Contraindications
Procedure
Interpretation of Results
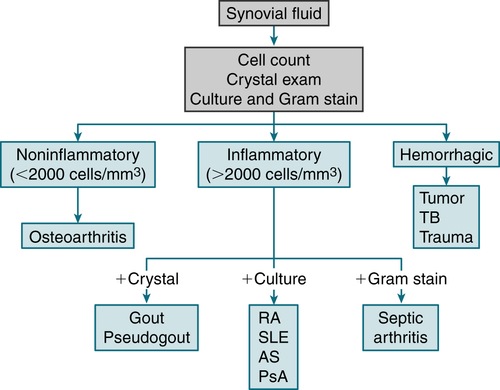
FIGURE 12-1 Algorithm for analysis of joint fluid. Examples of inflammatory arthritis are indicated, although many conditions can produce these findings. AS, ankylosing spondylitis; PsA, psoriatic arthritis. (From Goldman L, Schafer AI [eds]: Goldman’s Cecil Medicine, 24th edition. Philadelphia, Saunders, 2012.)
B. Arthritis, Monarticular and Oligoarticular
 Septic arthritis (Staphylococcus aureus, Neisseria gonorrhoeae, meningococci, streptococci, Streptococcus pneumoniae, enteric gram(−) bacilli)
Septic arthritis (Staphylococcus aureus, Neisseria gonorrhoeae, meningococci, streptococci, Streptococcus pneumoniae, enteric gram(−) bacilli) Crystalline-induced arthritis (gout, pseudogout, Ca oxalate, hydroxyapatite, and other basic Ca/phosphate crystals)
Crystalline-induced arthritis (gout, pseudogout, Ca oxalate, hydroxyapatite, and other basic Ca/phosphate crystals) Traumatic joint injury
Traumatic joint injury Hemarthrosis
Hemarthrosis Monarticular or oligoarticular flare of an inflammatory polyarticular rheumatic disease (RA, psoriatic arthritis, Reiter’s syndrome, SLE)
Monarticular or oligoarticular flare of an inflammatory polyarticular rheumatic disease (RA, psoriatic arthritis, Reiter’s syndrome, SLE)C. Arthritis, Polyarticular
 RA, juvenile (rheumatoid) polyarthritis
RA, juvenile (rheumatoid) polyarthritis SLE, other connective tissue diseases, erythema nodosum, palindromic rheumatism, relapsing polychondritis
SLE, other connective tissue diseases, erythema nodosum, palindromic rheumatism, relapsing polychondritis Psoriatic arthritis, ankylosing spondylitis
Psoriatic arthritis, ankylosing spondylitis Sarcoidosis
Sarcoidosis Lyme arthritis, bacterial endocarditis, Neisseria gonorrhoeae infection, rheumatic fever, Reiter’s disease
Lyme arthritis, bacterial endocarditis, Neisseria gonorrhoeae infection, rheumatic fever, Reiter’s disease Crystal deposition disease
Crystal deposition disease Hypersensitivity to serum or drugs
Hypersensitivity to serum or drugs Hepatitis B, HIV infection, rubella, mumps
Hepatitis B, HIV infection, rubella, mumps Other: serum sickness, leukemias, lymphomas, enteropathic arthropathy, Whipple’s disease, Behçet’s syndrome, Henoch-Schönlein purpura, familial Mediterranean fever, hypertrophic pulmonary osteoarthropathy
Other: serum sickness, leukemias, lymphomas, enteropathic arthropathy, Whipple’s disease, Behçet’s syndrome, Henoch-Schönlein purpura, familial Mediterranean fever, hypertrophic pulmonary osteoarthropathyD. Vasculitic Syndromes
E. Systemic Lupus Erythematosus (SLE)
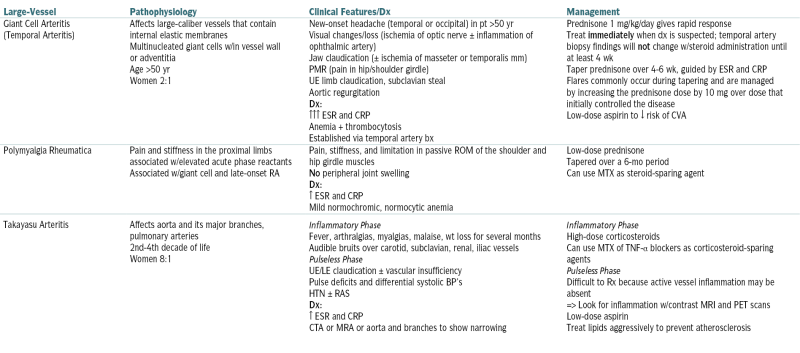
TABLE 12-1
Systemic Vasculitis∗
| Large-Vessel | Pathophysiology | Clinical Features/Dx | Management |
| Giant Cell Arteritis (Temporal Arteritis) |
Affects large-caliber vessels that contain internal elastic membranes Multinucleated giant cells w/in vessel wall or adventitia Age >50 yr Women 2:1 |
New-onset headache (temporal or occipital) in pt >50 yr Visual changes/loss (ischemia of optic nerve ± inflammation of ophthalmic artery) Jaw claudication (± ischemia of masseter or temporalis mm) PMR (pain in hip/shoulder girdle) UE limb claudication, subclavian steal Aortic regurgitation Dx: ↑↑↑ ESR and CRP Anemia + thrombocytosis Established via temporal artery bx |
Prednisone 1 mg/kg/day gives rapid response Treat immediately when dx is suspected; temporal artery biopsy findings will not change w/steroid administration until at least 4 wk Taper prednisone over 4-6 wk, guided by ESR and CRP Flares commonly occur during tapering and are managed by increasing the prednisone dose by 10 mg over dose that initially controlled the disease Low-dose aspirin to ↓ risk of CVA |
| Polymyalgia Rheumatica | Pain and stiffness in the proximal limbs associated w/elevated acute phase reactants Associated w/giant cell and late-onset RA |
Pain, stiffness, and limitation in passive ROM of the shoulder and hip girdle muscles No peripheral joint swelling Dx: ↑ ESR and CRP Mild normochromic, normocytic anemia |
Low-dose prednisone Tapered over a 6-mo period Can use MTX as steroid-sparing agent |
| Takayasu Arteritis | Affects aorta and its major branches, pulmonary arteries 2nd-4th decade of life Women 8:1 |
Inflammatory Phase Fever, arthralgias, myalgias, malaise, wt loss for several months Audible bruits over carotid, subclavian, renal, iliac vessels Pulseless Phase UE/LE claudication ± vascular insufficiency Pulse deficits and differential systolic BP’s HTN ± RAS Dx: ↑ ESR and CRP CTA or MRA or aorta and branches to show narrowing |
Inflammatory Phase High-dose corticosteroids Can use MTX of TNF-α blockers as corticosteroid-sparing agents Pulseless Phase Difficult to Rx because active vessel inflammation may be absent => Look for inflammation w/contrast MRI and PET scans Low-dose aspirin Treat lipids aggressively to prevent atherosclerosis |
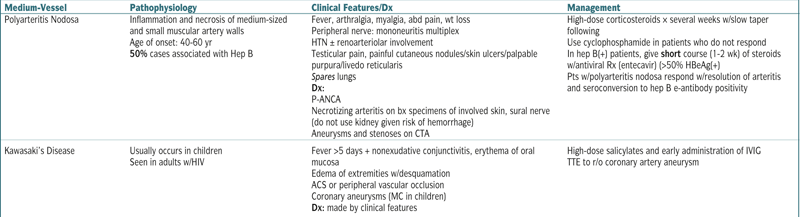
| Medium-Vessel | Pathophysiology | Clinical Features/Dx | Management |
| Polyarteritis Nodosa | Inflammation and necrosis of medium-sized and small muscular artery walls Age of onset: 40-60 yr 50% cases associated with Hep B |
Fever, arthralgia, myalgia, abd pain, wt loss Peripheral nerve: mononeuritis multiplex HTN ± renoarteriolar involvement Testicular pain, painful cutaneous nodules/skin ulcers/palpable purpura/livedo reticularis Spares lungs Dx: P-ANCA Necrotizing arteritis on bx specimens of involved skin, sural nerve (do not use kidney given risk of hemorrhage) Aneurysms and stenoses on CTA |
High-dose corticosteroids × several weeks w/slow taper following Use cyclophosphamide in patients who do not respond In hep B(+) patients, give short course (1-2 wk) of steroids w/antiviral Rx (entecavir) (>50% HBeAg(+) Pts w/polyarteritis nodosa respond w/resolution of arteritis and seroconversion to hep B e-antibody positivity |
| Kawasaki’s Disease | Usually occurs in children Seen in adults w/HIV |
Fever >5 days + nonexudative conjunctivitis, erythema of oral mucosa Edema of extremities w/desquamation ACS or peripheral vascular occlusion Coronary aneurysms (MC in children) Dx: made by clinical features |
High-dose salicylates and early administration of IVIG TTE to r/o coronary artery aneurysm |
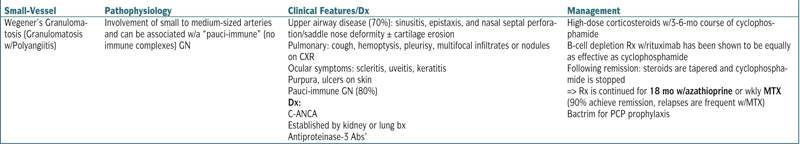
| Small-Vessel | Pathophysiology | Clinical Features/Dx | Management |
| Wegener’s Granulomatosis (Granulomatosis w/Polyangiitis) | Involvement of small to medium-sized arteries and can be associated w/a “pauci-immune” (no immune complexes) GN | Upper airway disease (70%): sinusitis, epistaxis, and nasal septal perforation/saddle nose deformity ± cartilage erosion Pulmonary: cough, hemoptysis, pleurisy, multifocal infiltrates or nodules on CXR Ocular symptoms: scleritis, uveitis, keratitis Purpura, ulcers on skin Pauci-immune GN (80%) Dx: C-ANCA Established by kidney or lung bx Antiproteinase-3 Abs’ |
High-dose corticosteroids w/3-6-mo course of cyclophosphamide B-cell depletion Rx w/rituximab has been shown to be equally as effective as cyclophosphamide Following remission: steroids are tapered and cyclophosphamide is stopped => Rx is continued for 18 mo w/azathioprine or wkly MTX (90% achieve remission, relapses are frequent w/MTX) Bactrim for PCP prophylaxis |
| Small-Vessel | Pathophysiology | Clinical Features/Dx | Management |
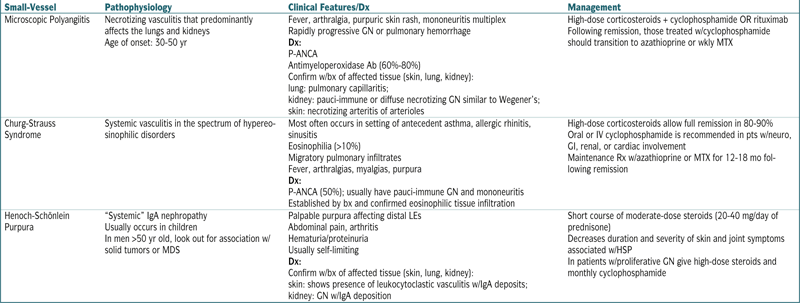
| Microscopic Polyangiitis | Necrotizing vasculitis that predominantly affects the lungs and kidneys Age of onset: 30-50 yr |
Fever, arthralgia, purpuric skin rash, mononeuritis multiplex Rapidly progressive GN or pulmonary hemorrhage Dx: P-ANCA Antimyeloperoxidase Ab (60%-80%) Confirm w/bx of affected tissue (skin, lung, kidney): lung: pulmonary capillaritis; kidney: pauci-immune or diffuse necrotizing GN similar to Wegener’s; skin: necrotizing arteritis of arterioles |
High-dose corticosteroids + cyclophosphamide OR rituximab Following remission, those treated w/cyclophosphamide should transition to azathioprine or wkly MTX |
| Churg-Strauss Syndrome | Systemic vasculitis in the spectrum of hypereosinophilic disorders | Most often occurs in setting of antecedent asthma, allergic rhinitis, sinusitis Eosinophilia (>10%) Migratory pulmonary infiltrates Fever, arthralgias, myalgias, purpura Dx: P-ANCA (50%); usually have pauci-immune GN and mononeuritis Established by bx and confirmed eosinophilic tissue infiltration |
High-dose corticosteroids allow full remission in 80-90% Oral or IV cyclophosphamide is recommended in pts w/neuro, GI, renal, or cardiac involvement Maintenance Rx w/azathioprine or MTX for 12-18 mo following remission |
| Henoch-Schönlein Purpura | “Systemic” IgA nephropathy Usually occurs in children In men >50 yr old, look out for association w/solid tumors or MDS |
Palpable purpura affecting distal LEs Abdominal pain, arthritis Hematuria/proteinuria Usually self-limiting Dx: Confirm w/bx of affected tissue (skin, lung, kidney): skin: shows presence of leukocytoclastic vasculitis w/IgA deposits; kidney: GN w/IgA deposition |
Short course of moderate-dose steroids (20-40 mg/day of prednisone) Decreases duration and severity of skin and joint symptoms associated w/HSP In patients w/proliferative GN give high-dose steroids and monthly cyclophosphamide |
| Small-Vessel | Pathophysiology | Clinical Features/Dx | Management |
| Essential Cryoglobulinemic Vasculitis | Immunoglobulins that precipitate from serum in the cold Type I cryoglobulins: monoclonal, self-aggregate, associated w/Waldenström’s and MM; associated w/hyperviscosity Type II cryoglobulins: monoclonal IgM or IgA rheumatoid factors that bind to Fc of IgG, associated w/hep C and HIV Type II cryoglobulins: seen in setting of autoimmune disorders (SLE, Sjögren’s, RA) |
Palpable purpura, mononeuritis multiplex LAD, HSM Renal failure ± GN Dx: ↓ C3, C4 (immune-complex GN) 80% of type-II cryoglobulinemic vasculitis is associated w/hep C |
Hep C–associated cryoglobulinemia responds to antiviral therapy w/interferon alfa + ribavirin Short course of corticosteroids In patients with renal failure, digital gangrene, neuro disease, 2-3-wk course of plasma exchange recommended |
| Cutaneous Leukocytoclastic Vasculitis | Seen in pts w/connective tissue diseases and as a reaction to drugs/viruses 40% idiopathic 60% ± autoimmune disease (SLE), drugs, infection, hematologic malignancy |
Palpable purpura, tender nodules, persistent urticaria, or shallow ulcers Lesions seen most commonly on distal LEs Dx: Neutrophils and mononuclear cells invading the walls of dermal capillaries, arterioles, and venules seen on bx |
Removal of offending drugs or treatment of the infectious etiology Favorably responds to NSAIDs, colchicine, or dapsone Manage urticarial lesions w/combining antihistamines (H1 and H2 blockers) |

∗ Vasculitis: Inflammation of blood vessel walls that causes vessel narrowing/occlusion, aneurysm, or rupture.
Etiology
 The exact etiology and pathogenesis are uncertain.
The exact etiology and pathogenesis are uncertain. Genetic susceptibility to lupus is likely inherited as a polygenic trait. Multiple genetic linkages including 1q23, 2q35-37, 6p21-11, and 12q24 show strong associations with SLE. Environmental factors such as ultraviolet light exposure and Epstein-Barr virus infection may have a triggering role. Autoantibodies can be present years before the diagnosis of SLE. Evidence supports the improper processing of nuclear proteins and nucleic acid from programmed cell death. This leads to the presentation of self-DNA to plasmacytoid dendritic cells. Plasmacytoid dendritic cells propagate antibody and immune complex production and other arms of specific autoimmunity.
Genetic susceptibility to lupus is likely inherited as a polygenic trait. Multiple genetic linkages including 1q23, 2q35-37, 6p21-11, and 12q24 show strong associations with SLE. Environmental factors such as ultraviolet light exposure and Epstein-Barr virus infection may have a triggering role. Autoantibodies can be present years before the diagnosis of SLE. Evidence supports the improper processing of nuclear proteins and nucleic acid from programmed cell death. This leads to the presentation of self-DNA to plasmacytoid dendritic cells. Plasmacytoid dendritic cells propagate antibody and immune complex production and other arms of specific autoimmunity.Diagnosis
H&P
 Constitutional: unexplained fever, fatigue, malaise
Constitutional: unexplained fever, fatigue, malaise Skin: malar rash sparing nasolabial folds (acute cutaneous lupus), annular or papulosquamous rash (subacute cutaneous lupus), raised erythematous patches with subsequent edematous plaques and adherent scales (discoid cutaneous lupus); alopecia, nasal, or oropharyngeal ulcerations; Raynaud’s phenomenon; petechiae, palpable purpura, skin ulceration, or digital ischemia (vasculitis); livedo reticularis or livedo racemosa (secondary antiphospholipid antibody syndrome)
Skin: malar rash sparing nasolabial folds (acute cutaneous lupus), annular or papulosquamous rash (subacute cutaneous lupus), raised erythematous patches with subsequent edematous plaques and adherent scales (discoid cutaneous lupus); alopecia, nasal, or oropharyngeal ulcerations; Raynaud’s phenomenon; petechiae, palpable purpura, skin ulceration, or digital ischemia (vasculitis); livedo reticularis or livedo racemosa (secondary antiphospholipid antibody syndrome) Musculoskeletal: arthritis (tenderness, swelling, effusion) typically affecting peripheral joints; myositis
Musculoskeletal: arthritis (tenderness, swelling, effusion) typically affecting peripheral joints; myositis Cardiac: pericardial rub (pericarditis), heart murmur (Libman-Sachs endocarditis and other valvular heart disease), congestive heart failure (myocarditis), premature atherosclerotic heart disease
Cardiac: pericardial rub (pericarditis), heart murmur (Libman-Sachs endocarditis and other valvular heart disease), congestive heart failure (myocarditis), premature atherosclerotic heart disease Pulmonary: pleuritis, pneumonitis, diffuse alveolar hemorrhage
Pulmonary: pleuritis, pneumonitis, diffuse alveolar hemorrhage Gastrointestinal: abdominal pain, intestinal vasculitis, ascites
Gastrointestinal: abdominal pain, intestinal vasculitis, ascites Neurologic: headache, psychosis, seizure, acute confusional states, peripheral or cranial neuropathy, transverse myelitis, CVA, chronic cognitive impairment
Neurologic: headache, psychosis, seizure, acute confusional states, peripheral or cranial neuropathy, transverse myelitis, CVA, chronic cognitive impairment Hematologic: anemia (hemolytic, anemia of chronic disease, aplastic anemia), thrombocytopenia, leukopenia, lymphadenopathy, secondary antiphospholipid antibody syndrome
Hematologic: anemia (hemolytic, anemia of chronic disease, aplastic anemia), thrombocytopenia, leukopenia, lymphadenopathy, secondary antiphospholipid antibody syndrome Renal: ARF, proteinuria, nephritic syndrome, nephrotic syndrome
Renal: ARF, proteinuria, nephritic syndrome, nephrotic syndromeW/Up
 The diagnosis of SLE is suspected when any four or more of the following 1997 American College of Rheumatology criteria are present:
The diagnosis of SLE is suspected when any four or more of the following 1997 American College of Rheumatology criteria are present:Labs
 Suggested initial laboratory evaluation of suspected SLE:
Suggested initial laboratory evaluation of suspected SLE: Consider additional laboratory testing:
Consider additional laboratory testing:Imaging
 CXR for evaluation of pulmonary involvement (pleural effusion, pulmonary infiltrates)
CXR for evaluation of pulmonary involvement (pleural effusion, pulmonary infiltrates) ECG for complaint of unexplained chest pain
ECG for complaint of unexplained chest pain Echo if unexplained murmur, evidence of new or unexplained congestive heart failure, or suspected pericarditis
Echo if unexplained murmur, evidence of new or unexplained congestive heart failure, or suspected pericarditisTreatment
 Defined courses of corticosteroids are useful for a variety of SLE symptoms.
Defined courses of corticosteroids are useful for a variety of SLE symptoms. Immunosuppressive drugs such as MTX or azathioprine are used as steroid-sparing drugs.
Immunosuppressive drugs such as MTX or azathioprine are used as steroid-sparing drugs. Joint pain and mild serositis are generally well controlled with NSAIDs or low-dose corticosteroids. Hydroxychloroquine and MTX are also effective for arthritis. Leflunomide or anti-TNF agents may be considered for difficult arthritis.
Joint pain and mild serositis are generally well controlled with NSAIDs or low-dose corticosteroids. Hydroxychloroquine and MTX are also effective for arthritis. Leflunomide or anti-TNF agents may be considered for difficult arthritis. Cutaneous manifestations
Cutaneous manifestations Hematologic manifestations
Hematologic manifestations Central nervous system manifestations
Central nervous system manifestations Renal disease (class III, IV, V lupus nephritis)
Renal disease (class III, IV, V lupus nephritis) Mycophenolate mofetil or azathioprine is used for maintenance Rx.
Mycophenolate mofetil or azathioprine is used for maintenance Rx. Severe nonrenal organ disease
Severe nonrenal organ disease New Rx
New RxF. Rheumatoid Arthritis (RA)
Etiology
 Unknown. It is likely that a combination of genetic and environmental factors lead to aberrant immune activation and inflammatory response in the joint. Stages of disease development presumably include:
Unknown. It is likely that a combination of genetic and environmental factors lead to aberrant immune activation and inflammatory response in the joint. Stages of disease development presumably include:Genetics
 Genetic factors account for more than 50% of risk of disease. RA is polygenic; identified genetic associations include HLA-DRB1, PTPN22, and PADI4.
Genetic factors account for more than 50% of risk of disease. RA is polygenic; identified genetic associations include HLA-DRB1, PTPN22, and PADI4.Diagnosis
 The American College of Rheumatology and the European League Against Rheumatism developed newer classification criteria for RA. Four variables constitute these criteria.
The American College of Rheumatology and the European League Against Rheumatism developed newer classification criteria for RA. Four variables constitute these criteria.H&P
 Initial presentation:
Initial presentation: Chronic long-standing disease:
Chronic long-standing disease: Extra-articular manifestations:
Extra-articular manifestations:
 Ocular disease
Ocular diseaseLabs
 RF: an immunoglobulin directed against the Fc region of the IgG
RF: an immunoglobulin directed against the Fc region of the IgG Anti–cyclic citrullinated peptide (anti-CCP) antibodies. More specific than RF for rheumatoid arthritis (up to 95% to 98%). Sensitivity similar to RF. The presence of either RF or anti-CCP (“sero[+] RA”) is associated with more severe RA.
Anti–cyclic citrullinated peptide (anti-CCP) antibodies. More specific than RF for rheumatoid arthritis (up to 95% to 98%). Sensitivity similar to RF. The presence of either RF or anti-CCP (“sero[+] RA”) is associated with more severe RA. ↑ ESR, ↑ CRP
↑ ESR, ↑ CRP CBC w/diff: possible mild anemia and leukocytosis
CBC w/diff: possible mild anemia and leukocytosis Synovial fluid: inflammatory, with >2000 PMNs
Synovial fluid: inflammatory, with >2000 PMNsImaging
 Plain radiography:
Plain radiography: MRI and musculoskeletal ultrasound are more sensitive for detecting erosive disease and joint effusion/synovitis.
MRI and musculoskeletal ultrasound are more sensitive for detecting erosive disease and joint effusion/synovitis.Treatment
 Early identification and treatment of RA with DMARDs are crucial. More than half of pts have radiographic joint damage within 2 yr of disease onset, but early aggressive treatment (with DMARDs) is associated with less damage.
Early identification and treatment of RA with DMARDs are crucial. More than half of pts have radiographic joint damage within 2 yr of disease onset, but early aggressive treatment (with DMARDs) is associated with less damage. NSAIDs: sometimes used initially to relieve pain and mild inflammation; may also be used later in the disease course for additional control of pain and inflammation. NSAIDs are not disease modifying.
NSAIDs: sometimes used initially to relieve pain and mild inflammation; may also be used later in the disease course for additional control of pain and inflammation. NSAIDs are not disease modifying. Corticosteroids: oral or intra-articular, frequently used initially to reduce inflammation rapidly until oral DMARD treatments take effect. They may also be used during acute flares or in low doses for additional control of inflammation. They have many side effects, including but not limited to weight gain and increased risk of diabetes, osteoporosis, and avascular necrosis.
Corticosteroids: oral or intra-articular, frequently used initially to reduce inflammation rapidly until oral DMARD treatments take effect. They may also be used during acute flares or in low doses for additional control of inflammation. They have many side effects, including but not limited to weight gain and increased risk of diabetes, osteoporosis, and avascular necrosis. DMARDs can be classified into “nonbiologic” and “biologic” treatments.
DMARDs can be classified into “nonbiologic” and “biologic” treatments. Nonbiologic DMARDs: commonly used agents are MTX, hydroxychloroquine, sulfasalazine, and leflunomide. Most of these are associated with potential toxicity and require close monitoring. They are also slow-acting drugs that require >8 wk to become fully effective.
Nonbiologic DMARDs: commonly used agents are MTX, hydroxychloroquine, sulfasalazine, and leflunomide. Most of these are associated with potential toxicity and require close monitoring. They are also slow-acting drugs that require >8 wk to become fully effective. MTX is the most commonly used DMARD worldwide for the treatment of RA.
MTX is the most commonly used DMARD worldwide for the treatment of RA. “Triple Rx”(MTX, hydroxychloroquine, and sulfasalazine) is superior to MTX alone.
“Triple Rx”(MTX, hydroxychloroquine, and sulfasalazine) is superior to MTX alone. Biologic DMARDs: newer biologically engineered therapies that target cytokines and cells involved in the RA inflammatory response. Major side effects include an increased risk of severe infection, most notably reactivation of TB with anti-TNF agents. A (−) PPD is prerequisite to initiate Rx. Biologic DMARDs are most effective when used in combination with a nonbiologic DMARD, usually MTX.
Biologic DMARDs: newer biologically engineered therapies that target cytokines and cells involved in the RA inflammatory response. Major side effects include an increased risk of severe infection, most notably reactivation of TB with anti-TNF agents. A (−) PPD is prerequisite to initiate Rx. Biologic DMARDs are most effective when used in combination with a nonbiologic DMARD, usually MTX. TNF-α inhibitors (TNFIs): include infliximab, etanercept, adalimumab, certolizumab pegol, and golimumab
TNF-α inhibitors (TNFIs): include infliximab, etanercept, adalimumab, certolizumab pegol, and golimumab Abatacept (CTLA-4Ig): a recombinant protein that prevents costimulatory binding of antigen presenting cell to T cell, thus preventing T-cell activation
Abatacept (CTLA-4Ig): a recombinant protein that prevents costimulatory binding of antigen presenting cell to T cell, thus preventing T-cell activation Tocilizumab (anti-IL6): a monoclonal antibody against the IL-6 receptor
Tocilizumab (anti-IL6): a monoclonal antibody against the IL-6 receptor Rituximab (anti-CD20): a monoclonal antibody against CD20 antigen on B lymphocytes
Rituximab (anti-CD20): a monoclonal antibody against CD20 antigen on B lymphocytesG. Spondyloarthropathies
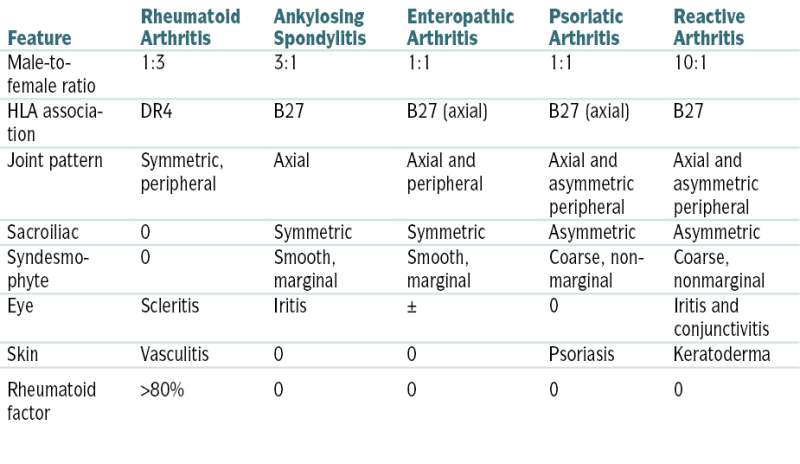
TABLE 12-2
Comparison of Rheumatoid Arthritis and the Spondyloarthropathies
| Feature | Rheumatoid Arthritis | Ankylosing Spondylitis | Enteropathic Arthritis | Psoriatic Arthritis | Reactive Arthritis |
| Male-to-female ratio | 1:3 | 3:1 | 1:1 | 1:1 | 10:1 |
| HLA association | DR4 | B27 | B27 (axial) | B27 (axial) | B27 |
| Joint pattern | Symmetric, peripheral | Axial | Axial and peripheral | Axial and asymmetric peripheral | Axial and asymmetric peripheral |
| Sacroiliac | 0 | Symmetric | Symmetric | Asymmetric | Asymmetric |
| Syndesmophyte | 0 | Smooth, marginal | Smooth, marginal | Coarse, nonmarginal | Coarse, nonmarginal |
| Eye | Scleritis | Iritis | ± | 0 | Iritis and conjunctivitis |
| Skin | Vasculitis | 0 | 0 | Psoriasis | Keratoderma |
| Rheumatoid factor | >80% | 0 | 0 | 0 | 0 |
From Goldman L, Schafer AI (eds): Goldman’s Cecil Medicine, 24th edition. Philadelphia, Saunders, 2012.
H. Systemic Sclerosis
Etiology
 The etiology of this condition is unknown. Genetic profiles show clustering of different alleles according to the subtype of SSc. There is abnormal selection of fibroblasts and aberrant control of connective tissue synthesis by fibroblasts and other cells. Although there are characteristic autoantibodies detected, it is not clear that they directly participate in the pathogenesis of the disease.
The etiology of this condition is unknown. Genetic profiles show clustering of different alleles according to the subtype of SSc. There is abnormal selection of fibroblasts and aberrant control of connective tissue synthesis by fibroblasts and other cells. Although there are characteristic autoantibodies detected, it is not clear that they directly participate in the pathogenesis of the disease. Extracellular connective tissue activation
Extracellular connective tissue activation Frequent immunologic abnormalities including autoantibodies
Frequent immunologic abnormalities including autoantibodies Inflammation in the early stages of disease
Inflammation in the early stages of disease Vasoconstriction
VasoconstrictionDiagnosis
Physical Findings
 Skin
Skin Musculoskeletal
Musculoskeletal Gastrointestinal involvement
Gastrointestinal involvement Pulmonary manifestations
Pulmonary manifestations Cardiac involvement
Cardiac involvement Renal involvement
Renal involvement Other organ involvement
Other organ involvement CREST syndrome (term now replaced by lcSSc)
CREST syndrome (term now replaced by lcSSc)Clinical Presentation
 Raynaud’s phenomenon: initial complaint in 70% of pts (The prevalence of Raynaud’s phenomenon is 5% to 10% in the general population; most cases do not progress to scleroderma.)
Raynaud’s phenomenon: initial complaint in 70% of pts (The prevalence of Raynaud’s phenomenon is 5% to 10% in the general population; most cases do not progress to scleroderma.) Finger or hand swelling that is sometimes associated with carpal tunnel syndrome
Finger or hand swelling that is sometimes associated with carpal tunnel syndrome Arthralgias/arthritis
Arthralgias/arthritis Internal organ involvement
Internal organ involvementLabs
 (+) Anti-Th/To antibodies with lcSSC (associated with PAH)
(+) Anti-Th/To antibodies with lcSSC (associated with PAH) Antinuclear antibodies (homogeneous, speckled, or nucleolar patterns) in both lcSSC and dcSSC
Antinuclear antibodies (homogeneous, speckled, or nucleolar patterns) in both lcSSC and dcSSC (−) antibody to native DNA
(−) antibody to native DNA (−) anti–smooth muscle antibody
(−) anti–smooth muscle antibody Autoantibodies against ribonucleoprotein (+) in 20% of pts, anti-U3RNP with dcSSC (associated with PAH, myositis)
Autoantibodies against ribonucleoprotein (+) in 20% of pts, anti-U3RNP with dcSSC (associated with PAH, myositis) RF (+) in 20% of pts
RF (+) in 20% of pts Anticentromere antibodies in one third of pts with lcSSc
Anticentromere antibodies in one third of pts with lcSSc (+) extractable nuclear antibody to Scl-70 in 40% of pts with dcSSc (also called anti-topoisomerase Ab, associated with ILD)
(+) extractable nuclear antibody to Scl-70 in 40% of pts with dcSSc (also called anti-topoisomerase Ab, associated with ILD) Routine biochemistry tests may indicate specific organ involvement (e.g., liver, kidney, muscle)
Routine biochemistry tests may indicate specific organ involvement (e.g., liver, kidney, muscle)Imaging
 Arthritis: joint radiographs
Arthritis: joint radiographs Gastrointestinal
Gastrointestinal Pulmonary
Pulmonary Heart
Heart Kidney: renal biopsy
Kidney: renal biopsy Skin: skin biopsy
Skin: skin biopsyTreatment
 No disease-modifying Rx available. Immunosuppressive agents used in individual pts. Prednisone should be used with extreme caution, especially in doses >20 mg/day
No disease-modifying Rx available. Immunosuppressive agents used in individual pts. Prednisone should be used with extreme caution, especially in doses >20 mg/day Raynaud’s syndrome:
Raynaud’s syndrome: Arthralgias: nonsteroidal anti-inflammatory drugs
Arthralgias: nonsteroidal anti-inflammatory drugs Skin: For extensive skin fibrosis, immunomodulatory drugs have been used such as MTX, mycophenolate, and cyclophosphamide but have not been proved to be beneficial.
Skin: For extensive skin fibrosis, immunomodulatory drugs have been used such as MTX, mycophenolate, and cyclophosphamide but have not been proved to be beneficial. Esophageal reflux
Esophageal reflux Pulmonary HTN and fibrosis
Pulmonary HTN and fibrosis Renal involvement
Renal involvementI. Enteropathic Arthritis
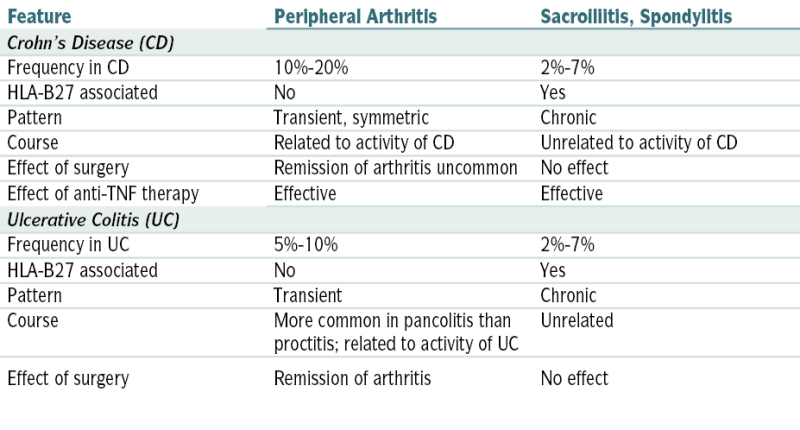
TABLE 12-3
Enteropathic Arthritis
| Feature | Peripheral Arthritis | Sacroiliitis, Spondylitis |
| Crohn’s Disease (CD) | ||
| Frequency in CD | 10%-20% | 2%-7% |
| HLA-B27 associated | No | Yes |
| Pattern | Transient, symmetric | Chronic |
| Course | Related to activity of CD | Unrelated to activity of CD |
| Effect of surgery | Remission of arthritis uncommon | No effect |
| Effect of anti-TNF therapy | Effective | Effective |
| Ulcerative Colitis (UC) | ||
| Frequency in UC | 5%-10% | 2%-7% |
| HLA-B27 associated | No | Yes |
| Pattern | Transient | Chronic |
| Course | More common in pancolitis than proctitis; related to activity of UC | Unrelated |
| Effect of surgery | Remission of arthritis | No effect |
From Goldman L, Schafer AI (eds): Goldman’s Cecil Medicine, 24th edition. Philadelphia, Saunders, 2012.
J. Crystal-Induced Arthritides
1. GOUT
Diagnosis
H&P
 The typical presentation is monarticular and characterized by sudden severe pain involving the first metatarsophalangeal joint (podagra), although the midtarsal and ankle are also frequently affected; any joint may be involved, but acute polyarthritis is uncommon.
The typical presentation is monarticular and characterized by sudden severe pain involving the first metatarsophalangeal joint (podagra), although the midtarsal and ankle are also frequently affected; any joint may be involved, but acute polyarthritis is uncommon.TABLE 12-4
Comparison of Crystal-Induced Arthritides
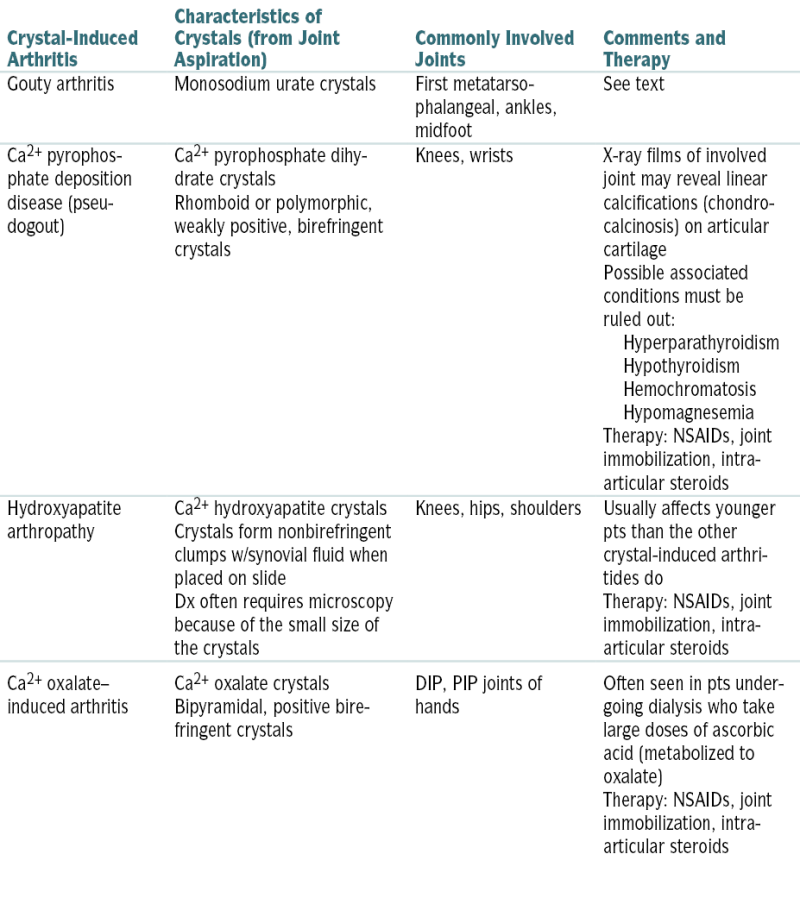
| Crystal-Induced Arthritis | Characteristics of Crystals (from Joint Aspiration) | Commonly Involved Joints | Comments and Therapy |
| Gouty arthritis | Monosodium urate crystals | First metatarsophalangeal, ankles, midfoot | See text |
| Ca2+ pyrophosphate deposition disease (pseudogout) | Ca2+ pyrophosphate dihydrate crystals Rhomboid or polymorphic, weakly positive, birefringent crystals |
Knees, wrists | X-ray films of involved joint may reveal linear calcifications (chondrocalcinosis) on articular cartilage Possible associated conditions must be ruled out: Hyperparathyroidism
Hypothyroidism
Hemochromatosis
Hypomagnesemia
Therapy: NSAIDs, joint immobilization, intra-articular steroids |
| Hydroxyapatite arthropathy | Ca2+ hydroxyapatite crystals Crystals form nonbirefringent clumps w/synovial fluid when placed on slide Dx often requires microscopy because of the small size of the crystals |
Knees, hips, shoulders | Usually affects younger pts than the other crystal-induced arthritides do Therapy: NSAIDs, joint immobilization, intra-articular steroids |
| Ca2+ oxalate–induced arthritis | Ca2+ oxalate crystals Bipyramidal, positive birefringent crystals |
DIP, PIP joints of hands | Often seen in pts undergoing dialysis who take large doses of ascorbic acid (metabolized to oxalate) Therapy: NSAIDs, joint immobilization, intra-articular steroids |
 PE reveals a warm, tender, swollen, erythematous joint; fever may be present, particularly if several joints are involved. Extensive soft tissue swelling, heat, and erythema extending to above and below the affected joint are frequently present and may be confused w/cellulitis.
PE reveals a warm, tender, swollen, erythematous joint; fever may be present, particularly if several joints are involved. Extensive soft tissue swelling, heat, and erythema extending to above and below the affected joint are frequently present and may be confused w/cellulitis.Labs
 Serum uric acid level may be ↑, but it is often nl during the acute attack, later rising when the sx resolve.
Serum uric acid level may be ↑, but it is often nl during the acute attack, later rising when the sx resolve. Aspiration and analysis of synovial fluid from the inflamed joint confirm the dx; examination of the fluid w/a polarized light microscope w/compensator reveals monosodium urate crystals (needle-shaped, strongly (−) birefringent crystals) w/synovial fluid leukocytes.
Aspiration and analysis of synovial fluid from the inflamed joint confirm the dx; examination of the fluid w/a polarized light microscope w/compensator reveals monosodium urate crystals (needle-shaped, strongly (−) birefringent crystals) w/synovial fluid leukocytes.Treatment
 NSAIDs: PO: indomethacin, naproxen, ibuprofen. Ketorolac may be given IM in NPO pts.
NSAIDs: PO: indomethacin, naproxen, ibuprofen. Ketorolac may be given IM in NPO pts. Colchicine can be given PO or IV; PO dose is 1.2 mg followed by a second dose of 0.6 mg 1 hr later. IV administration has been associated w/risk of bone marrow suppression and renal or hepatic cell damage. Extravasation can also cause tissue necrosis.
Colchicine can be given PO or IV; PO dose is 1.2 mg followed by a second dose of 0.6 mg 1 hr later. IV administration has been associated w/risk of bone marrow suppression and renal or hepatic cell damage. Extravasation can also cause tissue necrosis. Glucocorticoids (triamcinolone acetonide or ACTH): reserved for pts w/contraindications to NSAIDs or colchicine or if PO medication is precluded (e.g., postop). Triamcinolone acetonide, 60 mg IM; or ACTH, 40 IM or 25 by slow IV infusion. Intra-articular steroids may be used to treat a single inflamed joint (dexamethasone PO4-3 1-6 mg).
Glucocorticoids (triamcinolone acetonide or ACTH): reserved for pts w/contraindications to NSAIDs or colchicine or if PO medication is precluded (e.g., postop). Triamcinolone acetonide, 60 mg IM; or ACTH, 40 IM or 25 by slow IV infusion. Intra-articular steroids may be used to treat a single inflamed joint (dexamethasone PO4-3 1-6 mg).
Clinical Pearls
 Xanthine oxidase inhibitors (allopurinol, febuxostat) are used in pts w/frequent recurrent attacks. Hypouricemic Rx should not be started for at least 4 wk after the acute attack has resolved because it may prolong the acute attack and can also precipitate new attacks by rapidly lowering the serum uric acid level.
Xanthine oxidase inhibitors (allopurinol, febuxostat) are used in pts w/frequent recurrent attacks. Hypouricemic Rx should not be started for at least 4 wk after the acute attack has resolved because it may prolong the acute attack and can also precipitate new attacks by rapidly lowering the serum uric acid level. Colchicine, 0.6 mg PO bid, is indicated for acute gout prophylaxis before hypouricemic Rx is started. It is generally discontinued 6-8 wk after normalization of serum urate levels. Long-term colchicine Rx (0.6 mg qd or bid) may be necessary in pts w/frequent gout attacks despite the use of uricosuric agents. It can also be used as an alternative to uricosuric agents.
Colchicine, 0.6 mg PO bid, is indicated for acute gout prophylaxis before hypouricemic Rx is started. It is generally discontinued 6-8 wk after normalization of serum urate levels. Long-term colchicine Rx (0.6 mg qd or bid) may be necessary in pts w/frequent gout attacks despite the use of uricosuric agents. It can also be used as an alternative to uricosuric agents.K. Idiopathic Inflammatory Myopathies
TABLE 12-5
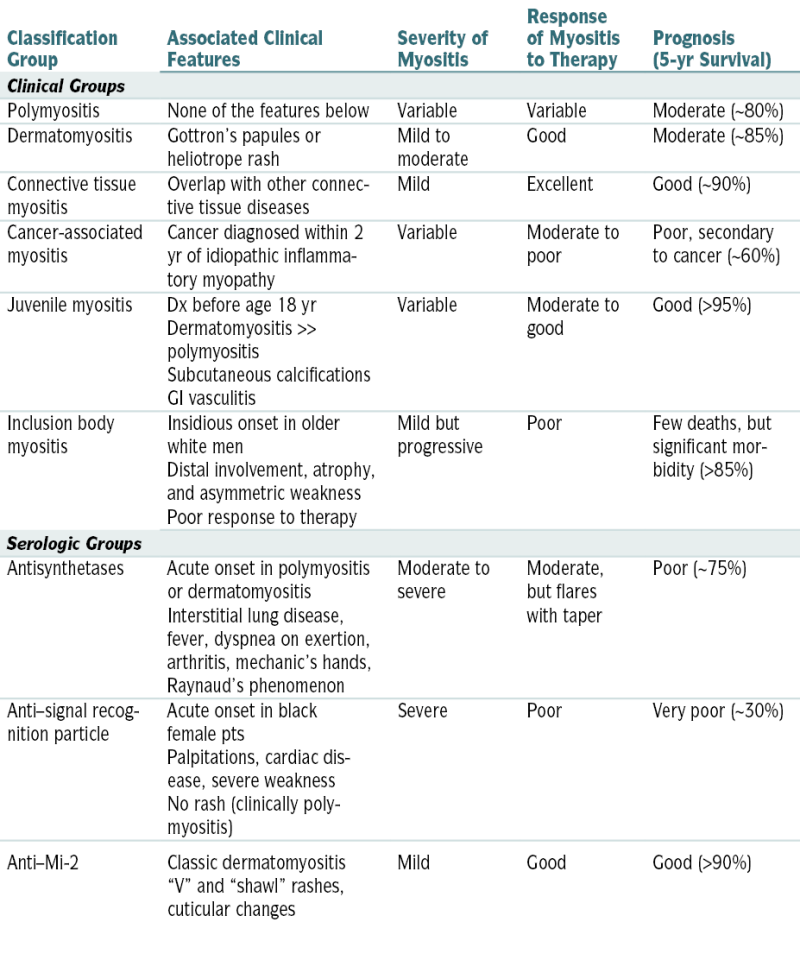
Classification of the Idiopathic Inflammatory Myopathies
| Classification Group | Associated Clinical Features | Severity of Myositis | Response of Myositis to Therapy | Prognosis (5-yr Survival) |
| Clinical Groups | ||||
| Polymyositis | None of the features below | Variable | Variable | Moderate (∼80%) |
| Dermatomyositis | Gottron’s papules or heliotrope rash | Mild to moderate | Good | Moderate (∼85%) |
| Connective tissue myositis | Overlap with other connective tissue diseases | Mild | Excellent | Good (∼90%) |
| Cancer-associated myositis | Cancer diagnosed within 2 yr of idiopathic inflammatory myopathy | Variable | Moderate to poor | Poor, secondary to cancer (∼60%) |
| Juvenile myositis | Dx before age 18 yr Dermatomyositis >> polymyositis Subcutaneous calcifications GI vasculitis |
Variable | Moderate to good | Good (>95%) |
| Inclusion body myositis | Insidious onset in older white men Distal involvement, atrophy, and asymmetric weakness Poor response to therapy |
Mild but progressive | Poor | Few deaths, but significant morbidity (>85%) |
| Serologic Groups | ||||
| Antisynthetases | Acute onset in polymyositis or dermatomyositis Interstitial lung disease, fever, dyspnea on exertion, arthritis, mechanic’s hands, Raynaud’s phenomenon |
Moderate to severe | Moderate, but flares with taper | Poor (∼75%) |
| Anti–signal recognition particle | Acute onset in black female pts Palpitations, cardiac disease, severe weakness No rash (clinically polymyositis) |
Severe | Poor | Very poor (∼30%) |
| Anti–Mi-2 | Classic dermatomyositis “V” and “shawl” rashes, cuticular changes | Mild | Good | Good (>90%) |
From Goldman L, Schafer AI (eds): Goldman’s Cecil Medicine, 24th edition. Philadelphia, Saunders, 2012.