389 |
Relapsing Polychondritis |
Relapsing polychondritis is an uncommon disorder of unknown cause characterized by inflammation of cartilage predominantly affecting the ears, nose, and laryngotracheobronchial tree. Other manifestations include scleritis, neurosensory hearing loss, polyarthritis, cardiac abnormalities, skin lesions, and glomerulonephritis. Relapsing polychondritis has been estimated to have an incidence of 3.5 per million population per year. The peak age of onset is between the ages of 40 and 50 years, but relapsing polychondritis may affect children and the elderly. It is found in all races, and both sexes are equally affected. No familial tendency is apparent. A significantly higher frequency of HLA-DR4 has been found in patients with relapsing polychondritis than in healthy individuals. A predominant subtype allele(s) of HLA-DR4 was not found. Approximately 30% of patients with relapsing polychondritis will have another rheumatologic disorder, the most frequent being systemic vasculitis, followed by rheumatoid arthritis, and systemic lupus erythematosus (SLE). Nonrheumatic disorders associated with relapsing polychondritis include Hashimoto’s thyroiditis, primary biliary cirrhosis, and myelodysplastic syndrome (Table 389-1). In most cases, these disorders antedate the appearance of relapsing polychondritis, usually by months or years; however, in other instances, the onset of relapsing polychondritis can accompany disease presentation.
|
DISORDERS ASSOCIATED WITH RELAPSING POLYCHONDRITISa |
aSystemic vasculitis is the most common association, followed by rheumatoid arthritis and systemic lupus erythematosus.
Source: Modified from CJ Michet et al: Ann Intern Med 104:74, 1986.
PATHOLOGY AND PATHOPHYSIOLOGY
The earliest abnormality of hyaline and elastic cartilage noted histologically is a focal or diffuse loss of basophilic staining indicating depletion of proteoglycan from the cartilage matrix. Inflammatory infiltrates are found adjacent to involved cartilage and consist predominantly of mononuclear cells and occasional plasma cells. In acute disease, polymorphonuclear white cells may also be present. Destruction of cartilage begins at the outer edges and advances centrally. There is lacunar breakdown and loss of chondrocytes. Degenerating cartilage is replaced by granulation tissue and later by fibrosis and focal areas of calcification. Small loci of cartilage regeneration may be present. Immunofluorescence studies have shown immunoglobulins and complement at sites of involvement. Extracellular granular material observed in the degenerating cartilage matrix by electron microscopy has been interpreted to be enzymes, immunoglobulins, or proteoglycans.
Immunologic mechanisms play a role in the pathogenesis of relapsing polychondritis. The accumulating data strongly suggest that both humoral and cell-mediated immunity play an important role in the pathogenesis of relapsing polychondritis. Immunoglobulin and complement deposits are found at sites of inflammation. In addition, antibodies to type II collagen and to matrilin-1 and immune complexes are detected in the sera of some patients. The possibility that an immune response to type II collagen may be important in the pathogenesis is supported experimentally by the occurrence of auricular chondritis in rats immunized with type II collagen. Antibodies to type II collagen are found in the sera of these animals, and immune deposits are detected at sites of ear inflammation. Humoral immune responses to type IX and type XI collagen, matrilin-1, and cartilage oligomeric matrix protein have been demonstrated in some patients. In a study, rats immunized with matrilin-1 were found to develop severe inspiratory stridor and swelling of the nasal septum. The rats had severe inflammation with erosions of the involved cartilage, which was characterized by increased numbers of CD4+ and CD8+ T cells in the lesions. The cartilage of the joints and ear pinna was not involved. All had IgG antibodies to matrilin-1. Matrilin-1 is a noncollagenous protein present in the extracellular matrix in cartilage. It is present in high concentrations in the trachea and is also present in the nasal septum but not in articular cartilage. A subsequent study demonstrated serum anti-matrilin-1 antibodies in approximately 13% of patients with relapsing polychondritis; approximately 70% of these patients had respiratory symptoms. Cell-mediated immunity may also be operative in causing tissue injury, since lymphocyte transformation can be demonstrated when lymphocytes of patients are exposed to cartilage extracts. T cells specific for type II collagen have been found in some patients, and CD4+ T cells have been observed at sites of cartilage inflammation.
CLINICAL MANIFESTATIONS
The onset of relapsing polychondritis is frequently abrupt, with the appearance of one or two sites of cartilaginous inflammation. The pattern of cartilaginous involvement and the frequency of episodes vary widely among patients. Noncartilaginous presentations may also occur. Systemic inflammatory features such as fever, fatigue, and weight loss occur and may precede the clinical signs of relapsing polychondritis by several weeks. Relapsing polychondritis may go unrecognized for several months or even years in patients who only initially manifest intermittent joint pain and/or swelling, or who have unexplained eye inflammation, hearing loss, valvular heart disease, or pulmonary symptoms.
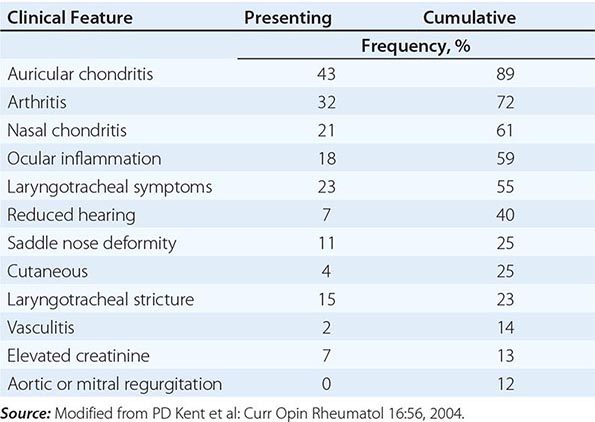
Auricular chondritis is the most frequent presenting manifestation of relapsing polychondritis, occurring in 40% of patients and eventually affecting about 85% of patients (Table 389-2). One or both ears are involved, either sequentially or simultaneously. Patients experience the sudden onset of pain, tenderness, and swelling of the cartilaginous portion of the ear (Fig. 389-1). This typically involves the pinna of the ears, sparing the earlobes because they do not contain cartilage. The overlying skin has a beefy red or violaceous color. Prolonged or recurrent episodes lead to cartilage destruction and result in a flabby or droopy ear. Swelling may close off the eustachian tube or the external auditory meatus, either of which can impair hearing. Inflammation of the internal auditory artery or its cochlear branch produces hearing loss, vertigo, ataxia, nausea, and vomiting. Vertigo is almost always accompanied by hearing loss.
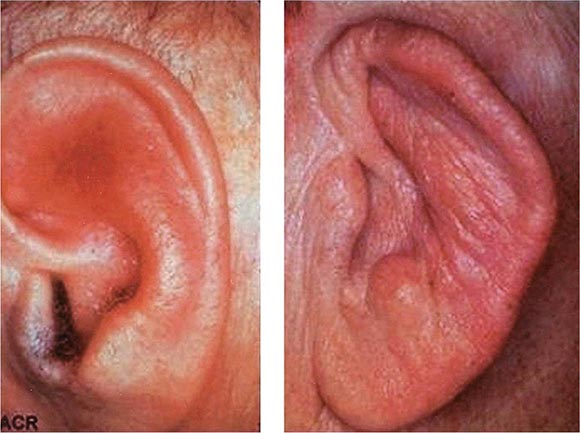
FIGURE 389-1 Left. The pinna is erythematous, swollen, and tender. Not shown is the ear lobule that is spared as there is no underlying cartilage. Right. The pinna is thickened and deformed. The destruction of the underlying cartilage results in a floppy ear. (Reprinted from the Clinical Slide Collection on the Rheumatic Diseases, ©1991, 1995, 1997, 1998, 1999. Used by permission of the American College of Rheumatology.)
|
CLINICAL MANIFESTATIONS OF RELAPSING POLYCHONDRITIS |
Approximately 61% of patients will develop nasal involvement, with 21% having this at the time of presentation. Patients may experience nasal stuffiness, rhinorrhea, and epistaxis. The bridge of the nose and surrounding tissue become red, swollen, and tender and may collapse, producing a saddle nose deformity (Fig. 389-2). In some patients, nasal deformity develops insidiously without overt inflammation. Saddle nose is observed more frequently in younger patients, especially in women.
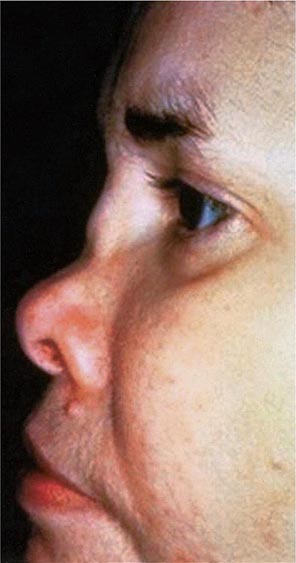
FIGURE 389-2 Saddle nose results from destruction and collapse of the nasal cartilage. (Reprinted from the Clinical Slide Collection on the Rheumatic Diseases, ©1991, 1995, 1997, 1998, 1999. Used by permission of the American College of Rheumatology.)
Joint involvement is the presenting manifestation in relapsing polychondritis in approximately one-third of patients and may be present for several months before other features appear. Eventually, more than one-half of the patients will have arthralgias or arthritis. The arthritis is usually asymmetric and oligo- or polyarticular, and it involves both large and small peripheral joints. An episode of arthritis lasts from a few days to several weeks and resolves spontaneously without joint erosion or deformity. Attacks of arthritis may not be temporally related to other manifestations of relapsing polychondritis. Joint fluid has been reported to be noninflammatory. In addition to peripheral joints, inflammation may involve the costochondral, sternomanubrial, and sternoclavicular cartilages. Destruction of these cartilages may result in a pectus excavatum deformity or even a flail anterior chest wall.
Eye manifestations occur in more than one-half of patients and include conjunctivitis, episcleritis, scleritis, iritis, uveitis, and keratitis. Ocular inflammation can be severe and visually threatening. Other manifestations include eyelid and periorbital edema, proptosis, optic neuritis, extraocular muscle palsies, retinal vasculitis, and renal vein occlusion.
Laryngotracheobronchial involvement occurs in ∼50% of patients and is among the most serious manifestations of relapsing polychondritis. Symptoms include hoarseness, a nonproductive cough, and tenderness over the larynx and proximal trachea. Mucosal edema, strictures, and/or collapse of laryngeal or tracheal cartilage may cause stridor and life-threatening airway obstruction necessitating tracheostomy. Involvement can extend into the lower airways resulting in tracheobronchomalacia. Collapse of cartilage in bronchi leads to pneumonia and, when extensive, to respiratory insufficiency.
Cardiac valvular regurgitation occurs in about 5–10% of patients and is due to progressive dilation of the valvular ring or to destruction of the valve cusps. Aortic regurgitation occurs in about 7% of patients, with the mitral and other heart valves being affected less often. Other cardiac manifestations include pericarditis, myocarditis, coronary vasculitis, and conduction abnormalities. Aneurysms of the proximal, thoracic, or abdominal aorta may occur even in the absence of active chondritis and occasionally rupture.
Renal disease occurs in about 10% of patients. The most common renal lesions include mesangial expansion or segmental necrotizing glomerulonephritis, which have been reported to have small amounts of electron-dense deposits in the mesangium where there is also faint deposition of C3 and/or IgG or IgM. Tubulointerstitial disease and IgA nephropathy have also been reported.
Approximately 25% of patients have skin lesions, which can include purpura, erythema nodosum, erythema multiforme, angioedema/urticaria, livedo reticularis, and panniculitis.
Features of vasculitis are seen in up to 25% of patients and can affect any size vessel. Large vessel vasculitis may present with aortic aneurysms, and medium vessel disease may affect the coronary, hepatic, mesenteric, or renal arteries or vessel supplying nerves. Skin vessel disease and involvement of the postcapillary venules can also occur. A variety of primary vasculitides have also been reported to occur in association with relapsing polychondritis (Chap. 385). One specific overlap is the “MAGIC” syndrome (mouth and genital ulcers with inflamed cartilage) in which patients present with features of both relapsing polychondritis and Behçet’s disease (Chap. 387).
LABORATORY FINDINGS AND DIAGNOSTIC IMAGING
There are no laboratory features that are diagnostic for relapsing polychondritis. Mild leukocytosis and normocytic, normochromic anemia are often present. Eosinophilia is observed in 10% of patients. The erythrocyte sedimentation rate and C-reactive protein are usually elevated. Rheumatoid factor and antinuclear antibody tests are occasionally positive in low titers, and complement levels are normal. Antibodies to type II collagen are present in fewer than one-half of the patients and are not specific. Circulating immune complexes may be detected, especially in patients with early active disease. Elevated levels of γ globulin may be present. Antineutrophil cytoplasmic antibodies (ANCA), either cytoplasmic (cANCA) or perinuclear (pANCA), are found in some patients with active disease. However, on target antigen–specific testing, there are only occasional reports of positive myeloperoxidase-ANCA, and proteinase 3-ANCA are very rarely found in relapsing polychondritis.
The upper and lower airways can be evaluated by imaging techniques such as computed tomography and magnetic resonance imaging (MRI). Bronchoscopy provides direct visualization of the airways but can be a high-risk procedure in patients with airway compromise. Pulmonary function testing with flow-volume loops can show inspiratory and/or expiratory obstruction. Imaging can also be useful to detect extracartilaginous disease. The chest film may show widening of the ascending or descending aorta due to an aneurysm, and cardiomegaly when aortic insufficiency is present. MRI can assess aortic aneurysmal dilatation. Electrocardiography and echocardiography can be useful in further evaluating for cardiac features of disease.
DIAGNOSIS
Diagnosis is based on recognition of the typical clinical features. Biopsies of the involved cartilage from the ear, nose, or respiratory tract will confirm the diagnosis but are only necessary when clinical features are not typical. Diagnostic criteria were suggested in 1976 by McAdam et al and modified by Damiani and Levine in 1979. These criteria continue to be generally used in clinical practice. McAdam et al proposed the following: (1) recurrent chondritis of both auricles; (2) nonerosive inflammatory arthritis; (3) chondritis of nasal cartilage; (4) inflammation of ocular structures, including conjunctivitis, keratitis, scleritis/episcleritis, and/or uveitis; (5) chondritis of the laryngeal and/or tracheal cartilages; and (6) cochlear and/or vestibular damage manifested by neurosensory hearing loss, tinnitus, and/or vertigo. The diagnosis is certain when three or more of these features are present along with a positive biopsy from the ear, nasal, or respiratory cartilage. Damiani and Levine later suggested that the diagnosis could be made when one or more of the above features and a positive biopsy were present, when two or more separate sites of cartilage inflammation were present that responded to glucocorticoids or dapsone, or when three or more of the above features were present.
The differential diagnosis of relapsing polychondritis is centered around its sites of clinical involvement. Patients with granulomatosis with polyangiitis (Wegener’s) may have a saddle nose and tracheal involvement but can be distinguished by the primary inflammation occurring in the mucosa at these sites, the absence of auricular involvement, and the presence of pulmonary parenchymal disease. Patients with Cogan’s syndrome have interstitial keratitis and vestibular and auditory abnormalities, but this syndrome does not involve the respiratory tract or ears. Reactive arthritis may initially resemble relapsing polychondritis because of oligoarticular arthritis and eye involvement, but it is distinguished in time by the appearance of urethritis and typical mucocutaneous lesions and the absence of nose or ear cartilage involvement. Rheumatoid arthritis may initially suggest relapsing polychondritis because of arthritis and eye inflammation. The arthritis in rheumatoid arthritis, however, is erosive and symmetric. In addition, rheumatoid factor titers are usually high compared with those in relapsing polychondritis, and anti-cyclic citrullinated peptide is usually not seen. Bacterial infection of the pinna may be mistaken for relapsing polychondritis but differs by usually involving only one ear, including the earlobe. Auricular cartilage may also be damaged by trauma or frostbite. Nasal destructive disease and auricular abnormalities can also be seen in patients using cocaine adulterated with levamisole. Ear involvement in this setting differs from relapsing polychondritis by typically manifesting as purpuric plaques with necrosis extending to the pinna, which does not contain cartilage.
PATIENT OUTCOME, PROGNOSIS, AND SURVIVAL
The course of relapsing polychondritis is highly variable. Some patients experience inflammatory episodes lasting from a few days to several weeks that then subside spontaneously or with treatment. Attacks may recur at intervals varying from weeks to months. In other patients, the disease has a chronic, smoldering course that may be severe. In one study, the 5-year estimated survival rate was 74% and the 10-year survival rate was 55%. In contrast to earlier series, only about one-half of the deaths could be attributed to relapsing polychondritis or complications of treatment. Airway complications accounted for only 10% of all fatalities. In general, patients with more widespread disease have a worse prognosis.
ACKNOWLEDGMENT
This chapter represents a revised version of the text authored by Dr. Bruce C. Gilliland that appeared in previous editions of Harrison’s Principles of Internal Medicine. Dr. Gilliland passed away on February 17, 2007, and had been a contributor to Harrison’s since the 11th edition.
390 |
Sarcoidosis |
DEFINITION
Sarcoidosis is an inflammatory disease characterized by the presence of noncaseating granulomas. The disease is often multisystem and requires the presence of involvement in two or more organs for a specific diagnosis. The finding of granulomas is not specific for sarcoidosis, and other conditions known to cause granulomas must be ruled out. These conditions include mycobacterial and fungal infections, malignancy, and environmental agents such as beryllium. Although sarcoidosis can affect virtually every organ of the body, the lung is most commonly affected. Other organs commonly affected are the liver, skin, and eye. The clinical outcome of sarcoidosis varies, with remission occurring in over one-half of patients within a few years of diagnosis; however, the remaining patients may develop a chronic disease that lasts for decades.
ETIOLOGY
Despite multiple investigations, the cause of sarcoidosis remains unknown. Currently, the most likely etiology is an infectious or noninfectious environmental agent that triggers an inflammatory response in a genetically susceptible host. Among the possible infectious agents, careful studies have shown a much higher incidence of Propionibacter acnes in the lymph nodes of sarcoidosis patients compared to controls. An animal model has shown that P. acnes can induce a granulomatous response in mice similar to sarcoidosis. Others have demonstrated the presence of a mycobacterial protein (Mycobacterium tuberculosis catalase-peroxidase [mKatG]) in the granulomas of some sarcoidosis patients. This protein is very resistant to degradation and may represent the persistent antigen in sarcoidosis. Immune response to this and other mycobacterial proteins has been documented by another laboratory. These studies suggest that a mycobacterium similar to M. tuberculosis could be responsible for sarcoidosis. The mechanism exposure/infection with such agents has been the focus of other studies. Environmental exposures to insecticides and mold have been associated with an increased risk for disease. In addition, health care workers appear to have an increased risk. Also, sarcoidosis in a donor organ has occurred after transplantation into a sarcoidosis patient. Some authors have suggested that sarcoidosis is not due to a single agent but represents a particular host response to multiple agents. Some studies have been able to correlate the environmental exposures to genetic markers. These studies have supported the hypothesis that a genetically susceptible host is a key factor in the disease.
INCIDENCE, PREVALENCE, AND GLOBAL IMPACT
![]() Sarcoidosis is seen worldwide, with the highest prevalence reported in the Nordic population. In the United States, the disease has been reported more commonly in African Americans than whites, with the ratio of African Americans to whites ranging from 3:1 to 17:0. Women appear to be slightly more susceptible than men. The higher incidence in African Americans may have been influenced by the fact that African Americans seem to develop more extensive and chronic pulmonary disease. Because most sarcoidosis clinics are run by pulmonologists, a selection bias may have occurred. Worldwide, the prevalence of the disease varies from 20–60 per 100,000 for many groups such as Japanese, Italians, and American whites. Higher rates occur in Ireland and Nordic countries. In one closely observed community in Sweden, the lifetime risk for developing sarcoidosis was 3%.
Sarcoidosis is seen worldwide, with the highest prevalence reported in the Nordic population. In the United States, the disease has been reported more commonly in African Americans than whites, with the ratio of African Americans to whites ranging from 3:1 to 17:0. Women appear to be slightly more susceptible than men. The higher incidence in African Americans may have been influenced by the fact that African Americans seem to develop more extensive and chronic pulmonary disease. Because most sarcoidosis clinics are run by pulmonologists, a selection bias may have occurred. Worldwide, the prevalence of the disease varies from 20–60 per 100,000 for many groups such as Japanese, Italians, and American whites. Higher rates occur in Ireland and Nordic countries. In one closely observed community in Sweden, the lifetime risk for developing sarcoidosis was 3%.
Sarcoidosis often occurs in young, otherwise healthy adults. It is uncommon to diagnose the disease in someone under age 18. However, it has become clear that a second peak in incidence develops around age 60. In a study of >700 newly diagnosed sarcoidosis patients in the United States, one-half of the patients were ≥40 years at the time of diagnosis.
Although most cases of sarcoidosis are sporadic, a familial form of the disease exists. At least 5% of patients with sarcoidosis will have a family member with sarcoidosis. Sarcoidosis patients who are Irish or African American seem to have a two to three times higher rate of familial disease.
PATHOPHYSIOLOGY AND IMMUNOPATHOGENESIS
The granuloma is the pathologic hallmark of sarcoidosis. A distinct feature of sarcoidosis is the local accumulation of inflammatory cells. Extensive studies in the lung using bronchoalveolar lavage (BAL) have demonstrated that the initial inflammatory response is an influx of T helper cells. In addition, there is an accumulation of activated monocytes. Figure 390-1 is a proposed model for sarcoidosis. Using the HLA-CD4 complex, antigen-presenting cells present an unknown antigen to the helper T cell. Studies have clarified that specific HLA haplotypes such as HLA-DRB1*1101 are associated with an increased risk for developing sarcoidosis. In addition, different HLA haplotypes are associated with different clinical outcomes.
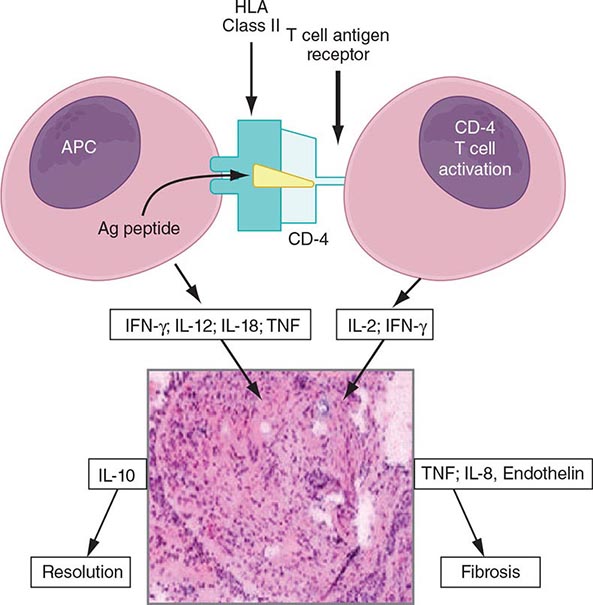
FIGURE 390-1 Schematic representation of initial events of sarcoidosis. The antigen-presenting cell and helper T cell complex leads to the release of multiple cytokines. This forms a granuloma. Over time, the granuloma may resolve or lead to chronic disease, including fibrosis. APC, antigen-presenting cell; HLA, human leukocyte antigen; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
The macrophage/helper T cell cluster leads to activation with the increased release of several cytokines. These include interleukin (IL)-2 released from the T cell and interferon γ and tumor necrosis factor (TNF) released by the macrophage. The T cell is a necessary part of the initial inflammatory response. In advanced, untreated HIV infection, patients who lack helper T cells rarely develop sarcoidosis. In contrast, several reports confirm that sarcoidosis becomes unmasked as HIV-infected individuals receive antiretroviral therapy, with subsequent restoration of their immune system. In contrast, treatment of established pulmonary sarcoidosis with cyclosporine, a drug that downregulates helper T cell responses, seems to have little impact on sarcoidosis.
The granulomatous response of sarcoidosis can resolve with or without therapy. However, in at least 20% of patients with sarcoidosis, a chronic form of the disease develops. This persistent form of the disease is associated with the secretion of high levels of IL-8. Also, studies have reported that patients with this chronic form of disease release excessive amounts of TNF in areas of inflammation. Specific gene signatures have been associated with more severe disease, such as cardiac, neurologic, and fibrotic pulmonary disease.
At diagnosis the natural history of the disease may be difficult to predict. One form of the disease, Löfgren’s syndrome, consists of erythema nodosum and hilar adenopathy on chest roentgenogram. In some cases, periarticular arthritis may be identified without erythema nodosum. Löfgren’s syndrome is associated with a good prognosis, with >90% of patients experiencing disease resolution within 2 years. Recent studies have demonstrated that the HLA-DRB1*03 was found in two-thirds of Scandinavian patients with Löfgren’s syndrome. More than 95% of those patients who were HLA-DRB1*03 positive had resolution of their disease within 2 years, whereas nearly one-half of the remaining patients had disease for more than 2 years. It remains to be determined whether these observations can be applied to a non-Scandinavian population.
CLINICAL MANIFESTATIONS
The presentation of sarcoidosis ranges from patients who are asymptomatic to those with organ failure. It is unclear how often sarcoidosis is asymptomatic. In countries where routine chest roentgenogram screening is performed, 20–30% of pulmonary cases are detected in asymptomatic individuals. The inability to screen for other asymptomatic forms of the disease would suggest that as many as one-third of sarcoidosis patients are asymptomatic.
Respiratory complaints including cough and dyspnea are the most common presenting symptoms. In many cases, the patient presents with a 2- to 4-week history of these symptoms. Unfortunately, due to the nonspecific nature of pulmonary symptoms, the patient may see physicians for up to a year before a diagnosis is confirmed. For these patients, the diagnosis of sarcoidosis is usually only suggested when a chest roentgenogram is performed.
Symptoms related to cutaneous and ocular disease are the next two most common complaints. Skin lesions are often nonspecific. However, because these lesions are readily observed, the patient and treating physician are often led to a diagnosis. In contrast to patients with pulmonary disease, patients with cutaneous lesions are more likely to be diagnosed within 6 months of symptoms.
Nonspecific constitutional symptoms include fatigue, fever, night sweats, and weight loss. Fatigue is perhaps the most common constitutional symptom that affects these patients. Given its insidious nature, patients are usually not aware of the association with their sarcoidosis until their disease resolves.
The overall incidence of sarcoidosis at the time of diagnosis and eventual common organ involvement are summarized in Table 390-1. Over time, skin, eye, and neurologic involvement seem more apparent. In the United States, the frequency of specific organ involvement appears to be affected by age, race, and gender. For example, eye disease is more common among African Americans. Under the age of 40, it occurs more frequently in women. However, in those diagnosed over the age of 40, eye disease is more common in men.
|
FREQUENCY OF COMMON ORGAN INVOLVEMENT AND LIFETIME RISKa |
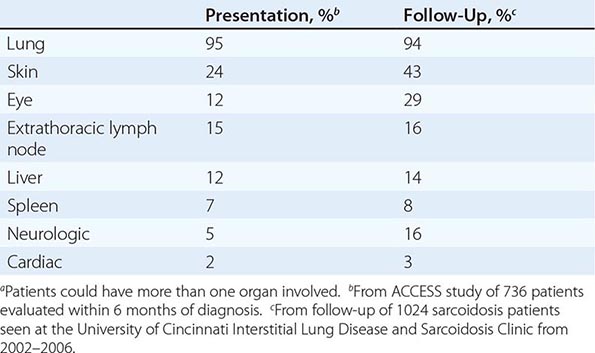
LUNG
Lung involvement occurs in >90% of sarcoidosis patients. The most commonly used method for detecting lung disease is still the chest roentgenogram. Figure 390-2 illustrates the chest roentgenogram from a sarcoidosis patient with bilateral hilar adenopathy. Although the computed tomography (CT) scan has changed the diagnostic approach to interstitial lung disease, the CT scan is not usually considered a monitoring tool for patients with sarcoidosis. Figure 390-3 demonstrates some of the characteristic CT features, including peribronchial thickening and reticular nodular changes, which are predominantly subpleural. The peribronchial thickening seen on CT scan seems to explain the high yield of granulomas from bronchial biopsies performed for diagnosis.
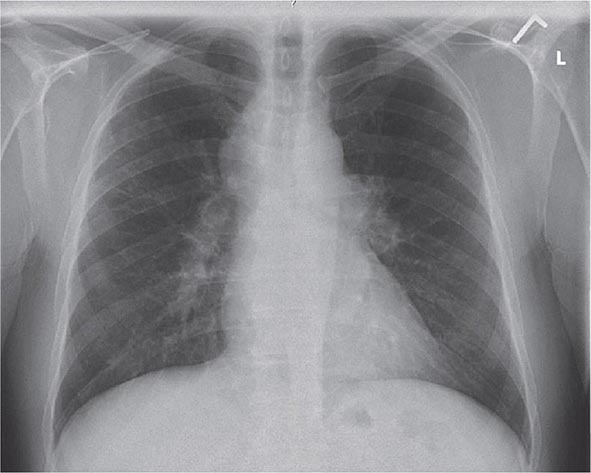
FIGURE 390-2 Posterior-anterior chest roentgenogram demonstrating bilateral hilar adenopathy, stage 1 disease.
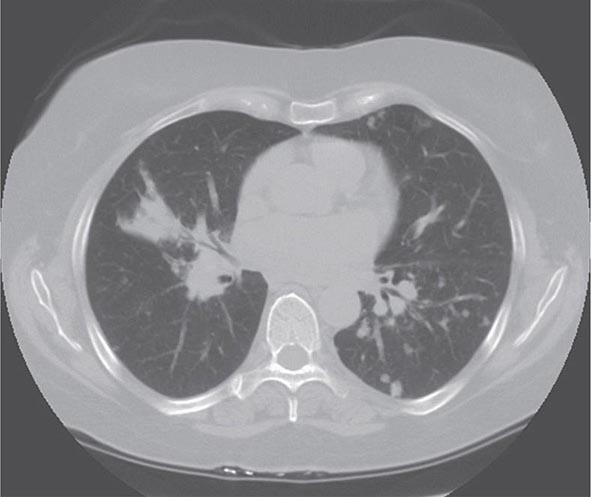
FIGURE 390-3 High-resolution computed tomography scan of chest demonstrating patchy reticular nodularity, including areas of confluence.
Although the CT scan is more sensitive, the standard scoring system described by Scadding in 1961 for chest roentgenograms remains the preferred method of characterizing the chest involvement. Stage 1 is hilar adenopathy alone (Fig. 390-2), often with right paratracheal involvement. Stage 2 is a combination of adenopathy plus infiltrates, whereas stage 3 reveals infiltrates alone. Stage 4 consists of fibrosis. Usually the infiltrates in sarcoidosis are predominantly an upper lobe process. Only in a few noninfectious diseases is an upper lobe predominance noted. In addition to sarcoidosis, the differential diagnosis of upper lobe disease includes hypersensitivity pneumonitis, silicosis, and Langerhans cell histiocytosis. For infectious diseases, tuberculosis and Pneumocystis pneumonia can often present as upper lobe diseases.
Lung volumes, mechanics, and diffusion are all useful in evaluating interstitial lung diseases such as sarcoidosis. The diffusion of carbon monoxide (DLCO) is the most sensitive test for an interstitial lung disease. Reduced lung volumes are a reflection of the restrictive lung disease seen in sarcoidosis. However, a third of the patients presenting with sarcoidosis still have lung volumes within the normal range, despite abnormal chest roentgenograms and dyspnea.
Approximately one-half of sarcoidosis patients present with obstructive disease, reflected by a reduced ratio of forced vital capacity expired in 1 second (FEV1/FVC). Cough is a very common symptom. Airway involvement causing varying degrees of obstruction underlies the cough in most sarcoidosis patients. Airway hyperreactivity, as determined by methacholine challenge, will be positive in some of these patients. A few patients with cough will respond to traditional bronchodilators as the only form of treatment. In some cases, high-dose inhaled glucocorticoids alone are useful. Airway obstruction can be due to large airway stenosis, which can become fibrotic and unresponsive to anti-inflammatory therapy.
Pulmonary arterial hypertension is reported in at least 5% of sarcoidosis patients. Either direct vascular involvement or the consequence of fibrotic changes in the lung can lead to pulmonary arterial hypertension. In sarcoidosis patients with end-stage fibrosis awaiting lung transplant, 70% will have pulmonary arterial hypertension. This is a much higher incidence than that reported for other fibrotic lung diseases. In less advanced, but still symptomatic, patients, pulmonary arterial hypertension has been noted in up to 50% of the cases. Because sarcoidosis-associated pulmonary arterial hypertension may respond to therapy, evaluation for this should be considered in persistently dyspneic patients.
SKIN
Skin involvement is eventually identified in over a third of patients with sarcoidosis. The classic cutaneous lesions include erythema nodosum, maculopapular lesions, hyper- and hypopigmentation, keloid formation, and subcutaneous nodules. A specific complex of involvement of the bridge of the nose, the area beneath the eyes, and the cheeks is referred to as lupus pernio (Fig. 390-4) and is diagnostic for a chronic form of sarcoidosis.
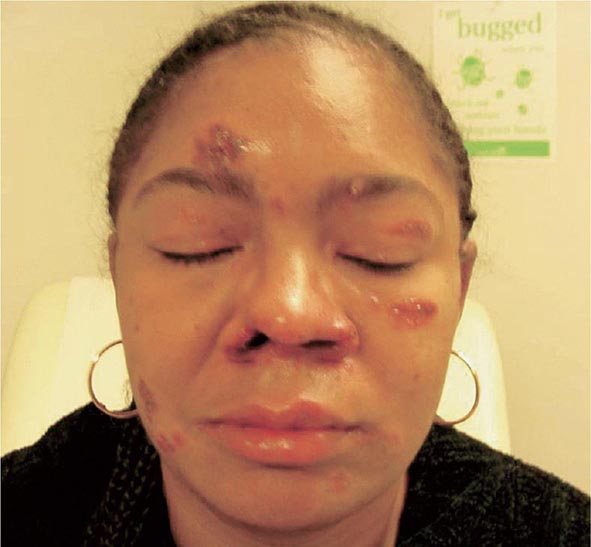
FIGURE 390-4 Chronic inflammatory lesions around nose, eyes, and cheeks, referred to as lupus pernio.
In contrast, erythema nodosum is a transient rash that can be seen in association with hilar adenopathy and uveitis (Löfgren’s syndrome). Erythema nodosum is more common in women and in certain self-described demographic groups including whites and Puerto Ricans. In the United States, the other manifestations of skin sarcoidosis, especially lupus pernio, are more common in African Americans than whites.
The maculopapular lesions from sarcoidosis are the most common chronic form of the disease (Fig. 390-5). These are often overlooked by the patient and physician, because they are chronic and not painful. Initially, these lesions are usually purplish papules and are often indurated. They can become confluent and infiltrate large areas of the skin. With treatment, the color and induration may fade. Because these lesions are caused by noncaseating granulomas, the diagnosis of sarcoidosis can be readily made by a skin biopsy.
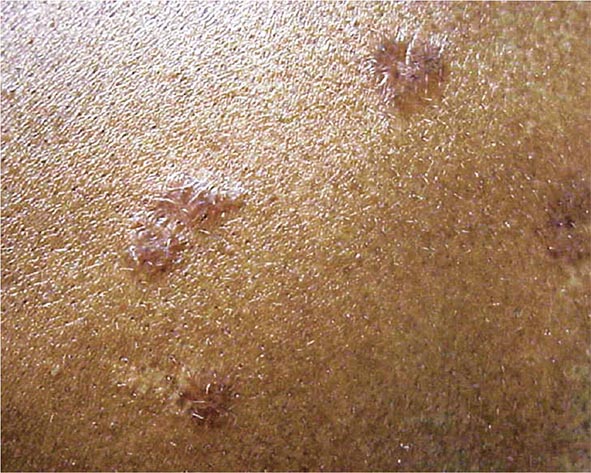
FIGURE 390-5 Maculopapular lesions on the trunk of a sarcoidosis patient.
EYE
The frequency of ocular manifestations for sarcoidosis varies depending on race. In Japan, >70% of sarcoidosis patients develop ocular disease, whereas in the United States only 30% have eye disease, with problems more common in African Americans than whites. Although the most common manifestation is an anterior uveitis, over a quarter of patients will have inflammation at the posterior of the eye, including retinitis and pars planitis. Although symptoms such as photophobia, blurred vision, and increased tearing can occur, some asymptomatic patients still have active inflammation. Initially asymptomatic patients with ocular sarcoidosis can eventually develop blindness. Therefore, it is recommended that all patients with sarcoidosis receive a dedicated ophthalmologic examination. Sicca is seen in over one-half of the chronic sarcoidosis patients. Dry eyes appear to be a reflection of prior lacrimal gland disease. Although the patient may no longer have active inflammation, the dry eyes may require natural tears or other lubricants.
LIVER
Using biopsies to detect granulomatous disease, liver involvement can be identified in over one-half of sarcoidosis patients. However, using liver function studies, only 20–30% of patients will have evidence of liver involvement. The most common abnormality of liver function is an elevation of the alkaline phosphatase level, consistent with an obstructive pattern. In addition, elevated transaminase levels can occur. An elevated bilirubin level is a marker for more advanced liver disease. Overall, only 5% of sarcoidosis patients have sufficient symptoms from their liver disease to require specific therapy. Although symptoms can be due to hepatomegaly, more frequently symptoms result from extensive intrahepatic cholestasis leading to portal hypertension. In this case, ascites and esophageal varices can occur. It is rare that a sarcoidosis patient will require a liver transplant, because even the patient with cirrhosis due to sarcoidosis can respond to systemic therapy. On a cautionary note, patients with both sarcoidosis and hepatitis C should avoid therapy with interferon a because of its association with the development or worsening of granulomatous disease.
BONE MARROW AND SPLEEN
One or more bone marrow manifestations can be identified in many sarcoidosis patients. The most common hematologic problem is lymphopenia, which is a reflection of sequestration of the lymphocytes into the areas of inflammation. Anemia occurs in 20% of patients, and leukopenia is less common. Bone marrow examination will reveal granulomas in about a third of patients. Although splenomegaly can be detected in 5–10% of patients, splenic biopsy reveals granulomas in 60% of patients. The CT scan can be relatively specific for sarcoidosis involvement of the spleen (Fig. 390-6). Both bone marrow and spleen involvement are more common in African Americans than whites. Although these manifestations alone are rarely an indication for therapy, on rare occasion, splenectomy may be indicated for massive symptomatic splenomegaly or profound pancytopenia. Nonthoracic lymphadenopathy can occur in up to 20% of patients.
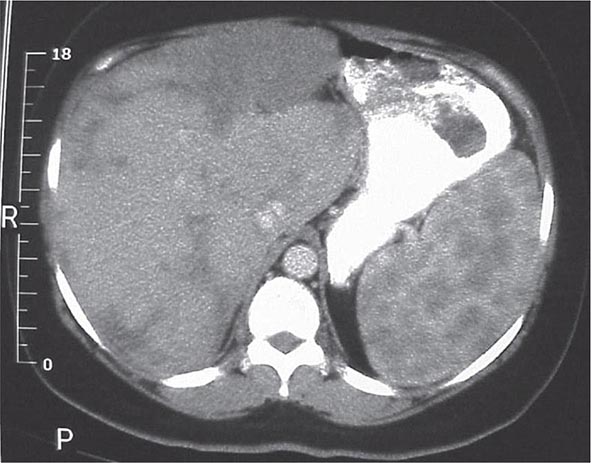
FIGURE 390-6 Computed tomography scan of the abdomen after oral and intravenous contrast. The stomach is compressed by the enlarged spleen. Within the spleen, areas of hypo- and hyperdensity are identified.
CALCIUM METABOLISM
Hypercalcemia and/or hypercalciuria occurs in about 10% of sarcoidosis patients. It is more common in whites than African Americans and in men. The mechanism of abnormal calcium metabolism is increased production of 1,25-dihydroxyvitamin D by the granuloma itself. The 1,25-dihydroxyvitamin D causes increased intestinal absorption of calcium, leading to hypercalcemia with a suppressed parathyroid hormone (PTH) level (Chap. 424). Increased exogenous vitamin D from diet or sunlight exposure may exacerbate this problem. Serum calcium should be determined as part of the initial evaluation of all sarcoidosis patients, and a repeat determination may be useful during the summer months with increased sun exposure. In patients with a history of renal calculi, a 24-h urine calcium measurement should be obtained. If a sarcoidosis patient with a history of renal calculi is to be placed on calcium supplements, a follow-up 24-h urine calcium level should be measured.
RENAL DISEASE
Direct kidney involvement occurs in <5% of sarcoidosis patients. It is associated with granulomas in the kidney itself and can lead to nephritis. However, hypercalcemia is the most likely cause of sarcoidosis-associated renal disease. In 1–2% of sarcoidosis patients, acute renal failure may develop as a result of hypercalcemia. Successful treatment of hypercalcemia with glucocorticoids and other therapies often improves but usually does not totally resolve the renal dysfunction.
NERVOUS SYSTEM
Neurologic disease is reported in 5–10% of sarcoidosis patients and appears to be of equal frequency across all ethnic groups. Any part of the central or peripheral nervous system can be affected. The presence of granulomatous inflammation is often visible on magnetic resonance imaging (MRI) studies. The MRI with gadolinium enhancement may demonstrate space-occupying lesions, but the MRI can be negative due to small lesions or the effect of systemic therapy in reducing the inflammation. The cerebral spinal fluid (CSF) findings include lymphocytic meningitis with a mild increase in protein. The CSF glucose is usually normal but can be low. Certain areas of the nervous system are more commonly affected in neurosarcoidosis. These include cranial nerve involvement, basilar meningitis, myelopathy, and anterior hypothalamic disease with associated diabetes insipidus (Chap. 404). Seizures and cognitive changes also occur. Of the cranial nerves, seventh nerve paralysis can be transient and mistaken for Bell’s palsy (idiopathic seventh nerve paralysis). Because this form of neurosarcoidosis often resolves within weeks and may not recur, it may have occurred prior to a definitive diagnosis of sarcoidosis. Optic neuritis is another cranial nerve manifestation of sarcoidosis. This manifestation is more chronic and usually requires long-term systemic therapy. It can be associated with both anterior and posterior uveitis. Differentiating between neurosarcoidosis and multiple sclerosis can be difficult at times. Optic neuritis can occur in both diseases. In some patients with sarcoidosis, multiple enhancing white matter abnormalities may be detected by MRI, suggesting multiple sclerosis. In such cases, the presence of meningeal enhancement or hypothalamic involvement suggests neurosarcoidosis, as does evidence of extraneurologic disease such as pulmonary or skin involvement, which also suggests sarcoidosis. Because the response of neurosarcoidosis to glucocorticoids and cytotoxic therapy is different from that of multiple sclerosis, differentiating between these disease entities is important.
CARDIAC
The presence of cardiac involvement is influenced by race. Although over a quarter of Japanese sarcoidosis patients develop cardiac disease, only 5% of sarcoidosis patients in the United States and Europe develop symptomatic cardiac disease. However, there is no apparent racial predilection between whites and African Americans. Cardiac disease, which usually presents as either congestive heart failure or cardiac arrhythmias, results from infiltration of the heart muscle by granulomas. Diffuse granulomatous involvement of the heart muscle can lead to profound dysfunction with left ventricular ejection fractions below 10%. Even in this situation, improvement in the ejection fraction can occur with systemic therapy. Arrhythmias can also occur with diffuse infiltration or with more patchy cardiac involvement. If the atrioventricular (AV) node is infiltrated, heart block can occur, which can be detected by routine electrocardiography. Ventricular arrhythmias and sudden death due to ventricular tachycardia are common causes of death. Arrhythmias are best detected using 24-h ambulatory monitoring, and electrophysiology studies may be negative. Other screening tests for cardiac disease include routine electrocardiography and echocardiography. The confirmation of cardiac sarcoidosis is usually performed with either MRI or positron emission tomography (PET) scanning. Because ventricular arrhythmias are usually multifocal due to patchy multiple granulomas in the heart, ablation therapy is not useful. Patients with significant ventricular arrhythmias should be considered for an implanted defibrillator, which appears to have reduced the rate of death in cardiac sarcoidosis. Although systemic therapy can be useful in treating the arrhythmias, patients may still have malignant arrhythmias up to 6 months after starting successful treatment, and the risk for recurrent arrhythmias occurs whenever medications are tapered.
MUSCULOSKELETAL SYSTEM
Direct granulomatous involvement of bone and muscle can be documented by radiography (x-ray, MRI, PET scan [Fig. 390-7], or gallium scan) or confirmed by biopsy in about 10% of sarcoidosis patients. However, a larger percentage of sarcoidosis patients complain of myalgias and arthralgias. These complaints are similar to those reported by patients with other inflammatory diseases, including chronic infections such as mononucleosis. Fatigue associated with sarcoidosis may be overwhelming for many patients. Recent studies have demonstrated a link between fatigue and small peripheral nerve fiber disease in sarcoidosis.
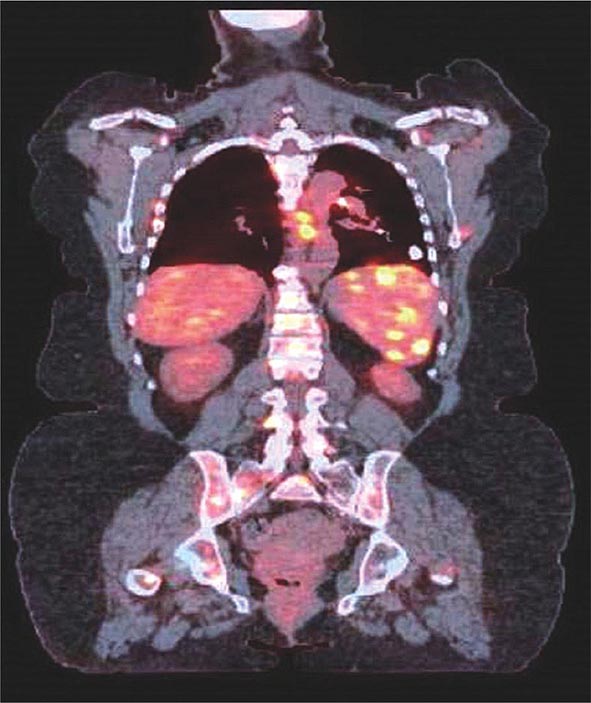
FIGURE 390-7 Positron emission tomography and computed tomography scan merged demonstrating increased activity in spleen, ribs, and spine of patient with sarcoidosis.
OTHER ORGAN INVOLVEMENT
Although sarcoidosis can affect any organ of the body, rarely does it involve the breast, testes, ovary, or stomach. Because of the rarity of involvement, a mass in one of these areas requires a biopsy to rule out other diseases including cancer. For example, in a study of breast problems in female sarcoidosis patients, a breast lesion was more likely to be a granuloma from sarcoidosis than from breast cancer. However, findings on the physical examination or mammogram cannot reliably differentiate between these lesions. More importantly, as women with sarcoidosis age, breast cancer becomes more common. Therefore, it is recommended that routine screening including mammography be performed along with other imaging studies (ultrasound, MRI) or biopsy as clinically indicated.
COMPLICATIONS
Sarcoidosis is usually a self-limited, non-life-threatening disease. However, organ-threatening disease can occur. These complications can include blindness, paraplegia, or renal failure. Death from sarcoidosis occurs in about 5% of patients seen in sarcoidosis referral clinics. The usual causes of death related to sarcoidosis are from lung, cardiac, neurologic, or liver involvement. In respiratory failure, an elevation of the right atrial pressure is a poor prognostic finding. Lung complications can also include infections such as mycetoma, which can subsequently lead to massive bleeding. In addition, the use of immunosuppressive agents can increase the incidence of serious infections.
LABORATORY FINDINGS
The chest roentgenogram remains the most commonly used tool to assess lung involvement in sarcoidosis. As noted above, the chest roentgenogram classifies involvement into four stages, with stages 1 and 2 having hilar and paratracheal adenopathy. The CT scan has been used increasingly in evaluating interstitial lung disease. In sarcoidosis, the presence of adenopathy and a nodular infiltrate is not specific for sarcoidosis. Adenopathy up to 2 cm can be seen in other inflammatory lung diseases such as idiopathic pulmonary fibrosis. However, adenopathy >2 cm in the short axis supports the diagnosis of sarcoidosis over other interstitial lung diseases.
The PET scan has increasingly replaced gallium-67 scanning to identify areas of granulomatous disease in the chest and other parts of the body (Fig. 390-7). Both tests can be used to identify potential areas for biopsy. Cardiac PET scanning has also proved useful in assessing cardiac sarcoidosis. The identification of hypermetabolic activity may be due to the granulomas from sarcoidosis and not to disseminated malignancy.
MRI has also proved useful in the assessment of extrapulmonary sarcoidosis. Gadolinium enhancement has been demonstrated in areas of inflammation in the brain, heart, and bone. MRI scans may detect asymptomatic lesions. Like PET scan, MRI changes appear similar to those seen with malignancy and infection. In some cases, biopsy may be necessary to determine the cause of the radiologic abnormality.
Serum levels of angiotensin-converting enzyme (ACE) can be helpful in the diagnosis of sarcoidosis. However, the test has somewhat low sensitivity and specificity. Elevated levels of ACE are reported in 60% of patients with acute disease and only 20% of patients with chronic disease. Although there are several causes for mild elevation of ACE, including diabetes, elevations of >50% of the upper limit of normal are seen in only a few conditions including sarcoidosis, leprosy, Gaucher’s disease, hyperthyroidism, and disseminated granulomatous infections such as miliary tuberculosis. Because the ACE level is determined by a biologic assay, the concurrent use of an ACE inhibitor such as lisinopril will lead to a very low ACE level.
DIAGNOSIS
The diagnosis of sarcoidosis requires both compatible clinical features and pathologic findings. Because the cause of sarcoidosis remains elusive, the diagnosis cannot be made with 100% certainty. Nevertheless, the diagnosis can be made with reasonable certainty based on history and physical features along with laboratory and pathologic findings.
Patients are usually evaluated for possible sarcoidosis based on two scenarios (Fig. 390-8). In the first scenario, a patient may undergo a biopsy revealing a noncaseating granuloma in either a pulmonary or an extrapulmonary organ. If the clinical presentation is consistent with sarcoidosis and there is no alternative cause for the granulomas identified, then the patient is felt to have sarcoidosis.
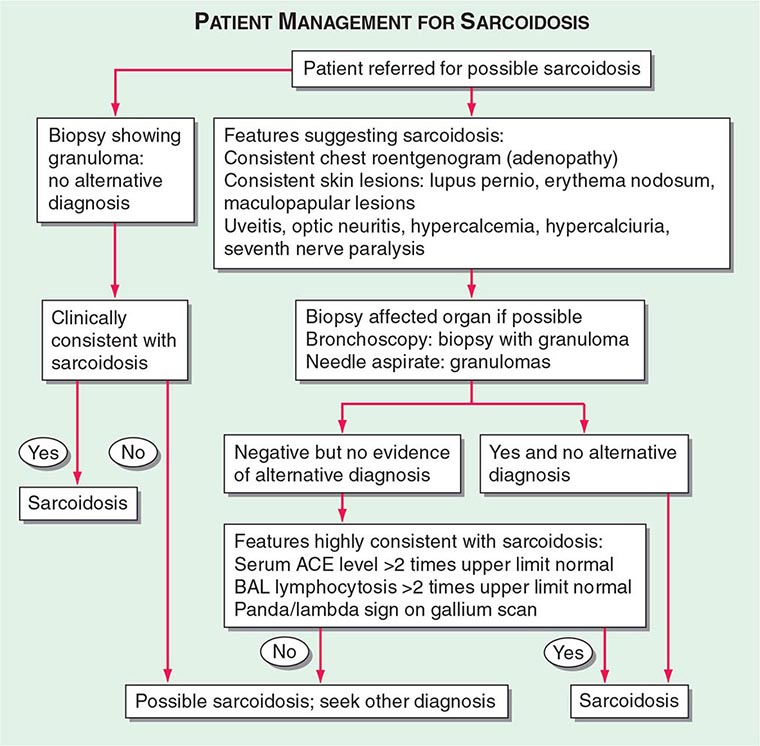
FIGURE 390-8 Proposed approach to management of patient with possible sarcoidosis. Presence of one or more of these features supports the diagnosis of sarcoidosis: uveitis, optic neuritis, hypercalcemia, hypercalciuria, seventh cranial nerve paralysis, diabetes insipidus. ACE, angiotensin-converting enzyme; BAL, bronchoalveolar lavage.
In the second scenario, signs or symptoms suggesting sarcoidosis such as the presence of bilateral adenopathy may be present in an otherwise asymptomatic patient or a patient with uveitis or a rash consistent with sarcoidosis. At this point, a diagnostic procedure should be performed. For the patient with a compatible skin lesion, a skin biopsy should be considered. Other biopsies to consider could include liver, extrathoracic lymph node, or muscle. In some cases, a biopsy of the affected organ may not be easy to perform (such as a brain or spinal cord lesion). In other cases, such as an endomyocardial biopsy, the likelihood of a positive biopsy is low. Because of the high rate of pulmonary involvement in these cases, the lung may be easier to approach by bronchoscopy. During the bronchoscopy, a transbronchial biopsy, bronchial biopsy, or transbronchial needle aspirate can be performed. The endobronchial ultrasonography-guided (EBUS) transbronchial needle aspirate can assist in diagnosing sarcoidosis in patients with mediastinal adenopathy (stage 1 or 2 radiographic pulmonary disease), whereas transbronchial biopsy has a higher diagnostic yield for those with only parenchymal lung disease (stage 3). These tests are complementary and may be performed together.
If the biopsy reveals granulomas, an alternative diagnosis such as infection or malignancy must be excluded. Bronchoscopic washings can be sent for cultures for fungi and tuberculosis. For the pathologist, the more tissue that is provided, the more comfortable is the diagnosis of sarcoidosis. A needle aspirate may be adequate in an otherwise classic case of sarcoidosis, but may be insufficient in a patient in whom lymphoma or fungal infection is a likely alternative diagnosis. Because granulomas can be seen on the edge of a lymphoma, the presence of a few granulomas from a needle aspirate may not be sufficient to clarify the diagnosis. Mediastinoscopy provides a larger sample to confirm the presence or absence of lymphoma in the mediastinum. Alternatively, for most patients, evidence of extrathoracic disease (e.g., eye involvement) may further support the diagnosis of sarcoidosis.
For patients with negative pathology, positive supportive tests may increase the likelihood of the diagnosis of sarcoidosis. These tests include an elevated ACE level, which can also be elevated in other granulomatous diseases but not in malignancy. A positive PET scan can support the diagnosis if multiple organs are affected. A BAL is often performed during the bronchoscopy. An increase in the percentage of lymphocytes supports the diagnosis of sarcoidosis. The use of the lymphocyte markers CD4 and CD8 can be used to determine the CD4/CD8 ratio of these increased lymphocytes in the BAL fluid. A ratio of >3.5 is strongly supportive of sarcoidosis but is less sensitive than an increase in lymphocytes alone. Although in general, an increase in BAL lymphocytes is supportive of the diagnosis, other conditions must be considered.
Supportive findings, when combined with commonly associated but nondiagnostic clinical features of the disease, improve the diagnostic probability of sarcoidosis. These clinical features include uveitis, renal stones, hypercalcemia, seventh cranial nerve paralysis, or erythema nodosum. The presence of one or more of these features in a patient suspected of having sarcoidosis increases the probability of sarcoidosis.
The Kviem-Siltzbach procedure is a specific diagnostic test for sarcoidosis. An intradermal injection of specially prepared tissue derived from the spleen of a known sarcoidosis patient is biopsied 4–6 weeks after injection. If noncaseating granulomas are seen, this is highly specific for the diagnosis of sarcoidosis. Unfortunately, there is no commercially available Kviem-Siltzbach reagent, and some locally prepared batches have lower specificity. Thus, this test is of historic interest and is rarely used in current clinical practice.
Because the diagnosis of sarcoidosis can never be certain, over time other features may arise that lead to an alternative diagnosis. Conversely, evidence for new organ involvement may eventually confirm the diagnosis of sarcoidosis.
PROGNOSIS
The risk of death or loss of organ function remains low in sarcoidosis. Poor outcomes usually occur in patients who present with advanced disease in whom treatment seems to have little impact. In these cases, irreversible fibrotic changes have frequently occurred. Over the past 20 years, the reported mortality from sarcoidosis has increased in the United States and England. Whether this is due to heightened awareness of the chronic nature of this disease or to other factors such as more widespread immunosuppressive therapy usage remains unclear.
For the majority of patients, initial presentation occurs during the granulomatous phase of the disease as depicted in Fig. 390-1. It is clear that many patients resolve their disease within 2–5 years. These patients are felt to have acute, self-limiting sarcoidosis. However, there is a form of the disease that does not resolve within the first 2–5 years. These chronic patients can be identified at presentation by certain risk factors at presentation such as fibrosis on chest roentgenogram, presence of lupus pernio, bone cysts, cardiac or neurologic disease (except isolated seventh nerve paralysis), and presence of renal calculi due to hypercalciuria. Recent studies also indicate that patients who require glucocorticoids for any manifestation of their disease in the first 6 months of presentation have a >50% chance of having chronic disease. In contrast, <10% of patients who require no systemic therapy in the first 6 months will require chronic therapy.
|
TREATMENT |
SARCOIDOSIS |
Indications for therapy should be based on symptoms or presence of organ- or life-threatening disease, including disease involving the eye, heart, or nervous system. The patient with asymptomatic elevated liver function tests or an abnormal chest roentgenogram probably does not benefit from treatment. However, these patients should be monitored for evidence of progressive, symptomatic disease.
One approach to therapy is summarized in Figs. 390-9 and 390-10. We have divided the approach into treating acute versus chronic disease. For acute disease, no therapy remains a viable option for patients with no or mild symptoms. For symptoms confined to only one organ, topical therapy is preferable. For multiorgan disease or disease too extensive for topical therapy, an approach to systemic therapy is outlined. Glucocorticoids remain the drugs of choice for this disease. However, the decision to continue to treat with glucocorticoids or to add steroid-sparing agents depends on the tolerability, duration, and dosage of glucocorticoids.Table 390-2 summarizes the dosage and monitoring of several commonly used drugs. According to the available trials, evidence-based recommendations are made. Most of these recommendations are for pulmonary disease because most of the trials were performed only in pulmonary disease. Treatment recommendations for extrapulmonary disease are usually similar with a few modifications. For example, the dosage of glucocorticoids is usually higher for neurosarcoidosis and lower for cutaneous disease. There was some suggestion that higher doses would be beneficial for cardiac sarcoidosis, but one study found that initial prednisone doses >40 mg/d were associated with a worse outcome because of toxicity.
|
COMMONLY USED DRUGS TO TREAT SARCOIDOSIS |
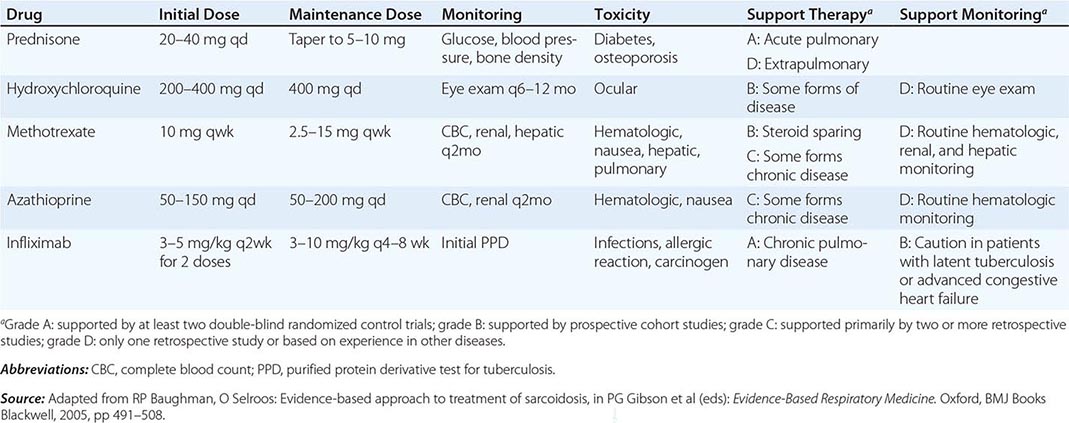
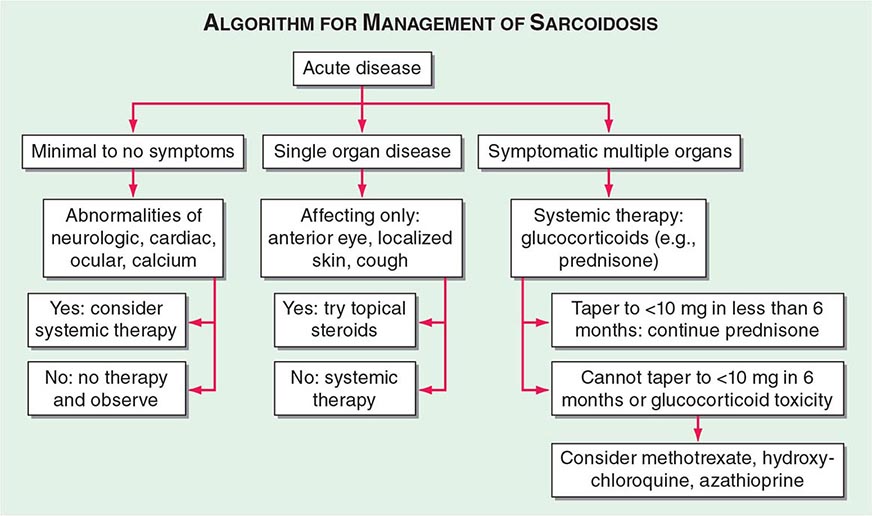
FIGURE 390-9 The management of acute sarcoidosis is based on level of symptoms and extent of organ involvement. In patients with mild symptoms, no therapy may be needed unless specified manifestations are noted.
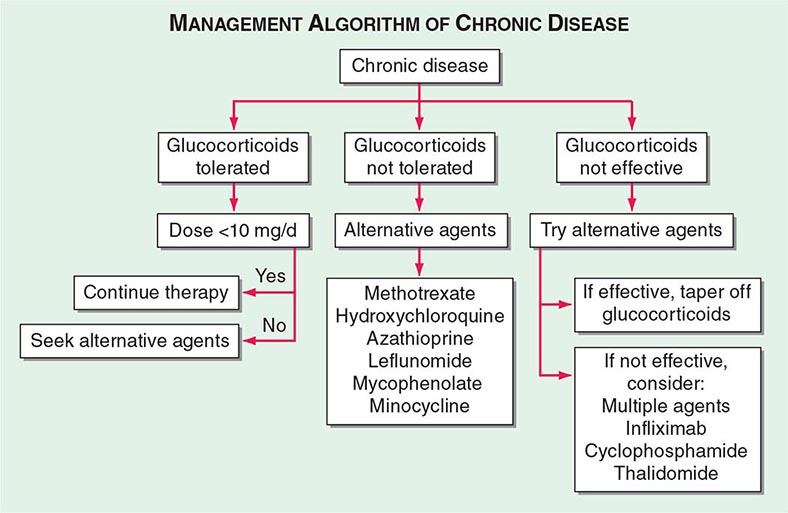
FIGURE 390-10 Approach to chronic disease is based on whether glucocorticoid therapy is tolerated or not.
Systemic therapies for sarcoidosis are usually immunosuppressive including glucocorticoids, cytotoxics, or biologics. Although most patients receive glucocorticoids as their initial systemic therapy, toxicity associated with prolonged therapy often leads to steroid-sparing alternatives. The antimalarial drugs such as hydroxychloroquine are more effective for skin than pulmonary disease. Minocycline may also be useful for cutaneous sarcoidosis. For pulmonary and other extrapulmonary disease, cytotoxic agents are often used. These include methotrexate, azathioprine, leflunomide, mycophenolate, and cyclophosphamide. The most widely studied cytotoxic agent has been methotrexate. This agent works in approximately two-thirds of sarcoidosis patients, regardless of the disease manifestation. In one retrospective study comparing methotrexate to azathioprine, both drugs were equally effective. However, methotrexate was associated with significantly less toxicity. As noted in Table 390-2, specific guidelines for monitoring therapy have been recommended. Cytokine modulators such as thalidomide and pentoxifylline have also been used in a limited number of cases.
The biologic anti-TNF agents have recently been studied in sarcoidosis, with prospective randomized trials completed for both etanercept and infliximab. Etanercept has a limited role as a steroid-sparing agent. Conversely, infliximab significantly improved lung function when administered to glucocorticoid and cytotoxic pretreated patients with chronic disease The difference in response between these two agents is similar to that observed in Crohn’s disease, where infliximab is effective and etanercept is not. However, there is a higher risk for reactivation of tuberculosis with infliximab compared to etanercept. The differential response rate could be explained by differences in mechanism of action because etanercept is a TNF receptor antagonist and infliximab is a monoclonal antibody against TNF. In contrast to etanercept, infliximab also binds to TNF on the surface of some cells that release TNF, which leads to cell lysis. This effect has been documented in Crohn’s disease. Adalimumab is a humanized monoclonal anti-TNF antibody that also appears effective for sarcoidosis when dosed at higher strengths, as recommended for the treatment of Crohn’s disease. The role of the newer therapeutic agents for sarcoidosis is still evolving. However, these targeted therapies confirm that TNF may be an important target, especially in the treatment of chronic disease. However, these agents are not a panacea, because sarcoidosis-like disease has occurred in patients treated with anti-TNF agents for nonsarcoidosis indications.
391e |
IgG4-Related Disease |
IgG4-related disease (IgG4-RD) is a fibroinflammatory condition characterized by a tendency to form tumefactive lesions. The clinical manifestations of this disease, however, are protean, and continue to be defined. IgG4-RD has now been described in virtually every organ system. Commonly affected organs are the biliary tree, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, and retroperitoneum. In addition, IgG4-RD involvement of the meninges, aorta, prostate, thyroid, pericardium, skin, and other organs is well described. The disease is believed to affect the brain parenchyma, the joints, the bone marrow, and the bowel mucosa only rarely (if ever).
The clinical features of IgG4-RD are numerous, but the pathologic findings are consistent across all affected organs. These findings include a lymphoplasmacytic infiltrate with a high percentage of IgG4-positive plasma cells; a characteristic pattern of fibrosis termed “storiform”; a tendency to target blood vessels, particularly veins, through an obliterative process (“obliterative phlebitis”); and a mild to moderate tissue eosinophilia.
IgG4-RD encompasses a number of conditions previously regarded as separate, organ-specific entities. A condition once known as “lymphoplasmacytic sclerosing pancreatitis” (among many other terms) became the paradigm of IgG4-RD in 2000, when Japanese investigators recognized that these patients had elevated serum concentrations of IgG4. This form of sclerosing pancreatitis is now termed type 1 (IgG4-related) autoimmune pancreatitis (AIP). By 2003, extrapancreatic disease manifestations had been identified in patients with type 1 AIP, and since then, the manifestations of IgG4-RD in many organs have been catalogued. Mikulicz’s disease, once considered to be a subset of Sjögren’s syndrome that affected the lacrimal, parotid, and submandibular glands, is now considered part of the IgG4-RD spectrum. Similarly, a subset of patients previously diagnosed as having primary sclerosing cholangitis was known to respond well to glucocorticoids, in contrast to the majority of patients with that diagnosis. This steroid-responsive subset is now explained by the fact that such patients actually have a separate disease, i.e., IgG4-related sclerosing cholangitis. In this manner, the understanding of IgG4-RD has extended to include nearly every specialty of medicine.
CLINICAL FEATURES
The major organ lesions are summarized in Table 391e-1. IgG4-RD usually presents subacutely, and most patients do not have severe constitutional symptoms. Fevers and dramatic elevations of C-reactive protein are unusual; however, some patients report substantial weight loss occurring over periods of months. Clinically apparent disease can evolve over months, years, or even decades before the manifestations within a given organ becomes sufficiently severe to bring the patient to medical attention. Some patients have disease that is marked by the appearance and then resolution or temporary improvement in symptoms within a particular organ. Other patients accumulate new organ involvement as their disease persists in previously affected organs. Many patients with IgG4-RD are misdiagnosed as having other conditions, particularly malignancies, or their findings are attributed initially to nonspecific inflammation. The disorder is often identified incidentally through radiologic findings or unexpectedly in pathology specimens.
|
ORGAN MANIFESTATIONS OF IGG4-RELATED DISEASE |
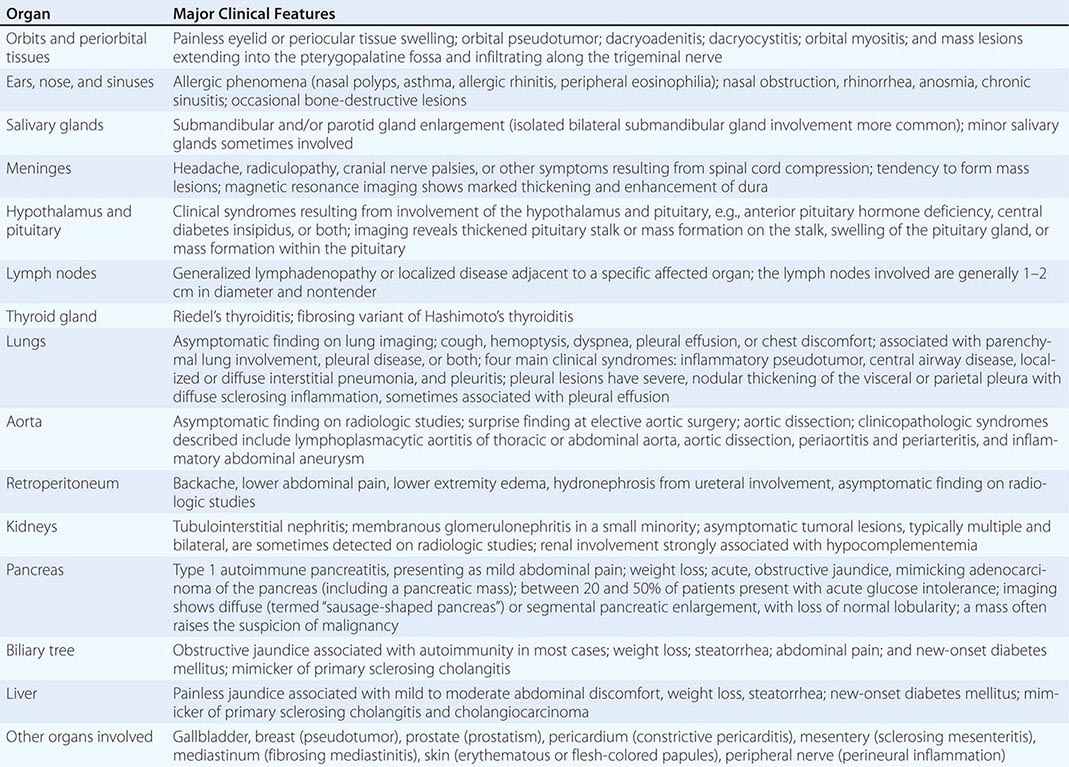
Multiorgan disease may be evident at diagnosis but can also evolve over months to years. Some patients have disease confined to a single organ for many years. Others have either known or subclinical organ involvement at the same time as the major clinical feature. Patients with type 1 AIP may have their major disease focus in the pancreas; however, thorough evaluations by history, physical examination, blood tests, urinalysis, and cross-sectional imaging may demonstrate lacrimal gland enlargement, sialoadenitis, lymphadenopathy, a variety of pulmonary findings, tubulointerstitial nephritis, hepatobiliary disease, aortitis, retroperitoneal fibrosis, or other organ involvement. Spontaneous improvement, sometimes leading to clinical resolution of certain organ system manifestations, is reported in a small percentage of patients.
Two common characteristics of IgG4-RD are allergic disease and the tendency to form tumefactive lesions that mimic malignancies (Fig. 391e-1). Many IgG4-RD patients have allergic features such as atopy, eczema, asthma, nasal polyps, sinusitis, and modest peripheral eosinophilia. IgG4-RD also appears to account for a significant proportion of tumorous swellings—pseudotumors—in many organ systems. Some patients undergo major surgeries (e.g., Whipple procedures or thyroidectomy) for the purpose of resecting malignancies before the correct diagnosis is identified. Frequent sites of pseudotumors are the major salivary glands, lacrimal glands, lungs, and kidneys; however, nearly all organs have been affected with this manifestation.
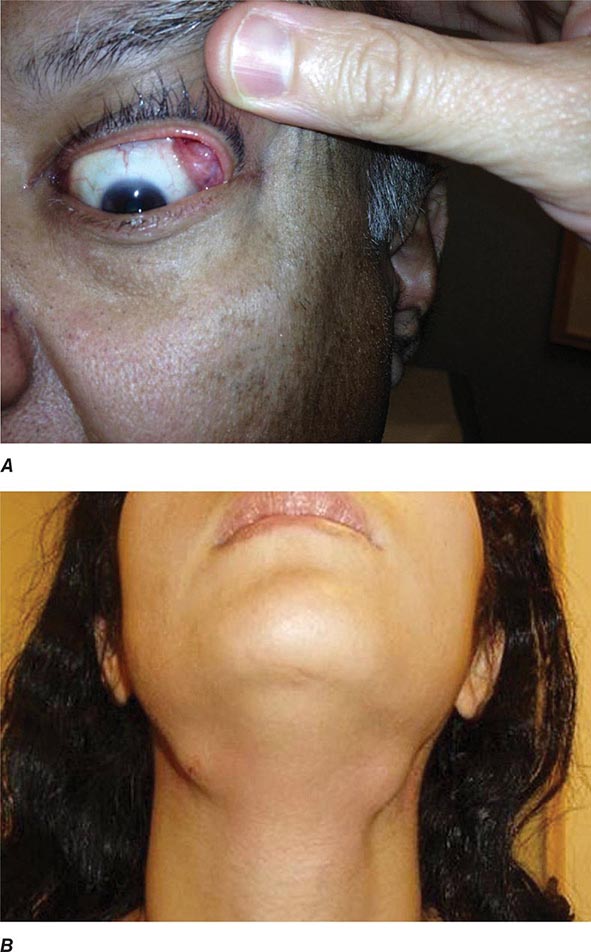
FIGURE 391e-1 A major clinical feature of IgG4-related disease is its tendency to form tumefactive lesions. Shown here are mass lesions of the lacrimal glands (A) and the submandibular glands (B).
IgG4-RD often causes major morbidity and can lead to organ failure; however, its general pattern is to cause damage in a subacute manner. Destructive bone lesions in the sinuses, head, and middle ear spaces that mimic granulomatous polyangiitis (formerly Wegener’s granulomatosis) also occur in IgG4-RD; less aggressive lesions are the rule in most organs. In regions such as the retroperitoneum, substantial fibrosis often occurs before the diagnosis is established, leading to ureteral entrapment, hydronephrosis, postobstructive uropathy, renal atrophy, and chronic pain, possibly resulting from the encasement of peripheral nerves by the inflammatory process. Undiagnosed or undertreated IgG4-related cholangitis can lead to hepatic failure within months. Similarly, IgG4-related aortitis, believed to be associated with between 10 and 50% of cases of inflammatory aortitis, can cause aneurysms and dissections. Substantial renal dysfunction and even renal failure can ensue from IgG4-related tubulointerstitial nephritis, and renal atrophy is a frequent sequel to this disease complication.
SEROLOGIC FINDINGS
The majority of patients with IgG4-RD have elevated serum IgG4 concentrations; however, the range of elevation varies widely. Serum concentrations of IgG4 as high as 30 or 40 times the upper limit of normal sometimes occur, usually in patients with disease that affects multiple organ systems simultaneously. Approximately 30% of patients have normal serum IgG4 concentrations despite classic histopathologic and immunohistochemical findings. Such patients tend to have disease that affects fewer organs. Patients with IgG4-related retroperitoneal fibrosis have a high likelihood of normal serum IgG4 concentrations, perhaps because the process has advanced to a fibrotic stage by the time the diagnosis is considered.
The correlation between serum IgG4 concentrations and disease activity and the need for treatment is imperfect. Serum IgG4 concentrations typically decline swiftly with the institution of therapy but often do not normalize completely. Patients can achieve clinical remissions yet have persistently elevated serum IgG4 concentrations. Rapidly rising serum IgG4 concentrations may identify patients at greatest risk for clinical flares and monitoring of serial IgG4 concentrations identifies early relapse in some patients; however, the temporal relationship between modest IgG4 elevations and the need for clinical treatment is poor. Clinical relapses occur in some patients despite persistently normal IgG4 concentrations.
IgG4 concentrations in serum are usually measured by nephelometry assays. These assays can lead to reports of spuriously low IgG4 values because of the prozone effect. This effect can be corrected by dilution of the serum sample in the laboratory. The prozone effect should be considered when the results of serologic testing for IgG4 concentrations appear to be at odds with the patient’s clinical features.
EPIDEMIOLOGY
The typical patient with IgG4-RD is a middle-aged to elderly man. This epidemiology stands in stark contrast to that of many classic autoimmune conditions, which tend to affect young women. Studies of AIP patients in Japan indicate that the male-to-female ratio in that disease subset is on the order of 3:1. Even more striking, male predominance has been reported in IgG4-related tubulointerstitial nephritis and IgG4-related retroperitoneal fibrosis. Among IgG4-RD manifestations that involve organs of the head and neck, the sex ratio may be closer to 1:1.
PATHOLOGY
The key histopathology characteristics of IgG4-RD are a dense lymphoplasmacytic infiltrate (Fig. 391e-2) that is organized in a storiform pattern (resembling a basket-weave), obliterative phlebitis, and a mild to moderate eosinophilic infiltrate. Lymphoid follicles and germinal centers are frequently observed. The infiltrate tends to aggregate around ductal structures when it affects glands such as the lacrimal, submandibular, and parotid glands or the pancreas. The inflammatory lesion often aggregates into tumefactive masses that destroy the involved tissue.
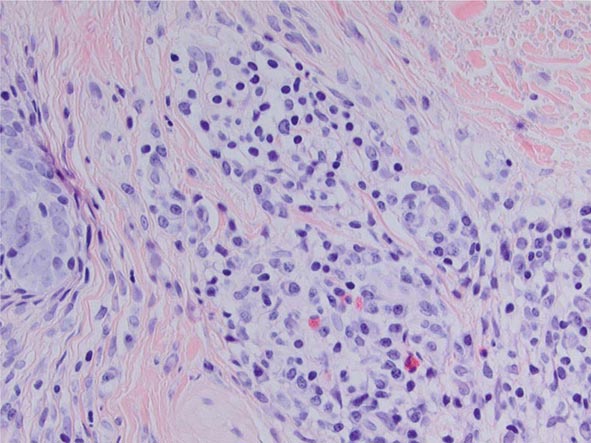
FIGURE 391e-2 Hallmark histopathology characteristics of IgG4-related disease (IgG4-RD) are a dense lymphoplasmacytic infiltrate and a mild to moderate eosinophilic infiltrate. The cellular inflammation is often encased in a distinctive type of fibrosis termed “storiform,” which often has a basket weave pattern. Abundant fibroblasts and strands of fibrosis accompany the lymphoplasmacytic infiltrate and eosinophils in this figure. This biopsy was taken from a nodular lesion on the cheek; however, the findings are identical to the pathology found in the pancreas, kidneys, lungs, salivary glands, and other organs affected by IgG4-RD.
Obliterative arteritis is observed in some organs, particularly the lung; however, venous involvement is more common (and is indeed a hallmark of IgG4-RD). Several histopathology features are uncommon in IgG4-RD and, when detected, mitigate against the diagnosis of IgG4-RD. These include intense neutrophilic infiltration, leukocytoclasis, granulomatous inflammation, multinucleated giant cells, and fibrinoid necrosis.
The inflammatory infiltrate is composed of an admixture of B and T lymphocytes. B cells are typically organized in germinal centers. Plasma cells staining for CD19, CD138, and IgG4 appear to radiate out from the germinal centers. In contrast, the T cells, usually CD4+, are distributed more diffusely throughout the lesion and generally represent the most abundant cell type. Fibroblasts, histiocytes, and eosinophils can all be observed in moderate numbers. Some biopsy samples are particularly enriched with eosinophils. In other samples, particularly from long-standing cases, fibrosis predominates.
The histologic appearance of IgG4-RD, although highly characteristic, requires immunohistochemical confirmation of the diagnosis with IgG4 immunostaining. IgG4-positive plasma cells predominate within the lesion, but plasma cells containing immunoglobulins from each subclass can be found. The number of IgG4-positive plasma cells can be quantified by either counting the number of cells per high-power field (HPF) or by calculating the ratio of IgG4- to IgG-bearing plasma cells. Tissue fibrosis predominates in the latter phases of organ involvement, and in this relatively acellular phase of inflammation, both the IgG4:total IgG ratio and the pattern of tissue fibrosis are more important than the number of IgG4-positive cells per HPF in establishing the diagnosis. In situ hybridization techniques are also now used to circumvent problems posed by increased background staining in conventional immunostaining techniques.
PATHOPHYSIOLOGY
The IgG4 molecule is believed to play an indirect role in the pathophysiology of disease in most organs. However, the molecule has properties that are unique among the immunoglobulin subclasses and that may contribute to tissue injury in some circumstances. As an example, IgG4 molecules have the ability to undergo Fab exchange, a phenomenon in which the two halves of the molecule dissociate from each other and reassociate with dissimilar hemi-molecules from other IgG4 molecules. This property is unique among the immunoglobulin subclasses. Partly as a result of Fab exchange, however, IgG4 antibodies bind antigen loosely. The molecules have low affinities for Fc receptors and C1q and are regarded generally as noninflammatory immunoglobulins. The low affinities for Fc receptors and C1q impair the ability of IgG4 antibodies to induce phagocyte activation, antibody-dependent cellular cytotoxicity, and complement-mediated damage. It is possible that the increased concentrations of IgG4 in serum and IgG4-bearing plasma cells in tissue are merely the result of other effector pathways, such as TH2/Treg cytokines, that are more central to the inflammation and tissue damage.
TREATMENT
Not every disease manifestation of IgG4-RD requires immediate treatment because the disease takes an indolent form in many patients. IgG4-related lymphadenopathy, for example, can be asymptomatic for years, without evolution to other disease manifestations. Thus, watchful waiting is prudent in some cases. Vital organ involvement must be treated aggressively, however, because IgG4-RD can lead to serious organ dysfunction and failure. Aggressive disease can lead quickly to end-stage liver disease, permanent impairment of pancreatic function, renal atrophy, aortic dissection or aneurysms, and destructive lesions in the sinuses and nasopharynx.
Glucocorticoids are the first line of therapy. Treatment regimens, extrapolated from experience with the management of type 1 AIP, generally begin with 40 mg/d of prednisone, with tapering to discontinuation or maintenance doses of 5 mg/d within 2 or 3 months. The clinical response to glucocorticoids is usually swift and striking; however, longitudinal data indicate that disease flares occur in more than 90% of patients within 3 years. Conventional steroid-sparing agents such as azathioprine and mycophenolate mofetil have been used in some patients; however, evidence for their efficacy is lacking.
For patients with relapsing or glucocorticoid-resistant disease, B cell depletion with rituximab is an excellent second-line therapy. Rituximab treatment (two doses of 1 g IV, separated by approximately 15 days) leads to a targeted, precipitous decline in serum IgG4 concentrations, suggesting that rituximab achieves its effects in part by preventing the repletion of short-lived plasma cells that produce IgG4. More important than its effects on IgG4 concentrations, however, may be the effect of B cell depletion on T cell function. Specific effects of rituximab on CD4+ effector T cells have been documented in IgG4-RD.
Rituximab may be an appropriate first-line therapy for some patients, particularly those at high risk for glucocorticoid toxicity and patients with immediately organ-threatening disease. The optimal approaches to remission maintenance, by either re-treatment with rituximab or continuous low-dose glucocorticoid therapy, require further study.
392 |
Familial Mediterranean Fever and Other Hereditary Autoinflammatory Diseases |
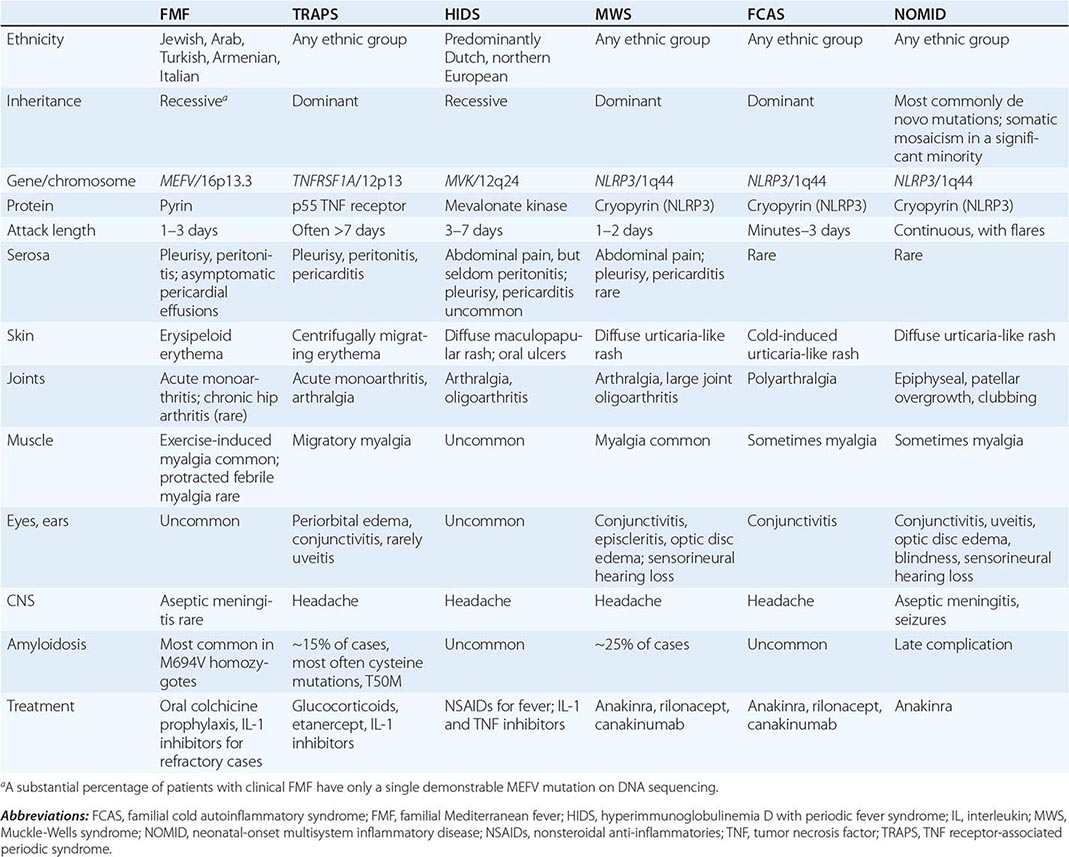
Familial Mediterranean fever (FMF) is the prototype of a group of inherited diseases (Table 392-1) that are characterized by recurrent episodes of fever with serosal, synovial, or cutaneous inflammation and, in some individuals, the eventual development of systemic AA amyloidosis (Chap. 137). Because of the relative infrequency of high-titer autoantibodies or antigen-specific T cells, the term autoinflammatory has been proposed to describe these disorders, rather than autoimmune. The innate immune system, with its myeloid effector cells and germline receptors for pathogen-associated molecular patterns and endogenous danger signals, plays a predominant role in the pathogenesis of the autoinflammatory diseases. Although the hereditary recurrent fevers comprise a major category of the autoinflammatory diseases, other inherited disorders of inflammation in which recurrent fever plays a less prominent role are now also considered to be autoinflammatory.
|
THE HEREDITARY RECURRENT FEVER SYNDROMES |
BACKGROUND AND PATHOPHYSIOLOGY
FMF was first recognized among Armenians, Arabs, Turks, and non-Ashkenazi (primarily North African and Iraqi) Jews. With the advent of genetic testing, FMF has been documented with increasing frequency among Ashkenazi Jews, Italians, and other Mediterranean populations, and occasional cases have been confirmed even in the absence of known Mediterranean ancestry. FMF is generally regarded as recessively inherited, but there is an increasing awareness of clear-cut clinical cases with only a single demonstrable genetic mutation, and, for certain relatively rare FMF mutations, there is strong evidence for dominant inheritance. Particularly in countries where families are small, a positive family history can only be elicited in ~50% of cases. DNA testing demonstrates carrier frequencies as high as 1:3 among affected populations, suggesting a heterozygote advantage.
The FMF gene encodes a 781-amino acid, ~95 kDa protein denoted pyrin (or marenostrin) that is expressed in granulocytes, eosinophils, monocytes, dendritic cells, and synovial and peritoneal fibroblasts. The N-terminal 92 amino acids of pyrin define a motif, the PYRIN domain, that is similar in structure to death domains, death effector domains, and caspase recruitment domains. PYRIN domains mediate homotypic protein-protein interactions and have been found in several other proteins, including cryopyrin (NLRP3), which is mutated in three other recurrent fever syndromes. Through a number of mechanisms, including the interaction of the PYRIN domain with an intermediary adaptor protein, pyrin regulates caspase-1 (interleukin [IL] 1β-converting enzyme), and thereby IL-1β secretion. Mice bearing FMF-associated pyrin mutations exhibit inflammation and excessive IL-1 production.
ACUTE ATTACKS
Febrile episodes in FMF may begin even in early infancy; 90% of patients have had their first attack by age 20. Typical FMF episodes generally last 24–72 h, with arthritic attacks tending to last somewhat longer. In some patients, the episodes occur with great regularity, but more often, the frequency of attacks varies over time, ranging from as often as once every few days to remissions lasting several years. Attacks are often unpredictable, although some patients relate them to physical exertion, emotional stress, or menses; pregnancy may be associated with remission.
If measured, fever is nearly always present throughout FMF attacks. Severe hyperpyrexia and even febrile seizures may be seen in infants, and fever is sometimes the only manifestation of FMF in young children.
Over 90% of FMF patients experience abdominal attacks at some time. Episodes range in severity from dull, aching pain and distention with mild tenderness on direct palpation to severe generalized pain with absent bowel sounds, rigidity, rebound tenderness, and air-fluid levels on upright radiographs. Computed tomography (CT) scanning may demonstrate a small amount of fluid in the abdominal cavity. If such patients undergo exploratory laparotomy, a sterile, neutrophil-rich peritoneal exudate is present, sometimes with adhesions from previous episodes. Ascites is rare.
Pleural attacks are usually manifested by unilateral, sharp, stabbing chest pain. Radiographs may show atelectasis and sometimes an effusion. If performed, thoracentesis demonstrates an exudative fluid rich in neutrophils. After repeated attacks, pleural thickening may develop.
FMF arthritis is most frequent among individuals homozygous for the M694V mutation, which is especially common in the non-Ashkenazi Jewish population. Acute arthritis in FMF is usually monoarticular, affecting the knee, ankle, or hip, although other patterns can be seen, particularly in children. Large sterile effusions rich in neutrophils are frequent, without commensurate erythema or warmth. Even after repeated arthritic attacks, radiographic changes are rare. Before the advent of colchicine prophylaxis, chronic arthritis of the knee or hip was seen in ~5% of FMF patients with arthritis. Chronic sacroiliitis can occur in FMF irrespective of the HLA-B27 antigen, even in the face of colchicine therapy. In the United States, FMF patients are much more likely to have arthralgia than arthritis.
The most characteristic cutaneous manifestation of FMF is erysipelas-like erythema, a raised erythematous rash that most commonly occurs on the dorsum of the foot, ankle, or lower leg alone or in combination with abdominal pain, pleurisy, or arthritis. Biopsy demonstrates perivascular infiltrates of granulocytes and monocytes. This rash is seen most often in M694V homozygotes and is relatively rare in the United States.
Exercise-induced (nonfebrile) myalgia is common in FMF, and a small percentage of patients develop a protracted febrile myalgia that can last several weeks. Symptomatic pericardial disease is rare, although some patients have small pericardial effusions as an incidental echocardiographic finding. Unilateral acute scrotal inflammation may occur in prepubertal boys. Aseptic meningitis has been reported in FMF, but the causal connection is controversial. Vasculitis, including Henoch-Schönlein purpura and polyarteritis nodosa (Chap. 385), may be seen at increased frequency in FMF. The M694V FMF mutation has recently been shown to be a risk factor for Behçet’s disease.
Laboratory features of FMF attacks are consistent with acute inflammation and include an elevated erythrocyte sedimentation rate, leukocytosis, thrombocytosis (in children), and elevations in C-reactive protein, fibrinogen, haptoglobin, and serum immunoglobulins. Transient albuminuria and hematuria may also be seen.
AMYLOIDOSIS
Before the advent of colchicine prophylaxis, systemic amyloidosis was a common complication of FMF. It is caused by deposition of a fragment of serum amyloid A, an acute-phase reactant, in the kidneys, adrenals, intestine, spleen, lung, and testes (Chap. 137). Amyloidosis should be suspected in patients who have proteinuria between attacks; renal or rectal biopsy is used most often to establish the diagnosis. Risk factors include the M694V homozygous genotype, positive family history (independent of FMF mutational status), the SAA 1 genotype, male gender, noncompliance with colchicine therapy, and having grown up in the Middle East.
DIAGNOSIS
For typical cases, physicians experienced with FMF can often make the diagnosis on clinical grounds alone. Clinical criteria sets for FMF have been shown to have high sensitivity and specificity in parts of the world where the pretest probability of FMF is high. Genetic testing can provide a useful adjunct in ambiguous cases or for physicians not experienced in FMF. Most of the more severe disease-associated FMF mutations are in exon 10 of the gene, with a smaller group of milder variants in exon 2. An updated list of mutations for FMF and other hereditary recurrent fevers can be found online at http://fmf.igh.cnrs.fr/infevers/.
Genetic testing has permitted a broadening of the clinical spectrum and geographic distribution of FMF and may be of prognostic value. Most studies indicate that M694V homozygotes have an earlier age of onset and a higher frequency of arthritis, rash, and amyloidosis. In contrast, the E148Q variant is quite common in certain populations and is more likely to affect overall levels of inflammation than to cause clinical FMF. E148Q is sometimes found in cis with exon 10 mutations, which may complicate the interpretation of genetic test results. Only ~70% of patients with clinically typical FMF have two identifiable mutations in trans. The inability to identify a second mutation even after intensive molecular analysis suggests that one FMF mutation may be sufficient to cause disease under some circumstances. In these cases clinical judgment is very important, and sometimes a therapeutic trial of colchicine may help to confirm the diagnosis. Genetic testing of unaffected individuals is usually inadvisable, because of the possibility of nonpenetrance and the potential impact of a positive test on future insurability.
If a patient is seen during his or her first attack, the differential diagnosis may be broad, although delimited by the specific organ involvement. After several attacks the differential diagnosis may include the other hereditary recurrent fever syndromes (Table 392-1); the syndrome of periodic fever with aphthous ulcers, pharyngitis, and cervical adenopathy (PFAPA); systemic-onset juvenile rheumatoid arthritis or adult Still’s disease; porphyria; hereditary angioedema; inflammatory bowel disease; and, in women, gynecologic disorders.
OTHER HEREDITARY RECURRENT FEVERS
Within 5 years of the discovery of the FMF gene, three additional genes causing five other hereditary recurrent fever syndromes were identified, catalyzing a paradigm shift in diagnosis and treatment of these disorders.
TNF RECEPTOR-ASSOCIATED PERIODIC SYNDROME (TRAPS)
TRAPS is caused by dominantly inherited mutations in the extracellular domains of the 55-kDa TNF receptor (TNFR1, p55). Although originally described in a large Irish family (and hence the name familial Hibernian fever), TRAPS has a broad ethnic distribution. TRAPS episodes often begin in childhood. The duration of attacks ranges from 1–2 days to as long as several weeks, and in severe cases symptoms may be nearly continuous. In addition to peritoneal, pleural, and synovial attacks similar to FMF, TRAPS patients frequently have ocular inflammation (most often conjunctivitis and/or periorbital edema), and a distinctive migratory myalgia with overlying painful erythema may be present. TRAPS patients generally respond better to glucocorticoids than to prophylactic colchicine. Untreated, about 15% develop amyloidosis. The diagnosis of TRAPS is based on the demonstration of TNFRSF1A mutations in the presence of characteristic symptoms. Two particular variants, R92Q and P46L, are common in certain populations and may act more as functional polymorphisms than as disease-causing mutations. In contrast, pathogenic TNFRSF1A mutations, including a number of substitutions at highly conserved cysteine residues, are associated with intracellular TNFR1 misfolding, aggregation, and retention, with consequent ligand-independent kinase activation, mitochondrial reactive oxygen species production, and proinflammatory cytokine release. Etanercept, a TNF inhibitor, ameliorates TRAPS attacks, but the long-term experience with this agent has been less favorable. Perhaps because of the ligand-independent signaling abnormalities in TRAPS, IL-1 inhibition has been beneficial in a large percentage of the patients in whom it has been used. Monoclonal anti-TNF antibodies should be avoided, because they may exacerbate TRAPS attacks.
HYPERIMMUNOGLOBULINEMIA D WITH PERIODIC FEVER SYNDROME (HIDS)
HIDS is a recessively inherited recurrent fever syndrome found primarily in individuals of northern European ancestry. It is caused by mutations in mevalonate kinase (MVK), encoding an enzyme involved in the synthesis of cholesterol and nonsterol isoprenoids. Attacks usually begin in infancy and last 3–5 days. Clinically distinctive features include painful cervical adenopathy, a diffuse maculopapular rash sometimes affecting the palms and soles, and aphthous ulcers; pleurisy is rare, as is amyloidosis. Although originally defined by the persistent elevation of serum IgD, disease activity is not related to IgD levels, and some patients with FMF or TRAPS may have modestly increased serum IgD. Moreover, occasional patients with MVK mutations and recurrent fever have normal IgD levels. For these reasons, some have proposed renaming this disorder mevalonate kinase deficiency (MKD). All patients with mutations have markedly elevated urinary mevalonate levels during their febrile attacks, although the inflammatory manifestations are likely to be due to a deficiency of isoprenoids rather than an excess of mevalonate. There is currently no established treatment for HIDS/MKD, although intermittent or continuous IL-1 inhibition and TNF inhibitors have been effective in small series.
THE CRYOPYRINOPATHIES, OR CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES (CAPS)
Three hereditary febrile syndromes, familial cold autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID), are all caused by mutations in NLRP3 (formerly known as CIAS1), the gene encoding cryopyrin (or NLRP3), and represent a clinical spectrum of disease. FCAS patients develop chills, fever, headache, arthralgia, conjunctivitis, and an urticaria-like rash in response to generalized cold exposure. In MWS, an urticarial rash is noted, but it is not usually induced by cold; MWS patients also develop fevers, abdominal pain, limb pain, arthritis, conjunctivitis, and, over time, sensorineural hearing loss. NOMID is the most severe of the three disorders, with chronic aseptic meningitis, a characteristic arthropathy, and rash. Like the FMF protein, pyrin, cryopyrin has an N-terminal PYRIN domain. Cryopyrin regulates IL-1β production through the formation of a macromolecular complex termed the inflammasome. Peripheral blood leukocytes from patients with FCAS, MWS, and NOMID release increased amounts of IL-1β upon in vitro stimulation, relative to healthy controls. Macrophages from cryopyrin-deficient mice exhibit decreased IL-1β production in response to certain gram-positive bacteria, bacterial RNA, and monosodium urate crystals. Patients with all three cryopyrinopathies show a dramatic response to injections of IL-1 inhibitors. Approximately one-third of patients with clinical manifestations of NOMID do not have germline mutations in NLRP3, but have been found to be mosaic for somatic NLRP3 mutations. Such patients also respond dramatically to IL-1 inhibition. Similarly, somatic mosaicism in NLRP3 has been found in Schnitzler’s syndrome, which presents in middle age with recurrent fever, urticarial rash, elevated acute phase reactants, monoclonal IgM gammopathy, and abnormal bone remodeling. IL-1 inhibition is the treatment of choice for Schnitzler’s syndrome.
OTHER INHERITED AUTOINFLAMMATORY DISEASES
There are a number of other Mendelian autoinflammatory diseases in which recurrent fevers are not a prominent clinical sign but that involve abnormalities of innate immunity. The syndrome of pyogenic arthritis with pyoderma gangrenosum and acne (PAPA) is a dominantly inherited disorder that presents with episodes of sterile pyogenic monoarthritis often induced by trauma, severe pyoderma gangrenosum, and severe cystic acne, usually beginning in puberty. It is caused by mutations in PSTPIP1, which encodes a pyrin-binding protein, and the arthritic manifestations often respond to IL-1 inhibition. Patients with the recessively inherited deficiency of the IL-1 receptor antagonist (DIRA) present with a generalized pustular rash and multifocal sterile osteomyelitis, and show dramatic clinical responses to anakinra, the recombinant form of the protein they lack. IL-36 is another member of the IL-1 family of cytokines that is regulated by an endogenous receptor antagonist. The recessively inherited deficiency of the IL-36 receptor antagonist (DITRA) presents with episodes of generalized pustular psoriasis and dramatic systemic inflammation.
Whereas PAPA, DIRA, and DITRA all involve mutations in IL-1-related molecules, other autoinflammatory diseases are caused by mutations in other components of innate immunity. Blau’s syndrome is caused by mutations in CARD15 (also known as NOD2), which regulates nuclear factor-κB activation. Blau’s syndrome is characterized by granulomatous dermatitis, uveitis, and arthritis; distinct CARD15 variants predispose to Crohn’s disease. Recessive mutations in one or more components of the proteasome lead to excessive interferon signaling and the syndrome of chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE), a severe form of generalized panniculitis. De novo gain-of-function mutations in TMEM173, encoding the stimulator of interferon genes (STING), cause severe vasculopathy and pulmonary fibrosis. Recessive loss-of-function mutations in CERCR1, encoding adenosine deaminase 2 (ADA2), cause a vasculopathy that can manifest as livedoid rash, early-onset lacunar strokes, or polyarteritis nodosa.
Finally, it should be noted that a number of common, genetically complex disorders are now sometimes considered autoinflammatory, because of evidence that components of the innate immune system, such as the inflammasome, may play a role in the pathogenesis. Two prominent examples are gout and atherosclerosis. Large clinical trials of IL-1 inhibitors have been initiated in both conditions.
SECTION 3 |
DISORDERS OF THE JOINTS AND ADJACENT TISSUES |
393 |
Approach to Articular and Musculoskeletal Disorders |
Musculoskeletal complaints account for >315 million outpatient visits per year and over 20% of all outpatient visits in the United States. The Centers for Disease Control and Prevention estimate that 22.7% (52.5 million) of the U.S. population has physician-diagnosed arthritis and 22 million have significant functional limitation. While many patients will have self-limited conditions requiring minimal evaluation and only symptomatic therapy and reassurance, specific musculoskeletal presentations or their persistence may herald a more serious condition that requires further evaluation or laboratory testing to establish a diagnosis. The goal of the musculoskeletal evaluation is to formulate a differential diagnosis that leads to an accurate diagnosis and timely therapy, while avoiding excessive diagnostic testing and unnecessary treatment (Table 393-1). There are several urgent conditions that must be diagnosed promptly to avoid significant morbid or mortal sequelae. These “red flag” diagnoses include septic arthritis, acute crystal-induced arthritis (e.g., gout), and fracture. Each may be suspected by its acute onset and monarticular or focal musculoskeletal pain (see below).
|
EVALUATION OF PATIENTS WITH MUSCULOSKELETAL COMPLAINTS |
Despite well-known links between certain disorders and laboratory testing, the majority of individuals with musculoskeletal complaints can be diagnosed with a thorough history and a comprehensive physical and musculoskeletal examination. The initial encounter should determine whether the musculoskeletal complaint signals a red flag condition (septic arthritis, gout, or fracture) or not. The evaluation should proceed to ascertain if the complaint is (1) articular or nonarticular in origin, (2) inflammatory or noninflammatory in nature, (3) acute or chronic in duration, and (4) localized (monarticular) or widespread (polyarticular) in distribution.
With such an approach and an understanding of the pathophysiologic processes, the musculoskeletal complaint or presentation can be characterized (e.g., acute inflammatory monarthritis or a chronic noninflammatory, nonarticular widespread pain) to narrow the diagnostic possibilities. A diagnosis can be made in the vast majority of individuals. However, some patients will not fit immediately into an established diagnostic category. Many musculoskeletal disorders resemble each other at the outset, and some may take weeks or months (but not years) to evolve into a recognizable diagnostic entity. This consideration should temper the desire to establish a definitive diagnosis at the first encounter.
ARTICULAR VERSUS NONARTICULAR
The musculoskeletal evaluation must discriminate the anatomic origin(s) of the patient’s complaint. For example, ankle pain can result from a variety of pathologic conditions involving disparate anatomic structures, including gonococcal arthritis, calcaneal fracture, Achilles tendinitis, plantar fasciitis, cellulitis, and peripheral or entrapment neuropathy. Distinguishing between articular and nonarticular conditions requires a careful and detailed examination. Articular structures include the synovium, synovial fluid, articular cartilage, intraarticular ligaments, joint capsule, and juxtaarticular bone. Nonarticular (or periarticular) structures, such as supportive extraarticular ligaments, tendons, bursae, muscle, fascia, bone, nerve, and overlying skin, may be involved in the pathologic process. Although musculoskeletal complaints are often ascribed to the joints, nonarticular disorders more frequently underlie such complaints. Distinguishing between these potential sources of pain may be challenging to the unskilled examiner. Articular disorders may be characterized by deep or diffuse pain, pain or limited range of motion on active and passive movement, and swelling (caused by synovial proliferation, effusion, or bony enlargement), crepitation, instability, “locking,” or deformity. By contrast, nonarticular disorders tend to be painful on active, but not passive (or assisted), range of motion. Periarticular conditions often demonstrate point or focal tenderness in regions adjacent to articular structures, are elicited with a specific movement or position, and have physical findings remote from the joint capsule. Moreover, nonarticular disorders seldom demonstrate swelling, crepitus, instability, or deformity of the joint itself.
INFLAMMATORY VERSUS NONINFLAMMATORY DISORDERS
In the course of a musculoskeletal evaluation, the examiner should determine the nature of the underlying pathologic process and whether inflammatory or noninflammatory findings exist. Inflammatory disorders may be infectious (Neisseria gonorrhoeae or Mycobacterium tuberculosis), crystal-induced (gout, pseudogout), immune-related (rheumatoid arthritis [RA], systemic lupus erythematosus [SLE]), reactive (rheumatic fever, reactive arthritis), or idiopathic. Inflammatory disorders may be identified by any of the four cardinal signs of inflammation (erythema, warmth, pain, or swelling), systemic symptoms (fatigue, fever, rash, weight loss), or laboratory evidence of inflammation (elevated erythrocyte sedimentation rate [ESR] or C-reactive protein [CRP], thrombocytosis, anemia of chronic disease, or hypoalbuminemia). Articular stiffness commonly accompanies chronic musculoskeletal disorders. The duration of stiffness may be prolonged (hours) with inflammatory disorders (such as RA or polymyalgia rheumatica) and may improve with activity. By contrast, intermittent stiffness (also known as gel phenomenon) is typical of noninflammatory conditions (such as osteoarthritis [OA]), shorter in duration (<60 min), and exacerbated by activity. Fatigue may accompany inflammation (as seen in RA and polymyalgia rheumatica) but may also be a consequence of fibromyalgia (a noninflammatory disorder), chronic pain, poor sleep, depression, anemia, cardiac failure, endocrinopathy, or malnutrition. Noninflammatory disorders may be related to trauma (rotator cuff tear), repetitive use (bursitis, tendinitis), degeneration or ineffective repair (OA), neoplasm (pigmented villonodular synovitis), or pain amplification (fibromyalgia). Noninflammatory disorders are often characterized by pain without synovial swelling or warmth, absence of inflammatory or systemic features, daytime gel phenomena rather than morning stiffness, and normal (for age) or negative laboratory investigations.
Identification of the nature of the underlying process and the site of the complaint will enable the examiner to characterize the musculoskeletal presentation (e.g., acute inflammatory monarthritis, chronic noninflammatory, nonarticular widespread pain), narrow the diagnostic considerations, and assess the need for immediate diagnostic or therapeutic intervention or for continued observation. Figure 393-1 presents an algorithmic approach to the evaluation of patients with musculoskeletal complaints. This approach relies on clinical and historic features, rather than laboratory testing, to diagnose many common rheumatic disorders.
FIGURE 393-1 Algorithm for the diagnosis of musculoskeletal complaints. An approach to formulating a differential diagnosis (shown in italics). CMC, carpometacarpal; CRP, C-reactive protein; DIP, distal interphalangeal; ESR, erythrocyte sedimentation rate; JIA, juvenile idiopathic arthritis; MCP, metacarpophalangeal; MTP, metatarsophalangeal; PIP, proximal interphalangeal; PMR, polymyalgia rheumatica; SLE, systemic lupus erythematosus.
A simpler, alternative approach would consider the most commonly encountered complaints first, based on frequency in younger versus older populations. The most prevalent causes of musculoskeletal complaints are shown in Fig. 393-2. Because trauma, fracture, overuse syndromes, and fibromyalgia are among the most common causes of joint pain, these should be considered during the initial encounter. If these possibilities are excluded, other frequently occurring disorders should be considered according to the patient’s age. Hence, those younger than 60 years are commonly affected by repetitive use/strain disorders, gout (men only), RA, spondyloarthritis, and uncommonly, infectious arthritis. Patients over age 60 years are frequently affected by OA, crystal (gout and pseudogout) arthritis, polymyalgia rheumatica, osteoporotic fracture, and uncommonly, septic arthritis. These conditions are between 10 and 100 times more prevalent than other serious autoimmune conditions, such as SLE, scleroderma, polymyositis, and vasculitis.
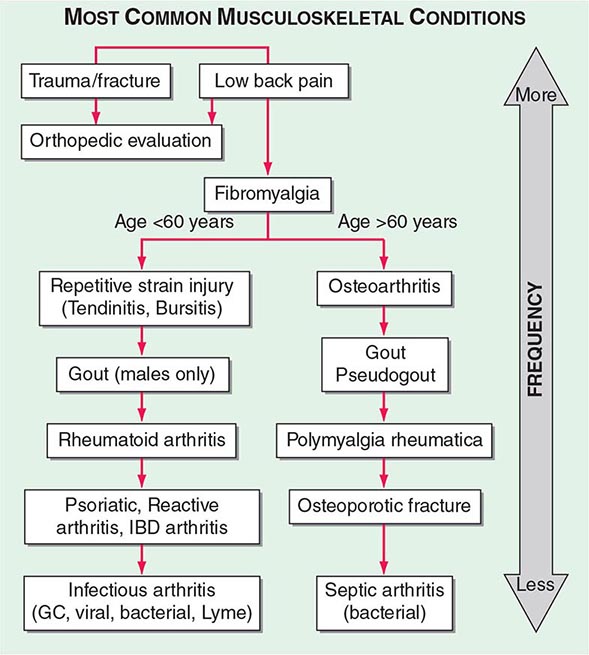
FIGURE 393-2 Algorithm for consideration of the most common musculoskeletal conditions. GC, gonococcal; IBD, inflammatory bowel disease.
CLINICAL HISTORY
Additional historic features may reveal important clues to the diagnosis. Aspects of the patient profile, complaint chronology, extent of joint involvement, and precipitating factors can provide important information. Certain diagnoses are more frequent in different age groups. SLE and reactive arthritis occur more frequently in the young, whereas fibromyalgia and RA are frequent in middle age, and OA and polymyalgia rheumatica are more prevalent among the elderly. Diagnostic clustering is also evident when sex and race are considered. Gout, spondyloarthritis, and ankylosing spondylitis are more common in men, whereas RA, fibromyalgia, and lupus are more frequent in women. Racial predilections may be evident. Thus, polymyalgia rheumatica, giant cell arteritis, and granulomatosis with polyangiitis (GPA; formerly called Wegener’s granulomatosis) commonly affect whites, whereas sarcoidosis and SLE more commonly affect African Americans. Familial aggregation is most common with ankylosing spondylitis, gout, and Heberden’s nodes of OA.
The chronology of the complaint is an important diagnostic feature and can be divided into the onset, evolution, and duration. The onset of disorders such as septic arthritis or gout tends to be abrupt, whereas OA, RA, and fibromyalgia may have more indolent presentations. The patients’ complaints may evolve differently and be classified as chronic (OA), intermittent (crystal or Lyme arthritis), migratory (rheumatic fever, gonococcal or viral arthritis), or additive (RA, psoriatic arthritis). Musculoskeletal disorders are typically classified as acute or chronic based on a symptom duration that is either less than or greater than 6 weeks, respectively. Acute arthropathies tend to be infectious, crystal-induced, or reactive. Chronic conditions include noninflammatory or immunologic arthritides (e.g., OA, RA) and nonarticular disorders (e.g., fibromyalgia).
The extent or distribution of articular involvement is often informative. Articular disorders are classified based on the number of joints involved, as either monarticular (one joint), oligoarticular or pauciarticular (two or three joints), or polyarticular (four or more joints). Although crystal and infectious arthritis are often mono- or oligoarticular, OA and RA are polyarticular disorders. Nonarticular disorders may be classified as either focal or widespread. Complaints secondary to tendinitis or carpal tunnel syndrome are typically focal, whereas weakness and myalgia, caused by polymyositis or fibromyalgia, are more widespread in their presentation. Joint involvement in RA tends to be symmetric and polyarticular. By contrast, spondyloarthritis, reactive arthritis, gout, and sarcoid are often asymmetric and oligoarticular. OA and psoriatic arthritis may be either symmetric or asymmetric and oligo- or polyarticular. The upper extremities are frequently involved in RA and OA, whereas lower extremity arthritis is characteristic of reactive arthritis and gout at their onset. Involvement of the axial skeleton is common in OA and ankylosing spondylitis but is infrequent in RA, with the notable exception of the cervical spine.
The clinical history should also identify precipitating events, such as trauma (osteonecrosis, meniscal tear), drug administration (Table 393-2), antecedent or intercurrent infection (rheumatic fever, reactive arthritis, hepatitis), or illnesses that may have contributed to the patient’s complaint. Certain comorbidities may have musculoskeletal consequences. This is especially so for diabetes mellitus (carpal tunnel syndrome), renal insufficiency (gout), depression or insomnia (fibromyalgia), myeloma (low back pain), cancer (myositis), and osteoporosis (fracture) or when using certain drugs such as glucocorticoids (osteonecrosis, septic arthritis) and diuretics or chemotherapy (gout) (Table 393-2).
|
DRUG-INDUCED MUSCULOSKELETAL CONDITIONS |
Abbreviations: ACE, angiotensin-converting enzyme; AZT, zidovudine; HCTZ, hydrochlorothiazide; IL-2, interleukin 2; TNF, tumor necrosis factor.
Lastly, a thorough rheumatic review of systems may disclose useful diagnostic information. A variety of musculoskeletal disorders may be associated with systemic features such as fever (SLE, infection), rash (SLE, psoriatic arthritis), nail abnormalities (psoriatic or reactive arthritis), myalgias (fibromyalgia, statin- or drug-induced myopathy), or weakness (polymyositis, neuropathy). In addition, some conditions are associated with involvement of other organ systems including the eyes (Behçet’s disease, sarcoidosis, spondyloarthritis), gastrointestinal tract (scleroderma, inflammatory bowel disease), genitourinary tract (reactive arthritis, gonococcemia), or nervous system (Lyme disease, vasculitis).
RHEUMATOLOGIC EVALUATION OF THE ELDERLY
The incidence of rheumatic diseases rises with age, such that 58% of those >65 years will have joint complaints. Musculoskeletal disorders in elderly patients are often not diagnosed because the signs and symptoms may be insidious, overlooked, or overshadowed by comorbidities. These difficulties are compounded by the diminished reliability of laboratory testing in the elderly, who often manifest nonpathologic abnormal results. For example, the ESR may be misleadingly elevated, and low-titer positive tests for rheumatoid factor and antinuclear antibodies (ANAs) may be seen in up to 15% of elderly patients. Although nearly all rheumatic disorders afflict the elderly, geriatric patients are particularly prone to OA, osteoporosis, osteoporotic fractures, gout, pseudogout, polymyalgia rheumatica, vasculitis, and drug-induced disorders (Table 393-2). The elderly should be approached in the same manner as other patients with musculoskeletal complaints, but with an emphasis on identifying the potential rheumatic consequences of medical comorbidities and therapies. The physical examination should identify the nature of the musculoskeletal complaint as well as coexisting diseases that may influence diagnosis and choice of treatment.
RHEUMATOLOGIC EVALUATION OF THE HOSPITALIZED PATIENT
Evaluation of a hospitalized patient with rheumatic complaints differs from that of an outpatient, owing to greater symptom severity, more acute presentations, and greater interplay of comorbidities. Patients with rheumatic disorders tend to be admitted for one of several reasons: (1) acute onset of inflammatory arthritis; (2) undiagnosed systemic or febrile illness; (3) musculoskeletal trauma; (4) exacerbation or deterioration of an existing autoimmune disorder (e.g., SLE); or (5) new medical comorbidities (e.g., thrombotic event, lymphoma, infection) arising in patients with an established rheumatic disorder. Notably, rheumatic patients are seldom if ever admitted because of widespread pain or serologic abnormalities or for the initiation of new therapies.
Acute monarticular inflammatory arthritis may be a “red flag” condition (e.g., septic arthritis, gout, pseudogout) that will require arthrocentesis and, on occasion, hospitalization if infection is suspected. However, new-onset inflammatory polyarthritis will have a wider differential diagnosis (e.g., RA, hepatitis-related arthritis, serum sickness, drug-induced lupus, polyarticular septic arthritis) and may require targeted laboratory investigations rather than synovial fluid analyses. Patients with febrile, multisystem disorders will require exclusion of crystal, infectious, or neoplastic etiologies and an evaluation driven by the dominant symptom/finding with the greatest specificity. Conditions worthy of consideration may include gout or pseudogout, vasculitis (giant cell arteritis in the elderly or polyarteritis nodosa in younger patients), adult-onset Still’s disease, SLE, antiphospholipid antibody syndrome, and sarcoidosis. Because the misdiagnosis of connective tissue disorders is common, patients who present with a reported preexisting rheumatic condition (e.g., SLE, RA, ankylosing spondylitis) should have their diagnosis confirmed by careful history, physical and musculoskeletal examination, and review of their medical records. It is important to note that when established rheumatic disease patients are admitted to the hospital, it is usually not for a medical problem related to their autoimmune disease, but rather because of either a comorbid condition or complication of drug therapy. Patients with chronic inflammatory disorders (e.g., RA, SLE, psoriasis) have an augmented risk of infection, cardiovascular events, and neoplasia.
Certain conditions, such as acute gout, can be precipitated in hospitalized patients by surgery, dehydration, or other events and should be considered when hospitalized patients are evaluated for the acute onset of a musculoskeletal condition. Lastly, overly aggressive and unfocused laboratory testing will often yield abnormal findings that are better explained by the patient’s preexisting condition(s) rather than a new inflammatory or autoimmune disorder.
PHYSICAL EXAMINATION
The goal of the physical examination is to ascertain the structures involved, the nature of the underlying pathology, the functional consequences of the process, and the presence of systemic or extraarticular manifestations. A knowledge of topographic anatomy is necessary to identify the primary site(s) of involvement and differentiate articular from nonarticular disorders. The musculoskeletal examination depends largely on careful inspection, palpation, and a variety of specific physical maneuvers to elicit diagnostic signs (Table 393-3). Although most articulations of the appendicular skeleton can be examined in this manner, adequate inspection and palpation are not possible for many axial (e.g., zygapophyseal) and inaccessible (e.g., sacroiliac or hip) joints. For such joints, there is a greater reliance on specific maneuvers and imaging for assessment.
|
GLOSSARY OF MUSCULOSKELETAL TERMS |
Examination of involved and uninvolved joints will determine whether pain, warmth, erythema, or swelling is present. The locale and level of pain elicited by palpation or movement should be quantified. One standard would be to count the number of tender joints on palpation of 28 easily examined joints (proximal interphalangeals, metacarpophalangeals, wrists, elbows, shoulders, and knees). Similarly, the number of swollen joints (0–28) can be counted and recorded. Careful examination should distinguish between true articular swelling (caused by bony hypertrophy, synovial effusion or proliferation) and nonarticular (or periarticular) involvement, which usually extends beyond the normal joint margins. Synovial effusion can be distinguished from synovial hypertrophy or bony hypertrophy by palpation or specific maneuvers. For example, small to moderate knee effusions may be identified by the “bulge sign” or “ballottement of the patellae.” Bursal effusions (e.g., effusions of the olecranon or prepatellar bursa) are often focal, periarticular, overlie bony prominences, and are fluctuant with sharply defined borders. Joint stability can be assessed by stabilizing the proximal joint, by palpation, and by the application of manual stress to the distal appendage. Subluxation or dislocation, which may be secondary to traumatic, mechanical, or inflammatory causes, can be assessed by inspection and palpation. Joint swelling or volume can be assessed by palpation. Distention of the articular capsule usually causes pain and evident enlargement or fluctuance. The patient will attempt to minimize the pain by maintaining the joint in the position of least intraarticular pressure and greatest volume, usually partial flexion. For this reason, inflammatory effusions may give rise to flexion contractures. Clinically, this may be detected as fluctuant or “squishy” swelling in larger joints and grape-like compressibility in smaller joints. Inflammation may result in fixed flexion deformities or diminished range of motion—especially on extension, when intraarticular pressure is increased. Active and passive range of motion should be assessed in all planes, with contralateral comparison. A goniometer may be used to quantify the arc of movement. Each joint should be passively manipulated through its full range of motion (including, as appropriate, flexion, extension, rotation, abduction, adduction, lateral bending, inversion, eversion, supination, pronation, medial/lateral deviation, and plantar- or dorsiflexion). Extreme range of motion may be seen with hypermobility syndrome, with joint pain and connective tissue laxity, often associated with Ehlers-Danlos or Marfan’s syndrome. Limitation of motion is frequently caused by inflammation, effusion, pain, deformity, contracture, or restriction from neuromyopathic causes. If passive motion exceeds active motion, a periarticular process (e.g., tendinitis, tendon rupture, or myopathy) should be considered. Contractures may reflect antecedent synovial inflammation or trauma. Minor joint crepitus is common during joint palpation and maneuvers but only indicates significant cartilage degeneration as it becomes coarser (e.g., OA). Joint deformity usually indicates a long-standing or aggressive pathologic process. Deformities may result from ligamentous destruction, soft tissue contracture, bony enlargement, ankylosis, erosive disease, subluxation, trauma, or loss of proprioception. Examination of the musculature will document strength, atrophy, pain, or spasm. Appendicular muscle weakness should be characterized as proximal or distal. Muscle strength should be assessed by observing the patient’s performance (e.g., walking, rising from a chair, grasping, writing). Strength may also be graded on a 5-point scale: 0 for no movement; 1 for trace movement or twitch; 2 for movement with gravity eliminated; 3 for movement against gravity only; 4 for movement against gravity and resistance; and 5 for normal strength. The examiner should assess for often-overlooked nonarticular or periarticular involvement, especially when articular complaints are not supported by objective findings referable to the joint capsule. The identification of soft tissue/nonarticular pain will prevent unwarranted and often expensive additional evaluations. Specific maneuvers may reveal common nonarticular abnormalities, such as a carpal tunnel syndrome (which can be identified by Tinel’s or Phalen’s sign). Other examples of soft tissue abnormalities include olecranon bursitis, epicondylitis (e.g., tennis elbow), enthesitis (e.g., Achilles tendinitis), and tender trigger points associated with fibromyalgia.
APPROACH TO REGIONAL RHEUMATIC COMPLAINTS
Although all patients should be evaluated in a logical and thorough manner, many cases with focal musculoskeletal complaints are caused by commonly encountered disorders that exhibit a predictable pattern of onset, evolution, and localization; they can often be diagnosed immediately on the basis of limited historic information and selected maneuvers or tests. Although nearly every joint could be approached in this manner, the evaluation of four common involved anatomic regions—the hand, shoulder, hip, and knee—are reviewed here.
HAND PAIN
Focal or unilateral hand pain may result from trauma, overuse, infection, or a reactive or crystal-induced arthritis. By contrast, bilateral hand complaints commonly suggest a degenerative (e.g., OA), systemic, or inflammatory/immune (e.g., RA) etiology. The distribution or pattern of joint involvement is highly suggestive of certain disorders (Fig. 393-3). Thus, OA (or degenerative arthritis) may manifest as distal interphalangeal (DIP) and proximal interphalangeal (PIP) joint pain with bony hypertrophy sufficient to produce Heberden’s and Bouchard’s nodes, respectively. Pain, with or without bony swelling, involving the base of the thumb (first carpometacarpal joint) is also highly suggestive of OA. By contrast, RA tends to cause symmetric, polyarticular involvement of the PIP, metacarpophalangeal (MCP), intercarpal, and carpometacarpal joints (wrist) with pain and palpable synovial tissue hypertrophy. Psoriatic arthritis may mimic the pattern of joint involvement seen in OA (DIP and PIP joints), but can be distinguished by the presence of inflammatory signs (erythema, warmth, synovial swelling), with or without carpal involvement, nail pitting, or onycholysis. Whereas lateral or medial subluxations at the PIP or DIP joints are most likely due to inflammatory OA or psoriatic arthritis, dorsal or ventral deformities (swan neck or boutonnière deformities) are typical of RA. Hemochromatosis should be considered when degenerative changes (bony hypertrophy) are seen at the second and third MCP joints with associated radiographic chondrocalcinosis or episodic, inflammatory wrist arthritis.
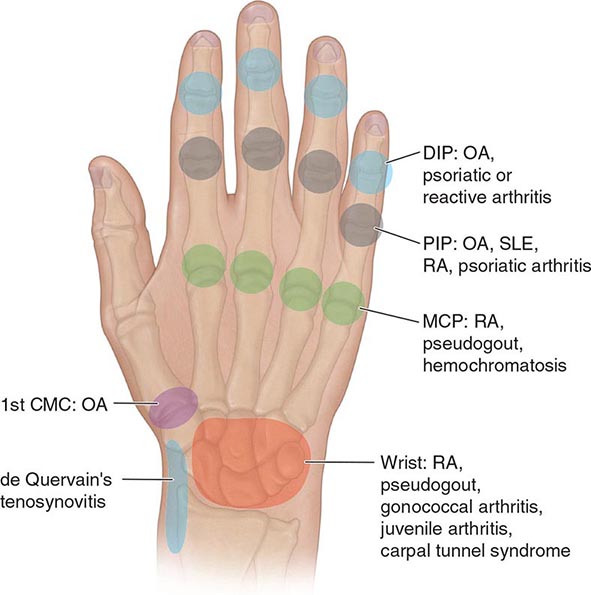
FIGURE 393-3 Sites of hand or wrist involvement and their potential disease associations. CMC, carpometacarpal; DIP, distal interphalangeal; MCP, metacarpophalangeal; OA, osteoarthritis; PIP, proximal interphalangeal; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus. (From JJ Cush et al: Evaluation of musculoskeletal complaints, in Rheumatology: Diagnosis and Therapeutics, 2nd ed, JJ Cush et al [eds]. Philadelphia, Lippincott Williams & Wilkins, 2005, pp 3–20. Used with permission from Dr. John J. Cush.)
Dactylitis manifests as soft tissue swelling of the whole digit and may have a sausage-like appearance. Common causes of dactylitis include psoriatic arthritis, spondyloarthritis, juvenile spondylitis, mixed connective tissue disease, scleroderma, sarcoidosis, and sickle cell disease. Soft tissue swelling over the dorsum of the hand and wrist may suggest an inflammatory extensor tendon tenosynovitis possibly caused by gonococcal infection, gout, or inflammatory arthritis (e.g., RA). Tenosynovitis is suggested by localized warmth, swelling, or pitting edema and may be confirmed when the soft tissue swelling tracks with tendon movement, such as flexion and extension of fingers, or when pain is induced while stretching the extensor tendon sheaths (flexing the digits distal to the MCP joints and maintaining the wrist in a fixed, neutral position).
Focal wrist pain localized to the radial aspect may be caused by de Quervain’s tenosynovitis resulting from inflammation of the tendon sheath(s) involving the abductor pollicis longus or extensor pollicis brevis (Fig. 393-3). This commonly results from overuse or follows pregnancy and may be diagnosed with Finkelstein’s test. A positive result is present when radial wrist pain is induced after the thumb is flexed and placed inside a clenched fist and the patient actively deviates the hand downward with ulnar deviation at the wrist. Carpal tunnel syndrome is another common disorder of the upper extremity and results from compression of the median nerve within the carpal tunnel. Manifestations include pain in the wrist that may radiate with paresthesia to the thumb, second and third fingers, and radial half of the fourth finger and, at times, atrophy of thenar musculature. Carpal tunnel syndrome is commonly associated with pregnancy, edema, trauma, OA, inflammatory arthritis, and infiltrative disorders (e.g., amyloidosis). The diagnosis may be suggested by a positive Tinel’s or Phalen’s sign. With each test, paresthesia in a median nerve distribution is induced or increased by either “thumping” the volar aspect of the wrist (Tinel’s sign) or pressing the extensor surfaces of both flexed wrists against each other (Phalen’s sign). The low sensitivity and moderate specificity of these tests may require nerve conduction velocity testing to confirm a suspected diagnosis.
SHOULDER PAIN
During the evaluation of shoulder disorders, the examiner should carefully note any history of trauma, fibromyalgia, infection, inflammatory disease, occupational hazards, or previous cervical disease. In addition, the patient should be questioned as to the activities or movement(s) that elicit shoulder pain. While arthritis is suggested by pain on movement in all planes, pain with specific active motion suggests a periarticular (nonarticular) process. Shoulder pain may originate in the glenohumeral or acromioclavicular joints, subacromial (subdeltoid) bursa, periarticular soft tissues (e.g., fibromyalgia, rotator cuff tear/tendinitis), or cervical spine (Fig. 393-4). Shoulder pain is referred frequently from the cervical spine but may also be referred from intrathoracic lesions (e.g., a Pancoast tumor) or from gallbladder, hepatic, or diaphragmatic disease. These same visceral causes may also manifest as focal scapular pain. Fibromyalgia should be suspected when glenohumeral pain is accompanied by diffuse periarticular (i.e., subacromial, bicipital) pain and tender points (i.e., trapezius or supraspinatus). The shoulder should be put through its full range of motion both actively and passively (with examiner assistance): forward flexion, extension, abduction, adduction, and internal and external rotation. Manual inspection of the periarticular structures will often provide important diagnostic information. Glenohumeral involvement is best detected by placing the thumb over the glenohumeral joint just medial and inferior to the coracoid process and applying pressure anteriorly while internally and externally rotating the humeral head. Pain localized to this region is indicative of glenohumeral pathology. Synovial effusion or tissue is seldom palpable but, if present, may suggest infection, RA, amyloidosis, or an acute tear of the rotator cuff. The examiner should apply direct manual pressure over the subacromial bursa that lies lateral to and immediately beneath the acromion (Fig. 393-4). Subacromial bursitis is a frequent cause of shoulder pain. Anterior to the subacromial bursa, the bicipital tendon traverses the bicipital groove. This tendon is best identified by palpating it in its groove as the patient rotates the humerus internally and externally. Direct pressure over the tendon may reveal pain indicative of bicipital tendinitis. Palpation of the acromioclavicular joint may disclose local pain, bony hypertrophy, or, uncommonly, synovial swelling. Whereas OA and RA commonly affect the acromioclavicular joint, OA seldom involves the glenohumeral joint, unless there is a traumatic or occupational cause.
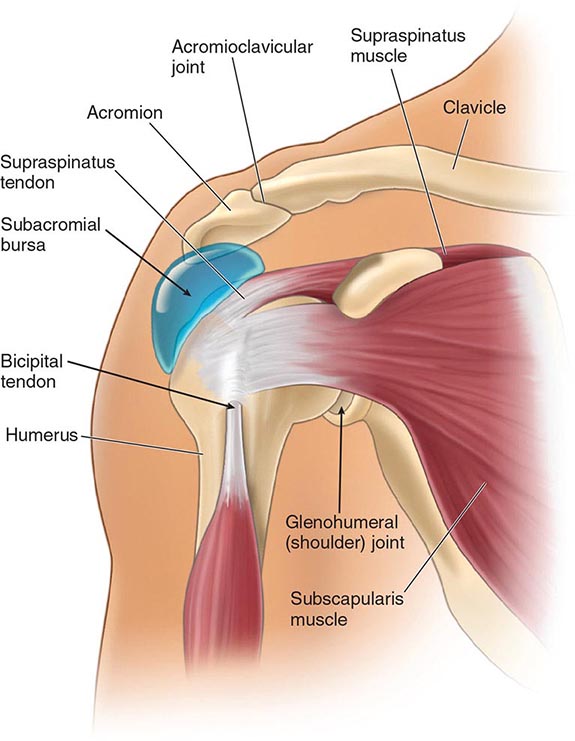
FIGURE 393-4 Origins of shoulder pain. The schematic diagram of the shoulder indicates with arrows the anatomic origins of shoulder pain.
Rotator cuff tendinitis or tear is a very common cause of shoulder pain. Nearly 30 percent of the elderly will have shoulder pain, with rotator cuff tendinitis or tear as the primary cause. The rotator cuff is formed by four tendons that attach the scapula to the proximal humerus (supraspinatus, infraspinatus, teres minor, and subscapularis tendons). Of these, the supraspinatus muscle is the most commonly damaged. Rotator cuff tendinitis is suggested by pain on active abduction (but not passive abduction), pain over the lateral deltoid muscle, night pain, and evidence of the impingement signs (pain with overhead arm activities). The Neer test for impingement is performed by the examiner raising the patient’s arm into forced flexion while stabilizing and preventing rotation of the scapula. A positive sign is present if pain develops before 180° of forward flexion. Tear of the rotator cuff is common in the elderly and often results from trauma; it may manifest in the same manner as tendinitis. The drop arm test is abnormal with supraspinatus pathology and is demonstrated by passive abduction of the arm to 90° by the examiner. If the patient is unable to hold the arm up actively or unable to lower the arm slowly without dropping, the test is positive. Tendinitis or tear of the rotator cuff is best confirmed by magnetic resonance imaging (MRI) or ultrasound.
KNEE PAIN
Knee pain may result from intraarticular (OA, RA) or periarticular (anserine bursitis, collateral ligament strain) processes or be referred from hip pathology. A careful history should delineate the chronology of the knee complaint and whether there are predisposing conditions, trauma, or medications that might underlie the complaint. For example, patellofemoral disease (e.g., OA) may cause anterior knee pain that worsens with climbing stairs. Observation of the patient’s gait is also important. The knee should be carefully inspected in the upright (weight-bearing) and supine positions for swelling, erythema, malalignment, visible trauma, muscle wasting, and leg length discrepancy. The most common malalignment in the knee is genu varum (bowlegs) or genu valgum (knock-knees) resulting from asymmetric cartilage loss medially or laterally. Bony swelling of the knee joint commonly results from hypertrophic osseous changes seen with disorders such as OA and neuropathic arthropathy. Swelling caused by hypertrophy of the synovium or synovial effusion may manifest as a fluctuant, ballotable, or soft tissue enlargement in the suprapatellar pouch (suprapatellar reflection of the synovial cavity) or regions lateral and medial to the patella. Synovial effusions may also be detected by balloting the patella downward toward the femoral groove or by eliciting a “bulge sign.” With the knee extended, the examiner should manually compress, or “milk,” synovial fluid down from the suprapatellar pouch and lateral to the patellae. The application of manual pressure lateral to the patella may cause an observable shift in synovial fluid (bulge) to the medial aspect. The examiner should note that this maneuver is only effective in detecting small to moderate effusions (<100 mL). Inflammatory disorders such as RA, gout, pseudogout, and psoriatic arthritis may involve the knee joint and produce significant pain, stiffness, swelling, or warmth. A popliteal or Baker’s cyst may be palpated with the knee partially flexed and is best viewed posteriorly with the patient standing and knees fully extended to visualize isolated or unilateral popliteal swelling or fullness.
Anserine bursitis is an often missed periarticular cause of knee pain in adults. The pes anserine bursa underlies the insertion of the conjoined tendons (sartorius, gracilis, semitendinosus) on the anteromedial proximal tibia and may be painful following trauma, overuse, or inflammation. It is often tender in patients with fibromyalgia, obesity, and knee OA. Other forms of bursitis may also present as knee pain. The prepatellar bursa is superficial and is located over the inferior portion of the patella. The infrapatellar bursa is deeper and lies beneath the patellar ligament before its insertion on the tibial tubercle.
Internal derangement of the knee may result from trauma or degenerative processes. Damage to the meniscal cartilage (medial or lateral) frequently presents as chronic or intermittent knee pain. Such an injury should be suspected when there is a history of trauma, athletic activity, or chronic knee arthritis, and when the patient relates symptoms of “locking” or “giving way” of the knee. With the knee flexed 90° and the patient’s foot on the table, pain elicited during palpation over the joint line or when the knee is stressed laterally or medially may suggest a meniscal tear. A positive McMurray test may also indicate a meniscal tear. To perform this test, the knee is first flexed at 90°, and the leg is then extended while the lower extremity is simultaneously torqued medially or laterally. A painful click during inward rotation may indicate a lateral meniscus tear, and pain during outward rotation may indicate a tear in the medial meniscus. Lastly, damage to the cruciate ligaments should be suspected with acute onset of pain, possibly with swelling, a history of trauma, or a synovial fluid aspirate that is grossly bloody. Examination of the cruciate ligaments is best accomplished by eliciting a drawer sign. With the patient recumbent, the knee should be partially flexed and the foot stabilized on the examining surface. The examiner should manually attempt to displace the tibia anteriorly or posteriorly with respect to the femur. If anterior movement is detected, then anterior cruciate ligament damage is likely. Conversely, significant posterior movement may indicate posterior cruciate damage. Contralateral comparison will assist the examiner in detecting significant anterior or posterior movement.
HIP PAIN
The hip is best evaluated by observing the patient’s gait and assessing range of motion. The vast majority of patients reporting “hip pain” localize their pain unilaterally to the posterior gluteal musculature (Fig. 393-5). Such pain tends to radiate down the posterolateral aspect of the thigh and may or may not be associated with complaints of low back pain. This presentation frequently results from degenerative arthritis of the lumbosacral spine or disks and commonly follows a dermatomal distribution with involvement of nerve roots between L4 and S1. Sciatica is caused by impingement of the L4, L5, or S1 nerve (i.e., from a herniated disk) and manifests as unilateral neuropathic pain extending from the gluteal region down the posterolateral leg to the foot. Some individuals instead localize their “hip pain” laterally to the area overlying the trochanteric bursa. Because of the depth of this bursa, swelling and warmth are usually absent. Diagnosis of trochanteric bursitis or enthesitis can be confirmed by inducing point tenderness over the trochanteric bursa. Gluteal and trochanteric pain are common findings in fibromyalgia. Range of movement may be limited by pain. Pain in the hip joint is less common and tends to be located anteriorly, over the inguinal ligament; it may radiate medially to the groin. Uncommonly, iliopsoas bursitis may mimic true hip joint pain. Diagnosis of iliopsoas bursitis may be suggested by a history of trauma or inflammatory arthritis. Pain associated with iliopsoas bursitis is localized to the groin or anterior thigh and tends to worsen with hyperextension of the hip; many patients prefer to flex and externally rotate the hip to reduce the pain from a distended bursa.
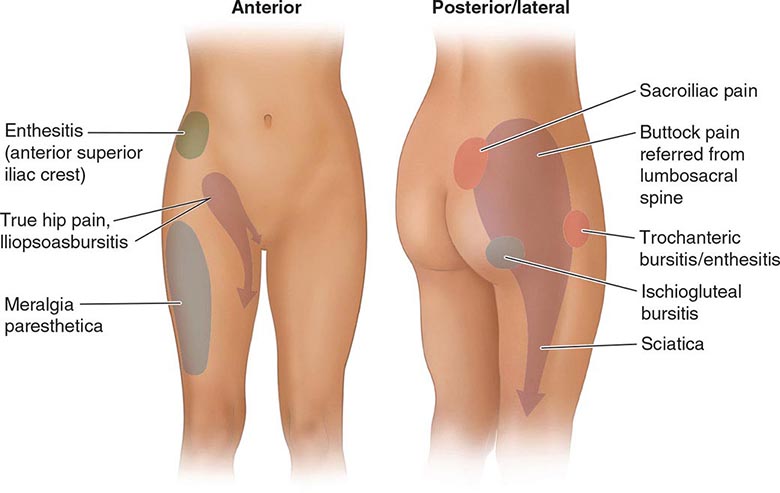
FIGURE 393-5 Origins of hip pain and dysesthesias. (From JJ Cush et al: Evaluation of musculoskeletal complaints, in Rheumatology: Diagnosis and Therapeutics, 2nd ed, JJ Cush et al [eds]. Philadelphia, Lippincott Williams & Wilkins, 2005, pp 3–20. Used with permission from Dr. John J. Cush.)
LABORATORY INVESTIGATIONS
The vast majority of musculoskeletal disorders can be easily diagnosed by a complete history and physical examination. An additional objective of the initial encounter is to determine whether additional investigations or immediate therapy is required. Additional evaluation is indicated with: (1) monarticular conditions; (2) traumatic or inflammatory conditions; (3) the presence of neurologic findings; (4) systemic manifestations; or (5) chronic symptoms (>6 weeks) and a lack of response to symptomatic measures. The extent and nature of the additional investigation should be dictated by the clinical features and suspected pathologic process. Laboratory tests should be used to confirm a specific clinical diagnosis and not be used to screen or evaluate patients with vague rheumatic complaints. Indiscriminate use of broad batteries of diagnostic tests and radiographic procedures is rarely a useful or cost-effective means to establish a diagnosis.
Besides a complete blood count, including a white blood cell (WBC) and differential count, the routine evaluation should include a determination of an acute-phase reactant such as the ESR or CRP, which can be useful in discriminating inflammatory from noninflammatory disorders. Both are inexpensive, easily obtained, and may be elevated with infection, inflammation, autoimmune disorders, neoplasia, pregnancy, renal insufficiency, advanced age, or hyperlipidemia. Extreme elevation of the acute-phase reactants (CRP, ESR) is seldom seen without evidence of serious illness (e.g., sepsis, pleuropericarditis, polymyalgia rheumatica, giant cell arteritis, adult Still’s disease).
Serum uric acid determinations are useful in the diagnosis of gout and in monitoring the response to urate-lowering therapy. Uric acid, the end product of purine metabolism, is primarily excreted in the urine. Serum values range from 238 to 516 μmol/L (4.0–8.6 mg/dL) in men; the lower values (178–351 μmol/L [3.0–5.9 mg/dL]) seen in women are caused by the uricosuric effects of estrogen. Urinary uric acid levels are normally <750 mg per 24 h. Although hyperuricemia (especially levels >535 μmol/L [9 mg/dL]) is associated with an increased incidence of gout and nephrolithiasis, levels may not correlate with the severity of articular disease. Uric acid levels (and the risk of gout) may be increased by inborn errors of metabolism (Lesch-Nyhan syndrome), disease states (renal insufficiency, myeloproliferative disease, psoriasis), or drugs (alcohol, cytotoxic therapy, thiazides). Although nearly all patients with gout will demonstrate hyperuricemia at some time during their illness, up to 50% of patients with an acute gouty attack will have normal serum uric acid levels. Monitoring serum uric acid may be useful in assessing the response to urate-lowering therapy or chemotherapy, with the target goal being a serum urate <6 mg/dL.
Serologic tests for rheumatoid factor (RF), cyclic anticitrullinated peptide (CCP or ACPA) antibodies, ANAs, complement levels, Lyme and antineutrophil cytoplasmic antibodies (ANCA), or antistreptolysin O (ASO) titer should be carried out only when there is clinical evidence to specifically suggest an associated diagnosis, because these have poor predictive value when used for screening, especially when the pretest probability is low. For most of these, there is no value to repeated or serial serologic testing. Although 4–5% of a healthy population will have positive tests for RF and ANAs, only 1% and <0.4% of the population will have RA or SLE, respectively. IgM RF (autoantibodies against the Fc portion of IgG) is found in 80% of patients with RA and may also be seen in low titers in patients with chronic infections (tuberculosis, leprosy, hepatitis); other autoimmune diseases (SLE, Sjögren’s syndrome); and chronic pulmonary, hepatic, or renal diseases. When considering RA, both serum RF and anti-CCP antibodies should be obtained as these are complementary. Both are comparably sensitive, but CCP antibodies are more specific than RF. In RA, the presence of anti-CCP and RF antibodies may indicate a greater risk for more severe, erosive polyarthritis. ANAs are found in nearly all patients with SLE and may also be seen in patients with other autoimmune diseases (polymyositis, scleroderma, antiphospholipid syndrome, Sjögren’s syndrome), drug-induced lupus (Table 393-2), chronic liver or renal disorders, and advanced age. Positive ANAs are found in 5% of adults and in up to 14% of elderly or chronically ill individuals. The ANA test is very sensitive but poorly specific for lupus, as only 1–2% of all positive results will be caused by lupus alone. The interpretation of a positive ANA test may depend on the magnitude of the titer and the pattern observed by immunofluorescence microscopy (Table 393-4). Diffuse and speckled patterns are least specific, whereas a peripheral, or rim, pattern (related to autoantibodies against double-strand [native] DNA) is highly specific and suggestive of lupus. Centromeric patterns are seen in patients with limited scleroderma (calcinosis, Raynaud’s phenomenon, esophageal involvement, sclerodactyly, telangiectasia [CREST] syndrome) or primary biliary sclerosis, and nucleolar patterns may be seen in patients with diffuse systemic sclerosis or inflammatory myositis.
|
ANTINUCLEAR ANTIBODY (ANA) PATTERNS AND CLINICAL ASSOCIATIONS |
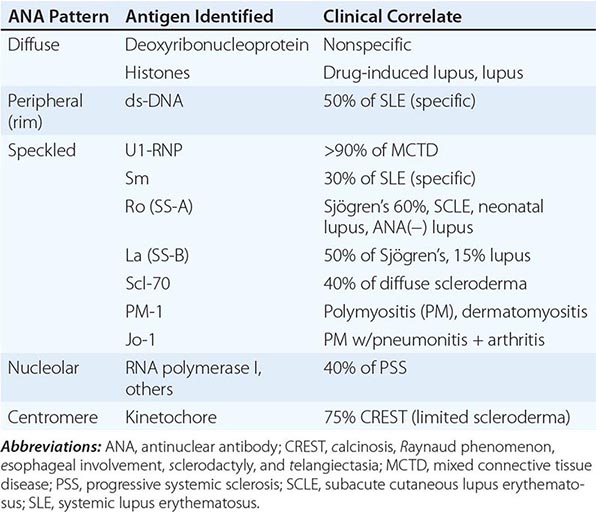
Aspiration and analysis of synovial fluid are always indicated in acute monarthritis or when an infectious or crystal-induced arthropathy is suspected. Synovial fluid may distinguish between noninflammatory and inflammatory processes by analysis of the appearance, viscosity, and cell count. Tests for synovial fluid glucose, protein, lactate dehydrogenase, lactic acid, or autoantibodies are not recommended because they have no diagnostic value. Normal synovial fluid is clear or a pale straw color and is viscous, primarily because of the high levels of hyaluronate. Noninflammatory synovial fluid is clear, viscous, and amber-colored, with a WBC count of <2000/μL and a predominance of mononuclear cells. The viscosity of synovial fluid is assessed by expressing fluid from the syringe one drop at a time. Normally, there is a stringing effect, with a long tail behind each synovial drop. Effusions caused by OA or trauma will have normal viscosity. Inflammatory fluid is turbid and yellow, with an increased WBC count (2000–50,000/μL) and a polymorphonuclear leukocyte predominance. Inflammatory fluid has reduced viscosity, diminished hyaluronate, and little or no tail following each drop of synovial fluid. Such effusions are found in RA, gout, and other inflammatory arthritides. Septic fluid is opaque and purulent, with a WBC count usually >50,000/μL, a predominance of polymorphonuclear leukocytes (>75%), and low viscosity. Such effusions are typical of septic arthritis but may also occur with RA or gout. In addition, hemorrhagic synovial fluid may be seen with trauma, hemarthrosis, or neuropathic arthritis. An algorithm for synovial fluid aspiration and analysis is shown in Fig. 393-6. Synovial fluid should be analyzed immediately for appearance, viscosity, and cell count. Monosodium urate crystals (observed in gout) are seen by polarized microscopy and are long, needle-shaped, negatively birefringent, and usually intracellular. In chondrocalcinosis and pseudogout, calcium pyrophosphate dihydrate crystals are usually short, rhomboid-shaped, and positively birefringent. Whenever infection is suspected, synovial fluid should be Gram stained and cultured appropriately. If gonococcal arthritis is suspected, nucleic acid amplification tests should be used to detect either Chlamydia trachomatis or N. gonorrhoeae infection. Synovial fluid from patients with chronic monarthritis should also be cultured for M. tuberculosis and fungi. Last, it should be noted that crystal-induced arthritis and septic arthritis occasionally occur together in the same joint.
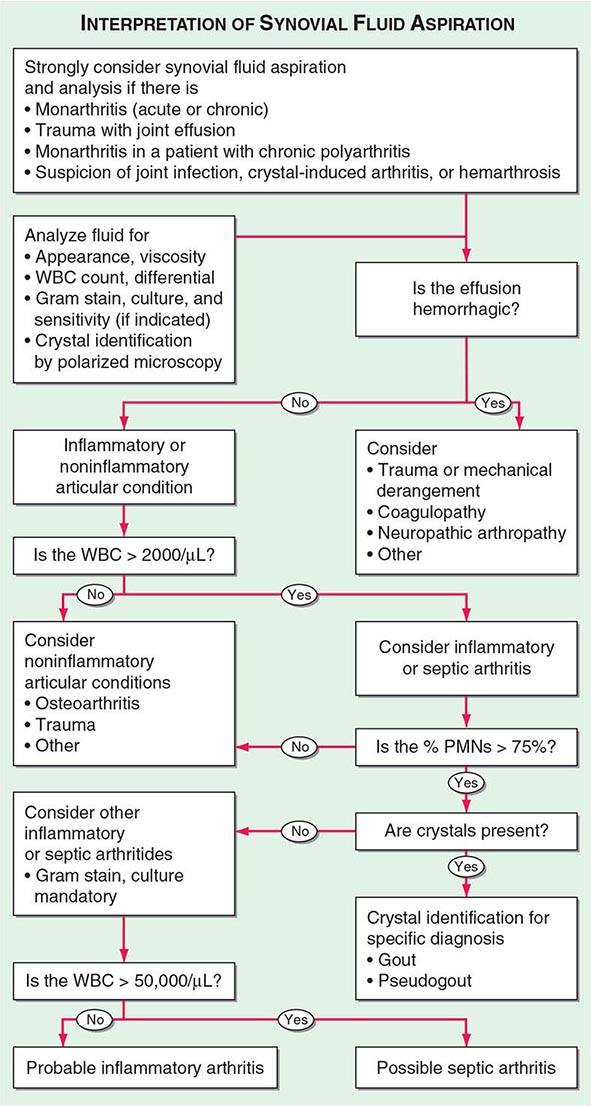
FIGURE 393-6 Algorithmic approach to the use and interpretation of synovial fluid aspiration and analysis. PMNs, polymorphonuclear (leukocytes); WBC, white blood cell (count).
DIAGNOSTIC IMAGING IN JOINT DISEASES
Conventional radiography has been a valuable tool in the diagnosis and staging of articular disorders. Plain x-rays are most appropriate and cost effective when there is a history of trauma, suspected chronic infection, progressive disability, or monarticular involvement; when therapeutic alterations are considered; or when a baseline assessment is desired for what appears to be a chronic process. However, in acute inflammatory arthritis, early radiography is rarely helpful in establishing a diagnosis and may only reveal soft tissue swelling or juxtaarticular demineralization. As the disease progresses, calcification (of soft tissues, cartilage, or bone), joint space narrowing, erosions, bony ankylosis, new bone formation (sclerosis, osteophytes, or periostitis), or subchondral cysts may develop and suggest specific clinical entities. Consultation with a radiologist will help define the optimal imaging modality, technique, or positioning and prevent the need for further studies.
Additional imaging techniques may possess greater diagnostic sensitivity and facilitate early diagnosis in a limited number of articular disorders and in selected circumstances and are indicated when conventional radiography is inadequate or nondiagnostic (Table 393-5). Ultrasonography is useful in the detection of soft tissue abnormalities, such as tendinitis, tenosynovitis, enthesitis, bursitis, and entrapment neuropathies. Wider use, lower cost, better technology, and enhanced site-specific transducers now allow for routine use in outpatient care. Owing to low cost, portability, and wider use, ultrasound use has grown and is the preferred method for the evaluation of synovial (Baker’s) cysts, rotator cuff tears, tendinitis and tendon injury, and crystal deposition on cartilage. Use of power Doppler allows for early detection of synovitis and bony erosions. Radionuclide scintigraphy is a very sensitive, but poorly specific, means of detecting inflammatory or metabolic alterations in bone or periarticular soft tissue structures. Scintigraphy is best suited for total-body assessment (extent and distribution) of skeletal involvement (neoplasia, Paget’s disease) and the assessment of patients with undiagnosed polyarthralgias, looking for occult arthritis. The use of scintigraphy has declined with greater use and declining cost of ultrasound and MRI. The limited tissue contrast resolution of scintigraphy may obscure the distinction between a bony or periarticular process and may necessitate the additional use of MRI. Scintigraphy using 99mTc, 67Ga, or 111In-labeled WBCs has been applied to a variety of articular disorders with variable success (Table 393-5). Although [99mTc] diphosphate scintigraphy may be useful in identifying osseous infection, neoplasia, inflammation, increased blood flow, bone remodeling, heterotopic bone formation, or avascular necrosis, MRI is preferred in most instances. Gallium scanning uses 67Ga, which binds serum and cellular transferrin and lactoferrin and is preferentially taken up by neutrophils, macrophages, bacteria, and tumor tissue (e.g., lymphoma). As such, it is primarily used in the identification of occult infection or malignancy. Scanning with 111In-labeled WBCs has been used to detect osteomyelitis and infectious or inflammatory arthritis. Despite their utility, 111In-labeled WBC or 67Ga scanning has largely been replaced by MRI, except when there is a suspicion of septic joint or prosthetic joint infections.
|
DIAGNOSTIC IMAGING TECHNIQUES FOR MUSCULOSKELETAL DISORDERS |
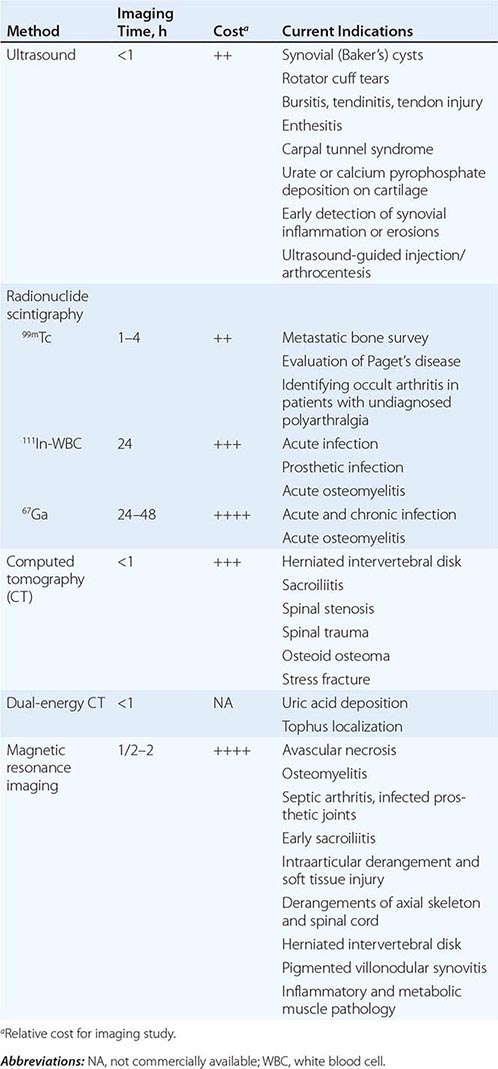
Computed tomography (CT) provides detailed visualization of the axial skeleton. Articulations previously considered difficult to visualize by radiography (e.g., zygapophyseal, sacroiliac, sternoclavicular, hip joints) can be effectively evaluated using CT. CT has been demonstrated to be useful in the diagnosis of low back pain syndromes (e.g., spinal stenosis vs herniated disk), sacroiliitis, osteoid osteoma, and stress fractures. Helical or spiral CT (with or without contrast angiography) is a novel technique that is rapid, cost effective, and sensitive in diagnosing pulmonary embolism or obscure fractures, often in the setting of initially equivocal findings. High-resolution CT can be advocated in the evaluation of suspected or established infiltrative lung disease (e.g., scleroderma or rheumatoid lung). The recent use of hybrid (positron emission tomography [PET] or single-photon emission CT [SPECT]) CT scans in metastatic evaluations has incorporated CT to provide better anatomic localization of scintigraphic abnormalities.
18F-Fluorodeoxyglucose (FDG) is the most commonly used radiopharmaceutical in PET scanning. FDG-PET/CT scans have been seldom used in the evaluation of septic or inflammatory arthritis. Dual-energy CT (DECT) scanning, developed in urology to identify urinary calculi, has been a highly sensitive and specific method used to identify and quantify uric acid deposition in tissues (Fig. 393-7).
FIGURE 393-7 Dual-energy computed tomography (DECT) scan from a 45-year-old woman with right ankle swelling around the lateral malleolus. Three-dimensional volume-rendered coronal reformatted DECT image shows that the mass is composed of monosodium urate (red) in keeping with tophus (arrow). (Used with permission from S Nicolaou et al: AJR 194:1072, 2010.)
MRI has significantly advanced the ability to image musculoskeletal structures. MRI has the advantages of providing multiplanar images with fine anatomic detail and contrast resolution (Fig. 393-8) that allows for the superior ability to visualize bone marrow and soft tissue periarticular structures. Although more costly with a longer procedural time than CT, the MRI has become the preferred technique when evaluating complex musculoskeletal disorders.
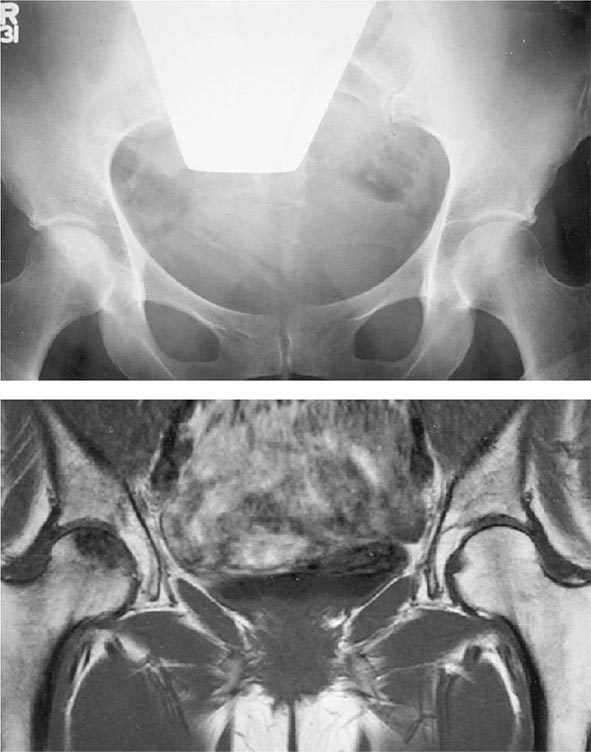
FIGURE 393-8 Superior sensitivity of magnetic resonance imaging (MRI) in the diagnosis of osteonecrosis of the femoral head. A 45-year-old woman receiving high-dose glucocorticoids developed right hip pain. Conventional x-rays (top) demonstrated only mild sclerosis of the right femoral head. T1-weighted MRI (bottom) demonstrated low-density signal in the right femoral head, diagnostic of osteonecrosis.
MRI can image fascia, vessels, nerve, muscle, cartilage, ligaments, tendons, pannus, synovial effusions, and bone marrow. Visualization of particular structures can be enhanced by altering the pulse sequence to produce either T1- or T2-weighted spin echo, gradient echo, or inversion recovery (including short tau inversion recovery [STIR]) images. Because of its sensitivity to changes in marrow fat, MRI is a sensitive but nonspecific means of detecting osteonecrosis, osteomyelitis, and marrow inflammation indicating overlying synovitis or osteitis (Fig. 393-8). Because of its enhanced soft tissue resolution, MRI is more sensitive than arthrography or CT in the diagnosis of soft tissue injuries (e.g., meniscal and rotator cuff tears); intraarticular derangements; marrow abnormalities (osteonecrosis, myeloma); and spinal cord or nerve root damage, synovitis, or cartilage damage or loss.
ACKNOWLEDGMENT
The author acknowledges the contributions of Dr. Peter E. Lipsky to this chapter in previous editions.
394 |
Osteoarthritis |
Osteoarthritis (OA) is the most common type of arthritis. Its high prevalence, especially in the elderly, and the high rate of disability related to disease make it a leading cause of disability in the elderly. Because of the aging of Western populations and because obesity, a major risk factor, is increasing in prevalence, the occurrence of OA is on the rise. In the United States, OA prevalence will increase by 66–100% by 2020.
OA affects certain joints, yet spares others (Fig. 394-1). Commonly affected joints include the cervical and lumbosacral spine, hip, knee, and first metatarsal phalangeal joint (MTP). In the hands, the distal and proximal interphalangeal joints and the base of the thumb are often affected. Usually spared are the wrist, elbow, and ankle. Our joints were designed, in an evolutionary sense, for brachiating apes, animals that still walked on four limbs. We thus develop OA in joints that were ill designed for human tasks such as pincer grip (OA in the thumb base) and walking upright (OA in knees and hips). Some joints, like the ankles, may be spared because their articular cartilage may be uniquely resistant to loading stresses.
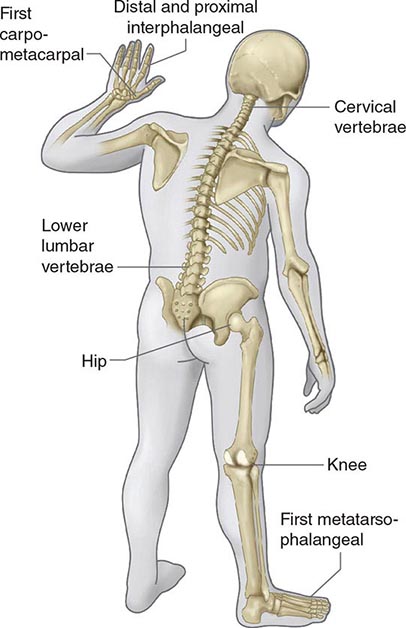
FIGURE 394-1 Joints commonly affected by osteoarthritis.
OA can be diagnosed based on structural abnormalities or on the symptoms these abnormalities evoke. According to cadaveric studies, by elderly years, structural changes of OA are nearly universal. These include cartilage loss (seen as joint space loss on x-rays) and osteophytes. Many persons with x-ray evidence of OA have no joint symptoms, and although the prevalence of structural abnormalities is of interest in understanding disease pathogenesis, what matters more from a clinical perspective is the prevalence of symptomatic OA. Symptoms, usually joint pain, determine disability, visits to clinicians, and disease costs.
Symptomatic OA of the knee (pain on most days of a recent month in a knee plus x-ray evidence of OA in that knee) occurs in ~12% of persons age ≥60 in the United States and 6% of all adults age ≥30. Symptomatic hip OA is roughly one-third as common as disease in the knee. Although radiographically evident hand OA and the appearance of bony enlargement in affected hand joints (Fig. 394-2) are extremely common in older persons, most cases are often not symptomatic. Even so, symptomatic hand OA occurs in ~10% of elderly individuals and often produces measurable limitation in function.
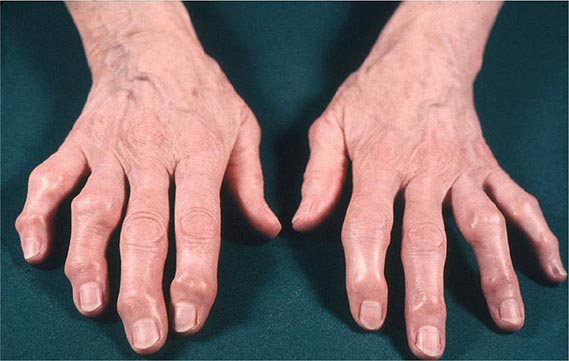
FIGURE 394-2 Severe osteoarthritis of the hands affecting the distal interphalangeal joints (Heberden’s nodes) and the proximal interphalangeal joints (Bouchard’s nodes). There is no clear bony enlargement of the other common site in the hands, the thumb base.
The prevalence of OA rises strikingly with age. Regardless of how it is defined, OA is uncommon in adults under age 40 and highly prevalent in those over age 60. It is also a disease that, at least in middle-aged and elderly persons, is much more common in women than in men, and sex differences in prevalence increase with age.
X-ray evidence of OA is common in the lower back and neck, but back pain and neck pain have not been tied to findings of OA on x-ray. Thus, back pain and neck pain are treated separately (Chap. 22).
DEFINITION
OA is joint failure, a disease in which all structures of the joint have undergone pathologic change, often in concert. The pathologic sine qua non of disease is hyaline articular cartilage loss, present in a focal and, initially, nonuniform manner. This is accompanied by increasing thickness and sclerosis of the subchondral bony plate, by outgrowth of osteophytes at the joint margin, by stretching of the articular capsule, by mild synovitis in many affected joints, and by weakness of muscles bridging the joint. In knees, meniscal degeneration is part of the disease. There are numerous pathways that lead to joint failure, but the initial step is often joint injury in the setting of a failure of protective mechanisms.
JOINT PROTECTIVE MECHANISMS AND THEIR FAILURE
Joint protectors include joint capsule and ligaments, muscle, sensory afferents, and underlying bone. Joint capsule and ligaments serve as joint protectors by providing a limit to excursion, thereby fixing the range of joint motion.
Synovial fluid reduces friction between articulating cartilage surfaces, thereby serving as a protector against friction-induced cartilage wear. This lubrication function depends on hyaluronic acid and on lubricin, a mucinous glycoprotein secreted by synovial fibroblasts whose concentration diminishes after joint injury and in the face of synovial inflammation.
The ligaments, along with overlying skin and tendons, contain mechanoreceptor sensory afferent nerves. These mechanoreceptors fire at different frequencies throughout a joint’s range of motion, providing feedback by way of the spinal cord to muscles and tendons. As a consequence, these muscles and tendons can assume the right tension at appropriate points in joint excursion to act as optimal joint protectors, anticipating joint loading.
Muscles and tendons that bridge the joint are key joint protectors. Their coordinated contractions at the appropriate time in joint movement provide the appropriate power and acceleration for the limb to accomplish its tasks. Focal stress across the joint is minimized by muscle contraction that decelerates the joint before impact and assures that when joint impact arrives, it is distributed broadly across the joint surface.
Failure of these joint protectors increases the risk of joint injury and OA. For example, in animals, OA develops rapidly when a sensory nerve to the joint is sectioned and joint injury induced. Similarly, in humans, Charcot’s arthropathy, a severe and rapidly progressive OA, develops when minor joint injury occurs in the presence of posterior column peripheral neuropathy. Another example of joint protector failure is rupture of ligaments, a well-known cause of the early development of OA.
CARTILAGE AND ITS ROLE IN JOINT FAILURE
In addition to being a primary target tissue for disease, cartilage also functions as a joint protector. A thin rim of tissue at the ends of two opposing bones, cartilage is lubricated by synovial fluid to provide an almost frictionless surface across which these two bones move. The compressible stiffness of cartilage compared to bone provides the joint with impact-absorbing capacity.
The earliest changes of OA may occur in cartilage, and abnormalities there can accelerate disease development. The two major macromolecules in cartilage are type 2 collagen, which provides cartilage its tensile strength, and aggrecan, a proteoglycan macromolecule linked with hyaluronic acid, which consists of highly negatively charged glycosaminoglycans. In normal cartilage, type 2 collagen is woven tightly, constraining the aggrecan molecules in the interstices between collagen strands, forcing these highly negatively charged molecules into close proximity with one another. The aggrecan molecule, through electrostatic repulsion of its negative charges, gives cartilage its compressive stiffness. Chondrocytes, the cells within this avascular tissue, synthesize all elements of the matrix and produce enzymes that break down the matrix. Synovium and chondrocytes synthesize and release cytokines and growth factors, which provide feedback that modulates synthesis of matrix molecules (Fig. 394-3). Cartilage matrix synthesis and catabolism are in a dynamic equilibrium influenced by the cytokine and growth factor environment. Mechanical and osmotic stress on chondrocytes induces these cells to alter gene expression and increase production of inflammatory cytokines and matrix-degrading enzymes. While chondrocytes synthesize numerous enzymes, matrix metalloproteinases (MMP) (especially collagenases and ADAMTS-5) are critical enzymes in the breakdown of cartilage matrix. Both collagenase and aggrecanases act primarily in the territorial matrix surrounding chondrocytes; however, as the osteoarthritic process develops, their activities and effects spread throughout the matrix, especially in the superficial layers of cartilage.
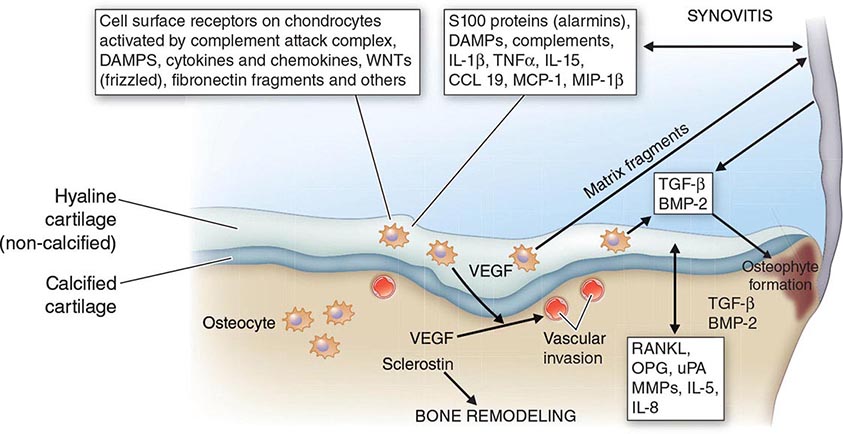
FIGURE 394-3 Selected factors involved in the osteoarthritic process including chondrocytes, bone, and synovium. Synovitis causes release of cytokines, alarmins, damage-associated molecular pattern (DAMP) molecules, and complement, which activate chondrocytes through cell surface receptors. Chondrocytes produce matrix molecules (collagen type 2, aggrecan) and the enzymes responsible for the degradation of the matrix (e.g., ADAMTS-5 and matrix metalloproteinases [MMPs]). Bone invasion occurs through the calcified cartilage, triggered by vascular endothelial growth factor (VEGF) and other molecules. IL, interleukin; TGF, transforming growth factor; TNF, tumor necrosis factor. (From RF Loeser et al: Arthritis Rheum 64:1697, 2012.)
The synovium, cartilage, and bone all influence disease development through cytokines, chemokines, and even complement activation (Fig. 394-3). These act on chondrocyte cell surface receptors and ultimately have transcriptional effects. Matrix fragments released from cartilage stimulate synovitis. Among the most important cytokines are interleukin (IL) 1β, which exerts transcriptional effects on chondrocytes, stimulating production of proteinases and suppressing cartilage matrix synthesis. Tumor necrosis factor (TNF) α may play a similar role to that of IL-1. These cytokines also induce chondrocytes to synthesize prostaglandin E2 and nitric oxide, which have complex effects on matrix synthesis and degradation. At early stages in the matrix response to injury and in the healthy response to loading, the net effect of cytokine stimulation may be matrix synthesis, but ultimately, the combination of effects on chondrocytes triggers matrix degradation. Enzymes in the matrix are held in check by activation inhibitors, including tissue inhibitor of metalloproteinase (TIMP). Growth factors are also part of this complex network, with BMP-2 and transforming growth factor β playing prominent roles in stimulating the development of osteophytes. Whereas healthy articular cartilage is avascular in part due to angiogenesis inhibitors present in cartilage, disease is characterized by the invasion of blood vessels into cartilage from underlying bone and proliferation of vessels within synovium. This is influenced by vascular endothelial growth factor (VEGF) synthesis in the cartilage and bone. With these blood vessels come nerves that may bring nociceptive innervation.
Probably as a result of chronic oxidative damage, articular chondrocytes exhibit an age-related decline in synthetic capacity while maintaining the ability to produce proinflammatory mediators and matrix-degrading enzymes, findings characteristic of a senescent secretory phenotype. These chondrocytes are unable to maintain tissue homeostasis (such as after insults of a mechanical or inflammatory nature). Thus, with age, cartilage is easily damaged by minor sometimes unnoticed injuries, including those that are part of daily activities.
OA cartilage is characterized by gradual depletion of aggrecan, an unfurling of the tightly woven collagen matrix, and loss of type 2 collagen. With these changes comes increasing vulnerability of cartilage, which loses its compressive stiffness.
RISK FACTORS
Joint vulnerability and joint loading are the two major factors contributing to the development of OA. On the one hand, a vulnerable joint whose protectors are dysfunctional can develop OA with minimal levels of loading, perhaps even levels encountered during everyday activities. On the other hand, in a young joint with competent protectors, a major acute injury or long-term overloading is necessary to precipitate disease. Risk factors for OA can be understood in terms of their effect either on joint vulnerability or on loading (Fig. 394-4).
FIGURE 394-4 Risk factors for osteoarthritis (OA) either contribute to the susceptibility of the joint (systemic factors or factors in the local joint environment) or increase risk by the load they put on the joint. Usually a combination of loading and susceptibility factors is required to cause disease or its progression.
SYSTEMIC RISK FACTORS
Age is the most potent risk factor for OA. Radiographic evidence of OA is rare in individuals under age 40; however, in some joints, such as the hands, OA occurs in >50% of persons over age 70. Aging increases joint vulnerability through several mechanisms. Whereas dynamic loading of joints stimulates cartilage matrix synthesis by chondrocytes in young cartilage, aged cartilage is less responsive to these stimuli. Partly because of this failure to synthesize matrix with loading, cartilage thins with age, and thinner cartilage experiences higher shear stress at basal layers and is at greater risk of cartilage damage. Also, joint protectors fail more often with age. Muscles that bridge the joint become weaker with age and also respond less quickly to oncoming impulses. Sensory nerve input slows with age, retarding the feedback loop of mechanoreceptors to muscles and tendons related to their tension and position. Ligaments stretch with age, making them less able to absorb impulses. These factors work in concert to increase the vulnerability of older joints to OA.
Older women are at high risk of OA in all joints, a risk that emerges as women reach their sixth decade. Although hormone loss with menopause may contribute to this risk, there is little understanding of the unique vulnerability of older women versus men to OA.
HERITABILITY AND GENETICS
OA is a highly heritable disease, but its heritability is joint specific. Fifty percent of the hand and hip OA in the community is attributable to inheritance, i.e., to disease present in other members of the family. However, the heritable proportion of knee OA is at most 30%, with some studies suggesting no heritability at all. Whereas many people with OA have disease in multiple joints, this “generalized OA” phenotype is rarely inherited and is more often a consequence of aging.
Emerging evidence has identified genetic mutations that confer a high risk of OA, the best replicated is a polymorphism within the growth differentiation factor 5 gene. This polymorphism diminishes the quantity of GDF5; GDF5 has its main influence on joint shape, and genes predisposing to OA are likely to increase risk of disease based on their effects on joint development and shape.
GLOBAL CONSIDERATIONS
![]() Hip OA is rare in China and in immigrants from China to the United States. However, OA in the knees is at least as common, if not more so, in Chinese than in whites from the United States, and knee OA represents a major cause of disability in China, especially in rural areas. Anatomic differences between Chinese and white hips may account for much of the difference in hip OA prevalence, with white hips having a higher prevalence of anatomic predispositions to the development of OA. Persons from Africa, but not African Americans, may also have a very low rate of hip OA.
Hip OA is rare in China and in immigrants from China to the United States. However, OA in the knees is at least as common, if not more so, in Chinese than in whites from the United States, and knee OA represents a major cause of disability in China, especially in rural areas. Anatomic differences between Chinese and white hips may account for much of the difference in hip OA prevalence, with white hips having a higher prevalence of anatomic predispositions to the development of OA. Persons from Africa, but not African Americans, may also have a very low rate of hip OA.
RISK FACTORS IN THE JOINT ENVIRONMENT
Some risk factors increase vulnerability of the joint through local effects on the joint environment. With changes in joint anatomy, for example, load across the joint is no longer distributed evenly across the joint surface, but rather shows an increase in focal stress. In the hip, three uncommon developmental abnormalities occurring in utero or in childhood, congenital dysplasia, Legg-Perthes disease, and slipped capital femoral epiphysis, leave a child with distortions of hip joint anatomy that often lead to OA later in life. Girls are predominantly affected by acetabular dysplasia, a mild form of congenital dislocation, whereas the other abnormalities more often affect boys. Depending on the severity of the anatomic abnormalities, hip OA occurs either in young adulthood (severe abnormalities) or middle age (mild abnormalities).
Major injuries to a joint also can produce anatomic abnormalities that leave the joint susceptible to OA. For example, a fracture through the joint surface often causes OA in joints in which the disease is otherwise rare such as the ankle and the wrist. Avascular necrosis can lead to collapse of dead bone at the articular surface, producing anatomic irregularities and subsequent OA.
Tears of ligamentous and fibrocartilaginous structures that protect the joints, such as the anterior cruciate ligament and the meniscus in the knee and the labrum in the hip, can lead to premature OA. Meniscal tears increase with age and when chronic are often asymptomatic but lead to adjacent cartilage damage and accelerated OA. Even injuries in which the affected person never received a diagnosis may increase risk of OA. For example, in the Framingham Study subjects, men with a history of major knee injury, but no surgery, had a 3.5-fold increased risk for subsequent knee OA.
Another source of anatomic abnormality is malalignment across the joint (Fig. 394-5). This factor has been best studied in the knee, which is the fulcrum of the longest lever arm in the body. Varus (bowlegged) knees with OA are at exceedingly high risk of cartilage loss in the medial or inner compartment of the knee, whereas valgus (knock-kneed) malalignment predisposes to rapid cartilage loss in the lateral compartment. Malalignment causes this effect by increasing stress on a focal area of cartilage, which then breaks down. There is evidence that malalignment in the knee not only causes cartilage loss but leads to underlying bone damage, producing bone marrow lesions seen on magnetic resonance imaging (MRI). Malalignment in the knee often produces such a substantial increase in focal stress within the knee (as evidenced by its destructive effects on subchondral bone) that severely malaligned knees may be destined to progress regardless of the status of other risk factors.
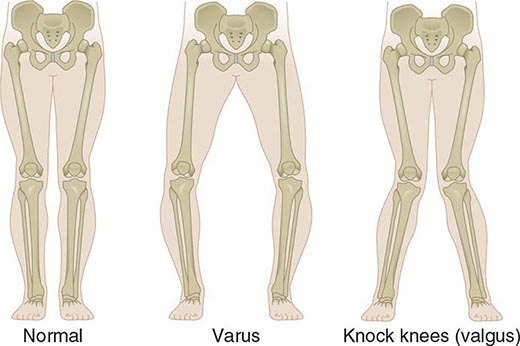
FIGURE 394-5 The two types of limb malalignment in the frontal plane: varus, in which the stress is placed across the medial compartment of the knee joint, and valgus, which places excess stress across the lateral compartment of the knee.
Weakness in the quadriceps muscles bridging the knee increases the risk of the development of painful OA in the knee.
Patients with knee OA have impaired proprioception across their knees, and this may predispose them to further disease progression. The role of bone in serving as a shock absorber for impact load is not well understood, but persons with increased bone density are at high risk of OA, suggesting that the resistance of bone to impact during joint use may play a role in disease development.
LOADING FACTORS
Obesity Three to six times body weight is transmitted across the knee during single-leg stance. Any increase in weight may be multiplied by this factor to reveal the excess force across the knee in overweight persons during walking. Obesity is a well-recognized and potent risk factor for the development of knee OA and, less so, for hip OA. Obesity precedes the development of disease and is not just a consequence of the inactivity present in those with disease. It is a stronger risk factor for disease in women than in men, and in women, the relationship of weight to the risk of disease is linear, so that with each increase in weight, there is a commensurate increase in risk. Weight loss in women lowers the risk of developing symptomatic disease. Not only is obesity a risk factor for OA in weight-bearing joints, but obese persons have more severe symptoms from the disease.
Obesity’s effect on the development and progression of disease is mediated mostly through the increased loading in weight-bearing joints that occurs in overweight persons. However, a modest association of obesity with an increased risk of hand OA suggests that there may be a systemic metabolic factor circulating in obese persons that affects disease risk also.
Repeated Use of Joint and Exercise There are two categories of repetitive joint use, occupational use and leisure time physical activities. Workers performing repetitive tasks as part of their occupations for many years are at high risk of developing OA in the joints they use repeatedly. For example, farmers are at high risk for hip OA, and miners have high rates of OA in knees and spine. Workers whose jobs require regular knee bending or lifting or carrying heavy loads have a high rate of knee OA. One reason why workers may get disease is that during long days at work, their muscles may gradually become exhausted, no longer serving as effective joint protectors.
It is widely recommended for people to adopt an exercise-filled lifestyle, and long-term studies of exercise suggest no consistent association of exercise with OA risk in the majority of persons. However, persons who already have injured joints may put themselves at greater risk by engaging in certain types of exercise. For example, persons who have already sustained major knee injuries are at increased risk of progressive knee OA as a consequence of running. In addition, compared to nonrunners, elite runners (professional runners and those on Olympic teams) have high risks of both knee and hip OA. Lastly, although recreational runners are not at increased risk of knee OA, studies suggest that they have a modest increased risk of disease in the hip.
PATHOLOGY
The pathology of OA provides evidence of the involvement of many joint structures in disease. Cartilage initially shows surface fibrillation and irregularity. As disease progresses, focal erosions develop there, and these eventually extend down to the subjacent bone. With further progression, cartilage erosion down to bone expands to involve a larger proportion of the joint surface, even though OA remains a focal disease with nonuniform loss of cartilage (Fig. 394-6).
FIGURE 394-6 Pathologic changes of osteoarthritis in a toe joint. Note the nonuniform loss of cartilage (arrowhead vs solid arrow), the increased thickness of the subchondral bone envelope (solid arrow), and the osteophyte (open arrow). (From the American College of Rheumatology slide collection.)
After an injury to cartilage, chondrocytes undergo mitosis and clustering. Although the metabolic activity of these chondrocyte clusters is high, the net effect of this activity is to promote proteoglycan depletion in the matrix surrounding the chondrocytes. This is because the catabolic is greater than the synthetic activity. As disease develops, collagen matrix becomes damaged, the negative charges of proteoglycans get exposed, and cartilage swells from ionic attraction to water molecules. Because in damaged cartilage proteoglycans are no longer forced into close proximity, cartilage does not bounce back after loading as it did when healthy, and cartilage becomes vulnerable to further injury. Chondrocytes at the basal level of cartilage undergo apoptosis.
With loss of cartilage come alterations in subchondral bone. Stimulated by growth factors and cytokines, osteoclasts and osteoblasts in the subchondral bony plate, just underneath cartilage, become activated. Bone formation produces a thickening and stiffness of the subchondral plate that occurs even before cartilage ulcerates. Trauma to bone during joint loading may be the primary factor driving this bone response, with healing from injury (including microcracks) producing stiffness. Small areas of osteonecrosis usually exist in joints with advanced disease. Bone death may also be caused by bone trauma with shearing of microvasculature, leading to a cutoff of vascular supply to some bone areas.
At the margin of the joint, near areas of cartilage loss, osteophytes form. These start as outgrowths of new cartilage, and with neurovascular invasion from the bone, this cartilage ossifies. Osteophytes are an important radiographic hallmark of OA. In malaligned joints, osteophytes grow larger on the side of the joint subject to most loading stress (e.g., in varus knees, osteophytes grow larger on the medial side).
The synovium produces lubricating fluids that minimize shear stress during motion. In healthy joints, the synovium consists of a single discontinuous layer filled with fat and containing two types of cells, macrophages and fibroblasts, but in OA, it can sometimes become edematous and inflamed. There is a migration of macrophages from the periphery into the tissue, and cells lining the synovium proliferate. Enzymes secreted by the synovium digest cartilage matrix that has been released from the surface of the cartilage.
Additional pathologic changes occur in the capsule, which stretches, becomes edematous, and can become fibrotic.
The pathology of OA is not identical across joints. In hand joints with severe OA, for example, there are often cartilage erosions in the center of the joint probably produced by bony pressure from the opposite side of the joint.
Basic calcium phosphate and calcium pyrophosphate dihydrate crystals are present microscopically in most joints with end-stage OA. Their role in osteoarthritic cartilage is unclear, but their release from cartilage into the joint space and joint fluid likely triggers synovial inflammation, which can, in turn, produce release of enzymes and trigger nociceptive stimulation.
SOURCES OF PAIN
Because cartilage is aneural, cartilage loss in a joint is not accompanied by pain. Thus, pain in OA likely arises from structures outside the cartilage. Innervated structures in the joint include the synovium, ligaments, joint capsule, muscles, and subchondral bone. Most of these are not visualized by the x-ray, and the severity of x-ray changes in OA correlates poorly with pain severity.
Based on MRI studies in osteoarthritic knees comparing those with and without pain and on studies mapping tenderness in unanesthetized joints, likely sources of pain include synovial inflammation, joint effusions, and bone marrow edema. Modest synovitis develops in many but not all osteoarthritic joints. Some diseased joints have no synovitis, whereas others have synovial inflammation that approaches the severity of joints with rheumatoid arthritis (Chap. 380). The presence of synovitis on MRI is correlated with the presence and severity of knee pain. Capsular stretching from fluid in the joint stimulates nociceptive fibers there, inducing pain. Increased focal loading as part of the disease not only damages cartilage but probably also injures the underlying bone. As a consequence, bone marrow edema appears on the MRI; histologically, this edema signals the presence of microcracks and scar, which are the consequences of trauma. These lesions may stimulate bone nociceptive fibers. Also, hemostatic pressure within bone rises in OA, and the increased pressure itself may stimulate nociceptive fibers, causing pain.
Pain may arise from outside the joint also, including bursae near the joints. Common sources of pain near the knee are anserine bursitis and iliotibial band syndrome.
Persons with chronic OA pain may develop nervous system alterations as a consequence of disease, changes which decrease inhibitory controls on nociception and its distribution. This may produce allodynia and hyperalgesia in some patients with OA.
CLINICAL FEATURES
Joint pain from OA is activity-related. Pain comes on either during or just after joint use and then gradually resolves. Examples include knee or hip pain with going up or down stairs, pain in weight-bearing joints when walking, and, for hand OA, pain when cooking. Early in disease, pain is episodic, triggered often by a day or two of overactive use of a diseased joint, such as a person with knee OA taking a long run and noticing a few days of pain thereafter. As disease progresses, the pain becomes continuous and even begins to be bothersome at night. Stiffness of the affected joint may be prominent, but morning stiffness is usually brief (<30 min).
In knees, buckling may occur, in part, due to weakness of muscles crossing the joint. Mechanical symptoms, such as buckling, catching, or locking, could also signify internal derangement, such as meniscal tears, and need to be evaluated. In the knee, pain with activities requiring knee flexion, such as stair climbing and arising from a chair, often emanates from the patellofemoral compartment of the knee, which does not actively articulate until the knee is bent ~35°.
OA is the most common cause of chronic knee pain in persons over age 45, but the differential diagnosis is long. Inflammatory arthritis is likely if there is prolonged morning stiffness and many other joints are affected. Bursitis occurs commonly around knees and hips. A physical examination should focus on whether tenderness is over the joint line (at the junction of the two bones around which the joint is articulating) or is outside of it. Anserine bursitis, medial and distal to the knee, is an extremely common cause of chronic knee pain that may respond to a glucocorticoid injection. Prominent nocturnal pain in the absence of end-stage OA merits a distinct workup. For hip pain, OA can be detected by loss of internal rotation on passive movement, and pain isolated to an area lateral to the hip joint usually reflects the presence of trochanteric bursitis.
No blood tests are routinely indicated for workup of patients with OA unless symptoms and signs suggest inflammatory arthritis. Examination of the synovial fluid is often more helpful diagnostically than an x-ray. If the synovial fluid white count is >1000/μL, inflammatory arthritis or gout or pseudogout is likely, the latter two being also identified by the presence of crystals.
X-rays are indicated to evaluate chronic hand pain and hip pain thought to be due to OA, as the diagnosis is often unclear without confirming radiographs. For knee pain, x-rays should be obtained if symptoms or signs are not typical of OA or if knee pain persists after inauguration of effective treatment. In OA, radiographic findings (Fig. 394-7) correlate poorly with the presence and severity of pain. Further, radiographs may be normal in early disease as they are insensitive to cartilage loss and other early findings.
FIGURE 394-7 X-ray of knee with medial osteoarthritis. Note the narrowed joint space on medial side of the joint only (white arrow), the sclerosis of the bone in the medial compartment providing evidence of cortical thickening (black arrow), and the osteophytes in the medial femur (white wedge).
Although MRI may reveal the extent of pathology in an osteoarthritic joint, it is not indicated as part of the diagnostic workup. Findings such as meniscal tears and cartilage and bone lesions occur in most patients with OA in the knee, but almost never warrant a change in therapy.
|
TREATMENT |
OSTEOARTHRITIS |
The goals of the treatment of OA are to alleviate pain and minimize loss of physical function. To the extent that pain and loss of function are consequences of inflammation, of weakness across the joint, and of laxity and instability, the treatment of OA involves addressing each of these impairments. Comprehensive therapy consists of a multimodality approach including nonpharmacologic and pharmacologic elements.
Patients with mild and intermittent symptoms may need only reassurance or nonpharmacologic treatments. Patients with on-going, disabling pain are likely to need both nonpharmacotherapy and pharmacotherapy.
Treatments for knee OA have been more completely evaluated than those for hip and hand OA or for disease in other joints. Thus, although the principles of treatment are identical for OA in all joints, we shall focus below on the treatment of knee OA, noting specific recommendations for disease in other joints, especially when they differ from those for the knee.
NONPHARMACOTHERAPY
Because OA is a mechanically driven disease, the mainstay of treatment involves altering loading across the painful joint and improving the function of joint protectors, so they can better distribute load across the joint. Ways of lessening focal load across the joint include:
1. avoiding activities that overload the joint, as evidenced by their causing pain;
2. improving the strength and conditioning of muscles that bridge the joint, so as to optimize their function; and
3. unloading the joint, either by redistributing load within the joint with a brace or a splint or by unloading the joint during weight bearing with a cane or a crutch.
The simplest effective treatment for many patients is to avoid activities that precipitate pain. For example, for the middle-aged patient whose long-distance running brings on symptoms of knee OA, a less demanding form of weight-bearing activity may alleviate all symptoms. For an older person whose daily constitutionals up and down hills bring on knee pain, routing the constitutional away from hills might eliminate symptoms.
Each pound of weight increases the loading across the knee three- to sixfold. Weight loss may have a commensurate multiplier effect, unloading both knees and hips and probably relieving pain in those joints.
In hand joints affected by OA, splinting, by limiting motion, often minimizes pain for patients with involvement especially in the base of the thumb. Weight-bearing joints such as knees and hips can be unloaded by using a cane in the hand opposite to the affected joint for partial weight bearing. A physical therapist can help teach the patient how to use the cane optimally, including ensuring that its height is optimal for unloading. Crutches or walkers can serve a similar beneficial function.
Exercise Osteoarthritic pain in knees or hips during weight bearing results in lack of activity and poor mobility, and because OA is so common, the inactivity that results represents a public health concern, increasing the risk of cardiovascular disease and obesity. Aerobic capacity is poor in most elders with symptomatic knee OA, worse than others of the same age.
Weakness in muscles that bridge osteoarthritic joints is multifactorial in etiology. First, there is a decline in strength with age. Second, with limited mobility comes disuse muscle atrophy. Third, patients with painful knee or hip OA alter their gait so as to lessen loading across the affected joint, and this further diminishes muscle use. Fourth, “arthrogenous inhibition” may occur, whereby contraction of muscles bridging the joint is inhibited by a nerve afferent feedback loop emanating in a swollen and stretched joint capsule; this prevents maximal attainment of voluntary maximal strength. Because adequate muscle strength and conditioning are critical to joint protection, weakness in a muscle that bridges a diseased joint makes the joint more susceptible to further damage and pain. The degree of weakness correlates strongly with the severity of joint pain and the degree of physical limitation. One of the cardinal elements of the treatment of OA is to improve the functioning of muscles surrounding the joint.
For knee and hip OA, trials have shown that exercise lessens pain and improves physical function. Most effective exercise regimens consist of aerobic and/or resistance training, the latter of which focuses on strengthening muscles across the joint. Exercises are likely to be effective, especially if they train muscles for the activities a person performs daily. Activities that increase pain in the joint should be avoided, and the exercise regimen needs to be individualized to optimize effectiveness. Range-of-motion exercises, which do not strengthen muscles, and isometric exercises that strengthen muscles, but not through range of motion, are unlikely to be effective by themselves. Low-impact exercises, including water aerobics and water resistance training, are often better tolerated by patients than exercises involving impact loading, such as running or treadmill exercises. A patient should be referred to an exercise class or to a therapist who can create an individualized regimen, and then an individualized home-based regimen can be crafted. In addition to conventional exercise regimens, tai chi may be effective for knee OA. However, there is no strong evidence that patients with hand OA benefit from therapeutic exercise.
Adherence over the long term is the major challenge to an exercise prescription. In trials involving patients with knee OA, who are engaged in exercise treatment, from a third to over half of patients stopped exercising by 6 months. Less than 50% continued regular exercise at 1 year. The strongest predictor of a patient’s continued exercise is a previous personal history of successful exercise. Physicians should reinforce the exercise prescription at each clinic visit, help the patient recognize barriers to ongoing exercise, and identify convenient times for exercise to be done routinely. The combination of exercise with calorie restriction and weight loss is especially effective in lessening pain.
Correction of Malalignment Malalignment in the frontal plane (varus-valgus) markedly increases the stress across the joint, which can lead to progression of disease and to pain and disability (Fig. 394-5). Correcting malalignment, either surgically or with bracing, may relieve pain in persons whose knees are malaligned. Malalignment develops over years as a consequence of gradual anatomic alterations of the joint and bone, and correcting it is often very challenging. One way is with a fitted brace, which takes an often varus osteoarthritic knee and straightens it by putting valgus stress across the knee. Unfortunately, many patients are unwilling to wear a realigning knee brace; in addition, in patients with obese legs, braces may slip with usage and lose their realigning effect. They are indicated for willing patients who can learn to put them on correctly and on whom they do not slip.
Other ways of correcting malalignment across the knee include the use of orthotics in footwear. Unfortunately, although they may have modest effects on knee alignment, trials have heretofore not demonstrated efficacy of a lateral wedge orthotic versus placebo wedges.
Pain from the patellofemoral compartment of the knee can be caused by tilting of the patella or patellar malalignment with the patella riding laterally or medially in the femoral trochlear groove. Using a brace to realign the patella, or tape to pull the patella back into the trochlear sulcus or reduce its tilt, has been shown, when compared to placebo taping in clinical trials, to lessen patellofemoral pain. However, patients may find it difficult to apply tape, and skin irritation from the tape is common. Commercial patellar braces may be a solution, but there is insufficient evidence on their efficacy to recommend them.
Although their effect on malalignment is questionable, neoprene sleeves pulled to cover the knee lessen pain and are easy to use and popular among patients. The explanation for their therapeutic effect on pain is unclear.
In patients with knee OA, acupuncture produces modest pain relief compared to placebo needles and may be an adjunctive treatment.
PHARMACOTHERAPY
Although nonpharmacologic approaches to therapy constitute its mainstay, pharmacotherapy serves an important adjunctive role in OA treatment. Available drugs are administered using oral, topical, and intraarticular routes.
Acetaminophen, Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), and Cyclooxygenase-2 (COX-2) Inhibitors Acetaminophen (paracetamol) is the initial analgesic of choice for patients with OA in knees, hips, or hands. For some patients, it is adequate to control symptoms, in which case more toxic drugs such as NSAIDs can be avoided. Doses up to 1 g three times daily can be used (Table 394-1).
|
PHARMACOLOGIC TREATMENT FOR OSTEOARTHRITIS |
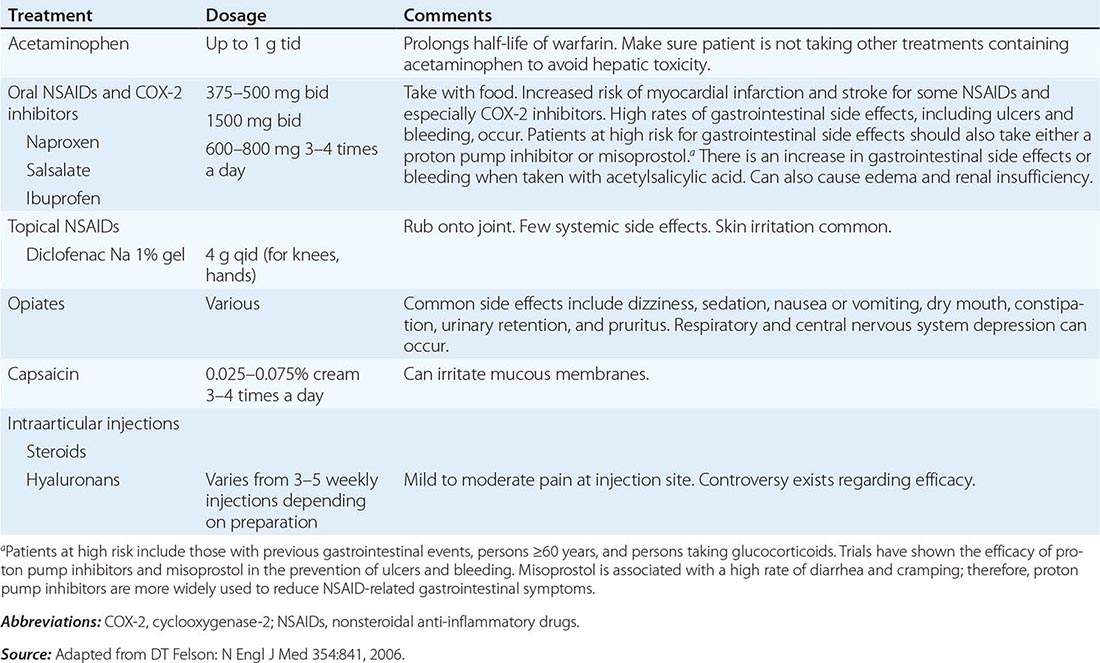
NSAIDs are the most popular drugs to treat osteoarthritic pain. They can be administered either topically or orally. In clinical trials, oral NSAIDs produce ~30% greater improvement in pain than high-dose acetaminophen. Occasional patients treated with NSAIDs experience dramatic pain relief, whereas others experience little improvement. Initially, NSAIDs should be administered topically or taken orally on an “as needed” basis because side effects are less frequent with low intermittent doses. If occasional medication use is insufficiently effective, then daily treatment may be indicated, with an anti-inflammatory dose selected (Table 394-1). Patients should be reminded to take low-dose aspirin and ibuprofen at different times to eliminate a drug interaction.
NSAIDs taken orally have substantial and frequent side effects, the most common of which is upper gastrointestinal toxicity, including dyspepsia, nausea, bloating, gastrointestinal bleeding, and ulcer disease. Some 30–40% of patients experience upper gastrointestinal (GI) side effects so severe as to require discontinuation of medication. To minimize the risk of nonsteroidal-related GI side effects, patients should not take two NSAIDs and should take medications after food; if risk is high, patients should take a gastroprotective agent, such as a proton pump inhibitor. Certain oral agents are safer to the stomach than others, including nonacetylated salicylates and nabumetone. Major NSAID-related GI side effects can occur in patients who do not complain of upper GI symptoms. In one study of patients hospitalized for GI bleeding, 81% had no premonitory symptoms.
Because of the increased rates of cardiovascular events associated with COX-2 inhibitors and with some conventional NSAIDs such as diclofenac, many of these drugs are not appropriate long-term treatment choices for older persons with OA, especially those at high risk of heart disease or stroke. The American Heart Association has identified rofecoxib and all other COX-2 inhibitors as putting patients at high risk, although low doses of celecoxib (≤200 mg/d) may not be associated with an elevation of risk. The only conventional NSAID that appears safe from a cardiovascular perspective is naproxen, but it does have GI toxicity.
There are other common side effects of NSAIDs, including the tendency to develop edema because of prostaglandin inhibition of afferent blood supply to glomeruli in the kidneys and, for similar reasons, a predilection toward reversible renal insufficiency. Blood pressure may increase modestly in some NSAID-treated patients. Oral NSAIDs should not be used in patients with stage IV or V renal disease and should be used with caution in those with stage III disease.
NSAIDs can be placed into a gel or topical solution with another chemical modality that enhances penetration of the skin barrier creating a topical NSAID. When absorbed through the skin, plasma concentrations are an order of magnitude lower than with the same amount of drug administered orally or parenterally. However, when these drugs are administered topically in proximity to a superficial joint (knees, hands, but not hips), the drug can be found in joint tissues such as the synovium and cartilage. Trial results have varied but generally have found that topical NSAIDs are slightly less efficacious than oral agents, but have far fewer GI and systemic side effects. Unfortunately, topical NSAIDs often cause local skin irritation where the medication is applied, inducing redness, burning, or itching in up to 40% of patients (see Table 394-1).
Intraarticular Injections: Glucocorticoids and Hyaluronic Acid Because synovial inflammation is likely to be a major cause of pain in patients with OA, local anti-inflammatory treatments administered intraarticularly may be effective in ameliorating pain, at least temporarily. Glucocorticoid injections provide such efficacy, but response is variable, with some patients having little relief of pain whereas others experience pain relief lasting several months. Glucocorticoid injections are useful to get patients over acute flares of pain and may be especially indicated if the patient has coexistent OA and crystal deposition disease, especially from calcium pyrophosphate dihydrate crystals (Chap. 395). There is no evidence that repeated glucocorticoid injections into the joint are dangerous.
Hyaluronic acid injections can be given for treatment of symptoms in knee and hip OA, but there is controversy as to whether they have efficacy versus placebo (Table 394-1).
Other Classes of Drugs and Nutraceuticals For patients with symptomatic knee or hip OA who have not had an adequate response to the treatments above and are either unwilling to undergo or are not candidates for total joint arthroplasty, opioid analgesics have shown modest efficacy and can be tried. Opioid management plans and patient selection are critical. Another option is the use of duloxetine, which has demonstrated modest efficacy in OA.
Recent guidelines recommend against the use of glucosamine or chondroitin for OA. Large publicly supported trials have failed to show that, compared with placebo, these compounds relieve pain in persons with disease.
Optimal nonsurgical therapy for OA is often achieved by trial and error, with each patient having idiosyncratic responses to specific treatments. When medical therapies have failed and the patient has an unacceptable reduction in their quality of life and ongoing pain and disability, then at least for knee and hip OA, total joint arthroplasty is indicated.
SURGERY
For knee OA, several operations are available. Arthroscopic debridement and lavage have diminished in popularity after randomized trials evaluating this operation have showed that its efficacy is no greater than that of sham surgery or no treatment for relief of pain or disability. Even mechanical symptoms such as buckling, which are extremely common in patients with knee OA, do not respond to arthroscopic debridement. Although arthroscopic meniscectomy is indicated for acute meniscal tears in which symptoms such as locking and acute pain are clearly related temporally to a knee injury that produced the tear, recent trials show that doing a partial meniscectomy in persons with OA and a symptomatic meniscal tear does not relieve knee pain or improve function.
For patients with knee OA isolated to the medial compartment, operations to realign the knee to lessen medial loading can relieve pain. These include a high tibial osteotomy, in which the tibia is broken just below the tibial plateau and realigned so as to load the lateral, nondiseased compartment, or a unicompartmental replacement with realignment. Each surgery may provide the patient with years of pain relief before a total knee replacement is required.
Ultimately, when the patient with knee or hip OA has failed medical treatment modalities and remains in pain, with limitations of physical function that compromise the quality of life, the patient should be referred for total knee or hip arthroplasty. These are highly efficacious operations that relieve pain and improve function in the vast majority of patients, although rates of success are higher for hip than knee replacement. Currently failure rates for both are ~1% per year, although these rates are higher in obese patients. The chance of surgical success is greater in centers where at least 25 such operations are performed yearly or with surgeons who perform multiple operations annually. The timing of knee or hip replacement is critical. If the patient suffers for many years until their functional status has declined substantially, with considerable muscle weakness, postoperative functional status may not improve to a level achieved by others who underwent operation earlier in their disease course.
Cartilage Regeneration Chondrocyte transplantation has not been found to be efficacious in OA, perhaps because OA includes pathology of joint mechanics, which is not corrected by chondrocyte transplants. Similarly, abrasion arthroplasty (chondroplasty) has not been well studied for efficacy in OA, but it produces fibrocartilage in place of damaged hyaline cartilage. Both of these surgical attempts to regenerate and reconstitute articular cartilage may be more likely to be efficacious early in disease when joint malalignment and many of the other noncartilage abnormalities that characterize OA have not yet developed.




