[level-membership-for-cardiovascular-category]47
Role of the Autonomic Nervous System in Atrial Fibrillation
The Cardiac Autonomic Nervous System (CANS)
The autonomic nervous system can be viewed as the interface between the central nervous system and the viscera, glands, and blood vessels. It is divided into three main components: sympathetic, parasympathetic, and enteric.1 Integration of neural trafficking among the afferent and efferent autonomic nerves as well as their associated autonomic neurons maintains a delicate homeostasis of the function of the viscera, vessels, and glands. In mammalian hearts, the efferent sympathetic preganglionic neurons are located in the intermediolateral columns of the gray matter of the spinal cord; the preganglionic fibers of these neurons pass through or synapse with the paravertebral ganglia (e.g., the stellate ganglia). The stellate ganglia, receiving neural inputs mainly from spinal nerves C6-T1, are the key neural structures for cardiac sympathetic innervation.1 The efferent parasympathetic preganglionic neurons are located in the motor nuclei of the vagus nerves (e.g., nucleus ambiguus) in the brain stem, from which the vagus nerves carry the preganglionic parasympathetic fibers to the heart. The parasympathetic postganglionic neurons are concentrated mainly in the ganglionated plexi embedded in epicardial fat pads, and the efferent postganglionic parasympathetic fibers are distributed over the entire heart. The afferent autonomic fibers, both sympathetic and parasympathetic, course along the cardiac plexus in the thorax and eventually reach the sensory neurons in the nodose ganglia at the base of the skull, as well as the dorsal root ganglia of the spinal cord. These afferent nerves and ganglia mediate important cardiorespiratory reflexes (e.g., baroreflex) and the pain sensation from the heart to the brain.2,3
The Extrinsic and Intrinsic CANS
The CANS regulates vascular tone, contractility, and electrophysiology by transducing and integrating afferent and efferent autonomic trafficking.2,3 Autonomic control of the heart is mediated by a highly integrated intrinsic and extrinsic CANS.2,3 The extrinsic CANS mainly consists of ganglia and their axons located outside the heart. The nucleus ambiguus, the dorsal vagal nucleus, and the vagus nerves constitute most of the parasympathetic limb of the extrinsic CANS, whereas the neurons in the intermediolateral column of the spinal cord, the stellate ganglia, and their axons en route to the heart make up most of the sympathetic limb of the extrinsic CANS. The intrinsic CANS is composed mainly of sympathetic and parasympathetic nerves, as well as ganglionated plexi (GP) on the heart itself or along the great vessels in the thorax such as the pulmonary artery, aorta, superior vena cava, and pulmonary veins (PVs). The stellate ganglia serve as the “head stage” for the sympathetic innervation of the heart. The postganglionic sympathetic fibers, mainly from the stellate ganglia, constitute the vast majority of the sympathetic innervation to both the atrium and the ventricle. The GP embedded in the epicardial fat pads contain up to several hundred autonomic neurons. The distribution of the major ventricular GP is limited to the proximal segments of the coronary arteries; they are in general small and not as extensive on the ventricles.2–5
The major atrial GP are located adjacent to the pulmonary vein (PV)-atrial junction, or the junction of the right atrium and the superior or inferior vena cava. Chiou et al discovered that the efferent parasympathetic fibers in the vagus nerves converge at a GP before innervating the heart.6 This GP, at the junction of the right pulmonary artery, aorta, and superior vena cava (SVC), was coined as the “head stage” GP because the bradycardia response induced by vagal stimulation in canine hearts was nearly abolished if the RPA-Ao GP was ablated.6
The different nomenclature used by anatomists introduced a great deal of confusion for scientific communication.2–5 In this chapter, we use the nomenclature based on clinical anatomy, for example, GP’s relation to PVs (Figure 47-1). The superior left GP (SLGP) and the inferior left GP (ILGP) are located adjacent to the PV-atrial junction of the left superior PV and the left inferior PV, respectively.7 The anterior right GP (ARGP) is situated at the caudal end of the sinoatrial node, near the right superior PV-atrial junction. The inferior right GP (IRGP) extends from the inferior right PV-atrial junction to the crux of the heart near the junction of the right atrium and inferior vena cava.
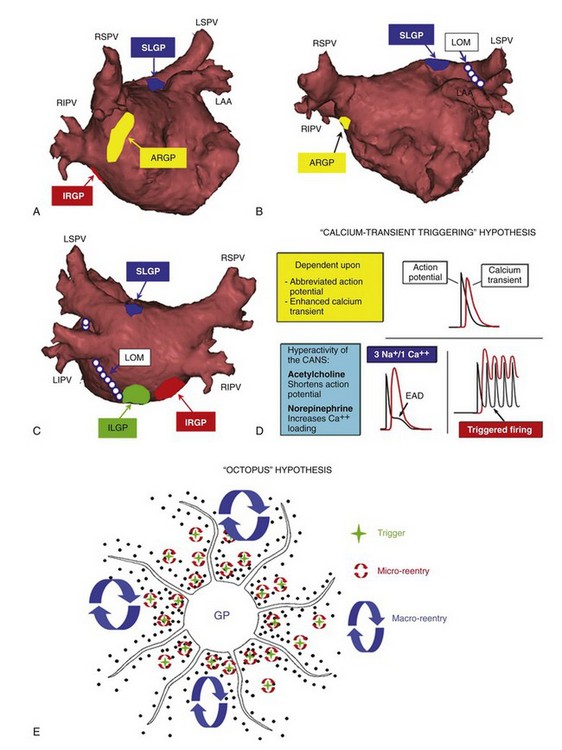
Figure 47-1 A-C, Location of the major atrial ganglionated plexi (GP) and the ligament of Marshall (LOM). SLGP, ILGP, ARGP, IRGP: superior left, inferior left, anterior right, and inferior right GP, respectively. LSPV, LIPV, RSPV, RIPV: left superior, left inferior, right superior, and right inferior pulmonary veins, respectively. A, Right anterior oblique (RAO) view. B, Anteroposterior (AP) caudal view. C, Posteroanterior (PA) view. D, Ca2+ transient hypothesis. Simultaneous activation of the sympathetic and parasympathetic systems induces early afterdepolarization and subsequent triggered firing (see text for detail). E, Octopus hypothesis. Hyperactivity of the autonomic neurons (head of the octopus) causes excessive release of the neurotransmitters of autonomic nerves (tentacles) at multiple sites. This leads to triggered firing and reentry at multiple sites to initiate and maintain atrial fibrillation (AF).
It was once thought that the ARGP specifically innervates the sinus node, while the IRGP at the crux of the heart innervates only the AV node. Recent studies indicate that the intrinsic CANS forms a complex neural network, and GP serve as “integration centers” to control the physiological functions of the heart.2,3,8 For example, high-frequency stimulation (HFS; 20 Hz) to the SLGP also markedly slowed the sinus rate (SR), proving that the ARGP is not the only GP that innervates the sinus node. Ablation of the ARGP greatly attenuated, but did not eliminate, the SR slowing response induced by SLGP stimulation, indicative of the role of ARGP as the gateway GP for the sinus node and the presence of other neural pathways bypassing the ARGP. Ablation of the four major atrial GP and the ligament of Marshall (LOM) exerts potent inhibitory effects on the activity of the CANS, supporting clinical implications targeting these GP to treat AF.9
The interplay between the intrinsic and extrinsic CANS is not well understood. The intrinsic CANS appears to function interdependently with as well as independently from the extrinsic CANS, as evidenced by its retaining nearly full control of cardiac physiology after autotransplantation.2,3 Armour elegantly described the intrinsic CANS as “the little brain on the heart.” Cooperative interaction between the extrinsic and intrinsic CANS maintains a homeostasis that facilitates balanced cardiac physiological functions.
Noncholinergic, Nonadrenergic Neurotransmitters in the Intrinsic CANS
Until the past two decades, it was believed that the sympathetic component of the intrinsic CANS is composed exclusively of postganglionic sympathetic fibers, and that all of the cardiac autonomic neurons are parasympathetic neurons expressing cholinergic markers. With advances in immunohistochemistry, subpopulations of cardiac autonomic neurons expressing various neurotransmitter markers have been identified.10 The presence of peptidergic, nitrergic, and noradrenergic neurons, along with their associated neurotransmitters such as neuropeptide-Y, vasoactive intestinal peptide (VIP), nitric oxide synthase, and angiotensin II, strongly indicates that autonomic control of cardiac physiology involves a milieu of neurotransmitters beyond acetylcholine and norepinephrine.10–12 Neuropeptide-Y co-released by prolonged sympathetic activation reduces acetylcholine release from the neighboring vagal nerve ending; this is a good example of sympathovagal cross-talk.11 These noncholinergic, nonadrenergic neurotransmitters often exert effects similar to those of cholinergic or adrenergic agonists or antagonists. Using cholinergic and adrenergic blockers to “eliminate” CANS control is an oversimplified approach.13,14 Liu et al demonstrated that until an antagonist of VIP ([Ac-Tyr1,D-phe2]-VIP) was administered, vagal stimulation continued to induce atrial fibrillation (AF) in canine hearts despite GP ablation+atropine+esmolol.12 A better understanding of the arrhythmogenic potential of these noncholinergic, nonadrenergic neurotransmitters may facilitate the development of new antiarrhythmic agents for treatment of AF.
Autonomic Mechanisms of AF Initiation and Maintenance
In 1978, Coumel et al described a group of patients with AF of vagal origin, manifested by the absence of structural heart disease and nocturnal onset of AF preceded by a slow SR.15 As the vast majority of AF patients appeared not to have the typical findings described by Coumel, vagal AF was viewed as a rarity. Landmark findings reported by Haïsseguerre et al demonstrated that paroxysmal AF, in most cases, originated from rapid focal firing in the PVs.16 Subsequent studies verified the pathophysiological roles of the PV muscle sleeve as an ideal substrate for reentry and discovered periodic acid–Schiff (PAS)-positive cells in the PV sleeves that appear to be reminiscent of Purkinje cells.17 However, these PAS-positive cells have not been shown to elicit rapid firing (300 to 600 beats/min [bpm]), and the PV sleeve, despite being an ideal reentrant substrate, cannot initiate reentry without a spontaneous, well-timed premature beat.
Over the past 15 years, AF ablation has evolved from eliminating the focal trigger(s) within the PVs (focal PV ablation) to circumferentially isolating the PV-atrial antrum (circumferential pulmonary vein isolation [CPVI]). However, the following fundamental questions have not been fully addressed: (1) Why do PVs elicit rapid firing? (2) How does PV firing initiate AF? (3) How does AF maintain itself, particularly in the first few hours before structural remodeling starts? The disappointing long-term outcome (<50% success, 5 years, single procedure) of the standard CPVI clearly indicates that a better understanding of the mechanisms underlying AF initiation and maintenance is crucial if more effective ablation targets are to be identified.18
The “Ca2+ Transient Triggering” Hypothesis
Clinical studies demonstrated that activation of both the sympathetic and the parasympathetic nervous system commonly preceded the initiation of paroxysmal AF.19,20 This finding was later corroborated by multiple basic studies.21–23 Patterson et al proposed a “Ca2+ transient triggering” hypothesis to explain the initiation of rapid PV firing (see Figure 47-1). This hypothesis states that norepinephrine (by sympathetic activation) augments the Ca2+ transient, and acetylcholine (by parasympathetic activation) shortens the PV activation potential duration (APD). The abbreviated APD makes the Ca2+ transient relatively prolonged, and the myocytes even more Ca2+ overloaded. This leads to activation of the forward mode of the Na+/Ca2+ exchanger, the formation of early afterdepolarization, and subsequent triggered firing from the PVs (Figure 47-1D). This hypothesis also helps explain the observation that PV firing often occurs at the distal segments of the PVs, where the APD is the shortest.24 Zhou et al proposed an “octopus hypothesis,” in which the autonomic neurons in the major atrial GP function like the head of an octopus, while their axons are analogous to the tentacles.25 When the head of the octopus becomes hyperactive, excessive release of neurotransmitters from its tentacles can initiate triggered firing and macro- and micro-reentry at multiple sites to initiate and maintain AF (Figure 47-1E). The octopus hypothesis also implies that targeting the head of the octopus (i.e., the GP) is perhaps the most effective means of mitigating a hyperactive state of the CANS and subsequent associated arrhythmias, i.e., paroxysmal AF.
Rapid Firing from Non-PV Sites: LOM and SVC
Although PV firing accounts for nearly 90% of initiation of paroxysmal AF, non-PV sites such as LOM and SVC are alternative sites for rapid firing and AF initiation.26,27 Notably, the initiation pattern of paroxysmal AF from the LOM, SVC, and PVs is remarkably similar. LOM itself is richly innervated and had been named the “left atrial neural fold” by some anatomists, signifying the abundance of its autonomic neural elements.5 However, substantial discrepancies in the relative abundance of sympathetic versus parasympathetic innervation have been reported.28–31 On the basis of both electrophysiological and immunohistochemical findings, Ulphani et al28 found LOM to be a parasympathetic conduit in normal dogs, whereas Doshi et al29 demonstrated sympathetic predominance within the LOM in dogs with chronic AF. In contrast, Makino et al found in human hearts that the LOM-LSPV (left superior pulmonary vein) junction was sympathetically predominant, whereas the LOM-coronary sinus junction was predominantly parasympathetic.30 Such an innervation gradient was later corroborated by Lin et al, who found that HFS at the LOM-coronary sinus junction mainly elicited AF, whereas HFS at the LOM-LSPV junction induced ventricular tachycardia, atrial tachycardia, or junctional tachycardia.31 However, it is unquestionable that the LOM provides an ideal substrate for triggered firing if sympathetic versus parasympathetic balance is altered, or if both become hyperactive.
Another common site for non-PV firing is the SVC-atrial junction, where high density autonomic innervation has been shown.6 In patients, the site of rapid firing and successful ablation was usually the posterior-septal aspect of the SVC-RA junction adjacent to the RPV-Ao GP, suggesting that this “head stage” GP may serve as the autonomic basis for SVC firing. Lu et al delivered HFS to this GP and elicited rapid firing and AF only at the SVC, not at the atrium or PV.32 Ablation of this GP only prolonged the refractory period of the SVC site. These findings indicate that the RPA-Ao GP may provide specific autonomic innervation to the SVC-atrial junction, and hyperactivity of this GP may serve as the basis for SVC firing.
Perpetuation of AF: the First Few Hours and Beyond
Electrical remodeling (e.g., shortening of the refractory period) and structural remodeling (e.g., fibrosis) are indispensable factors in the perpetuation of AF over a period of several weeks or months.33–35 However, how AF perpetuates itself within the first few minutes or hours after its initiation (e.g., before the occurrence of structural remodeling) is poorly understood. Any therapy that provides early termination of AF will have a great impact on AF therapy. Yu et al recorded the neural activity of the canine ARGP or SLGP during 6 hours of AF simulated by rapid atrial pacing.36 Rapid atrial pacing not only shortened the atrial refractory period but also progressively enhanced neural activity within the GP, providing direct evidence of autonomic remodeling during AF. Yu et al proposed that electrical remodeling and autonomic remodeling form a vicious cycle. A hyperactive state of the intrinsic CANS (e.g., GP) facilitates AF initiation. AF then shortens the refractory period further and augments GP activity to perpetuate AF itself. This “vicious cycle” hypothesis helps answer the fundamental questions described above: (1) Why do PVs elicit rapid firing? (2) How does PV firing initiate AF? (3) How does AF maintain itself?
Autonomic Basis for Complex Fractionated Atrial Electrograms (CFAEs)
In 2004, Nademanee et al described the technique for targeting CFAE to treat patients with paroxysmal and persistent AF.37 Although CFAE may simply be caused by physical properties of myocardial fibers such as anisotropic conduction or zones of slow conduction, two other hypotheses also help account for the formation of CFAE: rotor and autonomic hypotheses. The rotor hypothesis indicates that CFAE is formed when meandering rotors encounter heterogeneous substrates (e.g., dispersion of refractoriness).38 CFAE is therefore the by-product of the reentrant rotor, which breaks down at its boundary. Notably, infusion of acetylcholine into the animal heart is required to maintain a stable rotor, implying that the rotor hypothesis is also related to autonomic activity. The autonomic hypothesis stems from the locations of the CFAE originally described by Nadamanee et al, in which distribution of the CFAE correlates well with the locations of the major atrial GP. Lin et al showed in a canine model that CFAE can be produced by topical application of acetylcholine,39 and CFAE can be eliminated by ablation of the GP at a distance, indicating that activating the intrinsic CANS is critical in the formation of CFAE. Clinical studies later corroborated that CFAE tend to occur at presumed GP sites.40 Ablation of GP greatly reduced the extent of CFAE distribution around the GP, implying a possible causal relation between CANS activity and CFAE.7,40
Clinical studies have independently reported that cholinergic+adrenergic blockers failed to eliminate CFAE in patients undergoing AF ablation,13,14 casting doubt on the autonomic mechanism for CFAE. The CFAEs recorded in patients with paroxysmal AF were significantly more sensitive to autonomic blockers than the CFAEs in persistent AF patients, underlying the importance of other mechanisms (e.g., fibrosis) responsible for CFAE formation. Of note, a large milieu of noncholinergic, nonadrenergic neurotransmitters has been identified within the intrinsic CANS.10–12 Failure to eliminate CFAE by cholinergic and adrenergic blockade cannot exclude the autonomic mechanism underlying CFAE.
Ablating the CANS to Treat AF
In ablating the CANS to treat AF, the most practical targets are the major atrial GP (e.g., the head of the “octopus”). An accidental finding was reported by Pappone et al, who described a group of paroxysmal AF patients undergoing CPVI.41 When a vagal response was evoked by ablation, elimination of all evoked vagal responses around PV ostia resulted in outstanding success (99%) of freedom from AF. Scanavacca et al showed the feasibility of selective GP ablation using both endocardial and epicardial approaches, but the result of GP-only ablation was disappointing.42 When our understanding of the anatomy and physiology of the major atrial GP was advanced, the results of GP ablation improved substantially. A major limitation of GP ablation is that despite consistent location adjacent to the PV-atrial junction, the extent of each “hyperactive” GP that needs to be ablated to treat AF is largely unknown. Pokushalov et al randomly assigned 80 paroxysmal AF patients to two groups: (1) selective GP ablation guided by vagal responses induced by HFS (20 Hz), and (2) ablation of GP selected by presumed anatomical locations.43 At 13.1 ± 1.9 months, 42.5% of patients with HFS-guided GP ablation and 77.5% of patients with anatomic GP ablation were free of symptomatic AF (P = .02). This difference may be attributed to a significantly greater number of radiofrequency applications delivered in the latter group, covering a larger area for each GP. Although GP-only ablation appeared to produce results similar to those of standard CPVI, the combination of CPVI and GP ablation seems to lead to higher success rates than are achieved with CPVI alone.7,44 However, the value of GP ablation is not without debate. Adding GP ablation to CPVI introduces an arrhythmogenic substrate and increases the incidence of reentrant left atrial tachycardia by approximately 30%.7 This factor may lower the success rate compared with standard CPVI.45 Although the debate about adding GP ablation to CPVI continues, it cannot be overemphasized that CPVI transects the LOM, three of four major atrial GP at the PV-atrial junction (see Figure 47-1), and numerous autonomic nerves. The contribution of autonomic denervation to the efficacy of CPVI cannot be overlooked.
The disappointing long-term results of a single AF ablation raise further questions: What is the cause of AF recurrence after ablation? Is the isolated or destroyed myocardium the real trigger or substrate for AF? Although resumption of conduction between PV and atrium is considered the main cause for recurrence, mapping of the PV-atrial junction in patients without clinical recurrence after AF ablation is rarely done. Furlanello et al performed catheter ablation (PV isolation ± atrial flutter ablation) on 20 competitive athletes who had very symptomatic lone AF.46 Successful PV isolation was achieved in 19 of 20 patients in the first ablation procedure. When all patients were re-studied regardless of arrhythmia recurrence, 62 (81%) of the previously isolated PVs had resumed conduction. Most important, the incidence of conduction recurrence did not differ between patients with (82.5%) and without (74.6%) AF recurrence, leading to questions about the real cause of AF recurrence after ablation.
Modulation of CANS to Treat AF Without Destroying Autonomic Neural Elements
The consequences of neural degeneration and regeneration after ablation are poorly understood. If autonomic denervation and regeneration are critical elements in the efficacy and failure of CPVI, respectively, suppression of the hyperactive CANS by modulating, instead of destroying, the autonomic neural elements may be a more effective approach. Inspired by a clinical report by Tai et al47 showing that PV firing in paroxysmal AF patients could be inhibited by increased vagal reflex caused by phenylephrine-induced hypertension, the Oklahoma group hypothesized that by taking advantage of neural plasticity, low-level vagal stimulation (LL-VS) at voltages not slowing the SR or AV conduction may inhibit the CANS, and subsequently AF inducibility. A series of acute canine studies corroborated that LL-VS markedly lengthened the atrial and PV refractory period and inhibited AF inducibility.48–50 Notably, LL-VS was capable of preventing AF initiation and terminating AF. The antiarrhythmic effects were mediated by suppression of neural activity of the major atrial GP and the stellate ganglia.49 Long-term canine studies by Shen et al not only verified these findings but also discovered that suppression of stellate ganglion activity is responsible for the effects of LL-VS on AF.51 Notably, in acute canine studies, LL-VS of 80% below threshold voltage was as effective as 10% below threshold in suppressing AF, indicating that LL-VS may be a clinically feasible approach to the treatment of AF and other autonomically based diseases without permanent injury to the myocardium or to the intrinsic CANS.
Perspectives
Up to now, the clinical benefit of GP ablation in treating AF has been controversial, at best. In contrast, renal artery denervation that dramatically reduced blood pressure in patients with drug-resistant hypertension has refocused attention on the role of the autonomics in cardiovascular function.52 The suggested mechanism was that mitigation of sympathetic afferent signals to vasomotor centers in the brain reduced efferent sympathetic vasoconstriction of the renal arteries. Although it remains unclear which sympathetic pathways are modified by renal artery denervation, a more direct relationship between autonomic modulation and AF was reported in a clinical study by Pokushalov et al.53 Patients with concomitant AF and drug-resistant hypertension were randomly assigned to two groups: CPVI and CPVI plus renal artery denervation. At follow-up of 12 months, patients with CPVI alone were 29% AF-free, whereas among patients with pulmonary vein isolation plus renal artery denervation, 69% were AF-free (P = .03). The result that renal sympathetic denervation, rather than increased radiofrequency applications delivered to the PVs or the LA, markedly improved the AF ablation outcome highlights a paradigm change in the treatment of arrhythmia. Instead of solely targeting the myocardial substrates, modulation of CANS activity may alter both neural and myocardial substrates for arrhythmias and may mitigate the arrhythmias with minimal myocardial damage.
References
1. Goetz, CG. Autonomic nervous system. In Textbook of Clinical Neurology, ed 3, Philadelphia: Saunders; 2007:383–404.
2. Armour, JA. Functional anatomy of intrathoracic neurons innervating the atria and ventricles. Heart Rhythm. 2010; 7:994–996.
3. Armour, JA. The little brain on the heart. Cleveland Clin J Med. 2007; 74:S48–S51.
4. Saburkina, I, Rysevaite, K, Pauziene, N, et al. Epicardial neural ganglionated plexus of ovine heart: Anatomic basis for experimental cardiac electrophysiology and nerve protective cardiac surgery. Heart Rhythm. 2010; 7:942–950.
5. Pauza, DH, Skripka, V, Pauziene, N. Morphology of the intrinsic cardiac nervous system in the dog: A whole-mount study employing histochemical staining with acetylcholinesterase. Cells Tissues Organs. 2002; 172:297–320.
6. Chiou, CW, Eble, JN, Zipes, DP. Efferent vagal innervation of the canine atria and sinus and atrioventricular nodes: The third fat pad. Circulation. 1997; 95:2573–2584.
7. Po, SS, Nakagawa, H, Jackman, WM. Localization of left atrial ganglionated plexi in patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2009; 20:1186–1189.
8. Hou, Y, Scherlag, BJ, Zhou, J, et al. Interactive atrial neural network: Determining the connections between ganglionated plexi. Heart Rhythm. 2007; 4:56–63.
9. Lu, Z, Scherlag, BJ, Lin, J, et al. Autonomic mechanism for initiation of rapid firing from atria and pulmonary vein: Evidence by ablation of ganglionated plexi. Cardiovasc Res. 2009; 84:245–252.
10. Hoover, DB, Isaacs, ER, Jacques, F, et al. Localization of multiple neurotransmitters in surgically derived specimens of human atrial ganglia. Neuroscience. 2009; 164:1170–1179.
11. Herring, N, Paterson, DJ. Neuromodulators of peripheral cardiac sympatho-vagal balance. Exp Physiol. 2009; 94:46–53.
12. Liu, Y, Scherlag, BJ, Fan, Y, et al. Inducibility of atrial fibrillation after GP ablations and ‘autonomic blockade’: Evidence for the pathophysiological role of the non-adrenergic and non-cholinergic neurotransmitters. J Cardiovasc Electrophysiol. 2013; 24:188–195.
13. Knecht, S, Wright, M, Matsuo, S, et al. Impact of pharmacological autonomic blockade on complex fractionated atrial electrograms. J Cardiovasc Electrophysiol. 2010; 21:766–772.
14. Chaldoupi, SM, Linnenbank, AC, Wittkampf, FH, et al. Complex fractionated electrograms in the right atrial free wall and the superior/posterior wall of the left atrium are affected by activity of the autonomic nervous system. J Cardiovasc Electrophysiol. 2012; 23:26–33.
15. Coumel, P, Attuel, P, Lavallée, J, et al. The atrial arrhythmia syndrome of vagal origin. Arch Mal Coeur Vaiss. 1978; 71:645–656.
16. Haïssaguerre, M, Jaïs, P, Shah, DC, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998; 339:659–666.
17. Perez-Lugones, A, McMahon, JT, Ratliff, NB, et al. Evidence of specialized conduction cells in human pulmonary vein of patients with atrial fibrillation. J Cardiovasc Electrophysiol. 2003; 14:803–809.
18. Ouyang, F, Tilz, R, Chun, J, et al. Long-term results of catheter ablation in paroxysmal atrial fibrillation: Lessons from a 5-year follow-up. Circulation. 2010; 122:2368–2377.
19. Bettoni, M, Zimmermann, M. Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation. 2002; 105:2753–2759.
20. Amar, D, Zhang, H, Miodownik, S, et al. Competing autonomic mechanisms precede the onset of postoperative atirial fibrillation. J Am Coll Cardiol. 2003; 42:1262–1268.
21. Ogawa, M, Zhou, S, Tan, AY, et al. Left stellate ganglion and vagal nerve activity and cardiac arrhythmias in ambulatory dogs with pacing-induced congestive heart failure. J Am Coll Cardiol. 2007; 50:335–343.
22. Sharifov, OF, Fedorov, VV, Beloshapko, G, et al. Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J Am Coll Cardiol. 2004; 43:483–490.
23. Patterson, E, Lazzara, R, Szabo, B, et al. Sodium-calcium exchange initiated by the Ca2+ transient: An arrhythmia trigger within pulmonary vein. J Am Coll Cardiol. 2006; 47:1196–1206.
24. Po, SS, Li, Y, Tang, D, et al. Rapid and stable re-entry within the pulmonary vein as a mechanism initiating paroxysmal atrial fibrillation. J Am Coll Cardiol. 2005; 45:1871–1877.
25. Zhou, J, Scherlag, BS, Edwards, J, et al. Gradients of atrial refractoriness and inducibility of atrial fibrillation due to ganglionated plexi stimulation. J Cardiovasc Electrophysiol. 2007; 18:83–90.
26. Chang, HY, Lo, LW, Lin, YJ, et al. Long-term outcome of catheter ablation in patients with atrial fibrillation originating from the superior vena cava. J Cardiovasc Electrophysiol. 2013; 24:250–258.
27. Hwang, C, Fishbein, MC, Chen, PS. How and when to ablate the ligament of Marshall. Heart Rhythm. 2006; 3:1505–1507.
28. Ulphani, JS, Arora, R, Cain, JH, et al. The ligament of Marshall as a parasympathetic conduit. Am J Physiol Heart Circ Physiol. 2007; 293:H1629–H1635.
29. Doshi, RN, Wu, TJ, Yashima, M, et al. Relation between ligament of Marshall and adrenergic atrial tachyarrhythmia. Circulation. 1999; 100:876–883.
30. Makino, M, Inoue, S, Matsuyama, TA, et al. Diverse myocardial extension and autonomic innervation on ligament of Marshall in humans. J Cardiovasc Electrophysiol. 2006; 17:594–599.
31. Lin, J, Scherlag, BJ, Lu, Z, et al. Inducibility of atrial and ventricular arrhythmias along the ligament of Marshall: Role of autonomic factors. J Cardiovasc Electrophysiol. 2008; 19:955–962.
32. Lu, Z, Scherlag, BJ, Niu, G, et al. Functional properties of the superior vena cava (SVC)–aorta ganglionated plexi: Evidence suggesting an autonomic basis for rapid SVC firing. J Cardiovasc Electrophysiol. 2010; 21:1392–1399.
33. Wijffels, MC, Kirchhof, CJ, Dorland, R, et al. Atrial fibrillation begets atrial fibrillation: A study in awake chronically instrumented goats. Circulation. 1995; 92:1954–1968.
34. Everett, TH, 4th., Olgin, JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007; 4(3 Suppl):S24–S27.
35. Yue, L, Xie, J, Nattel, S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2011; 89:744–753.
36. Yu, L, Scherlag, BJ, Sha, Y, et al. Interactions between atrial electrical remodeling and autonomic remodeling: How to break the vicious cycle. Heart Rhythm. 2012; 9:804–809.
37. Nademanee, K, McKenzie, J, Kosar, E, et al. A new approach for catheter ablation of atrial fibrillation: Mapping of the electrophysiologic substrate. J Am Coll Cardiol. 2004; 43:2044–2053.
38. Kalifa, J, Tanaka, K, Zaitsev, AV, et al. Mechanisms of wave fractionation at boundaries of high-frequency excitation in the posterior left atrium of the isolated sheep heart during atrial fibrillation. Circulation. 2006; 113:626–633.
39. Lin, J, Scherlag, BJ, Lu, Z, et al. Autonomic mechanism for complex fractionated atrial electrograms. J Cardiovasc Electrophysiol. 2008; 19:835–842.
40. Katritsis, D, Sougiannis, D, Batsikas, K, et al. Autonomic modulation of complex fractionated atrial electrograms in patients with paroxysmal atrial fibrillation. J Interv Card Electrophysiol. 2011; 31:217–223.
41. Pappone, C, Santinelli, V, Manguso, F, et al. Pulmonary vein denervation enhances long-term benefit after circumferential ablation for paroxysmal atrial fibrillation. Circulation. 2004; 109:327–334.
42. Scanavacca, M, Pisani, CF, Hachul, D, et al. Selective atrial vagal denervation guided by evoked vagal reflex to treat patients with paroxysmal atrial fibrillation. Circulation. 2006; 114:876–885.
43. Pokushalov, E, Romanov, A, Shugayev, P, et al. Selective ganglionated plexi ablation for paroxysmal atrial fibrillation. Heart Rhythm. 2009; 6:1257–1264.
44. Katritsis, DG, Giazitzoglou, E, Zografos, T, et al. Rapid pulmonary vein isolation combined with autonomic ganglia modification: A randomized study. Heart Rhythm. 2011; 8:672–678.
45. Kron, J, Kasirajan, V, Wood, MA, et al. Management of recurrent atrial arrhythmias after minimally invasive surgical pulmonary vein isolation and ganglionic plexi ablation for atrial fibrillation. Heart Rhythm. 2010; 7:445–451.
46. Furlanello, F, Lupo, P, Pittalis, M, et al. Radiofrequency catheter ablation of atrial fibrillation in athletes referred for disabling symptoms preventing usual training schedule and sport competition. J Cardiovasc Electrophysiol. 2008; 19:457–462.
47. Tai, CT, Chiou, CW, Wen, ZC, et al. Effect of phenylephrine on focal atrial fibrillation originating in the pulmonary veins and superior vena cava. J Am Coll Cardiol. 2000; 36:788–793.
48. Li, S, Scherlag, BJ, Yu, L, et al. Low level vagosympathetic stimulation: A paradox and potential new modality for the treatment of focal atrial fibrillation. Circ Arrhythm Electrophysiol. 2009; 2:645–651.
49. Sha, Y, Scherlag, BJ, Yu, L, et al. Low-level right vagal stimulation: Anticholinergic and antiadrenergic effects. J Cardiovasc Electrophysiol. 2011; 10:1147–1153.
50. Xia, S, Scherlag, BJ, Yu, L, et al. Prevention and reversal of atrial fibrillation inducibility and autonomic remodeling by low-level vagosympathetic stimulation. J Am Coll Cardiol. 2011; 57:563–571.
51. Shen, MJ, Shinohara, T, Park, HW, et al. Continuous low-level vagus nerve stimulation reduces stellate ganglion nerve activity and paroxysmal atrial tachyarrhythmias in ambulatory canines. Circulation. 2011; 123:2204–2212.
52. Symplicity HTN-2 Investigators. Renal sympathetic denervation in patients with treatment-resistant hypertension: A randomised controlled trial. Lancet. 2010; 376:1903–1909.
53. Pokushalov, E, Romanov, A, Corbucci, G, et al. A randomized comparison of pulmonary vein isolation with versus without concomitant renal artery denervation in patients with refractory symptomatic atrial fibrillation and resistant hypertension. J Am Coll Cardiol. 2012; 60:1163–1170.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]47
Role of the Autonomic Nervous System in Atrial Fibrillation
The Cardiac Autonomic Nervous System (CANS)
The autonomic nervous system can be viewed as the interface between the central nervous system and the viscera, glands, and blood vessels. It is divided into three main components: sympathetic, parasympathetic, and enteric.1 Integration of neural trafficking among the afferent and efferent autonomic nerves as well as their associated autonomic neurons maintains a delicate homeostasis of the function of the viscera, vessels, and glands. In mammalian hearts, the efferent sympathetic preganglionic neurons are located in the intermediolateral columns of the gray matter of the spinal cord; the preganglionic fibers of these neurons pass through or synapse with the paravertebral ganglia (e.g., the stellate ganglia). The stellate ganglia, receiving neural inputs mainly from spinal nerves C6-T1, are the key neural structures for cardiac sympathetic innervation.1 The efferent parasympathetic preganglionic neurons are located in the motor nuclei of the vagus nerves (e.g., nucleus ambiguus) in the brain stem, from which the vagus nerves carry the preganglionic parasympathetic fibers to the heart. The parasympathetic postganglionic neurons are concentrated mainly in the ganglionated plexi embedded in epicardial fat pads, and the efferent postganglionic parasympathetic fibers are distributed over the entire heart. The afferent autonomic fibers, both sympathetic and parasympathetic, course along the cardiac plexus in the thorax and eventually reach the sensory neurons in the nodose ganglia at the base of the skull, as well as the dorsal root ganglia of the spinal cord. These afferent nerves and ganglia mediate important cardiorespiratory reflexes (e.g., baroreflex) and the pain sensation from the heart to the brain.2,3
The Extrinsic and Intrinsic CANS
The CANS regulates vascular tone, contractility, and electrophysiology by transducing and integrating afferent and efferent autonomic trafficking.2,3 Autonomic control of the heart is mediated by a highly integrated intrinsic and extrinsic CANS.2,3 The extrinsic CANS mainly consists of ganglia and their axons located outside the heart. The nucleus ambiguus, the dorsal vagal nucleus, and the vagus nerves constitute most of the parasympathetic limb of the extrinsic CANS, whereas the neurons in the intermediolateral column of the spinal cord, the stellate ganglia, and their axons en route to the heart make up most of the sympathetic limb of the extrinsic CANS. The intrinsic CANS is composed mainly of sympathetic and parasympathetic nerves, as well as ganglionated plexi (GP) on the heart itself or along the great vessels in the thorax such as the pulmonary artery, aorta, superior vena cava, and pulmonary veins (PVs). The stellate ganglia serve as the “head stage” for the sympathetic innervation of the heart. The postganglionic sympathetic fibers, mainly from the stellate ganglia, constitute the vast majority of the sympathetic innervation to both the atrium and the ventricle. The GP embedded in the epicardial fat pads contain up to several hundred autonomic neurons. The distribution of the major ventricular GP is limited to the proximal segments of the coronary arteries; they are in general small and not as extensive on the ventricles.2–5
The major atrial GP are located adjacent to the pulmonary vein (PV)-atrial junction, or the junction of the right atrium and the superior or inferior vena cava. Chiou et al discovered that the efferent parasympathetic fibers in the vagus nerves converge at a GP before innervating the heart.6 This GP, at the junction of the right pulmonary artery, aorta, and superior vena cava (SVC), was coined as the “head stage” GP because the bradycardia response induced by vagal stimulation in canine hearts was nearly abolished if the RPA-Ao GP was ablated.6
The different nomenclature used by anatomists introduced a great deal of confusion for scientific communication.2–5 In this chapter, we use the nomenclature based on clinical anatomy, for example, GP’s relation to PVs (Figure 47-1). The superior left GP (SLGP) and the inferior left GP (ILGP) are located adjacent to the PV-atrial junction of the left superior PV and the left inferior PV, respectively.7 The anterior right GP (ARGP) is situated at the caudal end of the sinoatrial node, near the right superior PV-atrial junction. The inferior right GP (IRGP) extends from the inferior right PV-atrial junction to the crux of the heart near the junction of the right atrium and inferior vena cava.
Figure 47-1 A-C, Location of the major atrial ganglionated plexi (GP) and the ligament of Marshall (LOM). SLGP, ILGP, ARGP, IRGP: superior left, inferior left, anterior right, and inferior right GP, respectively. LSPV, LIPV, RSPV, RIPV: left superior, left inferior, right superior, and right inferior pulmonary veins, respectively. A, Right anterior oblique (RAO) view. B, Anteroposterior (AP) caudal view. C, Posteroanterior (PA) view. D, Ca2+ transient hypothesis. Simultaneous activation of the sympathetic and parasympathetic systems induces early afterdepolarization and subsequent triggered firing (see text for detail). E, Octopus hypothesis. Hyperactivity of the autonomic neurons (head of the octopus) causes excessive release of the neurotransmitters of autonomic nerves (tentacles) at multiple sites. This leads to triggered firing and reentry at multiple sites to initiate and maintain atrial fibrillation (AF).
It was once thought that the ARGP specifically innervates the sinus node, while the IRGP at the crux of the heart innervates only the AV node. Recent studies indicate that the intrinsic CANS forms a complex neural network, and GP serve as “integration centers” to control the physiological functions of the heart.2,3,8 For example, high-frequency stimulation (HFS; 20 Hz) to the SLGP also markedly slowed the sinus rate (SR), proving that the ARGP is not the only GP that innervates the sinus node. Ablation of the ARGP greatly attenuated, but did not eliminate, the SR slowing response induced by SLGP stimulation, indicative of the role of ARGP as the gateway GP for the sinus node and the presence of other neural pathways bypassing the ARGP. Ablation of the four major atrial GP and the ligament of Marshall (LOM) exerts potent inhibitory effects on the activity of the CANS, supporting clinical implications targeting these GP to treat AF.9
The interplay between the intrinsic and extrinsic CANS is not well understood. The intrinsic CANS appears to function interdependently with as well as independently from the extrinsic CANS, as evidenced by its retaining nearly full control of cardiac physiology after autotransplantation.2,3 Armour elegantly described the intrinsic CANS as “the little brain on the heart.” Cooperative interaction between the extrinsic and intrinsic CANS maintains a homeostasis that facilitates balanced cardiac physiological functions.
Noncholinergic, Nonadrenergic Neurotransmitters in the Intrinsic CANS
Until the past two decades, it was believed that the sympathetic component of the intrinsic CANS is composed exclusively of postganglionic sympathetic fibers, and that all of the cardiac autonomic neurons are parasympathetic neurons expressing cholinergic markers. With advances in immunohistochemistry, subpopulations of cardiac autonomic neurons expressing various neurotransmitter markers have been identified.10 The presence of peptidergic, nitrergic, and noradrenergic neurons, along with their associated neurotransmitters such as neuropeptide-Y, vasoactive intestinal peptide (VIP), nitric oxide synthase, and angiotensin II, strongly indicates that autonomic control of cardiac physiology involves a milieu of neurotransmitters beyond acetylcholine and norepinephrine.10–12 Neuropeptide-Y co-released by prolonged sympathetic activation reduces acetylcholine release from the neighboring vagal nerve ending; this is a good example of sympathovagal cross-talk.11 These noncholinergic, nonadrenergic neurotransmitters often exert effects similar to those of cholinergic or adrenergic agonists or antagonists. Using cholinergic and adrenergic blockers to “eliminate” CANS control is an oversimplified approach.13,14 Liu et al demonstrated that until an antagonist of VIP ([Ac-Tyr1,D-phe2]-VIP) was administered, vagal stimulation continued to induce atrial fibrillation (AF) in canine hearts despite GP ablation+atropine+esmolol.12 A better understanding of the arrhythmogenic potential of these noncholinergic, nonadrenergic neurotransmitters may facilitate the development of new antiarrhythmic agents for treatment of AF.
Autonomic Mechanisms of AF Initiation and Maintenance
In 1978, Coumel et al described a group of patients with AF of vagal origin, manifested by the absence of structural heart disease and nocturnal onset of AF preceded by a slow SR.15 As the vast majority of AF patients appeared not to have the typical findings described by Coumel, vagal AF was viewed as a rarity. Landmark findings reported by Haïsseguerre et al demonstrated that paroxysmal AF, in most cases, originated from rapid focal firing in the PVs.16 Subsequent studies verified the pathophysiological roles of the PV muscle sleeve as an ideal substrate for reentry and discovered periodic acid–Schiff (PAS)-positive cells in the PV sleeves that appear to be reminiscent of Purkinje cells.17 However, these PAS-positive cells have not been shown to elicit rapid firing (300 to 600 beats/min [bpm]), and the PV sleeve, despite being an ideal reentrant substrate, cannot initiate reentry without a spontaneous, well-timed premature beat.
Over the past 15 years, AF ablation has evolved from eliminating the focal trigger(s) within the PVs (focal PV ablation) to circumferentially isolating the PV-atrial antrum (circumferential pulmonary vein isolation [CPVI]). However, the following fundamental questions have not been fully addressed: (1) Why do PVs elicit rapid firing? (2) How does PV firing initiate AF? (3) How does AF maintain itself, particularly in the first few hours before structural remodeling starts? The disappointing long-term outcome (<50% success, 5 years, single procedure) of the standard CPVI clearly indicates that a better understanding of the mechanisms underlying AF initiation and maintenance is crucial if more effective ablation targets are to be identified.18
The “Ca2+ Transient Triggering” Hypothesis
Clinical studies demonstrated that activation of both the sympathetic and the parasympathetic nervous system commonly preceded the initiation of paroxysmal AF.19,20 This finding was later corroborated by multiple basic studies.21–23 Patterson et al proposed a “Ca2+ transient triggering” hypothesis to explain the initiation of rapid PV firing (see Figure 47-1). This hypothesis states that norepinephrine (by sympathetic activation) augments the Ca2+ transient, and acetylcholine (by parasympathetic activation) shortens the PV activation potential duration (APD). The abbreviated APD makes the Ca2+ transient relatively prolonged, and the myocytes even more Ca2+ overloaded. This leads to activation of the forward mode of the Na+/Ca2+ exchanger, the formation of early afterdepolarization, and subsequent triggered firing from the PVs (Figure 47-1D
[/not-level-membership-for-cardiovascular-category]
