[level-membership-for-pathology-category]
Respiratory System
Tumors in the Lungs and the Pleura
TABLE 3-1
CONDITIONS THAT CAN CAUSE ARDS/DAD*
| Conditions | Causes |
| Infectious diseases | Septicemia with DAD and DIC (especially gram negative), diffuse pneumonitis by virus, mycoplasma, pneumocystis, tuberculosis (certain forms, e.g., typhobacillosis Landouzy) |
| Chemical injury and inhalants | Oxygen, irritant gases and inhaled chemicals, barbiturate overdose, salicylic acid, paraquat, heroin or methadone overdose, cytotoxic drugs, uremic pneumonitis, gastric aspiration |
| Physical injury | Trauma to lungs (contusion), head injury, fat embolism of various causes, air embolism, burns, ionizing radiation |
| Other | Shock of any cause, acute pancreatitis, near drowning aspiration |
TABLE 3-2
CLASSIFICATION OF IDIOPATHIC PULMONARY FIBROSIS*
| Feature | NSIP | UIP | DIP | AIP | LIP | COP |
| Interstitial inflammation | Prominent | Scant | Scant | Scant | Prominent | Scant |
| Interstitial fibrosis | ||||||
| Collagen | Variable, diffuse | Patchy | Variable, diffuse | No | Some areas | No |
| Fibroblast foci | Occasional | No | No | Yes, diffuse | No | No |
| BOOP | Occasional, focal | Occasional, focal | No | Occasional, focal | No | Prominent |
| Intraalveolar macrophages | Occasional, patchy | Occasional, patchy | Yes, diffuse | No | Patchy | No |
| Hyaline membranes | No | No | No | Yes, focal | No | No |
| Honeycombing | Rare | Yes | No | No | Sometimes | No |
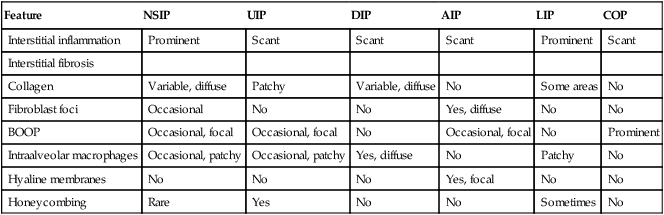
*AIP indicates acute interstitial pneumonitis; BOOP, bronchiolitis obliterans organizing pneumonitis; DIP, desquamative interstitial pneumonitis; NSIP, nonspecific interstitial pneumonitis; UIP, usual interstitial pneumonitis, LIP lymphocytic interstitial pneumonitis, COP cryptogenic organizing pneumonitis.
From Leslie KO, Wick MR. Practical Pulmonary Pathology. Philadelphia, 2005, Churchill Livingstone.
TABLE 3-3
CLINICAL AND PATHOLOGIC FEATURES OF PNEUMOCONIOSES*
| Entity | Clinical Appearance | Pathologic Changes |
| Coal miner’s lung | Black lung disease | Diffusely distributed, small focal anthracosilicosis, initially centriacinar and peribronchiolar with many carbon-laden macrophages and perifocal emphysema; extent of fibrosis depends on admixture of quartz |
| Silicosis | Acute silicosis (uncommon) | Alveolar lipoproteinosis and progressive diffuse interstitial fibrosis secondary to inhalation of small particulate silica crystals (e.g., after sand blasting) |
| Nodular silicosis (common) | Multiple growing silicotic nodules, usually 2 mm to 1 cm in diameter: fibrosing granulomas with concentric fibrous layering, some anthracotic pigment, small slitlike spaces, and needle-shaped crystalline spicules on polarization; perifocal emphysema | |
| Progressive massive silicosis | Multiple silicotic granulomas up to 10 cm in diameter, both lungs involved, massive and rapidly progressive fibrosis | |
| Asbestosis and asbestosrelated diseases | Asbestosis per se | Alveolitis with progressive interstitial fibrosis, deposition of asbestos bodies (golden-brown beaded rods consisting of asbestos fibers coated by ferroproteinaceous material); final stage: honeycombing lung |
| Pleural plaques and rounded atelectasis | Recurrent pleural fibrinous effusions, pleural fibrosis and pleural plaques (“sugar coating”), focal atelectasis secondary to pleural fibrosis | |
| Neoplasms | Malignant mesothelioma (↑ risk of bronchogenic carcinoma) | |
| Berylliosis | Berylliosis per se | Acute and recurrent pneumonitis, systemic sarcoidlike and fibrosing granulomas |
| Talcosis | Talcosis per se | Foreign body granulomas with birefringent talcum deposits, micronodular and diffuse interstitial fibrosis |
TABLE 3-4
INFECTIOUS AGENTS CAUSING PNEUMONIA
| Class | Etiologic Agent | Type of Pneumonia |
| Bacteria | Streptococcus pneumoniae | |
| Streptococcus pyogenes | ||
| Staphylococcus aureus | ||
| Klebsiella pneumoniae | ||
| Pseudomonas aeruginosa | ||
| Escherichia coli | ||
| Yersinia pestis | ||
| Legionella pneumophila | Legionnaires disease | |
| Peptostreptococcus, Peptococcus | Aspiration (anaerobic) pneumonia | |
| Bacteroides | ||
| Fusobacterium | ||
| Veillonella | Bacterial pneumonias | |
| Actinomycetes | Actinomyces israelii | Pulmonary nocardiosis |
| Nocardia asteroides | Pulmonary actinomycosis | |
| Fungi | Coccidioides immitis | Coccidioidomycosis |
| Histoplasma capsulatum | Histoplasmosis | |
| Blastomyces dermatitidis | Blastomycosis | |
| Aspergillus | Aspergillosis | |
| Phycomycetes | Mucormycosis | |
| Rickettsia | Coxiella burnetii | Q fever |
| Chlamydia | Chlamydia psittaci | Psittacosis |
| Ornithosis | ||
| Mycoplasma | Mycoplasma pneumoniae | Mycoplasmal pneumonia |
| Viruses | Influenza virus, adenovirus, respiratory syncytial virus, etc. | Viral pneumonia |
| Protozoa | Pneumocystis carinii | Pneumocystis pneumonia (plasma cell pneumonia) |
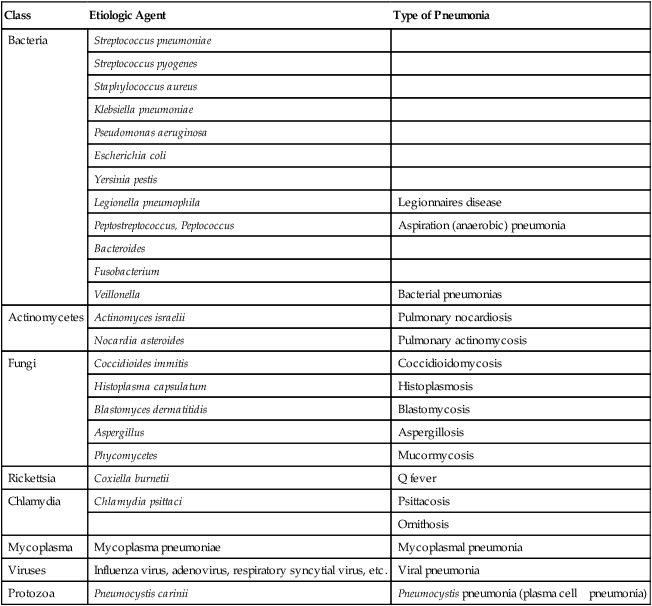
Infection of the lungs cause pneumonia (also “pneumonitis”). Following the rules of general inflammation (Chapter 1: Figures 1-5 to 1-8), pulmonary inflammation presents itself as alveolar pneumonia (common bacterial), interstitial lymphocytic pneumonitis (viral, immunological), granulomatous pneumonitis (Tb, allergic) or mixtures of the latter (certain viruses, protozoal, immunological). Table 3-4 summarizes common infectious agents causing pneumonia. Figures 3-14, 3-15, 3-16, and 3-17 present representative examples. Figures 3-5, 3-7, and 3-8 show examples of immunologic forms of pneumonitis. The prognosis of pneumonia depends upon the type of inflammatory reaction in the lungs: acute alveolar pneumonia (serofibrinous, neutrophilic) or pure interstitial lymphocytic pneumonitis (common cold virus) may resolve with complete recovery. Structural damage (e.g., abscess formation) or chronic infiltrative diseases (e.g., tuberculosis) always results in scarring, the extent of which will determine the persistence of clinical symptoms.
TABLE 3-5
PULMONARY FUNGAL INFECTIONS IN IMMUNOCOMPROMISED PATIENTS
| Organism | Pathologic Lesion |
| Candida albicans tropicalis | Participation of the lungs in systemic candidiasis: |

TABLE 3-6
FORMS AND FEATURES OF PULMONARY TUBERCULOSIS*
| Stage | Immune Reactivity | Clinicopathologic Features |
| Stage I (primary) | ||
| a. Initial infection | No immunity | Clinically inapparent nonspecific alveolitis |
| b. Initial infection | Developing immunity | Ghon’s primary affection: isolated granulomatous reaction, most commonly in right upper lobe |
| c. Primary lymphatic spread | Developing immunity | Lymphatic spread of infection to regional lymph nodes with respective granulomas: Ranke’s primary complex |
| Stage II (early postprimary generalization) | ||
| a. Lymphatic spread | Good | Isolated subpleural caseous granuloma, upper segments of right upper lobe |
| b. Bronchogenic spread | Intermediate | Acinar-nodular pulmonary tuberculosis with progressive caseous granulomas |
| Poor | Progressive caseous pneumonia without prominent granulomatous reaction | |
| c. Hematogenous spread | Intermediate | Systemic hematogenous caseous granulomas of different sizes (i.e., different ages) |
| (late postprimary generalization) | ||
| d. Hematogenous spread | Nonimmune → reactive | Miliary tuberculosis with systemic granulomas of uniform size (and age) |
| e. Hematogenous spread (also in early postprimary spread) | A-reactive | Miliary systemic necroses, tuberculosepsis acutissima, typhobacillosis Landouzy |
| Stage III (isolated organ tuberculosis) | High | Limited spread in isolated organs: cavernous pulmonary of renal tuberculosis, isolated tuberculomas (granulomas) in brain, spine, and other organs |
| Late generalization | High → low | Local or systemic spread of isolated organ tuberculosis (lymphatic, bronchogenic, or hematogenous) |
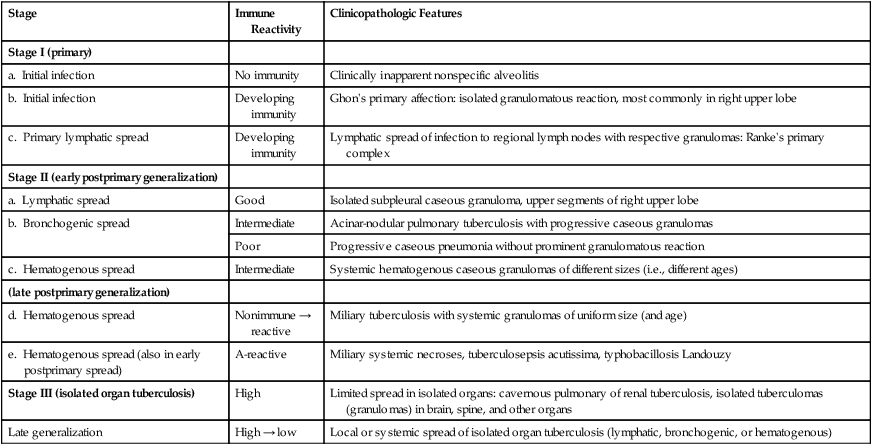
*The degree of immune reactivity (not resistance to disease) can be monitored by tuberculin skin testing; toxicity of tubercle bacteria is partly determined by “cord factor.”
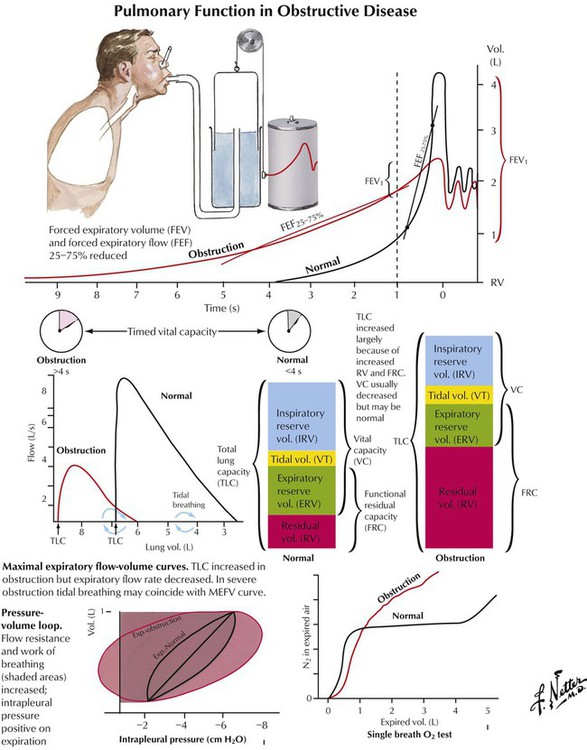
Chronic obstructive pulmonary disease (COPD) is characterized by reduced pulmonary airflow with normal or increased TLC and FVC combined with decreased FEV as determined by spirometric function tests. COPD follows increased resistance to airflow (by luminal narrowing of air ducts) or loss of elastic recoil (and by passive widening of air spaces). COPD is caused by a number of respiratory diseases, including chronic bronchitis, bronchiolitis and asthma, CF, bronchiectasis, and α1-antitrypsin deficiency. COPD results in a progressive and destructive emphysema and reduced intrapulmonary blood flow, pulmonary hypertension, and right heart insufficiency (cor pulmonale).
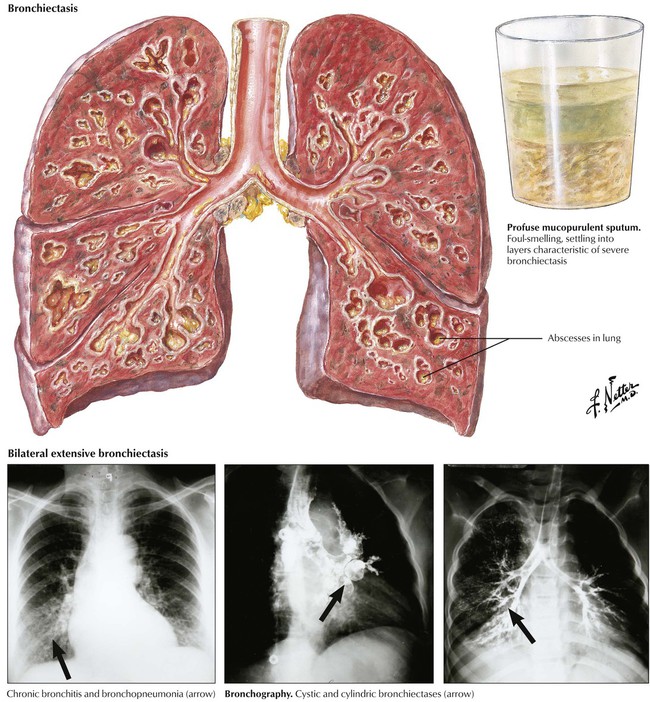
Chronic bronchitis with persistent and productive cough afflicts up to 25% of persons older than 40 years in smog-ridden cities and up to 15% in smokers. It is accompanied by repeated nonspecific infections, mucosal atrophy with mucoid metaplasia (1 goblet cell per 7 columnar cells changes to 1 goblet cell per 1 columnar cell), reduced dust clearance, and inflammatory destruction of elastic lamellae in the bronchial wall with reactive muscular hypertrophy. Finally, there is degeneration and cylindric bronchiectases, transient fibrotic narrowing of bronchioles (small airway disease), and emphysema.
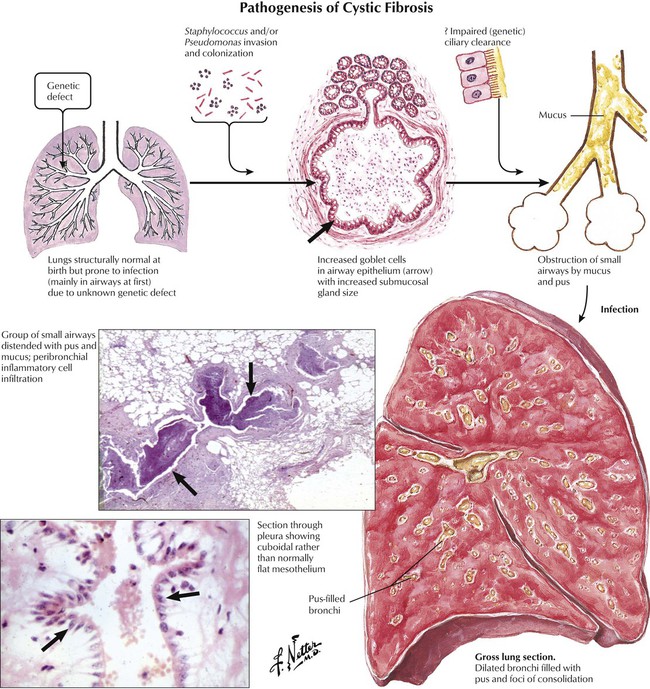
In cystic fibrosis (CF) (mucoviscidosis), a primary defect in chloride ion transport across epithelia results in the secretion of abnormally viscid mucus in all secretory glands, including the bronchial glands. Mucus inspissation blocks the airways, causing bronchiectasis and emphysema. Chronic abscess formation, which results from recurrent superinfections (Staphylococcus and Pseudomonas species) can complicate the course of CF. Although CF affects other organs, COPD is the cause of death in 80% to 90% of cases.
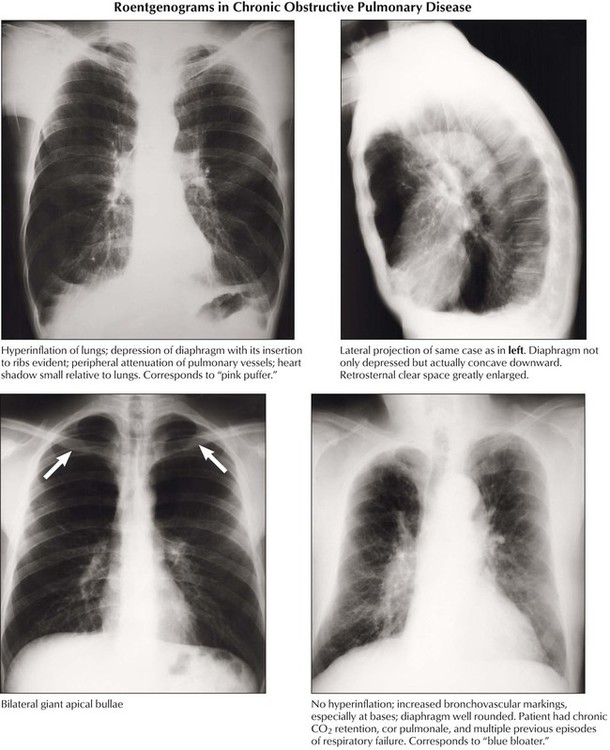
In emphysema, overinflation of the alveoli located distal to the terminal bronchioles is caused by the destruction of alveolar walls. The pathogenesis is thought to be an imbalance between increased (inflammatory) elastolysis and decreased antiprotease activity (e.g., in α1-antitrypsin deficiency). Emphysema is classified by anatomical nature and location in the lobule: centrilobular (centriacinar) emphysema affects the upper part of lungs after inhalation of toxic materials; panlobular (panacinar) emphysema is commonly found in the lower part of lungs, such as in α1-antitrypsin deficiency; paraseptal (distal acinar) emphysema frequently occurs subpleurally, adjacent to fibrosis; bullous emphysema results from enhanced focal destruction of air space walls with confluence of multiple air spaces. Interstitial emphysema expands in interstitial septae after acute overinflation of the lungs with rupture and perforation of air spaces into fibrous septae. It may spread to the mediastinum and subcutaneous tissues of the neck.
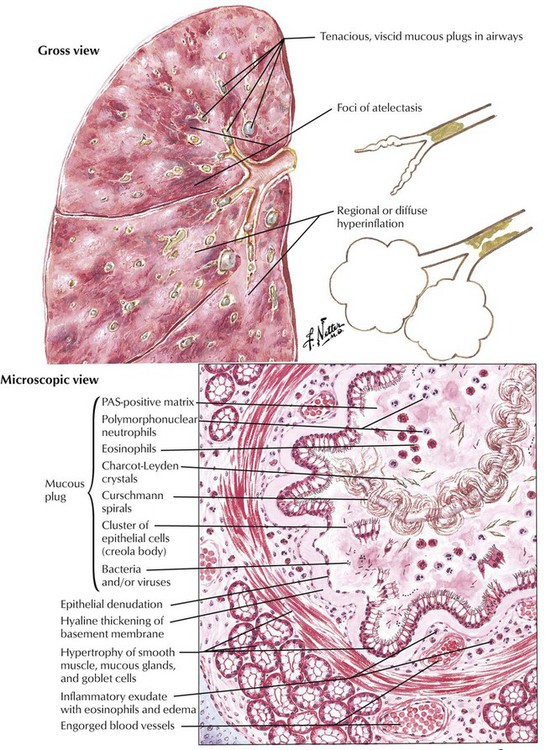
Asthma bronchiale is caused by an enhanced bronchoconstrictor response to type I (allergic) immune reaction to extrinsic or intrinsic stimuli. The etiology and pathogenesis is multifactorial and includes genetic conditions, psychologic stress, and allergic and infectious stimuli. Severe coughing with expectoration of a characteristic mucoid sputum with masses of eosinophils and their breakdown products (Charcot-Leyden crystals), gyrate mucus clumps (Curschmann spirals), and clusters of epithelial cells (Creola bodies) follows the acute phase of the attack. Histologically, asthmatic bronchitis appears as a mucoid metaplasia of bronchial epithelium, eosinophilic infiltration, hyaline thickening of the basement membrane, and muscular and glandular hypertrophy. Bronchial lumina are often occluded by mucous plugs. Status asthmaticus is a severe persistent bronchoconstriction that does not respond to treatment. It leads to severe hypoxia, acidosis, and hypercapnia and may be fatal.
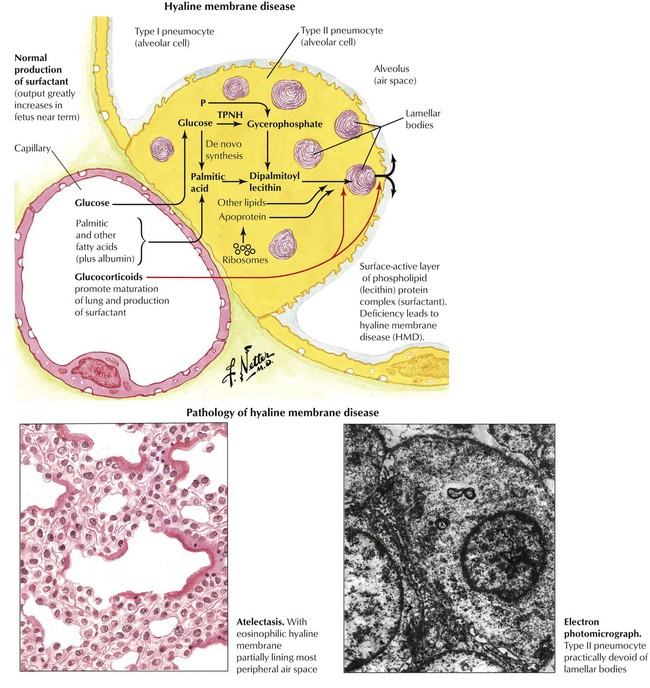
Adult respiratory distress syndrome is defined by reduced arterial oxygenation, decreased lung compliance, and diffuse noncardiogenic pulmonary infiltrates. The morphology is represented by diffuse alveolar damage (DAD). Alveolar and interstitial edema develops subsequent to diffuse alveolar epithelial and vascular endothelial injury with capillary congestion. Alveolar cell necrosis may occur with focal hemorrhage and capillary microthrombosis. The formation of hyaline membranes composed of plasma proteins, cellular debris, and fibrin precipitates is characteristic in ARDS. The decrease in functioning surfactant factor leads to a loss of type I alveolar cells and a reactive proliferation of type II alveolar cells. Progressive fibroblast proliferation leads to fibrosis. The etiology of ARDS/DAD is diverse and often cannot be identified from the morphologic substrate. Causes of ARDS/DAD are summarized in Table 3-1.
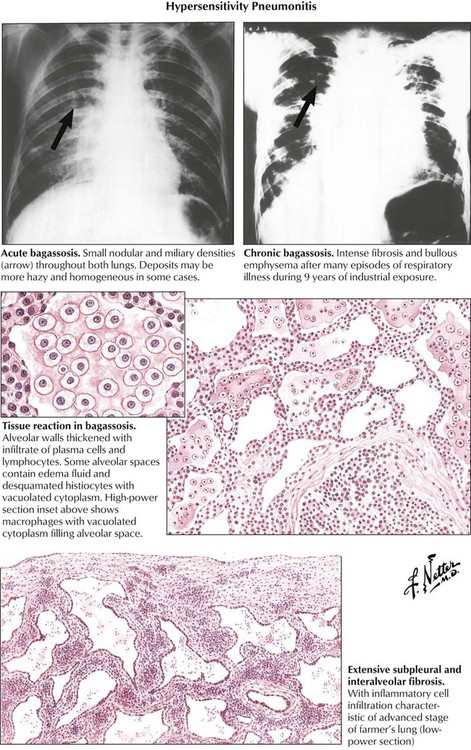
Hypersensitivity pneumonitis (extrinsic allergic alveolitis [EAA]), an acute immunologic reaction of the lung to inhaled antigens, is typically caused by occupational exposure to organic dusts (e.g., farmer’s disease bagassosis). Although EAA is classified as an acute RLD, its appearance is distinct from that of ARDS/DAD. EAA may run an acute or a chronic course with lymphoplasmacytic and monocytic interstitial infiltration (rarely eosinophils), mild alveolar exudate, and reactive alveolar cell proliferation (catarrh). There are noncaseating granulomas in approximately one third of cases. EAA is characterized by progressive fibroblast proliferation with interstitial and intraalveolar budding fibrosis and obliterative bronchiolitis. Inflammatory infiltrates usually recede in end-stage disease, leaving the nonspecific scenario of usual interstitial pneumonitis or fibrosis.
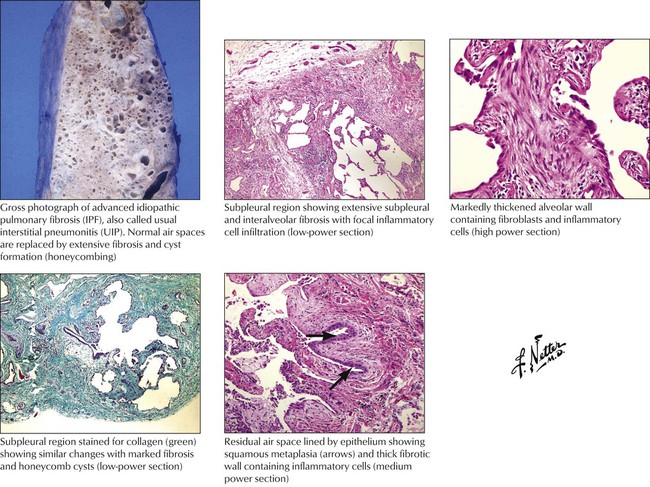
Idiopathic pulmonary fibrosis (IPF) refers to a poorly understood chronic inflammatory and progressive fibrosing disorder of the lung. It is not a single entity but a mixture of pathogenetically variable diseases, which may progress in part from an acute RLD (e.g., EAA). Hamman-Rich syndrome is the classic description of IPF. The etiology of IPF and its subtypes is often unclear. Potential causes include postinfectious syndromes (e.g., postadenovirus, parainfluenza virus, and influenza B virus infections). The pathogenesis seems to be (auto)immune because the bronchoalveolar lavage contains increased numbers of neutrophils, macrophages, T lymphocytes, and occasionally eosinophils. Tissue biopsy shows expression of major histocompatibility complex (MHC) II antigens in alveolar epithelial cells. Infiltrating lymphocytes show a predominance of CD4+ and CD8+ T cells. Classifications of IPF are shown in Table 3-2.
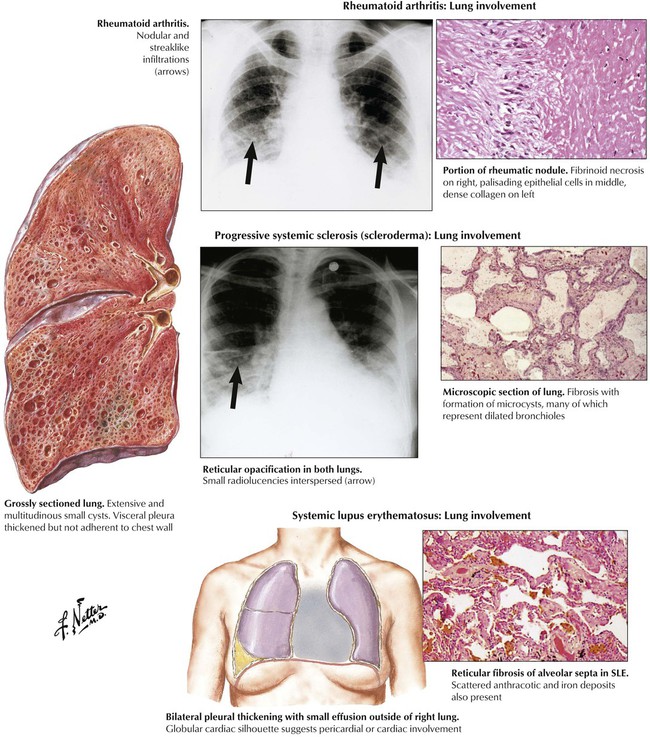
Restrictive pulmonary disease is evident in up to 70% of patients with systemic lupus erythematosus (SLE), approximately 50% of patients with progressive systemic sclerosis (PSS), and up to 20% of patients with rheumatoid arthritis (RA). Lung involvement in RA is characterized by diffuse interstitial fibrosis (fibrosing alveolitis), bronchiolitis obliterans, sclerosing granulomas and necrotizing nodules, and isolated fibrinous pleuritis. In SLE, characteristic pulmonary changes are DAD, nonspecific interstitial pneumonitis with focal atelectasis, nonspecific infections, leukocytoclastic vasculitis, and focal pulmonary hemorrhage. PSS presents as an IPF (cryptogenic fibrosing alveolitis) with vascular involvement and pleurisy.
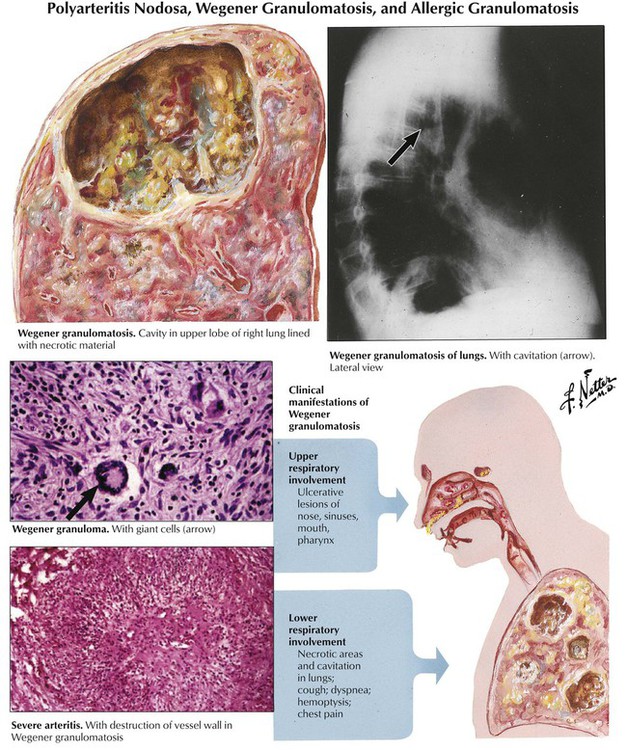
Several of the systemic vasculitis syndromes affect the lungs regularly or incidentally. The classic example is Wegener granulomatosis, which is characterized by necrotizing granulomatous lesions in the upper and lower respiratory tract accompanied by systemic vasculitis involving arteries and veins and focal necrotizing glomerulitis. Radiographs of the lungs reveal irregular and frequently multiple densities with or without cavitation, which may resemble metastatic disease. Bronchial disease may cause pulmonary atelectases.
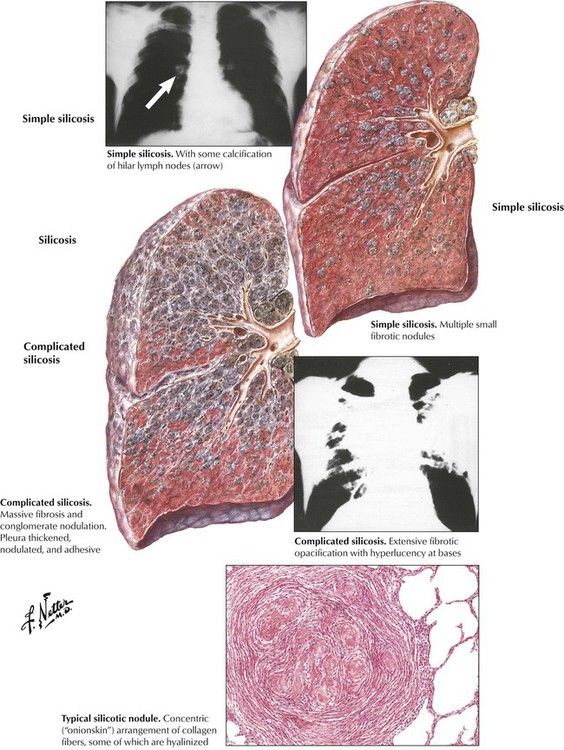
Pneumoconiosis describes a group of chronic RLDs that are caused by inhalation of mineral dusts. The most common forms of these occupational diseases are caused by coal dust, quartz, asbestos, and beryllium. Pneumoconioses are characterized by progressive pulmonary fibrosis, which reflects the dose, particle size, and fibroblastic potential of the individual dust. The dose is a function of dust concentration and duration of exposure. Silicosis, which is caused by inhalation of silica dust, is an example of a common pneumoconiosis. Characteristics of the more common pneumoconioses are shown in Table 3-3.
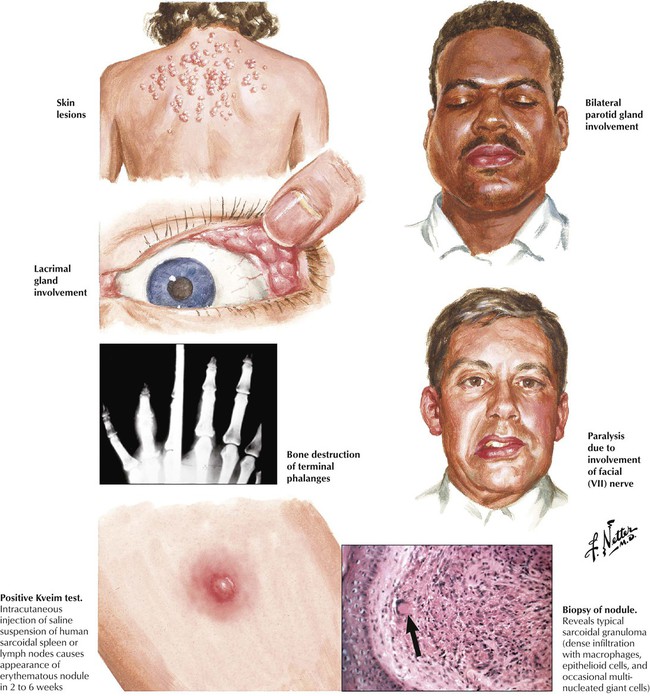
Sarcoidosis is a systemic disease of unknown origin characterized by the development of noncaseating epithelioid cell (EC) granulomas with subsequent fibrosis. On radiographs, pulmonary sarcoidosis shows a typical reticulonodular infiltrate with hilar lymphadenopathy. Histologically, multiple noncaseating granulomas are found in the bronchial or bronchiolar submucosa, along intralobular septae or the pleura. Granulomas undergo peripheral and “reticulated” fibrosis with final scarring. Multinucleated giant cells similar to Langhans cells may show intracytoplasmic star-shaped or laminar calcified inclusions (asteroid bodies, Schaumann bodies). The course may be acute or chronic persistent or progressive. Approximately one third of cases are complicated by sarcoid vasculitis. Approximately 20% of patients experience repeated recurrences and pulmonary dysfunction; 10% progress to pulmonary fibrosis and cor pulmonale.
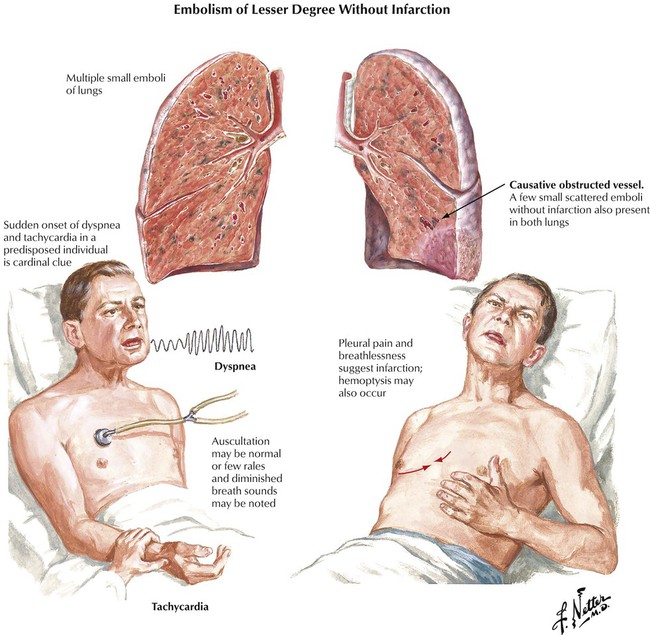
More than 90% of emboli in the pulmonary arteries arise from venous thromboses of the lower extremities. Approximately 60% to 80% of pulmonary embolisms (PEs) remain silent, presumably because of the small size of the thrombi. PE without infarction occurs without preexisting circulatory insufficiency. The tissue framework and collateral circulation remain intact because the dual arterial blood supply (pulmonary and bronchial arteries) prevents the thrombosis from occluding the pulmonary artery. The lung parenchyma shows severe congestions, with eventual intraalveolar hemorrhage. PE with pulmonary infarction occurs when blood supplied to the lung via the bronchial artery is insufficient, such as in chronic congestive heart failure (CHF) or in chronic pulmonary diseases with reduced vascularization. This hemorrhagic pulmonary infarction causes ischemic necrosis of lung tissue in addition to severe congestion. The infarction appears as a wedge-shaped, dark purple lung lesion with the base pointing toward the pleura and the occluded pulmonary artery at the tip.
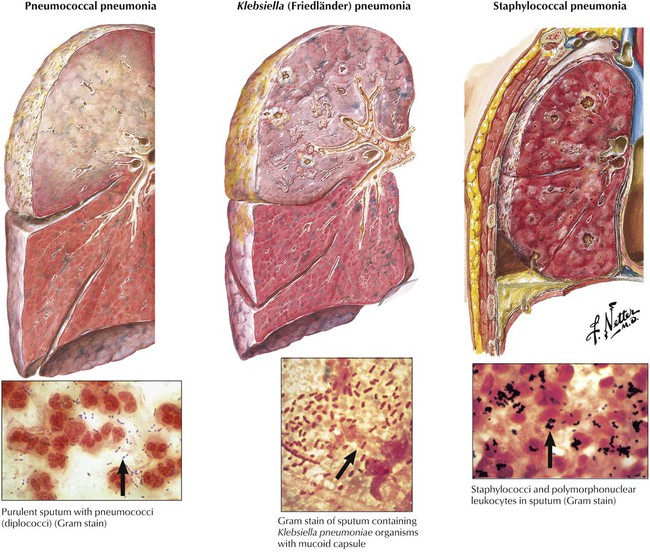
Bacterial pneumonias (Table 3-4) are caused by aerogenic infection (bronchopneumonia, BP) or hematogenous spread of infectious organisms (hematogenous pneumonia, HP). BP starts focally in one lobe with centrifugal spread (pneumococcal or Klebsiella BP), while HP affects both lungs in the peripheral mantle zone with centripetal spread. BP is accompanied by infectious bronchitis, HP rather by infectious (septic) vasculitis. The inflammatory reaction and its extent depend on the nature (toxicity) of the infectious organism and on the host defense status. Necrotizing or hemorrhagic reactions are caused by bacterial exo or endotoxins. Increased coagulation of the exudate may favor abscess formation (staphylococci) and decreased coagulation rapid spread (pneumococci). Segmental pneumonia indicates some defense deficiency. Lobar pneumonia is complicated by a local hypersensitivity reaction. Location, composition, and spread of the inflammatory reaction in pneumonia thus may permit some conclusion of nature and source of the infectious agent.
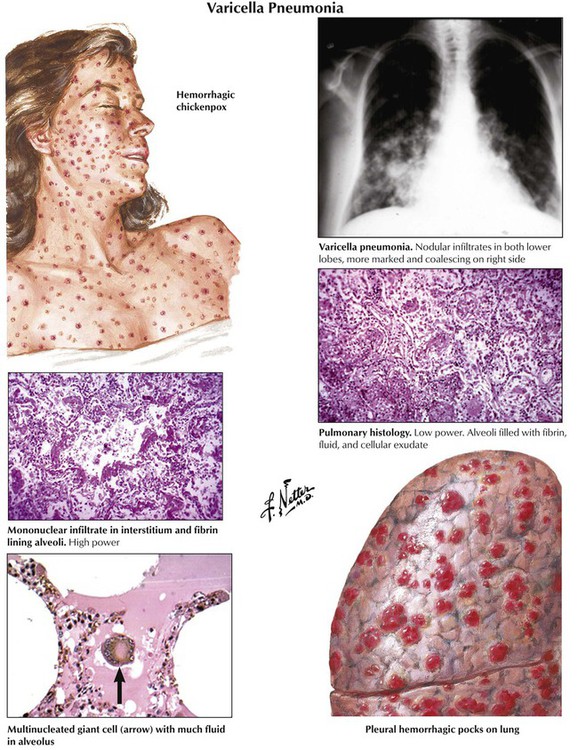
Viral pneumonitis is characterized by interstitial lymphocytic infiltration (nonspecific interstitial pneumonitis) combined with variable signs of DAD. Some cases are complicated by bronchiolitis obliterans (influenza virus, respiratory syncytial virus) or by focal necroses and eventual hemorrhage (herpes simplex virus [HSV], varicella-zoster and respiratory syncytial viruses, measles, influenza). Viruses that cause diagnostic cytopathic effects during some stages of infection include measles virus (Warthin-Finkeldey multinucleated giant cells), cytomegalovirus (giant cells with Cowdry type A intranuclear inclusions), other HSVs, and adenoviruses. Varicella pneumonia, a typical viral pneumonia, is caused by the varicella zoster virus.
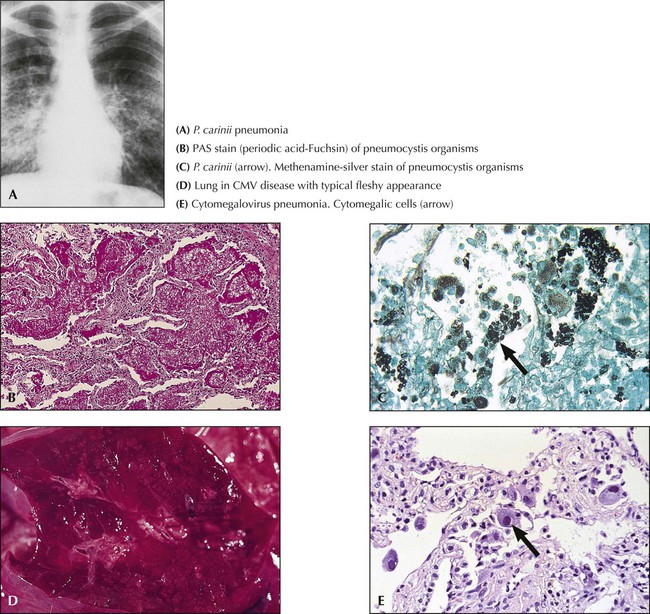
Fungal infections are a major cause for opportunistic pneumonitis in immunodeficient patients (Table 3-5). The most common lesions appear grossly as irregular yellowish-gray infiltrates with a granular dry and firm cut surface. Hemorrhage and occasional regional infarcts may occur in certain infections secondary to vascular involvement (e.g., in mucor mycosis). Some forms closely resemble tuberculosis (TB) (e.g., histoplasma capsulatum infections). Besides cytomegalovirus and various fungi, P carinii organisms are frequently isolated from “opportunistic” pneumonitis. Pathologic changes in P. carinii pneumonia show an interstitial plasmacellular (lymphoplasmacytic) pneumonia with alveolar foamy exudate, proliferating type II alveolar cells, and silver-stainable organisms. Certain fungal lung infections that occur in previously healthy persons may be accompanied by severe allergic reactions (e.g., allergic aspergillosis with bronchopulmonary infiltrates, eosinophilia, developing bronchiectasis, and eventual aspergilloma).
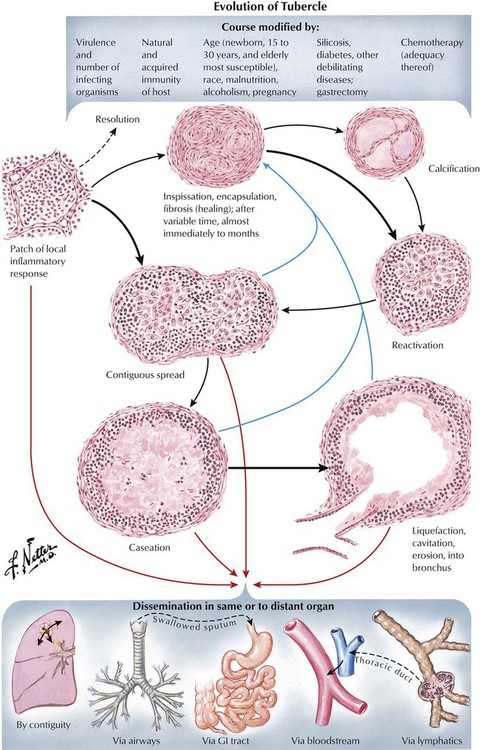
Pulmonary tuberculosis (TB) usually results from aerogenic infection with M. tuberculosis, typus humanus. The tuberculous granuloma is a classic epitheloid cell (EC) granuloma with palisading of EC around a central caseous necrosis. The EC layer contains multinucleated giant cells with peripherally located nuclei in a “string of pearls” pattern (Langhans–giant cells) surrounded by accumulations of lymphocytes. Tissue reaction to the state of immune reactivity forms for staging the clinicopathologic features of TB (Table 3-6). Infection in immunodeficient patients such as in human immunodeficiency virus (HIV) does not lead to typical tuberculous granulomas but to a nonspecific accumulation of macrophages at the sites of bacterial deposition (mycobacterial histiocytosis). Severe defense deficiencies cause tuberculosepsis acutissima (typhobacillosis Landouzy), a rapidly progressive and systemic form of TB with extensive caseous pneumonia in the lungs.
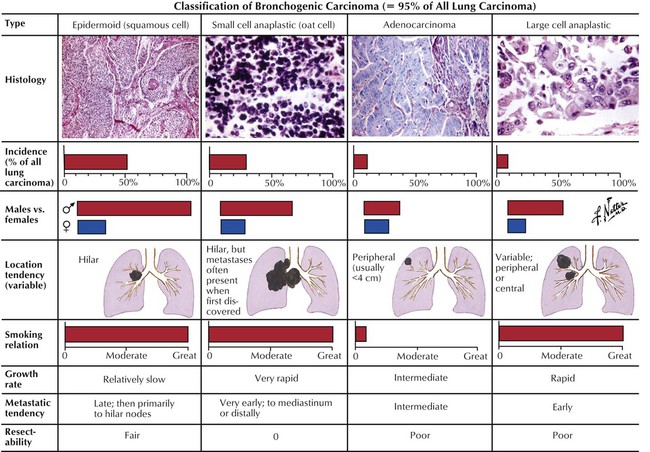
Carcinoma of the lung is the most frequent cause of cancer death worldwide (32% of cancer deaths in males, 25% in females). Bronchogenic carcinoma is classified into small-cell lung carcinoma ([SCLC] oat cell) and non–small-cell lung carcinoma (NSCLC), which includes squamous cell carcinoma (SCC), large-cell anaplastic carcinoma (LC), and adenocarcinoma (AC). Lung carcinomas vary in their primary location, spread, and overall biologic behavior. They frequently metastasize to regional lymph nodes (hilar, mediastinal) and to extralymphatic sites such as adrenal glands, brain, bone, and liver.
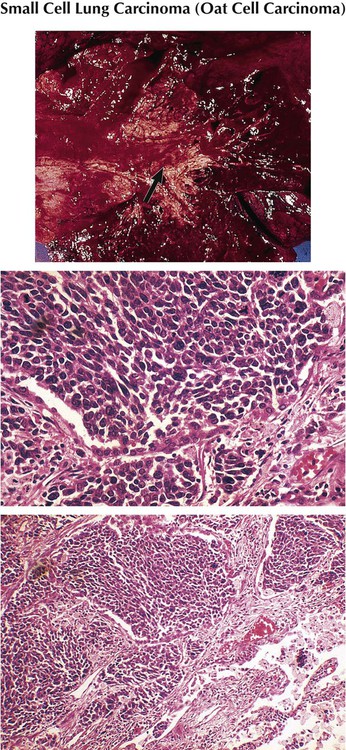
Small-cell lung cancer accounts for 20% of all lung cancers, with a male predominance and relation to cigarette smoking. It presents as a rapidly growing and metastasizing central lung mass occasionally accompanied by a paraneoplastic syndrome (myasthenia of Eaton-Lambert syndrome, ectopic corticotropin production, diabetes insipidus). The tumor consists histologically of sheets of small round or spindle cells with high mitotic index and scattered necroses. SCLC is essentially more sensitive to chemotherapy and therefore separated from all other lung cancers. However, it has the poorest 5-year survival rate (approximately 5%).
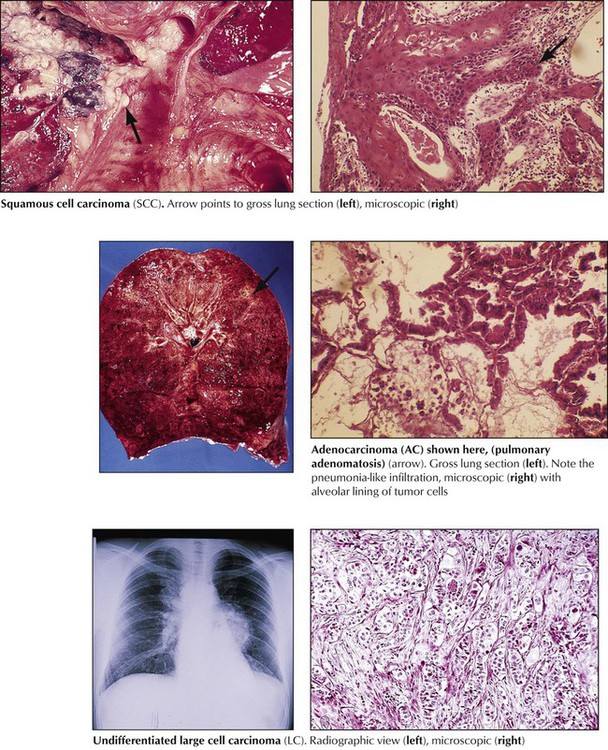
Squamous cell carcinoma (SCC), accounts for 30% of all lung cancers and is related to chemical carcinogens and cigarette smoking. SCC usually presents as a central lung mass invading the bronchial wall with rapid spread to local lymph nodes, brain, bone, and liver. Pancoast tumor is a variant of SCC in the apex of the lung that extends into adjacent thoracic and cervical nerves. Microscopically, tumor cells growing in sheets show variable degrees of squamous differentiation with keratinization. Adenocarcinoma (AC) accounts for another 30% of invasive lung cancers. It has a rather peripheral location and early lymphatic spread. There are several histologic subtypes (acinar, papillary bronchioloalveolar) and mixtures of these with variable degrees of differentiation (e.g., large-cell poorly differentiated AC). Large cell carcinoma (LC) accounts for approximately 10% of all lung cancers and may show features of squamous or glandular differentiation or both and pleomorphic or spindle cell variants. LC has the poorest 5-year survival rate of all NSCLCs (approximately 10%).
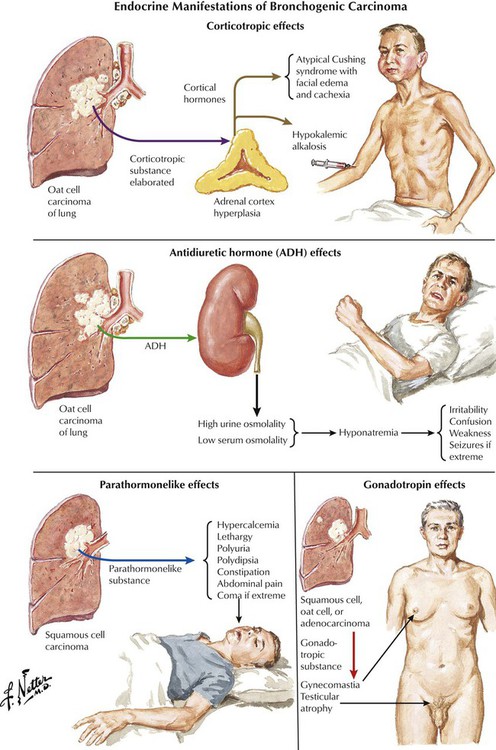
There are several nonmetastatic extrapulmonary manifestations of primary lung carcinoma, which are summarized as paraneoplastic syndromes. In addition to those pictured, these include skin changes, such as acanthosis nigricans, dermatomyositis/polymyositis, and myasthenia. Progressive multifocal leukoencephalopathy, occasionally also described as paraneoplastic syndrome, results from reactivation of latent polyomavirus infection (JC virus), and progressive focal demyelination in the central nervous system as can also be seen in other cases of immune deficiency (e.g., in HIV/acquired immunodeficiency virus [AIDS] and in certain cases of chronic lymphocytic leukemia).
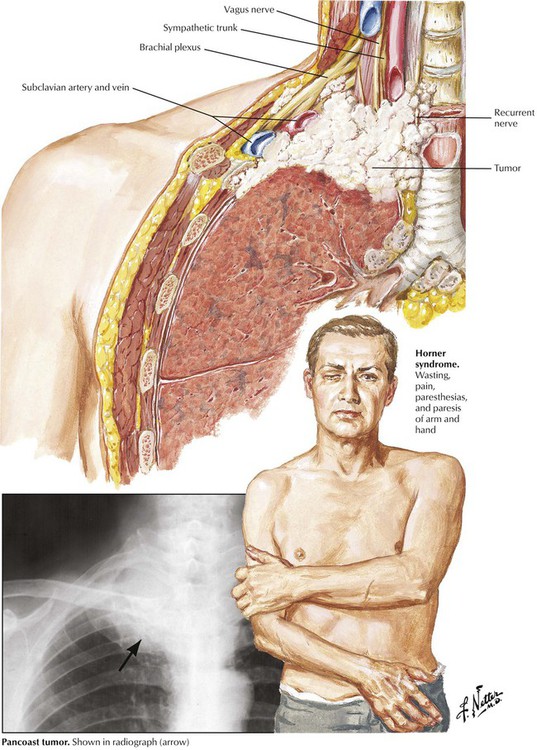
Pancoast tumor characterizes a special growth pattern of bronchogenic carcinoma with early invasion of homolateral soft tissues of the lower neck. The tumor subsequently grows into regional nerves (arm plexus, sympathicus, parasympathicus) and vessels, causing the clinical Horner syndrome: enophthalmos, ptosis, miosis, and anhydrosis (sunken-in eyeball, lowering of upper eyelid, narrowing of pupil, and loss of sweating).
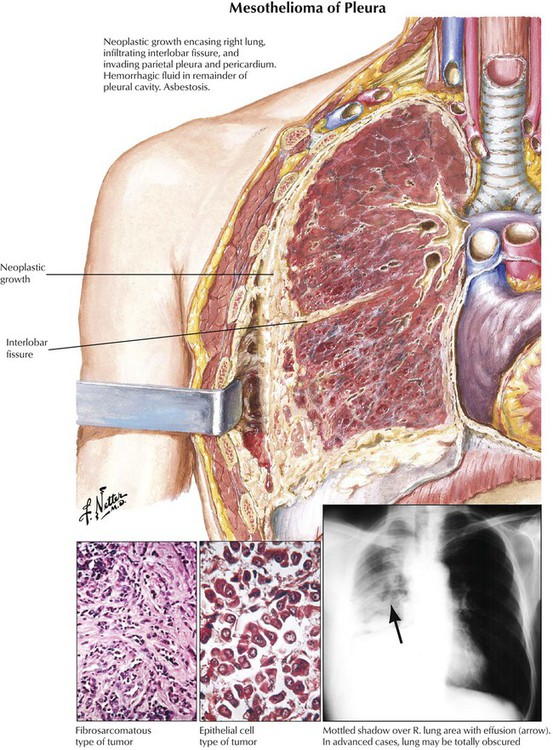
Other primary tumors of the lungs include carcinoids (a neuroendocrine tumor), mucoepidermoid and adenoid cystic carcinoma (counterparts to salivary gland tumors of bronchial glands), pulmonary blastoma, angiosarcoma and hemangioendothelioma, and malignant lymphoma. Malignant mesothelioma, a pleural fibrous tumor with adenoid mesothelial structures, may complicate pulmonary asbestosis. This firm tumor encases and compresses the lung (or both lungs) with limited direct invasion of peripheral lung parenchyma. Lymph nodes are rarely affected. The tumor extends locally within the thoracic cavity and along the mediastinum into bones, peritoneum, liver, and adrenals.
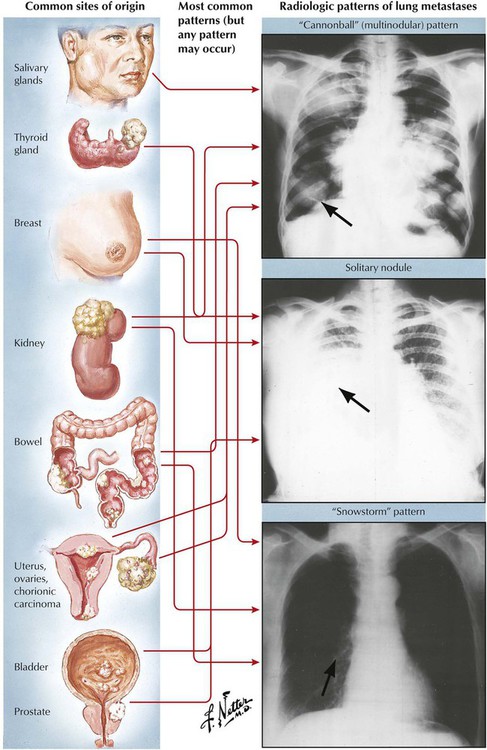
Metastases to the lungs occur in approximately 30% of all extrapulmonary malignant tumors. In the absence of a known primary tumor, clinical differential diagnosis may pose a problem. In contrast to a primary lung tumor, (hematogenous) metastases usually occur at multiple sites in both lungs. They are usually well-circumscribed foci, and cavitations occur rarely. Certain tumors, such as carcinoma of the pancreas and stomach, may show lymphangitic spread in the lungs, giving a characteristic appearance of fine-nodular and linear (reticulonodular) infiltrations. Diagnosis and search for the primary tumor is usually guided by biopsy of a lung metastasis with histologic and immunocytochemical investigation.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Respiratory System
Tumors in the Lungs and the Pleura
TABLE 3-1
CONDITIONS THAT CAN CAUSE ARDS/DAD*
| Conditions | Causes |
| Infectious diseases | Septicemia with DAD and DIC (especially gram negative), diffuse pneumonitis by virus, mycoplasma, pneumocystis, tuberculosis (certain forms, e.g., typhobacillosis Landouzy) |
| Chemical injury and inhalants | Oxygen, irritant gases and inhaled chemicals, barbiturate overdose, salicylic acid, paraquat, heroin or methadone overdose, cytotoxic drugs, uremic pneumonitis, gastric aspiration |
| Physical injury | Trauma to lungs (contusion), head injury, fat embolism of various causes, air embolism, burns, ionizing radiation |
| Other | Shock of any cause, acute pancreatitis, near drowning aspiration |
TABLE 3-2
CLASSIFICATION OF IDIOPATHIC PULMONARY FIBROSIS*
| Feature | NSIP | UIP | DIP | AIP | LIP | COP |
| Interstitial inflammation | Prominent | Scant | Scant | Scant | Prominent | Scant |
| Interstitial fibrosis | ||||||
| Collagen | Variable, diffuse | Patchy | Variable, diffuse | No | Some areas | No |
| Fibroblast foci | Occasional | No | No | Yes, diffuse | No | No |
| BOOP | Occasional, focal | Occasional, focal | No | Occasional, focal | No | Prominent |
| Intraalveolar macrophages | Occasional, patchy | Occasional, patchy | Yes, diffuse | No | Patchy | No |
| Hyaline membranes | No | No | No | Yes, focal | No | No |
| Honeycombing | Rare | Yes | No | No | Sometimes | No |
*AIP indicates acute interstitial pneumonitis; BOOP, bronchiolitis obliterans organizing pneumonitis; DIP, desquamative interstitial pneumonitis; NSIP, nonspecific interstitial pneumonitis; UIP, usual interstitial pneumonitis, LIP lymphocytic interstitial pneumonitis, COP cryptogenic organizing pneumonitis.
From Leslie KO, Wick MR. Practical Pulmonary Pathology. Philadelphia, 2005, Churchill Livingstone.
TABLE 3-3
CLINICAL AND PATHOLOGIC FEATURES OF PNEUMOCONIOSES*
| Entity | Clinical Appearance | Pathologic Changes |
| Coal miner’s lung | Black lung disease | Diffusely distributed, small focal anthracosilicosis, initially centriacinar and peribronchiolar with many carbon-laden macrophages and perifocal emphysema; extent of fibrosis depends on admixture of quartz |
| Silicosis | Acute silicosis (uncommon) | Alveolar lipoproteinosis and progressive diffuse interstitial fibrosis secondary to inhalation of small particulate silica crystals (e.g., after sand blasting) |
| Nodular silicosis (common) | Multiple growing silicotic nodules, usually 2 mm to 1 cm in diameter: fibrosing granulomas with concentric fibrous layering, some anthracotic pigment, small slitlike spaces, and needle-shaped crystalline spicules on polarization; perifocal emphysema | |
| Progressive massive silicosis | Multiple silicotic granulomas up to 10 cm in diameter, both lungs involved, massive and rapidly progressive fibrosis | |
| Asbestosis and asbestosrelated diseases | Asbestosis per se | Alveolitis with progressive interstitial fibrosis, deposition of asbestos bodies (golden-brown beaded rods consisting of asbestos fibers coated by ferroproteinaceous material); final stage: honeycombing lung |
| Pleural plaques and rounded atelectasis | Recurrent pleural fibrinous effusions, pleural fibrosis and pleural plaques (“sugar coating”), focal atelectasis secondary to pleural fibrosis | |
| Neoplasms | Malignant mesothelioma (↑ risk of bronchogenic carcinoma) | |
| Berylliosis | Berylliosis per se | Acute and recurrent pneumonitis, systemic sarcoidlike and fibrosing granulomas |
| Talcosis | Talcosis per se | Foreign body granulomas with birefringent talcum deposits, micronodular and diffuse interstitial fibrosis |
TABLE 3-4
INFECTIOUS AGENTS CAUSING PNEUMONIA
| Class | Etiologic Agent | Type of Pneumonia |
| Bacteria | Streptococcus pneumoniae | |
| Streptococcus pyogenes | ||
| Staphylococcus aureus | ||
| Klebsiella pneumoniae | ||
| Pseudomonas aeruginosa | ||
| Escherichia coli | ||
| Yersinia pestis | ||
| Legionella pneumophila | Legionnaires disease | |
| Peptostreptococcus, Peptococcus | Aspiration (anaerobic) pneumonia | |
| Bacteroides | ||
| Fusobacterium | ||
| Veillonella | Bacterial pneumonias | |
| Actinomycetes | Actinomyces israelii | Pulmonary nocardiosis |
| Nocardia asteroides | Pulmonary actinomycosis | |
| Fungi | Coccidioides immitis | Coccidioidomycosis |
| Histoplasma capsulatum | Histoplasmosis | |
| Blastomyces dermatitidis | Blastomycosis | |
| Aspergillus | Aspergillosis | |
| Phycomycetes | Mucormycosis | |
| Rickettsia | Coxiella burnetii | Q fever |
| Chlamydia | Chlamydia psittaci | Psittacosis |
| Ornithosis | ||
| Mycoplasma | Mycoplasma pneumoniae | Mycoplasmal pneumonia |
| Viruses | Influenza virus, adenovirus, respiratory syncytial virus, etc. | Viral pneumonia |
| Protozoa | Pneumocystis carinii | Pneumocystis pneumonia (plasma cell pneumonia) |
[/not-level-membership-for-pathology-category]
