Reciprocity of Cardiac Sodium and Potassium Channels in the Control of Excitability and Arrhythmias
Intermolecular Interactions Involving NaV1.5 and Kir2.x Channels
Reciprocal Regulation of NaV1.5 and Kir2.1 in Adult Rat Ventricular Myocytes
SAP97 and Syntrophin Are Involved in NaV1.5-Kir2.1 Interactions
NaV1.5 -Kir2.1 Interactions Are Posttranslational and Model-Independent
NaV1.5-Kir2.1 Interactions Involve Membrane Trafficking
Reciprocal NaV1.5 -Kir2.1 Interactions Control Reentry Frequency
Current understanding of the relationship between the sodium current (INa) and the inward rectifier potassium current (IK1), the two most important ionic currents controlling ventricular excitability, derives primarily from traditional electrophysiology. It derives also from basic and clinical studies on arrhythmogenesis in ion channel diseases and heart failure, which have demonstrated that modification in the peak density of either INa or IK1 changes cell excitability and conduction velocity (CV). However, the pathophysiologic consequences of a molecular interplay between the individual channels at the center of such diseases have not been investigated. In the heart, IK1 is the major current responsible for the maintenance of the resting membrane potential (RMP), whereas INa provides the largest fraction of the inward depolarizing current that flows during an action potential.1 It is well known that by controlling the RMP, IK1 modifies Na+ channel availability and therefore, cell excitability.2 In addition, IK1-INa interactions are important in stabilizing and controlling the frequency of the electrical rotors that are responsible for the most dangerous cardiac arrhythmias, including ventricular tachycardia (VT) and ventricular fibrillation (VF).3,4
Recent data obtained from adult transgenic mice, single adult rat ventricular myocytes (ARVMs), neonatal rat ventricular myocyte (NRVM) monolayers, and human embryonic kidney (HEK 293) cells, have demonstrated that the INa-IK1 interplay is much more complex than previously thought. It comprises a model independent, reciprocal modulation of expression of their respective channel proteins (Nav1.5 and Kir2.1) within a macromolecular complex that involves the MAGUK-type protein SAP97,5 and possibly additional scaffolding proteins. In adult transgenic mice overexpressing Kir2.1 (Kir2.1 OE), peak INa density is twice as large as that measured in cells from control hearts. In heterozygous Kir2.1 knockout mice (Kir2.1-/+), NaV1.5 protein and INa are significantly reduced. Similarly, in single ARVMs, IK1 increased significantly more upon adenoviral transfer of Kir2.1 plus NaV1.5 than when Kir2.1 was overexpressed alone. In NRVM monolayers, co-overexpression of NaV1.5 with Kir2.1 increased CV, abbreviated action potential duration (APD) and increased rotor frequency beyond those produced by Kir2.1 OE alone.5 Furthermore, recent data in the literature suggest that conditions that result in Nav1.5 protein reduction, such as that which occurs in dystrophin-deficient mdx5cv mice, are accompanied by a concomitant reduction in Kir2.1 protein levels.6 Importantly, the finding that coexpression of NaV1.5 can reduce internalization of Kir2.1 is a central mechanistic observation.5 The purpose of this chapter is to discuss those results in the context of cardiac excitability and mechanisms of reentrant arrhythmias. It will be shown that sodium and potassium channel interactions depend on more than membrane voltage alone. Altogether, the evidence that will be discussed suggests that cardiac cells undergo model-independent co-regulation involving post-translational mechanisms of Kir2.1 and NaV1.5, with important functional consequences for myocardial excitation, impulse velocity, and arrhythmogenesis. Moreover, the evidence suggests that similar interactions might apply to other sarcolemmal ion channels as well, which could themselves have unique effects on myocardial function.
Sodium Channels and Cardiac Excitation
From the clinical standpoint, multiple mutations in the SCN5A gene coding for NaV1.5 have been identified in association with the long QT syndrome (LQTS), Brugada syndrome, idiopathic ventricular fibrillation, cardiac conduction defects, and dilated cardiomyopathy associated with atrial fibrillation.7 Such mutations illustrate the pathophysiologic importance of these channels.
Homozygous knockout (KO) Scn5a-/- mouse embryos die during mid-gestation, most likely because of severe defects in ventricular morphogenesis,8 which provides evidence for an essential role of Nav1.5 in cardiac development.7 However, heterozygous KO mice (Scn5a+/−) show normal survival but exhibit slow atrial, atrioventricular (AV) and intraventricular conduction, prolonged ventricular refractoriness, and enhanced ventricular arrhythmia inducibility.7,8 Ventricular myocytes isolated from adult Scn5a+/− mice demonstrate an approximately 50% reduction in sodium conductance.8 However, an important question that has never been addressed, is whether, as has been demonstrated in other model systems, changes in Nav1.5 expression alter the level of functional expression of other membrane ion channels, particularly Kir2.1.5 Based on recent results,5 it would be reasonable to expect a significant reduction in Kir2.1 (and its respective ionic current, IK1) and in the RMP of adult ventricular cardiomyocytes as a consequence of one SCN5A allele being absent. Should that be the case, one would predict that IK1, the RMP and excitability would be rescued to normal levels by virally mediated gene transfer of Nav1.5 into adult cardiomyocytes isolated from these mice.
In a recent study using a transgenic mouse line overexpressing the human SCN5A gene,9 levels for the cardiac sodium mRNA transcript (Scn5a) and protein (Nav1.5) were each increased by approximately tenfold. However, no abnormalities were found in the electrophysiology of the ventricles. The QRS duration and the corrected QT interval (QTc) in SCN5A overexpressing mice were indistinguishable from their nontransgenic littermates. In addition, no ventricular arrhythmias were detected in the transgenic mice.9 The sodium current densities and APDs from transgenic ventricular cardiomyocytes were nearly identical to those of nontransgenic cells. Equally striking were the similarities between transgenic and normal hearts with respect to the sodium current densities and APDs found in atrial cells. However, baseline ECG recordings by telemetry revealed a shortened PR interval and P wave duration in the transgenic mice compared to their littermate controls, which the authors interpreted as being the result of altered AV nodal conduction.9 However, the possibility that His-Purkinje conduction was enhanced and thus caused the PR shortening was not considered. Nevertheless, it was clear that overexpression of SCN5A did not significantly increase the surface density of sodium channels in ventricular or atrial myocytes.9 This finding is different from what was found in the cultured neonatal rat myocyte. In these cells, INa is inherently relatively small, but viral transfer (and therefore overexpression) of Nav1.5 is accompanied by a significant increase in Nav1.5 and INa, excitability, and CV. As suggested by Abriel,7 one possible explanation to account for the disparate results of the two studies may be that the sodium channels are binding in a macromolecular complex. Based on this premise, and the assumption that there are a limited number of docking sites or complexes to which the sodium channel binds in the adult heart, then this complex (i.e., including its localization, stoichiometry, available components) could provide an upper limit as to how many functional sodium channels can be brought to the membrane in the adult mouse myocyte.7 However, such an idea might not be tenable in light of recent experiments demonstrating that transgenic overexpression of Kir2.1 increases INa density in the adult mouse heart (discussed later).
The Inward Rectifying Potassium Current (IK1)
Among the three strong inward-rectifying potassium channels (Kir2.1, 2.2 and 2.3) that are expressed in the heart, Kir2.1 is the most abundant in the ventricles. Kir channels are responsible for IK1, and they are involved in the depolarization, repolarization, and the resting phases of the cardiac action potential.10,11 It is usually accepted that, near the resting potential, the ventricular IK1 conductance is much larger than that of any other current, with the exception of the adenosine triphosphate (ATP)-sensitive potassium current (IKATP), which, however, is generally not present since the KATP channels are not open under normal conditions. It is thus likely that physiological and/or pathophysiological modulation of IK1 will have a significant effect on excitability. The Kir channels show strong rectification between –50 and 0 mV, which means that they remain closed during the AP plateau; they only open when the membrane potential repolarizes to levels between –30 and –80 mV, which occurs during the late phases of the action potential.12 Rectification is achieved by a voltage-dependent blockade by intracellular magnesium and/or one of the polyamines (putrescine, spermine, spermidine),12 which are known to interact with at least three amino acid residues located inside the pore of the channel complex.13 Investigators have used several strategies to modify and study Kir2.1, including a knockout mouse,14 antisense oligonucleotide targeting,15 and DNA transfection of a dominant-negative construct.16 These studies have helped to define the role of IK1 in cardiac excitability.17,18 As shown by Zartizky et al., ventricular myocytes from Kir2.1 knockout (Kir2.1−/−) mice lack IK1 in whole-cell recordings under physiologic conditions, which demonstrated that Kir2.1 is the major determinant of IK1.14 In that model, sustained outward K+ currents and Ba2+ currents through L- and T-type channels were not significantly altered by the mutation. However, the direct consequences of Kir2.1 disruption on Nav1.5 function were never studied in the homozygous Kir2.1 KO mice. Recently, the authors took advantage of the availability of the heterozygous Kir2.1 KO (Kir2.1−/+) mouse model to study the functional consequences of reducing Kir2.1, and therefore IK1, at both cellular and organismal levels. In particular, as discussed in detail below, the Kir2.1−/+ mouse was used to address the question of whether reduced Kir2.1 protein is associated with reduced expression of Nav1.5.5
Loss of function mutations in the KCNJ2 gene, which codes for Kir2.1, have been identified in patients affected by Andersen-Tawil syndrome (ATS), also known as LQTS type 7, which is characterized by prolonged repolarization.19 In addition to being expressed in the heart, Kir2.1 is expressed in other organs, such as skeletal muscle. As a result, ATS is associated with hypokalemic periodic paralysis and skeletal developmental abnormalities, including clinodactyly, low-set ears, mandibular hypoplasia, short stature, and scoliosis.19 In the heart, reduction of IK1 leads to QT prolongation and predisposes to arrhythmias; however, QT prolongation is less prominent in patients with ATS than in those with other types of LQTS.20 Moreover, although ATS patients develop ventricular tachyarrhythmias, including torsades de pointes, sudden cardiac death is rare.20
In patients with short QT syndrome type 3, a gain-of-function mutation (D172N) in the KCNJ2 gene was demonstrated.21 The D172N mutation causes a significant increase in the outward component of the I-V relation of IK1, and it is associated with an accelerated repolarization, which can be arrhythmogenic. However, the direct involvement of IK1 in arrhythmia mechanisms was not demonstrated in the affected patients; therefore, the mouse model of IK1 overexpression22 was used to study the effect of IK1 increase on VF at the molecular level.23 An increase of IK1 was shown to shorten repolarization and the QT interval and to exert a proarrhythmic effect in both the atria and the ventricles of this transgenic mouse model.22,23 Optical mapping and numerical studies in these mice demonstrated that, by increasing the RMP, IK1 overexpression enhances the availability of sodium channels during sustained reentry, which contributed to the observed increase in the frequency and stability of rotors and ventricular fibrillation.23 Recent experiments with the same Kir2.1 overexpressing mouse model have shown that Kir2.1, and therefore IK1, upregulation leads to a significant increase in the density of INa. These results strongly support the hypothesis that a change in the functional expression of Nav1.5 could be the result of protein-protein–type interactions with Kir2.1. Such interactions can be mediated through common partners in a macromolecular protein complex.5
Intermolecular Interactions Involving Nav1.5 and Kir2.x Channels
PDZ domain-mediated interactions are among the most frequently encountered protein-protein interactions in cell biology.24 PDZ stands for postsynaptic density protein (PSD-95), discs-large (the drosophila septate junction protein), and zona occludens-1 (the epithelial tight junction protein).25 The primary function of PDZ domains is to mediate protein-protein interactions by recognizing a consensus sequence (Thr/Ser-X-Val/Leu) usually located at the C-terminus of their target proteins.26–28 PDZs can combine with other interaction modules (such as WW, SH3, and PTB domains) and help to direct the specificity of receptor tyrosine kinases, establish and maintain cell polarity, direct protein trafficking, and coordinate synaptic signaling.29–32 Their pathophysiologic importance is highlighted by significant neuronal and developmental defects observed in PDZ knockout mice33–37 and by their implication in human inherited diseases.38–40
More than 70 PDZ domain–containing proteins (hereafter referred to as PDZ domain proteins) have been identified that interact with different ion channels, receptors, and signaling molecules.29 PDZ domain proteins are multidomain proteins that serve to link different proteins to form macromolecular complexes via interactions with their various domains. For example, the protein structure of synapse-associated protein 97 (SAP97) contains three PDZ domains and an SH3 domain, HOOK domain, I3 domain, and an inactive guanylate kinase (GK) domain. Interactions via its HOOK domain enable SAP97 to bind protein 4.1; and in so doing, SAP97 is able to link proteins bound to its PDZ domains to the actin–spectrin membrane cytoskeleton or to protein components of the actin–spectrin membrane cytoskeleton, such as protein 4.1.41 The complement of interacting proteins varies among the different PDZ domain proteins, and this provides a mechanism to recruit ion channel proteins into distinct macromolecular complexes depending on which scaffolding proteins they bind (e.g., see Tiffany et al.42). The cellular localization of PDZ domain proteins can also vary, and it has been suggested that ion channel–PDZ domain protein interactions might be an important mechanism for plasma membrane expression and distribution of ion channels.42–44 For example, ZO-1 and SAP97 are found in overlapping but distinct subcellular locations in the heart. ZO-1 is located exclusively in the intercalated disc, whereas SAP97 is found in the intercalated disc, lateral membranes,44–46 and at the T-tubules.5
Ankyrin-G and Nav1.5 Trafficking
A number of accessory proteins have been shown to interact and form a multiprotein complex with Nav1.5.7,47 Ankyrin-G and Nav1.5 are both localized at the intercalated disc and at T-tubule membranes in cardiomyocytes, and Nav1.5 coimmunoprecipitates with the 190-kDa ankyrin-G in detergent-soluble lysates from rat heart.47 The two proteins interact through a 9-amino acid motif in the Nav1.5 cytoplasmic loop II between DII and DIII, which helps to promote the localization of sodium channels to the cell membrane in cardiomyocytes.47 In 2004, Mohler et al.47 identified a point mutation (E1053K) in the ankyrin-binding motif of Nav1.5 that is associated with Brugada syndrome. The E1053K mutation abolishes binding of Nav1.5 to ankyrin-G and prevents the accumulation of Nav1.5 at cell surface sites in ventricular cardiomyocytes.47 Those data suggested that association of Nav1.5 with ankyrin-G is required for Nav1.5 localization, and therefore function, at excitable membranes in cardiomyocytes. Concerted ankyrin-G interaction with potassium (Kv7) and Nav channels has also been demonstrated in neurons,48 but Kir2.1 has not been shown to interact with ankyrin-G. There is still the possibility that a defect in ankyrin-mediated Nav1.5 trafficking and localization on the membrane could alter Kir2.1 functional expression. This idea has not yet been explored in any detail.
Nav1.5 Is Regulated by SAP97
The C-terminus of Nav1.5 contains 244 amino acid residues that have been shown to interact with a number of proteins, including fibroblast growth factor-homologous factor 1B, calmodulin, Nedd4-like ubiquitin-protein ligase, dystrophin and syntrophin, Fyn, and PTPH1 (protein-tyrosine phosphatase).49–51 Of importance, the three last three amino-acids (Ser-Ilc-Val, SIV) of the Nav1.5 C-terminus constitute a PDZ-domain binding motif that is known to interact with PDZ domains found in proteins of the membrane-associated guanylate kinase (MAGUK) family.52 MAGUK proteins act as scaffolding proteins involved in intermolecular interactions and protein trafficking to the cell membrane. Petitprez et al.53 reported their study on the interaction between SAP97, one of the cardiac MAGUK proteins, and Nav1.5, with the idea of demonstrating that the Nav1.5-SAP97 interaction is involved in the turnover and regulation of the biophysical properties of Nav1.5. Using Nav1.5 C-terminal fusion proteins in pull-down experiments with human and mouse heart protein extracts, these investigators demonstrated that the association between SAP97 and Nav1.5 depended on the PDZ-domain binding motif of Nav1.5. The interaction appeared to be specific for SAP97 and Nav1.5, because the Nav1.5 constructs did not pull down either PSD95 or ZO-1, two other MAGUK proteins that are expressed in the human heart.54 In patch-clamp experiments, Petitprez et al.53 further demonstrated that silencing SAP97 expression reduced the whole-cell sodium current without decreasing the total protein content in HEK293 cells stably expressing Nav1.5 channels. Taken together, with the demonstrated colocalization of NaV1.5 and SAP97 at both intercalated discs53 and T-tubules,5 these findings support the existence of an interaction between Nav1.5 and SAP97 in cardiac tissue. This interaction could have a role in determining the channel density at the plasma membrane. Therefore, Petitprez et al.53 have suggested that there are at least two pools of Nav1.5 channels in cardiomyocytes: one targeted to lateral membranes by the syntrophin-dystrophin complex, and another targeted to the intercalated discs by SAP97.
Nav1.5 Interacts with Syntrophins
Like other ion channels, Nav1.5 has been reported to be part of the dystrophin multiprotein complex.55 Gavillet et al.6 demonstrated that Nav1.5 interacts with dystrophin via syntrophin adaptor proteins through the PDZ domain-binding motif at the Nav1.5 C-terminus.50 Dystrophin-deficient mice (mdx5cv) have reduced protein levels of Nav1.5 in ventricular lysates, and this is directly associated with reduced INa in isolated cardiomyocytes and with conduction defects documented on an electrocardiogram. Immunostaining of frozen mouse heart slices demonstrated colocalization of Nav1.5 and dystrophin specifically at lateral membranes, but not at the intercalated discs.6 However, mechanisms by which a lack of dystrophin reduces Nav1.5 protein expression without altering the mRNA level6 have not been elucidated. A study by Ueda et al.56 demonstrated that α1-syntrophin (encoded by the SNTA1 gene), connects Nav1.5 to the neuronal nitric oxide synthase (nNOS) – plasma membrane Ca-ATPase (PMCA) complex in the heart. These results implicated SNTA1 as a susceptibility gene for inherited LQTS. The cardiac isoform of PMCA, PMCA4b, participates in the nNOS complex to inhibit nitric oxide synthesis.57 Furthermore, the study by Ueda et al.56 also identified a rare missense mutation (A390V-SNTA1) in α1-syntrophin that was found in an LQTS patient. The A390V mutation selectively disrupted association of α1-syntrophin with PMCA4b, increased Nav1.5 nitrosylation, and increased late sodium current, which all suggested that the SNTA1 mutation was pathogenic.
Kir2.1 Interacts with Both SAP97 and α-Syntrophin
Members of the Kir2.x subfamily (Kir2.1, Kir2.2 and Kir2.3) each have a C-terminal motif that enables interaction with PDZ domain proteins. However, the C-terminal motifs are not identical between the isoforms. RRESEI is found in Kir2.1 and Kir2.2, whereas RRESAI is found in Kir2.3.58 It is noteworthy that the PDZ binding motif sequence on Kir2 overlaps a consensus sequence (RRxS) for protein kinase A–dependent phosphorylation. Consistent with this finding, Kir2.3 binding to PDZ domain proteins is modulated in vitro by phosphorylation of Kir2.3 protein.59
A number of PDZ domain-containing proteins have already been shown to bind Kir2 channels. In two recent studies,60, 61 affinity purification was used to isolate PDZ domain proteins from cardiac and brain tissue extracts. Identification of these isolated proteins demonstrated that cardiac SAP97, CASK, Veli3, and Mint1 are found in a complex with Kir2.2 channels. For the most part, the consequences (e.g., channel expression and biophysics of the interactions between Kir2.x channels with the PDZ domain proteins) are unknown. An association of PDZ domain proteins with many other ion channels or receptors has been shown to affect various aspects of protein trafficking in the secretory and endocytic pathways. In general, PDZ domain–mediated protein-protein interactions can alter the rate of channel/receptor trafficking to the plasma membrane or alter the rate at which a given cell surface channel and/or receptor is endocytosed.62–64 It has also been suggested that binding to PDZ domain proteins is important for anchoring the Kir2 channels at the plasma membrane46; however, until recently, this hypothesis had not been tested directly, with the exception of the interaction of PSD-95 (a neuronal scaffolding protein) and Kir2.1 (discussed later).65
In 2004, Leonoudakis et al.61 used a proteomics approach to identify proteins associated with Kir2 channels via the channel’s C-terminal PDZ binding motif. Based on immunoaffinity purification and affinity chromatography from skeletal and cardiac muscle and brain, they demonstrated that, in addition to MAGUK-type proteins, Kir2 channels interact with various components of the dystrophin-associated protein complex, including α1-, β1-, and β2-syntrophin, dystrophin, and dystrobrevin. Their affinity pull-down experiments revealed that Kir2.1, Kir2.2, Kir2.3, and Kir4.1 all bind to scaffolding proteins but with different affinities for the dystrophin-associated protein complex, including dystrophin and dystrobrevin,60 as well as the MAGUK proteins such as SAP97, CASK, and Veli. Moreover, their immunofluorescent localization studies demonstrated that Kir2.2 colocalizes with syntrophin, dystrophin, and dystrobrevin at skeletal muscle neuromuscular junctions. Overall, the results of Leonoudakis et al.60,61 suggested that Kir2 channels associate with protein complexes that might be important to target and traffic channels to specific subcellular locales, as well as to anchor and stabilize channels in the plasma membrane.
Reciprocal Regulation of NaV1.5 and Kir2.1 in Adult Rat Ventricular Myocytes (ARVMs)
Voltage-clamp experiments presented in Figure 21-1 show a reciprocal regulation of Nav1.5 and Kir2.1 in adult rat ventricular myocytes (ARVMs). Nav1.5 was overexpressed using the adenoviral (Ad) construct (Ad-Nav1.5)5; a control group of myocytes was infected with an adenoviral construct encoding green fluorescence protein (Ad-GFP). Average INa-density–voltage (I-V) plots are shown in Figure 21-1, A. There was a significant increase (p < 0.005) in INa density compared with control cells. There was no significant difference for V1/2 values between control and Ad-Nav1.5–treated cells.5 Overexpression of Nav1.5 also resulted in a significant increase in IK1 density in both the inward (p < 0.01) and outward (p < 0.05) directions (see Figure 21-1, B). Next, the possibility was considered that Kir2.1 overexpression would increase Nav1.5 functional expression.5 Infection with Ad-Kir2.1 resulted in an increase of IK1 in ARVMs (Figure 21-1, C). Peak inward (–100 mV) and outward (–60 mV) currents were significantly larger (p < 0.01) than in control (Ad-GFP) cells. As hypothesized, peak INa density was also increased (p < 0.005; Figure 21-1, D) with no changes in voltage dependence of activation or inactivation.5 These results suggest that common molecular mechanisms are involved in the regulation of Kir2.1 and Nav1.5 functional expression.5
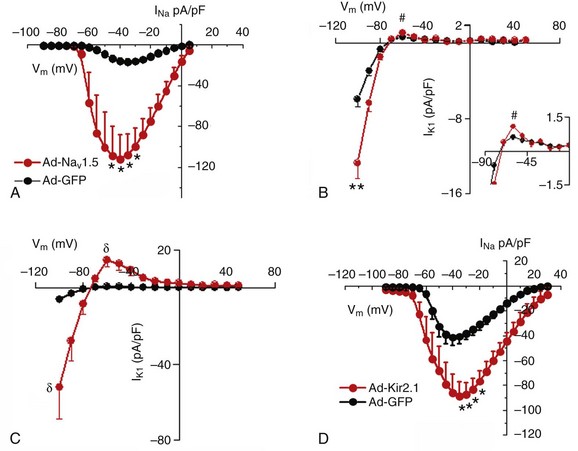
Figure 21-1 Reciprocal regulation of NaV1.5 and Kir2.1 in adult rat ventricular myocytes (ARVMs). A and B, NaV1.5 overexpression increases both INa and IK1 densities. A, Superimposed INa density–voltage relationships (5 mmol/L [Na+]o) for Ad-GFP (black; N = 1, n = 7) and Ad-NaV1.5 (red; N = 2, n = 6) infected cells. B, Superimposed IK1 density–voltage relationship for Ad-GFP (black; N = 3, n = 8) and Ad-NaV1.5 (red; N = 3, n = 8) infected cells. The inset shows the magnification of the outward component of the IK1 I-V relationship. C and D, Kir2.1 overexpression increases both IK1 and INa densities. C, Superimposed IK1 density–voltage relationships for Ad-GFP (black; N = 3, n = 8) and Ad-Kir2.1 (red; N = 2, n = 5). D, Superimposed INa density–voltage relationships (20 mmol/L [Na+]0) for Ad-GFP (black; N = 2, n = 6) and Ad-Kir2.1 (red; N = 4, n = 7). #p < 0.05; δp < 0.01; *p < 0.005 unpaired t test with Welch’s correction. (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
SAP97 and Syntrophin Are Involved in NaV1.5-Kir2.1 Interactions
SAP97 is able to link proteins bound to its PDZ domains, and because both Nav1.5 and Kir2.1 have PDZ binding domains at their respective C-terminus, the potential role of SAP97 in the reciprocal INa-IK1 interactions, discussed in the previous section, was examined. As described elsewhere in detail,5 adenoviral transfer was used to knock down SAP97 expression (Ad-shSAP97) in ARVMs, and then properties of IK1 and INa were studied under these conditions. As illustrated in Figure 21-2, an approximate 56% reduction in the relative levels of SAP97 (day 3 postinfection) (see Figure 21-2, B) was accompanied by an approximately 50% reduction in IK1 density compared with control cells (see Figure 21-2, C). Similarly, silencing SAP97 expression also reduced whole-cell INa density at several tested voltages (see Figure 21-2, D). Peak inward INa density was reduced by approximately 60% compared with the control cells (p < 0.005).
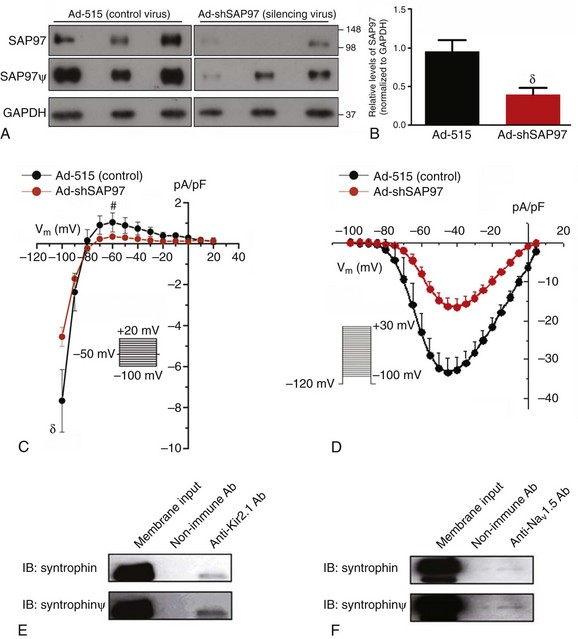
Figure 21-2 SAP97 and syntrophin are involved in NaV1.5-Kir2.1 interactions. A–D, Role of SAP97 in the reciprocal modulation of IK1 and INa. A–B, Ad-shSAP97 infection silences SAP97 expression in adult rat myocytes by day 3 in culture compared with control infected cells (Ad-515). A, Immunoblot showing immunodetection of SAP97 expression (top and middle) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (loading control; bottom) from cells infected with either a control (Ad-515) or shSAP97 (Ad-shSAP97) adenovirus. Middle (SAP97) shows a longer exposure of the same immunoblot shown in the top (denoted by ψ). The overexposure confirms expression of SAP97 in the Ad-SAP97–silenced cells and shows the contrasting intensities and apparent protein levels between control and SAP97-silenced ARVMs. B, Densitometric analysis of SAP97 expression normalized to GAPDH levels in control (black) and shSAP97-silenced (red) cells. Values represent data (mean ± SEM) from two different preparations harvested 3 days after infection. SAP97 expression was effectively knocked down by approximately 56% on day 3. C, IK1 density is reduced following SAP97 silencing in adult rat ventricular myocytes. Peak inward current density at –100 mV (control, –7.67 ± 1.52 pA/pF; Ad-shSAP97, –4.55 ± 0.47 pA/pF) and peak outward current density at –60 mV (control, 1.03 ± 0.46 pA/pF; ad-shSAP97, 0.34 ± 0.03 pA/pF) were significantly different (p = 0.02 and p = 0.04, respectively) between myocytes infected with shSAP97 (N = 4, n = 11) and those infected with Ad-515 (control; N = 2, n = 6). The inset shows the protocol used to measure the current. D, Effects of SAP97 knockdown on sodium channel current density in adult rat ventricular myocytes. Superimposed INa density–voltage relationships in control (Ad-515) and SAP97-silenced (Ad-shSAP97) cells. A significant reduction in peak INa was observed for SAP97-silenced cells at several tested voltages. For both control and silenced conditions, N = 2 and n = 11. The inset shows the voltage clamp step protocol. #p < 0.05; δp < 0.01; *p < 0.005 unpaired t test with Welch’s correction. E and F, Kir2.1 and NaV1.5 each associate with syntrophin in rat heart ventricle. Syntrophin is detected following immunoprecipitation with specific antibodies raised to Kir2.1 (E) or NaV1.5 (F). Coimmunoprecipitation reactions used membrane-enriched preparations generated from the ventricles of rat heart. (ψ images were digitally enhanced for clarity). IB, Antibody used for immunoblotting; IP, antibody used for immunoprecipitation. (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
An investigation of whether Kir2.1 and Nav1.5 also associate with syntrophin is demonstrated in Figure 21-2, E, F. Syntrophin was detected in ventricular membrane fractions following immunoprecipitation with antibodies raised to Kir2.1 (see Figure 21-2, E) or to Nav1.5 (see Figure 21-2, F).
NaV1.5-Kir2.1 Interactions Are Posttranslational and Model Independent
The availability of a transgenic mouse model overexpressing Kir2.123 gave us the opportunity to ensure that the interactions were not an artifact of adenoviral infection and to discard any species-related differences in the interactions. Therefore, additional patch-clamp experiments were conducted in ventricular myocytes isolated from these adult transgenic mice, which are known to have approximately twelvefold greater IK1 density than littermate wild type (WT) mice.66 A comparison of INa density in WT and Kir2.1 overexpressing (OE) mice is shown in Figure 21-3. Representative INa tracings from each genotype are presented in the left inset (see Figure 21-3, A). Compared with WT, the I-V relationship for INa in myocytes from Kir2.1 OE mice shows significantly larger INa density at several tested voltages (see Figure 21-3, A, right inset; p < 0.005). Consistent with the functional consequences observed with Kir2.1 overexpression (see Figure 21-3, A), increased Kir2.1 expression in the mouse heart leads to altered membrane protein expression of SAP97, syntrophin, and Nav1.5 in the transgenic Kir2.1 OE mouse heart (see Figure 21-3, B, C). Relative levels of SAP97, syntrophin, and Nav1.5 were significantly increased by factors of 1.6, 1.9, and 2.2 respectively, compared with levels in WT littermates (p < 0.01; see Figure 21-3, B, C). These data demonstrate that reciprocal regulation between Kir2.1 and Nav1.5 (and their respective currents) also occurs in the mouse heart, and is thus not model specific or unique to ARVMs. It also provides assurance that the observed reciprocal regulation in rat myocytes was not a virally mediated artifact of overexpression. However, real-time polymerase chain reaction (RT-PCR) analyses showed that although the gene encoding Kir2.1 (KCNJ2) was significantly elevated in the transgenic, Kir2.1 overexpressing mice, mRNA levels for the genes coding for SAP97 (DLG1), NaV1.5 (SCN5A), and syntrophin (SNTA1) were unchanged in the hearts of transgenic Kir2.1 overexpressing mice, compared with WT littermates (Figure 21-4, A).
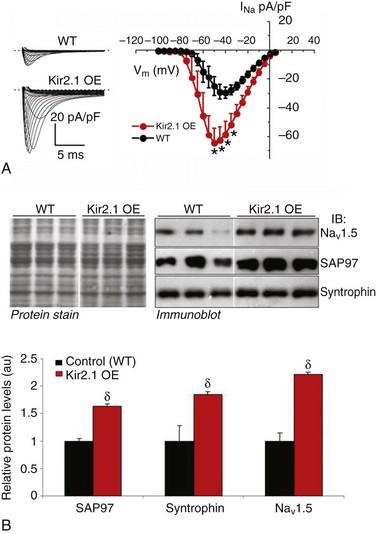
Figure 21-3 Reciprocal regulation of INa and NaV1.5 in Kir2.1 overexpressing mice. A, Superimposed INa density–voltage relationships in wild type (WT; black) and Kir2.1-overexpressing (OE, red) mice. The left inset shows representative examples of INa traces in each group. The dotted line denotes 0 pA. (WT N = 4, n = 10; Kir2.1 OE N = 2, n = 6. *p < 0.005, unpaired t test with Welch’s correction. B, Relative levels of syntrophin, SAP97, and NaV1.5 are significantly increased in hearts of transgenic mice overexpressing Kir2.1. Crude membrane vesicles were prepared from the ventricles of control (WT) and Kir2.1-OE mice. Samples (16 µg/lane) were analyzed with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted using specific antibodies for syntrophin, SAP97, or NaV1.5, as indicated. B, Representative immunoblots following detection of protein immunoreactivity with horseradish peroxidase–conjugated secondary antibodies and chemiluminescence (right). The corresponding Amido black nitrocellulose (protein stain) is shown on the left to demonstrate analysis of equal total protein. Protein concentrations were also verified by Lowry assay. (Bottom) Densitometric analysis of data shown in B (top) comparing relative protein levels between WT and Kir2.1-OE mice. Results are expressed as mean signal intensity and represent data from three animals for each genotype (N = 3 per genotype; δp < 0.01, mean ± SEM). (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
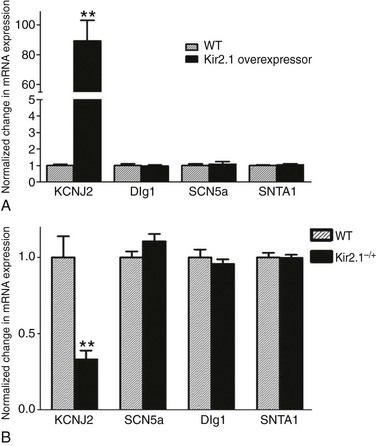
Figure 21-4 SAP97, syntrophin, and NaV1.5 mRNA levels are unchanged in the hearts of transgenic mouse models of Kir2.1 overexpression and underexpression. Total RNA was extracted from ventricular tissue that was harvested from Kir2.1-overexpressing (OE) mice, heterozygous Kir2.1 knockout (Kir2.1–/+), or the respective littermate wild type mice. The mRNA levels for the genes encoding for Kir2.1 (KCNJ2), SAP97 (Dlg1), and NaV1.5 (SCN5A) were determined by real-time polymerase chain reaction for all genotypes. Glyceraldehyde-3-phosphate dehydrogenase was used as an endogenous control for data normalization in each sample. Relative quantification of mRNA expression was calculated using the 2−ΔΔCt method. A, The bar graph compares messenger RNA (mRNA) expression for all genes between control and transgenic Kir2.1-OE ventricles. B, NaV1.5, SAP97, syntrophin mRNA levels are unchanged in the hearts of heterozygous Kir2.1 knockout (Kir2.1−/+) mice. N = 4 for all genotypes. **p < 0.005. (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
Additional RT-PCR experiments (see Figure 21-4, B) demonstrated that the expression levels of the genes encoding for NaV1.5, SAP97, and syntrophin mRNA were unchanged in ventricles of heterozygous KCNJ2 KO (Kir2.1–/+) mice14 compared with WT littermates. However, as shown in Figure 21-5, 50% allelic reduction of KCNJ2 gene expression by homologous recombination resulted in a significant decrease in relative membrane protein levels of NaV1.5, SAP97, and syntrophin (p < 0.05).
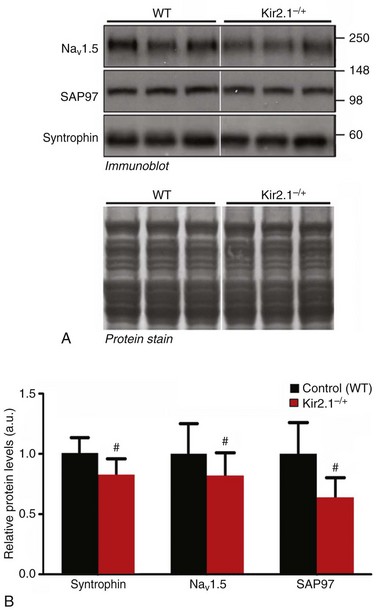
Figure 21-5 Transgenic reduction of Kir2.1 gene expression leads to a significant decrease in relative protein levels of NaV1.5 and SAP97, and syntrophin. Crude membrane vesicles were prepared from the ventricles of control (wild type [WT]) and Kir2.1−/+ mice. See Figure 21-3 for methods describing SDS-PAGE and immunoblotting. A, Representative immunoblots following detection of protein immunoreactivity with horseradish peroxidase–conjugated secondary antibodies and chemiluminescence (top). The corresponding Amido black nitrocellulose (protein stain) is shown on the bottom to demonstrate analysis of equal total protein. Protein concentrations were also verified by Lowry assay. B, Densitometric analysis comparing relative protein levels between WT and Kir2.1−/+ mice. Results are expressed as mean signal intensity (N = 7 per genotype. #p < 0.05, mean ± SEM. (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
NaV1.5-Kir2.1 Interactions Involve Membrane Trafficking
The results discussed in the previous section demonstrate that the synergistic and reciprocal effects of Kir2.1 and NaV1.5 expression on channel current density occur at posttranslational levels. Recently, it was suggested that these channel proteins share a common trafficking pathway where the synergistic effects act to modulate the surface levels of Kir2.1 and NaV1.5 channels. However, the mechanisms controlling Kir2.1 or NaV1.5 plasma membrane targeting or localization remain poorly explored. The balance between anterograde and retrograde trafficking pathways determines steady-state cell surface expression of channel proteins. Disease-associated mutations in both Kir2.1 and NaV1.5 have been shown to affect anterograde trafficking by inhibiting steps early in the secretory pathway to cause intracellular retention. However, it is unknown whether retention of Kir2.1 results in retention of NaV1.5, or vice versa. In contrast, once at the plasma membrane, endocytosis is the initial step in retrograde movement, after which internalized proteins can follow multiple routes to different intracellular fates.67 One well-recognized outcome is the targeting of internalized proteins to lysosomes followed by degradation. Alternatively, trafficking through recycling endosomes allows proteins to return to the plasma membrane and thereby protects them from degradation.68 Data presented recently in collaboration with Dr. Jeffrey Martens shows that Kir2.1 undergoes constitutive internalization in HL-1 myocytes, which contributes to the control of Kir2.1 steady-state surface density.5 In addition, it was demonstrated that NaV1.5 promotes cell surface expression of Kir2.1, at least in part, by reducing its internalization.5 However, it is unclear how NaV1.5 or macromolecular complex formation with SAP97 affect this process. Elucidation of the mechanisms controlling the surface expression of Kir2.1 and NaV1.5 will contribute significantly to the currently limited understanding of protein transport in the heart and to channelopathies involving altered trafficking.
Reciprocal NaV1.5 -Kir2.1 Interactions Control Reentry Frequency
The electrophysiological interplay between INa and IK1 is essential in the control of cardiac excitability and in determining the stability and frequency of reentry and VT/VF.3,23 In fact, IK1 upregulation is a substrate for very fast electrical rotors in the structurally normal ventricles.23 IK1 upregulation hyperpolarizes the resting membrane potential and thus increases sodium channel availability during reentry in a voltage-dependent manner. Recently, Milstein et al.5 demonstrated in neonatal rat ventricular myocyte (NRVM) monolayers that voltage dependency is not the only mechanism by which NaV1.5-Kir2.1 interactions control reentry dynamics and lead to faster rotors. Figure 21-6 shows reproduced phase maps and rotation frequency plots obtained from optical mapping experiments that investigated the electrophysiological consequences of the NaV1.5-Kir2.1 molecular interplay when one or both protein channels are overexpressed.5 A single stationary rotor maintained the electrical activity in each monolayer. It was previously shown in single NRVMs that Nav1.5 overexpression alone prolonged the APD, whereas Kir2.1 overexpression had the opposite effect.5 In Figure 21-6, A, APD prolongation induced by NaV1.5 overexpression in the monolayer was manifest as a lengthening of the wavelength and slowing of the reentry frequency. However, the shortened APD of Kir2.1 overexpressing monolayers significantly decreased the wavelength and increased the rotation frequency (see Figure 21-6, B). Most remarkable was the fact that combined overexpression of NaV1.5+Kir2.1 hyperpolarized the RMP, resulting in a shortened APD and a faster conduction velocity.5 Consequently, the frequency of reentry was even higher than that produced by Ad-Kir2.1 infection alone. These results suggest that the reciprocal intermolecular interplay of Kir2.1 and NaV1.5 could have profound consequences on the frequency and stability of reentry in the heart.
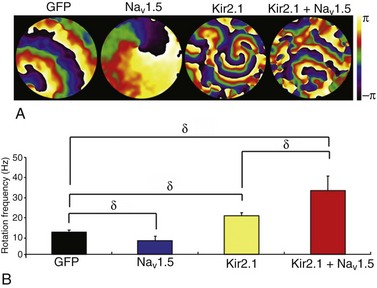
Figure 21-6 Molecular NaV1.5–Kir2.1 interactions modulate reentry frequency in neonatal rat ventricular myocyte (NRVM) monolayers. A, Phase maps for single rotations obtained from representative optical mapping movies of monolayers infected with Ad-GFP, Ad-NaV1.5, Ad-Kir2.1, or Ad-Kir2.1 plus Ad-NaV1.5. The color bar indicates the phase in the excitation–recovery cycle. B, Reentry frequencies in monolayers infected with Ad-GFP (black; n = 11), Ad-NaV1.5 (blue; n = 13), Ad-Kir2.1 (yellow; n = 11), or Ad-Kir2.1 plus Ad-NaV1.5 (red; n = 13). δp < 0.01, analysis of variance. (Modified from Milstein ML, Musa H, Balbuena DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A 109:E2134–E2143, 2012.)
In summary, the results discussed in this chapter provided the first evidence that two major ion channel proteins that control cardiac electrical function, NaV1.5 and Kir2.1, are part of a common macromolecular complex that involves at least two distinct scaffolding proteins, SAP97 and syntrophin (Figure 21-7). Most likely, their interactions provide a means for their reciprocal regulation, with vital functional consequences for myocardial excitation, conduction velocity, and arrhythmogenesis.5 It is tempting to speculate that the complex formed by these two major ion channels, along with their protein partners, represents a molecular sensing system in the cell that might enable it to monitor, and adjust accordingly, expression levels of proteins involved in generating and maintaining the cardiac impulse as well as adjusting to pathophysiological conditions. Most exciting, the demonstrated intermolecular interaction between these two essential channels controlling cardiac excitability opens a new pathway in the study of the molecular mechanisms underlying sudden cardiac death in highly prevalent heart diseases, including heart failure, and with inherited cardiac arrhythmias in which defects in the functional expression of Kir2.1 have been demonstrated clearly.69,70 Moreover, a mechanistic link between Kir2.1-NaV1.5 interactions, arrhythmogenesis and heart failure is further underscored by the demonstration that INa density decreases in chronic heart failure.71 The regulation of expression and mechanisms of interactions between Kir2.1 and NaV1.5 discussed here could contribute to the development of more efficacious treatments for arrhythmias in heart disease, particularly in heart failure and inherited arrhythmogenic diseases in which a defect in the membrane expression of NaV1.5 channel might affect the functional expression of Kir2.1, and vice versa.
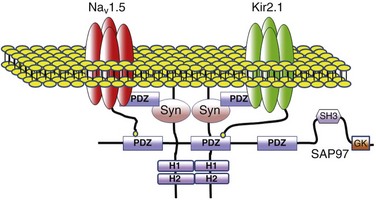
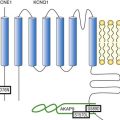
Figure 21-7 Schematic diagram of possible interactions between Kir2.1, NaV1.5, syntrophin, and SAP97 as well as their possible connection with the dystrophin-associated protein complex. The subcellular localization and channel activity of both NaV1.5 and Kir2.1 are regulated by protein–protein interactions via their respective C-terminal PDZ-binding motifs with such PDZ-domain–containing proteins as SAP97 and syntrophin. As shown here, the C-terminus of individual NaV1.5 and Kir2.1 channels may each bind to the same SAP97 or syntrophin molecule, but at different PDZ domains. It is possible that these interactions occur as part of a macromolecular complex (a “channelosome”) and result in changes in the expression of NaV1.5 or Kir2.1 and thereby influence their function in the cell membrane. GK, Guanylate kinase-like domain of SAP97; SH3, src kinase homology domain of SAP97.
References
1. Fozzard, HA, hanck, DA. Structure and function of voltage-dependent sodium channels: Comparison of brain II and cardiac isoforms. Physiol Rev. 1996; 76:887–926.
2. Lopatin, AN, Nichols, CG. Inward rectifiers in the heart: An update on i(k1). J Mol Cell Cardiol. 2001; 33:625–638.
3. Noujaim, SF, Berenfeld, O, Kalifa, J, et al. Universal scaling law of electrical turbulence in the mammalian heart. Proc Natl Acad Sci U S A. 2007; 104:20985–20989.
4. Noujaim, SF, Kaur, K, Milstein, M, et al. A null mutation of the neuronal sodium channel nav1. 6 disrupts action potential propagation and excitation-contraction coupling in the mouse heart. FASEB J. 2012; 26:63–72.
5. Milstein, ML, Musa, H, Balbuena, DP, et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012; 109(31):E2134–43.
6. Gavillet, B, Rougier, JS, Domenighetti, AA, et al. Cardiac sodium channel nav1. 5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006; 99:407–414.
7. Abriel, H. Roles and regulation of the cardiac sodium channel na v 1. 5: Recent insights from experimental studies. Cardiovasc Res. 2007; 76:381–389.
8. Papadatos, GA, Wallerstein, PM, Head, CE, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene scn5a. Proc Natl Acad Sci U S A. 2002; 99:6210–6215.
9. Zhang, T, Yong, SL, Tian, XL, et al. Cardiac-specific overexpression of scn5a gene leads to shorter p wave duration and pr interval in transgenic mice. Biochem Biophys Res Commun. 2007; 355:444–450.
10. Nichols, CG, Makhina, EN, Pearson, WL, et al. Inward rectification and implications for cardiac excitability. Circ Res. 1996; 78:1–7.
11. Shimoni, Y, Clark, RB, Giles, WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol. 1992; 448:709–727.
12. Nichols, CG, Lopatin, AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997; 59:171–191.
13. Yang, J, Jan, YN, Jan, LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier k+ channel. Neuron. 1995; 14:1047–1054.
14. Zaritsky, JJ, Redell, JB, Tempel, BL, et al. The consequences of disrupting cardiac inwardly rectifying k(+) current (i(k1)) as revealed by the targeted deletion of the murine kir2. 1 and kir2. 2 genes. J Physiol. 2001; 533:697–710.
15. Nakamura, TY, Artman, M, Rudy, B, et al. Inhibition of rat ventricular ik1 with antisense oligonucleotides targeted to kir2. 1 mrna. Am J Physiol. 1998; 274:H892–H900.
16. Preisig-Muller, R, Schlichthorl, G, Goerge, T, et al. Heteromerization of kir2. X potassium channels contributes to the phenotype of Andersen’s syndrome. PNAS. 2002; 99:7774–7779.
17. Miake, J, Marban, E, Nuss, HB. Functional role of inward rectifier current in heart probed by kir2. 1 overexpression and dominant-negative suppression. J Clin Invest. 2003; 111:1529–1536.
18. McLerie, M, Lopatin, A. Dominant-negative suppression of ik1 in the mouse heart leads to altered cardiac excitability. J Mol Cell Cardiol. 2003; 35:367–378.
19. Plaster, NM, Tawil, R, Tristani-Firouzi, M, et al. Mutations in kir2. 1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001; 105:511–519.
20. Anderson, CL, Delisle, BP, Anson, BD, et al. Most lqt2 mutations reduce kv11. 1 (herg) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006; 113:365–373.
21. Priori, SG, Pandit, SV, Rivolta, I, et al. A novel form of short qt syndrome (sqt3) is caused by a mutation in the kcnj2 gene. Circ Res. 2005; 96:800–807.
22. Li, J, McLerie, M, Lopatin, AN. Transgenic upregulation of ik1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol. 2004; 287:H2790–H2802.
23. Noujaim, SF, Pandit, SV, Berenfeld, O, et al. Up-regulation of the inward rectifier k+ current (i k1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007; 578:315–326.
24. Stiffler, MA, Grantcharova, VP, Sevecka, M, et al. Uncovering quantitative protein interaction networks for mouse pdz domains using protein microarrays. J Am Chem Soc. 2006; 128:5913–5922.
25. Stanfield, PR, Nakajima, S, Nakajima, Y. Constitutively active and g-protein coupled inward rectifier k+ channels: Kir2. 0 and kir3. 0. Rev Physiol Biochem Pharmacol. 2002; 145:47–179.
26. Kim, E, Niethammer, M, Rothschild, A, et al. Clustering of shaker-type k+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995; 378:85–88.
27. Kornau, HC, Schenker, LT, Kennedy, MB, et al. Domain interaction between nmda receptor subunits and the postsynaptic density protein psd-95. Science. 1995; 269:1737–1740.
28. Sato, T, Irie, S, Kitada, S, et al. Fap-1: A protein tyrosine phosphatase that associates with fas. Science. 1995; 268:411–415.
29. Garner, CC, Nash, J, Huganir, RL. Pdz domains in synapse assembly and signalling. Trends Cell Biol. 2000; 10:274–280.
30. Bilder, D. Pdz proteins and polarity: Functions from the fly. Trends Genet. 2001; 17:511–519.
31. Sheng, M, Sala, C. Pdz domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001; 24:1–29.
32. Nourry, C, Grant, SG, Borg, JP. Pdz domain proteins: Plug and play!. Sci STKE. 2003; 2003:RE7.
33. Hildebrand, JD, Soriano, P. Shroom, a pdz domain-containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell. 1999; 99:485–497.
34. Zhadanov, AB, Provance, DW, Jr., Speer, CA, et al. Absence of the tight junctional protein af-6 disrupts epithelial cell-cell junctions and cell polarity during mouse development. Curr Biol. 1999; 9:880–888.
35. Bladt, F, Tafuri, A, Gelkop, S, et al. Epidermolysis bullosa and embryonic lethality in mice lacking the multi-pdz domain protein grip1. Proc Natl Acad Sci U S A. 2002; 99:6816–6821.
36. Laverty, HG, Wilson, JB. Murine cask is disrupted in a sex-linked cleft palate mouse mutant. Genomics. 1998; 53:29–41.
37. Caruana, G, Bernstein, A. Craniofacial dysmorphogenesis including cleft palate in mice with an insertional mutation in the discs large gene. Mol Cell Biol. 2001; 21:1475–1483.
38. Boeda, B, El-Amraoui, A, Bahloul, A, et al. Myosin viia, harmonin and cadherin 23, three usher i gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002; 21:6689–6699.
39. Verpy, E, Leibovici, M, Zwaenepoel, I, et al. A defect in harmonin, a pdz domain-containing protein expressed in the inner ear sensory hair cells, underlies usher syndrome type 1c. Nat Genet. 2000; 26:51–55.
40. Boerkoel, CF, Takashima, H, Stankiewicz, P, et al. Periaxin mutations cause recessive dejerine-sottas neuropathy. Am J Hum Genet. 2001; 68:325–333.
41. Marfatia, SM, Byron, O, Campbell, G, et al. Human homologue of the drosophila discs large tumor suppressor protein forms an oligomer in solution. Identification of the self-association site. J Biol Chem. 2000; 275:13759–13770.
42. Tiffany, AM, Manganas, LN, Kim, E, et al. Psd-95 and sap97 exhibit distinct mechanisms for regulating k(+) channel surface expression and clustering. J Cell Biol. 2000; 148:147–158.
43. Melnyk, P, Zhang, L, Shrier, A, et al. Differential distribution of kir2. 1 and kir2. 3 subunits in canine atrium and ventricle. Am J Physiol Heart Circ Physiol. 2002; 283:H1123–H1133.
44. Murata, M, Buckett, PD, Zhou, J, et al. Sap97 interacts with kv1. 5 in heterologous expression systems. Am J Physiol Heart Circ Physiol. 2001; 281:H2575–H2584.
45. Tiffany, AM, Manganas, LN, Kim, E, et al. Psd-95 and sap97 exhibit distinct mechanisms for regulating k+ channel surface expression and clustering. J Cell Biol. 2000; 148:147–158.
46. Leonoudakis, D, Mailliard, W, Wingerd, K, et al. Inward rectifier potassium channel kir2. 2 is associated with synapse-associated protein sap97. J Cell Sci. 2001; 114:987–998.
47. Mohler, PJ, Rivolta, I, Napolitano, C, et al. Nav1. 5 e1053k mutation causing Brugada syndrome blocks binding to ankyrin-g and expression of nav1. 5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004; 101:17533–17538.
48. Pan, Z, Kao, T, Horvath, Z, et al. A common ankyrin-g-based mechanism retains kcnq and nav channels at electrically active domains of the axon. J Neurosci. 2006; 26:2599–2613.
49. Wu, L, Yong, SL, Fan, C, et al. Identification of a new co-factor, mog1, required for the full function of cardiac sodium channel nav 1. 5. J Biol Chem. 2008; 283:6968–6978.
50. Abriel, H, Kass, RS. Regulation of the voltage-gated cardiac sodium channel nav1. 5 by interacting proteins. Trends Cardiovasc Med. 2005; 15:35–40.
51. Jespersen, T, Gavillet, B, van Bemmelen, MX, et al. Cardiac sodium channel na(v)1. 5 interacts with and is regulated by the protein tyrosine phosphatase ptph1. Biochem Biophys Res Commun. 2006; 348:1455–1462.
52. Gardoni, F, Marcello, E, Di Luca, M. Postsynaptic density-membrane associated guanylate kinase proteins (psd-maguks) and their role in CNS disorders. Neuroscience. 2009; 158:324–333.
53. Petitprez, S, Zmoos, AF, Ogrodnik, J, et al. Sap97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels nav1. 5 in cardiomyocytes. Circ Res. 2011; 108:294–304.
54. Godreau, D, Vranckx, R, Maguy, A, et al. Expression, regulation and role of the maguk protein sap-97 in human atrial myocardium. Cardiovascular Research. 2002; 56:433–442.
55. Gee, SH, Madhavan, R, Levinson, SR, et al. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998; 18:128–137.
56. Ueda, K, Valdivia, C, Medeiros-Domingo, A, et al. Syntrophin mutation associated with long qt syndrome through activation of the nnos-scn5a macromolecular complex. Proc Natl Acad Sci U S A. 2008; 105:9355–9360.
57. Oceandy, D, Cartwright, EJ, Emerson, M, et al. Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation. 2007; 115:483–492.
58. Wang, L, Piserchio, A, Mierke, DF. Structural characterization of the intermolecular interactions of synapse-associated protein-97 with the nr2b subunit of n-methyl-d-aspartate receptors. J Biol Chem. 2005; 280:26992–26996.
59. Cohen, NA, Sha, Q, Makhina, EN, et al. Inhibition of an inward rectifier potassium channel (kir2. 3) by g-protein betagamma subunits. J Biol Chem. 1996; 271:32301–32305.
60. Leonoudakis, D, Conti, LR, Radeke, CM, et al. A multiprotein trafficking complex composed of sap97, cask, veli, and mint1 is associated with inward rectifier kir2 potassium channels. J Biol Chem. 2004; 279:19051–19063.
61. Leonoudakis, D, Conti, LR, Anderson, S, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (kir2. X)-associated proteins. J Biol Chem. 2004; 279:22331–22346.
62. Hruska-Hageman, AM, Benson, CJ, Leonard, AS, et al. Psd-95 and lin-7b interact with acid-sensing ion channel-3 and have opposite effects on h+- gated current. J Biol Chem. 2004; 279:46962–46968.
63. Lin, Y, Skeberdis, VA, Francesconi, A, et al. Postsynaptic density protein-95 regulates nmda channel gating and surface expression. J Neurosci. 2004; 24:10138–10148.
64. Jugloff, DG, Khanna, R, Schlichter, LC, et al. Internalization of the kv1. 4 potassium channel is suppressed by clustering interactions with psd-95. J Biol Chem. 2000; 275:1357–1364.
65. Leyland, ML, Dart, C. An alternatively spliced isoform of psd-93/chapsyn 110 binds to the inwardly rectifying potassium channel, kir2. 1. J Biol Chem. 2004. [M407575200].
66. Piao, L, Li, J, McLerie, M, et al. Transgenic upregulation of ik1 in the mouse heart is proarrhythmic. Basic Res Cardiol. 2007; 102:416–428.
67. Jordens, I, Marsman, M, Kuijl, C, et al. Rab proteins, connecting transport and vesicle fusion. Traffic. 2005; 6:1070–1077.
68. Mellman, I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996; 12:575–625.
69. Bendahhou, S, Donaldson, MR, Plaster, NM, et al. Defective potassium channel kir2. 1 trafficking underlies Andersen-Tawil syndrome. J Biol Chem. 2003; 278:51779–51785.
70. Li, GR, Lau, CP, Leung, TK, et al. Ionic current abnormalities associated with prolonged action potentials in cardiomyocytes from diseased human right ventricles. Heart Rhythm. 2004; 1:460–468.
71. Maltsev, VA, Sabbab, HN, Undrovinas, AI. Down-regulation of sodium current in chronic heart failure: Effect of long-term therapy with carvedilol. Cell Mol Life Sci. 2002; 59:1561–1568.