[level-membership-for-pulmolory-and-respiratory-category]
Chapter 59 Pulmonary Vasculitis and Hemorrhage
Pulmonary Vasculitis
Small Vessel Vasculitis
Granulomatosis with Polyangiitis and Microscopic Polyangiitis
Therapy
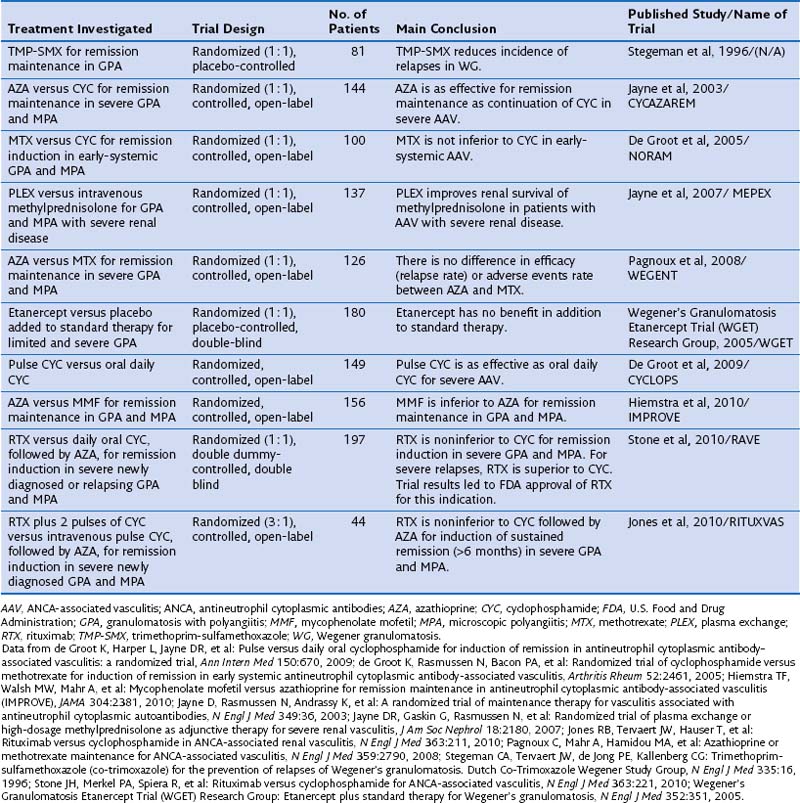
Standard therapy for GPA and MPA currently follows the same basic principles. Methotrexate (MTX) at a dose of up to 25 mg once a week in combination with oral prednisone is considered the standard of care for patients with limited GPA. However, only one prospective randomized trial has compared MTX and cyclophosphamide (CYC) for remission induction in such patients (Table 59-1). The trial, conducted by the European Vasculitis Study Group (EUVAS), showed that MTX is noninferior to CYC for remission induction, but the side effects were less frequent and less severe. The trial also documented that early discontinuation of immunosuppression in patients with ANCA-associated vasculitis is fraught with a high relapse rate. The largest reported group of patients with limited Wegener’s granulomatosis (WG) was treated with MTX for remission induction in the context of the Wegener’s Granulomatosis Trial (WGET). More than 90% of patients achieved remission with this regimen, and more than 70% achieved a sustained remission (lasting longer than 6 months). These rates are equivalent to those achieved with CYC in severe disease, as discussed next.
Alveolar Hemorrhage Syndromes
Cartin-Ceba R, Fervenza FC, Specks U. Treatment of antineutrophil cytoplasmic antibody-associated vasculitis with rituximab. Curr Opin Rheumatol. 2012;24:15–23.
De Groot K, Harper L, Jayne DR, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody–associated vasculitis: a randomized trial. Ann Intern Med. 2009;150:670.
Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349:36.
Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180.
Keogh KA, Specks U. Churg-Strauss syndrome. Clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med. 2003;115:284.
Lee AS, Specks U. Pulmonary capillaritis. Semin Respir Crit Care Med. 2004;25:547.
Polychronopoulos VS, Prakash UB, Golbin JM, et al. Airway involvement in Wegener’s granulomatosis. Rheum Dis Clin North Am. 2007;33:755.
Sinico RA, Di Toma L, Maggiore U, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005;52:2926.
Specks U. Methotrexate for Wegener’s granulomatosis: what is the evidence? Arthritis Rheum. 2005;52:2237.
Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 59 Pulmonary Vasculitis and Hemorrhage
Pulmonary Vasculitis
Small Vessel Vasculitis
Granulomatosis with Polyangiitis and Microscopic Polyangiitis
Therapy
Standard therapy for GPA and MPA currently follows the same basic principles. Methotrexate (MTX) at a dose of up to 25 mg once a week in combination with oral prednisone is considered the standard of care for patients with limited GPA. However, only one prospective randomized trial has compared MTX and cyclophosphamide (CYC) for remission induction in such patients (Table 59-1). The trial, conducted by the European Vasculitis Study Group (EUVAS), showed that MTX is noninferior to CYC for remission induction, but the side effects were less frequent and less severe. The trial also documented that early discontinuation of immunosuppression in patients with ANCA-associated vasculitis is fraught with a high relapse rate. The largest reported group of patients with limited Wegener’s granulomatosis (WG) was treated with MTX for remission induction in the context of the Wegener’s Granulomatosis Trial (WGET). More than 90% of patients achieved remission with this regimen, and more than 70% achieved a sustained remission (lasting longer than 6 months). These rates are equivalent to those achieved with CYC in severe disease, as discussed next.
Table 59-1 Randomized Controlled Trials Informing the Standard of Care in ANCA-Associated Vasculitis
[/not-level-membership-for-pulmolory-and-respiratory-category]
