Chapter 159 Sarcoidosis
Etiology
An autosomal dominant familial form of the disease typified by early onset of skin, eye, and joint involvement is described as Blau syndrome. Mutations in the CARD15/NOD2 gene on chromosome 16 have been found in affected family members and appear to be associated with development of sarcoidosis. Similar genetic mutations also have been found in individuals with early-onset sarcoidosis (rash, uveitis, arthritis) but without a family history of disease, suggesting that this nonfamilial disease and Blau syndrome are genetically and phenotypically identical (Chapter 157).
Clinical Manifestations
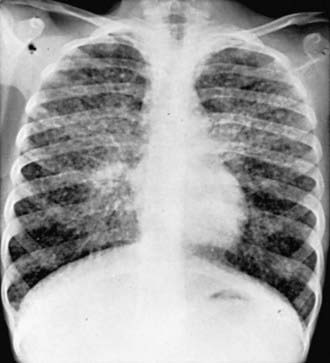
Sarcoidosis is a multisystem disease, and granulomatous lesions may occur in any organ of the body. The clinical manifestations depend on the extent and degree of granulomatous inflammation and are extremely variable. Children may present with nonspecific symptoms, such as fever, weight loss, and general malaise. In adults and older children, pulmonary involvement is most frequent, with infiltration of the thoracic lymph nodes and lung parenchyma. Isolated bilateral hilar adenopathy on chest radiograph is the most common finding, but parenchymal infiltrates and miliary nodules may also be seen (Fig. 159-1). Patients with lung involvement are commonly found to have restrictive changes on pulmonary function testing. Symptoms of pulmonary disease are seldom severe and generally consist of a dry, persistent cough.
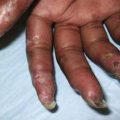
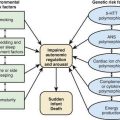
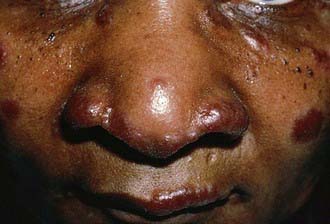
Extrathoracic lymphadenopathy and infiltration of the liver, spleen, and bone marrow also occur often. Infiltration of the liver and spleen typically leads to isolated hepatomegaly and splenomegaly, respectively, but actual organ dysfunction is rare. Cutaneous disease, such as plaques, nodules, erythema nodosum in acute disease, or lupus pernio in chronic sarcoidosis, appears in one quarter of cases and is usually present at onset. Red-brown to purple maculopapular lesions <1 cm on the face, neck, upper back, and extremities are the most common skin finding (Fig. 159-2). Ocular involvement is frequent and has variable manifestations, including anterior or posterior uveitis, conjunctival granulomas, eyelid inflammation, and orbital or lacrimal gland infiltration. The arthritis in sarcoidosis can be confused with juvenile rheumatoid arthritis. Central nervous system (CNS) involvement is rare in childhood but may manifest as seizures, cranial nerve involvement, intracranial mass lesions, and hypothalamic dysfunction. Kidney disease also occurs infrequently in children but typically manifests as renal insufficiency, proteinuria, transient pyuria, or microscopic hematuria as a result of either early monocellular infiltration or granuloma formation in kidney tissue. Only a small fraction of children have hypercalcemia or hypercalciuria, which is therefore an infrequent cause of kidney disease. Sarcoid granulomas can also infiltrate the heart and lead to cardiac arrhythmias and, rarely, sudden death. Other rare sites of disease involvement include blood vessels of any size, the gastrointestinal tract, muscles, bones, and testes.
Baumann RJ, Robertson WCJr. Neurosarcoid presents differently in children than in adults. Pediatrics. 2003;112:e480-e486.
Dempsey OJ, Paterson EW, Kerr KM, et al. Sarcoidosis. BMJ. 2009;339:620-625.
Ho LP, Urban BC, Thickett DR, et al. Deficiency of a subset of T cells with immunoregulatory properties in sarcoidosis. Lancet. 2005;365:1062-1072.
Hoffman AL, Milman N, Byg K-E. Childhood sarcoidosis in Denmark 1979–1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr. 2004;93:30-36.
Iannuzzi MC, Fontana JR. Sarcoidosis. JAMA. 2011;305(4):391-398.
Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357:2153-2165.
Milman N, Hoffman AL. Childhood sarcoidosis: long-term follow-up. European Respir J. 2008;31:592-598.
Newman LS, Rose CS, Bresnitz EA, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170:1324-1330.
Rosé CD, Wouters CH, Meiorin S, et al. Pediatric granulomatous arthritis. Arthritis Rheumatol. 2006;54:3337-3344.
Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
Spagnolo P, du Bois RM. Genetics of sarcoidosis. Clinics Dermatol. 2007;25:242-249.
Sverrild A, Backer V, Kyvik KO, et al. Heredity in sarcoidosis: a registry-based twin study. Thorax. 2008;63:894-896.