Chapter 30 Red blood cell disorders
Iron
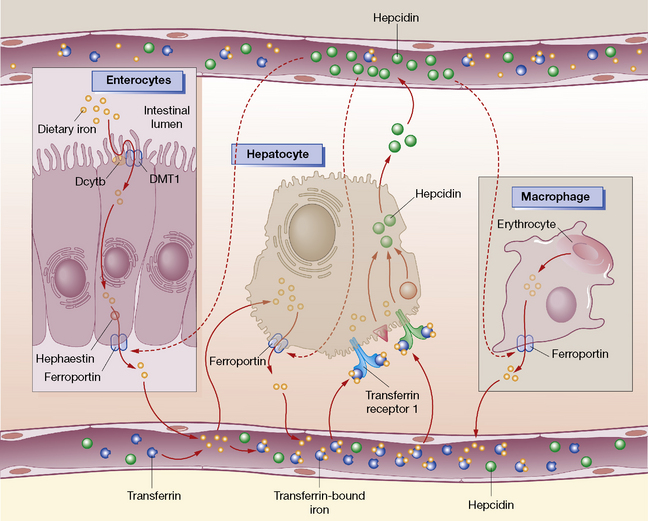
Dietary iron is reduced to the ferrous (Fe2 +) state at the brush border of the duodenum. Divalent metal transport 1 (DMT1), located on the membrane of duodenal enterocytes, then transports iron across the intestinal lumen; this is aided by an acidic environment. Ferroportin, facilitated by hephaestin, then releases iron into the bloodstream. Hepcidin is a key regulator of iron homeostasis and directly inhibits ferroportin release of iron (Fig. 30.1). In the plasma, iron binds to transferrin and delivers it to transferrin receptors on developing erythroblasts in the bone marrow. Alternatively, iron may be stored intracellularly, or excreted in faeces from shed mucosal cells. After 120 days red cells die and the haemoglobin from senescent erythrocytes is released, phagocytosed by macrophages and exported by ferroportin back to plasma transferrin. Excess iron is stored in the bone marrow, hepatocytes and spleen as ferritin (20–300 μg/L).
Iron deficiency
Iron deficiency is the commonest cause of anaemia worldwide. The major causes are:
• Inadequate dietary intake: young infants with inadequate intake of solids (18 months to 3 years), poverty and poor nutrition.
• Increased physiological iron requirements: increased iron demands for growth, i.e. prematurity, rapid growth in adolescence and pregnancy (especially third trimester).
• Reduced iron absorption: coeliac disease, post-gastrectomy and gluten-induced enteropathy.
• Blood loss: menstruation, menorrhagia, gastrointestinal malignancy, other causes of chronic haemorrhage (e.g. associated with salicylate and non-steroidal anti-inflammatory drugs), hookworm infection and chronic intravascular haemolysis.
Management of iron deficiency and prophylactic iron administration
Management of iron deficiency requires:
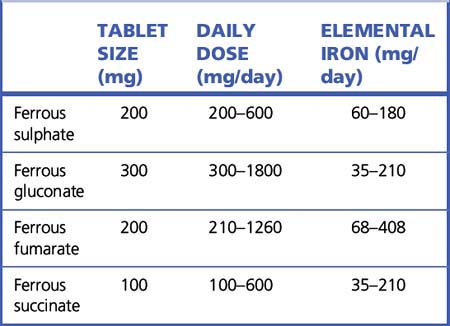
Oral iron preparations are the treatment of choice due to their effectiveness, safety and low cost. Iron can be administered as simple iron salts (e.g. ferrous sulphate, fumarate or gluconate) or in saccharated form (Table 30.1). Administration of 200 mg ferrous sulphate three times daily provides 180 mg elemental iron per day; up to 30% of the orally administered iron will be absorbed. The haemoglobin will increase by 1 g in the first week; a rise of 2 g/dL after 3 weeks’ therapy is evidence of adequate response. Daily administration for 1–3 months will correct anaemia due to iron deficiency. Therapy should be continued for a further 3 months and until the haemoglobin has normalised and iron stores replenished (i.e. serum ferritin > 50 μ/L).
Parenteral iron is rarely indicated and should only be administered if there is:
• Proven iron deficiency and oral iron cannot be tolerated.
• Ongoing blood loss so severe that oral therapy is insufficient to maintain iron stores.
Parenteral iron formulations include:
• Iron dextran (ferric hydroxide complexed with dextrans, 50 mg/mL) which can be administered by deep intramuscular injection, slow intravenous injection or infusion.
• Iron sucrose (ferric hydroxide complexed with sucrose, 20 mg/mL) which is delivered by slow intravenous injection or infusion (not recommended for children).
The dose of parenteral iron is based on body weight and the haemoglobin (Hb) deficit, as follows:
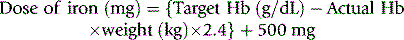
Adverse effects of intravenous iron include immediate, severe and potentially life-threatening anaphylactoid reactions, fever and arthropathy. Patients should therefore be closely monitored during administration and facilities for cardiopulmonary resuscitation should be available. A history of allergic disorders including asthma, eczema and anaphylaxis is a contraindication to parenteral iron. Intramuscular iron can be painful and may permanently stain the skin.1 Less severe manifestations include urticaria, rashes and nausea; delayed reactions such as arthralgia, myalgia and fever can also occur. Intramuscular iron has also been associated with soft tissue sarcomas. Oral iron preparations should not be given for 24 h prior to parenteral therapy or for 5 days after the last intravenous injection. This is to prevent adverse reactions as a result of saturation of transferrin binding capacity leading to a high, unbound plasma iron concentration.
Chronic iron overload
Severe tissue iron overload can result from excessive absorption (hereditary haemochromatosis), frequent or chronic red cell transfusion therapy (> 100 units as in thalassaemia or myelodysplasia2) leading to transfusion haemosiderosis and excessive parenteral iron therapy. Iron chelation is required to prevent irreversible end-organ (e.g. heart, liver) damage. In haemochromatosis iron is removed by weekly venesection (450 mL blood eliminates 200–250 mg iron) until the ferritin has normalised and thereafter, as required, to maintain the ferritin at < 50 μg/L.
Parenteral iron chelator
Desferrioxamine is administered by subcutaneous injection or intravenously (30–50 mg/kg/day) over an 8–12 h period, 5–7 nights per week. It has a half-life of 6 h. Compliance with therapy is a problem because of the slow parenteral administration. Desferrioxamine complexes with ferric iron to form ferrioxamine which is excreted in urine and in bile. Simultaneous administration of ascorbic acid should be avoided; although ascorbic acid increases the availability of free iron for chelation, it also mobilises iron from reticuloendothelial storage sites to a potentially toxic pool in parenchymal cells. Serious adverse effects of desferrioxamine are uncommon but do include anaphylactic reactions. Rapid infusion can result in hypotension, shock or urticaria. There is danger of potentially fatal adult respiratory distress syndrome if infusion proceeds beyond 24 h.3 Chronic use can result in hearing and visual disturbances (cataract and retinal damage).
Iron poisoning and acute overdose
Iron poisoning is commonest in children, is usually accidental and particularly dangerous. The phases in acute oral iron poisoning are shown in Box 30.1. Ferrous sulphate is the most toxic, while sustained-release iron preparations and multivitamins cause less severe poisoning. Poisoning is severe if the plasma iron concentration exceeds the total iron binding capacity (upper limit 75 mmol/L) or plasma becomes pink due to the formation of ferrioxamine. Treatment is urgent and involves chelating iron in plasma. As an immediate measure, raw egg and milk help bind iron in the stomach. Iron chelation therapy is required for severe toxicity. Desferrioxamine is administered by intravenous infusion (not exceeding 15 mg/kg/h and maximum 80 mg/kg in 24 h) to chelate serum free iron.
Vitamin B12
Vitamin B12 deficiency
The causes of vitamin B12 deficiency are listed in Table 30.2. The most common are:
• Inadequate dietary intake: the elderly and vegans.
• Pernicious anaemia. Autoimmune destruction of gastric parietal cells produces atrophic gastric mucosa and reduced secretion of intrinsic factor. Vitamin B12 deficiency results from failure to absorb cobalamin in the terminal ileum.
• Malabsorption syndromes. Intestinal disease affecting the terminal ileum can interrupt the normal enterohepatic circulation of vitamin B12 and result in vitamin B12 deficiency. Malabsorption can also result from poor release of vitamin B12 from food as a consequence of impaired secretion of acid and pepsin by the stomach.
• Drugs. A number of drugs can reduce vitamin B12 absorption, including metformin, aminosalicylic acid, nicotine, phenytoin and large doses of vitamin C.
| Deficiency | Cause | Abnormality |
|---|---|---|
| Vitamin B12 | Inadequate dietary intake | Veganism Breast-fed infants of vegan mothers |
| Reduced vitamin B12 absorption | Pernicious anaemia Achlorhydria Gastrectomy Gastric bypass surgery Ileal disease Malabsorption syndromes Stagnant loop syndrome Tropical sprue |
|
| Drugs | Nitrous oxide (prolonged exposure) Metformin Antacids Aminosalicylic acid Nicotine Phenytoin Zidovudine |
|
| Congenital defects | Transcobalamin deficiency Enzyme defects |
|
| Folate | Inadequate dietary intake | Poor diet: elderly, malnourished, poverty, alcoholics Psychiatrically disturbed patients Infants fed solely on goats’ milk |
| Malabsorption syndromes | Gluten-sensitive enteropathy Tropical sprue Salazopyrine therapy Partial gastrectomy Jejunal resection |
|
| Increased folate requirements | Pregnancy Prematurity Chronic haemolytic anaemia Psoriasis Exfoliative dermatitis Crohn’s disease Dialysis |
|
| Anti-folate drugs | Long-term antiepileptic use (phenytoin, primidone and phenobarbital) Methotrexate Trimethoprim Pyrimethamine |
Vitamin B12 deficiency can result in:
• Subclinical disease: mild anaemia without clinical symptoms or signs.
• Megaloblastic anaemia (macrocytic anaemia with oval macrocytes and hypersegmented neutrophils).
• Subacute combined degeneration of the brain, spinal cord and peripheral nerves.
• Abnormalities of epithelial tissue, particularly the alimentary tract, e.g. sore tongue and malabsorption.
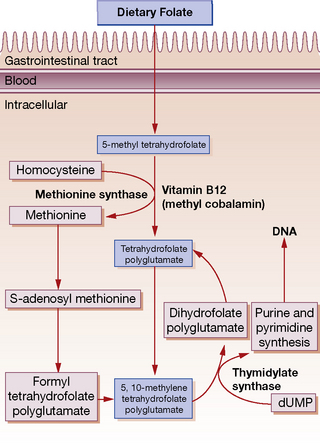
Cobalamin deficiency is diagnosed by measuring total plasma vitamin B12 (normal 150–450 pmol/L); this includes holotranscobalamin and holohaptocorrin. Studies now indicate that measuring holotranscobalamin (normal level 33–91 ng/L), the active portion of vitamin B12, is more sensitive and specific for vitamin B12 deficiency. In vitamin B12 deficiency the serum folate may be elevated and red cell folate reduced due to disturbance of normal absorption and metabolism (Fig. 30.2). Methylmalonic acid levels may be elevated and are relatively specific for vitamin B12 deficiency whereas elevated homocysteine levels are not.
Folic acid (pteroylglutamic acid)
Folic acid4 is one of the B group vitamins and is widely distributed, particularly in green vegetables, fruits, yeast and liver. A normal adult diet contains approximately 400 μg of folic acid (one-third in monoglutamate and two-thirds polyglutamate form) and 100–200 μg is absorbed. Daily requirements are 50–100 μg. Folate polyglutamate is deconjugated to the monoglutamate form prior to absorption in the proximal jejunum. Within the plasma folate is present mainly as 5-methyl tetrahydrofolate. This enters the cell and is demethylated to tetrahydrofolate, a process that requires vitamin B12 (see Fig. 30.2). Total folate content of the body is 6–10 mg, stores which will last for only 3–4 months on a folate-deficient diet. Folate is required for amino acid and DNA synthesis and cell division.
Folic acid deficiency
The most common causes of folic acid deficiency are those listed in Table 30.2.
Management of folic acid deficiency and prophylactic administration
In many countries foods such as flour are fortified to prevent neural tube defects. Assessment of folate status before conception should be considered and folate supplementation (400 μg/day orally) given if required. In women hoping to conceive who have had a previous neural tube defect fetus, 5 mg folate daily is recommended.5,6 Prophylactic folic acid (200–500 μg daily) should be taken throughout pregnancy; this is generally administered together with prophylactic iron.
Haemolytic anaemia
Drug-induced haemolytic anaemia
Many drugs can cause increased red cell destruction by immune or non-immune mechanisms (Table 30.3). Drug-induced immune-mediated haemolytic anaemia is most commonly secondary to penicillins, second- and third-generation cephalosporins (in particular cefotetan and ceftriaxone), quinine, quinidine and α-methyldopa. Other drugs and toxins can cause direct damage to the red cell membrane (e.g. copper, mitomycin C) or induce oxidative haemolysis (e.g. sulfonamides, dapsone). Drug withdrawal is usually sufficient but, if severe, red cell transfusions may be required.
Table 30.3 Some drugs and chemicals that can cause haemolytic anaemia
| Mechanism | Drug, chemical or toxin |
|---|---|
| Immune-mediated haemolytic anaemia | Cephalosporins Chlorpropamide Diclofenac Hydralazine Ibuprofen α-Interferon Isoniazid Mefenamic acid α-Methyldopa Penicillins Probenicid Procainamide Quinine Quinidine Sulfonamides Tetracyclines |
| Oxidative haemolytic anaemia | Aminosalicylic acid Dapsone Methylthioninium chloride (methylene blue) Nitrofurantoin Sulfonamides (e.g. sulfasalazine) Primaquine Pyridium (phenazopyridine) |
| Direct red cell membrane damage | Amphotericin Arsine Chlorate Cisplatin Ciclosporin Lead Mitomycin C |
Glucose-6-phosphate dehydrogenase deficiency
An 18-year-old male presented to a hospital emergency department febrile, dyspnoeic and jaundiced after eating a meal which included fava beans. He was anaemic with a Hb of 8.7 g/dL and the blood film showed ‘bite’ cells.7 A methyl violet stain for Heinz bodies was positive. Bilirubin and lactate dehydrogenase were elevated and his G6PD was reduced (20% activity). A diagnosis of G6PD-deficiency was made and classified as a ‘mild deficiency’. He was managed conservatively and did not require a transfusion. He was advised to avoid foods (broad beans) and drugs (i.e. primaquine, sulfonamides) that cause oxidative haemolysis, and to present early for medical attention at times of infections.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked inherited enzyme defect that causes haemolytic anaemia following an acute febrile illness, hypoxia or intake of drugs, foods or other substances that cause oxidation of haemoglobin (Table 30.4). Diagnosis is made on a blood film which shows ‘bite’ cells, resulting from splenic removal of oxidised haemoglobin, a positive Heinz body test and a G6PD enzyme assay. Treatment is supportive and avoidance of drugs and foods (e.g. fava beans) with oxidant potential.
Table 30.4 Drugs and substances that can cause oxidative haemolysis in G6PD deficiency
| Drug type | Drug name |
|---|---|
| Analgesics | Pyridium (phenazopyridine) Salicylate |
| Anti-bacterials | Dapsone Nalidixic acid Nitrofurantoin |
| Anti-malarials | Primaquine Pamaquine Pentaquine |
| Sulphonamides and sulphones | Sulphamethoxazole (including co-trimoxazole) Sulphasalazine Sulphacetamide Sulphanilamide Dapsone |
| Others | Ascorbic acid Flutemide Methylthioninium chloride (methylene blue) Naphthalene Probenicid Quinine Rasburicase |
Haemoglobinopathies
Sickle cell anaemia
In sickle cell disease, deoxygenated haemoglobin S (HbS) forms polymers resulting in erythrocytes becoming inflexible ‘sickle-shaped’ forms. Sickle red cells can obstruct blood flow and cause the clinical features of sickle cell disease, principally haemolytic anaemia, acute chest syndrome and painful crises. Hydroxycarbamide (hydroxyurea) can be used in sickle cell disease to increase the HbF levels in maturing erythrocytes, decrease HbS polymerisation and erythrocyte sickling, and reduce the frequency and severity of sickling complications. It also increases nitric oxide (promoting vasodilatation), causes a fall in leucocyte count and improves red cellular hydration, all of which result in a reduction in vaso-occlusive events.8 The indications for initiating hydroxycarbamide include frequent acute painful crises and acute chest syndrome. Beneficial effects of hydroxycarbamide have been seen in adults, children and infants, with a reduction in hospital admissions, pain, acute chest syndrome, blood transfusions and mortality. Neurological complications, e.g. stroke, may not be reduced. Long-term daily administration of hydroxycarbamide raises HbF levels to 15–20% (normally < 1% in adults). Hydroxycarbamide is relatively non-toxic, its myelosuppressive effects are reversible and the long-term risk of leukaemogenesis is negligible. There is no adverse effect on growth or development in children and it does not appear to increase the risk of malignancy.
Thalassaemia
Thalassaemias9 vary in their severity from clinically silent to transfusion-dependent with many being of intermediate severity. Severely affected thalassaemia major patients are likely to require regular red blood cell transfusions and iron chelation therapy (see p. 499). Iron chelation is usually started after 1 year of monthly blood transfusions. Patients are monitored regularly to assess growth, endocrine and cardiac function, iron status (serum ferritin, MRI for liver iron concentration) and adverse effects related to therapy. Patients with less severe thalassaemia (thalassaemia intermedia) generally only require intermittent transfusions; the need for iron chelation therapy is based on iron load (ferritin). Hydroxycarbamide can be used in these patients, in the same manner as for sickle cell disease, to increase HbF and total haemoglobin levels. Individuals with thalassaemia minor (i.e. carriers) are typically asymptomatic and do not require therapy.
Haemopoietic growth factors
Haemopoietic growth factors, such as ESA and granulocyte colony-stimulating factor (G-CSF), are produced by recombinant DNA technology. These can be administered to stimulate erythroid and myeloid lineages in the bone marrow and are potentially useful for secondary anaemias (e.g. chronic renal failure) and neutropenia due to disease or chemotherapy.10
Erythropoiesis-stimulating agents
Clinical uses of ESA
Anaemia due to cancer chemotherapy
ESA have been used in the management of chemotherapy-induced anaemia (Hb < 10 g/dL).11 ESA administration can reduce the need for red cell transfusions, improve quality of life and reduce symptoms related to anaemia. There is clinical trial evidence that ESA administration increases tumour progression, tumour recurrence, serious cardiac and thrombotic events and death. These serious safety concerns have restricted the use of ESA in oncology.
Granulocyte colony-stimulating factor
• Haemopoietic stem cell mobilisation into the peripheral blood for autologous or allogeneic transplantation. Blood mobilised progenitor cells are associated with earlier neutrophil and platelet recovery, fewer transfusions and shorter hospitalisation than those from bone marrow.
• To hasten neutrophil recovery following myelosuppressive chemotherapy, after autologous and allogeneic bone marrow transplantation, in aplastic anaemia and AIDS. G-CSF increases the neutrophil count four- to five-fold within hours of administration, shortens the duration of neutropenia and reduces infections in patients who have received cytotoxic myelosuppressive chemotherapy.
• To improve the neutrophil count in myelodysplastic syndromes, and congenital, cyclical and idiopathic neutropenia. G-CSF can reduce the risk of life-threatening infections and prolong survival.
Angelucci E., Barosi G., Camaschella C., et al. Italian Society of Haematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica. 2008;93:741–752.
Bennett C.L., Silver S.M., Djulbegovic B., et al. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anaemia. J. Am. Med. Assoc.. 2008;299:914–924.
Bohlius J., Schmidlin K., Brillant C., et al. Erythropoietin or darbepoetin for patients with cancer – meta-analysis based on individual patient data. Cochrane Database Syst Rev. 8(3), 2009. CD007303
Carmel R. Current concepts in cobalamin deficiency. Annu. Rev. Med.. 2000;51:357–375.
Davies J.K., Guinan E.C. An update on the management of severe idiopathic aplastic anaemia in children. Br. J. Haematol.. 2007;136:549–556.
Fleming R.E., Bacon B.R. Orchestration of iron homeostasis. N. Engl. J. Med.. 2005;352:1741–1744.
Kaushansky K. Lineage-specific haematopoietic growth factors. N. Engl. J. Med.. 2006;354:2034–2045.
Lanzkron S., Strouse J.J., Wilson R., et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann. Intern. Med.. 2008;148:939–955.
Lawler P.R., Filion K.B., Eisenberg M.J. Correcting anaemia heart failure: the efficacy and safety of erythropoiesis-stimulating agents. J. Card. Fail.. 2010;16:649–658.
McMullin M.F., Bareford D., Campbell P., et al. General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br. J. Haematol.. 2005;130:174–195.
Marsh J.C., Ball S.E., Cavenagh J., et al. Guidelines for the diagnosis and management of aplastic anaemia. British Committee for Standards in Haematology. Br. J. Haematol.. 2009;147:43–70.
Mehta A., Mason P.J., Vulliamy T.J. Glucose-6-phosphate dehydrogenase deficiency. Baillieres Best Pract. Res. Clin. Haematol.. 2000;13:21–38.
Ngo K., Kotecha D., Walters J.A. Erythropoiesis-stimulating agents for anaemia in chronic heart failure. Cochrane Database Syst. Rev. 8(3), 2009. CD007613
Rund D., Rachmilewitz E. β-Thalassemia. N. Engl. J. Med.. 2005;353:1135–1146.
Scheinberg P., Wu C.O., Nunez O., et al. Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br. J. Haematol.. 2009;144:206–216.
Vichinsky E. Clinical application of deferasirox: practical patient management. Am. J. Hematol.. 2008;83:398–402.
Wickramasinghe S.N. Diagnosis of megaloblastic anaemias. Blood Rev.. 2006;20:299–318.
1 Staining can be minimised by inserting the needle through the skin and then moving the subcutaneous tissue laterally before entering muscle, so that the needle track is disrupted when the needle is withdrawn (Z technique).
2 A 26-year-old subject with β-thalassaemia major had been transfused with 404 units of blood over his lifetime. His iron stores were so high (estimated at above 100 g) that he triggered a metal detector at an airport security checkpoint (Jim R T S 1979 Lancet ii:1028 [letter]).
3 Tenenbein M, Kowalski S, Sienko A et al 1992 Lancet 339:699–701.
5 Hernández-Díaz S et al 1991 Lancet 338:131–137.
6 A supplement of folic acid 5 mg/day is proposed for fuller risk reduction (Wald N J, Law M R, Morris J K, Walk D S 2001 Quantifying the effect of folic acid. Lancet 358:2069–2073).
7 The profile of some red cells carries a concave defect (like a bite), an appearance that is indicative of haemolytic anaemia.
8 Steinberg M H, Barton F, Castro O et al 2003 Effect of hydroxyurea on mortality and morbidity in adult sickle cell anaemia: risks and benefits up to 9 years of treatment. Journal of the American Medical Association 289:1645–1651.
9 The genetic defect results in reduced rate of synthesis or absent synthesis of one of the globin chains that make up haemoglobin.
10 Ozer H, Armitage J O, Bennett C L 2000 Update of recommendations for the use of haematopoietic colony-stimulating factors: evidence-based, clinical practice guidelines. Journal of Clinical Oncology 18:3558–3585.
11 Rizzo J D, Lichtin A E, Woolf S H et al 2002 Use of epoetin in patients with cancer: evidence-based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Haematology. Blood 100:2303–2320.