380 |
Rheumatoid Arthritis |
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown etiology marked by a symmetric, peripheral polyarthritis. It is the most common form of chronic inflammatory arthritis and often results in joint damage and physical disability. Because it is a systemic disease, RA may result in a variety of extraarticular manifestations, including fatigue, subcutaneous nodules, lung involvement, pericarditis, peripheral neuropathy, vasculitis, and hematologic abnormalities.
Insights gained by a wealth of basic and clinical research over the past two decades have revolutionized the contemporary paradigms for the diagnosis and management of RA. Serum antibodies to cyclic citrullinated peptides (anti-CCPs) are routinely used along with rheumatoid factor as a biomarker of diagnostic and prognostic significance. Advances in imaging modalities have improved our ability to detect joint inflammation and destruction in RA. The science of RA has taken a major leap forward with the identification of new disease-related genes and further deciphering of the molecular pathways of disease pathogenesis. The relative importance of these different mechanisms has been highlighted by the observed benefits of the new class of highly targeted biologic and small-molecule therapies. Despite these gains, incomplete understanding of the initiating pathogenic pathways of RA remains a sizable barrier to its cure and prevention.
The last two decades have witnessed a remarkable improvement in the outcomes of RA. The historic descriptions of crippling arthritis are currently encountered much less frequently. Much of this progress can be traced to the expanded therapeutic armamentarium and the adoption of early treatment intervention. The shift in treatment strategy dictates a new mind-set for primary care practitioners—namely, one that demands early referral of patients with inflammatory arthritis to a rheumatologist for prompt diagnosis and initiation of therapy. Only then will patients achieve their best outcomes.
CLINICAL FEATURES
The incidence of RA increases between 25 and 55 years of age, after which it plateaus until the age of 75 and then decreases. The presenting symptoms of RA typically result from inflammation of the joints, tendons, and bursae. Patients often complain of early morning joint stiffness lasting more than 1 h that eases with physical activity. The earliest involved joints are typically the small joints of the hands and feet. The initial pattern of joint involvement may be monoarticular, oligoarticular (≤4 joints), or polyarticular (>5 joints), usually in a symmetric distribution. Some patients with inflammatory arthritis will present with too few affected joints to be classified as having RA—so-called undifferentiated inflammatory arthritis. Those with an undifferentiated arthritis who are most likely to be diagnosed later with RA have a higher number of tender and swollen joints, test positive for serum rheumatoid factor (RF) or anti-CCP antibodies, and have higher scores for physical disability.
Once the disease process of RA is established, the wrists, metacarpophalangeal (MCP), and proximal interphalangeal (PIP) joints stand out as the most frequently involved joints (Fig. 380-1). Distal interphalangeal (DIP) joint involvement may occur in RA, but it usually is a manifestation of coexistent osteoarthritis. Flexor tendon tenosynovitis is a frequent hallmark of RA and leads to decreased range of motion, reduced grip strength, and “trigger” fingers. Progressive destruction of the joints and soft tissues may lead to chronic, irreversible deformities. Ulnar deviation results from subluxation of the MCP joints, with subluxation of the proximal phalanx to the volar side of the hand. Hyperextension of the PIP joint with flexion of the DIP joint (“swan-neck deformity”), flexion of the PIP joint with hyperextension of the DIP joint (“boutonnière deformity”), and subluxation of the first MCP joint with hyperextension of the first interphalangeal (IP) joint (“Z-line deformity”) also may result from damage to the tendons, joint capsule, and other soft tissues in these small joints. Inflammation about the ulnar styloid and tenosynovitis of the extensor carpi ulnaris may cause subluxation of the distal ulna, resulting in a “piano-key movement” of the ulnar styloid. Although metatarsophalangeal (MTP) joint involvement in the feet is an early feature of disease, chronic inflammation of the ankle and midtarsal regions usually comes later and may lead to pes planovalgus (“flat feet”). Large joints, including the knees and shoulders, are often affected in established disease, although these joints may remain asymptomatic for many years after onset.
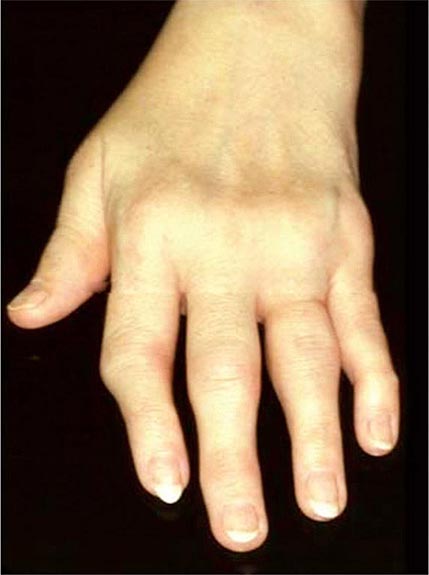
FIGURE 380-1 Metacarpophalangeal and proximal interphalangeal joint swelling in rheumatoid arthritis. (Courtesy of the American College of Rheumatology Image Bank.)
Atlantoaxial involvement of the cervical spine is clinically noteworthy because of its potential to cause compressive myelopathy and neurologic dysfunction. Neurologic manifestations are rarely a presenting sign or symptom of atlantoaxial disease, but they may evolve over time with progressive instability of C1 on C2. The prevalence of atlantoaxial subluxation has been declining in recent years, and occurs now in less than 10% of patients. Unlike the spondyloarthritides (Chap. 384), RA rarely affects the thoracic and lumbar spine. Radiographic abnormalities of the temporomandibular joint occur commonly in patients with RA, but they are generally not associated with significant symptoms or functional impairment.
Extraarticular manifestations may develop during the clinical course of RA, even prior to the onset of arthritis (Fig. 380-2). Patients most likely to develop extraarticular disease have a history of smoking, have early onset of significant physical disability, and test positive for serum RF. Subcutaneous nodules, secondary Sjögren’s syndrome, pulmonary nodules, and anemia are among the most frequently observed extraarticular manifestations. Recent studies have shown a decrease in the incidence and severity of at least some extraarticular manifestations, particularly Felty’s syndrome and vasculitis.
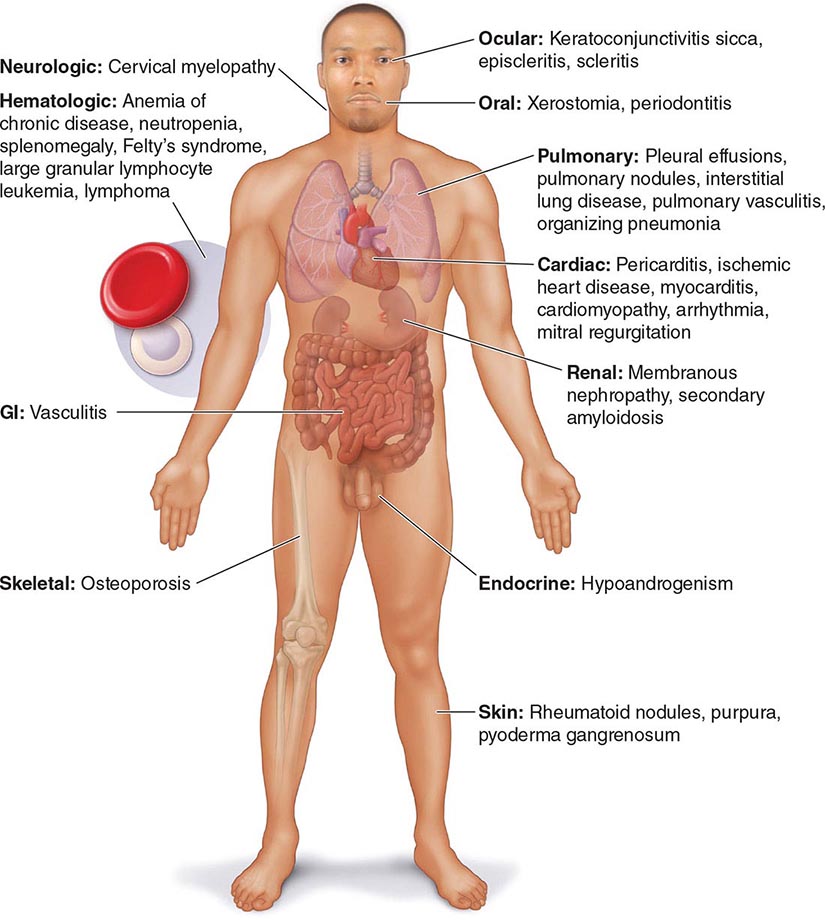
FIGURE 380-2 Extraarticular manifestations of rheumatoid arthritis.
The most common systemic and extraarticular features of RA are described in more detail in the sections below.
CONSTITUTIONAL
These signs and symptoms include weight loss, fever, fatigue, malaise, depression, and in the most severe cases, cachexia; they generally reflect a high degree of inflammation and may even precede the onset of joint symptoms. In general, the presence of a fever of >38.3°C (101°F) at any time during the clinical course should raise suspicion of systemic vasculitis (see below) or infection.
NODULES
Subcutaneous nodules occur in 30–40% of patients and more commonly in those with the highest levels of disease activity, the disease-related shared epitope (see below), a positive test for serum RF, and radiographic evidence of joint erosions. When palpated, the nodules are generally firm; nontender; and adherent to periosteum, tendons, or bursae; developing in areas of the skeleton subject to repeated trauma or irritation such as the forearm, sacral prominences, and Achilles tendon. They may also occur in the lungs, pleura, pericardium, and peritoneum. Nodules are typically benign, although they can be associated with infection, ulceration, and gangrene.
SJÖGREN’S SYNDROME
Secondary Sjögren’s syndrome (Chap. 383) is defined by the presence of either keratoconjunctivitis sicca (dry eyes) or xerostomia (dry mouth) in association with another connective tissue disease, such as RA. Approximately 10% of patients with RA have secondary Sjögren’s syndrome.
PULMONARY
Pleuritis, the most common pulmonary manifestation of RA, may produce pleuritic chest pain and dyspnea, as well as a pleural friction rub and effusion. Pleural effusions tend to be exudative with increased numbers of monocytes and neutrophils. Interstitial lung disease (ILD) may also occur in patients with RA and is heralded by symptoms of dry cough and progressive shortness of breath. ILD can be associated with cigarette smoking and is generally found in patients with higher disease activity, although it may be diagnosed in up to 3.5% of patients prior to the onset of joint symptoms. Diagnosis is readily made by high-resolution chest computed tomography (CT) scan. Pulmonary function testing shows a restrictive pattern (e.g., reduced total lung capacity) with a reduced diffusing capacity for carbon monoxide (DLCO). The presence of ILD confers a poor prognosis. The prognosis is not quite as poor as that of idiopathic pulmonary fibrosis (e.g., usual interstitial pneumonitis) because ILD secondary to RA responds more favorably than idiopathic ILD to immunosuppressive therapy (Chap. 315). Pulmonary nodules may be solitary or multiple. Caplan’s syndrome is a rare subset of pulmonary nodulosis characterized by the development of nodules and pneumoconiosis following silica exposure. Other less common pulmonary findings include respiratory bronchiolitis and bronchiectasis.
CARDIAC
The most frequent site of cardiac involvement in RA is the pericardium. However, clinical manifestations of pericarditis occur in less than 10% of patients with RA despite the fact that pericardial involvement may be detected in nearly one-half of the these patients by echocardiogram or autopsy studies. Cardiomyopathy, another clinically important manifestation of RA, may result from necrotizing or granulomatous myocarditis, coronary artery disease, or diastolic dysfunction. This involvement too may be subclinical and only identified by echocardiography or cardiac magnetic resonance imaging (MRI). Rarely, the heart muscle may contain rheumatoid nodules or be infiltrated with amyloid. Mitral regurgitation is the most common valvular abnormality in RA, occurring at a higher frequency than the general population.
VASCULITIS
Rheumatoid vasculitis (Chap. 385) typically occurs in patients with long-standing disease, a positive test for serum RF, and hypocomplementemia. The overall incidence has decreased significantly in the last decade to be less than 1% of patients. The cutaneous signs vary and include petechiae, purpura, digital infarcts, gangrene, livedo reticularis, and in severe cases large, painful lower extremity ulcerations. Vasculitic ulcers, which may be difficult to distinguish from those caused by venous insufficiency, may be treated successfully with immunosuppressive agents (requiring cytotoxic treatment in severe cases) as well as skin grafting. Sensorimotor polyneuropathies, such as mononeuritis multiplex, may occur in association with systemic rheumatoid vasculitis.
HEMATOLOGIC
A normochromic, normocytic anemia often develops in patients with RA and is the most common hematologic abnormality. The degree of anemia parallels the degree of inflammation, correlating with the levels of serum C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). Platelet counts may also be elevated in RA as an acute-phase reactant. Immune-mediated thrombocytopenia is rare in this disease.
Felty’s syndrome is defined by the clinical triad of neutropenia, splenomegaly, and nodular RA and is seen in less than 1% of patients, although its incidence appears to be declining in the face of more aggressive treatment of the joint disease. It typically occurs in the late stages of severe RA and is more common in whites than other racial groups. T cell large granular lymphocyte leukemia (T-LGL) may have a similar clinical presentation and often occurs in association with RA. T-LGL is characterized by a chronic, indolent clonal growth of LGL cells, leading to neutropenia and splenomegaly. As opposed to Felty’s syndrome, T-LGL may develop early in the course of RA. Leukopenia apart from these disorders is uncommon and most often due to drug therapy.
LYMPHOMA
Large cohort studies have shown a two- to fourfold increased risk of lymphoma in RA patients compared with the general population. The most common histopathologic type of lymphoma is a diffuse large B cell lymphoma. The risk of developing lymphoma increases if the patient has high levels of disease activity or Felty’s syndrome.
ASSOCIATED CONDITIONS
In addition to extraarticular manifestations, several conditions associated with RA contribute to disease morbidity and mortality rates. They are worthy of mention because they affect chronic disease management.
Cardiovascular Disease The most common cause of death in patients with RA is cardiovascular disease. The incidence of coronary artery disease and carotid atherosclerosis is higher in RA patients than in the general population even when controlling for traditional cardiac risk factors, such as hypertension, obesity, hypercholesterolemia, diabetes, and cigarette smoking. Furthermore, congestive heart failure (including both systolic and diastolic dysfunction) occurs at an approximately twofold higher rate in RA than in the general population. The presence of elevated serum inflammatory markers appears to confer an increased risk of cardiovascular disease in this population.
Osteoporosis Osteoporosis is more common in patients with RA than an age- and sex-matched population, with prevalence rates of 20–30%. The inflammatory milieu of the joint probably spills over into the rest of the body and promotes generalized bone loss by activating osteoclasts. Chronic use of glucocorticoids and disability-related immobility also contributes to osteoporosis. Hip fractures are more likely to occur in patients with RA and are significant predictors of increased disability and mortality rate in this disease.
Hypoandrogenism Men and postmenopausal women with RA have lower mean serum testosterone, luteinizing hormone (LH), and dehydroepiandrosterone (DHEA) levels than control populations. It has thus been hypothesized that hypoandrogenism may play a role in the pathogenesis of RA or arise as a consequence of the chronic inflammatory response. It is also important to realize that patients receiving chronic glucocorticoid therapy may develop hypoandrogenism owing to inhibition of LH and follicle-stimulating hormone (FSH) secretion from the pituitary gland. Because low testosterone levels may lead to osteoporosis, men with hypoandrogenism should be considered for androgen replacement therapy.
EPIDEMIOLOGY
RA affects approximately 0.5–1% of the adult population worldwide. There is evidence that the overall incidence of RA has been decreasing in recent decades, whereas the prevalence has remained the same because individuals with RA are living longer. The incidence and prevalence of RA varies based on geographic location, both globally and among certain ethnic groups within a country (Fig. 380-3). For example, the Native American Yakima, Pima, and Chippewa tribes of North America have reported prevalence rates in some studies of nearly 7%. In contrast, many population studies from Africa and Asia show lower prevalence rates for RA in the range of 0.2–0.4%.
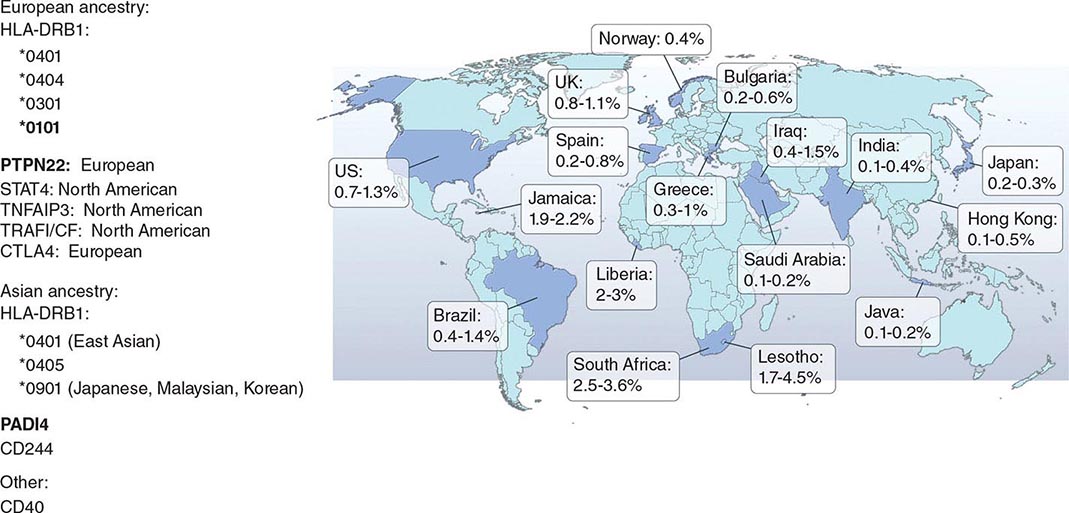
FIGURE 380-3 Global prevalence rates of rheumatoid arthritis (RA) with genetic associations. Listed are the major genetic alleles associated with RA. Although human leukocyte antigen (HLA)-DRB1 mutations are found globally, some alleles have been associated with RA in only certain ethnic groups.
Like many other autoimmune diseases, RA occurs more commonly in females than in males, with a 2–3:1 ratio. Interestingly, studies of RA from some of the Latin American and African countries show an even greater predominance of disease in females compared to males, with ratios of 6–8:1. Given this preponderance of females, various theories have been proposed to explain the possible role of estrogen in disease pathogenesis. Most of the theories center on the role of estrogens in enhancing the immune response. For example, some experimental studies have shown that estrogen can stimulate production of tumor necrosis factor a (TNF-α), a major cytokine in the pathogenesis of RA.
GENETIC CONSIDERATIONS
![]() It has been recognized for over 30 years that genetic factors contribute to the occurrence of RA as well as to its severity. The likelihood that a first-degree relative of a patient will share the diagnosis of RA is 2–10 times greater than in the general population. There remains, however, some uncertainty in the extent to which genetics plays a role in the causative mechanisms of RA. Although twin studies imply that genetic factors may explain up to 60% of the occurrence of RA, the more commonly stated estimate falls in the range of 10–25%. The estimate of genetic influence may vary across studies due to gene–environment interactions.
It has been recognized for over 30 years that genetic factors contribute to the occurrence of RA as well as to its severity. The likelihood that a first-degree relative of a patient will share the diagnosis of RA is 2–10 times greater than in the general population. There remains, however, some uncertainty in the extent to which genetics plays a role in the causative mechanisms of RA. Although twin studies imply that genetic factors may explain up to 60% of the occurrence of RA, the more commonly stated estimate falls in the range of 10–25%. The estimate of genetic influence may vary across studies due to gene–environment interactions.
The alleles known to confer the greatest risk of RA are located within the major histocompatibility complex (MHC). It has been estimated that one-third of the genetic risk for RA resides within this locus. Most, but probably not all, of this risk is associated with allelic variation in the HLA-DRB1 gene, which encodes the MHC II β-chain molecule. The disease-associated HLA-DRB1 alleles share an amino acid sequence at positions 70–74 in the third hypervariable regions of the HLA-DR β-chain, termed the shared epitope (SE). Carriership of the SE alleles is associated with production of anti-CCP antibodies and worse disease outcomes. Some of these HLA-DRB1 alleles bestow a high risk of disease (*0401), whereas others confer a more moderate risk (*0101, *0404, *1001, and *0901). Additionally, there is regional variation. In Greece, for example, where RA tends to be milder than in western European countries, RA susceptibility has been associated with the *0101 SE allele. By comparison, the *0401 or *0404 alleles are found in approximately 50–70% of northern Europeans and are the predominant risk alleles in this group. The most common disease susceptibility SE alleles in Asians, namely the Japanese, Koreans, and Chinese, are *0405 and *0901. Lastly, disease susceptibility of Native American populations such as the Pima and Tlingit Indians, where the prevalence of RA can be as high as 7%, is associated with the SE allele *1042. The risk of RA conferred by these SE alleles is less in African and Hispanic Americans than in individuals of European ancestry.
Genome-wide association studies (GWAS) have made possible the identification of several non-MHC-related genes that contribute to RA susceptibility. GWAS are based on the detection of single-nucleotide polymorphisms (SNPs), which allow for examination of the genetic architecture of complex diseases such as RA. There are approximately 10 million common SNPs within a human genome consisting of 3 billion base pairs. As a rule, GWAS identify only common variants, namely, those with a frequency of more than 5% in the general population.
Overall, several themes have emerged from GWAS in RA. First, the non-MHC loci identified as risk alleles for RA have only a modest effect on risk; they also contribute to the risk for developing other autoimmune diseases, such as type 1 diabetes mellitus, systemic lupus erythematosus, and multiple sclerosis. Second, although most of the non-HLA associations are described in patients with anti-CCP antibody-positive disease, there are several risk loci that are unique to anti-CCP antibody-negative disease. Third, risk alleles vary among ethnic groups. And fourth, the risk loci mostly reside in genes encoding proteins involved in the regulation of the immune response. However, the risk alleles identified by GWAS only account at present for approximately 5% of the genetic risk, suggesting that rare variants or other classes of DNA variants, such as variants in copy number, may be yet found that significantly contribute to the overall risk model.
Recently, imputation of SNP data from a GWAS meta-analysis shows amino acid substitutions in the MHC locus independently associated with the risk for RA are at position 11, 71, and 74 in HLA-DRβ1, position 9 of HLA-B, and position 9 of HLA-DPβ1. The amino acids at position 11, 71, and 74 are located in the antigen-binding grove of the HLA-DRβ1 molecule, highlighting positions 71 and 74 that form part of the original shared epitope.
Among the best examples of the non-MHC genes contributing to the risk of RA is the gene encoding protein tyrosine phosphatase non-receptor 22 (PTPN22). This gene varies in frequency among patients from different parts of Europe (e.g., 3–10%), but is absent in patients of East Asian ancestry. PTPN22 encodes lymphoid tyrosine phosphatase, a protein that regulates T and B cell function. Inheritance of the risk allele for PTPN22 produces a gain-of-function in the protein that is hypothesized to result in the abnormal thymic selection of autoreactive T and B cells and appears to be associated exclusively with anti-CCP-positive disease. The peptidyl arginine deiminase type IV (PADI4) gene is another risk allele that encodes an enzyme involved in the conversion of arginine to citrulline and is postulated to play a role in the development of antibodies to citrullinated antigens. A polymorphism in PADI4 has been associated with RA only in Asian populations.
Epigenetics is the study of heritable traits that affect gene expression but do not modify DNA sequence. It may provide a link between environmental exposure and predisposition to disease. The best-studied mechanisms include posttranslational histone modifications and DNA methylation. Although studies of epigenetic phenomena are limited, DNA methylation patterns have been shown to differ between RA patients and healthy controls, as well as patients with osteoarthritis.
ENVIRONMENTAL FACTORS
In addition to genetic predisposition, a host of environmental factors have been implicated in the pathogenesis of RA. The most reproducible of these environmental links is cigarette smoking. Numerous cohort and case control studies have demonstrated that smoking confers a relative risk for developing RA of 1.5–3.5. In particular, women who smoke cigarettes have a nearly 2.5 times greater risk of RA, a risk that persists even 15 years after smoking cessation. A twin who smokes will have a significantly higher risk for RA than his or her monozygotic co-twin, theoretically with the same genetic risk, who does not smoke. Interestingly, the risk from smoking is almost exclusively related to RF and anti-CCP antibody-positive disease. However, it has not been shown that smoking cessation, while having many health benefits, improves disease activity.
Researchers began to aggressively seek an infectious etiology for RA after the discovery in 1931 that sera from patients with this disease could agglutinate strains of streptococci. Certain viruses such as Epstein-Barr virus (EBV) have garnered the most interest over the past 30 years given their ubiquity, ability to persist for many years in the host, and frequent association with arthritic complaints. For example, titers of IgG antibodies against EBV antigens in the peripheral blood and saliva are significantly higher in patients with RA than the general population. EBV DNA has also been found in synovial fluid and synovial cells of RA patients. Because the evidence for these links is largely circumstantial, it has not been possible to directly implicate infection as a causative factor in RA.
PATHOLOGY
RA affects the synovial tissue and underlying cartilage and bone. The synovial membrane, which covers most articular surfaces, tendon sheaths, and bursae, normally is a thin layer of connective tissue. In joints, it faces the bone and cartilage, bridging the opposing bony surfaces and inserting at periosteal regions close to the articular cartilage. It consists primarily of two cell types—type A synoviocytes (macrophage-derived) and type B synoviocytes (fibroblast-derived). The synovial fibroblasts are the most abundant and produce the structural components of joints, including collagen, fibronectin, and laminin, as well as other extracellular constituents of the synovial matrix. The sublining layer consists of blood vessels and a sparse population of mononuclear cells within a loose network of connective tissue. Synovial fluid, an ultrafiltrate of blood, diffuses through the subsynovial lining tissue across the synovial membrane and into the joint cavity. Its main constituents are hyaluronan and lubricin. Hyaluronan is a glycosaminoglycan that contributes to the viscous nature of synovial fluid, which along with lubricin, lubricates the surface of the articular cartilage.
The pathologic hallmarks of RA are synovial inflammation and proliferation, focal bone erosions, and thinning of articular cartilage. Chronic inflammation leads to synovial lining hyperplasia and the formation of pannus, a thickened cellular membrane containing fibroblast-like synoviocytes and granulation-reactive fibrovascular tissue that invades the underlying cartilage and bone. The inflammatory infiltrate is made up of no less than six cell types: T cells, B cells, plasma cells, dendritic cells, mast cells, and, to a lesser extent, granulocytes. The T cells comprise 30–50% of the infiltrate, with the other cells accounting for the remainder. The topographical organization of these cells is complex and may vary among individuals with RA. Most often, the lymphocytes are diffusely organized among the tissue resident cells; however, in some cases, the B cells, T cells, and dendritic cells may form higher levels of organization, such as lymphoid follicles and germinal center–like structures. Growth factors secreted by synovial fibroblasts and macrophages promote the formation of new blood vessels in the synovial sublining that supply the increasing demands for oxygenation and nutrition required by the infiltrating leukocytes and expanding synovial tissue.
The structural damage to the mineralized cartilage and subchondral bone is mediated by the osteoclast. Osteoclasts are multinucleated giant cells that can be identified by their expression of CD68, tartrate-resistant acid phosphatase, cathepsin K, and the calcitonin receptor. They appear at the pannus-bone interface where they eventually form resorption lacunae. These lesions typically localize where the synovial membrane inserts into the periosteal surface at the edges of bones close to the rim of articular cartilage and at the attachment sites of ligaments and tendon sheaths. This process most likely explains why bone erosions usually develop at the radial sites of the MCP joints juxtaposed to the insertion sites of the tendons, collateral ligaments, and synovial membrane. Another form of bone loss is periarticular osteopenia that occurs in joints with active inflammation. It is associated with substantial thinning of the bony trabeculae along the metaphyses of bones, and likely results from inflammation of the bone marrow cavity. These lesions can be visualized on MRI scans, where they appear as signal alterations in the bone marrow adjacent to inflamed joints. Their signal characteristics show they are water-rich with a low fat content and are consistent with highly vascularized inflammatory tissue. These bone marrow lesions are often the forerunner of bone erosions.
The cortical bone layer that separates the bone marrow from the invading pannus is relatively thin and susceptible to penetration by the inflamed synovium. The bone marrow lesions seen on MRI scans are associated with an endosteal bone response characterized by the accumulation of osteoblasts and deposition of osteoid. Thus, in recent years, the concept of joint pathology in RA has been extended to include the bone marrow cavity. Finally, generalized osteoporosis, which results in the thinning of trabecular bone throughout the body, is a third form of bone loss found in patients with RA.
Articular cartilage is an avascular tissue comprised of a specialized matrix of collagens, proteoglycans, and other proteins. It is organized in four distinct regions (superficial, middle, deep, and calcified cartilage zones)—chondrocytes constitute the unique cellular component in these layers. Originally, cartilage was considered to be an inert tissue, but it is now known to be a highly responsive tissue that reacts to inflammatory mediators and mechanical factors, which in turn, alter the balance between cartilage anabolism and catabolism. In RA, the initial areas of cartilage degradation are juxtaposed to the synovial pannus. The cartilage matrix is characterized by a generalized loss of proteoglycan, most evident in the superficial zones adjacent to the synovial fluid. Degradation of cartilage may also take place in the perichondrocytic zone and in regions adjacent to the subchondral bone.
PATHOGENESIS
The pathogenic mechanisms of synovial inflammation are likely to result from a complex interplay of genetic, environmental, and immunologic factors that produces dysregulation of the immune system and a breakdown in self-tolerance (Fig. 380-4). Precisely what triggers these initiating events and what genetic and environmental factors disrupt the immune system remains a mystery. However, a detailed molecular picture is emerging of the mechanisms underlying the chronic inflammatory response and the destruction of the articular cartilage and bone.
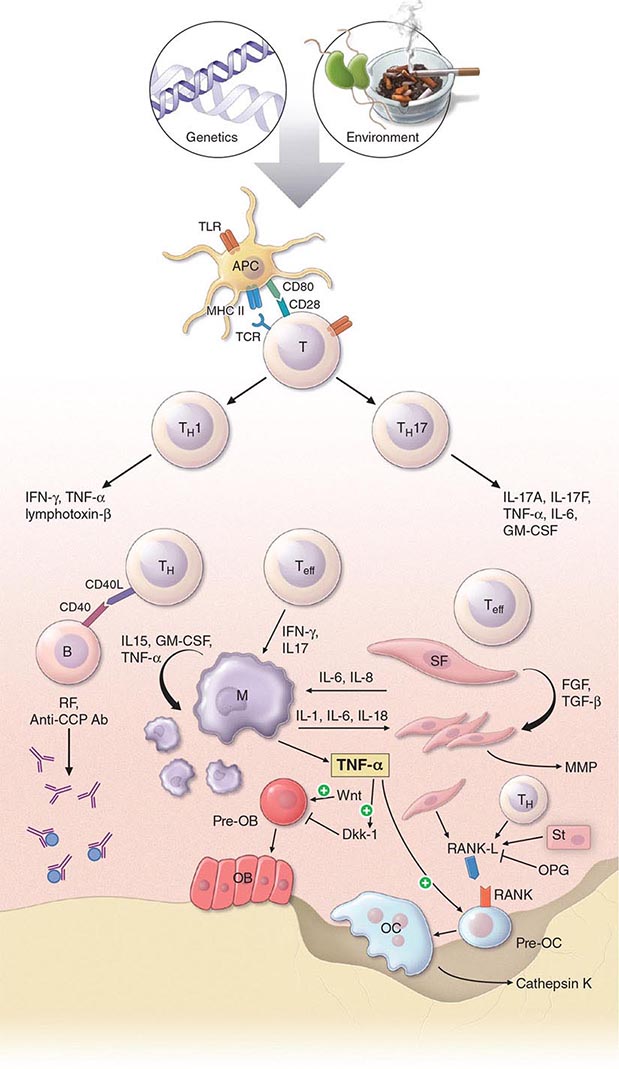
FIGURE 380-4 Pathophysiologic mechanisms of inflammation and joint destruction. Genetic predisposition along with environmental factors may trigger the development of rheumatoid arthritis (RA), with subsequent synovial T cell activation. CD4+ T cells become activated by antigen-presenting cells (APCs) through interactions between the T cell receptor and class II major histocompatibility complex (MHC)-peptide antigen (signal 1) with co-stimulation through the CD28-CD80/86 pathway, as well as other pathways (signal 2). In theory, ligands binding Toll-like receptors (TLRs) may further stimulate activation of APCs inside the joint. Synovial CD4+ T cells differentiate into TH1 and TH17 cells, each with their distinctive cytokine profile. CD4+ TH cells in turn activate B cells, some of which are destined to differentiate into autoantibody-producing plasma cells. Immune complexes, possibly comprised of rheumatoid factors (RFs) and anti–cyclic citrullinated peptides (CCP) antibodies, may form inside the joint, activating the complement pathway and amplifying inflammation. T effector cells stimulate synovial macrophages (M) and fibroblasts (SF) to secrete proinflammatory mediators, among which is tumor necrosis factor α (TNF-α). TNF-α upregulates adhesion molecules on endothelial cells, promoting leukocyte influx into the joint. It also stimulates the production of other inflammatory mediators, such as interleukin 1 (IL-1), IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF). TNF-α has a critically important function in regulating the balance between bone destruction and formation. It upregulates the expression of dickkopf-1 (DKK-1), which can then internalize Wnt receptors on osteoblast precursors. Wnt is a soluble mediator that promotes osteoblastogenesis and bone formation. In RA, bone formation is inhibited through the Wnt pathway, presumably due to the action of elevated levels of DKK-1. In addition to inhibiting bone formation, TNF-α stimulates osteoclastogenesis. However, it is not sufficient by itself to induce the differentiation of osteoclast precursors (Pre-OC) into activated osteoclasts capable of eroding bone. Osteoclast differentiation requires the presence of macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB (RANK) ligand (RANKL), which binds to RANK on the surface of Pre-OC. Inside the joint, RANKL is mainly derived from stromal cells, synovial fibroblasts, and T cells. Osteoprotegerin (OPG) acts as a decoy receptor for RANKL, thereby inhibiting osteoclastogenesis and bone loss. FGF, fibroblast growth factor; IFN, interferon; TGF, transforming growth factor.
In RA, the preclinical stage appears to be characterized by a breakdown in self-tolerance. This idea is supported by the finding that autoantibodies, such as RF and anti-CCP antibodies, may be found in sera from patients many years before clinical disease can be detected. However, the antigenic targets of anti-CCP antibodies and RF are not restricted to the joint, and their role in disease pathogenesis remains speculative. Anti-CCP antibodies are directed against deaminated peptides, which result from posttranslational modification by the enzyme PADI4. They recognize citrulline-containing regions of several different matrix proteins, including filaggrin, keratin, fibrinogen, and vimentin, and are present at higher levels in the joint fluid compared to the serum. Other autoantibodies have been found in a minority of patients with RA, but they also occur in the setting of other types of arthritis. They bind to a diverse array of autoantigens, including type II collagen, human cartilage gp-39, aggrecan, calpastatin, BiP (immunoglobulin binding protein), and glucose-6-phosphate isomerase.
In theory, environmental stimulants may synergize with other factors to bring about inflammation in RA. People who smoke display higher citrullination of proteins in bronchoalveolar fluid than those who do not smoke. Thus, it has been speculated that long-term exposure to tobacco smoke might induce citrullination of cellular proteins in the lung and stimulate the expression of a neoepitope capable of inducing self-reactivity, which in turns, leads to formation of immune complexes and joint inflammation. Exposure to silicone dust and mineral oil, which has adjuvant effects, has also been linked to an increased risk for anti-CCP antibody-positive RA.
How might microbes or their products be involved in the initiating events of RA? The immune system is alerted to the presence of microbial infections through Toll-like receptors (TLRs). There are 10 TLRs in humans that recognize a variety of microbial products, including bacterial cell-surface lipopolysaccharides and heat-shock proteins (TLR4), lipoproteins (TLR2), double-strand RNA viruses (TLR3), and unmethylated CpG DNA from bacteria (TLR9). TLR2, -3, and -4 are abundantly expressed by synovial fibroblasts in early RA and, when bound by their ligands, upregulate production of proinflammatory cytokines. Although such events could amplify inflammatory pathways in RA, a specific role for TLRs in disease pathogenesis has not been elucidated.
The pathogenesis of RA is built upon the concept that self-reactive T cells drive the chronic inflammatory response. In theory, self-reactive T cells might arise in RA from abnormal central (thymic) selection due to defects in DNA repair leading to an imbalance of T cell death and life, or defects in the cell signaling apparatus lowering the threshold for T cell activation. Similarly, abnormal selection of the T cell repertoire in the periphery might lead to a breakdown in T cell tolerance. The support for these theories comes mainly from studies of arthritis in mouse models. It has not been shown that patients with RA have abnormal thymic selection of T cells or defective apoptotic pathways regulating cell death. At least some antigen stimulation inside the joint seems likely, owing to the fact that T cells in the synovium express a cell-surface phenotype indicating prior antigen exposure and show evidence of clonal expansion. Of interest, peripheral blood T cells from patients with RA have been shown to display a fingerprint of premature aging that mostly affects inexperienced naïve T cells. In these studies, the most glaring findings have been the loss of telomeric sequences and a decrease in the thymic output of new T cells. Although intriguing, it is not clear how generalized T cell abnormalities might provoke a systemic disease dominated by synovitis.
There is substantial evidence supporting a role for CD4+ T cells in the pathogenesis of RA. First, the co-receptor CD4 on the surface of T cells binds to invariant sites on MHC class II molecules, stabilizing the MHC-peptide–T cell receptor complex during T cell activation. Because the SE on MHC class II molecules is a risk factor for RA, it follows that CD4+ T cell activation may play a role in the pathogenesis of this disease. Second, CD4+ memory T cells are enriched in the synovial tissue from patients with RA and can be implicated through “guilt by association.” Third, CD4+ T cells have been shown to be important in the initiation of arthritis in animal models. Fourth, some, but not all, T cell–directed therapies have shown clinical efficacy in this disease. Taken together, these lines of evidence suggest that CD4+ T cells play an important role in orchestrating the chronic inflammatory response in RA. However, other cell types, such as CD8+ T cells, natural killer (NK) cells, and B cells are present in synovial tissue and may also influence pathogenic responses.
In the rheumatoid joint, by mechanisms of cell-cell contact and release of soluble mediators, activated T cells stimulate macrophages and fibroblast-like synoviocytes to generate proinflammatory mediators and proteases that drive the synovial inflammatory response and destroy the cartilage and bone. CD4+ T cell activation is dependent on two signals: (1) T cell receptor binding to peptide-MHC on antigen-presenting cells; and (2) CD28 binding to CD80/86 on antigen-presenting cells. CD4+ T cells also provide help to B cells, which in turn, produce antibodies that may promote further inflammation in the joint. The previous T cell–centric model for the pathogenesis of RA was based on a TH1-driven paradigm, which came from studies indicating that CD4+ T helper (TH) cells differentiated into TH1 and TH2 subsets, each with their distinctive cytokine profiles. TH1 cells were found to mainly produce interferon γ (IFN-γ), lymphotoxin β, and TNF-α, whereas TH2 cells predominately secreted interleukin (IL)-4, IL-5, IL-6, IL-10, and IL-13. The recent discovery of another subset of TH cells, namely the TH17 lineage, has revolutionized our concepts concerning the pathogenesis of RA. In humans, naïve T cells are induced to differentiate into TH17 cells by exposure to transforming growth factor β (TGF-β), IL-1, IL-6, and IL-23. Upon activation, TH17 cells secrete a variety of proinflammatory mediators such as IL-17, IL-21, IL-22, TNF-α, IL-26, IL-6, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Substantial evidence now exists from both animal models and humans that IL-17 plays an important role not only in promoting joint inflammation, but also in destroying cartilage and subchondral bone
The immune system has evolved mechanisms to counterbalance the potential harmful immune-mediated inflammatory responses provoked by infectious agents and other triggers. Among these negative regulators are regulatory T (Treg) cells, which are produced in the thymus and induced in the periphery to suppress immune-mediated inflammation. They are characterized by the surface expression of CD25 and the transcription factor forkhead box P3 (FOXP3) and orchestrate dominant tolerance through contact with other immune cells and secretion of inhibitory cytokines, such as TGF-β, IL-10, and IL-35. Treg cells appear to be heterogeneous and capable of suppressing distinct classes (TH1, TH2, TH17) of the immune response. In RA, the data that Treg numbers are deficient compared to normal healthy controls are contradictory and inconclusive. Although some experimental evidence suggests that Treg suppressive activity is lost due to dysfunctional expression of cytotoxic T lymphocyte antigen 4 (CTLA-4), the nature of Treg defects in RA, if they exist, remains unclear.
Cytokines, chemokines, antibodies, and endogenous danger signals bind to receptors on the surface of immune cells and stimulate a cascade of intracellular signaling events that can amplify the inflammatory response. Signaling molecules and their binding partners in these pathways are the target of small-molecule drugs designed to interfere with signal transduction and block these reinforcing inflammatory loops. Examples of signal molecules in these critical inflammatory pathways include Janus kinase (JAK)/signal transducers and activators of transcription (STAT), spleen tyrosine kinase (Syk), mitogen-activated protein kinases (MAPKs), and nuclear factor-κB (NF-κB). These pathways exhibit significant cross-talk and are found in many cell types. Some signal transducers, such as JAK3, are primarily expressed in hematopoietic cells and play an important role in the inflammatory response in RA.
Activated B cells are also important players in the chronic inflammatory response. B cells give rise to plasma cells, which in turn, produce antibodies, including RF and anti-CCP antibodies. RFs may form large immune complexes inside the joint that contribute to the pathogenic process by fixing complement and promoting the release of proinflammatory chemokines and chemoattractants. In mouse models of arthritis, RF-containing immune complexes and anti-CCP-containing immune complexes synergize with other mechanisms to exacerbate the synovial inflammatory response.
RA is often considered to be a macrophage-driven disease because this cell type is the predominant source of proinflammatory cytokines inside the joint. Key proinflammatory cytokines released by synovial macrophages include TNF-α, IL-1, IL-6, IL-12, IL-15, IL-18, and IL-23. Synovial fibroblasts, the other major cell type in this microenvironment, produce the cytokines IL-1 and IL-6 as well as TNF-α. TNF-α is a pivotal cytokine in the pathobiology of synovial inflammation. It upregulates adhesion molecules on endothelial cells, promoting the influx of leukocytes into the synovial microenvironment; activates synovial fibroblasts; stimulates angiogenesis; promotes pain receptor sensitizing pathways; and drives osteoclastogenesis. Fibroblasts secrete matrix metalloproteinases (MMPs) as well as other proteases that are chiefly responsible for the breakdown of articular cartilage.
Osteoclast activation at the site of the pannus is closely tied to the presence of focal bone erosion. Receptor activator of nuclear factor-κB ligand (RANKL) is expressed by stromal cells, synovial fibroblasts, and T cells. Upon binding to its receptor RANK on osteoclast progenitors, RANKL stimulates osteoclast differentiation and bone resorption. RANKL activity is regulated by osteoprotegerin (OPG), a decoy receptor of RANKL that blocks osteoclast formation. Monocytic cells in the synovium serve as the precursors of osteoclasts and, when exposed to macrophage colony-stimulating factor (M-CSF) and RANKL, fuse to form polykaryons termed preosteoclasts. These precursor cells undergo further differentiation into osteoclasts with the characteristic ruffled membrane. Cytokines such as TNF-α, IL-1, IL-6, and IL-17 increase the expression of RANKL in the joint and thus promote osteoclastogenesis. Osteoclasts also secrete cathepsin K, which is a cysteine protease that degrades the bone matrix by cleaving collagen. Stimulation of osteoclasts also contributes to generalized bone loss and osteoporosis.
Increased bone loss is only part of the story in RA, as decreased bone formation plays a crucial role in bone remodeling at sites of inflammation. Recent evidence shows that inflammation suppresses bone formation. The proinflammatory cytokine TNF-α plays a key role in actively suppressing bone formation by enhancing the expression of dickkopf-1 (DKK-1). DKK-1 is an important inhibitor of the Wnt pathway, which acts to promote osteoblast differentiation and bone formation. The Wnt system is a family of soluble glycoproteins that bind to cell-surface receptors known as frizzled (fz) and low-density lipoprotein (LDL) receptor–related proteins (LRPs) and promote cell growth. In animal models, increased levels of DKK-1 are associated with decreased bone formation, whereas inhibition of DKK-1 protects against structural damage in the joint. Wnt proteins also induce the formation of OPG and thereby shut down bone resorption, emphasizing their key role in tightly regulating the balance between bone resorption and formation.
DIAGNOSIS
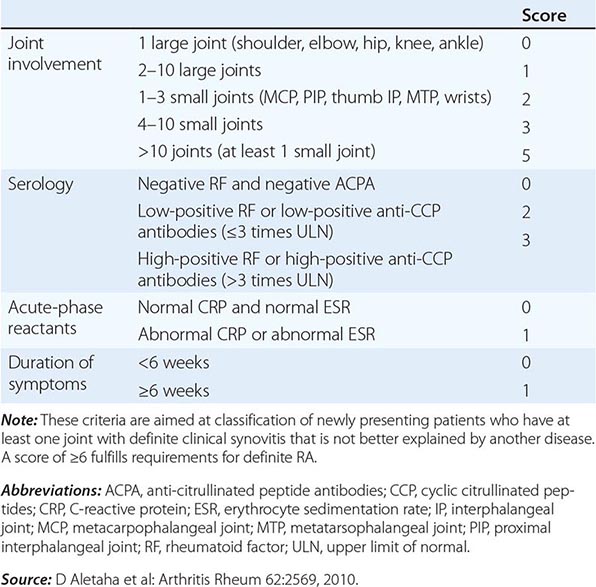
The clinical diagnosis of RA is largely based on signs and symptoms of a chronic inflammatory arthritis, with laboratory and radiographic results providing important supplemental information. In 2010, a collaborative effort between the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) revised the 1987 ACR classification criteria for RA in an effort to improve early diagnosis with the goal of identifying patients who would benefit from early introduction of disease-modifying therapy (Table 380-1). Application of the newly revised criteria yields a score of 0–10, with a score of ≥ 6 fulfilling the requirements for definite RA. The new classification criteria differ in several ways from the older criteria set. The new criteria include a positive test for serum anti-CCP antibodies (also termed ACPA, anti-citrullinated peptide antibodies) as an item, which carries greater specificity for the diagnosis of RA than a positive test for RF. The newer classification criteria also do not take into account whether the patient has rheumatoid nodules or radiographic joint damage because these findings occur rarely in early RA. It is important to emphasize that the new 2010 ACR-EULAR criteria are “classification criteria” as opposed to “diagnostic criteria” and serve to distinguish patients at the onset of disease who have a high likelihood of evolution to chronic disease with persistent synovitis and joint damage. The presence of radiographic joint erosions or subcutaneous nodules may inform the diagnosis in the later stages of the disease.
|
CLASSIFICATION CRITERIA FOR RHEUMATOID ARTHRITIS |
LABORATORY FEATURES
Patients with systemic inflammatory diseases such as RA will often present with elevated nonspecific inflammatory markers such as an ESR or CRP. Detection of serum RF and anti-CCP antibodies is important in differentiating RA from other polyarticular diseases, although RF lacks diagnostic specificity and may be found in association with other chronic inflammatory diseases in which arthritis figures in the clinical manifestations.
IgM, IgG, and IgA isotypes of RF occur in sera from patients with RA, although the IgM isotype is the one most frequently measured by commercial laboratories. Serum IgM RF has been found in 75–80% of patients with RA; therefore, a negative result does not exclude the presence of this disease. It is also found in other connective tissue diseases, such as primary Sjögren’s syndrome, systemic lupus erythematosus, and type II mixed essential cryoglobulinemia, as well as chronic infections such as subacute bacterial endocarditis and hepatitis B and C. Serum RF may also be detected in 1–5% of the healthy population.
The presence of serum anti-CCP antibodies has about the same sensitivity as serum RF for the diagnosis of RA. However, its diagnostic specificity approaches 95%, so a positive test for anti-CCP antibodies in the setting of an early inflammatory arthritis is useful for distinguishing RA from other forms of arthritis. There is some incremental value in testing for the presence of both RF and anti-CCP, as some patients with RA are positive for RF but negative for anti-CCP and visa versa. The presence of RF or anti-CCP antibodies also has prognostic significance, with anti-CCP antibodies showing the most value for predicting worse outcomes.
SYNOVIAL FLUID ANALYSIS
Typically, synovial fluid from patients with RA reflects an inflammatory state. Synovial fluid white blood cell (WBC) counts can vary widely, but generally range between 5000 and 50,000 WBC/μL compared to <2000 WBC/μL for a noninflammatory condition such as osteoarthritis. In contrast to the synovial tissue, the overwhelming cell type in the synovial fluid is the neutrophil. Clinically, the analysis of synovial fluid is most useful for confirming an inflammatory arthritis (as opposed to osteoarthritis), while at the same time excluding infection or a crystal-induced arthritis such as gout or pseudogout (Chap. 395).
JOINT IMAGING
Joint imaging is a valuable tool not only for diagnosing RA, but also for tracking progression of any joint damage. Plain x-ray is the most common imaging modality, but it is limited to visualization of the bony structures and inferences about the state of the articular cartilage based on the amount of narrowing of the joint space. MRI and ultrasound techniques offer the added value of detecting changes in the soft tissues such as synovitis, tenosynovitis, and effusions as well as greater sensitivity for identifying bony abnormalities. Plain radiographs are usually relied upon in clinical practice for the purpose of diagnosis and monitoring of affected joints. However, in selected cases, MRI and ultrasound can provide additional diagnostic information that may guide clinical decision making. Musculoskeletal ultrasound with power Doppler is increasingly used in rheumatology clinical practice for detecting synovitis and bone erosion.
Plain Radiography Classically in RA, the initial radiographic finding is periarticular osteopenia. Practically speaking, however, this finding is difficult to appreciate on plain films and, in particular, on the newer digitalized x-rays. Other findings on plain radiographs include soft tissue swelling, symmetric joint space loss, and subchondral erosions, most frequently in the wrists and hands (MCPs and PIPs) and the feet (MTPs). In the feet, the lateral aspect of the fifth MTP is often targeted first, but other MTP joints may be involved at the same time. X-ray imaging of advanced RA may reveal signs of severe destruction, including joint subluxation and collapse (Fig. 380-5).
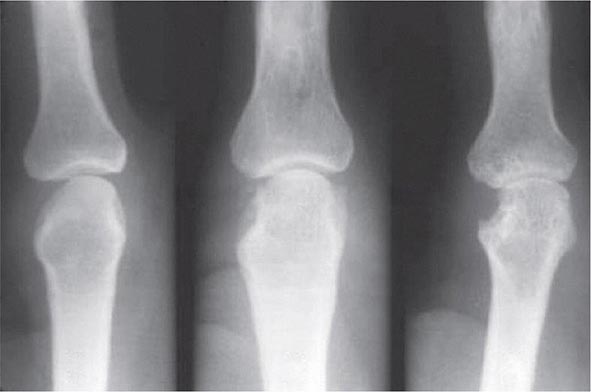
FIGURE 380-5 X-ray demonstrating progression of erosions on the proximal interphalangeal joint. (Courtesy of the American College of Rheumatology.)
MRI MRI offers the greatest sensitivity for detecting synovitis and joint effusions, as well as early bone and bone marrow changes. These soft tissue abnormalities often occur before osseous changes are noted on x-ray. Presence of bone marrow edema has been recognized to be an early sign of inflammatory joint disease and can predict the subsequent development of erosions on plain radiographs as well as MRI scans. Cost and availability of MRI are the main factors limiting its routine clinical use.
Ultrasound Ultrasound, including power color Doppler, has the ability to detect more erosions than plain radiography, especially in easily accessible joints. It can also reliably detect synovitis, including increased joint vascularity indicative of inflammation. The usefulness of ultrasound is dependent on the experience of the sonographer; however, it does offer the advantages of portability, lack of radiation, and low expense relative to MRI, factors that make it attractive as a clinical tool.
CLINICAL COURSE
The natural history of RA is complex and affected by a number of factors including age of onset, gender, genotype, phenotype (i.e., extraarticular manifestations or variants of RA), and comorbid conditions, which make for a truly heterogeneous disease. There is no simple way to predict the clinical course. It is important to realize that as many as 10% of patients with inflammatory arthritis fulfilling ACR classification criteria for RA will undergo a spontaneous remission within 6 months (particularly seronegative patients). However, the vast majority of patients will exhibit a pattern of persistent and progressive disease activity that waxes and wanes in intensity over time. A minority of patients will show intermittent and recurrent explosive attacks of inflammatory arthritis interspersed with periods of disease quiescence. Finally, an aggressive form of RA may occur in an unfortunate few with inexorable progression of severe erosive joint disease, although this highly destructive course is less common in the modern treatment era of biologics.
Disability, as measured by the Health Assessment Questionnaire (HAQ), shows gradual worsening of disability over time in the face of poorly controlled disease activity and disease progression. Disability may result from both a disease activity–related component that is potentially reversible with therapy and a joint damage–related component owing to the cumulative and largely irreversible effects of cartilage and bone breakdown. Early in the course of disease, the extent of joint inflammation is the primary determinant of disability, while in the later stages of disease, the amount of joint damage is the dominant contributing factor. Previous studies have shown that more than one-half of patients with RA are unable to work 10 years after the onset of their disease; however, increased employability and less work absenteeism has been reported recently with the use of newer therapies and earlier treatment intervention.
The overall mortality rate in RA is two times greater than the general population, with ischemic heart disease being the most common cause of death followed by infection. Median life expectancy is shortened by an average of 7 years for men and 3 years for women compared to control populations. Patients at higher risk for shortened survival are those with systemic extraarticular involvement, low functional capacity, low socioeconomic status, low education, and chronic prednisone use.
|
TREATMENT |
RHEUMATOID ARTHRITIS |
The amount of clinical disease activity in patients with RA reflects the overall burden of inflammation and is the variable most influencing treatment decisions. Joint inflammation is the main driver of joint damage and is the most important cause of functional disability in the early stages of disease. Several composite indices have been developed to assess clinical disease activity. The ACR 20, 50, and 70 improvement criteria (which corresponds to a 20%, 50%, and 70% improvement, respectively, in joint counts, physician/patient assessment of disease severity, pain scale, serum levels of acute-phase reactants [ESR or CRP], and a functional assessment of disability using a self-administered patient questionnaire) are a composite index with a dichotomous response variable. The ACR improvement criteria are commonly used in clinical trials as an endpoint for comparing the proportion of responders between treatment groups. In contrast, the Disease Activity Score (DAS), Simplified Disease Activity Index (SDAI), and the Clinical Disease Activity Index (CDAI) are continuous measures of disease activity. These scales are increasingly used in clinical practice for tracking disease status and, in particular, for documenting treatment response.
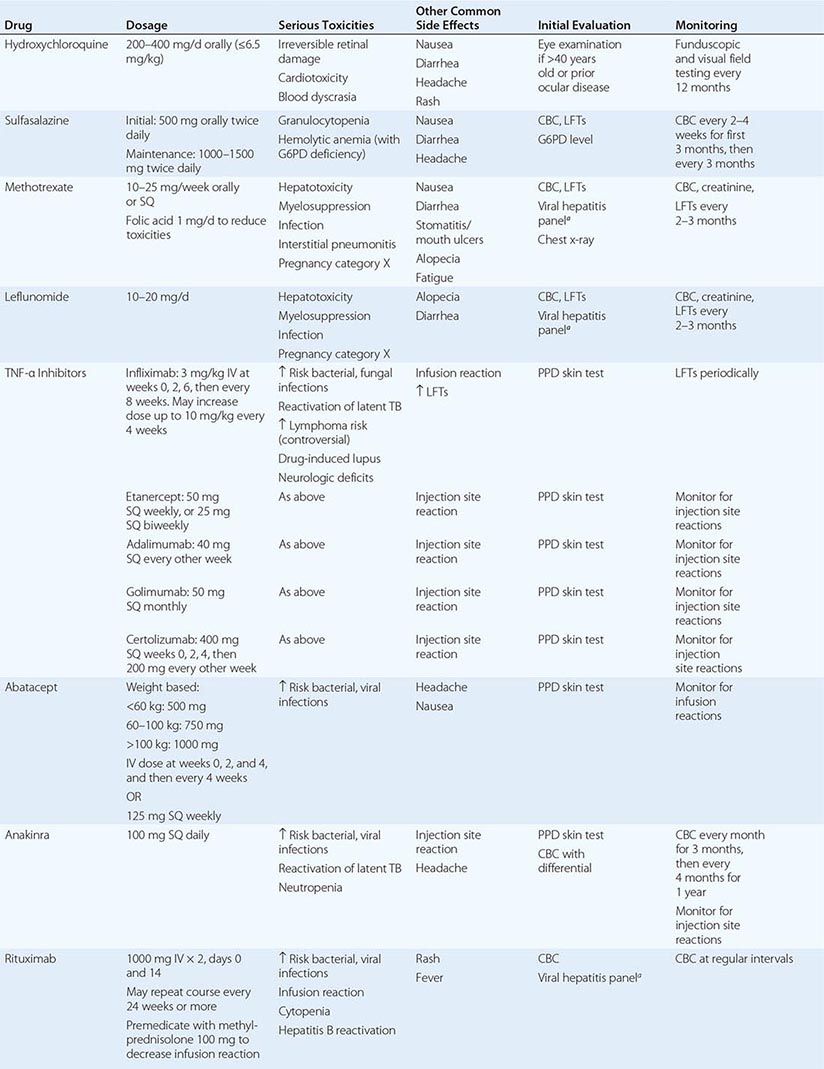
Several developments during the past two decades have changed the therapeutic landscape in RA. They include (1) the emergence of methotrexate as the disease-modifying antirheumatic drug (DMARD) of first choice for the treatment of early RA; (2) the development of novel highly efficacious biologicals that can be used alone or in combination with methotrexate; and (3) the proven superiority of combination DMARD regimens over methotrexate alone. The medications used for the treatment of RA may be divided into broad categories: nonsteroidal anti-inflammatory drugs (NSAIDs); glucocorticoids, such as prednisone and methylprednisolone; conventional DMARDs; and biologic DMARDs (Table 380-2). Although disease for some patients with RA is managed adequately with a single DMARD, such as methotrexate, the situation in most cases demands the use of a combination DMARD regimen that may vary in its components over the treatment course depending on fluctuations in disease activity and emergence of drug-related toxicities and comorbidities.
|
DMARDs USED FOR THE TREATMENT OF RHEUMATOID ARTHRITIS |
NSAIDs
NSAIDs were formerly viewed as the core of all other RA therapy, but they are now considered to be adjunctive therapy for management of symptoms uncontrolled by other measures. NSAIDs exhibit both analgesic and anti-inflammatory properties. The anti-inflammatory effects of NSAIDs derive from their ability to nonselectively inhibit cyclooxygenase (COX)-1 and COX-2. Although the results of clinical trials suggest NSAIDs are roughly equivalent in their efficacy, experience suggests that some individuals may preferentially respond to a particular NSAID. Chronic use should be minimized due to the possibility of side effects, including gastritis and peptic ulcer disease as well as impairment of renal function.
GLUCOCORTICOIDS
Glucocorticoids may serve in several ways to control disease activity in RA. First, they may be administered in low to moderate doses to achieve rapid disease control before the onset of fully effective DMARD therapy, which often takes several weeks or even months. Second, a 1- to 2-week burst of glucocorticoids may be prescribed for the management of acute disease flares, with dose and duration guided by the severity of the exacerbation. Chronic administration of low doses (5–10 mg/d) of prednisone (or its equivalent) may also be warranted to control disease activity in patients with an inadequate response to DMARD therapy. Low-dose prednisone therapy has been shown in prospective studies to retard radiographic progression of joint disease; however, the benefits of this approach must be carefully weighed against the risks. Best practices minimize chronic use of low-dose prednisone therapy owing to the risk of osteoporosis and other long-term complications; however, the use of chronic prednisone therapy is unavoidable in many cases. High-dose glucocorticoids may be necessary for treatment of severe extraarticular manifestations of RA, such as ILD. Finally, if a patient exhibits one or a few actively inflamed joints, the clinician may consider intraarticular injection of an intermediate-acting glucocorticoid such as triamcinolone acetonide. This approach may allow for rapid control of inflammation in the setting of a limited number of affected joints. Caution must be exercised to appropriately exclude joint infection, as it often mimics an RA flare.
Osteoporosis ranks as an important long-term complication of chronic prednisone use. The ACR recommends primary prevention of glucocorticoid-induced osteoporosis with a bisphosphonate in any patient receiving 5 mg/d or more of prednisone for greater than 3 months. Although prednisone use is known to increase the risk of peptic ulcer disease, especially with concomitant NSAID use, no evidence-based guidelines have been published regarding the use of gastrointestinal ulcer prophylaxis in this situation.
DMARDs
DMARDs are so named because of their ability to slow or prevent structural progression of RA. The conventional DMARDs include hydroxychloroquine, sulfasalazine, methotrexate, and leflunomide; they exhibit a delayed onset of action of approximately 6–12 weeks. Methotrexate is the DMARD of choice for the treatment of RA and is the anchor drug for most combination therapies. It was approved for the treatment of RA in 1986 and remains the benchmark for the efficacy and safety of new disease-modifying therapies. At the dosages used for the treatment of RA, methotrexate has been shown to stimulate adenosine release from cells, producing an anti-inflammatory effect. The clinical efficacy of leflunomide, an inhibitor of pyrimidine synthesis, appears similar to that of methotrexate; it has been shown in well-designed trials to be effective for the treatment of RA as monotherapy or in combination with methotrexate and other DMARDs.
Although similar to the other DMARDs in its slow onset of action, hydroxychloroquine has not been shown to delay radiographic progression of disease and thus is not considered to be a true DMARD. In clinical practice, hydroxychloroquine is generally used for treatment of early, mild disease or as adjunctive therapy in combination with other DMARDs. Sulfasalazine is used in a similar manner and has been shown in randomized, controlled trials to reduce radiographic progression of disease. Minocycline, gold salts, penicillamine, azathioprine, and cyclosporine have all been used for the treatment of RA with varying degrees of success; however, they are used sparingly now due to their inconsistent clinical efficacy or unfavorable toxicity profile.
BIOLOGICALS
Biologic DMARDs have revolutionized the treatment of RA over the past decade (Table 380-2). They are protein therapeutics designed mostly to target cytokines and cell-surface molecules. The TNF inhibitors were the first biologicals approved for the treatment of RA. Anakinra, an IL-1 receptor antagonist, was approved shortly thereafter; however, its benefits have proved to be relatively modest compared with the other biologicals and is rarely used for the treatment of RA with the availability of other more effective agents. Abatacept, rituximab, and tocilizumab are the newest members of this class.
Anti-TNF Agents The development of TNF inhibitors was originally spurred by the experimental finding that TNF is a critical upstream mediator of joint inflammation. Currently, five agents that inhibit TNF-α are approved for the treatment of RA. There are three different anti-TNF monoclonal antibodies. Infliximab is a chimeric (part mouse and human) monoclonal antibody, whereas adalimumab and golimumab are humanized monoclonal antibodies. Certolizumab pegol is a pegylated Fc-free fragment of a humanized monoclonal antibody with binding specificity for TNF-α. Lastly, etanercept is a soluble fusion protein comprising the TNF receptor 2 in covalent linkage with the Fc portion of IgG1. All of the TNF inhibitors have been shown in randomized controlled clinical trials to reduce the signs and symptoms of RA, slow radiographic progression of joint damage, and improve physical function and quality of life. Anti-TNF drugs are typically used in combination with background methotrexate therapy. This combination regimen, which affords maximal benefit in many cases, is often the next step for treatment of patients with an inadequate response to methotrexate therapy. Etanercept, adalimumab, certolizumab pegol, and golimumab have also been approved for use as monotherapy.
Anti-TNF agents should be avoided in patients with active infection or a history of hypersensitivity to these agents and are contraindicated in patients with chronic hepatitis B infection or class III/IV congestive heart failure. The major concern is the increased risk for infection, including serious bacterial infections, opportunistic fungal infection, and reactivation of latent tuberculosis. For this reason, all patients are screened for latent tuberculosis according to national guidelines prior to starting anti-TNF therapy (Chap. 202). In the United States, patients are skin tested using an intradermal injection of purified protein derivative (PPD); individuals with skin reactions of more than 5 mm are presumed to have had previous exposure to tuberculosis and are evaluated for active disease and treated accordingly. The QuantiFERON IFN-γ release assay may also be used in selected circumstances to screen for previous exposure to tuberculosis.
Anakinra Anakinra, the recombinant form of the naturally occurring IL-1 receptor antagonist. Although anakinra has seen limited use for the treatment of RA, it has enjoyed a resurgence of late as an effective therapy of some rare inherited syndromes dependent on IL-1 production, including neonatal-onset inflammatory disease, Muckle-Wells syndrome, and familial cold urticaria, as well as systemic juvenile-onset inflammatory arthritis and adult-onset Still’s disease. Anakinra should not be combined with an anti-TNF drug due to the high rate of serious infections as observed with this regimen in a clinical trial.
Abatacept Abatacept is a soluble fusion protein consisting of the extracellular domain of human CTLA-4 linked to the modified portion of human IgG. It inhibits the co-stimulation of T cells by blocking CD28-CD80/86 interactions and may also inhibit the function of antigen-presenting cells by reverse signaling through CD80 and CD86. Abatacept has been shown in clinical trials to reduce disease activity, slow radiographic progression of damage, and improve functional disability. Many patients receive abatacept in combination with methotrexate or another DMARD such as leflunomide. Abatacept therapy has been associated with an increased risk of infection.
Rituximab Rituximab is a chimeric monoclonal antibody directed against CD20, a cell-surface molecule expressed by most mature B lymphocytes. It works by depleting B cells, which in turn, leads to a reduction in the inflammatory response by unknown mechanisms. These mechanisms may include a reduction in autoantibodies, inhibition of T cell activation, and alteration of cytokine production. Rituximab has been approved for the treatment of refractory RA in combination with methotrexate and has been shown to be more effective for patients with seropositive than seronegative disease. Rituximab therapy has been associated with mild to moderate infusion reactions as well as an increased risk of infection. Notably, there have been isolated reports of a potentially lethal brain disorder, progressive multifocal leukoencephalopathy (PML), in association with rituximab therapy, although the absolute risk of this complication appears to be very low in patients with RA. Most of these cases have occurred on a background of previous or current exposure to other potent immunosuppressive drugs.
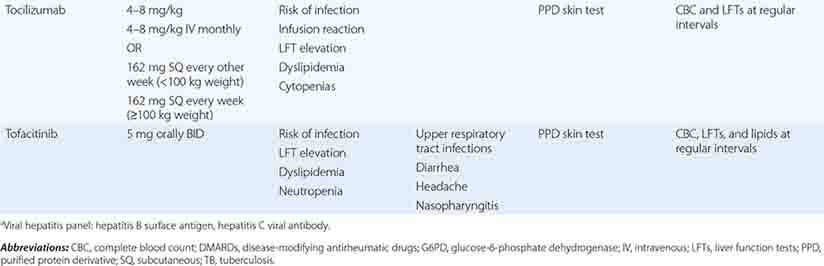
Tocilizumab Tocilizumab is a humanized monoclonal antibody directed against the membrane and soluble forms of the IL-6 receptor. IL-6 is a proinflammatory cytokine implicated in the pathogenesis of RA, with detrimental effects on both joint inflammation and damage. IL-6 binding to its receptor activates intracellular signaling pathways that affect the acute-phase response, cytokine production, and osteoclast activation. Clinical trials attest to the clinical efficacy of tocilizumab therapy for RA, both as monotherapy and in combination with methotrexate and other DMARDs. Tocilizumab has been associated with an increased risk of infection, neutropenia, and thrombocytopenia; however, the hematologic abnormalities appear to be reversible upon stopping the drug. In addition, this agent has been shown to increase LDL cholesterol; however, it is not known as yet if this effect on lipid levels increases the risk for development of atherosclerotic disease.
SMALL-MOLECULE INHIBITORS
Because some patients do not adequately respond to conventional DMARDS or biologic therapy, other therapeutic targets have been investigated to fill this gap. Recently, drug development in RA has focused attention on the intracellular signaling pathways that transduce the positive signals of cytokines and other inflammatory mediators that create the positive feedback loops in the immune response. These synthetic DMARDs aim to provide the same efficacy as biological therapies in an oral formulation.
Tofacitinib Tofacitinib is a small-molecule inhibitor that primarily inhibits JAK1 and JAK3, which mediate signaling of the receptors for the common γ-chain-related cytokines IL-2, -4, -7, -9, -15, and -21 as well as IFN-γ and IL-6. These cytokines all play roles in promoting T and B cell activation as well as inflammation. Tofacitinib, an oral agent, has been shown in randomized, placebo-controlled clinical trials to improve the signs and symptoms of RA significantly over placebo. Major adverse events include elevated serum transaminases indicative of liver injury, neutropenia, increased cholesterol levels, and elevation in serum creatinine. Its use is also associated with an increased risk of infections. Tofacitinib can be used as monotherapy or in combination with methotrexate.
|
ACR/EULAR PROVISIONAL DEFINITION OF REMISSION IN RHEUMATOID ARTHRITIS |
Source: Adapted from DT Felson et al: Arthritis Rheum 63:573, 2011.
GLOBAL CHALLENGES
![]() Developing countries are finding an increase in the incidence of noncommunicable, chronic diseases such as diabetes, cardiovascular disease, and RA in the face of ongoing poverty, rampant infectious disease, and poor access to modern health care facilities. In these areas, patients tend to have a greater delay in diagnosis and limited access to specialists, and thus greater disease activity and disability at presentation. In addition, infection risk remains a significant issue for the treatment of RA in developing countries because of the immunosuppression associated with the use of glucocorticoids and most DMARDs. For example, in some developing countries, patients undergoing treatment for RA have a substantial increase in the incidence of tuberculosis, which demands the implementation of far more comprehensive screening practices and liberal use of isoniazid prophylaxis than in developed countries. The increased prevalence of hepatitis B and C, as well as human immunodeficiency virus (HIV), in these developing countries also poses challenges. Reactivation of viral hepatitis has been observed in association with some of the DMARDs, such as rituximab. Also, reduced access to antiretroviral therapy may limit the control of HIV infection and therefore the choice of DMARD therapies.
Developing countries are finding an increase in the incidence of noncommunicable, chronic diseases such as diabetes, cardiovascular disease, and RA in the face of ongoing poverty, rampant infectious disease, and poor access to modern health care facilities. In these areas, patients tend to have a greater delay in diagnosis and limited access to specialists, and thus greater disease activity and disability at presentation. In addition, infection risk remains a significant issue for the treatment of RA in developing countries because of the immunosuppression associated with the use of glucocorticoids and most DMARDs. For example, in some developing countries, patients undergoing treatment for RA have a substantial increase in the incidence of tuberculosis, which demands the implementation of far more comprehensive screening practices and liberal use of isoniazid prophylaxis than in developed countries. The increased prevalence of hepatitis B and C, as well as human immunodeficiency virus (HIV), in these developing countries also poses challenges. Reactivation of viral hepatitis has been observed in association with some of the DMARDs, such as rituximab. Also, reduced access to antiretroviral therapy may limit the control of HIV infection and therefore the choice of DMARD therapies.
Despite these challenges, one should attempt to initiate early treatment of RA in the developing countries with the resources at hand. Sulfasalazine and methotrexate are all reasonably accessible throughout the world where they can be used as both monotherapy and in combination with other drugs. The use of biologic agents is increasing in the developed countries as well as in other areas around the world, although their use is limited by high cost; national protocols restrict their use, and concerns remain about the risk for opportunistic infections.
SUMMARY
Improved understanding of the pathogenesis of RA and its treatment has dramatically revolutionized the management of this disease. The outcomes of patients with RA are vastly superior to those of the prebiologic modifier era; more patients than in years past are able to avoid significant disability and continue working, albeit with some job modifications in many cases. The need for early and aggressive treatment of RA as well as frequent follow-up visits for monitoring of drug therapy has implications for our health care system. Primary care physicians and rheumatologists must be prepared to work together as a team to reach the ambitious goals of best practice. In many settings, rheumatologists have reengineered their practice in a way that places high priority on consultations for any new patient with early inflammatory arthritis.
The therapeutic regimens for RA are becoming increasingly complex with the rapidly expanding therapeutic armamentarium. Patients receiving these therapies must be carefully monitored by both the primary care physician and the rheumatologist to minimize the risk of side effects and identify quickly any complications of chronic immunosuppression. Also, prevention and treatment of RA-associated conditions such as ischemic heart disease and osteoporosis will likely benefit from a team approach owing to the value of multidisciplinary care.
Research will continue to search for new therapies with superior efficacy and safety profiles and investigate treatment strategies that can bring the disease under control more rapidly and nearer to remission. However, prevention and cure of RA will likely require new breakthroughs in our understanding of disease pathogenesis. These insights may come from genetic studies illuminating critical pathways in the mechanisms of joint inflammation. Equally ambitious is the lofty goal of biomarker discovery that will open the door to personalized medicine for the care of patients with RA.
381 |
Acute Rheumatic Fever |
Acute rheumatic fever (ARF) is a multisystem disease resulting from an autoimmune reaction to infection with group A streptococcus. Although many parts of the body may be affected, almost all of the manifestations resolve completely. The major exception is cardiac valvular damage (rheumatic heart disease [RHD]), which may persist after the other features have disappeared.
GLOBAL CONSIDERATIONS
![]() ARF and RHD are diseases of poverty. They were common in all countries until the early twentieth century, when their incidence began to decline in industrialized nations. This decline was largely attributable to improved living conditions—particularly less crowded housing and better hygiene—which resulted in reduced transmission of group A streptococci. The introduction of antibiotics and improved systems of medical care had a supplemental effect.
ARF and RHD are diseases of poverty. They were common in all countries until the early twentieth century, when their incidence began to decline in industrialized nations. This decline was largely attributable to improved living conditions—particularly less crowded housing and better hygiene—which resulted in reduced transmission of group A streptococci. The introduction of antibiotics and improved systems of medical care had a supplemental effect.
The virtual disappearance of ARF and reduction in the incidence of RHD in industrialized countries during the twentieth century unfortunately was not replicated in developing countries, where these diseases continue unabated. RHD is the most common cause of heart disease in children in developing countries and is a major cause of mortality and morbidity in adults as well. It has been estimated that between 15 and 19 million people worldwide are affected by RHD, with approximately one-quarter of a million deaths occurring each year. Some 95% of ARF cases and RHD deaths now occur in developing countries, with particularly high rates in sub-Saharan Africa, Pacific nations, Australasia, and South and Central Asia. The pathogenetic pathway from exposure to group A streptococcus followed by pharyngeal infection and subsequent development of ARF, ARF recurrences, and development of RHD and its complications is associated with a range of risk factors and, therefore, potential interventions at each point (Fig. 381-1). In affluent countries, many of these risk factors are well controlled, and where needed, interventions are in place. Unfortunately, the greatest burden of disease is found in developing countries, most of which do not have the resources, capacity, and/or interest to tackle this multifaceted disease. In particular, almost none of the developing countries has a coordinated, register-based RHD control program, which is proven to be cost effective in reducing the burden of RHD. Enhancing awareness of RHD and mobilizing resources for its control in developing countries are issues requiring international attention.
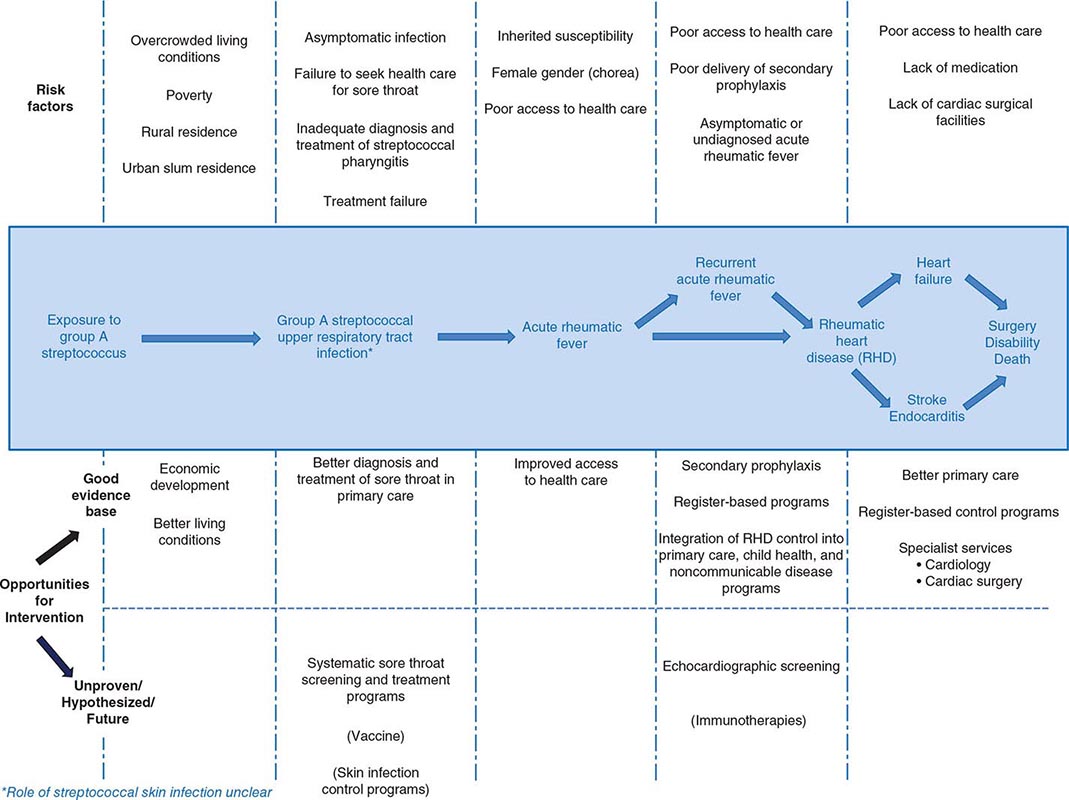
FIGURE 381-1 Pathogenetic pathway for acute rheumatic fever and rheumatic heart disease, with associated risk factors and opportunities for intervention at each step. Interventions in parentheses are either unproven or currently unavailable.
EPIDEMIOLOGY
ARF is mainly a disease of children age 5–14 years. Initial episodes become less common in older adolescents and young adults and are rare in persons age >30 years. By contrast, recurrent episodes of ARF remain relatively common in adolescents and young adults. This pattern contrasts with the prevalence of RHD, which peaks between 25 and 40 years. There is no clear gender association for ARF, but RHD more commonly affects females, sometimes up to twice as frequently as males.
PATHOGENESIS
ORGANISM FACTORS
Based on currently available evidence, ARF is exclusively caused by infection of the upper respiratory tract with group A streptococci (Chap. 173). Although classically, certain M-serotypes (particularly types 1, 3, 5, 6, 14, 18, 19, 24, 27, and 29) were associated with ARF, in high-incidence regions, it is now thought that any strain of group A streptococcus has the potential to cause ARF. The potential role of skin infection and of groups C and G streptococci is currently being investigated.
HOST FACTORS
Approximately 3–6% of any population may be susceptible to ARF, and this proportion does not vary dramatically between populations. Findings of familial clustering of cases and concordance in monozygotic twins—particularly for chorea—confirm that susceptibility to ARF is an inherited characteristic, with 44% concordance in monozygotic twins compared to 12% in dizygotic twins, and heritability more recently estimated at 60%. Most evidence for host factors focuses on immunologic determinants. Some human leukocyte antigen (HLA) class II alleles, particularly HLA-DR7 and HLA-DR4, appear to be associated with susceptibility, whereas other class II alleles have been associated with protection (HLA-DR5, HLA-DR6, HLA-DR51, HLA-DR52, and HLA-DQ). Associations have also been described with polymorphisms at the tumor necrosis factor α locus (TNF-α-308 and TNF-α-238), high levels of circulating mannose-binding lectin, and Toll-like receptors.
THE IMMUNE RESPONSE
The most widely accepted theory of rheumatic fever pathogenesis is based on the concept of molecular mimicry, whereby an immune response targeted at streptococcal antigens (mainly thought to be on the M protein and the N-acetylglucosamine of group A streptococcal carbohydrate) also recognizes human tissues. In this model, cross-reactive antibodies bind to endothelial cells on the heart valve, leading to activation of the adhesion molecule VCAM-1, with resulting recruitment of activated lymphocytes and lysis of endothelial cells in the presence of complement. The latter leads to release of peptides including laminin, keratin, and tropomyosin, which, in turn, activates cross-reactive T cells that invade the heart, amplifying the damage and causing epitope spreading. An alternative hypothesis proposes that the initial damage is due to streptococcal invasion of epithelial surfaces, with binding of M protein to type IV collagen RHD allowing it to become immunogenic, but not through the mechanism of molecular mimicry.
CLINICAL FEATURES
There is a latent period of ~3 weeks (1–5 weeks) between the precipitating group A streptococcal infection and the appearance of the clinical features of ARF. The exceptions are chorea and indolent carditis, which may follow prolonged latent periods lasting up to 6 months. Although many patients report a prior sore throat, the preceding group A streptococcal infection is commonly subclinical; in these cases, it can only be confirmed using streptococcal antibody testing. The most common clinical features are polyarthritis (present in 60–75% of cases) and carditis (50–60%). The prevalence of chorea in ARF varies substantially between populations, ranging from <2 to 30%. Erythema marginatum and subcutaneous nodules are now rare, being found in <5% of cases.
HEART INVOLVEMENT
Up to 60% of patients with ARF progress to RHD. The endocardium, pericardium, or myocardium may be affected. Valvular damage is the hallmark of rheumatic carditis. The mitral valve is almost always affected, sometimes together with the aortic valve; isolated aortic valve involvement is rare. Damage to the pulmonary or tricuspid valves is usually secondary to increased pulmonary pressures resulting from left-sided valvular disease. Early valvular damage leads to regurgitation. Over ensuing years, usually as a result of recurrent episodes, leaflet thickening, scarring, calcification, and valvular stenosis may develop (Fig. 381-2). See Videos 381-1 and 381-2 on the DVD. Therefore, the characteristic manifestation of carditis in previously unaffected individuals is mitral regurgitation, sometimes accompanied by aortic regurgitation. Myocardial inflammation may affect electrical conduction pathways, leading to P-R interval prolongation (first-degree atrioventricular block or rarely higher level block) and softening of the first heart sound.
FIGURE 381-2 Transthoracic echocardiographic image from a 5-year-old boy with chronic rheumatic heart disease. This diastolic image demonstrates leaflet thickening, restriction of the anterior mitral valve leaflet tip and doming of the body of the leaflet toward the interventricular septum. This appearance (marked by the arrowhead) is commonly described as a “hockey stick” or an “elbow” deformity. AV, aortic valve; LA, left atrium; LV, left ventricle; MV, mitral valve; RV, right ventricle. (Courtesy of Dr. Bo Remenyi, Department of Paediatric and Congenital Cardiac Services, Starship Children’s Hospital, Auckland, New Zealand.)
People with RHD are often asymptomatic for many years before their valvular disease progresses to cause cardiac failure. Moreover, particularly in resource-poor settings, the diagnosis of ARF is often not made, so children, adolescents, and young adults may have RHD but not know it. These cases can be diagnosed using echocardiography; auscultation is poorly sensitive and specific for RHD diagnosis in asymptomatic patients. Echocardiographic screening of school-aged children in populations with high rates of RHD is becoming more widespread and has been facilitated by improving technologies in portable echocardiography and the availability of consensus guidelines for the diagnosis of RHD on echocardiography (Table 381-1). Although a diagnosis of definite RHD on screening echocardiography should lead to commencement of secondary prophylaxis, the clinical significance of borderline RHD has yet to be determined.
|
WORLD HEART FEDERATION CRITERIA FOR ECHOCARDIOGRAPHIC DIAGNOSIS OF RHEUMATIC HEART DISEASE (RHD) IN INDIVIDUALS <20 YEARS OF AGEa |
aFor criteria in individuals >20 years of age, see source document.
Abbreviations: AR, aortic regurgitation; AV, aortic valve; MR, mitral regurgitation; MS, mitral stenosis; MV, mitral valve.
Source: Adapted from Remenyi B et al: World Heart Federation criteria for echocardiographic diagnosis of rheumatic heart disease-an evidence-based guideline. Nat Rev Cardiol 9:297–309, 2012.
JOINT INVOLVEMENT
The most common form of joint involvement in ARF is arthritis, i.e., objective evidence of inflammation, with hot, swollen, red, and/or tender joints, and involvement of more than one joint (i.e., polyarthritis). Polyarthritis is typically migratory, moving from one joint to another over a period of hours. ARF almost always affects the large joints—most commonly the knees, ankles, hips, and elbows—and is asymmetric. The pain is severe and usually disabling until anti-inflammatory medication is commenced.
Less severe joint involvement is also relatively common and has been recognized as a potential major manifestation in high-risk populations in diagnostic guidelines from Australia, but at the time of writing, this was not reflected in the Jones criteria. Arthralgia without objective joint inflammation usually affects large joints in the same migratory pattern as polyarthritis. In some populations, aseptic monoarthritis may be a presenting feature of ARF, which may, in turn, result from early commencement of anti-inflammatory medication before the typical migratory pattern is established.
The joint manifestations of ARF are highly responsive to salicylates and other nonsteroidal anti-inflammatory drugs (NSAIDs). Indeed, joint involvement that persists for more than 1 or 2 days after starting salicylates is unlikely to be due to ARF.
CHOREA
Sydenham’s chorea commonly occurs in the absence of other manifestations, follows a prolonged latent period after group A streptococcal infection, and is found mainly in females. The choreiform movements affect particularly the head (causing characteristic darting movements of the tongue) and the upper limbs (Chap. 448). They may be generalized or restricted to one side of the body (hemi-chorea). In mild cases, chorea may be evident only on careful examination, whereas in the most severe cases, the affected individuals are unable to perform activities of daily living. There is often associated emotional lability or obsessive-compulsive traits, which may last longer than the choreiform movements (which usually resolve within 6 weeks but sometimes may take up to 6 months).
SKIN MANIFESTATIONS
The classic rash of ARF is erythema marginatum (Chap. 24), which begins as pink macules that clear centrally, leaving a serpiginous, spreading edge. The rash is evanescent, appearing and disappearing before the examiner’s eyes. It occurs usually on the trunk, sometimes on the limbs, but almost never on the face.
Subcutaneous nodules occur as painless, small (0.5–2 cm), mobile lumps beneath the skin overlying bony prominences, particularly of the hands, feet, elbows, occiput, and occasionally the vertebrae. They are a delayed manifestation, appearing 2–3 weeks after the onset of disease, last for just a few days up to 3 weeks, and are commonly associated with carditis.
OTHER FEATURES
Fever occurs in most cases of ARF, although rarely in cases of pure chorea. Although high-grade fever (≥39°C) is the rule, lower grade temperature elevations are not uncommon. Elevated acute-phase reactants are also present in most cases.
EVIDENCE OF A PRECEDING GROUP A STREPTOCOCCAL INFECTION
With the exception of chorea and low-grade carditis, both of which may become manifest many months later, evidence of a preceding group A streptococcal infection is essential in making the diagnosis of ARF. Because most cases do not have a positive throat swab culture or rapid antigen test, serologic evidence is usually needed. The most common serologic tests are the anti-streptolysin O (ASO) and anti-DNase B (ADB) titers. Where possible, age-specific reference ranges should be determined in a local population of healthy people without a recent group A streptococcal infection.
CONFIRMING THE DIAGNOSIS
Because there is no definitive test, the diagnosis of ARF relies on the presence of a combination of typical clinical features together with evidence of the precipitating group A streptococcal infection, and the exclusion of other diagnoses. This uncertainty led Dr. T. Duckett Jones in 1944 to develop a set of criteria (subsequently known as the Jones criteria) to aid in the diagnosis. At the time of writing, the Jones criteria were undergoing revision but had not yet been released. The existing diagnostic guideline is a World Health Organization update of the 1992 Jones Criteria (Table 381-2), though it should be noted that other guidelines, including those from Australia and New Zealand, suggest more sensitive criteria for making the diagnosis in patients from settings or populations at high risk of ARF.
|
2002–2003 WORLD HEALTH ORGANIZATION CRITERIA FOR THE DIAGNOSIS OF RHEUMATIC FEVER AND RHEUMATIC HEART DISEASE (BASED ON THE 1992 REVISED JONES CRITERIA) |
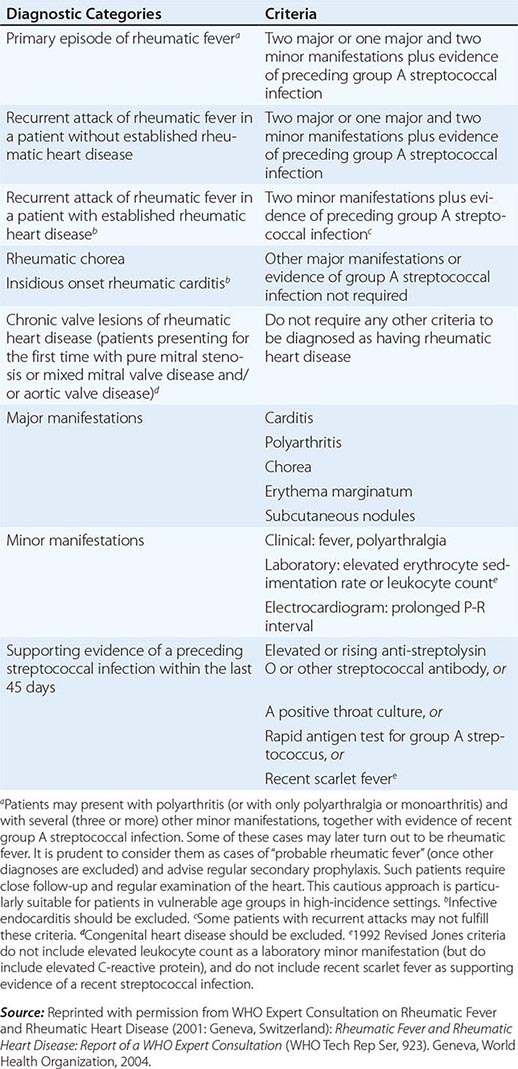
PROGNOSIS
Untreated, ARF lasts on average 12 weeks. With treatment, patients are usually discharged from hospital within 1–2 weeks. Inflammatory markers should be monitored every 1–2 weeks until they have normalized (usually within 4–6 weeks), and an echocardiogram should be performed after 1 month to determine if there has been progression of carditis. Cases with more severe carditis need close clinical and echocardiographic monitoring in the longer term.
Once the acute episode has resolved, the priority in management is to ensure long-term clinical follow-up and adherence to a regimen of secondary prophylaxis. Patients should be entered onto the local ARF registry (if present) and contact made with primary care practitioners to ensure a plan for follow-up and administration of secondary prophylaxis before the patient is discharged. Patients and their families should also be educated about their disease, emphasizing the importance of adherence to secondary prophylaxis.
PREVENTION
PRIMARY PREVENTION
Ideally, primary prevention would entail elimination of the major risk factors for streptococcal infection, particularly overcrowded housing. This is difficult to achieve in most places where ARF is common.
Therefore, the mainstay of primary prevention for ARF remains primary prophylaxis (i.e., the timely and complete treatment of group A streptococcal sore throat with antibiotics). If commenced within 9 days of sore throat onset, a course of penicillin (as outlined above for treatment of ARF) will prevent almost all cases of ARF that would otherwise have developed. In settings where ARF and RHD are common but microbiologic diagnosis of group A streptococcal pharyngitis is not available, such as in resource-poor countries, primary care guidelines often recommend that all patients with sore throat be treated with penicillin or, alternatively, that a clinical algorithm be used to identify patients with a higher likelihood of group A streptococcal pharyngitis. Although imperfect, such approaches recognize the importance of ARF prevention at the expense of overtreating many cases of sore throat that are not caused by group A streptococcus.
SECONDARY PREVENTION
The mainstay of controlling ARF and RHD is secondary prevention. Because patients with ARF are at dramatically higher risk than the general population of developing a further episode of ARF after a group A streptococcal infection, they should receive long-term penicillin prophylaxis to prevent recurrences. The best antibiotic for secondary prophylaxis is benzathine penicillin G (1.2 million units, or 600,000 units if ≤27 kg) delivered every 4 weeks. It can be given every 3 weeks, or even every 2 weeks, to persons considered to be at particularly high risk, although in settings where good compliance with an every-4-week dosing schedule can be achieved, more frequent dosing is rarely needed. Oral penicillin V (250 mg) can be given twice daily instead but is less effective than benzathine penicillin G. Penicillin-allergic patients can receive erythromycin (250 mg) twice daily.
The duration of secondary prophylaxis is determined by many factors, in particular the duration since the last episode of ARF (recurrences become less likely with increasing time), age (recurrences are less likely with increasing age), and the severity of RHD (if severe, it may be prudent to avoid even a very small risk of recurrence because of the potentially serious consequences) (Table 381-4). Secondary prophylaxis is best delivered as part of a coordinated RHD control program, based around a registry of patients. Registries improve the ability to follow patients and identify those who default from prophylaxis and to institute strategies to improve adherence.
|
AMERICAN HEART ASSOCIATION RECOMMENDATIONS FOR DURATION OF SECONDARY PROPHYLAXISa |
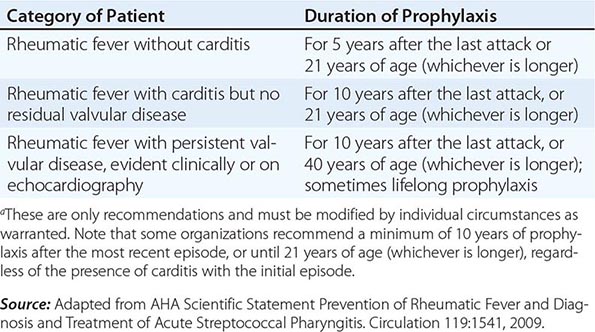
VIDEO 381-1A Transthoracic echocardiographic images of a 9-year-old girl with first episode of acute rheumatic fever. Images demonstrate the typical echocardiographic findings of acute rheumatic carditis. The valve leaflets are relatively thin and highly mobile. The failure of coaptation of the mitral valve leaflets is the result of chordal elongation and annular dilatation. The mitral valve regurgitation is moderate with a typical posterolaterally directed regurgitant jet of rheumatic carditis. A. Acute rheumatic carditis (apical four-chamber view echocardiogram).
VIDEO 381-1B Transthoracic echocardiographic images of a 9-year-old girl with first episode of acute rheumatic fever. Images demonstrate the typical echocardiographic findings of acute rheumatic carditis. The valve leaflets are relatively thin and highly mobile. The failure of coaptation of the mitral valve leaflets is the result of chordal elongation and annular dilatation. The mitral valve regurgitation is moderate with a typical posterolaterally directed regurgitant jet of rheumatic carditis. B. Acute rheumatic carditis (apical four-chamber view color Doppler echocardiogram).
VIDEO 381-1C Transthoracic echocardiographic images of a 9-year-old girl with first episode of acute rheumatic fever. Images demonstrate the typical echocardiographic findings of acute rheumatic carditis. The valve leaflets are relatively thin and highly mobile. The failure of coaptation of the mitral valve leaflets is the result of chordal elongation and annular dilatation. The mitral valve regurgitation is moderate with a typical posterolaterally directed regurgitant jet of rheumatic carditis. C. Acute rheumatic carditis (parasternal long-axis view echocardiogram).
VIDEO 381-1D Transthoracic echocardiographic images of a 9-year-old girl with first episode of acute rheumatic fever. Images demonstrate the typical echocardiographic findings of acute rheumatic carditis. The valve leaflets are relatively thin and highly mobile. The failure of coaptation of the mitral valve leaflets is the result of chordal elongation and annular dilatation. The mitral valve regurgitation is moderate with a typical posterolaterally directed regurgitant jet of rheumatic carditis. D. Acute rheumatic carditis (parasternal long-axis view color Doppler echocardiogram).
VIDEO 381-2A Transthoracic echocardiographic images are from a 5-year-old boy with chronic rheumatic heart disease with severe mitral valve regurgitation and moderate mitral valve stenosis. Images demonstrate the typical echocardiographic findings in advanced chronic rheumatic heart disease. Both the anterior and posterior mitral valve leaflets are markedly thickened. During diastole, the motion of the anterior mitral valve leaflet tip is restricted with doming of the body of the leaflet toward the interventricular septum. This appearance is commonly described as a “hockey stick” or an “elbow” deformity. A. Chronic rheumatic heart disease (parasternal long-axis view).
VIDEO 381-2B Transthoracic echocardiographic images are from a 5-year-old boy with chronic rheumatic heart disease with severe mitral valve regurgitation and moderate mitral valve stenosis. Images demonstrate the typical echocardiographic findings in advanced chronic rheumatic heart disease. Both the anterior and posterior mitral valve leaflets are markedly thickened. During diastole, the motion of the anterior mitral valve leaflet tip is restricted with doming of the body of the leaflet toward the interventricular septum. This appearance is commonly described as a “hockey stick” or an “elbow” deformity. B. Chronic rheumatic heart disease (apical two-chamber view echocardiogram).
382 |
Systemic Sclerosis (Scleroderma) and Related Disorders |
DEFINITION
Systemic sclerosis (SSc) is an uncommon connective tissue disorder characterized by multisystem involvement, heterogeneous clinical manifestations, a chronic and often progressive course, and significant disability and mortality. Multiple genes contribute to disease susceptibility; however, environmental exposures are likely to play a major role in causing SSc. The early stage of the disease is associated with prominent inflammatory features. Over time, functional and structural alterations in multiple vascular beds and progressive visceral organ dysfunction due to fibrosis dominate the clinical picture. Although thickened skin (scleroderma) is the distinguishing hallmark of SSc, skin induration can occur in localized forms of scleroderma and other disorders (Table 382-1). Patients with SSc can be broadly grouped into diffuse cutaneous and limited cutaneous subsets defined by the pattern of skin involvement, as well as clinical and laboratory features (Table 382-2). Diffuse cutaneous SSc (dcSSc) is associated with extensive skin induration, starting in the fingers and ascending from distal to proximal limbs and the trunk. These patients often have early interstitial lung disease and acute renal involvement. In contrast, in patients with limited cutaneous SSc (lcSSc), Raynaud’s phenomenon may precede other manifestations of SSc by years. In these patients, skin involvement remains limited to the fingers (sclerodactyly), distal limbs, and face, and the trunk is not affected. The constellation of calcinosis cutis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia, seen in some lcSSc patients, is termed the CREST syndrome. Visceral organ involvement in lcSSc tends to show insidious progression, and pulmonary arterial hypertension (PAH), interstitial lung disease, hypothyroidism, and primary biliary cirrhosis may occur as late complications. In some patients, Raynaud’s phenomenon and other characteristic features of SSc occur in the absence of skin thickening. This syndrome has been termed SSc sine scleroderma.
|
CONDITIONS ASSOCIATED WITH SKIN INDURATION |
|
SUBSETS OF SYSTEMIC SCLEROSIS (SSC): FEATURES OF LIMITED CUTANEOUS SSC VERSUS DIFFUSE CUTANEOUS SSC |
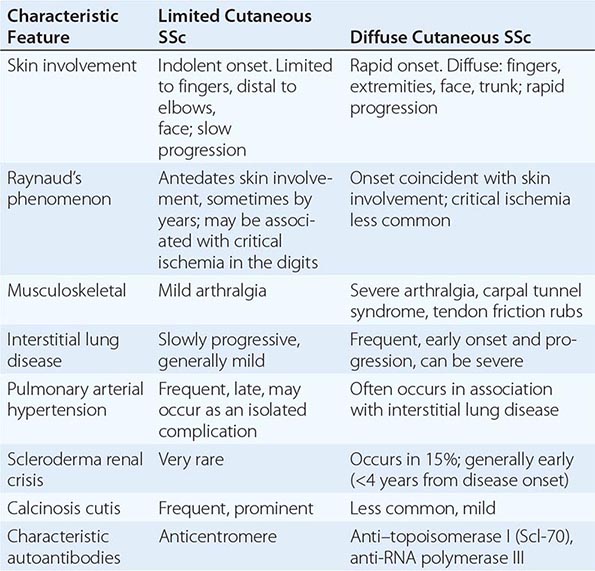
EPIDEMIOLOGY
SSc is an acquired sporadic disease with a worldwide distribution and affecting all races. In the United States, the incidence is estimated at 9–19 cases per million per year. The only community-based survey of SSc yielded a prevalence of 286 cases per million. There are an estimated 100,000 cases in the United States, although this number may be significantly higher if patients who do not meet strict classification criteria are also included. Rates of SSc in England, Australia, and Japan appear to be lower. Age, gender, and ethnicity are important in disease susceptibility. In common with other connective tissue diseases, SSc shows a strong female predominance (4.6:1), which is most pronounced in the childbearing years and declines after menopause. Although SSc can present at any age, the peak age of onset for both limited and diffuse cutaneous forms is 30–50 years. The incidence is higher in blacks than whites, and disease onset occurs at an earlier age. Furthermore, blacks with SSc are more likely to have diffuse cutaneous disease associated with interstitial lung involvement and a worse prognosis.
GENETIC CONSIDERATIONS
![]() In general, SSc shows modest heritability, and the genetic associations identified to date make only a small contribution to disease susceptibility. Concordance rates for SSc are low (4.7%) in monozygotic twins, although concordance for antinuclear antibody (ANA) positivity is significantly higher. On the other hand, evidence for genetic contribution to disease susceptibility is provided by the observation that 1.6% of SSc patients have a first-degree relative with SSc, a prevalence rate markedly increased compared to the general population. The risk of Raynaud’s phenomenon, interstitial lung disease, and other autoimmune diseases, including systemic lupus erythematosus (SLE) (Chap. 378), rheumatoid arthritis (Chap. 380), and autoimmune thyroiditis (Chap. 405), is also increased. Approaches to study the role of genetics in SSc use candidate gene single nucleotide polymorphism (SNP) analysis and genome-wide association studies (GWASs). Candidate gene studies in SSc have shown associations with multiple gene variants, many related to B and T lymphocyte activation and signaling (BANK1, BLK, CD247, CSK, IRAK1, IL2RA, PTPN22, and TNIP1). IRAK1, which codes for a gene involved in both innate and adaptive immunity, is the first X-linked gene associated with SSc and may contribute to female predominance. Other gene variants associated with SSc are involved in innate immunity and the interferon pathways (IRF5, IRF7, STAT4, TNFAIP3, and TLR2). In addition, candidate gene studies and GWAS both identified association with genes in the major histocompatibility complex (MHC), including NOTCH4 and PSORSC1. In addition to disease susceptibility, some of these genetic loci are associated with particular SSc disease manifestations or serologic subsets, including interstitial lung disease (ILD) (CTGF, CD226), PAH (TNIP1), and scleroderma renal crisis (HLA-DRB1*). Although the functional consequences of these gene variants are currently not well understood, they may result in altered immune function, leading to increased susceptibility to autoimmunity and inflammation. Of note, many of the genetic variants associated with SSc are also seen in other autoimmune disorders, including SLE, rheumatoid arthritis, and psoriasis, suggesting common pathways shared among these conditions. The genetic associations identified to date only explain a fraction of the heritability of SSc, and GWASs, fine mapping, and resequencing of DNA regions of interest to identify additional genetic susceptibility factors in SSc, particularly rare variants, are currently ongoing.
In general, SSc shows modest heritability, and the genetic associations identified to date make only a small contribution to disease susceptibility. Concordance rates for SSc are low (4.7%) in monozygotic twins, although concordance for antinuclear antibody (ANA) positivity is significantly higher. On the other hand, evidence for genetic contribution to disease susceptibility is provided by the observation that 1.6% of SSc patients have a first-degree relative with SSc, a prevalence rate markedly increased compared to the general population. The risk of Raynaud’s phenomenon, interstitial lung disease, and other autoimmune diseases, including systemic lupus erythematosus (SLE) (Chap. 378), rheumatoid arthritis (Chap. 380), and autoimmune thyroiditis (Chap. 405), is also increased. Approaches to study the role of genetics in SSc use candidate gene single nucleotide polymorphism (SNP) analysis and genome-wide association studies (GWASs). Candidate gene studies in SSc have shown associations with multiple gene variants, many related to B and T lymphocyte activation and signaling (BANK1, BLK, CD247, CSK, IRAK1, IL2RA, PTPN22, and TNIP1). IRAK1, which codes for a gene involved in both innate and adaptive immunity, is the first X-linked gene associated with SSc and may contribute to female predominance. Other gene variants associated with SSc are involved in innate immunity and the interferon pathways (IRF5, IRF7, STAT4, TNFAIP3, and TLR2). In addition, candidate gene studies and GWAS both identified association with genes in the major histocompatibility complex (MHC), including NOTCH4 and PSORSC1. In addition to disease susceptibility, some of these genetic loci are associated with particular SSc disease manifestations or serologic subsets, including interstitial lung disease (ILD) (CTGF, CD226), PAH (TNIP1), and scleroderma renal crisis (HLA-DRB1*). Although the functional consequences of these gene variants are currently not well understood, they may result in altered immune function, leading to increased susceptibility to autoimmunity and inflammation. Of note, many of the genetic variants associated with SSc are also seen in other autoimmune disorders, including SLE, rheumatoid arthritis, and psoriasis, suggesting common pathways shared among these conditions. The genetic associations identified to date only explain a fraction of the heritability of SSc, and GWASs, fine mapping, and resequencing of DNA regions of interest to identify additional genetic susceptibility factors in SSc, particularly rare variants, are currently ongoing.
ENVIRONMENTAL AND OCCUPATIONAL RISK FACTORS
Given the relatively modest genetic contribution to disease susceptibility, environmental factors, such as infectious agents, intestinal microbiota, and occupational, dietary, and drug exposures, are likely to play a major role in causing SSc. Patients with SSc show evidence of chronic infection of lesional tissue with Epstein-Barr virus (EBV). They also have increased antibodies to human cytomegalovirus (hCMV), and anti–topoisomerase I (Scl-70) autoantibodies recognize hCMV-associated antigenic epitopes, suggesting molecular mimicry as a possible mechanistic link between hCMV infection and SSc. An epidemic of a novel syndrome with features suggestive of SSc occurred in Spain in the 1980s. The outbreak, termed toxic oil syndrome, was linked to contaminated rapeseed oils used for cooking. Another epidemic outbreak, termed eosinophilia-myalgia syndrome (EMS), occurred a decade later and was linked to the consumption of L-tryptophan-containing dietary supplements. Although both of these novel toxic-epidemic syndromes were characterized by scleroderma-like chronic skin changes and variable visceral organ involvement, they were associated with clinical, pathologic, and laboratory features distinguishing them from SSc. Occupational exposures tentatively linked with SSc include silica dust in miners, polyvinyl chloride, epoxy resins, and aromatic hydrocarbons including toluene and trichloroethylene. Drugs implicated in SSc-like illnesses include bleomycin, pentazocine, and cocaine, and appetite suppressants linked with pulmonary hypertension. Although case reports and series describing SSc in women with silicone breast implants had raised concern regarding a possible causal role of silicone in SSc, large-scale epidemiologic investigations found no evidence of increased prevalence of SSc.
PATHOGENESIS
The following three cardinal pathophysiologic processes account for the protean clinical manifestations of SSc: (1) diffuse microangiopathy, (2) inflammation and autoimmunity, and (3) visceral and vascular fibrosis in multiple organs (Fig. 382-1). Autoimmunity and altered vascular reactivity are early manifestations. Complex and dynamic interplay between these processes initiates and then amplifies the fibrotic process.
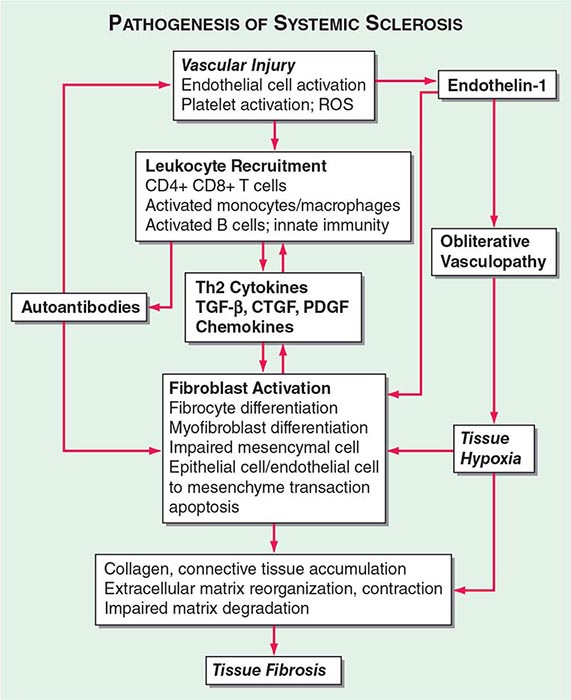
FIGURE 382-1 Initial vascular injury in a genetically susceptible individual leads to functional and structural vascular alterations, inflammation, and autoimmunity. The inflammatory and immune responses initiate and sustain fibroblast activation and differentiation, resulting in pathologic fibrogenesis and irreversible tissue damage. Vascular damage results in tissue ischemia that further contributes to progressive fibrosis and atrophy. C TGF, connective tissue growth factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor β.
ANIMAL MODELS OF DISEASE
No single animal model of SSc fully reproduces the three cardinal processes that underlie the pathogenesis, but some recapitulate selected aspects of the human disease, including fibrosis, microvascular involvement, and autoimmunity. Tight-skin mice (Tsk1) develop spontaneous skin thickening due to a mutation in the fibrillin-1 gene. The mutant fibrillin-1 protein disrupts extracellular matrix assembly and causes aberrant activation of transforming growth factor β (TGF-β). Fibrillin-1 mutations are associated with Marfan’s disease as well as the stiff skin syndrome but not SSc. Skin and lung fibrosis can be induced in mice by injection of bleomycin, HOCl, or double-stranded RNA or by transplantation of human leukocyte antigen (HLA)–mismatched bone marrow or spleen cells. Targeted genetic modifications in mice give rise to new disease models for dissecting the pathogenetic roles of individual molecules, cell types, and networks. For example, mice lacking Smad3, an intracellular TGF-β signal transducer, adiponectin, or the nuclear receptor peroxisome proliferator-activated receptor (PPAR) γ or overexpressing Wnt10b or adiponectin were either resistant or hypersensitive to chemically induced experimental scleroderma. These mouse models have potential utility in preclinical evaluation of potential therapies.
MICROANGIOPATHY
Involvement of small blood vessels in SSc affects multiple vascular beds and has important clinical sequelae including Raynaud’s phenomenon, ischemic digital ulcers, scleroderma renal crisis, and PAH. Raynaud’s phenomenon, an early disease manifestation, is characterized by an altered blood-flow response to cold challenge. This initially reversible functional abnormality is associated with autonomic and peripheral nervous system alterations, including impaired production of the neuropeptide calcitonin gene-related peptide from sensory afferent nerves and heightened sensitivity of α2-adrenergic receptors on vascular smooth-muscle cells. Isolated (primary) Raynaud’s phenomenon is extremely common and generally benign and nonprogressive. In contrast, SSc-associated Raynaud’s phenomenon is often progressive and complicated by irreversible structural changes, culminating in ischemic digital ulcers and loss of digits. Viruses, vascular cytotoxic factors, thrombogenic microparticles, complement and autoantibodies to phospholipids, β2 glycoprotein I (β2GPI), and endothelial cells are suspected triggers of endothelial cell injury in SSc. Endothelial injury results in dysregulated production of endothelium-derived vasodilatory (nitric oxide and prostacyclin) and vasoconstricting (endothelin-1) substances, as well as increased expression of intercellular adhesion molecule 1 (ICAM-1) and other surface adhesion molecules. Microvessels show enhanced permeability and transendothelial leukocyte diapedesis, abnormal activation of coagulation cascades, elevated thrombin production, and impaired fibrinolysis. Spontaneous platelet aggregation causes release of serotonin, platelet-derived growth factor (PDGF), and platelet alpha granules including thromboxane, a potent vasoconstrictor. Smooth-muscle cell–like myointimal cells proliferate, the basement membrane is thickened and reduplicated, and fibrosis of the adventitial layers develops. The vasculopathic process affects capillaries, as well as arterioles, and even large vessels in many organs, resulting in reduced blood flow and tissue ischemia. Progressive luminal occlusion due to intimal and medial hypertrophy, combined with persistent endothelial cell damage and adventitial fibrosis, establish a vicious cycle that culminates in the striking absence of blood vessels (rarefaction) in late-stage disease. Recurrent ischemia-reperfusion generates reactive oxygen species (ROS) that further damage the endothelium through peroxidation of membrane lipids. Paradoxically, the process of revascularization that normally reestablishes blood flow to ischemic tissue is defective in SSc despite elevated levels of vascular endothelial growth factor (VEGF) and other angiogenic factors. Moreover, the number of bone marrow–derived circulating endothelial progenitor cells is reduced. Thus, widespread capillary loss, obliterative vasculopathy of small and medium-sized arteries, and failure to repair damaged vessels are hallmarks of SSc.
IMMUNE DYSREGULATION
Cellular Immunity The following observations highlight the autoimmune nature of SSc: presence of circulating autoantibodies; familial clustering of SSc with other autoimmune diseases; detection of immune cells, including T cells with oligoclonal antigen receptors, in target organs; elevated circulating levels and spontaneous secretion from blood mononuclear cells of inflammatory cytokines and chemokines such as interleukin (IL) 1, IL-4, IL-10, IL-17, IL-33, CCL2, and CXCL4; and the association with variants in genes functionally implicated in immune responses. Genetic studies in SSc reveal strong and consistent associations with major histocompatibility locus alleles, as well as non-HLA-linked genes encoding mediators of both adaptive and innate immune responses (CD247, STAT4, IRF5, CD226, and TNFSF4). In early SSc, mononuclear inflammatory cell infiltrates comprised of activated T cells, monocytes/macrophages, and dendritic cells can be seen in skin, lungs, and other affected organs prior to appearance of fibrosis or vascular damage. Dendritic cells and T cells can often be found in close proximity to activated fibroblasts and myofibroblasts. Tissue-infiltrating T cells express CD45 and HLA-DR activation markers and display restricted T cell receptor signatures indicative of oligoclonal expansion in response to (unknown) antigen. Circulating T cells have elevated levels of chemokine receptors and α1 integrin adhesion molecules, accounting for their enhanced binding to endothelium and to fibroblasts. Endothelial cells express ICAM-1 and other adhesion molecules that facilitate leukocyte diapedesis. Activated macrophages and T cells show a TH2-polarized type 2 immune response driven by dendritic cells and thymic stromal lymphopoietin. TH2 cytokines such as IL-4 and IL-13 induce fibroblast activation and alternate M2 macrophage polarization, whereas the TH1 cytokine interferon γ (IFN-γ) blocks cytokine-mediated fibroblast activation. Alternately activated M2 macrophages produce TGF-β and promote fibrosis. Although the frequency of circulating regulatory T cells that enforce immune tolerance is elevated in SSc, their immunosuppressive function is defective. Molecular characterization of SSc skin biopsies using DNA microarrays identifies a subset showing markedly elevated expression of inflammation-associated genes, particularly chemokines and their receptors, interferon response genes, and mediators of innate immunity. Evidence of activated innate immunity and toll-like receptor signaling, indicative of activation by type 1 interferon produced by plasmacytoid dendritic cells, is prominent in peripheral blood cells.
Humoral Autoimmunity Circulating ANAs can be detected in virtually all patients with SSc. In addition, a number of SSc-specific autoantibodies have been described. These SSc-specific antibodies show strong association with distinct disease endophenotypes (Table 382-3). While most are directed against intracellular proteins associated with cell proliferation, such as topoisomerase I and RNA polymerases I, II, and III, others are directed against cell-surface antigens, receptors, or secreted proteins. Autoantibodies have clinical utility as diagnostic and prognostic biomarkers in SSc, and some, such as antibodies directed against the angiotensin II receptor or the PDGF receptor, may have a direct pathogenic role.
|
AUTOANTIBODIES AND ASSOCIATED FEATURES IN SYSTEMIC SCLEROSIS (SSC) |
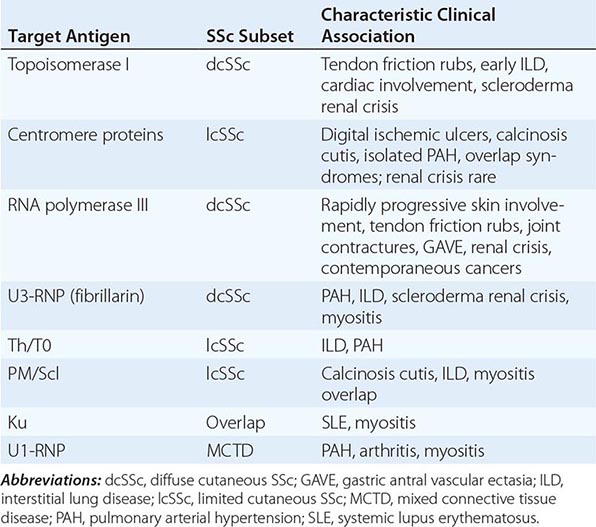
A variety of mechanisms have been proposed for the development of autoantibodies in SSc. Proteolytic cleavage, increased expression, or altered subcellular localization of certain cellular proteins in SSc could lead to their recognition as neoepitopes by the immune system, resulting in breakdown of immune tolerance. B cells are implicated in both the autoimmune and fibrotic process in SSc. In addition to antibody production, B cells also present antigen, secrete IL-6 and TGF-β, and modulate T cell and dendritic cell function.
FIBROSIS
Fibrosis affecting multiple organs, a distinguishing feature of SSc, is characterized by progressive replacement of normal tissue architecture with dense, stiff, and acellular connective tissue. Fibrosis characteristically follows, and is thought to be a consequence of, inflammation, autoimmunity, and microvascular damage. Fibroblasts are mesenchymal cells responsible for maintaining the functional and structural integrity of connective tissue. Upon activation by TGF-β and other extracellular cues, fibroblasts proliferate; migrate; secrete collagens, growth factors, chemokines, and cytokines; and transdifferentiate into contractile myofibroblasts. Under normal conditions, these fibrotic responses constitute self-limited physiologic remodeling necessary for tissue repair and regeneration. When these responses become sustained and amplified, pathologic fibrosis results. Autocrine stimulatory signaling by endogenously produced TGF-β and fibrotic mediators such as hypoxia, ROS, thrombin, Wnt ligands, connective tissue growth factor (CTGF), PDGF, lysophosphatidic acid, endothelin-1, mechanical forces, and endogenous ligands for toll-like receptors are responsible for maintaining sustained fibroblast activation underlying progressive fibrosis in SSc.
In addition to tissue-resident fibroblasts and transformation of epithelial cells into fibroblasts, bone marrow–derived circulating mesenchymal progenitor cells also contribute to fibrosis. The factors that regulate the differentiation of mesenchymal progenitor cells and their trafficking from the circulation into lesional tissue are unknown. Epithelial and endothelial cells, mesenchymal progenitor cells, and tissue fibroblasts can differentiate into smooth-muscle-like myofibroblasts. Although myofibroblasts can be detected transiently during normal wound healing, they persist in fibrotic tissue, possibly due to resistance to apoptosis, and contribute to scar formation via production of collagen and TGF-β and contraction of the surrounding extracellular matrix.
Explanted SSc fibroblasts may display an abnormally activated phenotype ex vivo, with variably increased rates of collagen gene transcription, spontaneous ROS generation, and constitutive expression of alpha smooth-muscle actin stress fibers. The persistence of the “scleroderma phenotype” of these cells during their serial passage in vitro may reflect autocrine TGF-β stimulatory loops, deregulated microRNA expressions, histone acetylation, and other epigenetic modifications.
PATHOLOGY
The distinguishing pathologic hallmark of SSc is the combination of widespread capillary loss and obliterative microangiopathy, together with fibrosis in the skin and internal organs. In early disease, perivascular inflammatory cell infiltrates composed of T lymphocytes, monocytes/macrophages, plasma cells, mast cells, and occasionally B cells may be detected in multiple organs. A bland noninflammatory obliterative vasculopathy as a late finding is prominent in the heart, lungs, kidneys, and intestinal tract. Fibrosis is found in the skin, lungs, gastrointestinal tract, heart, tendon sheaths, perifascicular tissue surrounding skeletal muscle, and some endocrine organs. In these tissues, accumulation of collagens, fibronectin, proteoglycans, tenascin, cartilage oligomeric matrix protein (COMP), and other structural macromolecules progressively disrupts normal architecture, resulting in impaired function of affected organs.
SKIN
In the skin, fibrosis causes dermal expansion and obliteration of the hair follicles, eccrine glands, and other appendages (Fig. 382-2A). Collagen fiber accumulation is most prominent in the reticular dermis, and the fibrotic process invades the subjacent adipose layer with entrapment of adipocytes. With disease progression, the intradermal adipose layer is diminished and may completely disappear. The epidermis is atrophic, and the rete pegs are effaced.
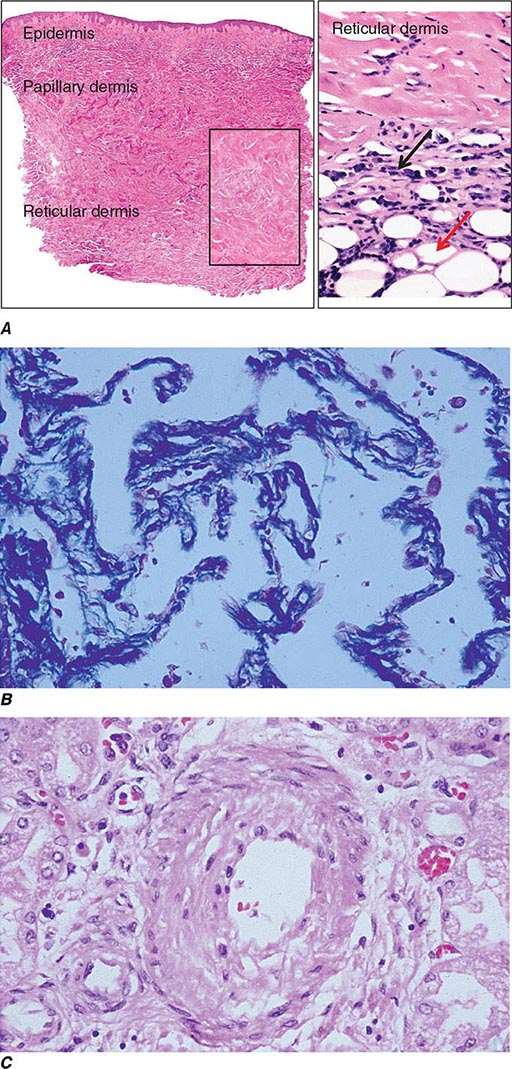
FIGURE 382-2 Pathologic findings in systemic sclerosis (SSc). A. Left panel: The skin is thickened due the marked expansion of the dermis. Inset, higher magnification showing thick hyalinized collagen bundles replace skin appendages. Right panel: Inflammation in the reticular dermis. Mononuclear inflammatory cells infiltrating the dermis and intradermal adipose tissue. B. Early interstitial lung disease. Diffuse fibrosis of the alveolar septae and a chronic inflammatory cell infiltrate. Trichrome stain. C. Pulmonary arterial obliterative vasculopathy. Striking intimal hyperplasia and narrowing of the lumen of a small pulmonary artery, with minimal interstitial fibrosis, in a patient with limited cutaneous SSc.
LUNGS
Patchy infiltration of the alveolar walls with T lymphocytes, macrophages, and eosinophils occurs in early disease. With progression, interstitial fibrosis and vascular damage dominate the pathologic picture, often coexisting within the same lesions in patients with dcSSc. Pulmonary fibrosis is characterized by expansion of the alveolar interstitium, with accumulation of collagen and other matrix proteins. The most common histologic pattern in SSc-associated ILD is nonspecific interstitial pneumonia (NSIP), distinct from the usual interstitial pneumonia (UIP) pattern characteristically seen in patients with idiopathic pulmonary fibrosis (Fig. 382-2B). Progressive thickening of the alveolar septae results in obliteration of the airspaces and loss of pulmonary blood vessels. This process impairs gas exchange and contributes to pulmonary hypertension. Intimal thickening of the pulmonary arteries, best seen with elastin stain, underlies pulmonary hypertension (Fig. 382-2C) and, at autopsy, is often associated with multiple pulmonary emboli and evidence of myocardial fibrosis.
GASTROINTESTINAL TRACT
Pathologic changes can be found at any level from the mouth to the rectum. The lower esophagus shows prominent atrophy of the muscular layers and characteristic vascular lesions; striated muscle in the upper third of the esophagus is generally spared. Replacement of the normal intestinal tract architecture results in diminished peristaltic activity, with gastroesophageal reflux, dysmotility, and small-bowel obstruction. Chronic reflux is associated with esophageal inflammation, ulcerations, and stricture formation and may lead to Barrett’s metaplasia.
KIDNEYS
In the kidneys, vascular lesions affecting the interlobular and arcuate arteries predominate. Chronic renal ischemia is associated with shrunken glomeruli. Acute scleroderma renal crisis is associated with a classic thrombotic microangiopathic pathology: reduplication of elastic lamina, marked intimal proliferation, and narrowing of the lumen in small renal arteries, commonly accompanied by thrombosis and hemolysis.
HEART
The heart is frequently affected, with prominent involvement of the myocardium and pericardium. The characteristic arteriolar lesions are concentric intimal hypertrophy and luminal narrowing, accompanied by contraction band necrosis reflecting ischemia-reperfusion injury and myocardial fibrosis. Fibrosis of the conduction system is common, especially at the sinoatrial node. Despite the prominent role of ischemia in SSc, the frequency of atherosclerotic coronary artery disease is comparable to the general population.
OTHER ORGANS
Synovitis may be found in early SSc; however, with disease progression, the synovium becomes fibrotic. Fibrosis of tendon sheaths and fascia produces palpable and sometimes audible tendon friction rubs. Inflammation and, in later stages, atrophy and fibrosis of the muscles are common findings. Fibrosis of the thyroid gland and of the minor salivary glands may be seen.
CLINICAL FEATURES
OVERVIEW
Virtually every organ can be clinically affected (Table 382-4). Most patients with SSc can be classified as lcSSc or dcSSc (Table 382-2). Although stratification of SSc patients into diffuse and limited cutaneous subsets is useful, disease expression is far more complex, and several distinct endophenotypes exist within each subset. For example, 10–15% of patients with lcSSc develop PAH without significant ILD. Other patients have systemic features of SSc without appreciable skin involvement (SSc sine scleroderma). Unique clinical phenotypes of SSc associate with specific autoantibodies (Table 382-3). Patients with “overlap” have typical SSc features coexisting with clinical and laboratory evidence of another autoimmune disease such as polymyositis, Sjögren’s syndrome, polyarthritis, autoimmune liver disease, or SLE.
|
FREQUENCY OF CLINICAL ORGAN INVOLVEMENT: LIMITED CUTANEOUS AND DIFFUSE CUTANEOUS FORMS OF SYSTEMIC SCLEROSIS (SSC) |
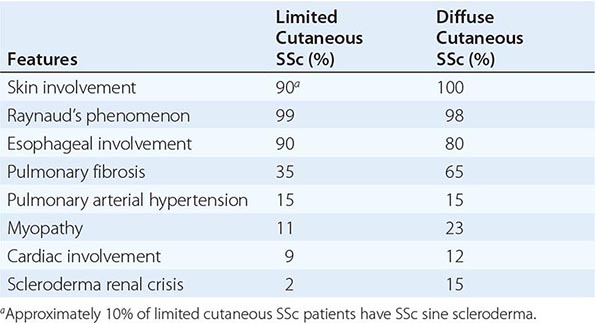
INITIAL CLINICAL PRESENTATION
The initial presentation is quite different in the diffuse and the limited cutaneous forms of the disease. In dcSSc, the interval between Raynaud’s phenomenon and onset of other disease manifestations is typically brief (weeks to months). Soft tissue swelling and intense pruritus are signs of the early inflammatory “edematous” phase. The fingers, hands, distal limbs, and face are usually affected first. Diffuse skin hyperpigmentation and carpal tunnel syndrome can occur. Arthralgias, muscle weakness, fatigue, and decreased joint mobility are common. During the ensuing weeks to months, the inflammatory edematous phase evolves into the “fibrotic” phase, with skin induration associated with hair loss, reduced production of skin oils, and a decline in sweating capacity. Progressive flexion contractures of the fingers ensue. The wrists, elbows, shoulders, hip girdles, knees, and ankles become stiff due to fibrosis of the supporting joint structures. While advancing skin involvement is the most visible manifestation of early dcSSc, important and frequently clinically silent internal organ involvement develops during this stage. The initial 4 years from disease onset is the period of rapidly evolving pulmonary and renal damage. If organ failure does not occur during this period, the systemic process may stabilize.
Compared to dcSSc, the course of lcSSc is characteristically more indolent. In these patients, the interval between Raynaud’s phenomenon and onset of manifestations such as gastroesophageal reflux, cutaneous telangiectasia, or soft tissue calcifications can be several years. On the other hand, scleroderma renal crisis and severe pulmonary fibrosis are uncommon in lcSSc. Clinically evident cardiac involvement and PAH develop in more than 15%. Overlap with keratoconjunctivitis sicca, polyarthritis, cutaneous vasculitis, and biliary cirrhosis is seen in some patients with lcSSc.
ORGAN INVOLVEMENT
RAYNAUD’S PHENOMENON
Raynaud’s phenomenon, the most frequent extracutaneous complication of SSc, is characterized by episodes of reversible vasoconstriction in the fingers and toes. Vasoconstriction may also affect the tip of the nose and earlobes. Attacks are triggered by a decrease in temperature, as well as emotional stress and vibration. Typical attacks start with pallor, followed by cyanosis of variable duration. Hyperemia ensues spontaneously or with rewarming of the digit. The progression of the three color phases reflects the underlying vasoconstriction, ischemia, and reperfusion.
As much as 3–5% of the general population has Raynaud’s phenomenon. In the absence of signs or symptoms of an underlying condition, Raynaud’s phenomenon is classified as primary and represents an exaggerated physiologic response to cold. Secondary Raynaud’s phenomenon can occur as a complication of SSc and other connective tissue diseases, hematologic and endocrine conditions, and occupational disorders, and can complicate the use of beta blockers and anticancer drugs such as cisplatin and bleomycin. Distinguishing primary versus secondary Raynaud’s phenomenon presents a diagnostic challenge. Primary Raynaud’s phenomenon is supported by the following: absence of an underlying cause; a family history of Raynaud’s phenomenon; absence of digital tissue necrosis, ulceration, or gangrene; and a negative ANA test. Secondary Raynaud’s phenomenon tends to develop at an older age (>30 years), is clinically more severe (episodes more frequent, prolonged, and painful), and is frequently associated with ischemic digital ulcers and loss of digits (Fig. 382-3). Nailfold capillaroscopy, where the cutaneous capillaries at the nail bed are viewed under a drop of grade B immersion oil using a low-power stereoscopic microscope, can be helpful in the evaluation of Raynaud’s phenomenon. Primary Raynaud’s phenomenon is associated with normal capillaries that appear as regularly spaced parallel vascular loops, whereas in patients with Raynaud’s associated with SSc and other connective tissue diseases, nailfold capillaries are distorted with widened and irregular loops, dilated lumen, and areas of vascular “dropout.” In addition to digits, cold-induced episodic Raynaud’s-like vasospasm has been documented in the pulmonary, renal, gastrointestinal, and coronary circulations in SSc.
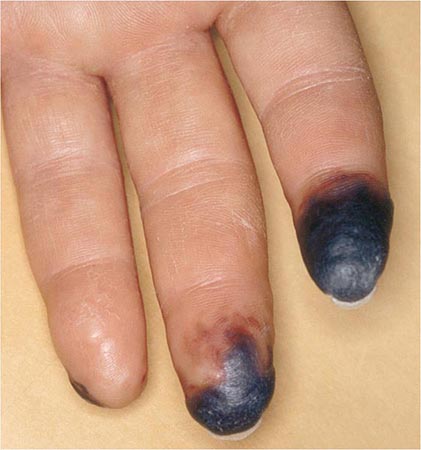
FIGURE 382-3 Digital necrosis. Sharply demarcated necrosis of the fingertip in a patient with limited cutaneous systemic sclerosis (SSc) associated with severe Raynaud’s phenomenon.
SKIN FEATURES
While early-stage SSc is associated with edematous skin changes, skin thickening is the hallmark that distinguishes SSc from other connective tissue diseases. The distribution of skin thickening is invariably symmetric and bilateral. It typically starts in the fingers and then characteristically advances from distal to proximal extremities in an ascending fashion. The involved skin is firm, coarse, and thickened, and the extremities and trunk may be darkly pigmented. In some patients, diffuse tanning in the absence of sun exposure is a very early manifestation of skin involvement. In dark-skinned patients, vitiligo-like hypopigmentation may occur. Because pigment loss spares the perifollicular areas, the skin may have a “salt-and-pepper” appearance, most prominently on the scalp, upper back, and chest. Dermal sclerosis due to collagen accumulation obliterates hair follicles, sweat glands, and eccrine and sebaceous glands, resulting in hair loss, decreased sweating, and dry skin. Transverse creases on the dorsum of the fingers disappear (Fig. 382-4). Fixed flexion contractures of the fingers cause reduced hand mobility and lead to muscle atrophy. Skin thickening in combination with fibrosis of the subjacent tendons accounts for contractures of the wrists, elbows, and knees. Thick ridges at the neck due to firm adherence of skin to the underlying platysma muscle interfere with neck extension. The face assumes a characteristic “mauskopf” appearance with taut and shiny skin, loss of wrinkles, and occasionally an expressionless facies due to reduced mobility of the eyelids, cheeks, and mouth. Thinning of the lips with accentuation of the central incisor teeth and fine wrinkles (radial furrowing) around the mouth complete the picture. Reduced oral aperture (microstomia) interferes with eating and oral hygiene. The nose assumes a pinched, beak-like appearance.
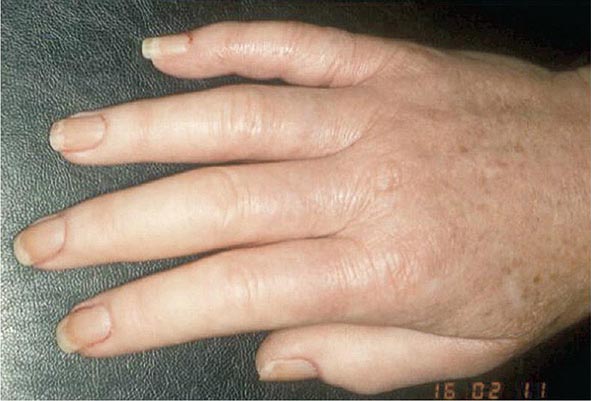
FIGURE 382-4 Sclerodactyly. Note skin induration on the fingers, and fixed flexion contractures at the proximal interphalangeal joints in a patient with limited cutaneous systemic sclerosis (SSc).
In established SSc, the skin is firmly bound to the subcutaneous fat (tethering) and undergoes thinning and atrophy. Telangiectasias are dilated skin capillaries 2–20 mm in diameter frequently seen in lcSSc. These lesions, reminiscent of hereditary hemorrhagic telangiectasia, are prominent on the face, hands, lips, and oral mucosa (Fig. 382-5). A greater number of telangiectasias correlates with the extent of microvascular complications, including PAH. Breakdown of atrophic skin leads to chronic ulcerations at the extensor surfaces of the proximal interphalangeal joints, the volar pads of the fingertips, and bony prominences such as the elbows and malleoli. Ulcers are painful and may become secondarily infected, resulting in osteomyelitis. Healing of ischemic fingertip ulcerations leaves characteristic fixed digital “pits.” Loss of soft tissue at the fingertips due to ischemia is frequent and may be associated with striking resorption of the terminal phalanges (acro-osteolysis) (Fig. 382-6).
FIGURE 382-5 Cutaneous vascular changes. A. Capillary changes at the nailfold in a patient with limited cutaneous systemic sclerosis (lcSSc). B. Telangiectasia on the face.
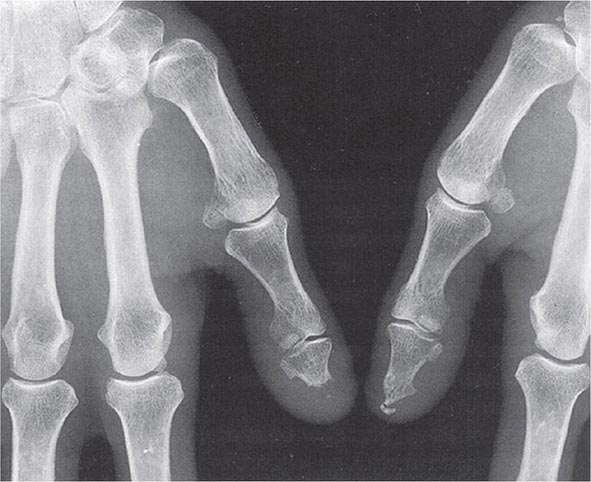
FIGURE 382-6 Acro-osteolysis. Note dissolution of terminal phalanges in a patient with long-standing limited cutaneous systemic sclerosis (lcSSc) and Raynaud’s phenomenon.
Calcium deposits (calcinosis) in the skin and soft tissues occur in patients with lcSSc who are positive for anticentromere antibodies. The deposits, varying in size from tiny punctate lesions to large conglomerate masses, are composed of calcium hydroxyapatite crystals and can be readily visualized on plain x-rays. Frequent locations include the finger pads, palms, extensor surfaces of the forearms, and the olecranon and prepatellar bursae (Fig. 382-7). They may occasionally ulcerate through the overlying skin, producing drainage of chalky white material, pain, and local inflammation. Paraspinal soft tissue calcifications may cause neurologic complications.
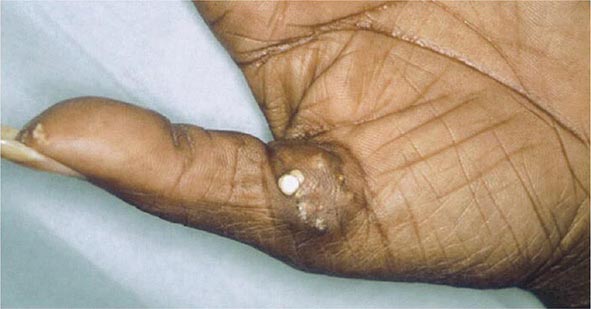
FIGURE 382-7 Calcinosis cutis. Note large calcific deposit breaking through the skin in a patient with limited cutaneous systemic sclerosis (lcSSc).
PULMONARY FEATURES
Pulmonary involvement is frequent in SSc and is the leading cause of death. The two principal forms are ILD and pulmonary vascular disease. Patients with SSc frequently develop some degree of both complications. Less frequent pulmonary manifestations include aspiration pneumonitis complicating chronic gastroesophageal reflux, pulmonary hemorrhage due to endobronchial telangiectasia, obliterative bronchiolitis, pleural reactions, restrictive ventilatory defect due to chest wall fibrosis, spontaneous pneumothorax, and drug-induced lung toxicity. The incidence of lung cancer is increased.
Interstitial Lung Disease (ILD) Evidence of ILD can be found in up to 90% of patients with SSc at autopsy and 85% by thin-section high-resolution computed tomography (HRCT). In contrast, clinically significant ILD develops in 16–43%; the frequency varies depending on the detection method used. Risk factors include male gender, African-American race, diffuse skin involvement, severe gastroesophageal reflux, and the presence of topoisomerase I autoantibodies, as well as a low forced vital capacity (FVC) or single-breath diffusing capacity of the lung for carbon monoxide (DLCO) at initial presentation. In these patients, the most rapid progression in lung disease occurs early in the course of the disease (within the first 3 years), when the FVC can decline by 30% per year.
Pulmonary involvement can remain asymptomatic until it is advanced. The most common presenting respiratory symptoms—exertional dyspnea, fatigue, and reduced exercise tolerance—are subtle and slowly progressive. A chronic dry cough may be present. Physical examination may reveal “Velcro” crackles at the lung bases. Pulmonary function testing (PFT) is a sensitive method for detecting early pulmonary involvement. The most common abnormalities are reductions in FVC, total lung capacity (TLC), and DLCO. A reduction in DLCO that is significantly out of proportion to the reduction in lung volumes should raise suspicion for pulmonary vascular disease, but may also be due to anemia. Oxygen desaturation with exercise is common.
Chest radiography can rule out infection and other causes of pulmonary involvement, but compared to HRCT, it is relatively insensitive for detection of early ILD. HRCT shows subpleural reticular linear opacities and ground-glass opacifications, predominantly in the lower lobes, even in asymptomatic patients (Fig. 382-8). Additional HRCT findings include mediastinal lymphadenopathy, pulmonary nodules, traction bronchiectasis, and uncommonly, honeycomb changes. The extent of pulmonary interstitial changes on HRCT is a predictor of mortality in SSc. Bronchoalveolar lavage (BAL) can demonstrate inflammation in the lower respiratory tract and may be useful for ruling out infection. Although an elevated proportion of neutrophils (>2%) and/or eosinophils (>3%) in the BAL fluid is correlated with more extensive lung disease on HRCT and is associated with more rapid decline in FVC and reduced survival, BAL is not useful for identifying reversible alveolitis. Lung biopsy is indicated only in patients with atypical findings on chest radiographs and should be thoracoscopically guided. The histologic pattern on lung biopsy may predict the risk of progression of ILD. The most common pattern in SSc, NSIP, carries a better prognosis than UIP.
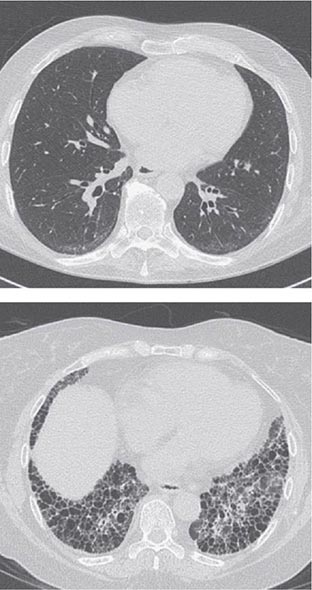
FIGURE 382-8 HRCT images of the chest from patients with systemic sclerosis. Top panel: Early interstitial lung disease. Mild changes with sub pleural reticulations and ground glass opacities in the lower lobes of the lung. Patient in supine position. Bottom panel: Extensive lung fibrosis with ground glass opacities, coarse reticular honeycombing, and traction bronchiectasis. (Courtesy of Rishi Agrawal, MD.)
Pulmonary Arterial Hypertension (PAH) PAH, defined as a mean pulmonary arterial pressure ≥25 mmHg with a pulmonary capillary wedge pressure ≤15 mmHg, develops in approximately 15% of patients with SSc and can occur in association with ILD or as an isolated abnormality. The natural history of SSc-associated PAH is variable, but in many patients, it follows a downhill course with development of right heart failure. The median survival of SSc patients with untreated PAH is 1 year following diagnosis. Risk factors for PAH include limited cutaneous disease, older age at disease onset, severe Raynaud’s phenomenon, and the presence of antibodies to centromere, U1-RNP, U3-RNP (fibrillarin), and B23.
The initial symptom of PAH is typically exertional dyspnea and reduced exercise capacity. With disease progression, angina, exertional near-syncope, and symptoms and signs of right-sided heart failure appear. Physical examination may show tachypnea, a prominent split S2 heart sound, palpable right ventricular heave, elevated jugular venous pressure, and dependent edema. Doppler echocardiography provides a noninvasive method for estimating the pulmonary arterial pressure. In light of the poor prognosis of untreated PAH, all SSc patients should be screened for its presence at initial evaluation. Echocardiographic estimates of pulmonary arterial systolic pressures >40 mmHg at rest suggest PAH. Pulmonary function testing may show a reduced DLCO in isolation or out of proportion with the severity of restriction. Right heart catheterization is the gold standard for diagnosing PAH. Because echocardiography can result in over- or underestimation of pulmonary arterial pressures in SSc, cardiac catheterization is always required to confirm the presence of PAH; accurately assess its severity, including the degree of right heart dysfunction; and rule out venoocclusive disease and other cardiac causes of pulmonary hypertension. Yearly echocardiographic screening for PAH is recommended in most patients with SSc; an isolated decline in DLCO may also be indicative of developing PAH. Serum levels of brain natriuretic peptide (BNP) and N-terminal pro-BNP correlate with the presence and severity of PAH in SSc, as well as survival. While BNP measurements can be useful in screening for PAH and in monitoring the response to treatment, elevated BNP levels are not specific for PAH and also occur in other forms of right and left heart disease. The prognosis of SSc-associated PAH is worse, and treatment response poorer, than in idiopathic PAH
GASTROINTESTINAL INVOLVEMENT
The gastrointestinal tract is affected in up to 90% of SSc patients with both limited and diffuse cutaneous forms of the disease. The pathologic features of atrophy of smooth muscle, intact mucosa, and obliterative small-vessel vasculopathy are similar throughout the length of the gastrointestinal tract.
Upper Gastrointestinal Tract Involvement Oropharyngeal manifestations due to a combination of xerostomia, reduced oral aperture, periodontal disease, and resorption of the mandibular condyles are frequent and cause much distress. The frenulum of the tongue may be shortened. Most patients have symptoms of gastroesophageal reflux disease (GERD): heartburn, regurgitation, and dysphagia. A combination of reduced lower esophageal sphincter pressure resulting in gastroesophageal reflux, impaired esophageal clearance of refluxed gastric contents due to diminished motility in the distal two-thirds of the esophagus, and delayed gastric emptying accounts for GERD. Manometry shows abnormal upper intestinal motility in most patients with SSc. Extraesophageal manifestations of GERD include hoarseness, chronic cough, and aspiration pneumonitis, which may aggravate underlying ILD. Chest computed tomography (CT) scan characteristically shows a dilated esophagus with intraluminal air. Endoscopy may be necessary to rule out opportunistic infections with Candida, herpes virus, and cytomegalovirus. Severe erosive esophagitis may be found on endoscopy in patients with minimal symptoms. Esophageal strictures and Barrett’s esophagus may complicate chronic GERD. Because Barrett’s esophagus is associated with an increased risk of adenocarcinoma, SSc patients with Barrett’s esophagus need periodic endoscopy and esophageal biopsy.
Gastroparesis with early satiety, abdominal distention, and aggravated reflux symptoms is common. The presence and severity of gastroparesis can be assessed by radionuclide gastric emptying studies. Gastric antral vascular ectasia (GAVE) in the antrum may occur. These subepithelial lesions, reflecting the diffuse small-vessel vasculopathy of SSc, are described as “watermelon stomach” due to their endoscopic appearance. Patients with GAVE can have recurrent episodes of gastrointestinal bleeding, resulting in chronic unexplained anemia.
Lower Gastrointestinal Tract Involvement Impaired intestinal motility may result in malabsorption and chronic diarrhea secondary to bacterial overgrowth. Fat and protein malabsorption and vitamin B12 and vitamin D deficiency ensue, sometimes culminating in severe malnutrition. Disturbed intestinal motor function can also cause intestinal pseudo-obstruction, with symptoms of nausea and abdominal distension that are indistinguishable from those of delayed gastric emptying. Patients present with recurrent episodes of acute abdominal pain, nausea, and vomiting. Radiographic studies show acute intestinal obstruction, and the major diagnostic challenge is to differentiate pseudoobstruction, which responds to supportive care and intravenous nutritional supplementation, from mechanical obstruction.
Colonic involvement may cause severe constipation, fecal incontinence, gastrointestinal bleeding from telangiectasia, and rectal prolapse. In late-stage SSc, wide-mouth sacculations or diverticula occur in the colon, occasionally causing perforation and bleeding. An occasional radiologic finding is pneumatosis cystoides intestinalis due to air trapping in the bowel wall that may rarely rupture and cause benign pneumoperitoneum. Although the liver is rarely affected, primary biliary cirrhosis may coexist with SSc.
RENAL INVOLVEMENT: SCLERODERMA RENAL CRISIS
Scleroderma renal crisis occurs in 10–15% of patients and generally within 4 years of the onset of the disease. Prior to the advent of angiotensin-converting enzyme (ACE) inhibitors, short-term survival in scleroderma renal crisis was <10%. The pathogenesis involves obliterative vasculopathy and luminal narrowing of the renal arcuate and interlobular arteries. Progressive reduction in renal blood flow, aggravated by vasospasm, leads to juxtaglomerular hyperplasia, increased renin secretion, and activation of angiotensin, with further renal vasoconstriction resulting in a vicious cycle that culminates in accelerated hypertension. Risk factors for scleroderma renal crisis include African-American race, male gender, and dcSSc with extensive and progressive skin involvement. Up to 50% of patients with scleroderma renal crisis have anti-RNA polymerase III antibodies. Palpable tendon friction rubs, pericardial effusion, new unexplained anemia, and thrombocytopenia may be harbingers of impending scleroderma renal crisis. High-risk patients with early SSc should be counseled to check their blood pressure daily. Patients with lcSSc or anticentromere antibodies rarely develop scleroderma renal crisis. Because there is an association between glucocorticoid use and scleroderma renal crisis, prednisone should be used in high-risk SSc patients only when absolutely required and at low doses (<10 mg/d).
Patients characteristically present with accelerated hypertension and progressive oliguric renal insufficiency. However, approximately 10% of patients with scleroderma renal crisis present with normal blood pressure. Normotensive renal crisis is generally associated with a poor outcome. Headache, blurred vision, and congestive heart failure may accompany elevation of blood pressure. Urinalysis typically shows mild proteinuria, granular casts, and microscopic hematuria; thrombocytopenia and microangiopathic hemolysis with fragmented red blood cells can be seen. Progressive oliguric renal failure over several days generally follows. In some cases, scleroderma renal crisis is misdiagnosed as thrombotic thrombocytopenic purpura or other forms of thrombotic microangiopathy. In these cases, a renal biopsy may be of some benefit. In addition, biopsy findings of vascular thrombosis and glomerular ischemic collapse predict poor renal outcomes. Oliguria or a creatinine >3 mg/dL at presentation predicts poor outcome, with permanent hemodialysis and high mortality. Rarely, crescentic glomerulonephritis occurs in the setting of SSc and may be associated with myeloperoxidase-specific antineutrophil cytoplasmic antibodies. Membranous glomerulonephritis may occur in patients treated with D-penicillamine. Asymptomatic renal function impairment occurs in up to half of SSc patients. Such subclinical renal involvement is associated with other vascular manifestations of SSc and rarely progresses.
CARDIAC INVOLVEMENT
Although it is often silent, cardiac involvement in SSc is frequently detected when patients are screened with sensitive diagnostic tools. Clinically evident cardiac involvement is associated with poor outcomes. Cardiac disease in SSc may be primary or secondary to PAH, ILD, or renal involvement. It occurs more frequently in patients with dcSSc than in those with lcSSc and generally develops within 3 years of the onset of skin thickening. Clinically evident cardiac involvement in SSc is a poor prognostic factor. The endocardium, myocardium, and pericardium may each be affected separately or together. Manifestations of pericardial involvement include acute pericarditis, pericardial effusions, constrictive pericarditis, and cardiac tamponade. Conduction system fibrosis occurs commonly and may be silent or manifested by atrial and ventricular tachycardias or heart block. Recurrent vasospasm and ischemia-reperfusion injury contribute to myocardial fibrosis, resulting in asymptomatic systolic or diastolic left ventricular dysfunction that may progress to overt heart failure. Systemic and pulmonary hypertension and lung and renal involvement may also impact on the heart. Despite the presence of widespread obliterative vasculopathy, the frequency of clinical or pathologic epicardial coronary artery disease in SSc is not increased. While conventional echocardiography has low sensitivity for detecting SSc preclinical heart involvement, newer modalities such as tissue Doppler echocardiography (TDE), cardiac magnetic resonance imaging (cMRI), thallium perfusion, and nuclear imaging (single photon emission CT [SPECT]) reveal a high prevalence of abnormal myocardial function or perfusion in SSc patients. The serum level of N-terminal pro-BNP, a ventricular hormone, is a marker for PAH in SSc, but may also have utility as a marker of primary cardiac involvement.
MUSCULOSKELETAL COMPLICATIONS
Carpal tunnel syndrome occurs frequently and may be a presenting manifestation. Generalized arthralgia and stiffness are prominent in early disease. Mobility of small and large joints is progressively impaired, especially in dcSSc. Most commonly affected are the hands. Contractures develop at the proximal interphalangeal joints and wrists. Large joint contractures can be accompanied by tendon friction rubs, characterized by leathery crepitation that can be heard or palpated upon passive movement, that are due to extensive fibrosis and adhesion of the tendon sheaths and fascial planes at the affected joint. Presence of tendon friction rubs is associated with increased risk for renal and cardiac complications and reduced survival. True joint inflammation is uncommon; however, occasional patients develop erosive polyarthritis in the hands. Muscle weakness is common and may indicate deconditioning, disuse atrophy, and malnutrition. Less commonly, inflammatory myositis indistinguishable from idiopathic polymyositis may occur. A chronic noninflammatory myopathy characterized by atrophy and fibrosis in the absence of elevated muscle enzyme levels can be seen in late-stage SSc. Bone resorption occurs most commonly in the terminal phalanges, where it causes loss of the distal tufts (acro-osteolysis) (Fig. 382-5). Resorption of the mandibular condyles can lead to bite difficulties. Osteolysis can also affect the ribs and distal clavicles.
OTHER DISEASE MANIFESTATIONS
Many SSc patients develop dry eyes and dry mouth (sicca complex). Biopsy of the minor salivary glands shows fibrosis rather than focal lymphocytic infiltration characteristic of primary Sjögren’s syndrome (Chap. 383). Hypothyroidism is common and generally due to fibrosis of the thyroid gland. The frequency of macrovascular involvement, including peripheral vascular and coronary artery disease, may be increased. Whereas the central nervous system is generally spared, sensory trigeminal neuropathy due to fibrosis or vasculopathy can occur, presenting with gradual onset of pain and numbness. Pregnancy in women with SSc may be associated with an increased rate of adverse fetal outcomes. Furthermore, cardiopulmonary involvement may worsen during pregnancy, and new onset of scleroderma renal crisis has been described. Erectile dysfunction is frequent in men with SSc and may be the initial disease manifestation. Inability to attain or maintain penile erection is due to vascular insufficiency and fibrosis.
Malignancy in SSc Epidemiologic studies indicate an increased risk of cancer in SSc. Lung cancer and esophageal adenocarcinoma typically occur in the setting of long-standing ILD or gastroesophageal reflux disease and may be caused by chronic inflammation and repair. In contrast, breast, lung, and ovarian carcinomas and lymphomas tend to occur in close temporal association with the clinical onset of SSc, particularly in patients who have autoantibodies to RNA polymerase III. In these cases, SSc may represent a paraneoplastic syndrome triggered by the antitumor immune response.
LABORATORY EVALUATION AND BIOMARKERS
A mild normocytic or microcytic anemia is frequent in patients with SSc and may indicate gastrointestinal bleeding caused by GAVE or chronic esophagitis. Macrocytic anemia may be caused by folate and vitamin B12 deficiency due to small-bowel bacterial overgrowth and malabsorption or by drugs such as methotrexate or alkylating agents. Microangiopathic hemolytic anemia caused by mechanical fragmentation of red blood cells during their passage through microvessels coated with fibrin or platelet thrombi is a hallmark of the thrombotic microangiopathy associated with scleroderma renal crisis. Thrombocytopenia and leukopenia may indicate drug toxicity. In contrast to other connective tissue diseases, the erythrocyte sedimentation rate (ESR) is generally normal; an elevation may signal coexisting myositis or malignancy.
Antinuclear autoantibodies are present in almost all patients with SSc and can be detected at disease onset. Autoantibodies against topoisomerase I (Scl-70) and centromere are mutually exclusive and quite specific for SSc (Table 382-3). Topoisomerase I antibodies are detected in 31% of patients with dcSSc, but in only 13% of patients with lcSSc. They are associated with increased risk of ILD and poor outcomes. Anticentromere antibodies are detected in 38% of patients with lcSSc, but in only 2% of patients with dcSSc and rarely in patients with Raynaud’s phenomenon and Sjögren’s syndrome. Anticentromere antibodies in SSc are associated with PAH, but only infrequently with significant cardiac or renal involvement or ILD. Nucleolar immunofluorescence pattern on serologic testing reflects antibodies to U3-RNP (fibrillarin), Th/To, or PM/Scl, whereas a speckled immunofluorescence pattern indicates antibodies to RNA polymerase III. Although antibodies to β2GPI occur in antiphospholipid antibody syndrome and are not specific for SSc, their presence in SSc is associated with an increased risk of ischemic lesions in the fingers.
DIAGNOSIS, STAGING, AND MONITORING
The diagnosis of SSc is made primarily on clinical grounds and is generally straightforward in patients with established disease. The presence of skin induration, with a characteristic symmetric distribution pattern associated with typical visceral organ manifestations, establishes the diagnosis with a high degree of certainty. Although the conditions listed in Table 382-1 can be associated with skin induration, the distribution pattern of skin lesions, together with the absence of Raynaud’s phenomenon or typical visceral organ manifestations or SSc-specific autoantibodies, differentiates these conditions from SSc. Occasionally, full-thickness biopsy of the skin is required for establishing the diagnosis of scleredema, scleromyxedema, or nephrogenic systemic fibrosis. In lcSSc, a history of antecedent Raynaud’s phenomenon and gastroesophageal reflux symptoms, coupled with the presence of sclerodactyly and capillary changes on nailfold capillaroscopy, often in combinations with cutaneous telangiectasia and calcinosis, helps to establish the diagnosis. The finding of digital tip pitting scars and radiologic evidence of pulmonary fibrosis in the lower lobes are particularly helpful diagnostically. Primary Raynaud’s phenomenon is a common benign condition that must be differentiated from early or limited SSc. Nailfold microscopy is particularly helpful in this situation, because in primary Raynaud’s phenomenon, the nailfold capillaries are normal, whereas in SSc, capillary abnormalities, as well as serum autoantibodies, can be detected even before other disease manifestations.
Establishing the diagnosis of SSc at an early stage of the disease may be a challenge. In dcSSc, initial symptoms are often nonspecific and relate to inflammation. Patients complain of fatigue, swelling, aching, and stiffness, and Raynaud’s phenomenon may initially be absent. Physical examination may reveal diffuse upper extremity edema and puffy fingers. Patients at this stage are sometimes diagnosed as early rheumatoid arthritis, SLE, myositis, or, most commonly, undifferentiated connective tissue disease. Within weeks to months, Raynaud’s phenomenon and characteristic clinical features appear accompanied by advancing induration of the skin. The presence of antinuclear and SSc-specific autoantibodies provides a high degree of diagnostic specificity. Raynaud’s phenomenon with fingertip ulcerations or other evidence of digital ischemia, coupled with telangiectasia, distal esophageal dysmotility, unexplained ILD or PAH, or accelerated hypertension with renal failure in the absence of clinically evident skin induration, suggests the diagnosis of SSc sine scleroderma. These patients may have anticentromere antibodies.
|
TREATMENT |
SYSTEMIC SCLEROSIS |
OVERVIEW: MANAGEMENT PRINCIPLES To date, no therapy has been shown to significantly alter the natural history of SSc. In contrast, multiple interventions are highly effective in alleviating the symptoms, slowing the progression of the cumulative organ damage, and reducing disability. A significant reduction in disease-related mortality has been noted during the past 25 years. In light of the marked heterogeneity in disease manifestations, organ complications, and natural history, treatment must be tailored to each individual patient’s unique needs.
A thorough investigation should be undertaken at baseline. Optimal management incorporates the following principles (Table 382-5): prompt and accurate diagnosis; classification and risk stratification based on clinical and laboratory evaluation; early recognition of organ-based complications and assessment of their extent, severity, and likelihood of deterioration; regular monitoring for disease progression, activity, new complications, and response to therapy; adjusting therapy; and continuing patient education. In order to minimize irreversible organ damage, the management of life-threatening complications must be proactive, with regular screening and initiation of appropriate intervention at the earliest possible opportunity. In light of the complex and multisystemic nature of the SSc, a team-based management approach integrating multiple specialists should be pursued whenever possible. Most patients are treated with combinations of drugs that impact different aspects of the disease. We encourage patients to become familiar with the spectrum of potential complications and understand therapeutic options and natural history, and empower them to partner with their treating physicians. This requires a long-term relationship between patient and physician, with ongoing counseling and encouragement.
|
KEY PRINCIPLES IN MANAGEMENT |
DISEASE-MODIFYING THERAPY: IMMUNOSUPPRESSIVE AGENTS Immunosuppressive agents used in the treatment of other autoimmune or connective tissue diseases have generally shown modest or no benefit in SSc. Glucocorticoids may alleviate stiffness and aching in early-stage dcSSc but do not influence the progression of skin or internal organ involvement, and their use is associated with an increased risk of scleroderma renal crisis. Therefore, glucocorticoids should be given only when absolutely necessary, at the lowest dose possible, and for brief periods only. The use of cyclophosphamide has been extensively studied in light of its efficacy in the treatment of vasculitis (Chap. 385), SLE (Chap. 378), and other autoimmune diseases (Chap. 377e).
Both oral and intermittent IV cyclophosphamide were shown to reduce the progression of SSc-associated ILD, with stabilization and, rarely, modest improvement of pulmonary function and HRCT findings after 1 year of treatment. Improvement in respiratory symptoms and skin induration was also noted. These beneficial effects wane upon discontinuation of therapy. The benefits of cyclophosphamide need to be balanced against its potential toxicity, including bone marrow suppression, opportunistic infections, hemorrhagic cystitis and bladder cancer, premature ovarian failure, and late secondary malignancies.
Methotrexate was associated with a modest skin improvement in small studies. Mycophenolate mofetil treatment was associated with improved skin induration in uncontrolled studies and was generally well tolerated. Small studies support the use of rituximab in SSc patients with skin involvement and ILD. The use of cyclosporine, azathioprine, extracorporeal photopheresis, thalidomide, rapamycin, imatinib, and IV immunoglobulin is currently not well supported by the literature. Intensive immune ablation using a conditioning regimen of high-dose chemotherapy with or without irradiation, followed by autologous stem cell reconstitution, has resulted in durable disease remission in some cases and is undergoing evaluation in randomized clinical trials. In light of its potential morbidity and mortality, as well as significant cost, autologous stem cell transplantation in SSc is still considered experimental.
Antifibrotic Therapy Because widespread tissue fibrosis in SSc causes progressive organ damage, drugs that interfere with the fibrotic process represent a rational therapeutic approach. D-Penicillamine has been extensively used as an antifibrotic agent. In retrospective studies, D-penicillamine stabilized and improved skin induration, prevented new internal organ involvement, and improved survival. However, a randomized controlled clinical trial in early active SSc found no difference in the extent of skin involvement between patients treated with standard-dose (750 mg/d) or very low-dose (125 mg every other day) D-penicillamine. Recent clinical trials show benefit of pirfenidone and of nintedanib in patients with idiopathic pulmonary fibrosis, with significant slowing of the loss of lung function. Whether these two new drugs will have comparable efficacy in the treatment of SSc-associated lung disease is still under investigation.
Vascular Therapy The goal of therapy is to control Raynaud’s phenomenon, prevent the development and enhance the healing of ischemic complications, and slow the progression of obliterative vasculopathy. Patients should dress warmly, minimize cold exposure or stress, and avoid drugs that precipitate or exacerbate vasospastic episodes. Some patients with Raynaud’s may respond to biofeedback therapy. Extended-release dihydropyridine calcium channel blockers such as nifedipine, amlodipine, or diltiazem can ameliorate Raynaud’s phenomenon, but their use is often limited by side effects (palpitations, dependent edema, worsening gastroesophageal reflux). While ACE inhibitors do not reduce the frequency or severity of episodes, angiotensin II receptor blockers such as losartan are effective and generally well tolerated. Patients with Raynaud’s phenomenon unresponsive to these therapies may require the addition of α1-adrenergic receptor blockers (e.g., prazosin), 5-phosphodiesterase inhibitors (e.g., sildenafil), serotonin reuptake inhibitors (e.g., fluoxetine), topical nitroglycerine, and intermittent infusions of IV prostaglandins. Low-dose aspirin and dipyridamole prevent platelet aggregation and may have a role as adjunctive agents. In patients with ischemic ulcers, the endothelin-1 receptor antagonist bosentan reduces the risk of new ulcers. Digital sympathectomy and local injections of botulinum type A (Botox) into the digits are options in patients with severe ischemia and impending loss of the digits. Empirical long-term therapy with statins and antioxidants may retard the progression of vascular damage and obliteration. Vasodilators such as ACE inhibitors, calcium channel blockers, and endothelin receptor blockers may also improve myocardial perfusion and left ventricular function.
TREATMENT OF GASTROINTESTINAL COMPLICATIONS Because oral problems including decreased oral aperture, decreased saliva production, gum recession and periodontal disease leading to teeth loss are common, regular dental care is recommended. Gastroesophageal reflux is very common and may occur in the absence of symptoms; therefore all patients with SSc should be treated. Patients should be instructed to elevate the head of the bed, eat frequent small meals, and avoid oral intake before bedtime. Proton pump inhibitors reduce acid reflux and may need to be given in relatively high doses. Prokinetic agents such as domperidone may be helpful, especially if delayed gastric emptying is present. Episodic gastrointestinal bleeding from gastric antral vascular ectasia (watermelon stomach) may be amenable to treatment with endoscopic laser photocoagulation, although recurrence can occur. Bacterial overgrowth due to small-bowel dysmotility causes abdominal bloating and diarrhea and may lead to malabsorption and severe malnutrition. Treatment with short courses of rotating broad-spectrum antibiotics such as metronidazole, erythromycin, and tetracycline can eradicate bacterial overgrowth. Parenteral hyperalimentation is indicated if malnutrition develops. Chronic hypomotility of the small bowel may respond to octreotide, but pseudo-obstruction is difficult to treat. Fecal incontinence, a frequently underreported complication of SSc, may respond to anti-diarrheal medication and biofeedback therapy.
TREATMENT OF PULMONARY ARTERIAL HYPERTENSION (PAH) In patients with SSc, PAH carries an extremely poor prognosis and accounts for 30% of deaths. Because PAH is asymptomatic until advanced, patients with SSc should be screened for its presence at initial evaluation, and on a yearly basis thereafter. Treatment is generally started with an oral endothelin-1 receptor antagonist such as bosentan or a phosphodiesterase type 5 inhibitor such as sildenafil. Patients may also require diuretics and digoxin when appropriate. If hypoxemia is documented, supplemental oxygen should be prescribed in order to avoid hypoxia-induced secondary pulmonary vasoconstriction. Prostacyclin analogues such as epoprostenol or treprostinil can be given by continuous IV or SC infusion, or via intermittent nebulized inhalations. Combination therapy with different classes of agents, such as an endothelin-1 antagonist and a phosphodiesterase inhibitor, is often necessary. Lung transplantation remains an option for selected patients who fail medical therapy.
TREATMENT OF RENAL CRISIS Scleroderma renal crisis is a medical emergency. Since the outcome is largely determined by the extent of renal damage present at the time that aggressive therapy is initiated, prompt recognition of impending or early scleroderma renal crisis is essential, and efforts should be made to avoid its occurrence. High-risk SSc patients with early disease, extensive and progressive skin involvement, tendon friction rubs, and anti-RNA polymerase III antibodies should be instructed to monitor their blood pressure daily and report significant alterations immediately. Potentially nephrotoxic drugs should be avoided, and glucocorticoids should be used only when absolutely necessary and at low doses. Patients presenting with scleroderma renal crisis should be immediately hospitalized. Once other causes of renal disease are excluded, treatment should be started promptly with titration of short-acting ACE inhibitors, with the goal of achieving rapid normalization of the blood pressure. In patients with hypertension persisting despite ACE inhibitor therapy, addition of angiotensin II receptor blockers, calcium channel blockers, and direct renin inhibitors should be considered. Anecdotal evidence indicates responses to endothelin-1 receptor blockers and prostacyclins. Up to two-thirds of patients with scleroderma renal crisis go on to dialysis. The outcome of scleroderma renal crisis is worse in patients with antibodies to topoisomerase I compared to those with antibodies to RNA polymerase III. Substantial renal recovery can occur following scleroderma renal crisis, and dialysis can be discontinued, in 30–50% of the patients. Kidney transplantation is appropriate for those unable to discontinue dialysis after 2 years. Survival of transplanted SSc patients is comparable to that of patients with other connective tissue diseases, and recurrence of renal crisis is rare.
SKIN CARE Because skin involvement in SSc is never life-threatening and because it stabilizes and may even regress spontaneously, over time, the management of SSc should not be dictated by its cutaneous manifestations. The inflammatory symptoms of early skin involvement can be controlled with antihistamines and cautious short-term use of low-dose glucocorticoids (<5 mg/d of prednisone). Retrospective studies have shown that D-penicillamine reduced the extent and progression of skin induration; however, these benefits could not be substantiated in a controlled prospective trial. Cyclophosphamide and methotrexate have modest effects on skin induration. Because the skin is dry, the use of hydrophilic ointments and bath oils is encouraged. Regular skin massage is helpful. Telangiectasia may present a cosmetic problem, especially on the face. Treatment with pulsed dye laser may have short-term benefit. Ischemic digital ulcers should be protected by occlusive dressing to promote healing and prevent infection. Infected skin ulcers are treated with topical antibiotics. Surgical debridement may be indicated. No therapy has been shown to be effective in preventing the formation of calcific soft tissue deposits or promoting their dissolution.
TREATMENT OF MUSCULOSKELETAL COMPLICATIONS Arthralgia and joint stiffness are common and distressing manifestations most prominent in early-stage disease. Short courses of nonsteroidal anti-inflammatory agents, weekly methotrexate, and cautious use of low-dose corticosteroids may alleviate these symptoms. Physical and occupational therapy can be effective for maintaining musculoskeletal function and improving long-term outcomes.
COURSE
The natural history of SSc is highly variable and difficult to predict, especially in early stages of the disease, when the specific subset—diffuse or limited cutaneous form—is not clear. Patient with dcSSc tend to have a more rapidly progressive course and worse prognosis than those with lcSSc.
In dcSSc, inflammatory symptoms such as fatigue, edema, arthralgia, and pruritus tend to subside, and the extent of skin thickening reaches a plateau at 2–4 years after disease onset, followed by slow regression. It is during the early edematous/inflammatory stage, generally lasting <3 years, that important visceral organ involvement occurs. While existing visceral organ involvement, such as pulmonary fibrosis, may progress even after skin involvement peaks, new organ involvement is rare. Scleroderma renal crisis almost invariably occurs within the first 4 years of disease. In late-stage disease (>6 years), the skin is usually soft and atrophic. Skin regression characteristically occurs in an order that is the reverse of initial involvement, with softening on the trunks followed by proximal and finally distal extremities; however, sclerodactyly and finger contractures generally persist. Relapse or recurrence of skin thickening after the peak of skin involvement has been reached is uncommon. Patients with lcSSc follow a clinical course that is markedly different than that of dcSSc. Raynaud’s phenomenon typically precedes other disease manifestations by years or even decades. Visceral organ complications such as PAH and ILD generally develop late and progress slowly.
PROGNOSIS
SSc confers a substantial increase in the risk of premature death. Age- and gender-adjusted mortality rates are fivefold to eightfold higher compared to the general population, and more than half of all patients with SSc die from their disease. In one population-based study of SSc, the median survival was 11 years. In patients with dcSSc, 5- and 10-year survival rates are 70% and 55%, respectively, whereas in patients with lcSSc, 5- and 10-year survival rates are 90% and 75%, respectively. The prognosis correlates with the extent of skin involvement, which itself is a surrogate for visceral organ involvement. Major causes of death are PAH, pulmonary fibrosis, gastrointestinal involvement, and cardiac disease. Scleroderma renal crisis is associated with a 30% 3-year mortality. Lung cancer and excess cardiovascular deaths also contribute to increased mortality. Markers of poor prognosis include male gender, African-American race, older age at disease onset, extensive skin thickening with truncal involvement, palpable tendon friction rubs, and evidence of significant or progressive visceral organ involvement. Laboratory predictors of increased mortality at initial evaluation include an elevated ESR, anemia, proteinuria and anti–topoisomerase I antibodies. In one study, SSc patients with extensive skin involvement, lung vital capacity <55% predicted, significant gastrointestinal involvement (pseudoobstruction or malabsorption), evidence of cardiac involvement (arrhythmias or congestive heart failure), or scleroderma renal crisis had a cumulative 9-year survival <40%. The severity of PAH is strongly associated with mortality, and SSc patients who had a mean pulmonary arterial pressure ≥45 mmHg had a 33% 3-year survival. The advent of ACE inhibitors in scleroderma renal crisis had a dramatic impact on survival, increasing from <10% at 1 year in the pre–ACE inhibitor era to >70% 3-year survival at the present time. Moreover, 10-year survival in SSc has improved from <60% in the 1970s to >66–78% in the 1990s, a trend that reflects both earlier detection and better management of complications.
LOCALIZED SCLERODERMA
The term scleroderma is commonly used to describe a group of localized skin disorders (Table 382-1). These occur more commonly in children than in adults. In contrast to SSc, localized scleroderma is rarely complicated by Raynaud’s phenomenon or significant internal organ involvement. Morphea presents as solitary or multiple circular patches of thickened skin or, rarely, as widespread induration (generalized or pansclerotic morphea); the fingers are spared. Linear scleroderma—streaks of thickened skin, typically in one or both lower extremities—may affect the subcutaneous tissues, leading to fibrosis and atrophy of supporting structures, muscle, and bone. In children, the growth of affected long bones can be retarded. When linear scleroderma lesions cross joints, significant contractures can develop.
MIXED CONNECTIVE TISSUE DISEASE
Patients who have lcSSc coexisting with features of SLE, polymyositis, and rheumatoid arthritis may have mixed connective tissue disease (MCTD). This overlap syndrome is generally associated with the presence of high titers of autoantibodies to U1-RNP. The characteristic initial presentation is Raynaud’s phenomenon associated with puffy fingers and myalgia. Gradually, lcSSc features of sclerodactyly, calcinosis, and cutaneous telangiectasia develop. Skin rashes suggestive of SLE (malar rash, photosensitivity) or of dermatomyositis (heliotrope rash on the eyelids, erythematous rash on the knuckles) occur. Arthralgia is common, and some patients develop erosive polyarthritis. Pulmonary fibrosis and isolated or secondary PAH may develop. Other manifestations include esophageal dysmotility, pericarditis, Sjögren’s syndrome, and renal disease, especially membranous glomerulonephritis. Laboratory evaluation indicates features of inflammation with elevated ESR and hypergammaglobulinemia. While anti-U1RNP antibodies are detected in the serum in high titers, SSc-specific autoantibodies are not found. In contrast to SSc, patients with MCTD often show a good response to treatment with glucocorticoids, and the long-term prognosis is better than that of SSc. Whether MCTD is a truly distinct entity or is, rather, a subset of SLE or SSc remains controversial.
EOSINOPHILIC FASCIITIS
Eosinophilic fasciitis is a rare idiopathic disorder associated with induration of the skin that generally develops rapidly. Adults are primarily affected. The skin has a coarse cobblestone “peau d’orange” appearance. In contrast to SSc, internal organ involvement is rare, and Raynaud’s phenomenon and SSc-associated autoantibodies are absent. Furthermore, skin involvement spares the fingers. Full-thickness excisional biopsy of the lesional skin reveals fibrosis of the subcutaneous fascia and is generally required for diagnosis. Inflammation and eosinophil infiltration in the fascia are variably present. In the acute phase of the illness, peripheral blood eosinophilia may be prominent. MRI appears to be a sensitive tool for the diagnosis of eosinophilic fasciitis. In some patients, eosinophilic fasciitis occurs in association with, or preceding, myelodysplastic syndromes or multiple myeloma. Treatment with glucocorticoids leads to prompt resolution of the eosinophilia. In contrast, skin changes generally show slow and variable improvement. The prognosis of patients with eosinophilic fasciitis is good.
383 |
Sjögren’s Syndrome |
DEFINITION, INCIDENCE, AND PREVALENCE
Sjögren’s syndrome is a chronic, slowly progressive autoimmune disease characterized by lymphocytic infiltration of the exocrine glands resulting in xerostomia and dry eyes. Approximately one-third of patients present with systemic manifestations; a small but significant number of patients develop malignant lymphoma. The disease presents alone (primary Sjögren’s syndrome) or in association with other autoimmune rheumatic diseases (secondary Sjögren’s syndrome) (Table 383-1).
|
ASSOCIATION OF SJÖGREN’S SYNDROME WITH OTHER AUTOIMMUNE DISEASES |
Middle-aged women (female-to-male ratio, 9:1) are primarily affected, although Sjögren’s syndrome may occur at any age, including childhood. The prevalence of primary Sjögren’s syndrome is ~0.5–1%, while 30% of patients with autoimmune rheumatic diseases suffer from secondary Sjögren’s syndrome.
PATHOGENESIS
Sjögren’s syndrome is characterized by both lymphocytic infiltration of the exocrine glands and B lymphocyte hyperreactivity. An oligomonoclonal B cell process, which is characterized by cryoprecipitable monoclonal immunoglobulins (IgMκ) with rheumatoid factor activity, is evident in up to 25% of patients.
Sera from patients with Sjögren’s syndrome often contain autoantibodies to non-organ-specific antigens such as immunoglobulins (rheumatoid factors) and extractable nuclear and cytoplasmic antigens (Ro/SS-A, La/SS-B). Ro/SS-A autoantigen consists of two polypeptides (52 and 60 kDa, respectively) in conjunction with cytoplasmic RNAs, whereas the 48-kDa La/SS-B protein is bound to RNA III polymerase transcripts. Autoantibodies to Ro/SS-A and La/SS-B antigens are usually detected at the time of diagnosis and are associated with earlier disease onset, longer disease duration, salivary gland enlargement, and more intense lymphocytic infiltration of minor salivary glands.
The major infiltrating cells in the affected exocrine glands are activated T and B lymphocytes. T cells predominate in mild lesions, whereas B cells are dominant in more severe lesions. Macrophages and dendritic cells also are found. The number of macrophages positive for interleukin (IL) 18 has been shown to correlate with parotid gland enlargement and low levels of the C4 component of complement, both of which are adverse predictors for lymphoma development.
Ductal and acinar epithelial cells appear to play a significant role in the initiation and perpetuation of autoimmune injury. These cells (1) express class II major histocompatibility complex (MHC) molecules, costimulatory molecules, and abberant expression of intracellular autoantigens on cell membranes and thus are able to provide signals essential for lymphocytic activation; (2) inappropriately produce proinflammatory cytokines and lymphoattractant chemokines necessary for sustaining the autoimmune lesion and allowing progression to more sophisticated ectopic germinal center formation, which occurs in one-fifth of patients; and (3) express functional receptors of innate immunity, particularly Toll-like receptors (TLRs) 3, 7, and 9, that may account for the perpetuation of the autoimmune response.
Both infiltrating T and B cells have a tendency to be resistant to apoptosis. Levels of B cell–activating factor (BAFF) have been found to be elevated in patients with Sjögren’s syndrome, especially those with hypergammaglobulinemia, and probably accounts for this antiapoptotic effect. Glandular epithelial cells seem to have an active role in the production of BAFF, which may be expressed and secreted after stimulation with type I interferon as well as with viral or synthetic double-stranded RNA. The triggering factor for epithelial activation appears to be a persistent enteroviral infection (possibly with coxsackievirus strains). Type I and type II interferon signatures have been described in ductal epithelial cells and T cells, respectively; their detection implies that interferons exert direct and cross-regulating effects on the pathogenic process.
A defect in cholinergic activity mediated through the M3 receptor and redistribution of the water-channel protein aquaporin 5, both leading to neuroepithelial dysfunction and diminished glandular secretions, have been proposed.
Molecular analysis of human leukocyte antigen (HLA) class II genes has revealed that Sjögren’s syndrome, regardless of the patient’s ethnic origin, is highly associated with the HLA DQA1*0501 allele. Genome-wide association studies have disclosed an increased prevalence of single-nucleotide polymorphisms in genes of IRF-5 and STAT-4, which participate in the activation of the type I interferon pathway.
CLINICAL MANIFESTATIONS
The majority of patients with Sjögren’s syndrome have symptoms related to diminished lacrimal and salivary gland function. In most patients, the primary syndrome runs a slow and benign course. The initial manifestations can be mucosal or nonspecific dryness, and 8–10 years may elapse from the initial symptoms to full-blown development of the disease.
The principal oral symptom of Sjögren’s syndrome is dryness (xerostomia). Patients report difficulty in swallowing dry food, an inability to speak continuously, a burning sensation, an increase in dental caries, and problems in wearing complete dentures. Physical examination shows a dry, erythematous, sticky oral mucosa. There is atrophy of the filiform papillae on the dorsum of the tongue, and saliva from the major glands is either not expressible or cloudy. Enlargement of the parotid or other major salivary glands occurs in two-thirds of patients with primary Sjögren’s syndrome but is uncommon in those with the secondary syndrome. Diagnostic tests include sialometry, sialography, and scintigraphy. Newer imaging techniques, including ultrasound, MRI, and magnetic resonance sialography of the major salivary glands, are also being used. Biopsy of the labial minor salivary gland permits histopathologic confirmation of focal lymphocytic infiltrates.
Ocular involvement is the other major manifestation of Sjögren’s syndrome. Patients usually describe a sandy or gritty feeling under the eyelids. Other symptoms include burning, accumulation of secretions in thick strands at the inner canthi, decreased tearing, redness, itching, eye fatigue, and increased photosensitivity. These symptoms, which define keratoconjunctivitis sicca, are attributed to the destruction of corneal and bulbar conjunctival epithelium. Diagnostic evaluation of keratoconjunctivitis sicca includes measurement of tear flow by Schirmer I test and determination of tear composition, with assessment of tear breakup time or tear lysozyme content. Slit-lamp examination of the cornea and conjunctiva after rose bengal staining reveals punctuate corneal ulcerations and attached filaments of corneal epithelium.
Involvement of other exocrine glands, which occurs less frequently, includes a decrease in mucous gland secretions of the upper and lower respiratory tree, resulting in dry nose, throat, and trachea (xerotrachea). In addition, diminished secretion of the exocrine glands of the gastrointestinal tract leads to esophageal mucosal atrophy, atrophic gastritis, and subclinical pancreatitis. Dyspareunia due to dryness of the external genitalia and dry skin also may occur.
Extraglandular (systemic) manifestations are seen in one-third of patients with Sjögren’s syndrome (Table 383-2) but are very rare in patients whose Sjögren’s syndrome is associated with rheumatoid arthritis. Patients with primary Sjögren’s syndrome more often report easy fatigability, low-grade fever, Raynaud’s phenomenon, myalgias, and arthralgias. Most patients with primary Sjögren’s syndrome experience at least one episode of non-erosive arthritis during the course of their disease. Manifestations of pulmonary involvement are frequently evident histologically but are rarely important clinically. Dry cough is the major manifestation that is attributed to small airway disease. Renal involvement includes interstitial nephritis, clinically manifested by hyposthenuria and renal tubular dysfunction with or without acidosis. Untreated acidosis may lead to nephrocalcinosis. Glomerulonephritis is a rare finding that occurs in patients with mixed cryoglobulinemia or with systemic lupus erythematosus overlapping with Sjögren’s syndrome. Vasculitis affects small and medium-sized vessels. The most common clinical features are purpura, recurrent urticaria, skin ulcerations, glomerulonephritis, and mononeuritis multiplex.
|
PREVALENCE OF EXTRAGLANDULAR MANIFESTATIONS IN PRIMARY SJÖGREN’S SYNDROME |
Different autoantibodies may determine the clinical expression of the disease. Patients positive for anticentromere autoantibody present with a clinical picture similar to that of limited scleroderma (Chap. 382). Antimitochondrial antibodies may connote liver involvement in the form of primary biliary cirrhosis (Chap. 369). Autoantibodies to 21-hydroxylate have recently been described in almost 20% of patients; their presence is associated with a blunted adrenal response.
Central nervous system involvement is rarely recognized. A few cases of myelitis associated with antibody to aquaporin 4 have been described.
Lymphoma is a well-known manifestation of Sjögren’s syndrome that usually presents later in the illness. Persistent parotid gland enlargement, purpura, leukopenia, cryoglobulinemia, low C4 complement levels, and ectopic germinal centers in minor salivary gland biopsy samples are manifestations suggesting the development of lymphoma. It is interesting that the same risk factors account for glomerulonephritis and lymphoma and that these risk factors are the ones that confer increased mortality risk. Most lymphomas are extranodal, low-grade marginal-zone B cell lymphomas and are usually detected incidentally during evaluation of the labial biopsy. The affected lymph nodes are usually peripheral. Survival rates are decreased in patients with B symptoms, lymph node mass >7 cm in diameter, and high or intermediate histologic grade.
Routine laboratory tests in Sjögren’s syndrome reveal mild normochromic, normocytic anemia. An elevated erythrocyte sedimentation rate is found in ~70% of patients.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
Primary Sjögren’s syndrome is diagnosed if (1) the patient presents with eye and/or mouth dryness, (2) eye tests disclose keratoconjunctivitis sicca, (3) mouth evaluation reveals the classic manifestations of the syndrome, and/or (4) the patient’s serum reacts with Ro/SS-A and/or La/SS-B autoantigens. Labial biopsy is needed when the diagnosis is uncertain or to rule out other conditions that may cause dry mouth or eyes or parotid gland enlargement (Tables 383-3 and 383-4). Validated diagnostic criteria have been established by a European study and have now been further improved by a European-American study group (Table 383-5). Hepatitis C virus infection should be ruled out since, apart from serologic tests, the clinicopathologic picture is almost identical to that of Sjögren’s syndrome. Enlargement of major salivary glands, particularly in seronegative patients, should raise the suspicion of IgG4-related syndrome, which may present also as chronic pancreatitis, interstitial nephritis, retroperitoneal fibrosis, and aortitis.
|
DIFFERENTIAL DIAGNOSIS OF SICCA SYMPTOMS |
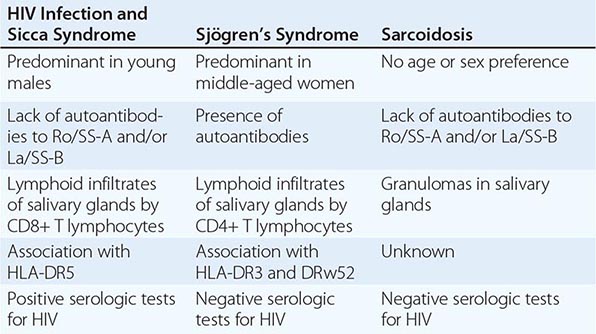
|
DIFFERENTIAL DIAGNOSIS OF SJÖGREN’S SYNDROME |
|
REVISED INTERNATIONAL CLASSIFICATION CRITERIA FOR SJÖGREN’S SYNDROMEa,b,c |
aExclusion criteria: past head and neck radiation treatment, hepatitis C infection, AIDS, preexisting lymphoma, sarcoidosis, graft-versus-host disease, use of anticholinergic drugs. bPrimary Sjögren’s syndrome: any four of the six items, as long as item IV (histopathology) or VI (serology) is positive; or any three of the four objective-criteria items (III, IV, V, VI). cIn patients with a potentially associated disease (e.g., another well-defined connective tissue disease), the presence of item I or item II plus any two from among items III, IV, and V may be considered indicative of secondary Sjögren’s syndrome.
Source: From C Vitali et al: Ann Rheum Dis 61:554, 2002. ©2002 with permission from BMJ Publishing Group Ltd.
|
TREATMENT |
SJÖGREN’S SYNDROME |
Treatment of Sjögren’s syndrome is aimed at symptom relief and limitation of the damaging local effects of chronic xerostomia and keratoconjunctivitis sicca through substitution for or stimulation of the missing secretions (Fig. 383-1).
FIGURE 383-1 Treatment algorithm for Sjögren’s syndrome. CHOP, cyclophosphamide, adriamycin (hydroxydaunorubicin), vincristine (oncovin), and prednisone.
to replace deficient tears, several ophthalmic preparations are readily available (hydroxypropyl methylcellulose; polyvinyl alcohol; 0.5% methylcellulose; Hypo Tears). If corneal ulcerations are present, eye patching and boric acid ointments are recommended. Certain drugs that may decrease lacrimal and salivary secretions, such as diuretics, antihypertensive drugs, anticholinergics, and antidepressants, should be avoided.
For xerostomia, the best replacement is water. Propionic acid gels may be used to treat vaginal dryness. To stimulate secretions, orally administered pilocarpine (5 mg thrice daily) or cevimeline (30 mg thrice daily) appears to improve sicca manifestations, and both are well tolerated. Hydroxychloroquine (200 mg) is helpful for arthralgias and mild arthritis.
Patients with renal tubular acidosis should receive sodium bicarbonate by mouth (0.5–2 mmol/kg in four divided doses). Glucocorticoids (1 mg/kg per day) and/or immunosuppressive agents (e.g., cyclophosphamide) are indicated only for the treatment of systemic vasculitis. Anti–tumor necrosis factor agents are ineffective. Monoclonal antibody to CD20 appears to be effective in patients with systemic disease, particularly in those with vasculitis, arthritis, and fatigability. Combination of anti-CD-20 with a classic CHOP regimen (cyclosporine, adriamycin [hydroxydaunorubicin], vincristine [oncovin], and prednisone) leads to increased survival rates among patients with high-grade lymphomas.




