Chapter 48 Rickets and Hypervitaminosis D
Rickets
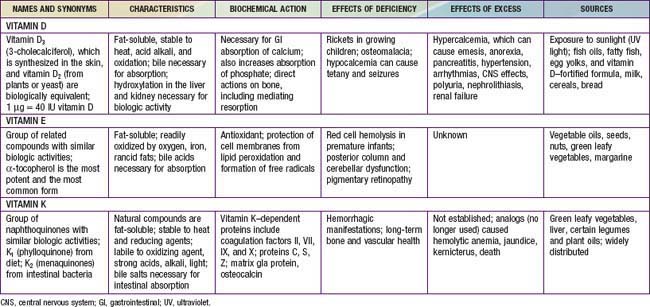
Rickets is principally due to vitamin D deficiency (Table 48-1) and was rampant in northern Europe and the USA during the early years of the 20th century. Although this problem was largely corrected through public health measures that provided children with adequate vitamin D, rickets remains a persistent problem in developed countries, with many cases still secondary to preventable nutritional vitamin D deficiency It remains a significant problem in developing countries as well, with some community-based and general hospital-based surveys among children in Africa finding the prevalence of rickets exceeds 10%. UNICEF has estimated that up to 25% of children in China have some evidence of rickets.
Etiology
There are many causes of rickets (Table 48-2), including vitamin D disorders, calcium deficiency, phosphorous deficiency, and distal renal tubular acidosis.
VITAMIN D DISORDERS
CALCIUM DEFICIENCY
PHOSPHORUS DEFICIENCY
RENAL LOSSES
Clinical Manifestations
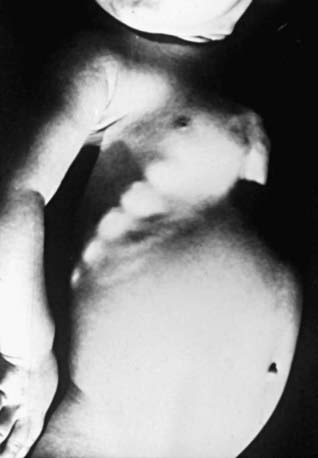
Most manifestations of rickets are due to skeletal changes (Table 48-3). Craniotabes is a softening of the cranial bones and can be detected by applying pressure at the occiput or over the parietal bones. The sensation is similar to the feel of pressing into a Ping-Pong ball and then releasing. Craniotabes may also be secondary to osteogenesis imperfecta, hydrocephalus, and syphilis. It is a normal finding in many newborns, especially near the suture lines, but it typically disappears within a few months of birth. Widening of the costochondral junctions results in a rachitic rosary, which feels like the beads of a rosary as the examiner’s fingers move along the costochondral junctions from rib to rib (Fig. 48-1). Growth plate widening is also responsible for the enlargement at the wrists and ankles. The horizontal depression along the lower anterior chest known as Harrison groove occurs from pulling of the softened ribs by the diaphragm during inspiration (Fig. 48-2). Softening of the ribs also impairs air movement and predisposes patients to atelectasis and pneumonia.
Table 48-3 CLINICAL FEATURES OF RICKETS
GENERAL
HEAD
CHEST
BACK
EXTREMITIES
HYPOCALCEMIC SYMPTOMS†
* These features are most commonly associated with the vitamin D deficiency disorders.
† These symptoms develop only in children with disorders that produce hypocalcemia (see Table 48-4).
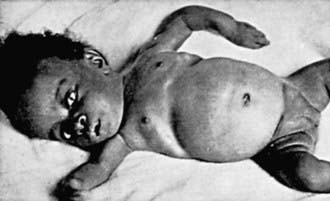
There is some variation in the clinical presentation of rickets based on the etiology. Changes in the lower extremities tend to be the dominant feature in X-linked hypophosphatemic rickets. Symptoms secondary to hypocalcemia occur only in those forms of rickets associated with decreased serum calcium (Table 48-4).
The chief complaint in a child with rickets is quite variable. Many children present because of skeletal deformities, whereas others have difficulty walking owing to a combination of deformity and weakness. Other common presenting complaints include failure to thrive and symptomatic hypocalcemia (Chapter 565).
Radiology
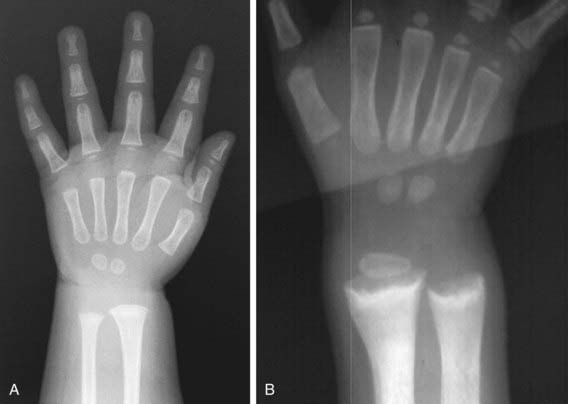
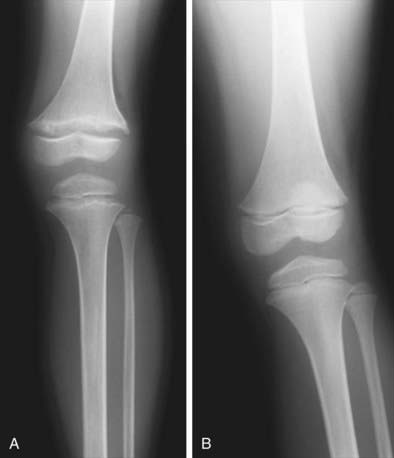
Rachitic changes are most easily visualized on posteroanterior radiographs of the wrist, although characteristic rachitic changes can be seen at other growth plates (Figs. 48-3 and 48-4). Decreased calcification leads to thickening of the growth plate. The edge of the metaphysis loses its sharp border, which is described as fraying. The edge of the metaphysis changes from a convex or flat surface to a more concave surface. This change to a concave surface is termed cupping and is most easily seen at the distal ends of the radius, ulna, and fibula. There is widening of the distal end of the metaphysis, corresponding to the clinical observation of thickened wrists and ankles as well as the rachitic rosary. Other radiologic features include coarse trabeculation of the diaphysis and generalized rarefaction.
Diagnosis
Most cases of rickets are diagnosed based on the presence of classic radiographic abnormalities. The diagnosis is supported by physical examination findings (see Table 48-3) and a history and laboratory test results that are consistent with a specific etiology.
Clinical Evaluation
Malabsorption of vitamin D is suggested by a history of liver or intestinal disease. Undiagnosed liver or intestinal disease should be suspected if the child has gastrointestinal (GI) symptoms, although occasionally rickets is the presenting complaint. Fat malabsorption is often associated with diarrhea or oily stools, and there may be signs or symptoms suggesting deficiencies of other fat-soluble vitamins (A, E, and K; Chapters 45, 49, and 50).
The physical examination focuses on detecting manifestations of rickets (see Table 48-3). It is important to observe the child’s gait, auscultate the lungs to detect atelectasis or pneumonia, and plot the patient’s growth. Alopecia suggests vitamin D-dependent rickets type 2.
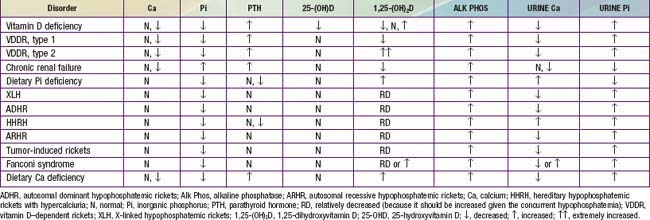
The initial laboratory tests in a child with rickets should include serum calcium, phosphorus, alkaline phosphatase, parathyroid hormone (PTH), 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D3, creatinine, and electrolytes (see Table 48-4 for interpretation). Urinalysis is useful for detecting the glycosuria and aminoaciduria (positive dipstick for protein) seen with Fanconi syndrome. Evaluation of urinary excretion of calcium (24 hr collection for calcium or calcium:creatinine ratio) is helpful if hereditary hypophosphatemic rickets with hypercalciuria or Fanconi syndrome is suspected. Direct measurement of other fat-soluble vitamins (A, E, and K) or indirect assessment of deficiency (prothrombin time for vitamin K deficiency) is appropriate if malabsorption is a consideration.
Vitamin D Disorders
Nutritional Vitamin D Deficiency
Clinical Manifestations
The clinical features are typical of rickets (see Table 48-3), with a significant minority presenting with symptoms of hypocalcemia; prolonged laryngospasm is occasionally fatal. These children have an increased risk of pneumonia and muscle weakness leading to a delay in motor development.
Laboratory Findings
Table 48-4 summarizes the principal laboratory findings. Hypocalcemia is a variable finding due to the actions of the elevated PTH to increase the serum calcium concentration. The hypophosphatemia is due to PTH-induced renal losses of phosphate, combined with a decrease in intestinal absorption.
Diagnosis and Differential Diagnosis
The diagnosis of nutritional vitamin D deficiency is based on the combination of a history of poor vitamin D intake and risk factors for decreased cutaneous synthesis, radiographic changes consistent with rickets, and typical laboratory findings (see Table 48-4). A normal PTH level almost never occurs with vitamin D deficiency and suggests a primary phosphate disorder.
Vitamin D–Dependent Rickets, Type 1
Children with vitamin D–dependent rickets type 1, an autosomal recessive disorder, have mutations in the gene encoding renal 1α-hydroxylase, preventing conversion of 25-D into 1,25-D. These patients normally present during the 1st 2 yr of life and can have any of the classic features of rickets (see Table 48-3), including symptomatic hypocalcemia. They have normal levels of 25-D but low levels of 1,25-D (see Table 48-4). Occasionally, 1,25-D levels are at the lower limit of normal, inappropriately low given the high PTH and low serum phosphorus levels, both of which should increase the activity of renal 1α-hydroxylase and cause elevated levels of 1,25-D. As in nutritional vitamin D deficiency, renal tubular dysfunction can cause a metabolic acidosis and generalized aminoaciduria.
Vitamin D–Dependent Rickets, Type 2
Patients with vitamin D–dependent rickets type 2 have mutations in the gene encoding the vitamin D receptor, preventing a normal physiologic response to 1,25-D. Levels of 1,25-D are extremely elevated in this autosomal recessive disorder (see Table 48-4). Most patients present during infancy, although rickets in less severely affected patients might not be diagnosed until adulthood. Less-severe disease is associated with a partially functional vitamin D receptor. Approximately 50-70% of children have alopecia, which tends to be associated with a more severe form of the disease and can range from alopecia areata to alopecia totalis. Epidermal cysts are a less common manifestation.
Chronic Renal Failure (Chapter 529.2)
With chronic renal failure, there is decreased activity of 1α-hydroxylase in the kidney, leading to diminished production of 1,25-D. In chronic renal failure, unlike the other causes of vitamin D deficiency, patients have hyperphosphatemia as a result of decreased renal excretion (see Table 48-4).
Calcium Deficiency
Clinical Manifestations
Children have the classic signs and symptoms of rickets (see Table 48-3). Presentation can occur during infancy or early childhood, although some cases are diagnosed in teenagers. Because calcium deficiency occurs after the cessation of breast-feeding, it tends to occur later than the nutritional vitamin D deficiency that is associated with breast-feeding. In Nigeria, nutritional vitamin D deficiency is most common at 4-15 mo of age, whereas calcium-deficiency rickets typically occurs at 15-25 mo of age.
Diagnosis
Laboratory findings include increased levels of alkaline phosphatase, PTH, and 1,25-D (see Table 48-4). Calcium levels may be normal or low, although symptomatic hypocalcemia is uncommon. There is decreased urinary excretion of calcium, and serum phosphorus levels may be low due to renal wasting of phosphate from secondary hyperparathyroidism, which can also cause aminoaciduria. In some children, there is coexisting nutritional vitamin D deficiency, with low 25-D levels.
Phosphorous Deficiency
Phosphatonin
Phosphatonin is a humoral mediator that decreases renal tubular reabsorption of phosphate and therefore decreases serum phosphorus. Phosphatonin also decreases the activity of renal 1α-hydroxylase, resulting in a decrease in the production of 1,25-D. Fibroblast growth factor–23 (FGF-23) is the most well characterized phosphatonin, but there are a number of other putative phosphatonins (discussed later). Increased levels of phosphatonin cause many of the phosphate-wasting diseases (see Table 48-2).
X-Linked Hypophosphatemic Rickets
Laboratory Findings
Patients have high renal excretion of phosphate, hypophosphatemia, and increased alkaline phosphatase; PTH and serum calcium levels are normal (see Table 48-4). Hypophosphatemia normally upregulates renal 1α-hydroxylase and should lead to an increase in 1,25-D, but these patients have low or inappropriately normal levels of 1,25-D.
Autosomal Dominant Hypophosphatemic Rickets
In ADHR, as in XLH, abnormal laboratory findings are hypophosphatemia, an elevated alkaline phosphatase level, and a low or inappropriately normal 1,25-D level (see Table 48-4). Treatment is similar to the approach used in XLH.
Hereditary Hypophosphatemic Rickets with Hypercalciuria
Clinical Manifestations
The dominant symptoms are rachitic leg abnormalities (see Table 48-3), muscle weakness, and bone pain. Patients can have short stature, with a disproportionate decrease in the length of the lower extremities. The severity of the disease varies, and some family members have no evidence of rickets but have kidney stones secondary to hypercalciuria.
Laboratory Findings
Laboratory findings include hypophosphatemia, renal phosphate wasting, elevated serum alkaline phosphatase levels, and elevated 1,25-D levels. PTH levels are low (see Table 48-4).
Overproduction of Phosphatonin
Tumor-induced osteomalacia is more common in adults than in children, where it can produce classic rachitic findings. Most tumors are mesenchymal in origin and are usually benign, small, and located in bone. These tumors secrete a number of different putative phosphatonins (FGF-23, frizzled-related protein 4, and matrix extracellular phosphoglycoprotein); different tumors secrete different phosphatonins or combinations of phosphatonins. These phosphatonins produce a biochemical phenotype that is similar to XLH, including urinary phosphate wasting, hypophosphatemia, elevated alkaline phosphatase levels, and low or inappropriately normal 1,25-D levels (see Table 48-4). Curative treatment is excision of the tumor. If the tumor cannot be removed, treatment is identical to that used for XLH.
Renal phosphate wasting leading to hypophosphatemia and rickets (or osteomalacia in adults) is a potential complication in McCune-Albright syndrome, an entity that includes the triad of polyostotic fibrous dysplasia, hyperpigmented macules, and polyendocrinopathy (Chapter 556.6). Affected patients have inappropriately low levels of 1,25-D and elevated levels of alkaline phosphatase. The renal phosphate wasting and inhibition of 1,25-D synthesis are related to the polyostotic fibrous dysplasia. Patients have elevated levels of the phosphatonin FGF-23, presumably produced by the dysplastic bone. Hypophosphatemic rickets can also occur in children with isolated polyostotic fibrous dysplasia. Although it is rarely possible, removal of the abnormal bone can cure this disorder in children with McCune-Albright syndrome. Most patients receive the same treatment as children with XLH. Bisphosphonate treatment decreases the pain and fracture risk associated with the bone lesions.
Rickets is an unusual complication of epidermal nevus syndrome (Chapter 643). Patients have hypophosphatemic rickets due to renal phosphate wasting and also have an inappropriately normal or low level of 1,25-D due to excessive production of FGF-23. The timing of presentation with rickets varies from infancy to early adolescence. Resolution of hypophosphatemia and rickets has occurred after excision of the epidermal nevi in some patients but not in others. In most cases, the skin lesions are too extensive to be removed, necessitating treatment with phosphorus supplementation and 1,25-D. Rickets due to phosphate wasting is an extremely rare complication in children with neurofibromatosis (Chapter 589.1), again presumably due to the production of a phosphatonin.
Fanconi Syndrome
Fanconi syndrome is secondary to generalized dysfunction of the renal proximal tubule (Chapter 523.1). There are renal losses of phosphate, amino acids, bicarbonate, glucose, urate, and other molecules that are normally reabsorbed in the proximal tubule. Some patients have partial dysfunction, with less generalized losses. The most clinically relevant consequences are hypophosphatemia due to phosphate losses and proximal renal tubular acidosis due to bicarbonate losses. Patients have rickets as a result of hypophosphatemia, with exacerbation from the chronic metabolic acidosis, which causes bone dissolution. Failure to thrive is a consequence of both rickets and renal tubular acidosis. Treatment is dictated by the etiology (Chapters 523.1 and 523.4).
Dent Disease (Chapter 525.3)
Dent disease is an X-linked disorder usually caused by mutations in the gene encoding a chloride channel that is expressed in the kidney. Some patients have mutations in the OCRL 1 gene, which can also cause Lowe syndrome (Chapter 523.1). Affected males have variable manifestations, including hematuria, nephrolithiasis, nephrocalcinosis, rickets, and chronic renal failure. Almost all patients have low molecular weight proteinuria and hypercalciuria. Other, less universal abnormalities are aminoaciduria, glycosuria, hypophosphatemia, and hypokalemia. Rickets occurs in approximately 25% of patients, and it responds to oral phosphorus supplements. Some patients also need 1,25-D, but this treatment should be used cautiously because it can worsen the hypercalciuria.
Rickets of Prematurity (Chapter 100)
Clinical Manifestations
Rickets of prematurity occurs 1-4 mo after birth. Infants can have nontraumatic fractures, especially of the legs, arms, and ribs. Most fractures are not suspected clinically. Because fractures and softening of the ribs lead to decreased chest compliance, some infants have respiratory distress due to atelectasis and poor ventilation. This rachitic respiratory distress usually develops >5 wk after birth, distinguishing it from the early-onset respiratory disease of premature infants. These infants have poor linear growth, with negative effects on growth persisting beyond 1 yr of age. An additional long-term effect is enamel hypoplasia. Poor bone mineralization can contribute to dolichocephaly. There may be classic rachitic findings, such as frontal bossing, rachitic rosary, craniotabes, and widened wrists and ankles (see Table 48-3). Most infants with rickets of prematurity have no clinical manifestations, and the diagnosis is based on radiographic and laboratory findings.
Distal Renal Tubular Acidosis (Chapter 523)
Distal renal tubular acidosis usually manifests with failure to thrive. Patients have a metabolic acidosis with an inability to acidify the urine appropriately. Hypercalciuria and nephrocalcinosis are typically present. There are many possible etiologies, including autosomal recessive and autosomal dominant forms. Rickets is variable, and it responds to alkali therapy (see Fig. 48-4).
Hypervitaminosis D
Etiology
Hypervitaminosis D is secondary to excessive intake of vitamin D. It can occur with long-term high intake or with a substantial, acute ingestion (see Table 48-1). Most cases are secondary to misuse of prescribed or over-the-counter vitamin D supplements, but other cases have been secondary to accidental overfortification of milk, contamination of table sugar, and inadvertent use of vitamin D supplements as cooking oil. The recommended upper limits for long-term vitamin D intake are 1,000 IU for children <1 year old and 2,000 IU for older children and adults. Hypervitaminosis D can also result from excessive intake of synthetic vitamin D analogs (25-D, 1,25-D). Vitamin D intoxication is never secondary to excessive exposure to sunlight, probably because ultraviolet irradiation can transform vitamin D3 and its precursor into inactive metabolites.
Abrams SA. Dietary guidelines for calcium and vitamin D: a new era. Pediatrics. 2011;127(3):566-568.
Ariceta G, Langman CB. Growth in X-linked hypophosphatemic rickets. Eur J Pediatr. 2007;166:303-309.
Cole CR, Grant FK, Tangpricha V, et al. 25-hydroxyvitamin D status of healthy, low-income, minority children in Atlanta, Georgia. Pediatrics. 2010;125:633-639.
DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev Suppl. 2007;4:440-445.
Doneray H, Ozkan B, Caner I, et al. Intragastric alendronate therapy in two infants with vitamin D intoxication: a new method. Clin Toxicol (Phila). 2008;46:300-302.
Dong Y, Pollock N, Stallman-Jorgensen IS, et al. Low 25-hydroxyvitamin D levels in adolescents: race, season, adiposity, physical activity, and fitness. Pediatrics. 2010;125:1104-1111.
Feng JQ, Ward LM, Liu S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310-1315.
Garabedian M. Regulation of phosphate homeostasis in infants, children, and adolescents, and the role of phosphatonins in this process. Curr Opin Pediatr. 2007;19:488-491.
Greer FR. Defining vitamin D deficiency in children: beyond 25-OH vitamin D serum concentrations. Pediatrics. 2009;124:1471-1473.
Grey V, Atkinson S, Drury D, et al. Prevalence of low bone mass and deficiencies of vitamins D and K in pediatric patients with cystic fibrosis from 3 Canadian centers. Pediatrics. 2008;122:1014-1020.
Iyer P, Diamond FBJr. Shedding light on hypovitaminosis D and rickets. Adv Pediatr. 2007;54:115-133.
Kumar J, Muntner P, Kaskel FJ, et al. Prevalence and associations of 25-hydroxyvitamin D deficiency in US children: NHANES 2001–2004. Pediatrics. 2009;124:e362-e370.
Lerch C, Meissner T: Interventions for the prevention of nutritional rickets in term born children, Cochrane Database Sys Rev (4):CD006164, 2007.
Misra M, Pacaud D, Petryk A, et al. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398-417.
Nguyen M, Boutignon H, Mallet E, et al. Infantile hypercalcemia and hypercalciuria: new insights into a vitamin D-dependent mechanism and response to ketoconazole treatment. J Pediatr. 2010;157:296-302.
Pearce SHS, Cheetham TD. Diagnosis and management of vitamin D deficiency. BMJ. 2010;340:142-146.
Perrine CG, Sharma AJ, Jefferds MED, et al. Adherence to vitamin D recommendations among US infants. Pediatrics. 2010;125:627-632.
Pettifor JM. What’s new in hypophosphataemic rickets? Eur J Pediatr. 2008;167:493-499.
Rosen CJ. Vitamin D insufficiency. N Engl J Med. 2011;364(3):248-254.
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53-58.
Sahota O. Reducing the risk of fractures with calcium and vitamin D. BMJ. 2010;340:109-110.
Saintonge S, Bang H, Gerber LM. Implications of a new definition of vitamin D deficiency in a multiracial us adolescent population: The National Health and Nutrition Examination Survey III. Pediatrics. 2009;123:797-803.
Shaikh A, Berndt T, Kumar R. Regulation of phosphate homeostasis by the phosphatonins and other novel mediators. Pediatr Nephrol. 2008;23:1203-1210.
Shaw N. Vitamin D and bone health in children. BMJ. 2011;342:239-240.
Shellhaas RA, Barks AK, Joshi SM. Prevalence and risk factors for vitamin D insufficiency among children with epilepsy. Pediatr Neurol. 2010;42:422-426.
Wagner CL, Greer FR, American Academy of Pediatrics Section on Breastfeeding; American Academy of Pediatrics Committee on Nutrition. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. 2008;122(5):1142-1152.
Winzenberg T, Powell S, Shaw KA, et al. Effects of vitamin D supplementation on bone density in healty children: systematic review and meta-analysis. BMJ. 2011;342:267.