5. Randomized controlled trials
Claudia M. Witt and George T. Lewith
Chapter contents
Confirmatory and exploratory studies102
Study hypothesis and hypothesis testing103
Determining the sample size104
The essential elements of a randomized controlled trial105
Randomization106
Blinding109
Controls110
The influence of expectation111
Pragmatic versus experimental studies114
Analyzing randomized controlled trials115
Conclusion116
Introduction
We plan to discuss the principles and concepts that underpin randomized controlled trials (RCTs). This research methodology can be employed in a number of different contexts but RCTs are usually used in clinical settings. A trial, holding other factors constant, can be run to compare performance about a number of criteria, such as: ‘which treatment works better than a placebo?’ or ‘which device is most energy-efficient?’ The fundamental principles of RCTs remain the same in whichever context they are applied and this chapter aims to outline the steps that are important in setting up an RCT and meeting the requirements of a research protocol. RCTs allow us to answer very specific questions. They set out to evaluate the effect of a particular treatment or strategy in a population where an intervention is introduced, often by comparing the outcome with a control group where no intervention, or a standard intervention, may have been used. The population must be well defined and carefully selected.
A number of implicit assumptions underpin the RCT as follows (adapted from Vickers et al. 1997):
• We have an incomplete understanding of the world and knowledge evolves and develops. It is contingent and never definitive.
• Research methods evolve and change as we continue to learn about the world. Thus any trial will be as good as we can make it at the time and the knowledge gained is likely to be modest and incremental.
• Logically, cause precedes effect, or put another way, A leads to B.
• Beliefs cannot influence random events.
• In a well-designed study, the researcher’s beliefs cannot influence the outcome.
• Good research aims to minimize the effects of bias, chance variation and confounding.
• Research that investigates whether treatments do more good than harm must be a priority.
These tenets lead to the claim that the RCT provides the ’gold standard’ for research. It is the best means of attributing real cause and effect and therefore adds to our stock of knowledge. Not all researchers necessarily believe all of these, but they provide a sound framework for conducting a RCT.
The research question
An RCT is challenging and researchers contemplating a trial must ask themselves:
• What is a good question?
• How can questions be matched to the research design?
• What is the best strategic approach to the research?
• How can we interpret the results appropriately?
The first prerequisite for refining a research question is to carry out a thorough literature search. A literature search is an iterative and developmental process that will contribute directly and indirectly to protocol development. It will help to identify whether the question one wants to ask has already been asked and also point out the strengths and weaknesses of previous research in addressing and answering the question.
A question is likely to be answerable if it is explicit, focused and feasible. In other words, it should be possible to link the effect of an intervention explicitly to a specific outcome. The research should be focused. There should be a very clear, simple primary question and a research method that will provide an answer – the trick is not to ask too many primary questions simultaneously, even in a complex study. If there are multiple questions, then the primary research question must be given priority. The primary research question must be framed so that it is both possible and practical to answer the question. Some examples of questions are given in Table 5.1.
| Category of question | Examples | Suitable for RCT |
|---|---|---|
| Attributing cause and clinical effect | Does homeopathically prepared grass pollen reduce symptoms of hayfever more than a non-active (placebo) treatment? | Y |
| Is polypharmacy more effective than a single-remedy approach in the homeopathic treatment of chronic hayfever? | Y | |
| What happens in clinical practice? | What is the cost-effectiveness of adding homeopathic treatment to a standard care package in hayfever? | Y |
| What are the patterns of cross-referral between conventional and CAM practitioners in a multidisciplinary pain clinic? | N | |
| How common are serious neurological complications following chiropractic cervical manipulation? | N | |
| What do people do? | How many people visit practitioners of CAM each year? | N |
| What do patients tell their primary care physician about usage of CAM? | N | |
| How many nurses practice complementary medicine? | N | |
| What do people believe and how do they explain it? | What do nurses believe about therapeutic touch? | N |
| What is the patient’s experience of the acupuncture consultation? | Y | |
| By what mechanisms does a therapy work? | What are the effects of needling the Hoku point on the production of endogenous opiates? | Y |
| Does something proposed in a therapy actually exist? | Does peppermint oil reduce histamine-induced contractions of tracheal smooth muscle? | Y |
| Does homeopathically prepared copper ameliorate the effects of copper poisoning in a plant model? | Y | |
| Is a diagnostic or prognostic test accurate? | How sensitive and specific is detection of gallbladder disease by examining photos of the iris? | N |
| Is tongue diagnosis reliable? | N |
The findings must be achievable within a reasonable period of time and within the bounds of the scientific and financial resources available. Randomization is designed to even out all the things we ‘don’t know’ about the groups we are comparing. These factors may allow for misinterpretation of the study’s findings and they are usually considered to be ‘confounding factors’. Randomization also involves minimizing ‘bias’: if either confounding factors or biases are known to the researcher, then they should be introduced into the study protocol at an early stage and may result in possibly modifying the research question or trial methodology, thus allowing an appropriate trial design to emerge.
Confirmatory and exploratory studies
In effect RCTs are mainly used to test a predefined hypothesis. When planning a confirmatory trial it is helpful to do a pilot trial with the aim of generating a hypothesis and to test the planned outcome measures and the study protocol for feasibility. In addition, pilot studies can be helpful in providing an idea of the effect size of the intervention and this in turn will inform the sample size for any larger, more definitive study. In these pilot trials statistics are used on an exploratory basis. The present published literature suggests that in CAM research confirmatory studies are often done without pilot trials and quite a large proportion of the confirmatory (definitive) studies seem like pilot studies and are too small to have enough power to detect a significant difference between groups. The reasons for this might be limited financial resources for CAM research and the fact that CAM has fewer qualified and experienced researchers than conventional medical research environments.
When planning a study it is important to clarify the study in more detail by considering the following aspects (Chow & Liu 2004):
• What aspects of the intervention are being studied?
• Is it important to investigate other issues that may have an impact on the intervention?
• Which control(s) or placebos might be used or considered?
Developing a hypothesis is an important issue when planning these studies. A hypothesis always consists of a null hypothesis (H0) which assumes no effect and an alternative hypothesis (HA) which holds the null hypothesis not to be true and consequently assumes an effect. The alternative hypothesis is more directly connected to the specific research question.
The aim of confirmatory studies is to answer research questions based on the hypothesis proposed by the researcher. Therefore, posing an adequate and answerable research question is essential as a clear, well-researched, thoughtful and specific question based on appropriate study design has a reasonable chance of providing a valid answer. One of the main reasons (other than failure to recruit) why the majority of research proposals are either unclear or fail to provide a useful answer is that the initial research question itself lacks clarity. The core elements of a precise research question for a confirmatory RCT can be summarized by PICO (patients, intervention, control intervention, outcome = primary endpoint; see example in Figure 5.1). Using PICO can be very helpful in developing your hypothesis.

Study hypothesis and hypothesis testing
The research question and the subsequent hypothesis build the basis for hypothesis testing. If the null hypothesis can be rejected based on a predefined significance level (e.g. 5% or 1%), the alternative hypothesis will be accepted. This means that, from the example shown in Figure 5.1, if there is a significant difference on the visual analogue scale (P < 0.05) between the acupuncture and the diclofenac group, the null hypothesis (H0: acupuncture = diclofenac) can be rejected and the alternative hypothesis (HA: acupuncture ≠ diclofenac) is applicable.
If there is no significant difference between both groups this does not necessarily mean that the null hypothesis is true as there may simply not be enough evidence to reject it. For example, if the sample size is too small the study may be underpowered. The chance of the study providing a statistically significant outcome is based on assuming that a 5% (or less) chance of this event occurring randomly is significant. This could mean that, with a P-value of 0.04 (a 4% chance of this happening randomly), the null hypothesis would be rejected, whereas with a P-value of 0.06 (a 6% chance of this happening randomly) there would not be enough evidence to reject the null hypothesis. The choice of a significance level of 5% is arbitrary but applicable throughout biological science and therefore the outcome of the study needs to be interpreted with caution, particularly with respect to the number of people entered. The greater sample size or number of people entered into the study, the more statistical ‘power’ it has and the more its statistical conclusions can be ‘trusted’.
However a significant difference between groups does not relate to the clinical importance of this finding as the statistical significance depends to a large extent on the sample size (number of people) and the variability of the condition within the study. Accordingly a large study might find a small but clinically unimportant difference between treatments, which is highly significant. Equally, a small study with few people might find a large difference that could be clinically important. Clinical importance describes a difference between two treatments that has a relevant effect size which is noticeable for, and valuable to, a patient with that condition.
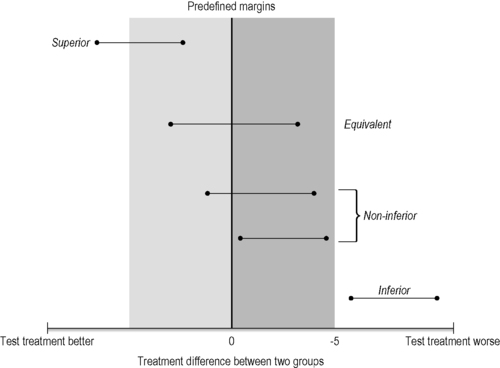
Most randomized controlled studies evaluate if one intervention is superior to another. This is generally the case for treatment comparisons with a waiting list or a placebo intervention as controls. However, for some comparisons we test for similarity between two treatments (equivalence) or non- inferiority, but these equivalence studies, which include both non-inferiority and non-superiority trials, are rare in CAM. A non-inferiority hypothesis could, however, be very useful when comparing a CAM treatment with a conventional treatment. Non-inferiority or equivalence trials have methodological features that differ from superiority trials. In an equivalence trial, the null and alternative hypotheses are reversed. This means the null hypothesis is that treatment A is different from treatment B, whereas the alternative hypothesis is that there is no difference between the treatments.
In addition the effect size of the reference treatment has to be known and the margin for the non-inferiority has to be predefined for the power calculation and subsequent sample size. This margin represents the smallest value for a clinically relevant effect and outcomes within this margin are defined as non-inferior (Piaggio et al. 2006). The interpretation of the results depends on where the confidence interval lies relative to both the margin of non-inferiority and the null effect. For two-sided evidence (equivalence trials) two margins are needed (one above zero and one below) (Figure 5.2).
Determining the sample size
When planning a study probably the most important question is how many patients are needed to have enough statistical power to detect a clinically meaningful difference between the treatment groups. If the budget is limited or the medical conditions allow only a small number of patients to be included in the study, the study may fail to reach adequate power and could be of very limited value. An appropriate sample size is essential for a worthwhile study. The sample size calculation requires that the hypothesis should be clearly stated and a valid design and appropriate test statistics should be used. In hypothesis testing two errors can occur: the type I error (α) takes place when the null hypothesis is rejected although it was true and the type II error (β) is when the null hypothesis is not rejected when it was false. The probability of making a type I error is called the level of significance whereas the type II error is called power. A type I error is usually considered to be the most important of the two items this approach primarily tries to control for. To determine the sample size the investigator should provide the following information:
• the significance level
• the power
• a clinically meaningful difference
• information about the standard deviation.
In clinical studies a significance level of 5% is usually chosen to reflect 95% confidence in the conclusions and usually a power of 80% or 90% is used in conjunction with this. Estimating a clinically meaningful difference and the standard deviation for the planned trial is often difficult, especially for CAM treatments, where there may be no previous data for the outcome for the intervention being evaluated. If the expected difference is large, fewer patients will be needed than if the expected difference is small. An important influence on the sample size is the variability of the patient outcomes for the primary outcome. If a high standard deviation is expected (lots of variability) more patients are needed to detect a significant difference between treatments than if the patient outcomes are more homogeneous.
Many CAM studies are conducted with small research budgets and consequently small sample sizes; therefore the sample size which would detect a significant difference between treatment groups may not be achieved. From a methodological perspective it is bad science to perform an underpowered study. If the budget is not sufficient to perform a trial of adequate size it is better to do no study, unless it is a pilot.
The essential elements of a randomized controlled trial
The RCT is the most reliable study design to detect whether the difference in the outcomes of two or more interventions is caused by a specific treatment. In effect it is the ‘gold standard’ for clinical research. With the aim for control for baseline differences, patients are allocated at random to receive one of several interventions. In addition, the treatment group is compared to at least one control group. Different control groups exist and the choice of control depends on the research question (Table 5.2). Placebo-controlled trials only tell us if the treatment is better than a placebo and we always need more information that that to inform clinical practice, where many treatments may be available for one condition.
| Research question | Adequate control group |
|---|---|
| Is the treatment effect specific? | Placebo |
| Is the treatment superior to no treatment? | Waiting list control |
| Is the treatment superior (or non-inferior) to standard therapy? | Standard treatment |
| Is the treatment superior (or non-inferior) to another treatment? | Other treatment |
| Is the treatment in addition to usual care superior to usual care alone? | Usual care |
Randomized trials can be open or blinded but it depends on the specific intervention as to which activities within a trial can be realistically blinded. Although RCTs are relatively straightforward, a lot of methodological errors have occurred in the past and no doubt will continue to appear in future literature. Guidelines for performing RCTs have been developed to improve their quality. These include good clinical practice, which regulates the functions and processes or the reporting (Altman et al. 2001) in RCTs as well as the inclusion of a standard reporting system for trial recruitment and compliance (Moher et al. 2001). Some of the CAMs that may be evaluated also have specific reporting requirements that are associated with Consolidated Standards of Reporting Trials (CONSORT); this includes Standards for Reporting Interventions in Controlled Trials of Acupuncture (STRICTA) (MacPherson et al. 2001) and for homeopathy RedHot (Dean 2006). These standards are of great importance and improve the quality of RCTs but they require more resources and funding in order to perform the RCT.
Randomization
Randomization is usually used to control conscious and unconscious bias as well as any unknown confounders in the allocation of patients to treatment groups. The purpose is to generate comparable patient groups with similar baseline characteristics. It is vital that the random allocation is performed properly and described in detail in the methodology, as inadequate or insecure randomization can be associated with very biased and usually positive findings (Shulz et al. 1995).
There are many examples where randomization is insufficiently performed and reported (Altman et al. 2001).
In order to avoid this criticism in planning a study the following issues need to be considered:
• Is randomization on patient or cluster level?
• How should the treatment groups be distributed (equal or unequal numbers)?
• Should you employ simple randomization or restricted randomization (blocking, stratification)?
• How will the randomization list be generated?
• How will the procedure operate so that it is appropriately concealed?
For instance, cluster randomization is used when whole social groups (families) or GP practices have to be assigned to different intervention groups. However, most trials use patient-level randomization and assignment is performed independently for each patient. In a two-armed trial with a 1:1 ratio the chance of a patient receiving either verum or control is 50%. RCTs with more arms (e.g. verum, control 1 and control 2) can also involve equal numbers in each group; for example in a three-armed trial with a 1:1:1 ratio the probability to be assigned to one of the groups is 33%. Unequal randomization with a higher chance of being assigned to the verum group can be helpful in an environment where the acceptance of the randomization process is poor from the patient’s or practitioner’s perspective. Patients with an interest in CAM often have clear preferences for CAM and this can be a major influence on their decision to participate in a randomized study. Therefore a 2:1 opportunity to receive their preferred treatment could improve the acceptability of randomization and improve recruitment and retention for the study. Ethics (e.g. in cancer) can also be an argument for unequal randomization. However, unequal randomization has disadvantages as the study may lose statistical power and blocking can be compromised, so it should only be used if there are very important reasons for doing so.
Simple or complete randomization is performed when a single random list is used and each patient is randomized regardless of the recruiting study centre and patient characteristics. In small trials or multicentre trials this can result in imbalances of group size and patient characteristics between the groups, but procedures for managing and restricting randomization help to solve this problem. The most frequently used methods are blocking and stratification. Both can be predefined and taken into account when generating the randomization list. Block randomization is used to ensure that the numbers of patients in the groups are comparable. In a two-armed trial with a randomization ratio of 1:1 and a block size of eight, four patients will be randomized into the verum and the other four into the control group. One important problem of blocked randomization is that allocation becomes predictable if the size of the blocks is known by the person who assigns and treats the patient, so this information must remain secure. This can be solved either by variable block size or blinding the study personnel to the block size. Stratified randomization is used to ensure that groups are comparable for important characteristics such as the provider of the treatment or the disease stage. This is done by doing separate randomization for ‘strata’ such as study centres or asthma staging. Stratification requires blocking within the ‘strata’, otherwise it too can become ineffective (Altman et al. 2001). Blocking and randomization should be used carefully. Figure 5.3 provides an example for stratification of study centres and using a variable block design.
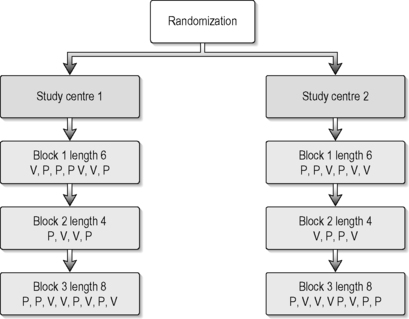 |
| FIGURE 5.3 |
One can adjust the probability of assignment of new patients to treatments during the study (Chow & Liu 2004). This is called adaptive randomization. Patient outcomes can be used as they become available to adjust the randomization assignment for future patients, perhaps assigning more of them to the ‘better, more effective treatment’. This allows one to improve expected patient outcomes during the experiment, while still being able to utilize the best available statistical decisions in a timely fashion. This type of randomization does not require the randomization list to be generated before the study starts. The random assignment is generated directly when the current patient is randomized, usually by a computer-based algorithm.
There are many ways to generate a random list prior to study start (Jadad & Enkin 2007). Older methods such as flipping a coin or throwing dice have been replaced by a number of validated software packages (e.g. SPSS, SAS, STATA) which ensure the quality and rigour of the random sequence.
Empirical investigations have shown that concealment is the most critical element of randomization (Schulz 1995). It is absolutely essential to ensure a concealed randomization procedure or the study loses rigour. The study personnel must not be able to predict the group to which a participant will be allocated and they should not be in a position to modify this allocation. The ideal strategy for concealment depends on the intervention. In drug trials identically packaged, consecutively numbered drug containers can be used based on the randomization list. For other interventions, central randomization, using a telephone hotline or an online database, is suitable. The physician who includes a patient contacts an external clinical trial unit by phone or via the web and they inform the physician about the group allocation if the trial is single-blind. In a double-blind trial neither the physician nor the patient should know which intervention the patients get. They might get the group code or in drug trials only the code number for the blinded drug containers.
If online databases are used, a safer approach is combining two independent databases (Vickers 2006) to ensure concealment. One database is used to register the new patient. The information is then forwarded to a second database which is only accessed by an independent person and the information is then sent to the appropriate person to ensure the patient has the correct, randomized treatment. In small studies where setting up an online database or a central telephone hotline is too complex and expensive, sequentially numbered, opaque, sealed envelopes can be used. However, here it should be ensured that the envelopes really are opaque and that they are sealed so that cheating is impossible.
Blinding
Randomization only ensures that patients in the groups are comparable for baseline characteristics, thus avoiding unknown confounders that may influence outcome, but will then be equally distributed to all the interventions in the study. This is vital and allows us to avoid bias. If patients were randomized to a group for which they had no preference they might be disappointed. This could influence how they rate the treatment outcome or result in more co-interventions. The same can occur in the provider rating; if they have special interest in one of the treatments their bias can influence the outcome. If a study does not use blinding it is classified as an open-label study, with all the disadvantages and bias that entails.
A single-blind trial is a study in which either the patient or the provider is unaware of the assignment of the patient. In reality it is mainly the patient who is blind. In a double-blind trial neither the patient nor the provider is aware of the patient’s treatment assignment. Sometimes trials are described as double-blind if patients and outcome assessors (not the provider) are blinded and this can be very misleading. Triple- and quadruple-blind trials are possible when, in addition to patients and providers, the outcome assessors and statisticians are blinded. To minimize potential biases treatment providers, patients and outcome assessors and even the statisticians should be blinded for the group allocation before the analysis is completed. This is possible in most pharmaceutical research; however if two clearly distinguishable treatments are compared patients and providers cannot be blinded. This occurs when comparing non-pharmacological intervention such as acupuncture or relaxation exercises with conventional treatment.
The issue of placebo is also much more complex and involved in CAM than it is in a simple drug trial; it is always more difficult to provide a real and convincing placebo which imitates the smell and the taste of a herbal medicine product or the visual and sensory effect of an acupuncture needle (Dube et al. 2007). However, even in CAM, when the verum and the placebo are indistinguishable, blinding might influence the providers’ treatment decisions. This is often discussed as a problem for trials of individualized homeopathy. In this type of homeopathy the provider prescribes an individual homeopathic remedy which should cover a whole range of symptoms for an individual patient (Witt et al. 2005). An option for the follow-up visits is usually to change the homeopathic remedy if no improvement is reported by the patient. This decision is based on an additional patient consultation, which can easily be influenced by the uncertainty of the provider (the homeopath) not knowing if there is no effect because the patient was allocated to the placebo group or whether the lack of response is due to the prescription of the wrong homeopathic remedy.
A vitally important factor for the success of blinding is to maintain it throughout the entire course of the trial. To assess if blinding is maintained throughout the study, patients, providers and assessors should be asked to guess the group allocation of each patient and the frequency of correct guesses can be compared between groups. However, if the verum is much more effective than the placebo, due to the effect of treatment, unblinding may happen. Asking these questions about blinding at an earlier stage does not solve the problem. We want to know if the patients were blinded at the time point when the primary outcome was assessed.
It is also helpful to assess the credibility of a treatment but this evaluates a slightly different dimension to blinding. Scales like this which have been used by Vincent (1990), are helpful and should be used in the early stages of the treatment.
Controls
In general there are four types of treatment that the patient can receive in the control group (Chow & Liu 2004): (1) placebo; (2) no treatment; (3) other active treatments; and (4) different dose or regime of the verum intervention.
Placebo controls
Placebo is used to detect the assumed specific effect of treatment, for example, in the case of pharmaceutical research, the true pharmacological activity of the active chemical treatments. Shapiro (1964) suggests that ‘A placebo is any therapy (or that component of any therapy) that is intentionally or knowingly used for its nonspecific, psychological, therapeutic effect, or that is used for a presumed specific therapeutic effect on a patient, symptom or illness but is without specific activity for the condition being treated […] The placebo effect is the nonspecific psychological or psycho-physiological therapeutic effect produced by a placebo.’ A full chapter in this text is devoted to a deeper understanding of the placebo effect and its implications for CAM research (see Chapter 16).
When using placebo controls in clinical trials, ethical aspects have to be taken into account. The decision to use a placebo control in a trial may be unethical but to some extent this depends on the severity of the disease and available options for conventional treatment. One of the key elements for the success of a placebo-controlled clinical trial is whether a placebo treatment can be made to match the active treatment in all aspects, such as size, colour, coating, texture and taste of a medicine or herbal decoction. This will be more difficult for herbal medicine, especially Chinese herbal medicine, where strong-tasting decoctions are used. It may be simpler for a homeopathic pill but may make the context of the homeopathic consultation very complicated and diminish the context validity or generalizability of homeopathic practice in this environment. For interventions such as massage, yoga and qigong, a true placebo intervention may be almost impossible as we have no real idea about the exact mechanisms involved in these therapies and therefore we cannot realistically design a specific placebo if the active ingredient is unknown. In acupuncture there is still an ongoing discussion as to whether there is an inert placebo intervention that is convincing and can be used in an RCT (see Chapter 11).
No-treatment control
A no-treatment group can quantify one part of non-specific effects that includes regression to the mean during the normal course of disease. This can be very helpful when performing studies in CAM interventions such as acupuncture, particularly if the sham-control response rates are high and well described. The response in the no-treatment group will always be lower than in a placebo group so the decision as to whether a no-treatment control group can be used requires both practical and ethical consideration as well as careful reference to the specific research question addressed by the study.
Active treatment control
The use of other active treatments as control groups can not only solve ethical problems, but is also helpful in providing data for decision-making in usual care. The active treatment control can be a standard treatment, or at least a treatment which already has a known efficacy. The advantage of this kind of control is that it should have shown superiority over placebo in previous well-designed trials so if the verum treatment is superior to the standard intervention, superiority over placebo might be assumed. However, we have limited evidence for many conventional treatments used in routine conventional medical care, so the choice of the active treatment control should be considered carefully. In spite of this, conventional routine medical care has been used as a control in several acupuncture studies (Vickers et al., 2004, Witt et al., 2006a, Witt et al., 2006b, Witt et al., 2006c and Witt et al., 2008) and can be very helpful in decision-making for health care purchasers who want to select the best, cheapest and safest treatment available. Superiority over placebo cannot be assumed in such a heterogenic active treatment control group so outcomes must be interpreted with caution.
Different doses or regimes of the verum intervention as controls
To characterize the dose–response relationship one really needs a phase II study in which different doses or regimes of the verum intervention are used. Phase II studies allow us to identify the minimum effective and the maximum tolerable dose. They are usually four-armed trials which involve three different doses of the intervention and a placebo group, but such studies are very rare in the area of CAM. The type of trial that we have been discussing in this chapter is usually referred to as a phase III clinical trial.
The influence of expectation
The influence of patient expectations on outcomes is related to both within-group changes (from baseline to follow-up) and between-group differences (between the ‘real’ treatment and placebo). These suggestions are supported by two systematic reviews (Crow et al., 1999 and Mondloch et al., 2001).
If patients included in a trial have higher expectations of a positive outcome than the ‘average’ patient then this could result in within-group changes which are larger than in a more representative sample. High expectations might also be associated with high response rates and improved outcomes in the placebo control group. This could result in a ceiling effect, making it more difficult to detect a significant difference between verum and placebo if everyone improves because they expect and believe the treatment will work for them. By pooling data from four acupuncture trials we were able to show a significant association between greater improvement and high outcome expectations (Linde et al. 2007).
In order to account for patient expectations and to relate this to the main outcomes, patient expectations should be assessed at baseline before randomization. A reliable and valid, yet simple, tool for measuring aspects of expectations in clinical trials is still needed. Meanwhile simple questions such as: ‘How effective do you consider the treatment in general?’ with responses such as ‘very effective, effective, slightly effective, not effective, don’t know’ should be used routinely in trials involving the assessment of chronic pain and other chronic benign conditions where expectation and belief may be important predictors of treatment outcome.
Study designs
Parallel group designs
The most commonly used study design for RCTs is the parallel group design in which each patient receives only one treatment in a randomized fashion. Compared to other designs, such as cross-over designs, a parallel group design is easy to implement, widely accepted and the analysis and interpretation of the results are less complicated. It is also the most appropriate for CAM as we are usually unsure about the duration of any treatment effect so designs like cross-over may result in confusion and carry-over effects when using interventions such as acupuncture. The simplest type of parallel group design is a two-arm study, as illustrated in Figure 5.4.
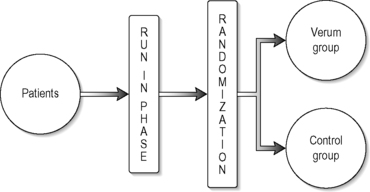 |
| FIGURE 5.4 |
A parallel group design can have three or four treatment (intervention) or comparison groups and patients can be allocated to the groups in equal or unequal numbers. A run-in period with no active treatment and perhaps an appropriate control can be useful to wash out the effects of previous therapy and encourage compliance (particularly with the trial outcome measures) as well as obtaining baseline data against which to compare posttreatment outcome.
Cross-over designs
A cross-over design is a modified randomized block design where patients receive more than one treatment at different preagreed time periods during the study. It is a complete cross-over design if each group in this design, in a randomized sequence, receives all treatments under investigation. The simplest cross-over design includes a two-group cross and two treatment periods (Figure 5.5).
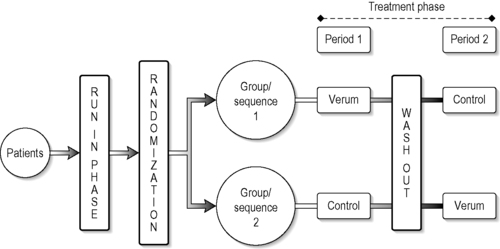
A cross-over can involve more treatment groups and the allocation to the groups can involve equal or unequal numbers of people.
The advantages of a cross-over design are that it allows within-patient comparison between treatments. Each patient in effect acts as his or her own control, which removes interpatient variability from the comparisons between treatments and provides good unbiased estimates for the difference between treatments (Chow & Liu 2004). To prevent carry-over effects, washout phases can be included in the design but the main problem with CAM treatments is that the washout periods are debatable and unclear for almost all the CAM interventions that have been studied. These designs are almost impossible to interpret if treatment effects are maintained for an indefinite period. Consequently this design is more suitable for drug trials than for interventions such as acupuncture. A good example for a cross-over design in CAM is that of Frei et al. (2005), who investigated the use of homeopathy in children with attention deficit hyperactivity disorder. In this study, patients received verum or placebo for 6 weeks and were then crossed over to receive placebo or verum for a second period of 6 weeks.
Group sequential designs
We may be investigating a treatment that is of possible benefit but may also be harmful to the patients entered. In these situations a group sequential design is valuable as it divides patient entry into a number of equal-sized groups. This allows for some interim analysis and the early identification of adverse reactions so that any decision to stop the trial is based on repeated significance tests of the accumulated data after each group is evaluated (Pocock 1977). These designs are based on sequential testing and other adaptive rules. The literature in this area is huge (Chow & Liu 2004) and the problem of multiple testing has to be taken into account.
Pragmatic versus experimental studies
Pragmatic studies aim to evaluate whether an intervention is effective under the sort of conditions that a patient might find in a clinic. These trials have model validity as far as the intervention is concerned. For instance, an acupuncture or herbal treatment may be adapted to suit some trial conditions in a very rigorous, placebo-controlled experiment and this may not really reflect good clinical practice. The resulting study may be of high scientific quality, but in effect is of little use to clinicians and health care providers, as one may not be able to generalize to the wider population from the results obtained. Pragmatic studies use wide inclusion criteria, allow flexibility in the treatment regimen and often focus on patient-centred outcomes such as quality of life or self-recorded pain (Zwarenstein 2008). Experimental or explanatory studies address whether an intervention works under controlled conditions. These studies typically use placebos or standardized interventions as controls. Experimental studies can have a high internal validity whereas pragmatic studies may have less internal validity but are more generalizable and relevant to clinical practice (see Chapter 1). The external validity of a study depends on the following aspects: (1) study participants; (2) interventions; and (3) outcome measures.
The generalizability of the study results depends on whether the group of study participants is representative of the usual type of patients with this specific condition. In experimental studies homogeneous patient groups are preferred, with the aim of reducing bias and enlarging internal validity. These patients are rarely representative of the people suffering from the disease under investigation, for example elderly patients over 70 years of age often suffer from cardiovascular diseases but because of their comorbidity they are often excluded from experimental studies on cardiovascular drugs. Within the context of health service research and decision-making it makes sense to investigate a treatment in a representative manner and as it is used in routine clinical care (Witt 2009). Therefore if you wished to evaluate whether acupuncture should be reimbursed by health insurance companies it would be better to assess the broad range of acupuncture styles likely to be found in practice instead of using a standardized treatment protocol.
This tension remains unresolved within the context of clinical trials in CAM and indeed conventional medicine; there are many studies in which CAM treatments have been applied in an inadequate manner and without model validity in order to improve rigour, reduce bias or increase feasibility. Such trials are very difficult to interpret and may be very of little value in the context of clinical practice and health care decision-making.
Using patient-centred outcomes should include taking relevant and valid measures that are appropriate to the condition being studied and ideally incorporating follow-up periods which are long enough to provide information about long-term effects. In pragmatic studies more subjective symptom-oriented outcomes such as symptom diaries or disease-specific quality-of-life questionnaires are often used. In contrast, more conventionally structured experimental studies may involve ‘hard’ outcome parameters such as laboratory measurements, if the condition within the study allows these measures to be usefully employed.
Most CAM trials in chronic benign diseases such as migraine and osteoarthritis have study observation periods of about 6 months at the most. Long-term follow-up in these trials is rare. This is unfortunate as health care providers would ideally like to be able to make thoughtful decisions that take into account the long-term effects of an intervention.
In reality there is often a compromise between internal and external validity in the design of any trial and this is usually and sensibly based on the study or research question. If the aim of the study is to give guidance to health service purchasers, a more population-based and ‘pragmatic’ approach is needed. If the aim is to develop a new intervention and determine its efficacy over placebo, an experimental study is more likely to produce the most valuable results. As mentioned in Chapter 1, CAM treatments are in general very diverse, widely used and widely available before they are evaluated scientifically. Consequently pragmatic studies can provide helpful information enabling medical decision-making. Ideally it would be best to have information from both experimental and pragmatic studies for all treatments, but this is not usually possible.
Analysing randomized controlled trials
Study populations
There are different types of study populations. The intention-to-treat (ITT) population includes all patients who were randomized at the beginning of the trial with the intention that they should receive one of the study interventions. This means that even data from patients who did not follow the protocol, or who dropped out without receiving any treatment, need to be analyzed. The ITT analysis represents a conservative but widely used statistical approach and enhances the external validity of the study. The per protocol population (PP) represents the other side of this equation and includes only patients who followed the study protocol; this group of people should be predefined before analysing the data. For trials with a superiority hypothesis the ITT analysis is the primary analysis and the PP analysis can be performed as a secondary, but much less convincing, dataset. However in studies that have a non-inferiority hypothesis the converse may be true.
Attrition rates and missing data
In studies with longer follow-up periods patients may drop out prematurely or not follow the study protocol, perhaps by not completing the outcome questionnaires. The outcomes for these patients might differ considerably from those remaining in the study. Patients who dropped out might have been non-responders or might have experienced more severe side-effects. It is therefore important to keep attrition rates as low as possible in order to increase the study rigour. There are statistical methods that can reduce the impact of missing data, but these are far from perfect. If the amount of missing data is small, statistical methods are helpful in estimating the impact of the missing data and several such statistical methods exist. The commonest one is the ‘last value carried forward’. This means that the values from an earlier measurement are transferred to the following one. However, if a disease is progressive, these values will be positive and a more appropriate method is to use imputation techniques. These substitute a missing data point or a missing component of a data point with some value. Once all missing values have been imputed, the dataset can be analysed using standard techniques for complete data. There are different imputation techniques available (Twisk and de Vente, 2002 and Donders et al., 2006).
Baseline differences between groups
Randomization does not always generate groups which are comparable for all baseline characteristics. If studies are small baseline differences between groups can often occur just by chance. A treatment is only independently associated with the outcome after all confounders have been taken into account. There is some evidence that the baseline value of the outcome can influence the outcome after treatment (Vickers 2004). Because of this, the use of analysis of covariance (ANCOVA) with the baseline values as a covariate is the most simple recommended analytical approach for all RCTs, even if baseline differences are not found. This method accounts for any variability in baseline values between the two or more groups in the study.
Conclusion
This chapter describes the main principles of RCT design, particularly with respect to CAM. RCTs were developed for the pharmaceutical industry, ideally to test the specific effects of drugs. They can usefully be adapted to evaluate a variety of different CAM techniques but this needs to be done thoughtfully and appropriately. The most important issue with respect to the design of an RCT is the research question. If you want to consider an RCT design, do you have a specific question that is best answered through an RCT? The advent of the RCT is both a blessing and a curse; it has certainly improved our understanding of medical evidence, but inevitably it is limited in its scope. Good science and clinical relevance do not always go hand in hand, and attempting to achieve both simultaneously requires clear thinking and the ability to work within a research team.
References
Altman, D.G.; Schulz, K.F.; Moher, D.; et al., The revised CONSORT statement for reporting randomized trials: explanation and elaboration, Ann. Intern. Med. 134 (8) (2001) 663–694.
Chow, S.C.; Liu, J.P., In: Design and Analysis of Clinical Trialssecond ed. (2004) John Wiley, Hoboken, US, p. 101; 142, 167, 179, 422.
Crow, R.; Gage, H.; Hampson, S.; et al., The role of expectancies in the placebo effect and their use in the delivery of health care: a systematic review, Health Technol. Assess. 3 (3) (1999) 1–96.
Donders, A.R.; van der Heijden, G.J.; Stijnen, T.; et al., Review: a gentle introduction to imputation of missing values, J. Clin. Epidemiol. 59 (10) (2006) 1087–1091.
Dube, A.; Manthata, L.N.; Syce, J.A., The design and evaluation of placebo material for crude herbals: Artemisia afra herb as a model, Phytother. Res. 21 (5) (2007) 448–451.
Dean, E.M.; Coulter, M.K.; Fisher, P.; et al., Report data on homeopathic treatments (RedHot): a supplement to CONSORT, Forsch. Komplementarmed. (13) (2006) 368–371.
Frei, H.; Everts, R.; von, A.K.; et al., Homeopathic treatment of children with attention deficit hyperactivity disorder: a randomised, double blind, placebo controlled crossover trial, Eur. J. Pediatr. 164 (12) (2005) 758–767.
Jadad, A.; Enkin, M.W., Randomised controlled trials. second ed. (2007) Blackwell BMJ Books, London.
Linde, K.; Witt, C.M.; Streng, A.; et al., The impact of patient expectations on outcomes in four randomized controlled trials of acupuncture in patients with chronic pain, Pain 128 (3) (2007) 264–271.
MacPherson, H.; White, A.; Cummings, M.; et al., Standards for reporting interventions in controlled trials of acupuncture: the STRICTA recommendations, Complement. Ther. Med. (9) (2001) 246–249.
Moher, D.; Schulz, K.F.; Altman, D., The CONSORT Statement: revised recommendations for improving the quality of reports of parallel group randomised trials, JAMA 285 (15) (2001) 1987–1991.
Mondloch, M.V.; Cole, D.C.; Frank, J.W., Does how you do depend on how you think you’ll do? A systematic review of the evidence for a relation between patients’ recovery expectations and health outcomes, CMAJ 165 (2) (2001) 174–179.
Piaggio, G.; Elbourne, D.R.; Altman, D.G.; et al., Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement, JAMA 295 (10) (2006) 1152–1160.
Pocock, S.J., Group sequential methods in the design and analysis of clinical trials, Biometrika 64 (2) (1977) 191–199.
Schulz, K.F., Subverting randomization in controlled trials, JAMA 274 (18) (1995) 1456–1458.
Schulz, K.F.; Chalmers, I.; Hayes, R.J.; et al., Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials, JAMA 273 (5) (1995) 408–412.
Shapiro, A.K., Etiological factors in placebo effect, JAMA 187 (1964) 712–714.
Twisk, J.; de Vente, W., Attrition in longitudinal studies. How to deal with missing data, J. Clin. Epidemiol. 55 (4) (2002) 329–337.
Vincent, C., Credibility assessments in trials of acupuncture, Complement. Med. Res. 4 (1990) 8–11.
Vickers, A.J., Statistical reanalysis of four recent randomized trials of acupuncture for pain using analysis of covariance, Clin. J. Pain 20 (5) (2004) 319–323.
Vickers, A.J., How to randomize, J. Soc. Integr. Oncol. 4 (4) (2006) 194–198.
Vickers, A.; Cassileth, B.; Ernst, E.; et al., How should we research unconventional therapies? A panel report from the conference on Complementary and Alternative Medicine Research Methodology, National Institutes of Health, Int. J. Techno. Assess. Health Care 13 (1) (1997) 111–121.
Vickers, A.J.; Rees, R.W.; Zollman, C.E.; et al., Acupuncture for chronic headache in primary care: large, pragmatic, randomised trial, BMJ 328 (7442) (2004) 744.
Witt, C.M., Efficacy, effectiveness, pragmatic trials–guidance on terminology and the advantages of pragmatic trails, Forsch Komplementmed 16 (5) (2009) 292–294.
Witt, C.M.; Lüdtke, R.; Baur, R.; et al., Homeopathic medical Practice: Long-term results of a cohort study with 3981 patients, BMC Public Health 5 (2005) 115.
Witt, C.M.; Jena, S.; Selim, D.; et al., Pragmatic randomized trial of effectiveness and cost-effectiveness of acupuncture for chronic low back pain, Am. J. Epidemiol. 164 (2006) 487–496.
Witt, C.M.; Jena, S.; Brinkhaus, B.; et al., Acupuncture in Patients with Osteoarthritis of the Knee and the Hip, Arthritis Rheum. 54 (11) (2006) 3485–3493.
Witt, C.M.; Jena, S.; Brinkhaus, B.; et al., Acupuncture for patients with chronic neck pain, Pain 125 (2006) 98–106.
Witt, C.M.; Jena, S.; Brinkhaus, B.; et al., Acupuncture for patients with chronic neck pain, Pain 125 (2006) 98–106.
Witt, C.M.; Reinhold, T.; Brinkhaus, B.; et al., Acupuncture in patients with dysmenorrhea: a randomized study on clinical effectiveness and cost-effectiveness in usual care, Am. J. Obstet. Gynecol. 198 (2) (2008) 166–168.
Zwarenstein, M.; Treweek, S.; Gagnier, J.J.; et al., CONSORT group; Pragmatic Trials in healthcare (Practic) group. Improving the reporting of pragmatic trials: an extension of the CONSORT statement, BMJ 337 (2008) 2390; a.
Further reading
In: (Editors: Saks, M.; Allsop, J.) Researching Health (2007) Sage Publications, London.
Schulz, K.F.; Schulz, D.A.; Grimes, Allocation concealment in randomised trials, defending against deciphering, Epidemiology SeriesLancet (2002) pp. 359.