Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is a soft tissue tumor originating from immature mesenchymal cells that form any tissue except bone. It is the most common soft tissue sarcoma in children and adolescents, accounting for approximately 50% of soft tissue sarcomas. The overall survival (OS) of RMS patients has improved to 71% as a result of the Intergroup Rhabdomyosarcoma Study Group (IRSG; established in 1972), which is now continued by the Children’s Oncology Group Soft Tissue Sarcoma (COG-STS) committee.1,2
RMS is the third most common childhood extracranial solid tumor, after neuroblastoma and Wilms tumor.3 Approximately 350 new cases of RMS are diagnosed annually in the USA, corresponding to an incidence of 4.5 cases per million children aged 20 years or younger.4,5 RMS has a bimodal distribution with approximately 65% of cases occurring in children <6 years and the remaining cases developing in children aged 10–18 years.6 More than 80% of cases are diagnosed before the age of 14 years.7 Outcomes are worse in adolescents who usually present with alveolar RMS, lymph node involvement, delay in diagnosis, and metastases.8 Poor outcomes are also seen in infants usually due to failure to achieve adequate local control.9
Approximately one-third of RMS occurs in the head and neck (including parameningeal locations). Genitourinary locations are found in 22–31% of cases; trunk and extremities are the next most common sites.1,5
RMS is classified as a small, round, blue cell tumor of childhood, a category including neuroblastoma, Ewing sarcoma, small cell osteogenic sarcoma, non-Hodgkin lymphoma, and leukemia. Six major pathologic subtypes of RMS are outlined by the International Classification of RMS in order of decreasing 5-year survival): (1) embryonal (botryoid); (2) embryonal (spindle cell); (3) embryonal, not otherwise specified (NOS); (4) alveolar, NOS or solid variant; (5) anaplasia, diffuse; and (6) undifferentiated sarcoma.10 Botryoid RMS is associated with a superior 5-year survival rate of 95% and most commonly occurs in the bladder or vagina in infants and young children, and in the nasopharynx in older children.11 Spindle cell RMS is commonly found in the paratesticular area, head and neck, extremities, or orbit.12 Embryonal and embryonal variants account for approximately 60–70% of all pediatric RMS cases in younger patients.5,11,13,14 Alveolar, anaplastic, and undifferentiated variants of RMS account for approximately 35% of all new cases, and generally have the worst prognosis. The incidence of alveolar RMS is increasing with an annual per cent change (APC) of 4.09%.5 The alveolar subtype is seen more commonly in older patients.11 A ‘solid alveolar’ variant lacks the characteristic alveolar septations but behaves similarly to the alveolar subtype.15 The prognostic value of each specific histologic subtype has varied somewhat in different studies.14–16 However, in IRS-IV, 3-year failure-free survival (FFS) was 83% for embryonal, 66% for alveolar, 55% for undifferentiated, and 66% for unclassified sarcoma (P < 0.001).1
Staging
Two classification schemes have been used to categorize RMS. The clinical grouping system was devised by the IRSG and is a surgicopathologic system based on the initial operative assessment (Table 70-1).11 Stratification of survival was based on the ability for complete resection; thus, an initial surgical approach was required, which varied by institution.13 Clinical grouping has consistently been an independent prognostic indicator as shown in Figure 70-1.
TABLE 70-1
Intergroup Rhabdomyosarcoma Study Group Clinical Grouping Classification of Rhabdomysarcoma
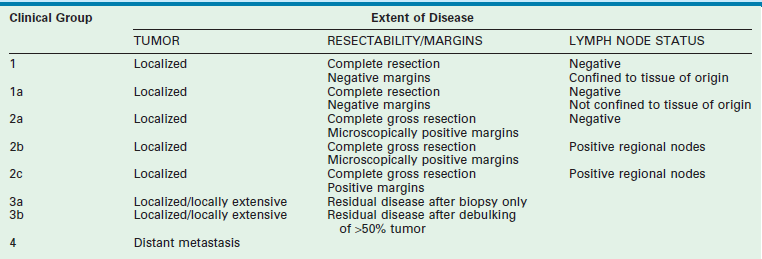
From Qualman SJ, Coffin CM, Newton WA, et al. Intergroup Rhabdomyosarcoma Study: Update for pathologists. Pediatr Dev Pathol 1998;1:550–61.
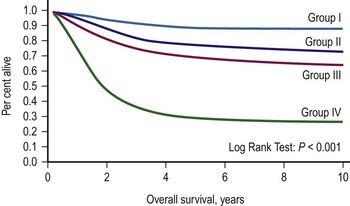
FIGURE 70-1 Overall survival according to clinical group assignment (Intergroup Rhabdomyosarcoma Study [IRS] I to IV). Survival of patients treated on IRS-I, IRS-II, IRS-III/IV-P (IV pilot), and IRS-IV by clinical group at diagnosis is shown. A significant difference is seen in outcome by the extent of initial surgical resection, with the best outcome among the patients with completely resected tumors (group I), followed by those with microscopic residual (group II) and gross residual (group III) disease. Patients with metastatic disease (group IV) at diagnosis fare poorly. (Anderson JR. Personal communication, 2000; from Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2002.)
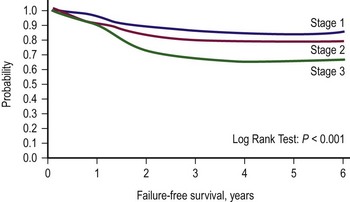
The second classification scheme is a pretreatment staging system based on the tumor/node/metastasis (TNM) system, which was introduced by the COG-STS committee (Table 70-2).17 The system includes primary tumor site, lymph node involvement, distant metastatic disease, and size. Favorable primary sites include the orbit, eyelid, other nonparameningeal head/neck sites, and nonbladder, nonprostate genitourinary structures. Unfavorable primary sites include extremities (including buttock), trunk, retroperitoneum, perineum, urinary bladder and prostate, and cranial parameningeal sites. IRS-IV was the first study to use this staging system to classify patients prospectively. FFS data for patients with local or regional tumors, according to stage, are shown in Figure 70-2. For these tumors, pretreatment staging is a more accurate predictor of outcome than the clinical grouping classification system.
TABLE 70-2
Rhabdomyosarcoma Tumor/Node/Metastasis (TNM) Pretreatment Staging Classification
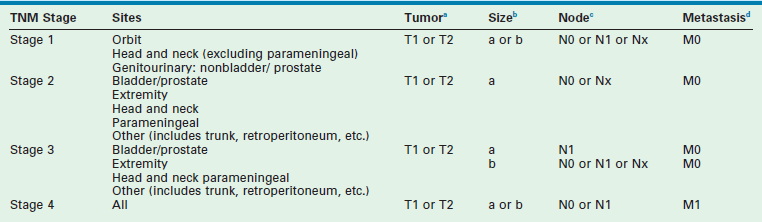
aTumor: T1, confined to anatomic site of origin. T2, extension and/or fixation to surrounding tissue.
ba, <5 cm in diameter; b, ≥5 cm in diameter.
cRegional nodes: N0, regional nodes not clinically involved; N1, regional nodes clinically involved by neoplasm; Nx, clinical status of regional nodes unknown (especially sites that preclude lymph node evaluation).
dMetastasis: M0, no distant metastases present; M1, distant metastases.
Molecular Biology
Mutations in certain tumor suppressor genes, including TP53, predispose individuals to RMS. Patients with Li–Fraumeni syndrome and neurofibromatosis type 1 are more likely to develop RMS, and approximately 10% of patients with RMS have one of these syndromes.18,19
Embryonal RMS is characterized by loss of heterozygosity (LOH) with loss of maternal genetic information and duplication of paternal genetic information at the 11p15 locus.20 This genetic locus is the site of the insulin-like growth factor-2 (IGF-2) gene and LOH results in overexpression, which can lead to RMS through several different possible pathways.13,18 The etiology of RMS can also be linked to the loss of 9q22, which corresponds to a tumor suppressor gene in embryonal RMS patients.20
In approximately 70% of alveolar RMS, a common translocation between chromosomes 2 and 13, t(2;13)(q35;q14), is present.21 This translocation usually involves the PAX3 gene that regulates transcription driving neuromuscular development and the FOXO1 gene that is involved in the differentiation process of myoblasts.22 Less commonly, the translocation involves the PAX7 gene located at 1p36 to the same location on chromosome 13.23 PAX3/FOXO1 expression can increase IGF-2 expression and an IGF-binding protein, providing a common pathway for both embryonal and alveolar RMS. These known molecular disturbances are now being adopted and studied clinically. Several studies have demonstrated a worse prognosis with the presence of a PAX3/FOXO1 fusion gene in alveolar subtypes of RMS, and an improved prognosis with PAX7/FOXO1.18,24,25 Additionally, the t(2;13) translocation has been shown to characterize alveolar RMS with a poor prognosis, whereas the t(1;13) translocation is associated with improved outcome.26 The ELMO1 gene has been implicated in alveolar RMS metastasis.27 Another recent study has correlated a 34-metagene with COG risk classification groups.28 The exact molecular pathway leading to the development of alveolar RMS potentially caused or exacerbated by these translocations remains to be found.
Clinical Presentation
The clinical presentation of RMS is variable and depends largely on tumor site, patient age, and the presence of distant metastases. Most symptoms are secondary to the local mass effect. RMSs involving the head and neck region including orbits, parameningeal tissues (middle ear, nasal cavity, paranasal sinuses, nasopharynx, and infratemporal fossa), and nonparameningeal tissues (scalp, face, oral cavity, oropharynx, hypopharynx and neck) are most commonly embryonal subtype and rarely involve regional lymph nodes. Symptoms can include proptosis, ophthalmoplegia, nasal/sinus obstruction with or without discharge, cranial nerve palsies, meningeal symptoms, or a painless, enlarging mass. Paratesticular RMS may present as a painless swelling in the scrotum or inguinal canal, and have a high rate of lymph node spread to the retroperitoneum.
Tumors involving the bladder, urinary tract, or prostate can cause obstruction, hematuria, constipation, and/or urinary frequency. Vaginal tumors are more common in very young patients and may present as vaginal bleeding, discharge, or a mass. Tumors involving the uterus more commonly present in older girls with extensive tumor at diagnosis. RMSs involving the extremity usually present as a mass, tend to be more aggressive, and most are alveolar subtype. They have an approximately 50% incidence of regional lymph node involvement and around 15% of patients present with metastatic disease. Presentation in the neonate is extremely rare with most babies having the embryonal/botryoid/undifferentiated subtypes (0.4%).14,29–34
Diagnosis
The diagnosis of RMS may be suspected clinically, but needs to be confirmed by biopsy. Depending on location, this may require endoscopy, needle biopsy, or excisional/incisional biopsies. Biopsy incisions should be made so that complete excision is possible if a subsequent wide local excision is necessary. Prior to the definitive operation, a full evaluation including imaging, laboratory studies, and bone marrow evaluation should be performed (Box 70-1).7
Surgical Treatment
Primary Resection
Prognosis in rhabdomyosarcoma patients is linked to the amount of residual disease present after resection (see Fig. 70-1). Complete tumor resection, with no microscopic residual disease, offers the best chance for cure. However, in many sites such as the orbit, bladder, prostate, vagina, uterus, complete tumor resection is not feasible while still preserving function of vital organs and avoiding a mutilating procedure. Over the last 35 years, organ salvage rates have increased without adversely affecting OS. The operative approach depends on the primary tumor site, size, presence or absence of lymph node involvement, and distant metastases. These decisions must be made in the context of a multidisciplinary team, including the surgeon, oncologist, pediatric radiologist, and radiotherapist to allow optimal patient care. A tissue biopsy and biologic studies are recommended for definitive diagnosis if resection of the primary tumor site with negative margins is not possible.
Primary Re-excision
Primary re-excision (PRE) of a tumor includes any repeat attempt at complete resection before the initiation of any other form of therapy. As with the primary tumor resection, PRE is performed to allow a clinical group I classification without sacrificing vital structures or organs causing impaired function or poor cosmesis. PRE is recommended in patients whose initial resection had positive or unknown margin status, or was performed for presumed benign disease. If not feasible, reliance on adjuvant irradiation and chemotherapy is advisable. PRE has been shown to improve survival by converting a significant proportion of patients from group IIa to group I in patients with extremity tumors and perineal RMS.17,35,36 The Italian Cooperative Study Group concluded that PRE is the treatment of choice for children with RMS and nonrhabdomyosarcoma soft tissue sarcoma (NRSTS) who are age 3 years or younger and cannot receive radiation therapy (RT). It is also preferred for paratesticular sites as well.35 PRE and postoperative irradiation showed equivalent results in achieving local control in extremity and trunk sites. PRE was not effective for tumors >5 cm.
Lymph Node Evaluation
RMS frequently spreads to regional lymph nodes early in the disease and lymph node involvement significantly worsens the prognosis. However, involved nodes can go undetected by sophisticated imaging techniques. Any clinically or radiographically suspicious lymph node requires histologic confirmation. It is important to determine regional lymph node status to determine if more aggressive treatments such as RT are needed. Pediatric surgeons may be asked to evaluate the regional lymph node status, but lymphadenectomy serves a diagnostic purpose only.7
Specific recommendations regarding regional lymph node evaluation will be reviewed according to the specific site. However, in general, it is recommended that RMS patients with primary tumors of the extremity, primary tumors of the perineum, and paratesticular tumors in children older than 10 years old undergo operative evaluation of regional lymph node status even if there is no clinically apparent disease.19,37 If disease is found, then distal lymph nodes should be sampled to determine metastatic disease.19 Additionally, during the course of tumor resection for genitourinary and retroperitoneal tumors, lymph node sampling should be performed if feasible.
Lymphatic mapping with sentinel lymph node biopsy may allow adequate staging while limiting operative morbidity.37,38 Most experience with this technique has been gained with extremity and truncal tumors, and with adult breast cancer and melanoma.19 Typically, lymphoscintigraphic mapping is performed before the planned operation. Intraoperatively, vital blue dye is injected at the primary tumor site and a detector is used to identify the sentinel lymph node. Through a small incision and limited dissection, a blue sentinel lymph node(s) with evidence of the radioactive tracer is excised. These techniques often limit the extent and morbidity of the operation when compared with formal lymph node dissection as was previously performed.
Second-Look Operations
At the initial operation, the size, invasion, or location of RMS tumors frequently prohibits complete resection to achieve group I or II status. In IRS-IV, among all patients without metastatic disease (n = 883), 62% were classified as group III.1 After intensive multiagent chemotherapy with or without irradiation, these patients usually benefit from a second-look operation. Clinical and radiographic assessments are often inaccurate. The goals of the second-look operation are to remove residual tumor and determine response prior to additional therapy. This approach has been shown to improve patient survival and/or to classify patients as complete responders.19,39,40 Alternatively, a negative biopsy (i.e., no tumor) at second-look operation does not exclude or diminish the possibility of recurrence.39,41
Surgical Treatment for Recurrent Disease
Despite the successes of primary therapy for RMS, survival after relapse remains very poor. Approximately 30% of RMS patients will experience relapse, and between 50% and 95% of these will die of progressive disease.42 Results from the Cooperative Weichteilsarkom Studiengruppe identified certain factors that may predict relapse including increased age (>10 years), histology (alveolar), tumor size (>5 cm), tumor site, stage after surgery, and lack of RT.43
In a large review of relapsed RMS patients, 95% of all failures occurred within 3 years from treatment initiation.42 Patterns of relapse were as follows: local, 35%; regional, 16%; distant, 41%; and unknown, 8%. Overall, the median survival time from first recurrence or progression was 0.8 year, and the estimated 5-year survival after recurrence was 17%. Higher survival postrelapse was associated with botryoid histology and initial clinical group I assignment. For embryonal RMS, local recurrence predicted improved survival compared to those patients with regional or distant recurrences. Factors associated with a better prognosis with recurrent disease include embryonal/botryoid histology, stage or group I disease, and relapse >1 year after completing primary therapy.7,44 RMS patients with local relapse compose a very challenging patient population, and the results of repeated attempts at surgical resection are not fully known. Many RMS patients with locally recurrent disease are those with primary tumors of the extremities, pelvis, retroperitoneum, and other sites at which repeated operations are difficult, and often mutilating.
The IRSG recommends that patients with locally recurrent disease be treated according to risk stratification. For more favorably rated relapse patients, intensive multiagent chemotherapy followed by RT and/or surgical treatment, and for less favorably rated patients, initial dose-intensified chemotherapy and maintenance therapy or experimental therapies may be offered.42 In a smaller study of RMS and NRSTS patients with first relapse (n = 44), repeat resection appeared to improve survival with embryonal RMS, but not with alveolar RMS or other soft tissue sarcoma histologic subtypes.45 A retrospective review of patients treated at MD Anderson Cancer Center for recurrent RMS over an 11-year period (n = 32) demonstrated a 37% disease-free survival with a mean follow-up period of 4.9 years.46 Morbidity was 35%, and 52% of the operations were considered aggressive. Several factors prevalent in survivors included embryonal/botryoid histology and RMS of vaginal/paratesticular origin. Factors associated with those who died included alveolar histology and extremity and bladder/prostate disease.
Surgical Treatment for Metastatic Rhabdomyosarcoma
Approximately 30% of RMS patients present with distant metastatic disease, most commonly involving lung (39%), bone marrow (32%), lymph nodes (30%), or bones (27%).5,47 Patients with distant metastatic disease have a median survival of 20 months. The OS at 5, 15, and 30 years is 33%, 29%, and 23%, respectively, and FFS is <30%.2,5 Operation combined with neoadjuvant or adjuvant RT improves survival by 20 months, and significantly increases 5-year survival compared to operation or RT alone.5
In the IRS-IV patient cohort, 24% had metastases isolated to the lung.47 Most patients underwent a diagnostic biopsy followed by intensive salvage multimodality therapy. There was no difference in survival between patients who underwent operation with histologic confirmation of lung metastases and those diagnosed only by imaging. The value of aggressive resection of RMS pulmonary metastases remains unclear as opposed to other soft tissue sarcomas in children and adults where aggressive resection of isolated pulmonary metastases is thought to be beneficial for longer survival.47,48
Site-Specific Surgical Guidelines
Head/Neck Tumors
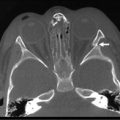
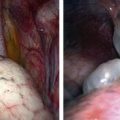
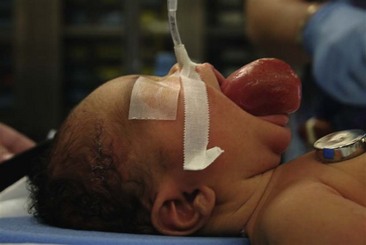
Nonparameningeal orbital and other head and neck RMS tumors are considered favorable sites and are classified as stage 1, regardless of size or nodal status. Operative management for orbital and other head and neck RMS tumors is largely restricted to biopsy followed by adjuvant chemotherapy and RT (Fig. 70-3). Parameningeal tumors have slightly worse prognosis due to the presence of abundant lymphatics and a delay in diagnosis because of the hidden tumor. Previously, mutilating operations were performed. However, with more effective systemic therapy, these operations are rarely needed. Metastatic disease to regional nodes is present in <3% of cases.30 Unless there is clinical evidence of lymph node involvement, routine nodal sampling is not warranted. An aggressive operative approach may be justified with recurrent disease. Additionally, surgical intervention may be warranted when there is evidence of persistent disease after adjuvant therapy. These tumors are optimally managed by oncologic otolaryngologists and neurosurgeons. Superficial lesions such as parotid tumors and small eyelid lesions may benefit from primary resection especially when wide surgical excision is feasible (Fig. 70-4).
Tumors of the Extremities
Extremity RMS constitutes 15–20% of all pediatric RMS, with the median age of 6 years.19,47,49 Extremity tumors are considered to be an unfavorable site, and 71% have alveolar histology. Initial staging of extremity RMS begins at stage 2, but can be upstaged based on size, nodal involvement, and metastasis (see Table 70-2). Evaluation of extremity RMS starts with careful physical examination and is followed by computed tomography (CT) to evaluate for bony erosion and magnetic resonance imaging (MRI) to determine tumor size and involvement of surrounding structures.50 F-fluorodeoxyglucose positron emission tomography (FDG-PET) is also helpful for clinically negative nodes or distant metastases that are equivocal by conventional CT or MRI.51
The operative goal for primary extremity tumors <5 cm is to obtain circumferential disease-free margins while preserving form and function. In one study, FFS and OS were not affected by utilizing conservative surgery and adjuvant RT versus primary amputation in RMS of the hands and feet.52 The required resection margin is unclear based on current evidence, but a 5 mm circumferential margin is currently recommended.50 Moreover, there is no clear evidence that a larger margin decreases the chance of recurrence. After distortion of the tissues associated with resection and histopathologic processing, the actual margin measured in pathology is usually smaller.53 Unfortunately, complete resection with negative margins is rarely achieved in extremity RMS patients.14 Removal of tumor in a piecemeal fashion is considered group II even if the surgeon is confident all tumor is removed. When gross or microscopic tumor is left in the tumor bed, titanium clips should be placed to aid in RT and possible re-excision.50
A high rate of regional lymph node involvement is found with extremity RMS. Lymph node disease status is a critical component of the preoperative staging classification scheme (see Table 70-2) and has a direct impact on the plan of care. Therefore, evaluation of the regional lymph drainage basin is mandatory, regardless of clinical node status.19 In IRS-IV (n = 139), 37% of all extremity RMS patients had positive regional lymph nodes. Positive regional lymph nodes were found in 50% of the patients who underwent surgical evaluation (n = 76). An additional 13 patients had regional node disease diagnosed by imaging alone. Among patients with a negative clinical and radiologic examination, 17% had positive nodes found on biopsy.14 These findings imply that some patients with negative imaging who did not undergo lymph node biopsy probably had undetected positive lymph nodes. Undetected positive nodal disease will lead to inaccurate staging and perhaps less than optimal treatment. However, complete nodal dissection offers no survival benefit and discovery of a positive node does not warrant nodal dissection. Sentinel node biopsy is a viable option for the evaluation of the draining nodal basin.38,54,55 If the nodal basin is found to harbor a positive node, more proximal nodal evaluation is warranted to exclude in-transit disease to further clarify tumor stage and group.52
Survival with multimodality therapy now approaches 75%, with a significantly lower rate for patients with nodal disease.19 As reviewed previously, for extremity RMS with microscopically positive or indeterminant margins, or after an initial resection was performed for presumed benign disease, PRE is recommended and has been shown to improve survival.36
Genitourinary Tumors
Within the genitourinary system, RMS can arise from the vagina, vulva, uterus, paratesticular region, prostate, or bladder. Rarely, the kidney or ureter may be involved. In the IRS-IV study, genitourinary RMS accounted for 12% of all patients and >30% of nonmetastatic tumor patients.47 Bladder/prostate genitourinary primary tumors were most common.
Bladder/Prostate Tumors
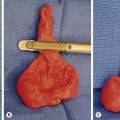
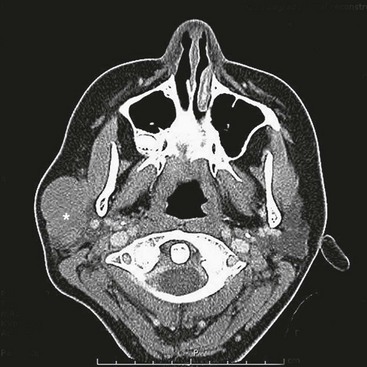
RMS of the bladder and prostate occur in 2% and 4%, respectively.5 Ultrasound is usually performed at initial diagnosis, and serial CT or MRI are used to track the efficacy of primary chemotherapy.53 Secondary to the tumor location, the initial operative approach is usually limited to biopsy, which can usually be obtained using endoscopy. However, the surgeon should be aware of the amount of tissue taken for biopsy as the endoscopic biopsy forceps take very small samples. Also, artifact from using cautery can mimic a spindle cell appearance or destroy the entire sample. Open biopsy is performed when endoscopy is not possible. Pelvic and retroperitoneal lymph nodes also should be sampled for disease in these patients. Internal ureteral stents and/or percutaneous nephrostomy tubes may be necessary with urinary obstruction. Suprapubic catheters are not advised secondary to the risk of seeding the tract with tumor.53 Historically, aggressive initial resection was performed with good local control rates, but with significant morbidity and low bladder salvage rates. Survival has improved significantly since the IRSG trials, and bladder salvage is now the primary goal in these patients. However, complete resection may be feasible when the primary tumor involves only the dome of the bladder. Most bladder and prostate tumors are treated with chemotherapy and RT, followed by a more limited and conservative resection.56 More aggressive tumor resection including anterior or total pelvic exenteration may be indicated to achieve local control if the tumor fails to respond to primary chemotherapy. The rate of exenterative cystectomy with current treatment is approximately 30% (Figs 70-5 and 70-6).57–59 Many patients will develop bladder dysfunction even with bladder salvage.19
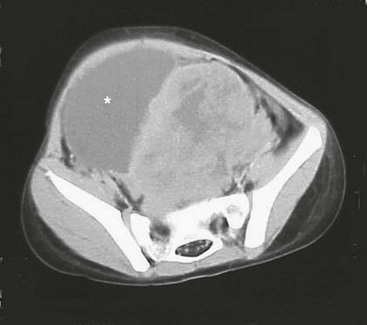
FIGURE 70-5 This CT scan shows a large mass emanating from the pelvis. Biopsy confirmed this mass to be a rhabdomyosarcoma. The bladder has been marked with an asterisk.
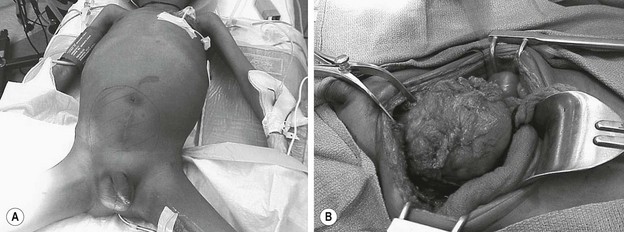
FIGURE 70-6 (A) The patient presented with lower abdominal distention and a large mass that was palpable. (B) The mass (visualized in the operative photograph) was not responsive to chemotherapy, and an attempt at complete resection was made. Most, but not all, of the mass could be excised. The patient eventually died of metastatic disease. This patient’s CT scan is seen in Figure 70-5.
Vagina, Vulva, or Uterus
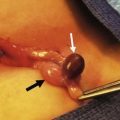
Tumors of the female genitourinary tract are considered a favorable site (stage 1). They have a 5-year OS >80% and comprise 3.5% all RMS tumors with half originating in the vagina. They are typically embryonal or botryoid histologic subtype (Fig. 70-7) and lymph node involvement is uncommon.60 Management begins with a biopsy, often transvaginally, and relies on effective chemotherapy such as systemic high dose cyclophosphamide as primary treatment. There is no role for initial aggressive resection such as vaginectomy or hysterectomy.61 Some physicians recommend avoidance of RT in group II and III vaginal RMS in patients <24 months old due to the high morbidity associated with external beam radiation.62 These patients are followed with routine abdominal and pelvic MRI to document tumor response.63,64 A second-look operation with cystoscopy and re-biopsy is common. As with other pelvic RMS, relapsed or persistent disease in the vagina, vulva, and uterus is associated with a very poor outcome. In such cases, aggressive attempts at local control, including RT, partial or total vaginectomy, hysterectomy, or pelvic exenteration, are all viable options. Unless there is direct involvement of the ovary by advanced/recurrent disease, there is no role for oophorectomy.
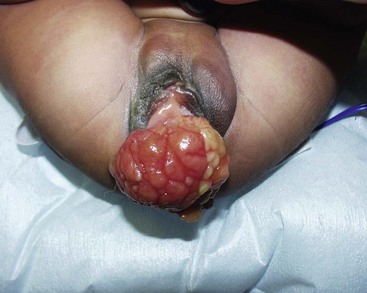
Paratesticular Tumors
Paratesticular RMS is considered a favorable site, with all paratesticular patients classified preoperatively as having stage 1 disease regardless of tumor size, invasiveness, or lymph node status. The overall FFS rate for paratesticular RMS is > 80%, and is 90% in patients younger than 10 years old.1
The recommended initial operative approach for paratesticular tumors is radical orchiectomy with proximal ligation of the spermatic cord at the internal inguinal ring via an inguinal incision. A trans-scrotal incision for biopsy or resection is contraindicated. If done, a subsequent hemiscrotectomy may be required to resect the contaminated tumor bed. Meticulous operative technique is extremely important. Tumor spillage upstages patients to clinical group IIa regardless of the completeness of resection.65 When paratesticular tumors are fixed to the scrotal skin or invade the overlying scrotal skin, hemiscrotectomy is indicated. With this approach, 90% to 95% of patients are able to have complete resection (groups I or II).37
Paratesticular RMS has a high incidence (~30%) of spread to the retroperitoneal lymph nodes (RPLNs). Therefore, careful evaluation of these nodes is an important part of the staging workup for these patients. All patients should have thin-cut (3.8–5.0 mm) abdominal and pelvic CT scans to assess the retroperitoneum. The treatment guidelines vary by age since the outcome and the frequency of RPLN positivity with paratesticular RMS is partially age dependent. For patients younger than 10 years who are clinical group I, and do not have lymph node enlargement on CT scan, RPLN dissection or sampling is not recommended. These patients are followed with CT scans every 3 months. Suggestive or positive CT scans should prompt ipsilateral retroperitoneal node dissection with further therapy depending on the histologic findings. For all patients 10 years or older, or with tumor >5 cm, ipsilateral RPLN dissection is recommended up to the level of the renal hilum. A systematic approach is used to remove lymph nodes from the internal inguinal ring, along the iliac vessels and aorta, up to the renal hilum. Positive suprarenal nodes are considered metastatic. Thus, patients are considered group IV.50 RPLN dissection is associated with significant morbidity, including loss of ejaculatory function and lower extremity lymphedema. Thus, risk-based guidelines are used to limit unnecessary patient morbidity secondary to RPLN dissection.66 However, involved lymph nodes are missed in 15–20% of patients by CT scan, and a high index of suspicion is necessary.14,17,29
Other Sites
RMS occurring in other sites is rare. Primary tumors arising in the thorax, abdomen, pelvis, and perineum were found in only 12% of all IRS-IV patients.47 Truncal RMS tumors usually have an alveolar subtype. Primary resection is preferred in truncal tumors <5 cm when a negative operative margin can be realistically anticipated. For larger primary tumors, initial incisional biopsy is preferred. Additionally, postoperative reconstructive procedures may be necessary, especially with larger tumors, using prosthetic meshes or myocutaneous flaps.19 Paraspinal masses need to be differentiated from extraosseous Ewing’s sarcoma. No data are available from which to make definitive guidelines regarding regional lymph node evaluation. For many truncal lesions, the primary lymphatic drainage basin can be equivocal and preoperative lymphoscintigraphy may be helpful.
Retroperitoneal and nongenitourinary pelvic tumors can be difficult to manage because of their relatively hidden location and subsequent late presentation. More than 90% present with large invasive tumors as clinical group III or IV.67 These patients generally undergo initial biopsy, aggressive multiple-agent chemotherapy, RT, and second-look operation, if indicated. The role for debulking large retroperitoneal or nongenitourinary pelvic tumors is unclear. However, data have demonstrated survival benefit in a subset of patients who underwent debulking.68,69
Biliary RMS generally has a good prognosis without the need for aggressive resection. Most patients have botryoid RMS that responds well to chemotherapy. Biopsy followed by neoadjuvant chemotherapy frequently results in resolution of symptoms, including jaundice. Operative intervention is fraught with complications, including the inability to resect all disease in most cases and infections associated with external biliary drains.50,70
Perineal and perianal RMSs also are sites that have an overall poor outcome partially due to late presentation.71 More than one-third of patients are seen initially with presumed benign disease (usually infections).47 Improved survival is associated with tumors <5 cm, low clinical group and stage, negative nodes, and age younger than 10 years. Outcomes are not related to the tumor histology in this site.50 PRE frequently lowers the clinical group assignment, which improves outcome. From 1972 to 1997, 71 patients had perineal and perianal tumors, with a five-year FFS rate of 45%. In this group, 46% of patients had regional lymph node involvement with five-year OS of 33% and 71% for N1 and N0 respectively.17
Current/Future Research
Data from IRS-III and IRS-IV were used to develop the risk stratification groupings used for IRS-V and current studies (Table 70-3). Low-risk patients are estimated to have three-year FFS rates of 88%.72 The low-risk patient group is broken down into subset A with a 90% FFS and subset B with an 87% FFS. This subgroup is limited to patients with localized embryonal, botryoid, or spindle cell tumors. Patients receive either VAC (vincristine, actinomycin D, cyclophosphamide) or VA (vincristine, actinomycin D) chemotherapy and RT to residual tumor after resection. Current research (ARST0331) is focused on reducing the dose and subsequent systemic side effects of cyclophosphamide therapy while evaluating the impact on survival.2
TABLE 70-3
Intergroup Rhabdomyosarcoma Study Group Studies Based on Risk-Based Protocol Assignment

FFS, Failure-free survival; IRS, Intergroup Rhabdomyosarcoma Study.
Intermediate-risk patients have a predicted FFS rate of 65–73% and include those with localized alveolar or undifferentiated sarcoma, stages 1 to 3, or embryonal RMS stages 2 and 3 with gross residual disease, or stage 4 embryonal RMS patients younger than 10 years old. In IRS-V, these patients were randomized to VAC and RT or VAC alternating with vincristine, topotecan, and cyclophosphamide and RT, but there was no survival benefit seen.2 Multiple chemotherapy regimens were evaluated with other IRS studies and vincristine with irinotecan (VI) showed the best response rate. Thus, in the most recent trial (ARST0531), patients are being randomized to either VAC or VAC alternating with VI.2
High-risk patients have a 3-year FFS of <30% and include stage 4 or group IV alveolar or undifferentiated tumors and embryonal RMS patients older than 10 years old. These patients receive chemotherapy with irinotecan followed by VAC and RT. For patients responding to irinotecan, treatment continues with four additional cycles of irinotecan and vincristine, in addition to VAC. Current research (ARST08P1) is designed to evaluate whether or not the addition of cixutumumab and/or temozolomide to the current chemotherapeutic regimen will improve outcomes.2
References
1. Crist, WM, Anderson, JR, Meza, JL, et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J Clin Oncol. 2001; 19:3091–3102.
2. Malempati, S, Hawkins, DS. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) soft-tissue Sarcoma committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012; 59:5–10.
3. Kramer, S, Meadows, AT, Jarrett, P, et al. Incidence of childhood cancer: Experience of a decade in a population-based registry. J Natl Cancer Inst. 1983; 70:49–55.
4. Gurney, JG, Young, JL, Jr., Roffers, SD, et al. Soft tissue sarcomas. In: Ries LAG, Smith MA, Gurney JG, et al, eds. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995. Bethesda, MD: National Cancer Institute, SEER Program, 1999.
5. Perez, EA, Kassira, N, Cheung, MC, et al. Rhabdomyosarcoma in children: A SEER population based study. J Surg Res. 2011; 170(2):e243–e251.
6. Dagher, R, Helman, L. Rhabdomyosarcoma: An overview. Oncologist. 1999; 4:34–44.
7. Rodeberg, D, Paidas, C. Childhood rhabdomyosarcoma. Semin Pediatr Surg. 2006; 15:57–62.
8. Bisogno, G, Compostella, A, Ferrari, A, et al. Rhabdomyosarcoma in adolescents: A report from the AIEOP Soft Tissue Sarcoma Committee. Cancer. 2012; 118:821–827.
9. Malempati, S, Rodeberg, DA, Donaldson, SS, et al. Rhabdomyosarcoma in infants younger than 1 year: A report from the Children’s Oncology Group. Cancer. 2011; 117:3493–3501.
10. Newton, WA, Jr., Soule, EH, Hamoudi, AB, et al. Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies I and II: Clinicopathologic correlation. J Clin Oncol. 1988; 6:67–75.
11. Qualman, SJ, Coffin, CM, Newton, WA, et al. Intergroup Rhabdomyosarcoma Study: Update for pathologists. Pediatr Dev Pathol. 1998; 1:550–561.
12. Leuschner, I, Newton, WA, Jr., Schmidt, D, et al. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993; 17:221–230.
13. Wexler, LH, Crist, LM, Helman, LJ. Rhabdomyosarcoma and the undifferentiated sarcomas. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2002:939–971.
14. Neville, HL, Andrassy, RJ, Lobe, TE, et al. Preoperative staging, prognostic factors, and outcome for extremity rhabdomyosarcoma: A preliminary report from the Intergroup Rhabdomyosarcoma Study IV (1991–1997). J Pediatr Surg. 2000; 35:317–321.
15. Tsokos, M, Webber, BL, Parham, DM, et al. Rhabdomyosarcoma. A new classification scheme related to prognosis. Arch Pathol Lab Med. 1992; 116:847–855.
16. Kodet, R, Newton, WA, Jr., Hamoudi, AB, et al. Orbital rhabdomyosarcomas and related tumors in childhood: Relationship of morphology to prognosis–an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol. 1997; 29:51–60.
17. Blakely, ML, Andrassy, RJ, Raney, RB, et al. Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: A report of Intergroup Rhabdomyosarcoma Studies I through IV, 1972 through 1997. J Pediatr Surg. 2003; 38:347–353.
18. Slater, O, Shipley, J. Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol. 2007; 60:1187–1194.
19. Leaphart, Rodeberg, D. Pediatric surgical oncology: Management of rhabdomyosarcoma. Surg Oncol. 2007; 16:173–185.
20. Bridge, JA, Liu, J, Weibolt, V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: An intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer. 2000; 27:337–344.
21. Turc-Carel, C, Lizard-Nacol, S, Justrabo, E, et al. Consistent chromosomal translocation in alveolar rhabdomyosarcoma. Cancer Genet Cytogenet. 1986; 19:361–362.
22. Shapiro, DN, Sublett, JE, Li, B, et al. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 1993; 53:5108–5112.
23. Davis, RJ, D’Cruz, CM, Lovell, MA, et al. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994; 54:2869–2872.
24. Khan, J, Bittner, ML, Saal, LH, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci U S A. 1999; 96:13264–13269.
25. Duan, F, Smith, LM, Gustafson, DM, et al. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: A report from the Children’s Oncology Group. Genes Chromosomes Cancer. 2012; 51:662–674.
26. Kelly, KM, Womer, RB, Sorensen, PH, et al. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol. 1997; 15:1831–1836.
27. Rapa, E, Hill, SK, Morten, KJ, et al. The over-expression of cell migratory genes in alveolar rhabdomyosarcoma could contribute to metastatic spread. Clin Exp Metastasis. 2012; 29:419–429.
28. Davicioni, E, Anderson, JR, Buckley, JD, et al. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: A report from the children’s oncology group. J Clin Oncol. 2010; 28:1240–1246.
29. Wiener, ES, Anderson, JR, Ojimba, JI, et al. Controversies in the management of paratesticular rhabdomyosarcoma: Is staging retroperitoneal lymph node dissection necessary for adolescents with resected paratesticular rhabdomyosarcoma? Semin Pediatr Surg. 2001; 10:146–152.
30. Crist, W, Gehan, EA, Ragab, AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clinl Oncol. 1995; 13:610–630.
31. Raney, RB, Jr. Rhabdomyosarcoma and related tumors of the head and neck in childhood. In: Maurer HM, Ruymann FB, Pochedly C, eds. Rhabdomyosarcoma and Related Tumors in Children and Adolescents. Boca Raton, FL: CRC, 1991.
32. Raney, RB, Jr., Tefft, M, Newton, WA, et al. Improved prognosis with intensive treatment of children with cranial soft tissue sarcomas arising in nonorbital parameningeal sites. A report from the Intergroup Rhabdomyosarcoma Study. Cancer. 1987; 59:147–155.
33. Rodeberg, D, Arndt, C, Breneman, J, et al. Characteristics and outcomes of rhabdomyosarcoma patients with isolated lung metastases from IRS-IV. J Pediatr Surg. 2005; 40:256–262.
34. Lobe, TE, Wiener, ES, Hays, DM, et al. Neonatal rhabdomyosarcoma: The IRS experience. J Pediatr Surg. 1994; 29:1167–1170.
35. Cecchetto, G, Carli, M, Sotti, G, et al. Importance of local treatment in pediatric soft tissue sarcomas with microscopic residual after primary surgery: Results of the Italian Cooperative Study RMS-88. Med Pediatr Oncol. 2000; 34:97–101.
36. Hays, DM, Lawrence, W, Jr., Wharam, M, et al. Primary reexcision for patients with ‘microscopic residual’ tumor following initial excision of sarcomas of trunk and extremity sites. J Pediatr Surg. 1989; 24:5–10.
37. Wiener, ES, Lawrence, W, Hays, D, et al. Retroperitoneal node biopsy in paratesticular rhabdomyosarcoma. J Pediatr Surg. 1994; 29:171–177.
38. Neville, HL, Andrassy, RJ, Lally, KP, et al. Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg. 2000; 35:961–964.
39. Hays, DM, Raney, RB, Crist, WM, et al. Secondary surgical procedures to evaluate primary tumor status in patients with chemotherapy-responsive stage III and IV sarcomas: A report from the Intergroup Rhabdomyosarcoma Study. J Pediatr Surg. 1990; 25:1100–1105.
40. Schalow, EL, Broecker, BH. Role of surgery in children with rhabdomyosarcoma. Med Pediatr Oncol. 2003; 41:1–6.
41. Godzinski, J, Flamant, F, Rey, A, et al. Value of postchemotherapy bioptical verification of complete clinical remission in previously incompletely resected (stage I and II pT3) malignant mesenchymal tumors in children: International Society of Pediatric Oncology 1984 Malignant Mesenchymal Tumors Study. Med Pediatr Oncol. 1994; 22:22–26.
42. Pappo, AS, Anderson, JR, Crist, WM, et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J Clin Oncol. 1999; 17:3487–3493.
43. Dantonello, TM, Int-Veen, C, Winkler, P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008; 26:406–413.
44. Mattke, AC, Bailey, EJ, Schuck, A, et al. Does the time-point of relapse influence outcome in pediatric rhabdomyosarcomas? Pediatr Blood Cancer. 2009; 52:772–776.
45. Klingebiel, T, Pertl, U, Hess, CF, et al. Treatment of children with relapsed soft tissue sarcoma: Report of the German CESS/CWS REZ 91 trial. Med Pediatr Oncol. 1998; 30:269–275.
46. Hayes-Jordan, A, Doherty, DK, West, SD, et al. Outcome after surgical resection of recurrent rhabdomyosarcoma. J Pediatr Surg. 2006; 41:633–638.
47. Breneman, JC, Lyden, E, Pappo, AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma–a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003; 21:78–84.
48. Hacker, FM, von Schweinitz, D, Gambazzi, F. The relevance of surgical therapy for bilateral and/or multiple pulmonary metastases in children. Eur J Pediatr Surg. 2007; 17:84–89.
49. Sultan, I, Qaddoumi, I, Yaser, S, et al. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2,600 patients. J Clin Oncol. 2009; 27:3391–3397.
50. Dasgupta, R, Rodeberg, DA. Update on rhabdomyosarcoma. Semin Pediatr Surg. 2012; 21:68–78.
51. Kumar, R, Shandal, V, Shamim, SA, et al. Clinical applications of PET and PET/CT in pediatric malignancies. Expert Rev Anticancer Ther. 2010; 10:755–768.
52. La, TH, Wolden, SL, Su, Z, et al. Local therapy for rhabdomyosarcoma of the hands and feet: Is amputation necessary? A report from the Children’s Oncology Group. Int J Radiat Oncol Biol Phys. 2011; 80:206–212.
53. Ferrer, FA, Isakoff, M, Koyle, MA. Bladder/prostate rhabdomyosarcoma: Past, present and future. J Urol. 2006; 176:1283–1291.
54. Gow, KW, Rapkin, LB, Olson, TA, et al. Sentinel lymph node biopsy in the pediatric population. J Pediatr Surg. 2008; 43:2193–2198.
55. De Corti, F, Dall’Igna, P, Bisogno, G, et al. Sentinel node biopsy in pediatric soft tissue sarcomas of extremities. Pediatr Blood Cancer. 2009; 52:51–54.
56. Lobe, TE, Wiener, E, Andrassy, RJ, et al. The argument for conservative, delayed surgery in the management of prostatic rhabdomyosarcoma. J Pediatr Surg. 1996; 31:1084–1087.
57. Corpron, CA, Andrassy, RJ, Hays, DM, et al. Conservative management of uterine pediatric rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study III and IV pilot. J Pediatr Surg. 1995; 30:942–944.
58. Hays, DM, Lawrence, W, Jr., Crist, WM, et al. Partial cystectomy in the management of rhabdomyosarcoma of the bladder: A report from the Intergroup Rhabdomyosarcoma Study. J Pediatr Surg. 1990; 25:719–723.
59. Hays, DM, Raney, RB, Ragab, A, et al. Retention of functional bladders among patients with vesicle/prostatic sarcomas in the intergroup rhabdomyosarcoma studies (IRS) (1978–1990) [Abstract]. Med Pediatr Oncol. 423, 1991.
60. Arndt, CA, Donaldson, SS, Anderson, JR, et al. What constitutes optimal therapy for patients with rhabdomyosarcoma of the female genital tract? Cancer. 2001; 91:2454–2468.
61. Andrassy, RJ, Wiener, ES, Raney, RB, et al. Progress in the surgical management of vaginal rhabdomyosarcoma: A 25-year review from the Intergroup Rhabdomyosarcoma Study Group. J Pediatr Surg. 1999; 34:731–734.
62. Walterhouse, DO, Meza, JL, Breneman, JC, et al. Local control and outcome in children with localized vaginal rhabdomyosarcoma: A report from the Soft Tissue Sarcoma committee of the Children’s Oncology Group. Pediatr Blood Cancer. 2011; 57:76–83.
63. Fletcher, BD, Kaste, SC. Magnetic resonance imaging for diagnosis and follow-up of genitourinary, pelvic, and perineal rhabdomyosarcoma. Urol Radiol. 1992; 14:263–272.
64. Finelli, A, Babyn, P, Lorie, GA, et al. The use of magnetic resonance imaging in the diagnosis and followup of pediatric pelvic rhabdomyosarcoma. J Urol. 2000; 163:1952–1953.
65. Hamilton, CR, Pinkerton, R, Horwich, A. The management of paratesticular rhabdomyosarcoma. Clin Radiol. 1989; 40:314–317.
66. Heyn, R, Raney, RB, Jr., Hays, DM, et al. Late effects of therapy in patients with paratesticular rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol. 1992; 10:614–623.
67. Blakely, ML, Spurbeck, WW, Pappo, AS, et al. The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg. 1999; 34:672–675.
68. Blakely, ML, Lobe, TE, Anderson, JR, et al. Does debulking improve survival rate in advanced-stage retroperitoneal embryonal rhabdomyosarcoma? J Pediatr Surg. 1999; 34:736–741.
69. Raney, RB, Stoner, JA, Walterhouse, DO, et al. Results of treatment of fifty-six patients with localized retroperitoneal and pelvic rhabdomyosarcoma: A report from The Intergroup Rhabdomyosarcoma Study-IV, 1991–1997. Pediatr Blood Cancer. 2004; 42:618–625.
70. Spunt, SL, Lobe, TE, Pappo, AS, et al. Aggressive surgery is unwarranted for biliary tract rhabdomyosarcoma. J Pediatr Surg. 2000; 35:309–316.
71. Hill, DA, Dehner, LP, Gow, KW, et al. Perianal rhabdomyosarcoma presenting as a perirectal abscess: A report of 11 cases. J Pediatr Surg. 2002; 37:576–581.
72. Raney, RB, Anderson, JR, Barr, FG, et al. Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades of life: A selective review of intergroup rhabdomyosarcoma study group experience and rationale for Intergroup Rhabdomyosarcoma Study V. J Pediatr Hematol Oncol. 2001; 23:215–220.