcoma: report of three cases. Chin Med J. 1996;11:191-194.
185 Attanoos R.L., Appleton M.A., Gibbs A.R. Primary sarcomas of the lung: a clinicopathological and immunohistochemical study of 14 cases. Histopathology. 1996;29:29-36.
186 Morgan P.GM., Ball J. Pulmonary leiomyosarcomas. Br J Dis Chest. 1980;74:245-252.
187 Wick M.R., Scheithauer B.W., Piehler J.M., et al. Primary pulmonary leiomyosarcomas: a light and electron microscopic study. Arch Pathol Lab Med. 1982;106:510-514.
188 Chaudhuri M.R. Primary leiomyosarcoma of the lung. Br J Dis Chest. 1973;67:75-80.
189 Koizumi N., Fukuda T., Ohnishi Y., et al. Pulmonary myxoid leiomyosarcoma. Pathol Int. 1995;45:879-884.
190 Keel S.B., Bacha E., Mark E.J., et al. Primary pulmonary sarcoma: a clinicopathologic study of 26 cases. Mod Pathol. 1999;12:1124-1131.
191 Yu H., Ren H., Miao Q., et al. Pulmonary leiomyosarcoma—report of three cases. Chin Med Sci J. 1996;11:191-194.
192 Hummel P., Cangiarella J.F., Cohen J.M., et al. Transthoracic fine-needle aspiration biopsy of pulmonary spindle cell and mesenchymal lesions: a study of 61 cases. Cancer. 2001;93:187-198.
193 Yamaguchi T., Imamura Y., Nakayama K., et al. Primary pulmonary leiomyosarcoma. Report of a case diagnosed by fine needle aspiration cytology. Acta Cytol. 2002;46:912-916.
194 Dail D.H., Liebow A.A. Intravascular bronchioloalveolar tumor. Am J Pathol. 1975;78:6a. [abstract]
195 Dail D.H., Liebow A.A., Gmelich J.T., et al. Intravascular, bronchiolar, and alveolar tumor of the lung (IVBAT). Cancer. 1983;51:452-464.
196 Corrin B., Manners B., Millard M., et al. Histogenesis of so-called “intravascular bronchioloalveolar tumour”. J Pathol. 1979;128:163-167.
197 Weiss S.W., Enzinger F.M. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer. 1982;50:970-981.
198 Schattenberg T., Kam R., Klopp M., et al. Pulmonary epithelioid hemangioendothelioma: report of three cases. Surg Today. 2008;38:844-849.
199 Weiss S.W., Ishak K.G., Dail D.H., et al. Epithelioid hemangioendothelioma and related lesions. Semin Diagn Pathol. 1986;3:259-287.
200 Kitaichi M., Nagai S., Nishimura K., et al. Pulmonary epithelioid hemangioendothelioma in 21 patients, including three with partial spontaneous regression. Eur Respir J. 1998;12:89-96.
201 Rock M.J., Kaufman R.A., Lobe T.E., et al. Epithelioid hemangioendothelioma of the lung (intravascular bronchioloalveolar tumor) in a young girl. Pediatr Pulmonol. 1991;11:181-186.
202 Carter E.J., Bradburne R.M., Jhung J.W., et al. Alveolar hemorrhage with epithelioid hemangioendothelioma: a previously unreported manifestation of a rare tumor. Am Rev Respir Dis. 1990;142:700-701.
203 Briens E., Caulet-Maugendre S., Desrues B., et al. Alveolar hemorrhage revealing epithelioid hemangioendothelioma. Respir Med. 1997;91:111-114.
204 Yi E.S., Auger W.R., Friedman P.J., et al. Intravascular bronchioloalveolar tumor of the lung presenting as pulmonary thromboembolic disease and pulmonary hypertension. Arch Pathol Lab Med. 1995;119:255-260.
205 Ross G.J., Violi L., Friedman A.C., et al. Intravascular bronchioloalveolar tumor: CT and pathologic correlation. J Comput Assist Tomogr. 1989;13:240-243.
206 Einsfelder B., Kuhnen C. Epithelioid hemangioendothelioma of the lung (IVBAT)—clinicopathological and immunohistochemical analysis of 11 cases. Pathologe. 2006;27(2):106-115.
207 Saqi A., Nisbet L., Gagneja P., et al. Primary pleural epithelioid hemangioendothelioma with rhabdoid phenotype: report and review of the literature. Diagn Cytopathol. 2007;35:203-208.
208 Bhagavan B.S., Murthy M.SN., Dorfman H.D., et al. Intravascular bronchiolo-alveolar tumor (IVBAT): a low-grade sclerosing epithelioid angiosarcoma of lung. Am J Surg Pathol. 1982;6:41-52.
209 Corrin B., Harrison W.J., Wright D.H. The so-called intravascular bronchioloalveolar tumour of lung (low grade sclerosing angiosarcoma). Diagn Histopathol. 1983;6:229-237.
210 Corrin B., Dewar A., Simpson C.G. Epithelioid hemangioendothelioima of the lung. Ultrastruct Pathol. 1996;20:345-347.
211 Buggage R.R., Soudi N., Olson J.L., et al. Epithelioid hemangioendothelioma of the lung: pleural effusion cytology, ultrastructure, and brief literature review. Diagn Cytopathol. 1995;13:54-60.
212 Bollinger B.K., Laskin W.B., Knight C.B. Epithelioid hemangioendothelioma with multiple site involvement: literature review and observations. Cancer. 1994;73:610-615.
213 Weinreb I., Cunningham K.S., Perez-Ordoñez B., et al. CD10 is expressed in most epithelioid hemangioendotheliomas: a potential diagnostic pitfall. Arch Pathol Lab Med. 2009;133:1965-1968.
214 Carretero A., Elmberger P.G., Sköld C.M., et al. Pulmonary epithelioid hemangioendothelioma: report of a case with fine needle aspiration biopsy. Acta Cytol. 2006;50:455-459.
215 Celikel C., Yumuk P.F., Basaran G., et al. Epithelioid hemangioendothelioma with multiple organ involvement. APMIS. 2007;115:881-888.
216 Ohori N.P., Yousem S.A., Sonmez-Alpan E., et al. Estrogen and progesterone receptors in lymphangioleiomyomatosis, epithelioid hemangioendothelioma, and sclerosing hemangioma of the lungs. Am J Clin Pathol. 1991;96:529-535.
217 Stout A.P., Murray M.R. Hemangiopericytoma: a vascular tumor featuring Zimmerman’s pericytes. Ann Surg. 1942;116:26-33.
218 Meade J.B., Whitwell F., Bickford B.J., et al. Primary haemangiopericytoma of lung. Thorax. 1974;29:1-15.
219 Murphey M.D. World Health Organization classification of bone and soft tissue tumors: modifications and implications for radiologists. Semin Musculoskelet Radiol. 2007;11:201-214.
220 Sakurai H., Tanaka W., Kaji M., et al. Intrapulmonary localized fibrous tumor of the lung: a very unusual presentation. Ann Thorac Surg. 2008;86:1360-1362.
221 Enzinger F.M., Weiss S.W. Hemangiopericytoma, 2nd ed. Soft Tissue Tumors. CV Mosby, St. Louis, 1988;596-613.
222 Shin M.S., Ho K.J. Primary hemangiopericytoma of lung: radiography and pathology. AJR. 1979;133:1077-1083.
223 Van Damme H., Dekoster G., Creemers E., et al. Primary pulmonary hemangiopericytoma: early local recurrence after perioperative rupture of the giant tumor mass (two cases). Surgery. 1990;108:105-109.
224 Yousem S.A., Hochholzer L. Primary pulmonary hemangiopericytoma. Cancer. 1987;59:549-555.
225 Yaghmai I. Angiographic manifestations of soft-tissue and osseous hemangiopericytomas. Radiology. 1978;126:653-659.
226 Halle M., Blum U., Dinkel E., et al. CT and MR features of primary pulmonary hemangiopericytomas. J Comput Assist Tomogr. 1993;17:51-55.
227 Rusch V.W., Shuman W.P., Schmidt R., et al. Massive pulmonary hemangiopericytoma: an innovative approach to evaluation and treatment. Cancer. 1989;64:1928-1936.
228 Liu C.C., Wang H.W., Li F.Y., et al. Solitary fibrous tumors of the pleura: clinicopathological characteristics, immunohistochemical profiles, and surgical outcomes with long-term follow-up. Thorac Cardiovasc Surg. 2008;56:291-297.
229 Kiefer T., Wertzel H., Freudenberg N., et al. Long-term survival after repetitive surgery for malignant hemangiopericytoma of the lung with subsequent systemic metastases: case report and review of the literature. Thorac Cardiovasc Surg. 1997;45:307-309.
230 Wong P.P., Yagoda A. Chemotherapy of malignant hemangiopericytoma. Cancer. 1978;41:1256-1260.
231 Jha N., McNeese M., Barkley H.T., et al. Does radiotherapy have a role in hemangiopericytoma management? Report of 14 new cases and a review of the literature. Int J Radiat Oncol Biol Phys. 1987;13:1399-1402.
232 Mira J.G., Chu F.CH., Fortner J.G. The role of radiotherapy in the management of malignant hemangiopericytoma: report of eleven new cases and review of the literature. Cancer. 1977;39:1254-1259.
233 Hansen C.P., Francis D., Bertelsen S. Primary hemangiopericytoma of the lung: case report. Scand J Thorac Cardiovasc Surg. 1990;24:89-92.
234 Weiss S.W., Enzinger F.M. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978;41:2250-2266.
235 Yousem S.A., Hochholzer L. Malignant fibrous histiocytoma of the lung. Cancer. 1987;60:2532-2541.
236 McDonnell T., Kyriakos M., Roper C., et al. Malignant fibrous histiocytoma of the lung. Cancer. 1988;61:137-145.
237 Lee J.T., Shelburne J.D., Linder J. Primary malignant fibrous histiocytoma of the lung: a clinicopathologic and ultrastructural study of five cases. Cancer. 1984;53:1124-1130.
238 Bedrossian C.W., Verani R., Unger K.M., et al. Pulmonary malignant fibrous histiocytoma: light and electron microscopic studies of one case. Chest. 1979;75:186-189.
239 Kern W.H., Hughes R.K., Meyer B.W., et al. Malignant fibrous histiocytoma of the lung. Cancer. 1979;44:1793-1801.
240 Chowdhury L.N., Swerdlow M.A., Jao W., et al. Postirradiation malignant fibrous histiocytoma of the lung: demonstration of alpha-1-antitrypsin-like material in neoplastic cells. Am J Clin Pathol. 1980;74:820-826.
241 Kimizuka G., Okuzawa K., Yarita T. Primary giant cell malignant fibrous histiocytoma of the lung: a case report. Pathol Int. 1999;49:342-346.
242 Barbas C.S., Capelozzi V.L., Takagaki T.Y., et al. Primary malignant fibrous histiocytoma of the lung: report of a case with bronchial brushing cytologic features. Acta Cytol. 1997;41:919-923.
243 Halyard M.Y., Camoriano J.K., Culligan J.A., et al. Malignant fibrous histiocytoma of the lung: report of four cases and review of the literature. Cancer. 1996;78:2492-2497.
244 Shah S.J., Craver R.D., Yu L.C. Primary malignant fibrous histiocytoma of the lung in a child: a case report and review of literature. Pediatr Hematol Oncol. 1996;13:531-538.
245 Corpa-Rodríguez M.E., Mayoralas-Alises S., García-Sánchez J., et al. Postoperative course in 7 cases of primary sarcoma of the lung. Arch Bronconeumol. 2005;41:634-637.
246 Juettner F.M., Popper H., Sommersgutter K., et al. Malignant fibrous histiocytoma of the lung: prognosis and therapy of a rare disease: report of two cases and review of the literature. Thorac Cardiovasc Surg. 1987;35:226-231.
247 Higashiyama M., Doi O., Kodama K., et al. Successful surgery of malignant fibrous histiocytoma in the lung with gross extension into the right main pulmonary artery. Thorac Cardiovasc Surg. 1993;41:73-76.
248 McDermott V.G., MacKenzie S., Hendry G.M. Case report: primary intrathoracic rhabdomyosarcoma: a rare childhood malignancy. Br J Radiol. 1993;66:937-941.
249 Schiavetti A., Dominici C., Matrunola M., et al. Primary pulmonary rhabdomyosarcoma in childhood: clinicobiologic features in two cases with review of the literature. Med Pediatr Oncol. 1996;26:201-207.
250 Noda T., Todani T., Watanabe Y., et al. Alveolar rhabdomyosarcoma of the lung in a child. J Pediatr Surg. 1995;30:1607-1608.
251 Hancock B.J., DiLorenzo M., Youssef S., et al. Childhood primary pulmonary neoplasms. J Pediatr Surg. 1993;28:1133-1136.
252 Choi J.S., Choi J.S., Kim E.J. Primary pulmonary rhabdomyosarcoma in an adult with neurofibromatosis-1. Ann Thorac Surg. 2009;88:1356-1358.
253 Allan B.T., Day D.L., Dehner L.P. Primary pulmonary rhabdomyosarcoma of the lung in children: report of two cases presenting with spontaneous pneumothorax. Cancer. 1987;59:1005-1011.
254 Murphy J.J., Blair G.K., Fraser G.C., et al. Rhabdomyosarcoma arising within congenital pulmonary cysts: report of three cases. J Pediatr Surg. 1992;27:1364-1367.
255 Lee S.H., Rengaciary S.S., Paramesh J. Primary pulmonary rhabdomyosarcoma: a case report and review of the literature. Hum Pathol. 1981;12:92-96.
256 Eriksson A., Thunell M., Lundquist G. Pedunculated endobronchial rhabdomyosarcoma with fatal asphyxia. Thorax. 1982;37:390-391.
257 Triche T.J., Askin F.B., Kissane J.M. Neuroblastoma, Ewing’s sarcoma, and the differential diagnosis of small round blue cell tumors. In: Finegold M., editor. Pathology of Neoplasia in Children and Adolescents. Philadelphia: WB Saunders; 1986:145-195.
258 Wick M.R., Swanson P.E., Manivel J.C. Immunohistochemical analysis of soft tissue sarcomas: comparisons with electron microscopy. Appl Pathol. 1988;6:169-196.
259 Stock N., Chibon F., Binh M.B., et al. Adult-type rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol. 2009;33:1850-1859.
260 Morgenstern D.A., Rees H., Sebire N.J., et al. Rhabdomyosarcoma subtyping by immunohistochemical assessment of myogenin: tissue array study and review of the literature. Pathol Oncol Res. 2008;14:233-238.
261 Dehner L.P. Soft tissue, peritoneum, and retroperitoneum. In: Dehner L.P., editor. Pediatric Surgical Pathology. 2nd ed. Baltimore: Williams & Wilkins; 1987:869-938.
262 Sun C.CJ., Kroll M., Miller J.E. Primary chondrosarcoma of the lung. Cancer. 1982;50:1864-1866.
263 Kurotaki H., Tateoka H., Takeuchi M., et al. Primary mesenchymal chondrosarcoma of the lung: a case report with immunohistochemical and ultrastructural studies. Acta Pathol Jpn. 1992;42:364-371.
264 Hayashi T., Tsuda N., Iseki M., et al. Primary chondrosarcoma of the lung: a clinicopathologic study. Cancer. 1993;72:69-74.
265 Morgan A.D., Salama F.D. Primary chondrosarcoma of the lung: case report and review of the literature. J Thorac Cardiovasc Surg. 1972;64:460-466.
266 Fallahnejad M., Harrell D., Tucker J., et al. Chondrosarcoma of the trachea: report of a case and five-year followup. J Thorac Cardiovasc Surg. 1973;65:210-213.
267 Parker L.A., Molina P.L., Bignault A.G., et al. Primary pulmonary chondrosarcoma mimicking bronchogenic cyst on CT and MRI. Clin Imaging. 1996;20:181-183.
268 Boueiz A., Abougergi M.S., Noujeim C., et al. Primary dedifferentiated chondrosarcoma of the lung. South Med J. 2009;102:861-863.
269 Steurer S., Huber M., Lintner F. Dedifferentiated chondrosarcoma of the lung: case report and review of the literature. Clin Lung Cancer. 2007;8:439-442.
270 Zeren H., Moran C.A., Suster S., et al. Primary pulmonary sarcomas with features of monophasic synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of 25 cases. Hum Pathol. 1995;26:474-480.
271 Yoon G.S., Park S.Y., Kang G.H., et al. Primary pulmonary sarcoma with morphologic features of biphasic synovial sarcoma: a case report. J Korean Med Sci. 1998;13:71-76.
272 Kaplan M.A., Goodman M.D., Satish J., et al. Primary pulmonary sarcoma with morphologic features of monophasic synovial sarcoma and chromosome translocation t(X;18). Am J Clin Pathol. 1996;105:195-199.
273 Roberts C.A., Seemayer T.A., Neff J.R., et al. Translocation t(X;18) in primary synovial sarcoma of the lung. Cancer Genet Cytogenet. 1996;88:49-52.
274 Sekeres M., Vasconcelles M.J., McMenamin M., et al. Two patients with sarcoma. Case 1: Synovial cell sarcoma of the lung. J Clin Oncol. 2000;18:2341-2342.
275 Zaring R.A., Roepke J.E. Pathologic quiz case: pulmonary mass in a patient presenting with a hemothorax. Diagnosis: primary pulmonary biphasic synovial sarcoma. Arch Pathol Lab Med. 1999;123:1287-1289.
276 Bacha E.A., Wright C.D., Grillo H.C., et al. Surgical treatment of primary pulmonary sarcomas. Eur J Cardiothorac Surg. 1999;15:456-460.
277 Hisaoka M., Hashimoto H., Iwamasa T., et al. Primary synovial sarcoma of the lung: report of two cases confirmed by molecular detection of SYT-SSX fusion gene transcripts. Histopathology. 1999;34:205-210.
278 Bégueret H., Galateau-Salle F., Guillou L., et al. Primary intrathoracic synovial sarcoma: a clinicopathologic study of 40 t(X;18)-positive cases from the French Sarcoma Group and the Mesopath Group. Am J Surg Pathol. 2005;29:339-346.
279 Fisher C. Synovial sarcoma. Ann Diagn Pathol. 1998;2:401-421.
280 Essary L.R., Vargas S.O., Fletcher C.D. Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer. 2002;94:459-469.
281 Sawamura K., Hashimoto T., Nanjo S., et al. Primary liposarcoma of the lung. J Surg Oncol. 1982;19:243-246.
282 Achir A., Ouadnouni Y., Smahi M., et al. Primary pulmonary liposarcoma—a case report. Thorac Cardiovasc Surg. 2009;57:119-120.
283 Loddenkemper C., Pérez-Canto A., Leschber G., et al. Primary dedifferentiated liposarcoma of the lung. Histopathology. 2005;46:710-712.
284 Yousem S.A. Angiosarcoma presenting in the lung. Arch Pathol Lab Med. 1986;110:112-115.
285 Bartley T.D., Arean V.M. Intrapulmonary neurogenic tumors. J Thorac Cardiovasc Surg. 1965;50:114-123.
286 Manabe H., Umemoto T., Takagi H., et al. Primary neurogenous sarcoma of the lung; report of a case. Jpn J Thorac Surg. 2005;58:337-340.
287 Reingold I.M., Amromin G.D. Extraosseous osteosarcoma of the lung. Cancer. 1971;28:491-498.
288 Ren L., Guo S.P., Zhou X.G., et al. Angiomatoid fibrous histiocytoma: first report of primary pulmonary origin. Am J Surg Pathol. 2009;33:1570-1574.
289 Trabelsi A., Ben Abdelkrim S., Taher Yacoubi M., et al. Primary alveolar soft part sarcoma of the lung. Rev Mal Respir. 2009;26:329-332. [article in French]
290 Kim Y.D., Lee C.H., Lee M.K., et al. Primary alveolar soft part sarcoma of the lung. J Korean Med Sci. 2007;22:369-372.
291 Wakely P.E.Jr, McDermott J.E., Ali S.Z. Cytopathology of alveolar soft part sarcoma: a report of 10 cases. Cancer Cytopathol. 2009;117:500-507.
292 Ladanyi M., Lui M.Y., Antonescu C.R., et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:48-57.
293 Wilson R.W., Moran C.A. Primary melanoma of the lung: a clinicopathologic and immunohistochemical study of eight cases. Am J Surg Pathol. 1997;21:1196-1202.
294 Ost D., Joseph C., Sogoloff H., et al. Primary pulmonary melanoma: case report and literature review. Mayo Clin Proc. 1999;74:62-66.
295 Jensen O.A., Egedorf J. Primary malignant melanoma of the lung. Scand J Respir Dis. 1967;48:127-135.
296 Salm R. A primary malignant melanoma of the bronchus. J Pathol Bacteriol. 1963;85:121-126.
297 Bagwell S.P., Flynn S.D., Cox P.M., et al. Primary malignant melanoma of the lung. Am Rev Respir Dis. 1989;139:1543-1547.
298 Carstens P.HB., Kuhns J.G., Ghazi C. Primary malignant melanomas of the lung and adrenal. Hum Pathol. 1984;15:910-914.
299 Reid J.D., Mehta V.T. Melanoma of the lower respiratory tract. Cancer. 1966;19:627-631.
300 Robertson A.J., Sinclair D.JM., Sutton P.P., et al. Primary melanocarcinoma of the lower respiratory tract. Thorax. 1980;35:158-159.
301 Reed R.J., Kent E.M. Solitary pulmonary melanomas: two case reports. J Thorac Cardiovasc Surg. 1964;48:226-231.
302 Gephardt G.N. Malignant melanoma of the bronchus. Hum Pathol. 1981;12:671-673.
303 Ozdemir N., Cangir A.K., Kutlay H., et al. Primary malignant melanoma of the lung in an oculocutaneous albino patient. Eur J Cardiothorac Surg. 2001;20:864-867.
304 Hashimoto T., Oka K., Hakozaki H., et al. Benign clear cell tumor of the lung. Ultrastruct Pathol. 2001;25:479-483.
305 Gaffey M.J., Mills S.E., Askin F.B., et al. Clear cell tumor of the lung: a clinicopathologic, immunohistochemical, and ultrastructural study of eight cases. Am J Surg Pathol. 1990;14:248-259.
306 Kuhnen C., Preisler K., Muller K.M. Pulmonary lymphangioleiomyomatosis: morphologic and immunohistochemical findings. Pathologe. 2001;22:197-204. [article in German]
307 Ferrans V.J., Yu Z.X., Nelson W.K., et al. Lymphangioleiomyomatosis (LAM): a review of clinical and morphological features. J Nippon Med Sch. 2000;67:311-329.
308 Ito M., Sugamura Y., Ikari H., et al. Angiomyolipoma of the lung. Arch Pathol Lab Med. 1998;122:1023-1025.
309 Hynes J.K., Smith H.C., Holmes D.R. Pulmonary artery sarcoma: preoperative diagnosis noninvasively by two-dimensional echocardiography. Circulation. 1983;67:459-477.
310 Lyerly H.K., Reves J.G., Sabiston D.C. Management of primary sarcomas of the pulmonary artery and reperfusion intrabronchial hemorrhage. Surg Gynecol Obstet. 1986;163:291-298.
311 Gaissert H.A., Mark E.J. Tracheobronchial gland tumors. Cancer Control. 2006;13:286-294.
312 Banerjee S.S., Eyden B. Divergent differentiation in malignant melanomas: a review. Histopathology. 2008;52:119-129.
313 Maeda R., Isowa N., Onuma H., et al. Primary malignant melanoma of the lung with rapid progression. Gen Thorac Cardiovasc Surg. 2009;57:671-674.
314 Yamada G., Tanaka Y., Otsuka M., et al. Metastatic malignant melanoma mimicking primary lung adenocarcinoma. Intern Med. 2006;45:1255-1256.
315 Saint-Blancard P., Vaylet F., Jancovici R. A rare pulmonary tumour, primary malignant melanoma. Rev Mal Respir. 2009;26:57-61.
316 Reddy V.S., Mykytenko J., Giltman L.I., et al. Primary malignant melanoma of the lung: review of literature and report of a case. Am Surg. 2007;73:287-289.
317 Kundranda M.N., Clark C.T., Chaudhry A.A., et al. Primary malignant melanoma of the lung: a case report and review of the literature. Clin Lung Cancer. 2006;7:279-281.
318 de Wilt J.H., Farmer S.E., Sarah E.J., et al. Isolated melanoma in the lung where there is no known primary site: metastatic disease or primary lung tumor? Melanoma Res. 2005;15:531-537.
319 McGlennen R.C., Manivel J.C., Stanley S.J., et al. Pulmonary artery trunk sarcoma: a clinicopathologic, ultrastructural, and immunohistochemical study of four cases. Mod Pathol. 1989;2:486-494.
320 Tanaka I., Masuda R., Inoue M., et al. Primary pulmonary artery sarcoma: report of a case with complete resection and graft replacement, and review of 47 surgically-treated cases reported in the literature. Thorac Cardiovasc Surg. 1994;42:64-68.
321 Al-Robaish A., Lien D.C., Slatnik J., et al. Sarcoma of the pulmonary artery trunk: report of a case complicated with hemopericardium and cardiac tamponade. Can J Cardiol. 1995;11:707-709.
322 Timmers L., Bové T., De Pauw M. Intimal sarcoma of the pulmonary artery: a report of two cases. Acta Cardiol. 2009;64:677-679.
323 Austin B.A., Griffin B.P. Pulmonary artery intimal sarcoma: a brief case series. J Am Soc Echocardiogr. 2008;21:978.e5-978.e7.
324 Sethi G.K., Slaven J.E., Kepes J.J. Primary sarcoma of the pulmonary artery. J Thorac Cardiovasc Surg. 1972;63:587-596.
325 Baker P.B., Goodwin R.A. Pulmonary artery sarcoma. Arch Pathol Lab Med. 1985;109:35-40.
326 Weijmer M.C., Kummer J.A., Thijs L.G. Case report of a patient with an intimal sarcoma of the pulmonary trunk presenting as a pulmonary embolism. Neth J Med. 1999;55:80-83.
327 Pagni S., Passik C.S., Riordan C., et al. Sarcoma of the main pulmonary artery: an unusual etiology for recurrent pulmonary emboli. J Cardiovasc Surg. 1999;40:457-461.
328 Babatasi G., Massetti M., Agostini D., et al. Leiomyosarcoma of the heart and great vessels. Ann Cardiol Angiol. 1998;47:451-458.
329 Fujii H., Osako M., Otani H., et al. Primary pulmonary artery sarcoma. Jpn Circ J. 1998;62:379-381.
330 Babatasi G., Massetti M., Galateau F., et al. Pulmonary artery trunk leiomyosarcoma. Thorac Cardiovasc Surg. 1998;46:45-47.
331 Mazzucco A., Luciani G.B., Bertolini P., et al. Primary leiomyosarcoma of the pulmonary artery: diagnostic and surgical implications. Ann Thorac Surg. 1994;57:222-225.
332 Johansson L., Carien B. Sarcoma of the pulmonary artery: report of four cases with electron microscopic and immunohistochemical examinations, and review of the literature. Virchows Arch A. 1994;424:217-224.
333 Ramp U., Gerharz C.D., Iversen S., et al. Sarcoma of the pulmonary artery: report of two cases and a review of the literature. J Cancer Res Clin Oncol. 1992;118:551-556.
334 Mayer F., Aebert H., Rudert M., et al. Primary malignant sarcomas of the heart and great vessels in adult patients—a single-center experience. Oncologist. 2007;12:1134-1142.
335 Huo L., Moran C.A., Fuller G.N., et al. Pulmonary artery sarcoma: a clinicopathologic and immunohistochemical study of 12 cases. Am J Clin Pathol. 2006;125:419-424.
336 Tavora F., Miettinen M., Fanburg-Smith J., et al. Pulmonary artery sarcoma: a histologic and follow-up study with emphasis on a subset of low-grade myofibroblastic sarcomas with a good long-term follow-up. Am J Surg Pathol. 2008;32:1751-1761.
337 Bleisch V.R., Kraus F.T. Polypoid sarcoma of the pulmonary trunk. Cancer. 1980;46:314-321.
338 Law M.R., Gregor A., Hodson M.E., et al. Malignant mesothelioma of the pleura: a study of 52 treated and 54 untreated patients. Thorax. 1984;39:25-259.
339 Adams V.I., Unni K.K., Muhm J.R., et al. Diffuse malignant mesothelioma of pleura: diagnosis and survival in 92 cases. Cancer. 1986;58:1540-1551.
340 Grondin S.C., Sugarbaker D.J. Malignant mesothelioma of the pleural space. Oncology. 1999;13:919-932.
341 Coffin C.M., Dehner L.P. Mesothelial and related neoplasms in children and adolescents: a clinicopathologic and immunohistochemical analysis of eight cases. Pediatr Pathol. 1992;12:333-347.
342 Gentiloni N., Febbraro S., Barone C., et al. Peritoneal mesothelioma in recurrent familial peritonitis. J Clin Gastroenterol. 1997;24:276-279.
343 Walz R., Koch H.K. Malignant pleural mesothelioma: some aspects of epidemiology, differential diagnosis, and prognosis. Histological and immunohistochemical evaluation and followup of mesotheliomas diagnosed from 1964 to January 1985. Pathol Res Pract. 1990;186:124-134.
344 Bianchi C., Brollo Ramani L., Zuch C. Pleural plaques as risk indicators for malignant pleural mesothelioma: a necropsy-based study. Am J Ind Med. 1997;32:445-449.
345 Sanden A., Jarvholm B. A study of possible predictors of mesothelioma in shipyard workers exposed to asbestos. J Occup Med. 1991;33:770-773.
346 Hasan F.M., Nash G., Kazemi H. The significance of asbestos exposure in the diagnosis of mesothelioma: a 28-year experience from a major urban hospital. Am Rev Respir Dis. 1977;115:761-768.
347 Hasan F.M., Nash G., Kazemi H. Asbestos exposure and related neoplasia: the 28-year experience of a major urban hospital. Am J Med. 1978;65:649-654.
348 Grant D.C., Seltzer S.E., Antman K.H., et al. Computed tomography of malignant pleural mesothelioma. J Comput Assist Tomogr. 1983;7:626-632.
349 Kishimoto T., Ono T., Okada K., et al. Relationship between number of asbestos bodies in autopsy lung and pleural plaques on chest x-ray film. Chest. 1989;95:549-552.
350 Navratil M., Trippe F. Prevalence of pleural calcification in persons exposed to asbestos dust, and in the general population in the same district. Environ Res. 1972;5:210-216.
351 Peterson J.T., Greenberg S.D., Buffler P. Non-asbestos-related malignant mesothelioma: a review. Cancer. 1984;54:951-960.
352 Cavazza A., Travis L.B., Travis W.D., et al. Post-irradiation malignant mesothelioma. Cancer. 1996;77:1379-1385.
353 Strickler H.D., Goedert J.J., Fleming M., et al. Simian virus 40 and pleural mesothelioma in humans. Cancer Epidemiol Biomarkers Prev. 1996;5:473-475.
354 Wick M.R., Mills S.E. Mesothelial proliferations: an increasing morphological spectrum. Am J Clin Pathol. 2000;113:619-622.
355 Law M.R., Hodson M.E., Heard B.E. Malignant mesothelioma of the pleura: relation between histological type and clinical behavior. Thorax. 1982;37:810-815.
356 Gaertner E., Zeren E.H., Fleming M.V., et al. Biphasic synovial sarcomas arising in the pleural cavity: a clinicopathologic study of five cases. Am J Surg Pathol. 1996;20:36-45.
357 Argani P., Zakowski M.F., Klimstra D.S., et al. Detection of the SYT-SSX chimeric RNA of synovial sarcoma in paraffin-embedded tissue and its application in problematic cases. Mod Pathol. 1998;11:65-71.
358 Miettinen M., Limon J., Niezabitowski A., et al. Calretinin and other mesothelioma markers in synovial sarcoma: analysis of antigenic similarities and differences with malignant mesothelioma. Am J Surg Pathol. 2001;25:610-617.
359 Attanoos R.L., Biggs A.R. Pathology of malignant mesothelioma. Histopathology. 1997;30:403-418.
360 Andrion A., Mazzuco G., Bernardi P., et al. Sarcomatous tumor of the chest wall with osteochondroid differentiation: evidence of mesothelial origin. Am J Surg Pathol. 1989;13:707-712.
361 Yousem S.A., Hochholzer L. Malignant mesothelioma with osseous and cartilaginous differentiation. Arch Pathol Lab Med. 1987;111:62-66.
362 Okamoto T., Yokota S., Shinkawa K., et al. Pleural malignant mesothelioma with osseous, cartilaginous, and rhabdomyogenic differentiation. Nihon Kokyuki Gakkai Zasshi. 1998;36:696-701.
363 Ordonez N.G., Tornos C. Malignant peripheral nerve sheath tumor of the pleura with epithelial and rhabdomyoblastic differentiation: report of a case clinically simulating mesothelioma. Am J Surg Pathol. 1997;21:1515-1521.
364 Corson J.M. Pathology of diffuse malignant pleural mesothelioma. Semin Thorac Cardiovasc Surg. 1997;9:347-355.
365 Avellini C., Alampi G., Cocchi V., et al. Malignant sarcomatoid mesothelioma of the pleura: a histological and immunohistochemical study of a case. Pathologica. 1991;83:335-340.
366 Cagle P.T., Truong L.D., Roggli V.L., et al. Immunohistochemical differentiation of sarcomatoid mesotheliomas from other spindle cell neoplasms. Am J Clin Pathol. 1989;92:566-571.
367 Carter D., Otis C.N. Three types of spindle cell tumors of the pleura: fibroma, sarcoma, and sarcomatoid mesothelioma. Am J Surg Pathol. 1988;12:747-753.
368 Montag A.G., Pinkus G.S., Corson J.M. Keratin protein immunoreactivity of sarcomatoid and mixed types of diffuse malignant mesothelioma: an immunoperoxidase study of 30 cases. Hum Pathol. 1988;19:336-342.
369 Hammar S.P., Bolen J.W. Sarcomatoid pleural mesothelioma. Ultrastruct Pathol. 1985;9:337-343.
370 Weinbreck N., Vignaud J.M., Begueret H., et al. SYT-SSX fusion is absent in sarcomatoid mesothelioma allowing its distinction from synovial sarcoma of the pleura. Mod Pathol. 2007;20:617-621.
371 Khalidi H.S,., Medeiros L.J,., Battifora H. Lymphohistiocytoid mesothelioma: an often misdiagnosed variant of sarcomatoid malignant mesothelioma. Am J Clin Pathol. 2000;113(5):649-654.
372 Henderson D.W., Attwood H.D., Constance T.J., et al. Lymphohistiocytoid mesothelioma: a rare lymphomatoid variant of predominantly sarcomatoid mesothelioma. Ultrastruct Pathol. 1988;12:367-384.
373 Mangano W.E., Cagle P.T., Churg A., et al. The diagnosis of desmoplastic malignant mesothelioma and its distinction from fibrous pleurisy: a histologic and immunohistochemical analysis of 31 cases including p53 immunostaining. Am J Clin Pathol. 1998;110:191-199.
374 Wilson G.E., Hasleton P.S., Chatterjee A.K. Desmoplastic malignant mesothelioma: a review of 17 cases. J Clin Pathol. 1992;45:295-298.
375 Crotty T.B., Colby T.V., Gay P.C., et al. Desmoplastic malignant mesothelioma masquerading as sclerosing mediastinitis: a diagnostic dilemma. Hum Pathol. 1992;23:79-82.
376 Epstein J.I., Budin R.E. Keratin and epithelial membrane antigen immunoreactivity in nonneoplastic fibrous pleural lesions: implications for the diagnosis of desmoplastic mesothelioma. Hum Pathol. 1986;17:514-519.
377 Cantin R., Al-Jabi M., McCaughey W.T. Desmoplastic diffuse mesothelioma. Am J Surg Pathol. 1982;6:215-222.
378 Colby T.V. The diagnosis of desmoplastic malignant mesothelioma. Am J Clin Pathol. 1998;110:135-136.
379 Colby T.V. Malignancies in the lung and pleura mimicking benign processes. Semin Diagn Pathol. 1995;12:30-44.
380 Center R., Lukeis R., Dietzsch E., et al. Molecular deletion of 9p sequences in non-small cell lung cancer and malignant mesothelioma. Genes Chromosomes Cancer. 1993;7:47-53.
381 Cheng J.Q., Jhanwar S.C., Lu Y.Y., et al. Homozygous deletions within 9p21-p22 identify a small critical region of chromosomal loss in human malignant mesotheliomas. Cancer Res. 1993;53:4761-4763.
382 Brown R.W., Clark G.M., Tandon A.K., et al. Multiple-marker immunohistochemical phenotypes distinguishing malignant pleural mesothelioma from pulmonary adenocarcinoma. Hum Pathol. 1993;24:347-354.
383 Riera J.R., Astengo-Osuna C., Longmate J.A., et al. The immunohistochemical diagnostic panel for epithelial mesothelioma: a reevaluation following heat-induced epitope retrieval. Am J Surg Pathol. 1997;21:1409-1419.
384 Ordonez N.G. Role of immunohistochemistry in differentiating epithelial mesothelioma from adenocarcinoma: review and update. Am J Clin Pathol. 1999;112:75-89.
385 Burns T.R., Greenberg D., Mace M.L., et al. Ultrastructural diagnosis of epithelial malignant mesothelioma. Cancer. 1985;56:2036-2040.
386 Cury P.M., Butcher D.N., Corrin B., et al. The use of histological and immunohistochemical markers to distinguish pleural malignant mesothelioma and in situ mesothelioma from reactive mesothelial hyperplasia and reactive pleural fibrosis. J Pathol. 1999;189:251-257.
387 Kung I.T., Thallas V., Spencer E.J., et al. Expression of muscle actins in diffuse mesotheliomas. Hum Pathol. 1995;26:565-570.
388 Hurliman J. Desmin and neural marker expression in mesothelial cells and mesotheliomas. Hum Pathol. 1994;25:753-757.
389 Klima M., Bossart M.I. Sarcomatous type of malignant mesothelioma. Ultrastruct Pathol. 1983;4:349-358.
390 Allen T.C., Cagle P.T., Churg A.M., et al. Localized malignant mesothelioma. Am J Surg Pathol. 2005;29:866-873.
391 Flint A., Weiss S.W. CD34 and keratin expression distinguishes solitary fibrous tumor (fibrous mesothelioma) of pleura from desmoplastic mesothelioma. Hum Pathol. 1995;26:428-431.
392 Huncharek M., Kelsey K., Mark E.J., et al. Treatment and survival in diffuse malignant pleural mesothelioma: a study of 83 cases from the Massachusetts General Hospital. Anticancer Res. 1996;16(3A):1265-1268.
393 Machin T., Mashiyama E.T., Henderson J.A., et al. Bony metastases in desmoplastic pleural mesothelioma. Thorax. 1988;43:155-156.
394 Chahinian A.P., Antman K., Goutsou M., et al. Randomized phase II trial of cisplatin with mitomycin or doxorubicin for malignant mesothelioma by the Cancer & Leukemia Group B. J Clin Oncol. 1993;11:1559-1565.
395 Sugarbaker D.J., Garcia J.P., Richards W.G., et al. Extrapleural pneumonectomy in the multimodality therapy of malignant pleural mesothelioma results in 120 consecutive patients. Ann Surg. 1996;224:288-296.
396 DaValle M.J., Faber L.P., Kittle C.F., et al. Extrapleural pneumonectomy for diffuse malignant mesothelioma. Ann Thorac Surg. 1986;42:612-618.
397 Lee M.J., Grogan L., Meehan S., et al. Pleural granulocytic sarcoma: CT characteristics. Clin Radiol. 1991;43:57-59.
398 Luppi G., Cesinaro A.M., Zoboli A., et al. Mesenchymal chondrosarcoma of the pleura. Eur Respir J. 1996;9:840-843.
399 Goetz S.P., Robinson R.A., Landas S.K. Extraskeletal myxoid chondrosarcoma of the pleura: report of a case clinically simulating mesothelioma. Am J Clin Pathol. 1992;97:498-502.
400 Okby N.T., Travis W.D. Liposarcoma of the pleural cavity: clinical and pathologic features of 4 cases with a review of the literature. Arch Pathol Lab Med. 2000;124:699-703.
401 Wong W.W., Pluth J.R., Grado G.L., et al. Liposarcoma of the pleura. Mayo Clin Proc. 1994;69:882-885.
402 Stark P., Smith D.C., Watkins G.E., et al. Primary intrathoracic extraosseous osteogenic sarcoma: report of three cases. Radiology. 1990;174:725-726.
403 Matono R., Maruyama R., Ide S., Kitagawa D., et al. Extraskeletal osteosarcoma of the pleura: a case report. Gen Thorac Cardiovasc Surg. 2008;56:180-182.
404 Moran C.A., Suster S., Koss M.N. The spectrum of histologic growth patterns in benign and malignant fibrous tumors of the pleura. Semin Diagn Pathol. 1992;9:169-180.
405 Kim S.Y., Kim M.Y., Hwang Y.J., et al. Low-grade fibromyxoid sarcoma: CT, sonography, and MR findings in 3 cases. J Thorac Imaging. 2005;20:294-297.
406 Miyoshi N., Takami K., Okami J., et al. Extrapleural pneumonectomy for relapsed solitary fibrous tumors of the pleura with pleural dissemination. Jpn J Thorac Surg. 2007;60:800-805.
407 Krishnadas R., Froeschle P.O., Berrisford R.G. Recurrence and malignant transformation in solitary fibrous tumour of the pleura. Thorac Cardiovasc Surg. 2006;54:65-67.
408 Ali S.Z., Hoon V., Hoda S., et al. Solitary fibrous tumor: a cytologic–histologic study with clinical, radiologic, and immunohistochemical correlations. Cancer. 1997;81:116-121.
409 Hanau C.A., Miettinen M. Solitary fibrous tumor: histological and immuno histochemical spectrum of benign and malignant variants presenting at different sites. Hum Pathol. 1995;26:440-449.
410 Mezzetti M., Augusti A. Fibrosarcoma of the pleura: circumscribed primary pleural neoplasms. Arch Ital Chir. 1969;95:544-555.
411 Meyer M., Krause U. Solitary fibrous tumors of the pleura. Chirurg. 1999;70:979-1952.
412 Vallat-Decouvelaere A.V., Dry S.M., Fletcher C.DM. Atypical and malignant solitary fibrous tumors in extrathoracic locations: evidence of their comparability to intrathoracic tumors. Am J Surg Pathol. 1998;22:1501-1511.
413 Yokoi T., Tsuzuki T., Yatabe Y., et al. Solitary fibrous tumor: significance of p53 and CD34 immunoreactivity in its malignant transformation. Histopathology. 1998;32:423-432.
414 Myoui A., Aozasa K., Iuchi K., et al. Soft tissue sarcoma of the pleural cavity. Cancer. 1991;68:1550-1554.
415 England D.M., Hochholzer L., McCarthy M.J. Localized benign and malignant fibrous tumors of the pleura. Am J Surg Pathol. 1989;13:640-658.
416 Fukasawa Y., Takada A., Tateno M., et al. Solitary fibrous tumor of the pleura causing recurrent hypoglycemia by secretion of insulin-like growth factor II. Pathol Int. 1998;48:47-52.
417 Theegarten D., Meisel M. Malignant fibrous histiocytoma of the thoracic wall in the area of a tuberculous pleural callosity. Pneumonologie. 1993;47:458-460.
418 Watanabe S., Hitomi S., Nakamura T., et al. A clinical study of six surgically-treated patients with malignant tumors arising from chronic pleuritis and pyothorax. Nippon Kyobu Geka Gakkai Zasshi. 1989;37:281-286.
419 Saiffudin A., DaCosta P., Chalmers A.G., et al. Primary malignant localized fibrous tumors of the pleura: clinical, radiological, and pathological features. Clin Radiol. 1992;45:13-17.
420 Toochika H., Kiminok K., Tagawa Y., et al. Malignant fibrous histiocytoma of the chest cavity: report of four resected cases. Nippon Kyobu Geka Gakkai Zasshi. 1990;38:647-653.
421 Moran C.A., Suster S., Koss M.N. Smooth muscle tumors presenting as pleural neoplasms. Histopathology. 1995;27:227-234.
422 Gibbs A.R. Smooth muscle tumors of the pleura. Histopathology. 1995;27:295-296.
423 Al-Daraji W.I., Salman W.D., Nakhuda Y., et al. Primary smooth muscle tumor of the pleura: a clinicopathological case report with ultrastructural observations and a review of the literature. Ultrastruct Pathol. 2005;29:389-398.
424 Askin F.B., Rosai J., Sibley R.K., et al. Malignant small cell tumor of the thoracopulmonary region in childhood. Cancer. 1979;43:2438-2451.
425 Israel M.A. Peripheral neuroepithelioma. In: Williams C.J., Krikorian J.G., Green M.R., Rhagavan D., editors. Textbook of Uncommon Cancer. New York: John Wiley & Sons; 1988:683-690.
426 Jurgens H., Bier V., Harms D., et al. Malignant peripheral neuroectodermal tumors. Cancer. 1988;61:349-357.
427 Dang N.C., Siegel S.E., Phillips J.D. Malignant chest wall tumors in children and young adults. J Pediatr Surg. 1999;34:1773-1778.
428 Promnitz S., Petri F., Schulz H.J., et al. Askin’s tumor: a rare entity. Case report with references to the literature. Pneumonologie. 1999;53:393-399.
429 Liptay M.J., Fry W.A. Malignant bone tumors of the chest wall. Semin Thorac Cardiovasc Surg. 1999;11:278-284.
430 Kabiri H., El-Fakir Y., Mahassini N., et al. Malignant small cell thoracic pulmonary tumor. Rev Pneumonol Clin. 1999;55:21-25.
431 Taneli C., Genc A., Erikci V., et al. Askin tumors in children: a report of four cases. Eur J Pediatr Surg. 1998;8:312-314.
432 Sallustio G., Pirronti T., Lasorella A., et al. Diagnostic imaging of primitive neuroectodermal tumor of the chest wall (Askin tumor). Pediatr Radiol. 1998;28:697-702.
433 Askin F.B., Perlman E.J. Neuroblastoma and peripheral neuroectodermal tumors. Am J Clin Pathol. 1998;109(4 suppl 1):S23-S30.
434 Sabate J.M., Franquet T., Parellada J.A., et al. Malignant neuroectodermal tumor of the chest wall (Askin tumor): CT and MR findings in eight patients. Clin Radiol. 1994;49:634-638.
435 Shamberger R.C., Tarbell N.J., Perez-Atayde A.R., et al. Malignant small round cell tumor (Ewing’s-PNET) of the chest wall in children. J Pediatr Surg. 1994;29:179-185.
436 Winer-Muram H.T., Kauffman W.M., Gronemeyer S.A., et al. Primitive neuroectodermal tumors of the chest wall (Askin tumors): CT and MR findings. Am J Roentgenol. 1993;161:265-268.
437 Dehner L.P. Soft tissue sarcomas of childhood. Natl Cancer Inst Monogr. 1981;56:43-59.
438 Sapi Z., Szentirmay Z., Orosz Z. Desmoplastic small round cell tumor of the pleura: a case report with further cytogenetic and ultrastructural evidence of “mesothelioblastemic” origin. Eur J Surg Oncol. 1999;25:633-634.
439 Parkash V., Gerald W.L., Parma A., et al. Desmoplastic small round cell tumor of the pleura. Am J Surg Pathol. 1995;19:659-665.
440 Bian Y., Jordan A.G., Rupp M., et al. Effusion cytology of desmoplastic small round cell tumor of the pleura: a case report. Acta Cytol. 1993;37:77-82.
441 Sarkar M.R., Bahr R. The Askin tumor. Chirurg. 1992;63:973-976.
442 Contesso G., Llombart-Bosch A., Terrier P., et al. Does malignant small round cell tumor of the thoracopulmonary region (Askin tumor) constitute a clinicopathologic entity? Cancer. 1992;69:1012-1020.
443 Takahashi K., Dambara T., Uekusa T., et al. Massive chest wall tumor diagnosed as Askin tumor: successful treatment by intensive combined modality therapy in an adult. Chest. 1993;104:287-288.
444 Fitzgibbons J.F., Feldhaus S.J., McNamara L.F., et al. Diagnostic features and treatment of the Askin tumor—malignant small cell tumor of the thoracopulmonary region: a case report. Nebr Med J. 1993;78:2-6.
445 Kushner B.H., LaQuaglia M.P., Cheung N.K., et al. Clinically critical impact of molecular genetic studies in pediatric solid tumors. Med Pediatr Oncol. 1999;33:530-535.
446 Dehner L.P. Peripheral neuroectodermal tumor and Ewing’s sarcoma. Am J Surg Pathol. 1993;17:1-13.
447 Lae M.E., Roche P.C., Jin L., Lloyd R.V., et al. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol. 2002;26:823-835.
448 Gautam U., Srinivasan R., Rajwanshi A., et al. Comparative evaluation of flow-cytometric immunophenotyping and immunocytochemistry in the categorization of malignant small round cell tumors in fine-needle aspiration cytologic specimens. Cancer. 2008;114:494-503.
449 Barnoud R., Sabourin J.C., Pasquier D., et al. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol. 2000;24:830-836.
450 Hill D.A., Pfeifer J.D., Marley E.F., et al. WT1 staining reliably differentiates desmoplastic small round cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am J Clin Pathol. 2000;114:345-353.
451 Miser J.S., Kinsella T.J., Triche T.J., et al. Treatment of peripheral neuroepithelioma in children and young adults. J Clin Oncol. 1987;5:1752-1758.
452 Farhi D.C., Odell C.A., Shurin S.B. Myelodysplastic syndrome and acute myeloid leukemia after treatment for solid tumor of childhood. Am J Clin Pathol. 1993;100:270-275.
453 Cohen M., Kaschula R.O. Primary pulmonary tumors in childhood: a review of 31 years’ experience and the literature. Pediatr Pulmonol. 1992;14:222-232.
454 Hill D.A., Sadeghi S., Schultz M.Z., et al. Pleuropulmonary blastoma in an adult: an initial case report. Cancer. 1999;85:2368-2374.
455 Szczesny T., Hussein N., Szczesna A. Pulmonary blastoma and related primary malignant pulmonary neoplasms. Pneumonol Alergol Pol. 1999;67:263-270.
456 Romeo C., Impellizzeri P., Grosso M., et al. Pleuropulmonary blastoma: long-term survival and literature review. Med Pediatr Oncol. 1999;33:372-376.
457 Benouachane T., El-Khorassani M., Nachef M.N., et al. Pleuropulmonary blastoma: report of 4 cases. Rev Mal Respir. 1999;16:390-394.
458 Baraniya J., Desai S., Kane S., et al. Pleuropulmonary blastoma. Med Pediatr Oncol. 1999;32:52-56.
459 Priest J.R., McDermott M.B., Bhatia S., et al. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer. 1997;80:147-161.
460 Priest J.R., Watterson J., Strong L., et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr. 1996;128:220-224.
461 Lopez-Andreu J.A., Ferris-Tortajada J., Gomez J. Pleuropulmonary blastoma and congenital cystic malformations. J Pediatr. 1996;129:773-775.
462 Tagge E.P., Mulvihill D., Chandler J.C., et al. Childhood pleuropulmonary blastoma: caution against nonoperative management of congenital lung cysts. J Pediatr Surg. 1996;31:187-190.
463 Wright J.R. Pleuropulmonary blastoma: a case report documenting transition from type I (cystic) to type III (solid). Cancer. 2000;88:2853-2858.
464 Piastra M., Ruggiero A., Caresta E., et al. Critical presentation of pleuropulmonary blastoma. Pediatr Surg Int. 2005;21:223-226.
465 Priest J.R., Williams G.M., Hill D.A., et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44:14-30.
466 Hill D.A., Ivanovich J., Priest J.R., et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325(5943):965.
467 MacSweeney F., Papagiannopoulos K., Goldstraw P., et al. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol. 2003;27:1139-1146.
468 Papagiannopoulos K., Hughes S., Nicholson A.G., et al. Cystic lung lesions in the pediatric and adult population: surgical experience at the Brompton Hospital. Ann Thorac Surg. 2002;73:1594-1598.
469 Dehner L.P. Pleuropulmonary blastoma is THE pulmonary blastoma of childhood. Semin Diagn Pathol. 1994;11:144-151.
470 Hill D.A., Jarzembowski J.A., Priest J.R., et al. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am J Surg Pathol. 2008;32:282-295.
471 Priest J.R., Magnuson J., Williams G.M., et al. Cerebral metastasis and other central nervous system complications of pleuropulmonary blastoma. Pediatr Blood Cancer. 2007;49:266-273.
472 Lin B.T., Colby T.V., Gown A.M., et al. Malignant vascular tumors of the serous membranes mimicking mesothelioma: a report of 14 cases. Am J Surg Pathol. 1996;20:1431-1439.
473 Crotty E.J., McAdams H.P., Erasmus J.J., et al. Epithelioid hemangioendothelioma of the pleura: clinical and radiologic features. Am J Roentgenol. 2000;175:1545-1549.
474 Del Frate C., Mortele K., Zanardi R., et al. Pseudomesotheliomatous angiosarcoma of the chest wall and pleura. J Thorac Imaging. 2003;18:200-203.
475 Kurtz J.E., Serra S., Duclos B., et al. Diffuse primary angiosarcoma of the pleura: a case report and review of the literature. Sarcoma. 2004;8:103-106.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
14 Sarcomas and Sarcomatoid Neoplasms of the Lungs and Pleural Surfaces
This chapter summarizes the clinicopathologic information pertaining to such lesions. It devotes exclusive attention to malignant lesions; benign mesenchymal neoplasms of the lung and pleura are discussed in Chapter 19. Pseudotumors are likewise considered in another portion of this book. Nevertheless, those pathologic categories and other lesions will indeed be mentioned here in the context of differential diagnosis. The entities that are discussed are arranged in order of frequency, to give the reader a sense of their relative incidences.
Sarcomatoid Carcinoma of the Lung
Recent changes in the classification of lung tumors have included sarcoma-like tumors. Five subtypes of those lesions are now codified; pleomorphic carcinoma, spindle cell carcinoma, giant cell carcinoma, carcinosarcoma, and pulmonary blastoma are predicated on the particulars of their microscopic appearances.1,2 We consider all of these neoplasms to be part of the same tumor family, that of sarcomatoid carcinomas (SCs), as discussed in more detail subsequently. Although SCs are rare in an absolute sense, they represent the most common mesenchymal-like malignancies of the airways.3,4 True sarcomas are very infrequently seen in the tracheobronchial tree.4–13 Therefore, one usually considers a cytologically atypical spindle cell tumor of the lung to be an SC unless thorough immunohistological and ultrastructural studies indicate otherwise.5 This review will discuss neoplasms in this general category, using information taken from the pertinent literature as well as the personal experience of the authors.
Historical and Terminologic Considerations
Over time, morphologically similar tumors have been given a variety of designations in the upper and lower respiratory tracts. These diagnostic terms have included blastoma, sarcomatoid carcinoma, spindle cell carcinoma, squamous cell carcinoma with pseudosarcomatous stroma, pseudosarcoma, and carcinosarcoma, based largely on the specific microscopic attributes of the lesions in question and the conceptual leanings of the authors describing them.14–43
More than 70 years ago, Saphir and Vass44 assessed the literature then extant on carcinosarcomas and concluded that they represented primary epithelial malignancies that had undergone divergent differentiation (tumor metaplasia). Their paper cited several lesions of the lung. Thereafter, opposing publications on histogenesis espoused the opinion that biphasic neoplasms of the airways were “collision” tumors, or that they reflected the proliferation of non-neoplastic mesenchymal tissue components that were induced by the carcinomatous elements.45–47 At the turn of the last century, Krompecher48 and others49 had held to the same theories as those of Saphir and Vass. In the past two decades, results of studies using electron microscopy, immunohistology, and “molecular” assays of clonality have tended to support the latter foresighted views of those pioneers convincingly. Hence, it is believed currently that blastomas, carcinosarcomas, carcinomas with pseudosarcomatous stroma, and SCs comprise a single morphological spectrum of basically epithelial tumors, regardless of their anatomic locations.34–36 Biphasic sarcomatoid carcinoma and monophasic sarcomatoid carcinoma have been proposed as replacements for the former designations of carcinosarcoma and spindle cell carcinoma, respectively.33,34,37 In the penultimate iteration of the World Health Organization nosologic scheme, such lesions were included in the category designated “carcinoma with pleomorphic, sarcomatoid, or sarcomatous elements.”36
Clinicopathologic Features of Pulmonary Sarcomatoid Carcinomas
Clinical Attributes
SCs arise in the large bronchi and peripheral lung fields much more often than in the trachea, although the authors have indeed seen some lesions that originated above the carina. The majority of individuals with pulmonary SC are men, and most have a history of heavy smoking.14,23,24 The average patient is 60 years of age.33 Clinical signs and symptoms produced by these tumors are directly associated with the tumor’s specific location. Endoluminal lesions in large tubular airways characteristically cause refractory or recurrent pneumonia in the corresponding distal parenchyma, or they are associated with progressive dyspnea, cough, hemoptysis, and audible expiratory rhonchi over the affected lung field.14–17,23,45–47,50,51 In contrast, SC in the peripheral lung often manifests no symptoms or, alternatively, leads to chest pain caused by invasion of the pleura and extrapulmonary soft tissue.23 As might be expected, central endobronchial tumors are smaller than peripheral SCs; their average sizes are 6 cm and greater than 10 cm, respectively46,52 (Fig. 14-1).
Despite their anaplastic nature, SCs of the lung are surgically resectable in roughly 90% of cases,23,33 and approximately one half of patients with such neoplasms present with stage I disease. Paradoxically, however, the prognosis of pulmonary SC is still dismal. Overall 5-year survival is 20%, with a slightly better figure being associated with small, central endobronchial lesions.23,33,52 Metastatic SC of the lung involves the same organ sites that are affected by more usual forms of lung cancer: namely, the opposite lung, liver, bones, adrenal glands, and brain.46,51 The metastases may exhibit either carcinomatous or sarcoma-like histologic configurations, or both.46 Adjuvant radiation treatment and chemotherapy have been used in many cases of pulmonary SC, but these measures have provided little benefit in general.14,33,38
Macroscopic Features
Grossly, the lesions of pulmonary SC that are greater than 5 cm in size tend to exhibit central necrosis and hemorrhage, and they also demonstrate irregular permeation of the surrounding lung parenchyma33,34 (Fig. 14-2). Tumors that are smaller and located within bronchial confines often exhibit a polypoid appearance and are attached to the subjacent mucosa by a relatively narrow stalk of tissue.52 SCs in the peripheral lung parenchyma may have the gross appearance of conventional adenocarcinomas.34
Histologic Characteristics
Homologous Biphasic Sarcomatoid Carcinoma
Variants of SC that are also called spindle cell carcinomas are constituted microscopically by a predominance of nondescript spindled and pleomorphic cells, admixed with a minor, obviously carcinomatous, component. The latter portion of such lesions is generally inconspicuous and variably distributed; in roughly 40% of cases, such foci are very rare and require extensive sampling to document their presence. The general appearance of the carcinomatous elements is that of a well- to moderately differentiated squamous malignancy in most instances, whereas adenosquamous, adenocarcinomatous, large cell undifferentiated, or neuroendocrine carcinoma is seen in a minority of cases14,53–56 (Fig. 14-3). Rare examples of this tumor type show mixtures of several carcinoma morphotypes.54 Zones of transition between epithelial and sarcoma-like components are usually evident, at least focally.
The sarcomatoid elements of this subtype of SC lack specialized differentiation into identifiable myogenic, chondro-osseous, or vasoformative tissues by standard light microscopy, and, as such, are homologous (organ-appropriate) to the lung. They are composed of markedly heterogeneous cells with nuclear atypia and variable growth patterns. The corresponding microscopic images range from those of fibromatosis-like or low-grade fibrosarcoma-like areas, with relatively bland nuclear features, sparse mitoses, moderate-to-rich matrical collagen deposition, and a “herringbone” pattern, to others in which pleomorphic giant cells are mixed with fusiform elements showing dense cellularity, coarse chromatin, prominent nucleoli, and numerous mitoses14–16,23,33,45 (Fig. 14-4). The last of these descriptions is closely similar to that attending spindle cell–pleomorphic malignant fibrous histiocytoma (MFH) of the soft tissues.54 Neoplastic spindle cells often infiltrate the submucosa of small- and medium-sized bronchi, which nonetheless tend to retain their mural cartilage plates and mucosal integrity.33,34
Sarcoma-like elements in some biphasic spindle cell carcinomas may include cytologically bland, osteoclast-like giant cells52,57,58 (Fig. 14-5). The latter are admixed with atypical fusiform cells or more uniform polygonal tumor cells. Another variant histologic pattern is that in which bluntly fusiform tumor cells surround discrete zones of necrosis, producing a necrotizing granuloma-like image. Rare examples of SC may demonstrate extravasated erythrocytes between relatively bland spindled tumor cells, simulating the characteristics of Kaposi sarcoma (KS) or even nodular fasciitis.33
Heterologous Biphasic Sarcomatoid Carcinoma
Other biphasic sarcomatoid neoplasms differ from the descriptions just given, in regard to their content of focal myogenous, vasogenic, chondro-osseous, or lipogenic differentiation.59 Thus, they are analogous to the heterologous form of malignant mixed müllerian tumors of the uterus, ovaries, and other female genital sites.60,61 Those neoplasms may exhibit microscopic foci that simulate embryonal or adult-type pleomorphic rhabdomyosarcoma, which contain proliferations of closely apposed compact round cells with a slightly myxoid background, or large “strap” cells with cytoplasmic eosinophilia and cross-striations, respectively14–16,33,34,46,52 (Fig. 14-6). Other heterologous SCs contain components that closely imitate the histologic features of osteosarcoma or chondrosarcoma.23,34 In light of this information, it is easy to understand why lesions with such microscopic features were believed to be carcinosarcomas in the past and are still so designated by some observers today. The obviously carcinomatous elements in these lesions usually take the form of squamous carcinoma, but lesions with glandular or neuroendocrine differentiation have also been reported.14,54,55 Transitional zones between obvious epithelial foci, nondescript sarcomatoid areas, and myosarcoma-like components are often evident.
With regard to the relationship between biologic behavior and histologic appearance, there is no difference in the clinical evolution of homologous and heterologous biphasic pulmonary SCs. A distinction is made between those lesions only to reflect their synonymity with sarcomatoid epithelial tumors in other body sites.60,61
Monophasic Sarcomatoid Carcinomas
Some SCs display no conventional light microscopic evidence of epithelial differentiation whatsoever. A carcinomatous nature for these neoplasms is discerned only after immunohistochemical or ultrastructural evaluations have been done, but it is usually suspected beforehand because of the clinical and gross characteristics of the lesions.33
Most tumors in this category are constituted exclusively of cell populations like those in the sarcoma-like components of biphasic SCs. These potentially include foci that have an unremarkable spindle cell or pleomorphic image (Fig. 14-7) as well as areas imitating rhabdomyosarcoma, osteosarcoma, or other morphologic appearances that do not correspond to native tissues in the non-neoplastic lung. Because of the monomorphic nature of the tumor variants under discussion here, which lack any attributes of conventional lung carcinomas histologically, the corresponding diagnosis suggested by the World Health Organization criteria for pulmonary neoplasms62 (based only on hematoxylin and eosin stains and conventional histologic examination) would be that of a primary pulmonary sarcoma. The latter point has made the existence of monophasic SC of the lung somewhat contentious. Nevertheless, we have no doubt of its validity as a reproducible pathologic entity, and other authors appear to concur.35
Indeed, one might go so far as to state that virtually all malignant pulmonary tumors that are exclusively composed of heterologous (organ-inappropriate) elements—such as osteoblastic tissues63,64—are, in reality, monophasic SCs. That statement pertains even if no immunohistochemical evidence of epithelial differentiation can be found, because the ultimate biological evolution of such lesions is identical to that of conventional lung cancers.
Special Variants of Sarcomatoid Carcinoma of the Lung
Pulmonary Blastoma
Since its initial description by Barnett and Barnard65 and a later discussion by Spencer,66 pulmonary blastoma (PB) has been regarded by some observers as the pulmonary counterpart of primitive childhood tumors of other organs.67–73 This view has been fostered in part by confusion of PB with pleuropulmonary blastoma (PPB; discussed subsequently), the latter of which is primarily seen in adolescent patients.74–80 PB is a biphasic neoplasm, containing a mixture of tubular epithelial cell profiles and compact groupings of nondescript bluntly fusiform cells with a blastema-like configuration39,74,77 (Fig. 14-8). These resemble the elements of renal Wilms tumors.81 On the other hand, PPB altogether lacks epithelial differentiation and may instead show divergent mesenchymal differentiation into myogenous or chondro-osseous tissues.77 Moreover, PB shows no particular disease associations, whereas PPB is linked in a familial fashion to a number of other malignant neoplasms and non-neoplastic disorders.79
If one carefully excludes examples of PPB from consideration, it becomes clear that PB is seen overwhelmingly in adults, and its clinical characteristics are superimposable on those of ordinary lung cancers and other pulmonary SCs. This realization allows one to more easily embrace an alternative view of the nature of PB that was advanced in the past by Souza and colleagues,82 Stackhouse and associates,14 and Millard,83 among others. Those authors held the opinion that PB is merely a special, usually peripheral form of pulmonary SC (carcinosarcoma), rather than a blastemal neoplasm that contains truly embryonal tissues. We also espouse the latter premise.
Returning to the particular microscopic attributes of PB, it should be noted that this tumor may demonstrate the same range of epithelial and mesenchymoid differentiation that is seen in other biphasic pulmonary SCs.39,69,84 The epithelial elements in PB resemble fetal pulmonary pseudoglands (a misnomer), composed of stratified columnar cells with glycogen-rich clear cytoplasm and high nuclear-to-cytoplasmic ratios.69,84–86 Luminal mucin may be present in those cellular arrays, and squamous morules are sometimes also evident.69 Interestingly, “occult” neuroendocrine differentiation is a rather common finding in the epithelial components of PB, with potential histochemical argyrophilia and immunoreactivity for neuroendocrine markers.87,88 There is a significant sharing of microscopic features between classic PB and the tumor described as “pulmonary endodermal tumor resembling fetal lung” or alternatively as “well-differentiated adenocarcinoma simulating fetal lung” (Fig. 14-9). It differs in its relative lack of a malignant stromal component and more frequent synthesis of a particular oncofetal polypeptide—α-fetoprotein.39,85,87–90
The elements of PB showing mesenchymoid differentiation are, as stated earlier, usually nondescript morphologically and blastema-like or fibroblast-like. However, examples of this tumor have been documented in which heterologous rhabdomyoblastoid, leiomyosarcomatoid, or apparent chondro-osseous tissues were present.69,84 This observation serves to further solidify the linkage of PB to other sarcomatoid pulmonary carcinomas, as do reports of some tumors in which “typical” PB was admixed with homologous or heterologous biphasic SC, as described earlier.82,91–93
The clinical behavior of PB is difficult to determine with certainty, because of the aforementioned contamination of some series with cases of PPB. However, mortality figures of 30% to 70% have been reported, with death usually being due to distant metastases.67,69,84 Secondary deposits of PB may have a purely epithelial, purely mesenchymal-like, or biphasic appearance, as is true of other SCs.
Pseudoangiosarcomatous (Pseudovascular) Carcinoma
The authors have studied several lung tumors in which obvious squamous cell carcinoma was admixed with areas demonstrating interanastomosing channels mantled by anaplastic, plump, epithelioid cells, focally grouped into pseudopapillae. Because the open spaces in these areas contained erythrocytes and focally formed blood lakes, the histologic appearance was that of biphasic SC in which an angiosarcomatoid component was admixed with overt squamous carcinoma.94 (Fig. 14-10). These neoplasms are believed to represent the pulmonary counterparts of pseudovascular adenoid squamous cell carcinoma, as seen in the skin, breast, thyroid gland, and other organs.34,94–99 This is a tumor type that is known to simulate true angiosarcoma but lacks actual endothelial differentiation. Thus, pseudoangiosarcomatous carcinoma (PASC) would be an apt synonym.96
Primary pulmonary angiosarcoma is, comparatively, a very rare lesion, comprising only 10% of true sarcomas of the lung in one report from the Mayo Clinic.2 Mainly anecdotal reports of this tumor exist in the remaining literature, and not all of them satisfy rigorous diagnostic criteria.100–108 Metastases to the lung from angiosarcomas arising in extrapulmonary sites are much more common, including examples that have originated in the heart, great vessels, or extrathoracic viscera.100
Similar to reports on previously cited pseudovascular carcinomas in other body sites, two publications have specifically considered squamous cell carcinomas of the lung that imitated angiosarcomas. The first, by Banerjee and coworkers,96 showed that such tumors produced clinical symptoms and signs resembling those of ordinary types of lung cancer. They presented in the fifth to seventh decades of life; were associated with cigarette smoking, complaints of cough, weight loss, and dyspnea; and were visible on chest radiographs as well-defined central or peripheral parenchymal masses. In another series by Nappi and colleagues,94 the tumors were essentially identical microscopically to pseudovascular squamous carcinomas of the breast and skin. Important differences between PASCs and true pleuropulmonary angiosarcomas include an absence of atypical endothelial cells in stromal blood vessels surrounding the tumor mass in PASCs; less infiltrative growth through the interstitium of the lung; and, perhaps most importantly, the presence of small foci of morphologically obvious squamous cell carcinoma in most PASCs.94
Inflammatory Sarcomatoid Carcinoma
Much attention has been given to a group of space-occupying lesions in the lung that carry the popular but inaccurate designation of inflammatory pseudotumors (IPs).101–114 These proliferations may occur in children or adults and have been divided into fibrohistiocytic, plasma cell granulomatous, and focal organizing pneumonia types, based on their individual clinicopathological features. The nomenclature used for this group of lesions has been well summarized by Koss114 and Matsubara and associates.106 There is still some controversy over whether all such lesions are neoplastic, or whether some might be reactive in nature.115 However, the bulk of available information indicates that IPs are relatively innocuous clinical imitators of overtly malignant neoplasms.114 Occasional cases have been linked causally to specific infectious organisms,116,117 but the etiologic factors associated with most pulmonary IPs are uncertain.
In contrast, primary SC is generally regarded as a neoplasm that may simulate pleuropulmonary mesenchymal malignancies, and it is typically not mentioned in discussions on the pathologic differential diagnosis of IPs. This is so because SC usually demonstrates obvious cytologic anaplasia and lacks a significant component of inflammatory cells. Indeed, the morphologic distinction between IPs and all bronchogenic carcinomas (including SC) has been portrayed by some authors as an uncomplicated process.105,108 However, we have observed several examples of pulmonary SC that exhibited surprisingly bland morphologic appearances, and which, as a result, were separable from IP only by thorough study and adjunctive pathologic techniques. These have been designated as examples of inflammatory sarcomatoid carcinoma (ISC).118,119
Examples of ISC are composed of variably densely apposed spindle cells with only modest pleomorphism, arranged haphazardly or in fascicular and storiform patterns. The stroma is at least partially myxoid in some cases and may be prominently so. The tumors demonstrate an irregular, spiculated interface with the surrounding lung. The adjacent parenchyma exhibits interstitial fibrosis, and small nodular infiltrates of mature lymphocytes are admixed with dense collagenous tissue at the periphery of ISCs (Fig. 14-11). Focally hyalinized, keloidal-type collagen is admixed with the tumor cells in the central portions of some of these tumors, with or without small foci of central necrosis. Vascular invasion and luminal obliteration by neoplastic cells may be apparent. Similarly, bronchial submucosal infiltration is another potential observation. ISCs do not contain appreciable stromal neutrophils, eosinophils, or xanthoma cells, but a moderate number of lymphocytes and plasma cells can be seen.
Cytologically, the nuclei of the tumor cells in ISCs are relatively uniform in size and spindle-shaped, with coarse chromatin and occasional small nucleoli (Fig. 14-12). Cytoplasm is moderate in amount and amphophilic. Generally, there are no more than 2 mitoses per 10 high-power fields (×400) on average, and pathologic division figures are absent. Thorough sampling of the tumor tissue in cases of ISC typically demonstrates minute foci of cohesive epithelioid cells, suggesting the diagnosis of squamous carcinoma on conventional histologic grounds. However, some ISCs lack such foci and are recognizable as carcinomas only with special pathologic evaluations (see Fig. 14-12B).118
Pleurotropic (Pseudomesotheliomatous) Sarcomatoid Carcinoma
Occasional examples of SC are distinctive not because of their histologic attributes but because of their macroscopic appearances. In particular, a small subset of these neoplasms arises in the very periphery of the pulmonary parenchyma and grows preferentially into the pleura that encases the lungs.120,121 This produces clinical symptoms and signs that are indistinguishable from those of malignant mesothelioma; hence, the names pleurotropic or pseudomesotheliomatous carcinoma.121,122 Moreover, the microscopic features of pleurotropic SC are basically superimposable with those of biphasic or sarcomatoid mesotheliomas.
Results of Adjunctive Pathologic Studies
Accounts of the electron microscopic and immunohistologic characteristics of pulmonary SC have not been altogether uniform. Some authors have preferred the view that such data in fact confirm the existence of true carcinosarcomas,123–125 whereas others have thought that this information instead supports the concept of a pathologic continuum that is predicated on carcinoma in pure form.15,17,38,126–130 We strongly prefer the second of these opinions.
It is true that the sarcoma-like elements in SC of the airways do not uniformly exhibit the ultrastructural presence of intercellular junctions and tonofibrils, or immunoreactivity for keratin or epithelial membrane antigen (EMA) in fusiform and pleomorphic tumor cells. In fact, these generic markers of epithelial differentiation may be seen only extremely focally in such neoplastic components, and we have even seen isolated examples in which cell membrane–based EMA reactivity was obvious, but keratin positivity was altogether absent. Humphrey and coworkers observed cytoplasmic tonofibrils or keratin positivity in the sarcomatoid elements of only three of eight pulmonary SCs.15 However, it should be noted that the latter study was performed with a single heteroantiserum to high-molecular-weight keratin, representing a relatively insensitive means of immunodetection. In our previously published experience with SCs of the respiratory tract,33 81% were ultrastructurally or immunohistochemically proven (with a mixture of monoclonal antikeratin antibodies [AE1/AE3/CAM5.2/MAK-6]) to be wholly epithelial in nature, and other authors have recorded similar findings at immunohistochemical and genetic levels of investigation.126–132
The fact that features of epithelial differentiation are present at all in the sarcomatoid elements of these neoplasms strongly supports the premise that respiratory tract SC is a basically carcinomatous lesion “in transition.”133 This concept has been well-accepted in reference to dedifferentiated sarcomas of the soft tissue, in which clonal evolution is thought to account for a change in the morphology as well as the immunophenotype of the progenitor lesion.134 Lessons learned in the latter sphere—as well as molecular biologic assessments of clonality in SCs135—have direct corollaries in the context under discussion here. We have observed the coexpression of vimentin (a “primordial” intermediate filament) in all examples of keratin-positive SC of the airways, and a minority of these lesions are additionally labeled for desmin (the intermediate filament of myogenous cells) and muscle-specific actin in the same cells that contain the other two filament proteins (Fig. 14-13).33 Collagen type IV is also seen surrounding individual tumor cells in most instances. Markers of neuroendocrine differentiation, such as chromogranin-A, CD57, and synaptophysin, likewise may be seen in selected lesions in their overtly epithelial components,53,55 and S100 protein is apparent in foci resembling chondroid tissue by conventional microscopy.4 In contrast, Friend leukemia-virus integration protein-1 (FLI-1) and CD31 are typically absent in PASC, whereas one or both of those endothelial determinants would be expected in true pleuropulmonary angiosarcomas.95,136
These accrued observations coincide with ultrastructural findings reported by Battifora in two cases of SC, which demonstrated the coincidence of desmosomes, tonofibrils, and collagen production in the same neoplastic cells, implying the presence of multilinear differentiation.137 Thus, SC can be viewed basically as an epithelial neoplasm with divergent mesenchymal differentiation, in which carcinoma cells acquire the potential to express a mesenchymal phenotype at light microscopic, ultrastructural, and immunohistologic levels. The pathogenetic bases for this peculiarity are currently unknown, but the practical deduction to be gleaned from this construct is that all SCs of the respiratory tract should be treated clinically as poorly differentiated carcinomas.
Wakely has reviewed the characteristics of pulmonary spindle cell tumors as seen in fine needle aspiration biopsy specimens138 (see Fig. 14-7). He concluded that adjunctive pathologic studies, such as those discussed above, were virtually mandatory before definitive diagnoses could be reached in that context.
Differential Diagnosis of Sarcomatoid Carcinoma
The differential diagnosis of SCs of the airways principally centers on the exclusion of true sarcomas, which are discussed later in this chapter. As a particular word of caution, it should be noted that synovial sarcoma (SS) and sarcomatoid mesothelioma may be very closely similar to SC as seen with the electron microscope or in immunophenotypic evaluations. The marked propensity for SS to affect children, adolescents, and young adults, its typical t(X;18) (p11.2;q11.2) cytogenetic aberration,139,140 and nuclear labeling for transducin-like enhancer [of Split]-1 (TLE1) protein141–144 (not seen in carcinomas) are crucial points in its distinction from SC of the upper airways. Of course, nuances of histologic appearances and radiographic characteristics are also valuable in this specific differential diagnostic setting. Similarly, roentgenologic findings are more helpful than morphologic observations in making the distinction between SC and spindle cell or biphasic mesothelioma. The ultrastructural profiles of the latter two lesions are again very similar,1,4,145 and, aside from selective reactivity for calretinin and podoplanin146 in mesotheliomas, the same comment applies to their immunophenotypes.
True Primary Sarcomas of the Lung
Kaposi Sarcoma
The natural history of KS is a sad testimony to the global impact of acquired immunodeficiency syndrome (AIDS). Before the 1980s, KS was a relatively rare neoplasm outside of Africa and the Mediterranean basin. Moreover, with relatively uncommon exceptions, this lesion was a cutaneous proliferation that uncommonly involved the viscera.147 However, today, with particular regard to the intrathoracic organs, KS is—in most large metropolitan areas of the world—the most common of all pulmonary sarcomas.148 Whereas initial presentation of this tumor in the bronchopulmonary tract was an almost-unknown phenomenon prior to the advent of AIDS, it is currently a well-recognized variation of the latter disease.149
Clinical Summary
In the context just mentioned, many patients with KS of the lung are homosexual men,150–156 but they also include other high-risk groups for AIDS such as intravenous drug abusers. Most individuals with KS generally have other symptoms and signs of AIDS, such as weight loss, fever, night sweats, fatigue, lymphadenopathy, and opportunistic infections. However, fever may be directly caused by KS in the lungs. There has been a case report of an AIDS patient with persistent pyrexia for which no source of infection was found but that finally resolved after radiation therapy.157 Cutaneous KS is usually detected early in its clinical evolution, but identical tumors of the bronchial mucosa and lung parenchyma typically have grown to a volume sufficient to produce symptoms and therefore are relatively advanced at the time of diagnosis.158 Presenting complaints specific to the neoplasm include dyspnea, stridor (when endobronchial lesions are present), cough, and hemoptysis, which may be massive.149
On bronchoscopic examination, nodular or flat bluish-red discolorations in the mucosa are seen, some of which may be actively bleeding. This bronchoscopic appearance is usually considered diagnostic, and endobronchial lesions are not generally biopsied. The diagnostic yield of transbronchial biopsies is usually low, and unless they are deep enough, KS of the lung will be missed because the mucosa itself is uninvolved.159 Open lung biopsies are more productive, but they are not absolutely sensitive.
Radiographic findings on chest x-rays may be nonspecific, showing only ill-defined interstitial infiltrates (Fig. 14-14). An alveolar filling pattern is usually evident only if the patient has suffered hemoptysis and aspirated blood, but pleural effusions or pneumothorax may be seen in cases where the lesion involves the serosal surfaces as well as the lung parenchyma.160,161 Mediastinal adenopathy is not common, but, if it is present, this can be very helpful in separating KS from Pneumocystis jiroveci infection, because the latter does not cause adenopathy. Computed tomography (CT) scans and magnetic resonance imaging (MRI) generally provide no more information than chest radiography. In summary, the presence of bilateral pleural effusions and bilateral interstitial infiltrates with ill-defined nodularity is suggestive of pulmonary KS, especially in a patient with known tumor elsewhere.150,151
Pathologic Findings
As alluded to above, it is distinctly uncommon for the pathologist to be able to make a definitive diagnosis of KS of the lung on a transbronchial biopsy specimen. Usually, a wedge biopsy is necessary, as obtained via video-guided thoracoscopy or a limited thoracotomy.159 On gross examination, this type of specimen exhibits numerous hemangiomatoid or ecchymosis-like zones of bluish-red discoloration in the parenchyma, with ill-defined borders.
In the lung, KS shows a tendency to grow along pre-existing fibrous intrapulmonary septa, and it also concentrates around small tubular airways and blood vessels (Fig. 14-15). The tumor comprises a mixture of ectatic, thin-walled blood vessels that “dissect” or push through the pulmonary interstitial collagen, together with haphazardly arranged fascicles of spindle cells that show only modest nuclear atypia and may contain cytoplasmic vacuoles.149,159 Extravasated erythrocytes and hemosiderin pigment are common in and around the tumor masses (Fig. 14-16). Pleural KS “layers” itself over the submesothelial mantle of connective tissue, effacing the mesothelium itself in doing so.
The differential diagnosis of KS from vascular granulation tissue and other spindle cell proliferations in the lung is greatly enhanced by immunohistochemical analyses. KS is reactive for latent nuclear antigen-1 of human herpesvirus-8 in approximately 85% of cases162 (Fig. 14-17). It also labels for FLI-1163 and podoplanin.164
Therapy and Prognosis
Regardless of its occurrence in AIDS or in non–human immunodeficiency virus (HIV)-related cases, the presence of KS in the lung is prognostically ominous. Virtually all patients with visceral disease die within 2 years, from infection if not from KS itself.148,154,156 Because of the multiplicity of KS, surgical resection is not a realistic option in the management of patients with this neoplasm. Chemotherapy is considered the treatment of choice, with a relatively good response rate and relatively rapid improvement within 2 to 4 weeks.25 Chemotherapy regimens in the few published therapeutic trials designed specifically for pulmonary KS have primarily included adriamycin, bleomycin, and vincristine.149,165–167 Gill and colleagues found an 85% response with combination chemotherapy in a group of 13 patients.166 Patients who benefited from this treatment included those who achieved at least partial responses. Complete response is defined by the following three criteria:
A partial response is characterized by the same three points, except that the degree of resolution is not total.165,166
Despite fairly good results with combination chemotherapy, patients with pulmonary KS do not show long survival. In the trial reported by Gill and colleagues, the median survival for responders was slightly but significantly longer than that of nonresponders (10 versus 6 months, respectively). However, considerable overlap between the two groups was present.166
Other more experimental (and inconclusive) approaches have included the administration of zidovudine, interferon, and other antiviral compounds.147,153,154 Radiotherapy may provide palliation of symptoms but is noncurative. The most important piece of data in prognosticating cases of KS of the lung is the serologic HIV status of the patient, inasmuch as AIDS is currently a uniformly lethal, albeit chronic, illness.
Fibrosarcoma
Primary fibrosarcoma of the lung (FSL), like its soft tissue counterpart, is defined as a fibroblastic spindle cell neoplasm without any evidence of specialized cellular differentiation. Although it has been cited in the past—along with leiomyosarcoma—as the most common primary pulmonary sarcoma,168 FSL was, and probably still is, overdiagnosed.169 Two separate studies from the Mayo Clinic cited two different time-dependent incidence figures for FSL. From 1950 to 1978, it constituted 50% of all primary pulmonary sarcomas170 but only 20% in the decade 1980 to 1990.171 As considered previously, it is our belief that the great majority of pulmonary “fibrosarcomas” are actually SCs. Only those tumors that have been subjected to rigorous and specialized pathologic examination should be accepted as bona fide examples of this rare sarcoma variant.
Clinical Summary
Guccion and Rosen studied 13 cases of FSL, which were divided into endobronchial and intrapulmonary types.168 This classification scheme was said to have clinical and prognostic importance. In conjunction with a review of 48 reported cases in the literature, the authors just cited found that the majority of endobronchial FSLs occurred in children and young adults, whereas parenchymal tumors predominated in middle-aged and elderly patients. In contrast, Pettinato and associates reported three parenchymal tumors in two newborns and a 6-month old infant.172 There was roughly an equal distribution by gender among endobronchial lesions; however, most intraparenchymal neoplasms occurred in men. All cases of endobronchial FSL in a series reported from the Armed Forces Institute of Pathology (AFIP) had symptoms of cough, hemoptysis, or chest pain; some of the parenchymal cases did as well.168 Thoracic imaging studies of FSL usually show discrete, homogenous masses. However, one reported pulmonary fibrosarcoma simulated a bronchogenic cyst clinically and radiographically.173 Gladish and coworkers13 have observed that fibrosarcoma is much more likely to arise in the soft tissue of the chest wall and secondarily involve the lung than it is to show the converse of that relationship.
Pathologic Findings
FSL is histologically identical to its soft tissue counterparts and characteristically shows sheets and intertwining fascicles of spindle-shaped cells with a typical interdigitating growth pattern and discernible stromal collagenogenesis (Fig. 14-18). The tumor cells contain oval to elongated, hyperchromatic nuclei, and scant amphophilic cytoplasm with ill-defined cellular borders. In addition, some areas may have a slightly epithelioid appearance, in which the tumor cells are more ovoid than spindled, and others may show significant pleomorphism that merges with the image of MFH (Fig. 14-19). Mitotic activity is variable.
Electron microscopic and immunohistochemical studies are required to confirm the fibroblastic nature of these neoplasms. The tumor cells in FSL are characterized by abundant rough endoplasmic reticulum and free ribosomes, as well as the production of extracellular collagen fibers that may be aligned at right angles to the tumor cell membranes. There should be no detectable myofilaments, pericellular basal lamina, or intercellular junctions in lesions thought to represent FSL. Because there are no specific immunologic markers for fibroblasts, the diagnosis of fibrosarcoma is one of ultimate immunohistologic exclusion. Tumor cells in FSL generally stain only for vimentin, a primitive intermediate filament protein, and they lack all epithelial, myogenous, neural, and endothelial markers.172,174,175
Therapy and Prognosis
Although resection is the treatment of choice, many surgically treated fibrosarcomas of the lungs do recur, and survival after this event is short, with patient fatality usually occurring within 2 years.168 Three cases seen at the Mayo Clinic between 1980 and 1990 occurred in young women whose lesions all recurred within 15 months following surgical excision.171 In contrast, primary bronchopulmonary fibrosarcomas in children appear to have a relatively favorable prognosis and behave only as low-grade malignancies.172,173 The five patients with pediatric FSL reported by Pettinato and colleagues all had complete surgical removal of their tumors, and four were disease-free after 4 to 9 years. The fifth case in that series had insignificant follow-up.172 The efficacy of adjunctive chemotherapy and irradiation has not yet been proven.
Primary Pulmonary Hyalinizing Spindle Cell Tumor with Giant Rosettes
A rare lesion that is probably biologically related to FSL is one called hyalinizing spindle cell tumor with giant rosettes (HSCT). This entity was originally documented as a low-grade sarcoma in the deep soft tissues of adults,176 but at least two cases have been reported as primary pulmonary examples.177,178 HSCT has a distinctive morphologic appearance, featuring the multifocal presence of large rosette-like structures amid a bland spindle cell proliferation (Fig. 14-20). The lesion has infiltrative borders, with no necrosis and only limited mitotic activity.
HSCT has a partial kinship with Evans tumor (low-grade fibromyxoid sarcoma)179 of soft tissue, on behavioral, morphologic, and cytogenetic grounds. Both of those tumors manifest a t(7;16) (q33;p11) chromosomal translocation, producing fusion of the FUS and CREB3L2 genes.178 Unlike FSL, HSCT demonstrates some immunophenotypic variability, often labeling for alpha-isoform actin in its spindle cell population. The cells in the giant rosettes may manifest immunoreactivity for S100 protein and CD45RO,176 the latter of which is more typically a hematopoietic determinant. The precise biologic potential of HSCT in the lung is uncertain, because of the anecdotal nature of reported cases.
Primary Pulmonary Leiomyosarcoma
The most common anatomic locations for leiomyosarcomas in general are the uterus, gastrointestinal tract, and soft tissue, in order of relative frequency. The extremely uncommon primary pulmonary leiomyosarcoma (PPLMS) presumably originates from bronchial or pulmonary vascular smooth muscle. Only three cases of leiomyosarcoma were found among roughly 10,000 primary malignancies of the lung at one large American medical center between 1980 and 1990.171 Because secondary pulmonary involvement by malignant smooth muscle tumors is a relatively frequent event, the diagnosis of PPLMS absolutely requires exclusion of an occult extrathoracic neoplasm presenting with a single “herald” metastasis to the lung.
Clinical Summary
A series of 19 PPLMSs seen at the AFIP was divided into those neoplasms that were predominantly endobronchial and others that were intraparenchymal, in analogy to pulmonary fibrosarcomas.168 The majority of these tumors in children are endobronchial in nature,180,181 whereas those in adults are not. In contrast to leiomyosarcomas of the soft tissue, which occur most commonly in women, patients in the aforementioned report from the AFIP were almost exclusively men. However, another survey that reviewed 92 cases of PPLMS in the literature found a male-to-female ratio of 2.5, suggesting that the paramilitary study just cited was biased demographically by its affiliation with the armed services.182 In contrast to carcinoma of the lung, leiomyosarcoma is not associated with cigarette smoking or other potential inhalant carcinogens. Most patients with PPLMS (particularly its endobronchial form) are symptomatic, often complaining of cough, hemoptysis, or chest pain. However, intraparenchymal lesions may be discovered incidentally on chest radiographs. Roentgenographically, PPLMS usually takes the form of a discrete mass (Fig. 14-21), sometimes with cavitation or cyst formation that is best seen by CT of the thorax.168,183,184
Pathologic Findings
Parenchymal tumors range from 3 to 15 cm in maximum dimension. They are well circumscribed, white to yellowish tan, and variably firm. Cut surfaces of these neoplasms commonly show hemorrhagic and necrotic areas. Endobronchial tumors are often smaller than the intraparenchymal lesions, presumably because of confinement by the bronchial walls.185
Microscopy discloses histologic features that mirror those of leiomyosarcomas elsewhere in the body. On low-power magnification, there are interlacing fascicles of spindled cells arranged haphazardly, yielding a “whorled” appearance (Fig. 14-22). The neoplastic cells have cigar-shaped nuclei with blunt ends, a moderate amount of cytoplasm, and indistinct cell borders (Fig. 14-23). Fascicles cut in cross section demonstrate characteristic intracellular perinuclear lucencies.185–188 Prominent myxoid stromal change may be observed.189
The differential diagnosis of PPLMS includes fibrosarcoma and malignant peripheral nerve sheath tumor, as well as SC. Electron microscopy and immunohistochemistry are again helpful in confirming the smooth muscle nature of a spindle cell neoplasm.187 Ultrastructural features of leiomyogenous differentiation include cytoplasmic dense bodies punctuating skeins of thin filaments, subplasmalemmal dense plaques, plasmalemmal pinocytotic vesicles, and pericellular basal lamina (Fig. 14-24). Immunoreactivity for desmin, muscle-specific actin, calponin, caldesmon, or smooth muscle actin is also characteristic of the tumor cells in PPLMS.190
Transthoracic fine needle aspiration of possible PPLMS can be attempted if the lesion is large and peripherally located. The cytologic preparations from that procedure typically show a dyshesive population of relatively monotonous spindle cells, with blunt-ended fusiform nuclei. Mitotic figures and nuclear pleomorphism are variably seen; some lesions also may exhibit a more epithelioid cytologic image191–193 (Fig. 14-25).
Therapy and Prognosis
The natural history of PPLMS and its responses to various therapeutic regimens are difficult to predict because of the rarity of this neoplasm. However, there is generally a consensus that surgical resection is the treatment of choice182,185–187 and that it produces a survival rate of 45% to 50% at 5 years. Survival for as long as 15 to 30 years has been documented in PPLMS cases.168,170 However, pulmonary leiomyosarcomas appear to be relatively resistant to irradiation and chemotherapy.182,188 Various prognostic variables have been discussed in connection with these lesions.168,170 Endobronchial tumors are thought to be less aggressive than parenchymal neoplasms, largely because the former tend to be smaller and are diagnosed earlier. It follows, therefore, that tumor size is an important indicator of biologic behavior for all pulmonary leiomyosarcomas. The scope of mitotic activity may affect prognosis as well. In an AFIP series on PPLMS, a mitotic rate of 8 or less per 10 high-power fields was associated with infrequent metastasis and a generally favorable clinical outcome.168
Epithelioid Hemangioendothelioma
In 1975, Dail and Liebow reported the first cases of an unusual pulmonary neoplasm that they termed intravascular bronchioloalveolar tumor (IVBAT). This name reflected their original hypothesis that the lesion in question was an epithelial tumor—specifically, a bronchioloalveolar carcinoma variant showing prominent vascular invasion.194,195 Four years thereafter, Corrin and associates alternatively proposed an endothelial origin for this tumor based on the results of ultrastructural studies.196 Subsequent evaluations by other authors have confirmed the vascular histogenesis of the IVBAT. Indeed, in 1982, Weiss and Enzinger described a series of soft tissue tumors that were histologically identical to IVBAT, and these authors were the first to use the term epithelioid hemangioendothelioma (EH) to emphasize their distinctively epithelioid (or histiocytoid) cytologic features.197 In addition to the lungs and soft tissues, EH also primarily occurs in the bone and liver.198
Clinical Summary
EH of the lung is a neoplasm that arises predominantly in female patients; women account for roughly 80% of all cases.195,199,200 It primarily occurs in young adults, with approximately 50% of affected individuals being younger than 40 years of age; only 10% are older than 50 years of age at diagnosis.198,201 Many affected persons are asymptomatic, and their tumors are detected incidentally on chest radiographs. Patients who have tumor-related complaints usually present with pleuritic pain, dyspnea, and cough. Case reports have also documented alveolar hemorrhage as a presenting sign of pulmonary EH,202,203 and it may simulate thromboembolic disease symptomatically as well.204 Chest radiographs commonly show numerous, small nodular lesions throughout both lung fields (Fig. 14-26). Therefore, EH enters the roentgenographic differential diagnosis of multiple pulmonary nodules in asymptomatic young women, together with metastatic germ cell tumors, chondroid pulmonary hamartomas, multiple arteriovenous malformations of the lung, deposits of “benign metastasizing leiomyoma,” and malignant lymphoma.205
Pathologic Findings
The pathologic diagnosis of EH is almost always made by open lung biopsy, inasmuch as transbronchial biopsy is usually ineffectual because of sampling constraints. Most nodules of EH are discrete and usually measure less than 2 cm. They are grayish-white to tan and have a chondroid macroscopic consistency. More nodules are typically seen on histologic examination than are apparent grossly. Microscopically, EH is typified by multiple oval or round nodules with hypocellular, sclerotic, or necrotic centers195,199 (Fig. 14-27). These are surrounded by rims of viable, more cellular tissue that is associated with a myxohyaline fibrous stroma; exceptionally, metaplastic bone formation may be apparent.206 The neoplastic cell population is composed of plump, epithelioid cells, which are the histologic hallmark of EH (Fig. 14-28). They have centrally located, round-to-oval nuclei, with ample amounts of eosinophilic cytoplasm. Often, intracytoplasmic vacuoles are evident, which should raise the possibility of endothelial differentiation on light microscopy. Saqi and colleagues have described “rhabdoid” cellular differentiation in EH as well.207
Ultrastructural and immunohistochemical evaluation can be of great assistance in confirming the endothelial origin of EH.196,198,208–211 Briefly, ultrastructural features of this tumor include cytoplasmic vacuoles, Weibel-Palade bodies (Fig. 14-29), cell membranous pinocytotic vesicles, and pericellular basal lamina.210 Histochemical and immunologic markers of endothelial differentiation—such as Ulex europaeus I lectin, anti-CD31, anti-FLI-1, and anti-CD34—are helpful in labeling the neoplastic cells in virtually all examples of EH.206,212 Weinreb and coworkers have also described labeling for CD10 in a majority of EH cases.213
Fine needle aspiration biopsy of EH yields a loosely cohesive population of epithelioid cells that may be binucleated or multinucleated. Variably sized cytoplasmic vacuoles are demonstrable in a proportion of the tumor cells (Fig. 14-30).214
Therapy and Prognosis
Surgery is usually not feasible as effective treatment for EH because of its tendency to show intrapulmonary multicentricity. Unfortunately, irradiation and chemotherapy likewise have been of little benefit.195,199 Nevertheless, EH is generally an indolent neoplasm that is classified as a borderline malignancy. It is associated with a protracted clinical course and potential survival of several years after diagnosis.208 Most patients do eventually succumb to the tumor and die of respiratory failure secondary to progressive parenchymal replacement; in one series reported by Einsfelder and Kuhnen, 36% of patients were dead or likely to die of their tumors after 52 months’ follow-up.206 Adverse prognostic factors that predict a more rapid decline in pulmonary function include prominent symptoms at the time of presentation; radiographic demonstration of extensive intravascular, endobronchial, or pleural spread of the tumor;195 and the presence of fusiform tumor cells.200
A particularly vexing clinical problem is represented by those patients who have EH synchronously in several organs, including the lungs. In such cases, one is never certain whether tumor multifocality or metastasis is operative. Pragmatically, each involved organ is usually treated as if it harbors an independent primary tumor in this scenario.215
The general predilection of EH for women, a reported association of primary hepatic EH with oral contraceptives, and the lack of effective therapy for this tumor have prompted some investigators to explore the possibility of treatment involving hormonal modulation. These tumors have been examined for possible expression of estrogen and progesterone receptor proteins as well as other estradiol-binding moieties. Ohori and colleagues analyzed five cases of pulmonary EH for steroid hormone receptors by immunohistochemical methods, using paraffin-embedded material.216 Only one case showed apparent binding of estradiol. Our own unpublished experience with the immunohistologic characteristics of pulmonary EH has disclosed no reactivity with monoclonal antibodies against estrogen and progesterone receptor proteins. Thus, we believe that hormonal therapies are unlikely to produce significant results in this setting.
Hemangiopericytoma and Intrapulmonary Solitary Fibrous Tumor
As first described in 1942 by Stout and Murray,217 hemangiopericytoma (HPC) is an uncommon, potentially malignant neoplasm that shows apparent differentiation toward the phenotype of pericytes. These are cells with long cytoplasmic processes that surround capillaries and serve a vasoregulatory function. HPC occurs most commonly in the deep muscles of the thigh, the pelvic fossa, and the retroperitoneum. However, 5% to 10% of all hemangiopericytomas are said to present as primary pulmonary tumors.185,218 It must be remembered that the lungs and the bones are the anatomic sites that most frequently harbor metastases of HPC,219 and therefore a primary extrapulmonary tumor must be excluded before a diagnosis of a primary HPC can be rendered safely.
It should also be understood that the currently recommended classification scheme for mesenchymal neoplasms has merged HPC with solitary fibrous tumor (SFT).220,221 Hence, for all intents and purposes, the two entities are considered to be closely related if not identical, and the abbreviation of HPC-SFT will be used in reference to that tumor group.
Clinical Summary
Pulmonary HPC-SFT affects men and women equally, and most commonly arises in middle adulthood. The peak incidence of this lesion is in the fifth decade of life, although individuals as young as 4 years of age and as old as 73 years of age have been reported.222,223 Some tumors are detected incidentally on radiographic studies without causing pulmonary symptoms; 6 of 18 cases in one series fit this scenario.224 Alternatively, presenting symptoms may include hemoptysis, chest pain, cough, and dyspnea, and, more rarely, pulmonary osteoarthropathy.224
Various radiographic imaging studies have been used in studying this neoplasm. Although angiography has usually not been performed, vascular contrast studies of HPC-SFT generally show a characteristic intralesional “blush.”225 No other pathognomonic features are evident in chest x-rays, CT scans, and MRIs of the lung.223,226 Plain film x-rays typically show a discrete, homogeneously dense mass with lobulated contours. On CT images, however, HPC-SFT is heterogeneous. Central low-density areas are evident that correspond to necrotic foci, and an apparent capsule may be seen at the interface with surrounding lung parenchyma. MRIs also show intratumoral heterogeneity with respect to tissue density and are apparently more sensitive in depicting intralesional hemorrhage. These images were found to be the most useful in delineating the potential plane of surgical separation between an HPC-SFT and surrounding soft tissue in one report.227 In summary, a radiologic diagnosis of pulmonary HPC-SFT may be suspected in a middle-aged person lacking pulmonary symptoms, but whose radiographic imaging studies reveal a large, lobulated, sharply marginated, variably dense mass (Fig. 14-31) that does not cause compression atelectasis.226
Pathologic Findings
HPC-SFTs of the lung can attain large sizes, and lesions measuring up to 18 cm have been documented. The typical gross appearance of this tumor is that of a well-circumscribed, yellow to tan-brown mass with a pseudocapsule, as well as areas of internal necrosis and hemorrhage. Histologic sections typically show a relatively monomorphous cellular proliferation surrounding thin-walled, anastomosing vascular channels lined by a single endothelial layer. These blood vessels often (but not always) assume gaping, “staghorn,” or “antler-like” configurations (Figs. 14-32 and 14-33). The population of neoplastic cells is uniform, with oval compact nuclei and ill-defined cytoplasm.185 Mitotic activity and areas of necrosis and hemorrhage are noted frequently. Vascular invasion of large pulmonary vessels, however, is uncommon. With regard to the latter features, some pathologists have, in the past, rendered a diagnosis of benign hemangiopericytoma if necrosis, hemorrhage, and mitoses were absent. In our view, this approach is highly inadvisable. We have seen cases of pulmonary HPC-SFT with exceedingly bland histologic profiles in which metastasis nonetheless supervened. Accordingly, it is advised that each report on this tumor should carry the statement that HPC-SFT is at least potentially malignant behaviorally.219,224
Pulmonary HPC-SFT has sometimes been overdiagnosed because other neoplasms may show foci that resemble the former lesion. In this regard, it is notable that in their seminal report, Stout and Murray admonished others to make the diagnosis of hemangiopericytoma by ultimate exclusion.217 The histologic differential diagnosis includes SC, SS, fibrous histiocytomas, leiomyosarcoma, and mesenchymal chondrosarcoma.219 Distinctions among these neoplasms are best made by ancillary studies.
Electron microscopy can confirm the pericytic nature of HPC through the demonstration of polygonal cells with cytoplasmic processes, pinocytotic vesicles, basal lamina, and a paucity of other organelles.174,219 HPC-SFT is a neoplasm that demonstrates a relatively restricted group of immunoreactants; these include vimentin, collagen type IV, CD34, CD99, CD57, and bcl-2 protein54,228 (Fig. 14-34). Endothelial stains such as Ulex europaeus I, CD31, and FLI-1 highlight the lining of intralesional vascular spaces but do not label the surrounding tumor cells. Silver impregnation techniques highlight a complex reticulin matrix with individual cell investment.
Therapy and Prognosis
As in the management of soft tissue HPC-SFT, complete surgical excision is the mainstay of therapy for primary pulmonary tumors of this type. However, it is known that intraoperative rupture of pulmonary HPC-SFT may occur (especially those tumors that are adherent to the chest wall). As expected, this complication results in early local recurrence, as reported by Van Damme and associates, and should therefore be avoided at all costs.223 Chemotherapy and irradiation have not been shown to be consistently effective adjuvant modalities, but they may have some role to play in management.229,230 A study by Jha and coworkers on the general role of radiation therapy in treating HPC-SFT showed that postoperative irradiation was useful for local tumor control, salvage therapy after local recurrence, and palliation.231 Tumors less than 5 cm in maximum dimension exhibit a better response than those that are larger than 10 cm.232
As mentioned above, hemangiopericytomas in general are well known for their unpredictable biologic behavior. Postoperative survival has ranged from 10 weeks to 18 years.223,232 Even with apparently complete surgical resection, HPC-SFT recurs locally within 2 years in approximately 50% of cases,224,227,233 and later recurrences are seen as well; distant metastasis, however, is uncommon.228
Clinical and histologic features that have been cited as prognostically useful219,222,227 include the presence of symptoms at presentation, mitotic activity of four or more mitoses per 10 high-power microscopic fields, spontaneous tumor necrosis, vascular invasion, and tumor size greater than 5 cm. In one series, metastases were seen in one third of tumors that measured greater than 5 cm, and in two thirds of those greater than 10 cm.222 However, Yousem and Hochholzer did not find any single histologic or clinical feature that was statistically significant in reliably predicting the clinical course of primary pulmonary HPC-SFT.224
Malignant Fibrous Histiocytoma
MFH (now commonly called pleomorphic sarcoma, not further specified) is a common, extensively studied soft tissue sarcoma of older adults that develops most frequently in the extremities and the retroperitoneum. In a series of 200 cases by Weiss and Enzinger,234 the lungs were the most common site of metastases. Thus, exclusion of an occult soft tissue tumor is once again necessary before a diagnosis of primary pulmonary malignant fibrous histiocytoma (PPMFH) can be made. A review of Mayo Clinic cases found only four examples among 10,134 tumors arising in the lung.171 Currently, there are fewer than 75 reported cases of PPMFH in the English literature.234–245
Clinical Summary
In general, MFH is a neoplasm of patients who are in late middle age, with a median of 54 years. However, its occasional occurrence in children and young adults has also been reported.235,244 No consistent predilection for either gender is seen. Previous irradiation is a pathogenetic risk factor for tumors arising in soft tissue, and the literature similarly contains sporadic reports of PPMFH presenting in patients who have received radiation therapy previously. Clinical and radiographic features of this tumor are nonspecific, and a distinction from the much more common epithelial tumors of the lungs absolutely requires tissue examination. The majority of patients present with symptoms of cough, chest pain, hemoptysis, or dyspnea. Chest x-rays generally show a solitary mass with a nondescript appearance and a relatively homogeneous density on CT or MRI studies.243
Pathologic Findings
Most examples of PPMFH are intraparenchymal, but occasional endobronchial lesions have also been observed.235 There is apparently no predilection for any particular lobe of either lung. These tumors are usually large, ranging up to 25 cm in maximum dimension,246 with an average size of 6 to 7 cm. They are well circumscribed, lobular, and white-tan, and not uncommonly they contain central necrosis or cavitation on macroscopic examination.
Histologically, PPMFH is characterized by fusiform and pleomorphic elements that are arranged in storiform, fascicular, or medullary patterns (Fig. 14-35). As the name “MFH” implies, this tumor was originally thought to be composed of malignant fibroblast-like and the histiocytoid cells; however, it now appears that there is little if any relationship between the neoplastic elements and true histiocytes. Fusiform tumor cells contain elongated nuclei and relatively scant cytoplasm, and the histiocytoid cells have round-to-oval nuclei with a moderate quantity of amphophilic cytoplasm (Fig. 14-36). A hallmark of most lesions in this category is the presence of large, bizarre, often multinucleated cells with irregular contours. Mitoses, including atypical forms, are easily found and number from 5 to 30 per 10 high-power microscopic fields.174
The differential diagnosis of PPMFH by light microscopy includes primary or secondary pleomorphic sarcomas (e.g., dedifferentiated leiomyosarcoma and pleomorphic rhabdomyosarcoma), metastatic malignant melanoma, and SC.13 Immunohistochemical and electron microscopic studies can be used to separate these pathologic entities.236–240,246 Ultrastructurally, PPMFH shows fibroblastic and histiocyte-like differentiation, with abundant rough endoplasmic reticulum, numerous lysosomes, and a variable number of small cytoplasmic lipid droplets. Desmosomes, tonofibrils, elongated cell processes, myogenous filament skeins, and cytoplasmic dense bodies are absent. PPMFH expresses vimentin but is devoid of other specialized markers of myogenous, neural, or epithelial differentiation on immunohistologic analyses.174
Therapy and Prognosis
The rarity of PPMFH again serves as an impediment to assessments of optimal treatment for this tumor. Surgical resection is currently the recommended treatment of choice, even if the lesion in question shows limited extrapulmonary spread to the intrathoracic great vessels or soft tissue.247 Adjunctive chemotherapy and irradiation have not proven to be effective in the few published cases of PPMFH in which these treatments have been used.235,245,246 In one series of 22 examples,235 7 of 15 patients who underwent radical surgical resection suffered relapses and died from metastatic disease. There was recurrence in the lungs and pleura, as well as metastasis to the liver and brain; almost all of these events occurred within 12 months of diagnosis. However, survival for as long as 5 to 10 years has been documented in a few patients with PPMFH.235,237
[/not-level-membership-for-pulmolory-and-respiratory-category]
