Chapter 69 Retinoblastoma
Retinoblastoma is the most frequent neoplasm of the eye in childhood, representing 2.5% to 4% of all pediatric cancers. The average age-adjusted incidence rate of retinoblastoma in the United States and Europe is 2 to 5 per million children (approximately 1 in 14,000 to 18,000 live births).1,2 Retinoblastoma is a cancer of the very young; two thirds of all cases are diagnosed before 2 years of age, and 95% of cases are diagnosed before 5 years of age.1
Etiology and Epidemiology
The incidence of retinoblastoma is higher (6 to 10 cases per million children) in Africa, India, and children of Native American descent in North America. The increased incidence occurs primarily in unilateral cases. In industrialized countries, an increased incidence of retinoblastoma is associated with poverty and low levels of maternal education, that suggests environmental factors in its etiology.3,4 Recent studies have suggested a role for human papillomaviruses in the pathogenesis of retinoblastoma. The viral oncoprotein E7 of high-risk human papillomavirus types has been shown to bind to and inactivate the RB1 gene product (pRB), acting functionally to create the biallelic loss of RB1.5,6 High-risk human papillomavirus sequences have been detected in 28% to 36% of tumors.6
Biology
In 1971, based on the mathematical analysis of the age at presentation of bilateral (hereditary) and unilateral (mostly nonhereditary) cases of retinoblastoma, Knudson7 proposed the “two-hit hypothesis,” in which two mutational events in a developing retinal cell lead to the development of retinoblastoma. This hypothesis was subsequently extended to suggest that the two events could be mutations of both alleles of the RB1 gene, located on chromosome 13q14 and identified in 1986.8,9 Its product, pRb, is required for the transition of the cell through the G1 phase of mitosis. pRb appears to function as a tumor suppressor at least in part by inhibiting cell cycle progression past the G1-S restriction point; on entry to the S phase, cells become irreversibly committed to division. pRb is the major gatekeeper to control this critical point in growth regulation; lack of pRb activity removes the pRb constraint on cell cycle control, resulting in deregulated cell proliferation.
The RB1 gene is a large gene, containing 27 exons over about 200 kilobases of DNA, and mutations have been described in almost every exon.10 New germline mutations have an overwhelming preference for the paternal allele. Penetrance of the trait is greater than 90%.11
In both hereditary and nonhereditary retinoblastoma the second tumorigenic event is usually chromosomal, often as a result of mitotic recombination errors.12 This second hit occurs at a much higher frequency than the first hit, and it is more sensitive to environmental factors such as ionizing radiation, perhaps explaining the increased risk of radiation-induced malignancies in survivors of retinoblastoma.
Prevention, Early Detection, and Genetic Counseling
The successful management of retinoblastoma depends on the ability to detect the disease while it is still intraocular. Disease stage correlates with delay in diagnosis.14 In developing countries, late diagnosis is associated with orbital and metastatic disease.15 Eye assessment is encouraged in all neonates and at subsequent health visits. Mass screening is being considered, especially where the tumor is common in areas of South America and Asia.
Retinoblastoma is a unique neoplasm in that the genetic form imparts a predisposition to developing tumor in an autosomal dominant fashion with almost complete penetrance (85% to 95%).16 The majority of such children acquire the first mutation as a new germline mutation, with only 15% to 25% having a positive family history. However, some families display an inheritance pattern characterized by reduced penetrance and expressivity. Also, the RB1 gene mutation can occur at a late stage of embryogenesis, resulting in a variable expression depending on the tissue, causing mosaicisms in 10% to 15% of the patients or their progenitors.17 Recognizing the inheritance pattern and the existence of mosaicisms, the following familial risk estimates can be made16:
Genetic counseling is of the utmost importance to assist parents in understanding the genetic consequences of each form of retinoblastoma and to estimate the risk in relatives. With the refinement in methods of mutational analysis over the past decade, detection rates have increased to more than 90%.10
Pathology and Pathways of Spread
Macroscopically, retinoblastoma is soft and friable, tending to outgrow its blood supply with resultant necrosis and calcification. Dissemination within the vitreous and retina in the form of small, white nodules (seeds) is common, sometimes making it difficult to distinguish a multicentric primary tumor from a disseminated tumor.18 The microscopic appearance depends on the degree of differentiation. Undifferentiated retinoblastoma is composed of small, round, densely packed cells with hypochromatic nuclei and scant cytoplasm. Several degrees of photoreceptor differentiation have been described, characterized by distinctive cellular arrangements. Flexner-Wintersteiner rosettes (clusters of low columnar cells arranged around central lumens bounded by eosinophilic membranes analogous to the external membrane of the normal retina) are specific for retinoblastoma and are noted in 70% of tumors. Fleurettes (composed of larger cells with abundant eosinophilic cytoplasm arranged in a distinctive fleur-de-lis pattern) are less frequent; cells exhibit ultrastructural characteristics of photoreceptor differentiation. Especially well-differentiated tumors composed almost entirely of fleurettes have been called retinomas or retinocytomas. Ultrastructurally, retinoblastoma cells demonstrate photoreceptor differentiation with the presence of the 9-0 microtubule doublet pattern, abundant cytoplasmic microtubules, synaptic ribbons, and neurosecretory granules.
Clinical Manifestations, Patient Evaluation, and Staging
Retinoblastoma is a tumor of young children. Bilateral retinoblastoma presents at a younger age (usually before 1 year of age) than patients with unilateral disease (typically at age 1 to 3 years).16,18,19 Half the cases of retinoblastoma diagnosed during the first year are bilateral, compared with fewer than 10% when diagnosed after 1 year of age.
Although most patients with bilateral retinoblastoma carry a germline mutation of the RB1 gene, only 5% carry a deletion involving the 13q14 locus large enough to be detected by karyotyping. In those cases, retinoblastoma is part of a more complex syndrome resulting from the loss of additional genetic material. Patients with the 13q− syndrome are characterized by typical facial dysmorphic features, subtle skeletal abnormalities, and varying degrees of mental retardation and motor impairment.20 The severity of deficits correlate with the size of the deletion; normal psychomotor development may be seen in those patients in whom the deletion is restricted to the 13q14 band.
Trilateral retinoblastoma refers to the association of bilateral retinoblastoma with a typically asynchronous brain tumor.21 The majority of tumors are pineal region primitive neuroectodermal tumors (PNETs, pineoblastomas); 20% to 25% of the tumors are suprasellar or parasellar. The PNETs exhibit varying degrees of neuronal or photoreceptor differentiation, suggesting an origin from the germinal layer of primitive cells. Trilateral disease occurs in 3% to 9% of patients with the genetic form of retinoblastoma and appears to be more common in familial cases. The prognosis until recently has been almost uniformly fatal. The median age at diagnosis of trilateral retinoblastoma is 23 to 48 months; the interval between initial diagnosis of bilateral retinoblastoma and the diagnosis of the brain tumor is usually more than 20 months.22 In recent years, with the more widespread use of chemoreductive therapy and less use of radiation therapy (RT) for patients with bilateral retinoblastoma, the incidence of trilateral retinoblastoma has decreased dramatically.23 Approximately 5% of patients with bilateral disease develop pineal cysts, which appear to be a forme fruste of trilateral retinoblastoma.24
The diagnosis of intraocular retinoblastoma is usually made without pathologic confirmation. An examination under anesthesia with a maximally dilated pupil and scleral indentation is required to examine the entire retina (Fig. 69-1). Endophytic tumors are those that grow inward to the vitreous cavity and may seed the vitreous cavity. Exophytic retinoblastoma grows into the subretinal space, causing progressive retinal detachment and subretinal seeding. A very detailed documentation of the number, location, and size of tumors, of the presence of retinal detachment and subretinal fluid, and of the presence of vitreous and subretinal seeds must be performed. Wide-angle real-time retinal imaging systems such as RetCam (Clarity Medical Systems, Pleasanton, Calif.) provide a 130-degree field of view and digital recording, thus facilitating diagnosis and follow-up.
Imaging studies that aid in the diagnosis include bidimensional ultrasonography, computed tomography, and magnetic resonance imaging (MRI). Imaging is particularly important to evaluate extraocular extension and to differentiate retinoblastoma from other causes of leukocoria. Evaluation for the presence of metastatic disease also needs to be considered in a subgroup of patients. Metastatic disease occurs in 10% to 15% of patients, usually in association with advanced intraocular features, such as deep choroidal and scleral invasion, or involvement of the iris or ciliary body or of the optic nerve beyond the lamina cribrosa.13 In such cases, additional staging should include bone scintigraphy, bone marrow aspiration and biopsy, and lumbar puncture.
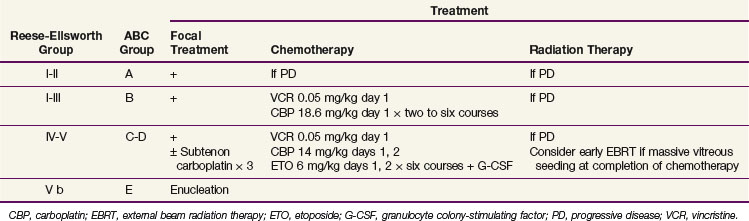
The Reese-Ellsworth grouping system has been the standard for intraocular disease. This system was designed to predict the outcome after external beam radiation therapy (EBRT). It divides eyes into five groups on the basis of the size, location, and number of lesions and on the presence of vitreous seeding25 (Table 69-1). The changed paradigm for “conservative” chemoreduction strategies for intraocular retinoblastoma has made the Reese-Ellsworth system less predictive of ocular preservation. A new staging system (i.e., the International Classification of Retinoblastoma) has been developed to provide a simpler, more user-friendly classification applicable to current therapies. The new system is based on the extent of tumor seeding within the vitreous cavity and subretinal space and seems to be a better predictor of treatment success26 (Table 69-2).
TABLE 69-1 Reese-Ellsworth Grouping for Suitability for Treatment of Retinoblastoma by Radiation Therapy
| Group | Description |
|---|---|
| I (Very Favorable) | |
| Ia | Solitary tumor smaller than 4 dd at or behind the equator |
| Ib | Multiple tumors, none larger than 4 dd, all at or behind equator |
| II (Favorable) | |
| IIa | Solitary tumor 4-10 dd, at or behind equator |
| IIb | Multiple tumors 4-10 dd, at or behind equator |
| III (Doubtful) | |
| IIIa | Any lesion anterior to equator |
| IIIb | Solitary tumor larger than 10 dd behind equator |
| IV (Unfavorable) | |
| IVa | Multiple tumors, some larger than 10 dd |
| IVb | Any lesion extending anteriorly to the ora serrata |
| V (Very Unfavorable) | |
| Va | Massive tumors involving more than half the retina |
| Vb | Vitreous seeding |
dd, disc diameter (1.5 mm).
From Reese AB, Ellsworth RM: The evaluation and current concept of retinoblastoma therapy, Trans Am Acad Ophthalmol Otolaryngol 67:164-172, 1963.
TABLE 69-2 International Classification of Retinoblastoma
| Group A |
Modified from Shields CL, Mashayekhi A, Au AK, et al: The International Classification of Retinoblastoma predicts chemoreduction success, Ophthalmology 113:2276-2280, 2006.
For patients undergoing enucleation, pathologic staging incorporates features known to influence outcome, guiding the therapeutic approach; such factors include choroidal and scleral involvement, optic nerve extension, and presence of metastatic disease. A third proposed staging system developed by an international consortium of ophthalmologists and pediatric oncologists incorporates the most important elements of the older systems27 (Table 69-3). Growth and invasion occur as a sequence of events, and extraretinal extension occurs only after the tumor has reached large intraocular dimensions. As part of this process, retinoblastoma extends into the ocular coats (choroids and sclera), the optic nerve, and the anterior segment. Extraocular disease is the next step in this progression; locoregional dissemination occurs by direct extension through the sclera into the orbital contents and preauricular lymph nodes. Extraorbital disease manifests as intracranial tumor, leptomeningeal dissemination, or hematogenous metastasis.
TABLE 69-3 International Retinoblastoma Staging System, Proposed
| Stage | Treatment |
|---|---|
| 0 | Patients treated conservatively |
| I | Eye enucleated, completely resected histologically |
| II | Eye enucleated, microscopic residual tumor |
| III | Regional extension |
| IIIa | Overt orbital disease |
| IIIb | Preauricular or cervical lymph node extension |
| IV | Metastatic disease |
| IVa | Hematogenous metastasis (without CNS involvement) |
| IVb | CNS extension (with or without any other site of regional or metastatic disease) |
From Chantada G, Doz F, Antonelli CB, et al: A proposal for an international retinoblastoma staging system, Pediatr Blood Cancer 47:801-805, 2006.
Primary Therapy
Principles of Treatment
Treatment of retinoblastoma aims to save life and preserve useful vision, with attention to late functional and carcinogenic effects. Contemporary care requires coordination among the patient’s ophthalmologist, pediatric oncologist, and radiation oncologist.28
Surgery
Enucleation is indicated for large tumors filling the vitreous when there is little or no likelihood of restoring vision and when tumor is present in the anterior chamber or associated with neovascular glaucoma. The eye must be removed intact, without seeding tumor cells into the orbit.29 For optimal staging, a long segment (10 to 15 mm) of the optic nerve is removed with the globe. An orbital implant is usually fitted during the same procedure, and the extraocular muscles are attached to it. A ceramic false eye is later fitted in the orbital socket. Orbital exenteration is very seldom indicated. For patients presenting with orbital (extraocular) disease, judicious use of chemotherapy, surgery, and RT provides tumor control and often orbital exenteration can be avoided.
Focal Surgical Therapies
Focal treatment is used for small tumors (<3-6 mm), classically in patients with bilateral disease and in combination with chemotherapy. Focal therapies are central to ongoing studies utilizing conservative therapy (chemotherapy and focal treatment) to avoid enucleation and EBRT in unilateral intraocular disease. Photocoagulation with the argon laser is used for the treatment of tumors situated at or posterior to the equator of the eye and for the treatment of retinal neovascularization occasionally seen after RT.30 This technique is limited to tumors measuring no greater than 4.5 mm at its base and no greater than 2.5 mm in height. The treatment aims to obliterate blood supply to the tumor. Two or three monthly sessions are usually required, and local tumor control is achieved in approximately 70% of cases. Cryotherapy is used for the treatment of small equatorial and peripheral lesions that measure no more than 3.5 mm at the base and no more than 2 mm in height.31 One or two monthly sessions of triple freeze and thaw are performed; the resultant tumor control is usually excellent. A more recently introduced focal therapy is transpupillary thermotherapy, which applies focused heat at sub-photocoagulation levels, usually with a diode laser.32 In thermotherapy, the goal is to deliver a temperature of 42° C to 60° C for 5 to 20 minutes to the tumor, relatively sparing retinal vessels from photocoagulation. The use of focal treatments is especially important in conjunction with chemotherapy, when both treatment modalities appear to have synergistic effects. Sequential thermotherapy and carboplatin enhances the antitumor effect by increasing the platinum-DNA adducts; thus, thermochemotherapy is becoming an important component of therapy for intraocular retinoblastoma.33 In general, local control rates of 70% to 80% can be achieved. Complications of focal treatments include transient serous retinal detachment, retinal traction and tears, and localized fibrosis.
Chemotherapy
Chemotherapy is indicated in patients with extraocular disease, in the subgroup of patients with intraocular disease with high-risk histologic features, and in patients with bilateral disease in conjunction with aggressive focal therapies. In patients with early-stage unilateral disease, chemotherapy is central to conservation therapy when combined with focal therapy. Agents effective in the treatment of retinoblastoma include platinum compounds, etoposide, cyclophosphamide, doxorubicin, vincristine, and ifosfamide.28,34
Radiation Therapy
The spectrum of radiation techniques includes local brachytherapy plaques, focal external beam irradiation (intensity-modulated radiation therapy [IMRT] or stereotactic photon RT, or proton beam), full ocular EBRT (with the same technical options as just mentioned, recent reports documenting techniques to limit dose to the bony orbit and contralateral eye), and orbital irradiation.35–47 Recognizing the balance of potential secondary carcinogenesis (particularly in bilateral or hereditary retinoblastoma) and orbital growth changes, while noting the long documented efficacy of RT in this disease, challenges the radiation oncologist as to indications and timing for management, optimal treatment planning and delivery, and requisite follow-up.48–50
Intraocular Retinoblastoma
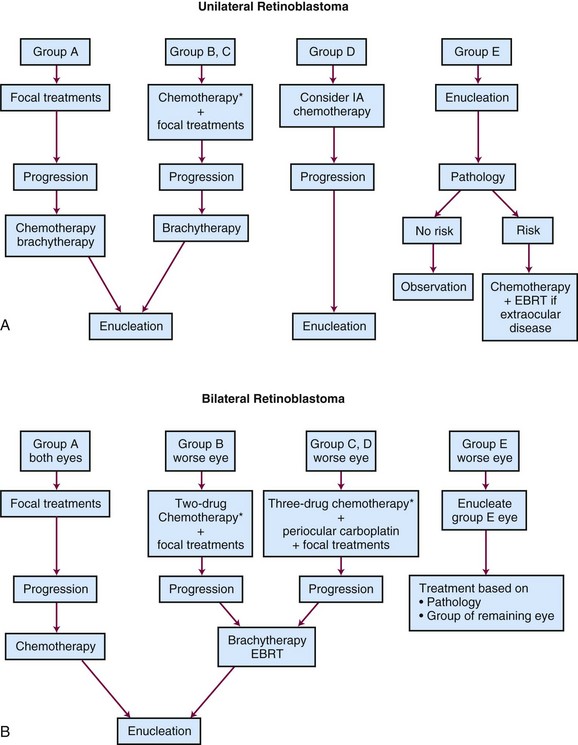
Unilateral Retinoblastoma
For unilateral intraocular presentations, enucleation is curative for 85% to 90% of children. Outcome after enucleation is excellent, with good functional results and minimal long-term effects.48 In view of the apparent success in treating bilateral intraocular disease with chemotherapy and focal therapy, a similar “conservative” approach may be appropriate in unilateral intraocular disease, especially in very young children who may later evidence contralateral disease.48 For patients with early intraocular disease (Reese-Ellsworth groups I to III, International Classification of Retinoblastoma group B), a nonintense two-drug regimen of vincristine and carboplatin had been suggested, based on overall results with three-drug chemotherapy (also including etoposide) documented at nearly 100% for ocular and visual preservation.28 A Children’s Oncology Group trial in moderately advanced intraocular disease was discontinued when the event-free survival rate with the two-drug regimen fell below the monitoring guidelines.* Variations on the three-drug “standard regimen” are under investigation. In patients with advanced intraocular tumors (groups C and D), ocular preservation rates are typically at 50% to 70% and EBRT may be required.36,50
Postoperative chemotherapy and RT are indicated with findings of scleral invasion, microscopic tumor at the transection line of the optic nerve, or overt invasion of the optic nerve (see later under treatment of extraocular retinoblastoma).51,52 Postoperative chemotherapy may be beneficial for selected patients with higher risk for extraocular dissemination (i.e., with concurrent retrolaminar and choroidal involvement and possibly with massive choroidal involvement).28,53,54,55,56
Bilateral Retinoblastoma
Bilateral disease typically presents as advanced disease in one eye and relatively more localized disease contralaterally. The classic treatment has been enucleation of the eye with more advanced disease (when little or no visual potential is apparent) and EBRT for the remaining eye. The radiation oncologist has argued to irradiate both eyes when one has tumor burden, clearly obviating any likelihood of useful vision. Two major sequelae led to changing the classic approach: irradiation of the orbit during a period of rapid growth in very young children results in significant reduction in orbital and zygomatic facial growth, whether symmetrical (after opposed lateral radiation techniques) or asymmetrical (with unilateral orbital irradiation), with dysmorphic changes apparent before puberty.43,46,48 Of greater importance, the risk for development of secondary cancers approaches 40% in hereditary forms of retinoblastoma after irradiation; the standard incidence ratio for largely infield sarcomas exceeds 100 times the expected rate, with late development of carcinomas (e.g., breast, thyroid, lung, head, and neck) at or above 10 times the expected rate. This risk may be age related and is less in older children.49,57
The treatment of patients with bilateral retinoblastoma now incorporates upfront chemotherapy, intended to achieve maximum chemoreduction of the intraocular tumor burden, followed by aggressive focal therapies (see Fig. 69-1). Relatively effective carboplatin-based multiple-agent chemotherapy and planned consolidative local therapies have resulted in increased ocular and visual preservation and a decrease (and delay) in the necessity for enucleation or EBRT. The best results have followed combinations of vincristine, carboplatin, and etoposide* (Tables 69-4 and 69-5).
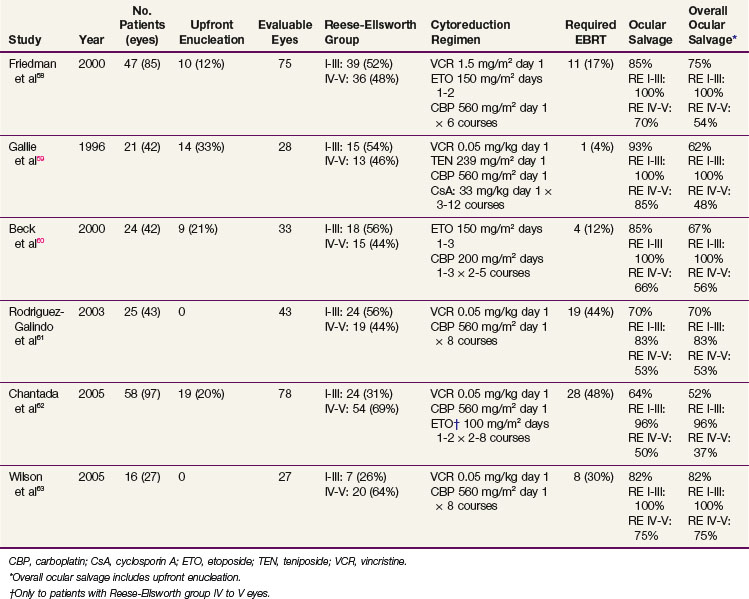
With more advanced disease, a major proportion of failures represent tumor growth as vitreous seeding and/or subretinal implants. Carboplatin diffuses well into the vitreous, but recent interest has stimulated a large prospective Children’s Oncology Group trial including systemic and subtenon administration (injected posteriorly between a thin membrane that surrounds the globe posterior to the limbus or beneath the conjunctiva), based on preclinical data documenting intraocular carboplatin concentrations 7 to 10 times higher when administered subconjunctivally.64,65,66 Improvement in the outcome of advanced ocular disease after subtenon instillation has not yet been documented clinically; there is recent interest in intra-arterial carboplatin administration.67,68
New agents with better intraocular penetration are being investigated. Topotecan is of interest and has excellent intraocular penetration and preclinical efficacy.69
The recent description of MDM4 amplification as the mechanism of inactivation of the TP53 pathway in retinoblastoma has opened the door to the use of specific targeted molecular therapies. Nutlin-3 is a small-molecule inhibitor of the MDM2-TP53 interaction.70 MDM4 and MDM2 bind TP53 with similar affinities, and studies have shown that nutlin-3 prevents the MDM4-T53 interaction in retinoblastoma cells, inducing TP53-mediated apoptosis in vitro and in vivo. Subconjunctival injection of nutlin-3 to mice with intraocular retinoblastoma resulted in significant tumor responses. The combination of nutlin-3 with a DNA-damaging agent such as topotecan resulted in a synergistic effect.13
Intravitreal and Intra-arterial Chemotherapy for Intraocular Retinoblastoma
Japanese investigators have pioneered the administration of intravitreal and intra-arterial melphalan for patients with advanced or recurrent intraocular retinoblastoma.71 Clinical responses have been documented after intravitreal melphalan and hyperthermia.71 A small, localized retinoblastoma would seem an excellent candidate for chemotherapy directly into the tumor vasculature. However, cannulation of the retinal artery is required, limiting the applicability of this procedure.67,71 Abramson and associates reported that direct cannulation of the ophthalmic artery using a microcatheter to instill melphalan resulted in high ocular salvage rates.68,72
Treatment of Extraocular Retinoblastoma
Extraocular dissemination of retinoblastoma usually reflects delayed diagnosis and treatment. In Europe and the United States, fewer than 5% of patients present with extraocular disease, in contrast to more than 20% to 40% in less developed countries.53,54 Four patterns of extraocular disease have been recognized: (1) locoregional, including orbital infiltration, tumor extending into the optic nerve or to the cut end of the nerve, and lymphatic spread to preauricular lymph nodes; (2) CNS dissemination; (3) trilateral retinoblastoma; and (4) metastatic retinoblastoma.
Orbital and Locoregional Retinoblastoma
Orbital retinoblastoma occurs as a result of progression of the tumor through the emissary vessels and sclera; scleral disease is considered to be extraocular and is treated as such. Orbital retinoblastoma is isolated in 60% to 70% of cases; lymphatic, hematogenous, and CNS metastases occur in 30% to 40% of patients with metastasis.55 Treatment includes systemic chemotherapy and RT; with this approach, 60% to 85% of patients are cured. Failures occur predominantly in the CNS, highlighting the importance of drugs with well-documented CNS penetration. Different chemotherapy regimens have proved to be effective and have included vincristine, cyclophosphamide, and doxorubicin; platinum- and epipodophyllotoxin-based regimens; or combinations thereof.28 For patients with macroscopic orbital disease, initial chemotherapy is followed by surgery, usually obviating the need for orbital exenteration. Postoperative chemotherapy and orbital irradiation (40 to 45 Gy) consolidate therapy.56 Similar management should be followed for patients with scleral disease, including RT, although good outcomes without RT also have been reported.53 Patients with isolated involvement of the optic nerve at the transection level should also receive similar systemic treatment with irradiation to the entire orbit (typically to 36 Gy) and a 9- to 10-Gy boost to the chiasm (total: 45 to 46 Gy).51,52 The preauricular and cervical lymph nodes should be explored carefully because 20% of patients with orbital extension have lymphatic metastases.55 Lymphatic dissemination may not carry a worse prognosis, provided that the involved lymph nodes are also irradiated.
Central Nervous System Disease
Intracranial dissemination occurs by direct extension through the optic nerve, and its prognosis is dismal.53,56 Treatment includes platinum-based intensive systemic chemotherapy and CNS-directed therapy. Although intrathecal chemotherapy has been traditionally used, there is no preclinical or clinical evidence to support its use. The use of irradiation in these patients is controversial, yet standard therapy includes craniospinal irradiation (23.4 to 36 Gy to the neuraxis and a boost to 45 Gy to sites of overt or nodular disease). Therapeutic intensification with high-dose, marrow ablative chemotherapy and autologous hematopoietic progenitor cell rescue has been explored, but its role is not yet clear.73,74 Reports of survivors are anecdotal.
Trilateral Retinoblastoma
The prognosis for patients with trilateral retinoblastoma is very poor; most patients die of disseminated neuraxis disease in less than 9 months.22 The rare survivors are usually asymptomatic (diagnosed by screening imaging) and treated with intensive chemotherapy with or without craniospinal irradiation.22,75 Given the short interval between the diagnosis of retinoblastoma and the occurrence of trilateral retinoblastoma, routine screening might detect the majority of cases within 2 years. Current strategies include avoiding irradiation and using intensive chemotherapy followed by consolidation with myeloablative chemotherapy and autologous hematopoietic progenitor cell rescue, an approach similar to that being used in the treatment of brain tumors in infants.76 Success has been noted anecdotally and in a series from Memorial Sloan-Kettering Cancer Center.75,77
Extracranial Metastatic Retinoblastoma
Hematogenous metastases are most common in the bones, bone marrow, and, less frequently, liver. Although long-term survivors have been reported after treatment with conventional chemotherapy, cures are anecdotal. Recent reports show that metastatic retinoblastoma can be cured using high-dose, marrow-ablative chemotherapy and autologous hematopoietic progenitor cell rescue73,74,78,79,80 (Table 69-6). The approach is similar to that for metastatic neuroblastoma; patients receive short and intensive induction regimens usually containing alkylating agents, anthracyclines, etoposide, and platinum compounds and are then consolidated with marrow-ablative chemotherapy and autologous hematopoietic cell rescue.74 In the series from Memorial Sloan-Kettering Cancer Center,74 patients with hematogenous bone metastases who responded to induction chemotherapy did not always require RT.
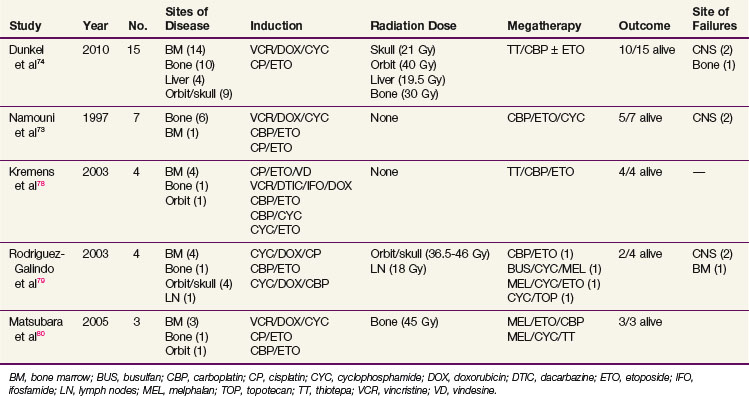
Irradiation Techniques/tolerances
Episcleral Plaque Brachytherapy
The indications for radioactive plaques have evolved along with progress in technology and physics from the introduction of radon seed implants in the 1920s, through broad experience with cobalt-60 plaques in the 1950s, and more recent use of iodine-125 (125I), iridium-192 (192Ir), and ruthenium-106 (106Ru).38–42 Primary plaque therapy is an option for localized retinoblastoma that is typically limited to one or two foci not involving the optic disc or macula. As an option among focal therapies in conjunction with chemoreduction therapy, radioactive plaques can be curative with small or moderate-sized lesions, including those with focal vitreous seeding immediately overlying the tumor(s).41
Most plaques today use 125I, a low-energy gamma ray emitter (27 to 35 keV; half-life, 60 days) that is readily available in encapsulated form, allowing custom fabrication of gold plaques of varying sizes and configured either as round or notched templates, the latter to allow placement adjacent to the optic nerve.39,41,42 One can differentially load 125I seeds within the wells of the plaque to achieve the dose distribution appropriate for the tumor volume (based on diameter and height). One typically uses 125I plaques for tumor height up to 10 mm and diameter less than or equal to 16 mm. Figure 69-2 shows some current and prototype plaques; Figure 69-3 demonstrates a case using a collimating plaque to treat retinoblastoma. 192Ir is an alternative radioisotope (a higher-energy gamma ray emitter at 295 to 612 keV; half-life, 74 days), also available as sealed sources with somewhat less desirable dosimetric characteristics. Experience with 106Ru has primarily been reported from Europe.39,42 This pure beta emitter provides a spectrum of beta particles with energies up to 3.5 MeV; the half-life is 374 days, which is important because 106Ru is an integral part of the plaque that can be used for up to a year. The dosimetric profile of 106Ru limits utilization to tumors with height of 6 mm or less; the uniformly round plaques are typically not applied within 2 mm of the optic nerve.39 Both 125I and 106Ru limit radiation exposure to patient and medical personnel while allowing diminished dosing to the underlying orbital bones and the optic lens.

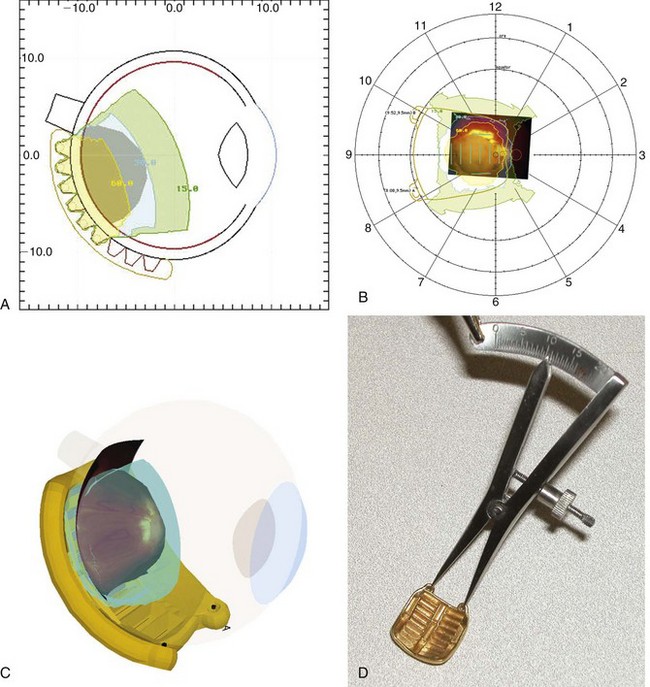
The dose is prescribed at the tumor apex or extended to include overlying localized vitreous seeds if present. The target dose is usually 40 Gy at the apex for 125I at 40 to 80 cGy/hr.38,40,41 Most of the experience with 106Ru reports apical doses of 50 Gy, sometimes 70 Gy.39,42 The dose at the tumor base (scleral dose) approximates 120 Gy for 125I and 150 to 200 Gy with 106Ru.
A review from both the Wills Eye Hospital and Hahnemann University Hospital in Philadelphia showed use of episcleral plaques in 15% of 900 cases treated between 1976 and 1999.38 Of the 208 tumors treated largely with 125I plaques, 29% used plaques as primary intervention and 71% after progression (prior EBRT and/or chemoreduction therapy). Ocular control was documented in 83% of eyes treated; recurrence occurred in 12% of those without prior therapy and in 20% after initial or subsequent progression. Ocular complications included nonproliferative retinopathy (27% at 5 years), papillopathy (26%), and nonproliferative maculopathy (25%).38 A subsequent report from the same institutions updates 125I plaques used at the time of progression after chemoreduction therapy (1994-2005).41 Eighty-four tumors were treated in 71 eyes of 64 patients with dosimetric factors as outlined earlier; average area at the base was 9 mm, and average height was 4 mm. Plaques were round (83%) or notched to allow placement adjacent to the optic nerve (17%); the average dose received at the fovea was 45 Gy, and at the optic nerve it was 22 Gy. Tumor control was documented in 95% of patients after chemoreduction therapy with or without earlier EBRT; failures occurred at a median of 4 months post implant.
Experience with 175 106Ru plaques (134 patients, 119 with bilateral disease) at the Jules-Gonin Eye Hospital, Lausanne, Switzerland, between 1992 and 2006 indicated tumor control in 94% of lesions and ocular retention in 89% of patients at 5 years.39 Enucleation was ultimately required in 13% of eyes for tumor, with intraocular hemorrhage obscuring visual follow-up, or because of radiation neuropathy or retinopathy (in equal proportions).39
External Beam Irradiation
The classic presentation requiring full orbital irradiation has been the young child with bilateral disease, in which the more advanced eye was enucleated and irradiation indicated for contralateral ocular disease or, less commonly, irradiation was administered for both eyes when reasonable likelihood of disease control and visual preservation were both present bilaterally. The single ipsilateral or bilateral opposed “D” fields provided coverage to the posterior retina, with acknowledged underdosage to the eye anterior to the equator associated with attempts to spare the ipsilateral lens and the contralateral eye when uninvolved or the disease was under control (e.g., after brachytherapy). Whether through straight lateral or coronally oblique fields to miss the contralateral eye, the retina between the ora serrata anteriorly and the equator of the globe was relatively underdosed. Hence, the Reese-Ellsworth staging system that includes not only disease volume and centricity but also location—where anterior disease resulted in higher stage designation—reflects the difficulty in achieving radiation control through then standard approaches.43,44,47
Initial indications for focal therapies (especially cryotherapy) were in the setting of disease recurrence after EBRT, ultimately achieving “success” in controlling tumor while avoiding enucleation and, where disease did not preclude such with massive retinal involvement or macular extension, useful vision.26,44 EBRT alone or in combination with subsequent focal therapy is successful (see earlier discussion) in 80% to 100% of children in Reese-Ellsworth groups I to III. With adequate attention to anterior beam placement, ocular preservation has been documented in 50% to 80% of cases even with group IV or V disease.26,43
In the setting of primary chemoreduction therapy, low-dose consolidative EBRT has been tested at a reduced “prophylactic” dose of 26 Gy when initially advanced disease (International Classification of Retinoblastoma group E) is yet responsive to induction chemotherapy. “Prophylactic” full ocular irradiation (26 Gy) resulted in ocular disease control without enucleation or requirement for full “therapeutic” doses of EBRT) in 80% of patients at 5 years compared with 33% with somewhat less advanced disease treated with the same chemoreduction technique alone.36
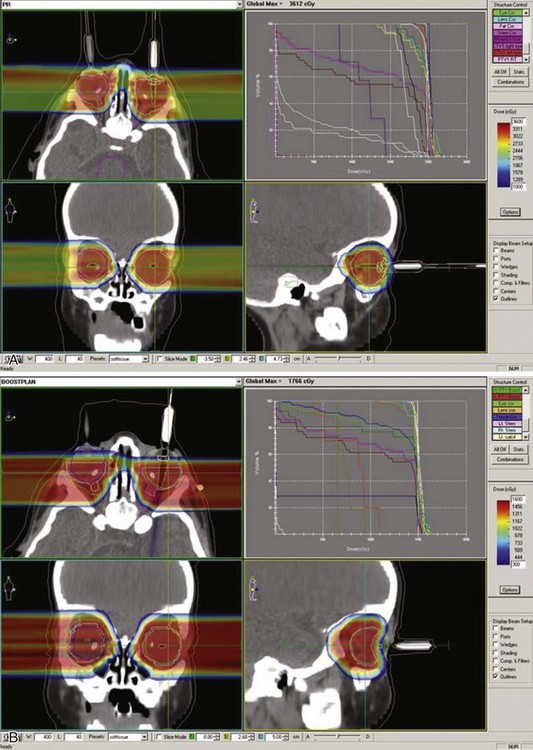
Reports over the past 5 years document more sophisticated approaches to either full ocular or focal radiation techniques. Stereotactic conformal RT has been used to mimic the focal dose distribution of episcleral implants for locally progressive disease or as focal consolidative therapy.35,37,43 Ocular suction systems provide precision localization and immobilization of the eye during anesthesia for stereotactic techniques35,37 (Fig. 69-4). Such techniques allow full therapeutic doses to focal posterior tumors (40 to 42 Gy) while limiting dose to the remainder of the globe, the optic nerve, and the ipsilateral lens.43,45 IMRT techniques document coverage of the entire retina/vitreous while limiting dose to the lacrimal gland and the bones of the orbit.43,47,50 The potential superiority of proton beam therapy, utilizing relatively simple scatter or scanning proton plans (e.g., single lateral field [Fig. 69-5]) to encompass the entire retina or for focal therapy at recurrence, has been reported with impressive dosimetry, if clinical experience is yet evolving.43,45 Figure 69-6 shows a patient’s plan with bilateral disease with frontal lobe sparing using active proton scanning.
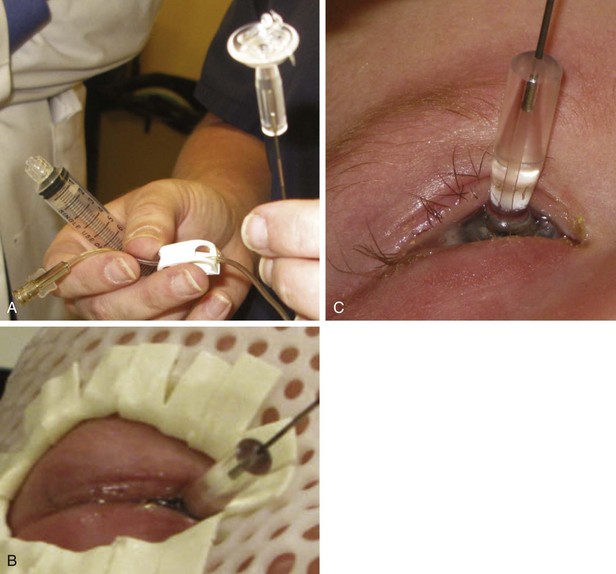
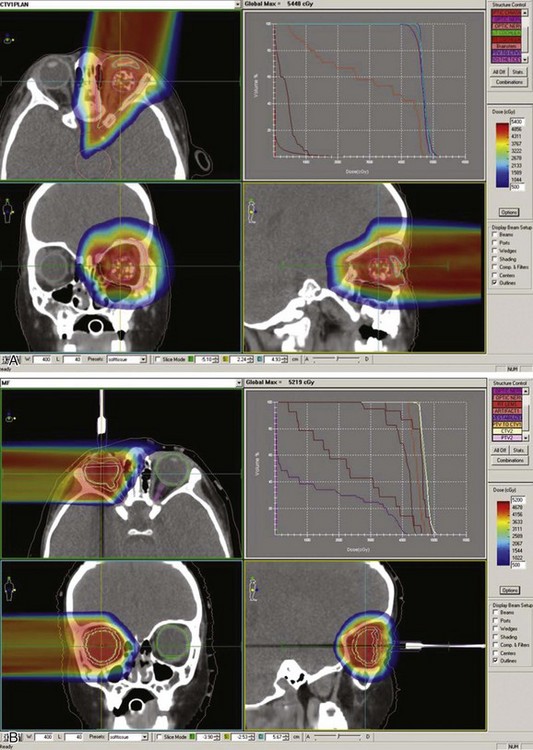
When there is overt disease involving the optic nerve (MRI at diagnosis or histology showing tumor at the cut end of the nerve after initial enucleation), the standard therapy is to combine aggressive chemotherapy with enucleation and postoperative orbital RT, the posterior aspect of the target volume encompassing the optic nerve to the chiasm. Recent data from Argentina and Children’s Hospital of Los Angeles show disease control in 70% or more of cases; seven of nine disease-free children from the Children’s Hospital of Los Angeles had not received RT.51,52 The use of photon IMRT or proton beam irradiation shows significant advantages in dose distribution, with full orbital RT targeting soft tissues and optic nerve extension.43,47,50
The prescription dose for retinoblastoma is classically 40 to 45 Gy at 1.8 to 2.0 Gy per fraction. Foote and colleagues44 reviewed the Mayo Clinic experience, documenting a lack of dose response above 45 Gy. A similar review by Merchant and associates46 showed similar rates of ocular preservation and potentially fewer treatment-related toxicities with 36 Gy in 18 fractions, with apparent diminution in disease control for more advanced disease (Reese-Ellsworth groups III to V). There is some evidence suggesting a lower threshold for postirradiation retinopathy when full ocular RT follows chemotherapy.43 The most recent dose relationship from Wills Eye Hospital in Philadelphia shows excellent disease control and ocular preservation in advanced intraocular disease (International Classification of Retinoblastoma group E) with 26-Gy consolidative full-eye irradiation when given on completion of chemotherapy, at a time when there is no evidence of recurrent or “active” tumor; toxicities appear to be less than when full dose (40 Gy) is required for overt residual/progressive tumor.36
Long-Term Effects of Retinoblastoma and Its Treatment
Children with retinoblastoma have the highest rate of secondary cancers of any cancer cohort. The risk is largely in patients with germline mutations of the RB1 gene, identified in long-term survivor cohorts as those with hereditary retinoblastoma (a family history—10% of children presenting with retinoblastoma; and those with bilateral disease absent a family history—30%). Patients with nonhereditary retinoblastoma are not inherently at increased risk. Exposure to RT increases the risk of second malignant neoplasms by over threefold.57,81,82 The largest cohort study combines data from major pediatric centers in Boston and New York City to report standard incidence ratios of subsequent cancers in 1601 cases surviving over 1 year post diagnosis treated between 1914 and 1984.57 The cumulative incidence of second malignant neoplasms at 50 years after diagnosis is 36% with the hereditary form and 5.7% in the sporadic/nonhereditary cohort. Within the hereditary cohort, the incidence of second malignant neoplasms is over 38% after RT and 21% without RT; the standard incidence ratios for second malignant neoplasms range from 22 times the expected incidence after RT in the hereditary population to 7 times without RT.57 Within the sporadic cohort, the incidence of second malignant neoplasms exceeds the normal population only for breast cancer (2.8 times), especially among those with radiation exposure (10 times).57 Second cancers correlate as well with timing of RT; children receiving RT during their first year may have a higher risk of developing a second cancer within the field of irradiation.81
The most common second malignant neoplasm is osteogenic sarcoma, which arises both inside and outside the radiation fields and accounts for nearly one third of all second cancers.57,81,82,83 Among the hereditary retinoblastoma cohort, 75% of second malignant neoplasms occurring within 25 years of initial therapy are sarcomas; osteosarcomas usually develop during the growth spurt years and occur with increased frequency even among the nonirradiated population, noting the proximity of genetic aberrations associated with osteosarcoma and the RB1 gene.83 Soft tissue sarcomas and melanomas are second in frequency, accounting for 20% to 25% of cases. Patients with hereditary retinoblastoma are also at risk of developing epithelial cancers late in adulthood; the most common such tumors are lung, breast, and colon carcinomas.57,82 Finally, an interesting observation is the increased incidence of lipomas in survivors of hereditary retinoblastoma. The incidence of a second neoplasm appears to be higher in patients with lipomas, suggesting that the presence of lipomas could be a clinical marker of susceptibility to second neoplasms.84
Treatment Algorithms, Controversies, Challenges, and Future Directions
Once properly staged, it is important to review the most appropriate treatment approach, which may range from upfront enucleation to an eye-preserving approach with systemic or intra-arterial chemotherapy and aggressive focal consolidation or RT in its different modalities. Decisions must be made in the context of a multidisciplinary team with expertise in the disease. Ideally, patients should be managed at a state of the art retinoblastoma treatment center. The newer techniques of potential value in retinoblastoma are relatively unique: subtenon chemotherapy administration, intra-arterial chemotherapy, and other aspects of the disease make a simple chart of what to do relatively inadequate to the task. Current therapeutic algorithms are presented in Table 69-4 and Figure 69-7.
1 Young JL, Smith MA, Roffers SD, et al. Retinoblastoma. NIH publication No. 99-4649]. Ries LAG, Smith MA, Gurney JG, et al, editors. Cancer incidence and survival among children and adolescents: united states SEER program 1975-1995. Bethesda, MD: National Cancer Institute, SEER Program, 1999.
3 Bunin GR, Meadows AT, Emanuel BS, et al. Pre- and postconception factors associated with sporadic heritable and nonheritable retinoblastoma. Cancer Res. 1989;49:5730-5735.
4 Orjuela MA, Titievsky L, Liu X, et al. Fruit and vegetable intake during pregnancy and risk for development of sporadic retinoblastoma. Cancer Epidemiol Biomarkers Prev. 2005;14:1433-1440.
8 Lee WH, Bookstein R, Hong F, et al. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235:1394-1399.
9 Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643-646.
10 Richter S, Vandezande K, Chen N, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253-269.
16 Draper GJ, Sanders BM, Brownhill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br J Cancer. 1992;66:211-219.
19 Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505-508.
22 Kivela T. Trilateral retinoblastoma. A meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829-1837.
26 Shields CL, Mashayekhi A, Au AK, et al. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113:2276-2280.
27 Chantada G, Doz F, Antonelli CBG, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801-805.
28 Rodriguez-Galindo C, Chantada GL, et al. Retinoblastoma. Current treatment and future perspectives. Curr Treat Options Neurol. 2007;9:294-307.
29 Shields CL, Shields JA. Recent developments in the management of retinoblastoma. J Pediatr Ophthalmol Strabismus. 1999;35:8-18.
35 Eldebawy E, Patrocinio H, Evans M, et al. Stereotactic radiotherapy as an alternative to plaque brachytherapy in retinoblastoma. Pediatr Blood Cancer. 2010;55:1210-1212.
36 Shields CL, Ramasubramanian A, Thangappan A, et al. Chemoreduction for group E retinoblastoma. Comparison of chemoreduction alone versus chemoreduction plus low-dose external radiotherapy in 76 eyes. Ophthalmology. 2009;116:544-551.
37 Sahgal A, Millar BA, Michaels H, et al. Focal stereotactic external beam radiotherapy as a vision-sparing method for the treatment of peripapillary and perimacular retinoblastoma. Preliminary results. Clin Oncol (R Coll Radiol). 2006;18:628-634.
38 Shields CL, Shields JA, Cater J, et al. Plaque radiotherapy for retinoblastoma: Long-term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116-2121.
39 Schueler AO, Fluhe D, Anastassiou G, et al. β-Ray brachytherapy with 106ruthenium plaques for retinoblastoma. Int J Radiat Oncol Biol Phys. 2006;65:1212-1221.
40 Freire JE, De Potter P, Brady LW, Longton WA. Brachytherapy in primary ocular tumors. Semin Surg Oncol. 1997;13:167-176.
41 Shields CL, Mashayekhi A, Sun H, et al. Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology. 2006;113:2087-2092.
42 Abouzeid H, Moeckli R, Gaillard MC, et al. 106Ruthenium brachytherapy for retinoblastoma. Int J Radiat Oncol Biol Phys. 2008;71:821-828.
43 Munier FL, Verwey J, Verwey J, et al. New developments in external beam radiotherapy for retinoblastoma. From lens to normal tissue-sparing techniques. Clin Exp Ophthalmol. 2008;36:78-89.
44 Foote RL, Garretson BR, Schomberg PJ, et al. External beam irradiation for retinoblastoma. Patterns of failure and dose-response analysis. Int J Radiat Oncol Biol Phys. 1989;16:823-830.
45 Lee CT, Bilton SD, Famiglietti RM, et al. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma. How do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362-372.
46 Merchant TE, Gould CJ, Hilton NE, et al. Ocular preservation after 36 Gy external beam radiation therapy for retinoblastoma. J Pediatr Hematol Oncol. 2002;24:246-249.
47 Reisner ML, Viegas CM, Grazziotin RZ, et al. Retinoblastoma—comparative analysis of external radiotherapy techniques, including an IMRT technique. Int J Radiat Oncol Biol Phys. 2007;67:933-941.
55 Doz F, Khelfaoui F, Mosseri V, et al. The role of chemotherapy in orbital involvement of retinoblastoma. Cancer. 1994;74:722-732.
56 Chantada G, Fandiño A, Casak S, et al. Treatment of overt extraocular retinoblastoma. Med Pediatr Oncol. 2003;40:158-161.
57 Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma. An extended follow-up. J Clin Oncol. 2005;23:2272-2279.
58 Friedman DL, Himelstein B, Shields CL, et al. Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma. J Clin Oncol. 2000;18:12-17.
61 Rodriguez-Galindo C, Wilson MW, Haik BG, et al. Treatment of intraocular retinoblastoma with vincristine and carboplatin. J Clin Oncol. 2003;21:2019-2025.
62 Chantada GL, Fandiño A, Raslawski EC, et al. Experience with chemoreduction and focal therapy for intraocular retinoblastoma in a developing country. Pediatr Blood Cancer. 2005;44:455-460.
63 Wilson MW, Haik BG, Liu T, et al. Effect on ocular survivalo of adding early intensive focal treatments to a two-drug chemotherapy regimen in patients with retinoblastoma. Am J Ophthalmol. 2005;140:397-406.
66 Abramson D, Frank CM, Dunkel IJ. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. Ophthalmology. 1999;106:1947-1950.
67 Mohri M. The technique of selective ophthalmic arterial infusion for conservative treatment of recurrent intraocular retinoblastoma. Keio Igaku. 1993;70:679-687.
68 Abramson DH, Dunkel IJ, Brodie SE. Bilateral superselective ophthalmic artery chemotherapy for bilateral retinoblastoma: Tandem therapy. Arch Ophthalmol. 2010;128:370-372.
69 Laurie NA, Gray JK, Zhang J, et al. Topotecan combination chemotherapy in two new rodent models of retinoblastoma. Clin Cancer Res. 2005;11:7569-7578.
71 Kaneko A, Suzuki S. Eye-preservation treatment of retinoblastoma with vitreous seeding. Jpn J Clin Oncol. 2003;33:601-607.
72 Abramson DH, Dunkel IJ, Brodie SE, et al. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma. Initial results. Ophthalmology. 2008;115:1398-1404.
73 Namouni F, Doz F, Tanguy ML, et al. High-dose chemotherapy with carboplatin, etoposide and cyclophosphamide followed by a haematopoietic stem cell rescue in patients with high-risk retinoblastoma. A SFOP and SFGM study. Eur J Cancer. 1997:2368-2375.
74 Dunkel IJ, Khakoo Y, Kernan NA, et al. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatric Blood Cancer. 2010;55:55-59.
75 Dunkel IJ, Jubran RF, Gururangan S, et al. Trilateral retinoblastoma. Potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54:384-387.
82 Fletcher O, Easton D, Anderson K, et al. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357-363.
83 Kleinerman RA, Tucker MA, Abramson DH, et al. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24-31.
1 Young JL, Smith MA, Roffers SD, et al. Retinoblastoma. NIH publication No. 99-4649]. Ries LAG, Smith MA, Gurney JG, et al, editors. Cancer incidence and survival among children and adolescents: united states SEER program 1975-1995. Bethesda, MD: National Cancer Institute, 1999.
2 Parkin DM, Stiller CA, Draper GJ, Bieber CA. The international incidence of childhood cancer. Int J Cancer. 1988;42:511-520.
3 Bunin GR, Meadows AT, Emanuel BS, et al. Pre- and postconception factors associated with sporadic heritable and nonheritable retinoblastoma. Cancer Res. 1989;49:5730-5735.
4 Orjuela MA, Titievsky L, Liu X, et al. Fruit and vegetable intake during pregnancy and risk for development of sporadic retinoblastoma. Cancer Epidemiol Biomarkers Prev. 2005;14:1433-1440.
5 Griep A, Krawcek J, Lee D, et al. Multiple genetic loci modify risk for retinoblastoma in transgenic mice. Invest Ophthalmol Vis Sci. 1998;39:2723-2732.
6 Orjuela M, Castaneda VP, Ridaura C, et al. Presence of human papillomavirus in tumor tissue from children with retinoblastoma. An alternative mechanism for tumor development. Clin Cancer Res. 2000;6:4010-4016.
7 Knudson AG. Mutation and cancer. Statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820-823.
8 Lee WH, Bookstein R, Hong F, et al. Human retinoblastoma susceptibility gene. Cloning, identification, and sequence. Science. 1987;235:1394-1399.
9 Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643-646.
10 Richter S, Vandezande K, Chen N, et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet. 2003;72:253-269.
11 Harbour JW. Molecular basis of low-penetrance retinoblastoma. Arch Ophthalmol. 2001;119:1699-1704.
12 Zhu X, Dunn JM, Goddard AD, et al. Mechanisms of loss of heterozygosity in retinoblastoma. Cytogenet Cell Genet. 1992;59:248-252.
13 Reference deleted in proofs.
14 Goddard AG, Kingston JE, Hungerford JL. Delay in diagnosis of retinoblastoma. Risk factors and treatment outcome. Br J Ophthalmol. 1999;83:1320-1323.
15 Chantada G, Fandiño A, Manzitti J, et al. Late diagnosis of retinoblastoma in a developing country. Arch Dis Child. 1999;80:171-174.
16 Draper GJ, Sanders BM, Brownhill PA, Hawkins MM. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br J Cancer. 1992;66:211-219.
17 Sippel KC, Fraioli RE, Smith GD, et al. Frequency of somatic and germ-line mosaicism in retinoblastoma. Implications for genetic counseling. Am J Hum Genet. 1998;62:610-619.
18 Sang DN, Albert DM. Retinoblastoma. Clinical and histopathologic features. Hum Pathol. 1982;13:133-147.
19 Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505-508.
20 Baud O, Cormier-Daire V, Lyonnet S, et al. Dysmorphic phenotype and neurological impairment in 22 retinoblastoma patients with constitutional cytogenetic 13q deletion. Clin Genet. 1999;55:478-482.
21 Holladay DA, Holladay A, Montebello JF, Redmond KP. Clinical presentation, treatment, and outcome of trilateral retinoblastoma. Cancer. 1991;67:710-715.
22 Kivela T. Trilateral retinoblastoma. A meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829-1837.
23 Shields CL, Meadows AT, Shields JA, et al. Chemoreduction for retinoblastoma may prevent intracranial neuroblastic malignancy (trilateral retinoblastoma). Arch Ophthalmol. 2001;119:1269-1272.
24 Beck-Popovic M, Balmer A, Maeder P, et al. Benign pineal cysts in children with bilateral retinoblastoma. A new variant of trilateral retinoblastoma? Pediatr Blood Cancer. 2006;46:755-761.
25 Reese AB, Ellsworth RM. The evaluation and current concept of retinoblastoma therapy. Trans Am Acad Ophthalmol Otolaryngol. 1963;67:164-172.
26 Shields CL, Mashayekhi A, Au AK, et al. The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology. 2006;113:2276-2280.
27 Chantada G, Doz F, Antonelli CB, et al. A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer. 2006;47:801-805.
28 Rodriguez-Galindo C, Chantada GL, Haik B, Wilson MW. Retinoblastoma. Current treatment and future perspectives. Curr Treat Options Neurol. 2007;9:294-307.
29 Shields CL, Shields JA. Recent developments in the management of retinoblastoma. J Pediatr Ophthalmol Strabismus. 1999;35:8-18.
30 Shields JA, Shields CL, De Potter P. Photocoagulation of retinoblastoma. Int Ophthalmol Clin. 1993;33:95-99.
31 Shields JA, Parsons H, Shields CL, Giblin ME. The role of cryotherapy in the management of retinoblastoma. Am J Ophthalmol. 1989;108:260-264.
32 Shields CL, Santos MCM, Diniz W, et al. Thermotherapy for retinoblastoma. Arch Ophthalmol. 1999;117:885-893.
33 Murphree AL, Villablanca JG, Deegan WF, et al. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch Ophthalmol. 1996;114:1348-1356.
34 Schouten-van Meeteren AY, Moll AC, Imhof SM, Veerman AJP. Overview: Chemotherapy for retinoblastoma. An expanding area for clinical research. Med Pediatr Oncol. 2002;38:428-438.
35 Eldebawy E, Patrocinio H, Evans M, et al. Stereotactic radiotherapy as an alternative to plaque brachytherapy in retinoblastoma. Pediatr Blood Cancer. 2010;55:1210-1212.
36 Shields CL, Ramasubramanian A, Thangappan A, et al. Chemoreduction for group E retinoblastoma: Comparison of chemoreduction alone versus chemoreduction plus low-dose external radiotherapy in 76 eyes. Ophthalmology. 2009;116:544-551.
37 Sahgal A, Millar BA, Michaels H, et al. Focal stereotactic external beam radiotherapy as a vision-sparing method for the treatment of peripapillary and perimacular retinoblastoma. Preliminary results. Clin Oncol (R Coll Radiol). 2006;18:628-634.
38 Shields CL, Shields JA, Cater J, et al. Plaque radiotherapy for retinoblastoma. Long-term tumor control and treatment complications in 208 tumors. Ophthalmology. 2001;108:2116-2121.
39 Schueler AO, Fluhe D, Anastassiou G, et al. β-Ray brachytherapy with 106ruthenium plaques for retinoblastoma. Int J Radiat Oncol Biol Phys. 2006;65:1212-1221.
40 Freire JE, De Potter P, Brady LW, Longton WA. Brachytherapy in primary ocular tumors. Semin Surg Oncol. 1997;13:167-176.
41 Shields CL, Mashayekhi A, Sun H, et al. Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology. 2006;113:2087-2092.
42 Abouzeid H, Moeckli R, Gaillard MC, et al. 106Ruthenium brachytherapy for retinoblastoma. Int J Radiat Oncol Biol Phys. 2008;71:821-828.
43 Munier FL, Verwey J, Verwey J, et al. New developments in external beam radiotherapy for retinoblastoma. From lens to normal tissue-sparing techniques. Clin Exp Ophthalmol. 2008;36:78-89.
44 Foote RL, Garretson BR, Schomberg PJ, et al. External beam irradiation for retinoblastoma: Patterns of failure and dose-response analysis. Int J Radiat Oncol Biol Phys. 1989;16:823-830.
45 Lee CT, Bilton SD, Famiglietti RM, et al. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma. How do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys. 2005;63:362-372.
46 Merchant TE, Gould CJ, Hilton NE, et al. Ocular preservation after 36 Gy external beam radiation therapy for retinoblastoma. J Pediatr Hematol Oncol. 2002;24:246-249.
47 Reisner ML, Viegas CM, Grazziotin RZ, et al. Retinoblastoma—comparative analysis of external radiotherapy techniques, including an IMRT technique. Int J Radiat Oncol Biol Phys. 2007;67:933-941.
48 Ross G, Lipper EG, Abramson D, Preiser L. The development of young children with retinoblastoma. Arch Pediatr Adolesc Med. 2001;155:80-83.
49 Abramson DH, Frank CM. Second nonocular tumors in survivors of bilateral retinoblastoma. A possible age effect on radiation-related risk. Ophthalmology. 1998;105:573-580.
50 Krasin MJ, Crawford BT, Zhu Y, et al. Intensity-modulated radiation therapy for children with intraocular retinoblastoma. Potential sparing of the bony orbit. Clin Oncol (R Coll Radiol). 2004;16:215-222.
51 Chantada GL, Guitter MR, Fandiño AC, et al. Treatment results in patients with retinoblastoma and invasion to the cut end of the optic nerve. Pediatr Blood Cancer. 2009;52:218-222.
52 Armenian SH, Panigrahy A, Murphee AL, Jubran R. Management of retinoblastoma with proximal optic nerve enhancement on MRI at diagnosis. Pediatr Blood Cancer. 2008;51:479-484.
53 Antoneli CB, Steinhorst F, de Cássia Braga Ribeiro K, et al. Extraocular retinoblastoma. A 13-year experience. Cancer. 2003;98:1292-1298.
54 Menon BS, Reddy SC, Maziah W, et al. Extraocular retinoblastoma. Med Pediatr Oncol. 2000;35:75-76.
55 Doz F, Khelfaoui F, Mosseri V, et al. The role of chemotherapy in orbital involvement of retinoblastoma. Cancer. 1994;74:722-732.
56 Chantada G, Fandiño A, Casak S, et al. Treatment of overt extraocular retinoblastoma. Med Pediatr Oncol. 2003;40:158-161.
57 Kleinerman RA, Tucker MA, Tarone RE, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma. An extended follow-up. J Clin Oncol. 2005;23:2272-2279.
58 Friedman DL, Himelstein B, Shields CL, et al. Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma. J Clin Oncol. 2000;18:12-17.
59 Gallie BL, Budning A, DeBoer G, et al. Chemotherapy with focal therapy can cure intraocular retinoblastoma without radiotherapy. Arch Ophthalmol. 1996;114:1321-1328.
60 Beck MN, Balmer A, Dessing C, et al. First-line chemotherapy with local treatment can prevent external-beam irradiation and enucleation in low-stage intraocular retinoblastoma. J Clin Oncol. 2000;18:2881-2887.
61 Rodriguez-Galindo C, Wilson MW, Haik BG, et al. Treatment of intraocular retinoblastoma with vincristine and carboplatin. J Clin Oncol. 2003;21:2019-2025.
62 Chantada GL, Fandiño A, Raslawski EC, et al. Experience with chemoreduction and focal therapy for intraocular retinoblastoma in a developing country. Pediatr Blood Cancer. 2005;44:455-460.
63 Wilson MW, Haik BG, Liu T, et al. Effect on ocular survival of adding early intensive focal treatments to a two-drug chemotherapy regimen in patients with retinoblastoma. Am J Ophthalmol. 2005;140:397-406.
64 Mendelsohn ME, Abramson DH, Madden T, et al. Intraocular concentrations of chemotherapeutic agents after systemic or local administration. Arch Ophthalmol. 1998;116:1209-1212.
65 Hayden BH, Murray TG, Scott IU, et al. Subconjunctival carboplatin in retinoblastoma. Impact of tumor burden and dose schedule. Arch Ophthalmol. 2000;118:1549-1554.
66 Abramson D, Frank CM, Dunkel IJ. A phase I/II study of subconjunctival carboplatin for intraocular retinoblastoma. Ophthalmology. 1999;106:1947-1950.
67 Mohri M. The technique of selective ophthalmic arterial infusion for conservative treatment of recurrent intraocular retinoblastoma. Keio Igaku. 1993;70:679-687.
68 Abramson DH, Dunkel IJ, Brodie SE. Bilateral superselective ophthalmic artery chemotherapy for bilateral retinoblastoma. Tandem therapy. Arch Ophthalmol. 2010;128:370-372.
69 Laurie NA, Gray JK, Zhang J, et al. Topotecan combination chemotherapy in two new rodent models of retinoblastoma. Clin Cancer Res. 2005;11:7569-7578.
70 Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-888.
71 Kaneko A, Suzuki S. Eye-preservation treatment of retinoblastoma with vitreous seeding. Jpn J Clin Oncol. 2003;33:601-607.
72 Abramson DH, Dunkel IJ, Brodie SE, et al. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma. Initial results. Ophthalmology. 2008;115:1398-1404.
73 Namouni F, Doz F, Tanguy ML, et al. High-dose chemotherapy with carboplatin, etoposide and cyclophosphamide followed by a haematopoietic stem cell rescue in patients with high-risk retinoblastoma. A SFOP and SFGM study. Eur J Cancer. 1997;33:2368-2375.
74 Dunkel IJ, Khakoo Y, Kernan NA, et al. Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer. 2010;55:55-59.
75 Dunkel IJ, Jubran RF, Gururangan S, et al. Trilateral retinoblastoma. Potentially curable with intensive chemotherapy. Pediatr Blood Cancer. 2010;54:384-387.
76 Fangusaro J, Finlay J, Sposto R, et al. Intensive chemotherapy followed by consolidative myeloablative chemotherapy with autologous hematopoietic cell rescue (AuHCR) in young children with newly diagnosed supratentorial primitive neuroectodermal tumors (sPNETs). Report of the Head Start I and II experience. Pediatr Blood Cancer. 2008;50:312-318.
77 Wright KD, Qaddoumi I, Patay Z, et al. Successful treatment of early detected trilateral retinoblastoma using standard infant brain tumor therapy. Pediatr Blood Cancer. 2010;55:570-572.
78 Kremens B, Wieland R, Reinhard H, et al. High-dose chemotherapy with autologous stem cell rescue in children with retinoblastoma. Bone Marrow Transplant. 2003;31:281-284.
79 Rodriguez-Galindo C, Wilson MW, Haik BG, et al. Treatment of metastatic retinoblastoma. Ophthalmology. 2003;110:1237-1240.
80 Matsubara H, Makimoto A, Higa T, et al. A multidisciplinary treatment strategy that includes high-dose chemotherapy for metastatic retinoblastoma without CNS involvement. Bone Marrow Transplant. 2005;35:763-766.
81 Abramson DH. Second nonocular cancers in retinoblastoma. A unified hypothesis. The Franceschetti Lecture. Ophthalmic Genet. 1999;20:193-204.
82 Fletcher O, Easton D, Anderson K, et al. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357-363.
83 Kleinerman RA, Tucker MA, Abramson DH, et al. Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst. 2007;99:24-31.
84 Li FP, Abramson DH, Tarone RE, et al. Hereditary retinoblastoma, lipoma, and second primary cancers. J Natl Cancer Inst. 1997;89:83-84.