Chapter 73 Rare Pediatric Tumors
Primary Liver Tumors of Childhood
Etiology and Epidemiology
Primary malignant liver tumors (PMLTs) of childhood are very rare malignancies, comprising between 0.5% and 2% of pediatric tumors.1 The incidence in the United States is approximately 1.6 per million.2 The most common forms are hepatoblastoma (HBL) and hepatocellular carcinoma (HCC), but benign vascular tumors, mesenchymal hamartomas, adenomas, nodular hyperplasia, and hepatobiliary tumors are reported as well.1 The median age at presentation for HBL is 1 year, and it predominates in males with a ratio of 1.5 : 1. HCC presents at a median age of 12 and is more prevalent in males with ratios varying from 2 : 1 to 11 : 1.
HBL has been associated with prematurity, low birth weight, Beckwith-Wiedemann syndrome, familial adenomatous polyposis, maternal ingestion of oral contraceptives, and fetal alcohol syndrome.3 HCC has a strong association with hepatitis B virus (HBV) and is also associated with α1-antitrypsin deficiency, hereditary tyrosinemia, extrahepatic biliary atresia, Fanconi anemia, ataxia-telangiectasia, Sotos syndrome, and glucose-6-phosphatase deficiency.
Biologic Characteristics and Molecular Biology
HBL can be associated with several genetic aberrations. These include Beckwith-Wiedemann syndrome, most prominently, with loss of heterozygosity (LOH) at 11p15, as well as familial adenomatous polyposis and Li-Fraumeni syndrome. The most common chromosome aberrations are extra copies of 1q, 2q, 7q, 8, 17q, and 20. LOH of 11p15 is seen in a third of patients with HBL, and LOH of chromosome 1p is seen in an additional third.3 Mutations of β-catenin and activation of Wnt/β-catenin signaling have been associated with HBL. Wilms’ tumor and rhabdomyosarcoma have also been associated with HBL.
Pathology and Patterns of Spread
HBL is the most common PMLT of childhood, representing 60% to 75% of cases.3 The most widely used classification system was proposed by Ishak and Glunz.3 It distinguishes two forms of HBL, the epithelial type and the mixed type. The epithelial type encompasses a poorly differentiated embryonal type and a highly differentiated fetal type. The pure fetal hepatoblastoma (PFH) has normal hepatocytes with rare mitoses and is associated with a favorable prognosis. Mixed-type HBL contains both epithelial elements and mesenchymal tissue. The small cell undifferentiated (or anaplastic) histologic subtype of epithelial HBL has a very poor prognosis.
HCC accounts for 25% to 40% of childhood PMLTs. There is a strong relationship with preexistent hepatic disease or cirrhosis. HCC is divided into traditional HCC and a fibrolamellar histologic variant. The fibrolamellar variant tends to occur in noncirrhotic livers of adolescents and is characterized by large polygonal neoplastic cells with lamellar collagen bundles. Historically, this variant was thought to have a higher resection rate and superior outcome compared with traditional HCC. However, a report from the Pediatric Intergroup Hepatoma Protocol INT-0098 found that fibrolamellar histology was not associated with superior response to standard therapies or prognosis.5
Clinical Manifestations, Patient Evaluation, and Staging
Workup begins with an ultrasound evaluation of the abdomen, which typically reveals a solid hepatic mass. Magnetic resonance imaging (MRI) and computed tomography (CT) are useful to delineate the mass, its vascularity, and the potential for resection. Surgeons may require an angiogram to evaluate the hepatic arteries before determination of resectability. Imaging of the chest with CT should be performed; lung metastases are apparent at diagnosis in 20% of children with HBL and 30% of children with HCC.6–8 Laboratory evaluation includes routine chemistries as well as determination of the serum alpha-fetoprotein level, which is elevated in over 90% of children with HBL and 60% to 80% of children with HCC.1,3,7–9 An alpha-fetoprotein value less than 100 ng/mL is associated with a poor prognosis.
The most common staging systems for PMLT are the PRETEXT presurgical system and the Children’s Oncology Group postoperative system (Table 73-1). The PRETEXT system (Fig. 73-1) was created in 1990 by the International Society of Pediatric Oncology Liver Study Group (SIOPEL) and is based on preoperative imaging and the number of affected liver segments.6
TABLE 73-1 Children’s Oncology Group Staging System for Hepatoblastoma
| Stage | Description |
|---|---|
| I | Completely resected localized tumors |
| II | Grossly resected tumors with microscopic residual tumor |
| III | Unresectable tumors (measurable residual tumor or abdominal lymph node involvement) |
| IV | Distant metastases |
From Schnater JM, Kohler SE, Lamers WH, et al: Where do we stand with hepatoblastoma? A review. Cancer 98:668-678, 2003.
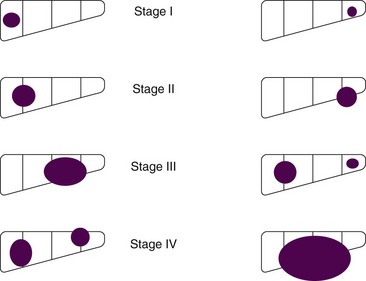
Figure 73-1 PRETEXT staging system for hepatoblastoma.
Adapted from Schnater JM, Aronson DC, Plaschkes J, et al: Surgical view of the treatment of patients with hepatoblastoma. Results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer 94:1111-1120, 2001.
Primary and Adjuvant Therapy and Results
The SIOPEL-1 trial reported by the International Society of Paediatric Oncology (SIOP) tested four to six courses of preoperative cisplatin and doxorubicin. Eighty-two percent of patients showed at least a partial response based on imaging and alpha-fetoprotein levels; 83% underwent delayed surgery, and in 77% of cases surgery achieved complete resection. The 5-year event-free survival and overall survival (OS), were 66% and 75%, respectively.8 The SIOPEL-3 trial compared cisplatin alone with cisplatin plus doxorubicin for three cycles preoperatively, followed by two postoperative cycles in children with HBL involving three or fewer liver segments with an alpha-fetoprotein level less than 100 ng/mL. Outcomes at 3 years were identical: 95% and 93% underwent complete resection in the respective chemotherapy arms, and OS was 95% and 93%, respectively. Grade 3 and 4 toxicities were significantly more common in the combination arm.10
Children with HCC on the SIOPEL-1 trial tended to have more advanced disease at presentation and fared significantly worse than patients with HBL. Partial response was seen in 49%, and complete resection was achieved in only 36%; 5-year OS and event-free survival were 28% and 17%, respectively.7 The Pediatric Intergroup Hepatoma Protocol INT-0098 randomized patients with HCC to postoperative cisplatin/vincristine/5-fluorouracil versus cisplatin and doxorubicin. There was no difference in results between the treatment regimens, and the overall 5-year event-free survival was 17% (75% for stage I, 8% for stage II, and 0% for stage IV).5 Subsequent SIOPEL studies evaluated a regimen of cisplatin and carboplatin, with no substantial improvement in outcome.7
Locally Advanced Disease and Palliation
Children with locally advanced and metastatic disease are treated with aggressive preoperative chemotherapy followed by surgery if possible. Pediatric Oncology Group study 9345 evaluated children with unresectable or metastatic HBL. Neoadjuvant therapy with carboplatin and carboplatin/vincristine/5-fluorouracil was followed by surgery when feasible or with high-dose cisplatin and etoposide. Resection was possible in 68% of patients with stage III disease and 36% of patients with stage IV disease; the 5-year event-free survival for the small number of children achieving resection was 79%.11 Current approaches include orthotopic liver transplantation when disease is unresectable, with documented long-term survival in a limited number of patients.6,11 The ongoing Children’s Oncology Group trial for locally advanced disease was highlighted previously; for “high-risk” patients with metastatic tumor or alpha-fetoprotein value less than 100 ng/mL, a national trial is testing the addition of irinotecan.
Metastatic disease is treated with systemic therapy followed by resection when possible. If local control can be achieved, metastasectomy is recommended. In rare patients with unresectable primary tumors, response to chemotherapy, and complete removal of pulmonary metastases, the SIOPEL-1 trial showed a limited number of children were long-term survivors.6
Irradiation Techniques
RT is not routinely used for PMLTs. An earlier series of 15 children with incompletely resected disease reported outcome after preoperative or postoperative irradiation.4 Doses ranged from 25 to 40 Gy in conjunction with chemotherapy. Six of eight patients treated postoperatively were disease free at 4 to 83 months, and one of four children with preoperative irradiation had histologic tumor control. The current Children’s Oncology Group HBL study specifically excludes use of RT. There are few current indications for hepatic irradiation or treatment beyond palliation to metastatic sites with HBL or HCC. RT has several technical challenges. Future attempts to incorporate RT for PMLTs in children would need to take advantage of modern radiotherapy techniques, including respiratory gating, given the critical organs surrounding the liver and the impact of combined therapeutic approaches.12
Pediatric Extracranial Germ Cell Tumors
Etiology and Epidemiology
Germ cell tumors (GCTs) are rare tumors in childhood, accounting for approximately 3% of pediatric malignancies.13–15 Teilium has described these tumors as arising from primordial germ cells that escape normal developmental influences. In the embryo, germ cells migrate from the allantoic stalk to the genital ridge and finally to the appropriate genital site. Therefore, GCTs can be found in the ovary or testis and aberrant migration accounts for the location of these tumors in midline structures such as the sacrococcygeal region, retroperitoneum, mediastinum, and pineal gland.
GCTs are more common in females than males.16 They have a bimodal age distribution, with extragonadal and testicular tumors occurring most frequently in children younger than 3 years old and gonadal tumors occurring most commonly in pubertal adolescents.
Prevention and Early Detection
GCTs are associated with intersex disorders, including pseudohermaphroditism, androgen insensitivity, and 5α-reductase deficiency. Undescended testis is highly associated with an increased risk of testicular malignancy, with the highest risk occurring in the intra-abdominal testis. This risk approaches 30 to 50 times the normal incidence of testicular cancer and can occur in either testis.14
Biologic Characteristics and Molecular Biology
Chromosomal abnormalities are common in malignant GCTs. The most frequent is isochromosome 12p [i(12p), which occurs in 75% to 80% of malignant ovarian or testicular GCTs]; other aberrations include loss of chromosome 13 and gain of chromosomes 21, 8, or 1q.17
Pathology and Pathways of Spread
GCTs span a spectrum of entities from benign to malignant, with elements of multiple histologic types in 25% of cases.16 The most common pediatric GCT is the teratoma, which is composed of tissue from more than one embryonic layer. Teratomas can be either mature, consisting of well-differentiated tissues, or immature, consisting of immature elements (most commonly neuroepithelial tissue).
Yolk sac or endodermal sinus tumors are highly malignant, occur most commonly in the ovary, testis, and sacrococcygeal region, and secrete alpha-fetoprotein. Histologically they show classic perivascular formations called Schiller-Duval bodies. Embryonal carcinomas are composed of large, pleomorphic undifferentiated cells. They often occur in combination with endodermal sinus tumors. Choriocarcinomas are rare tumors composed of malignant cytotrophoblasts and syncytiotrophoblasts. They typically secrete beta–human chorionic gonadotropin. All three of these GCT types are highly undifferentiated tumors with the propensity to metastasize to lung, liver, bone, and lymph nodes. In a series of 95 patients with sacrococcygeal endodermal sinus tumors,16 15% had positive lymph nodes at diagnosis and 35% had distant metastases.
Clinical Manifestations, Patient Evaluation, and Staging
The most common location for pediatric GCTs is the sacrococcygeal region, followed by the ovary, testis, and mediastinum. Unusual locations include the retroperitoneum, neck, stomach, and vagina. Sacrococcygeal tumors are classified according to Altman’s classification. Type I are predominately external, and type IV are entirely presacral. Neonates typically present with large, external, protruding sacral masses, which are commonly benign teratomas. Older children more commonly present with large pelvic masses, with malignant degeneration commonly apparent in children older than age 6 to 12 months.14
Initial evaluation should include a careful history and physical examination. Laboratory studies should include a complete blood cell count, renal and hepatic function tests, and evaluation of alpha-fetoprotein, beta–human chorionic gonadotropin, and lactate dehydrogenase levels; determination of CA-125 may also be of value, especially in sacrococcygeal teratomas. Tumor marker profiles for different histologies are listed in Table 73-2. Imaging includes ultrasonography of the primary site, CT of the abdomen and pelvis to evaluate for lymphadenopathy and resectability as appropriate, and CT of the chest to evaluate for metastatic disease. For secreting tumors, primary resection may be performed for both diagnosis and treatment if it is deemed that a complete resection can be performed. In nonsecreting and unresectable tumors, an open biopsy should be performed before starting definitive therapy.
GCTs are staged according to the site of origin using the Children’s Cancer Group and Pediatric Oncology Group staging system. The staging systems for ovarian, testicular, and extragonadal sites are outlined in Table 73-3.
TABLE 73-3 Children’s Cancer Group and Pediatric Oncology Group Staging System for Pediatric Germ Cell Tumors
| Stage | Description |
|---|---|
| Ovarian | |
| I | Limited to ovary (ovaries), peritoneal washings negative, tumor markers normal after appropriate half-life |
| II | Microscopic residual or positive lymph nodes (<2 cm), peritoneal washing negative, tumor markers positive or negative |
| III | Lymph node involvement >2 cm, gross residual disease, biopsy only, contiguous visceral involvement, peritoneal washings positive, tumor markers positive or negative |
| IV | Distant metastases |
| Testicular | |
| I | Limited to testis, tumor marker normal after appropriate half-life, completely resected with high inguinal orchiectomy |
| II | Transscrotal orchiectomy, microscopic disease in scrotum or high in spermatic cord, retroperitoneal lymph node <2 cm, increased tumor marker after appropriate half-life |
| III | Retroperitoneal lymph node >2 cm, no visceral of extra-abdominal involvement |
| IV | Distant metastases |
| Extragonadal | |
| I | Complete resection at any site, negative margins, coccygectomy for sacrococcygeal sites |
| II | Microscopic residual, lymph nodes negative |
| III | Gross residual or biopsy only, regional lymph nodes positive or negative |
| IV | Distant metastases |
Primary Therapy, Adjuvant Therapy, and Results
The optimal surgery for ovarian tumors has been debated, and historically principles of resection were based on the adult epithelial ovarian tumor experience. Recently the Children’s Oncology Group reported a series of children with ovarian GCTs who underwent conservative surgical resection and platinum-based chemotherapy; the 6-year survival rate approximated 95%. These researchers concluded that surgical guidelines should be changed to favor a more conservative approach including collection of ascites; examination and palpation of peritoneal surfaces, retroperitoneal lymph nodes, omentum, and the contralateral ovary with biopsy or excision of suspicious nodules; and complete resection of the ipsilateral tumor with potential sparing of a normal fallopian tube.18
Sacrococcygeal tumors are often resected using an inverted-V incision to spare the levator ani muscle and external sphincter. The entire coccyx must be resected because local recurrences of up to 37% are reported with inadequate surgery.15
Adjuvant therapy is based on the stage, degree of resection, and histologic subtype (Table 73-4). Currently there is no indication for adjuvant chemotherapy in mature or immature teratomas, and these patients are followed with close observation after surgery. Reported survival with surgery alone varies in the literature from 82% to 100%.14 In the Pediatric Oncology Group 9048/CCG 8891 trial, children with extracranial immature teratomas (32% with a malignant element) were treated with surgery alone with a 3-year overall event-free survival of 93%. Four of five patients with recurrence were disease free after platinum-based salvage chemotherapy.19
TABLE 73-4 General Treatment Guidelines for Extracranial Germ Cell Tumors
| Low-Risk Patients | |
| All teratomas | Surgery and observation |
| Stage I gonadal | |
| Stage I extragonadal | |
| Intermediate-Risk Patients | |
| Stage II-IV gonadal | Surgery with adjuvant chemotherapy |
| Stage II extragonadal | |
| High-Risk Patients | |
| Stage III-IV extragonadal | Surgery with adjuvant chemotherapy |
| Initial Unresectable or Biopsy Only | Neoadjuvant chemotherapy followed by second surgery, followed by adjuvant chemotherapy for residual pathologic disease |
There has been dramatic improvement in survival for children with stage II or greater malignant GCTs with the addition of cisplatin-based chemotherapy. The most commonly used regimen is PEB (cisplatin/etoposide/bleomycin). Using this regimen, OS has increased to more than 80% for all stages of GCTs.16,18,20,21
Locally Advanced Disease and Palliation
Bulky, locally invasive, and metastatic disease require treatment with neoadjuvant chemotherapy followed by second-look surgery; additional chemotherapy is utilized for residual tumor at the time of surgery. Even in this setting the OS is excellent owing to the chemosensitivity of these tumors. For high-risk patients, a high-dose PEB regimen (HDPEB) has been compared with standard-dose PEB. The Pediatric Oncology Group 9040/CCG 8882 trial20 randomized patients with stage III and IV gonadal and stage I to IV extragonadal GCTs to HDPEB versus PEB, reporting 6-year event-free survival of 89.6% versus 80.5% but no benefit in OS; there were more toxic deaths in the high-dose arm of the trial. Surgery for metastatic foci is usually not indicated because these foci tend to respond to chemotherapy. For relapsed or refractory tumors, chemotherapy with ifosfamide added to platinum and etoposide is recommended.
Irradiation Techniques
The role of RT for GCTs has diminished significantly owing to the effectiveness of current chemotherapy regimens. Historically, RT was used in conjunction with surgery and chemotherapy, with OS rates in the range of 48% to 62%.13,22,23 Seminomas and dysgerminomas were often cured with limited surgery and RT. Currently, the role of RT is limited to cases with unresectable, refractory, or recurrent disease unresponsive to chemotherapy. Classically, disease control for malignant GCTs has required radiation doses of 40 to 45 Gy or more for residual disease or disease that is resistant to chemotherapy.
Treatment Algorithms, Controversies, Challenges, and Future Possibilities
Treatment is based on tumor site, type, and grade and is detailed in Table 73-4. Surgical resection is necessary for local control and is preceded and/or followed by PEB chemotherapy. In patients with locally advanced or metastatic disease, survival still approaches 75% owing to the sensitivity of these tumors to platinum-based chemotherapy. With the high curability of these tumors, current protocols are using risk-adapted algorithms based on histology, site, stage, and genetic aberrations to decrease toxicity associated with chemotherapy.
Juvenile Nasopharyngeal Angiofibroma
Etiology and Epidemiology
Juvenile nasopharyngeal angiofibroma (JNA) is a highly vascular tumor that is histologically benign but locally invasive. JNA occurs almost exclusively in adolescent boys and young adult men, suggesting a prominent hormonal role in the tumor’s etiology; the average age at diagnosis is 17 years.24,25 There is an increased incidence of JNA in patients with familial adenomatous polyposis, suggesting a connection to the β-catenin pathway.26
Biologic Characteristics and Molecular Biology I
Both androgen and estrogen receptors have been demonstrated in JNAs.27,28 Recently, estrogen beta-adrenergic receptors have been found in a high percentage of JNAs. Schlauder and associates28 have postulated that the presence of aromatase in tumor cells converts endogenous androgens to estrogens, causing tumor growth via an autocrine-like mechanism.29 Most tumors stain for vascular endothelial growth factor.30
Pathology and Pathways of Spread
JNA is a benign tumor, although its exact nature is controversial. Some have suggested that it is a vascular hamartoma and similar to a hemangioma,31 but others believe it is neoplastic.32 Histologically, tumors are composed of fibrous connective tissue with abundant endothelium-lined vascular spaces.31 Localization of β-catenin to tumor stromal cells suggests these may be the neoplastic component rather than the endothelial cells.33 Tumors typically arise from the superior margin of the sphenopalatine foramen and invade laterally through the pterygomaxillary fissure toward the infratemporal fossa.24 Intracranial extension is seen in up to a third of cases, although actual dural invasion is uncommon.34,35 Tumors can be locally invasive of bone and extend into the parapharyngeal spaces, paranasal sinuses, orbit, and base of skull. This pattern of spread predicts for a high risk of local recurrence.35 Blood supply is primarily from the internal maxillary arteries of the external carotid system.
Clinical Manifestations, Patient Evaluation, and Staging
Presenting symptoms include recurrent painless spontaneous epistaxis, nasal obstruction, nasal discharge, a reduced sense of smell, snoring, headache, cranial nerve palsies, and facial swelling.25 Angiography is essential to define the tumor’s blood supply for planning surgery and, typically, for preoperative embolization to decrease surgical blood loss. There are usually multiple tortuous feeding vessels with a dense, homogeneous blush in the capillary phase.36 CT and MRI help define the anatomic extent of the enhancing tumor. Distant metastases do not occur, so systemic evaluation is not required. Biopsy can be hazardous owing to the tumor’s vascularity; not all authors require biopsy confirmation before treatment if clinical and radiographic data are consistent with the diagnosis.37
Several staging systems have been proposed (Table 73-5). Most are designed to guide decisions regarding the resectability and optimal surgical approach to the tumor rather than to predict prognosis.37,38–41
TABLE 73-5 Staging Systems for Juvenile Nasopharyngeal Angiofibroma
| Andrews Staging System39 | |
| Stage I | Tumor limited to the nasal cavity and nasopharynx |
| Stage II | Tumor extension into the pterygopalatine fossa, or maxillary, sphenoidal, or ethmoidal sinuses |
| Stage IIIa | Extension into the orbit or infratemporal fossa without intracranial extension |
| Stage IIIb | Stage IIIa with minimal extradural intracranial extension |
| Stage IVa | Extensive extradural intracranial or intradural extension |
| Stage IVb | Extension into cavernous sinus, pituitary, or optic chiasm |
| Carrillo Staging System37 | |
| Stage I | Tumor limited to nasopharynx, nasal fossae, maxillary antrum, anterior ethmoid cells and sphenoidal sinus |
| Stage IIa | Invasion to pterygomaxillary fossae or infratemporal fossae anterior to pterygoid plates, with major diameter <6 cm |
| Stage IIb | Invasion to pterygomaxillary fossae or infratemporal fossae anterior to pterygoid plates, with major diameter ≥6 cm |
| Stage III | Invasion to infratemporal fossae posterior to pterygoid plates or posterior ethmoid cells |
| Stage IV | Extensive skull base invasion >2 cm or intracranial invasion |
| Chandler Staging System40 | |
| Stage I | Tumor confined to the nasopharynx |
| Stage II | Tumor extending into the nasal cavity and/or sphenoidal sinus |
| Stage III | Tumor involvement of one or more of the maxillary or ethmoidal sinuses, pterygomaxillary and infratemporal fossae, and orbit and/or cheek |
| Stage IV | Tumor extending into the cranial cavity |
| Fisch Staging System38 | |
| Type I | Tumor limited to the nasopharynx and nasal cavity with no bone destruction |
| Type II | Tumors invading the pterygomaxillary fossa and the maxillary, ethmoidal, and sphenoidal sinuses with bone destruction |
| Type III | Tumors invading the infratemporal fossa, orbit, and parasellar region remaining lateral to the cavernous sinus |
| Type IV | Tumors with massive invasion of the cavernous sinus, optic chiasmal region, or pituitary fossa |
Primary and Adjuvant Therapy and Results
Surgical removal, often preceded by tumor embolization, is the primary treatment for JNA. A craniofacial approach is used for locally advanced tumors. Endoscopic techniques have less morbidity and are effective for earlier-stage lesions.24 Gross total removal is usually curative.34 A number of cases prove not to be amenable to complete resection; depending on case selection, there is a rate of local recurrence after surgery that approximates 20% to 40%, with most recurrences occurring in large, incompletely resected lesions.24,25 Moderate doses of RT may be indicated for postoperative residual tumor, although observation is more often considered because spontaneous involution of tumor is a well-recognized phenomenon.42,43 Most recurrences present within a year of surgery. The risk of recurrence is greatest in patients with large tumors that erode the skull base, young age at presentation, and irradiated tumors that are slow to regress.36,44 There are currently no indications for adjuvant chemotherapy after initial resection.
Locally Advanced Disease and Palliation
RT can provide effective control for recurrent or large, unresectable tumors. Objective tumor response after RT is typically slow, but ultimate control rates range from 75% to 92%45–48; 90% of responders have no residual tumor 3 years after treatment.49 Given the strong association of JNA with hormonal receptors, there has been interest in hormonal manipulation for patients with advanced disease. Diethylstilbestrol and flutamide have been used, but results have been inconsistent.36,50,51 Case reports of chemotherapy for recurrent tumor show efficacy for doxorubicin, dactinomycin, vincristine, cyclophosphamide, and cisplatin in selected patients.52,53,54
Techniques of Irradiation
No radiation dose-response curve has been reported for JNA. Local control rates greater than 80% are seen with doses ranging from 30 to 50 Gy,37,55,56–59 and a dose 36 Gy is commonly used. Intensity-modulated RT (IMRT) techniques provide high conformality for sparing of normal structures (Fig. 73-2). Stereotactic radiosurgery using single doses of 17 to 20 Gy and hypofractionated RT using 45 Gy in three fractions have been reported effective in small series of patients.60–62
Pleuropulmonary Blastoma
Etiology and Epidemiology
Pleuropulmonary blastoma (PPB) is a dysontogenetic neoplasm of childhood that originates in the lung and/or pleura.63 Although rare, it is the most common primary lung tumor in children.64 It is analogous to other dysontogenetic tumors such as Wilms’ tumor, neuroblastoma, and hepatoblastoma.65
PPB is thought to progress through a distinct sequence of clinical and pathologic changes beginning as a relatively nonaggressive cystic lesion and subsequently developing into the more malignant mixed cystic/solid and purely solid morphologies.66 Median age at presentation is 3 years.67 Younger children typically present with predominantly cystic tumors, whereas older children are more likely to have significant solid components.68 Boys and girls are equally affected.68 Siblings of patients with PPB have a higher incidence of PPB than the general population, although a genetic cause has not yet been found.69 There is also an increased incidence of other types of dysplasia and neoplasia in relatives of children with PPB.70,71
Prevention and Early Detection
Cystic PPB is clinically indistinguishable from benign lung cysts, and some authors recommend excision of all such lesions.69 Further supporting this approach is evidence that PPB can arise de novo from lung cysts, so that resection of these “precancerous” lesions is indicated.72,73 Others advocate a policy of watchful waiting for purely cystic lesions, only intervening if radiographic changes suggest progression.
Biologic Characteristics and Molecular Biology
TP53 mutations have been described in PPB and may portend a worse prognosis.74 Polysomy of chromosome 8 has also been noted.75
Pathology and Pathways of Spread
Histologically, PPB has small, primitive blastemal cells separated by an uncommitted sarcomatous stroma.65,68,76 Tumor cells stain with vimentin and may show myogenic differentiation.64 Rhabdomyoblasts and cartilage nodules are reported in 40% to 50% of patients.69
Tumors usually arise in the lung parenchyma. Spread to contiguous structures such as the mediastinum and pleura is associated with a poorer prognosis. Invasion into the chest wall is uncommon.77 Hematogenous metastases occur, most often to the brain. In one series,63 the incidence of brain metastases was 11% in mixed cystic/solid tumors and 54% in purely solid tumors.
Clinical Manifestations, Patient Evaluation, and Staging
Presenting symptoms of PPB include cough, dyspnea, wheezing, symptoms of respiratory infection, and, occasionally, spontaneous pneumothorax. Chest CT usually reveals a heterogeneous low-attenuation mass with pleural effusion and mediastinal shift.77 Differential diagnosis includes rhabdomyosarcoma, Askin tumor, and nonrhabdoid sarcomas; examination of the cystic fluid or solid tumor is required to make a diagnosis. Bone scintigraphy and MRI of the brain should be performed for staging, especially for tumors with a significant solid component.
PPB has no anatomic staging system but is classified into three morphologic types that have prognostic significance. Type I consists of purely cystic tumors, type III lesions are solid tumors, and type II have mixed cystic and solid components.70 Both type II and type III tumors have significantly poorer prognosis than type I, with an incidence of distant metastases at diagnosis approximating 30%.67 Recently, a fourth type, type Ir (type I—regressed), has been described for cystic lesions without a neoplastic component, hypothesized to have “regressed” from an earlier type I PPB or, alternatively, represent a genetically determined lung cyst that has not yet evolved along a dysplastic path.78
Primary and Adjuvant Therapy and Results
Surgery is the cornerstone of management, although most patients cannot undergo gross total resections owing to involvement of critical structures.67,76 Neoadjuvant chemotherapy can be used in large tumors to render the tumor resectable; adjuvant chemotherapy is often given postoperatively to patients with type II and III tumors. Chemotherapy may improve survival in type I tumors,66,78 although it is not widely recommended if the tumor is completely resected.67 Drug regimens parallel those used for other pediatric sarcomas and include ifosfamide/carboplatin/etoposide (ICE regimen), and ifosfamide/vincristine/dactinomycin (Actinomycin-D)/doxorubicin (IVADo regimen). Other active drugs include irinotecan79 and cisplatin.68 RT may be indicated in unresectable tumors, although large treatment volumes and young patient age often hinder the use of this modality.80 Five-year survival rates are approximately 80% for patients with type I tumors and 45% for patients with types II and III.67,68,76
Locally Advanced Disease and Palliation
In patients who experience a recurrence, local relapse is somewhat more common than distant failure.67 When type I tumors recur, they usually recur as types II or III.66,69 Re-resection should be performed when possible, but second-line chemotherapy is often the primary treatment owing to the extent of disease. RT may be useful for brain metastases and palliation of painful bone lesions.
Irradiation Techniques
Fractionated external beam RT using a dose of 44 Gy has been recommended for incompletely resected tumors, although its role in local control is difficult to assess.67,76 Normal tissue tolerances may preclude this amount of irradiation; use of a lesser dose may be required in circumstances in which volume and dose exceed organ tolerance. Microscopic margins were controlled in one case report using 36 Gy,68 but local recurrence was reported after 30 Gy in 10 fractions in a patient with gross tumor.81 Intracavitary phosphorus-32 has also been used and may contribute to local control in selected patients.76
Hemangiomas and Lymphangiomas
Etiology and Epidemiology
Hemangiomas occur in about 1% of infants, most often during the first few months of life, with about a third of these presenting in the head and neck.82 Lymphangiomas are much less common, with an incidence between 1 in 6,000 and 1 in 16,000 live births.83 Half are congenital; 90% are evident before the age of 2 years. Three fourths of lymphangiomas occur in the neck or head areas.
Pathology and Pathways of Spread
Hemangiomas consist of thin-walled vessels lined by endothelial cells and a discontinuous layer of pericytes and reticular fibers.84 They have been reported to occur in a wide variety of locations, including the skin, liver, bone, central nervous system, bladder, trachea, and other soft tissues.85 Lymphangiomas are dilated endothelial-lined lymph channels with thick walls containing smooth muscle.86 Lymphangiomas can involve the skin, skeletal tissue, spleen, liver, mediastinum, or lung.87 Hemangiomas and lymphangiomas classically are malformations or low-grade/benign neoplasms with no capacity for metastasis.
Clinical Manifestations, Patient Evaluation, and Staging
Hemangiomas are classified as capillary, cavernous, or mixed, depending on the size of the vessels involved. When they involve the skin, capillary hemangiomas are often raised, circumscribed red lesions. They usually present early in the first year of life, and the great majority undergo spontaneous involution over the subsequent years. They are distinct from vascular malformations such as port-wine stains, which do not regress.82 Clinical manifestations of hemangiomas are largely dependent on the location of the lesion and can include cosmetic deformity, functional impairment, airway obstruction, bleeding, infection, and high-output cardiac failure. Kasabach-Merritt syndrome describes a giant hemangioma causing thrombocytopenia due to consumption coagulopathy.
Lymphangiomas are also categorized according to the size of the abnormal lymph vessels. Lymphangioma circumscriptum contains relatively small-caliber lymph channels and is usually superficial in location. Cystic hygroma or cavernous lymphangioma consists of large, cystic areas of lymph fluid arising in deeper tissues. Unlike hemangiomas, lymphangiomas do not tend to regress spontaneously and may enlarge during adolescence.88 Symptoms of lymphangiomas can include disfigurement, pain, infection, and complications of chronic lymph secretion. Lymphangiomas occurring in the mediastinum may cause chylothorax, which can be life threatening.
Classification schemes have been proposed for these lesions, but there is no widely accepted staging system.84,89
Primary and Adjuvant Therapy and Results
RT was the mainstay of therapy for both hemangiomas and lymphangiomas through the early 1950s and, in some centers, into the 1970s. Subsequently, the use of RT has been all but abandoned as the natural history of spontaneous involution of hemangiomas was recognized and the late morbidities of RT were reported.90
Spontaneous involution will occur in 95% of capillary hemangiomas over the course of several years; most require no treatment.91 Cavernous hemangiomas do not usually regress spontaneously and require treatment if symptomatic.85 For symptomatic hemangiomas, topical or intralesional corticosteroids are often the first line of treatment.92 Systemic corticosteroids are also effective.82 Surgical resection is an effective treatment in appropriate circumstances. Other treatments include interferon, laser ablation, cryotherapy, compression, electrocautery, and embolization. Other pharmacologic interventions include antiangiogenic and antifibrinolytic agents. Irradiation for hemangiomas with life-threatening complications can be performed and is often effective, with improvement or resolution occurring in more than 80% of patients.85,93
Surgical removal of lymphangiomas is curative, although can be cosmetically or functionally morbid in many anatomic sites. Sclerotherapy using agents such as OK-432 or bleomycin is a common nonsurgical treatment, with complete responses seen in about 40% of patients and partial responses in another 40% to 45%.83 Other nonsurgical therapies include diathermy, cryotherapy, and interferon alfa-2b.87 RT has also been used with some efficacy in selected patients.88,94–96
Locally Advanced Disease and Palliation
Mortality from Kasabach-Merritt syndrome can exceed 10%. Large hemangiomas causing coagulopathy or cardiac failure require aggressive treatment.97 RT may be useful when other modalities fail.98 Fractionated RT to doses of 10 to 20 Gy has been effective in these life-threatening situations.85,93,97,98 Very rare malignant tumors such as lymphangiosarcoma and malignant hemangioendothelioma are highly aggressive neoplasms and require multimodality treatment.99,100
Irradiation Techniques
Although the efficacy of RT for these conditions is well established, the availability of other effective treatments and the concern for late effects of irradiation in this young patient population appropriately limit RT to a seldom-used option today.91,101 No definite dose-response relationship has been established for the treatment of hemangiomas, and fractionated radiation doses ranging from 3 to 40 Gy have been recommended.85,97 Braun-Falco and colleagues91 reported a large series of patients with hemangiomas; most were treated with fractionated radiation doses between 12 and 25 Gy. Responses were seen in 73% of patients, with complete responses in 50%. Some have reported good results, with lower doses of 3 to 11 Gy,102 although others recommend a dose of at least 25 Gy.103 Partial improvement after RT can often be noted within a few weeks, but maximum response can take several months to years.
Lymphangioma has been successfully treated with 10 to 20 Gy of fractionated RT.88,95,96 Responses can be seen much more quickly in lymphangiomas than in hemangiomas.
Desmoplastic Small Round Cell Tumor
Etiology and Epidemiology
Desmoplastic small round cell tumor (DSRCT) was first characterized by Gerald and associates in 1991104 as an extremely aggressive neoplasm, usually presenting as an intra-abdominal mass in adolescent males. Age at diagnosis ranges from 6 to 49 years, with a median of 22 years; male-to-female ratios range from 5 : 1 to 20 : 1.105
Biologic Characteristics and Molecular Biology I
Although the cell of origin remains unknown, DSRCT arises along the serosal lining of body cavities and visceral organs. It demonstrates divergent differentiation and is immunohistochemically reactive with markers for epithelium (epithelial membrane antigen), mesenchyme (vimentin), myogen (desmin), and neurons (neuron-specific enolase, CD56).106 Grossly, the outer surface of these typically large tumors is bosselated, with the tumor showing areas of necrosis and hemorrhage. Microscopically, it is composed of well-defined nests of small uniform cells with sparse cytoplasm within a predominantly desmoplastic stroma.107,108
Cytogenetically, DSRCT is characterized by a t(11;22)(p13;q12) translocation. This leads to the fusion of the Ewing’s sarcoma gene (EWS) on chromosome 22 with the Wilms’ tumor gene (WT1) on chromosome 11.107,109 This EWS-WT1 chimeric fusion is postulated to be the cause of the tumor’s multiply antigenic nature.110,111
Clinical Manifestations, Patient Evaluation, and Staging
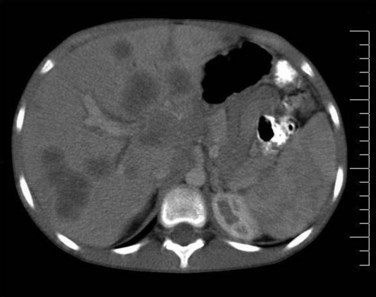
DSRCT is an extremely aggressive neoplasm with a very poor prognosis. It most often presents as a large intra-abdominal mass, although it has been reported to arise in other sites, including paratestes,112 central nervous system,113 kidney,114 pleura,115 and bone.111 Symptoms of the abdominal manifestation of the disease include abdominal pain/cramping, distention, constipation/diarrhea, dysphagia, weight loss, and jaundice. Pathways of spread include direct extension, hematogenous seeding, and nodal involvement (Fig. 73-3). Significant amounts of peritoneal seeding and studding are usually present, and ascites is common. A majority of patients have disease beyond the primary tumor at diagnosis, with lymph node, visceral (liver, lung), or bone metastases.116–118
CT and/or MRI of the chest, abdomen, and pelvis should be performed for evaluation; combined imaging with CT and positron emission tomography (CT/PET) may be helpful. Initial open or imaged-guided biopsy is suggested, because extension of disease usually precludes complete resection of disease at presentation.116 There is no accepted staging system for this tumor.
Primary and Adjuvant Therapy and Results
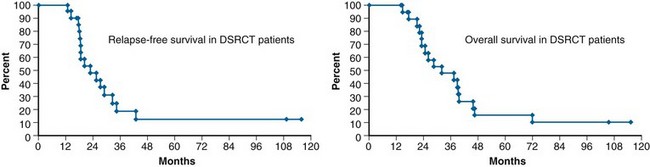
There are no uniform treatment recommendations for DSRCT, and a wide range of treatment regimens have been used. The most common treatment approach is aggressive multimodality therapy with neoadjuvant chemotherapy (typically cyclophosphamide, doxorubicin, and vincristine, with or without ifosfamide and etoposide), second-look surgery with a goal of complete or near-complete (maximal debulking) resection, and whole-abdominopelvic irradiation.116,118,119 In one series, 3-year survival was 58% in patients who underwent aggressive tumor debulking versus 0% for those who did not.118 Other reported treatments include high-dose chemotherapy with autologous bone marrow rescue and continuous hyperthermic peritoneal perfusion with cisplatin.120 Despite favorable response and intermediate 2- to 3-year survival, the prognosis is poor, with survival at 5 years rarely above 15%118,122 (Fig. 73-4).
Irradiation Techniques
RT plays an important role in the trimodality treatment of DSRCT. In a series of patients from Memorial Sloan-Kettering Cancer Center (MKSCC), 3-year survival was 55% for those receiving chemotherapy, surgery, and RT versus 27% when fewer than three modalities were used.118 Because of the extensive serosal spread of DSRCT, whole-abdominopelvic irradiation is indicated for consolidation of primary therapy. After the initial MSKCC reports, wide abdominal irradiation is typically delivered in 1.5-Gy fractions to a total of 30 Gy; the large series from MSKCC was based on twice-daily fractionation.105 Kidneys and liver should be shielded at 15 to 17 Gy and 23 to 25 Gy, respectively, to respect normal tissue tolerances. Whole-abdominal RT has also been delivered using IMRT to spare bone marrow.121 If the primary disease is intrapelvic or occupies a reasonably confined area of the abdomen, reduced volume “boost” may be continued to 45 Gy as permitted by surrounding structure tolerance.
Treatment Algorithms, Controversies, Challenges, and Future Possibilities
The current approach to treatment after biopsy consists of neoadjuvant chemotherapy, second-look debulking surgery, and whole-abdominal RT. Several of these tumors are immunohistochemically positive for markers such as placental alkaline phosphatase, ERBB2, androgen receptor, and c-KIT, which may present options for targeted biologic therapies.109 Additionally, there are case reports of treatment with bevacizumab122 and trastuzumab.109 Other targeted therapies based on the downstream effects of the EWS-WT1 fusion product are under investigation.123
Langerhans Cell Histiocytosis
Etiology and Epidemiology
Langerhans cell histiocytosis (LCH) refers to the spectrum of diseases arising from clonal proliferation of the CD1a- and CD207-positive reticuloendothelial Langerhans cell. The historical name “histiocytosis X” was coined in the 1950s to indicate the histiocytic origin of the disease, with the “X” reflecting the mysterious clinical spectrum of disease presentation.124 This designation was replaced by the term LCH in 1987.125 Incidence is usually quoted as one to five cases per million children per year. More than 50% of children are diagnosed between 1 and 15 years of age, with a peak incidence between 1 and 4 years of age.126–128
Biologic Characteristics and Molecular Biology
There has been considerable debate as to whether LCH is a reactive or immune dysregulatory process versus a true neoplasm.129 A clonal disorder, it is known to spontaneously regress in some patients and prove fatal in others. Most cases arise spontaneously, although there are rare genetic forms of the disease associated with specific cytokine gene variants that tend to occur early with multifocal disease.130
Pathology and Pathways of Spread
The basic lesion of LCH is formed by collections of pathologic Langerhans cells, indeterminate cells, interdigitating cells, and macrophages accompanied by T lymphocytes, with variable numbers of multinucleated giant histiocytes and eosinophils, as well as dendritic cells. Granulocytes and plasma cells, when present, represent reactants.131 For definitive diagnosis, the Langerhans morphology must be present and Birbeck granules must be visualized by electron microscopy or CD1a expressed on the cell surface. (More recently, CD207 is the most sensitive and specific antibody.)
Clinical Manifestations, Patient Evaluation, and Staging
The mildest form of LCH, previously known as eosinophilic granuloma of bone, is usually seen in children between 5 and 15 years of age. Presentation is a purely lytic lesion of bone, most often affecting the skull but occurring anywhere in the skeleton, including the spine, ribs, pelvis, and long bones. Presenting symptoms include pain, swelling, and pathologic fracture.132 Classic Hand-Schüller-Christian disease is intermediate in severity, presenting as a triad of exophthalmos, skull lesions, and diabetes insipidus; it generally presents in somewhat younger children between the ages of 2 and 15 years.133 The most aggressive form of LCH, classically identified as Letterer-Siwe disease, usually presents in children younger than age 3 years and carries a very poor prognosis. This form typically involves multiple organ systems, with presentations including splenomegaly, lymphadenopathy, anemia, and blood dyscrasias. The skeleton is frequently diffusely involved, and children may have otitis media, fever, papular rash, and cachexia.
There is no uniform staging system for LCH, thus underscoring the hesitancy of many to associate LCH with a true malignancy. In studies, children are generally grouped by burden of disease or number of systems involved such as single or multiple sites, single or multiple organs, and “non-risk sites” (skin, bone, lymph nodes) versus “risk organs” (liver, spleen, lung, bone marrow).134–136 Those with “risk organs” involved are now often considered as having “high-risk” LCH versus others with “low-risk” disease. A study of 217 patients from Turkey reported 41.5% with single-system disease, 34.1% with multiple-system disease, and 24.4% with multiple-system disease with organ dysfunction.132 Overall, approximately 30% of patients present with unifocal lesions and 70% present with more extensive disease.135
Primary and Adjuvant Therapy and Results
For patients with a low burden of disease the goal is to give as little treatment as possible to preserve quality of life with good function. Children with minimal involvement often have spontaneous resolution of their disease and require no active treatment. In cases such as solitary tumor of bone, a simple surgical curettage or injection of corticosteroids may be adequate to control the disease.133,137
Locally Advanced Disease and Palliation
For patients with multifocal disease (with or without organ involvement), chemotherapy is clearly indicated and is risk adapted based on disease severity. Most regimens include vinblastine and prednisone. The purine analogue cladribine is effective for visceral organ involvement, potentially replacing mercaptopurine as a “standard” agent. In nonresponders and refractory disease, stem cell transplantation has been used.138,139
Irradiation Techniques
LCH is markedly radiosensitive; RT was frequently used in the past for local disease control. Today, RT is used much less frequently because of the good results obtained with other modalities and the risk of late effects after RT in children (primarily the low risk of secondary neoplasms after low-dose irradiation). In cases in which surgical resection would cause loss of function, when a lesion is recurrent or refractory to other local treatment, or a single vertebral body is involved, low-dose RT can be considered.134 One series reported 88% long-term local control of bone lesions using RT doses of 6 to 15 Gy (median, 9 Gy).140 Local control was somewhat less when soft tissues were involved. A German study reported local control of 96% for single osseous lesions and 92% for bone lesions in patients with multiple-organ disease.141 RT using 15 to 20 Gy to the sellar/hypothalamic area has been used in cases of diabetes insipidus secondary to LCH, with modest—in fact, controversial—efficacy.142,143 In several retrospective series, when RT has been delivered within 1 to 2 weeks of onset, 25% to 35% of patients show improvement in diabetes insipidus.142,144
Treatment Algorithms, Controversies, Challenges, and Future Possibilities
Overall, most patients enjoy a favorable prognosis. Unfortunately, there remain a minority of children (up to 20% to 30%) with extensive organ involvement who succumb to LCH within 5 years. Targeted therapies for LCH are being investigated, and several monoclonal antibodies have been proposed or used in small case studies.145,146,147 The Histiocyte Society148 serves as a clearinghouse for information regarding LCH as well as for administration of prospective clinical trials.
Nasopharyngeal Carcinoma
Etiology and Epidemiology
Comparisons are often made between the adult and juvenile variants of nasopharyngeal carcinoma, and indeed much of the treatment regimens for PNC has been extrapolated from adult data owing to the rarity of the pediatric tumors. The diseases are similar in their relationship to Epstein-Barr virus infection, to diets containing nitrites and salted fish, and to the male predominance. However, in children, the greatest incidence occurs in the Mediterranean and North America rather than China, Southeast Asia, and Alaska; undifferentiated histology is the most common type; and patients present with significantly more advanced disease at diagnosis. In one retrospective series, 92% of pediatric patients had stage III to IV disease versus 67% of adults with nasopharyngeal carcinoma. Additionally, data show despite higher stage disease that children have a better overall prognosis than adults in both OS and locoregional disease control.149,150,151
Biologic Characteristics and Molecular Biology
Epstein-Barr virus (EBV) is the best established etiologic factor and is associated with 90% of PNCs, particularly the undifferentiated and nonkeratinizing types. The association with EBV was initially based on serologic studies that found high titers of IgG and IgA against the early antigen (D) or viral capsid antigen (VCA). EBV titers of IgG and IgA to these antigens have been shown to be an early predictor of clinical relapse or resistance to treatment in patients in the United States.152 The relationship between EBV and nasopharyngeal cancer was confirmed when EBV DNA was found in neoplastic epithelial cells but was absent in the surrounding lymphocytes.153
Clonal genomes have also been found in early dysplastic lesions, leading to the hypothesis that infection is an early event in carcinogenesis. The genome is a double-stranded DNA virus encoding 100 genes. However, only 10 of these are expressed in vitro; six are nuclear proteins (EBNAs), two are latent membrane proteins (LMPs), and there are two nontranslated RNA molecules (EBER) that likely aid in cell growth. In nasopharyngeal carcinoma, LMP1 acts as an oncogene, changing growth patterns and up-regulating proteins that inhibit apoptosis, including BCL2.154 It also binds to various proteins associated with tumor necrosis factor receptors and is involved in activation of transcription factors, cellular adhesion molecules, and cytokines. LMP2 is also expressed in nasopharyngeal carcinoma and acts by preventing reactivation of the virus by blocking phosphorylation of tyrosine kinases. EBERs are not transcribed as proteins but play a role in oncogenesis and resistance to apoptosis.154,155,156
There is also evidence for genetic predisposition based on human leukocyte antigen (HLA) subtypes. The HLA-A2 Bsin2 haplotype and HLA-Aw19, HLA-Bw46, and HLA-B17 types have been shown to have increased risk of nasopharyngeal carcinoma.157,158 Several studies have demonstrated deletions of chromosomes 3p and 16q; other aberrations include 9p, 11q, 13q, and 14q. These findings suggest that tumor suppressor genes are located in these regions and that loss of a normal copy of these genes can lead to promotion of PNC.154
Other well-known molecular changes include inactivation of TP53, rearrangement of the retinoblastoma tumor suppressor genes, and polymorphism of CYP2E1. Genetic gains on chromosome 12 as well as losses on 11q, 13q, and 16q have been associated with invasiveness, whereas TP53 mutation and altered expression of cadherins are associated with metastasis.157,159–161 Clinically, molecular markers have been pursued as a means to predict for prognosis and survival. Although there was no predictor for survival found in a study of TP53, BCL2, Ki67, and c-KIT, there was a trend toward a reduced recurrence rate; however, on multivariate analysis, only early stage and young age predicted for good prognosis.151 A small proportion of PNC exhibit t(15;19) with the BRD4-NUT oncogene; the typically midline NUT-rearranged carcinomas are particularly aggressive PNCs.
Thus, the development of PNC is thought to be a multiple-step process involving EBV infection resulting in the expression of LMP1 and overexpression of TP53 in the epithelial tumor cells. BCL2 has been studied in this population and, although it may be significant in adult nasopharyngeal carcinoma, it seems unrelated to the development of the pediatric disease.162
Pathology and Pathways of Spread
The World Health Organization (WHO) classification of nasopharyngeal carcinoma includes three histiotypes: keratinizing squamous cell carcinoma (type I), nonkeratinizing squamous cell carcinoma (type II), and undifferentiated carcinoma (type III, earlier called lymphoepithelioma). Lymphoepithelioma (or Schmincke’s tumor) refers to an undifferentiated tumor with prominent nonmalignant lymphocytic infiltration. Type I shows significant keratin production, is usually seen in adults, and is not usually associated with EBV. In type II there can be groups of cells with oval or fusiform nuclei and scant cytoplasm. The Cologne modification of the WHO classification further subdivides types II and III by degree of lymphoid infiltration; type IIa has pleomorphism without lymphoid infiltration, whereas type IIb has significant lymphoid infiltrate. Type IIIa is characterized by larger, eosinophilic nuclei, whereas type IIIb has smaller basophilic nuclei; virtually all PNC cases are type III undifferentiated carcinomas.154,163
Nasopharyngeal carcinoma typically originates in the fossa of Rosenmüller, the space posterolateral to the eustachian tube orifice. Routes of direct spread include the oropharynx or perineural spread along cranial nerves extending to the skull base. The cervical lymph nodes are typically involved early, but the spinal accessory chain and retropharyngeal nodes are at risk as well. Distant metastases at presentation are rare but when they occur are most commonly seen in the bones, lungs, liver, bone marrow, and mediastinum.164
Clinical Manifestations, Patient Evaluation, and Staging
Painless cervical lymphadenopathy is the most common presenting symptom, occurring in up to 90% of patients. The rich lymphatic supply of the nasopharynx explains this high rate of early nodal metastasis; regional lymphatics include the internal jugular nodes, the spinal accessory nodes, and the retropharyngeal nodes. The midline location of the nasopharynx puts the bilateral nodal basins at risk. Other common symptoms at presentation include nasal obstruction or bleeding, hearing loss, or otitis from obstruction of the eustachian tubes, which can lead to ear pain and tinnitus, headache, facial pain, neck pain, and various cranial neuropathies from invasion of the base of skull.154,157,158,165 Symptom duration before diagnosis ranges from 1 to 24 months, with an average of 5 months.166 The syndrome of inappropriate antidiuretic hormone secretion has been reported and can cause severe hyponatremia.154,164,167
Local extension in PNC is explained by anatomy; the pharyngobasilar fascia acts as a barrier and directs the spread of tumor toward the skull base anterior to the clivus. Indications of local bone and marrow involvement on MRI include decreased T1-weighted signal, increased T2-weighted signal, and postcontrast enhancement. The petroclival fissure may be widened from tumor invasion and is seen best on CT bone windows. In later stages of disease, tumor may cross the pharyngobasilar fascia to invade the parapharyngeal space.168
Staging is based on the American Joint Committee on Cancer staging system for nasopharyngeal carcinoma for adults (Table 73-6). The nodal staging for nasopharyngeal tumors takes into account the anatomic level of nodal involvement, an important prognostic factor; retropharyngeal lymph nodes, regardless of their laterality, are considered N1.169,170
TABLE 73-6 Nasopharyngeal Carcinoma Staging: American Joint Committee on Cancer
| Primary Tumor | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| Tis | Carcinoma in situ |
| T1 | Primary tumor confined to nasopharynx, extending to oropharynx, and/or nasal cavity without parapharyngeal space extension |
| T2 | Tumor extends to soft tissues |
| T2a | Tumor extends to oropharynx and/or nasal cavity (no parapharyngeal extension) |
| T2b | Tumor within parapharyngeal space* |
| T3 | Invasion of bone of skull base and/or paranasal sinuses |
| T4 | Tumor with intracranial extension and/or cranial nerve involvement, extension to hypopharynx, orbit, or infratemporal fossa/masticator space |
| Regional Lymph Nodes | |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node involvement |
| N1 | Unilateral metastasis ≤6 cm above the supraclavicular fossa and/or unilateral or bilateral retropharyngeal lymph nodes ≤6 cm |
| N2 | Bilateral lymph node metastasis, ≤6 cm, above the supraclavicular fossa |
| N3 | Metastasis in a lymph node >6 cm and/or extension to supraclavicular fossa |
| N3a | >6 cm |
| N3b | Extension to supraclavicular fossa |
| Distant Metastases | |
| M0 | No distant metastasis |
| M1 | Distant metastasis |
| AJCC Stage | |
| Stage 0 | TisN0M0 |
| Stage I | T1N0M0 |
| Stage IIA | T2aN0M0 |
| Stage IIB | T1-2aN1M0 T2bN0-1M0 |
| Stage III | T1-2a-2bN2M0 T3N0-1-2M0 |
| Stage IVA | T4N0-1-2M0 |
| Stage IVB | T1-4N3M0 |
| Stage IVC | M1 |
* Parapharyngeal extension denotes posterolateral spread of tumor.
From Edge SB, Byrd DR, Compton CC, et al, editors: AJCC Cancer Staging Handbook, ed 7, New York, 2010, Springer.
Primary and Adjuvant Therapy and Results
Locally recurrent or metastatic disease presents within 1 to 2 years; survival rates approximate 50% to 60%.154 Systemic therapies have thus been pursued, in the form of neoadjuvant, concurrent, and adjuvant chemotherapy, especially for the majority of patients with locally and/or regionally advanced disease.
Given the rarity of PNC, data have often been extrapolated from adult trials. Caution must be employed when extrapolating these data because PNCs are largely distinct biologic entities. The Intergroup 0099 trial compared chemoradiation with RT alone in adult patients with nasopharyngeal carcinoma, delivering 70 Gy with or without cisplatin, followed by three cycles of adjuvant cisplatin and 5-fluorouracil.171 Three-year progression-free survival was 69% versus 24% favoring the chemotherapy arm of the trial; 3-year OS was 78% versus 47%, also favoring the chemotherapy arm. Additionally, a recent meta-analysis by Langendijk and colleagues172 reported a significant survival benefit for the group who received concurrent chemoradiation: a 20% survival benefit with chemoradiation versus irradiation alone.
The earliest series of PNCs were published in the 1980s and generally included small numbers of patients treated with RT alone,173–175,176 noting OS of 50% to 55% with relapse-free survival in the range of 36% to 75%, following irradiation to 35 to 86 Gy.
Reports of PNCs published in the past 2 decades used better staging and improved RT techniques, including evolution of three-dimensional and IMRT volume-based therapy. Cisplatin-based chemotherapy regimens were also introduced in this era. Studies by Ayan and associates177 and Wolden and colleagues178 showed improved outcome after chemoradiation, particularly in the neoadjuvant setting; data suggested a benefit primarily when radiation doses exceeded 60 Gy, with OS in the most aggressively treated cohort as high as 76%.
Laskar and colleagues179 explored prognostic factors correlated with disease-free survival (DFS) and noted age, nodal stage, TNM stage, node size, node fixation, radiation dose, and response to chemotherapy to be significant. In comparing concurrent cyclophosphamide versus concurrent modified vincristine, cyclophosphamide, epirubicin, and dactinomycin (Actinomycin-D) versus neoadjuvant cisplatin in conjunction with 46 to 76 Gy to the nasopharynx and regional lymph nodes, Kupeli and colleagues180 documented the benefit of cisplatin, with OS 80% versus 63% with AVAC and 31% with cyclophosphamide. Parallel to the experience in adults, 10-year survival was 88% without lymph node involvement compared with 39% with nodes larger than 3 cm and 0% with supraclavicular node involvement. Ozyar and colleagues150 reported a large, multi-institutional international review of PNC over a 25-year interval documenting overall 5-year survival of 77.4% and DFS of 68.8% after combined irradiation and chemotherapy; the only significant negative prognostic factor for OS and DFS was N3 disease. RT doses less than 66 Gy adversely affected locoregional control, and RT without chemotherapy decreased locoregional control and DFS.
A German prospective pediatric trial tested three cycles of cisplatin, 5-fluorouracil, methotrexate, and leucovorin followed by RT (59.4 Gy) for stage III/IV PNC; patients were then maintained on interferon-beta for 6 months. The 108-month DFS was 91%; OS was 95%. The excellent outcome was hypothesized as due, in part, to the administration of interferon-beta during a period of relative immune suppression in EBV-positive nasopharyngeal carcinoma.163,181
The Pediatric Oncology Group 9486 study showed 4-year event-free and OS of 77% and 75%, respectively, following neoadjuvant chemotherapy (cisplatin, 5-fluorouracil, methotrexate, and leucovorin) and RT (50.4 Gy to the upper neck, 45 Gy to the lower neck, and a boost to the primary tumor to a total dose of 61.2 Gy).182
There is significant toxicity using doses of 50 to 70 Gy in children and adolescents. Xerostomia, neck fibrosis, dental caries, chronic sinusitis, chronic serous otitis, trismus, hypopituitarism, stunted growth, hypothyroidism, hypoplasia of facial bones or clavicles, and second malignancies have been well documented154,178,180 (Table 73-7).
TABLE 73-7 Incidence of Treatment-Related Toxicity in Nasopharyngeal Cancer
| Complication | Percentage |
|---|---|
| Xerostomia | 3.5-84 |
| Soft tissue fibrosis | 23-61 |
| Dental caries | 7-33 |
| Chronic otitis | 18 |
| Hypothyroidism | 9.5-12 |
| Trismus | 7.1-9 |
| Chronic sinusitis | 7 |
| Second malignancy | 2-6 |
| Osteoradionecrosis | 3 |
| Optic neuritis | 3 |
The optimal dose of RT for disease control has been controversial, especially in the setting of effective systemic therapy. Although earlier reports suggested improved locoregional control with doses greater than 60 Gy, recent studies have explored lower-dose, response-adapted RT. Habrand and associates183 reported outcome after sequential cisplatin-based regimens and response-adapted RT (50 Gy after marked response and 65 to 70 Gy after poor chemoresponse).177,183 Five-year event-free survival and OS were 64% and 68%, respectively; locoregional failures were similar between the groups, but the low RT dose group actually had significantly improved OS and event-free survival, with apparent reduction in late toxicities. Orbach and colleagues184 reported results following one of three chemotherapy protocols (both neoadjuvant and adjuvant chemotherapy). Objective response to preirradiation chemotherapy was apparent in 78% of cases; those with complete response or very good partial response received an attenuated RT (median dose 47 Gy to the clinically negative cervical nodes and 59.4 Gy to the primary tumor); those with less response received a 60-Gy median dose to all involved sites. OS for the entire group was 73% at 5 years, and local failure rate was 10% with regional failure rate of 3% and distant failure rate of 18%. The 5-year OS did not correlate with radiation dose; no patient demonstrated isolated cervical nodal relapse.
Irradiation Techniques
The principles of RT for PNC require delivering a tumoricidal dose to the primary tumor and clinically involved nodes, as well as a prophylactic dose to nodal areas clinically at risk. The optimal doses for tumor eradication are not yet known, but there is emerging evidence that radiation dose can be adapted based on clinical response to neoadjuvant chemotherapy.183,184 Recent data suggest that IMRT with sparing of normal tissues offers reduced toxicity and superior target coverage; experience with both acute and long-term tolerance suggest IMRT may be the treatment of choice, recognizing concerns regarding an increased risk of secondary malignancies in a pediatric population due to an increase in integral dose with IMRT. This risk has been projected to be 2% for patients treated with IMRT compared with 1% for patients treated with conventional radiation techniques.185
6 Schnater JM, Aronson DC, Plaschkes J, et al. Surgical view of the treatment of patients with hepatoblastoma. Results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer. 2002;94:1111-1120.
7 Czauderna P, Mackinlay G, Perilongo G, et al. Hepatocellular carcinoma in children. Results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol. 2002;20:2798-2804.
8 Pritchard J, Brown J, Shafford E, et al. Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma. A successful approach. Results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol. 2000;18:3819-3828.
9 Fuchs J, Rydzynski J, Von Schweinitz D, et al. Pretreatment prognostic factors and treatment results in children with hepatoblastoma. A report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer. 2002;95:172-182.
10 Perilongo G, Maibach R, Shafford E, et al. Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med. 2009;361:1662-1670.
11 Katzenstein HM, London WB, Douglass EC, et al. Treatment of unresectable and metastatic hepatoblastoma. A Pediatric Oncology Group phase II study. J Clin Oncol. 2002;20:3438-3444.
14 Rescorla FJ. Pediatric germ cell tumors. Semin Surg Oncol. 1999;16:144-158.
16 Gobel U, Schneider DT, Calaminus G, et al. Germ-cell tumors in childhood and adolescence. GPOH MAKEI and the MAHO study groups. Ann Oncol. 2000;11:263-271.
18 Billmire D, Vinocur C, Rescorla F, et al. Outcome and staging evaluation in malignant germ cell tumors of the ovary in children and adolescents, An intergroup study. J Pediatr Surg. 2004;39:424-429. discussion 429
20 Cushing B, Giller R, Cullen JW, et al. Randomized comparison of combination chemotherapy with etoposide, bleomycin, and either high-dose or standard-dose cisplatin in children and adolescents with high-risk malignant germ cell tumors. A pediatric intergroup study—Pediatric Oncology Group 9049 and Children’s Cancer Group 8882. J Clin Oncol. 2004;22:2691-2700.
24 Bleier BS, Kennedy DW, Palmer JN, et al. Current management of juvenile nasopharyngeal angiofibroma. A tertiary center experience 1999-2007. Am J Rhinol Allergy. 2009;23:328-330.
26 Valanzano R, Curia MC, Aceto G, et al. Genetic evidence that juvenile nasopharyngeal angiofibroma is an integral FAP tumour. Gut. 2005;54:1046-1047.
28 Schlauder SM, Knapp C, Steffensen TS, Bui MM. Aromatase may play a critical role in the pathogenesis of juvenile nasopharyngeal angiofibroma. Fetal Pediatr Pathol. 2009;28:232-238.
37 Carrillo JF, Maldonado F, Albores O, et al. Juvenile nasopharyngeal angiofibroma. Clinical factors associated with recurrence, and proposal of a staging system. J Surg Oncol. 2008;98:75-80.
42 Spielmann PM, Adamson R, Cheng K, Sanderson RJ. Juvenile nasopharyngeal angiofibroma. Spontaneous resolution. Ear Nose Throat J. 2008;87:521-523.
44 Reddy KA, Mendenhall WM, Amdur RJ, et al. Long-term results of radiation therapy for juvenile nasopharyngeal angiofibroma. Am J Otolaryngol. 2001;22:172-175.
50 Labra A, Chavolla-Magana R, Lopez-Ugalde A, et al. Flutamide as a preoperative treatment in juvenile angiofibroma (JA) with intracranial invasion. Report of 7 cases. Otolaryngol Head Neck Surg. 2004;130:466-469.
53 Goepfert H, Cangir A, Lee YY. Chemotherapy for aggressive juvenile nasopharyngeal angiofibroma. Arch Otolaryngol. 1985;111:285-289.
55 Lee JT, Chen P, Safa A, et al. The role of radiation in the treatment of advanced juvenile angiofibroma. Laryngoscope. 2002;112:1213-1220.
63 Priest JR, Magnuson J, Williams GM, et al. Cerebral metastasis and other central nervous system complications of pleuropulmonary blastoma. Pediatr Blood Cancer. 2007;49:266-273.
66 Priest JR, Hill DA, Williams GM, et al. Type I pleuropulmonary blastoma. A report from the International Pleuropulmonary Blastoma Registry. J Clin Oncol. 2006;24:4492-4498.
67 Indolfi P, Bisogno G, Casale F, et al. Prognostic factors in pleuro-pulmonary blastoma. Pediatr Blood Cancer. 2007;48:318-323.
68 Parsons SK, Fishman SJ, Hoorntje LE, et al. Aggressive multimodal treatment of pleuropulmonary blastoma. Ann Thorac Surg. 2001;72:939-942.
69 Hill DA, Jarzembowski JA, Priest JR, et al. Type I pleuropulmonary blastoma. Pathology and biology study of 51 cases from the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol. 2008;32:282-295.
73 Tagge EP, Mulvihill D, Chandler JC, et al. Childhood pleuropulmonary blastoma. Caution against nonoperative management of congenital lung cysts. J Pediatr Surg. 1996;31:187-189. discussion 190
76 Priest JR, McDermott MB, Bhatia S, et al. Pleuropulmonary blastoma. A clinicopathologic study of 50 cases. Cancer. 1997;80:147-161.
78 . Pleuropulmonary Blastoma Registry. Available online at www.ppbregistry.org
105 Goodman KA, Wolden SL, La Quaglia MP, Kushner BH. Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys. 2002;54:170-176.
116 Quaglia MP, Brennan MF. The clinical approach to desmoplastic small round cell tumor. Surg Oncol. 2000;9:77-81.
117 Al Balushi Z, Bulduc S, Mulleur C, Lallier M. Desmoplastic small round cell tumor in children. A new therapeutic approach. J Pediatr Surg. 2009;44:949-952.
118 Lal DR, Su WT, Wolden SL, et al. Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg. 2005;40:251-255.
123 Gerald WL, Rosai J, Ladanyi M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci U S A. 1995;92:1028-1032.
124 Lichtenstein L. Histiocytosis X. Integration of eosinophilic granuloma of bone, “Letterer-Siwe disease” and “Schuller-Christian disease” as related manifestations of a single nosologic entity. Arch Pathol. 1953;56:84-102.
125 Broadbent V, Egeler RM, Nesbit MEJr. Langerhans cell histiocytosis—clinical and epidemiological aspects. Br J Cancer Suppl. 1994;23:S11-S16.
131 Schmitz L, Favara BE. Nosology and pathology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12:221-246.
132 Yagci B, Varan A, Caglar M, et al. Langerhans cell histiocytosis. Retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol. 2008;25:399-408.
133 Buckwalter JA, Brandser E, Robinson RA. The variable presentation and natural history of Langerhans cell histiocytosis. Iowa Orthop J. 1999;19:99-105.
140 Selch MT, Parker RG. Radiation therapy in the management of Langerhans cell histiocytosis. Med Pediatr Oncol. 1990;18:97-102.
141 Olschewski T, Seegenschmiedt MH. Radiotherapy for bony manifestations of Langerhans cell histiocytosis. Review and proposal for an international registry. Strahlenther Onkol. 2006;182:72-79.
142 Demiral AN. The role of radiotherapy in the management of diabetes insipidus caused by Langerhans cell histiocytosis. J BUON. 2002;7:217-219.
147 Windebank K, Nanduri V. Langerhans cell histiocytosis. Arch Dis Child. 2009;94:904-908.
148 The Histiocyte Society. Available at www.histiocytesociety.org
149 Downing NL, Wolden S, Wong P, et al. Comparison of treatment results between adult and juvenile nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys. 2009;75:1064-1070.
150 Ozyar E, Selek U, Laskar S, et al. Treatment results of 165 pediatric patients with non-metastatic nasopharyngeal carcinoma. A Rare Cancer Network study. Radiother Oncol. 2006;81:39-46.
154 Ayan I, Kaytan E, Ayan N. Childhood nasopharyngeal carcinoma. From biology to treatment. Lancet Oncol. 2003;4:13-21.
176 Ingersoll L, Shiao Y, Donaldson S. Nasopharyngeal carcinoma in the young. A combined M.D. Anderson and Stanford experience. Int J Radiat Oncol Biol Phys. 1990;19:881-887.
177 Ayan I, Altun M. Nasopharyngeal carcinoma in children. Retrospective review of 50 patients. Int J Radiat Oncol Biol Phys. 1996;35:485-492.
181 Mertens R, Granzen B, Lassay L, et al. Treatment of nasopharyngeal carcinoma in children and adolescents. Definitive results of a multicenter study (NPC-91-GPOH). Cancer. 2005;104:1083-1089.
184 Orbach D, Brisse H, Helfre S, et al. Radiation and chemotherapy combination for nasopharyngeal carcinoma in children. Radiotherapy dose adaptation after chemotherapy response to minimize late effects. Pediatr Blood Cancer. 2008;50:849-853.
1 Bellani FF, Massimino M. Liver tumors in childhood. Epidemiology and clinics. J Surg Oncol Suppl. 1993;3:119-121.
2 Fraumeni JFJr, Miller RW, Hill JA. Primary carcinoma of the liver in childhood. An epidemiologic study. J Natl Cancer Inst. 1968;40:1087-1099.
3 Schnater JM, Kohler SE, Lamers WH, et al. Where do we stand with hepatoblastoma? A review. Cancer. 2003;98:668-678.
4 Habrand JL, Nehme D, Kalifa C, et al. Is there a place for radiation therapy in the management of hepatoblastomas and hepatocellular carcinomas in children? Int J Radiat Oncol Biol Phys. 1992;23:525-531.
5 Katzenstein HM, Krailo MD, Malogolowkin MH, et al. Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer. 2003;97:2006-2012.
6 Schnater JM, Aronson DC, Plaschkes J, et al. Surgical view of the treatment of patients with hepatoblastoma. Results from the first prospective trial of the International Society of Pediatric Oncology Liver Tumor Study Group. Cancer. 2002;94:1111-1120.
7 Czauderna P, Mackinlay G, Perilongo G, et al. Hepatocellular carcinoma in children. Results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol. 2002;20:2798-2804.
8 Pritchard J, Brown J, Shafford E, et al. Cisplatin, doxorubicin, and delayed surgery for childhood hepatoblastoma. A successful approach. Results of the first prospective study of the International Society of Pediatric Oncology. J Clin Oncol. 2000;18:3819-3828.
9 Fuchs J, Rydzynski J, Von Schweinitz D, et al. Pretreatment prognostic factors and treatment results in children with hepatoblastoma. A report from the German Cooperative Pediatric Liver Tumor Study HB 94. Cancer. 2002;95:172-182.
10 Perilongo G, Maibach R, Shafford E, et al. Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med. 2009;361:1662-1670.
11 Katzenstein HM, London WB, Douglass EC, et al. Treatment of unresectable and metastatic hepatoblastoma. A Pediatric Oncology Group phase II study. J Clin Oncol. 2002;20:3438-3444.
12 Cheng JC, Wu JK, Huang CM, et al. Dosimetric analysis and comparison of three-dimensional conformal radiotherapy and intensity-modulated radiation therapy for patients with hepatocellular carcinoma and radiation-induced liver disease. Int J Radiat Oncol Biol Phys. 2003;56:229-234.
13 Brodeur GM, Howarth CB, Pratt CB, et al. Malignant germ cell tumors in 57 children and adolescents. Cancer. 1981;48:1890-1898.
14 Rescorla FJ. Pediatric germ cell tumors. Semin Surg Oncol. 1999;16:144-158.
15 Rescorla FJ, Breitfeld PP. Pediatric germ cell tumors. Curr Probl Cancer. 1999;23:257-303.
16 Gobel U, Schneider DT, Calaminus G, et al. Germ-cell tumors in childhood and adolescence. GPOH MAKEI and the MAHO study groups. Ann Oncol. 2000;11:263-271.
17 Bussey KJ, Lawce HJ, Olson SB, et al. Chromosome abnormalities of eighty-one pediatric germ cell tumors. Sex-, age-, site-, and histopathology-related differences. A Children’s Cancer Group study. Genes Chromosomes Cancer. 1999;25:134-146.
18 Billmire D, Vinocur C, Rescorla F, et al. Outcome and staging evaluation in malignant germ cell tumors of the ovary in children and adolescents, An intergroup study. J Pediatr Surg. 2004;39:424-429. discussion 429
19 Marina NM, Cushing B, Giller R, et al. Complete surgical excision is effective treatment for children with immature teratomas with or without malignant elements. A Pediatric Oncology Group/Children’s Cancer Group Intergroup Study. J Clin Oncol. 1999;17:2137-2143.
20 Cushing B, Giller R, Cullen JW, et al. Randomized comparison of combination chemotherapy with etoposide, bleomycin, and either high-dose or standard-dose cisplatin in children and adolescents with high-risk malignant germ cell tumors. A pediatric intergroup study—Pediatric Oncology Group 9049 and Children’s Cancer Group 8882. J Clin Oncol. 2004;22:2691-2700.
21 Billmire D, Vinocur C, Rescorla F, et al. Malignant retroperitoneal and abdominal germ cell tumors. An intergroup study. J Pediatr Surg. 2003;38:315-318. discussion 318
22 Ablin AR, Krailo MD, Ramsay NK, et al. Results of treatment of malignant germ cell tumors in 93 children. A report from the Children’s Cancer Study Group. J Clin Oncol. 1991;9:1782-1792.
23 Jereb B, Wollner N, Exelby P. Radiation in multidisciplinary treatment of children with malignant ovarian tumors. Cancer. 1979;43:1037-1042.
24 Bleier BS, Kennedy DW, Palmer JN, et al. Current management of juvenile nasopharyngeal angiofibroma. A tertiary center experience 1999-2007. Am J Rhinol Allergy. 2009;23:328-330.
25 Tang IP, Shashinder S, Gopala Krishnan G, Narayanan P. Juvenile nasopharyngeal angiofibroma in a tertiary centre. Ten-year experience. Singapore Med J. 2009;50:261-264.
26 Valanzano R, Curia MC, Aceto G, et al. Genetic evidence that juvenile nasopharyngeal angiofibroma is an integral FAP tumour. Gut. 2005;54:1046-1047.
27 Hwang HC, Mills SE, Patterson K, Gown AM. Expression of androgen receptors in nasopharyngeal angiofibroma. An immunohistochemical study of 24 cases. Mod Pathol. 1998;11:1122-1126.
28 Schlauder SM, Knapp C, Steffensen TS, Bui MM. Aromatase may play a critical role in the pathogenesis of juvenile nasopharyngeal angiofibroma. Fetal Pediatr Pathol. 2009;28:232-238.
29 Montag AG, Tretiakova M, Richardson M. Steroid hormone receptor expression in nasopharyngeal angiofibromas. Consistent expression of estrogen receptor beta. Am J Clin Pathol. 2006;125:832-837.
30 Saylam G, Yucel OT, Sungur A, Onerci M. Proliferation, angiogenesis and hormonal markers in juvenile nasopharyngeal angiofibroma. Int J Pediatr Otorhinolaryngol. 2006;70:227-234.
31 Liang J, Yi Z, Lianq P. The nature of juvenile nasopharyngeal angiofibroma. Otolaryngol Head Neck Surg. 2000;123:475-481.
32 Abraham SC, Wu TT. Nasopharyngeal angiofibroma. Hum Pathol. 2001;32:455.
33 Abraham SC, Montgomery EA, Giardiello FM, Wu TT. Frequent beta-catenin mutations in juvenile nasopharyngeal angiofibromas. Am J Pathol. 2001;158:1073-1078.
34 Margalit N, Wasserzug O, De-Row A, et al. Surgical treatment of juvenile nasopharyngeal angiofibroma with intracranial extension. J Neurosurg Pediatr. 2009;4:113-117.
35 Herman P, Lot G, Chapot R, et al. Long-term follow-up of juvenile nasopharyngeal angiofibromas. Analysis of recurrences. Laryngoscope. 1999;109:140-147.
36 Grybauskas V, Parker J, Friedman ML. Juvenile nasopharyngeal angiofibroma. Otolaryngol Clin North Am. 1986;19:647-657.
37 Carrillo JF, Maldonado F, Albores O, et al. Juvenile nasopharyngeal angiofibroma. Clinical factors associated with recurrence, and proposal of a staging system. J Surg Oncol. 2008;98:75-80.
38 Fisch U. The infratemporal fossa approach for nasopharyngeal tumors. Laryngoscope. 1983;93:36-44.
39 Andrews JC, Fisch U, Valavanis A, et al. The surgical management of extensive nasopharyngeal angiofibromas with the infratemporal fossa approach. Laryngoscope. 1989;99:429-437.
40 Chandler JR, Goulding R, Moskowitz L, Quencer RM. Nasopharyngeal angiofibromas. Staging and management. Ann Otol Rhinol Laryngol. 1984;93:322-329.
41 Radkowski D, McGill T, Healy GB, et al. Angiofibroma. Changes in staging and treatment. Arch Otolaryngol Head Neck Surg. 1996;122:122-129.
42 Spielmann PM, Adamson R, Cheng K, Sanderson RJ. Juvenile nasopharyngeal angiofibroma. Spontaneous resolution. Ear Nose Throat J. 2008;87:521-523.
43 Weprin LS, Siemers PT. Spontaneous regression of juvenile nasopharyngeal angiofibroma. Arch Otolaryngol Head Neck Surg. 1991;117:796-799.
44 Reddy KA, Mendenhall WM, Amdur RJ, et al. Long-term results of radiation therapy for juvenile nasopharyngeal angiofibroma. Am J Otolaryngol. 2001;22:172-175.
45 Economou TS, Abemayor E, Ward PH. Juvenile nasopharyngeal angiofibroma. An update of the UCLA experience, 1960-1985. Laryngoscope. 1988;98:170-175.
46 Jereb B, Anggard A, Baryd I. Juvenile nasopharyngeal angiofibroma. A clinical study of 69 cases. Acta Radiol Ther Phys Biol. 1970;9:302-310.
47 Sinha PP, Aziz HI. Juvenile nasopharyngeal angiofibroma. A report of seven cases. Radiology. 1978;127:501-503.
48 Vadivel SP, Bosch A, Jose B. Juvenile nasopharyngeal angiofibroma. J Surg Oncol. 1980;15:323-326.
49 Cummings BJ, Blend R, Keane T, et al. Primary radiation therapy for juvenile nasopharyngeal angiofibroma. Laryngoscope. 1984;94:1599-1605.
50 Labra A, Chavolla-Magana R, Lopez-Ugalde A, et al. Flutamide as a preoperative treatment in juvenile angiofibroma (JA) with intracranial invasion. Report of 7 cases. Otolaryngol Head Neck Surg. 2004;130:466-469.
51 Gates GA, Rice DH, Koopmann CFJr, Schuller DE. Flutamide-induced regression of angiofibroma. Laryngoscope. 1992;102:641-644.
52 Schick B, Kahle G, Hassler R, Draf W. [Chemotherapy of juvenile angiofibroma—an alternative?]. HNO. 1996;44:148-152.
53 Goepfert H, Cangir A, Lee YY. Chemotherapy for aggressive juvenile nasopharyngeal angiofibroma. Arch Otolaryngol. 1985;111:285-289.
54 Goepfert H, Cangir A, Ayala AG, Eftekhari F. Chemotherapy of locally aggressive head and neck tumors in the pediatric age group. Desmoid fibromatosis and nasopharyngeal angiofibroma. Am J Surg. 1982;144:437-444.
55 Lee JT, Chen P, Safa A, et al. The role of radiation in the treatment of advanced juvenile angiofibroma. Laryngoscope. 2002;112:1213-1220.
56 McAfee WJ, Morris CG, Amdur RJ, et al. Definitive radiotherapy for juvenile nasopharyngeal angiofibroma. Am J Clin Oncol. 2006;29:168-170.
57 Fields JN, Halverson KJ, Devineni VR, et al. Juvenile nasopharyngeal angiofibroma. Efficacy of radiation therapy. Radiology. 1990;176:263-265.
58 Cummings BJ. Juvenile nasopharyngeal angiofibroma. Control rates and treatment costs. Head Neck Surg. 1980;3:169-170.
59 Cummings BJ. The treatment of juvenile nasopharyngeal angiofibroma. The case for radiation therapy. J Laryngol Otol Suppl. 1983;8:101-102.
60 Park CK, Kim DG, Paek SH, et al. Recurrent juvenile nasopharyngeal angiofibroma treated with gamma knife surgery. J Korean Med Sci. 2006;21:773-777.
61 Dare AO, Gibbons KJ, Proulx GM, Fenstermaker RA. Resection followed by radiosurgery for advanced juvenile nasopharyngeal angiofibroma. Report of two cases. Neurosurgery. 2003;52:1207-1211. discussion 1211
62 Deguchi K, Fukuiwa T, Saito K, Kurono Y. Application of cyberknife for the treatment of juvenile nasopharyngeal angiofibroma. A case report. Auris Nasus Larynx. 2002;29:395-400.
63 Priest JR, Magnuson J, Williams GM, et al. Cerebral metastasis and other central nervous system complications of pleuropulmonary blastoma. Pediatr Blood Cancer. 2007;49:266-273.
64 Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population. A review and 25-year experience at a large children’s hospital. Arch Pathol Lab Med. 2008;132:1079-1103.
65 Manivel JC, Priest JR, Watterson J, et al. Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood. Cancer. 1988;62:1516-1526.
66 Priest JR, Hill DA, Williams GM, et al. Type I pleuropulmonary blastoma. A report from the International Pleuropulmonary Blastoma Registry. J Clin Oncol. 2006;24:4492-4498.
67 Indolfi P, Bisogno G, Casale F, et al. Prognostic factors in pleuro-pulmonary blastoma. Pediatr Blood Cancer. 2007;48:318-323.
68 Parsons SK, Fishman SJ, Hoorntje LE, et al. Aggressive multimodal treatment of pleuropulmonary blastoma. Ann Thorac Surg. 2001;72:939-942.
69 Hill DA, Jarzembowski JA, Priest JR, et al. Type I pleuropulmonary blastoma. Pathology and biology study of 51 cases from the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol. 2008;32:282-295.
70 Fosdal MB. Pleuropulmonary blastoma. J Pediatr Oncol Nurs. 2008;25:295-302.
71 Priest JR, Watterson J, Strong L, et al. Pleuropulmonary blastoma. A marker for familial disease. J Pediatr. 1996;128:220-224.
72 Senac MOJr, Wood BP, Isaacs H, Weller M. Pulmonary blastoma. A rare childhood malignancy. Radiology. 1991;179:743-746.
73 Tagge EP, Mulvihill D, Chandler JC, et al. Childhood pleuropulmonary blastoma. Caution against nonoperative management of congenital lung cysts. J Pediatr Surg. 1996;31:187-189. discussion 190
74 Kusafuka T, Kuroda S, Inoue M, et al. P53 gene mutations in pleuropulmonary blastomas. Pediatr Hematol Oncol. 2002;19:117-128.
75 Vargas SO, Nose V, Fletcher JA, Perez-Atayde AR. Gains of chromosome 8 are confined to mesenchymal components in pleuropulmonary blastoma. Pediatr Dev Pathol. 2001;4:434-445.
76 Priest JR, McDermott MB, Bhatia S, et al. Pleuropulmonary blastoma. A clinicopathologic study of 50 cases. Cancer. 1997;80:147-161.
77 Naffaa LN, Donnelly LF. Imaging findings in pleuropulmonary blastoma. Pediatr Radiol. 2005;35:387-391.
78 . Pleuropulmonary Blastoma Registry. Available online at www.ppbregistry.org
79 Ohta Y, Fujishima M, Hasegawa H, et al. High therapeutic effectiveness of postoperative irinotecan chemotherapy in a typical case of radiographically and pathologically diagnosed pleuropulmonary blastoma. J Pediatr Hematol Oncol. 2009;31:355-358.
80 Pinarli FG, Oguz A, Ceyda K, et al. Type II pleuropulmonary blastoma responsive to multimodal therapy. Pediatr Hematol Oncol. 2005;22:71-76.
81 Lal P, Sharma DN, Biswal BM, et al. Role of radiotherapy in conjunction with chemotherapy in giant pleuropulmonary blastoma. Indian Pediatr. 1998;35:186-188.
82 Bartlett JA, Riding KH, Salkeld LJ. Management of hemangiomas of the head and neck in children. J Otolaryngol. 1988;17:111-120.
83 Acevedo JL, Shah RK, Brietzke SE. Nonsurgical therapies for lymphangiomas. A systematic review. Otolaryngol Head Neck Surg. 2008;138:418-424.
84 Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children. A classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412-422.
85 Schild SE, Buskirk SJ, Frick LM, Cupps RE. Radiotherapy for large symptomatic hemangiomas. Int J Radiat Oncol Biol Phys. 1991;21:729-735.
86 Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473-486.
87 Ozeki M, Funato M, Kanda K, et al. Clinical improvement of diffuse lymphangiomatosis with pegylated interferon alfa-2b therapy. Case report and review of the literature. Pediatr Hematol Oncol. 2007;24:513-524.
88 Johnson DW, Klazynski PT, Gordon WH, Russell DA. Mediastinal lymphangioma and chylothorax. The role of radiotherapy. Ann Thorac Surg. 1986;41:325-328.
89 de Serres LM, Sie KC, Richardson MA. Lymphatic malformations of the head and neck. A proposal for staging. Arch Otolaryngol Head Neck Surg. 1995;121:577-582.
90 Costello SA, Seywright M. Postirradiation malignant transformation in benign haemangioma. Eur J Surg Oncol. 1990;16:517-519.
91 Braun-Falco O, Schultze U, Meinhof W, Goldschmidt H. Contact radiotherapy of cutaneous hemangiomas. Therapeutic effects and radiation sequelae in 818 patients. Arch Dermatol Res. 1975;253:237-247.
92 Musumeci ML, Schlecht K, Perrotta R, et al. Management of cutaneous hemangiomas in pediatric patients. Cutis. 2008;81:315-322.
93 Ogino I, Torikai K, Kobayasi S, et al. Radiation therapy for life- or function-threatening infant hemangioma. Radiology. 2001;218:834-839.
94 O’Cathail S, Rostom AY, Johnson ML. Successful control of lymphangioma circumscriptum by superficial X-rays. Br J Dermatol. 1985;113:611-615.
95 Denton AS, Baker-Hines R, Spittle MF. Radiotherapy is a useful treatment for lymphangioma circumscriptum. A report of two patients. Clin Oncol (R Coll Radiol). 1996;8:400-401.
96 Yildiz F, Atahan IL, Ozyar E, et al. Radiotherapy in congenital vulvar lymphangioma circumscriptum. Int J Gynecol Cancer. 2008;18:556-559.
97 Hesselmann S, Micke O, Marquardt T, et al. Case report: Kasabach-Merritt syndrome. A review of the therapeutic options and a case report of successful treatment with radiotherapy and interferon alpha. Br J Radiol. 2002;75:180-184.
98 Mitsuhashi N, Furuta M, Sakurai H, et al. Outcome of radiation therapy for patients with Kasabach-Merritt syndrome. Int J Radiat Oncol Biol Phys. 1997;39:467-473.
99 Lezama-del Valle P, Gerald WL, Tsai J, et al. Malignant vascular tumors in young patients. Cancer. 1998;83:1634-1639.
100 Costa MA, Sousa A, Vieira E. Hemangioendothelioma. A rare vascular tumor in childhood and adolescence. Pediatr Hematol Oncol. 1996;13:333-337.
101 Lindberg S. Radiotherapy of childhood haemangiomas. From active treatment to radiation risk estimates. Radiat Environ Biophys. 2001;40:179-189.
102 Chang B, Gu H, Qian H, et al. Treatment of cutaneous hemangiomas with low-dose soft x-ray. Chin Med J (Engl). 1997;110:893-894.
103 Sealy R, Barry L, Buret E, et al. Cavernous haemangioma of the head and neck in the adult. J R Soc Med. 1989;82:198-201.
104 Gerald WL, Miller HK, Battifora H, et al. Intra-abdominal desmoplastic small round-cell tumor. Report of 19 cases of a distinctive type of high-grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol. 1991;15:499-513.
105 Goodman KA, Wolden SL, La Quaglia MP, Kushner BH. Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys. 2002;54:170-176.
106 Nathan JD, Gingalewsik C, Salem RR. Intra-abdominal desmoplastic small round cell tumor. Yale J Biol Med. 1999;72:287-293.
107 Takekawa Y, Ugajin W, Koide H, et al. Pathologic, cytologic and immunohistochemical findings of an intra-abdominal desmoplastic small round cell tumor in a 15-year-old male. Pathol Int. 2000;50:417-420.
108 Chan AS, MacNeill S, Thorner P, et al. Variant EWS-WT1 chimeric product in the desmoplastic small round cell tumor. Pediatr Dev Pathol. 1999;2:188-192.
109 Chalasani P, Shtivelband MI, Cranmer LD. Advanced desmoplastic small round cell tumor. Near complete response with trastuzumab-based chemotherapy. Clin Adv Hematol Oncol. 2009;7:473-475.
110 Parkash V, Gerald WL, Parma A, et al. Desmoplastic small round cell tumor of the pleura. Am J Surg Pathol. 1995;19:659-665.
111 Murphy AJ, Bishop K, Pereira C, et al. A new molecular variant of desmoplastic small round cell tumor. Significance of WT1 immunostaining in this entity. Hum Pathol. 2008;39:1763-1770.
112 Cummings TJ, Brown NM, Stenzel TT. TaqMan junction probes and the reverse transcriptase polymerase chain reaction. Detection of alveolar rhabdomyosarcoma, synovial sarcoma, and desmoplastic small round cell tumor. Ann Clin Lab Sci. 2002;32:219-224.
113 Neder L, Scheithauer BW, Turel KE, et al. Desmoplastic small round cell tumor of the central nervous system. Report of two cases and review of the literature. Virchows Arch. 2009;454:431-439.
114 Collardeau-Frachon S, Ranchere-Vince D, Delattre O, et al. Primary desmoplastic small round cell tumor of the kidney. A case report in a 14-year-old girl with molecular confirmation. Pediatr Dev Pathol. 2007;10:320-324.
115 Karavitakis EM, Moschovi M, Stefanaki K, et al. Desmoplastic small round cell tumor of the pleura. Pediatr Blood Cancer. 2007;49:335-338.
116 Quaglia MP, Brennan MF. The clinical approach to desmoplastic small round cell tumor. Surg Oncol. 2000;9:77-81.
117 Al Balushi Z, Bulduc S, Mulleur C, Lallier M. Desmoplastic small round cell tumor in children. A new therapeutic approach. J Pediatr Surg. 2009;44:949-952.
118 Lal DR, Su WT, Wolden SL, et al. Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg. 2005;40:251-255.
119 Kushner BH, LaQuaglia MP, Wollner N, et al. Desmoplastic small round-cell tumor. Prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol. 1996;14:1526-1531.
120 Hayes-Jordan A, Anderson P, Curley S, et al. Continuous hyperthermic peritoneal perfusion for desmoplastic small round cell tumor. J Pediatr Surg. 2007;42:E29-E32.
121 Jahraus CD, Glisson SDSt. Clair WH: Treatment of desmoplastic small round cell tumor with image-guided intensity modulated radiation therapy as a component of multimodality treatment. Tumori. 2005;91:253-255.
122 Saab R, Khoury JD, Krasin M, et al. Desmoplastic small round cell tumor in childhood. The St. Jude Children’s Research Hospital Experience. Pediatr Blood Cancer. 2008;49:274-279.
123 Gerald WL, Rosai J, Ladanyi M. Characterization of the genomic breakpoint and chimeric transcripts in the EWS-WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci U S A. 1995;92:1028-1032.
124 Lichtenstein L. Histiocytosis X. Integration of eosinophilic granuloma of bone, “Letterer-Siwe disease” and “Schuller-Christian disease” as related manifestations of a single nosologic entity. Arch Pathol. 1953;56:84-102.
125 Broadbent V, Egeler RM, Nesbit MEJr. Langerhans cell histiocytosis—clinical and epidemiological aspects. Br J Cancer Suppl. 1994;23:S11-S16.
126 Grifo AH. Langerhans cell histiocytosis in children. J Pediatr Oncol Nurs. 2009;26:41-47.
127 Alston RD, Tatevossian RG, McNally RJ, et al. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007;48:555-560.
128 Carstensen H, Ornvold K. [Langerhans-cell histiocytosis (histiocytosis X) in children]. Ugeskr Laeger. 1993;155:1779-1783.
129 Ladisch S. Langerhans cell histiocytosis. Curr Opin Hematol. 1998;5:54-58.
130 De Filippi P, Badulli C, Cuccia M, et al. Specific polymorphisms of cytokine genes are associated with different risks to develop single-system or multi-system childhood Langerhans cell histiocytosis. Br J Haematol. 2006;132:784-787.
131 Schmitz L, Favara BE. Nosology and pathology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am. 1998;12:221-246.
132 Yagci B, Varan A, Caglar M, et al. Langerhans cell histiocytosis. Retrospective analysis of 217 cases in a single center. Pediatr Hematol Oncol. 2008;25:399-408.
133 Buckwalter JA, Brandser E, Robinson RA. The variable presentation and natural history of Langerhans cell histiocytosis. Iowa Orthop J. 1999;19:99-105.
134 Allen CE, McClain KL. Langerhans cell histiocytosis. A review of past, current and future therapies. Drugs Today (Barc). 2007;43:627-643.
135 Morimoto A, Ikushima S, Kinugawa N, et al. Improved outcome in the treatment of pediatric multifocal Langerhans cell histiocytosis. Results from the Japan Langerhans Cell Histiocytosis Study Group-96 protocol study. Cancer. 2006;107:613-619.
136 Gasent Blesa JM, Alberola Candel V, Solano Vercet C, et al. Langerhans cell histiocytosis. Clin Transl Oncol. 2008;10:688-696.
137 Titgemeyer C, Grois N, Minkov M, et al. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL-HX 83- and 90-study. Med Pediatr Oncol. 2001;37:108-114.
138 Kudo K, Ohga S, Morimoto A, et al. Improved outcome of refractory Langerhans cell histiocytosis in children with hematopoietic stem cell transplantation in Japan. Bone Marrow Transplant. 2010;45:901-906.
139 Kesik V, Citak C, Kismet E, et al. Hematopoietic stem cell transplantation in Langerhans cell histiocytosis. Case report and review of the literature. Pediatr Transplant. 2009;13:371-374.
140 Selch MT, Parker RG. Radiation therapy in the management of Langerhans cell histiocytosis. Med Pediatr Oncol. 1990;18:97-102.
141 Olschewski T, Seegenschmiedt MH. Radiotherapy for bony manifestations of Langerhans cell histiocytosis. Review and proposal for an international registry. Strahlenther Onkol. 2006;182:72-79.
142 Demiral AN. The role of radiotherapy in the management of diabetes insipidus caused by Langerhans cell histiocytosis. J BUON. 2002;7:217-219.
143 Isoo A, Ueki K, Ishida T, et al. Langerhans cell histiocytosis limited to the pituitary-hypothalamic axis—two case reports. Neurol Med Chir (Tokyo). 2000;40:532-535.
144 Rosenzweig KE, Arceci RJ, Tarbell NJ. Diabetes insipidus secondary to Langerhans’ cell histiocytosis: is radiation therapy indicated? Med Pediatr Oncol. 1997;29:36-40.
145 Dina A, Zahava V, Iness M. The role of vascular endothelial growth factor in Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2005;27:62-66.
146 Jordan MB, McClain KL, Yan X, et al. Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005;44:251-254.
147 Windebank K, Nanduri V. Langerhans cell histiocytosis. Arch Dis Child. 2009;94:904-908.
148 . The Histiocyte Society. Available at www.histiocytesociety.org
149 Downing NL, Wolden S, Wong P, et al. Comparison of treatment results between adult and juvenile nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys. 2009;75:1064-1070.
150 Ozyar E, Selek U, Laskar S, et al. Treatment results of 165 pediatric patients with non-metastatic nasopharyngeal carcinoma. A Rare Cancer Network study. Radiother Oncol. 2006;81:39-46.
151 Bar-Sela G, Ben Arush MW, Sabo E, et al. Pediatric nasopharyngeal carcinoma. Better prognosis and increased c-Kit expression as compared to adults. Pediatr Blood Cancer. 2005;45:291-297.
152 Naegele RF, Champion J, Murphy S, et al. Nasopharyngeal carcinoma in American Children. Epstein-Barr virus-specific antibody titers and prognosis. Int J Cancer. 1982;29:209-212.
153 Hawkins EP, Krischer JP, Smith BE, et al. Nasopharyngeal carcinoma in children—a retrospective review and demonstration of Epstein-Barr viral genomes in tumor cell cytoplasm. A report of the Pediatric Oncology Group. Hum Pathol. 1990;21:805-810.
154 Ayan I, Kaytan E, Ayan N. Childhood nasopharyngeal carcinoma. From biology to treatment. Lancet Oncol. 2003;4:13-21.
155 Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481-492.
156 Vasef MA, Ferlito A, Weiss LM. Nasopharyngeal carcinoma, with emphasis on its relationship to Epstein-Barr virus. Ann Otol Rhinol Laryngol. 1997;106:348-356.
157 Vokes EE, Liebowitz DN, Weichselbaum RR. Nasopharyngeal carcinoma. Lancet. 1997;350:1087-1091.
158 Cvitkovic E, Bachouchi M, Armand JP. Nasopharyngeal carcinoma. Biology, natural history, and therapeutic implications. Hematol Oncol Clin North Am. 1991;5:821-838.
159 Claudio PP, Howard CM, Fu Y, et al. Mutations in the retinoblastoma-related gene RB2/p130 in primary nasopharyngeal carcinoma. Cancer Res. 2000;60:8-12.
160 Sheu LF, Chen A, Meng CL, et al. Analysis of bcl-2 expression in normal, inflamed, dysplastic nasopharyngeal epithelia, and nasopharyngeal carcinoma. Association with p53 expression. Hum Pathol. 1997;28:556-562.
161 Hildesheim A, Anderson LM, Chen CJ, et al. CYP2E1 genetic polymorphisms and risk of nasopharyngeal carcinoma in Taiwan. J Natl Cancer Inst. 1997;89:1207-1212.
162 Preciado MV, Chabay PA, De Matteo EN, et al. Epstein Barr virus associated pediatric nasopharyngeal carcinoma. Its correlation with p53 and bcl-2 expression. Med Pediatr Oncol. 2002;38:345-348.
163 Mertens R, Granzen B, Lassay L, et al. Nasopharyngeal carcinoma in childhood and adolescence. Concept and preliminary results of the cooperative GPOH study NPC-91. Gesellschaft fur Padiatrische Onkologie und Hamatologie. Cancer. 1997;80:951-959.
164 Douglass E. Management of infrequent cancers of childhood. In: Pizzo P, editor. Principles and Practice of Pediatric Oncology. ed 3. Philadelphia: Lippincott-Raven; 1997:977-1003.
165 Polychronopoulou S, Kostaridou S, Panagiotou JP, et al. Nasopharyngeal carcinoma in childhood and adolescence. A single institution’s experience with treatment modalities during the last 15 years. Pediatr Hematol Oncol. 2004;21:393-402.
166 Afqir S, Ismaili N, Alaoui K, et al. Nasopharyngeal carcinoma in adolescents. A retrospective review of 42 patients. Eur Arch Otorhinolaryngol. 2009;266:1767-1773.
167 Krmar RT, Ferraris JR, Ruiz SE, et al. Syndrome of inappropriate secretion of antidiuretic hormone in nasopharynx carcinoma. Pediatr Nephrol. 1997;11:502-503.
168 Stambuk HE, Patel SG, Mosier KM, et al. Nasopharyngeal carcinoma. Recognizing the radiographic features in children. AJNR Am J Neuroradiol. 2005;26:1575-1579.
169 American Joint Committee on Cancer. AJCC Cancer Staging Manual, 7th ed. New York: Springer; 2010.
170 Cooper JS, Cohen R, Stevens RE. A comparison of staging systems for nasopharyngeal carcinoma. Cancer. 1998;83:213-219.
171 Al-Sarraf M, LeBlanc M, Giri PG, et al. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer. Phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16:1310-1317.
172 Langendijk JA, Leemans CR, Buter J, et al. The additional value of chemotherapy to radiotherapy in locally advanced nasopharyngeal carcinoma. A meta-analysis of the published literature. J Clin Oncol. 2004;22:4604-4612.
173 Lombardi F, Gasparini M, Gianni C, et al. Nasopharyngeal carcinoma in childhood. Med Pediatr Oncol. 1982;10:243-250.
174 Gasparini M, Lombardi F, Rottoli L, et al. Combined radiotherapy and chemotherapy in stage T3 and T4 nasopharyngeal carcinoma in children. J Clin Oncol. 1988;6:491-494.
175 Jenkin RD, Anderson JR, Jereb B, et al. Nasopharyngeal carcinoma—a retrospective review of patients less than thirty years of age. A report of Children’s Cancer Study Group. Cancer. 1981;47:360-366.
176 Ingersoll L, Shiao Y, Donaldson S. Nasopharyngeal carcinoma in the young. A combined M.D. Anderson and Stanford experience. Int J Radiat Oncol Biol Phys. 1990;19:881-887.
177 Ayan I, Altun M. Nasopharyngeal carcinoma in children. Retrospective review of 50 patients. Int J Radiat Oncol Biol Phys. 1996;35:485-492.
178 Wolden SL, Steinherz PG, Kraus DH, et al. Improved long-term survival with combined modality therapy for pediatric nasopharynx cancer. Int J Radiat Oncol Biol Phys. 2000;46:859-864.
179 Laskar S, Sanghavi V, Muckaden MA, et al. Nasopharyngeal carcinoma in children. Ten years’ experience at the Tata Memorial Hospital, Mumbai. Int J Radiat Oncol Biol Phys. 2004;58:189-195.
180 Kupeli S, Varan A, Ozyar E, et al. Treatment results of 84 patients with nasopharyngeal carcinoma in childhood. Pediatr Blood Cancer. 2006;46:454-458.
181 Mertens R, Granzen B, Lassay L, et al. Treatment of nasopharyngeal carcinoma in children and adolescents. Definitive results of a multicenter study (NPC-91-GPOH). Cancer. 2005;104:1083-1089.
182 Rodriguez-Galindo C, Wofford M, Castleberry RP, et al. Preradiation chemotherapy with methotrexate, cisplatin, 5-fluorouracil, and leucovorin for pediatric nasopharyngeal carcinoma. Cancer. 2005;103:850-857.
183 Habrand J, Guillot Valls D, Petras S, et al. Carcinoma of the nasopharynx in children and adolescents treated with initial chemotherapy followed by adapted doses of radiotherapy. Int J Radiat Oncol Biol Phys. 2004;60:S247.
184 Orbach D, Brisse H, Helfre S, et al. Radiation and chemotherapy combination for nasopharyngeal carcinoma in children. Radiotherapy dose adaptation after chemotherapy response to minimize late effects. Pediatr Blood Cancer. 2008;50:849-853.
185 Varan A, Ozyar E, Corapcioglu F, et al. Pediatric and young adult nasopharyngeal carcinoma patients treated with preradiation cisplatin and docetaxel chemotherapy. Int J Radiat Oncol Biol Phys. 2009;73:1116-1120.