Chapter 58 Pulmonary Hypertension
Classification
Pulmonary hypertension was previously classified as either primary or secondary, depending on the absence or the presence, respectively, of identifiable causes or risk factors. The diagnosis of primary pulmonary hypertension was one of exclusion after ruling out all other causes for PH. Subsequent classification schemes have attempted to create categories of PH that share pathologic and clinical features, as well as similar therapeutic options. These classification schemes have allowed investigators to conduct clinical trials in well-defined patient groups with a shared underlying pathogenesis for their PH, resulting in the development of new targeted drug therapies; consequently, improvements in both quality of life and survival can now be expected in this otherwise deadly disease. This more inclusive category of PAH also has afforded increased opportunities for treatment of some less common forms of the disorder that were previously too rare for individual treatment studies. The most recent classification scheme was the product of the aforementioned Fourth World Symposium on Pulmonary Hypertension (Box 58-1).
Box 58-1
Updated Clinical Classification of Pulmonary Hypertension
Group 1: Pulmonary Arterial Hypertension
PAH is a subset of PH defined as a mPAP greater than 25 mm Hg determined with the patient at rest and a normal pulmonary capillary wedge pressure (PCWP) and/or left ventricular end-diastolic pressure (LVEDP) and a lesion localized to the pulmonary arteriole (Figure 58-1, A to C). Unfortunately, a limitation of these classification schemes is the fact that many of these patients have “multifactorial pulmonary hypertension.” The clinician is thus faced with treating PH in a variety of clinical scenarios that often include features from more than one of the World Health Organization (WHO) classification categories (i.e., groups 1 to 5, with an additional 1′ grouping as described later on). For example, the clinical presentation may include somewhat elevated pulmonary venous pressures, mild to moderate obstructive or restrictive lung disease, or a form of valvular heart disease that typically would not account for pulmonary hypertension severity. Patients with such “out of proportion” PH are not included in clinical trials; therefore, data pertaining to the safety and efficacy of conventional PAH therapies in this population are extremely limited.
Heritable Pulmonary Arterial Hypertension
Sporadic mutations in BMPR2 have been identified in approximately 11% to 40% of patients with presumably the idiopathic form of PAH and are seen in 70% to 80% of patients with familial PAH but are relatively uncommon in patients with so-called associated PAH (i.e., category 1.4 in Box 58-1). Although penetrance is low and only approximately 25% of carriers will go on to develop PAH, genetic anticipation also has been demonstrated (i.e., in affected families, each successive generation has more severe PAH developing at an earlier age). BMPR2 has been localized to chromosomal region 2q31-32, and inheritance occurs in an autosomal dominant fashion. Recently, it has been suggested that patients with PAH associated with BMPR2 mutations may represent a subgroup with more severe disease who are less likely to demonstrate vasoreactivity than those with IPAH. Because this mutation can occur sporadically in as many as 25% of patients with PAH and does not occur in all patients with so-called familial PAH, the term heritable is now favored over the designation familial.
Drug- and Toxin-Induced Pulmonary Arterial Hypertension
A number of risk factors for the development of PAH have been identified (Box 58-2). Risk factors for PAH include “any factor or condition that is suspected to play a predisposing or facilitating role in the development of the disease.” Such risk factors have been categorized as “definite, very likely, possible, or unlikely, based on the strength of their association with [pulmonary hypertension] and their probable causal role.” As a result of the Dana Point symposium, methamphetamine use was reclassified as a very likely risk factor for the development of PAH.
Group 1′: Pulmonary Arterial Hypertension Associated with Pulmonary Venous or Capillary Abnormalities
Pathobiology
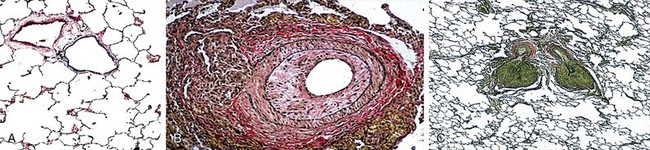
Normal pulmonary arteries have a thin medial layer of circular muscle whose thickness is less than 5% of the diameter of the vessel (see Figure 58-1, A). Consequently, under physiologic conditions, the pulmonary circulation is characterized by high flow, low pressure, and low vascular resistance. The histopathologic findings in PAH are characterized by variable intimal hyperplasia, medial hypertrophy, adventitial proliferation, and fibrosis culminating in concentric obliterative lesions (see Figure 58-1, B) that occur in close proximity to plexiform lesions (see Figure 58-1, C). The plexiform lesion results from neointimal proliferation and progresses from a cellular to a fibrotic lesion with advancing disease. It is made up of a predominance of endothelial cells in different stages of vascular organization, suggesting an abnormal form of angiogenesis. Pulmonary vascular remodeling also has been associated with in situ thrombosis and infiltration by inflammatory and progenitor cells.
Unfortunately, what is known about the pathobiology of PAH largely stems from research in patients with IPAH or from animal models that are meant to represent IPAH. Nevertheless, the pathobiologic features of PAH are thought to result from a multiple-hit hypothesis involving the interaction of a predisposing state with an inciting stimulus (Figure 58-2). Consequently, the resulting imbalance favors vasoconstriction, thrombosis, and mitogenesis. Restoration of this balance by inhibition of endothelin and thromboxane or augmentation of nitric oxide and prostacyclin forms the basis of today’s current therapies.
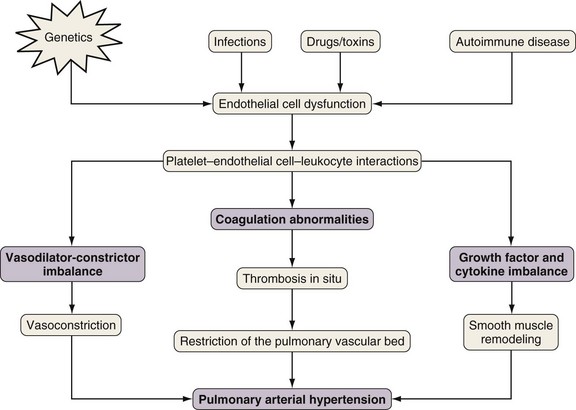
Figure 58-2 Proposed pathogenesis of pulmonary arterial hypertension (PAH).
(From Reed AK, Evans TW, Wort SJ: Pulmonary hypertension. In Albert RK, Spiro SG, Jett JR, editors: Clinical respiratory medicine, ed 3, Philadelphia, Mosby, 2008.)
Ongoing research on other mediators and pathways (Table 58-1) promises new targets for novel therapies.
Table 58-1 Mediators and Pathways in Pulmonary Arterial Hypertension
| Increased Activity | Decreased Activity |
|---|---|
Diagnostic Approach
Signs and Symptoms
There are no early signs or symptoms of PH. Therefore, annual screening should be performed in high-risk populations (Table 58-2). A diagnosis of PH should be considered in any patient who presents with breathlessness in the absence of specific cardiac or pulmonary disease, or in patients who have underlying cardiac or pulmonary disease and present with increasing breathlessness that is not explained by the underlying disease itself. The initial symptoms and signs are nonspecific and may include fatigue, progressive dyspnea on exertion, palpitations, chest pain, dizziness, and cough. Unfortunately, the mean duration of symptoms before diagnosis reported in most registries approaches 2 years. Exertional dizziness and syncope are suggestive of an inadequate cardiac output and should raise clinical suspicion for PH. As PH progresses, patients go on to develop symptoms and signs of right-sided heart failure including jugular venous distention, right ventricular heave, tricuspid regurgitation, right ventricular gallops, ascites, and edema. Establishing an accurate diagnosis has important implications for therapy.
Table 58-2 Recommendation for Screening for Pulmonary Arterial Hypertension
| Risk Factor | Recommendation |
|---|---|
| Family history of PAH | Yes |
| Connective tissue disease | |
| Scleroderma | Yes |
| Other | No |
| Congenital heart disease | |
| Large ASD, nonoperated | Yes |
| Large VSD, nonoperated | Yes |
| HIV infection | No |
| Portal hypertension | No |
| Consideration for liver transplantation | Yes |
| Use of appetite-suppressant drugs | No |
| Previous pulmonary embolism | No |
| Increasing dyspnea | Yes |
| Massive/submassive PE | Yes |
ASD, atrial septal defect; HIV, human immunodeficiency virus; PE, pulmonary embolism; VSD, ventricular septal defect.
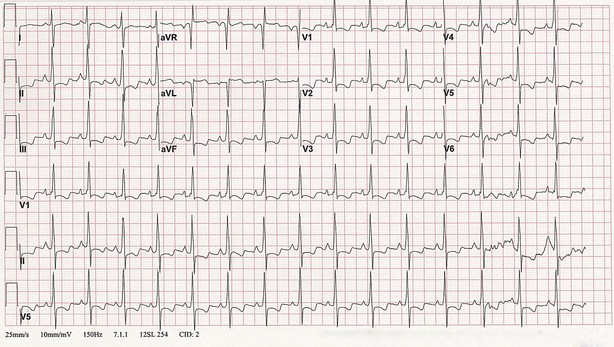
Electrocardiogram
The electrocardiogram shows abnormalities in 85% of patients with established PH but is not adequately sensitive as a screen for PH. Typical changes include right axis deviation with evidence of right ventricular or right atrial hypertrophy and right ventricular strain (Figure 58-3). The degree of these changes does not always reflect the severity of disease, and a normal ECG appearance does not eliminate the diagnosis of PH. Nevertheless, the following ECG findings have negative prognostic implications: (1) right axis deviation, (2) a tall R wave and small S wave with R/S ratio greater than 1 in lead V1, (3) qR complex in lead V1, (4) rSR′ pattern in lead V1, (5) a large S wave and small R wave with R/S ratio less than 1 in lead V5 or V6, or (6) S1-S2-S3 pattern (see Figure 58-3).
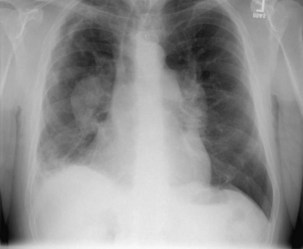
Chest Radiograph
The chest radiograph also is not particularly sensitive for detecting PH. However, incidental findings of enlarged main and hilar pulmonary arterial shadows (right descending greater than 1.6 cm, left greater than 1.8 cm), with concomitant attenuation of peripheral pulmonary vascular markings (“pruning”), and right ventricular hypertrophy as evidenced by a reduced retrosternal clear space on the lateral projection should prompt a workup for PH. Chest radiographic findings also may lead to the diagnosis of an underlying pulmonary process (Figure 58-4).
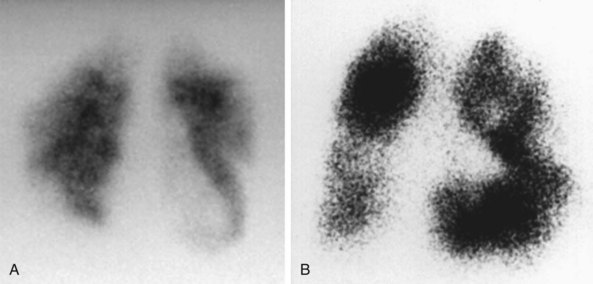
Ventilation-Perfusion Scan
PH caused by chronic thromboembolic disease is a potentially surgically curable condition that should be considered in all patients with unexplained PH. A ventilation-perfusion scan is essential to rule out chronic thromboembolic disease in all patients with unexplained pulmonary hypertension; a normal scan effectively excludes a diagnosis of chronic thromboembolic disease. In patients with PAH, the ventilation-perfusion scan typically is normal or displays only minor, patchy perfusion defects, whereas in patients with CTEPH, at least one but often several major segmental or subsegmental mismatches in ventilation and perfusion will be evident (Figure 58-5, A and B). Unmatched perfusion defects also may be seen in PVOD. The “gold standard” diagnostic modality remains contrast angiography performed at the time of catheterization, which is able to characterize the nature and extent of any thromboembolic disease.
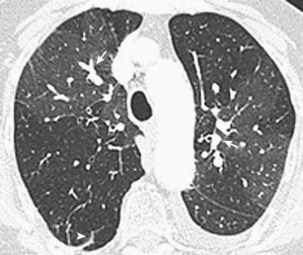
Chest Computed Tomography
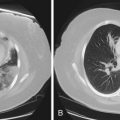
A wide spectrum of abnormalities on computed tomography (CT) scanning has been described in patients with chronic thromboembolic disease. Such abnormalities may include right ventricular enlargement, dilated central pulmonary arteries, presence of persistent thromboembolic material within central pulmonary arteries, increased bronchial artery collateral flow, variability in the size and distribution of pulmonary arteries, parenchymal abnormalities consistent with previous infarcts, or mosaic attenuation of the pulmonary parenchyma (Figure 58-6). These are nonspecific findings, however, and their absence does not exclude the presence of surgically accessible disease, because only sixth- or seventh-generation pulmonary arteries can be visualized with confidence. Therefore, contrast-enhanced CT should not be used to exclude the diagnosis of chronic thromboembolic disease.
Echocardiogram
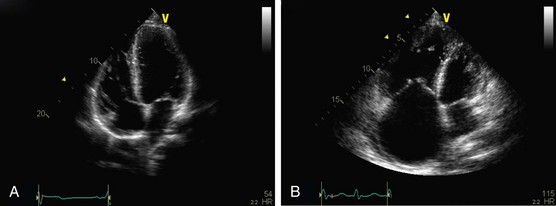
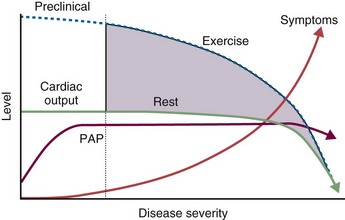
Signs indicative of PH on echocardiogram include increased sPAP or tricuspid regurgitant jet, right atrial and ventricular hypertrophy, flattening of the intraventricular septum, small left ventricular dimension, and a dilated pulmonary artery (Figure 58-7, A and B). A pericardial effusion in the setting of PH carries a poor prognosis. Although echocardiography has been found to correlate with right-sided heart catheterization, Doppler echocardiography generally underestimates systolic PAP in patients with severe PH and overestimates systolic PAP in populations consisting mostly of subjects with normal pressures. Furthermore, the systolic PAP may fall in decompensated right ventricular heart failure (Figure 58-8). Echocardiography also is particularly inaccurate in persons with advanced lung disease and both underestimates and overestimates the degree of PH in these patients. With echocardiographic evidence of significant PH, a right-sided heart catheterization should always be undertaken at least once to provide an accurate baseline assessment of the pulmonary hemodynamics and to permit reversibility studies. Echocardiography is, however, useful in following disease progression, removing the need for repeated pulmonary artery catheterizations.
Approach to Therapy
Controversies and Pitfalls
• Combination therapy: Combination therapy that uses drugs with different therapeutic mechanisms is an attractive option for patients who fail to improve or deteriorate with first-line treatment (monotherapy), but trials to confirm the value of this approach are ongoing and may be limited by the expense of these therapies. Many experts are interested in the concept of induction therapy followed by tailoring to a maintenance regimen.
• Out-of-proportion magnitude of pulmonary hypertension: Most patients present with features of more than one WHO group. Unfortunately, clinical trials are lacking in patients with PH that is out of proportion to their parenchymal lung disease or left heart dysfunction. Decisions on how to treat these patients are limited to anecdotal reports and the clinician’s own experience.
• Peripheral/nonoperable CTEPH: Data on use of PAH therapies for patients with peripheral or nonoperable CTEPH are evolving, but large clinical trials showing a benefit in this population also are lacking
• Exercise-induced pulmonary hypertension: Convincing data show that patients should be treated early for their PH; however the exercise definition (greater than 30 mm Hg) for PH was removed at the most recent symposium in Dana Point. Further studies regarding exercise hemodynamics are sorely needed to allow for earlier detection and treatment, with consequent better prognosis.
• Associated PAH: Most of what is known about PAH comes from IPAH; likewise, clinical trials largely comprise patients with IPAH, with a minority of patients with scleroderma spectrum of disease and occasionally diet drugs–related PAH or congenital heart disease. Caution should be taken before applying these data to all forms of PAH.
Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S56–S66.
Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78–S84.
Hoeper MM, Barberà JA, Channick RN, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S85–S96.
Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–894.
McLaughlin VV, Archer SL, Badesh DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. J Am Coll Cardiol. 2009;53:1573–1619.
McLaughlin VV, McGoon MD. Pulmonary arterial hypertension. Circulation. 2006;114:1417–1431.
McLaughlin VV, Presberg KW, Doyle RL, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):78S–92S.
Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54.
Tuder RM. Pathology of pulmonary arterial hypertension. Semin Respir Crit Care Med. 2009;30:376–385.
Tuder RM, Abman SH, Braun T, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9.