[level-membership-for-cardiovascular-category]4
REGULATION OF CARDIAC FUNCTION
Introduction
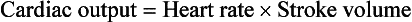
When the textbook person (see Chapter 1) is at rest their cardiac output, the volume of blood pumped by each side of the heart, is about 5 L per minute. In understanding how this is achieved the ‘rule of 70’ might be helpful. The heart typically beats at about 70 bpm and, when the ventricle empties it goes from a volume of about 140 mL at the end of the filling phase, diastole, to about 70 mL at the end of the contraction phase, systole. The ‘stroke volume’ is therefore about 70 mL and about 70 mL of blood remains in the ventricle at the end of each cardiac contraction. The ejection fraction of the heart, the stroke volume expressed as a percentage of end-diastolic volume, is thus typically about 50%. When appropriate an increase in the heart’s output can be achieved by a combination of an increase in heart rate and stroke volume. The maximum effective heart rate in the adult is about 180 bpm and the stroke volume can be increased both by filling the ventricle more during diastole and emptying it further during systole. In a very fit textbook person the maximum cardiac output is of the order of 25 L/min.
Venous return
When a person is upright, their head and shoulders are above the level of the heart but most of the rest of the body is below that level and thus gravity is largely a negative factor in determining venous return of blood to the heart. In a horizontal posture however the venous return will not be significantly opposed by gravity. For this reason if a person faints, it is usually best to leave them horizontal to aid the flow of blood back to the heart. Conversely, if a person has poor cardiac function a sitting position will decrease the venous return and hence the work load of the heart. Many people with cardiac problems sleep either in a chair or propped up in bed. The effect of gravity on venous pressure is further discussed in Chapter 10.
The skeletal muscle pump
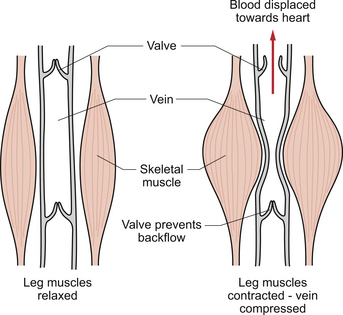
The veins, especially in the limbs, are often to be found between major blocks of muscle. If muscle activity is increased, then the alternate compression and release of the veins will result in blood being propelled back towards the heart (Fig. 4.1). This mechanism is dependent on the presence of valves in veins in the lower limbs. This aspect of venous return is useful for guardsmen on parade. If they are standing still and upright over long periods of time there is a danger they will faint because their cardiac output, limited by the poor venous return, is not enough to supply their tissue’s needs especially on hot summer days when skin blood flow may be increased. They are told to move their toes in their boots and thus use the alternate contraction and relaxation of their leg muscles to aid venous return.
Control of cardiac output
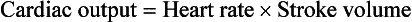
The respiratory system has ‘dead space’ areas such as the nasal passages, trachea and bronchi which are ventilated but where no gas exchange takes place. This means that during exercise there is a functional advantage in keeping the respiratory rate relatively low and breathing deeply. The heart however is a through flow system and, within limits, an increase in heart rate will produce a proportionate rise in cardiac output, as long as the venous return is increased to provide the necessary blood to pump. This is true in an adult up to a heart rate of about 180 bpm. If the heart rate goes much above this value then cardiac output falls precipitously because there is an encroachment on the ‘rapid filling time’—that period of time which immediately follows the end of ventricular systole and when most of the ventricular filling occurs (see Chapter 3). The result is that excessively high heart rates are associated with a reduced stroke volume. Small minute to minute changes in cardiac output, as might be associated with mild exercise such as walking, tend to be achieved largely by changes in both heart rate and stroke volume (see Chapter 13). Changes in stroke volume reach maximum levels at fairly moderate exercise intensity and the further increases during heavy exercise are achieved just with heart rate changes. The effects of exercise on cardiac function are considered in more detail in Chapter 13.
Regulation of heart rate
There is considerable variation in the resting heart rate in the adult population with a normal range of 50–100 bpm. As explained in Chapter 2 the rate of beating of the heart is normally determined by the pacemaker at the sinoatrial node (SAN). The rate of this pacemaker is modulated by both the sympathetic and parasympathetic nervous systems. At rest the parasympathetic inhibition dominates so that the typical resting heart rate of about 70 bpm is less than the natural intrinsic rate of the SAN which is about 110–120 bpm (see Chapter 2). Small increases in heart rate, such as might occur during walking, are produced by inhibition of the parasympathetic tone. If the heart rate is to go above about 110 bpm, then an increased sympathetic nerve activity is also required.
The role of the central nervous system control centres and the function of the baroreceptor reflex in the control of heart rate are further discussed in Chapter 10.
The sympathetic innervation of the heart is routed via the cervical and stellate sympathetic ganglia. It is the sympathetic fibres on the right side of the body which have the major effect on heart rate (Fig. 4.2). The fibres from the left side of the sympathetic nervous system are more concerned with the regulation of cardiac contractility. The noradrenaline (norepinephrine) released at the nerve endings acts on β1 receptors in the SAN and increases the Ca++ current in phase 4 of the SAN action potential. This accelerates the rate of firing of the SAN cells (see Chapter 2).
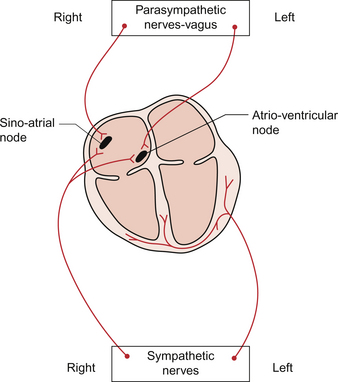
The parasympathetic innervation of the pacemaker tissue is via the left and right branches of the vagus nerve but, like the sympathetic innervation, the fibres are distributed differentially (Fig. 4.2). The right vagus goes mainly to the SAN with a small innervation of the atrioventricular node (AVN) whereas the left vagus goes primarily to the AVN with only a small outflow to the SAN. The vagal fibres to the heart originate in the dorsal motor nucleus of the vagus or in the nucleus ambiguus (see Chapter 10). The parasympathetic ganglia, the location of the nerve cell bodies, are on the cardiac surface or within the heart itself and the short postganglionic fibres release acetylcholine as their neurotransmitter. This acetylcholine acts on muscarinic (M2) receptors in the SAN and, via an inhibitory G protein mechanism, reduces the production of cAMP within the cell. Cyclic AMP activates populations of Na+ channels and L-type Ca++ channels causing depolarization of the pacemaker cells. Inhibiting cAMP formation therefore reduces the rate of depolarization and slows the heart rate. There is also a hyperpolarizing effect produced following binding of acetylcholine to M2 receptors. This is mediated by activation of a population of K++ channels. As a result the membrane potential is moved closer to the potassium equilibrium potential (EK) of −94 mV and therefore further away from the threshold at which an action potential is triggered (see Chapter 2). The right and left vagus nerves have little effect on cardiac contractility.
Regulation of stroke volume
The heart is an unusual form of pump in that not only is it able to change the rate at which it pumps but, if necessary, it is also able to change the volume of blood pumped at each stroke. Under resting conditions, the ventricles in the adult heart typically fill to about 140 mL (the end-diastolic volume) and empty to about 70 mL (the end-systolic volume) giving rise to a stroke volume of about 70 mL.However as the normal range of heart rate is quite wide at 50–100 bpm, individuals with a slow resting heart rate will operate with a stroke volume greater than 70 mL and people with a fast heart rate will have a correspondingly smaller resting stroke volume. Resting cardiac output will therefore be comparable between individuals with the same body size irrespective of resting heart rate. An outline of the case history of a man with increased stroke volume in his heart is described in Case 4.1:1.
Preload effects on the heart
The cumulative effect of the factors which determine ventricular end-diastolic volume or end-diastolic fibre length of the ventricular muscle is called a ‘preload’ effect on the heart. This is effectively the resting length from which the heart muscle contracts. Preload effects are often conveniently quantified as the right or left atrial pressure, the filling pressure which determines how much blood enters the ventricle during diastole. Although for most purposes this is a useful concept it is not entirely satisfactory. A ventricle stiffened by fibrotic changes for example will need a higher filling pressure than normal in order to reach a certain end-diastolic volume. Similarly, stenosis of a valve between the atria and ventricles will require a higher atrial pressure to ensure adequate ventricular filling.
Figure 4.3 shows the relationship between atrial pressure and stroke volume for the right and left sides of the heart. In both cases an increase in filling pressure (atrial pressure) leads to increased stretch of the ventricular muscle and results in an increase in stroke volume (stroke work). This is called Starling’s law of the heart. The background to Starling’s law and the mechanisms involved are explained in Box 4.1. Eventually as atrial pressure increases a maximum stroke volume is reached, imposed fundamentally by the physical size of the heart and by the presence of the pericardium as a potentially restricting bag around the outside of the heart. Under resting conditions the heart is typically operating at about halfway up the Starling curve, hence the ability to double stroke volume during exercise. Figure 4.3 also shows that the filling pressure of the left ventricle is higher than for the right ventricle. This partly reflects the thicker ventricular muscle wall on the left which requires a higher filling pressure.
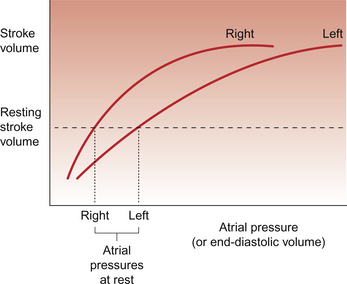
During diastole the right ventricle fills as a result of the pressure gradient between the atrium and ventricle. As there is no valve between the venous system and the right atrium blood can flow from the vena cavae across the right atrium and into the ventricle. The contraction of the right atrium only ‘tops up’ the ventricle with a small addition of blood and in a normal heart, this is not a major component of preload effects on the heart. The relationship between cardiac output and right atrial pressure means that, to some extent, an increase in venous return can produce an increase in cardiac output (as happens when a person lies down) and a fall in venous return (such as might happen on standing up) will cause a fall in cardiac output. However the main importance of the relationship described by Starling’s law is that it is the mechanism by which ventricular balance is achieved. This is of immense physiological importance—if the right ventricle output exceeded that of the left by only 0.1 mL per beat there would be 7 mL of blood trapped in the lung after a minute and 420 mL after an hour. What actually happens if right output exceeds left is that the pressure in the pulmonary circulation rises, left ventricular preload increases and, as a result, the left ventricle pumps more strongly, keeping the two sides of the heart in balance.
There are many aspects of cardiovascular function which determine right atrial pressure including pressures within the thorax and the physical characteristics of the ventricles. However, the three most important variables are blood volume, venous return and venous tone, the state of smooth muscle contraction in the walls of the veins (Fig. 4.4). Some of these key variables are discussed below. Venous return mechanisms have already been covered earlier in this chapter.
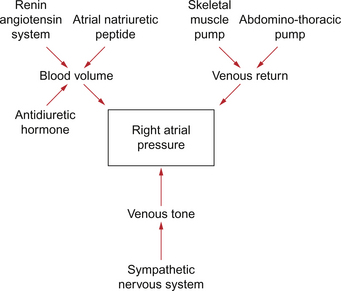
Blood volume
In the textbook person blood volume is about 5 L but it is not evenly distributed throughout the cardiovascular system. The large and small veins act as a reservoir for blood within the body and accommodate about two thirds of the total blood volume (see Chapter 14). If a person donates blood or suffers a haemorrhage then central venous pressure and preload on the heart will both be decreased. Similarly if the blood volume is expanded by infusing blood, saline or a plasma volume expander such as dextran or albumin then the central venous pressure and preload will be increased (see Chapter 14).
A family of hormones, the natriuretic peptides, of which the first to be discovered was atrial natriuretic peptide (ANP), are involved in blood volume regulation. An increase in stretch in the wall of the atria, as would occur following expansion of blood volume, leads to increased natriuretic hormone release and increased loss of salt and water via the kidneys to bring the blood volume back to normal (see Chapters 6 and 14). The major actions of ANP are firstly to increase glomerular filtration rate in the kidney. This is achieved by afferent arteriolar dilatation and efferent arteriolar constriction. In addition, ANP inhibits the actions of aldosterone. The renin-angiotensin-aldosterone system is of central importance in blood volume regulation as it is a sodium- and water-retaining system (see Chapters 6, 9 and 14). Renin secretion is promoted in response to a fall in blood pressure or a fall in blood volume. The baroreceptor reflex and activation of the sympathetic nervous system is particularly important in this context.
Regulation of blood volume is fundamental to the maintenance of a normal cardiac output. Changes in blood volume are a major factor in the compensation reactions which accompany heart failure (see Chapter 6).
Venomotor tone
The blood contained within the venous reservoir vessels is under a pressure which depends upon the blood volume but also on the extent to which the walls of the veins resist being stretched. The veins contain some smooth muscle (see Chapter 1). It is a relatively small amount compared to the smooth muscle in the walls of the arteries or arterioles but none the less if the activity in the sympathetic nerves to the veins, which act through α1-adrenoceptors, is increased then central venous pressure will be raised and preload increased. This mechanism is activated for example when moving from a horizontal to an upright posture as this tends to redistribute about 500 mL of blood from the thoracic venous system to the lower limbs. Venomotor control opposes the tendency to venous pooling in the lower half of the body imposed by the effects of gravity. Loss of venomotor tone is frequently a clinical problem particularly in old people and results in postural hypotension, a fall in cardiac output and arterial blood pressure as a result of standing up. This results in dizziness or fainting. The effects of gravity on hydrostatic pressure in the circulation are discussed in Chapter 10.
Residual pressure
This is the pressure ‘left over’ from the pressure in the aorta after the blood has been forced through the peripheral tissues. However, most of the potential energy stored as arterial pressure is dissipated as the blood passes through the arterioles (see Chapter 9). The pressure in the capillaries is only about 25 mm Hg and this is reduced even further by the time the blood reaches the great veins. Thus the residual pressure, or ‘vis a tergo’ as it is sometimes called, contributes little to venous return.
Ventricular suction
When the right ventricle relaxes at the end of systole there is a tendency for it to ‘spring open’ and thus to ‘suck’ blood in from the atrium—the so-called ‘vis a fronte’. However any attempt to drink from a glass using a floppy drinking straw will soon convince you that suction alone is not sufficient to produce a flow through thin-walled unsupported vessels such as the veins. Remember that the trachea, a body tube in which suction does occur, is held open by rings of cartilage producing a structure rather like a vacuum cleaner hose. Veins do not have this advantage and collapse if the pressure inside is less than that outside the vein. This is the normal state of the jugular vein in a person who is upright and it only becomes inflated when pressure rises in the vein, as in heart failure (see Chapter 6).
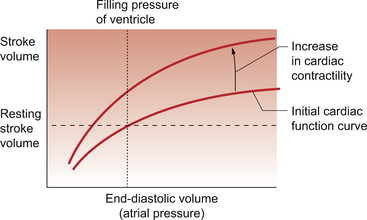
Contractility effects on the heart
It is possible to increase the force of ventricular contraction without increasing the end-diastolic volume and stretching the muscle fibres. When this happens the heart empties more and there is said to be an increase in contractility (Fig. 4.5). Physiological alterations in contractility are associated with changes in the [Ca++] inside the cardiac myocyte (see Chapter 2). Activation of the left branch of the sympathetic nerve supply to the ventricular muscle will increase contractility through a mechanism linked to a β1 adrenoceptor.
Many pathological mechanisms result in a decrease in contractility, often associated with an intracellular acidosis as a result of myocardial ischaemia. This is a common characteristic of heart failure (see Chapter 6). The strongest natural negative inotrope is the H+ ion and any episode of acidosis, such as might occur with cardiac ischaemia, will have a detrimental effect on cardiac performance. The effect is seen more quickly in the case of respiratory rather than metabolic acidosis (see Chapter 1) because the entry of CO2 into cardiac myocytes with the subsequent generation of H+ inside the cells is faster than the entry of the polar H+ ions from the extracellular fluid. The mechanism by which acidic conditions inside cardiac myocytes lead to decreased force of contraction is interesting. A doubling of [H+] can halve the strength of contraction of the heart. Surprisingly there is an increase in the intracellular [Ca++] which would normally be expected to increase the force of cardiac muscle contraction. The complex mechanisms that actually lead to a reduced force of contraction include reduced binding of Ca++ to troponin and a decrease in the force developed by the cross-bridges that do form.
Pharmacological agents which increase contractility are called positive inotropes although they tend to be compounds which also have a range of other physiological actions. Many of these drugs have a sympathomimetic action resembling endogenous noradrenaline (norepinephrine) and adrenaline (epinephrine), compounds which have actions via all types of adrenoceptors (see Chapter 9). Isoproterenol (isoprenaline) is a non-selective agonist for both β1 and β2 receptors and it increases both contractility of the ventricles and heart rate. Dopamine has actions at several types of receptor including specific dopamine receptors and also both β1 and β2 receptors. It therefore has a positive inotropic action which is accompanied by an intrarenal vasodilator action. The latter effect may be beneficial in circumstances where there is intense sympathetically driven intrarenal vasoconstriction as a result of a fall in arterial blood pressure and activation of the baroreceptor reflex. This would occur for example in many cases of heart failure and is potentially a cause of acute renal failure which may progress to chronic renal failure. Dobutamine is a dopamine analogue which has a powerful positive inotropic action but causes relatively little change in heart rate.
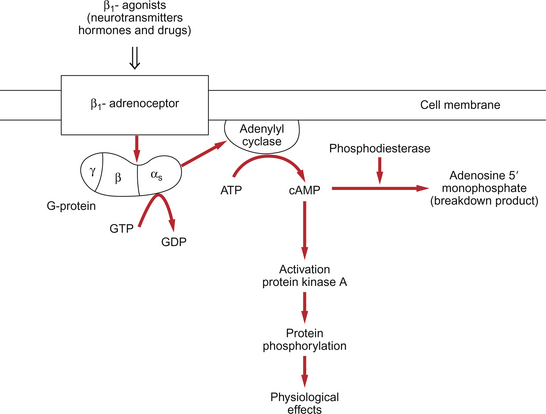
The inotropic actions of sympathetic nerve stimulation act through β-receptors which are coupled to cyclic AMP formation (Fig. 4.6). This second messenger activates protein kinases leading to increased transmembrane flux of Ca++ and increased affinity of the myofilaments for Ca++. In addition there is increased reuptake of Ca++ into the sarcoplasmic reticulum, an effect which shortens systole and prolongs diastole with consequent beneficial effects on coronary blood flow (see Chapter 5). Cyclic AMP mediated pathways can also be manipulated using blockers of the cAMP breakdown enzyme, phosphodiesterase. Milrinone and enoximone are phosphodiesterase III inhibitor drugs which are used as positive inotropes.
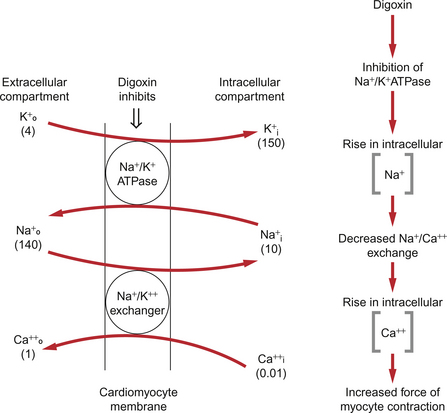
Digitalis glycosides increase contractility by inhibition of the Na+/K+-ATPase, a mechanism which is clearly different to the sympathomimetic drugs. The mechanism by which intracellular Ca++ is increased is illustrated in Figure 4.7. Inhibition of the Na+/K+-ATPase will lead to an increase in intracellular [Na+]. There is also a 3 Na+/Ca++ antiporter protein in the cell membrane. A rise in [Na+] inside the cell will therefore reduce Ca++ expulsion from the cell and this results in retention of Ca++ inside the cell. This extra Ca++ is stored in the sarcoplasmic reticulum of the cell and is available for release when the cell is activated, hence providing a positive inotropic action.
Negative inotropes
In some clinical situations such as hypertension or angina, it may be appropriate to decrease the force of contraction of the heart muscle. This will reduce the cardiac output and the work load carried out by the heart and also reduce the oxygen demand of the heart. In this case a drug with a negative inotropic action might be used. These drugs include β1-receptor antagonists such as atenolol (see Chapter 10) which suppress the effects of activating the sympathetic nerves to the heart. They have the effect of slowing the heart rate as well as decreasing the force of ventricular contraction. Slowing the heart rate may also improve coronary blood flow by prolonging diastole (see Chapter 5).
Calcium-channel-blocking drugs (calcium antagonists) act on voltage-gated ‘L-type’ calcium channels. There are different types of calcium channel blockers, some that have predominant effects on calcium channels in the smooth muscle of blood vessels (see Chapter 10) and others that have a greater effect on cardiac muscle cells. Nifedipine and amlodipine dilate arterioles in the periphery and hence reduce blood pressure by lowering the peripheral vascular resistance. The work required of the heart to overcome the afterload (see below) is therefore reduced although there may be a reflex tachycardia. Verapamil and diltiazem tend to act more directly on the SAN and AVN to slow the heart rate and the rate of impulse conduction to the ventricle. Most calcium-channel-blocking drugs will reduce cardiac contractility and, in some cases, coronary blood flow may be improved following the relief of coronary artery spasm (see Chapter 5).
Afterload effects on the heart
The aortic valve will open and allow blood to pass into the aorta when pressure in the left ventricle exceeds pressure in the aorta. If pressure rises in the aorta this means that the work load on the ventricle will increase. The combined effect of factors which determine ventricular wall stress (primarily chamber radius, wall thickness and the resistance to outflow of blood) are called the afterload effects on the heart. Hypertension therefore imposes an increased afterload on the left ventricle as does aortic valve stenosis. The pathological response to increased afterload is cardiac myocyte hypertrophy which may progress to cardiac failure (see Chapter 6).
In a normally functioning heart the acute changes in arterial blood pressure which occur during everyday life have little effect on the stroke volume of the heart unless blood pressure rises to very high levels (Fig. 4.8). Sustained increases in blood pressure however lead to a reduction in stroke volume and this is part of the rationale for antihypertensive therapy. In this circumstance reducing blood pressure will increase stroke volume of the heart. In cardiac failure the performance of the ventricles may be much more sensitive to afterload changes than a normal heart (see Chapter 6).
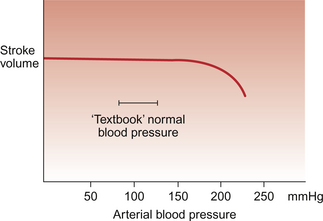
Fig. 4.8 Afterload effects on the normal heart. The stroke volume of the left ventricle remains quite constant over a wide range of arterial blood pressure. Only when blood pressure rises beyond the normal range encountered under physiological conditions does afterload on the heart start to reduce stroke volume. In a failing heart (see Chapter 6) blood pressure increases can have a much more marked adverse effect on stroke volume.
Summary
During exercise, such as running, all the different ways of increasing cardiac output will come into play. The heart rate will rise as a result of increased sympathetic and decreased parasympathetic drive. The movement of the limbs and the increase in ventilation will increase the venous return, and hence ventricular end-diastolic volume, so that the heart will ‘move up a Starling curve’. The increased sympathetic outflow will produce an increase in contractility. In very fit athletes the heart may even shrink during exercise (i.e. the end-diastolic volume decreases) as a result of the large increase in contractility and better emptying. Peripheral resistance falls during dynamic exercise and mean arterial blood pressure changes are small. Afterload effects are not therefore a major factor determining cardiac performance during dynamic exercise. This is not the case in static exercise (see Chapter 13).
Allen, D. G., Kentish, J. C. The cellular basis of the length–tension relation in cardiac muscle. J. Mol. Cell Cardiol.. 1985; 17:821–840.
Brady, A. J. Mechanical properties of isolated cardiac myocytes. Physiol. Rev.. 1991; 71:413–427.
Fuchs, F., Smith, S. S. Calcium, cross-bridges and the Frank–Starling relationship. News Physiol. Sci.. 2001; 16:5–10.
Levick, J. R. An Introduction to Cardiovascular Physiology, fifth ed. London: Arnold; 2009.
McDonald, K. S., Herron, T. J. It takes ‘heart’ to win: What makes the heart powerful? News Physiol. Sci.. 2002; 17:185–190.
Molkentin, J. D., Dorn, G. W. Cytoplasmic signalling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol.. 2001; 63:391–426.
Orchard, C. H., Kentish, J. C. Effects of changes in pH on the contractile function of cardiac muscle. Am. J. Physiol.. 1990; 358:C967–C981.
Waller, D. G., Renwick, A. G., Hillier, K. Medical Pharmacology and Therapeutics, third ed. Edinburgh: Saunders; 2009.
Zimmer, H. -G. Who discovered the Frank–Starling mechanism? News Physiol. Sci.. 2002; 17:181–184.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]4
REGULATION OF CARDIAC FUNCTION
Introduction
When the textbook person (see Chapter 1) is at rest their cardiac output, the volume of blood pumped by each side of the heart, is about 5 L per minute. In understanding how this is achieved the ‘rule of 70’ might be helpful. The heart typically beats at about 70 bpm and, when the ventricle empties it goes from a volume of about 140 mL at the end of the filling phase, diastole, to about 70 mL at the end of the contraction phase, systole. The ‘stroke volume’ is therefore about 70 mL and about 70 mL of blood remains in the ventricle at the end of each cardiac contraction. The ejection fraction of the heart, the stroke volume expressed as a percentage of end-diastolic volume, is thus typically about 50%. When appropriate an increase in the heart’s output can be achieved by a combination of an increase in heart rate and stroke volume. The maximum effective heart rate in the adult is about 180 bpm and the stroke volume can be increased both by filling the ventricle more during diastole and emptying it further during systole. In a very fit textbook person the maximum cardiac output is of the order of 25 L/min.
Venous return
When a person is upright, their head and shoulders are above the level of the heart but most of the rest of the body is below that level and thus gravity is largely a negative factor in determining venous return of blood to the heart. In a horizontal posture however the venous return will not be significantly opposed by gravity. For this reason if a person faints, it is usually best to leave them horizontal to aid the flow of blood back to the heart. Conversely, if a person has poor cardiac function a sitting position will decrease the venous return and hence the work load of the heart. Many people with cardiac problems sleep either in a chair or propped up in bed. The effect of gravity on venous pressure is further discussed in Chapter 10.
The skeletal muscle pump
The veins, especially in the limbs, are often to be found between major blocks of muscle. If muscle activity is increased, then the alternate compression and release of the veins will result in blood being propelled back towards the heart (Fig. 4.1). This mechanism is dependent on the presence of valves in veins in the lower limbs. This aspect of venous return is useful for guardsmen on parade. If they are standing still and upright over long periods of time there is a danger they will faint because their cardiac output, limited by the poor venous return, is not enough to supply their tissue’s needs especially on hot summer days when skin blood flow may be increased. They are told to move their toes in their boots and thus use the alternate contraction and relaxation of their leg muscles to aid venous return.
Control of cardiac output
The respiratory system has ‘dead space’ areas such as the nasal passages, trachea and bronchi which are ventilated but where no gas exchange takes place. This means that during exercise there is a functional advantage in keeping the respiratory rate relatively low and breathing deeply. The heart however is a through flow system and, within limits, an increase in heart rate will produce a proportionate rise in cardiac output, as long as the venous return is increased to provide the necessary blood to pump. This is true in an adult up to a heart rate of about 180 bpm. If the heart rate goes much above this value then cardiac output falls precipitously because there is an encroachment on the ‘rapid filling time’—that period of time which immediately follows the end of ventricular systole and when most of the ventricular filling occurs (see Chapter 3). The result is that excessively high heart rates are associated with a reduced stroke volume. Small minute to minute changes in cardiac output, as might be associated with mild exercise such as walking, tend to be achieved largely by changes in both heart rate and stroke volume (see Chapter 13). Changes in stroke volume reach maximum levels at fairly moderate exercise intensity and the further increases during heavy exercise are achieved just with heart rate changes. The effects of exercise on cardiac function are considered in more detail in Chapter 13.
Regulation of heart rate
There is considerable variation in the resting heart rate in the adult population with a normal range of 50–100 bpm. As explained in Chapter 2 the rate of beating of the heart is normally determined by the pacemaker at the sinoatrial node (SAN). The rate of this pacemaker is modulated by both the sympathetic and parasympathetic nervous systems. At rest the parasympathetic inhibition dominates so that the typical resting heart rate of about 70 bpm is less than the natural intrinsic rate of the SAN which is about 110–120 bpm (see Chapter 2). Small increases in heart rate, such as might occur during walking, are produced by inhibition of the parasympathetic tone. If the heart rate is to go above about 110 bpm, then an increased sympathetic nerve activity is also required.
The role of the central nervous system control centres and the function of the baroreceptor reflex in the control of heart rate are further discussed in Chapter 10.
The sympathetic innervation of the heart is routed via the cervical and stellate sympathetic ganglia. It is the sympathetic fibres on the right side of the body which have the major effect on heart rate (Fig. 4.2). The fibres from the left side of the sympathetic nervous system are more concerned with the regulation of cardiac contractility. The noradrenaline (norepinephrine) released at the nerve endings acts on β1 receptors in the SAN and increases the Ca++ current in phase 4 of the SAN action potential. This accelerates the rate of firing of the SAN cells (see Chapter 2).
The parasympathetic innervation of the pacemaker tissue is via the left and right branches of the vagus nerve but, like the sympathetic innervation, the fibres are distributed differentially (Fig. 4.2). The right vagus goes mainly to the SAN with a small innervation of the atrioventricular node (AVN) whereas the left vagus goes primarily to the AVN with only a small outflow to the SAN. The vagal fibres to the heart originate in the dorsal motor nucleus of the vagus or in the nucleus ambiguus (see Chapter 10). The parasympathetic ganglia, the location of the nerve cell bodies, are on the cardiac surface or within the heart itself and the short postganglionic fibres release acetylcholine as their neurotransmitter. This acetylcholine acts on muscarinic (M2) receptors in the SAN and, via an inhibitory G protein mechanism, reduces the production of cAMP within the cell. Cyclic AMP activates populations of Na+ channels and L-type Ca++ channels causing depolarization of the pacemaker cells. Inhibiting cAMP formation therefore reduces the rate of depolarization and slows the heart rate. There is also a hyperpolarizing effect produced following binding of acetylcholine to M2 receptors. This is mediated by activation of a population of K++ channels. As a result the membrane potential is moved closer to the potassium equilibrium potential (EK) of −94 mV and therefore further away from the threshold at which an action potential is triggered (see Chapter 2). The right and left vagus nerves have little effect on cardiac contractility.
Regulation of stroke volume
The heart is an unusual form of pump in that not only is it able to change the rate at which it pumps but, if necessary, it is also able to change the volume of blood pumped at each stroke. Under resting conditions, the ventricles in the adult heart typically fill to about 140 mL (the end-diastolic volume) and empty to about 70 mL (the end-systolic volume) giving rise to a stroke volume of about 70 mL.However as the normal range of heart rate is quite wide at 50–100 bpm, individuals with a slow resting heart rate will operate with a stroke volume greater than 70 mL and people with a fast heart rate will have a correspondingly smaller resting stroke volume. Resting cardiac output will therefore be comparable between individuals with the same body size irrespective of resting heart rate. An outline of the case history of a man with increased stroke volume in his heart is described in Case 4.1:1.
[/not-level-membership-for-cardiovascular-category]
