Renal Failure
Evaluation of Renal Function
Diagnostic Strategies
Protein
The dipstick test for protein, which uses the color change of tetrabromophenol blue, can detect protein at concentrations of 10 to 15 mg/dL but does not yield reliably positive results until the concentration is greater than 30 mg/dL. Moreover, the relation between color intensity and protein concentration is only approximate. The dipstick reagent is three to five times more sensitive to albumin than to globulins and immunoglobulin light chains (e.g., Bence Jones protein)—an important limitation.1 False-positive results are caused by alkaline urine, hematuria, or prolonged immersion of the dipstick in the urine. False-negative results are seen with dilute urine.
Microscopic Examination
After dipstick testing of the urine has been completed, 10 mL of urine is placed in a conical test tube and spun at 2000 revolutions per minute for 5 minutes (higher speeds may break up casts). The supernatant is discarded. The sediment is resuspended in the residual urine, and a drop is placed on a slide and covered with a coverslip. Observations are recorded as the number of cells per high-power field. A level of two to three RBCs per high-power field in adult men or two to four RBCs per high-power field in adult women is commonly accepted as normal; in many studies a finding of five RBCs per high-power field is considered to represent the threshold of abnormality.2
Serum and Urine Chemical Analysis
Creatinine and Blood Urea Nitrogen.: The normal range for the serum creatinine level extends from 0.5 mg/dL in thin people to 1.5 mg/dL in muscular persons. Spurious elevations (up to 2 mg/dL) can be caused by acetoacetate (which cross-reacts with creatinine in commonly used assays) as well as by certain medications that either cross-react in the assay or reversibly inhibit tubular creatinine secretion despite a normal GFR (generally less than 0.5 mg/dL). Serum creatinine concentration is a function of the amount of creatinine entering the blood from muscle, its volume of distribution, and its rate of excretion. Because the first two are usually constant, changes in serum creatinine concentration generally reflect changes in GFR. The creatinine clearance is commonly estimated by the Cockcroft-Gault equation3:
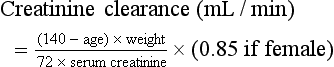
Urine Sodium and Fractional Excretion of Sodium.: Measurement of the urine sodium concentration provides information on the integrity of tubular reabsorptive function. Normally, urine sodium concentration parallels sodium intake. Low urine sodium concentration thus indicates not only intact reabsorptive function but also the presence of a stimulus to conserve sodium. The urine sodium concentration, as well as the fractional excretion of sodium (FENa), an additional measure of tubular sodium handling, helps distinguish between the two most common causes of AKI: prerenal azotemia and ATN (Table 97-1).
Table 97-1
Typical Urinary Findings in Prerenal Azotemia and Acute Tubular Necrosis
| LABORATORY TEST FINDING | PRERENAL AZOTEMIA | ACUTE TUBULAR NECROSIS |
| Urinalysis | Normal or hyaline casts | Brown granular casts, cellular debris |
| Urine sodium concentration (mEq/L) | <20 | >40 |
| Fractional excretion of sodium (%) | <1 | >1 |
| Urine-to-plasma creatinine ratio | >40 | <20 |
Urinary indices are most helpful in oliguric patients.4 In general, an oliguric patient with a urine sodium concentration below 20 mEq/L and an FENa below 1% should be considered to have prerenal azotemia, whereas urine sodium concentration above 40 mEq/L and FENa above 1% suggest ATN. Values in patients with prerenal azotemia overlap somewhat with those in patients with nonoliguric ATN, particularly if the renal injury is mild and some capability to retain sodium has been preserved. Thus, intermediate values for urine sodium concentration and FENa are of little help in differentiating between the two conditions. The administration of mannitol or a loop diuretic within the several hours preceding urine collection also may make interpretation of urine values difficult because the urinary sodium will tend to be higher and the urine less concentrated, causing the results in prerenal azotemia to resemble those in intrinsic renal failure (Box 97-1).
Renal Imaging
Renal imaging is often helpful in evaluation of the patient with kidney dysfunction, particularly when obstruction is suspected. Contrast-enhanced computed tomography (CT) scanning provides an anatomic image of the urinary tract but does not provide an evaluation of renal function. The classic CT findings of obstruction are kidneys that are normal to large in size, nephrograms that become increasingly dense, and delayed opacification of dilated collecting systems. However, contrast-enhanced CT subjects the kidneys of an already azotemic patient to the risk of an additional potential insult from the contrast agent. Thus techniques such as ultrasonography and CT that do not involve contrast administration are much preferred in patients with preexisting renal insufficiency (Fig. 97-1).
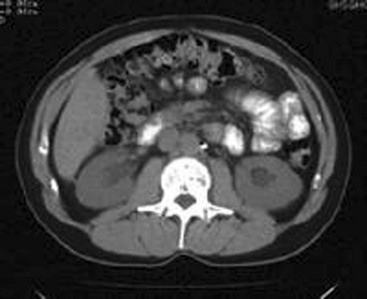
Figure 97-1 Computed tomography scan of bilateral hydronephrotic kidneys without intravenous contrast medium.
Computed Tomography.: Noncontrast CT may be useful in evaluating some azotemic patients. Hydronephrosis can be recognized without the use of contrast material. Often, dilated ureters also can be seen without contrast enhancement, and the level of obstruction can be determined. Moreover, the cause of obstruction (e.g., bilateral stones, lymphoma, retroperitoneal hemorrhage, metastatic cancer, retroperitoneal fibrosis) often can be delineated as well. Occasionally, obstruction severe enough to result in renal failure may not cause detectable proximal dilation of the urinary tract. Bilateral ureteral obstruction produced by malignancy or retroperitoneal fibrosis is the most important cause of this nondilated obstructive uropathy. When noninvasive studies yield negative results, the diagnosis of obstruction can be made by retrograde pyelography or by antegrade pyelography performed via a percutaneous nephrostomy.
Ultrasonography.: Ultrasonography allows accurate measurement of renal dimensions and is a safe and reasonably reliable method of excluding obstruction as a cause of AKI. The normal kidney shows an echo-free renal parenchyma surrounding the echogenic central urothelium of the renal pelvis and calices. The sonographic appearance of the kidney in obstruction is that of an enlarged central sonolucent area that spreads the normal central echo densities. A similar pattern may be produced by renal cysts, but without associated ureteral dilation. Dilation of the collecting system generally is apparent within 24 to 36 hours of the onset of obstruction, but obstruction may not be evident in patients who are evaluated early in the development of obstructive AKI.
Hematuria and Proteinuria
Principles of Disease
Microscopic hematuria often is discovered incidentally on routine urinalysis, but as little as 1 mL of blood in 1 L of urine can cause grossly appreciable hematuria. Although not invariably a sign of disease, the finding of hematuria calls for an effort to rule out any treatable underlying disorder. Both gross and microscopic hematuria are caused by similar disorders, but the amount of blood in the urine does not correlate with the severity or the seriousness of the underlying condition.2
The causes of hematuria can be divided into hematologic, renal, and postrenal; renal causes may be further classified as glomerular or nonglomerular (Box 97-2). Overall, the most common causes of nontraumatic hematuria, in roughly descending order of occurrence, are kidney stones, urinary tract infection (UTI), carcinoma of the kidney or bladder, urethritis, benign prostatic hypertrophy, and glomerulonephritis. The scope of the differential diagnosis can be narrowed by taking into account the patient’s age and sex and by distinguishing between upper and lower urinary tract sources. When gross hematuria is present, cystoscopy can determine whether blood is emerging from one or both ureteral orifices, thereby defining a source in the upper tract. Red cell casts indicate a renal source, as does associated proteinuria (excretion of more than 500 mg of albumin in 24 hours). In differentiating between proteinuria from renal parenchymal disease and that simply produced by admixture of urine with extravasated blood, a useful rule of thumb is that 1 mL of whole blood contains approximately 5 billion RBCs and approximately 50 mg of albumin.
Other clues to the cause are obtained by careful questioning. Because glomerulonephritis or interstitial nephritis may be caused by a variety of bacterial, viral, and parasitic infections, a history of recent infection is important. Symptoms suggestive of a multisystem disorder (e.g., systemic lupus erythematosus) also should be sought, as should a history of human immunodeficiency virus infection.5 Because drugs may cause acute interstitial nephritis (AIN), papillary necrosis, or hemorrhagic cystitis, a complete medication history is elicited. When hematuria is associated with anticoagulant use, significant underlying disease can be identified in about one third of patients.6 The family history may provide a clue to the presence of polycystic or other familial kidney disease, sickle cell disease, or renal calculi. A history of recent strenuous exercise is important to identify; 15 to 20% of normal persons exhibit hematuria after strenuous exercise. The mechanism is unclear, but the hematuria resolves spontaneously within a few days.
Radiography and Ultrasonography
The role of urinary tract imaging studies in the immediate evaluation of hematuria also is limited. Visualization of the urinary tract generally is helpful only when the history suggests renal colic or other disorders of the upper urinary tract (e.g., polycystic kidney disease, tumor, obstruction). CT without contrast is the imaging modality of choice.7 Ultrasonography can be used to determine kidney size and shape and to detect renal masses or obstruction. Further imaging studies, if indicated, should be planned after urologic consultation.
Proteinuria
Patients with the nephrotic syndrome are at increased risk for thromboembolic events, including deep vein thrombosis of the lower extremity, renal vein thrombosis, and pulmonary embolism. The reason for this propensity appears to be a hypercoagulable state that is complex and incompletely understood.8 Hyperlipidemia is another typical feature of the nephrotic syndrome; the mechanism is thought to be related indirectly to hypoalbuminemia and decreased oncotic pressure or viscosity. The major clinical significance of the nephrotic syndrome, however, is that it indicates the presence of an underlying renal process or systemic disease affecting the glomerulus (Box 97-3).
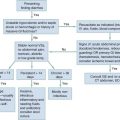
Acute Kidney Injury
The hallmark of AKI (formerly known as acute renal failure [ARF]) is progressive azotemia, which commonly is accompanied by a wide range of other disturbances, depending on the severity and duration of renal dysfunction. These include metabolic derangements (e.g., metabolic acidosis and hyperkalemia), disturbances of body fluid balance (particularly volume overload), and a variety of effects on almost every organ system (Box 97-4).
The causes of AKI may be divided into those that decrease renal blood flow (prerenal), produce a renal parenchymal insult (intrarenal), or obstruct urine flow (obstructive or postrenal). Identification of either a prerenal or a postrenal cause of AKI generally makes it possible to initiate specific corrective therapy; if these two broad categories of AKI can be excluded, an intrarenal cause is implicated. The renal parenchymal causes of AKI can be usefully subdivided into those primarily affecting the glomeruli, the intrarenal vasculature, or the renal interstitium. The term acute tubular necrosis denotes another broad category of intrinsic renal failure that cannot be attributed to specific glomerular, vascular, or interstitial causes (Fig. 97-2).4
Principles of Disease
Decreased renal perfusion that is sufficient to cause a decrease in the GFR results in azotemia. The possible causes can be grouped into entities causing intravascular volume depletion, volume redistribution, or decreased cardiac output (Box 97-5). Patients who have preexisting renal disease are particularly sensitive to the effects of diminished renal perfusion.
Patients who have congestive heart failure (CHF) or cirrhosis form an important subset of those with prerenal azotemia. These patients often are salt-overloaded and water-overloaded, yet their effective intra-arterial volume is decreased. Administration of diuretics has the potential to decrease intravascular volume further, resulting in decreased glomerular filtration and prerenal azotemia. For some patients with advanced CHF or hepatic disease, a state of chronic stable prerenal azotemia may be the best achievable compromise between symptomatic volume overload and severe renal hypoperfusion.9
Glomerular perfusion also may be decreased in patients with normal intravascular volume and normal renal blood flow who take angiotensin-converting enzyme (ACE) inhibitors or, more commonly, prostaglandin inhibitors. All nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, inhibit prostaglandin synthesis. Renal vasodilator prostaglandins are critical in maintaining glomerular perfusion in patients with conditions such as CHF, chronic renal insufficiency, and cirrhosis, in which elevated circulating levels of renin and angiotensin II decrease renal blood flow and GFR. In this setting, a decrease in the production of vasodilator prostaglandins may result in acute intrarenal hemodynamic changes and a reversible decrease in renal function.10 This phenomenon also is seen with the selective cyclooxygenase-2 inhibitor class of NSAIDs.11,12 Other risk factors include advanced age, diuretic use, renovascular disease, and diabetes. This entity is distinct from other renal complications of NSAIDs, including interstitial nephritis and papillary necrosis.
Postrenal (Obstructive) Acute Renal Failure
Obstruction is an eminently reversible cause of AKI and should be considered in every patient with newly discovered azotemia or worsening renal function. Obstruction may occur at any level of the urinary tract but most commonly is produced by prostatic hypertrophy or by functional bladder neck obstruction (e.g., secondary to medication side effects or neurogenic bladder) (Box 97-6). Intrarenal obstruction may result from intratubular precipitation of uric acid crystals (e.g., with tumor lysis), oxalic acid (as in ethylene glycol ingestion), phosphates, myeloma proteins, methotrexate, sulfadiazine, acyclovir, or indinavir.13 Bilateral ureteral obstruction (or obstruction of the ureter of a solitary kidney) may be caused by retroperitoneal fibrosis, tumor, surgical misadventure, stones, or blood clots. A sudden deterioration in renal function in the setting of diabetes mellitus, analgesic nephropathy, or sickle cell disease should suggest papillary necrosis.
Intrinsic Acute Renal Failure
Of the specific intrarenal disorders that cause AKI, glomerulonephritis, interstitial nephritis, and abnormalities of the intrarenal vasculature are amenable to specific therapy and are important to consider as possible causes. These entities are responsible for only 5 to 10% of cases of AKI in adult inpatients; most are caused by ATN. In adults in whom AKI develops outside the hospital, the incidence of glomerular, interstitial, and small-vessel disease is much greater. In children, these entities account for approximately one half of the cases of AKI (Box 97-7).4
Glomerular Disease.: Acute glomerulonephritis may represent a primary renal process or may be the manifestation of any of a wide range of other disease entities (see Box 97-7). Patients may have dark urine, hypertension, edema, or CHF (secondary to volume overload) or may be completely asymptomatic, in which case the diagnosis rests on an incidental finding on urinalysis. The hematuria associated with glomerular disease may be microscopic or gross and may be persistent or intermittent. Proteinuria, although often in the range of 500 mg/day to 3 g/day, not uncommonly is in the nephrotic range. The presence of hematuria, proteinuria, or red cell casts is highly suggestive of glomerulonephritis. In fact, red cell casts are essentially diagnostic of active glomerular disease, although occasionally they are seen with other types of renal disease. Conversely, the absence of red cell casts, proteinuria, and hematuria essentially excludes glomerulonephritis as the cause of AKI.
Interstitial Disease.: AIN most commonly is precipitated by drug exposure or by infection.14 Drug-induced AIN is poorly understood, but the absence of a clear relationship to the dose and the recurrence of the syndrome on rechallenge with the offending agent suggests that an immunologic mechanism is responsible. The most commonly incriminated drugs are the penicillins, diuretics, and NSAIDs. AIN has been reported in association with bacterial, fungal, protozoan, and rickettsial infections.
Patients with AIN classically have rash, fever, eosinophilia, and eosinophiluria, but it is common for one or more of these cardinal signs to be absent.15 Pyuria, gross or microscopic hematuria, and mild proteinuria are observed in some cases. A definite diagnosis sometimes can be made only on renal biopsy. Treatment of AIN is directed at removing the presumed cause; infections should be treated and offending drugs discontinued. Renal function generally returns to baseline over several weeks, although chronic renal failure has been reported to occur.
Intrarenal Vascular Disease.: Vascular disease of the kidney can be classified according to the size of the vessel that is affected. Disorders such as renal arterial thrombosis or embolism, which affect large blood vessels, must be bilateral (or affect a single functioning kidney) to produce AKI. Whether to attribute such cases of AKI to prerenal or intrarenal vascular causes is a matter of semantics. The most common cause of thrombosis probably is trauma; thrombosis also may occur after angiography or may be secondary to aortic or renal arterial dissection. Renal atheroembolism is thought to occur commonly—at least on a microscopic level—after arteriography but is an uncommon cause of AKI. Similarly, patients with chronic atrial fibrillation or infective endocarditis may experience embolization of the kidney but rarely develop AKI as a result. Renal arterial embolism can cause acute renal infarction, generally manifested by sudden flank, back, chest, or upper abdominal pain. Urinary findings, including hematuria, are variable. Fever, nausea, and vomiting are not uncommon; in some cases, evidence of embolization to other vessels provides a useful clue. The diagnosis usually is made by renal flow scanning or arteriography. Surgical embolectomy has been reported to restore function when undertaken within several hours of occlusion, but significant return of function has been documented in patients operated on as long as 6 weeks after total occlusion. This outcome presumably is possible because collateral circulation has developed in association with a preexisting partial occlusion.
Several diseases that affect the smaller intrarenal vessels can cause AKI (see Box 97-7). Patients whose disease is severe enough to cause ARF also generally are found to have hypertension, microangiopathic hemolytic anemia, and other systemic and organ-specific manifestations. Infection with Escherichia coli O157:H7 has emerged as a major cause of hemolytic uremic syndrome, an important cause of AKI in children.16
Malignant hypertension, although much less common since the advent of more effective antihypertensive therapy, has by no means disappeared. Patients with scleroderma (systemic sclerosis) may have “scleroderma renal crisis,”17,18 characterized by malignant hypertension and rapidly progressive renal failure. Whereas vasculitis associated with glomerular capillary inflammation typically causes gross or microscopic hematuria and formation of red cell casts, vascular involvement of the medium-size vessels, such as that produced by scleroderma, often spares the preglomerular vessels and tends not to produce an active urine sediment. Extrarenal manifestations (rash, fever, arthritis, pulmonary symptoms) are usually evident.
For malignant hypertension, both as a separate entity and as a part of scleroderma renal crisis, appropriate treatment can produce a gratifying remission of AKI. Patients with malignant hypertension have been reported to recover renal function after aggressive antihypertensive therapy, with temporary maintenance with dialysis if necessary.19 In patients with scleroderma renal crisis, specific therapy with ACE inhibitors has been shown to result in improvement in renal function in a significant proportion of cases.18
Acute Tubular Necrosis
The most common precipitant of ATN is renal ischemia occurring during surgery or after trauma and sepsis.20 The remainder of cases occur in the setting of medical illness, most commonly as a result of the administration of nephrotoxic aminoglycoside antibiotics or radiocontrast agents or in association with rhabdomyolysis. Multiple causes can be identified in some cases; in others a definitive cause is never established.
ATN is common in postoperative patients, although not all cases can be attributed to intraoperative hypotension or hemorrhage. Concomitant sepsis, increased age, preexisting renal disease, and other comorbid conditions are associated with a worse outcome.20,21
Nephrotoxins constitute the other major cause of ATN. Among the most prominent of these are the endogenous pigments myoglobin and hemoglobin. Rhabdomyolysis and ARF resulting from crush injuries first received widespread attention after their description in survivors of the London blitz during World War II, but many other causes of pigment nephropathy have been reported (Box 97-8). Hypotension secondary to fluid loss into damaged muscle is thought to worsen the effects of myoglobinuria on the renal tubule, as does acidemia. Hemolysis, resulting in the release of hemoglobin into the circulation and hemoglobinuria, can cause ATN but usually only in the presence of coexisting dehydration, acidosis, or other causes of decreased renal perfusion. ATN may be associated with the hemolysis of as little as 100 mL of blood.
ATN associated with rhabdomyolysis is often oliguric; it is characterized by rapid increases in the serum creatinine, potassium, phosphorus, and uric acid levels.22,23 Creatine released from muscle is metabolized to creatinine, which may result in serum creatinine increases of more than 2 mg/dL/day, in contrast to the increase of 0.5 to 1.0 mg/dL/day typically seen in other forms of AKI. The BUN/creatinine ratio often is less than 10 : 1. Intracellular potassium released from damaged muscle may raise the serum potassium by 1 to 2 mEq/L in several hours. Likewise, phosphate released from muscle may cause dramatic increases in the serum phosphate level. Uric acid, produced by metabolism of purines released from damaged muscle, may accumulate to levels high enough to cause acute uric acid nephropathy.
No biochemical parameter can be used to predict which patients who have rhabdomyolysis will experience AKI. In one classic study of patients in whom alcoholism, muscle compression, and seizures were the most common causes of rhabdomyolysis, AKI developed in only one third.22 Neither the degree of serum CK elevation, the presence (or absence) of myoglobinuria, nor the degree of hyperkalemia correlates well with the development of AKI.
Antibiotics and radiographic contrast agents are other nephrotoxins that commonly are implicated in the development of ATN. Aminoglycosides are the most commonly implicated antibiotics. Higher doses and longer duration of therapy are associated with higher serum drug levels, leading to greater accumulation of drug in the renal parenchyma and a greater likelihood of nephrotoxicity. Increased age, impaired renal function, dehydration, and exposure to other nephrotoxins are additional risk factors. Once-daily administration of a somewhat higher dose is associated with less nephrotoxicity but equal effectiveness.24,25
The most important risk factors for radiocontrast agent–induced ATN are preexisting renal insufficiency, diabetes mellitus, multiple myeloma, age older than 60 years, volume depletion, and higher doses of contrast material. Among these, preexisting renal insufficiency is the most important.26 Diabetic patients with a serum creatinine level less than 1.5 mg/dL are at low risk for the development of radiocontrast agent–induced ATN, whereas those whose serum creatinine is greater than 1.5 mg/dL are at significant risk. Multiple myeloma, particularly in patients who are dehydrated, is another reasonably well-documented risk factor. Advanced age also appears to make ATN more likely, possibly because of decreased renal mass and cortical blood flow. Volume depletion appears to be an independent risk factor, and aggressive volume expansion before contrast exposure has been shown to have a protective effect.27,28 Finally, large doses and repeated doses of contrast material are associated with increased risk of ATN, particularly if a second study is performed within 72 hours of the first.
N-acetylcysteine in combination with periprocedural hydration appears to decrease the incidence of contrast-associated ATN,29 although this has not been a consistent finding in published studies.30 Modest volumes of intravenous normal saline (3 mL/kg over 1 hour, followed by 1.5 mL/kg/hr for 4 hours after contrast exposure) appear to be effective in decreasing the likelihood of nephrotoxicity; sodium bicarbonate does not appear to offer an advantage over normal saline.30,31 Recent reviews continue to emphasize the importance of appropriately limiting exposure to contrast agents as well as ensuring preprocedural hydration.27
Clinical Features
The area of the bladder is palpated and percussed. A distended bladder is percussible when it contains 150 mL of urine, and the dome is palpable abdominally when it contains 500 mL. Ultrasonography can be used to detect bladder distention or postvoid residual volume if there is a question of urinary retention.32
Diagnostic Strategy
Radiography and Ultrasonography
Significant hydronephrosis usually is readily demonstrable by ultrasonography and may indicate either upper or lower tract obstruction. In questionable cases, or if bilateral ureteral obstruction is strongly suspected on the basis of clinical findings, the next step is retrograde urography performed by a urologist.33 CT imaging is less useful in this setting; in fact, intravenous radiocontrast material may compound the injury to the kidney.
Management
ED management of AKI is directed at reversing decreases in GFR and urine output (if possible) while minimizing further hemodynamic and toxic insults, maintaining normal fluid and electrolyte balance, and managing other complications of AKI as required. Because renal failure alters the metabolism and action of many drugs, often in ways that are not predictable, great care should be exercised in prescribing all medications. A compendium of guidelines for drug dosage in renal failure, such as the one by Aronoff and colleagues,34 is of great help for this purpose.
When prerenal and postrenal factors have been ruled out, the challenge to the emergency physician is to identify the cause of intrinsic renal AKI, keeping in mind the multitude of known possible causes (see Box 97-7). The clinical setting and physical and laboratory findings often allow the scope of the differential diagnosis to be considerably narrowed. The clinical picture is often most consistent with the broad category of ATN.
It has been noted repeatedly that patients who have oliguric AKI have a significantly higher mortality rate and a much greater risk of complications than those who are not oliguric. The difference in prognosis may simply reflect a more severe renal insult in patients who are oliguric, however, and it is not clear that interventions aimed at converting oliguric to nonoliguric AKI have an effect on renal function or mortality.20 Nevertheless, because nonoliguric patients are easier to manage, an attempt to increase urine flow is warranted.
Use of loop diuretics or mannitol often is effective in increasing urine flow when intravascular volume deficits have been corrected. Furosemide has not been shown to shorten the clinical course or affect mortality. Mannitol appears to be most useful when given at the time of or shortly after the renal insult; the recommended dose is 12.5 to 25 g given intravenously (IV). If urine output does not increase, further doses may cause hyperosmolality and clinically significant intravascular volume overload in patients with impaired renal function.35
Mannitol has been shown to prevent AKI in experimental models of myoglobinuria, presumably by inducing osmotic diuresis and decreasing intratubular deposition of pigment. Furosemide, on the other hand, has not consistently shown a beneficial effect. Other studies have suggested that myoglobin precipitates in an acid urine but not in an alkaline urine. Thus, aggressive volume repletion, alkalinization, and mannitol infusion have traditionally been recommended after crush injuries to reduce the likelihood or severity of AKI.23 This regimen also helps control hyperkalemia. Other evidence, however, suggests that aggressive volume resuscitation alone may be equally effective.23 When AKI has occurred, management is similar to that of other forms of AKI, but early dialysis may be required to control rapidly developing hyperkalemia, hyperphosphatemia, and hyperuricemia.
Patients who have radiocontrast agent–induced ATN usually require only supportive therapy but are often hospitalized and seen by a nephrologist. A more significant aspect of ED management is preventing the occurrence of contrast nephropathy, particularly by identification of risk factors in patients for whom contrast studies are being considered. BUN and serum creatinine levels should be checked before contrast exposure in patients with risk factors. Moreover, before contrast medium is administered to a high-risk patient, it should be established that there is a compelling reason to perform the contrast study and that there is no adequate alternative to use of a contrast agent. The patient should be volume repleted before the study, the administered dose of contrast agent should be kept as low as possible, and multiple studies should be avoided, as should concomitant use of other nephrotoxins. As noted previously, N-acetylcysteine given before and after contrast administration appears to decrease the incidence of contrast nephropathy. Intravenous saline given before and after contrast administration appears to be at least as important in prevention.27–31
Volume and Metabolic Complications
Hyperkalemia results in serious disturbances in cardiac electrophysiology that may culminate in cardiac arrest. Although some hyperkalemic patients note muscular weakness, most generally are asymptomatic until major manifestations of cardiotoxicity appear. Accordingly, detection of hyperkalemia is a primary consideration in these patients. ECG changes correlate only roughly with the serum potassium level. Mild hyperkalemia (serum potassium less than 6.0 mEq/L) may be cautiously observed without specific treatment while all exogenous sources of potassium are eliminated. If the serum potassium level is greater than 6.5 mEq/L, and particularly if ECG changes are present, urgent intervention is necessary. When cardiotoxicity must be reversed immediately (e.g., when there is hemodynamic compromise), intravenous calcium (10 mL of 10% calcium gluconate or calcium chloride infused over 2 minutes and repeated after 5 minutes if necessary) is the treatment of choice. Calcium directly antagonizes the membrane effects of hyperkalemia. Intravenous insulin (given with glucose to prevent hypoglycemia) temporarily shifts potassium to the intracellular space. Bicarbonate appears to be less effective in shifting potassium into cells than once thought.36 It should be used with caution in patients with renal failure because of its potential to cause volume overload and to provoke hypocalcemic tetany or seizures. The safety and efficacy of beta-agonists in hyperkalemic patients have been well documented; like insulin, inhaled albuterol (in a dose of 10-20 mg) causes potassium to move into cells, thereby controlling hyperkalemia for 2 hours or more.37,38 Elimination of potassium from the body can be promoted by use of a potassium-binding ion exchange resin (sodium polystyrene sulfonate [Kayexalate]) or, much more effectively, by enhancing urinary potassium excretion or by initiating dialysis.36 Hypocalcemia is a common feature of AKI and can develop rapidly after its onset. Vitamin D–dependent intestinal absorption of calcium is decreased in AKI because of decreased renal synthesis of 1,25-dihydroxyvitamin D. Another factor promoting hypocalcemia is the complexing of calcium with retained phosphate. Rhabdomyolysis-associated AKI in particular often is associated with the deposition of complexed calcium in muscle and other tissues. Asymptomatic hypocalcemia requires no immediate treatment, but subtle or frank tetany should be treated with intravenous calcium (10-20 mL of 10% calcium gluconate infused over several minutes).
Hyperphosphatemia resulting from decreased renal elimination of phosphate is another common feature of AKI. The serum phosphorus level usually ranges from 6 to 8 mg/dL but may be much higher with rhabdomyolysis or in catabolic states. A calcium-phosphate product greater than 70 may result in metastatic soft tissue calcification. Hyperphosphatemia often is treated with oral calcium-based antacids that bind ingested phosphate in the gut.39
Organ System Effects
Impaired erythropoiesis, shortened RBC survival, hemolysis, hemodilution, and GI blood loss all play a role in the normocytic normochromic anemia that usually accompanies AKI. Although mild thrombocytopenia may be present, it is the qualitative defect in platelet function, which is felt to be caused by the effect of circulating uremic toxins, that is more significant and that contributes to these patients’ bleeding tendencies. In patients with active bleeding or those in whom an invasive procedure is being contemplated, the prolonged bleeding time can be corrected pharmacologically.40 Administration of 1-deamino-8-D-arginine vasopressin (DDAVP) shortens the bleeding time within 30 minutes. Infusion of 10 units of cryoprecipitate normalizes the bleeding time in 1 to 2 hours, with a return to baseline in 24 hours.
Chronic Kidney Disease
Principles of Disease
The standard terminology for chronic renal failure has changed.41 Chronic kidney disease denotes kidney damage or decreased renal function for 3 months or more and is characterized by irreversible nephron loss and scarring. Chronic renal insufficiency, which denotes a condition in which GFR has been moderately reduced but not to a degree sufficient to cause clear-cut clinical symptoms, has been replaced by an indication of the degree to which GFR is reduced. End-stage renal disease, now termed kidney failure, describes a condition in which renal function has diminished to a low level and in which serious, life-threatening manifestations can be expected to occur without dialysis or transplantation. At this stage the kidneys often are shrunken and diffusely scarred to such a degree that it may be impossible to make an etiologic diagnosis, even on pathologic examination.
The causes of CKD are numerous; their relative frequency depends primarily on the population studied. As with AKI, they can be conveniently classified (Box 97-9) as prerenal (vascular), intrinsic renal (glomerular and tubulointerstitial), or postrenal (obstructive). Glomerular disease accounts for approximately one third to one half of the cases of CKD; in the United States, diabetic nephropathy forms the largest group of these. Hypertensive nephrosclerosis is another important cause, particularly among blacks, in whom it may be the cause of 25% or more of cases of CKD. Among children and adolescents, reflux nephropathy is the most common cause of CKD. Renal failure related to intravenous drug use or to human immunodeficiency virus disease is a major consideration in some populations. Clues to other specific causes may be gained from elements of the history, physical examination, or laboratory and imaging studies. Although determining the underlying cause of CKD can permit the underlying disease to be treated and make possible some improvement in renal function in some cases, this is the exception rather than the rule.
Clinical Features
Cardiovascular System
The cardiovascular system is perhaps most dramatically affected in CKD.42–44 Many of the manifestations can be attributed to the effects of chronic volume overload, anemia, hyperlipidemia, alterations in calcium and phosphorus metabolism, and volume- and hormonally mediated hypertension.44–47 Pericarditis, with or without pericardial fluid accumulation, also is common in CKD, particularly among patients who have not had dialysis.
Pulmonary Effects
Similarly, uremic pleuritis, with or without associated pleural fluid collections, may develop in some patients. So-called “uremic lung,” manifested radiographically by “batwing” perihilar infiltrates, represents pulmonary edema and is almost always caused by volume overload or myocardial dysfunction. Noninflammatory pleural effusion caused by volume overload also is fairly common. Of special importance in the ED evaluation is the fact that the radiographic appearance in pulmonary edema may at times be misleading, simulating an infectious lobar infiltrate or even assuming a nodular appearance in some cases.48
Dermatologic Features
The skin of patients with CKD has a characteristic yellowish tinge. Uremic frost, the result of deposition of urea from evaporated sweat on the skin, is a classic finding that, like “uremic fetor,” is seen only rarely now with the widespread use of dialysis (Fig. 97-3). Diffuse pruritus is often a major source of discomfort for the patient with CKD; in some cases it may be caused by calcium deposition in the skin secondary to derangements in calcium metabolism.
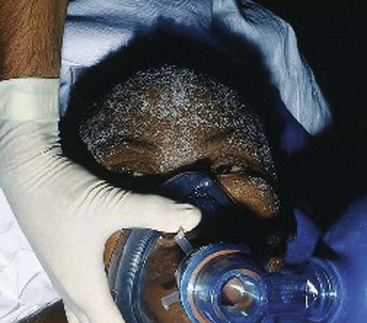
Figure 97-3 Uremic frost. Note the fine white powder on the skin of this patient with kidney failure.
The use of gadolinium-based contrast agents for magnetic resonance imaging (MRI) has been associated with the development of nephrogenic systemic fibrosis, a potentially fatal disorder that occurs in patients with chronic kidney failure.49
Diagnostic Strategies
Once it has been determined that the patient is in no immediate danger, the ED evaluation can establish that renal failure is indeed chronic rather than acute. An explicit history to that effect obtained from previous medical records or from the patient or family provides the most straightforward and reliable confirmation, as does the presence of a dialysis access device on physical examination. If such a history is unavailable, the finding of bilaterally small kidneys (readily detected on plain abdominal films or by ultrasonography) constitutes equally good evidence. However, the converse is not necessarily true—a finding of normal-sized or large kidneys does not rule out CKD (Box 97-10). In such cases, additional diagnostic steps are required to establish the diagnosis. A convincing history of the long-standing presence of the presenting symptoms or of symptoms such as nocturia may be helpful in suggesting chronicity, as may a history of familial kidney disease such as polycystic kidney disease or Alport’s syndrome. Laboratory abnormalities such as anemia, acidosis, hyperuricemia, hypocalcemia, and hyperphosphatemia can occur in patients with acute kidney failure as early as 10 days after onset. Although urinary findings likewise tend not to be helpful, the presence of broad waxy casts on microscopic examination is suggestive of chronic disease, whereas the finding of an “active” sediment (e.g., red cell casts) is good evidence for an acute process.
Although as a rule chronic kidney failure is irreversible and slowly progressive, an essential component of the ED evaluation is to exclude the possibility of potentially reversible factors (in effect, ruling out “acute on chronic” renal failure) and to ensure that treatable causes of CKD—disorders that if treated might allow for some return of renal function—have not been overlooked. These potentially reversible factors and treatable causes of CKD are important to keep in mind because they represent the only potential opportunity to reverse the patient’s disease rather than simply to manage its results (Box 97-11).
Primary among superimposed reversible factors are those that lead to decreased renal perfusion. Of these, the most common is volume depletion. Regardless of the initiating cause, the process is exacerbated by the diseased kidney’s impaired ability to conserve sodium and to concentrate the urine appropriately. Decreased renal perfusion caused by cardiac dysfunction of any cause is another extremely common and potentially reversible factor. An uncommonly encountered but important vascular cause of reversible deterioration of renal function is scleroderma renal crisis, a syndrome of accelerated hypertension and severe vasoconstriction in patients with underlying scleroderma that can be reversed by timely treatment with ACE inhibitors.18
Drugs and toxins constitute another important group of reversible factors. Not only may these agents exacerbate renal insufficiency by causing intravascular volume depletion (diuretics), decreased renal perfusion (antihypertensive agents), or increased catabolism (tetracycline); they also can cause ATN (radiographic contrast material), AIN (many drugs), or inhibition of renal prostaglandin synthesis (NSAIDs). Particularly noteworthy is the dramatic decrease in renal function produced when an ACE inhibitor is administered to a patient with renal insufficiency caused by bilateral renal artery stenosis (or renal artery stenosis in a solitary kidney).50
Management
Patients with CKD also are uniquely susceptible to iatrogenic illness. First, they are less able to handle fluid and solute loads than are normal persons. Just as important, the presence of renal failure significantly alters the metabolism and action of many drugs, often in ways that are not predictable (Box 97-12). Thus the dose and schedule of every administered agent, even apparently innocuous ones such as antacids, laxatives, antiemetics, or multivitamin preparations, should be carefully considered.51 For this purpose, ready access to a compendium such as the one by Aronoff and colleagues or availability of a hospital pharmacist for frequent consultations is invaluable.34 In general, consultation with the patient’s nephrologist is recommended on completion of the initial ED evaluation, because management and follow-up monitoring after the patient has left the ED often are complex.
Hyperkalemia
An ECG should be obtained whenever hyperkalemia is a possibility, and if signs of hyperkalemia are noted, appropriate therapy should be started immediately, even before laboratory confirmation of a high serum potassium level. ECG changes may be completely absent even when hyperkalemia is severe, however.52 Thus a normal ECG does not preclude the need for laboratory confirmation of a normal serum potassium level. A potassium level of 6 mEq/L should be considered potentially dangerous, even though many patients with CKD chronically tolerate levels somewhat above this threshold without ECG changes. A patient with CKD who is in cardiac arrest should be assumed to be hyperkalemic and treated accordingly while the usual resuscitative measures are taken.
The most rapidly effective treatment for hyperkalemia is intravenous calcium, which transiently reverses the cardiac manifestations of hyperkalemia without altering the serum potassium level or total-body potassium (Table 97-2). Calcium should be given to buy time until more definitive measures can take effect. It makes little sense to administer calcium in response to an elevated serum potassium level in the absence of manifestations of hyperkalemia on the ECG.
Table 97-2
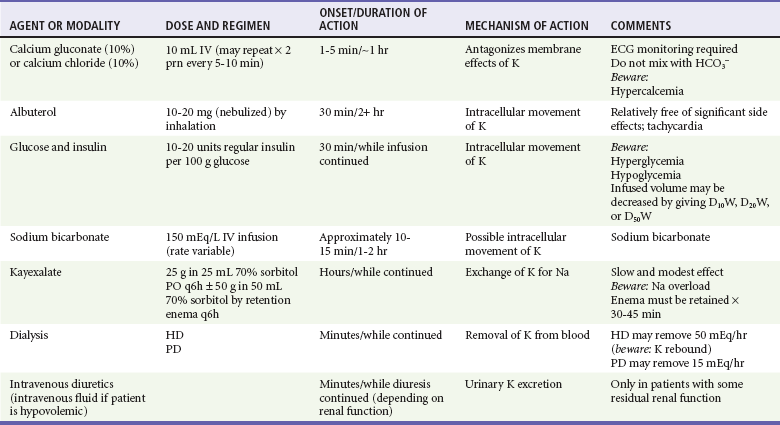
In treating hyperkalemia, it also is important to keep in mind the CKD patient’s limited ability to tolerate volume and solute loads (see Table 97-2). Thus, sodium bicarbonate infusion risks causing volume overload and precipitating pulmonary edema. Intravenous glucose and insulin treatment also entails some volume administration but is much more effective in any case, and hyperkalemia can be controlled for as long as the infusion is continued. Another effective temporizing measure is the administration of inhaled albuterol to promote movement of potassium into cells while more definitive maneuvers are being instituted.37,38 To remove potassium from the body, dialysis remains the most rapid and effective intervention. Sodium polystyrene sulfonate (Kayexalate), a resin that exchanges sodium for potassium ions, can be administered orally or rectally but is slow acting and only somewhat effective as a temporizing measure until dialysis (if necessary) can be instituted. In patients who still retain some renal function, the most effective way to treat hyperkalemia may be to administer an intravenous diuretic such as furosemide (if the patient is not hypovolemic) and to give volume if necessary. Large doses of diuretic may be necessary for a satisfactory diuresis to be induced. In light of the potential for ototoxicity with the use of loop-active diuretics, these drugs should be administered by slow infusion rather than by bolus and may be contraindicated in patients who also are receiving other potentially ototoxic agents. During the course of any of these therapeutic interventions, both the ECG and serum potassium levels must be monitored frequently.
Pulmonary Edema
Perhaps the most common ED problem in patients with CKD is pulmonary edema secondary to volume overload. Surprisingly, the diagnosis is not always straightforward. A history of increasing dyspnea on exertion or paroxysmal nocturnal dyspnea may be suggestive, but physical examination may not reveal the expected signs of CHF, and even chest radiography may be deceptive (Fig. 97-4).48 Recent weight gain or a body weight considerably over “dry weight” (typically more than 5 pounds) is the most reliable clue, and in the absence of convincing evidence of another cause for dyspnea, volume overload should be assumed to be the cause, and the patient should be treated accordingly.
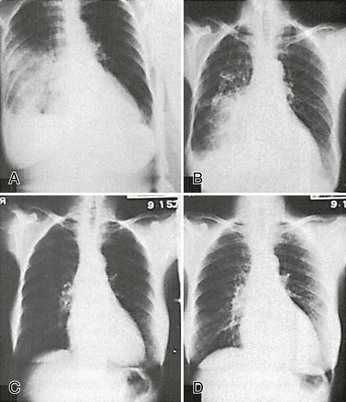
Treatment of pulmonary edema in the patient with CKD is of necessity somewhat different from that in other patients (Box 97-13).53,54 Arrangements for initiation of dialysis should be made as soon as possible because it is the most rapidly effective means to decrease intravascular volume in the absence of renal function. Other immediate measures should be instituted in the meantime. Although such measures may occasionally prove to be effective enough to avoid dialysis temporarily in patients who possess some residual renal function, it should nevertheless be anticipated that the response to even extremely aggressive medical therapy short of dialysis will be inadequate.
The CKD patient with pulmonary edema is placed in the sitting position, and high-flow oxygen is administered by mask. The use of continuous or bilevel positive airway pressure (CPAP or BL-PAP) delivered by face mask has been reported to be a useful adjunct in patients with CKD, as it is in patients without renal failure.55,56 Sublingual or topical nitroglycerin, or both, can be administered immediately and functions rapidly to reduce both preload and afterload; an intravenous infusion can be begun promptly and titrated to effect. Intravenous nitroprusside may have an advantage in producing more arteriolar dilation if the patient is hypertensive. Diuretics are not expected to be helpful unless the patient has retained a significant level of renal function.
Infection
Because infection is a major contributor to morbidity and mortality among patients with CKD, the possibility of serious infection should be entertained even when the expected classic findings are not all present.51,57–59 For example, bacteremia may manifest with fever alone, just as in other patients with impaired immunity. Patients with pneumonia may have only vague dyspnea or malaise, symptoms that may be attributed to volume overload or uremia. Thus, all diagnostic possibilities should be pursued, and empirical broad-spectrum antibiotic coverage often is advisable until infection has been ruled out in the hospital. Bacteremia resulting from vascular access infection is quite common in patients receiving hemodialysis, as is peritonitis in patients undergoing peritoneal dialysis.
UTI can occur even in patients with minimal urine output or those with long-standing renal failure. Urinary stasis is undoubtedly a predisposing factor.59 Upper UTI associated with a clinical picture typical of pyelonephritis or renal colic is seen most commonly in patients with polycystic kidney disease and requires parenteral treatment. A clinical diagnosis can be made presumptively in the ED, but invasive measures sometimes are necessary to document infection and guide therapy. For infected cysts, lipid-soluble antibiotics (e.g., clindamycin, trimethoprim-sulfamethoxazole) offer the best antibiotic penetration; surgical intervention for refractory infection sometimes becomes necessary, however.61
Dialysis
Indications for Dialysis.: The decision to initiate chronic dialysis in the patient with CKD generally is made by the patient’s nephrologist in the setting of gradually decreasing GFR and slowly progressive manifestations of renal failure. The absolute value of the BUN or serum creatinine generally is used only as a rough guide to when chronic dialysis should be instituted. Provision of vascular or peritoneal access usually has been arranged weeks to months before the anticipated initiation of dialysis, to allow the access site to mature and to minimize any mechanical complications of the procedure.
For patients who come to the ED with AKI, however, as well as for patients with CKD in whom acute problems have developed, it is the emergency physician who must be prepared to make the decision to arrange for dialysis to be provided emergently (Box 97-14). How urgently dialysis must be initiated depends not only on the severity and acuteness of the presenting problem but also on the availability of technical facilities and trained dialysis personnel and the effectiveness of available temporizing measures for the problem at hand.
Other severe electrolyte and acid-base disturbances, including diabetic ketoacidosis,61 may sometimes necessitate emergent dialysis. Occasional patients with renal failure and severe hypercalcemia uncontrollable by other modalities (e.g., patients with multiple myeloma causing both renal failure and hypercalcemia) may require dialysis. The occasional patient with renal failure in whom severe hypermagnesemia develops after inappropriate therapy or magnesium ingestion may require immediate dialysis to reverse life-threatening paralysis or cardiac dysrhythmia. Severe metabolic acidosis in the setting of renal failure is another indication for emergent dialysis, particularly if volume overload precludes the administration of reasonable amounts of bicarbonate. Of note, bicarbonate also can precipitate tetany and convulsions if administered IV (e.g., to treat acidosis or hyperkalemia) to a patient with hypocalcemia.
The occurrence of uremic symptoms or signs such as nausea, vomiting, lethargy, or twitching indicates a need for dialysis but does not necessitate immediate initiation of dialysis unless symptoms are severe. Pericarditis, even in the absence of cardiac tamponade, often is considered an indication for urgent dialysis,62 but it is not uncommon for pericarditis to occur in well-dialyzed CKD patients as well.63 In a previously undialyzed patient with progressive renal insufficiency, the appearance of pericarditis indicates that it is time to initiate dialysis, although not necessarily on an emergency basis.
Complications of Dialysis Therapy.: Optimal ED management of acute problems referred from the dialysis unit, or those occurring at home or in the interdialytic period, requires knowledge of and familiarity with the particular problems associated with CKD and dialysis.51,57–59 Consultation with the nephrologist or dialysis nurse is important in arranging a consistent care plan for the dialysis patient with an emergent condition and in ensuring appropriate further acute care or follow-up.
Complications of Hemodialysis:
Vascular Access–Related Complications: The performance of hemodialysis depends on reliable vascular access, and it is the vascular access device that is responsible for complications of dialysis that most often require evaluation in the ED setting. These problems must be attended to promptly to minimize the risk of losing the patient’s dialysis “lifeline.”
Similarly, if the patient reports that the thrill in the access has been lost, a vascular surgeon is consulted immediately. Although thrombolytic agents sometimes are used, definitive treatment generally is surgical revision.57,58 The access device should not be forcefully manipulated or irrigated because rupture of the vessel or venous embolization may result.
Infection of the vascular access is not uncommon and can result in persistent or recurrent bacteremia as well as loss of the access. Infection appears to be a consequence of contamination at the time of puncture for dialysis; most infections are caused by staphylococci typical of skin flora. Infections are more likely to occur in grafts than in native fistulae.64,65 The signs and symptoms of an access infection—redness, warmth, and tenderness over the site—often are obvious, but in many cases, localizing findings are absent and the patient has only fever or a history of recurrent episodes of fever and documented bacteremia. For this reason, it is common practice to obtain blood cultures for all patients on hemodialysis who have a fever without an obvious source of infection and to treat them presumptively for an access infection.66 A careful search for other sources of infection is done, however, before nonapparent access infection is concluded to be the cause. Infections such as odontogenic abscess, extremity cellulitis (particularly in diabetics), and perirectal abscess can easily be missed.
Although some nephrologists prefer to admit all dialysis patients with fever to the hospital, management of these patients on an outpatient basis often is possible, provided that they otherwise feel well and do not appear to be septic and provided that they can care for themselves at home and return promptly if their condition worsens. This course is made more practicable by the fact that they can be loaded with intravenous antibiotics that dependably maintain adequate blood levels until the time of the next scheduled dialysis treatment, at which time the culture and sensitivity test results can be checked and therapy adjusted accordingly. Vancomycin 1 to 1.5 g given IV as a single loading dose is the drug of choice in this situation because most access infections are staphylococcal and because this drug is only minimally hemodialyzable and needs to be given only every 5 to 7 days in the chronic dialysis patient. If a gram-negative infection also is thought to be likely, as in a patient who has had recent episodes of gram-negative bacteremia, a loading dose of a second drug (e.g., a third-generation cephalosporin or an aminoglycoside) also can be administered.66 Patients can be reloaded with these drugs at the end of their next hemodialysis session if culture results prove to be positive.
Non–Vascular Access–Related Complications: The hemodialysis procedure itself, which entails invasion of the vasculature, anticoagulation, and significant shifts of fluid and solutes, often is associated with acute complications such as hypotension, shortness of breath, chest pain, and neurologic abnormalities.
Hypotension: Hypotension that occurs after dialysis most commonly is the result of an acute reduction in circulating intravascular volume and the failure of the patient’s homeostatic mechanisms to compensate for it. Because hemodialysis is episodic, each treatment must remove the excess fluid that has accumulated over the period since the last dialysis (generally 2 to 3 days), and patients often are relatively volume-overloaded at the beginning of each treatment. With rapid removal of extracellular fluid, there is inadequate time for transcellular fluid shifts to replace intravascular volume. Antihypertensive medications, particularly beta-blockers, that are required when the patient is in a volume-expanded state can contribute to the hypotension when intravascular volume is normalized.
Most episodes of hypotension that occur during hemodialysis will resolve spontaneously or can be readily managed by either a decrease in blood flow rate or the infusion of small volumes of saline (to effect transient volume expansion) or hypertonic solutions (to reverse transient acute hypo-osmolality). Patients with significant hypotension who do not respond to these maneuvers often are brought to the ED for further evaluation. Patients on dialysis should be considered to be at risk for acute myocardial infarction, acute dysrhythmias, and sepsis. These are common causes of hypotension among all patients in the ED, and consideration should first be given to these entities (Box 97-15).51
Dialysis patients are commonly treated with epoetin or darbepoetin to prevent severe anemia, but untreated patients typically have low baseline hemoglobin levels, and acute blood loss may result in symptomatic angina or CHF. Serum levels of clotting factors are normal in CKD, but patients are routinely anticoagulated for each hemodialysis treatment. Although transient thrombocytopenia may occur during the dialysis procedure, the qualitative platelet defect characteristic of renal failure represents the most important factor in bleeding that continues beyond the peridialytic period.67 This abnormality is only partially reversed by dialysis but can be corrected by administration of DDAVP, which increases release of factor VIII–von Willebrand factor (vWF) polymers from vascular endothelium. DDAVP has been used successfully to normalize the bleeding time in preparation for surgery in patients with CKD. Cryoprecipitate and conjugated estrogen both have been shown to produce similar effects for a longer period.40
Similarly, an elevated central venous pressure is of little use in differentiating tamponade from underlying right-sided heart failure. Even a bedside ultrasonographic examination that shows pericardial fluid, although suggestive, is not proof that tamponade is present, because many dialysis patients chronically have pericardial effusions that do not cause hemodynamic compromise.63 Ultrasonographic demonstration of right ventricular diastolic collapse is more specific, but a definitive diagnosis of tamponade depends on the direct demonstration of equal pressures in the right and left atria on cardiac catheterization.
Shortness of Breath: Shortness of breath in dialysis patients generally is caused by volume overload. In the patient who becomes short of breath while being dialyzed, however, other causes must be sought—primarily sudden cardiac failure, pericardial tamponade, pleural effusion, or pleural hemorrhage. Air embolism and anaphylactoid reactions are unusual causes. Often, pneumonia or underlying reactive airway disease is responsible.
Chest Pain: Chest pain during dialysis must be taken seriously because cardiovascular disease is a leading cause of death in patients with CKD, and most episodes of chest pain occurring during dialysis are likely to be ischemic in origin.68,69 Most dialysis patients have risk factors for coronary artery disease, related to either CKD itself or the underlying condition that led to renal failure, and many have well-documented coronary artery disease.42,43 CKD is commonly associated with hypertension, hyperlipidemia, carbohydrate intolerance, and disturbances of calcium and phosphorus metabolism. In addition, dialysis patients may be anemic, and many are chronically volume-overloaded. During hemodialysis, these underlying factors may be added to acute physiologic stresses such as transient hypotension and hypoxemia, which often are associated with the dialysis procedure, thereby increasing myocardial oxygen demand while decreasing oxygen delivery.
The presence of renal failure and its associated electrolyte and acid-base disturbances does not in general obscure the usual ECG changes of angina or acute myocardial infarction. The pattern of the change of serum cardiac enzymes with acute infarction also is not altered by CKD, although the baseline level of these enzymes may be higher than in the general population. Troponin appears to perform best as a marker of infarction in patients with CKD.70–72 Treatment of ischemic chest pain is the same as for other populations.
Among nonischemic causes of chest pain, pericarditis should always be a consideration, even in the well-dialyzed patient. The presentation is essentially the same as in nonrenal patients; fever, a friction rub, or atrial dysrhythmias may be associated findings, and signs of pericardial effusion or early tamponade should be sought. Indomethacin often is effective in relieving pain, but some patients eventually require further measures, such as pericardiocentesis with corticosteroid instillation or pericardial stripping. Patients with pericarditis often receive more frequent or intensified dialysis because pericarditis is thought to be a marker for inadequate dialysis.62
Neurologic Dysfunction: Neurologic dysfunction manifesting during or immediately after hemodialysis often is caused by disequilibrium syndrome, a constellation of symptoms and signs that is thought to result from rapid changes in body fluid composition and osmolality during hemodialysis. It usually occurs only in patients with high BUN levels who are just starting hemodialysis. The syndrome does not occur with peritoneal dialysis. Typically, patients have headache, malaise, nausea, vomiting, and muscle cramps, but features in more severe cases may include altered mental status, seizures, or coma. Symptoms resolve over several hours as fluid and solutes are redistributed across cell membranes.
It is dangerous, however, to attribute an altered mental status to disequilibrium syndrome unless other potential causes have been ruled out (Box 97-16), particularly when symptoms persist, fluctuate, or worsen during a reasonable period of observation. Likewise, when seizures occur during dialysis, it is tempting but unwise to attribute them to disequilibrium syndrome without considering other, potentially serious causes, even in patients who have had seizures in the past. In particular, the finding of any new focal neurologic abnormality calls for, at a minimum, an immediate head CT scan to detect intracranial hemorrhage. Similarly, if fever or other evidence of infection is present, meningitis must be a serious consideration. Other considerations include hyperglycemia and hypoglycemia (especially in the diabetic patient), electrolyte abnormalities, hypoxic states, hypotension of any cause, and other toxic or metabolic causes. The treatment of seizures in patients with CKD is essentially the same as in other populations.
Complications of Peritoneal Dialysis.: As with hemodialysis, most of the complications of peritoneal dialysis are related to the dialysis access device, in this case the peritoneal catheter.51,57 In contrast to hemodialysis, however, the dialytic process in peritoneal dialysis occasions few immediate difficulties. Whatever volume or metabolic problems develop often are a consequence of the fact that the typical patient maintained on peritoneal dialysis is seen by a doctor or nurse only once a month.
Peritonitis is the most common complication of peritoneal dialysis. Fortunately, it is in general much less severe than other types of peritonitis and can be treated readily on an outpatient basis despite the continued presence of a foreign body—the Tenckhoff catheter—in the peritoneal cavity.57,73 Occasionally, when an episode of peritonitis responds poorly to antimicrobial therapy or when a patient has repeated episodes of peritonitis caused by the same organism, the catheter must be removed and the patient sustained with hemodialysis until the infection is completely cleared and a new catheter can be placed. Repeated infections do, however, carry the risk of permanently altering peritoneal permeability or effective surface area and necessitating a permanent switch to hemodialysis.
Peritonitis in patients on peritoneal dialysis presumably is caused by inadvertent bacterial contamination of the dialysate or tubing during an exchange, or by extension of an infection of the exit site or the subcutaneous tunnel into the peritoneal cavity. A majority of cases of peritonitis are caused by Staphylococcus aureus or Staphylococcus epidermidis, and most of the remainder (approximately 30%) by gram-negative enteric organisms.73 Fungal infections are uncommon but generally are refractory to medical therapy and are often considered an indication for catheter removal. Polymicrobial infection suggests direct contamination from the GI tract and mandates a search for the site of perforation or fistula, although such a source is identified in only a minority of cases. No organism is identified in approximately 10 to 20% of cases of peritoneal dialysis–associated peritonitis.
A common treatment regimen is a loading dose of vancomycin 30 mg/kg given IP, followed by further intraperitoneal doses every 5 to 7 days, plus ceftazidime or cefepime 1 g IP or gentamicin 0.6 mg/kg IP. The last two regimens are given as a loading dose followed by maintenance doses administered IP once daily at the time of an exchange.73 Heparin 500 to 1000 units also may be added to each bag of dialysate for the first few days of treatment to help reduce the formation of fibrin strands that may obstruct the catheter. Patients should be seen by the dialysis nurse in 24 to 48 hours for assessment of the response to therapy and adjustment of antibiotic therapy as necessary after review of the results of culture and sensitivity testing.
Perhaps the most serious potential pitfall in caring for the patient maintained on peritoneal dialysis with abdominal pain or other signs of peritonitis is to overlook other serious intra-abdominal conditions whose presentation may mimic that of peritonitis. Patients on peritoneal dialysis are at increased risk for abdominal wall or inguinal hernia because of chronically increased intra-abdominal pressures; previous abdominal surgery also places them at risk for hernia, as well as for obstruction secondary to adhesions. The manifestations of serious disorders unrelated to dialysis (e.g., acute appendicitis, diverticulitis, cholecystitis, acute pancreatitis, ischemic bowel, perforated viscus) also may be attributed to ordinary peritoneal dialysis–associated peritonitis, with the potential for disastrous consequences. The accessibility of the peritoneal fluid for examination may prove to be helpful in documenting the presence of an inflammatory process, but it also has the potential to mislead ED investigation of its cause. A finding of brownish or fecal material in the peritoneal drainage should suggest a ruptured viscus until proven otherwise, and immediate surgical consultation should be sought. Detection of localized tenderness, a palpable mass, or an incarcerated hernia on physical examination can be extremely helpful in making the diagnosis. Abdominal radiography may be useful for demonstrating the presence of ileus, but pneumoperitoneum may reflect only the introduction of air during a recent fluid exchange rather than a perforated viscus.74 Thus it is important to keep in mind the possibility that disorders other than peritonitis may underlie the patient’s symptoms, and astute clinical judgment rather than specific criteria should dictate requests for surgical consultation or decisions regarding hospitalization of the patient for observation.
Infection of the catheter exit site is another relatively common problem for which the patient on chronic peritoneal dialysis may seek care in the ED.73 This infection tends to be caused by typical skin flora and manifests with the usual local signs of infection. Although not serious in themselves, exit site infections should be taken seriously because they may lead to infection of the subcutaneous tunnel, which can cause repeated episodes of peritonitis and may ultimately necessitate removal of the catheter. Any visible exudate is cultured and Gram stained, and therapy with an oral antibiotic such as trimethoprim-sulfamethoxazole is started, pending the results of culture and sensitivity testing. The patient is instructed to cleanse the site meticulously several times a day using povidone-iodine or peroxide solution.
Key Concepts






References
1. Orth, SR, Ritz, E. The nephrotic syndrome. N Engl J Med. 1998;338:1202.
2. Cohen, RA, Brown, RS. Microscopic hematuria. N Engl J Med. 2003;348:2330.
3. Cockcroft, DW, Gault, MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31.
4. Singri, N, Ahya, SN, Levin, ML. Acute renal failure. JAMA. 2003;289:7473.
5. Kimmel, P, Barisoni, L, Kopp, JB. Pathogenesis and treatment of HIV-associated renal diseases: Lessons from clinical and animal studies, molecular pathologic correlations, and genetic investigations. Ann Intern Med. 2003;139:214.
6. Thaller, TR, Wang, LP. Evaluation of asymptomatic microscopic hematuria in adults. Am Fam Physician. 1999;60:1143.
7. Vieweg, J, et al. Unenhanced helical computerized tomography for the evaluation of patients with acute flank pain. J Urol. 2000;160:1465.
8. Singhal, R, Brimble, KS. Thromboembolic complications in the nephrotic syndrome: Pathophysiology and clinical management. Thromb Res. 2006;118:397.
9. Dagher, L, Moore, K. The hepatorenal syndrome. Gut. 2001;49:729.
10. Ulinski, T, Guigonis, V, Dunan, O, Bensman, A. Acute renal failure after treatment with non-steroidal anti-inflammatory drugs. Eur J Pediatr. 2004;163:148.
11. Perazella, MA, Tray, K. Selective cyclooxygenase-2 inhibitors: A pattern of nephrotoxicity similar to traditional nonsteroidal anti-inflammatory drugs. Am J Med. 2001;111:64.
12. Whelton, A, et al. Effects of celecoxib and naproxen on renal function in the elderly. Arch Intern Med. 2000;160:1465.
13. Perazella, MA. Drug-induced renal failure: Update on new medications and unique mechanisms of nephrotoxicity. Am J Med Sci. 2003;325:349.
14. Baker, RJ, Pusey, CD. The changing profile of acute tubulointerstitial nephritis. Nephrol Dial Transplant. 2004;19:8.
15. Fletcher, A. Eosinophiluria and acute interstitial nephritis. N Engl J Med. 2008;358:1760.
16. Garg, AX, et al. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: A systematic review, meta-analysis, and meta-regression. JAMA. 2003;290:1360.
17. Adams, BD. Scleroderma renal crisis [letter]. Ann Emerg Med. 2003;42:713.
18. Steen, VD, Costantino, JP, Shapiro, AP, Medsger, TA, Jr. Outcome of renal crisis in systemic sclerosis: Relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med. 1990;113:352.
19. Bakir, AA, Bazilinski, N, Dunea, G. Transient and sustained recovery from renal shutdown in accelerated hypertension. Am J Med. 1986;80:172.
20. Esson, ML, Schrier, RW. Diagnosis and treatment of acute tubular necrosis. Ann Intern Med. 2002;137:744.
21. Nash, K, Hafeez, A, Hou, S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39:930.
22. Gabow, PA, Kaehny, WD, Kelleher, SP. The spectrum of rhabdomyolysis. Medicine (Baltimore). 1982;61:141.
23. Bosch, X, Poch, E, Grau, JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361:62.
24. Barza, M, Ioannidis, JP, Cappelleri, JC, Lau, J. Single or multiple daily doses of aminoglycosides: A meta-analysis. BMJ. 1996;312:338.
25. Hatala, R, Dinh, T, Cook, DJ. Once-daily aminoglycoside dosing in immunocompetent adults: A meta-analysis. Ann Intern Med. 1996;124:717.
26. McCullough, PA. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419.
27. Sinert, R, Doty, CI. Update: Prevention of contrast-induced nephropathy in the emergency department. Ann Emerg Med. 2009;54:e1.
28. Mueller, C, et al. Prevention of contrast media-associated nephropathy: Randomized comparison of 2 hydration regimens in 1620 patients undergoing coronary angioplasty. Arch Intern Med. 2002;162:329.
29. Kelly, AM, Dwamena, B, Cronin, P, Bernstein, SJ, Carlos, RC. Meta-analysis: Effectiveness of drugs for preventing contrast-induced nephropathy. Ann Intern Med. 2008;148:284.
30. Zoungas, S, et al. Systematic review: Sodium bicarbonate treatment regimens for the prevention of contrast-induced nephropathy. Ann Intern Med. 2009;151:631.
31. Brar, SJ, et al. Sodium bicarbonate vs. sodium chloride for the prevention of contrast medium-induced nephropathy in patients undergoing coronary angiography. JAMA. 2008;300:1038.
32. Aquilera, PA, Choi, T, Durham, BA. Ultrasound-guided suprapubic cystostomy catheter placement in the emergency department. J Emerg Med. 2004;26:319.
33. Lyons, K, Matthews, P, Evans, C. Obstructive uropathy without dilatation: A potential diagnostic pitfall. Br Med J (Clin Res Ed). 1988;296:1517.
34. Aronoff, GR, Bennett, WM, Berns, JS. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults and Children, 5th ed. Philadelphia: American College of Physicians; 2007.
35. Better, OS, Rubinstein, I, Winaver, JM, Knochel, JP. Mannitol therapy revisited (1940-1997). Kidney Int. 1997;52:886.
36. Weisberg, LS. Management of severe hyperkalemia. Crit Care Med. 2008;36:3246.
37. Allon, M, Dunlay, R, Copkney, C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426.
38. Montoliu, J, Almirall, J, Ponz, E, Campistol, JM, Revert, L. Treatment of hyperkalemia in renal failure with salbutamol inhalation. J Intern Med. 1990;228:35.
39. Kestenbaum, B. Mineral metabolism disorders in chronic kidney disease. JAMA. 2011;305:1138.
40. Hedges, SJ, Dehoney, SB, Hooper, JS, Amanzadeh, J, Busti, AJ. Evidence-based treatment recommendations for uremic bleeding. Nat Clin Pract Nephrol. 2007;3:138.
41. Levey, AS, et al. National Kidney Foundation practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Ann Intern Med. 2003;139:137.
42. Luke, RG. Chronic renal failure—a vasculopathic state. N Engl J Med. 1998;339:841.
43. Shlipak, MG, et al. Cardiovascular mortality risk in chronic kidney disease. JAMA. 2005;293:1737.
44. Sarnak, MJ, et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation. 2003;108:2154.
45. Goodman, WG, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478.
46. Cheung, AK, et al. Atherosclerotic cardiovascular disease risks in chronic hemodialysis patients. Kidney Int. 2000;58:353.
47. Collins, AJ, et al. Chronic kidney disease and cardiovascular disease in the Medicare population. Kidney Int. 2003;64:524.
48. Kohen, JA, Opsahl, JA, Kjellstrand, CM. Deceptive patterns of uremic pulmonary edema. Am J Kidney Dis. 1986;7:456.
49. Agarwal, R, et al. Gadolinium-based contrast agents and nephrogenic systemic fibrosis: A systematic review and meta-analysis. Nephrol Dial Transplant. 2009;24:856.
50. Hricik, DE, et al. Captopril-induced functional renal insufficiency in patients with bilateral renal-artery stenoses or renal-artery stenosis in a solitary kidney. N Engl J Med. 1983;308:373.
51. Wolfson, AB, Singer, I. Hemodialysis-related emergencies: Part I. J Emerg Med. 1987;5:533.
52. Szerlip, HM, Weiss, J, Singer, I. Profound hyperkalemia without electrocardiographic manifestations. Am J Kidney Dis. 1986;7:461.
53. Gehm, L, Propp, DA. Pulmonary edema in the renal failure patient. Am J Emerg Med. 1989;7:336.
54. Venkat, A, Kaufmann, KR, Venkat, K. Care of the end-stage renal disease patient on dialysis in the ED. Am J Emerg Med. 2006;24:847.
55. Huff, JS, Whelan, TV. CPAP as adjunctive treatment of severe pulmonary edema in patients with ESRD. Am J Emerg Med. 1994;12:388.
56. Sacchetti, A, McCabe, J, Torres, M, Harris, RL. ED management of acute congestive heart failure in renal dialysis patients. Am J Emerg Med. 1993;11:644.
57. Pastan, S, Bailey, J. Dialysis therapy. N Engl J Med. 1998;338:1428.
58. Ifudu, O. Care of patients undergoing hemodialysis. N Engl J Med. 1998;339:1054.
59. Chaudhry, A, Stone, WJ, Breyer, JA. Occurrence of pyuria and bacteriuria in asymptomatic hemodialysis patients. Am J Kidney Dis. 1993;21:180.
60. Gabow, PA, Bennett, WM. Renal manifestations: Complication management and long-term outcome of autosomal dominant polycystic kidney disease. Semin Nephrol. 1991;11:643.
61. Blicker, J, Herd, AM, Talbot, J. Diabetic ketoacidosis in the dialysis-dependent patient: Two case reports and recommendations for treatment. Can J Emerg Med. 2004;6:281.
62. Lundin, AP. Recurrent uremic pericarditis: A marker of inadequate dialysis. Semin Dial. 1990;3:5.
63. Frommer, JP, Young, JB, Ayus, JC. Asymptomatic pericardial effusion in uremic patients: Effect of long-term dialysis. Nephron. 1985;39:296.
64. Nassar, GM, Ayus, JC. Infectious complications of the hemodialysis access. Kidney Int. 2001;60:1.
65. Allon, M. Dialysis catheter-related bacteremia: Treatment and prophylaxis. Am J Kidney Dis. 2004;44:779.
66. Mermel, LA, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:1.
67. Eberst, ME, Berkowitz, LR. Hemostasis in renal disease: Pathophysiology and management. Am J Med. 1994;96:168.
68. Bleyer, AJ, Russell, GB, Satko, SG. Sudden and cardiac death rates in hemodialysis patients. Kidney Int. 1999;55:1553.
69. Herzog, CA. Acute myocardial infarction in patients with end-stage renal disease. Kidney Int. 1999;56:S130.
70. Watnick, S, Perazella, MA. Cardiac troponins: Utility in renal insufficiency and end-stage renal disease. Semin Dial. 2002;15:66.
71. McCullough, PA, et al. Performance of multiple cardiac biomarkers measured in the emergency department in patients with chronic kidney disease and chest pain. Acad Emerg Med. 2002;9:1389.
72. Donnino, MW, et al. Prevalence of elevated troponin I in end-stage renal disease patients receiving hemodialysis. Acad Emerg Med. 2004;11:979.
73. Li, PK, et al. Peritoneal dialysis-related infections recommendations: 2010 update. Perit Dial Int. 2010;30:393.
74. Kiefer, T, Schenk, U, Weber, J, Hübel, E, Kuhlmann, U. Incidence and significance of pneumoperitoneum in continuous ambulatory peritoneal dialysis. Am J Kidney Dis. 1993;22:30.