CHAPTER 99 Pulmonary Hypertension
PULMONARY HYPERTENSION
Definition
Pulmonary hypertension (PH) is defined as a resting mean pulmonary arterial pressure more than 25 mm Hg at rest or more than 30 mm Hg with exercise. Among the various subclassifications of PH, the subgroup referred to as pulmonary arterial hypertension adds the criterion that the pulmonary capillary wedge pressure should be 15 mm Hg or less.2
Etiology and Pathophysiology
Anatomy and Physiology of the Pulmonary Circulation
The pulmonary circulation consists of two parallel networks, the pulmonary arterial circulation and bronchial arterial circulation.1 The pulmonary arterial circulation consists of a series of branching arteries, arterioles, capillaries, venules, and veins. Pulmonary arteries travel adjacent to the lobar, segmental, and subsegmental airways to the level of the terminal bronchioles. The small pulmonary arteries from the subsegmental level to the terminal bronchioles possess a thick muscular media. At the level of the respiratory bronchioles and alveolar ducts, the small pulmonary arteries have lost much of the muscle within the arteriolar media as well as their external elastic membranes, and are now termed pulmonary arterioles, ranging in size from 10 to 150 µm. These vessels ramify further within the alveolar walls and form a rich capillary network. Capillary blood collects in venules, which progressively coalesce to form pulmonary veins. Pulmonary veins course within interlobular septa, eventually emptying into the left atrium.
Pathogenesis
The pulmonary vascular endothelium actively responds to changes in oxygen tension, transmural pressure, and pulmonary blood flow, and actively participates in the regulation of pulmonary arterial pressure through the elaboration of a number of vasoactive substances, including prostacyclin, nitrous oxide, and endothelin.3 These agents have direct effects on pulmonary vascular smooth muscle tone, promoting relaxation or vasodilation, and also may directly affect platelet function.
Various pathologic derangements may be seen in patients with PH, varying somewhat depending on the cause of PH. In general, regardless of the cause of PH, the pulmonary arteries become dilated,1 occasionally to the point of being considered aneurysmal. Pulmonary arterial atherosclerosis, typically absent in the pulmonary arteries of normal adults, may be extensive and frequently involves smaller vessels in patients with PH. PH-related pulmonary arterial atherosclerosis histopathologically appears similar to atherosclerosis occurring in systemic arteries, although complicating features, such as necrosis, ulceration, and calcification, are relatively uncommon.
Plexogenic pulmonary arteriopathy is the term applied to a constellation of vascular changes often encountered in patients with primary PH, but may also be seen in patients with PH of other causes, including hepatic disease, connective tissue disorders, congenital cardiovascular disease, and some anorexigenic medications.2
Classification
There have been a number of proposed classification schemes to categorize the causes of PH. In 1998, during the Second World Symposium on Pulmonary Hypertension held in Evian, France, a clinical classification for PH was proposed (Box 99-1).3–5 The aim of the Evian classification was to provide categories for causes of PH that shared similar pathophysiologic mechanisms, clinical presentations, and therapeutic considerations. Similar to oncologic staging considerations, such a classification was designed to allow standardization of diagnosis and treatment for patients with PH, conduction of trials with similar patient cohorts, and investigation of pathobiologic abnormalities in patient populations that are highly characterized. In 2003, the Third World Symposium on Pulmonary Arterial Hypertension, held in Venice, evaluated the usefulness of the Evian classification system and recommended some minor modifications. The Venice classification was adopted by the World Health Organization (WHO) and has been referred to as the Revised World Health Organization Classification of Pulmonary Hypertension (Box 99-2). More recently, the revised WHO-Venice classification underwent revision at the Fourth World Symposium on Pulmonary Hypertension held in Dana Point, California. The new classification system is referred to as the 2008 Dana Point Pulmonary Hypertension classification system.6 Traditionally, pulmonary hypertension has been classified into primary versus secondary causes or precapillary and postcapillary causes; this classification scheme is still in common use, particularly among interpreting physicians, and is included here as well. The pre- and postcapillary PH classification scheme is commonly reviewed in radiology texts (Box 99-3),1 but the 2008 Dana Point classification system is in more widespread use among clinicians and investigators dealing with PH. The 2008 Dana Point classification system will be used for this chapter.
BOX 99-1 Classification of Pulmonary Hypertension: Evian Classification
BOX 99-2 Classification of Pulmonary Hypertension: Venice-Revised WHO Classification
I PULMONARY ARTERIAL HYPERTENSION
BOX 99-3 Classification of Pulmonary Hypertension: Precapillary Versus Postcapillary Etiologies








In the 2008 Dana Point classification schemes, the category of pulmonary arterial hypertension (PAH) includes a subgroup of patients with PH without any identifiable cause, or idiopathic PAH,6 previously referred to as primary pulmonary hypertension (PPH). Other subgroups in the category of PAH include disorders that share localization of the lesions to small, muscular pulmonary arterioles, including heritable causes of PH (e.g., patients with mutations in bone morphogenetic protein receptor 2 and activin receptor-like kinase 1), drug and toxin exposures, and associations with certain conditions, such as collagen vascular diseases, HIV-related PH, portopulmonary hypertension, congenital systemic to pulmonary artery shunts, schistosomiasis, and chronic hemolytic anemias. The morphologic and clinical characteristics of these disorders are similar, and they also share a clinical response to treatment with continuous infusion of epoprostenol.5 Most patients previously diagnosed with primary pulmonary hypertension would now be classified as patients with idiopathic pulmonary arterial hypertension (IPAH) or, less commonly, heritable PH or anorexigen-induced PH (Box 99-4).7 Persistent PH of the newborn was also recognized as a cause of PAH in the 2008 Dana Point PH classification system. The Venice-revised WHO classification system also includes another subcategory in the pulmonary arterial hypertension group, referred to as “pulmonary arterial hypertension associated with significant venous or capillary involvement.” This subcategory includes pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. In the 2008 Dana Point system, this subcategory remains a group 1 (PAH) condition, but is now referred to as “pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis” in recognition of the similarities between these two disorders (see Box 99-4). Both entities were classified differently in the Evian system.
BOX 99-4 Classification of Pulmonary Hypertension: 2008 Dana Point Classification
The category referred to as pulmonary venous hypertension in the Evian system and as PH with left heart disease in the Venice-revised WHO system is now called pulmonary hypertension secondary to left heart disease (see Box 99-4). It includes systolic and diastolic dysfunction as well as left-sided valvular and myocardial conditions that require therapies directed at improving cardiac function or repairing or reducing valvular mechanical dysfunction, as opposed to treatment with vasodilator therapy.5,6 In fact, epoprostenol infusion in these patients can be harmful. Pulmonary veno-occlusive disease and lesions producing extrinsic compression on pulmonary veins are included in this category in the Evian system, but were reclassified in the Venice-revised WHO system in 2003 and again in the 2008 Dana Point system.
PH secondary to hypoxia and/or lung disease (see Box 99-4) results from inadequate arterial blood oxygenation caused by chronic obstructive pulmonary disease, interstitial lung diseases, sleep-disordered breathing, alveolar hypoventilation, and residence at high altitude. Usually, the elevated pulmonary arterial pressures in these patients are fairly modest (<35 mm Hg).
Group 4 in the 2008 Dana Point PH classification system is chronic thromboembolic pulmonary hypertension (see Box 99-4).6 In many patients with chronic thromboembolic PH, the central pulmonary arteries show emboli and/or thrombi, and such patients may benefit from pulmonary endarterectomy.1 In other patients, emboli or thrombi are relatively distally located and are not amenable to surgical treatment. These patients may respond to chronic pulmonary vasodilator therapy, similar to patients with IPAH. Both subgroups of patients are treated with lifelong anticoagulation.
PH caused by disorders of the pulmonary vasculature in the Evian system was simply referred to as the miscellaneous category in the Venice-revised WHO system, and is now called pulmonary hypertension with unclear multifactorial mechanisms in the 2008 Dana Point classification. Included in this group are a variety of disorders such as myeloproliferative diseases, splenectomy, cystic lung diseases, sarcoidosis, and metabolic disorders. External vascular compression, particularly fibrosing mediastinitis, previously considered a subcategory of pulmonary arterial hypertension in the Evian system, was moved to the miscellaneous category in the Venice-revised WHO system and is now classified as a group 5 lesion in the 2008 Dana Point system.6
Manifestations
Imaging Techniques and Findings
The characteristic finding of pulmonary arterial hypertension on chest radiography, CT, or MRI is dilation of the central pulmonary arteries, with rapid tapering of the pulmonary vessels as they course peripherally.1 This pattern is present regardless of the cause of the PH.
Radiography
Chest radiography may show enlargement of the main pulmonary artery segment and dilation of the right and left interlobar pulmonary arteries in patients with PH, regardless of cause (Fig. 99-1).
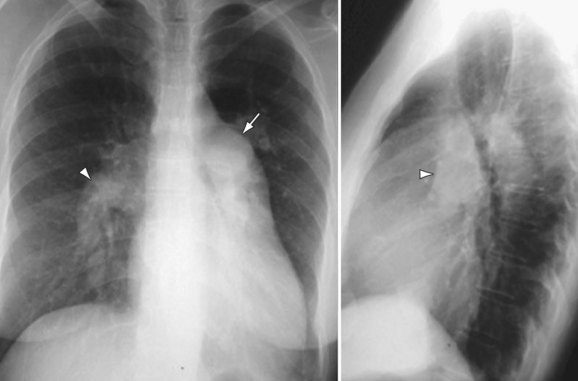
 FIGURE 99-1
FIGURE 99-1Computed Tomography
The main pulmonary arterial segment cannot be directly measured on chest radiography, but is easily measured on CT or MRI. When the main pulmonary artery segment exceeds 3 cm (Fig. 99-2), PH is often present. However, PH may be present in patients with a normal-sized main pulmonary arterial segment. The main pulmonary artery–to–ascending aortic ratio is also a useful internal indicator that can suggest enlargement of the main pulmonary artery. When the ratio of the main pulmonary artery to the aorta exceeds 1, elevated pulmonary pressures are usually present.1
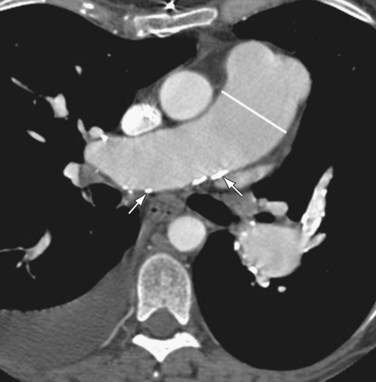
 FIGURE 99-2
FIGURE 99-2When PH is prolonged and severe, calcification of the pulmonary arteries, usually affecting the main, right, and/or left pulmonary arteries and, less commonly, the lobar pulmonary arteries, may be present (see Fig. 99-2). This finding is usually, but not invariably, associated with irreversible pulmonary vascular disease.
Patients with elevated pulmonary arterial pressures may also show right ventricular enlargement and right ventricular hypertrophy (Fig. 99-3) on cross-sectional imaging studies.1 The right atrium and inferior vena cava may also be enlarged, and contrast reflux into the infradiaphragmatic inferior vena cava is also commonly seen.
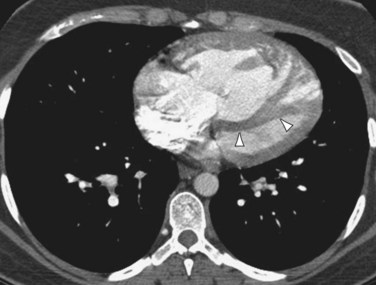
 FIGURE 99-3
FIGURE 99-3Magnetic Resonance Imaging
MRI will show all the vascular abnormalities that thoracic CT detects in patients with PH, such as main pulmonary artery enlargement and right ventricular enlargement and hypertrophy.1 Additionally, MRI can provide functional information equivalent to echocardiography, such as direction and velocity of blood flow, in addition to specific anatomic information. MRI techniques are well suited to the evaluation of patients with PH because they allow a detailed anatomic and extensive functional examination of the entire cardiovascular system.
IDIOPATHIC PULMONARY ARTERIAL HYPERTENSION
Definition
The term primary pulmonary hypertension was first used in 1951 to describe hypertensive vasculopathy in the pulmonary arteries without an identifiable cause.5 More recently, it has been recognized that patients with heritable forms of PAH and those with various conditions associated with PH, including connective tissue diseases, the use of appetite suppressant medications, portopulmonary hypertension, and HIV infection, among others, may share pathologic and clinical features similar to PPH, and were previously collectively referred to as secondary PH. The disorders encompassed by the term secondary PH are a fairly heterogeneous group of conditions that also include conditions affecting the pulmonary venous circulation or pulmonary parenchyma as well. The use of secondary PH has been largely abandoned because it does not provide a useful framework for the diagnosis or treatment of patients with PH. Therefore, the Venice group recommended changing the term primary PH to idiopathic pulmonary arterial hypertension; this nomenclature was adopted in the Venice-revised WHO classification system and is maintained in the 2008 Dana Point classification system.
Etiology and Pathophysiology
The cause of IPAH is unknown. The prevalence of PAH is estimated at about 15/million.7 The disease affects females more commonly than males. The Venice-revised WHO classification system previously recognized two forms of IPAH, familial and idiopathic (sporadic). The 2008 Dana Point classification system now refers to familial PAH as heritable PAH, and considers IPAH as a separate subcategory of PAH (see Box 99-4).6 Heritable forms of PAH account for approximately 10% of PAH cases, show autosomal dominant inheritance with incomplete penetrance, and have been localized to chromosome 2 at the locus of the bone morphogenic protein receptor type II gene.5 Heritable PAH associated with mutations in activin receptor-like kinase type 1 may be associated with hereditary hemorrhagic telangiectasia.
Manifestations of Disease
Clinical Presentation
Patients with IPAH usually present with dyspnea on exertion. The mean age of diagnosis is approximately 37 years.7 Other presenting symptoms include fatigue, chest pain, syncope, and occasionally cough.
Imaging Techniques and Findings
Radiography
Chest radiography in patients with IPAH typically shows enlargement of the main, right, and left pulmonary arteries (see Fig. 99-1), often with enlargement of the right ventricle (see Fig. 99-3) and right atrium.1 CT and black blood MRI techniques will show these same findings to advantage. Occasionally, black blood MRI will show increased signal within the pulmonary arteries resulting from slow flow.
Computed Tomography
High-resolution CT (HRCT) in patients with IPAH may show inhomogeneous lung opacity resulting from differential pulmonary parenchymal perfusion (Fig. 99-4).1 The regions of decreased pulmonary parenchymal attenuation represent areas of mosaic perfusion, and the vessels in these regions of lung may be visibly smaller than their counterparts in the regions of relatively increased lung attenuation. Although airway diseases may result in a similar pattern of mosaic perfusion, vascular and airway causes of mosaic perfusion may be distinguished using postexpiratory imaging. When caused by an obstructive airway process, regional differences in lung attenuation become accentuated with postexpiratory imaging, whereas a proportional increase in attenuation in areas of both increased and decreased attenuation will be seen in patients with pulmonary vascular disease.
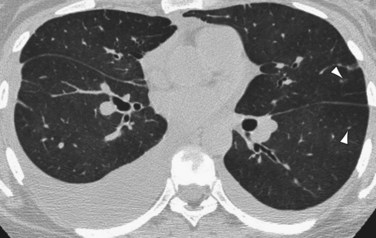
 FIGURE 99-4
FIGURE 99-4Occasionally, HRCT will show centrilobular ground-glass opacities in patients with IPAH (Fig. 99-5), representing foci of hemorrhage, plexiform lesions, or cholesterol granulomas.8
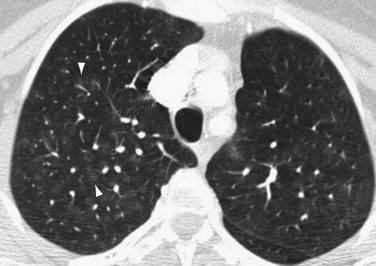
 FIGURE 99-5
FIGURE 99-5PULMONARY ARTERIAL HYPERTENSION RELATED TO RISK FACTORS OR ASSOCIATED WITH CONDITIONS
Definition
Pulmonary arterial hypertension related to associated conditions (APAH) includes PH in patients with connective tissue diseases, HIV infection, portal hypertension, congenital cardiovascular systemic to pulmonary shunts, schistosomiasis, and chronic hemolytic anemias.6 The term risk factors in the category of PAH indicates any condition suspected to play a role in predisposing or facilitating the development of PAH, whereas the term associated conditions is used when it is impossible to determine whether a predisposing factor was present prior to the development of PAH.5 The Evian conference in 1998 explored a number of potential risk factors and classified them according to the strength of association with PAH and the likelihood of a causal role in the development of PAH as definite, very likely, possible, or unlikely, according to the strength of available evidence in the literature (Box 99-5).
Etiology and Pathophysiology
PAH associated with connective tissue disease usually occurs in patients with systemic sclerosis, particularly those with limited forms of this disorder, such as CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia).7,9 Although most patients with systemic sclerosis do not develop clinical evidence of PAH, histopathologic changes of PAH are common in patients with systemic sclerosis. Echocardiographic studies have suggested that mild or moderate PAH is not uncommon in patients with systemic sclerosis.
Most patients with congenital systemic to pulmonary vascular shunts are corrected during infancy or early childhood, before the onset of severe PAH. For the infrequent patient who escapes surgical repair of the causative lesion during childhood, lung biopsy may be performed to assess the potential success for reversing the vasculopathy following surgical intervention.1 This histopathologic grading system is called the Heath-Edwards grading system, and was originally described using a six-point scale. More recently, a three-point system has been proposed. The Heath-Edwards grading system allows prediction of disease reversibility. Grades I and II (medial hypertrophy, intimal proliferation, and neomuscularization) represent mild reversible disease. Grade III is considered borderline and is characterized by intimal fibrosis and luminal obliteration. Higher grades, corresponding with plexiform lesions, aneurysm, and necrotizing arteritis, are considered irreversible changes, and surgical correction of the abnormality is not warranted.
In prior PH classification systems, PH with schistosomiasis was previously thought to be the result of embolic obstruction of the pulmonary arteries by the organism’s eggs. However, recent data suggest that PH related to schistosomiasis shares a clinical presentation and histopathologic findings similar to those of IPAH.6
PAH is increasingly recognized as a complication of chronic hemolytic anemias, particularly sickle cell disease; estimates of the prevalence of PAH in patients with sickle cell disease range from 10% to 30%.7 The pathophysiology of the development of PAH in patients with sickle cell disease is likely multifactorial, including a number of disparate mechanisms, such as asplenia, chronic thromboembolic disease, pulmonary parenchymal disease from repeated episodes of inflammation and scarring, vasculopathy associated with sickle cell disease, and a relative paucity of vasodilating substances, such as nitrous oxide with an abundance of vasconstricting agents, such as endothelin-1.
Manifestations of Disease
Clinical Presentation
PAH associated with liver disease is typically encountered in the setting of cirrhosis and very rarely in patients with noncirrhotic portal hypertension caused by portal fibrosis or multifocal nodular hyperplasia. Most patients with cirrhosis do not develop pulmonary arterial hypertension; the frequency of this association based on hemodynamic studies is 2% to 6% of cirrhotic patients.7 However, the frequency may be higher when considering a subset of patients with severe cirrhosis, such as those awaiting liver transplantation. Hepatic disease-related pulmonary arterial hypertension slowly improves following liver transplantation.
Many patients with congenital cardiovascular disease and systemic to pulmonary vascular shunts are asymptomatic. When symptoms are present, palpitations, shortness of breath, fatigue, cyanosis, and dyspnea on exertion are common. Heart failure may occur in some patients. Physical examinations may reveal suggestive cardiac murmurs.10
Imaging Techniques and Findings
Radiography
The various conditions associated with PAH show similar features on chest radiography- enlarged main and central pulmonary arteries, with rapid tapering of more peripheral vessels.1 Right atrial and ventricular enlargement may be present. Patients with connective tissue disease–associated PAH may show evidence of fibrotic and/or inflammatory lung disease, although the latter is not invariably present.
Computed Tomography
Cross-sectional imaging studies show findings similar to those of patients with IPAH, including pulmonary arterial enlargement, right atrial and ventricular enlargement, and inhomogeneous lung opacity.1 However, in patients with collagen vascular disease–associated PAH, fibrotic lung disease may be more readily visible on CT than chest radiography. Similarly, whereas chest radiography provides few clues to the hepatic cause in patients with portopulmonary PAH, cross-sectional imaging studies readily show features of cirrhosis in these patients. Imaging findings of PAH related to HIV are almost identical to those of IPAH and other causes of PAH.
Unlike many of the other conditions seen in patients with PAH, imaging studies in patients with PAH related to congenital systemic to pulmonary vascular shunts may show features that allow a specific diagnosis to be made. Furthermore, cross-sectional imaging studies may also provide methods that allow quantification of the degree of vascular shunting and thereby provide direction for treatment. The chronic increase in pulmonary flow causes the characteristic radiographic changes associated with pulmonary hypertension—increased size of the pulmonary trunk and central pulmonary arteries, diminished peripheral vessel caliber, and right ventricular chamber dilation. Often, the pulmonary arterial enlargement is striking, greater than that seen in other causes of PH (Fig. 99-6). It is important to note, however, that normal chamber size may represent increasing pulmonary pressures with progression toward Eisenmenger syndrome.
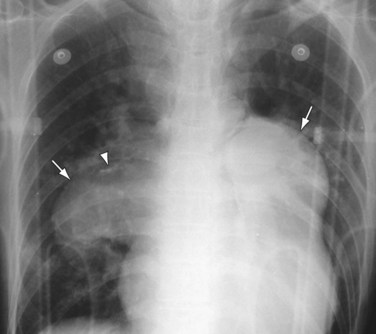
 FIGURE 99-6
FIGURE 99-6Thoracic CT may show calcification1 and thrombus in the main pulmonary arteries resulting from high pressures and turbulent flow in the affected vessels. CT, particularly multislice CT pulmonary angiography, may also show abnormal vascular connections directly, such as atrial septal defects and patent ductus arteriosus. In the case of a patent ductus arteriosus, CT or MR imaging (Fig. 99-7) may reveal dilation of the patent ductus, possibly with aneurysm formation or mural calcification.
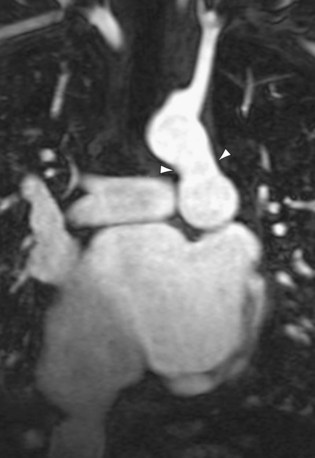
 FIGURE 99-7
FIGURE 99-7PULMONARY VENO-OCCLUSIVE DISEASE AND/OR PULMONARY CAPILLARY HEMANGIOMATOSIS
Definition
Pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH) were placed in separate groups in the Evian system (see Box 99-1), distinct from PAH. In the Venice-revised WHO system (see Box 99-2), PVOD and PCH were classified together as a subcategory of PAH in recognition of similarities in the clinical presentation, histopathologic features, and risk factors for disease of these two disorders. Such risk factors include use of anorexic medications, HIV infection, and scleroderma.6 PVOD and PCH also share similarities with PAH, including reports of a familial association, but there are important differences between PAH and PVOD-PCH, especially in regard to imaging findings and treatment. Therefore, PVOD and PCH are included as group 1 lesions in the Dana Point classification of PH, but in a subcategory separate from other causes of PAH (1′; see Box 99-4).
Etiology and Pathophysiology
Pulmonary veno-occlusive disease is an idiopathic condition but has been associated with factors such as pregnancy, medications (e.g., chemotherapeutic agents [bleomycin and carmustine] and oral contraceptives), toxic ingestion, systemic lupus erythematosus, some collagen vascular diseases, and bone marrow transplantation.11 Mutations in the bone morphogenic protein receptor-2 have also been detected in patients with PVOD. Immunologic mechanisms and viral infections have also been implicated as potential causes.
The characteristic hemodynamic feature of PVOD is normal to low pulmonary capillary wedge pressures, with normal left atrial and ventricular function and pressures.11 PVOD affects the venous bed in a patchy distribution, accounting for the variability in the wedge pressure.
Histopathologic specimens in patients with PVOD show fibrous obliteration of small pulmonary veins and venules.11,12 Plexiform lesions are absent, but capillary proliferation may be seen, which suggests the presence of angiogenesis. Prior episodes of infarction and hemosiderosis may be encountered. Acute or recanalized thrombi may be present.
Manifestations of Disease
Clinical Presentation
PVOD is a very uncommon disease, with an estimated annual incidence of 0.1 to 0.2 patients/million persons. The age of patients affected by PVOD is wide, ranging from newborns to 70 years, but it is most commonly encountered in children and young adults.11,13 In children and adolescents, no known gender predilection exists, but men are more commonly affected than women when PVOD manifests in young adults. Patients with PVOD present with chronic, progressive dyspnea, fatigue, and malaise, usually superimposed on other findings suggestive of PH, such as right heart failure. It has been suggested that PVOD is misdiagnosed as IPAH or chronic thromboembolic disease in a significant number of patients, which may lead to incorrect treatment.
It is important to distinguish patients with PVOD from those with interstitial lung disease because therapy for interstitial lung disease–induced pulmonary hypertension is vasodilation, which could precipitate pulmonary edema in patients with PVOD.11 The prognosis for PVOD is poor, with most patients succumbing within 2 to 3 years of diagnosis. Treatment typically rests on heart-lung transplantation.
Imaging Techniques and Findings
Radiography
Chest radiography in patients with PVOD shows typical features of PH, but evidence of pulmonary edema is also often present (Fig. 99-8A), which is a distinguishing feature for PVOD compared with many other causes of PH.11 Left atrial enlargement and redistribution of blood flow into the upper lobes are typically absent, allowing distinction from various causes of left-sided cardiac disease, particularly mitral stenosis.
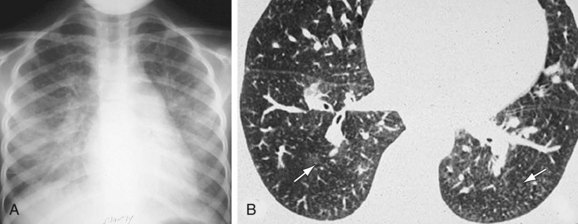
 FIGURE 99-8 Pulmonary arterial hypertension with significant venous or capillary involvement—pulmonary veno-occlusive disease. A, Frontal chest radiograph shows bilateral interstitial opacity and interlobular septal thickening suggestive of hydrostatic pulmonary edema. Note enlargement of the main pulmonary artery, suggesting elevated pulmonary pressures. B, Axial CT scan shows poorly defined areas of ground-glass opacity, some of which are vaguely centrilobular (arrows) in morphology. A few thickened interlobular septa were seen on other images. Unless interlobular septal thickening is pronounced, differentiation from other causes of pulmonary hypertension, especially pulmonary capillary hemangiomatosis (see Fig. 99-9), is difficult.
FIGURE 99-8 Pulmonary arterial hypertension with significant venous or capillary involvement—pulmonary veno-occlusive disease. A, Frontal chest radiograph shows bilateral interstitial opacity and interlobular septal thickening suggestive of hydrostatic pulmonary edema. Note enlargement of the main pulmonary artery, suggesting elevated pulmonary pressures. B, Axial CT scan shows poorly defined areas of ground-glass opacity, some of which are vaguely centrilobular (arrows) in morphology. A few thickened interlobular septa were seen on other images. Unless interlobular septal thickening is pronounced, differentiation from other causes of pulmonary hypertension, especially pulmonary capillary hemangiomatosis (see Fig. 99-9), is difficult.
Computed Tomography
Thoracic CT and HRCT will show normal or small central pulmonary veins, patchy dependent ground-glass opacity, smoothly thickened interlobular septa (see Fig. 99-8B), and pleural effusions.11,13 Ground-glass opacity centrilobular nodules may be seen.14 Mild lymphadenopathy may be present. Right-sided cardiac chambers are often enlarged, but the left atrium should not be enlarged. Multifocal air space opacities caused by pulmonary hemorrhage, air space edema, or venous infarction are uncommon. Inhomogeneous lung opacity consistent with mosaic perfusion may be present.
Nuclear Medicine and Angiography
V/Q scintigraphy in patients with PVOD will show patchy perfusion, most likely secondary to superimposed pulmonary arterial hypertension. Pulmonary angiography shows features of PH, including enlarged pulmonary arteries with peripheral pruning, but with delayed filling of the central pulmonary veins and a prolonged parenchymal enhancement phase.11 These latter findings are secondary to the patchy obstruction of the venous system.
PULMONARY CAPILLARY HEMANGIOMATOSIS
Etiology and Pathophysiology
Various causes have been suggested for PCH, including congenital factors, vascular neoplasia, and autoimmune disease, and as a response to PVOD. Similar to PVOD, PCH has been noted to occur in association with various other conditions, including systemic lupus erythematosus, Takayasu arteritis, scleroderma, Kartagener syndrome, and hypertrophic cardiomyopathy. Lung biopsies in patients with PCH typically show proliferation of circumscribed capillary channels within the walls of alveoli. Intervening lung tissue is often normal.11 The capillary proliferation results in the appearance of densely cellular alveolar walls. The proliferating capillaries compress the walls of pulmonary veins and venules, producing intimal fibrosis and secondary venous occlusion. Progressive scarring with in situ thrombosis develops, with pulmonary infarcts occurring as a result of vascular obliteration. Compensatory muscular hypertrophy of pulmonary arteries results from the foregoing processes. Like PVOD, histopathologic specimens in patients with PCH show hemosiderosis, interstitial edema, dilation of the lymphatic system, and vascular intimal fibrosis, with arterial medial hypertrophy.
Manifestations of Disease
Clinical Presentation
PCH is a rare disorder, occurring in approximately 4/million individuals.15 The age range of patients affected by PCH is broad, 2 to 71 years, with a mean of 29 years. Males and females are affected equally. Patients typically present with fatigue and progressive shortness of breath. Cough may occasionally be present and chest pain, syncope, and digital clubbing have been reported. The clinical presentation of PCH is similar to that of PVOD, although hemoptysis may occur in patients with PCH but usually does not occur in patients with PVOD.11 Similarly, hemorrhagic pleural effusions have been noted in patients with PCH, but typically do not occur in patients with PVOD. Other findings typically associated with PH may be present as well. As with PVOD, patients with PCH have elevated pulmonary arterial pressures but normal or near-normal pulmonary capillary wedge pressures. The prognosis of patients with PCH is rather poor, with a median survival of 3 years after diagnosis.
Imaging Techniques and Findings
Radiography
Chest radiographs in patients with PCH shows the usual findings of PH associated with bilateral, basilar reticular, and nodular opacities.11 Unlike PVOD, interlobular septal thickening and pleural effusions are unusual and, when present, are usually less pronounced in patients with PCH compared with patients with PVOD. Enlargement of mediastinal lymph nodes has been reported in patients with PCH.
Computed Tomography
Thoracic CT shows enlargement of the main and central pulmonary arteries and right ventricle. Poorly defined centrilobular or lobular ground-glass opacities are also often present in patients with PCH (Fig. 99-9).11 The presence of centrilobular ground-glass opacities tends to favor PCH, whereas smooth interlobular septal thickening and pleural effusions tend to favor the diagnosis of PVOD. Reports of interlobular septal thickening, pleural effusions, and lymphadenopathy in patients with PCH are noted, however, and ground-glass opacity centrilobular nodules may occur in patients with PVOD.14 Areas of mosaic perfusion reflecting oligemia may be present. Overall differentiation of PCH from PVOD by imaging is difficult.
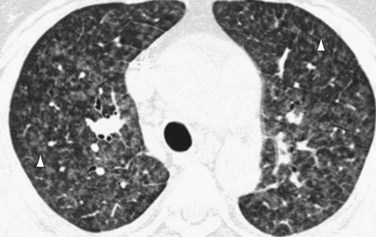
 FIGURE 99-9
FIGURE 99-9Nuclear Medicine and Angiography
V/Q typically does not show findings that specifically suggest the diagnosis of PCH. V/Q scans are often abnormal in patients with PCH,11 but may even be interpreted as normal. Pulmonary angiography in patients with PCH is often normal or only nonspecifically abnormal.
PULMONARY HYPERTENSION WITH LEFT HEART DISEASE (PULMONARY VENOUS HYPERTENSION)
Definition
The most frequently encountered causes of PH with left heart disease, previously referred to as pulmonary venous hypertension in the Evian system and now referred to as pulmonary hypertension caused by left heart disease in the 2008 Dana Point classification of PH, include left ventricular systolic and diastolic dysfunction and valvular heart disease, such as aortic stenosis, aortic regurgitation, and mitral stenosis. An obstructing intra-atrial tumor or thrombus is an uncommon cause of elevated pulmonary arterial pressure.1 In the case of patients with left-sided tumor or thrombus, the size of the obstructing lesion correlates with the degree of PH. Overall, left-sided cardiac disease, sometimes also referred to as nonpulmonary arterial pulmonary hypertension,7 is probably the most common cause of pulmonary hypertension.
Etiology and Pathophysiology
Secondary PH arises when increased venous pressure requires elevated arterial pressure to allow forward flow of blood. The histopathologic changes in the arterial system are therefore secondarily caused by the increased venous pressure. Histopathologic specimens from the venous system of patients with PH related to left heart disease show vascular medial hypertrophy, interstitial thickening and edema, hemosiderosis and, occasionally, venous infarction.1 Typically, no significant gradient is seen between the elevated pulmonary arterial pressure and pulmonary capillary wedge pressure, and pulmonary vascular resistance is normal or near normal. Occasionally, in some patients, pulmonary arterial pressure rises disproportionately to left atrial pressure in patients with pulmonary hypertension caused by left heart disease, probably as a result of vascular remodeling and/or increased pulmonary vasomotor tone.
Manifestations of Disease
Imaging Techniques and Findings
Radiography
Chest radiographs and CT scans show the characteristic findings of pulmonary venous hypertension, including interlobular septal thickening (Kerley A and B lines), pleural effusion, and air space opacity representing alveolar edema (Fig. 99-10).
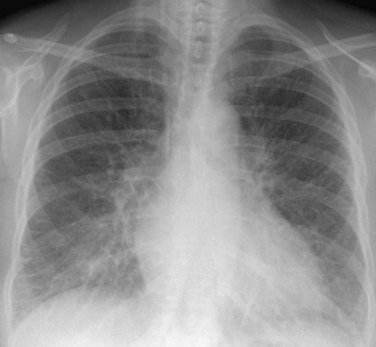
 FIGURE 99-10
FIGURE 99-10Computed Tomography
If pulmonary arterial hypertension has developed, the central pulmonary arteries will also be enlarged. In addition to the foregoing findings, mitral stenosis may result in ossified nodules within the pulmonary parenchyma. Atrial myxomas, if calcified, may be occasionally seen within the left atrium on chest radiographs. On contrast-enhanced thoracic CT (Fig. 99-11) or MRI scans, myxomas appear as filling defects within a cardiac chamber, most commonly the left atrium and often attached to the interatrial septum or anterior leaflet of the mitral valve.
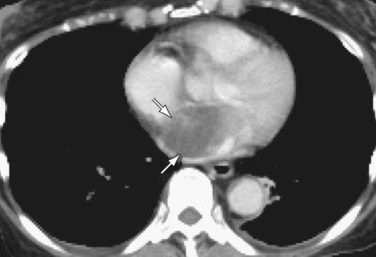
 FIGURE 99-11
FIGURE 99-11Synopsis of Treatment Options
Medical Treatment
Typically, treatment of PH caused by left heart disease is directed at the cause of left heart failure; the best example of success with this strategy is balloon valvotomy for mitral stenosis.9 When right heart failure develops in patients with PH caused by left heart disease, the prognosis is poor. In these patients, prognosis is most closely associated with the degree of pulmonary artery pressure elevation and right ventricular, rather than left ventricular, ejection fraction.
PULMONARY HYPERTENSION CAUSED BY LUNG DISEASES AND/OR HYPOXIA
Prevalence and Epidemiology
The prevalence of pulmonary hypertension in patients with COPD is uncertain, but is related to disease severity.7 In most patients with COPD, pulmonary hypertension is mild (mean pulmonary arterial pressure [mPAP] < 35 mm Hg) and treatment is directed at the COPD itself rather than pulmonary hypertension. The onset of PH in the COPD patient is associated with a poor prognosis, with a 5-year survival of only 10% in patients with pulmonary artery pressures higher than 45 mm Hg.
PH is fairly common among patients with interstitial lung diseases, including those with idiopathic pulmonary fibrosis. The exact prevalence of PH in patients with idiopathic pulmonary fibrosis is uncertain, but has been estimated to be as high as 40% using echocardiographic assessment.7
Etiology and Pathophysiology
The PH that results from the aforementioned mechanisms that produce hypoxia is exacerbated by the alveolar capillary destruction that is also present in patients with COPD. Various biologic mechanisms influence the development of pulmonary hypertension in patients with COPD, and some researchers think that severe pulmonary hypertension may be a disorder that occasionally manifests in patients with COPD as much as it is a condition resulting from COPD.7
As with COPD, the degree of PH in patients with idiopathic pulmonary fibrosis is typically modest although, again like COPD, a small subset of patients with idiopathic pulmonary fibrosis may have more marked pulmonary arterial pressure elevations.7 The correlation between measurements of lung variables and severity of pulmonary hypertension is poor in patients with idiopathic pulmonary fibrosis, suggesting that there may be more to the development of PH than the mere degree of pulmonary fibrotic disease would predict.
The ability of thoracic CT to predict PH (mPAP > 25 mm Hg) in patients with idiopathic pulmonary fibrosis has been questioned. Considering a main pulmonary artery measurement of 29 mm or higher as abnormal, the sensitivity of this measurement for the detection of elevated pulmonary arterial pressure on right heart catheterization may exceed 85%,16 but at the expense of insufficient specificity and negative predictive value.17,18
Sleep-disordered breathing is a term that includes a number of sleep-related breathing disorders, such as central sleep apnea, obstructive sleep apnea, and nocturnal desaturation (>10% of total time spent sleeping with arterial oxygenation <90%). The prevalence of sleep-disordered breathing may be as high as 4% in middle-aged men. Sleep-disordered breathing and other alveolar hypoventilation syndromes result in chronically depressed Po2 and elevated Pco2. The chronic hypoxemia produces pulmonary arterial vasoconstriction, vascular remodeling,3 and increased pulmonary arterial resistance, resulting in PH.
CHRONIC THROMBOEMBOLIC PULMONARY HYPERTENSION
Definition
In the 2003 Venice PH classification (see Box 99-2), chronic thromboembolic pulmonary hypertension (CTEPH) included a heterogeneous group of disorders producing pulmonary arterial obstruction, such as chronic thromboemboli, foreign bodies, and tumors.6 However, it is now recognized that there are significant differences in the clinical presentation, imaging findings, and management of patients with chronic thromboembolic disease compared with patients with nonthrombotic pulmonary arterial embolic disease. Additionally, the Venice classification system (see Box 99-2) recognized two subgroups of CTEPH: (1) proximal disease, accessible to pulmonary thromboendarterectomy; and (2) distal disease, which is not accessible to surgical therapy. However, the distinction between proximal and distal chronic thromboembolic disease is not always clear and general agreement regarding what constitutes proximal and distal disease is lacking. Therefore, in the 2008 Dana Point PH classification, CTEPH is regarded as resulting from thromboemboli and has been maintained as a group 4 lesion, but nonthrombotic emboli are now considered group 5 lesions (PH with Unclear Multifactorial Etiologies; see Box 99-4). Additionally, the distinction of proximal and distal disease in the Venice PH classification (see Box 99-2) has been abandoned. From a radiologist’s viewpoint, however, it nevertheless remains useful to consider nonthrombotic emboli at the same sitting as CTEPH, because often nonthrombotic emboli will be encountered as pulmonary arterial filling defects during pulmonary CTA studies. Therefore, recognizing that nonthrombotic emboli are classified as group 5 lesions in the Dana Point PH classification system, particulate, tumor, and parasitic emboli will be discussed here.
Etiology and Pathophysiology
Thromboemboli have numerous sources, including the deep veins of the pelvis and thigh, the right atrium, indwelling catheters, or septic thromboemboli in patients with endocarditis involving the tricuspid or pulmonic valves.1 Acute thromboembolic disease can produce transient pulmonary arterial pressure elevations, but sustained elevations are more likely a consequence of chronic thromboembolic disease, which may occur in up to 4% of patients with acute emboli.6,7
Thromboemboli induce pulmonary arterial hypertension by occluding the pulmonary vascular bed. This process may occur through numerous repeated small thromboembolic episodes, a few large embolic episodes that fail to resolve completely, or the development of in situ thrombosis in small vessels and proximal migration of pulmonary arterial thrombosis, with secondary endothelial changes and cellular hyperplasia, vascular webs, and incomplete remodeling of thromboemboli.19 There are data to suggest that the latter mechanism plays some role in the development of CTEPH. The common event leading to the development of elevated pulmonary arterial pressures is cytokine-mediated pulmonary arterial scarring resulting from lysis of pulmonary thromboemboli.
Histopathologically, chronic emboli may organize, resulting in vascular channels interspersed with connective tissue. Fibrous bands and webs, representing organizing thrombi, are present,1 often in association with fresh thromboemboli. The elevated pulmonary pressures also produce the characteristic histopathologic changes of medial hypertrophy and intimal proliferation and luminal obliteration, often in association with atherosclerosis. Plexogenic lesions are occasionally present in patients with chronic thromboembolic PH.7
Manifestations of Disease
Clinical Presentation
Patients at high risk for the development of CTEPH include oncologic patients, patients with chronic cardiac or pulmonary disease, and patients with clotting disorders. Patients with CTEPH typically present with dyspnea on exertion, cough, chest pain, and syncope.1 Anticardiolipin antibody may be found in 10% of patients with CTEPH,7 but the only other consistent abnormality of the coagulation and fibrinolytic systems identified in patients with CTEPH is an elevated level of factor VIII. The onset of PH in patients with chronic thromboembolic disease generally portends a poor prognosis.
Imaging Techniques and Findings
Computed Tomography
Helical CT pulmonary angiography is the study of choice for the evaluation of central CTEPH. The main pulmonary artery is often enlarged. Eccentric filling defects adjacent to the vessel wall, representing organizing thrombi, are characteristic of chronic thromboembolic disease. Such thrombi may calcify. The eccentric nature of organizing thrombi may be shown to advantage with multiplanar reformatted imaging. Organizing thrombi may undergo recanalization, in which case CT will show small foci of contrast within an occluded vessel. Linear intraluminal filling defects, representing intravascular webs, may be also seen. Abrupt narrowing of pulmonary arteries with reduction in arterial diameter is common in patients with chronic thromboembolic PH, and pulmonary CT angiography (CTA) may also show markedly hypertrophied bronchial arteries in these patients.1 Enlargement of the right ventricular and right atrium is common.
HRCT may show bilateral, geographically distributed inhomogeneous lung opacity in patients with CTEPH, representing mosaic perfusion.1 The vessels in the regions of decreased pulmonary parenchymal attenuation are often visibly smaller than their counterparts in the areas of normal or increased parenchymal attenuation. Small foci of subpleural consolidation, representing areas of prior pulmonary infarction, may also be evident.
Magnetic Resonance Imaging
Findings of central CTEPH on MRI and MRA are similar to those encountered on CT. Chronic, organized thrombi appear as very low signal foci adjacent to the vascular wall on T1-weighted or black blood images1 as well as flow-sensitive sequences, such as gradient echo or balanced fast-field echo (FFE) images. Vascular webs and stenoses and hypertrophied bronchial arteries may be visible. MRI provides the additional benefit of accurate assessment of right ventricular performance, and improvements in right ventricular ejection fraction and peak systolic pulmonary arterial flow velocity may be seen in surgically treated patients with CTEPH. Velocity-encoded cine phase contrast MRI has been used to assess the degree of bronchopulmonary shunt volume through hypertrophied bronchial arteries before and after surgical pulmonary endarterectomy, and it has been reported that reduction of bronchopulmonary shunt volume represents a favorable response to surgical therapy in patients with CTEPH.20
Nuclear Medicine and Positron Emission Tomography
V/Q scanning is often interpreted as high probability in patients with chronic thromboembolic PH, although scintigraphy may underestimate the degree of hemodynamic derangement associated with CTEPH. Furthermore, in the original PIOPED series,21 the positive predictive value of a high-probability V/Q scan interpretation decreased from 91% to 74% for patients with a history of thromboembolic disease, representing a loss of specificity in the high-probability scan interpretation. Other studies have shown high sensitivity and specificity for V/Q scintigraphy for the detection of chronic thromboembolic disease, but the limitations of this technique often favor helical CT pulmonary angiography as the test of choice in the diagnostic evaluation of these patients. Nevertheless, expert consensus recommendations have usually indicated that V/Q scintigraphy may be used to screen patients for suspected CTEPH as a result of reports indicating that V/Q scintigraphy is more sensitive for the detection of CTEPH amenable to surgical therapy.22 A normal or very low-probability scan result excludes CTEPH, whereas a high-probably scan result usually prompts catheter pulmonary angiography.7 Investigators at the University of California, San Diego, who have the most experience in the evaluation and treatment of patients with CTEPH, use a combination of transthoracic echocardiography, V/Q scintigraphy, right heart catheterization, and catheter pulmonary angiography in the preoperative assessment of patients suspected of CTEPH.19
Synopsis of Treatment Options
Medical Treatment
The treatment of CTEPH depends on the extent of the thrombus burden in the pulmonary circulation. If thromboemboli are shown within the lobar arteries or more proximally, the patient is considered an appropriate candidate for surgical thromboembolectomy.1 Thromboemboli distal to the proximal segmental vessels are usually not amenable to surgical resection, and medical management is the preferred treatment. Medical therapies typically used for patients with CTEPH who are not suitable candidates for surgery include oral anticoagulation, digitalis, calcium channel blockers, and PAH-specific medical therapy (e.g., prostacyclin analogues, endothelin receptor antagonists). In some cases, balloon angioplasty of affected pulmonary arteries or lung transplantation may be performed for inoperable patients with CTEPH.23
Surgical or Interventional Therapy
Surgical pulmonary thromboendarterectomy is an effective treatment for properly selected patients with CTEPH, and often provides significant immediate improvement in symptoms and pulmonary hemodynamics. However, surgical pulmonary thromboendarterectomy can be associated with significant morbidity and mortality, and another 10% to 15% of patients who undergo the procedure may fail to have substantial reductions in pulmonary vascular resistance.23 In the hands of experienced surgeons, the mortality related to surgical pulmonary thromboendarterectomy is approximately 4% to 7%.24 The best outcomes following surgical pulmonary thromboendarterectomy are seen with patients treated at centers with recognized experience in the surgical management of CTEPH, concordance between preoperative pulmonary vascular resistance and anatomic extent of disease, preoperative pulmonary vascular resistance less than 1000 to 1200, absence of a number of comorbid conditions (e.g., splenectomy) and a significant decrease in pulmonary vascular resistance following surgery. It has been suggested that a postoperative residual pulmonary vascular resistance of more than 500 dyn · s · cm−5 is associated with a mortality of just over 30% in patients undergoing surgical pulmonary thromboendarterectomy, whereas the mortality for these patients is less than 1% when the postoperative pulmonary vascular resistance falls below 500 dyn · s · cm−5 following surgery. Preoperative imaging assessment is essential for segregating those patients who stand a might benefit from surgical pulmonary thromboendarterectomy from those who will not. When visible central thromboemboli are present on multislice computed tomography pulmonary angiography (msCTPA) or catheter pulmonary angiography, it is more likely that patients will have improvements in pulmonary vascular resistance after pulmonary thromboendarterectomy. However, the amount of central thromboembolic material does not necessarily correlate with mPAP; some patients with central disease will not respond well to surgical pulmonary thromboendarterectomy, whereas others without central disease may have favorable results with surgery. The cause of this discrepancy is uncertain but suggests that factors other then mere vascular obstruction are involved in the development of chronic thromboembolic pulmonary hypertension.
NONTHROMBOTIC EMBOLIZATION
Etiology and Pathophysiology
Tumor Embolization
As many as 25% of patients with solid malignancies may have microemboli that ultimately lodge in the pulmonary circulation.1 The most frequent cause of tumor microembolization is gastric carcinoma, but breast, ovarian, lung, renal, hepatocellular, and prostate malignancies may also produce tumor emboli. Most emboli preferentially occlude small arteries and arterioles, with the exception of atrial myxomas and renal carcinomas, which may form larger, more centrally located thromboemboli.
Parasitic Embolization
In the Venice PH classification system (see Box 99-2), PH resulting from schistosomiasis was classified as a group V lesion, PH caused by chronic thrombotic or embolic disease.6 However, in the 2008 Dana Point PH classification scheme (see Box 99-4), PH caused by schistosomiasis has been reclassified as a group 1 lesion. Nevertheless, pulmonary schistosomiasis will be discussed here because it is convenient to consider the various causes of nonthrombotic pulmonary emboli together.25
Pulmonary schistosomiasis is most commonly the result of infection with Schistosoma mansoni, which is endemic in the Middle East, Africa, and South America.1 A latency period of 5 years or more after the onset of infection typically occurs before cardiopulmonary disease is seen. Secreted ova migrate into the lungs via portal-systemic collaterals and lodge in medium-sized muscular pulmonary arteries and arterioles. In the pulmonary circulation, the ova elicit an inflammatory reaction that results in medial hypertrophy, granuloma formation, intimal hyperplasia, collagen deposition and fibrosis, and eventually obliterative arteritis. An associated eosinophilic alveolitis may be present.
Particulate Embolization
Pulmonary arterial embolization with talc most commonly occurs in intravenous drug users. When intravenous drug users abuse medications containing talc, they inject a suspension containing crushed tablets, which are intended for oral use, and the injected talc embolizes small pulmonary arterioles.1 The talc may then migrate from the pulmonary vasculature into the surrounding interstitium, where its presence elicits a foreign body granulomatous response. Vascular thrombosis with recanalization, intimal hyperplasia, medial arterial hypertrophy, fibrosis, and refractile talc particles are present in histopathologic specimens.
Manifestations of Disease
Clinical Presentation
Tumor Embolization
Because tumor emboli are quite small, symptoms directly related to tumor embolization are uncommon. If the embolic load is high enough, patients may present with dyspnea on exertion, chest pain, hypoxemia, cough, syncope, and even cor pulmonale.1
Parasitic Embolization
Presenting symptoms of parasitic embolization include hepatosplenomegaly, right heart symptoms, dyspnea, and cough. Patients with cardiopulmonary schistosomiasis always have cirrhosis and portal hypertension.1
Particulate Embolization
Pulmonary talcosis may produce few symptoms, but patients may present with chronic, progressive shortness of breath, cough, and dyspnea on exertion.1 Symptoms may progress even after cessation of drug abuse.
Mercury may be injected into the vascular system accidentally or intentionally.1 Intentional mercury injection most often occurs as the consequence of a suicide attempt.
Imaging Techniques and Findings
Radiography
Tumor Embolization
Chest radiographs in patients with intravascular tumor embolization are often normal. When abnormal, findings resembling those of pulmonary lymphangitic carcinomatosis are commonly seen.1
Parasitic Embolization
Chest radiography in patients with parasitic embolization shows findings consistent with pulmonary arterial hypertension.1 Small nodules, representing parasitic granulomas, may be seen. Pulmonary infarction is rarely seen as a result of parasitic embolization. Contrast-enhanced thoracic CT may show embolized organisms directly in the pulmonary arteries or right-sided cardiac chambers (Fig. 99-12). Thoracic CT or MRI can also show the presence, location, and morphology of cardiac or pericardial parasitic infection directly, as may occur with echinococcosis.
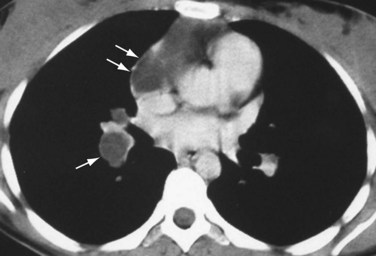
 FIGURE 99-12
FIGURE 99-12Particulate Embolization
The chest radiographic findings of mercury embolization include fine, branching, symmetric nodules that result from intra-arterial mercury deposits. Metal deposits may also collect within the heart, particularly at the apex of the right ventricle.1
Chest radiographs in patients with talc embolization show diffuse bilateral small nodular (2 to 3 mm) opacities throughout the lung parenchyma.1 Perihilar conglomerate masses associated with fibrosis, producing retraction of lung parenchyma and relative hyperlucency in the lower lung, may be seen in some patients; these findings may resemble progressive massive fibrosis.
Computed Tomography
Tumor Embolization
In addition to the characteristic cross-sectional imaging findings of PH and thromboembolism noted, thoracic CT scans in patients with tumor emboli may reveal lymphadenopathy, findings consistent with lymphangitic carcinomatosis, and peripheral wedge-shaped opacities representing pulmonary infarction.1 When emboli affect larger vessels, such as subsegmental pulmonary arteries, thoracic CT may reveal a beaded appearance of these vessels (Fig. 99-13). When smaller vessels are affected, such as at the centrilobular level, beading and nodularity may be observed, and the affected vessels may assume a branching configuration, resembling “tree in bud” opacity.
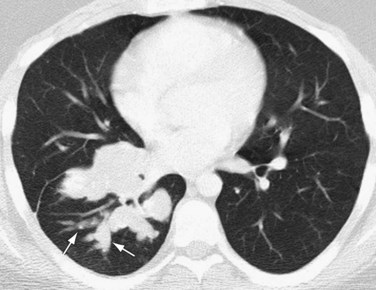
 FIGURE 99-13
FIGURE 99-13Parasitic Embolization
HRCT in patients with parasitic embolization may show nodules, interstitial prominence and thickening, and ground-glass opacity in combination with the classic findings of PH.1
Particulate Embolization
Thoracic CT scans in patients with pulmonary talcosis often show numerous micronodules, with or without patchy ground-glass opacity. Upper lobe fibrotic opacities, resembling progressive massive fibrosis, may be present.1 These fibrotic opacities may show high attenuation caused by the presence of talc.
PULMONARY HYPERTENSION WITH UNCLEAR MULTIFACTORIAL MECHANISMS
Definition
In the 2003 Venice-revised WHO classification of PH (see Box 99-2), a number of inflammatory and idiopathic causes that may be associated with PH were classified under the category “miscellaneous.” Included in this category are various conditions such as sarcoidosis, lymphangiomatosis, and extrinsic pulmonary venous compression. The latter may be caused by malignancy, such as mediastinal and hilar lymphadenopathy or tumor, but inflammatory causes, in particular fibrosing mediastinitis, may also elevate pulmonary pressures through pulmonary arterial and/or venous compression or obliteration. These conditions have been categorized as group 5 lesions in the 2008 Dana Point PH classification system (see Box 99-4). Other conditions that may be associated with PH, including hematologic abnormalities and metabolic abnormalities previously classified as group I lesions in the 2003 Venice system, as well as a number of systemic disorders, have been included in this group as well.6
The first subcategory of the group 5 2008 Dana Point classification of PH (see Box 99-4) includes hematologic conditions, such as myeloproliferative diseases, essential thrombocytosis, polycythemia vera, chronic myeloid leukemia, and splenectomy.6 Various mechanisms may result in PH in patients with these conditions, including congestive heart failure, high cardiac output, asplenia, or mechanical pulmonary arterial obstruction caused by circulating megakaryocytes. Several other systemic conditions in group 5 include sarcoidosis, Langerhans cell histiocytosis, lymphangioleiomyomatosis, pulmonary vasculitis, and neurofibromatosis. The mechanisms postulated to produce PH in patients with sarcoidosis include capillary bed destruction resulting from fibrosis, pulmonary artery and venous compression secondary to lymphadenopathy, and granulomatous inflammation directly involving the pulmonary vessels, especially the veins. PH in patients with end-stage Langerhans cell histiocytosis is common and may be related to a combination of pulmonary parenchymal destruction, hypoxemia, and vasculopathy. PH is relatively uncommon in patients with lymphangioleiomyomatosis but, when present, may be the result of hypoxemia caused by capillary destruction produced by the cystic lesions characteristic of this condition.
Also in group 5 of the 2008 Dana Point classification of PH (see Box 99-4) are a number of metabolic disorders, such as Gaucher disease, glycogen storage disease, and thyroid conditions such as hyper- and hypothyroidism.6 Various mechanisms for the development of PH in these conditions include hypoxemia, pulmonary parenchymal disease, portocaval shunts (in the case of type Ia glycogen storage disease), and capillary blockade by Gaucher cells. Precisely how thyroid disorders produce PH are unclear, but an association between autoimmune thyroid disease and PAH raises the possibility of a linked genetic pathogenesis.
The last subcategory of group 5 in the 2008 Dana Point classification (see Box 99-4) includes a number of disorders that produce mechanical obstruction of the pulmonary arteries and/or veins, including central obstructing tumors, metastatic microvascular obstruction, and fibrosing mediastinitis.6 Also included in this category is PH associated with end-stage renal disease in those on long-term hemodialysis. Tumor microvascular obstruction has been reviewed previously, and centrally obstructing tumors are usually readily apparent as a cause of symptoms suggesting PH. Fibrosing mediastinitis may present with suggestive thoracic imaging findings and will be discussed in detail in the next sections.
Fibrosing Mediastinitis
Etiology and Pathophysiology
Fibrosing mediastinitis (FM) represents a progressive proliferation of collagenous and fibrous tissue within the mediastinum, producing encasement and compression of mediastinal structures. Granulomatous infections, particularly Histoplasma capsulatum and Mycobacterium tuberculosis, are among the most common causes of FM.1
The deposition of fibrous tissue in FM commonly affects relatively soft and deformable structures, such as the systemic veins within the mediastinum, trachea and central airways, esophagus, pulmonary arteries, and pulmonary veins. PH may result when fibrous encasement of the pulmonary arteries or draining pulmonary veins occurs, with the former often mistaken for chronic thromboembolic disease. Pulmonary venous obstruction occurs in a patchy distribution, producing wide variations in pulmonary capillary wedge pressure measurements,1 although usually the wedge pressure is elevated. Cardiac catheterization studies typically show normal left heart size and pressures.
Manifestations of Disease
Clinical Presentation
Common symptoms in patients with FM depend on the structures most severely involved. Patients often present with nonspecific symptoms of pulmonary venous hypertension, including dyspnea and hemoptysis.1 The treatment for FM is limited. If localized pulmonary venous involvement predominates, surgical treatment may be effective. Steroids have shown minor efficacy.
Imaging Techniques and Findings
Radiography
Chest radiography often shows widening of the mediastinum, with hilar prominence and calcified lymph nodes. Findings of pulmonary venous hypertension, including interlobular septal thickening and air space consolidation representing alveolar edema, may be present. FM may result in airway stenoses that present with lobar volume loss.1
Computed Tomography
Thoracic CT will reveal extensive infiltration of the mediastinum with abnormal soft tissue. FM caused by Histoplasmosis capsulatum usually produces extensive lymph node calcification, readily identifiable by CT. Often, the abnormal mediastinal and hilar soft tissue resembles lymph nodes, and the diagnosis of FM may be mistaken for lymphadenopathy. Similarly, FM and chronic thromboembolic disease may superficially resemble one another. Contrast-enhanced CT in patients with FM may show extensive collateral vein formation resulting from fibrotic involvement of systemic thoracic veins, such as the superior vena cava or azygos vein. Pulmonary artery compression may also be directly visualized. Pulmonary venous infarcts, appearing as subpleural wedge-shaped consolidations may be seen in patients with pulmonary venous involvement.1
Angiography
Catheter pulmonary angiography findings depend on the location of obstruction. If the arterial circulation is primarily affected, asymmetric narrowing of the pulmonary arteries will be present. When pulmonary veins are primarily affected, venous phase angiography will show stenoses, dilation, or obstruction in combination, often near the junction of the affected vein and the left atrium.1
KEY POINTS
 Pulmonary hypertension is classified according to the 2008 Dana Point classification system, which recognizes five major categories—pulmonary arterial hypertension, pulmonary hypertension secondary to left heart disease, pulmonary hypertension secondary to hypoxia and/or lung disease, chronic thromboembolic pulmonary hypertension, and pulmonary hypertension with unclear multifactorial mechanisms.
Pulmonary hypertension is classified according to the 2008 Dana Point classification system, which recognizes five major categories—pulmonary arterial hypertension, pulmonary hypertension secondary to left heart disease, pulmonary hypertension secondary to hypoxia and/or lung disease, chronic thromboembolic pulmonary hypertension, and pulmonary hypertension with unclear multifactorial mechanisms.

Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation. 2007;115:1039-1050.
Frazier AA, Galvin JR, Franks TJ, et al. From the archives of the AFIP: pulmonary vasculature: hypertension and infarction. Radiographics. 2000;20:491-524.
Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol. 2009;54:S67-S77.
McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573-1619.
Reddy GP, Gotway MB, Araoz PA. Imaging of chronic thromboembolic pulmonary hypertension. Semin Roentgenol. 2005;40:41-47.
Rossi SE, Goodman PC, Franquet T. Nonthrombotic pulmonary emboli. AJR Am J Roentgenol. 2000;174:1499-1508.
Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43-S54.
1 Frazier AA, Galvin JR, Franks TJ, et al. From the archives of the AFIP: pulmonary vasculature: hypertension and infarction. Radiographics. 2000;20:491-524.
2 Badesch DB, Abman SH, Simonneau G, et al. Medical therapy for pulmonary arterial hypertension: updated ACCP evidence-based clinical practice guidelines. Chest. 2007;131:1917-1928.
3 Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655-1665.
4 Galie N, Torbicki A, Barst R, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J. 2004;25:2243-2278.
5 Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S-12S.
6 Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43-54.
7 McLaughlin VV, Archer SL, Badesch DB, et al. American College of Cardiology Foundation Task Force on Expert Consensus Documents; American Heart Association; American College of Chest Physicians; American Thoracic Society, Inc; Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension. A report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53:1573-1619.
8 Nolan RL, McAdams HP, Sporn TA, et al. Pulmonary cholesterol granulomas in patients with pulmonary artery hypertension: chest radiographic and CT findings. AJR Am J Roentgenol. 1999;172:1317-1319.
9 Chin KM, Rubin LJ. Pulmonary arterial hypertension. J Am Coll Cardiol. 2008;51:1527-1538.
10 Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation. 2007;115:1039-1050.
11 Frazier AA, Franks TJ, Mohammed TL, et al. From the archives of the AFIP: pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Radiographics. 2007;27:867-882.
12 Resten A, Maitre S, Humbert M, et al. Pulmonary hypertension: CT of the chest in pulmonary venoocclusive disease. AJR Am J Roentgenol. 2004;183:65-70.
13 Swensen SJ, Tashjian JH, Myers JL, et al. Pulmonary venoocclusive disease: CT findings in eight patients. AJR Am J Roentgenol. 1996;167:937-940.
14 Resten A, Maitre S, Humbert M, et al. Pulmonary arterial hypertension: thin-section CT predictors of epoprostenol therapy failure. Radiology. 2002;222:782-788.
15 El-Gabaly M, Farver CF, Budev MA, Mohammed TL. Pulmonary capillary hemangiomatosis imaging findings and literature update. J Comput Assist Tomogr. 2007;31:608-610.
16 Tan RT, Kuzo R, Goodman LR, et al. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Medical College of Wisconsin Lung Transplant Group. Chest. 1998;113:1250-1256.
17 Zisman DA, Karlamangla AS, Ross DJ, et al. High-resolution chest CT findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132:773-779.
Chen H, De Marco T, Golden JA, Gould MK. Utility of CT for predicting pulmonary hypertension in patients with parenchymal lung disease: Similar results, different conclusion? Chest. 2008;133:1053-1054.
19 Thistlethwaite PA, Kaneko K, Madani MM, Jamieson SW. Technique and outcomes of pulmonary endarterectomy surgery. Ann Thorac Cardiovasc Surg. 2008;14:274-282.
20 Reddy GP, Gotway MB, Araoz PA. Imaging of chronic thromboembolic pulmonary hypertension. Semin Roentgenol. 2005;40:41-47.
21 The PIOPED Investigators. Value of the ventilation/perfusion scan in acute pulmonary embolism. Results of the prospective investigation of pulmonary embolism diagnosis (PIOPED). JAMA. 1990;263:2753-2759.
22 Tunariu N, Gibbs SJ, Win Z, et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med. 2007;48:680-684.
23 Rubin LJ, Hoeper MM, Klepetko W, et al. Current and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responses. Proc Am Thorac Soc. 2006;3:601-607.
24 Keogh AM, Mayer E, Benza RL, et al. Interventional and surgical modalities of treatment in pulmonary hypertension. J Am Coll Cardiol. 2009;54:S67-S77.
25 Rossi SE, Goodman PC, Franquet T. Nonthrombotic pulmonary emboli. AJR Am J Roentgenol. 2000;174:1499-1508.
