[level-membership-for-critical-care-medicine-category]
13 Renal Disorders
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• Urine output will decrease if cardiac output and renal blood flow are compromised; as a result, urine output is a sensitive indicator of cardiac output.
• The normal hourly volume of urine output in children is small, and a small compromise in urine volume may indicate a significant compromise in renal perfusion or function.
• A child’s glomerular filtration rate will approach adult values by about 3 years of age.
• Renal failure may be present in a child with a low, normal, or high volume of urine output.
• Accurate measurement of urine volume and composition provide fundamental data on which clinical decisions are made.
• The modified pRIFLE is a classification system to standardize the definition of acute kidney injury (AKI) in pediatric patients. The acronym, pRIFLE stands for: pediatric risk of renal dysfunction, injury to the kidney, failure of kidney function, loss of kidney function, and end-stage kidney disease. Studies have shown that use of the pRIFLE enhances the classification of AKI epidemiology and the course of AKI in critically ill pediatric patients.
• Renal replacement therapies differ significantly in their effectiveness in treating AKI and its complications. It is therefore helpful for the nurse to be aware of the advantages and disadvantages of each therapy so that the nurse can participate in decisions regarding the best treatment modality for each patient.
Essential anatomy and physiology
Kidney Structure
Gross Anatomy
The kidneys lie anterior and lateral to the twelfth thoracic and first, second, and third lumbar vertebrae and behind the abdominal peritoneum; therefore they are retroperitoneal structures. The kidneys are embedded in a mass of fatty tissue called the adipose capsule, and each capsule is enclosed in the renal fascia (Fig. 13-1). The kidneys are not secured to the abdominal wall, but are held in position by the renal fascia and the large renal arteries and veins. The adipose capsule and the pararenal fat help to protect the kidney and keep it in place.
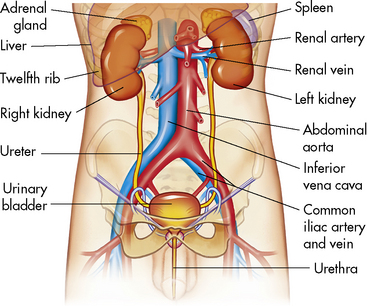
Fig. 13-1 Components of the urinary system.
(From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7. St Louis, 2010, Mosby.)
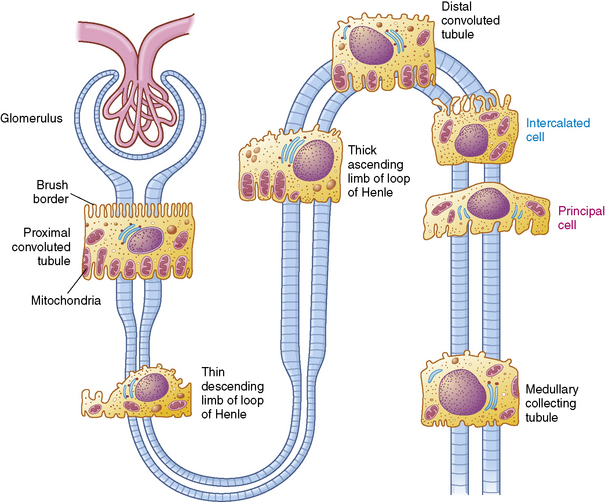
A longitudinal section of the kidney shows the three general areas of renal structure: the cortex, the medulla, and the pelvis (Fig. 13-2). The renal cortex is the outer portion of the kidney. It has a granular appearance and extends in fingerlike projections into the medullary areas. The cortex contains most of the nephrons, the smallest functioning unit of the kidney. The cortex also contains all glomeruli, the proximal and distal convoluted tubules, and the first parts of the loop of Henle and the collecting ducts.
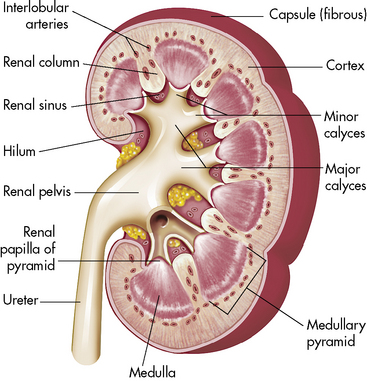
Fig. 13-2 Cross-section of the kidney.
(From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7. St Louis, 2010, Mosby.)
The functioning unit of the kidney is the nephron, which consists of a vascular component and a tubular component (Fig. 13-3). Each kidney contains approximately 1 million distinct nephrons. Eighty-five percent of all nephrons originate in the outermost area of the cortex. The remaining nephrons are the juxtamedullary nephrons that originate in the inner cortical area. The long loops of Henle from the juxtamedullary nephrons that extend deep into the medulla lie parallel to the medullary collecting ducts and play an important role in the concentration of urine (see Evolve Fig 13-1 in the Chapter 13 Supplement on the Evolve Website).
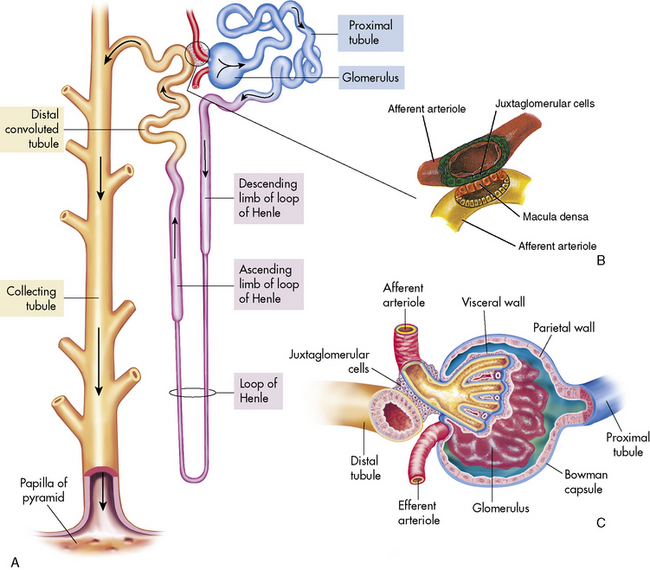
Renal Vasculature
The afferent arteriole enters the glomerular capsule and divides to form the glomerulus, a tuft of capillaries that allows filtration of plasma through the capillary membranes. The glomerular capillaries do not recombine into venous channels, but instead recombine into a second arteriole called the efferent arteriole (Fig. 13-4). Because arterioles are present at either end of the glomerular capillary system, constriction or dilation of these arterioles will alter the resistance to flow through the glomerular capillaries and thus will regulate glomerular filtration.
Renal Tubules and Collecting Ducts
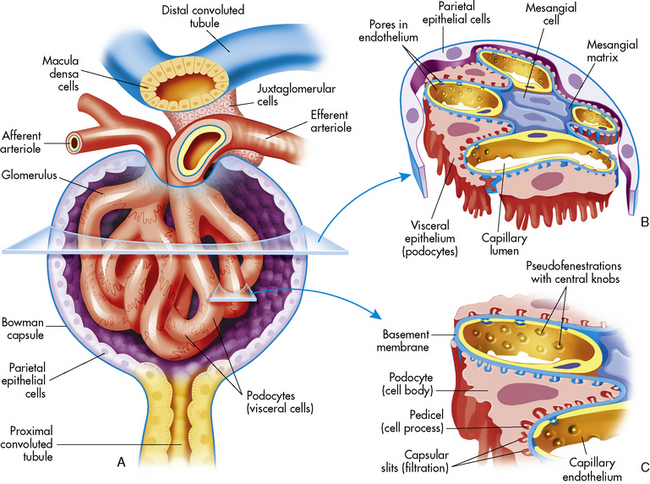
The tubular component of the nephron begins as a single layer of flat epithelial cells surrounding the glomerulus. This layer is known as Bowman’s capsule (Fig. 13-4, B). Filtered plasma from the glomerular capillaries will enter Bowman’s capsule and flow into a coiled tubule called the proximal tubule, also known as the proximal convoluted tubule (Fig. 13-5).
Ureters
The two ureters conduct urine from the renal pelvis to the urinary bladder; they are located behind the peritoneum and they descend through the pelvic cavity, crossing over the common iliac arteries. The ureters are attached to the bladder at an oblique angle; they enter the bladder laterally and tunnel between the bladder mucosa and detrusor muscle, creating a flap-valve design that normally prevents reflux of urine into the ureters during bladder contraction.30
Each ureteral wall has three layers: an inner epithelial lining, a middle muscular layer, and the outer fibrous layer that is continuous with the renal capsule. The middle muscular portion of the ureter consists of both a circular and a longitudinal muscle layer. The circular muscles propel the urine toward the bladder by peristaltic contraction, and they generate enough pressure to overcome the resistance caused by the oblique ureteral insertions into the bladder. Contraction of the longitudinal fibers opens the lumen of the ureter. These ureteral muscle fibers are innervated by fibers from the aortic, spermatic or ovarian, and hypogastric plexuses. Peristalsis persists even when the ureter is denervated, allowing ureters to be transplantable.55
The Bladder and Urethra
The urinary bladder is a hollow, muscular organ that stores urine. There are three openings in the bladder wall: the entrances of the two ureters and the exit of the urethra. These openings form the corners of a triangle, called the trigone. There is a dense area of smooth (involuntary) muscle around the neck of the bladder at the orifice of the urethra; this muscle constitutes the internal sphincter. The urethra extends from the urinary bladder to the body surface. At the point where the urethra passes through the muscles of the pelvic floor, striated (voluntary) circular muscles form an external sphincter.55
If the spinal cord is damaged above the sacral spinal level, the patient initially loses all micturation reflexes because inhibitory and facilitory reflexes from the brain cannot be transmitted through the injured spinal cord. Later, however, simple spinal reflexes can return and the patient can void when bladder distension is sufficient. In this case, the bladder reflex will be initiated at the volume of urine that is usually present in the bladder during the patient’s convalescent period.45
Glomerular Function
Filtration Physiology
Intravascular colloid osmotic pressure, or oncotic pressure, is the pressure opposing free water movement out of the vascular space. It is generated by dissolved proteins, ions, and other particles that are normally present in the blood. Larger particles such as proteins cannot move readily across a capillary membrane; therefore they remain in the vascular space, exerting an osmotic pressure of approximately 35 mm Hg. This oncotic pressure opposes hydrostatic filtration from the vascular space.55
The hydrostatic pressure present in Bowman’s capsule is the pressure exerted on the glomerulus by fluid in the Bowman’s capsule and collecting ducts. This pressure is normally 10 to 15 mm Hg and opposes fluid filtration from the glomerulus. Because tubular fluid is normally protein-free, the oncotic pressure in Bowman’s capsule is normally negligible. (See Evolve Fig. 13-2 in the Chapter 13 Supplement on the Evolve Website for a diagram of these pressures.)

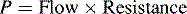
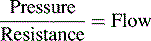
Glomerular Filtration Rate (GFR)
Renal function can be evaluated by calculating the GFR. The GFR, in turn, is roughly equivalent to the creatinine clearance; therefore it can be estimated by calculating the creatinine clearance. This estimate should not, however, be the sole means of determining renal function.56
Creatinine is a small molecule byproduct of skeletal muscle creatine metabolism. Creatinine is released at a near constant rate into the bloodstream, is filtered freely at the glomerulus, and is not broken down, reabsorbed, or synthesized by the renal tubules. Only a tiny amount of creatinine is secreted by the renal tubules. In effect, all of the creatinine that is filtered from the vascular space at the glomerulus remains in the urine and can be measured, and the creatinine clearance mirrors the GFR.62

It is important to note that laboratory determination of serum creatinine concentration may be affected by some cephalosporin antibiotics. For this reason the blood sample for analysis of the serum creatinine level should be obtained when antibiotic drug levels are at their lowest.7
Tubular Function
Reabsorption
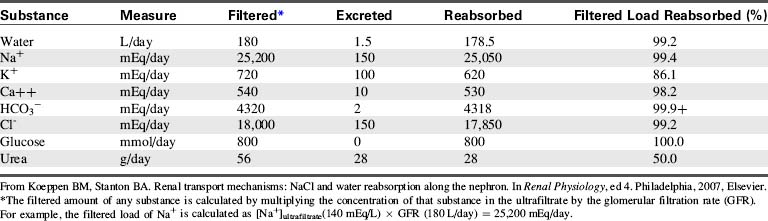
Table 13-1 summarizes the work of the renal tubular cells in the process of reabsorption. An average of 180 L of water (protein-free plasma) is filtered through the glomerulus of an adolescent per day, and yet the average urine output is 1.5 L/day. This means that 178.5 L of water is reabsorbed out of the tubular lumen back into the body’s circulation per day.
Table 13-1 Filtration, Excretion, and Reabsorption of Water, Electrolytes, and Solutes by the Kidneys
Transport Maximum and Thresholds
If the renal threshold and transport maximum are approximately equal to the daily filtered load of a substance, then the kidneys participate in regulation of the serum concentration of the substance. In such a case, a slight increase or decrease in plasma and filtered concentration of the substance changes its rate of renal reabsorption and excretion, so the serum concentration returns to normal. The renal threshold and transport maximum for phosphate are close to the normal daily filtered load of phosphate, so the serum phosphate concentration is regulated by kidney tubular function. Phosphate transport and reabsorption also will be affected by the serum calcium concentration, parathyroid hormone (PTH), and adrenal cortical hormones.63
Reabsorption and Secretion in the Proximal Tubule
The selective reabsorption of solute begins in the proximal tubules. Approximately 67% of the filtered water, Na+, Cl−, K+, and other solutes such as bicarbonate are reabsorbed in the proximal tubule. In addition, the proximal tubules normally reabsorb all filtered amino acids and glucose.63
Sodium
The primary mechanism for regulation of intracellular and extracellular fluid volume involves renal sodium excretion.63 Sodium is filtered freely at the glomerulus, so its concentration in the proximal glomerular filtrate is identical to its plasma concentration. Sodium is reabsorbed by an active transport mechanism; the mechanism is carrier-mediated and requires energy so the sodium can move against a gradient. Sodium is not secreted into the tubules.
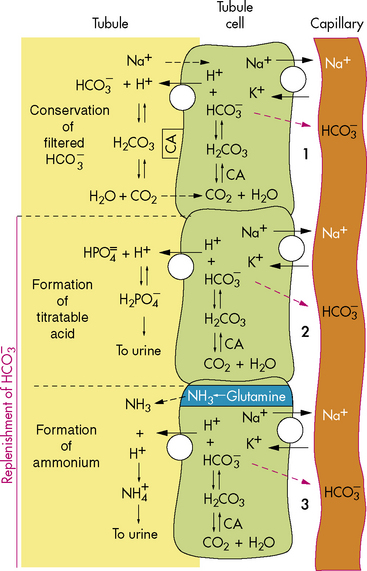
Bicarbonate and Hydrogen Ions
As a result of this process, for every bicarbonate ion that combines with a hydrogen ion in the lumen of the tubule, a bicarbonate ion ultimately will diffuse into the peritubular capillaries (Fig. 13-6). This secretion of hydrogen ions and reabsorption of bicarbonate ions occurs along the length of the renal tubules, but 90% of bicarbonate reabsorption occurs in the proximal tubule. (See section, Regulation of Acid-Base Balance.)
Potassium
Nearly all filtered potassium is reabsorbed by the proximal tubule, and the remaining potassium is reabsorbed in the ascending limb. The proximal reabsorption of the filtered potassium occurs at a constant rate and does not alter, despite the presence of hyperkalemia or hypokalemia. Potassium is also secreted by the distal convoluted tubule and cortical collecting duct, and the rate of K+ reabsorption or secretion in these tubular segments depends on a variety of hormones and factors. Overall, the rate of renal K+ excretion is determined by the distal convoluted tubule and the cortical collecting duct.63 The net result is normally a continuous loss of potassium in the urine.
Calcium
Calcium reabsorption is controlled by parathyroid hormone. Just as for sodium reabsorption, 80% to 90% of filtered calcium is reabsorbed in the proximal tubule and loop of Henle; only 10% of filtered calcium enters the distal tubule for concentration adjustments. When sodium reabsorption is inhibited by loop diuretics, calcium excretion is enhanced.79 Thiazide diuretics increase calcium reabsorption.
Drugs
The glomerulus is nonselective in its filtration of solutes, because the glomerular membrane does not restrict the passage of small molecules. Most drugs are of a small molecular size, and only a fraction of any drug is bound to serum albumin; most will filter into the tubular fluid. Changes in the GFR or in the degree of protein binding will alter the amount of drug present in the glomerular filtrate. Protein binding of a drug can be influenced by competition between drugs for the same protein binding sites (see Chapter 4).
The Loop of Henle
The loop of Henle, located within the renal cortex and the medulla, provides a countercurrent mechanism for urine concentration. The descending limb of the loop of Henle does not transport sodium or chloride actively, but it is highly permeable to sodium and water. Thus, as the filtrate passes through the descending limb of the loop, it becomes progressively more concentrated. The osmolality can increase from 300 to 1200 mOsm/L between the beginning of the descending limb and the tip of the loop of Henle (Fig. 13-7).
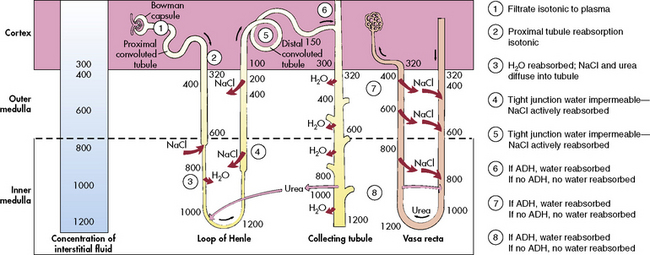
The blood vessels surrounding the loop of Henle form a hairpin loop structure, called the vasa recta. The vasa recta consists of capillaries that run parallel to the loop of Henle and the collecting ducts (for an illustration, see Evolve Fig. 13-1 in the Chapter 13 Supplement on the Evolve Website). As these capillaries follow the loop of Henle into the interstitium of the renal medulla, where osmolality is high (as the result of the tubular countercurrent mechanism), water shifts out of capillaries into the interstitial fluid, and sodium and chloride move from the interstitial fluid into the capillaries.
The Distal Tubule and Collecting Ducts
The distal tubule arises from the ascending limb of the loop of Henle; its thick cellular structure is distinct from the thin cells of the ascending loop. Thick cuboidal cells continue up through the renal cortical area to a point where the distal tubule is in direct contact with the afferent arteriole of its glomerulus. At this junction, the distal tubule cells become more densely packed and more columnar, and the muscle cells of the arteriole enlarge and take on a granular appearance. This point of contact between the distal tubule and the glomerular afferent arteriole is called the juxtaglomerular apparatus (see Fig. 13-3).
Renin, Aldosterone, and Antidiuretic Hormone
Renin is secreted from the polkissen cells of the afferent arteriole in the juxtaglomerular apparatus. In turn, renin forms angiotensin I from renin substrate (a circulating peptide from the liver). The amounts of renin released and angiotensin formed are determined by the renal perfusion pressure, sympathetic nervous system stimulation, circulating vasoactive substances, and changes in electrolyte concentration.55
Angiotensin I circulates to the lung and is converted enzymatically to angiotensin II. Angiotensin II produces peripheral vasoconstriction and an increase in aldosterone secretion, which increases renal sodium and water reabsorption. These effects should increase intravascular volume (Fig. 13-8). Angiotensin I and II are destroyed by angiotensinase, an enzyme that is present in plasma and secreted by a variety of organs, such as the kidney, intestine, and liver.
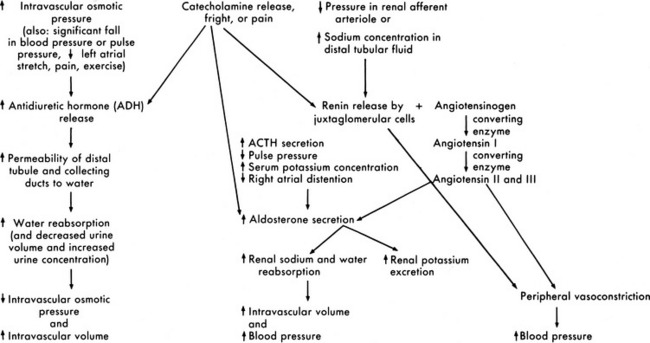
Fig. 13-8 Renal response to changes in extracellular fluid volume and electrolyte concentration or stress.
Aldosterone is secreted by the adrenal cortex in response to pituitary adrenal corticotropic hormone (ACTH) secretion and a variety of other stimuli. A fall in the pulse pressure, decreased stretch of the right atrium, and an increased serum potassium concentration all stimulate aldosterone secretion.63 An important stimulus for aldosterone is formation of angiotensin from renin released by the juxtaglomerular apparatus. Aldosterone stimulates epithelial cell transport of sodium in the renal tubular epithelium, along the intestinal lumen, and in sweat and saliva. Increased aldosterone levels increase the active reabsorption of sodium and decrease potassium reabsorption. The increased sodium reabsorption produces water reabsorption; this increases intravascular volume and reduces the juxtamedullary secretion of renin. The reduction in potassium tubular reabsorption increases potassium excretion in the urine and should result in a fall in the serum potassium concentration. These responses to aldosterone should in turn reduce the stimulus for aldosterone secretion (see Fig. 13-8).
Antidiuretic hormone (ADH), or arginine vasopressin (AVP), secretion also affects the final concentration of urine. ADH is produced by the supraoptic and paraventricular nuclei in the hypothalamus and is transported to the posterior lobe of the pituitary, where it is released in response to an increase in serum osmolality. ADH secretion is stimulated by serum osmolality greater than 280 to 285 mOsm/L (or a rise in serum osmolality of 2% or more). It also is secreted in response to significant (10%-15%) volume depletion, a fall in blood pressure, painful stimuli, fear, and exercise. Hemoconcentration, diabetic ketoacidosis,90 and mannitol administration increase ADH secretion, and administration of hypertonic glucose often inhibits ADH secretion.34,54 The predominant stimulus for ADH secretion is a rise in serum osmolality sensed by osmoreceptors in and around the supraoptic nucleus of the hypothalamus.
Regulation of Acid-Base Balance
Buffering Systems
The Bicarbonate-Carbonic Acid Buffering System
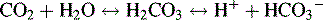
The increase in hydrogen ion concentration will result in a fall in serum pH unless or until CO2 elimination by the lungs is enhanced and/or hydrogen ion excretion and bicarbonate ion reabsorption by the kidneys is increased (see section, Interpretation of Blood Gas Values).
Additional Plasma Buffers
Proteins present in the blood can also act as buffers. Hemoglobin is the most important nonbicarbonate buffer. It binds with hydrogen ions and transports CO2 from the tissues to the lungs for elimination.34
Renal Hydrogen Ion Excretion and Bicarbonate Reabsorption
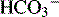 concentration through hydrogen ion secretion and bicarbonate reabsorption and reclamation. Renal compensation for respiratory acidosis requires several hours to begin and will not be fully effective for several days; it requires reabsorption of all filtered bicarbonate and generation of new bicarbonate through the formation of titratable acids.
concentration through hydrogen ion secretion and bicarbonate reabsorption and reclamation. Renal compensation for respiratory acidosis requires several hours to begin and will not be fully effective for several days; it requires reabsorption of all filtered bicarbonate and generation of new bicarbonate through the formation of titratable acids.New bicarbonate can be formed when CO2 combines with water, yielding carbonic acid. The carbonic acid then dissociates into hydrogen ions and bicarbonate; the hydrogen ion is bound to phosphate buffers or ammonia to form hydrogen phosphate or ammonium (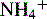 ). Hydrogen phosphate and ammonium are nonreabsorbable, and they are excreted unchanged in the urine. When hydrogen ions are excreted in this way, a quantity of acid can be measured in the urine; this buffering mechanism results in the formation of titratable acids (see Fig. 13-6). The amount of hydrogen ion excreted in the urine is limited, because the kidney cannot secrete urine with a pH lower than approximately 4.4. In addition, the formation of titratable acid will be limited by the amount of ammonia, phosphate, and other inorganic buffers available.
). Hydrogen phosphate and ammonium are nonreabsorbable, and they are excreted unchanged in the urine. When hydrogen ions are excreted in this way, a quantity of acid can be measured in the urine; this buffering mechanism results in the formation of titratable acids (see Fig. 13-6). The amount of hydrogen ion excreted in the urine is limited, because the kidney cannot secrete urine with a pH lower than approximately 4.4. In addition, the formation of titratable acid will be limited by the amount of ammonia, phosphate, and other inorganic buffers available.
Interpretation of Blood Gas Values
Evaluation of the pH and PaCO2
Blood gas analysis requires evaluation of the pH, the PaCO2, the calculated base deficit or excess, and the serum bicarbonate. The first step is evaluation of the pH. If the pH is less than 7.35, acidosis is present; if the pH is greater than 7.45, alkalosis is present. The second step is evaluation of the PaCO2 in light of the pH to determine whether any existing change in pH can be explained by the alteration in PaCO2. For every uncompensated torr unit rise in PaCO2 above 45, the pH should fall 0.008 units below 7.35, and for every uncompensated torr unit fall in PaCO2 below 35, the pH should rise 0.008 units above 7.45. Acidosis or alkalosis in excess of that predicted from the PaCO2 must be metabolic in origin (Box 13-1).
Box 13-1 Evaluation of pH and PaCO2 and Calculation of Base Deficit or Excess
1. Subtract the child’s PaCO2 from 45 (result can be a negative or positive number)
2. Multiply difference obtained in Step 1 by 0.008 (result can be a negative or positive number).
3. Add the product obtained in Step 2 and 7.35; this yields the pH predicted from the PaCO2 alone. If child’s pH is lower than predicted, metabolic acidosis is present; if the pH is higher than predicted from the PaCO2, metabolic alkalosis is present.
4. To calculate base deficit, subtract the predicted pH (calculated in Step 3) from the child’s actual pH and multiply this difference by 0.66. A base deficit more negative than (larger than) −2 indicates the presence of metabolic acidosis, and a base excess greater than 2 indicates the presence of metabolic alkalosis.
PaCO2, Partial pressure of carbon dioxide.
The Base Deficit or Excess
The base deficit or excess is a calculated number that is indicative of acidosis (base deficit) or metabolic alkalosis (base excess). The normal base deficit or excess is −2 to 2. A number more negative than (larger than) −2 indicates a base deficit and the presence of metabolic acidosis. A positive number higher than 2 indicates a base excess and the presence of metabolic alkalosis. The base deficit or excess is calculated by subtracting the pH predicted from the PaCO2 (see Box 13-1) from the actual pH and multiplying the result by 0.66.
Rules to Assess Effectiveness of Compensation
A few rules are helpful in the evaluation of compensation for acid-base disorders. These are summarized in Box 13-2.
Box 13-2 Rules for Assessment of Respiratory and Renal Compensatory Responses in Acid-Base Disturbances
Compensatory mechanisms bring pH toward but not to a normal level.
If respiratory compensation is intact in metabolic disturbances:
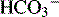 ] + 15 = 2 digits of pH after the decimal point, or
] + 15 = 2 digits of pH after the decimal point, orIf metabolic compensation is intact in respiratory disturbances:
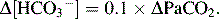
In chronic respiratory acidosis
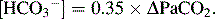
In acute respiratory alkalosis
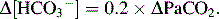
In chronic respiratory alkalosis
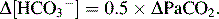
here Δ indicates the degree of deviation from normal value.
From Ichikawa I, Narins RG, Harris HW Jr: Regulation of acid-base homeostasis. In Ichikawa I, editor: Pediatric textbook of fluids and electrolytes, Baltimore, 1990, Williams and Wilkins, p. 84.PaCO2, Partial pressure of carbon dioxide.
Acidosis
Metabolic Acidosis
Spontaneous respiratory compensation is the most effective method of treating metabolic acidosis. However, if respiratory effort or function is compromised or if profound acidosis is present, administration of a buffering agent may be required. Before administration of sodium bicarbonate, providers should assess and support the patient’s airway and ventilation, because the buffering action of the sodium bicarbonate will result in the formation of CO2. The sodium bicarbonate dose that should correct acidosis to a total CO2 of 15 mEq/L is estimated using the following formula76:
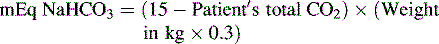
An alternative formula for determination of the sodium bicarbonate dose utilizes the base deficit. This formula estimates that the bicarbonate deficit equals the base deficit, distributed chiefly in the extracellular space (one third of the total body weight) as follows66:
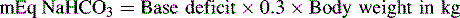
Calcium Regulation
The normal total serum calcium concentration is 9 to 11 mg/dL, and normal ionized calcium concentration is approximately 4.4 to 5.3 mg/dL.80 The serum calcium concentration is evaluated in light of the serum protein concentration. An increase in the serum albumin and globulin will increase the amount of calcium bound to proteins, so will reduce the amount of the serum calcium that is present in the ionized form. For each 1 g/dL increase in serum albumin, 0.8 mg/dL of calcium is removed from its ionized state and is bound to the albumin. Increases in serum globulin level, however, will lower the ionized calcium concentration by only 0.16 mg/dL. If serum albumin and globulin concentrations are reduced, a relatively greater portion of the patient’s total serum calcium will be present in the ionized form. As a result, the patient with a low total serum calcium concentration and a reduction in serum albumin may have a normal serum ionized calcium concentration.
Changes in the serum pH also will affect the amount of calcium bound by proteins. An increase of 0.1 in serum pH will increase protein-bound calcium by 0.12 mg/dL. Conversely, when the serum pH falls, more calcium is removed from the protein binding sites and is ionized, and available to participate in chemical reactions. Thus, when a decreased total serum calcium concentration is present in a patient with alkalosis, the serum ionized calcium concentration is probably extremely low. Alternatively, if the total serum calcium concentration is low in a patient with acidosis, the serum ionized calcium concentration may not be reduced significantly.103
Calcium homeostasis requires regulation of the amount of calcium filtered and reabsorbed by the kidneys, the amount of calcium absorbed from and excreted by the gastrointestinal tract, and the mobilization or deposition of calcium phosphate and other minerals in the bone matrix. These three methods of calcium regulation are controlled by parathyroid hormone (PTH), which is secreted by the four parathyroid glands. When serum ionized calcium levels fall, PTH is released, increasing the renal reabsorption of calcium and the gastrointestinal absorption of calcium. PTH enhances the movement of calcium and phosphate from the bone into the extracellular fluid. In addition, PTH decreases renal tubular reabsorption of phosphate, resulting in excretion of the phosphate that was released when calcium was mobilized from the bone.54
Gastrointestinal Absorption of Calcium
The intestinal absorption of calcium occurs through an active transport system controlled by PTH. When the serum calcium concentration falls and PTH is released, intestinal absorption of calcium will increase somewhat. More importantly, PTH will stimulate renal activation of vitamin D3. The presence of activated vitamin D3 will greatly accelerate gastrointestinal absorption of ingested calcium (see Chapters 12 and 14).
Prenatal and Postnatal Development of Renal Function
During fetal life, the placenta performs many of the functions of the kidney, so congenital renal malformations may not cause fetal distress. Urine secretion into the amniotic fluid begins during the ninth through twelfth weeks of gestation. Most kidney growth occurs during the last 20 weeks of gestation, and the GFR increases rapidly between the twenty-eighth and thirty-fifth weeks of gestation. All nephrons of the mature kidney are formed by the twenty-eighth week of gestation.40
After birth, kidney size increases in proportion to body length. Kidney weight doubles in the first 10 months of life, more as the result of proximal tubular growth than from an increase in glomerular size. The GFR also increases significantly after birth. The GFR of the full-term neonate (per square meter of body surface area) is approximately one third the GFR of an adult. Renal blood flow and the GFR double during the first 2 weeks of life, and GFR is nearly equal to adult values within the first 3 years of life.40
Immediately after birth, the neonate normally has a high urine volume with low osmolality, thought to be the result of immaturity of renal sodium and fluid regulatory mechanisms. Because increases in systemic arterial pressure and systemic vascular resistance also result in an increase in renal blood flow and GFR during this time, these factors also may be responsible for the high urine volume. Beyond the first several hours of life, urine volume normally falls and urine concentration gradually rises.40
The newborn kidney is able to excrete amino acids and conserve sodium and glucose as well as the adult kidney. However, the newborn kidney is less able to excrete free water and to concentrate urine than is the adult kidney.40 As a result, the infant kidney may be less able to excrete a large water load and may be unable to concentrate urine in response to dehydration.
Factors Influencing Body Fluid Composition and Distribution
Serum Osmolality
Osmotic pressure in a fluid is measured in milliosmoles and is the force exerted by particles in solution that will draw water across a semipermeable membrane. The normal serum osmolality is approximately 275 to 295 mOsm/L. Sodium and its chief anions, chloride and bicarbonate, account for 90% of the total osmolality of the plasma. The serum osmolality can be calculated by adding the concentrations of the solutes (including sodium, potassium, calcium, magnesium, sulfate, creatinine, glucose, protein, and urea) per unit of solvent. For simplicity, the total serum osmolality is estimated using the serum concentrations of sodium, glucose, and blood urea nitrogen (BUN) as follows86:
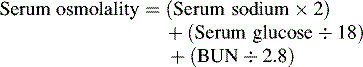
Factors Influencing Water Movement Between Body Compartments
Under normal conditions, free water is distributed within the intracellular and the extracellular (including intravascular and interstitial) compartments, so that osmolality is equal within all spaces.34 Acute changes in the osmolality of any one body fluid compartment will result in free water movement across the semipermeable vascular and cell membranes until osmotic equilibrium is restored. Acute changes in serum sodium and osmolality can cause acute water shifts between the intracellular and extracellular spaces. Sudden, significant water shifts are tolerated poorly.
An acute fall in serum sodium and the resulting intracellular water shift can cause cerebral edema. If neurologic symptoms develop, urgent treatment is needed. An acute rise in serum sodium and osmolality can cause a water shift from the cells to the extracellular space. A significant extracellular water shift can cause cerebral dysfunction and intracranial bleeding. For further information, see Chapter 12.
Changes in Body Fluid Composition and Distribution During Critical Illness
Critically ill patients have a tendency to retain fluids, because antidiuretic hormone and aldosterone secretion are typically increased. Catecholamine release, hypotension, fright, or pain can stimulate antidiuretic hormone (ADH), renin, and aldosterone release. ADH release also is known to be stimulated by any condition that reduces left atrial pressure (including hemorrhage, positive pressure ventilation, and severe pulmonary hypertension), and the administration of general anesthetics, morphine, or barbiturates.54
Postoperative fluid administration must be tailored to prevent fluid overload or sodium imbalance. Typical fluid and electrolyte losses in the urine most closely resemble 0.45% sodium chloride, whereas insensible losses through the skin and respiratory tract are more similar to 0.2% sodium chloride. For this reason, 0.2% or 0.45% sodium chloride with 5% or 10% glucose may be administered during the postoperative period to replace insensible and urine losses only, but not for the provision of maintenance fluids. Recent reports of hyponatremia in critically ill children have led to caution in the use of hypotonic fluids, and greater use of isotonic crystalloids (see Chapter 12).
The stressed patient will tend to retain sodium and water as a result of renin, aldosterone, and ADH secretion; therefore administration of hypotonic solutions (e.g., 5% dextrose and water) should be avoided unless sodium intake is restricted (e.g., in postoperative patients with congenital heart disease). Excessive gastrointestinal fluid and electrolyte losses should be replaced with a solution approximating the electrolyte concentration in the gastrointestinal fluid lost.43 These recommendations should serve only as guidelines and must, of course, be adjusted to meet the patient’s individual requirements.
Potential Effects of Drugs and Solutions on Renal Function
Some drugs and solutions used in the evaluation or treatment of critically ill patients may be nephrotoxic. Antibiotics such as the cephalosporins, the aminoglycosides, and the sulfonamides may be nephrotoxic in infants and children.23,99 Alpha adrenergic medications that produce renal vasoconstriction can result in decreased renal perfusion and oliguric renal failure. Indomethacin administration to promote constriction of the neonatal ductus arteriosus can produce a fall in GFR or a decrease in urine output.
The use of hypertonic angiographic contrast agents can result in renal vein thrombosis, medullary hypoperfusion, renal ischemia, and renal insufficiency. Because these agents have an osmolality of 1300 to 1940 mOsm/L, they should be administered carefully in low doses and only to well-hydrated infants. If possible, low-osmolality radiocontrast agents should be used, and other nephrotoxic drugs should be avoided.23 Use of nephrotoxic drugs in children also should be restricted if circulatory compromise or renal insufficiency is present.
Diuretics
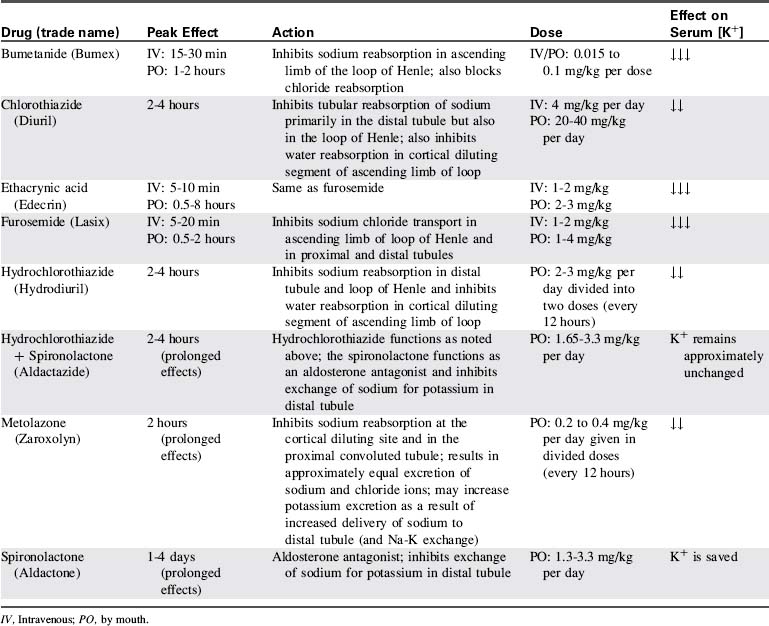
Diuretics can be classified according to their renal site of action and their chemical groups.31 The osmotic agents and carbonic anhydrase inhibitors are proximal tubule diuretics that are less commonly used than the more popular thiazides and sulfonamide derivatives. Potassium-sparing diuretics act in the distal tubule. Furosemide (Lasix) and ethacrynic acid exert their effect on the loop of Henle (Box 13-3).
The classifications, effects, doses, and side effects of the most frequently used pediatric diuretics are included in Table 13-2.
Proximal Tubule Diuretics
Osmotic Agents
Hypertonic glucose and mannitol are not the drugs of choice for routine diuresis. However, they can be extremely useful in promoting diuresis in children with marginal renal function, because their effect is produced as soon as they are filtered through the glomerulus, and they do not depend on renal tubular excretory or reabsorptive functions. In the setting of rhabdomyolysis, radiocontrast administration, and cardiopulmonary bypass, osmotic diuretics have been used in an attempt to prevent acute kidney injury.3
Intravenous administration of any osmotic agent can produce a temporary but significant increase in intravascular volume, because the intravenous osmotic agent produces an intravascular fluid shift. Use of these agents typically is contraindicated in patients with congestive heart failure or hypervolemia, because they can cause further increase intravascular volume. However, these drugs can be extremely useful in the treatment of patients with increased intracranial pressure (see Chapter 11).
Distal Tubule Diuretics
Most agents that act on the distal convoluted tubule will be ineffective if the GFR less than 40 mL/min or less than 40% of normal.31
Potassium-Sparing Diuretics
Potassium-sparing diuretics constitute a separate class of diuretics and include spironolactone, triamterene, and amiloride. These drugs inhibit distal tubule sodium reabsorption and potassium secretion. Consequently, sodium and water are excreted, and potassium is spared. This action is the most common reason for the use of these drugs—preserving the serum potassium. These diuretics are often administered in combination with other more potent diuretics such as furosemide.31
Loop of Henle Diuretics
The loop diuretics, furosemide (Lasix) and ethacrynic acid (Edecrin) are the most potent and popular diuretics used in the care of critically ill children. Both drugs inhibit sodium chloride transport in the ascending limbs of the loop, so that natriuresis and diuresis result. These drugs are often effective in patients responding maximally to other diuretics, and they will be effective despite a decrease in glomerular filtration rate.31 Both drugs can be administered intravenously, and they have a rapid onset.
Large diuresis can decrease plasma volume, causing a contraction alkalosis. The increased hydrogen ion excretion that results from the administration of loop diuretics can further contribute to the alkalotic state (Box 13-4).
Common clinical conditions
Hyponatremia
Hyponatremia can develop in patients with renal dysfunction after aggressive use of diuretics. A pseudohyponatremia may be reported in patients with significant hyperlipidemia or hypoproteinemia,96 and severe hyperglycemia may depress the serum sodium concentration (see Hyponatremia in Chapter 12). An acute fall in the serum sodium and osmolality is problematic because it can produce an acute intracellular shift of water, causing cerebral edema.
In any patient with renal failure, serum electrolytes and serum and urine osmolality will be assessed frequently; hyponatremia or other electrolyte imbalances should be promptly detected and treated. Hyponatremia producing neurologic symptoms is treated urgently with hypertonic (3%) saline (2-4 mL/kg). Hyponatremia not associated with neurologic symptoms is treated more slowly, raising the serum sodium at a rate of approximately 0.5 mEq/L per hour (see Chapter 12).14,103
Hypokalemia
Severe hypokalemia can produce ventricular irritability and arrhythmias such as premature ventricular contractions, ventricular tachycardia and ventricular fibrillation. In addition, the electrocardiogram may reveal flattened or inverted T waves, ST-segment depression, and the appearance of a U wave. The child may also develop vomiting and a paralytic ileus.85
If hypokalemia develops, small doses of potassium can be added to the dialysate, or small doses of potassium chloride can be administered intravenously (0.5-1.0 mEq/kg, administered over several hours). If the hypokalemia is related to prolonged use of 4.25% glucose dialysate, and the child’s condition permits it, the treatment of choice is to change the dialysate solution.42,50,89 For further information, see Chapter 12.
Hyperkalemia
Clinical Signs and Symptoms
Hyperkalemia can produce characteristic changes in the electrocardiogram. Initially a tall, peaked T wave develops; this is followed by a widening of the QRS complex, ST-segment depression, and decreasing amplitude of the R wave. As the serum potassium concentration continues to rise, the P-R interval is prolonged, the amplitude of the P wave decreases, and finally the P wave disappears. Ventricular arrhythmias or ventricular fibrillation can develop at any point during this progression (Fig. 12-3).
Management
Once the child’s serum potassium concentration exceeds 6.5-7.0 mEq/L, there is a high risk of serious cardiac arrhythmias, and immediate intervention is required. Although the evidence supporting emergent therapies for hyperkalemia is largely anecdotal, widely accepted goals are: stabilize the myocardial cellular membrane, expand the extracellular fluid volume, shift potassium into the cells, and remove potassium from the body (see “Acute Kidney Injury” later in this chapter and Chapter 12, Table 12-4 for more information).
Calcium chloride or calcium gluconate is administered to transiently counteract adverse effects of hyperkalemia on cardiac cells. Calcium chloride (20 mg/kg) is infused intravenously over 1 to 5 minutes (calcium gluconate is used in neonates). During the calcium infusion, the nurse should monitor for bradycardia. In the presence of arrhythmias, an immediate improvement in the patient’s electrocardiogram (ECG) may be observed as the calcium is given; these effects last approximately 30 min.50 During this 30-minute window, additional therapy is provided to reduce the serum potassium concentration.
Cellular uptake of potassium from the extracellular (including intravascular) fluid is enhanced with the infusion of glucose plus insulin, one of the most effective methods of reducing hyperkalemia in critically ill children. A hypertonic glucose solution is administered (1-2 mL/kg of 25% dextrose [1 g/kg]) with a dose of regular insulin (0.1 U/kg).50 The serum potassium concentration should begin to fall within 15 to 30 minutes, and effects may be apparent for about an hour.
The half-life of regular insulin is longer than the half-life of hypertonic glucose. Therefore late hypoglycemia often is observed after administration of the glucose and insulin. The child’s serum glucose concentration should be monitored and point-of-care glucose testing should be used (if available), especially 1 to 2 hours following administration. Supplementary glucose should be administered as needed.5
Sodium polystyrene sulfonate is administered as a 20% suspension in a 5% glucose solution. The dose of 1.0 g/kg can be repeated as often as two or three times in 24 hours.52 Because the greatest exchange of sodium for potassium occurs in the large intestine, an oral or nasogastric dose of 1.0 g/kg (given in divided doses) can reduce the serum potassium 1 mEq/L over a 24-hour period. The enema dose is somewhat higher and usually totals 2 g/kg, given once or twice per day. The serum sodium will rise following sodium polystyrene sulfonate administration, and fluid retention may develop.
Inhaled albuterol causes rapid movement of potassium into the cells by stimulating the beta 2 adrenergic receptors, which in turn increase activity of the sodium-potassium-adenosine triphosphatase pump in the cell membrane, increasing cellular uptake of potassium. Albuterol stimulates the release of insulin, which also promotes cellular uptake of potassium.83 Albuterol may provide rapid but unpredictable lowering of the serum potassium.
If all other measures to reduce serum potassium concentration fail, then hemofiltration, exchange transfusion, or hemodialysis may be required (see section, Care of the Child During Dialysis, Hemoperfusion, and Hemofiltration).
Acute Renal Failure and Acute Kidney Injury
Acute renal failure is a sudden reduction in renal function characterized by progressive accumulation of nitrogenous waste products of protein metabolism, urea, and creatinine in the blood. This condition is called azotemia. Oliguria is present in 30% to 70% of patients with acute renal failure (ARF).47
ARF is a multifaceted and often severe disorder that results in complicated imbalances in fluid and electrolytes, acid-base status, and the function of other organ systems. Critically ill patients further complicated with ARF continue to have a high mortality rate despite treatment and technological advances. The occurrence of ARF (depending on the definition used) is estimated to range from 1% to 25% of critically ill patients.2,60
An extremely important milestone regarding the definition of ARF was published in 2004 as a result of the Second International Consensus Conference of the Acute Dialysis Quality Initiative Group. One of the goals of this conference was to arrive at a consensus definition of ARF, because more than 30 different definitions existed in the literature.11 The multiple definitions confounded research studies, their comparisons, and any meaningful interpretation of best treatment practices.
The Acute Dialysis Quality Initiative Group defined ARF for clinical research as “an abrupt and sustained decrease in glomerular function, urine output, or both.”2 The group then developed consensus criteria for essential features of ARF. The consensus criteria are known collectively as the RIFLE criteria, using the acronym indicating: risk of renal dysfunction, injury to the kidney, failure of kidney function, loss of kidney function, and end-stage kidney disease. These criteria have gained wide acceptance.53 In this classification, there are separate criteria for creatinine and urine output to enable accurate and reproducible staging of the progression of renal dysfunction.11 Changes in serum creatinine or changes in urine output, or both, will place a patient in a classification level of acute kidney injury (AKI).
Akcan-Arikan et al. published modified RIFLE criteria (pRIFLE) “for use in critically ill children (Table 13-3), assessing acute kidney injury incidence, and course along with renal and/or nonrenal comorbidities.”4 The pediatric criteria are based on (1) the patient’s estimated creatinine clearance using the Schwartz formula, (2) the constant k values4 determined by age (Box 13-5), and (3) urine output (see Evolve Box 13-1 for an example of the calculation of the pediatric RIFLE score in the Chapter 13 Supplement on the Evolve Website).
Table 13-3 Pediatric-Modified Acute Renal Failure (pRIFLE) Criteria
| Criteria | Estimated Creatinine Clearance | Urine Output |
| Risk | Decreased by 25% | <0.5 L/kg per hour × 8 hours |
| Injury | Decreased by 50% | <0.5 mL/kg per hour × 16 hours |
| Failure | Decreased by 75% or <35 mL/min per 1.73 m2 | <0.3 mL/kg per hour × 24 hours or anuric for 12 hours |
| Loss | Persistent failure >4 weeks | |
| End-stage | Persistent failure >3 months |
pRIFLE, Pediatric modified RIFLE.
Reproduced from Akcan-Arikan A, et al: Modified RIFLE criteria in critically ill children with acute kidney injury, Kidney Int 71:1028-1035, 2007.
Box 13-5 Schwartz Formula and K Values for Calculating Estimated Creatinine Clearance
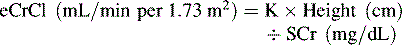
That is, estimated creatinine clearance (eCrCl) in milliliters per minute per 1.73 meter squared is equal to K—a constant value that varies with age*—times the height in centimeters divided by serum creatinine (SCr) in milligrams per deciliter
The pediatric modified pRIFLE criteria provide a systematic, standard method of defining and classifying acute kidney injury in children. The criteria have been shown to reliably predict increased cost, length of stay, mortality, and need for renal replacement therapy.8
In this chapter, the term AKI is used “to represent the entire spectrum of acute renal failure” as proposed by the Acute Kidney Injury Network in their report of an initiative to improve outcomes in acute kidney injury.70 The use of a standard definition of AKI and the pRIFLE classification help focus on early changes and recognition, which may lead to prevention.
Etiology
There are many causes of AKI in critically ill children. Critically ill neonates can develop AKI as a result of asphyxia, hypoxia, shock, and sepsis.6 AKI also can occur as a complication of umbilical artery or vein catheterization and subsequent thrombosis of the aorta, the renal artery, or the renal vein. Critically ill children most commonly develop AKI as a complication of major surgical procedures, multisystem organ failure, drug toxicity, or toxic ingestions.9,27
Postrenal Failure
Congenital disorders such as posterior urethral valves and bilateral ureteropelvic junction strictures are the most common postrenal causes of AKI in neonates and infants.98 Blood clots, calculi, inflammation or edema, tumors, and hemorrhagic cystitis are a few of the additional conditions that can prevent urine flow into the bladder or prevent adequate bladder evacuation.
Intrinsic Renal Failure
Intrinsic forms of AKI include all causes of renal dysfunction associated with damage to renal parenchymal cells. The most common causes of intrinsic AKI are associated with acute tubular necrosis (ATN) from ischemia and nephrotoxic substances or drugs. ATN is the most frequent cause of intrinsic renal failure in children.27
AKI in the neonate can be associated with renal structural anomalies. Because the placenta performs the excretory functions of the kidneys, neonates with significant renal malformations can have normal plasma electrolyte concentrations at birth. Ninety-five percent of all preterm and term infants excrete urine within the first 24 hours of life85; therefore failure of micturation in the first 2 days of life is strongly suggestive of severe congenital renal anomalies. A history of oligohydramnios, limb deformities, and characteristic facial features suggest the presence of Potter’s syndrome, which includes renal dysplasia or bilateral renal agenesis. Many neonates with oligohydramnios also have associated pulmonary hypoplasia.
Pathophysiology
Obstruction and damage to the renal vasculature can complicate umbilical artery or vein catheterization in the neonate; however, this is an uncommon cause of ARF in children. Renal vasculature pathology in children is most frequently the result of hemolytic uremic syndrome and other pathologies, including sickle cell disease, Kawasaki disease, and Henoch-Schonlein Purpura.58
The unpredictability of tubular healing makes it impossible to predict the rate of recovery of nephron function after ischemic or nephrotoxic injury.27 Renal failure can be complicated by the development of backleak of fluid through the damaged tubular basement membrane. Backleak prevents elimination of the filtrate and results in reabsorption of the creatinine and other nitrogenous wastes back into the circulation.79
Although the precise pathophysiology of AKI is not understood, almost all theories include a severe reduction in renal blood flow by 25% to 50%. This reduction in renal blood flow often occurs despite a normal systemic arterial pressure and is thought to result from intense renal vasoconstriction27 that reduces GFR and renal cortical blood flow. This reduction stimulates renin and aldosterone secretion and produces sodium and water retention and decreased urine volume.
Clinical Signs and Symptoms
Clinical signs produced by uremia include an altered level of consciousness, seizures, anorexia, nausea, vomiting, abnormal platelet function, diminished white blood cell function, and pericarditis.71 Once AKI develops, the ability of the kidneys to regulate fluid volume and potassium, calcium, and glucose concentrations is severely impaired. In addition, renal regulation of acid-base balance is reduced. Finally, many patients with AKI develop anemia and coagulopathies, and they are at risk for the development of gastrointestinal hemorrhage and infection.71 As a result, assessment of children with AKI must include assessment of the reversible causes of renal failure as well as the recognition and management of its complications.
Disorders of Electrolyte and Acid-Base Balance
Hyperkalemia is one of the most serious complications of acute kidney injury, because it can result in fatal cardiac arrhythmias. Hyperkalemia develops because distal tubular injury impairs potassium secretion, and reduced glomerular filtration limits the formation of urine; therefore potassium secretion in the cortical collecting tubule is reduced.77
Hyperphosphatemia develops as a result of a reduction in the GFR. The tubular maximum for phosphate reabsorption varies inversely with the GFR; as the GFR falls, the tubular maximum rises, and more phosphate is transported actively out of the tubules and returned to the circulation. If chronic kidney disease develops, hyperparathyroidism will partially compensate for this hyperphosphatemia by increasing calcium mobilization from bone so that phosphate is precipitated. Although hyperphosphatemia itself may produce no symptoms until the phosphate level is extremely high (e.g., much higher than 5.5 mg/dL), it will produce hypocalcemia that can result in neuromuscular or cardiovascular complications.77
Evaluation of Renal Function
During initial assessment of the child with AKI, it is important to attempt to differentiate between reversible prerenal or postrenal AKI and renal injury resulting from renal parenchymal damage. The tests to differentiate between prerenal and intrinsic renal injury basically evaluate the ability of the kidney to conserve sodium and concentrate urine (Table 13-4).
Table 13-4 Laboratory Tests in Differential Diagnosis of Prerenal and Intrinsic Renal Failure
| Characteristic | Prerenal | Intrinsic Renal |
| Urine specific gravity | >1.020 | ≤1.010 |
| Urine osmolality (mOsm/L) | >500 | <400 |
| Urine creatinine (mg/dL) | >100 | <70 |
| Creatinine urine:plasma ratio | >30 | <20 (<10 in neonates) |
| Urea urine:plasma ratio | >14 | <6 |
| Urine urea (mEq/L) | >2000 | <400 |
| Urine sodium (mEq/L) | <10 mEq/L | >30 mEq/L (>25 mEq/L in neonates) |
| Urine potassium (mEq/L) | 30-70 | <20-40 |
| Urine Na:K ratio | <1.0 | 0.8-1.0 |
| Urine appearance (microscopic) | Hyaline casts | Cellular casts |
| Serum BUN:creatinine ratio | >20:l | <10:l |
| FENa | <1% (<2.5% in neonates) | >1% to 3% (>3.5 in neonates) |
| Fluid status | Dry, hypovolemic or inadequately perfused | Euvolemic |
BUN, Blood urea nitrogen; FENa, fractional excretion of filtered sodium.

When prerenal azotemia is present, the FENa is less than 1% (<2.5% in neonates)40; when AKI is caused by renal damage the FENa is greater than 1% to 3%.82 It is important to note that evaluation of renal function using the FENa is not reliable if the urine sample is collected after diuretic therapy, because diuretics increase the urine sodium concentration.
When AKI results from renal damage, the child’s urine usually is not concentrated, and it often contains casts of renal tubular cells. If the test result for blood in the urine is positive with a bedside reagent strip, but the urine contains no RBCs on microscopic examination, providers should suspect the presence of hemoglobinuria or myoglobinuria.44
Management
Fluid balance and Renal Perfusion
Assessment of Systemic Perfusion
If the child has pale mucous membranes or nail beds and cool extremities with sluggish capillary refill, cardiac output may be inadequate and systemic and renal perfusion may be compromised. Inadequate cardiac output also results in tachycardia and tachypnea (unless the child’s respiratory rate is controlled with mechanical ventilation), diminished intensity of peripheral pulses, metabolic acidosis and oliguria. Hypotension is often a late sign of poor perfusion in children (see “Shock” in Chapter 6).
If clinically significant hypovolemia is present, the child’s CVP usually is less than 5 mm Hg. Patients with AKI may require administration of 20 mL/kg boluses of isotonic crystalloid (normal saline or Ringer’s lactate); the fluid bolus is repeated (up to three times) based on the patient’s response, with careful monitoring to prevent fluid overload.27 If hypovolemia results from hemorrhage, isotonic crystalloid may be administered initially, but blood products will be required.
If the child’s systemic perfusion improves after fluid administration, but urine output does not increase, furosemide (or mannitol [0.2-0.5 g/kg] in rhabdomyolysis) can be prescribed early in the course of AKI in an attempt to convert from oliguric to nonoliguric renal failure; however, this approach has not reduced mortality. Because persistent vasoconstriction is often present in AKI, larger doses of furosemide (e.g., 2-5 mg/kg) should be administered intermittently followed by an infusion of furosemide.71
Cardiovascular Support
The drug of choice for oliguria with cardiovascular dysfunction is dopamine, because it dilates the renal vasculature, increasing renal blood flow (20%-40%) and GFR (5%-20%) when it is administered in low (0.5-2 mcg/kg per minute) doses.20,59 There is no evidence to support the use of low-dose dopamine in the prevention of AKI,26,95 but it is used once AKI is present. Higher doses of dopamine (>8-10 mcg/kg per minute) should be avoided, because they can produce alpha-adrenergic effects, including renal vasoconstriction and decreased renal blood flow, and can result in decreased urine output.
An additional sympathomimetic drug such as dobutamine (2-20 mcg/kg per minute) may also be administered. If systemic perfusion remains poor, systemic vasodilators such as sodium nitroprusside (0.5-8 mcg/kg per min) or nitroglycerin (0.25-10 mcg/kg per min) may be required (see Management of Shock in Chapter 6).
Fluid Therapy
When oliguric AKI is present, the child’s fluid intake should be restricted to insensible water losses (approximately 300-400 mL/m2 body surface area per day) plus 24-hour urine output.71 Too often, repeated boluses of fluid are administered in an unsuccessful attempt to increase urine output, and they produce hypervolemia. Repeated administration of osmotic diuretics also should be avoided once the patient has failed to respond to them, because these agents will increase intravascular volume and serum osmolality.
Electrolyte and Acid-Base Balance
Potassium Balance
1. Intravenous infusion of 10% calcium chloride (0.1-0.2 mL/kg [10-20 mg/kg]) over 1 to 5 minutes, or 10% calcium gluconate (0.5-1.0 mL/kg [50-100 mg/kg]) over 2 to 4 minutes. Give calcium slowly while monitoring the ECG carefully for bradycardia. The administration of calcium is thought to reduce cardiotoxicity of high serum potassium, counteracting the adverse effects of hyperkalemia on neuromuscular membranes.
2. Intravenous infusion of a sodium bicarbonate (1-3 mEq/kg [average of 2.5 mEq/kg] over 30 min will raise the serum pH so that potassium shifts from the vascular and interstitial spaces into cells (in exchange for hydrogen ions that move from the cells). The bicarbonate solution generally is diluted 1:1 with sterile water to reduce osmolality.
3. Intravenous infusion of concentrated glucose or glucose and insulin (1-2 mL/kg of 25% glucose plus 0.1 units/kg of regular insulin) to enhance intracellular movement of potassium.
4. Nebulized albuterol (rapid nebulizer or continuous nebulizer of 0.1-0.3 mg/kg) or salbutamol (IV dose of 4-5 mcg/kg over 20 min and repeated after 2 hours)103: as a beta-2 adrenergic agonist, albuterol activates the sodium-potassium pump and stimulates the pancreas to secrete insulin; these actions shift potassium into the cells.81
5. The previous treatments do not remove potassium from the body; they merely transiently lower the serum potassium concentration by shifting potassium into cells. Potassium must be removed either through the use of sodium polystyrene sulfonate or through hemodialysis or hemofiltration before the serum potassium concentration reaches critical levels.
Box 13-6 Administration of Glucose and Insulin to Reduce Critical Hyperkalemia
Standard dose: 1-2 mL 25% glucose/kg body weight + 0.1 unit regular insulin/kg body weight
Phosphorus and Calcium Therapy
Oral phosphate binders will bind ingested phosphate before it is absorbed.98 Calcium carbonate (Tums) tablets will bind phosphate before absorption. Antacid solutions containing magnesium (e.g., Maalox) are avoided because the magnesium can lower calcium levels. Aluminum hydroxide solutions (Amphojel) are no longer used because aluminum deposition in bone tissue has been reported with prolonged use.
Metabolic Acidosis
The typical dose of the sodium bicarbonate is 1 mEq/kg, but a buffering dose also may be determined by the calculated base deficit or the child’s bicarbonate or serum CO2. The formula for calculating the sodium bicarbonate (NaHCO3) dose using the base deficit is as follows66:
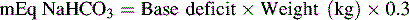
Another formula for calculation of the NaHCO3 dose is based on the patient’s serum bicarbonate as follows66:
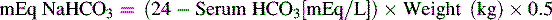
Glucose
The infant’s serum glucose concentration should be checked frequently with point-of-care testing (e.g., heelstick glucose evaluation) so that hypoglycemia can be detected and treated promptly. When hypoglycemia is present, treatment with a continuous glucose infusion (2-4 mL/kg per hour of 5% dextrose solution or 1-2 mL/kg per hour of 10% dextrose solution) is preferable to intermittent bolus infusion of hypertonic glucose, because repeated bolus therapy can cause intermittent increases in the serum osmolality and contribute to the risk of intracranial hemorrhage.84 If the serum glucose is extremely low, an initial bolus dose of glucose (2-4 mL/kg of 25% dextrose, diluted for infants and for peripheral infusion) may be necessary to establish an adequate serum glucose concentration. The continuous glucose infusion must, of course, be considered when totaling fluid intake.
Hematologic Complications
The healthcare team should evaluate the child’s platelet count, prothrombin time, and activated partial thromboplastin time on a regular basis, and administer appropriate blood components as needed (see Chapter 15 for doses of and cautions regarding blood component therapy).
If bleeding develops, desmopressin or l-deamino-8-arginine vasopressin (DDAVP) will correct uremic platelet dysfunction, although the mechanism of action is unclear. The effects of an intravenous dose of approximately 0.3-0.4 mcg/kg should be apparent within several hours. Side effects are minimal, although fluid retention may be exacerbated by this drug. DDAVP also may be administered prophylactically to patients with uremia to prevent bleeding during surgery.51
Treatment of Hypertension
Antihypertensives will be prescribed if the infant or child demonstrates severe hypertension or moderate hypertension with symptoms (Box 13-7). The use of angiotensin converting enzyme inhibitors (e.g., captopril) is controversial in patients with AKI, because it can decrease renal blood flow and cause potential ischemic injury.71,73 The doses of all drugs should be evaluated and adjusted as needed in the presence of reduced GFR (see section, Adjustment of Medication Dosages).
Nutrition
Any form of nutrition should provide calories in the form of glucose or essential amino acids to minimize the accumulation of metabolic waste products.98 The child’s daily caloric requirements will total approximately 50% to 75% of normal daily maintenance requirements when AKI is present, because a large portion of the daily maintenance calories are used for basal requirements and growth (Table 13-5).
Table 13-5 Estimated Normal Maintenance Caloric Requirements for Infants and Children
| Age | Kcal/kg per 24 hours | |
| 0-6 months | 90-110 | |
| 6-12 months | 80-100 | |
| 12-36 months | 75-90 | |
| 4-10 years | 65-75 | |
| >10 years, male | 40-45 | |
| >10 years, female | 38-30 | |
| Nutrient | Percent of total daily calories | |
| Carbohydrates Fat |
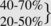 |
Combined 85-88 |
| Protein | 7-15 | |
Adjustment of Medication Dosages
When the child develops renal failure, the healthcare team should reevaluate the doses of all drugs the child is receiving, especially drugs excreted by the kidney. The drug dose can be reduced or the interval between drug doses can be increased in light of the child’s reduced GFR. A review of guidelines for drug therapy in renal failure should be consulted to determine the relative portion of renal and nonrenal modes of excretion of specific drugs.7
Psychosocial Aspects
When the child develops AKI, the child and the family are usually frightened. At the same time that the nurse must provide the most thorough observations and skilled care, the child and family are most in need of reassurance and support. If the child’s physical care requires the nurse’s undivided attention, the nurse should request assistance from a colleague or additional supportive staff (e.g., chaplain, social worker, patient ombudsman). The child requires explanations and preparation for uncomfortable treatments or procedures (as age appropriate), gentle handling, and soothing verbal and nonverbal interaction. (See Chapter 2 for further information.) Box 13-8 summarizes the care of the child with AKI.
Box 13-8 Nursing Care of the Child with Acute Kidney Injury (Acute Renal Failure)
1 Potential Acute Prerenal Failure Related to:
Expected Patient Outcomes
• Patient will demonstrate effective systemic perfusion.
• Patient will demonstrate normal urine output with good renal concentrating ability (specific gravity greater than 1.005) when urine volume is reduced.
• Patient will demonstrate adequate, but not excessive, intravascular volume.
• Patient will demonstrate normal serum electrolyte concentration (including BUN and creatinine).
Nursing Interventions
• Record urine volume and total fluid intake hourly and notify provider if urine output <l-2 mL/kg per hour or if fluid intake greatly exceeds output.
• Insert and maintain urinary catheter—ensure that catheter is functioning properly. Irrigate per physician (or other on-call provider) order or unit policy if patency is questionable. Maintain aseptic technique when manipulating catheter.
• Ensure that catheter tubing is placed to facilitate gravity drainage of urine.
• Record urine osmolality and specific gravity every 2-4 hours (or per orders or unit policy); notify on-call provider if urine osmolality and specific gravity do not rise when urine volume falls.
• Monitor color of urine; notify on-call provider of cloudy or rusty urine. Cloudy urine can indicate infection or the presence of cell casts in the urine. Rusty urine can indicate hemolysis.
• Assess patient’s systemic perfusion: skin should be warm, peripheral pulses should be strong, capillary refill should be brisk, and mucous membranes should be pink. If the skin is cool, peripheral pulses are difficult to palpate, and capillary refill is sluggish, or color is pale or mottled, notify the on-call provider. Monitor urine output; a fall in urine output in the presence of poor systemic perfusion may indicate inadequate renal perfusion, and on-call provider should be notified.
• Support cardiovascular function as needed (and ordered) to maintain urine output greater than 1 mL/kg per hour. Fluid challenge of 20 mL/kg isotonic crystalloid or colloid initially may be ordered to improve systemic perfusion. If systemic perfusion does not improve despite the presence of a CVP greater than 5-10 mm Hg and signs of adequate intravascular volume (see Box 13-11), administration of inotropic agents or vasodilators may be necessary.
• Administer diuretic agents (furosemide [1-2 mg/kg IV] or mannitol [0.25-0.5 g/kg]) as ordered; monitor patient response and notify on-call provider if response is inadequate.
• Obtain urine samples as ordered for laboratory analysis of osmolality, sodium concentration, BUN and creatinine. Simultaneous serum samples must also be obtained.
• If prerenal failure is present, serum BUN will usually begin to rise before serum creatinine.
• When hypovolemia produces prerenal failure, urine sodium content will fall to less than 10 mEq/L (sodium is actively reabsorbed by functioning kidneys in presence of hypovolemia), and urine osmolality will exceed 500 mOsm/L (kidneys conserving water).
• After urine and serum electrolytes are obtained, calculate FENa
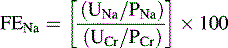
2 Potential Hypervolemia/Fluid Volume Excess Related to:
Expected Patient Outcomes
• Patient will not demonstrate signs of hypervolemia, including high CVP, hepatomegaly, systemic edema, tachycardia, hypertension, tachypnea or increased requirement for ventilation support, pulmonary edema, high pulmonary artery wedge pressure, full (tense) fontanelle in infants, hyponatremia.
• Patient will not demonstrate excessive weight gain (greater than 50 g/24 hours in infants, greater than 200 g/24 hours in children, greater than 500 g/24 hours in adolescents).
Nursing Interventions
• Measure and record all fluid intake and output hourly; notify on-call provider immediately if urine output falls or positive fluid balance is present.
• Limit fluid intake (as ordered) once AKI is suspected. Typically, fluid intake is restricted to 300-400 mL/m2 BSA per day plus urine output if renal failure is present. Minimize fluid used to flush monitoring lines and dilute medications. Closely supervise oral intake, if allowed or tolerated.
• Administer diuretic therapy as ordered. Monitor patient’s urine response and monitor electrolyte balance closely.
• Monitor for signs of hypervolemia including tachycardia, high CVP or PAWP, systemic and pulmonary edema and hypertension, possible signs of congestive heart failure.
• Measure child’s weight daily or twice daily (as ordered or per unit policy); utilize same scale each time and ensure consistent weighing time. Use bed scales for unstable patients. Notify provider of excessive weight gain.
• Be prepared to assist in initiation of CRRT if clinically significant hypervolemia develops.
3 Potential Electrolyte Imbalance Related to:
• Decreased renal potassium excretion
• Increased renal sodium excretion
• Decreased renal excretion of phosphate, calcium phosphate precipitation and decreased renal activation of vitamin D
Nursing Interventions
• Monitor patient’s serum electrolytes closely; notify on-call provider of abnormal results, including high or rapidly rising potassium concentration.
• Limit potassium intake (as ordered).
• Monitor for clinical signs of hyperkalemia, including peaked T wave, arrhythmias, diarrhea, and muscle weakness. If hyperkalemia is suspected, notify on-call provider, obtain serum sample for measurement of potassium concentration, and prepare to institute emergency measures to reduce serum potassium concentration.
• If significant hyperkalemia is present, administer (as ordered):


 Concentrated glucose: 1-2 mL/kg of 25% dextrose or glucose plus insulin (0.1 U/kg): combine 6 units regular insulin with 25% Dextrose to total 100 mL; administer 1-2 mL/kg (for further information, see Box 13-6)
Concentrated glucose: 1-2 mL/kg of 25% dextrose or glucose plus insulin (0.1 U/kg): combine 6 units regular insulin with 25% Dextrose to total 100 mL; administer 1-2 mL/kg (for further information, see Box 13-6)
• If symptomatic hyperkalemia persists, prepare to assist with initiation of hemofiltration or dialysis.
• Be prepared to support cardiovascular function as needed.
• Monitor serum sodium concentration; notify provider of hyponatremia or hypernatremia.
• Monitor for clinical evidence of hyponatremia, including change in level of consciousness, muscle cramps, anorexia, abnormal reflexes, Cheyne-Stokes respiration, or seizures. Notify on-call provider if these are observed, obtain serum sample to evaluate sodium concentration, and prepare to administer 3% saline if ordered (2-4 mEq/kg).
• Monitor serum calcium concentration; notify physician of reduced total or ionized calcium concentration.
• Monitor for clinical signs of hypocalcemia, including muscle tingling or change in muscle tone, seizures, tetany, positive Chvostek sign (twitching of the side of the face when the facial nerve is tapped in front of the ear), or compromised cardiovascular function. If these signs are observed, notify the on-call provider, draw a serum sample for measurement of total and ionized serum calcium concentrations, and prepare to administer calcium chloride (20 mg/kg of 10% CaCl; use calcium gluconate in infant).
• Administer antacid-phosphate binders (as ordered) to reduce serum phosphate levels.
• Administer vitamin D as ordered to enhance calcium absorption.
4 Potential Metabolic Acidosis Related to:
Nursing Interventions
• Monitor arterial blood gases and serum lactate, and notify provider of development of acidosis.
• If metabolic acidosis develops, administer sodium bicarbonate as ordered (1 mEq/kg or mEq dose to correct part or all of the calculated deficit: base deficit × Weight in kg × 0.3).
• Administer sodium bicarbonate only if ventilation is effective or effectively supported.
• Monitor serum potassium concentration, because acidosis will produce a rise in serum potassium concentration.
5 Potential Drug Toxicity Related to:
Nursing Interventions
• When renal failure develops, review patient’s drug doses and drug administration schedule, and discuss these with on-call provider (doses of drugs excreted by kidneys will be reduced as renal function is reduced).
• Note clinical signs of toxicity of all drugs child receives in the nursing care plan, and monitor for these signs; notify on-call provider of any signs of drug toxicity.
6 Potential Infection, Related to:
Nursing Interventions
• Perform meticulous hand washing before and after patient contact; ensure that hand washing is performed by every member of the healthcare team.
• Change all dressings and IV tubing per unit policy.
• Monitor patient temperature and white blood cell count, and notify physician (or on-call provider) of fever, leukocytosis, or leukopenia.
• Assess all skin puncture sites and notify provider of any erythemia, drainage, or fluctuance of wound edges, obtain cultures of urine, skin puncture sites, and blood (as ordered) if infection or sepsis are suspected.
• Monitor appearance of urine and peritoneal dialysis fluid; notify physician of cloudiness and obtain cultures as ordered.
• Administer antibiotics as ordered (check dose in light of compromised renal function).
7 Potential Patient/Family Anxiety Related to:
Nursing Interventions
• Provide the child (as age-appropriate) and family with consistent explanations and support (see Chapter 2).
• Provide the child with positive reinforcement and encouragement.
• Provide explanations before any treatments are performed, and prepare the child for painful therapy with truthful information.
• Plan activities that will allow the child (as age-appropriate) to demonstrate anger, frustration, or sadness; encourage expression of these feelings if the child is willing to discuss them.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
13 Renal Disorders
Pearls
• Urine output will decrease if cardiac output and renal blood flow are compromised; as a result, urine output is a sensitive indicator of cardiac output.
• The normal hourly volume of urine output in children is small, and a small compromise in urine volume may indicate a significant compromise in renal perfusion or function.
• A child’s glomerular filtration rate will approach adult values by about 3 years of age.
• Renal failure may be present in a child with a low, normal, or high volume of urine output.
• Accurate measurement of urine volume and composition provide fundamental data on which clinical decisions are made.
• The modified pRIFLE is a classification system to standardize the definition of acute kidney injury (AKI) in pediatric patients. The acronym, pRIFLE stands for: pediatric risk of renal dysfunction, injury to the kidney, failure of kidney function, loss of kidney function, and end-stage kidney disease. Studies have shown that use of the pRIFLE enhances the classification of AKI epidemiology and the course of AKI in critically ill pediatric patients.
• Renal replacement therapies differ significantly in their effectiveness in treating AKI and its complications. It is therefore helpful for the nurse to be aware of the advantages and disadvantages of each therapy so that the nurse can participate in decisions regarding the best treatment modality for each patient.
Essential anatomy and physiology
Kidney Structure
Gross Anatomy
The kidneys lie anterior and lateral to the twelfth thoracic and first, second, and third lumbar vertebrae and behind the abdominal peritoneum; therefore they are retroperitoneal structures. The kidneys are embedded in a mass of fatty tissue called the adipose capsule, and each capsule is enclosed in the renal fascia (Fig. 13-1). The kidneys are not secured to the abdominal wall, but are held in position by the renal fascia and the large renal arteries and veins. The adipose capsule and the pararenal fat help to protect the kidney and keep it in place.
Fig. 13-1 Components of the urinary system.
(From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7. St Louis, 2010, Mosby.)
A longitudinal section of the kidney shows the three general areas of renal structure: the cortex, the medulla, and the pelvis (Fig. 13-2). The renal cortex is the outer portion of the kidney. It has a granular appearance and extends in fingerlike projections into the medullary areas. The cortex contains most of the nephrons, the smallest functioning unit of the kidney. The cortex also contains all glomeruli, the proximal and distal convoluted tubules, and the first parts of the loop of Henle and the collecting ducts.
Fig. 13-2 Cross-section of the kidney.
(From Patton KT, Thibodeau GA: Anatomy and physiology, ed 7. St Louis, 2010, Mosby.)
The functioning unit of the kidney is the nephron, which consists of a vascular component and a tubular component (Fig. 13-3). Each kidney contains approximately 1 million distinct nephrons. Eighty-five percent of all nephrons originate in the outermost area of the cortex. The remaining nephrons are the juxtamedullary nephrons that originate in the inner cortical area. The long loops of Henle from the juxtamedullary nephrons that extend deep into the medulla lie parallel to the medullary collecting ducts and play an important role in the concentration of urine (see Evolve Fig 13-1 in the Chapter 13 Supplement on the Evolve Website).
Renal Vasculature
The afferent arteriole enters the glomerular capsule and divides to form the glomerulus, a tuft of capillaries that allows filtration of plasma through the capillary membranes. The glomerular capillaries do not recombine into venous channels, but instead recombine into a second arteriole called the efferent arteriole (Fig. 13-4). Because arterioles are present at either end of the glomerular capillary system, constriction or dilation of these arterioles will alter the resistance to flow through the glomerular capillaries and thus will regulate glomerular filtration.
Renal Tubules and Collecting Ducts
The tubular component of the nephron begins as a single layer of flat epithelial cells surrounding the glomerulus. This layer is known as Bowman’s capsule (Fig. 13-4, B). Filtered plasma from the glomerular capillaries will enter Bowman’s capsule and flow into a coiled tubule called the proximal tubule, also known as the proximal convoluted tubule (Fig. 13-5).
Ureters
The two ureters conduct urine from the renal pelvis to the urinary bladder; they are located behind the peritoneum and they descend through the pelvic cavity, crossing over the common iliac arteries. The ureters are attached to the bladder at an oblique angle; they enter the bladder laterally and tunnel between the bladder mucosa and detrusor muscle, creating a flap-valve design that normally prevents reflux of urine into the ureters during bladder contraction.30
Each ureteral wall has three layers: an inner epithelial lining, a middle muscular layer, and the outer fibrous layer that is continuous with the renal capsule. The middle muscular portion of the ureter consists of both a circular and a longitudinal muscle layer. The circular muscles propel the urine toward the bladder by peristaltic contraction, and they generate enough pressure to overcome the resistance caused by the oblique ureteral insertions into the bladder. Contraction of the longitudinal fibers opens the lumen of the ureter. These ureteral muscle fibers are innervated by fibers from the aortic, spermatic or ovarian, and hypogastric plexuses. Peristalsis persists even when the ureter is denervated, allowing ureters to be transplantable.55
The Bladder and Urethra
The urinary bladder is a hollow, muscular organ that stores urine. There are three openings in the bladder wall: the entrances of the two ureters and the exit of the urethra. These openings form the corners of a triangle, called the trigone. There is a dense area of smooth (involuntary) muscle around the neck of the bladder at the orifice of the urethra; this muscle constitutes the internal sphincter. The urethra extends from the urinary bladder to the body surface. At the point where the urethra passes through the muscles of the pelvic floor, striated (voluntary) circular muscles form an external sphincter.55
If the spinal cord is damaged above the sacral spinal level, the patient initially loses all micturation reflexes because inhibitory and facilitory reflexes from the brain cannot be transmitted through the injured spinal cord. Later, however, simple spinal reflexes can return and the patient can void when bladder distension is sufficient. In this case, the bladder reflex will be initiated at the volume of urine that is usually present in the bladder during the patient’s convalescent period.45
Glomerular Function
Filtration Physiology
Intravascular colloid osmotic pressure, or oncotic pressure, is the pressure opposing free water movement out of the vascular space. It is generated by dissolved proteins, ions, and other particles that are normally present in the blood. Larger particles such as proteins cannot move readily across a capillary membrane; therefore they remain in the vascular space, exerting an osmotic pressure of approximately 35 mm Hg. This oncotic pressure opposes hydrostatic filtration from the vascular space.55
The hydrostatic pressure present in Bowman’s capsule is the pressure exerted on the glomerulus by fluid in the Bowman’s capsule and collecting ducts. This pressure is normally 10 to 15 mm Hg and opposes fluid filtration from the glomerulus. Because tubular fluid is normally protein-free, the oncotic pressure in Bowman’s capsule is normally negligible. (See Evolve Fig. 13-2 in the Chapter 13 Supplement on the Evolve Website for a diagram of these pressures.)
Glomerular Filtration Rate (GFR)
Renal function can be evaluated by calculating the GFR. The GFR, in turn, is roughly equivalent to the creatinine clearance; therefore it can be estimated by calculating the creatinine clearance. This estimate should not, however, be the sole means of determining renal function.56
Creatinine is a small molecule byproduct of skeletal muscle creatine metabolism. Creatinine is released at a near constant rate into the bloodstream, is filtered freely at the glomerulus, and is not broken down, reabsorbed, or synthesized by the renal tubules. Only a tiny amount of creatinine is secreted by the renal tubules. In effect, all of the creatinine that is filtered from the vascular space at the glomerulus remains in the urine and can be measured, and the creatinine clearance mirrors the GFR.62
It is important to note that laboratory determination of serum creatinine concentration may be affected by some cephalosporin antibiotics. For this reason the blood sample for analysis of the serum creatinine level should be obtained when antibiotic drug levels are at their lowest.7
Tubular Function
Reabsorption
Table 13-1 summarizes the work of the renal tubular cells in the process of reabsorption. An average of 180 L of water (protein-free plasma) is filtered through the glomerulus of an adolescent per day, and yet the average urine output is 1.5 L/day. This means that 178.5 L of water is reabsorbed out of the tubular lumen back into the body’s circulation per day.
Table 13-1 Filtration, Excretion, and Reabsorption of Water, Electrolytes, and Solutes by the Kidneys
Transport Maximum and Thresholds
If the renal threshold and transport maximum are approximately equal to the daily filtered load of a substance, then the kidneys participate in regulation of the serum concentration of the substance. In such a case, a slight increase or decrease in plasma and filtered concentration of the substance changes its rate of renal reabsorption and excretion, so the serum concentration returns to normal. The renal threshold and transport maximum for phosphate are close to the normal daily filtered load of phosphate, so the serum phosphate concentration is regulated by kidney tubular function. Phosphate transport and reabsorption also will be affected by the serum calcium concentration, parathyroid hormone (PTH), and adrenal cortical hormones.63
Reabsorption and Secretion in the Proximal Tubule
The selective reabsorption of solute begins in the proximal tubules. Approximately 67% of the filtered water, Na+, Cl−, K+, and other solutes such as bicarbonate are reabsorbed in the proximal tubule. In addition, the proximal tubules normally reabsorb all filtered amino acids and glucose.63
Sodium
The primary mechanism for regulation of intracellular and extracellular fluid volume involves renal sodium excretion.63 Sodium is filtered freely at the glomerulus, so its concentration in the proximal glomerular filtrate is identical to its plasma concentration. Sodium is reabsorbed by an active transport mechanism; the mechanism is carrier-mediated and requires energy so the sodium can move against a gradient. Sodium is not secreted into the tubules.
Bicarbonate and Hydrogen Ions
As a result of this process, for every bicarbonate ion that combines with a hydrogen ion in the lumen of the tubule, a bicarbonate ion ultimately will diffuse into the peritubular capillaries (Fig. 13-6). This secretion of hydrogen ions and reabsorption of bicarbonate ions occurs along the length of the renal tubules, but 90% of bicarbonate reabsorption occurs in the proximal tubule. (See section, Regulation of Acid-Base Balance.)
Potassium
Nearly all filtered potassium is reabsorbed by the proximal tubule, and the remaining potassium is reabsorbed in the ascending limb. The proximal reabsorption of the filtered potassium occurs at a constant rate and does not alter, despite the presence of hyperkalemia or hypokalemia. Potassium is also secreted by the distal convoluted tubule and cortical collecting duct, and the rate of K+ reabsorption or secretion in these tubular segments depends on a variety of hormones and factors. Overall, the rate of renal K+ excretion is determined by the distal convoluted tubule and the cortical collecting duct.63 The net result is normally a continuous loss of potassium in the urine.
Calcium
Calcium reabsorption is controlled by parathyroid hormone. Just as for sodium reabsorption, 80% to 90% of filtered calcium is reabsorbed in the proximal tubule and loop of Henle; only 10% of filtered calcium enters the distal tubule for concentration adjustments. When sodium reabsorption is inhibited by loop diuretics, calcium excretion is enhanced.79 Thiazide diuretics increase calcium reabsorption.
Drugs
The glomerulus is nonselective in its filtration of solutes, because the glomerular membrane does not restrict the passage of small molecules. Most drugs are of a small molecular size, and only a fraction of any drug is bound to serum albumin; most will filter into the tubular fluid. Changes in the GFR or in the degree of protein binding will alter the amount of drug present in the glomerular filtrate. Protein binding of a drug can be influenced by competition between drugs for the same protein binding sites (see Chapter 4).
The Loop of Henle
The loop of Henle, located within the renal cortex and the medulla, provides a countercurrent mechanism for urine concentration. The descending limb of the loop of Henle does not transport sodium or chloride actively, but it is highly permeable to sodium and water. Thus, as the filtrate passes through the descending limb of the loop, it becomes progressively more concentrated. The osmolality can increase from 300 to 1200 mOsm/L between the beginning of the descending limb and the tip of the loop of Henle (Fig. 13-7).
The blood vessels surrounding the loop of Henle form a hairpin loop structure, called the vasa recta. The vasa recta consists of capillaries that run parallel to the loop of Henle and the collecting ducts (for an illustration, see Evolve Fig. 13-1 in the Chapter 13 Supplement on the Evolve Website). As these capillaries follow the loop of Henle into the interstitium of the renal medulla, where osmolality is high (as the result of the tubular countercurrent mechanism), water shifts out of capillaries into the interstitial fluid, and sodium and chloride move from the interstitial fluid into the capillaries.
The Distal Tubule and Collecting Ducts
The distal tubule arises from the ascending limb of the loop of Henle; its thick cellular structure is distinct from the thin cells of the ascending loop. Thick cuboidal cells continue up through the renal cortical area to a point where the distal tubule is in direct contact with the afferent arteriole of its glomerulus. At this junction, the distal tubule cells become more densely packed and more columnar, and the muscle cells of the arteriole enlarge and take on a granular appearance. This point of contact between the distal tubule and the glomerular afferent arteriole is called the juxtaglomerular apparatus (see Fig. 13-3).
Renin, Aldosterone, and Antidiuretic Hormone
Renin is secreted from the polkissen cells of the afferent arteriole in the juxtaglomerular apparatus. In turn, renin forms angiotensin I from renin substrate (a circulating peptide from the liver). The amounts of renin released and angiotensin formed are determined by the renal perfusion pressure, sympathetic nervous system stimulation, circulating vasoactive substances, and changes in electrolyte concentration.55
Angiotensin I circulates to the lung and is converted enzymatically to angiotensin II. Angiotensin II produces peripheral vasoconstriction and an increase in aldosterone secretion, which increases renal sodium and water reabsorption. These effects should increase intravascular volume (Fig. 13-8). Angiotensin I and II are destroyed by angiotensinase, an enzyme that is present in plasma and secreted by a variety of organs, such as the kidney, intestine, and liver.
Fig. 13-8 Renal response to changes in extracellular fluid volume and electrolyte concentration or stress.
Aldosterone is secreted by the adrenal cortex in response to pituitary adrenal corticotropic hormone (ACTH) secretion and a variety of other stimuli. A fall in the pulse pressure, decreased stretch of the right atrium, and an increased serum potassium concentration all stimulate aldosterone secretion.63 An important stimulus for aldosterone is formation of angiotensin from renin released by the juxtaglomerular apparatus. Aldosterone stimulates epithelial cell transport of sodium in the renal tubular epithelium, along the intestinal lumen, and in sweat and saliva. Increased aldosterone levels increase the active reabsorption of sodium and decrease potassium reabsorption. The increased sodium reabsorption produces water reabsorption; this increases intravascular volume and reduces the juxtamedullary secretion of renin. The reduction in potassium tubular reabsorption increases potassium excretion in the urine and should result in a fall in the serum potassium concentration. These responses to aldosterone should in turn reduce the stimulus for aldosterone secretion (see Fig. 13-8).
Antidiuretic hormone (ADH), or arginine vasopressin (AVP), secretion also affects the final concentration of urine. ADH is produced by the supraoptic and paraventricular nuclei in the hypothalamus and is transported to the posterior lobe of the pituitary, where it is released in response to an increase in serum osmolality. ADH secretion is stimulated by serum osmolality greater than 280 to 285 mOsm/L (or a rise in serum osmolality of 2% or more). It also is secreted in response to significant (10%-15%) volume depletion, a fall in blood pressure, painful stimuli, fear, and exercise. Hemoconcentration, diabetic ketoacidosis,90 and mannitol administration increase ADH secretion, and administration of hypertonic glucose often inhibits ADH secretion.34,54 The predominant stimulus for ADH secretion is a rise in serum osmolality sensed by osmoreceptors in and around the supraoptic nucleus of the hypothalamus.
Regulation of Acid-Base Balance
Buffering Systems
The Bicarbonate-Carbonic Acid Buffering System
The increase in hydrogen ion concentration will result in a fall in serum pH unless or until CO2 elimination by the lungs is enhanced and/or hydrogen ion excretion and bicarbonate ion reabsorption by the kidneys is increased (see section, Interpretation of Blood Gas Values).
Additional Plasma Buffers
Proteins present in the blood can also act as buffers. Hemoglobin is the most important nonbicarbonate buffer. It binds with hydrogen ions and transports CO2 from the tissues to the lungs for elimination.34
Renal Hydrogen Ion Excretion and Bicarbonate Reabsorption
New bicarbonate can be formed when CO2 combines with water, yielding carbonic acid. The carbonic acid then dissociates into hydrogen ions and bicarbonate; the hydrogen ion is bound to phosphate buffers or ammonia to form hydrogen phosphate or ammonium (). Hydrogen phosphate and ammonium are nonreabsorbable, and they are excreted unchanged in the urine. When hydrogen ions are excreted in this way, a quantity of acid can be measured in the urine; this buffering mechanism results in the formation of titratable acids (see Fig. 13-6). The amount of hydrogen ion excreted in the urine is limited, because the kidney cannot secrete urine with a pH lower than approximately 4.4. In addition, the formation of titratable acid will be limited by the amount of ammonia, phosphate, and other inorganic buffers available.
