16 Retinal Degenerations
Retinal Dystrophies
Age-related Macular Degeneration
Treatment of Age-related Choroidal Neovascularization
Idiopathic Polypoidal Chorioretinopathy (IPCV)
Inherited Central Receptor (Macular) Dystrophies
Inherited Retinal Dystrophies with Generalized Retinal Involvement
Progressive Retinal Dystrophies
Normal vision depends on the proper functioning of the macula which, when disturbed by disease, affects central vision and often causes the patient to present for the first time. Central distortion of vision (metamorphopsia) or minification of the image are pathognomonic for macular disease. Other symptoms apart from loss of acuity include photophobia, glare and dazzle (which can be severe), poor colour perception, night-blindness and a central or paracentral scotoma. Macular disease in infants frequently presents as congenital nystagmus.
Classifying macular disease by the anatomical layer that is primarily affected—choriocapillaris, Bruch’s membrane, retinal pigment epithelium (RPE), photoreceptors or neuroretina—is a commonly used and helpful way to conceptualize and diagnose retinal problems. Stereoscopic examination of the fundus with an indirect biomicroscopy lens through a dilated pupil is essential to assess the location of a lesion within the retina and its relationship to the superficial retinal vessels. However, although examination can infer the initial site of the lesion in many retinal diseases the primary defect cannot be precisely localized to a single cell layer as defects in one retinal layer produce changes in adjacent structures. For example, retinitis pigmentosa is associated with photoreceptor degeneration and also RPE and choriocapillaris atrophy with later neuroretinal degeneration and vascular attenuation. Our understanding of the pathogenesis of various retinal diseases has improved in recent years with advances in molecular genetics and cell biology and some diseases that were thought to affect, for example, primarily the photoreceptors have now been shown to have their defect in the RPE. Fundus photography, fundus fluorescein angiography (FFA), indocyanine green (ICG) angiography, optical coherence tomography (OCT) and autofluorescence (AF) imaging are valuable investigations for diagnosis and documentation. Many macular lesions are small and produce scotomas that are difficult to demonstrate; an Amsler grid can help in these cases. Electrodiagnostic tests and psychophysical testing are time consuming but very useful for diagnosing, documenting and assessing the prognosis of inherited retinal disease (see Ch. 1).
Occasionally deciding whether visual loss is caused by macular or optic nerve disease can be difficult as both reduce acuity and colour vision and can cause central scotomas. Retinal lesions tend to produce a ‘positive’ scotoma in which patients are aware of dark obscuration or something in front of their vision in contrast to the ‘negative’ scotoma of neurological disease (for example, patients do not notice a hemianopic field defect as ‘dark’). An afferent pupillary defect indicates optic nerve disease unless the macular lesion is extensive. Another useful and easily performed test is the photostress test in which recovery of visual acuity following dazzle with a bright light is delayed with macular lesions whereas vision recovers normally with optic nerve disease. Retinal lesions produce a blue-yellow pattern of colour deficit in contrast to the red-green pattern of optic nerve disease (see Ch. 19). These clinical tests are subjective, however, and less reliable than pattern electroretinography (PERG) which in most cases can distinguish macular from optic nerve disease (see Ch. 1).
AGE-RELATED MACULAR DEGENERATION
PATHOGENESIS OF AMD
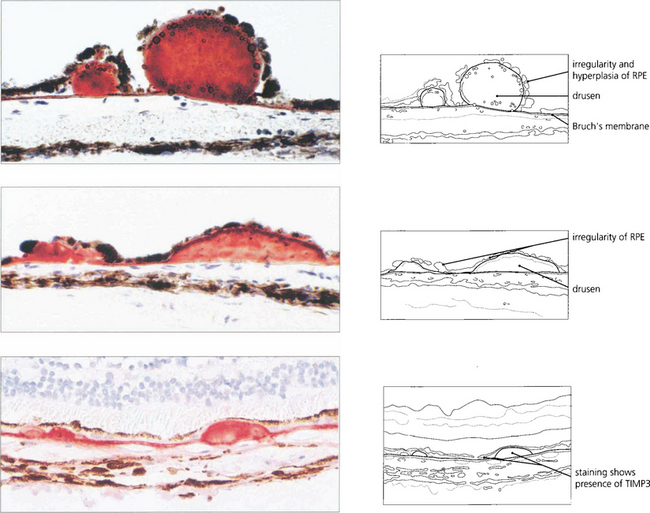
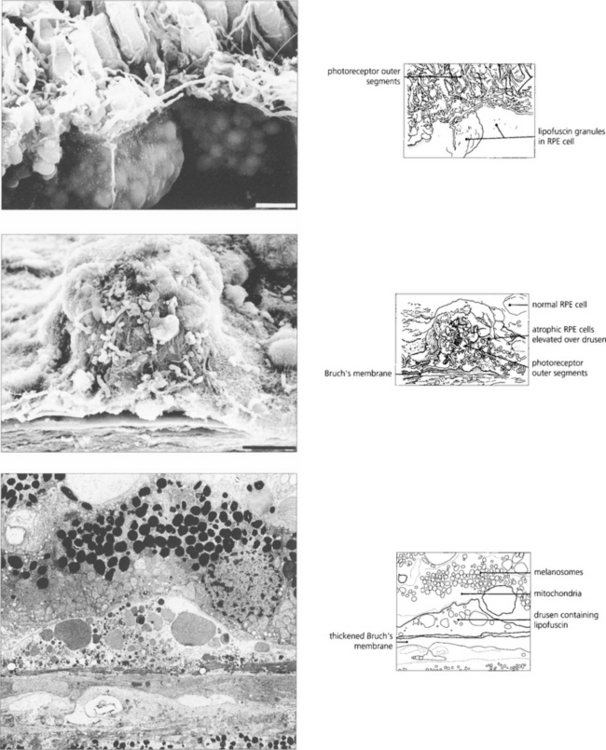
AMD appears to result from defects in the RPE cell–Bruch’s membrane–choriocapillaris complex and is thought to be a collection of heterogeneous disorders associated with multiple genetic and environmental factors. Much current research centres on defects in the RPE cell and Bruch’s membrane (see Ch. 13). In normal ageing progressive thickening of Bruch’s membrane occurs with accumulation of lipids, noncollagenous proteins and other extracellular material such as advanced glycation endproducts, collagen and elastin changes and increased calcification. By the fourth decade all eyes show accumulation of membranous debris within both collagenous layers. With time, a second series of changes is seen with deposition of diffuse material both between the RPE and its basement membrane (basal linear deposit) and between the basement membrane and the inner collagenous layer (basal laminar deposit). The latter can become extensive with a complex morphology and may be confused with diffuse or soft drusen, drusen being focal accumulations between the RPE basement membrane and the inner collagenous layer of Bruch’s membrane. Lipofuscin accumulates with age in the RPE cell from metabolism of photoreceptor outer segments from oxidative damage or RPE overloading by failed photoreceptors. The specific mechanisms underlying these changes are not fully understood, but it is thought that the resultant increase in thickness and reduction in molecular transport through Bruch’s membrane and the RPE cell may contribute to photoreceptor cell death and AMD. In addition, anatomical changes and delayed filling on fluorescein angiography are seen in the choroidal circulation.
DRUSEN AND ATROPHIC MACULAR CHANGES
Drusen lie in Bruch’s membrane and stain progressively during fluorescein angiography. They are assessed more easily by fluorescein angiography than by fundoscopy as angiography shows RPE atrophy more clearly. Different drusen staining patterns emerge during angiography. Conventionally, drusen that stain readily are taken to be ‘hydrophilic’, whereas those that do not are ‘hydrophobic’. Hydrophilic drusen are more proteinaceous and are associated with an increased risk of choroidal neovascularization; hydrophobic drusen have increased lipid and are more associated with PED. Other factors, such as drusen size and extracellular matrix characteristics probably influence staining too. However, it is the extent of RPE atrophy seen as window defects (see Ch. 13) on angiography or as blackness on autofluorescence imaging—rather than drusen—that better reflects visual loss.
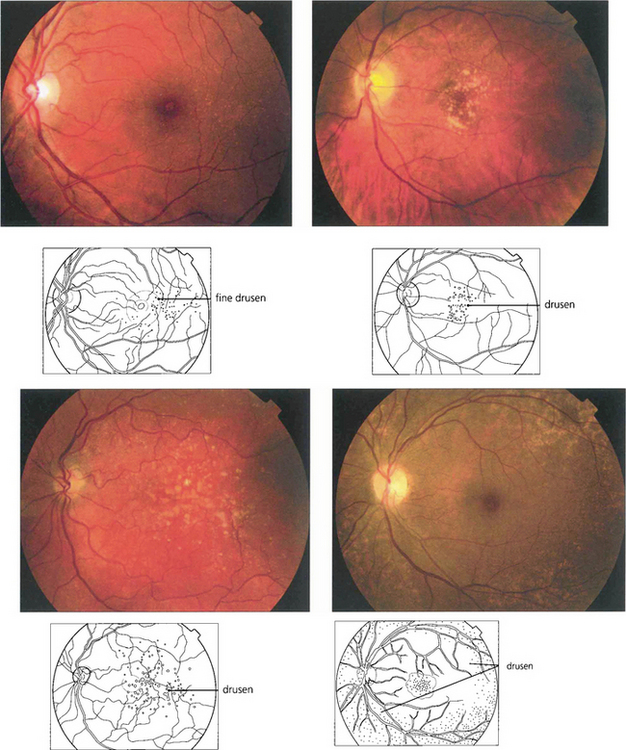
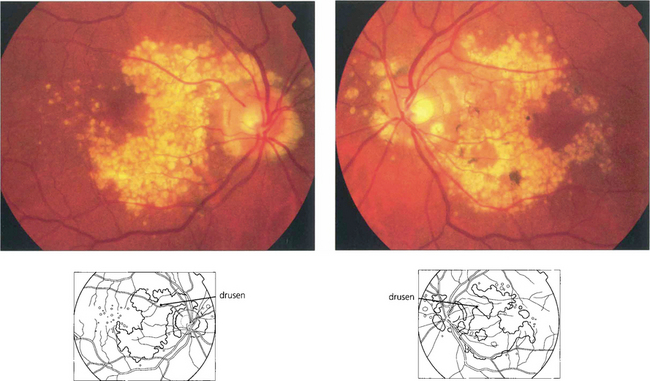
Fig. 16.3 Drusen come in various patterns, shapes and sizes with variable amounts of RPE change. Some patients develop widespread drusen throughout the posterior pole.
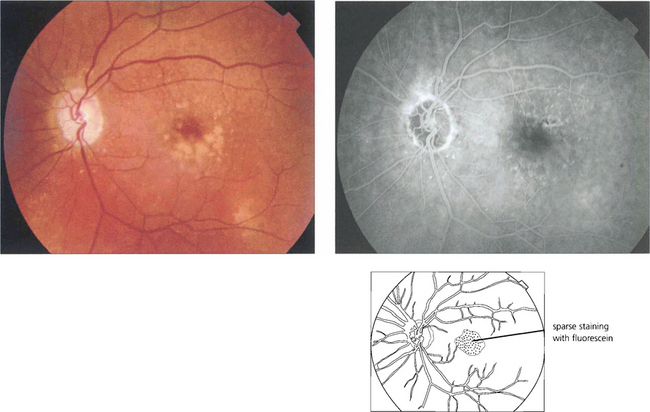
Fig. 16.4 The drusen in this patient’s macular area are relatively hydrophobic, failing to stain as angiography progresses.
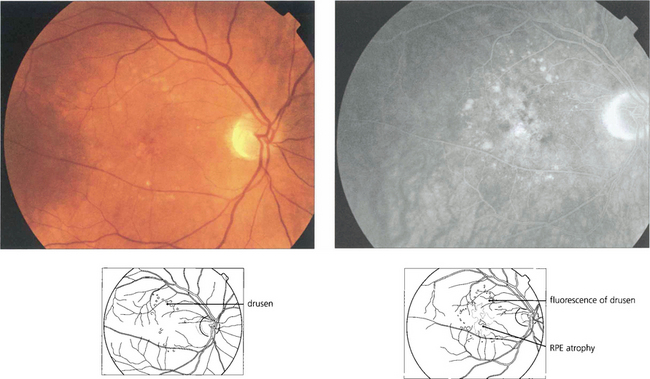
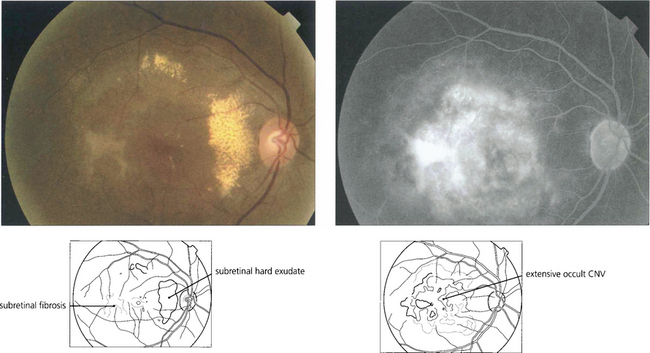
Fig. 16.5 This patient has more hydrophilic drusen. Note the submacular hyperfluorescence, which probably denotes early occult choroidal neovascularization.
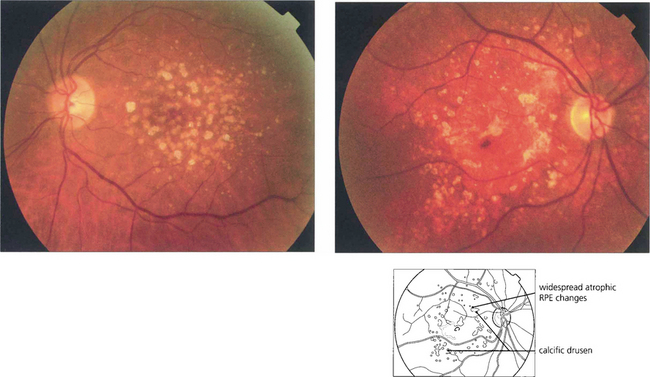
Fig. 16.6 Calcific drusen have a flat, glistening, white and refractile appearance (left). These eyes tend to lose vision from RPE atrophy rather than neovascularization, as seen in the more gross example (right).
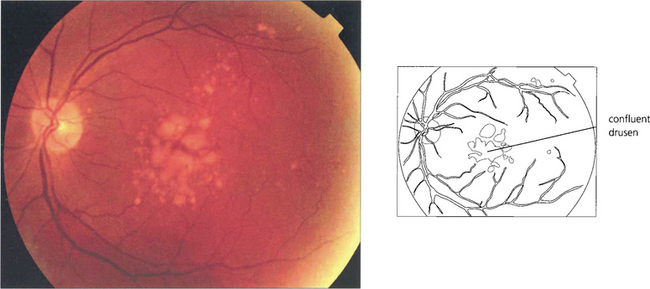
Fig. 16.7 This patient has a large area of confluent drusen under the macula. Such patients have a considerable risk of developing choroidal neovascular lesions.
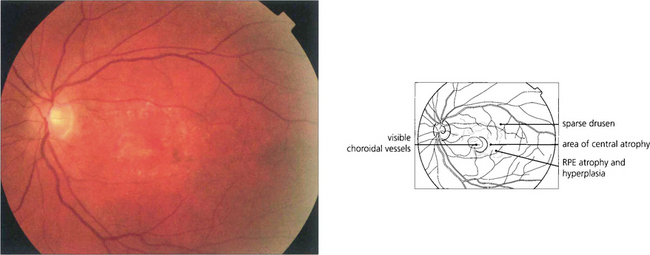
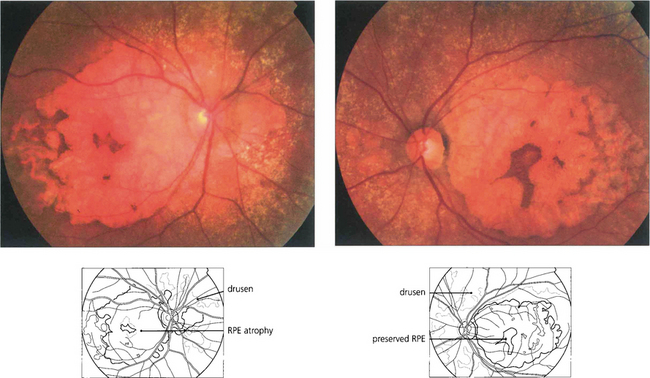
Fig. 16.8 The degree of RPE atrophy varies considerably. This patient has small, sparse drusen, subretinal pigmentation from RPE hyperplasia and a central–paracentral RPE defect, accounting for an acuity of 20/100 and a paracentral scotoma.
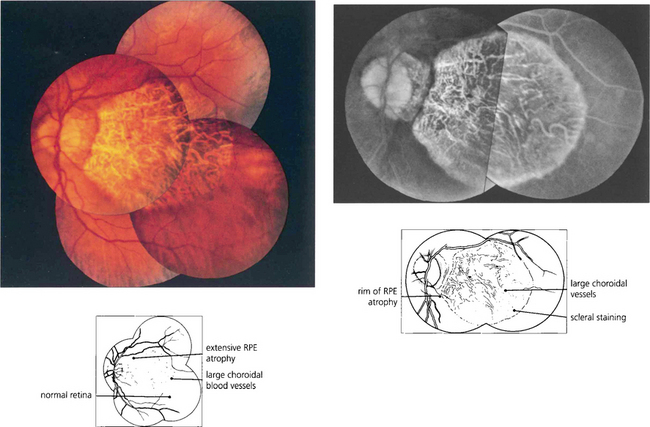
Fig. 16.9 Central areolar choroidal atrophy can be dominantly inherited but it is also a phenotypic variant of AMD in which there is a sharp circumscribed demarcation line between normal and abnormal retina with central atrophy of the choriocapillaris, RPE and photoreceptors revealing large choroidal vessels in the base (left). These lesions start in the macular area and gradually increase in extent over time with severe loss of visual acuity. Drusen are not normally seen. (right) Angiogram of the same patient shows the large window defect due to loss of the RPE and choriocapillaris. Large choroidal vessels are seen in the base and fill normally. Note the relative hyperfluorescence of the rim of the lesion from scleral staining (see Ch. 13).
RETINAL PIGMENT EPITHELIAL DETACHMENT
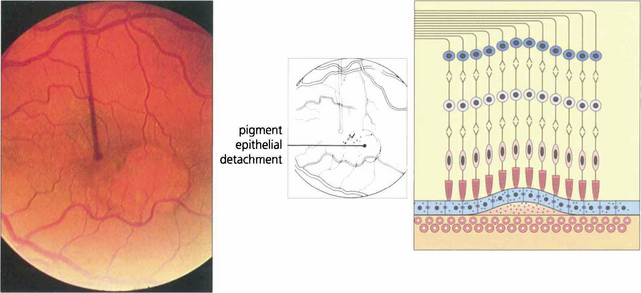
Fig. 16.10 Colour photograph showing the well circumscribed elevation of the macula, which has a more prominent and thicker appearance than that seen with central serous retinopathy (see Fig. 16.38). In elderly patients other signs of macular degeneration such as drusen, pigmentation and atrophy are frequently seen. Any surrounding lipid exudate or haemorrhage strongly suggests choroidal neovascularization within the lesion.
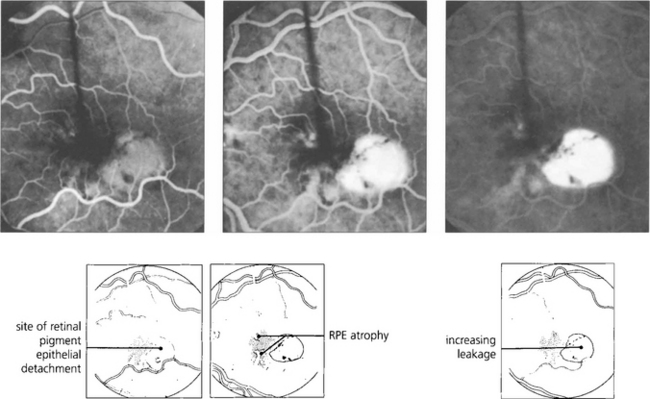
Fig. 16.11 The fluorescein angiogram shows early diffuse leakage which increases in intensity as the run progresses but remains localized to the area of the lesion. Other degenerative changes in the RPE are often seen.
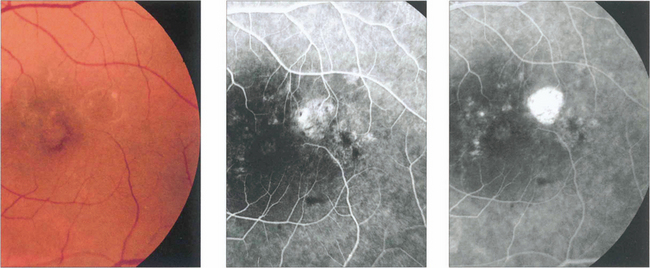
Fig. 16.12 Elderly patients may have an underlying neovascular membrane, which is indicated by any unevenness of staining or hyperfluorescence within the lesion.
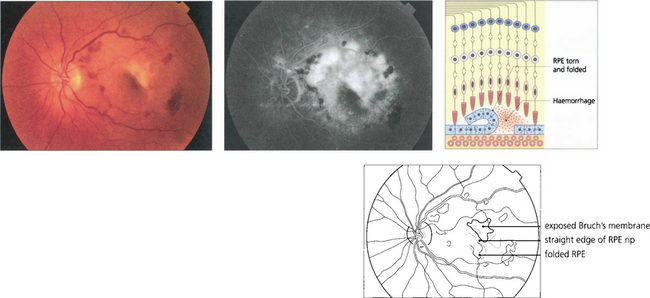
Fig. 16.13 Excessive tension within the PED causes it to rip and fold back on itself. This is often associated with haemorrhage into the lesion and acute loss of acuity in the eye. Characteristically RPE rips show a dark area where the RPE has folded back on itself separated by a straight line from the area of denuded RPE.
By courtesy of Ms H Jackson.
CHOROIDAL NEOVASCULAR MEMBRANES
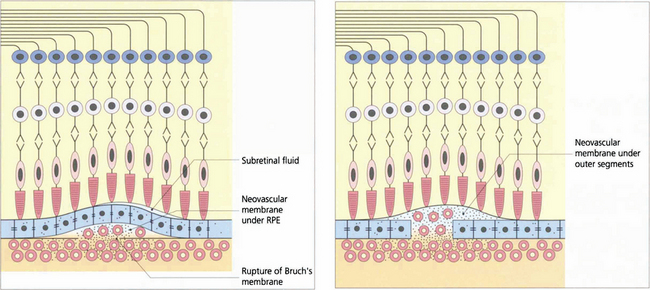
Fig. 16.14 CNV can grow between Bruch’s membrane and the RPE (type 1) or in the subretinal space between the RPE and photoreceptors (type 2); combinations of these occur as well. The appearance of CNV on angiography depends on the amount of haemorrhage, pigment hyperplasia and RPE atrophy. Based on FFA findings, choroidal neovascular membranes (CNV) are divided into classical and occult forms. Classical CNV is defined as well demarcated areas of hyperfluorescence that can be seen in the early phase of fluorescein angiography with progressive leakage of dye obscuring the boundaries of the CNV as angiography progresses; these may correspond to type 2 membranes. Occult CNV is either a fibrovascular PED with areas of irregular elevation of RPE producing an area of stippled hyperfluorescence as the angiogram progresses or late leakage from an undetermined source; these may correspond to type 1 membranes. An alternative explanation of the different angiographic appearances is that a classical CNV represents a type 1 membrane with overlying RPE atrophy. Further clinical pathological studies are needed.
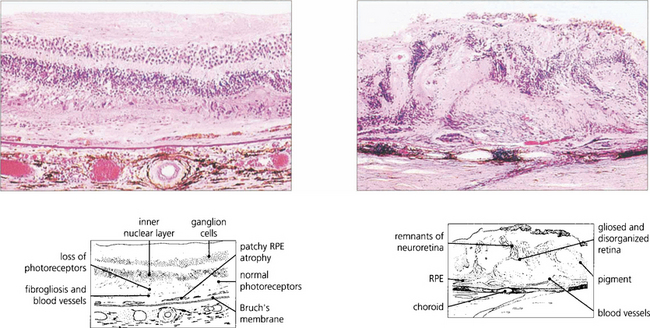
Fig. 16.15 Serial sections of eyes with drusen have shown that neovascularization of drusen in the posterior pole is not uncommon and that only a minority of these lesions progress to a clinically apparent CNV. Pathology of end-stage disciform macular degeneration shows a layer of vascularized fibrous tissue replacing the photoreceptors (left). In a gross example (right) the retina is completely destroyed by an elevated mass of vascularized fibroglial tissue.
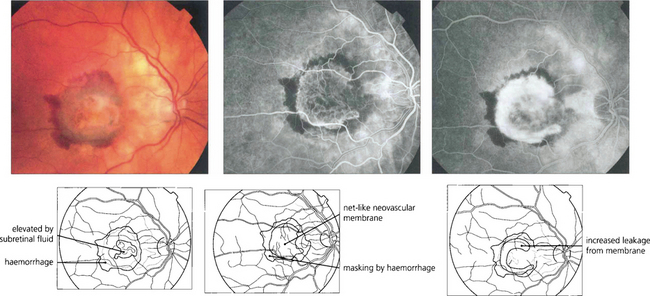
Fig. 16.16 The typical classical CNV lesion, no matter what the aetiology, consists of subretinal elevation either beneath or adjacent to the fovea. Recent lesions often have a pale, raised appearance with variable amounts of lipid or haemorrhage. A subretinal haemorrhage in the macular area should always raise the suspicion of a neovascular cause. Presenting symptoms are either metamorphopsia or blurring of vision, depending on the size and site of the lesion. Early-phase fluorescein angiography of the same lesion shows the classical appearance of a neovascular complex lying beneath the retina which, because it is derived from the choroid, fills early in angiography. Individual vessels in the neovascular membrane may be identified in the earliest stages as a net-like membrane. In common with other neovascular tissue, the capillaries in the neovascular membrane have loose endothelial cell junctions and in late stages the membrane shows intense leakage into the adjacent retina, blurring the membrane. These lesions are amenable to photodynamic treatment.
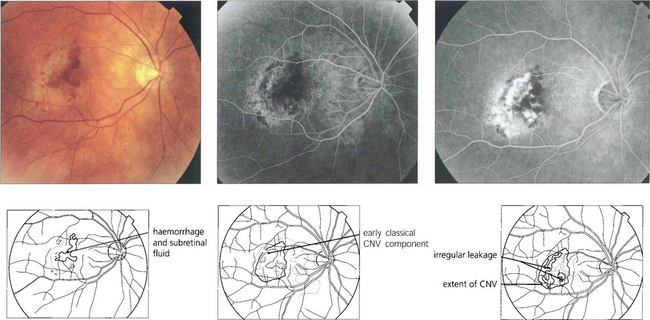
Fig. 16.17 Occult CNV membranes are more common than classical lesions. This patient presented with distortion and blurring of vision to 20/80. Angiography shows an irregular leakage in the lesion, staining intensively in the later phases. These lesions do not benefit as much from photodynamic treatment.
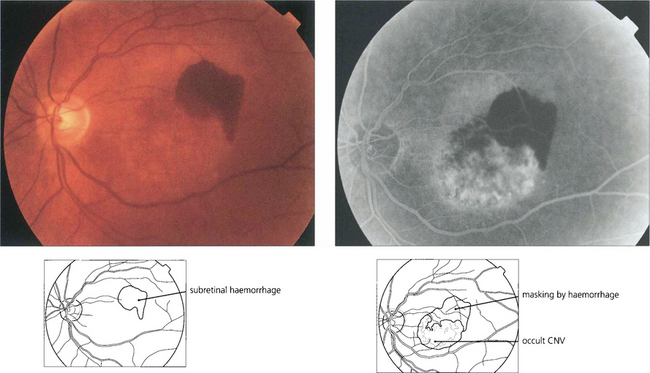
Fig. 16.18 CNV is often associated with subretinal haemorrhage which may produce a sudden drop in visual acuity causing the patient to present. In this situation haemorrhage may mask the true extent of the membrane and fluorescein angiography should be postponed until sufficient haemorrhage has been absorbed for visualization. ICG angiography may help in these circumstances as it reveals the extent of the membrane despite the overlying haemorrhage.
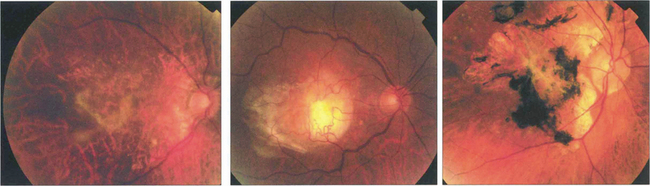
OTHER CAUSES OF CHOROIDAL NEOVASCULAR MEMBRANES
ANGIOID STREAKS
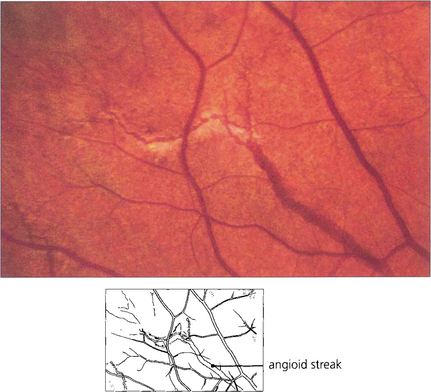
Fig. 16.21 Angioid streaks are splits in Bruch’s membrane. They appear as linear fractures extending from the optic disc and ramifying towards the periphery mimicking retinal blood vessels. They are usually reddish brown but may show degenerative pigmentary changes. Their appearance on angiography depends on the degree of associated pigmentary disturbance and RPE loss. The majority of streaks are hyperfluorescent, although early in the run they may mask background choroidal fluorescence. Subretinal neovascularization in the papillomacular region or alongside the optic disc is a frequent complication. Angioid streaks occur as an isolated ocular finding or with systemic associations which include elastic tissue disorders such as pseudoxanthoma elasticum and Ehlers–Danlos syndrome.
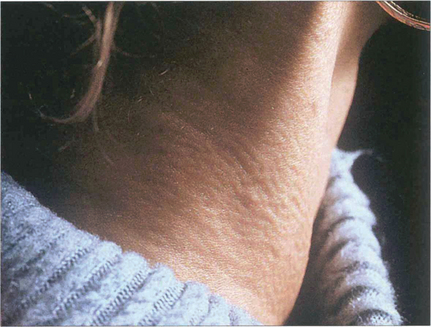
Fig. 16.22 Pseudoxanthoma elasticum is a recessively inherited disorder affecting connective tissue elastic fibres. Patients have cutaneous, ocular and vascular manifestations and suffer premature hypertension, atheroma, cardiac valve disorders and gastrointestinal haemorrhage. This patient has the characteristic ‘chicken skin’ changes on her neck, which she disguises by wearing a polo-neck shirt.
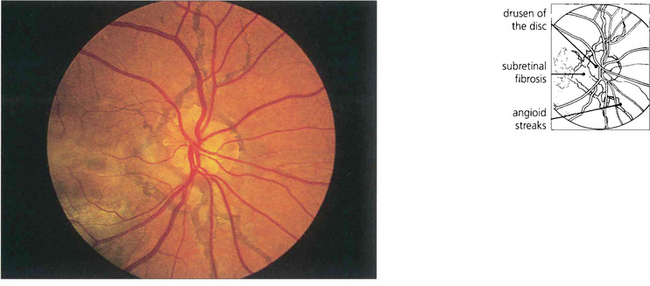
Fig. 16.23 Remarkably conspicuous angioid streaks radiate from the patient’s optic disc which also has the well recognized association of pronounced optic disc drusen. Juxtapapillary and subretinal fibrosis from neovascularization can be seen underlying the macula. This patient has widespread RPE changes within and adjacent to areas of subretinal neovascularization; visual prognosis is poor.
By courtesy Mr R K Blach.
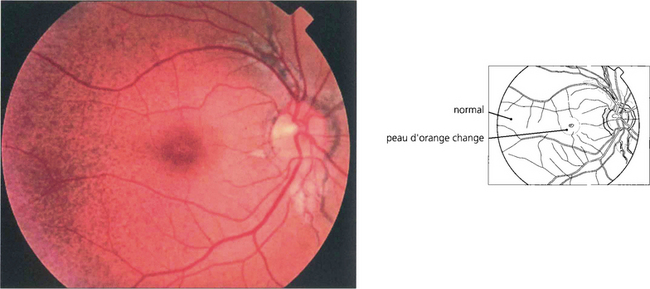
Fig. 16.24 In patients with pseudoxanthoma elasticum, Bruch’s membrane and the RPE may also be altered, giving the fundus a slightly mottled, yellowish and refractile appearance known as ‘peau d’orange’. This is most noticeable in the posterior pole and contrasts with the normal appearing periphery.
CHOROIDAL RUPTURE
‘Choroidal rupture’ is caused by severe blunt injury to the eye but the appearance is in fact due to rupture of Bruch’s membrane (see Ch. 14). CNV can develop some time later in lesions extending into the macular area.
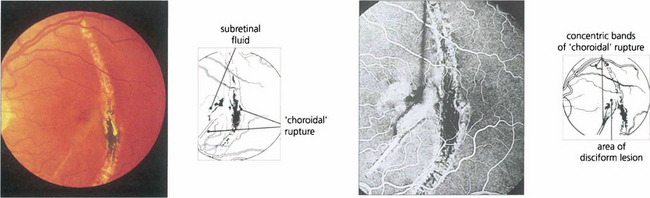
Fig. 16.25 In this patient, rupture is seen as a typical chorioretinal scar lying concentric to the optic disc with fresh, pale, submacular fluid that has caused recent onset of blurring and distorted vision. Fluorescein angiography demonstrates the extent of the RPE and choriocapillaris disruption with a subfoveal CNV. Photodynamic therapy has a potential role in the managment of these patients.
OCULAR HISTOPLASMOSIS AND PUNCTATE INNER CHOROIDOPATHY
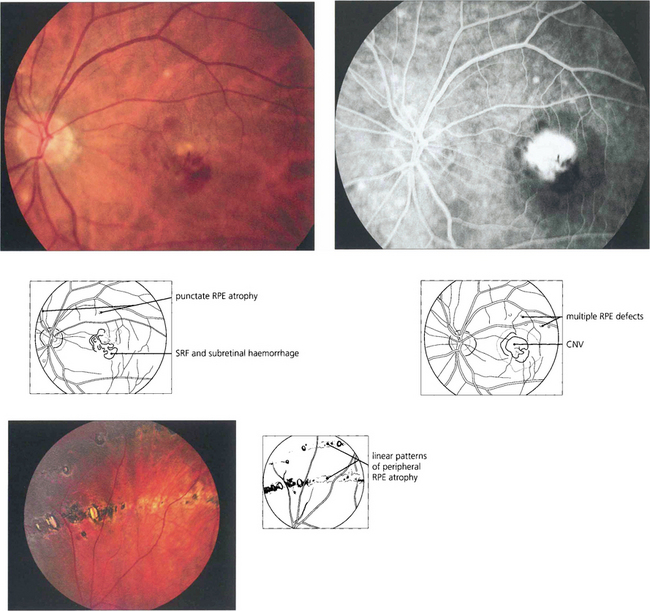
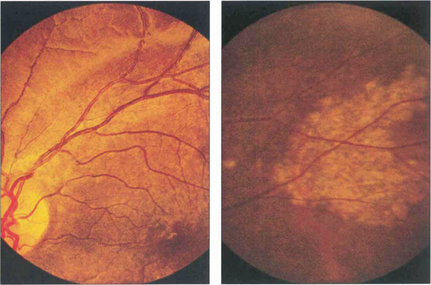
Fig. 16.26 The typical appearance of the fundus comprises the triad of atrophic changes around the optic disc, localized punched-out areas of pigment epithelial atrophy in the posterior pole and atrophic, usually linear, RPE lesions in the equatorial retina (bottom left). Atrophic changes in the macular region carry a substantial risk of developing CNV, typically in early middle age.
PERIPAPILLARY CNV
Choroidal neovascularization may develop adjacent to the optic disc, either spontaneously or in association with disc abnormalities such as tilting or chronic pathological disc swelling or drusen of the disc (see Ch. 17).
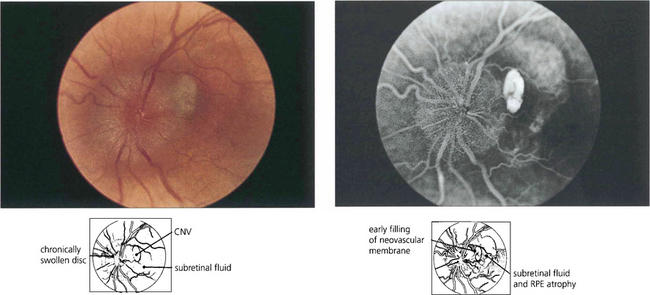
MYOPIC MACULAR DEGENERATION
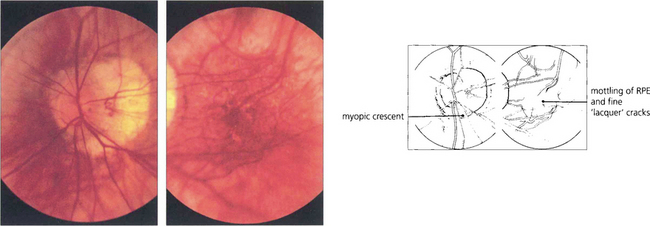
Fig. 16.29 High myopes have an increased risk of developing atrophic macular degeneration—seen as chorioretinal thinning or atrophy, posterior staphylomata and splits in Bruch’s membrane termed ‘lacquer’ cracks—and also peripheral lattice degeneration. This patient shows a large myopic crescent on the temporal side of the optic disc. Retinal vessels are pulled straight as they enter the enlarged posterior pole. In the macula there is patchy pigmentary disturbance and fine cracks in Bruch’s membrane.
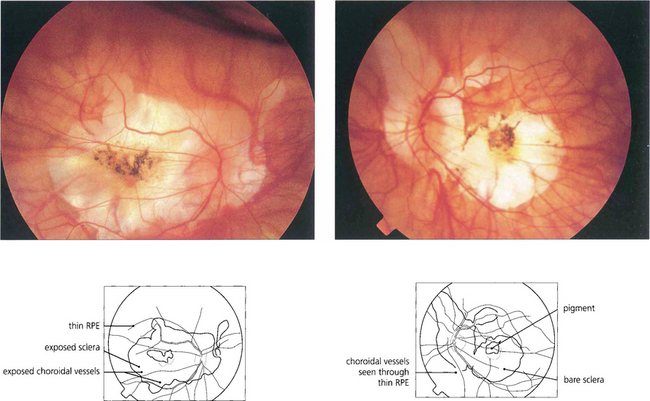
Fig. 16.30 This patient shows marked myopic chorioretinal atrophy in both eyes with exposed sclera at each posterior pole.
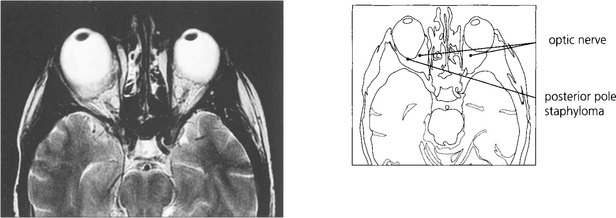
Fig. 16.31 Magnetic resonance imaging demonstrates a large posterior pole staphyloma extending posteriorly to the optic disc in the right eye.
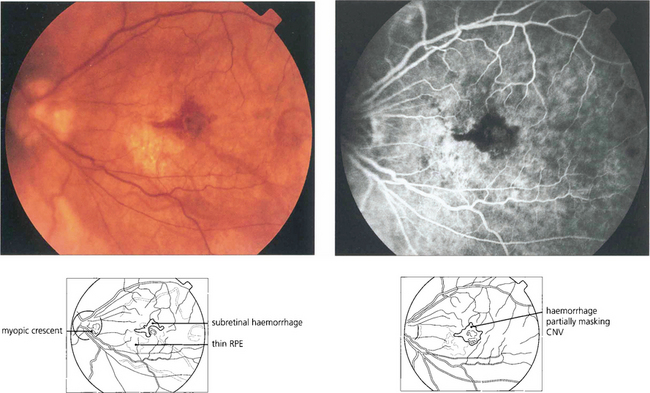
Fig. 16.32 High myopes have an increased risk of developing CNV in early adulthood, usually presenting with a fairly small localized haemorrhage (Foster–Fuchs’ spot). Extensive exudate or serous detachment does not occur and the condition resolves to leave a small pigmented scar. Visual recovery is much better than with other types of CNV lesion. Photodynamic therapy may improve the prognosis.
TREATMENT OF AGE-RELATED CHOROIDAL NEOVASCULARIZATION
Thermal burns produced by argon laser will destroy choroidal neovascularization and limit, to some extent, further retinal destruction and visual loss. Lesions can be treated up to the edge of the foveal avascular zone but care must be taken with juxtafoveal lesions as damage from the burn can spread outside the treated area. A problem with lesions adjacent to the fovea is that the macular yellow luteal pigment absorbs energy which may then thermally injure the overlying neuroretina and enlarge the scotoma. If light is restricted to the green bands of the argon spectrum the laser energy is taken up by blood and the RPE, sparing the neuroretina; long-wavelength krypton or diode laser energy is also taken up by RPE and choroidal pigment but not by the luteal pigment and so can be useful in this situation. Subfoveal CNV cannot be treated by thermal laser without destroying central vision. Although successful treatment improves the short-term visual prognosis this is not sustained in the long term and studies show that after 3–4 years vision has fallen in 50–60 per cent of patients from further macular degeneration.
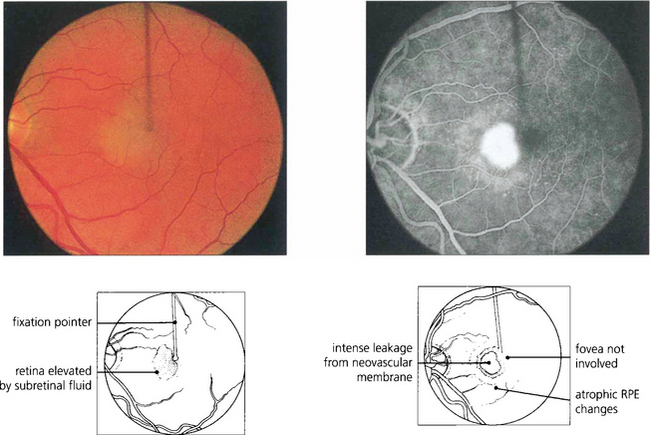
Fig. 16.33 This patient has a fresh parafoveal CNV with serous detachment extending into the fovea and an acuity of 20/40. Fluorescein angiography is essential prior to treatment to assess the full extent of the membrane. The most important images are taken in the earliest arterial phases of angiography, before leakage of dye into the retina has obscured the fine vascular detail. Fixation must be identified.
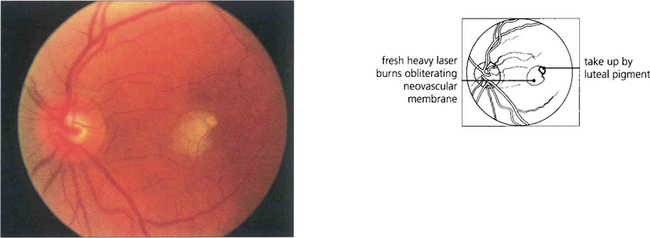
Fig. 16.34 Lesions must be treated heavily and completely; partial or incomplete photocoagulation carries the risk of provoking a rapid extension of the membrane. Immediately after treatment by argon laser an opaque reaction can be seen deep in the outer retina. The more intense parafoveal burn is due to energy absorption by luteal pigment. Visual acuity fell to 20/80 immediately after treatment.
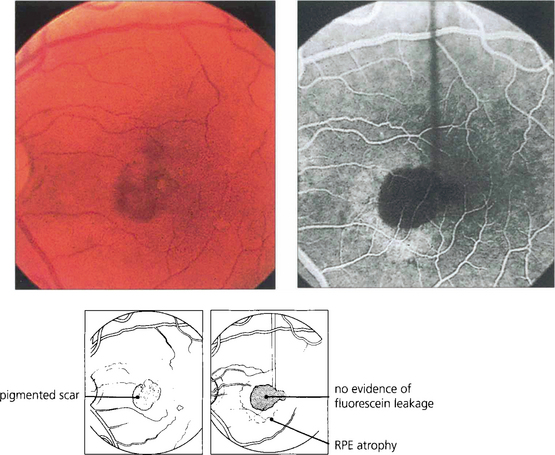
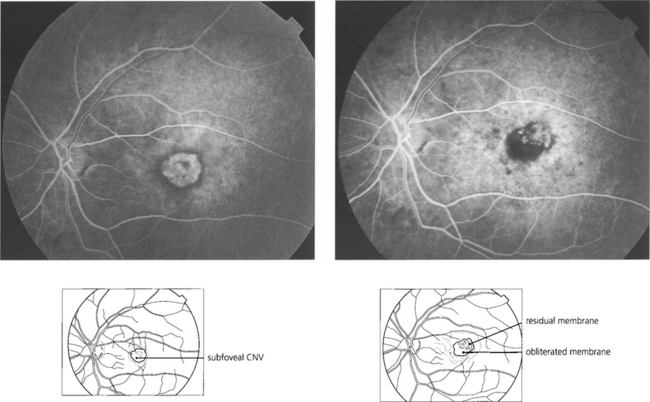
Fig. 16.35 Four weeks’ later a pigmented scar is forming in the region of the burns. Fluorescein angiography shows complete obliteration of the membrane. Visual acuity improved to 20/30 with a small paramacular scotoma which the patient easily compensated for. Risk of recurrence in eyes with thermally treated CNV is approximately 10% a year; the fellow eye carries a similar risk of developing CNV formation.
IDIOPATHIC POLYPOIDAL CHORIORETINOPATHY (IPCV)
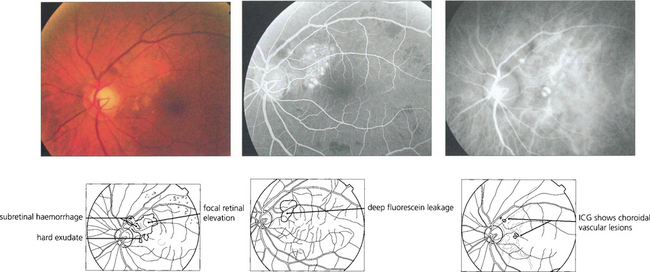
Fig. 16.37 This black patient has subretinal haemorrhage and hard exudates adjacent to the disc. Fluorescein angiography shows deep subretinal leakage and ICG angiography shows two foci of polypoid change underlying these which probably represent herniation of choroidal vessels through Bruch’s membrane.
CENTRAL SEROUS RETINOPATHY
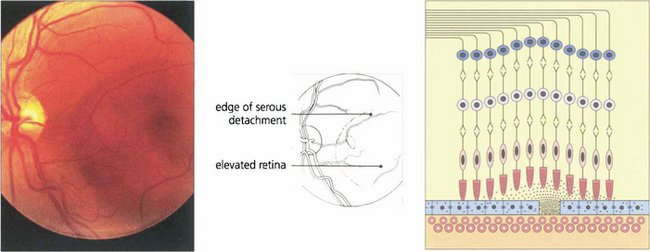
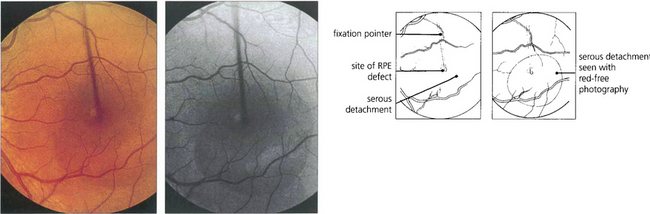
Fig. 16.38 The typical fundus appearance is of a thin translucent blister underlying the macula. This may be difficult to see by direct ophthalmoscopy and is more readily visible by biomicroscopy. The diagram illustrates the small break in the RPE and serous retinal detachment elevating the macula.
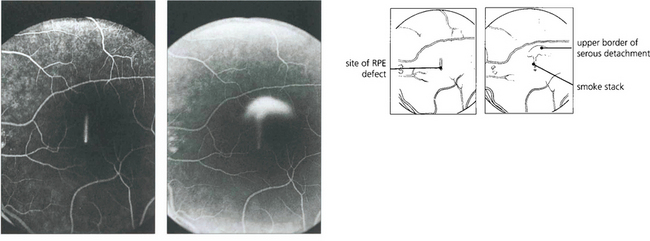
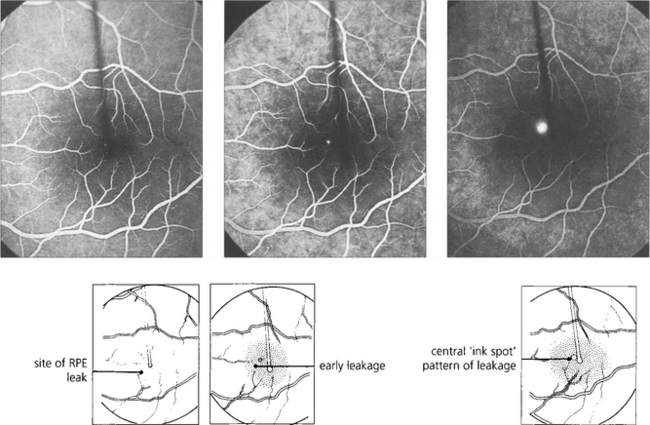
Fig. 16.39 Fluorescein angiography in the early stages of the disease shows a focal defect in the RPE with a typical ‘smoke stack’ pattern of leakage. The leakage appears as a small hyperfluorescent spot at the site of the breech in the RPE and flows upwards in a column, influenced, presumably, by local convection. When the dye reaches the upper margin of the detachment it spreads laterally along its circumference and eventually fills the whole detachment.
INHERITED RETINAL DYSTROPHIES
CLASSIFICATION OF INHERITED RETINAL DISEASE
Clinically inherited retinal dystrophies are classified by natural history (stationary or progressive), mode of inheritance (autosomal dominant, autosomal recessive, X-linked recessive, mitochondrial or, less commonly, multiallelic inheritance) and putative site of dysfunction within the retina. The latter may be inferred from the results of psychophysical and electrophysiological testing (see Ch. 1) and the various methods of fundus imaging such as fluorescein angiography, autofluorescence imaging and optical coherence tomography. This approach has limitations and a more robust classification which more accurately reflects disease pathogenesis awaits the identification of the many genetic mutations associated with retinal disease. More than 90 genes and 140 loci associated with inherited retinal degeneration have now been identified with many more yet to be discovered. At present, routine molecular diagnostic testing is available for only a few disorders but the numbers will increase as advances are made in the technology of molecular genetic analysis.
INHERITED CENTRAL RECEPTOR (MACULAR) DYSTROPHIES
AUTOSOMAL RECESSIVE INHERITANCE
Stargardt disease and fundus flavimaculatus (FFM)
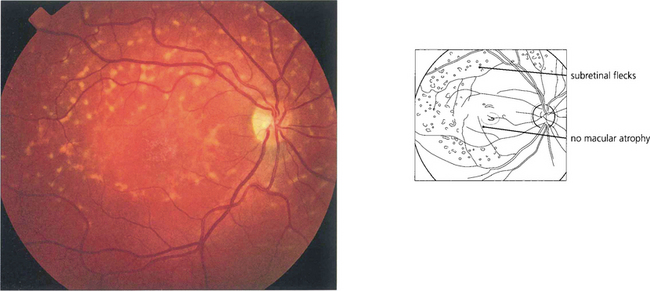
Fig. 16.42 The term fundus flavimaculatus (FFM) is often used to describe a phenotype in which there are multiple white flecks without significant macular atrophy. It appears that STGD and FFM are caused by mutations in the same gene and both patterns may be seen within the same family.
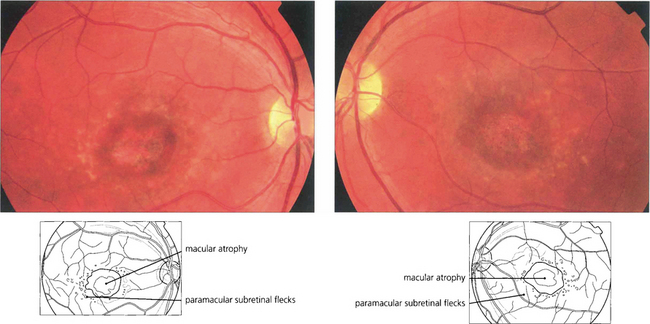
Fig. 16.43 Typically there is macular atrophy with white flecks at the level of the RPE at the posterior pole. The flecks may be fish tail-shaped (pisciform), round, oval or semilunar. The area of macular atrophy may in the early stages have a ‘beaten bronze’ appearance.
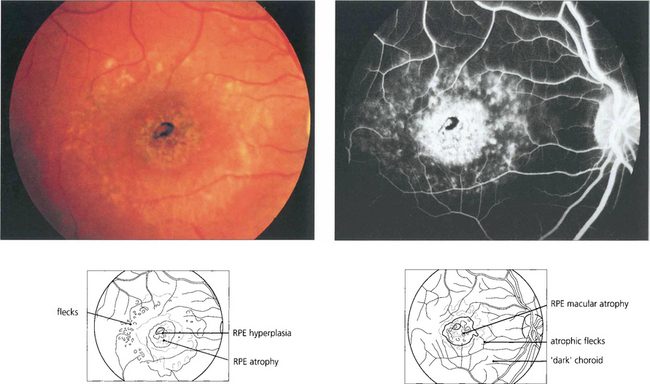
Fig. 16.44 Fluorescein angiography classically reveals a dark or masked choroid. The reduced visualization of the choroidal circulation in the early phase of fundus fluorescein angiography (FFA) is believed to be secondary to excess lipofuscin accumulation in the RPE obscuring the fluorescence emanating from choroidal capillaries. The retinal flecks appear hypofluorescent on FFA early in their evolution but at a later stage they appear hyperfluorescent as a result of RPE atrophy.
AUTOSOMAL DOMINANT INHERITANCE
Best disease (vitelliform macular dystrophy)
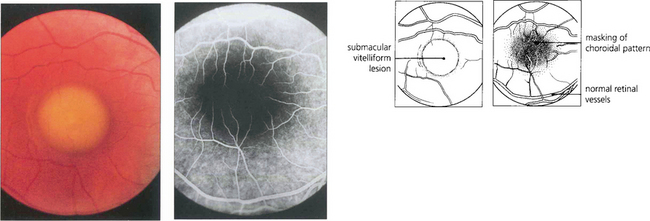
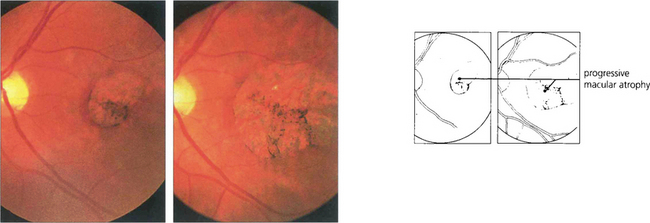
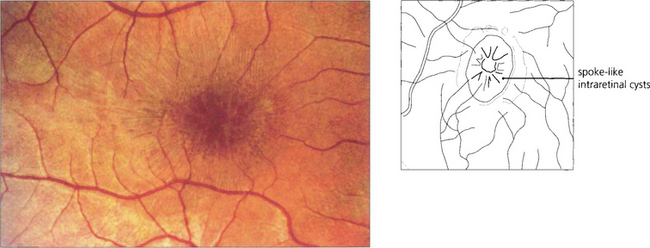
Fig. 16.46 The phenotype has been classified into five stages. Stage 0 (previtelliform) is characterized by a normal fundus appearance in an asymptomatic gene carrier with an abnormal EOG. In stage I, minor RPE changes are seen. The classical vitelliform lesion (stage II) is characterized by the ‘egg yolk’ macular lesion that masks the underlying choroidal fluorescence. This appearance is usually seen during the first or second decades, often associated with normal, or slightly reduced, visual acuity.
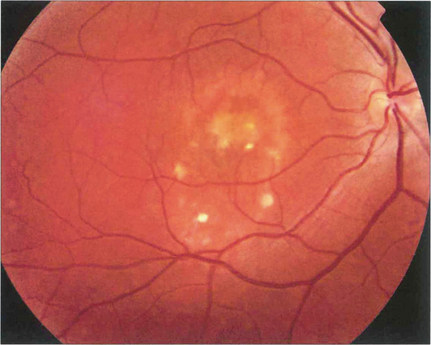
Fig. 16.47 The ‘egg yolk’ begins to break up secondary to resorption of the yellow material lying between the RPE and sensory retina (stage IIa) with visual impairment usually being noticeable at this stage.
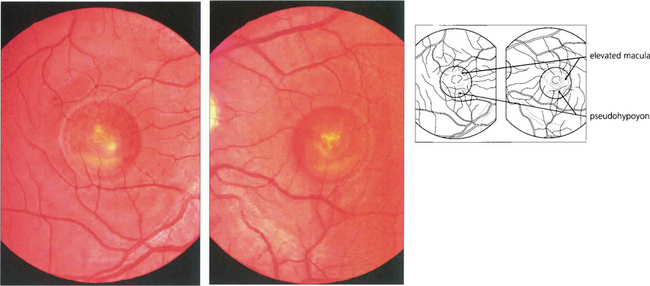
Fig. 16.48 Stage III (pseudohypoyon) is seen when part of the lesion is resorbed, leaving the appearance of a ‘fluid level’ at the macula. The yellow material is completely resorbed over time, leaving an area of RPE atrophy (stage IVa) and often subretinal fibrosis (IVb). Choroidal neovascularization is an established complication leading to severe visual loss (IVc).
Adult vitelliform macular dystrophy
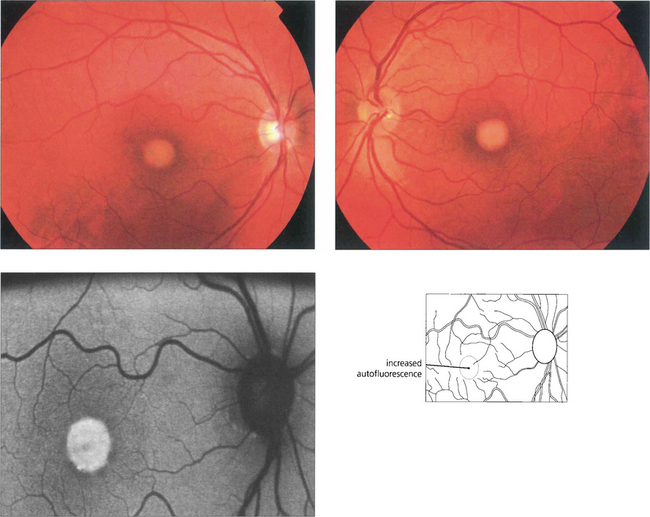
Fig. 16.49 The typical clinical appearance is of bilateral, round or oval, yellow, symmetrical, subretinal lesions, typically one-third to one-half optic disc diameter in size. The autofluorescence image shows increased autofluorescence corresponding to the macular lesion suggesting that this is due to accumulated lipofuchsin.
By courtesy of Mr Andrew Webster.
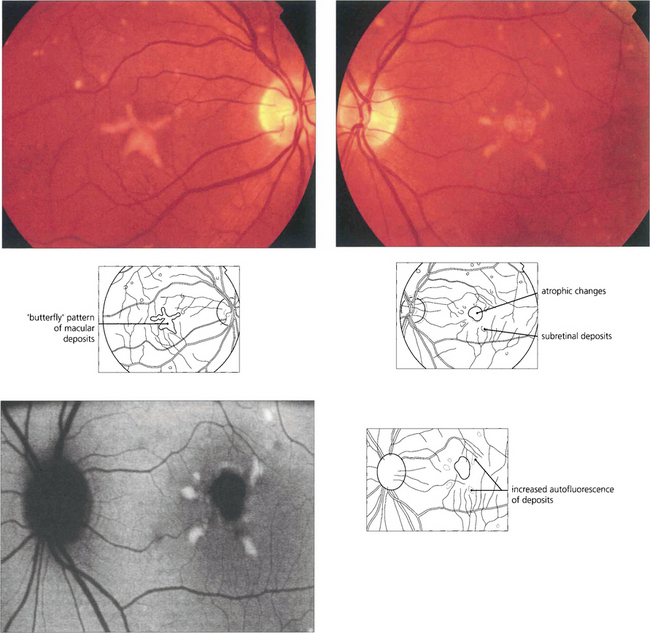
North Carolina macular dystrophy
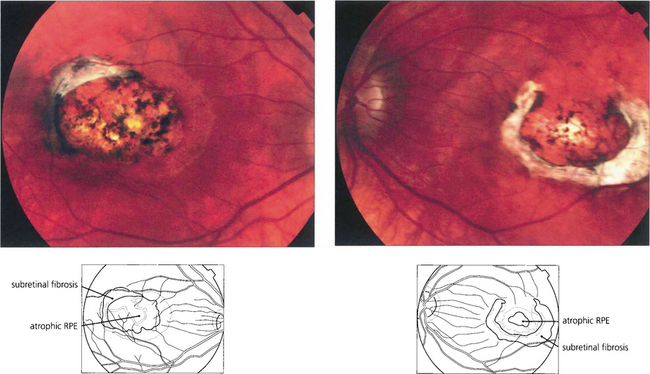
Fig. 16.52 Bilaterally symmetrical fundus appearances in MCDR1 range from a few small (less than 50μm) yellow drusen-like lesions in the central macula (grade 1) to larger confluent lesions (grade 2) and macular colobomatous lesions (grade 3). Occasionally MCDR1 is complicated by SRNVM at the macula.
X-LINKED INHERITANCE
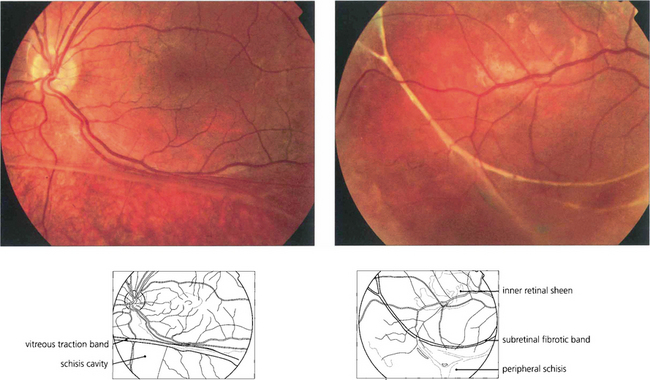
MITOCHONDRIAL INHERITANCE
Maternally inherited diabetes and deafness (MIDD)
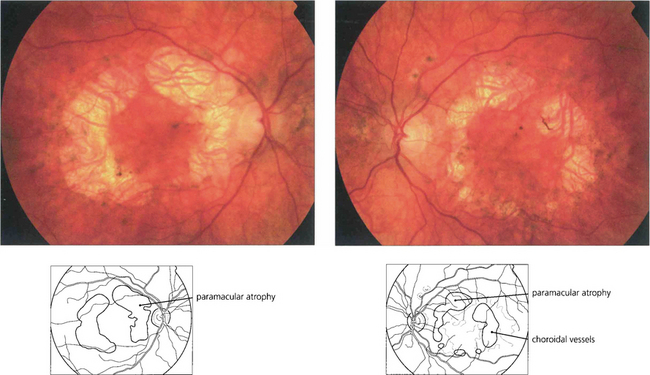
Fig. 16.56 A bilateral macular dystrophy is present in the majority of patients with MIDD characterized by RPE atrophy that can surround the macula or be more extensive and encompass the optic disc. In advanced cases areas RPE atrophy encircling the macula may coalesce and involve the fovea at a late stage.
INHERITED RETINAL DYSTROPHIES WITH GENERALIZED RETINAL INVOLVEMENT
STATIONARY RETINAL DYSTROPHIES
Congenital stationary night-blindness
XL CSNB is further subdivided into complete and incomplete forms. Patients with complete CSNB are invariably myopic and have more pronounced night-blindness. Both complete and incomplete CSNB show a negative type of ERG in that the photoreceptor derived a-wave in the maximal response is normal but there is selective reduction in the inner nuclear derived b-wave so that it is smaller than the a-wave. In complete CSNB there is no detectable rod-specific ERG. Cone ERGs show subtle abnormalities now known to reflect ON pathway dysfunction (see Ch. 1). In contrast there is a detectable rod-specific ERG in incomplete CSNB and cone ERGs are much more abnormal than in complete CSNB, reflecting involvement of both ON and OFF pathways.
Mutations in genes encoding three components of the rod phototransduction cascade have been reported in association with the dominant form of CSNB; namely rhodopsin, the α-subunit of rod transducin and the rod cyclic guanosine monophosphate (cGMP) phosphodiesterase β-subunit. Two genes (CACNA1F and NYX) have now been implicated in XL CSNB. Incomplete CSNB is associated with mutation in CACNA1F, which encodes the retina-specific α1F-subunit of the voltage-gated L-type calcium channel expressed in the outer nuclear layer, inner nuclear layer and ganglion cell layer. The loss of functional channels impairs the calcium flux into rod and cone photoreceptors required to sustain tonic neurotransmitter release from presynaptic terminals. This may result in an inability to maintain the normal transmembrane potential of bipolar cells, such that the retina remains in a partially light-stimulated state, unable to respond to changes in light levels. Complete CSNB is associated with mutation in NYX, the gene encoding the leucine-rich proteoglycan, nyctalopin. It has been suggested that nyctalopin plays a role in the development and function of the ON pathway within the retina.
Fundus albipunctatus
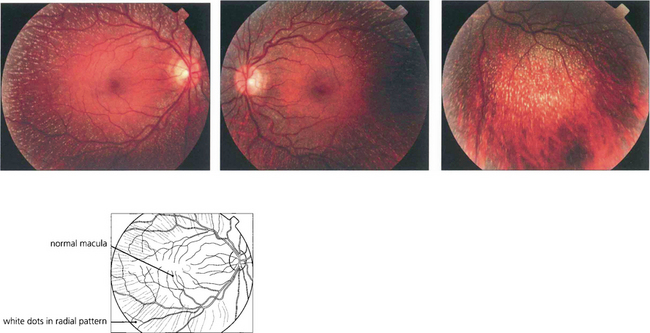
Fig. 16.57 There is a characteristic fundus appearance with multiple white dots scattered throughout the retina at the level of the RPE. The dots are most numerous in the mid-periphery and are usually absent at the macula. Patients present either with night-blindness or because the abnormal retinal appearance is noted on routine fundoscopy.
PROGRESSIVE RETINAL DYSTROPHIES
ROD–CONE DYSTROPHIES
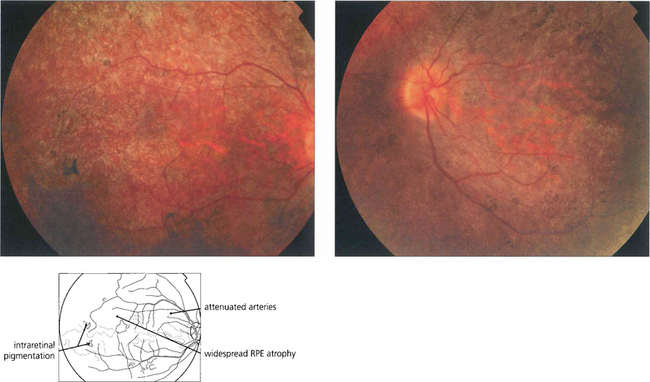
Retinitis pigmentosa
Genetics of RP
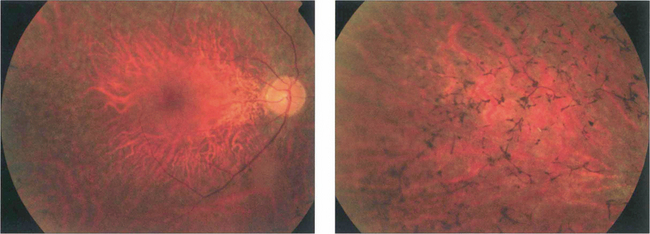
Fig. 16.59 The appearance of the fundus in the early stages of RP is variable but may include mild pigment epithelial atrophy in the mid-periphery often with small white dots at the level of the RPE. Later typical bone spicule pigment deposition is seen in the equatorial retina with arteriolar narrowing and optic disc pallor.
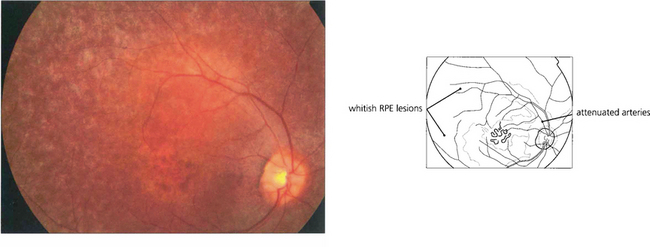
Fig. 16.60 Retinitis punctata albescens, a variant of RP. Fundus photograph showing multiple white deposits scattered throughout the retina with macular atrophy.
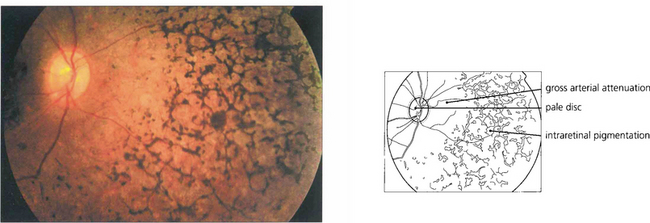
Fig. 16.61 The classical fundus appearance of optic disc pallor, retinal arteriolar attenuation and peripheral retinal pigment epithelial atrophy with ‘bone corpuscle’ pigmentation is seen in advanced disease. The typical intraretinal bone corpuscular pigmentation is produced by RPE migration into the inner retina following photoreceptor loss. Other changes include vitreous cells, posterior subcapsular cataract, optic disc drusen and macular oedema. Occasionally, retinovascular changes similar to those in Coats’ disease are seen. In sector RP, the disease may remain confined to the lower retina and this is reflected in upper field loss. This form of the disease is usually inherited as an AD trait and has an excellent long-term visual prognosis.
X-linked RP
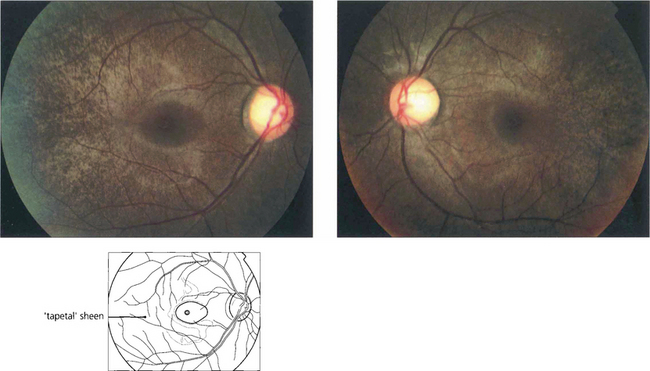
Fig. 16.62 Fundus abnormalities are common in XL heterozygotes. A prominent tapetal reflex may be seen at the posterior pole or mild pigment epithelial thinning and pigmentation may be present in the equatorial retina. The ERG is usually abnormal in heterozygotes with delay in the 30-Hz flicker response probably being the most frequently found abnormality, although reduced rod amplitude may also occur.
Syndromic RP
The mitochondrial cytopathies are an uncommon group of multisystem disorders in which there is biochemical, histopathological or genetic evidence of mitochondrial dysfunction. Clinical abnormalities often begin in childhood and may include lactic acidosis, anaemia, myopathy, neurological abnormalities, endocrine disturbance, renal disease, neurosensory hearing loss and an RP-like dystrophy. A number of syndromes have been recognized including Kearns–Sayre syndrome or chronic progressive external ophthalmoplegia (see Ch. 2).
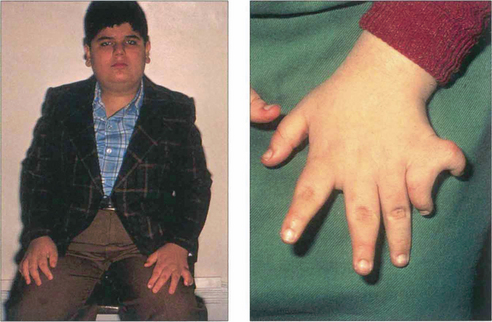
Fig. 16.63 The Bardet–Biedl syndrome (BBS) is an autosomal recessive disorder characterized by obesity, intellectual impairment, polydactyly, hypogonadism and a progressive RP-like dystrophy. Renal failure, due to ureteric reflux, and hypertension are common and are the main causes of death. Early macular involvement is common; a ‘bull’s eye’ maculopathy may be seen. To date, seven loci and five genes have been identified. The Laurence–Moon syndrome is a closely related rare autosomal recessive disorder in which there is no obesity or polydactyly but affected patients have a short stature, hypogonadism, intellectual impairment, ataxia, spinal paraparesis and RP-like dystrophy.
CONE AND CONE–ROD DYSTROPHIES
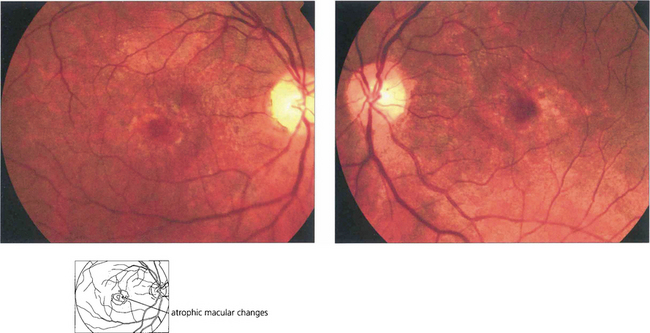
Fig. 16.64 In cone dystrophies fundus examination may show a typical ‘bull’s eye’ maculopathy with annular RPE atrophy and central sparing.
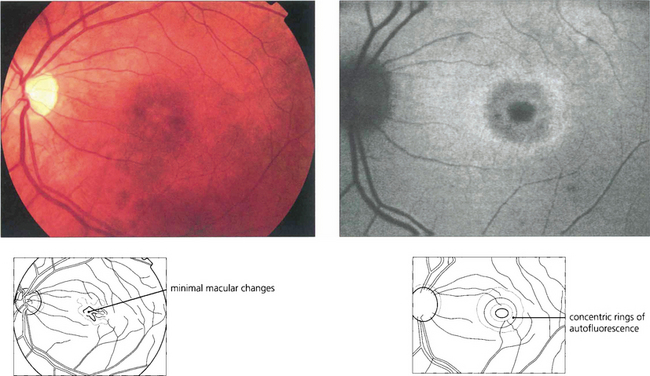
Fig. 16.65 In some cases there may only be minor macular RPE atrophy. The optic discs show a variable degree of temporal pallor and these patients can be misdiagnosed as having optic nerve disease. In this patient autofluorescence imaging shows RPE changes more clearly. The retinal periphery is usually normal in pure cone dystrophies although rarely white flecks similar to those seen in fundus flavimaculatus may be seen.
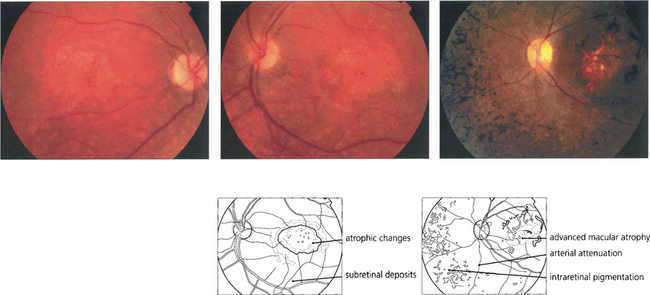
Fig. 16.66 In patients with CORD fundus examination shows macular atrophy or a bull’s eye maculopathy in the early stages (left, middle). Peripheral RPE atrophy, retinal pigmentation, arteriolar attenuation and optic disc pallor are often seen in the late stages of the disease process and can resemble advanced RP (right).
INHERITED CHORIORETINAL DYSTROPHIES
CHOROIDERAEMIA
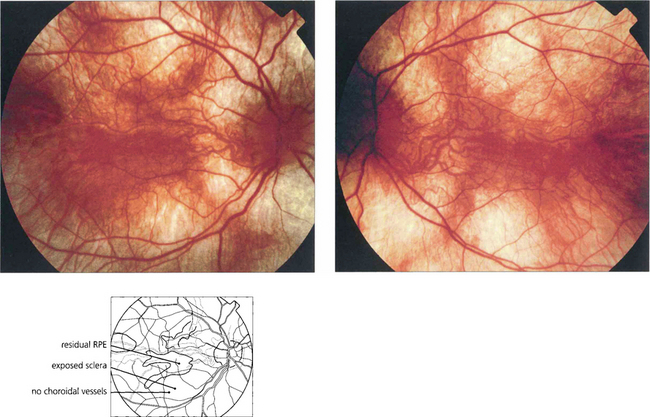
Fig. 16.67 Focal areas of atrophy of the RPE and choriocapillaris develop as the disease progresses. These areas coalesce to a widespread atrophic appearance throughout the equatorial retina. This later spreads to involve the peripheral and more posterior retina; the macula is spared until late in the disease. The ERG is markedly abnormal at an early stage and is usually undetectable in adult life.
GYRATE ATROPHY
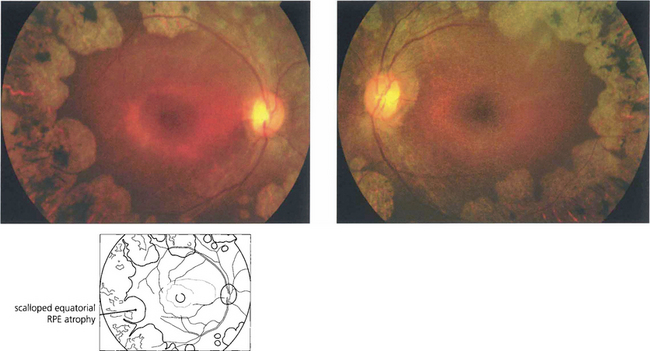
Fig. 16.69 The earliest fundus changes are seen as small discrete areas of choroidal and RPE atrophy in the mid and far peripheral fundus. The atrophic areas subsequently coalesce and enlarge towards the posterior pole, with a characteristic scalloped appearance at the leading edge.
By courtesy of Professor Alan Bird.
NEURODEGENERATIVE DISORDERS
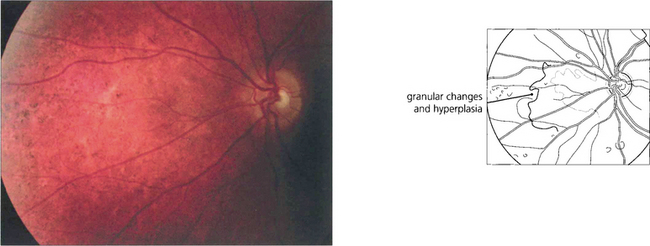
BATTEN DISEASE
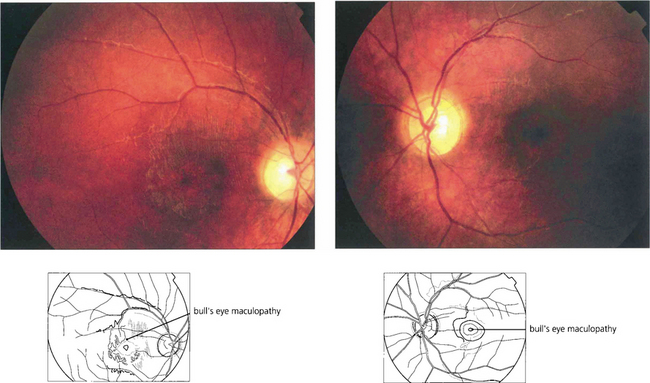
Fig. 16.70 Fundoscopy usually initially shows macular atrophy with later arteriolar attenuation and peripheral retinal atrophy and pigmentation. The ERG is substantially abnormal at an early stage. This diagnosis should be excluded in children aged between 5 and 8 years who present with visual loss and show evidence of macular atrophy and an abnormal full-field ERG by looking for the presence of vacuolated lymphocytes in the peripheral blood. The diagnosis can be confirmed by molecular genetic testing.
DRUGS CAUSING RETINAL TOXICITY
ANTIMALARIALS
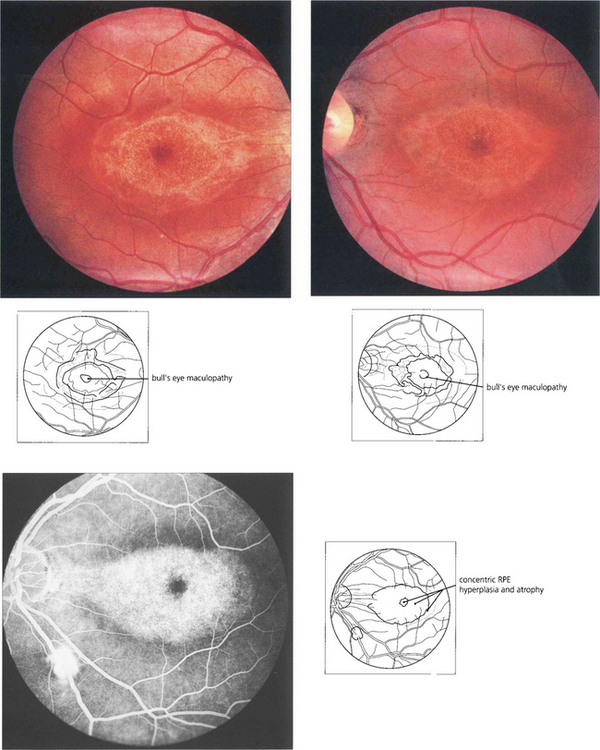
Chloroquine and hydroxychloroquine are used in malarial prophylaxis and treatment and as disease-modifying drugs in connective tissue disorders such as systemic lupus erythematosus (SLE). Retinal toxicity can occur with both drugs but hydroxychloroquine is much less toxic and for this reason has now replaced chloroquine in rheumatic therapy. Although the drugs bind to melanin, their action appears to be related to their effect on protein synthesis. Toxicity is related to daily dosage in milligrams per kilogram and to duration of treatment. Patients can take chloroquine for many years for malaria prophylaxis without retinal toxicity provided the dosage regimen is not exceeded. Patients with SLE can take hydroxychlorqine in dosages of 6.5–7 mg/day (200–400 mg/day) for up to 10 years without problem provided they have normal renal and liver function. The first signs of toxicity are perifoveal granularity at the level of the RPE and loss of the foveal reflex; continued use may lead to a bull’s eye maculopathy and eventually to a retinitis pigmentosa-like appearance. Initial symptoms include decreased visual acuity and paracentral scotomata. Once toxicity is established, progressive retinal damage may continue despite ceasing therapy. Corneal epithelial whorls are seen with chloroquine but are of no visual significance (see Ch. 6).
THIORIDAZINE (MELLERIL)
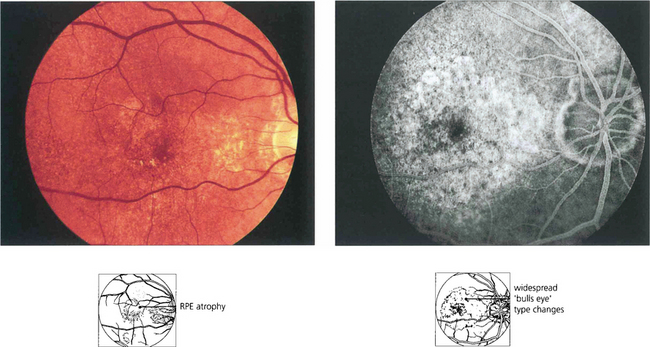
Fig. 16.73 Phenothiazines bind to melanin and concentrate in the pigment of the uveal tract and RPE. Thioridazine (Melleril) is used as a psychotrophic drug in schizophrenia. It will produce a pigmentary retinopathy in dosages in excess of 600 mg/day for over 8 weeks’ duration. Early toxicity is associated with fine granular RPE changes that become more pronounced with progression. Later, hyperpigmentary changes resembling a salt-and-pepper retinopathy may develop as may chorioretinal atrophy. Some recovery follows drug withdrawal although in some cases retinal changes will progress.
TAMOXIFEN
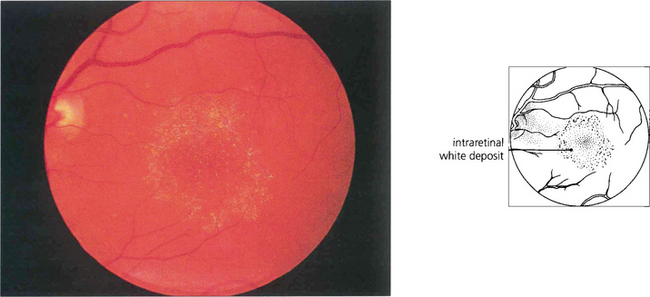
Fig. 16.74 Tamoxifen is an oestrogen receptor antagonist widely used in the treatment of breast carcinoma. Retinopathy is very rare at normal dosages but deposition of a metabolite in the inner retina is occasionally seen in patients who have taken high dosages. Cystoid macular oedema is the main cause of visual loss.
QUININE
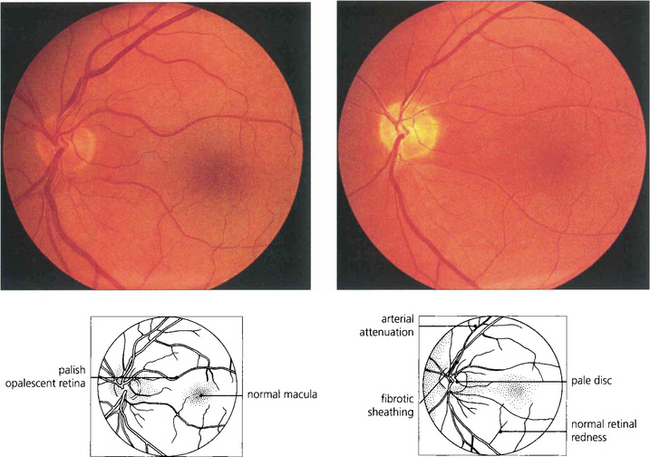
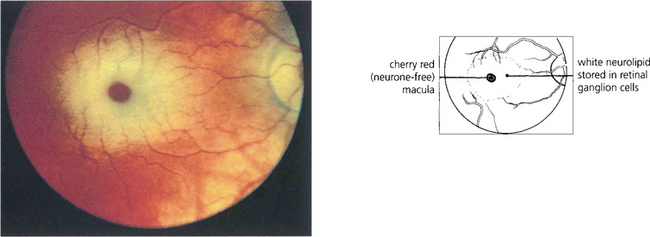
Fig. 16.75 Quinine is toxic to the neuroretina. With acute poisoning it produces a similar retinal appearance to central retina artery occlusion, with cloudy intracellular oedema of the neuronal layer and a cherry-red spot. This is due to neuronal toxicity and swelling rather than vascular infarction. Acute poisoning can be treated by extracorporal haemoperfusion to remove the drug from the blood. The changes resolve over some weeks, leaving a pale disc and marked arterial attenuation. This patient took quinine as an attempted suicide. He had a final acuity of 20/20 with grossly constricted visual fields and night-blindness.