[level-membership-for-cardiovascular-category]
7 Restenosis and Drug-Eluting Stents
Restenosis
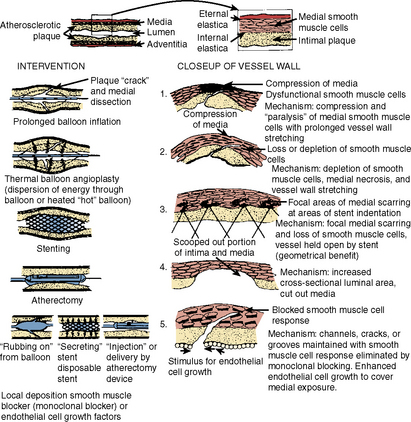
The second component of restenosis, recoil and negative remodeling of the arterial wall is inhibited by stents. Compared to balloon angioplasty, stents have reduced restenosis from 40% to 50% after percutaneous transluminal coronary angioplasty (PTCA) to 20% after bare-metal stenting. Drug-eluting stents (DESs) have brought restenosis rates to <10% in most patient subgroups. Restenosis still occurs inside stents (called in-stent restenosis [ISR]) mostly, if not exclusively, as a result of endothelial cell proliferation. Rarely does vascular recoil make a contribution, but it must be considered in treating the ISR lesion. Restenosis is not device-specific but rather a function of the anatomic substrate and the type of injury produced (Fig. 7-1).
Definitions of Restenosis
Angiographic Restenosis
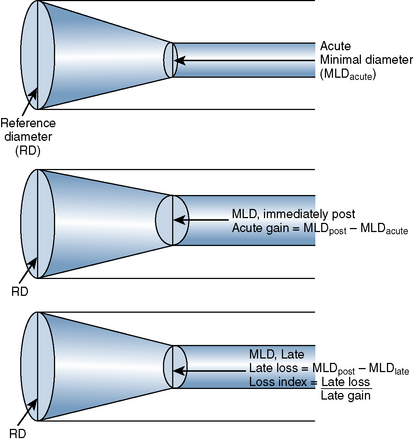
The late loss index is the loss at the lesion site divided by the amount of acute gain (Fig. 7-2). The loss index is accepted as the most sensitive measure of the effectiveness of the technique and should range from 0.4 to 0.6 mm for balloon angioplasty. The lower the loss index is, the more effective the antirestenosis treatment will be.
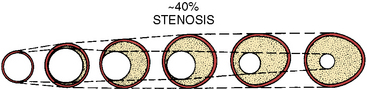
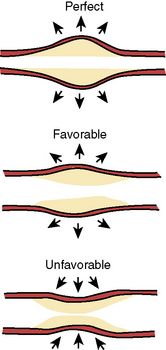
Intravascular ultrasound (IVUS) imaging is superior to angiography for anatomic and morphologic restenosis definitions. Recent IVUS studies have shown that an important component of restenosis is vessel recoil, a feature prevented by stenting. Normal vessel modeling maintains the coronary lumen. Late negative remodeling of the injured vessel is also prevented by stenting (Figs. 7-3 and 7-4).
Time Course
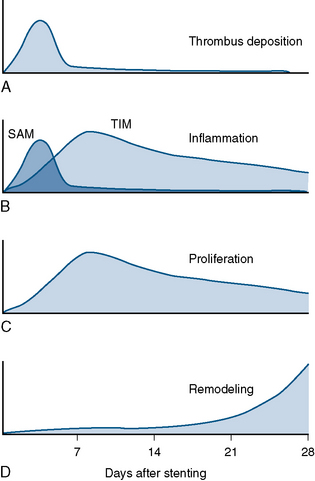
Different mechanisms produce restenosis in a time-dependent manner. Early restenosis is due to thrombus, whereas late restenosis is related more to remodeling (Fig. 7-5).
Patient Subsets at Higher Risk of Restenosis
In-Stent Restenosis
There are two major categories of in-stent restenosis—focal and diffuse (Fig. 7-6)—and within each category several subtypes of responses relative to the proliferation within and/or adjacent to the stent are noted. Figure 7-7 suggests a scheme for the treatment of ISR. Note that brachytherapy is now rarely used.
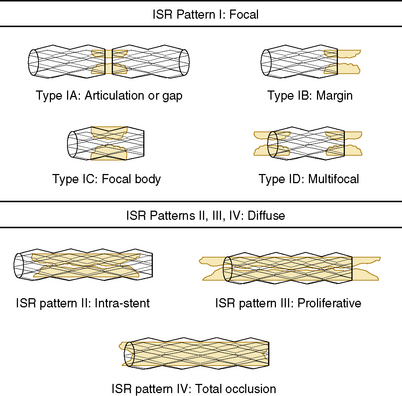
Figure 7-6 Classification system proposed for in-stent restenosis (ISR).
(From Mehran R, Dangas G, Abizaid AS, et al. Angiographic patterns of in-stent restenosis classification and implications for long-term outcome. Circulation 1999;100:1872–1878.)
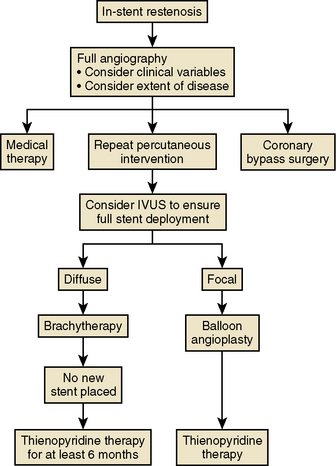
Figure 7-7 Algorithm for treatment of in-stent restenosis.
(Modified from Topol EJ. Textbook of interventional cardiology, 4th ed. Philadelphia: Saunders, 2003: 468.)
Stent fracture is associated with in-stent restenosis in Drug-Eluting Stents.
Brachytherapy
The adverse effects of brachytherapy included the following:
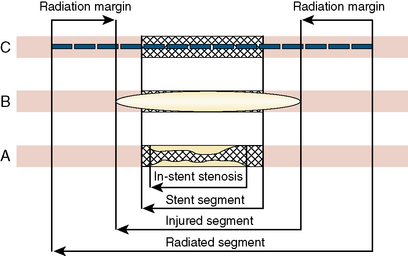
Of all the adverse effects, late thrombosis occurring more than 30 days after radiation therapy was one of the most feared major complications of vascular brachytherapy. Late thrombosis in early clinical trials was reported in up to 14% of patients. Late thrombosis also occurs with other vascular brachytherapy and relates to the healing arrest and lack of stent re-endothelialization. An effective strategy to prevent late rethrombosis is limiting restenting at the time of radiation treatment. It is essential to administer at least 12 months of antiplatelet therapy, preferably clopidogrel in addition to aspirin, for all radiation cases, both beta and gamma emitters. Definitions of artery and stent segments address by brachytherapy are shown on Figure 7-8.
Drug-Eluting Stents
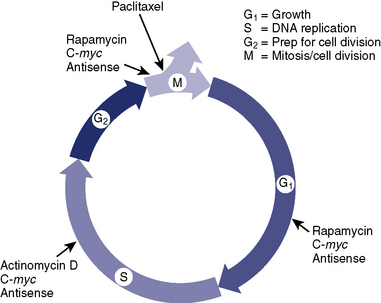
Current DESs available in the United States include drug compounds that contain antiproliferative agents such as paclitaxel, sirolimus, everolimus, or zotarolimus (Fig. 7-9).

Drugs for Coated Stents (Fig. 7-10)
Azar R.R., Prpic R., Ho K.K., et al. Impact of end-stage renal disease on clinical and angiographic outcomes after coronary stenting. Am J Cardiol. 2000;86(5):485–489.
Colombo A., Drzewiecki J., Banning A. Randomized study to assess the effectiveness of slow- and moderate-release polymer-based paclitaxel-eluting stents for coronary artery lesions. Circulation. 2003;108(7):788–794.
Cypher Family of Stents Instructions for Use. Cordis Corporation, a Johnson & Johnson Company, September 2009.
Dangas G., Claessen B.E., Caixeta A., et al. In-stent restenosis in the drug-eluting stent era. J Am Coll Cardiol. 2010;56:1897–1907.
Endeavor Zotarolimus-Eluting Coronary Stent System Instructions for Use. Santa Rosa, CA: Medtronic Vascular, 2009.
Fajadet J., Wijns W., Laarman G.J., et al. Randomized, double-blind, multicenter study of the Endeavor zotarolimus-eluting phosphorylcholine-encapsulated stent for the treatment of native coronary artery lesions: clinical and angiographic results of the ENDEAVOR II Trial. Circulation. 2006;114:798–806.
Garg S., Serruys P.W. Coronary stents: current status. J Am Coll Cardiol. 2010;56:S1–S42.
Garg S., Serruys P.W. Coronary stents: looking forward. J Am Coll Cardiol. 2010;56:S43–S78.
Gupta A., Mancini D., Kirtane A.J., et al. Value of drug-eluting stents in cardiac transplant recipients. Am J Cardiol. 2009;103(5):659–662.
Holmes D.R.Jr, Kereiakes D., Garg S., et al. Stent thrombosis. J Am Coll Cardiol. 2010;56:1357–1365.
Kandzari D., Leon M., Popma J., et al. Comparison of zotarolimus-eluting and sirolimus-eluting stents in patients with native coronary artery disease: a randomized controlled trial. JACC Cardiovasc Interv. 2006 De;48(12):2440–2447.
Kandzari D.E., Rao S.V., Moses J.W., et al. Clinical and angiographic outcomes with sirolimus-eluting stents in total coronary occlusions: the ACROSS/TOSCA-4 trial. JACC Cardiovasc Interv. 2009;2(2):97–106.
Kastrati A., Pache J., Dirschinger J., et al. Primary intracoronary stenting in acute myocardial infarction: long-term clinical and angiographic follow-up and risk factor analysis. Am Heart J. 2000;139:208–216.
Keeley E.C., Boura J.A., Grines C.L. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomized trials. Lancet. 2003;361:13–20.
Khot U.N., Friedman D.T., Pettersson G., et al. Radial artery bypass grafts have an increased occurrence of angiographically severe stenosis and occlusion compared with left LIMA and SVGs. Circulation. 2004;109:2086–2089.
Lee M.S., Jurewitz D., Aragon J., et al. Stent fracture associated with drug-eluting stents: clinical characteristics and implications. Catheter Cardiovasc Interv. 2007;69:387–394.
Lee M.S., Kobashigawa J., Tobis J. Comparison of percutaneous coronary intervention with bare-metal and drug-eluting stents for cardiac allograft vasculopathy. JACC Cardiovasc Interv. 2008;1:710–715.
Lee T.T., Feinberg L., Baim D.S., et al. Effect of diabetes mellitus on five-year clinical outcomes after single-vessel coronary stenting. Am J Cardiol. 2006;98(6):718–721.
Mehran R., Dangas G., Abizaid A., et al. Treatment of focal in-stent restenosis with balloon angioplasty alone versus stenting: short- and long-term results. Am Heart J. 2001;141:610–614.
Morice M.C., Serruys P.W., Sousa J.E., et al. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346:1773–1780.
Moses J.W., Leon M.B., Popma J.J., et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N Engl J Med. 2003;34:1315–1323.
Ormiston J.A., Serruys P.W., Regar E., et al. A bioabsorbable everolimus-eluting coronary stent system for patients with single de-novo coronary artery lesions (ABSORB): a prospective open-label trial. Lancet. 2008;371(9616):899–907.
Sabik J.F.III, Lytle B.W., Blackstone E.H., et al. Comparison of saphenous vein and internal thoracic artery graft patency by coronary system. Ann Thorac Surg. 2005;79:544–551.
Sadeghi H.M., Stone G.W., Grines C.L., et al. Impact of renal insufficiency in patients undergoing primary angioplasty for acute myocardial infarction. Circulation. 2003;108(22):2769–2775.
Serruys P.W., Ormiston J.A., Onuma Y., et al. A bioabsorbable everolimus-eluting coronary stent system (ABSORB): 2-year outcomes and results from multiple imaging methods. Lancet. 2009;373(9667):897–910.
Sousa J.E., Costa M.A., Abizaid A.C., et al. Sustained suppression of neointimal proliferation by sirolimus-eluting stents: one-year angiographic and intravascular ultrasound follow-up. Circulation. 2001;104:2007–2011.
Stone G.W., Ellis S.G., Cox D.A. A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease. N Engl J Med. 2004 Jan 15;350(3):221–231.
Stone G.W., Midei M., Newman W., et al. Comparison of an everolimus-eluting stent and a paclitaxel-eluting stent in patients with coronary artery disease: a randomized trial. JAMA. 2008;299(16):1903–1913.
Stone G.W., Midei M., Newman W., et al. Randomized comparison of everolimus-eluting and paclitaxel-eluting stents: Two-year clinical follow-up from the clinical evaluation of the Xience V Everolimus Eluting Coronary Stent System in the Treatment of Patients with de novo Native Coronary Artery Lesions (SPIRIT) III trial. Circulation. 2009;119:680–686.
Taxus Liberté Paclitaxel-eluting Coronary Stent System Instructions for Use. Natick, MA: Boston Scientific Corporation, August 2009.
West N.E., Ruygrok P.N., Disco C.M., et al. Clinical and angiographic predictors of restenosis after stent deployment in diabetic patients. Circulation. 2004;109(7):867–873.
Xience Everolimus Coronary Stent System Instructions for Use. Santa Clara, CA: Abbott Vascular; July 2008.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
7 Restenosis and Drug-Eluting Stents
Restenosis
The second component of restenosis, recoil and negative remodeling of the arterial wall is inhibited by stents. Compared to balloon angioplasty, stents have reduced restenosis from 40% to 50% after percutaneous transluminal coronary angioplasty (PTCA) to 20% after bare-metal stenting. Drug-eluting stents (DESs) have brought restenosis rates to <10% in most patient subgroups. Restenosis still occurs inside stents (called in-stent restenosis [ISR]) mostly, if not exclusively, as a result of endothelial cell proliferation. Rarely does vascular recoil make a contribution, but it must be considered in treating the ISR lesion. Restenosis is not device-specific but rather a function of the anatomic substrate and the type of injury produced (Fig. 7-1).
Definitions of Restenosis
Angiographic Restenosis
The late loss index is the loss at the lesion site divided by the amount of acute gain (Fig. 7-2). The loss index is accepted as the most sensitive measure of the effectiveness of the technique and should range from 0.4 to 0.6 mm for balloon angioplasty. The lower the loss index is, the more effective the antirestenosis treatment will be.
Intravascular ultrasound (IVUS) imaging is superior to angiography for anatomic and morphologic restenosis definitions. Recent IVUS studies have shown that an important component of restenosis is vessel recoil, a feature prevented by stenting. Normal vessel modeling maintains the coronary lumen. Late negative remodeling of the injured vessel is also prevented by stenting (Figs. 7-3 and 7-4).
Time Course
Different mechanisms produce restenosis in a time-dependent manner. Early restenosis is due to thrombus, whereas late restenosis is related more to remodeling (Fig. 7-5).
