RESISTANCE BLOOD VESSELS
Introduction
Failure to match blood flow to metabolic need will lead to loss of function of a tissue, to the development of pain and subsequently to tissue death. The case history of a lady with a problem in the regulation of the blood flow to her fingers is introduced in Case 9.1:1.
Resistance to blood flow
Where the blood flows to when it leaves the aorta depends on the relative resistance to flow in each part of the circulation. In each case, the blood flows through a series of blood vessels, arteries, arterioles, capillaries, venules and veins. The structure of the walls of all these vessels is described in Chapter 1 (see Figs 1.7, 1.8).
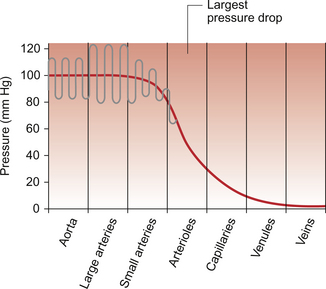
As noted in Chapter 8, there has to be a pressure gradient to achieve blood flow. Mean pressure in the arterial tree is typically close to 100 mm Hg and pressure in the right atrium is about 0 mm Hg (i.e. close to atmospheric pressure). Figure 9.1 shows the pressure drop going round the systemic circulation and it can be seen that the population of blood vessels through which there is the largest drop in pressure is the arterioles. These vessels, therefore, must be the segment of the circulation that have the highest resistance to blood flow. This concept is important because it means that by regulating the arterioles, we can:
The product of the cardiac output and the peripheral resistance to blood flow determines the arterial blood pressure (see Chapter 10). As the arterial pressure provides the driving force to perfuse tissues, physiological control systems act to keep arterial pressure relatively constant from moment to moment and from day to day. Indeed, sustained raised arterial pressure can cause serious damage to many parts of the body (see Chapter 10). The consequence of a fall in arterial pressure is often poor brain blood flow which results in syncope (fainting).
Adjustment of the arteriolar resistance is achieved by altering the state of contraction of vascular smooth muscle. The mechanisms of smooth muscle contraction are now described.
Vascular smooth muscle
Smooth muscle is located in the walls of the hollow structures of the body including blood vessels, airways, gut and bladder. The cells are spindle-shaped with a central nucleus and this is the first way in which smooth muscle cells differ from skeletal or cardiac muscle cells (see Chapter 6).
Source of Ca++ for smooth muscle contraction
As with the other two types of muscle, contraction of smooth muscle is triggered by a rise in intracellular [Ca++]. The source of the calcium is, however, different in the three types of muscle. The calcium involved in skeletal muscle contraction is stored intracellularly. It is released from the sarcoplasmic reticulum and is pumped back into these stores during muscle relaxation. In cardiac muscle, most of the calcium used in contraction derives from intracellular stores but some enters the cardiac muscle cell down a concentration gradient from the extracellular fluid via plasma membrane calcium ion channels (see page 20).
The membrane potential of vascular smooth muscle studied in vitro is close to −60 mV but in vivo it is only about −40 mV. This is because the pressure inside blood vessels stretches the smooth muscle and this stretch leads to the opening of a population of ion channels which result in partial depolarization of the cell and hence partial contraction of the smooth muscle. This is the basis for what has long been known as the ‘Bayliss Effect’. Basically, if you stretch vascular smooth muscle it responds by contracting. An advantage of having partially contracted vascular smooth muscle is that physiological mediators (locally released chemicals, hormones or neurotransmitters) can either cause further contraction or relaxation of smooth muscle as appropriate. Some physiological mediators (see section ‘Metabolite control of local blood flow’ on p. 104) act via a population of ATP-sensitive K+ channels. A decrease in [ATP] inside the smooth muscle cell increases the probability that this population of K+ channels will be open. This leads to hyperpolarization and hence relaxation of smooth muscle.
Relaxation of smooth muscle requires that intracellular [Ca++] is reduced. This can be achieved either by pumping the calcium back into intracellular stores or by expelling it outside the cell (see page 20 for a description of the equivalent mechanisms in cardiac muscle).
The use of different sources of calcium for contraction in the three types of muscle is illustrated by the pharmacological effects of calcium channel blocking drugs. Drugs such as nifedipine, diltiazem and verapamil will, to varying degrees, reduce heart rate and the contractility of the heart (see Chapter 5). These drugs may also be used to achieve peripheral vasodilatation as part of antihypertensive therapy (see Chapter 10). Their side effects are fairly predictable. These include facial flushing, headache and dizziness as a result of their effects on vascular smooth muscle but also constipation is a common side effect because of the effects of calcium channel blocking drugs on gut smooth muscle. Calcium channel blocking drugs have no effect on skeletal muscle function because all the calcium needed for contraction is stored within the sarcoplasmic reticulum.
The calcium channel blocking drug nifedipine was tried as therapy for the patient with Raynaud’s disease described in Case 9.1:2, the aim being to cause vasodilation and improve the blood flow to the fingers.
Contraction of smooth muscle
The contractile mechanism for smooth muscle is different to the two other types of muscle. In skeletal and cardiac muscle the contractile proteins, actin and myosin, are arranged in parallel layers and this is the origin of the striated (striped) appearance when these muscles are viewed under the polarized light microscope. Contraction of striated muscle (see Chapter 2) is initiated by the binding of Ca++ to the control protein troponin. This has the effect of moving another protein, tropomyosin, out of a groove on the bundle of actin filaments. Formation of a ‘cross-bridge’ is then achieved by the myosin head having access to a binding site on the actin filament. Muscle contraction takes place with the hydrolysis of ATP to provide the energy.
Smooth muscle does have actin and myosin as contractile proteins but does not have troponin. The Ca++ released into the cytosol of smooth muscle cells binds to the protein calmodulin. The calcium–calmodulin complex activates the enzyme myosin light chain kinase and this promotes phosphorylation of the myosin filament. Once this has been achieved, interaction between actin and myosin phosphate generates contraction of the smooth muscle cell. Figure 9.2 summarizes the events associated with cross-bridge formation and hence contraction of smooth muscle. When intracellular [Ca++] decreases, myosin is dephosphorylated by myosin light chain phosphatase. Even when dephosphorylated myosin can retain its interaction with actin. These attachments are called latch-bridges. They only detach slowly and so they maintain a level of muscle tension with little consumption of ATP.
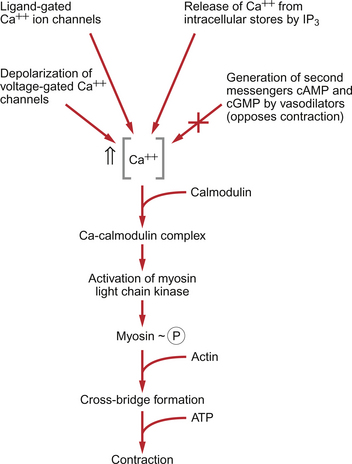
There are several broad types of mechanism which contribute to the overall regulation of intracellular [Ca++]. These mechanisms are illustrated in Figures 9.3 and 9.4. Some vasoconstrictor agents such as noradrenaline (norepinephrine) act through more than one mechanism:
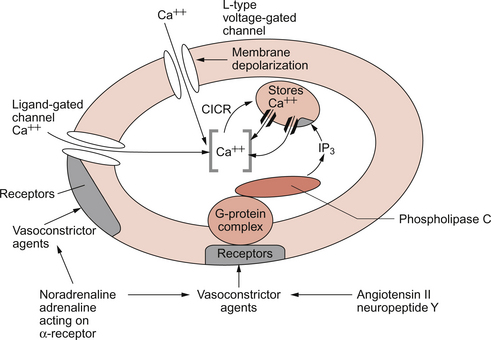
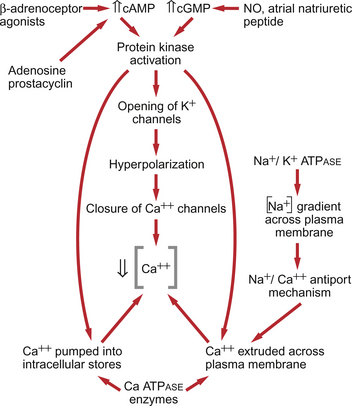
Fig. 9.4 Main pathways leading to a decrease in intracellular [Ca++] as part of vasodilatation mechanisms.
• Vasoconstrictor hormones such as noradrenaline, angiotensin II, endothelins, vasopressin and thromboxane A2 bind to G-protein coupled receptors. Subsequent generation of the second messenger inositol trisphosphate (IP3) leads to the opening of channels in intracellular calcium stores and release of Ca++ (Fig. 9.3).
• Vasoconstrictors also lead to membrane depolarization by several mechanisms. These include opening of ligand gated ion channels in the plasma membrane which permits influx of Na+ and Ca++ accompanied by inhibition of K+ channels (Fig. 9.3).
• Intracellular [Ca++] also depends on the Ca++ removal mechanisms. These include pumping Ca++ back into intracellular stores and active extrusion of Ca++ across the plasma membrane both of which involve Ca-ATPase enzymes. There is also a Na+/Ca++ antiport exchanger. Entry of Na+ into the cell down its concentration gradient is coupled to extrusion of Ca++ against its concentration gradient. The low intracellular [Na+] is of course maintained by the sodium pump (Na+/K+ ATPase) (Fig. 9.4).
• Vasodilator agents act via production of either cAMP (e.g. adenosine, prostacyclin, β-adrenoceptor agonists) or cGMP (nitric oxide, atrial natriuretic peptide) as second messengers. Both cAMP and cGMP activate protein kinases and hence lead to protein phosphorylation. A reduction in plasma [Ca++] may then be secondary to cell hyperpolarization following opening of K+ channels. The hyperpolarization closes Ca++ channels. An alternative mechanism for vasodilatation is the activation of Ca++ pumps leading to either extrusion of Ca++ from the cell or sequestration of Ca++ into intracellular stores (Fig. 9.4).
Inappropriate spasm of vascular smooth muscle is the diagnosis suggested in the case study in Case 9.1:2.
Local control of vascular smooth muscle
Endothelial factors in the control of local blood flow
The adult human circulation consists of about 60 000 miles of tubing (see Chapter 1). It is lined by a thin monolayer of endothelial cells. These cells not only provide a barrier between the blood and the other cells of the body (see Chapter 11), but they are also the source of a range of vasoactive agents which cause relaxation or contraction of underlying blood vessel smooth muscle (Figs 9.5, 9.6). One of these compounds, before it was chemically identified, was initially named endothelium-derived relaxing factor (EDRF). It is now thought that most, but not necessarily all, of the vascular effects of EDRF can be attributed to nitric oxide. Other factors produced by the endothelium and which also affect vascular smooth muscle contraction have been identified (see p. 103).
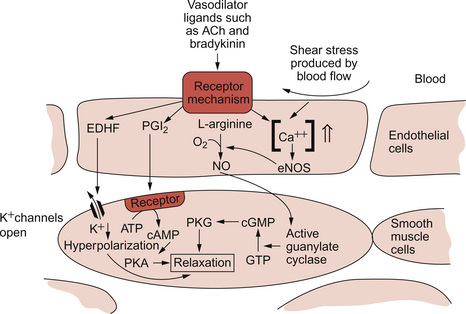
Nitric oxide (NO)
NO is synthesized from the amino acid l-arginine by the action of nitric oxide synthase (NOS) enzymes (Fig. 9.5). The terminology for these enzymes is a little confusing as it reflects the original site of discovery rather than current opinion of their site of importance. Endothelial NOS (eNOS) and neuronal NOS (nNOS) are both constitutively expressed in a wide range of cells, including many cell types in the cardiovascular system. These enzymes generate NO continuously. Inducible NOS (iNOS) is synthesized by cells exposed to inflammatory cytokines such as tumour necrosis factor alpha (TNFα), interleukin 1β (IL1β) and interferon alpha (IFNα). A range of other cytokines have the opposite effect and suppress iNOS expression. Overall the balance of local cytokines determines the expression of iNOS and the rate of NO production as required. NO produced in this way in macrophages has cytotoxic actions. Excessive production of NO by iNOS also occurs in some forms of septic shock and will lead to peripheral vasodilatation and a fall in arterial blood pressure (see Chapter 14). It is assumed that iNOS generated NO does not contribute to the normal physiological control of blood vessel diameter.
NO generated constitutively by eNOS and nNOS is part of moment to moment normal vascular control mechanisms. As the physiological half-life of NO is short (a few seconds), it must be generated continuously and is available to contribute to short-term changes in blood vessel diameter. The main site of action of NO is on large diameter arterioles. The single most important trigger for NO production in relation to circulatory control is increased shear stress on the blood vessel wall (Fig. 9.5). If blood flow velocity in an artery increases, this leads to increased NOS activity in the endothelial cells and hence vessel dilatation. The existence of flow dependent vasodilatation was first reported by Schretzenmayr in 1923 but the mechanisms involved only became apparent when NO was recognized as an important vasoactive compound in 1986. NO synthesis can also be triggered by blood borne agonists such as bradykinin, acetylcholine and thrombin acting via receptors on the endothelial cell (Fig. 9.5). This has applied importance, for example, when considering the action of the angiotensin converting enzyme inhibitor (ACEI) group of drugs (see Chapter 10). The ACE enzyme is responsible for the inactivation of bradykinin as well as for the activation of angiotensin II. ACEI drugs, therefore, lead to increased bradykinin concentration and so increased NO synthesis. Part of the antihypertensive action of ACEI drugs is, therefore, related to NO induced vasodilatation.
NO produced in endothelial cells diffuses out of these cells and through the plasma membrane of underlying smooth muscle cells. Within smooth muscle NO binds to the haem part of guanylate cyclase and thereby increases the rate of production of cyclic GMP. In smooth muscle cells this mediator, via activation of protein kinase G, leads to a reduction in intracellular [Ca++] and hence smooth muscle relaxation (Fig. 9.5).
Endothelial dysfunction is important in many disease mechanisms including diabetes, hypertension (see Chapter 10) and atherosclerosis. This dysfunction is not, however, simply explained by alterations in the rate of NO production by endothelial cells. The superoxide anion O−2 inactivates NO and forms the peroxynitrite anion (ONOO−). This highly reactive anion has many different adverse effects on cell function. Countering the effects of superoxide anion is the strategic basis for many new approaches to therapy for chronic vascular disease. In the absence of NO, superoxide anion leads to the production of hydroxyl anion (OH−) which can also have damaging effects.
Drugs which are sources of NO
The group of drugs known as organic nitrates or nitrovasodilators have been in clinical use since the nineteenth century. They either spontaneously release NO (e.g. sodium nitroprusside) or are enzymically degraded and release NO (e.g. glyceryl trinitrate (GTN), amyl nitrate, isosorbide mononitrate and isosorbide dinitrate). These drugs therefore mimic the effects of endogenous NO. Nitric oxide and the nitrosothiols which are produced by the interaction of NO with sulphydryl compounds such as glutathione activate guanylate cyclase (see above) and lead to vasodilatation. The rationale for the use of these drugs in the management of angina is further discussed in Chapter 5 but the fundamentals are as follows. The drugs have their actions on three particular populations of blood vessels.
1. Capacitance vessels (mainly small veins): dilatation of these vessels leads to venous pooling of blood and a reduction in preload on the heart (see Chapter 4).
2. Arterial resistance vessels (mainly arterioles): vasodilatation in this case leads to a reduction in arterial blood pressure and consequently a drop in afterload effects on the heart (see Chapter 4).
3. Coronary arteries are often, mistakenly, thought to be the major site of action of nitrovasodilator drugs. In an ischaemic heart these vessels may well be nearly maximally dilated as a result of the accumulation of local metabolites (see p. 104). There is therefore little remaining vasoconstriction which could be reversed by nitric oxide donor drugs. However, nitrovasodilators may improve blood supply within the heart by actions on collateral blood vessels and can also be useful in the relief of coronary artery spasm (see Chapter 5).
The side effects of nitrovasodilator drugs are fairly predictable. Effects on the venous capacitance vessels will lead to pooling of blood in the veins and hence to postural hypotension, dizziness, fainting (syncope) and a reflex increase in heart rate (see Chapter 10). Arterial dilatation leads to headache as a result of dilatation of cerebral blood vessels and raised intracranial pressure (see p. 108) and to increased skin blood flow, which appears particularly as facial flushing.
Other endothelium derived relaxing factors
The chemical structure and precise physiological role of endothelium derived hyperpolarizing factor (EDHF) is as yet unknown. It is thought to cause smooth muscle relaxation by promoting the opening of K+ channels (Fig. 9.4).
Prostacyclin (prostaglandin I2, PGI2) is released from endothelial cells and is a potent inhibitor of platelet aggregation. PGI2 is also a vasodilator and acts by increasing the production of cyclic AMP and hence activating protein kinase A (PKA) in smooth muscle cells (Fig. 9.5).
Endothelins
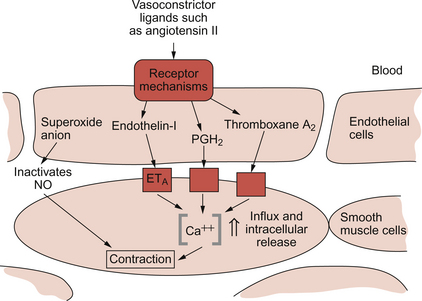
The endothelins are a group of peptides which were first discovered in 1988. There are three endothelins designated ET-1, ET-2 and ET-3. Of these three compounds, ET-1 is considered the most important in humans and it has its action predominantly on the ETA type receptor but it also has actions on the ETB receptor. Interaction of ET-1 with ETB receptor on the endothelial cell leads to increased NO production (a vasodilator), but the dominant action of ET-1 is vasoconstriction following binding to an ETA receptor on vascular smooth muscle (Fig. 9.6).
In addition to a role in normal vascular control, there is much interest in the involvement of endothelins in disease mechanisms. Sustained endothelin induced vasoconstriction has been implicated in the mechanisms of essential hypertension, congestive heart failure and chronic renal failure amongst others. Intermittent vasoconstriction produced by endothelins is thought to occur in a range of conditions including unstable angina, acute renal failure, subarachnoid haemorrhage, Raynaud’s disease and migraine. In addition to the intrinsic vasoconstrictor effects, endothelins augment the effects of other vasoconstrictors such as angiotensin II, noradrenaline (norepinephrine) and serotonin (Fig. 9.6).
Other endothelium derived constricting factors
Endothelial cells can synthesize the vasoconstrictor prostanoids thromboxane A2 and prostaglandin H2 (Fig. 9.6). Other contributions of endothelial cells to blood pressure elevation include the production of superoxide anions which inhibit the dilator actions of nitric oxide (see p. 102) and the activation of the physiologically inert peptide angiotensin I to the potent pressor agent angiotensin II. The enzyme involved, angiotensin-converting enzyme (ACE), is present on the surface of the endothelial cells (see p. 106).
Metabolite control of local blood flow
A major rationale for endothelial involvement in vascular control mechanisms (described above) appears to be to adjust arterial and arteriolar diameter to match changes in blood flow velocity. Increased flow velocity leads to increased shear stress between blood and the endothelial wall. This is an important factor regulating production of both NO and ET-1. The equivalent rationale for metabolite control of blood vessel size is that it enables matching of blood flow to the metabolic needs of a tissue. A local increase in metabolic rate will lead to an accumulation of metabolites which will in turn cause vasodilatation. The principle of metabolite induced vasodilatation is outlined in Figure 9.7. Metabolite effects, therefore, provide the major mechanism for regulating the distribution of blood flow. Metabolite-based regulation appears to be particularly important both in tissues, such as the brain, which require a fairly constant blood flow and in tissues in which metabolic needs may fluctuate widely, such as the heart or skeletal muscle. The kidney, which has excretion and fluid volume regulation as its main functions, has a high blood flow compared to the tissue’s metabolic needs. Metabolites therefore play little role in blood flow regulation in the kidney. The major target vessels for metabolite based control are arterioles and precapillary sphincters. The main site of metabolite effects is on small, rather than large, arterioles.
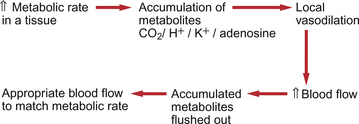
Adenosine is a potent vasodilator in skeletal and cardiac muscle cells. It is formed in the muscle cells either by the complete dephosphorylation of ATP to adenosine or in a parallel pathway which involves the intermediate formation of S-adenosyl methionine from ATP (see Fig 5.5). It should be remembered that the normal [ATP] inside a muscle cell is of the order of 5×10−3 mol/L whereas [adenosine] outside the myocyte is about 1×10−8 mol/L, a 500 000-fold difference. Significant accumulation of adenosine, therefore, only requires the use of a minute proportion of the available ATP. Adenosine, a non-polar molecule, can cross myocyte membranes. The action of adenosine as a dilator is partly mediated by ATP-sensitive K+ channels on the endothelial cells which increase NO production and partly by similar actions on K+ channels leading to hyperpolarization of the underlying smooth muscle.
K+ ions leave skeletal and cardiac muscle cells and also neurons during the repolarization phase of action potentials. Changes in extracellular [K+] contribute to the initiation of the cardiovascular responses to exercise (see Chapter 13).
Hormonal control of blood vessel diameter
• Local hormones (autocoids) are mainly involved in local responses as part of pathological events. Specific examples (see this page) include histamine, bradykinin and serotonin but the definition could arguably also include the endothelium-derived mediators NO, PGI2 and the endothelins described above.
• Systemic hormones circulate in the blood and have an effect all round the circulation. The most important examples here are the renin-angiotensin system and the catecholamines adrenaline (epinephrine) and noradrenaline (norepinephrine) released from the adrenal medulla.
Local hormones
Histamine
The major vascular responses to H1 receptor stimulation include a transient increase in capillary permeability which can lead to oedema (see Chapter 11). If this occurs to a substantial extent, it can lead to a reduction in circulating blood volume and, consequently, hypotension. This may be accompanied by histamine induced arteriole and capillary dilatation which will also contribute to blood pressure lowering. In severe allergic reactions, such as an anaphylactic reaction to a bee sting, these responses may be life threatening. Treatment includes the rapid use of antihistamine drugs, together with glucocorticoids as anti-inflammatory agents and adrenaline (epinephrine) as a vasoconstrictor (see p. 107). Actions of neurally released histamine in the skin contribute to the weal and flare response which is part of local allergic responses.
Bradykinin
The nine amino acid peptide bradykinin causes dilatation of arterioles and an increase in venule permeability. It is generated by the enzyme kallikrein from kininogen during inflammatory responses. Bradykinin also binds to receptors on the endothelial cells and increases the production of nitric oxide (see p. 102). Interest in this area has been stimulated by the development of angiotensin converting enzyme (ACE) inhibitor drugs (see p. 107). ACE inactivates bradykinin and so ACE inhibitor drugs increase the physiological half-life of bradykinin. Bradykinin is also the most potent autocoid in pain responses, an action shared by histamine (acting on H3 receptors) and by serotonin.
Systemic hormones
Renin-angiotensin system
Renin is a systemic hormone secreted by the kidney. It is an enzyme which generates angiotensin I as shown in Figure 9.8.
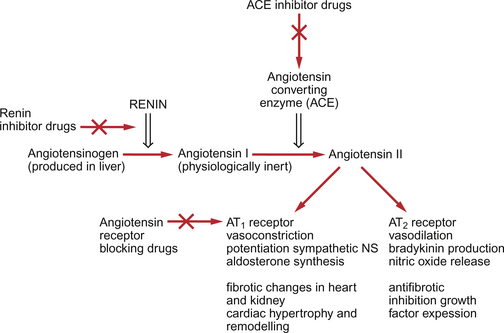
• Direct pressor action via receptors on vascular smooth muscle. In addition Ang II promotes the formation in endothelial cells of vasoconstrictors such as ET-1 and thromboxane A2.
• Potentiation of sympathetic nervous system activity by several mechanisms. This contributes to the pressor effect of Ang II.
• Stimulation of aldosterone synthesis by actions on the zona glomerulosa of the adrenal cortex. This role of Ang II contributes to blood volume regulation (see Chapter 14).
• Increase in antidiuretic hormone (ADH) secretion.
• Potentiation of thirst responses (dipsogenic actions) by effects within the brain.
The full range of responses to a second receptor type, the AT2 receptor, is not yet clear but it appears to oppose the pressor actions of Ang II mediated by the AT1 receptor.
Ang II has an important role in pathological events as well. It can promote cell hypertrophy and hyperplasia and also promotes fibrotic changes in many tissues. An example is in the series of changes referred to as ‘remodelling of the heart’ during progressive cardiac failure (see Chapter 6).
Pharmacological blockade of the renin-angiotensin system
What drugs are available to block the renin angiotensin system? β-adrenoceptor blocking drugs (see p. 106) provided the earliest, but rather non-specific, form of renin blockade. Renin secretion from the juxtaglomerular cells in the kidney is partly regulated by circulating catecholamines and by sympathetic nerves acting through β-adrenoceptors. However, beta-blockers have many other components to their pharmacological spectrum of activity.
Angiotensin converting enzyme inhibitors (ACEI) were developed during the 1980s. There are now over 30 drugs in this class available internationally. The first drug clinically available was captopril and the second was enalapril. All of the drug names in this class end in -pril. They not only block the formation of the active hormone Ang II but also block the breakdown of bradykinin to inactive peptides (see p. 106).
The antihypertensive actions of ACEI therefore include blockade of the direct vasoconstrictor effects of Ang II and a modest diuretic action mediated by reduced aldosterone production. The drugs are also described as being sympatholytic as Ang II potentiation of the sympathetic nervous system is blocked. A further component of the antihypertensive action may be linked to increased levels of the vasodilator bradykinin. Certainly, increased [bradykinin] is the basis for the major side effect of these drugs, a dry cough in 10–30% of patients. Bradykinin is also an agonist on endothelial cells for the production of nitric oxide (Fig. 9.5). This is also thought to contribute to the hypotensive action of these drugs.
Adrenal medullary hormones
Receptors for catecholamines were originally divided into two types, α and β, classified on the basis of agonist potency as indicated in Figure 9.9. Subsequently, following the development of selective antagonist drugs, the receptor types were subdivided into α1 and α2, β1, β2 and β3. Sometimes further subdivisions of this basic classification based on pharmacological and gene cloning studies are used.
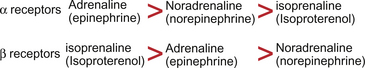
The main receptor type on the heart is the β1 receptor. Stimulation leads to an increase in the force and rate of cardiac contraction (see p. 45). The dominant catecholamine receptor on blood vessels is the α1 receptor and this mediates vasoconstriction. In some parts of the peripheral circulation postsynaptic α2 receptors also exist on vascular smooth muscle and stimulation leads to vasoconstriction. Presynaptic α2 receptors modulate the release of neurotransmitters into the synaptic cleft. Thus, a rise in transmitter concentration in the synapse stimulates presynaptic α2 receptors and shuts off further transmitter release. Blood vessels also have a limited distribution of β1 and β2 receptors which, when activated, lead to vasodilatation.
Autonomic nervous system and peripheral circulation control
In summary, the major characteristics of the nerve supply to blood vessels are that they are sympathetic in origin releasing noradrenaline (norepinephrine) onto α1 receptors and resulting in vasoconstriction. Regulation of these nerves is further discussed in relation to blood pressure regulation in Chapter 10. A possible role for sympathetically induced vasoconstriction in patients with Raynaud’s disease is discussed in the case history in Case 9.1:3.
Special circulations
The gross distribution of blood flow to the various parts of the body is discussed in Chapter 13. The regulation of some specific vascular beds is discussed here. Coronary blood flow regulation is described in Chapter 5.
Brain (cerebral) circulation
The brain receives about 15% of resting cardiac output, for the textbook 70 kg person a flow rate of about 750 mL/min. This is a relatively high flow rate as the brain, which typically weighs 1.4 kg in a textbook adult, only represents about 2% of body weight. Flow rate is substantially higher to the grey matter (mainly cell bodies) than to the white matter (mainly nerve axons) of the brain. The brain has a high oxygen consumption rate and a high heat generation rate. Interruption of the blood supply to the brain results in loss of consciousness within a few seconds and permanent damage within a few minutes.
Brain blood flow is autoregulated (Fig. 9.10) such that flow is kept fairly constant at about 55 mL/min/100 g tissue and this is independent of fluctuations in mean arterial pressure across the range 60–175 mm Hg. Although the brain has a rich sympathetic nerve supply, stimulation of these nerves makes little difference to autoregulation. However, the autoregulation mechanism is very sensitive to the Pco2 of arterial blood. If a subject hyperventilates and therefore reduces arterial Pco2, then brain blood flow will decrease substantially. This is the reason behind the feeling of ‘light headedness’ following a period of voluntary hyperventilation. The autoregulation response is abolished by hypercapnia (high Pco2) and brain blood flow then increases in proportion to arterial pressure (Fig. 9.10). As the brain is encased in the cranium and cannot expand, retention of CO2, as in chronic obstructive pulmonary disease, results in cerebral vasodilatation and raised intracranial pressure. The patient may therefore complain of headache. Increased metabolic activity in specific parts of the brain will lead to local increases in blood flow (functional hyperaemia).
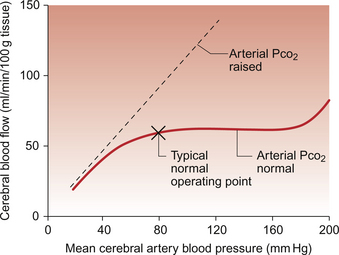
As humans have an erect posture, the brain is above the level of the heart. Mean cerebral artery pressure is therefore typically of the order of 77 mm Hg. Figure 9.10 shows that if arterial pressure falls below about 50 mm Hg the autoregulation mechanism fails. If the perfusion pressure of the brain falls below about 40 mm Hg then syncope (fainting) occurs. Such a fall in blood pressure may occur as a result of a sudden drop in cardiac output (e.g. because of venous pooling of blood in the limbs) or because of excessive peripheral vasodilatation (e.g. because of the effects of a high ambient temperature). However, when fainting occurs in response to psychological stress associated with fear, pain or shock, the mechanisms are less well understood. In the period immediately before a faint the subject becomes pale and sweats profusely. They tend to hyperventilate, leading to hypocapnia, and then often yawn. Loss of consciousness follows a sudden increase in vagal outflow leading to a slowing of heart rate (bradycardia). This is accompanied by dilatation of peripheral vascular vessels, particularly in skeletal muscle. This is attributed to an inhibition of sympathetic vasoconstriction outflow which originates in the hypothalamus. This series of events is sometimes referred to as a ‘vasovagal attack’.
Skeletal muscle blood flow
Under resting conditions, skeletal muscle receives about 20% of the cardiac output even though muscle accounts for 50% of body weight. Only a relatively small proportion, about one third, of capillaries are being fully perfused at any one time in resting muscle. Access of blood to the remaining capillaries is limited by the closure of precapillary sphincters. These sphincters have no nerve supply but are sensitive to changes in local metabolite concentration and so will open during exercise. Terminal arterioles are also dilated by metabolite accumulation. During vigorous exercise, muscle blood flow can increase more than 20-fold and may account for 80–90% of the increased cardiac output. This can mean an increase in muscle blood flow from about 1 L/min at rest to 20–22 L/min in intense exercise for the textbook person (see Chapter 13).
There is a rich sympathetic vasoconstrictor nerve supply to blood vessels in skeletal muscles. Under resting conditions, this maintains muscle blood flow at a relatively low level. Cutting the sympathetic nerves to a resting muscle leads to a doubling of blood flow. During exercise, the sympathetic vasoconstrictor nerves continue to be active but their effects on vascular tone are opposed by local accumulation of metabolites. Interstitial [K+] is particularly important as a metabolite in skeletal muscle. During muscle action potentials, K+ ions leave the muscle and local interstitial fluid [K+] may rise from a resting value of about 4 mmol/L to as high as 9 mmol/L at the start of exercise before blood flow has fully increased. A concurrent rise in local osmolarity of up to 10% and a rise in inorganic [phosphate] also contribute to vascular regulation. The magnitude of any adenosine-mediated effects is related to the extent of local tissue hypoxia.
Skin (cutaneous) blood flow
A major feature of the cutaneous circulation is its role in body temperature regulation. Heat loss is promoted by increasing the blood flow through capillary loops which run close to the surface of the skin. Shunting of blood towards or away from these capillary loops is achieved by opening and closing arteriovenous anastomoses, thick wall coiled vessels which link arterioles and veins in the skin. These anastomoses, together with cutaneous arterioles and veins, are controlled by the sympathetic nerve supply acting through α1 receptors. Central control of these nerves originates in the hypothalamus, the location of the body’s thermostat. Aspects of the regulation of skin blood flow in a person with Raynaud’s disease are described in Box 9.1 in this chapter.
Kidney (renal) blood flow
The renal vasculature has an intense sympathetic vasoconstrictor nerve supply. At times of activation of the sympathetic nervous system, as in exercise (see Chapter 13) and during circulatory shock (see Chapter 14), renal blood flow is reduced substantially below resting levels. If this period of reduced blood flow is prolonged during circulatory shock situations and GFR is reduced, this may lead to pathological changes within the nephron (acute tubular necrosis—ATN) which can compromise patient survival.
Splanchnic blood supply
Under resting conditions the splanchnic blood supply (gastrointestinal tract and liver) receives about 24% of the cardiac output. The venous drainage from most of the gastrointestinal (GI) tract enters the hepatic portal vein and this supplies 70% of the hepatic blood flow. The remaining 30% is provided by the hepatic artery. During exercise and other situations when the baroreceptor reflex is activated, sympathetically mediated constriction of the veins and venules displaces blood from the splanchnic beds so that more blood volume is available for use in other parts of the circulation. This constriction which raises the local resistance to flow also contributes to the maintenance of arterial blood pressure.
Born, G. V. R., Schwartz, C. J. Vascular Endothelium. Physiology, Pathology and Therapeutic Opportunities. Stuggart: Schattauer; 1997.
Braddock, M., Schwachtgen, J. -L., Houston, P., et al. Fluid shear stress modulation of gene expression in endothelial cells. News Physiol. Sci.. 1998; 13:241–246.
Hobbs, A. J., Higgs, A., Moncada, S. Inhibition of nitric oxide synthase as a potential therapeutic target. Annu. Rev. Pharmacol. Ther.. 1999; 39:191–220.
Kelm, M. Flow-mediated dilatation in human circulation: diagnostic and therapeutic aspects. Am. J. Physiol.. 2002; 282:H1–H5.
Ledoux, J., Werner, M. E., Brayden, J. E., Nelson, M. T. Calcium activated potassium channels and the regulation of vascular tone. Physiology. 2006; 21:69–79.
Levick, J. R. An Introduction to Cardiovascular Physiology, fifth ed. New York: Arnold; 2009.
Pohl, U., de Wit, C. A unique role of NO in the control of blood flow. NIPS: News Physiol. Sci.. 1999; 14:74–80.
Tomita T., Bolton T. B., Bolton T. B., eds. Smooth Muscle Excitation. London: Academic Press, 1996.
Waller, D. G., Renwick, A. G., Hillier, K. Medical Pharmacology and Therapeutics, third ed. Edinburgh: WB Saunders; 2009.
Wolfe, J. H. N. ABC of Vascular Diseases. London: BMJ Books; 1992.